Overview
No other class of pharmacologic agents is more central to the practice of anesthesiology than the intravenous sedatives and hypnotics. It is this group of agents that we rely upon to provide everything through the spectrum from anxiolysis to light and deep sedation then on to general anesthesia. The term sedative refers to a drug that induces a state of calm or sleep. The term hypnotic refers to drug that induces hypnosis or sleep. There is significant overlap in the two terms as well as with the related term anxiolytic, which refers to any agent that reduces anxiety as nearly all such substances have sedation as a side effect. For practical purposes, we generally combine the terms and refer to all of these drugs as sedative-hypnotics, drugs that reversibly depress the activity of the central nervous system. Depending on the specific agent, the dose, and the rate of administration, the many sedative-hypnotics can be used to allay anxiety with minimal sedation, produce varying degrees of sedation, or rapidly induce the state of drug-induced unconsciousness we call general anesthesia. We will review the pharmacology of these important agents in this chapter.
γ-Aminobutyric Acid Agonists
Propofol
Propofol is a substituted isopropylphenol (2,6-diisopropylphenol) that is administered intravenously as 1% solution in an aqueous solution of 10% soybean oil, 2.25% glycerol, and 1.2% purified egg phosphatide.1–3 This drug is chemically distinct from all other drugs that act as intravenous (IV) sedative-hypnotics. Administration of propofol, 1.5 to 2.5 mg/kg IV (equivalent to thiopental, 4 to 5 mg/kg IV, or methohexital, 1.5 mg/kg IV) as a rapid IV injection (<15 seconds), produces unconsciousness within about 30 seconds. Awakening is more rapid and complete than that after induction of anesthesia with all other drugs used for rapid IV induction of anesthesia. The more rapid return of consciousness with minimal residual central nervous system (CNS) effects is one of the most important advantages of propofol compared with alternative drugs administered for the same purpose.
Commercial Preparations
Propofol is an insoluble drug that requires a lipid vehicle for emulsification. Current formulations of propofol use a soybean oil as the oil phase and egg lecithin as the emulsifying agent that is composed of long chain triglycerides.4This formulation supports bacterial growth and causes increased plasma triglyceride concentrations when prolonged IV infusions are used. Diprivan and generic propofol differ with respect to the preservatives used and the pH of the formulation. Diprivan uses the preservative disodium edetate (0.005%) with sodium hydroxide to adjust the pH to 7 to 8.5. A generic formulation of propofol incorporates sodium metabisulfite (0.25 mg/mL) as the preservative and has a lower pH (4.5 to 6.4). Propofol, unlike thiopental, etomidate, and ketamine, is not a chiral compound.
The mixing of propofol with any other drug is not recommended although lidocaine has been frequently added to propofol in attempts to prevent pain with IV injection. However, mixing of lidocaine with propofol may result in coalescence of oil droplets, which may pose the risk of pulmonary embolism.5
A low-lipid emulsion of propofol (Ampofol) contains 5% soybean oil and 0.6% egg lecithin but does not require a preservative or microbial growth retardant.6 This formulation is equipotent to Diprivan but is associated with a higher incidence of pain on injection.
An alternative to emulsion formulations of propofol and associated side effects (pain on injection, risk of infection, hypertriglyceridemia, pulmonary embolism) is creation of a prodrug (Aquavan) by cleaving groups to the parent compound that increase its water solubility (phosphate monoesters, hemisuccinates). Propofol is liberated after hydrolysis by endothelial cell surface alkaline phosphatases. In this regard, injection of the water-soluble propofol phosphate prodrug results in propofol and dose-dependent sedative effects.7,8 However, although the absence of lipid emulsion obviates pain on injection, the release of a small amount of formaldehyde byproduct causes an unpleasant dysesthesia or burning sensation often in the genital area. Compared with propofol, this prodrug has a slower onset, larger volume of distribution, and higher potency.9
Another nonlipid formulation of propofol uses cyclodextrins as a solubilizing agent.10 Cyclodextrins are ring sugar molecules that form guest (propofol)–host complexes migrating between the hydrophilic center of the cyclodextrin molecule and the water-soluble phase. This allows propofol, which is poorly soluble in water, to be presented in an injectable form. After injection, propofol migrates out of the cyclodextrin into the blood. This preparation is in clinical trials and has not been released for general human use.
Mechanism of Action
Propofol is a relatively selective modulator of γ-aminobutyric acid (GABAA) receptors although it also has activity at glycine receptors. Propofol is presumed to exert its sedative-hypnotic effects through a GABAA receptor interaction.11 GABA is the principal inhibitory neurotransmitter in the brain. When GABAA receptors are activated, transmembrane chloride conductance increases, resulting in hyperpolarization of the postsynaptic cell membrane and functional inhibition of the postsynaptic neuron. The interaction of propofol (also etomidate and barbiturates) with specific components of GABAAreceptors appears to decrease the rate of dissociation of the inhibitory neurotransmitter, GABA from the receptor, thereby increasing the duration of the GABA-activated opening of the chloride channel with resulting hyperpolarization of cell membranes.
