The term resuscitation for many people means resuscitation from cardiac arrest, so much so that the term cardiopulmonary resuscitation and its acronym “CPR” is widely used not only by health care professionals but by laypeople as well. This is understandable as cardiovascular disease is the number one cause of death in the United States, and in 2010, it is estimated that 746,000 Americans died of cardiovascular disease.1 Unfortunately, the first manifestation of cardiovascular disease is often sudden death due to ventricular tachycardia, ventricular fibrillation, or asystole. In order to improve outcome from cardiac arrest, the American College of Cardiology, American Heart Association, and many other organizations have joined together to educate health care professionals and laypeople on how to resuscitate patients who have had a cardiac arrest by promoting the American Heart Association’s Basic Life Support and Advanced Cardiac Life Support courses.
However, resuscitation is also applied to resuscitation from other near-death events. Death from traumatic injury is the third leading cause of death overall in the United States and the primary cause of death in individuals younger than the age of 45 years.2 These individuals most often die because of hemorrhage. As such, the American College of Surgeons Committee on Trauma has developed the Advanced Trauma Life Support course to standardize proven treatment and improve the outcome of patients who have sustained traumatic injury.
For the neonatologist, resuscitation might bring to mind the resuscitation of neonates whose cardiorespiratory function is depressed at birth.3 Primary and secondary apnea is the most common etiology of the need for resuscitation of the neonate and the most common antecedent of cardiac arrest (see Chapter 44, Physiology of the Newborn). In adults, respiratory arrest can precede cardiac arrest and vice versa. If either is not recognized in a timely manner and treated effectively, one will but quickly precipitate the other.
Common to all these scenarios and the actual mechanism by which patients die is inadequate oxygen delivery to tissues. Oxygen delivery is defined as:
![]() o2 = CO × CaO2
o2 = CO × CaO2
where ![]() O2 is equal to oxygen delivery, CO represents cardiac output, and CaO2 is the O2 content of arterial blood.
O2 is equal to oxygen delivery, CO represents cardiac output, and CaO2 is the O2 content of arterial blood.
In a 70-kg person, oxygen delivery is assumed to be 1 L per minute, derived by multiplying cardiac output (5 L per minute) times arterial oxygen content (200 mL O2/L). Arterial oxygen content can be calculated by multiplying 15 g hemoglobin/dL (or 150g/L) times 1.39 (the amount of oxygen each gram of hemoglobin can hold when fully saturated) times the arterial oxygen saturation. For this calculation, we are assuming a hemoglobin oxygen saturation of 100%.
From these equations, one can gain insight into the three mechanisms from which death might occur from cardiac and brain ischemia if the patient is not resuscitated in a timely and effective fashion. (a) During ventricular tachycardia, ventricular fibrillation, or during asystole, cardiac output decreases to zero as will oxygen delivery to the tissues. (b) During hemorrhagic shock, as hemoglobin levels decrease, the arterial oxygen content decreases and cardiac output falls because of decreased intravascular volume resulting in decreased left ventricular end diastolic volume. (c) During apnea from whatever cause, as whatever oxygen is contained in the functional residual capacity of the lung decreases, the amount of oxygen available to bind with hemoglobin decreases, which also results in a steadily decreasing oxygen delivery and eventually, cell death.
For the purposes of this chapter then, the focus will be on resuscitation from cardiac events, traumatic injury, and decreased fractional inspired concentration of oxygen (FIO2).
Pathophysiology
The basic physiology of death due to decreased oxygen delivery is in some ways straightforward but at the same time, quite complex. As oxygenated hemoglobin is delivered to peripheral tissues, O2dissociates from hemoglobin because of the concentration gradient between the oxygenated hemoglobin in red blood cells and the capillary blood perfusing the tissue. The dissociation of O2 from hemoglobin is facilitated by the respiratory acidosis created by CO2 released through cellular respiration. Oxygen diffuses out of red blood cells through the plasma, across cell membranes, and into the cytoplasm where it is taken up by mitochondria. In oxidative metabolism, O2 breaks down pyruvate, derived from glucose, amino acids, and fatty acids in the Krebs cycle. Water and CO2 are the byproducts and the energy produced is used by the coenzymes nicotinamide, adenine, dinucleotide, and flavin adenine dinucleotide to add a phosphate molecule to adenosine diphosphate to create adenosine triphosphate (ATP). The third phosphate bond is readily broken and the energy that is released is used by cells for protein synthesis, for endocytosis and exocytosis, to provide energy for other transporter processes, for maintenance of the electrical potential across the membrane, and to maintain the integrity of the cell membrane itself. Oxygen is the molecule that serves as the electron acceptor by which nicotinamide adenine dinucleotide and flavin adenine dinucleotide produce more ATP than can be created by anaerobic metabolism. Based strictly on the byproducts of the chemical reactions involved, anaerobic metabolism of pyruvate leads to the production of 4 molecules of ATP, whereas the metabolism of pyruvate in the presence of O2 can potentially produce 38 molecules of ATP. Anaerobic metabolism is insufficient in the long term to produce enough ATP molecules to maintain cell integrity. In the absence of oxygen, cells die (Fig. 47-1) at a variable rate depending on their metabolic rate. Typically, neurons are the most sensitive to lack of O2 and will develop irreversible damage within 3 to 5 minutes; myocytes and hepatocytes on the other hand can survive 1 to 2 hours in the absence of O2; muscle cells may survive for several hours. By decreasing metabolic rate, hypothermia can prolong the “safe” ischemic time. Neurons for example decrease their metabolic rate by approximately 7% for every 1°C decrease in temperature.4 There is a limit, however, to how much hypothermia can delay a cell’s demise. For example, during circulatory arrest while a patient is on cardiopulmonary bypass, at 15°C to 20°C, neurons do not display irreversible cell damage for as long as 30 to 60 minutes, but after 60 minutes, the risk for potential irreversible neuronal damage increases markedly. Hypothermia has the same effect on other cells such as myocytes and hepatocytes, as well as cells in other organs. This factor allows transplant surgeons to prolong the amount of time between when an organ is harvested and when it is transplanted, thus improving the matching potential for transplantable organs.
