α- and β-Adrenergic Receptor Antagonists
α- and β-Adrenergic receptor antagonists prevent the interaction of the endogenous neurotransmitter norepinephrine or sympathomimetics with the corresponding adrenergic receptors.1 Interference with normal adrenergic receptor function attenuates sympathetic nervous system homeostatic mechanisms and evokes predictable pharmacologic responses. Presynapthic agonsim of α2 receptors will result in a similar attenuation of sympathetic outflow.
α-Adrenergic Receptor Antagonists
α-Adrenergic receptor antagonists bind selectively to α-adrenergic receptors and interfere with the ability of catecholamines or other sympathomimetics to provoke α responses. Drug-induced α-adrenergic blockade prevents the effects of catecholamines and sympathomimetics on the heart and peripheral vasculature. The inhibitory action of epinephrine on insulin secretion is prevented. Orthostatic hypotension, baroreceptor-mediated reflex tachycardia, and impotence are invariable side effects of α-adrenergic blockade. Furthermore, absence of β-adrenergic blockade permits maximum expression of cardiac stimulation from norepinephrine, typically leading to tachycardia. These side effects prevent the use of nonselective α-adrenergic antagonists in the management of ambulatory essential hypertension.
Mechanism of Action
Phentolamine, prazosin, and yohimbine are competitive (reversible binding with receptors) α-adrenergic antagonists. In contrast, phenoxybenzamine binds covalently to α-adrenergic receptors to produce an irreversible and insurmountable type of α receptor blockade. Once α blockade has been established with phenoxybenzamine, even massive doses of sympathomimetics are ineffective until the effect of phenoxybenzamine is terminated by metabolism.
Phentolamine and phenoxybenzamine are nonselective α antagonists acting at postsynaptic α1 receptors as well as presynaptic α2 receptors. Prazosin is selective for α1 receptors, whereas yohimbine is selective for α2 receptors.
Phentolamine
Phentolamine is a substituted imidazoline derivative that produces transient nonselective α-adrenergic blockade (Fig. 19-1). Administered intravenously (IV), phentolamine produces peripheral vasodilation and a decrease in systemic blood pressure that manifests within 2 minutes and lasts 10 to 15 minutes. This vasodilation reflects α1 receptor blockade and a direct action of phentolamine on vascular smooth muscle. Decreases in blood pressure elicit baroreceptor-mediated increases in sympathetic nervous system activity manifesting as cardiac stimulation. In addition to reflex stimulation, phentolamine-induced α2 receptor blockade permits enhanced neural release of norepinephrine manifesting as increased heart rate and cardiac output. Indeed, cardiac dysrhythmias and angina pectoris may accompany the administration of phentolamine. Hyperperistalsis, abdominal pain, and diarrhea may be caused by a predominance of parasympathetic nervous system activity. Phentolamine undergoes principally hepatic metabolism and only about 10% of the drug is excreted unchanged in the urine.

Clinical Uses
The principal use of phentolamine is the treatment of acute hypertensive emergencies, as may accompany intraoperative manipulation of a pheochromocytoma or autonomic nervous system hyperreflexia. Administration of phentolamine, 30 to 70 µg/kg IV (1 to 5 mg), produces a prompt but transient decrease in systemic blood pressure. A continuous infusion of phentolamine (0.1 to 2 mg per minute) may be used to maintain normal blood pressure during the intraoperative resection of a pheochromocytoma. Local infiltration with a phentolamine-containing solution (5 to 15 mg in 10 mL of normal saline) is appropriate when a sympathomimetic is accidentally administered extravascularly.
Phenoxybenzamine
Phenoxybenzamine is a haloalkylamine derivative that acts as a nonselective α-adrenergic antagonist by combining covalently with α-adrenergic receptors (Fig. 19-2). Blockade at postsynaptic α1 receptors is more intense than at α2receptors.
Pharmacokinetics
Absorption of phenoxybenzamine from the gastrointestinal tract is incomplete. Onset of α-adrenergic blockade is slow, taking up to 60 minutes to reach peak effect even after IV administration. This delay in onset is due to the time required for structural modification of the phenoxybenzamine molecule, which is necessary to render the drug pharmacologically active. The elimination half-time of phenoxybenzamine is about 24 hours, emphasizing the likelihood of cumulative effects with repeated doses.
Cardiovascular Effects
Phenoxybenzamine administered to a supine, normovolemic patient in the absence of increased sympathetic nervous system activity produces little change in systemic blood pressure. Orthostatic hypotension, however, is prominent, especially in the presence of preexisting hypertension or hypovolemia. In addition, impairment of compensatory vasoconstriction results in exaggerated blood pressure decreases in response to blood loss or vasodilating drugs such as volatile anesthetics. Despite decreases in blood pressure, cardiac output is often increased and renal blood flow is not greatly altered unless preexisting renal vasoconstriction is present. Cerebral and coronary vascular resistances are not changed. α-Adrenergic blockade produced by maternal treatment may result in neonatal hypotension and respiratory distress in the first 72 hours of life.2
Noncardiac Effects
Phenoxybenzamine prevents the inhibitory action of epinephrine on the secretion of insulin. Catecholamine-induced glycogenolysis in skeletal muscles or lipolysis is not altered. Stimulation of the radial fibers of the iris is prevented, and miosis is a prominent component of the response to phenoxybenzamine. Sedation may accompany chronic phenoxybenzamine therapy. Nasal stuffiness is due to unopposed vasodilation in mucous membranes in the presence of α-adrenergic blockade.
Clinical Uses
Phenoxybenzamine, 0.5 to 1.0 mg/kg orally (prazosin is an alternative) is administered preoperatively to control blood pressure in patients with pheochromocytoma. Chronic α-adrenergic blockade, by relieving intense peripheral vasoconstriction, permits expansion of intravascular fluid volume as reflected by a decrease in the hematocrit. Excessive vasoconstriction with associated tissue ischemia, as accompanies hemorrhagic shock, may be reversed by phenoxybenzamine but only after intravascular fluid volume has been replenished.
Treatment of peripheral vascular disease characterized by intermittent claudication is not favorably influenced by α-adrenergic blockade because cutaneous rather than skeletal muscle blood flow is increased. The most beneficial clinical responses to α-adrenergic blockade are in diseases with a large component of cutaneous vasoconstriction, such as Raynaud’s disease, where smaller arteries that supply blood to skin narrow, limiting blood circulation to affected areas.
Yohimbine
Yohimbine is a selective antagonist at presynaptic α2 receptors, leading to enhanced release of norepinephrine from nerve endings. As a result, this drug may be useful in the treatment of the rare patient suffering from idiopathic orthostatic hypotension. In the past, impotence had been successfully treated with yohimbine in male patients with vascular, diabetic, and psychogenic origins. Yohimbine readily crosses the blood–brain barrier and may be associated with increased skeletal muscle activity and tremor. Excessive doses of yohimbine may produce tachycardia, hypertension, rhinorrhea, paresthesias, and dissociative states. Observations that α2-adrenergic agonists can decrease anesthetic requirements by actions on presynaptic α2 receptors in the central nervous system (CNS) suggest a possible interaction of yohimbine with volatile anesthetics.
Doxazosin
Doxazosin is approved for both treatment of hypertension and benign prostatic hypertrophy. It is a selective postsynaptic α1 receptor antagonist that is 65% bioavailable with oral administration. Peak levels of doxazosin are seen 2 to 3 hours following oral administration and effectively relaxes prostatic and vascular smooth muscle. Doxazosin is primarily metabolized in the liver by O-demethylation and excreted in the feces. The terminal elimination life of doxazosin is 22 hours and is recommended as a single daily dose in the morning.
Prazosin
Prazosin is a selective postsynaptic α1 receptor antagonist that leaves intact the inhibiting effect of α2 receptor activity on norepinephrine release from nerve endings. As a result, prazosin is less likely than nonselective α-adrenergic antagonists to evoke reflex tachycardia. Prazosin dilates both arterioles and veins. Following oral administration, the onset of action is approximately 30 minutes and the duration of action is about 4 to 6 hours. Elimination of prazosin is principally by hepatic metabolism.
Terazosin
α Blocker therapy of benign prostatic hypertrophy is based on α1-mediated innervation of prostatic smooth muscle that controls contraction of the prostate and obstruction of the bladder outlet. Terazosin is a long-acting orally effective α1-adrenergic antagonist that may be useful in the treatment of benign prostatic hyperplasia by virtue of its ability to relax prostatic smooth muscle.
Tamsulosin
Tamsulosin is an orally effective α1a-adrenergic antagonist that is indicated for the treatment of the signs and symptoms of benign prostatic hyperplasia. Side effects may include orthostatic hypotension, vertigo, and syncope. The clearance of tamsulosin is decreased in the presence of cimetidine.
Tolazoline
Tolazoline is a competitive nonselective α-adrenergic receptor antagonist. This drug has been used to treat persistent pulmonary hypertension of the newborn but its use for this purpose has been largely replaced by nitric oxide. Side effects of tolazoline include systemic hypotension with reflex tachycardia, cardiac dysrhythmias, and pulmonary and gastrointestinal hemorrhages. Tolazoline is excreted mainly unchanged by the kidneys.
α-Adrenergic Receptor Agonists
α2-Adrenergic receptor agonists bind selectively to presynaptic α2-adrenergic receptors and, by a negative feedback mechanism, decrease the release of norepinephrine from presynaptic nerve terminals and reduce sympathetic outflow with similar decreases in blood pressure as α1 antagonists. Most α2 receptors are found in the central nervous system especially in the brainstem and the locus ceruleus. Peripherial inhibition of α2 receptors can result in inhibition of insulin release and induction of glucagon from the pancreas. Clinical pharmacologic effects include hypotension, bradycardia, and central sedation with some mild effects of analgesia all related to the sympatholytic effects.
Mechanism of Action
α2-Adrenergic receptor agonists have selective affinity for α2-adrenergic receptors and act competitively. Binding of α2 agonists can be displaced from binding sites in the central nervous system resulting in reversal of the CNS effects. Withdrawal after even short-term use can result in a rebound effect with a dramatic increase in sympathetic outflow causing elevations in heart rate and hypertension to even dangerous levels.
Clonidine
Administration results in dose-dependent decreases in heart rate and blood pressure and is used clinically to treat resistant hypertension and tremors from central stimulant medications. Clonidine is a partial agonist of α2 receptors with a 400:1 α2:α1 receptor preference. Clonidine is available in an intravenous, oral, and transdermal preparation and is metabolized in the liver but is excreted mostly unchanged in the urine and to a lesser extent in the bile and feces. Terminal half-life is approximately 12 to 16 hours but can be extremely variable with any liver or kidney dysfunction.
Dexmedetomidine
Dexmedetomidine is a selective α2 agonist with a 1,600:1 preference for α2 receptors. It is intravenously administered as an infusion from 0.1 to 1.5 µg/kg/minute with a terminal elimination half-life of 2 hours. Most often, this α2agonist is used in the intensive care and operating room settings as a sedative and analgesic due to its central sympatholytic effects. Dexmedetomidine undergoes extensive biotransformation in the liver and is excreted mostly in the urine; liver impairment can dramatically increase plasma levels and duration of action due to significantly decreased metabolism during infusion. It’s potent binding and short half-life can induce physiologic dependence and result in the aforementioned withdrawal phenomenon after only days of administration resulting in tachycardia, hypertension, and anxiety. Interestingly, large intravenous boluses (0.25 to 1 µg/kg over 3 to 5 minutes) result in a paradoxical hypertension with a decrease in heart rate and resembles phenylephrine and is the resultant effect of crossover α1 stimulation.
β-Adrenergic Receptor Antagonists
β-Adrenergic receptor antagonists bind selectively to β-adrenergic receptors and interfere with the ability of catecholamines or other sympathomimetics to provoke β responses. Drug-induced β-adrenergic blockade prevents the effects of catecholamines and sympathomimetics on the heart and smooth muscles of the airways and blood vessels. β Antagonist therapy should be continued throughout the perioperative period to maintain desirable drug effects and to avoid the risk of sympathetic nervous system hyperactivity associated with abrupt discontinuation of these drugs. Propranolol is the standard β-adrenergic antagonist drug to which all other β-adrenergic antagonists are compared.
Mechanism of Action
β-Adrenergic receptor antagonists exhibit selective affinity for β-adrenergic receptors, where they act by competitive inhibition. Binding of antagonist drugs to β-adrenergic receptors is reversible such that the drug can be displaced from the occupied receptors if sufficiently large amounts of agonist become available. Competitive antagonism causes a rightward displacement of the dose-response curve for the agonist, but the slope of the curve remains unchanged, emphasizing that sufficiently large doses of the agonist may still exert a full pharmacologic effect. Chronic administration of β-adrenergic antagonists is associated with an increase in the number of β-adrenergic receptors.
β-Adrenergic receptors are G protein–coupled receptors and their occupancy by agonists (norepinephrine, epinephrine) stimulates G proteins that in turn activate adenylate cyclase to produce cyclic adenosine monophosphate (cAMP). Protein kinases are activated by cAMP, which phosphorylates proteins including L-type voltage-dependent calcium channels and troponin C in a variety of tissues (especially myocardium). The net effect of β-adrenergic stimulation in the heart is to produce positive chronotropic, inotropic, and dromotropic effects and these are the responses that are blunted by β-adrenergic receptor antagonists. It is estimated that about 75% of β receptors in the myocardium are β1, whereas β2 receptors account for about 20% of β receptors.
Structure–Activity Relationships
β-Adrenergic antagonists are derivatives of the β agonist drug isoproterenol (Fig. 19-3). Substitutions on the benzene ring determine whether the drug acts on β-adrenergic receptors as an antagonist or agonist. The levorotatory forms of β antagonists and agonists are more potent than the dextrorotatory forms. For example, the dextrorotatory isomer of propranolol has less than 1% of the potency of the levorotatory isomer for blocking β-adrenergic receptors.