In contrast to volatile anesthetics, spinal motor neuron excitability, as measured by H reflexes, is not altered by propofol, suggesting that immobility during propofol anesthesia is not caused by drug-induced spinal cord depression.12
Pharmacokinetics
Clearance of propofol from the plasma exceeds hepatic blood flow, emphasizing that tissue uptake (possibly into the lungs), as well as hepatic oxidative metabolism by cytochrome P450, is important in removal of this drug from the plasma (Fig. 5-1) (Table 5-1).13 Hepatic metabolism is rapid and extensive, resulting in inactive, water-soluble sulfate and glucuronic acid metabolites that are excreted by the kidneys.14 Propofol may also undergo ring hydroxylation by cytochrome P450 to form 4-hydroxypropofol which is then glucuronidated or sulfated. Although the glucuronide and sulfate conjugates of propofol appear to be pharmacologically inactive, 4-hydroxypropofol has about one-third the hypnotic activity of propofol. Less than 0.3% of a dose is excreted unchanged in urine. The elimination half-time is 0.5 to 1.5 hours, but more important, the context-sensitive half-time for propofol infusions lasting up to 8 hours is less than 40 minutes.15 The context-sensitive half-time of propofol is only minimally influenced by the duration of the infusion at times relevant for surgery because of rapid metabolic clearance when the infusion is discontinued so that the drug that returns from tissue storage sites to the circulation is not available to retard the decrease in plasma concentrations of the drug. However, when used as a sedative for prolonged intensive care unit (ICU) care, the context-sensitive half-time is highly relevant and should be considered. Propofol, like thiopental and alfentanil, has a short effect-site equilibration time such that effects on the brain occur promptly after IV administration.


The fact that total body clearance of propofol exceeds hepatic blood flow is consistent with extrahepatic clearance (pulmonary uptake and first-pass elimination, renal excretion) of propofol.14,16 Pulmonary uptake of propofol is significant and influences the initial availability of propofol. Although propofol can be transformed in the lungs to 2,6-diisopropyl-1,4-quiniol, most of the drug that undergoes pulmonary uptake during the first pass is released back into the circulation.17,18 Glucuronidation is the major metabolic pathway for propofol and uridine 5′-diphospho-glucuronosyltransferase isoforms are expressed in the kidneys and brain.
Despite the rapid clearance of propofol by metabolism, there is no evidence of impaired elimination in patients with cirrhosis of the liver. Plasma concentrations of propofol at the time of awakening are similar in alcoholic and normal patients.19 Extrahepatic elimination of propofol occurs during the anhepatic phase of orthotopic liver transplantation. Renal dysfunction does not influence the clearance of propofol despite the observation that nearly three-fourths of propofol metabolites are eliminated in urine in the first 24 hours.20 Patients older than 60 years of age exhibit a decreased rate of plasma clearance of propofol compared with younger adults. The rapid clearance of propofol confirms this drug can be administered as a continuous infusion during surgery without an excessive cumulative effect. Propofol readily crosses the placenta but is rapidly cleared from the neonatal circulation.21 The effect of instituting cardiopulmonary bypass on the plasma propofol concentration is unpredictable, with some studies reporting a decrease, whereas other observations fail to document any change.22
Clinical Uses
Propofol has become the induction drug of choice for many forms of anesthesia, especially when rapid and complete awakening is considered desirable.3 Continuous IV infusion of propofol, with or without other anesthetic drugs, has become a commonly used method for producing IV “conscious” sedation or as part of a balanced or total IV anesthetic.1,3 Administration of propofol as a continuous infusion may be used for sedation of patients in the ICU.2 In this regard, a 2% solution may be useful to decrease the volume of lipid emulsion administered with long-term sedation. A computer-controlled infusion pump is available to allow the clinician to select the propofol target concentration and the computer calculates the infusion rates that are necessary to achieve this target concentration based on the pharmacokinetics of propofol.23
Induction of Anesthesia
The induction dose of propofol in healthy adults is 1.5 to 2.5 mg/kg IV, with blood levels of 2 to 6 µg/mL producing unconsciousness depending on associated medications and the patient’s age. As with barbiturates, children require higher induction doses of propofol on a milligram per kilogram basis, presumably reflecting a larger central distribution volume and higher clearance rate. Elderly patients require a lower induction dose (25% to 50% decrease) as a result of a smaller central distribution volume and decreased clearance rate and increased pharmacodynamic activity.3 Awakening typically occurs at plasma propofol concentrations of 1.0 to 1.5 µg/mL. The complete awakening without residual CNS effects that is characteristic of propofol is the principal reason this drug has replaced thiopental for induction of anesthesia in many clinical situations. Thiopental is not currently available for use in the United States.
Intravenous Sedation
The short context-sensitive half-time of propofol, combined with the short effect-site equilibration time, make this a readily titratable drug for production of IV sedation.1 The prompt recovery without residual sedation and low incidence of nausea and vomiting make propofol particularly well suited to ambulatory conscious sedation techniques. The typical conscious sedation dose of 25 to 100 µg/kg/minute IV produces minimal analgesic and amnestic effects.3 In selected patients, midazolam or an opioid may be added to propofol for continuous IV sedation. A sense of well-being may accompany recovery from conscious sedation with propofol. When compared with anesthesia based on isoflurane, patients anesthetized with propofol reported less early postoperative pain.24 A conventional patient-controlled analgesia delivery system set to deliver 0.7 mg/kg doses of propofol with a 3-minute lockout period has been used as an alternative to continuous IV sedation techniques. Propofol has emerged as the agent of choice for sedation for brief gastrointestinal endoscopy procedures. So reliable are the pharmacologic properties of propofol that extensive design and testing have gone in to creation of a computer-assisted personalized sedation for upper endoscopy and colonoscopy, called SEDASYS. A comparative, multicenter randomized study concluded that this system could provide endoscopist/nurse teams a safe and effective means to administer propofol to effect minimal to moderate sedation during routine colonoscopy and esophagogastroduodenoscopy without the need for a trained anesthesia provider.25 The SEDASYS system received approval from the United States Food and Drug Administration in 2014 and is expected to be introduced in 2014.