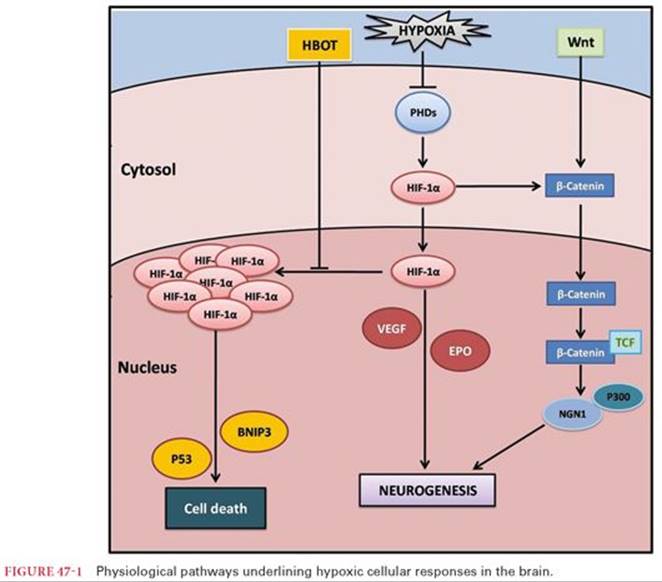
Although temperature has an impact on survivability of ischemic cells, time is the factor that most directly and importantly impacts the reversibility of ischemia—the safe ischemic time. Independent of all other factors, the number of minutes that cardiac function is arrested, the amount of time that the hemoglobin concentration is below 6 to 7 g/dL, or the duration of apnea or asphyxia all directly correlate with morbidity and mortality. For cardiac arrest, evidence from assessment of the effect of duration of the arrest on survivability suggests that mortality increases by 8% to 10% for each minute for which there is no cardiac output.5 From a physiologic perspective, the most vital factor then in determining outcome is the timely restoration of normal cardiac, pulmonary, and hemostatic function. Drugs play a limited, but very important, role in the return of the aforementioned functions in certain circumstances as will be described later.
Cardiac Arrest
The National Library of Medicine in its MeSH (Medical Subject Headings) terms defines “heart arrest” as the cessation of effective cardiac function, which if not recognized and treated within minutes will lead to “sudden cardiac death.” Sudden cardiac death in turn is the unexpected rapid natural death due to cardiovascular collapse within 1 hour of the onset of symptoms usually caused by the worsening of existing heart disease.
Ventricular tachyarrhythmias account for many cardiac arrests; 6% to 14% of the time, no known structural lesions of the heart can be identified.6 If no structural cause can be identified, individuals who have a prolonged QT interval are at increased risk of developing polymorphic ventricular tachycardia, that is, torsades de pointes especially if they receive certain drugs that are also known to prolong the QT. Mutations in genes that code for ion channels that generate the cardiac action potential have been identified in patients with the long QT syndrome.7,8 There are more than 150 drugs that have been reported to prolong the QT interval; list the drugs that are most relevant to anesthesiologists. Drugs that prolong the QT interval are thought to do so by modifying the activity of the same ion channels responsible for the long QT syndrome. The effects of the different drugs range from mild to severe with the latter having the greatest probability of precipitating torsades de pointes.9 The ability of a drug to precipitate a malignant tachyarrhythmia is related not just to the drug itself but also to the dose, to drug interactions, to genetic factors, to the gender of the patient, and to the type and severity of preexisting cardiac disease.
Ventricular fibrillation is the cause for most of the other instances of sudden cardiac arrest with a relatively small proportion of people developing asystole as the initial manifestation of sudden cardiac arrest. The true incidence of the precipitating arrhythmia is not known because most sudden cardiac arrests occur out of hospital and typically, several minutes pass before the rhythm can be assessed. The cardiac diseases that lead to the genesis of ventricular fibrillation resulting in cardiac collapse are varied, and the association with sudden death in some cases is not well understood. Similar to patients who have cardiovascular collapse from ventricular tachyarrhythmias, there are patients who develop ventricular fibrillation who do not have identifiable structural heart disease. There is a correlation between what is normally considered to be benign, early repolarization (defined as an elevation of the QRS–ST segment of at least 0.1 mV in the inferior or lateral leads on the electrocardiogram) and the development of ventricular fibrillation.10,11 As such, early repolarization may be a sign of electrochemical imbalance within the myocardium.