Classification
β-Adrenergic receptor antagonists are classified as nonselective for β1 and β2 receptors (propranolol, nadolol, timolol, pindolol) and cardioselective (metoprolol, atenolol, acebutolol, betaxolol, esmolol, bisoprolol) for β1 receptors (Tables 19-1 and 19-2). It is important to recognize that β receptor selectivity is dose dependent and is lost when large doses of the antagonist are administered. This emphasizes that selectivity should not be interpreted as specificity for a specific type of β-adrenergic receptor. β-Adrenergic antagonists are further classified as partial or pure antagonists on the basis of the presence or absence of intrinsic sympathomimetic activity (see Tables 19-1 and 19-2). Drugs that exhibit cardiac selectivity for β1 receptors (cardioselective) are better suited for administration to patients with asthma and reactive airway disease. Theoretically, cardioselective drugs are better suited for treatment of patients with essential hypertension as these drugs lack inhibition of peripheral β2 receptors that produce vasodilation.


β1 Receptor blockade is associated with slowing of the sinus rate, slowing of conduction of cardiac impulses through the atrioventricular node, and a decrease in inotropy. These effects are relatively greater during activity than during rest. The result is a decrease in myocardial oxygen demand, with a subsequent decrease in the occurrence of myocardial ischemia during exercise. The decrease in heart rate also increases diastolic perfusion time, which may enhance myocardial perfusion. β2 Receptor blockade increases the risk of bronchospasm in patients with reactive airway disease and may worsen the clinical symptoms of peripheral vascular disease.
β-Adrenergic antagonists may produce some degree of membrane stabilization in the heart (inhibition of propagation of action potentials across the cell membrane similar to sodium channel blockers that are class I antiarrhythmics) and thus resemble quinidine. This membrane stabilization effect, however, is detectable only at plasma concentrations that are far higher than needed to produce clinically adequate β-adrenergic blockade.
Pharmacokinetics
The principal difference in pharmacokinetics between all the β-adrenergic receptor antagonists is the elimination half-time ranging from brief for esmolol (about 10 minutes) to hours for the other drugs (see Table 19-1). Elimination half-time is considered in the perioperative period when redosing intervals are being developed or when conversion to another β-adrenergic receptor drug is planned. Among the β-adrenergic receptor antagonists, only propranolol is highly protein bound. The volume of distribution of these drugs is high and they are rapidly distributed following IV administration.
β-Adrenergic receptor antagonists are eliminated by several different pathways and this must be considered in the presence of renal and/or hepatic dysfunction (see Table 19-1). The therapeutic plasma concentration varies greatly among these drugs and between patients (interpatient variability). Explanations for interpatient variability include differences in basal sympathetic nervous system tone, flat dose-response curves for the drug so changes in plasma concentrations evoke minimal changes in pharmacologic effects, impact of active metabolites, and genetic differences in β-adrenergic receptors that influence how an individual patient responds to a given drug and plasma concentration.
Propranolol
Propranolol is a nonselective β-adrenergic receptor antagonist that lacks intrinsic sympathomimetic activity and thus is a pure antagonist (see Table 19-1). Antagonism of β1 and β2 receptors produced by propranolol is about equal. As the first β-adrenergic antagonist introduced clinically, propranolol is the standard drug to which all β-adrenergic antagonists are compared. Typically, propranolol is administered in stepwise increments until physiologic plasma concentrations have been attained, as indicated by a resting heart rate of 55 to 60 beats per minute.
Cardiac Effects
The most important pharmacologic effects of propranolol are on the heart. Because of β1-receptor blockade, propranolol decreases heart rate and myocardial contractility, resulting in decreased cardiac output. These effects on heart rate and cardiac output are especially prominent during exercise or in the presence of increased sympathetic nervous system activity. Heart rate slowing induced by propranolol lasts longer than the negative inotropic effects, suggesting a possible subdivision of β1 receptors. Concomitant blockade of β2 receptors by propranolol increases peripheral vascular resistance, including coronary vascular resistance. Although prolongation of systolic ejection and dilatation of the cardiac ventricles caused by propranolol increases myocardial oxygen requirements, the oxygen-sparing effects of decreased heart rate and myocardial contractility predominate. As a result, propranolol may relieve myocardial ischemia, even though drug-induced increases in coronary vascular resistance oppose coronary blood flow. Sodium retention associated with propranolol therapy most likely results from intrarenal hemodynamic changes that accompany drug-induced decreases in cardiac output.
Pharmacokinetics
Propranolol is rapidly and almost completely absorbed from the gastrointestinal tract, but systemic availability of the drug is limited by extensive hepatic first-pass metabolism, which may account for 90% to 95% of the absorbed dose. There is considerable individual variation in the magnitude of hepatic first-pass metabolism, accounting for up to 20-fold differences in plasma concentrations of propranolol in patients after oral administration of comparable doses.3 Hepatic first-pass metabolism is the reason the oral dose of propranolol (40 to 800 mg per day) must be substantially greater than the IV dose (0.05 mg/kg given in increments of 0.5 to 1.0 mg every 5 minutes). Propranolol is not effective when administered intramuscularly.
Protein Binding
Propranolol is extensively bound (90% to 95%) to plasma proteins. Heparin-induced increases in plasma concentrations of free fatty acids due to increased lipoprotein lipase activity result in decreased plasma protein binding of propranolol (Fig. 19-4).4 In addition, hemodilution that occurs when cardiopulmonary bypass is initiated may alter protein binding of drugs because of the nonphysiologic protein concentration in the pump prime.

Metabolism
Clearance of propranolol from the plasma is by hepatic metabolism. An active metabolite, 4-hydroxypropranolol, is detectable in the plasma after oral administration of propranolol. Indeed, cardiac β-blocking activity after equivalent doses of propranolol is greater after oral than after IV administration, presumably reflecting the effects of this metabolite, which is equivalent in activity to the parent compound. The elimination half-time of propranolol is 2 to 3 hours, whereas that of 4-hydroxypropranolol is even briefer. The plasma concentration of propranolol or the total dose does not correlate with its therapeutic effects. Furthermore, the assay for propranolol may not detect 4-hydroxypropranolol.
Elimination of propranolol is greatly decreased when hepatic blood flow decreases. In this regard, propranolol may decrease its own clearance rate by decreasing cardiac output and hepatic blood flow. Alterations in hepatic enzyme activity may also influence the rate of hepatic metabolism. Renal failure does not alter the elimination half-time of propranolol, but accumulation of metabolites may occur.
Clearance of Local Anesthetics
Propranolol decreases clearance of amide local anesthetics by decreasing hepatic blood flow and inhibiting metabolism in the liver.5 For example, in humans, propranolol causes clearance to be decreased to a much greater extent (46%) than would be predicted from a maximum 25% decrease in hepatic blood flow, implying that drug metabolism in the liver has also been affected.6 Bupivacaine clearance is relatively insensitive to changes in hepatic blood flow (low-extraction drug), suggesting that the 35% decrease in clearance of this local anesthetic reflects propranolol-induced decreases in metabolism (Fig. 19-5).5 Because clearance of drugs with low extraction ratios is inversely related to plasma protein binding, an increase in bupivacaine binding to α1-acid glycoprotein (responsible for 90% binding of bupivacaine) caused by propranolol could explain a decrease in clearance. Nevertheless, propranolol does not alter α1-acid glycoprotein concentrations.6 It is conceivable that systemic toxicity of bupivacaine could be increased by propranolol and presumably other β antagonists that interfere with the clearance of this and other amide local anesthetics.

Clearance of Opioids
Pulmonary first-pass uptake of fentanyl is substantially decreased in patients being treated chronically with propranolol.7 As a result, two to four times as much injected fentanyl enters the systemic circulation in the time period immediately after injection. This response most likely reflects the ability of one basic lipophilic amine (propranolol) to inhibit the pulmonary uptake of a second basic lipophilic amine (fentanyl).
Nadolol and Pindolol
Nadolol and pindolol are nonselective β-adrenergic receptor antagonists; nadolol is unique in that its long duration of action permits once daily administration.
Pharmacokinetics
Nadolol is slowly and incompletely absorbed (an estimated 30%) from the gastrointestinal tract. Metabolism does not occur, with about 75% of the drug being excreted unchanged in urine and the remainder in bile. Therefore, wide individual variations in plasma concentrations that occur with nadolol cannot be attributed to differences in metabolism, as occur with propranolol. The elimination half-time is 20 to 40 hours, accounting for the need to administer this drug only once a day. The elimination half-time of pindolol is 3 to 4 hours, and this is increased to longer than 11 hours in patients with renal failure.
Timolol
Timolol is a nonselective β-adrenergic receptor antagonist that is as effective as propranolol for various therapeutic indications. In addition, timolol is effective in the treatment of glaucoma because of its ability to decrease intraocular pressure, presumably by decreasing the production of aqueous humor. Timolol is administered as eyedrops in the treatment of glaucoma, but systemic absorption may be sufficient to cause resting bradycardia and increased airway resistance. Indeed, bradycardia and hypotension that are refractory to treatment with atropine have been observed during anesthesia in pediatric and adult patients receiving topical timolol with or without pilocarpine.8 Timolol may be associated with impaired control of ventilation in neonates, resulting in unexpected postoperative apnea.9 Immaturity of the neonate’s blood–brain barrier may facilitate access of this drug to the CNS.
Pharmacokinetics
Timolol is rapidly and almost completely absorbed after oral administration. Nevertheless, extensive first-pass hepatic metabolism limits the amount of drug reaching the systemic circulation to about 50% of that absorbed from the gastrointestinal tract. Protein binding of timolol is not extensive. The elimination half-time is about 4 hours.
Metoprolol
Metoprolol is a selective β1-adrenergic receptor antagonist that prevents inotropic and chronotropic responses to β-adrenergic stimulation. Conversely, bronchodilator, vasodilator, and metabolic effects of β2receptors remain intact such that metoprolol is less likely to cause adverse effects in patients with chronic obstructive airway disease or peripheral vascular disease and in patients vulnerable to hypoglycemia. It is important to recognize, however, that selectivity is dose related, and large doses of metoprolol are likely to become nonselective, exerting antagonist effects at β2 receptors as well as β1 receptors. Indeed, airway resistance may increase in asthmatic patients treated with metoprolol, although the magnitude of increase will be less than that evoked by propranolol. Furthermore, metoprolol-induced increases in airway resistance are more readily reversed with β2-adrenergic agonists such as terbutaline.
Pharmacokinetics
Metoprolol is readily absorbed from the gastrointestinal tract, but this is offset by substantial hepatic first-pass metabolism such that only about 40% of the drug reaches the systemic circulation. Protein binding is low; it is estimated to account for about 10% of the drug. None of the hepatic metabolites have been identified as active. A small amount (<10%) of the drug appears unchanged in urine. There are two available oral formulation of metoprolol: metoprolol tartrate and metoprolol succinate. The elimination half-time of metoprolol tartrate is 2 to 3 hours and correlates to a need for at least a twice daily dosing strategy with a thrice daily or four times daily strategy providing a more reliable control of heart rate. Metoprolol succinate results in a significantly extended time to peak concentrations and overall decreased plasma concentrations compared to metoprolol tartrate at equal daily doses. Metoprolol succinate elimination half-time is 5 to 7 hours and can be used in once daily dosing regimens but in some patients can still result in β-blocker withdrawal tachycardia at 24 hours necessitating twice daily dosing. Overall, plasma concentrations of metoprolol do not correlate with therapeutic effects of the drug.
Atenolol
Atenolol is the most selective β1-adrenergic antagonist that may have specific value in patients in whom the continued presence of β2 receptor activity is desirable. In patients at risk for coronary artery disease who must undergo noncardiac surgery, treatment with IV atenolol before and immediately after surgery, followed by oral therapy during the remainder of the hospitalization, decreases mortality and the incidence of cardiovascular complications for as long as 2 years.10 Perioperative administration of atenolol to patients at high risk for coronary artery disease significantly decreases the incidence of postoperative myocardial ischemia.11
The antihypertensive effect of atenolol is prolonged, permitting this drug to be administered once daily for the treatment of hypertension. Like nadolol, atenolol does not enter the CNS in large amounts, but fatigue and mental depression still occur. Unlike nonselective β-adrenergic antagonists, atenolol does not appear to potentiate insulin-induced hypoglycemia and can thus be administered with caution to patients with diabetes mellitus whose hypertension is not controlled by other antihypertensives.
Pharmacokinetics
About 50% of an orally administered dose of atenolol is absorbed from the gastrointestinal tract, with peak concentrations occurring 1 to 2 hours after oral administration. Atenolol undergoes little or no hepatic metabolism and is eliminated principally by renal excretion. The elimination half-time is 6 to 7 hours; this may increase to more than 24 hours in patients with renal failure. Therapeutic plasma concentrations of atenolol are 200 to 500 ng/mL.
Betaxolol
Betaxolol is a cardioselective β1-adrenergic antagonist with no intrinsic sympathomimetic activity and weak membrane-stabilizing activity. High doses can be expected to produce some β2-adrenergic antagonist effects on bronchial and vascular smooth muscle. Absorption after an oral dose is nearly complete. Its elimination half-time is 11 to 22 hours, making it one of the longest acting β-adrenergic antagonists. Clearance is primarily by metabolism, with renal elimination contributing less to overall removal of the drug from the plasma. A single oral dose daily is useful for the treatment of hypertension. A topical preparation is used as an alternative to timolol for treatment of chronic open-angle glaucoma. The risk of bronchoconstriction in patients with airway hyperreactivity may be less with betaxolol than with timolol.
Bisoprolol
Bisoprolol is a β1 selective antagonist drug without significant intrinsic agonist activity. The elimination half-time is 9 to 12 hours. Bisoprolol is eliminated equally by renal and nonrenal mechanisms. Metabolites are pharmacologically inactive. The most prominent pharmacologic effect of bisoprolol is a negative chronotropic effect. Bisoprolol is useful in the treatment of essential hypertension and has been shown to improve survival in patients with mild to moderate congestive heart failure (see Table 19-2).
Nebivolol
Nebivolol is a very potent and selective β1-antagonist drug12 and is 3.5 times more selective than bisoprolol13 without significant intrinsic agonist activity. Above doses of 10 mg, or in those patients with certain CYP polymorphisms, Nebivolol can exhibit low β2 antagonism as well. With an elimination half-time of 12 to 19 hours, it is a once daily regimen that allows accidental delay in subsequent doses without withdrawal. Nebivolol is equally eliminated in the urine unchanged and in the feces as a inactive metabolite. The most prominent pharmacologic effect of bisoprolol is a negative chronotropic effect and is currently approved and useful in the treatment of essential hypertension.
Esmolol
Esmolol is a rapid-onset and short-acting selective β1-adrenergic receptor antagonist that is administered only IV (see Fig. 19-3). After a typical initial dose of 0.5 mg/kg IV over about 60 seconds, the full therapeutic effect is evident within 5 minutes, and its action ceases within 10 to 30 minutes after administration is discontinued. These characteristics make esmolol a useful drug for preventing or treating adverse systemic blood pressure and heart rate increases that occur intraoperatively in response to noxious stimulation, as during tracheal intubation. For example, esmolol, 150 mg IV, administered about 2 minutes before direct laryngoscopy and tracheal intubation provides reliable protection against increases in both heart rate and systolic blood pressure, which predictably accompanies tracheal intubation (Fig. 19-6).14