Propofol has been administered as a sedative during mechanical ventilation in the ICU in a variety of patient populations including postoperative patients (cardiac surgery, neurosurgery) and patients with head injury.2 Propofol also provides control of stress responses and has anticonvulsant and amnestic properties. After cardiac surgery, propofol sedation appears to modulate postoperative hemodynamic responses by decreasing the incidence and severity of tachycardia and hypertension.26 Increasing metabolic acidosis, lipemic plasma, bradycardia, and progressive myocardial failure has been described, particularly in children who were sedated with propofol during management of acute respiratory failure in the ICU.27
Maintenance of Anesthesia
The typical dose of propofol for maintenance of anesthesia is 100 to 300 µg/kg/minute IV, often in combination with a short-acting opioid.3 General anesthesia that includes propofol is typically associated with minimal postoperative nausea and vomiting, and awakening is prompt, with minimal residual sedative effects.
Nonhypnotic Therapeutic Applications
In addition to its clinical application as an IV induction drug, propofol has been shown to have beneficial effects that were not anticipated when the drug was initially introduced in 1989.28
Antiemetic Effects
The incidence of postoperative nausea and vomiting is decreased when propofol is administered, regardless of the anesthetic technique.28 Subhypnotic doses of propofol (10 to 15 mg IV) may be used in the postanesthesia care unit to treat nausea and vomiting, particularly if it is not of vagal origin. In the postoperative period, the advantage of propofol is its rapid onset of action and the absence of serious side effects. Propofol is generally efficacious in treating postoperative nausea and vomiting at plasma concentrations that do not produce significant sedation. Simulations indicate that antiemetic plasma concentrations of propofol are achieved by a single IV dose of 10 mg followed by 10 µg/kg/minute.29 Propofol in subhypnotic doses is effective against chemotherapy-induced nausea and vomiting. When administered to induce and maintain anesthesia, it is more effective than ondansetron in preventing postoperative nausea and vomiting.30
Propofol has a profile of CNS depression that differs from other anesthetic drugs. In contrast to thiopental, for example, propofol uniformly depresses CNS structures, including subcortical centers. Most drugs of known antiemetic efficacy exert this effect via subcortical structures, and it is possible that propofol modulates subcortical pathways to inhibit nausea and vomiting or produces a direct depressant effect on the vomiting center. Nevertheless, the mechanisms mediating the antiemetic effects of propofol remain unknown. An antiemetic effect of propofol based on inhibition of dopaminergic activity is unlikely given that subhypnotic doses of propofol fail to increase plasma prolactin concentrations. A rapid and distinct increase in plasma prolactin concentrations is characteristic of drugs that block the dopaminergic system.31 Subhypnotic doses of propofol that are effective as an antiemetic do not inhibit gastric emptying and propofol is not considered a prokinetic drug.32
Antipruritic Effects
Propofol, 10 mg IV, is effective in the treatment of pruritus associated with neuraxial opioids or cholestasis.33 The mechanism of the antipruritic effect may be related to the drug’s ability to depress spinal cord activity. In this regard, there is evidence that intrathecal opioids produce pruritus by segmental excitation within the spinal cord.