Independent of the etiology of the cardiac collapse, for resuscitation to be successful, CPR must be initiated as soon as possible. Weisfeldt and Becker12 have proposed a three-phase model of resuscitation: the electrical phase, the circulatory phase, and the metabolic phase. The electrical phase begins with the cardiac collapse and lasts for approximately 4 minutes; the circulatory phase represents an approximation of 6 minutes window from 4 to 10 minutes; and the metabolic window which commences at approximately 10 minutes. The best outcomes are achieved if the heart is defibrillated within the first 4 minutes after the cardiac arrest, during the electrical phase. This observation has been borne out by studies of patients who have an implantable cardioverter defibrillator. One meta-analysis of three large such studies that compared the administration of amiodarone to implantation of an implantable cardioverter defibrillator demonstrated decreased arrhythmias and improved survivability in patients who had an internal defibrillator.13 In a canine model, Yakaitis and colleagues14 demonstrated that for defibrillation per se to be successful, the electroshock had to be delivered within 3 minutes of the onset of ventricular fibrillation. Once the circulatory phase has begun, the most important intervention to achieve the return of spontaneous circulation (ROSC) is to commence chest compressions that will deliver some oxygenated blood to tissues. Another animal study showed that once the circulatory phase has begun, a threefold improvement in results is obtained if chest compressions are performed first, along with the administration of epinephrine and then defibrillation attempted compared to performing the defibrillation first and then performing chest compressions.15
Once the patient transitions into the metabolic phase, the chances of ROSC and more importantly, the chances of a full neurologic recovery are quite low—neither immediate countershock nor chest compressions followed by countershock are associated with good outcomes. Investigators have speculated that even with ROSC, 10 minutes or more of global ischemia induces cells to release cytotoxins,16and bacteria are more able to translocate across the intestinal endothelial barrier.17 Cytotoxins and endotoxin may further injure an already damaged heart, and worse, may disrupt mechanisms maintaining cellular integrity.18 As circulation returns, the hyperoxia that is often present will exacerbate the reperfusion syndrome and has been associated with increased mortality.19
In those patients with ROSC, the enthusiasm clinicians had for the ability of hypothermia to attenuate the neurologic injury that many of these patients developed has been tempered by a more recent well-performed study that did not demonstrate an improvement in neurologic outcome with hypothermia (33°C vs. 36°C).20
Once patients enter the metabolic phase after 10 minutes of untreated cardiac arrest, the chances of meaningful recovery are slim. Improvement in outcome in patients who do have ROSC will require development of therapies to attenuate the ischemia-reperfusion syndrome.
Hemorrhagic Shock
In patients who sustain traumatic injury, hemorrhage accounts for 30% to 40% of the deaths. Of these, a majority occur before the patients arrive at a hospital. Among traumatized patients who arrive at the hospital, mortality in the first several hours correlates with inadequate resuscitation and the presence of coagulopathy.21 During the recent combat experiences in Iraq and Afghanistan, particularly during the later years of the combat in Afghanistan when dismounted complex blast injury was recognized, there was a strong correlation between the degree of hemorrhage, the severity of the injury, and the overall mortality.22 However, service members who were alive upon arrival at a level III medical facility had a greater than 95% chance of survival. These results were obtained because of the military’s emphasis on controlling hemorrhage at the point of injury, recognition and treatment of acute traumatic coagulopathy (ATC), and the use of damage control surgery. In both civilians and wounded warriors, it is impossible to quantify the amount of blood that is lost from the intravascular circulation, but an estimate can be made from the amount of blood transfused, the degree of lactic acidosis, and the alterations and vital signs. In an observational study of patients who refused blood transfusions because of religious beliefs, there was no 30-day mortality among patients whose hemoglobin was greater than 7.1 g/dL. However, as the hemoglobin level decreased below 6 g/dL, mortality increased significantly.23
Although the volume of blood lost cannot be accurately measured, the number of units of blood given and changes in coagulation factors are readily recorded and analyzed. Improvements in early hemorrhage control and resuscitation and the prevention and aggressive treatment of coagulopathy appear to have the greatest potential to improve outcomes in severely injured trauma patients. A cascade of life-threatening medical problems can begin with severe hemorrhage, and many of these occur simultaneously: (a) hemorrhage, (b) impaired resuscitation, (c) shock, (d) inflammation, and (e) coagulopathy. The severity of each problem is commonly associated with the extent of overall blood loss. Low blood pressure due to blood loss indicates immediate complications, including the incidence of multiple organ failure and life-threatening infections.