Lidocaine or fentanyl is effective in blunting the increase in systolic blood pressure associated with laryngoscopy and tracheal intubation, but heart rate is not influenced (see Fig. 19-6).14 Other reports describe prevention of perioperative tachycardia and hypertension with esmolol, 100 to 200 mg IV, administered over 15 seconds before the induction of anesthesia.15,16 Prior administration of esmolol, 500 µg/kg/minute IV, to patients undergoing electroconvulsive therapy with anesthesia induced by methohexital plus succinylcholine results in attenuation of the heart rate increase and a decrease in the length of the electrically induced seizures.17 Esmolol has been used during resection of pheochromocytoma and may be useful in the perioperative management of thyrotoxicosis, pregnancy-induced hypertension, and epinephrine- or cocaine-induced cardiovascular toxicity.18–22 Conversely, treatment of excessive sympathetic nervous system activity produced by cocaine or systemic absorption of topical or subcutaneous epinephrine with β-adrenergic receptor antagonists has been associated with fulminant pulmonary edema and irreversible cardiovascular collapse.23 It is possible that acute drug-induced β-receptor antagonism removes the ability of the heart to increase heart rate and myocardial contractility to compensate for catecholamine-induced increases in left ventricular afterload. In this regard, persistent symptomatic systemic hypertension owing to catecholamine-induced sympathetic nervous system stimulation is more safely treated with a peripheral vasodilator drug such as sodium nitroprusside or nitroglycerin. Detrimental effects of catecholamine release during anesthesia in patients with hypertrophic obstructive cardiomyopathy and in patients experiencing hypercyanotic spells associated with tetralogy of Fallot may be blunted by administration of esmolol.24
The β1 selectivity of esmolol may unmask β2-mediated vasodilation by epinephrine-secreting tumors. Administration of esmolol to patients chronically treated with β-adrenergic antagonists has not been observed to produce additional negative inotropic effects.25 The presumed reason for this observation is that esmolol, in the dose used, does not occupy sufficient additional β receptors to produce detectable increases in β blockade. Likewise, esmolol infused during cardiopulmonary bypass is not associated with adverse effects after discontinuation of cardiopulmonary bypass.26
Esmolol (1 mg/kg IV followed by 250 µg/kg/minute V) significantly decreases the plasma concentration of propofol required to prevent patient movement in response to a surgical skin incision.27 This effect does not seem to be explained by a pharmacokinetic interaction between the two drugs.
Pharmacokinetics
Esmolol is available for IV administration only. The only other β-adrenergic antagonists that may be administered IV are propranolol and metoprolol. The commercial preparation of esmolol is buffered to pH 4.5 to 5.5, which may be one of the factors responsible for pain on injection. The drug is compatible with commonly used IV solutions and nondepolarizing neuromuscular blocking drugs. The elimination half-time of esmolol is about 9 minutes, reflecting its rapid hydrolysis in the blood by plasma esterases that is independent of renal and hepatic function.28 Less than 1% of the drug is excreted unchanged in urine, and about 75% is recovered as an inactive acid metabolite. Clinically insignificant amounts of methanol also occur from the hydrolysis of esmolol. Plasma esterases responsible for the hydrolysis of esmolol are distinct from plasma cholinesterase, and the duration of action of succinylcholine is not predictably prolonged in patients treated with esmolol.29 Evidence of the short duration of action of esmolol is return of the heart rate to predrug levels within 15 minutes after discontinuing the drug. Indeed, plasma concentrations of esmolol are usually not detectable 15 minutes after discontinuing the drug. Poor lipid solubility limits transfer of esmolol into the CNS or across the placenta.19
Side Effects
The side effects of β-adrenergic antagonists are similar for all available drugs, although the magnitude may differ depending on their selectivity and the presence or absence of intrinsic sympathomimetic activity. β-Adrenergic antagonists exert their most prominent pharmacologic effects as well as side effects on the cardiovascular system. These drugs may also alter airway resistance, carbohydrate and lipid metabolism, and the distribution of extracellular ions. β-Adrenergic antagonists may cause hypoglycemia.30 Additive effects between drugs used for anesthesia and β-adrenergic antagonists may occur. β-Adrenergic antagonists penetrate the blood–brain barrier and cross the placenta. Gastrointestinal side effects include nausea, vomiting, and diarrhea. Fever, rash, myopathy, alopecia, and thrombocytopenia have been associated with chronic β-adrenergic antagonist treatment. β-Adrenergic antagonists have been reported to decrease plasma concentrations of high-density lipoproteins and to increase triglyceride and uric acid levels.
The principal contraindication to administration of β-adrenergic antagonists is preexisting atrioventricular heart block or cardiac failure not caused by tachycardia. Administration of β-adrenergic antagonists to hypovolemic patients with compensatory tachycardia may produce profound hypotension.31 Nonselective β-adrenergic antagonists or high doses of selective β-adrenergic antagonists are not recommended for administration to patients with chronic obstructive airway disease. In patients with diabetes mellitus, there is the risk that β-adrenergic blockade may mask the signs of hyperglycemia and thus delay its clinical recognition.
Cardiovascular System
β-Adrenergic antagonists produce negative inotropic and chronotropic effects. In addition, the conduction speed of cardiac impulses through the atrioventricular node is slowed and the rate of spontaneous phase 4 depolarization is decreased. Preexisting atrioventricular heart block due to any cause may be accentuated by β-adrenergic antagonists.
The cardiovascular effects of β-adrenergic blockade reflect removal of sympathetic nervous system innervation to the heart (β1 blockade) and not membrane stabilization, which occurs only at high plasma concentrations of the antagonist drug. In addition, nonselective β-adrenergic blockade resulting in β2-adrenergic receptor antagonism may impede left ventricular ejection due to unopposed α-adrenergic receptor–mediated peripheral vasoconstriction. The magnitude of cardiovascular effects produced by β-adrenergic antagonists is greatest when preexisting sympathetic nervous system activity is increased, as during exercise or in patients in cardiac failure. Indeed, the tachycardia of exercise is consistently attenuated by β-adrenergic antagonists. Furthermore, administration of a β antagonist may precipitate cardiac failure in a patient who was previously compensated. Resting bradycardia is minimized and cardiac failure is less likely to occur when a partial β-adrenergic antagonist with intrinsic sympathomimetic activity is administered. Acute cardiac failure is rare with oral administration of β-adrenergic antagonists.
Classically, β-adrenergic antagonists prevent inotropic and chronotropic effects of isoproterenol as well as baroreceptor-mediated increases in heart rate evoked by decreases in systemic blood pressure in response to peripheral vasodilator drugs. Conversely, the influence of β-adrenergic antagonists on the cardiac-stimulating effects of calcium, glucagon, and digitalis preparations is not detectable. Likewise, β-adrenergic antagonists do not alter the response to α-adrenergic agonists such as epinephrine or phenylephrine. Indeed, the pressor effect of epinephrine is enhanced because the nonselective β antagonists prevent the β2 vasodilating effect of epinephrine and leave unopposed its α-adrenergic effect. The presence of unopposed α-adrenergic–induced vasoconstriction may provoke paradoxical hypertension and may even precipitate cardiac failure in the presence of diseased myocardium that cannot respond to sympathetic nervous system stimulation because of β-adrenergic blockade. Unexpected hypertension has occurred in patients receiving clonidine who subsequently receive a nonselective β-adrenergic antagonist.32 Presumably, blockade of the vasodilating effect normally produced by activity of β2 receptors leaves unopposed α-adrenergic effects to provoke peripheral vasoconstriction with resulting hypertension.
Patients with peripheral vascular disease do not tolerate well the peripheral vasoconstriction associated with β2 receptor blockade produced by nonselective β-adrenergic antagonists. Indeed, the development of cold hands and feet is a common side effect of β blockade. Vasospasm associated with Raynaud’s disease is accentuated by propranolol.
The principal antidysrhythmic effect of β-adrenergic blockade is to prevent the dysrhythmogenic effect of endogenous or exogenous catecholamines or sympathomimetics. This reflects a decrease in sympathetic nervous system activity. Membrane stabilization is probably of little importance in the antidysrhythmic effects produced by usual doses of β-adrenergic antagonists.
Treatment of Excess Myocardial Depression
The usual clinical manifestations of excessive myocardial depression produced by β-adrenergic blockade include bradycardia, low cardiac output, hypotension, and cardiogenic shock.33 Bronchospasm and depression of ventilation may also be associated with an overdose of β-adrenergic antagonist drugs. Seizures and prolonged intraventricular conduction of cardiac impulses are thought to be the result of local anesthetic properties of certain β-adrenergic antagonists (see Table 19-1). Hypoglycemia is a rare manifestation of β-adrenergic antagonist overdose.
Excessive bradycardia and/or decreases in cardiac output due to drug-induced β blockade should be treated initially with atropine in incremental doses of 7 µg/kg IV. Atropine is likely to be effective by blocking vagal effects on the heart and thus unmasking any residual sympathetic nervous system innervation. If atropine is ineffective, drugs to produce direct positive chronotropic and inotropic effects are indicated. For example, continuous infusion of the nonselective β-adrenergic agonist isoproterenol, in doses sufficient to overcome competitive β blockade, is appropriate. The necessary dose of isoproterenol may be 2 to 25 µg per minute IV (60 µg per minute IV was not effective in one report), which is 5 to 20 times the necessary dose in the absence of β blockade.33 When a pure β-adrenergic antagonist is responsible for excessive cardiovascular depression, a pure β1-adrenergic agonist such as dobutamine is recommended because isoproterenol, with β1- and β2-adrenergic agonist effects, could produce vasodilation before its inotropic effect develops. Dopamine is not recommended because α-adrenergic–induced vasoconstriction is likely to occur with the high doses required to overcome β blockade.
Glucagon administered to adults, 1 to 10 mg IV followed by 5 mg per hour IV, effectively reverses myocardial depression produced by β-adrenergic antagonists at normal doses because these drugs do not exert their effects by means of β-adrenergic receptors. For example, glucagon stimulates adenylate cyclase and increases intracellular cAMP concentrations independent of β-adrenergic receptors.33 Calcium chloride, 250 to 1,000 mg IV, may also act independent of β-adrenergic receptors to offset excessive cardiovascular depression produced by β-adrenergic antagonists. Glucagon appears to be particularly effective in the presence of life-threatening bradycardia and has been described as the drug of choice to treat massive β-adrenergic antagonist overdose.33
In the presence of bradycardia that is unresponsive to pharmacologic therapy, it may be necessary to place a transvenous artificial cardiac pacemaker. Hemodialysis should be reserved to remove minimally protein-bound, renally excreted β-adrenergic antagonists in patients refractory to pharmacologic therapy.
Airway Resistance
Nonselective β-adrenergic antagonists such as propranolol consistently increase airway resistance as a manifestation of bronchoconstriction due to blockade of β2 receptors. These airway resistance effects are exaggerated in patients with preexisting obstructive airway disease. Because bronchodilation is a β2-adrenergic agonist response, selective β1-adrenergic antagonists such as metoprolol and esmolol are less likely than propranolol to increase airway resistance.
Metabolism
β-Adrenergic antagonists alter carbohydrate and fat metabolism. For example, nonselective β-adrenergic antagonists such as propranolol interfere with glycogenolysis that ordinarily occurs in response to release of epinephrine during hypoglycemia. This emphasizes the need for β2 receptor activity in glycogenolysis. Furthermore, tachycardia, which is an important warning sign of hypoglycemia in insulin-treated diabetics, is blunted by β-adrenergic antagonists. For this reason, nonselective β-adrenergic antagonists are not recommended for administration to patients with diabetes mellitus who may be at risk for developing hypoglycemia because of treatment with insulin or oral hypoglycemics. Altered fat metabolism is evidenced by failure of sympathomimetics or sympathetic nervous system stimulation to increase plasma concentrations of fatty-free acids in the presence of β-adrenergic blockade.
Distribution of Extracellular Potassium
Distribution of potassium across cell membranes is influenced by sympathetic nervous system activity as well as insulin. Specifically, stimulation of β2-adrenergic receptors seems to facilitate movement of potassium intracellularly. As a result, β-adrenergic blockade inhibits uptake of potassium into skeletal muscles, and the plasma concentration of potassium may be increased. Indeed, increases in the plasma concentration of potassium associated with infusion of this ion are greater in the presence of β-adrenergic blockade produced by propranolol (Fig. 19-7).34 In animals, increases in the plasma concentration of potassium after administration of succinylcholine last longer when β-adrenergic blockade is present.35 In view of the speculated role of β2 receptors in regulating plasma concentrations of potassium, it is likely that selective β1-adrenergic antagonists would impair skeletal muscle uptake of potassium less than nonselective β-adrenergic antagonists.