Anticonvulsant Activity
Propofol possesses antiepileptic properties, presumably reflecting GABA-mediated presynaptic and postsynaptic inhibition of chloride ion channels. In this regard, propofol in doses of greater than 1 mg/kg IV decreases seizure duration 35% to 45% in patients undergoing electroconvulsive therapy.34
Attenuation of Bronchoconstriction
Compared with thiopental, propofol decreases the prevalence of wheezing after induction of anesthesia and tracheal intubation in healthy and asthmatic patients (Fig. 5-2).35 However, a newer formulation of propofol uses metabisulfite as a preservative. Metabisulfite may cause bronchoconstriction in asthmatic patients. In an animal model, propofol without metabisulfite attenuated vagal nerve stimulation–induced bronchoconstriction, whereas propofol with metabisulfite did not attenuate vagally or methacholine-induced bronchoconstriction and metabisulfite alone caused increases in airway responsiveness.36Following tracheal intubation, in patients with a history of smoking, airway resistance was increased more following the administration of propofol containing metabisulfite than ethylenediaminetetraacetic acid (EDTA).37 Therefore, the preservative used for propofol can have effects on its ability to attenuate bronchoconstriction. Nevertheless, propofol-induced bronchoconstriction has been described in patients with allergy histories. The formulation of propofol administered to these patients was Diprivan containing soybean oil, glycerin, yolk lecithin, and sodium edetate.38

Analgesia
Propofol does not relieve acute nociceptive pain. However in animal models, low-dose propofol equivalent to antiemetic concentrations earlier was highly effective in relieving nociceptive responses to neuropathic pain.39
Effects on Organ Systems
Central Nervous System
Propofol decreases cerebral metabolic rate for oxygen (CMRO2), cerebral blood flow, and intracranial pressure (ICP).40,41 Administration of propofol to produce sedation in patients with intracranial space-occupying lesions does not increase ICP.42 However, large dose propofol may decrease systemic blood pressure sufficiently to also decrease cerebral perfusion pressure. Cerebrovascular autoregulation in response to changes in systemic blood pressure and reactivity of the cerebral blood flow to changes in PaCO2 are not affected by propofol. Cerebral blood flow velocity changes in parallel with changes in PaCO2 in the presence of propofol and midazolam (Fig. 5-3).43 Propofol produces cortical electroencephalographic (EEG) changes that are similar to those of thiopental, including the ability of high doses to produce burst suppression.44 Cortical somatosensory evoked potentials as used for monitoring spinal cord function are not significantly modified in the presence of propofol alone but the addition of nitrous oxide or a volatile anesthetic results in decreased amplitude.45 Propofol does not interfere with the adequacy of electrocorticographic recordings during awake craniotomy performed for the management of refractory epilepsy, provided administration is discontinued at least 15 minutes before recording.46 At equal levels of sedation, propofol produces the same degree of memory impairment as midazolam, whereas thiopental has less memory effect and fentanyl has none.47

Development of tolerance to drugs that depress the CNS is a common finding, occurring with repeated exposure to opioids, sedative-hypnotic drugs, ketamine, and nitrous oxide. However, tolerance to propofol does not develop in children undergoing repeated exposure to the drug during radiation therapy.48
Cardiovascular System
Propofol produces decreases in systemic blood pressure, which are greater than those evoked by comparable doses of thiopental (Fig. 5-4).49 These decreases in blood pressure are often accompanied by corresponding changes in cardiac output and systemic vascular resistance. The relaxation of vascular smooth muscle produced by propofol is primarily due to inhibition of sympathetic vasoconstrictor nerve activity.50 A negative inotropic effect of propofol may result from a decrease in intracellular calcium availability secondary to inhibition of trans-sarcolemmal calcium influx. Stimulation produced by direct laryngoscopy and intubation of the trachea reverses the blood pressure effects of propofol. Propofol also effectively blunts the hypertensive response to placement of a laryngeal mask airway. The impact of propofol on desflurane-mediated sympathetic nervous system activation is unclear. In one report, propofol 2 mg/kg IV blunted the increase in epinephrine concentration, which accompanied a sudden increase in the delivered desflurane concentration but did not attenuate the transient cardiovascular response.51 Conversely, in another report, induction of anesthesia with propofol, but not etomidate, blunted the sympathetic nervous system activation and systemic hypertension associated with the introduction of rapidly increasing inhaled concentrations of desflurane.52 The blood pressure effects of propofol may be exaggerated in hypovolemic patients, elderly patients, and patients with compromised left ventricular function. Adequate hydration before rapid IV administration of propofol is recommended to minimize the blood pressure reduction.
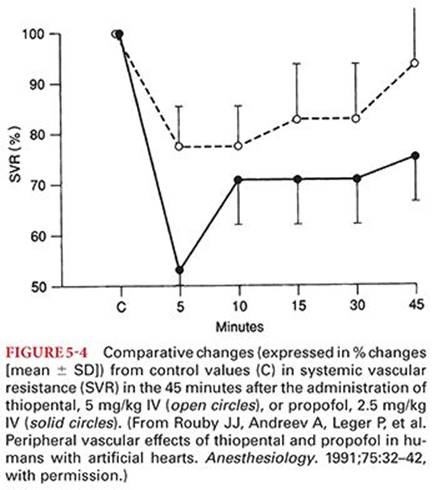
Addition of nitrous oxide does not alter the cardiovascular effects of propofol. The pressor response to ephedrine is augmented by propofol (Fig. 5-5).53

Despite decreases in systemic blood pressure, heart rate typically remains unchanged. Baroreceptor reflex control of heart rate may be depressed by propofol.54 However, bradycardia and asystole have been observed after induction of anesthesia with propofol, resulting in the occasional recommendation that anticholinergic drugs be administered when vagal stimulation is likely to occur in association with administration of propofol (see the section “Bradycardia-Related Death”). Propofol may decrease sympathetic nervous system activity to a greater extent than parasympathetic nervous system activity, resulting in a predominance of parasympathetic activity.1 Propofol does not alter sinoatrial or atrioventricular node function in normal patients or in patients with Wolff-Parkinson-White syndrome, thus making it an acceptable drug to administer during ablative procedures.55,56 Nevertheless, there is a case report of a patient with Wolff-Parkinson-White syndrome in whom δ waves on the electrocardiogram disappeared during infusion of propofol.57 Unlike sevoflurane, propofol does not prolong the QTc interval on the electrocardiogram.58
Bradycardia-Related Death
Profound bradycardia and asystole after administration of propofol have been described in healthy adult patients, despite prophylactic anticholinergics.59–62 The risk of bradycardia-related death during propofol anesthesia has been estimated to be 1.4 in 100,000. Propofol anesthesia, compared with other anesthetics, increases the incidence of the oculocardiac reflex in pediatric strabismus surgery, despite prior administration of anticholinergics.63
Heart rate responses to IV administration of atropine are attenuated in patients receiving propofol compared with awake patients (Fig. 5-6).64 This decreased responsiveness to atropine cannot be effectively overcome by larger doses of atropine suggesting that propofol may induce suppression of sympathetic nervous system activity. Treatment of propofol-induced bradycardia may require treatment with a direct β agonist such as isoproterenol.