Respiratory Arrest
Respiratory and cardiac arrest are distinct, but if untreated, one inevitably leads to the other. There are a multitude of etiologies of respiratory arrest, and respiratory arrest per se implies any process that inhibits the delivery of sufficient oxygen to the mitochondria to maintain aerobic metabolism. The respiratory centers in the brainstem can be injured by penetrating injury and by increased intracranial pressure due to infection or intraventricular hemorrhage. Even blunt trauma to the head of sufficient force can produce a similar result. Atkinson et al.,24 using a mechanical blow to the head, demonstrated in a rodent model that as the force of the blow incrementally increased, there came a point that the animal became completely apneic. There are a number of drugs that can depress the respiratory centers, most notably opioids, sedatives, and hypnotics. These drugs can suppress ventilation or induce complete apnea even at low doses in patients with central sleep apnea,25 in patients with chronic obstructive pulmonary disease (COPD) and hypercarbia, and in geriatric patients with comorbid conditions. Although it is true that opioids suppress ventilation, some have used this relationship to justify withholding opioids from patients with severe COPD at the end of their lives.26 One study of over 2,000 patients with severe COPD found that there was a correlation between the dose of opioids and mortality at high doses, but at lower doses, the effect was not seen, and yet at lower doses, the patients’ dyspnea was atenuated.27
Whatever the cause, be it through a central nervous system mechanism, or a peripheral mechanism such as glottic edema from anaphylactic shock, or airway disruption from trauma or a foreign body, death does not occur immediately. The hemoglobin in the circulating blood carries enough O2 for maintenance of aerobic metabolism for 1 to 3 minutes. In addition, there is O2 that has already entered the lungs. During normal breathing at end expiration, there is O2 in the functional residual capacity (FRC) that can be calculated as the FIO2 times the volume of air in the FRC which in turn is equal to the person’s weight in kilograms times 15 mL/kg. For a 70-kg person, breathing room air the FRC would contain approximately 220 mL of O2 ([70 kg × 15mL/kg] × .21), enough to meet metabolic demand. At 3 to 4 minutes of complete apnea, evidence of tissue ischemia would become apparent and at longer than 5 minutes, irreversible damage would occur, especially in the brain. Cardiac arrest would soon follow unless oxygenation and ventilation were immediately rapidly restored.
Other causes of hypoxemic death are related to the neuromuscular system. A spinal cord transection above C4 would result in apnea because the phrenic nerves arise from C1 through C4. Likewise, patients with amyotrophic lateral sclerosis in which upper and lower motor neurons in the motor cortex, in the brainstem, and in the spinal cord die over time are unable to maintain respiratory effort. The cause is not understood but genetic factors play a role.28 Neuromuscular blocking agents produce the same effect albeit more quickly. Any process that decreases the alveolar to arterial O2 gradient can likewise interfere with the delivery of O2 to tissues, for example, drowning or acute respiratory distress syndrome. Cyanide poisoning is the most well-known cause of inhibition of O2 use within mitochondria by inhibiting cytochrome C oxidase. Adequate oxygen is supplied but cannot be used for aerobic metabolism.
All the causes of hypoxemic death discussed so far come about from inhibition or destruction of the body’s normal physiologic mechanisms. There are also environmental factors that play a role as well, the most important of which would be any factor that decreases the FIO2 would interrupt aerobic metabolism. The best example for anesthesiologists would be a helium quench from a magnetic resonance imaging scanner. If the required safety valves in the room were not working correctly, the helium could displace all the O2 in the room.
Pharmacology
Cardiopulmonary Resuscitation
The primary goal when resuscitating a patient from cardiac arrest is the ROSC, which is best achieved with effective chest compressions delivered immediately by a bystander, and if the patient has ventricular fibrillation, the delivery of electroshock therapy to defibrillate the heart. However, if these maneuvers are unsuccessful in restoring cardiac function and an advanced practitioner is present, drug therapy has been shown to increase the rate of ROSC. Recommendations for advanced cardiac life support (ACLS) are continually updated and provided by the American Heart Association. Although the drugs currently recommended for ACLS improve the likelihood of ROSC, none has improved long-term outcome.29
Epinephrine
Epinephrine is a catecholamine (vasoactive compound with a catechol—benzene ring with two hydroxyl groups [Fig. 47-2] in its structure). Dopamine is its precursor, and in turn, epinephrine is the precursor for norepinephrine. Chromaffin cells in the adrenal medulla and the terminal boutons of certain nerves produce and release epinephrine, which gains its biologic activity by binding to α- and β-adrenergic receptors. Among its many effects, epinephrine activation of α receptors produces vasoconstriction in the peripheral vasculature, whereas activation of β receptors within the heart increases chronotropy, inotropy, dromotropy, and lusitropy. The majority of epinephrine produced is metabolized within the same cells; it was synthesized simply because there is so much leakage of the catecholamine from the vesicles where it is stored into the cytoplasm. There is a very dynamic equilibrium between vesicular and cytoplasmic epinephrine.30

When administered intravenously during a cardiac arrest, epinephrine is thought to increase the ROSC primarily by binding to and activating α-adrenergic receptors.31 Activation of these receptors on the venules32 in the periphery increases venous return to the heart, allowing chest compressions to better increase cardiac output. On the arterial side, the vasoconstriction caused by α receptor activation constricts the peripheral arterial system further centralizing the circulation to perfuse vital organs with the highest oxygen requirement. The increase in cardiac output with an increased systemic vascular resistance is hypothesized to improve coronary artery and carotid artery blood flow.33 By activating β receptors within the heart, epinephrine augments cardiac output even further, with additional benefit to the patient. However, some believe that epinephrine’s effect on β receptors is more detrimental than beneficial because of increased metabolic demand.