Interaction with Anesthetics
Myocardial depression produced by inhaled or injected anesthetics could be additive with depression produced by β-adrenergic antagonists. Nevertheless, clinical experience and controlled studies in patients and animals have confirmed that additive myocardial depression with β-adrenergic antagonists and anesthetics is not excessive, and treatment with β-adrenergic antagonists may therefore be safely maintained throughout the perioperative period.1 An exception may be patients treated with timolol in whom profound bradycardia has been observed in the presence of inhaled anesthetics.
Additive cardiovascular effects with inhaled anesthetics and β-adrenergic antagonists seem to be greatest with enflurane and least with isoflurane.1 Sevoflurane and desflurane, like isoflurane, do not seem to be associated with significant additive cardiovascular effects when administered to patients being treated with β-adrenergic antagonists. Cardiac output and systemic blood pressure are similar with or without β-adrenergic blockade in the presence of one or two minimum alveolar concentration isoflurane.36 Even acute hemorrhage does not alter the interaction between isoflurane and β-adrenergic antagonists.37,38 In contrast, cardiac depression is more likely to occur in the presence of β blockade when acute hemorrhage occurs in animals anesthetized with enflurane.39 Cardiovascular responses to even high doses of opioids such as fentanyl are not altered by preexisting β-adrenergic blockade. In the presence of anesthetic drugs that increase sympathetic nervous system activity (ketamine), or when excessive sympathetic nervous system activity is present because of hypercarbia, the acute administration of a β-adrenergic antagonist may unmask direct negative inotropic effects of concomitantly administered anesthetics, with resulting decreases in systemic blood pressure and cardiac output.40
Nervous System
β-Adrenergic antagonists may cross the blood–brain barrier to produce side effects. For example, fatigue and lethargy are commonly associated with chronic propranolol therapy. Vivid dreams are frequent, but psychotic reactions are rare. Memory loss and mental depression have been alleged to occur, although β-adrenergic antagonist therapy has not been shown to produce these effects.41 Peripheral paresthesias have been described. Atenolol and nadolol are less lipid soluble than other β-adrenergic antagonists and thus may be associated with a lower incidence of CNS effects.
Fetus
β-Adrenergic antagonists can cross the placenta and cause bradycardia, hypotension, and hypoglycemia in newborn infants of mothers who are receiving the drug. Breast milk is also likely to contain β-adrenergic antagonists administered to the mother.
Withdrawal Hypersensitivity
Acute discontinuation of β-adrenergic antagonist therapy can result in excess sympathetic nervous system activity that manifests in 24 to 48 hours. Presumably, this enhanced activity reflects an increase in the number of β-adrenergic receptors (upregulation) during chronic therapy with β-adrenergic antagonists. Continuous infusion of propranolol, 3 mg per hour IV, is effective in maintaining therapeutic plasma concentrations in adult patients who cannot take drugs orally during the perioperative period.42
Clinical Uses
Clinical uses of β-adrenergic antagonists are multiple and in equivalent doses, all β-adrenergic antagonists seem to be equally effective in producing desired therapeutic effects (Table 19-3). It is accepted that patients being treated with β-adrenergic receptor antagonists should have their medication continued uninterrupted through the perioperative period. It is also recommended that patients at high risk for myocardial ischemia and presenting for major surgery should be treated with β-adrenergic receptor antagonists beginning preoperatively and continuing into the postoperative period.

Treatment of Essential Hypertension
Chronic therapy with β-adrenergic antagonists results in gradual decreases in systemic blood pressure. The antihypertensive effect of β-adrenergic blockade is largely dependent on decreases in cardiac output due to decreased heart rate. Large doses of β-adrenergic antagonists may decrease myocardial contractility as well. In many patients, systemic vascular resistance remains unchanged. An important advantage in the use of β-adrenergic antagonists for the treatment of essential hypertension is the absence of orthostatic hypotension. Often, a β-adrenergic antagonist is used in combination with a vasodilator to minimize reflex baroreceptor–mediated increases in heart rate and cardiac output produced by the vasodilator. All orally administered β-adrenergic antagonists appear to be equally effective antihypertensive drugs. Release of renin from the juxtaglomerular apparatus that occurs in response to stimulation of β2 receptors is prevented by nonselective β-adrenergic antagonists such as propranolol. This may account for a portion of the antihypertensive effect of propranolol, especially in patients with high circulating plasma concentrations of renin. Because drug-induced decreases in secretion of renin will lead to decreased release of aldosterone, β-adrenergic antagonists will also prevent the compensatory sodium and water retention that accompanies treatment with a vasodilator.
Management of Angina Pectoris
Orally administered β-adrenergic antagonists are equally effective in decreasing the likelihood of myocardial ischemia manifesting as angina pectoris. This desirable response reflects drug-induced decreases in myocardial oxygen requirements secondary to decreased heart rate and myocardial contractility. The effective dose usually decreases resting heart rate to less than 60 beats per minute. A more important measure is the heart rate during exercise, which should not exceed 75% of the heart rate at which myocardial ischemia occurs.
The concept that β-adrenergic antagonists and calcium channel blockers act on different determinants of the myocardial oxygen supply-to-demand ratio suggests combined uses of these drugs would be beneficial in the management of patients with coronary artery disease. Nevertheless, the evidence from clinical studies suggests that patients managed with combined therapy do not experience greater beneficial therapeutic effects but may experience more adverse effects than if they had received optimal treatment with a single drug.43
Treatment of Acute Coronary Syndrome
It is recommended that all patients who experience an acute myocardial infarction receive IV β-adrenergic antagonists (assuming no contraindications are present) as early as possible, whether or not they receive reperfusion therapy. Treatment with β-adrenergic antagonists is contraindicated in the presence of severe bradycardia, unstable left ventricular failure, and atrioventricular heart block. Relative contraindications to treatment with β-adrenergic antagonists include asthma or reactive airway disease, mental depression, and peripheral vascular disease. Diabetes mellitus is not a contraindication to treatment with β-adrenergic antagonists recognizing that signs of hypoglycemia may be masked.
A growing body of literature is rising against the use of β blockers in patients who present with an acute coronary syndrome within the first 8 hours, especially if they present with ST elevation myocardial infarction (STEMI) or in cardiogenic shock.44 STEMI patients had a greater risk of developing cardiogenic shock after β-blocker administration and those in shock had a greater risk of decompensating even further.45 However, the incidence of nonfatal reinfarction and recurrent myocardial ischemia is decreased compared with patients in whom oral metoprolol was initiated 6 days following myocardial infarction.46 β-Adrenergic antagonist prophylaxis after acute myocardial infarction is considered to be one of the most scientifically substantiated, cost-effective preventive medical treatments. Whether β-adrenergic antagonists can decrease mortality in patients with angina pectoris who have not yet experienced a myocardial infarction is unknown.
The cardioprotective effect of β-adrenergic antagonists is present with both cardioselective and nonselective drugs (see Tables 19-1 and 19-2). The mechanism of the cardioprotective effect is uncertain, but antidysrhythmic actions may be important. A nonselective β-adrenergic antagonist that prevents epinephrine-induced decreases in plasma potassium concentrations (a β2-mediated response) may be useful in decreasing the incidence of ventricular dysrhythmias.
Perioperative β-Adrenergic Receptor Blockade
Perioperative β-adrenergic receptor blockade is recommended for patients considered at risk for myocardial ischemia (known coronary artery disease, positive preoperative stress tests, diabetes mellitus treated with insulin, left ventricular hypertrophy) during high-risk surgery (vascular surgery, thoracic surgery, intraperitoneal surgery, anticipated large blood loss).47,48 The goal of preoperative therapy is a resting heart rate between 65 and 80 beats per minute.49 All β-adrenergic receptor antagonists except those with intrinsic sympathetic nervous system activity decrease mortality. Perioperative myocardial ischemia is the single most important potentially reversible risk factor for mortality and cardiovascular complications after noncardiac surgery. Administration of atenolol for 7 days before and after noncardiac surgery in patients at risk for coronary artery disease may decrease mortality and the incidence of cardiovascular complications for as long as 2 years after surgery.10 In another report, administration of atenolol IV before induction of anesthesia and every 12 hours after noncardiac surgery for 7 days to patients at high risk for coronary artery disease reduced the incidence of postoperative myocardial ischemia by 30% to 50%.11Decreases in perioperative myocardial ischemia are associated with reductions in the risk for death at 24 months. The incidence of bronchospasm, hypotension, bradycardia, and cardiac dysrhythmias was not increased in treated patients. The mechanism for the beneficial effects of perioperative β-adrenergic receptor blockade is not known but is most likely multifactorial (Table 19-4). It is not known if patients with cardiac risk factors but no signs of underlying coronary artery disease will benefit from perioperative administration of a β-adrenergic antagonist.50