Lungs
Propofol produces dose-dependent depression of ventilation, with apnea occurring in 25% to 35% of patients after induction of anesthesia with propofol.65 Opioids administered with the preoperative medication enhances ventilatory depressant. Painful surgical stimulation is likely to counteract the ventilatory depressant effects of propofol. A maintenance infusion of propofol decreases tidal volume and frequency of breathing. The ventilatory response to arterial hypoxemia are also decreased by propofol due to an effect at the central chemoreceptors.66 Likewise, propofol at sedative doses significantly decreases the slope and causes a downward shift of the ventilatory response curve to hypoxia.67 Hypoxic pulmonary vasoconstriction seems to remain intact in patients receiving propofol.
Hepatic and Renal Function
Propofol does not normally affect hepatic or renal function as reflected by measurements of liver transaminase enzymes or creatinine concentrations. Prolonged infusions of propofol have been associated with hepatocellular injury accompanied by lactic acidosis, bradydysrhythmias, and rhabdomyolysis as part of the propofol infusion syndrome described in the following texts. In rare instances, presumed propofol-induced hepatocellular injury following uneventful anesthesia and surgery has been described.68 Prolonged infusions of propofol may also result in excretion of green urine, reflecting the presence of phenols in the urine. This discoloration does not alter renal function. Urinary uric acid excretion is increased after administration of propofol and may manifest as cloudy urine when the uric acid crystallizes in the urine under conditions of low pH and temperature.20 This cloudy urine is not considered to be detrimental or indicative of adverse renal effects of propofol.
Intraocular Pressure
Laparoscopic surgery is associated with increased intraocular pressure and some consider laparoscopic surgery with the head down position a risk in the presence of preexisting ocular hypertension. In this regard, propofol is associated with significant decreases in intraocular pressure that occur immediately after induction of anesthesia and are sustained during tracheal intubation.1 Total IV anesthesia with propofol for laparoscopic surgery was associated with lower intraocular pressures than in patients undergoing similar surgery with isoflurane anesthesia (Fig. 5-7).69

Coagulation
Propofol does not alter tests of coagulation or platelet function. This is reassuring because the emulsion in which propofol is dispensed resembles intralipid, which has been associated with alterations in blood coagulation. However, propofol inhibits platelet aggregation that is induced by proinflammatory lipid mediators including thromboxane A2 and platelet-activating factor.70
Other Side Effects
Side effects of propofol may reflect the parent drug or actions attributed to the oil-in-water emulsion formulation. For example, some of the side effects of propofol (bradycardia, risk of infection, pain on injection, hypertriglyceridemia with prolonged administration, potential for pulmonary embolism) are believed to be due in large part to the lipid emulsion formulation.7,8
Allergic Reactions
Allergenic components of propofol include the phenyl nucleus and diisopropyl side chain.71 Patients who develop evidence of anaphylaxis on first exposure to propofol may have been previously sensitized to the diisopropyl radical, which is present in many dermatologic preparations. Likewise, the phenol nucleus is common to many drugs. Indeed, anaphylaxis to propofol during the first exposure to this drug has been observed, especially in patients with a history of other drug allergies, often to neuromuscular blocking drugs.72 Propofol-induced bronchoconstriction has been described in patients with allergy histories.38 The formulation of propofol administered to these patients was Diprivan containing soybean oil, glycerin, yolk lecithin, and sodium edetate.
Lactic Acidosis
Lactic acidosis (“propofol infusion syndrome”) has been described in pediatric and adult patients receiving prolonged high-dose infusions of propofol (>75 µg/kg/ minute) for longer than 24 hours.73,74 Severe, refractory, and fatal bradycardia in children in the ICU has been observed with long-term propofol sedation.75,76 Even short-term infusions of propofol (Diprivan) for surgical anesthesia have been associated with development of metabolic acidosis.77,78Unexpected tachycardia occurring during propofol anesthesia should prompt laboratory evaluation for possible metabolic (lactic) acidosis. Measurement of arterial blood gases and serum lactate concentrations is recommended. Documentation of an increased ion gap is useful but will take time and delay treatment, which includes prompt discontinuation of propofol administration.79 Metabolic acidosis in its early stages is reversible with discontinuation of propofol administration although cardiogenic shock requiring assistance with extracorporeal membrane oxygenation has been described in a patient receiving a prolonged propofol infusion (Diprivan) for a craniotomy.80
The mechanism for sporadic propofol-induced metabolic acidosis is unclear but may reflect poisoning (cytopathic hypoxia) of the electron transport chain and impaired oxidation of long chain fatty acids by propofol or a propofol metabolite in uniquely susceptible patients.81 Indeed, this propofol infusion syndrome mimics the mitochondrial myopathies, in which there are specific defects in the mitochondrial respiratory chain associated with specific mitochondrial DNA abnormalities, resulting in abnormal lipid metabolism in cardiac and skeletal muscles. These individuals, who are probably genetically susceptible, remain asymptomatic until a triggering event (sepsis, malnutrition) intervenes.