Epinephrine has been repeatedly demonstrated to improve ROSC when administered to patients in cardiac arrest but without an improvement in overall outcome. Ditchey and Lindenfeld34 observed that even with an improvement in coronary perfusion pressure in a canine model of cardiac arrest 10 minutes after the administration of epinephrine during chest compression, myocytes had decreased ATP compared to controls and an accompanying lactic acidosis. In a clinical study, Olasveengen and colleagues observed that patients arriving at the hospital following a cardiac arrest who had received epinephrine as part of their CPR regimen had no improvement in tissue perfusion nor was there any difference in long-term survival compared to case-matched controls.35 Other studies, one in rodents36 and one in patients,37demonstrated decreased survival in subjects who had received epinephrine following cardiac arrest that was treated with CPR. The difficulty with interpreting clinical studies is that patients who receive epinephrine are more likely to have had a longer period without cardiac activity and therefore greater duration of CPR—and therefore less likely to have a good outcome no matter the intervention.
If the effects of epinephrine are attributed to its effect on α-adrenergic receptors, then drugs with even more α receptor specificity might be indicated. However, studies of epinephrine compared to either norepinephrine38 or phenylephrine39 during cardiac arrest with CPR showed no benefit to the more α receptor–specific drugs. If it is vasoconstriction that explains the better ROSC of epinephrine but its β receptor properties that offset the benefit, then perhaps the administration of a drug such as vasopressin that works through a different mechanism than does epinephrine might result in better outcomes. However, a study comparing vasopressin to epinephrine for out-of-hospital CPR following cardiac arrest found no difference in outcomes between the two drugs.40
The most recent guidelines of the American Heart Association recommend 1 mg of epinephrine administered intravenously or by the intraosseous injection every 3 to 5 minutes with recognition of the fact that although there may be ROSC, there are no prospective randomized controlled trials that demonstrate an improvement in outcome.
Vasopressin
Vasopressin (antidiuretic hormone) is an oligopeptide composed of nine amino acids and is a nonapeptide synthesized in the hypothalamus. The axons of the neuronal cells that produce vasopressin extend into the posterior pituitary where it is released.41 Because there is no blood brain barrier in the capillaries in the posterior pituitary, the vasopressin can be readily released into blood.42 Vasopressin is released in response to a decrease in mean arterial pressure, a decrease in end diastolic left ventricular pressure, or an increase in plasma osmolality.43 In the periphery, vasopressin can bind to one of three subtypes of vasopressin receptors, V1, V2, or V3, all of which are transmembrane proteins, but the G proteins to which these receptors are coupled are unique to each receptor.44 Activation of these receptors results in a number of actions, but most importantly in the kidney, activation of these receptors results in an antidiuretic effect.
When administered intravenously, vasopressin distributes from the plasma into the extracellular fluid rapidly, exerting its effects within just a few minutes. The plasma t1/2 is between 4 and 20 minutes. Vasopressin is metabolized in the liver and kidneys, with a small amount excreted unchanged in the urine.45
The use of vasopressin to treat sudden cardiac arrest came about from the results of several reports. In one study of patients who had a sudden cardiac arrest in whom vasopressin levels were measured before the initiation of CPR, it was found that the levels were quite high compared to control. Of most interest to investigators was the observation that patients who had ROSC had higher levels of vasopressin in their blood than those patients who were not successfully resuscitated.46,47 These results served as a stimulus for additional studies. Vasopressin administered during CPR to patients who had had a sudden cardiac arrest improved coronary perfusion pressure, even in some patients whose coronary artery perfusion pressure was unresponsive to epinephrine.47 In three prospective studies of patients in whom CPR was initiated to treat cardiac arrest, either vasopressin or epinephrine were administered intravenously as the initial pharmacologic intervention. In one study of 40 patients who had had out-of-hospital cardiac arrest and who had continuing ventricular fibrillation despite electric defibrillation, either epinephrine (1 mg) or vasopressin (40 units) were administered intravenously. Significantly more patients who received vasopressin had ROSC and significantly more patients were alive at 24 hours.48 In a second larger study of 200 hospitalized patients who were administered either epinephrine (1 mg) or vasopressin (40 units) intravenously during CPR for cardiac arrest, there were no differences in the ROSC.49 In a third even larger study, vasopressin compared to epinephrine for the treatment of out-of-hospital cardiac arrest in over 1,000 patients, there was no significant difference between vasopressin and epinephrine in ROSC or survival in patients with ventricular tachycardia or pulseless electrical activity but significantly more patients with asystole who received vasopressin had ROSC.40
Independent of these findings, the current guidelines of the American Heart Association for ACLS state that vasopressin (40 units) can be administered intravenously or by the intraosseous route to instead or either the first or second dose of epinephrine.