Preoperatively, oral therapy can be initiated with atenolol 50 mg or bisoprolol 5 to 10 mg daily or metoprolol 25 to 50 mg twice daily. If the patient is seen the morning of surgery, atenolol 5 to 10 mg IV or metoprolol 5 to 10 mg IV can be titrated. Esmolol is an acceptable drug to achieve β-adrenergic receptor blockade during surgery and postoperatively in the intensive care unit where continuous IV infusions can be monitored. Alternatively, IV atenolol or metoprolol can be administered until the patient can take oral atenolol or bisoprolol. β-adrenergic receptor antagonists with sympathomimetic actions are not likely selections to produce perioperative β blockade. The Perioperative Ischemic Evaluation (POISE) trial has raised concerns over regimens that start β blockers in the acute preoperative setting due to an all-cause increased mortality that is driven by an increase in cerebrovascular events. Criticisms of the POISE trial51 point to its very aggressive escalation regimen (as much as 400 mg metoprolol succinate dosed in 24 hours) without regard to patient’s baseline hemodynamics (systolic and mean pressure reductions of >30% below preoperative levels) which is well known to cause cerebrovascular hypoperfusion and infarction. This has prompted many practitioners to only start low-dose β-blocker regimens in the preoperative period or to hold initiation until postoperatively.
Treatment of Intraoperative Myocardial Ischemia
Appearance of evidence of myocardial ischemia on the electrocardiogram or as wall motion abnormalities on the transesophageal echocardiogram may benefit from treatment with a β-adrenergic receptor blocking drug, assuming the absence of contraindications (severe reactive airway disease, shock, left ventricular failure) and the presence of an adequate concentration of inhaled anesthetic drugs. The drug selected should be titrated IV to the desired heart rate (about 60 beats per minute) to attenuate myocardial ischemia. Treatment options include esmolol (1 to 1.5 mg/kg IV followed by a continuous infusion of 50 to 300 µg/kg/minute), metoprolol (5 mg IV), atenolol (5 to 10 mg IV), or propranolol (1 to 10 mg IV). The advantage of esmolol is the ability to titrate its effects to the desired heart rate. Nitroglycerin is often added to this treatment regimen.
Suppression of Cardiac Dysrhythmias
β-Adrenergic receptor blocking drugs are effective in the treatment of cardiac dysrhythmias as a result of enhanced sympathetic nervous system stimulation (thyrotoxicosis, pheochromocytoma, perioperative stress). Esmolol and propranolol are effective for controlling the ventricular response rate to atrial fibrillation and atrial flutter. These drugs are also effective for controlling atrial dysrhythmias following cardiac surgery. Propranolol may be effective for controlling torsades de pointes in patients with prolonged QTc intervals on the electrocardiogram. Acebutolol, metoprolol, atenolol, propranolol, and timolol are approved for prevention of sudden death following acute myocardial infarction.
Management of Congestive Heart Failure
Controlled studies have demonstrated that metoprolol, carvedilol, and bisoprolol improve ejection fraction and increase survival in patients in chronic heart failure (see Table 19-2). Sustained-release metoprolol is associated with improved survival.52 Carvedilol, a nonselective β blocker with vasodilator and antioxidant properties, has been shown to decrease mortality associated with congestive heart failure.53 When β blocking drugs are used to treat congestive heart failure, the initial doses of β blockers should be minimal and gradually increased.
Prevention of Excessive Sympathetic Nervous System Activity
β-Adrenergic blockade is associated with attenuated heart rate and blood pressure changes in response to direct laryngoscopy and tracheal intubation.1,54 Hypertrophic obstructive cardiomyopathies are often treated with β-adrenergic antagonists. Tachycardia and cardiac dysrhythmias associated with pheochromocytoma and hyperthyroidism are effectively suppressed by propranolol. The likelihood of cyanotic episodes in patients with tetralogy of Fallot is minimized by β blockade. Propranolol has been used intraoperatively to prevent reflex baroreceptor–mediated increases in heart rate evoked by vasodilators administered to produce controlled hypotension. Even anxiety states as caused by public speaking have been treated with propranolol.
Preoperative Preparation of Hyperthyroid Patients
Thyrotoxic patients can be prepared for surgery in an emergency by IV administration of propranolol or esmolol or electively by oral administration of propranolol (40 to 320 mg daily).55,56 Advantages of β-adrenergic antagonists include rapid suppression of excessive sympathetic nervous system activity and elimination of the need to administer iodine or antithyroid drugs.
Combined α- and β-Adrenergic Receptor Antagonists
Labetalol
Labetalol is a unique parenteral and oral antihypertensive drug that exhibits selective α1- and nonselective β1- and β2-adrenergic antagonist effects (Fig. 19-8).57,58 Presynaptic α2 receptors are spared by labetalol such that released norepinephrine can continue to inhibit further release of catecholamines via the negative feedback mechanism resulting from stimulation of α2 receptors. Labetalol is one-fifth to one-tenth as potent as phentolamine in its ability to block α receptors and is approximately one-fourth to one-third as potent as propranolol in blocking β receptors. In humans, the β to α blocking potency ratio is 3:1 for oral labetalol and 7:1 for IV labetalol.

Pharmacokinetics
Metabolism of labetalol is by conjugation of glucuronic acid, with 5% of the drug recovered unchanged in urine. The elimination half-time is 5 to 8 hours and is prolonged in the presence of liver disease and unchanged by renal dysfunction.
Cardiovascular Effects
Administration of labetalol lowers systemic blood pressure by decreasing systemic vascular resistance (α1 blockade), whereas reflex tachycardia triggered by vasodilation is attenuated by simultaneous β blockade. Cardiac output remains unchanged. In addition to producing vasodilation by α1 blockade, labetalol may cause vasodilation that is mediated by β2-adrenergic agonist activity.59 The maximum systemic blood pressure–lowering effect of an IV dose of labetalol (0.1 to 0.5 mg/kg) is present in 5 to 10 minutes.
Clinical Uses
Labetalol is a safe and effective treatment for hypertensive emergencies. For example, labetalol has been administered IV to control severe hypertension that may be associated with an epinephrine overdose as may occur during submucosal injection to produce surgical hemostasis.60 Conversely, caution has been urged in using β-adrenergic blockers to treat phenylephrine and epinephrine overdose resulting from systemic absorption following topical application.21 Large bolus doses of labetalol (2 mg/kg IV) administered to treat hypertensive emergencies may result in excessive decreases in blood pressure, whereas smaller doses (20 to 80 mg IV) are less likely to produce undesirable decreases in blood pressure.61,62 Repeated doses of labetalol, 20 to 80 mg IV, may be administered about every 10 minutes until the desired therapeutic response is achieved.62 Rebound hypertension after withdrawal of clonidine therapy and hypertensive responses in patients with pheochromocytoma can be effectively treated with labetalol. Labetalol is also effective in the treatment of angina pectoris. Availability of both an oral (100 to 600 mg twice a day) and IV preparation is useful for converting a patient with a hypertensive crisis to oral therapy after initial control with IV therapy.
Labetalol, 0.1 to 0.5 mg/kg IV, can be administered to anesthetized patients to attenuate increases in heart rate and blood pressure that are presumed to result from abrupt increases in the level of surgical stimulation. It is possible that existing depressant effects of the anesthetic drugs could accentuate the blood pressure–lowering properties of labetalol. In contrast to the results with nitroprusside, controlled hypotension produced with intermittent injections of labetalol, 10 mg IV, is not associated with increases in heart rate, intrapulmonary shunt, or cardiac output.63
Side Effects
Orthostatic hypotension is the most common side effect of labetalol therapy. Bronchospasm is possible in susceptible patients, reflecting the β-adrenergic antagonist effects of labetalol. Other adverse effects associated with β-adrenergic antagonists (congestive heart failure, bradycardia, heart block) are a potential risk of labetalol therapy, but the likely incidence and severity is substantially decreased. Incomplete α-adrenergic blockade in the presence of more complete β blockade could result in excessive α stimulation.60 Fluid retention in patients treated chronically with labetalol is the reason for combining this drug with a diuretic during prolonged therapy.
Carvedilol
Carvedilol is a nonselective β-adrenergic receptor antagonist with α1 blocking activity. This drug has no intrinsic β-adrenergic agonist effect. Following oral administration, carvedilol is extensively metabolized to products with pharmacologic activity possessing weak vasodilator actions. The elimination half-time is 7 to 10 hours and protein binding is extensive. Carvedilol is indicated for the treatment of mild to moderate congestive heart failure owing to ischemia or cardiomyopathy (see Table 19-2). This drug is also useful for the treatment of essential hypertension.
Calcium Channel Blockers
Calcium channel blockers (also known as calcium entry blockers and calcium antagonists) are a diverse group of structurally unrelated compounds that selectively interfere with inward calcium ion movement across myocardial and vascular smooth muscle cells.64,65 Calcium ions play a key role in the electrical excitation of cardiac cells and vascular smooth muscle cells.
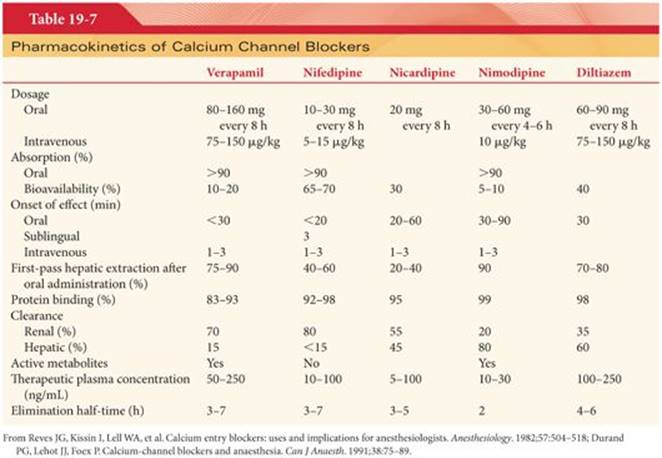
Commercially available calcium channel blockers are classified based on chemical structure as phenylalkylamines, dihydropyridines, and benzothiazepines (Table 19-5 and Fig. 19-9). The phenylalkylamines and benzothiazepines are selective for the atrioventricular node, whereas the dihydropyridines are selective for the arteriolar beds.66 Common side effects of calcium channel blockers include systemic hypotension, peripheral edema, flushing, and headache. The various calcium channel blockers differ in terms of side effects, usual doses, metabolism, and duration of action (Tables 19-6 and 19-7).



Mechanism of Action
Calcium channel blockers bind to receptors on voltage-gated calcium ion channels (L, long-lasting; N, neural; and T, transient opening subtypes) resulting in maintenance of these channels in an inactive (closed) state. As a result, calcium influx is decreased and there is a reduction in intracellular calcium. The L-type channel (slow channel) has five subunits: α1, α2, β, γ, and δ. The α1 subunit forms the central part of the channel and provides the main pathway for calcium ion entry into cells. All clinically used calcium channel blockers bind to a unique site on the α1 subunit of the L-type calcium channels and thus diminish entry of calcium ions into cells. These structurally diverse drugs differ in their tissue selectivity, their binding site location on the α1 subunit, and their mechanism of calcium blockade.
Voltage-gated calcium ion channels are present in the cell membranes of skeletal muscle, vascular smooth muscle, cardiac muscle, mesenteric muscle, glandular cells, and neurons (Fig. 19-10).66 Calcium ion influx through L-type calcium channels is responsible for phase 2 of the cardiac action potential, which is important in excitation/contraction coupling in cardiac and vascular smooth muscle and depolarization in sinoatrial and atrioventricular nodal tissue. Thus, blockade of slow calcium channels by calcium channel blockers predictably results in slowing of the heart rate, reduction in myocardial contractility, decreased speed of conduction of cardiac impulses through the atrioventricular node, and vascular smooth muscle relaxation.67

Direct activation of the vascular smooth muscle cell voltage-gated channels by neural impulses initiates an action potential, calcium ion influx, and myofilament contraction (see Fig. 19-10).66 This process is known as excitation-contraction coupling. The intracellular calcium combines with calmodulin, the calcium-binding protein, to form the calcium-calmodulin complex. This complex activates myosin and causes the formation of cross-bridges with actin. These cross-bridges begin the process of muscular contraction.
Pharmacologic Effects
The pharmacologic effects of calcium channel blockers may be predicted by considering the normal role of calcium ions in the production of action potentials, especially in cardiac cells. It is predictable that calcium channel blockers will produce decreased myocardial contractility, decreased heart rate, decreased activity of the sinoatrial node, decreased rate of conduction of cardiac impulses through the atrioventricular node, and vascular smooth muscle relaxation with associated vasodilation and decreases in systemic blood pressure.67,68 Calcium channel blockers produce these effects to varying degrees (see Table 19-6).
All of the calcium channel blockers are effective for the treatment of coronary artery spasm. Calcium channel blockers decrease vascular smooth muscle contractility, thereby increasing coronary blood flow and causing peripheral vasodilation with reductions in systemic vascular resistance and systemic blood pressure. These drug-induced responses contribute to the antiischemic effects characteristic of calcium channel blockers. Because calcium channel blockers dilate the coronary arteries via a mechanism that is different from that of nitrates, the two classes of drugs complement each other in the treatment of coronary artery spasm. Calcium channel blockers are also effective for the treatment of chronic stable angina pectoris caused by fixed obstructive coronary artery lesions and for the treatment of unstable angina pectoris.
All calcium channel blockers exert negative inotropic effects, which are most significant with verapamil and diltiazem.
Phenylalkylamines
The phenylalkylamines bind to the intracellular portion of the L-type channel α1 subunit when the channel is in an open state and conceptually occlude the channel (Fig. 19-11).66