The differential diagnosis when propofol-induced lactic acidosis is suspected includes hyperchloremic metabolic acidosis associated with large volume infusions of 0.9% saline and metabolic acidosis associated with excessive generation of organic acids, such as lactate and ketones (diabetic acidosis, release of a tourniquet). Measurement of the anion gap and individual measurements of anions and organic acids will differentiate hyperchloremic metabolic acidosis from lactic acidosis.
Proconvulsant Activity
The majority of reported propofol-induced “seizures” during induction of anesthesia or emergence from anesthesia reflect spontaneous excitatory movements of subcortical origin.28 These responses are not thought to be due to cortical epileptic activity. Prolonged myoclonus associated with meningismus has been associated with propofol administration.82 The incidence of excitatory movements and associated ECG changes are low after the administration of propofol.83 Propofol resembles thiopental in that it does not produce seizure activity on the EEG when administered to patients with epilepsy, including those undergoing cortical resection.44 There appears to be no reason to avoid propofol for sedation, induction, and maintenance of anesthesia in patients with known seizures.10
Abuse Potential
Intense dreaming activity, amorous behavior, and hallucinations have been reported during recovery from low-dose infusions of propofol.28 Addiction to virtually all opioids and hypnotics, including propofol, has been described.84,85The death of music pop star Michael Jackson in 2009 from an overdose of propofol he was receiving as a sleep aid has recently brought the dangers of propofol misuse to public attention.86
Bacterial Growth
Propofol strongly supports the growth of Escherichia coli and Pseudomonas aeruginosa, whereas the solvent (Intralipid) appears to be bactericidal for these same organisms and bacteriostatic for Candida albicans.87 Clusters of postoperative surgical infections manifesting as temperature elevations have been attributed to extrinsic contamination of propofol.88,89 For this reason, it is recommended that (a) an aseptic technique be used in handling propofol as reflected by disinfecting the ampule neck surface or vial rubber stopper with 70% isopropyl alcohol; (b) the contents of the ampule containing propofol should be withdrawn into a sterile syringe immediately after opening and administered promptly; and (c) the contents of an opened ampule must be discarded if they are not used within 6 hours. In the ICU, the tubing and any unused portion of propofol must be discarded after 12 hours. Despite these concerns, there is evidence that when propofol is aseptically drawn into an uncapped syringe, it will remain sterile at room temperature for several days.90 Given the cost of propofol, some have questioned the logic of discarding unused drug at the end of an anesthetic or 6 hours, whichever occurs sooner.3
Antioxidant Properties
Propofol has potent antioxidant properties that resemble those of the endogenous antioxidant vitamin E.91,92 Like vitamin E, propofol contains a phenolic hydroxyl group that scavenges free radicals and inhibits lipid peroxidation. A neuroprotective effect of propofol may be at least partially related to the antioxidant potential of propofol’s phenol ring structure. For example, propofol reacts with lipid peroxyl radicals and thus inhibits lipid peroxidation by forming relatively stable propofol phenoxyl radicals. In addition, propofol also scavenges peroxynitrite, which is one of the most potent reactive metabolites for the initiation of lipid peroxidation. Because peroxynitrite is a potent bactericidal agent, it is likely that the peroxynitrite-scavenging activity of propofol contributes to this anesthetic’s known ability to suppress phagocytosis.93 Conversely, propofol might be beneficial in disease states, such as acute lung injury, in which peroxynitrite formation is thought to play an important role.94
Reintroduction of molecular oxygen into previously ischemic tissues (removal of an aortic cross-clamp) can further damage partially injured cells (reperfusion injury). Oxygen leads to the formation of free oxygen radicals, which react with polyunsaturated fatty acids of cell membranes resulting in disruption of cell membranes. Myocardial cell injury can cause postischemic dysfunction, myocardial stunning, and reperfusion cardiac dysrhythmias. Propofol strongly attenuates lipid peroxidation during coronary artery bypass graft surgery.95
Pain on Injection
Pain on injection is the most commonly reported adverse event associated with propofol administration to awake patients. This unpleasant side effect of propofol occurs in less than 10% of patients when the drug is injected into a large vein rather than a dorsum vein on the hand. Preceding the propofol with (using the same injection site as for propofol) 1% lidocaine or by prior administration of a potent short-acting opioid decreases the incidence of discomfort experienced by the patient. The incidence of thrombosis or phlebitis is usually less than 1%. Changing the composition of the carrier fat emulsion for propofol to long and medium chain triglycerides decreases the incidence of pain on injection.96
Accidental intraarterial injection of propofol has been described as producing severe pain but no vascular compromise.97 In an animal model, propofol-exposed arteries showed no changes in the vascular smooth muscle, and the endothelium was not damaged.98
Airway Protection
Inhaled and injected anesthetic drugs alter pharyngeal function with the associated risk of impaired upper airway protection and pulmonary aspiration. Subhypnotic concentrations of propofol, isoflurane, and sevoflurane decrease pharyngeal contraction force.99
Miscellaneous Effects
Propofol does not trigger malignant hyperthermia and has been administered to patients with hereditary coproporphyria without incident.100–102 Secretion of cortisol is not influenced by propofol, even when administered for prolonged periods in the ICU. Temporary abolition of tremors in patients with Parkinson’s disease may occur after the administration of propofol.103 For this reason, propofol may not be ideally suited for patients undergoing stereotactic neurosurgery during which the symptom is required to identify the correct anatomic location.