Amiodarone
Electric defibrillation is first-line treatment for ventricular fibrillation. Amiodarone is the second pharmacologic treatment (after either epinephrine or vasopressin) in patients who have ventricular fibrillation refractory to electric defibrillation. Administered intravenously at a dose of 300 mg50 or 5 mg/kg,51 amiodarone compared to lidocaine has been shown to be superior for ROSC in patients with ventricular fibrillation refractory to electric defibrillation. However, no study has yet demonstrated that long-term outcome in terms of morbidity or mortality is improved. The American Heart Association has recommended amiodarone to treat refractory ventricular fibrillation or pulseless ventricular tachycardia.52
Following intravenous administration, amiodarone enters the central compartment and from there it undergoes extensive tissue redistribution. The distribution half-life of amiodarone from the central compartment (t1/2α) may be as short as 4 hours. The terminal half-life (t1/2β) is lengthy and also variable (9 to 77 days) because of prolonged release of amiodarone out of adipocytes due to the lipophilicity of the drug.53,54
Hemodynamic effects of orally administered amiodarone are usually negligible. When administered as an intravenous bolus, there have been reports of hypotension that was thought due to the vasoactive solvents (polysorbate and benzyl alcohol) in which it was compounded. When administered with a different diluent, rapid administration of amiodarone intravenously is not associated with hypotension.55
Effects on the thyroid gland and the liver are of more significance.56,57 The similarity between the chemical structures of amiodarone and thyroid hormone explain the former, and the latter is most likely due to direct injury to lipid bilayers of the hepatocyte cell membrane and inhibition of lysosomal function.58 However, these adverse effects are only seen in patients taking the drug in high dose for extended periods of time, not in patients who are administered one or two doses of the drug during sudden cardiac arrest.
Hemorrhage
The most critical intervention in hemorrhage is to stop the bleeding—either by applying a tourniquet in the field to an extremity or if there is massive internal bleeding from a crush injury or ruptured aortic aneurysm—by transporting the patient as quickly as possible to a level I trauma hospital and from the emergency department directly to the operating room. The importance of surgical intervention as quickly as possible was underscored by Maddox and colleagues in a study of patients who were transported by emergency medical personnel randomized to the (at the time) conventional therapy of placing an intravenous cannula and initiating the infusion of crystalloid during transport to the hospital or to essentially no therapy, the “scoop and run” approach. Emergency personnel evaluated the patient, treated the ABCs (airway, breathing, and circulation), and then placed the patient in the ambulance with immediate transport back to the hospital. The latter group not only received less crystalloid but had better survival than the conventionally treated group.59
Other components of damage control resuscitation include maintaining normothermia, limiting intravenous crystalloid, using a hemoglobin target of 7 to 8 g/dL, and identifying patients at risk of developing ATC. As with resuscitation from cardiac arrest, there is a limited role for drugs in improving outcome in patients who sustained traumatic injury with possibly one exception. In the CRASH-2 trial, such patients were randomized to a tranexamic acid group or control. The group that received tranexamic acid within an hour of admission had a small reduction in mortality (1.6% in all comers—a recent subgroup analysis suggested 2.5%). Beyond 3 hours, there appeared to be an increase in mortality.60
Tranexamic Acid
Tranexamic acid, a synthetic analog of the amino acid lysine, is an antifibrinolytic that has found widespread use following the removal of aprotinin from the market. By binding to specific sites on plasminogen and plasmin, it inhibits the transformation of plasminogen to plasmin. Circulating plasmin degrades fibrin, an integral part of blood clots. The role of tranexamic acid in managing patients with traumatic injury has not yet been determined, but there are a number of level I trauma centers in the United States who administer it routinely to their patients. It is also commonly used in massive hemorrhage during cancer surgery, removal of invasive placenta, and major orthopedic surgery.
Oxygenation/Ventilation
In patients with pulmonary failure or respiratory arrest, immediate assisted ventilation and oxygenation is the intervention most likely to increase the chances of survival. There are no drugs that have been demonstrated in large prospective randomized trials to improve the chances of successful resuscitation in this setting. Respiratory arrest due to specific drug overdose is an exception. Respiratory arrest secondary to an opioid is reversed with naloxone. Less commonly, for respiratory arrest secondary to or contributed to by benzodiazepine overdose, flumazenil antagonizes the drug effects. Likewise, for patients with cyanide toxicity and respiratory arrest at the cellular level, sodium thiosulfate is indicated.
As stated at the onset, resuscitation is simple in some ways. In patients with cardiac arrest, defibrillation in the first 3 minutes and CPR/defibrillation during the next 5 to 7 minutes achieves the best results. In patients with profound hemorrhage, immediate control of bleeding and infusion of red blood cells has the biggest impact on outcome. In patients with pulmonary or respiratory arrest, immediate assisted ventilation and oxygenation is the most likely intervention to increase the chances of survival. The difficulty is in the absence of therapies that can preserve tissue integrity until an energy source can be reestablished.
References
1. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127(1):e6–e245.
2. Murphy SL, Kochanek KD. Deaths: Final Data for 2010. Vol 61. Hyattsville, MD: National Center for Health Statistics; 2013.
3. Finer NN, Rich W, Wang C, et al. Airway obstruction during mask ventilation of very low birth weight infants during neonatal resuscitation. Pediatrics. 2009;123(3):865–869.
4. Mrozek S, Vardon F, Geeraerts T. Brain temperature: physiology and pathophysiology after brain injury. Anesthesiol Res Pract. 2012;2012:989487.
5. Valenzuela TD, Roe DJ, Cretin S, et al. Estimating effectiveness of cardiac arrest interventions: a logistic regression survival model. Circulation. 1997;96(10):3308–3313.