Verapamil
Verapamil is a synthetic derivative of papaverine that is supplied as a racemic mixture. The dextroisomer of verapamil is devoid of activity at slow calcium channels and instead acts on fast sodium channels, accounting for the local anesthetic effects of verapamil (1.6 times as potent as procaine).69 The levoisomer of verapamil is specific for slow calcium channels, and the predominance of this action accounts for the classification of verapamil as a calcium-blocking drug.
Side Effects
Verapamil has a major depressant effect on the atrioventricular node, a negative chronotropic effect on the sinoatrial node, a negative inotropic effect on cardiac muscle, and a moderate vasodilating effect on coronary and systemic arteries. The negative inotropic effects of verapamil seem to be exaggerated in patients with preexisting left ventricular dysfunction. For these reasons, verapamil should not be administered to patients in heart failure or patients with severe bradycardia, sinus node dysfunction, or atrioventricular nodal block. Likewise, this drug’s negative inotropic and chronotropic effects may be enhanced in the presence of concomitant treatment with β-adrenergic antagonists. Isoproterenol may be useful to increase heart rate in the presence of drug-induced heart block. Verapamil may precipitate ventricular dysrhythmias in patients with Wolff-Parkinson-White syndrome.
Clinical Uses
Verapamil is effective in the treatment of supraventricular tachydysrhythmias, reflecting its primary site of action on the atrioventricular node (see Chapter 21). The mild vasodilating effects produced by verapamil make this drug useful in the treatment of vasospastic angina pectoris and essential hypertension. Indeed, calcium channel blockers are as effective as β blockers in relieving angina pectoris. Verapamil is not as active as nifedipine in its effects on vascular smooth muscle and therefore causes a less pronounced decrease in systemic blood pressure and less reflex peripheral sympathetic nervous system activity. Verapamil is effective in the treatment of symptomatic hypertrophic cardiomyopathy with or without left ventricular outflow obstruction.70 Calcium channel antagonists should not be routinely administered for acute myocardial infarction as postinfarction mortality is not decreased.71
Verapamil may be useful in the treatment of maternal and fetal tachydysrhythmias as well as premature labor.72 Administered IV to parturients, verapamil prolongs atrioventricular conduction of the fetus despite limited placental transport of the drug. Fetal hepatic extraction of verapamil is substantial, as evidenced by a plasma concentration in the fetal carotid artery that is less than that in the umbilical vein. Verapamil may decrease uterine blood flow, suggesting caution in the administration of this drug to parturients with impaired uteroplacental perfusion.73
Pharmacokinetics
Oral verapamil is almost completely absorbed, but extensive hepatic first-pass metabolism limits bioavailability to 10% to 20% (see Table 19-7). As a result, the oral dose (80 to 160 mg three times daily) is about 10 times the IV dose. The therapeutic plasma concentration of verapamil is 100 to 300 ng/mL. The activity of oral verapamil peaks in 30 to 45 minutes compared with about 15 minutes following IV administration. Pharmacologic effects following IV administration of verapamil appear within 2 to 3 minutes and may last 6 hours.
Demethylated metabolites of verapamil predominate, with norverapamil possessing sufficient activity to contribute to the antidysrhythmic properties of the parent drug. In view of the nearly complete hepatic metabolism of verapamil, almost none of the drug appears unchanged in the urine. Conversely, an estimated 70% of an injected dose of verapamil is recovered in urine as metabolites and about 15% is excreted via bile. Chronic oral administration of verapamil or the presence of renal dysfunction leads to the accumulation of norverapamil.
The elimination half-time of verapamil is 6 to 12 hours, and this may be prolonged in patients with liver disease. In this regard, chronic treatment with verapamil has rarely been associated with increased plasma concentrations of transaminase enzymes. Like nifedipine, verapamil is highly protein bound (90%), and the presence of other drugs (lidocaine, diazepam, propranolol) can increase the pharmacologically active, unbound portion of the drug.
Dihydropyridines
The dihydropyridines prevent calcium entry into the vascular smooth cells by extracellular allosteric modulation of the L-type voltage-gated calcium ion channels (see Fig. 19-11).66 The primary affinity of the dihydropyridines nifedipine, nicardipine, isradipine, felodipine, and amlodipine is for the peripheral arterioles, whereas nimodipine favors cerebral vessels. The vasodilating effects of these drugs on venous capacitance vessels are minimal. As with other peripheral vasodilators, a reflex tachycardia attributed to sympathetic nervous system activity or baroreceptor reflexes may be observed with the acute administration of dihydropyridines.
Nifedipine
Nifedipine is a dihydropyridine derivative with greater coronary and peripheral arterial vasodilator properties than verapamil. There is minimal effect on venous capacitance vessels. Unlike verapamil, nifedipine has little or no direct depressant effect on sinoatrial or atrioventricular node activity. Peripheral vasodilation and the resulting decrease in systemic blood pressure produced by nifedipine activate baroreceptors, leading to increased peripheral sympathetic nervous system activity most often manifesting as an increased heart rate. This increased sympathetic nervous system activity counters the direct negative inotropic, chronotropic, and dromotropic effects of nifedipine. Nevertheless, nifedipine may produce excessive myocardial depression, especially in patients with preexisting left ventricular dysfunction or concomitant therapy with a β-adrenergic antagonist drug. The presence of aortic stenosis may also exaggerate the cardiac depressant effects of nifedipine.
Clinical Uses
Nifedipine is administered orally with a 10- to 30-mg dose producing an effect in about 20 minutes with a peak effect between 60 and 90 minutes. Nifedipine is used to treat patients with angina pectoris, especially that due to coronary artery vasospasm.
Pharmacokinetics
Absorption of an oral or sublingual dose of nifedipine is about 90%, with onset of an effect being detectable within about 20 minutes after administration (see Table 19-7). It is likely that most of the absorption of sublingual nifedipine is via the gastrointestinal tract from swallowed saliva. Protein binding approaches 90%. Hepatic metabolism is nearly complete, with elimination of inactive metabolites principally in urine (about 80%) and, to a lesser extent, in bile. The elimination half-time is 3 to 7 hours.
Side Effects
The side effects of nifedipine include flushing, vertigo, and headache. Less common side effects include peripheral edema (venodilation), hypotension, paresthesias, and skeletal muscle weakness. Glucose intolerance and hepatic dysfunction occur rarely. Nifedipine may induce renal dysfunction. Abrupt discontinuation of nifedipine has been associated with coronary artery vasospasm.
Nicardipine
Nicardipine lacks effects on the sinoatrial node and atrioventricular node and has minimal myocardial depressant effects. This drug has the greatest vasodilating effects of all the calcium entry blockers, with vasodilation being particularly prominent in the coronary arteries. Combination with a β-adrenergic antagonist for the treatment of angina is a consideration, as dihydropyridine calcium channel blockers do not significantly depress the sinoatrial node. Of all the antianginal drugs, the dihydropyridine calcium channel blockers produce the greatest dilatation of the peripheral arterioles. Therefore, either nifedipine or nicardipine may be particularly useful in patients who have residual hypertension despite β-adrenergic blockade.
Nicardipine is available in IV and oral preparations (other dihydropyridine calcium channel blockers are available only as oral preparations). A long elimination half-time is the basis for the recommendation that about 72 hours should elapse before increasing the oral dose. Nicardipine is metabolized in the liver and is highly protein bound (about 95%). The side effects of nicardipine are similar to nifedipine.
Clinical Uses
Nicardipine is used as a tocolytic drug having a similar tocolytic effect as salbutamol but with fewer side effects. When nicardipine is administered as a tocolytic, it binds to the inside of myometrial L-type voltage-dependent calcium ion channels, causing them to remain closed, and thus inhibits uterine contractility. Pulmonary edema associated with salbutamol used as a tocolytic has also been reported in a parturient treated with nicardipine.74 Nicardipine, 40 µg/kg IV administered immediately before performing electroconvulsive therapy is effective in blunting acute hemodynamic responses to the treatment.75
Nimodipine
Nimodipine is a highly lipid-soluble analogue of nifedipine. Lipid solubility facilitates its entrance into the CNS, where it blocks the influx of extracellular calcium ions necessary for contraction of large cerebral arteries.
Clinical Uses
The lipid solubility of nimodipine and its ability to cross the blood–brain barrier is responsible for the potential value of this drug in treating patients with subarachnoid hemorrhage.
Cerebral Vasospasm
The vasodilating effect of nimodipine on cerebral arteries is uniquely valuable in preventing or attenuating cerebral vasospasm that often accompanies subarachnoid hemorrhage.76 The initial event in the development of vasospasm may be an intracellular influx of calcium ions that cause contraction of smooth muscle cells in large cerebral arteries. Administration of nimodipine, 0.7 mg/kg orally as an initial dose followed by 0.35 mg/kg every 4 hours for 21 days, is associated with a decreased incidence of neurologic deficits due to cerebral vasospasm in patients who had experienced subarachnoid hemorrhage.77Blood and cerebrospinal fluid levels of nimodipine with this dosing regimen were 6.9 ng/mL and 0.77 ng/mL, respectively. In comatose patients who cannot take oral medications, the recommendation is to extract the contents of the nimodipine capsule into a syringe and administer the drug into a nasogastric tube. To ensure delivery of the drug into the stomach via a nasogastric tube, it is necessary to add up to 30 mL of saline to the nimodipine-containing solution.78 Side effects have not been observed with the oral administration of nimodipine. Symptoms of excessive nimodipine effect would be expected to be related to cardiovascular effects such as peripheral vasodilation with associated systemic hypotension. Theoretically, drug-induced cerebral vasodilation could evoke increases in intracranial pressure, particularly in patients with preexisting decreases in intracranial compliance.
Cerebral Protection
Nimodipine has also been evaluated for cerebral protection after global ischemia as associated with cardiac arrest. The theoretical basis for considering calcium channel blockers for this purpose is the observation that lack of oxygen interferes with maintenance of the normal calcium ion gradient across cell membranes, leading to a massive increase (at least 200-fold) in the intraneuronal concentrations of this ion. In this regard, nimodipine is associated with improved neurologic outcome when administered to primates within 5 minutes after experiencing 17 minutes of cerebral ischemia.79 The dose of nimodipine used (10 µg/kg IV followed by 1 µg/kg/minute IV) was associated with decreases in blood pressure that responded to infusion of fluids and/or dopamine.
Amlodipine
Amlodipine is a dihydropyridine derivative that is available for only oral administration (5 to 10 mg) resulting in a peak plasma concentration in 6 to 12 hours. Elimination half-time is 30 to 40 hours and about 90% of the drug undergoes hepatic metabolism to inactive products. Amlodipine appears to have minimal detrimental effects on myocardial contractility and provides antiischemic effects comparable to β blockers in patients with acute coronary syndrome.71 The combination of amlodipine and β blockers may be more effective in the treatment of myocardial ischemia than either drug alone.
Benzothiazepines
Benzothiazepines act at the L-type channel α1 subunit, although the mechanism of action is not well understood (see Fig. 19-11).66 The benzothiazepine diltiazem may have two additional effects: It may act on the sodium–potassium pump, decreasing the amount of intracellular sodium available for exchange with extracellular calcium, and it may inhibit calcium-calmodulin binding.
Diltiazem
Diltiazem, like verapamil, blocks predominantly the calcium channels of the atrioventricular node and is therefore a first-line medication for the treatment of supraventricular tachydysrhythmias (see Chapter 21). It may also be used for the chronic control of essential hypertension. The effects of diltiazem on the sinoatrial and atrioventricular nodes and its vasodilating properties appear to be intermediate between those of verapamil and the dihydropyridines. Diltiazem exerts minimal cardiodepressant effects and is unlikely to interact with β-adrenergic blocking drugs to decrease myocardial contractility.43
Clinical Uses
The clinical use and drug interactions for diltiazem are similar to those of verapamil. Diltiazem is available as an oral capsule and can also be administered IV, especially for the management of angina pectoris. The recommended IV dose is 0.25 to 0.35 mg/kg over 2 minutes and is repeated in 15 minutes, if needed. After the initial IV dose, diltiazem can be given by continuous infusion of about 10 mg per hour for up to 24 hours.
Pharmacokinetics
Oral absorption of diltiazem is excellent with an onset of action in 15 minutes and a peak effect in about 30 minutes (see Table 19-7). The drug is 70% to 80% bound to proteins and is excreted as inactive metabolites principally in bile (about 60%) and, to a lesser extent, in urine (about 35%). Active metabolites of diltiazem include desacetyldiltiazem and desmethyldiltiazem. The elimination half-time for the parent drug is 4 to 6 hours and about 20 hours for metabolites. As with verapamil, liver disease may necessitate a decrease in the dosage of diltiazem.
Drug Interactions
The known pharmacologic effects of calcium channel blockers on cardiac, skeletal, and vascular smooth muscle, as well as on the conduction velocity of cardiac impulses, make drug interactions possible.65Verapamil and diltiazem have depressant effects on the generation of cardiac action potentials at the sinoatrial node and slow the movement of cardiac impulses through the atrioventricular node. Therefore, patients with preexisting cardiac conduction abnormalities may experience greater degrees of atrioventricular heart block with concurrent administration of β blockers or digoxin. Myocardial depression and peripheral vasodilation produced by volatile anesthetics could be exaggerated by similar actions of calcium channel blockers. Vasodilating effects of calcium channel blockers could result in exaggerated systemic hypotension should these drugs be administered to hypovolemic patients.
The likelihood of adverse circulatory changes due to interactions between calcium channel blockers and anesthetic drugs would seem to be greater in patients with preexisting atrioventricular heart block or left ventricular dysfunction. Nevertheless, treatment with calcium channel blockers can be continued until the time of surgery without risk of significant drug interactions, especially with respect to conduction of cardiac impulses.80 Toxicity reflecting an overdose of calcium channel blockers may be partially reversed with IV administration of calcium or dopamine.
Anesthetic Drugs
Calcium channel blockers are vasodilators and myocardial depressants. In fact, the negative inotropic effects, depressant effects on sinoatrial node function, and peripheral vasodilating effects of these drugs and those of volatile anesthetics are similar, and there is evidence that volatile anesthetics have blocking effects on calcium channels.81 For these reasons, calcium channel blockers must be administered with caution to patients with impaired left ventricular function or hypovolemia. Patients treated with a combination of β-adrenergic blockers and nifedipine tolerate high-dose fentanyl anesthesia and do not show evidence of additive depression of cardiac function when verapamil is infused.82 Conversely, in anesthetized patients with preexisting left ventricular dysfunction, administration of verapamil is associated with myocardial depression and decreased cardiac output.83 Furthermore, IV administration of verapamil or diltiazem during open chest surgery in patients with depressed ventricular function and anesthetized with a volatile anesthetic may be associated with further decreases in ventricular function.81
Treatment of cardiac dysrhythmias with calcium channel blockers in anesthetized patients produces only transient decreases in systemic blood pressure and infrequent prolongation of the P-R interval on the electrocardiogram. Because of the tendency to produce atrioventricular heart block, verapamil should be used cautiously in patients being treated with digitalis or β-adrenergic blocking drugs. Nevertheless, in patients with preoperative evidence of cardiac conduction abnormalities, the chronic combined administration of calcium channel blockers and β-adrenergic antagonists is not associated with cardiac conduction abnormalities in the perioperative period (Table 19-8).80 β-Adrenergic agonists increase the number of functioning slow calcium channels in myocardial cell membranes through a cAMP mechanism and readily counter the effects of calcium channel blockers. Nevertheless, there is no evidence that patients being treated chronically with calcium channel blockers are at increased risk for anesthesia.