Etomidate
Etomidate is a carboxylated imidazole–containing compound that is chemically unrelated to any other drug used for the IV induction of anesthesia.104 The imidazole nucleus renders etomidate, like midazolam, water soluble at an acidic pH and lipid soluble at physiologic pH.
Commercial Preparation
The original formulation of etomidate included 35% propylene glycol (pH 6.9) contributing to a high incidence of pain during IV injection and occasional venous irritation. This has been changed to a fat emulsion, which has virtually abolished pain on injection and venous irritation, whereas the incidence of myoclonus remains unchanged. An oral formulation of etomidate for transmucosal delivery has been shown to produce dose-dependent sedation.105Administration through the oral mucosa results in direct systemic absorption while bypassing hepatic metabolism. As a result, higher blood concentrations are achieved more rapidly compared with drug that is administered by mouth.
Mechanism of Action
Etomidate is unique among injected and inhaled anesthetics in being administered as a single isomer.104 The anesthetic effect of etomidate resides predominantly in the R(+) isomer, which is approximately five times as potent as the S(−) isomer. In contrast to barbiturates, etomidate appears to be relatively selective as a modulator of GABAA receptors. Stereoselectivity of etomidate supports the concept that GABAA receptors are the site of action of etomidate. Etomidate exerts its effects on GABAA receptors by binding directly to a specific site or sites on the protein and enhancing the affinity of the inhibitory neurotransmitter (GABA) for these receptors.106 Antagonism of steroid-induced psychosis by etomidate is consistent with enhancement of GABA receptor function by this anesthetic drug.107 Etomidate is not known to modulate other ligand-gated ion channels in the brain at clinically relevant concentrations.
Pharmacokinetics
The volume of distribution (Vd) of etomidate is large, suggesting considerable tissue uptake (see Table 5-1). Distribution of etomidate throughout body water is favored by its moderate lipid solubility and existence as a weak base (pK 4.2, pH 8.2, 99% unionized at physiologic pH). Etomidate penetrates the brain rapidly, reaching peak levels within 1 minute after IV injection. About 76% of etomidate is bound to albumin independently of the plasma concentration of the drug. Decreases in plasma albumin concentrations, however, result in dramatic increases in the unbound pharmacologically active fraction of etomidate in the plasma. Prompt awakening after a single dose of etomidate principally reflects the redistribution of the drug from brain to inactive tissue sites. Rapid metabolism is also likely to contribute to prompt recovery.
Metabolism
Etomidate is rapidly metabolized by hydrolysis of the ethyl ester side chain to its carboxylic acid ester, resulting in a water-soluble, pharmacologically inactive compound. Hepatic microsomal enzymes and plasma esterases are responsible for this hydrolysis. Hydrolysis is nearly complete, as evidenced by recovery of less than 3% of an administered dose of etomidate as unchanged drug in urine. About 85% of a single IV dose of etomidate can be accounted for as the carboxylic acid ester metabolite in urine, whereas another 10% to 13% is present as this metabolite in the bile. Overall, the clearance of etomidate is about five times that for thiopental; this is reflected as a shorter elimination half-time of 2 to 5 hours. Likewise, the context-sensitive half-time of etomidate is less likely to be increased by continuous infusion as compared with thiopental.
Cardiopulmonary Bypass
Institution of hypothermic cardiopulmonary bypass causes an initial decrease of about 34% in the plasma etomidate concentration that then returns to within 11% of the prebypass value only to be followed by a further decrease with rewarming.22 The return of the plasma concentration toward prebypass levels is attributed to decreased metabolism, and the subsequent decrease on rewarming is attributed to increased metabolism. In addition, hepatic blood flow changes during cardiopulmonary bypass may alter metabolism, as etomidate is a high–hepatic extraction drug.
Clinical Uses
Etomidate may be viewed as an alternative to propofol or barbiturates for the IV induction of anesthesia, especially in the presence of an unstable cardiovascular system. After a standard induction dose of 0.2 to 0.4 mg/kg IV, the onset of unconsciousness occurs within one arm-to-brain circulation time. Involuntary myoclonic movements are common during the induction period as a result of alteration in the balance of inhibitory and excitatory influences on the thalamocortical tract. The frequency of this myoclonic-like activity can be attenuated by prior administration of an opioid. Awakening after a single IV dose of etomidate is more rapid than after barbiturates, and there is little or no evidence of a hangover or cumulative drug effect. Recovery of psychomotor function after administration of etomidate is intermediate between that of methohexital and thiopental. The duration of action is prolonged by increasing the dose of etomidate or administering the drug as a continuous infusion. As with barbiturates, analgesia is not produced by etomidate. For this reason, administration of an opioid before induction of anesthesia with etomidate may be useful to blunt the hemodynamic responses evoked by direct laryngoscopy and tracheal intubation. Etomidate, 0.15 to 0.3 mg/kg IV, has minimal effects on the duration of electrically induced seizures and thus may serve as an alternative to drugs that decrease the duration of seizures (propofol, thiopental) in patients undergoing electroconvulsive therapy.34
The principal limiting factor in the clinical use of etomidate for induction of anesthesia is the ability of this drug to transiently depress adrenocortical function (see the section “Adrenocortical Suppression”). It is widely viewed that postoperative nausea and vomiting is increased in patients receiving etomidate for induction of anesthesia.108 Nevertheless, comparison of etomidate with propofol did not document an increased incidence of nausea and vomiting in the first 24 hours after surgery in patients receiving etomidate.109
Side Effects
Central Nervous System
Etomidate is a potent direct cerebral vasoconstrictor that decreases cerebral blood flow and CMRO2 35% to 45%.110 As a result, previously increased ICP is lowered by etomidate. These effects of etomidate are similar to those changes produced by comparable doses of thiopental. Suppression of adrenocortical function limits the clinical usefulness for long-term treatment of intracranial hypertension (see the section “Adrenocortical Suppression”).