6. Cobb LA, Fahrenbruch CE, Olsufka M, et al. Changing incidence of out-of-hospital ventricular fibrillation, 1980–2000. JAMA. 2002;288(23):3008–3013.
7. Newton-Cheh C, Guo CY, Larson MG, et al. Common genetic variation in KCNH2 is associated with QT interval duration: the Framingham Heart Study. Circulation. 2007;116(10):1128–1136.
8. Splawski I, Timothy KW, Tateyama M, et al. Variant of SCN5A sodium channel implicated in risk of cardiac arrhythmia. Science. 2002;297(5585):1333–1336.
9. Heist EK, Ruskin JN. Drug-induced arrhythmia. Circulation. 2010;122(14):1426–1435.
10. Haissaguerre M, Derval N, Sacher F, et al. Sudden cardiac arrest associated with early repolarization. N Engl J Med. 2008;358(19):2016–2023.
11. Kim SH, Kim do Y, Kim HJ, et al. Early repolarization with horizontal ST segment may be associated with aborted sudden cardiac arrest: a retrospective case control study. BMC Cardiovasc Disord. 2012;12:122.
12. Weisfeldt ML, Becker LB. Resuscitation after cardiac arrest: a 3-phase time-sensitive model. JAMA. 2002;288(23):3035–3038.
13. Connolly SJ, Hallstrom AP, Cappato R, et al. Meta-analysis of the implantable cardioverter defibrillator secondary prevention trials. Eur Heart J. 2000;21(24):2071–2078.
14. Yakaitis RW, Ewy GA, Otto CW, et al. Influence of time and therapy on ventricular defibrillation in dogs. Crit Care Med. 1980;8(3):157–163.
15. Niemann JT, Cairns CB, Sharma J, et al. Treatment of prolonged ventricular fibrillation. Immediate countershock versus high-dose epinephrine and CPR preceding countershock. Circulation. 1992;85(1):281–287.
16. White BC, Grossman LI, Krause GS. Brain injury by global ischemia and reperfusion: a theoretical perspective on membrane damage and repair. Neurology. 1993;43(9):1656–1665.
17. Cerchiari EL, Safar P, Klein E, et al. Visceral, hematologic and bacteriologic changes and neurologic outcome after cardiac arrest in dogs. The visceral post-resuscitation syndrome. Resuscitation. 1993;25(2):119–136.
18. Adrie C, Laurent I, Monchi M, et al. Postresuscitation disease after cardiac arrest: a sepsis-like syndrome? Curr Opin Crit Care. 2004;10(3):208–212.
19. Kilgannon JH, Jones AE, Shapiro NI, et al. Association between arterial hyperoxia following resuscitation from cardiac arrest and in-hospital mortality. JAMA. 2010;303(21):2165–2171.
20. Nielsen N, Wetterslev J, Cronberg T, et al. Targeted temperature management at 33°C versus 36°C after cardiac arrest. N Engl J Med. 2013;369(23):2197–2206.
21. Kauvar DS, Lefering R, Wade CE. Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma. 2006;60(6)(suppl):S3–S11.
22. Andersen RC, Fleming M, Forsberg JA, et al. Dismounted complex blast injury. J Surg Orthop Adv. 2012;21(1):2–7.
23. Carson JL, Noveck H, Berlin JA, et al. Mortality and morbidity in patients with very low postoperative Hb levels who decline blood transfusion. Transfusion. 2002;42(7):812–818.
24. Atkinson JL, Anderson RE, Murray MJ. The early critical phase of severe head injury: importance of apnea and dysfunctional respiration. J Trauma. 1998;45(5):941–945.
25. Mogri M, Khan MI, Grant BJ, et al. Central sleep apnea induced by acute ingestion of opioids. Chest. 2008;133(6):1484–1488.
26. Goodridge D, Lawson J, Rocker G, et al. Factors associated with opioid dispensation for patients with COPD and lung cancer in the last year of life: a retrospective analysis. Int J Chron Obstruct Pulmon Dis. 2010;5:99–105.
27. Ekstrom MP, Bornefalk-Hermansson A, Abernethy AP, et al. Safety of benzodiazepines and opioids in very severe respiratory disease: national prospective study. BMJ. 2014;348:g445.
28. Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477(7363):211–215.
29. Papastylianou A, Mentzelopoulos S. Current pharmacological advances in the treatment of cardiac arrest. Emerg Med Int. 2012;2012:815857.
30. Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: a contemporary view with implications for physiology and medicine. Pharmacol Rev. 2004;56(3):331–349.
31. Yakaitis RW, Otto CW, Blitt CD. Relative importance of alpha and beta adrenergic receptors during resuscitation. Crit Care Med. 1979;7(7):293–296.
32. Greenway CV, Lawson AE. The effects of adrenaline and noradrenaline on venous return and regional blood flows in the anaesthetized cat with special reference to intestinal blood flow. J Physiol. 1966;186(3):579–595.
33. Michael JR, Guerci AD, Koehler RC, et al. Mechanisms by which epinephrine augments cerebral and myocardial perfusion during cardiopulmonary resuscitation in dogs. Circulation. 1984;69(4):822–835.
34. Ditchey RV, Lindenfeld J. Failure of epinephrine to improve the balance between myocardial oxygen supply and demand during closed-chest resuscitation in dogs. Circulation. 1988;78(2):382–389.