Neuromuscular Blocking Drugs
Calcium channel blockers alone do not produce a skeletal muscle relaxant effect (Fig. 19-12).84 Conversely, these drugs potentiate the effects of depolarizing and nondepolarizing neuromuscular blocking drugs (see Fig. 19-12).84,85This potentiation resembles that produced by mycin antibiotics in the presence of neuromuscular blocking drugs. The local anesthetic effects of verapamil and diltiazem, reflecting inhibition of sodium ion flux via fast sodium channels, may also contribute to the potentiation of neuromuscular blocking drugs. Observations of skeletal muscle weakness after administration of verapamil to a patient with muscular dystrophy are inconsistent with diminished release of neurotransmitter.86 Therefore, the neuromuscular effects of verapamil may be more likely to manifest in patients with a compromised margin of safety of neuromuscular transmission.

Antagonism of neuromuscular blockade may be impaired because of diminished presynaptic release of acetylcholine in the presence of a calcium channel blocker.87 Indeed, calcium ions are necessary for the release of acetylcholine at the neuromuscular junction. In one report, edrophonium but not neostigmine was effective in antagonizing nondepolarizing neuromuscular blockade that was potentiated by verapamil.88
Local Anesthetics
Verapamil and diltiazem have potent local anesthetic activity, which may increase the risk of local anesthetic toxicity when regional anesthesia is administered to patients being treated with this drug.89
Potassium-Containing Solutions
Calcium channel blockers slow the inward movement of potassium ions. For this reason, hyperkalemia in patients being treated with verapamil may occur after much smaller amounts of exogenous potassium infusion as associated with the use of potassium chloride to treat hypokalemia or administration of stored whole blood.90 In animals, however, pretreatment with verapamil does not alter the increases in plasma potassium concentrations that follow the administration of succinylcholine.91
Dantrolene
The ability of both verapamil and dantrolene to inhibit intracellular calcium ion flux and excitation-contraction coupling would suggest this combination might be useful in the treatment of malignant hyperthermia. In swine, however, the administration of dantrolene in the presence of verapamil or diltiazem results in hyperkalemia and cardiovascular collapse (Fig. 19-13).92 A patient receiving verapamil developed hyperkalemia and myocardial depression within 1.5 hours of being treated with dantrolene administered IV.93 This same patient did not experience hyperkalemia when nifedipine was substituted for verapamil before pretreatment with dantrolene.

Whenever calcium channel blockers, especially verapamil or diltiazem, and dantrolene must be administered concurrently, invasive hemodynamic monitoring and frequent measurement of the plasma potassium concentration are recommended. It is speculated that verapamil alters normal homeostatic mechanisms for regulation of plasma potassium concentrations and may result in hyperkalemia from dantrolene-induced potassium release. Furthermore, there is evidence that verapamil does not influence the ability of known triggering drugs to evoke malignant hyperthermia in susceptible animals.94
Platelet Function
Calcium channel blockers may interfere with calcium-mediated platelet function.
Digoxin
Calcium channel blockers may increase the plasma concentration of digoxin, presumably by decreasing its plasma clearance.
H2 Antagonists
Cimetidine and ranitidine by altering hepatic enzyme activity and/or hepatic blood flow may increase the plasma concentrations of calcium channel blockers.
Risks of Chronic Treatment
Despite the popularity of calcium channel blockers in the treatment of cardiovascular diseases (essential hypertension, angina pectoris), there is increasing concern about the long-term safety of these drugs, especially the short-acting dihydropyridine derivatives.95 For example, the risk of developing cardiovascular complications has been described as being greater in patients treated with nifedipine compared with those receiving placebo or conventional therapy.96 Increased perioperative bleeding and an increased incidence of gastrointestinal hemorrhage have been reported in patients receiving a dihydropyridine derivative.97,98 The risk of developing cancer may be increased in those treated with calcium channel blockers compared with β-adrenergic antagonists or angiotensin-converting enzyme inhibitors.99 For these reasons, treatment with calcium channel blockers, especially short-acting dihydropyridine derivatives, should generally be reserved for second-step rather than initial therapy.95
Cytoprotection
Drug-induced calcium channel blockade may provide cytoprotection against ischemic reperfusion injury. By decreasing calcium ion entry into cells and the conversion of xanthine dehydrogenase to xanthine oxidase (dependent on the calcium-calmodulin complex), calcium channel blockers may limit the accumulation of oxygen free radicals. Calcium channel blockers may attenuate renal injury from nephrotoxic drugs such as cisplatinum and iodinated radiographic contrast media. The vasodilator effects of calcium channel blockers and resultant control of systemic hypertension may result in increases in renal blood flow and glomerular filtration rate, thus favoring natriuresis.
References
1. Foex P. A- and β-adrenoceptor antagonists. Br J Anaesth. 1984;56:751–765.
2. Aplin SC, Yee KF, Cole MJ. Neonatal effects of long-term maternal phenoxybenzamine therapy. Anesthesiology. 2004;100:1608–1610.
3. Shand DG. Drug therapy—propranolol. N Engl J Med. 1975;293:280–285.
4. Wood M, Shand DG, Wood AJ. Propranolol binding in plasma during cardiopulmonary bypass. Anesthesiology. 1979;51:512–516.
5. Bowdle TA, Freund PR, Slattery JT. Propranolol reduces bupivacaine clearance. Anesthesiology. 1987;66:36–38.
6. Conrad KA, Beyers JM, Finley PR, et al. Lidocaine elimination: effects of metoprolol and propranolol. Clin Pharmacol Ther. 1983;33:133–138.
7. Roerig DL, Kotryl KJ, Ahlf SB, et al. Effect of propranolol on the first pass uptake of fentanyl in the human and rat lung. Anesthesiology. 1989;71:62–68.
8. Mishra P, Calvey TN, Williams NE, et al. Intraoperative bradycardia and hypotension associated with timolol and pilocarpine eye drops. Br J Anaesth. 1983;55:897–899.
9. Bailey PL. Timolol and postoperative apnea in neonates and young infants. Anesthesiology. 1984;61:622.
10. Mangano DT, Layug EL, Wallace A, et al. Effect of atenolol on mortality and cardiovascular morbidity after noncardiac surgery. N Engl J Med. 1996;335:1713–1720.
11. Wallace A, Layug B, Tateo I, et al. Prophylactic atenolol reduces postoperative myocardial ischemia. Anesthesiology. 1998;88:7–17.
12. Nuttall SL, Routledge HC, Kendall MJ. A comparison of the beta1-selectivity of three beta1-selective beta-blockers. J Clin Pharm Ther. 2003:28(3):179–186.
13. Bundkirchen A, Brixius K, Bölck B, et al. Beta1-adrenoceptor selectivity of nebivolol and bisoprolol. A comparison of [3H]CGP 12.177 and [125l]iodocyanopindolol binding studies. Eur J of Pharmacol. 2003:460(1):19–26.
14. Helfman SM, Gold MI, DeLisser EA, et al. Which drug prevents tachycardia and hypertension associated with tracheal intubation: lidocaine, fentanyl, or esmolol? Anesth Analg. 1991;72:482–486.
15. Oxorn D, Knox JWD, Hill J. Bolus doses of esmolol for the prevention of perioperative hypertension and tachycardia. Can J Anaesth. 1990;37:206–209.
16. Sheppard S, Eagle CJ, Strunin L. A bolus dose of esmolol attenuates tachycardia and hypertension after tracheal intubation. Can J Anaesth. 1990;37:202–205.
17. Howie MB, Black HA, Zvara D, et al. Esmolol reduces autonomic hypersensitivity and length of seizures induced by electroconvulsive therapy. Anesth Analg. 1990;71:384–388.
18. Nicholas E, Deutschman CS, Allo M, et al. Use of esmolol in the intraoperative management of pheochromocytoma. Anesth Analg. 1988;67:1114–1117.
19. Ostman PL, Chestnut DH, Robillard JE, et al. Transplacental passage and hemodynamic effects of esmolol in the gravid ewe. Anesthesiology. 1988;69:738–741.
20. Pollan S, Tadjziechy M. Esmolol in the management of epinephrine and cocaine-induced cardiovascular toxicity. Anesth Analg. 1989;69:663–664.
21. Thorne AC, Bedford RF. Esmolol for perioperative management of thyrotoxic goiter. Anesthesiology. 1989;71:291–294.
22. Zakowski M, Kaufman B, Berguson P, et al. Esmolol use during resection of pheochromocytoma: report of three cases. Anesthesiology. 1989;20:875–877.
23. Gourdine SB, Hollinger I, Jones J, et al. New York state guidelines on the topical use of phenylephrine in the operating room. Anesthesiology. 2000;92:859–864.
24. Ooi LG, O’Shea PJ, Wood AJ. Use of esmolol in the postbypass management of hypertrophic obstructive cardiomyopathy. Br J Anaesth. 1993;70:104–106.
25. de Bruijn NP, Croughwell N, Reves JG. Hemodynamic effects of esmolol in chronically β-blocked patients undergoing aortocoronary bypass surgery. Anesth Analg. 1987;66:137–141.
26. Cork RC, Kramer TH, Dreischmeier B, et al. The effect of esmolol given during cardiopulmonary bypass. Anesth Analg. 1995;80:28–40.
27. Johansen JW, Flaishon R, Sebel PS. Esmolol reduces anesthetic requirement for skin incision during propofol/nitrous oxide/morphine anesthesia. Anesthesiology. 1997;86:364–371.
28. Hall RI. Esmolol—just another β blocker? Can J Anaesth. 1992;39:757–764.
29. McCammon RL, Hilgenberg JC, Sandage BW, et al. The effect of esmolol on the onset and duration of succinylcholine-induced neuromuscular blockade. Anesthesiology. 1985;63:A317.
30. Brown DR, Brown MJ. Hypoglycemia associated with preoperative metoprolol administration. Anesth Analg. 2004;99:1427–1428.
31. Ramsey JG. Esmolol. Can J Anaesth. 1991;38:155–158.
32. Nies AS, Shand DG. Clinical pharmacology of propranolol. Circulation. 1975;52:6–15.
33. DeLima LGR, Kharasch ED, Butler S. Successful pharmacologic treatment of massive atenolol overdose: sequential hemodynamics and plasma atenolol concentrations. Anesthesiology. 1995;83:204–207.
34. Rosa RM, Silva P, Young JB, et al. Adrenergic modulation of extrarenal potassium disposal. N Engl J Med. 1980;302:431–434.
35. McCammon RL, Stoelting RK. Exaggerated increase in serum potassium following succinylcholine in dogs with β blockade. Anesthesiology. 1984;61:723–725.
36. Philbin DM, Lowenstein E. Lack of β-adrenergic activity of isoflurane in the dog: a comparison of circulatory effects of halothane and isoflurane after propranolol administration. Br J Anaesth. 1976;48:1165–1170.
37. Horan BF, Prys-Roberts C, Hamilton WK, et al. Haemodynamic responses to enflurane anaesthesia and hypovolaemia in the dog, and their modification by propranolol. Br J Anaesth. 1977;49:1189–1197.
38. Roberts JG, Foex P, Clarke TNS, et al. Haemodynamic interactions of high-dose propranolol pretreatment and anaesthesia in the dog. III. The effects of haemorrhage during halothane and trichloroethylene anaesthesia. Br J Anaesth. 1976;48:411–418.
39. Horan BF, Prys-Roberts C, Roberts JG, et al. Haemodynamic responses to isoflurane anaesthesia and hypovolaemia in the dog, and their modification by propranolol. Br J Anaesth. 1977;49:1179–1187.
40. Foex P, Ryder WA. Interactions of adrenergic β-receptor blockade (oxprenolol) and PCO2 in the anesthetized dog: influence of intrinsic sympathomimetic activity. Br J Anaesth. 1981;53:19–26.
41. Bright RA, Everitt DE. β-blockers and depression: evidence against an association. JAMA. 1992;267:1783–1787.
42. Smulyan H, Weinberg SE, Howanitz PJ. Continuous propranolol infusion following abdominal surgery. JAMA. 1982;247:2539–2542.
43. Packer M. Combined β-adrenergic and calcium-entry blockade in angina pectoris. N Engl J Med. 1989;320:709–717.
44. Brandler E, Paladino L, Sinert R. Does the early administration of beta-blockers improve the in-hospital mortality rate of patients admitted with acute coronary syndrome? Acad Emerg Med. 2010:17(1)1–10.
45. Chen ZM, Pan HC, Chen YP, et al. Early intravenous then oral metoprolol in 45,852 patients with acute myocardial infarction: randomized placebo-controlled trial. Lancet. 2005: 366:1622–1632.
46. The TIMI Study Group. Immediate versus delayed catheterization and angioplasty following thrombolytic therapy for acute myocardial infarction. JAMA. 1988;260:2849–2856.
47. London MJ. Beta-adrenergic-receptor blockade and myocardial ischemia: something old, something new. J Cardiothorac Vasc Anesth. 2002;16(6):667–669.
48. Shjojania KG, Duncan BW, McDonald KM, et al. Safe but sound: patient safety meets evidence-based medicine. JAMA. 2002;288:508–513.
49. Auerbach AD, Goldman L. β-blockers and reduction of cardiac events in noncardiac surgery. JAMA. 2002;287:1435–1444.
50. Eagle KA, Froehlich JB. Reducing cardiovascular risk in patients undergoing noncardiac surgery. N Engl J Med. 1996;335:1761–1763.
51. Devereaux PJ, Yang H, Yusuf S, et al. Effects of extended-release metoprolol succinate in patients undergoing non-cardiac surgery (POISE trial): a randomised controlled trial. Lancet. 2008:371:1839–1847.
52. Hjalmarson A, Goldstein S, Gagerberg B, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF). JAMA. 2000;283:1295–1302.
53. Colucci WS, Packer M, Bristow MR, et al. Carvedilol inhibits clinical progression in patients with mild symptoms of heart failure. Circulation. 1996;94:1199–1205.
54. Prys-Roberts C, Foex P, Biro GP, et al. Studies of anaesthesia in relation to hypertension. V. Adrenergic β-receptor blockade. Br J Anaesth. 1973;45:671–681.
55. Lee TC, Coffey RJ, Currier BM, et al. Propranolol and thyroidectomy in the treatment of thyrotoxicosis. Ann Surg. 1982;195:766–772.
56. Lennquits S, Jortso E, Anderberg B, et al. β blockers compared with antithyroid drug as preoperative treatment in hyperthyroidism: drug tolerance, complications, and postoperative thyroid function. Surgery. 1985;98:1141–1146.
57. MacCarthy EP, Bloomfield SS. Labetalol: a review of its pharmacology, pharmacokinetics, clinical uses and adverse effects. Pharmacotherapy. 1983;3:193–219.
58. Wallin JD, O’Neill WM. Labetalol: current research and therapeutic status. Arch Intern Med. 1983;143:485–490.
59. Baum T, Watkins RW, Sybertz EJ, et al. Antihypertensive and hemodynamic actions of SCH 19927, the R,R-isomer of labetalol. J Pharmacol Exp Ther. 1981;218:441–452.
60. Larsen LS. Labetalol in the treatment of epinephrine overdose. Ann Emerg Med. 1990;19:680–682.
61. Lebel M, Langlois S, Belleau LJ, et al. Labetalol infusion in hypertensive emergencies. Clin Pharmacol Ther. 1985;37:615–618.
62. Wilson DJ, Wallin JD, Vlachakis ND, et al. Intravenous labetalol in the treatment of severe hypertensive and hypertensive emergencies. Am J Med. 1983;75:95–102.
63. Goldberg ME, McNulty SE, Azad SS, et al. A comparison of labetalol and nitroprusside for inducing hypotension during major surgery. Anesth Analg. 1990;70:537–542.
64. Durand PG, Lehot JJ, Foex P. Calcium-channel blockers and anaesthesia. Can J Anaesth. 1991;38:75–89.
65. Kaplan NM. Calcium entry blockers in the treatment of hypertension: current status and future prospects. N Engl J Med. 1989;262:817–823.
66. Kanneganti M, Halpern NA. Acute hypertension and calcium-channel blockers. New Horiz. 1996;4:19–25.
67. Reves JG. The relative hemodynamic effects of calcium entry blockers. Anesthesiology. 1984;61:3–5.
68. Reves JG, Kissin I, Lell WA, et al. Calcium entry blockers: uses and implications for anesthesiologists. Anesthesiology. 1982;57:504–518.
69. Kraynack BJ, Lawson NW, Gintautas J. Local anesthetic effect of verapamil in vitro. Reg Anesth. 1982;7:114–117.
70. Spirito P, Seidman CE, McKenna WJ, et al. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–780.
71. Opie LH. First line drugs in chronic stable effort angina: the case for newer, longer-acting calcium channel blocking drugs. J Am Coll Cardiol. 2000;36:1967–1971.
72. Murad SHN, Tabsh KMA, Conklin KA, et al. Verapamil: placental transfer and effects on maternal and fetal hemodynamics and atrioventricular conduction in the pregnant ewe. Anesthesiology. 1985;62:49–53.
73. Murad SHN, Tabsh KMA, Shilyanski G, et al. Effects of verapamil on uterine blood flow and maternal cardiovascular function in the awake pregnant ewe. Anesth Analg. 1985;64:7–10.
74. Bal L, Thierry S, Brocas E, et al. Pulmonary edema induced by calcium-channel blockade for tocolysis. Anesth Analg. 2004;99:910–911.
75. Zhang Y, White PF, Thornton L, et al. The use of nicardipine for electroconvulsive therapy: a dose-ranging study. Anesth Analg. 2005;100(2):378–381.
76. Verma A. Opportunities for neuroprotection in traumatic brain injury. J Head Trauma Rehabil. 2000;15:1149–1156.
77. Allen GS, Ahn HS, Preziosi TJ, et al. Cerebral arterial spasm: a controlled trial of nimodipine in patients with subarachnoid hemorrhage. N Engl J Med. 1983;308:619–624.
78. Gelmers HJ, Gorter K, deWeerdt CJ, et al. A controlled trial of nimodipine in acute ischemic stroke. N Engl J Med. 1988;318:303–307.
79. Steen PA, Gisvold SE, Milde JH, et al. Nimodipine improves outcome when given after complete cerebral ischemia in primates. Anesthesiology. 1985;62:406–414.
80. Henling CE, Slogoff S, Kodali SV, et al. Heart block after coronary artery bypass—effect of chronic administration of calcium-entry blockers and β-blockers. Anesth Analg. 1984;63:515–520.
81. Merin RG. Calcium channel blocking drugs and anesthetics: is the drug interaction beneficial or detrimental? Anesthesiology. 1987;66:111–113.
82. Kapur PA, Norel EJ, Dajee H, et al. Hemodynamic effects of verapamil administration after large doses of fentanyl in man. Can Anaesth Soc J. 1986;33:138–144.
83. Chew CYC, Hecht HS, Collett JT, et al. Influence of severity of ventricular dysfunction on hemodynamic responses to intravenously administered verapamil in ischemic heart disease. Am J Cardiol. 1981;47:917–922.
84. Durant NN, Nguyen N, Katz RL. Potentiation of neuromuscular blockade by verapamil. Anesthesiology. 1984;60:298–303.
85. van Poorten JE, Chasmana KM, Kuypers SM, et al. Verapamil and reversal of vecuronium neuromuscular blockade. Anesth Analg. 1984;63:155–157.
86. Zalman F, Perloff JK, Durant NW, et al. Acute respiratory failure following intravenous verapamil in Duchenne’s muscular dystrophy. Am Heart J. 1983;105:510–511.
87. Lawson NW, Kraynack BJ, Gintautas J. Neuromuscular and electrocardiographic responses to verapamil in dogs. Anesth Analg. 1983;62:50–54.
88. Jones RM, Cashman JN, Casson WR, et al. Verapamil potentiation of neuromuscular blockade: failure of reversal with neostigmine but prompt reversal with edrophonium. Anesth Analg. 1985;64:1021–1025.
89. Rosenblatt RM, Weaver JM, Want Y, et al. Verapamil potentiates the toxicity of local anesthetics. Anesth Analg. 1984;63:269.
90. Nugent M, Tinker JH, Moyer TP. Verapamil worsens rate of development and hemodynamic effects of acute hyperkalemia in halothane-anesthetized dogs. Effects of calcium therapy. Anesthesiology. 1984;60:435–439.
91. Roth JL, Nugent M, Gronert GA. Verapamil does not alter succinylcholine-induced increases in serum potassium during halothane anesthesia in normal dogs. Anesth Analg. 1985;64:1202–1204.
92. Saltzman LS, Kates RA, Corke BC, et al. Hyperkalemia and cardiovascular collapse after verapamil and dantrolene administration in swine. Anesth Analg. 1984;63:473–478.
93. Rubin AS, Zablocki AD. Hyperkalemia, verapamil, and dantrolene. Anesthesiology. 1987;66:246–249.
94. Gallant EM, Foldes FF, Rempel WE, et al. Verapamil is not a therapeutic adjunct to dantrolene in porcine malignant hyperthermia. Anesth Analg. 1985;64:601–606.
95. Chobanian AV. Calcium-channel blockers: lessons learned from MIDAS and other clinical trials. JAMA. 1996;276:829–830.
96. Furberg CD, Posaty BM, Meyer JV. Nifedipine: dose-related increase in mortality in patients with coronary heart disease. Circulation. 1995;92:1326–1331.
97. Pahor M, Guralnik JM, Furberg CD, et al. Risk of gastrointestinal hemorrhage with calcium antagonists in hypertensive patients over 67. Lancet. 1996;347:1061–1066.
98. Wagenknecht LE, Furberg CD, Hammon JW, et al. Surgical bleeding: unexpected effect of a calcium antagonist. BMJ. 1995;310:776–777.
99. Pahor M, Guralnik JM, Salive ME, et al. Do calcium-channel blockers increase the risk of cancer? Am J Hypertens. 1996;9:695–699.