35. Olasveengen TM, Wik L, Sunde K, et al. Outcome when adrenaline (epinephrine) was actually given vs. not given—post hoc analysis of a randomized clinical trial. Resuscitation. 2012;83(3):327–332.
36. McCaul CL, McNamara PJ, Engelberts D, et al. Epinephrine increases mortality after brief asphyxial cardiac arrest in an in vivo rat model. Anesth Analg. 2006;102(2):542–548.
37. Holmberg M, Holmberg S, Herlitz J. Low chance of survival among patients requiring adrenaline (epinephrine) or intubation after out-of-hospital cardiac arrest in Sweden. Resuscitation. 2002;54(1):37–45.
38. Callaham M, Madsen CD, Barton CW, et al. A randomized clinical trial of high-dose epinephrine and norepinephrine vs standard-dose epinephrine in prehospital cardiac arrest. JAMA. 1992;268(19):2667–2672.
39. Silfvast T, Saarnivaara L, Kinnunen A, et al. Comparison of adrenaline and phenylephrine in out-of-hospital cardiopulmonary resuscitation. A double-blind study. Acta Anaesthesiol Scand. 1985;29(6):610–613.
40. Wenzel V, Krismer AC, Arntz HR, et al. A comparison of vasopressin and epinephrine for out-of-hospital cardiopulmonary resuscitation. N Engl J Med. 2004;350(2):105–113.
41. Treschan TA, Peters J. The vasopressin system: physiology and clinical strategies. Anesthesiology. 2006;105(3):599–612; quiz 639–540.
42. Leng G, Brown CH, Russell JA. Physiological pathways regulating the activity of magnocellular neurosecretory cells. Prog Neurobiol. 1999;57(6):625–655.
43. Goldsmith SR. Baroreceptor-mediated suppression of osmotically stimulated vasopressin in normal humans. J Appl Physiol. 1988;65(3):1226–1230.
44. Thibonnier M, Berti-Mattera LN, Dulin N, et al. Signal transduction pathways of the human V1-vascular, V2-renal, V3-pituitary vasopressin and oxytocin receptors. Prog Brain Res. 1998;119:147–161.
45. Baumann G, Dingman JF. Distribution, blood transport, and degradation of antidiuretic hormone in man. J Clin Invest. 1976;57(5):1109–1116.
46. Lindner KH, Haak T, Keller A, et al. Release of endogenous vasopressors during and after cardiopulmonary resuscitation. Heart. 1996;75(2):145–150.
47. Morris DC, Dereczyk BE, Grzybowski M, et al. Vasopressin can increase coronary perfusion pressure during human cardiopulmonary resuscitation. Acad Emerg Med. 1997;4(9):878–883.
48. Lindner KH, Dirks B, Strohmenger HU, et al. Randomised comparison of epinephrine and vasopressin in patients with out-of-hospital ventricular fibrillation. Lancet. 1997;349(9051):535–537.
49. Stiell IG, Hebert PC, Wells GA, et al. Vasopressin versus epinephrine for inhospital cardiac arrest: a randomised controlled trial. Lancet. 2001;358(9276):105–109.
50. Kudenchuk PJ, Cobb LA, Copass MK, et al. Amiodarone for resuscitation after out-of-hospital cardiac arrest due to ventricular fibrillation. N Engl J Med. 1999;341(12):871–878.
51. Dorian P, Cass D, Schwartz B, et al. Amiodarone as compared with lidocaine for shock-resistant ventricular fibrillation. N Engl J Med. 2002;346(12):884–890.
52. Neumar RW, Otto CW, Link MS, et al. Part 8: adult advanced cardiovascular life support: 2010 American Heart Association guidelines for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2010;122(18)(suppl 3):S729–767.
53. Freedman MD, Somberg JC. Pharmacology and pharmacokinetics of amiodarone. J Clin Pharmacol. 1991;31(11):1061–1069.
54. Chow MS. Intravenous amiodarone: pharmacology, pharmacokinetics, and clinical use. Ann Pharmacother. 1996;30(6):637–643.
55. Somberg JC, Timar S, Bailin SJ, et al. Lack of a hypotensive effect with rapid administration of a new aqueous formulation of intravenous amiodarone. Am J Cardiol. 2004;93(5):576–581.
56. Narayana SK, Woods DR, Boos CJ. Management of amiodarone-related thyroid problems. Ther Adv Endocrinol Metab. 2011;2(3):115–126.
57. Varma RR, Troup PJ, Komorowski RA, et al. Clinical and morphologic effects of amiodarone on the liver. Gastroenterology. 1985;88(4):1091–1093.
58. Poucell S, Ireton J, Valencia-Mayoral P, et al. Amiodarone-associated phospholipidosis and fibrosis of the liver. Light, immunohistochemical, and electron microscopic studies. Gastroenterology.1984;86(5)(pt 1):926–936.
59. Bickell WH, Wall MJ Jr, Pepe PE, et al. Immediate versus delayed fluid resuscitation for hypotensive patients with penetrating torso injuries. N Engl J Med. 1994;331(17):1105–1109.
60. Shakur H, Roberts I, Bautista R, et al; CRASH-2 Trial Collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet. 2010;376(9734):23–32.