Etomidate produces a pattern on the EEG that is similar to thiopental. However, the frequency of excitatory spikes on the EEG is greater with etomidate than with thiopental and methohexital, suggesting caution in administration of etomidate to patients with a history of seizures.83 Like methohexital, etomidate may activate seizure foci, manifesting as fast activity on the EEG.111 For this reason, etomidate should also be used with caution in patients with focal epilepsy. Conversely, this characteristic has been observed to facilitate localization of seizure foci in patients undergoing cortical resection of epileptogenic tissue. Etomidate also possesses anticonvulsant properties and has been used to terminate status epilepticus. Etomidate has been observed to augment the amplitude of somatosensory evoked potentials, making monitoring of these responses more reliable.112
Cardiovascular System
Cardiovascular stability is characteristic of induction of anesthesia with 0.3 mg/kg IV of etomidate. After this dose of etomidate, there are minimal changes in heart rate, stroke volume, or cardiac output, whereas mean arterial blood pressure may decrease up to 15% because of decreases in systemic vascular resistance. The decrease in systemic blood pressure in parallel with changes in systemic vascular resistance suggests that administration of etomidate to acutely hypovolemic patients could result in sudden hypotension. When an induction dose of etomidate is 0.45 mg/kg IV, significant decreases in systemic blood pressure and cardiac output may occur.113 The cardiovascular effects of etomidate and thiopental are similar when continuously infused in patients with severe valvular heart disease.114
Effects of etomidate on myocardial contractility are important to consider, as this drug has been proposed for induction of anesthesia in patients with little or no cardiac reserve. It is difficult to document anesthetic-induced negative inotropic effects in vivo because of concurrent changes in preload, afterload, sympathetic nervous system activity, and baroreceptor reflex activity. Therefore, direct effects of anesthetics on intrinsic myocardial contractility may be more accurately assessed in vitro. In this regard, etomidate causes dose-dependent decreases in developed tension in isolated cardiac muscle obtained from patients undergoing coronary artery bypass graft operations or cardiac transplantation (Fig. 5-8).115 This depression was reversible with β-adrenergic stimulation. Nevertheless, concentrations required to produce these negative inotropic effects are in excess of those achieved with clinical use. In this regard, etomidate may differ from most other IV anesthetics in that depressive effects on myocardial contractility are minimal at concentrations needed for the production of anesthesia. Hepatic and renal functions tests are not altered by etomidate. Intraocular pressure is decreased by etomidate to a similar degree as by thiopental. Etomidate does not result in detrimental effects when accidentally injected into an artery.

Ventilation
The depressant effects of etomidate on ventilation seem to be less than those of barbiturates, although apnea may occasionally accompany a rapid IV injection of the drug.116 In the majority of patients, etomidate-induced decreases in tidal volume are offset by compensatory increases in the frequency of breathing. These effects on ventilation are transient, lasting only 3 to 5 minutes. Etomidate may stimulate ventilation independently of the medullar centers that normally respond to carbon dioxide. For this reason, etomidate may be useful when maintenance of spontaneous ventilation is desirable. Depression of ventilation may be exaggerated when etomidate is combined with inhaled anesthetics or opioids during continuous infusion techniques.
Pain on Injection
Pain on injection and venous irritation has been virtually eliminated with use of etomidate in a lipid emulsion vehicle rather than propylene glycol.
Myoclonus
Commonly administered IV anesthetics can cause excitatory effects that may manifest as spontaneous movements, such as myoclonus, dystonia, and tremor. These spontaneous movements, particularly myoclonus, occur in 50% to 80% of patients receiving etomidate in the absence of premedication.83 In one report, 87% of patients receiving etomidate developed excitatory effects, of which 69% were myoclonic. Multiple spikes appeared on the EEG of 22% of these patients.83 In this same report, the frequency of excitatory effects was 17% after thiopental, 13% after methohexital, and 6% after propofol, and none of the patients treated with other drugs developed myoclonus with spike activity on the EEG.83 Inclusion of atropine in the preoperative medication may suppress spike activity on the EEG associated with the administration of etomidate. Prior administration of an opioid (fentanyl, 1 to 2 µg/kg IV) or a benzodiazepine may decrease the incidence of myoclonus associated with administration of etomidate. Furthermore, the incidence and intensity of myoclonus following the administration of etomidate is dose-related and suppressed by pretreatment with small doses of etomidate (0.03 to 0.075 mg/kg IV) before administration of the induction dose.117
The mechanism of etomidate-induced myoclonus appears to be disinhibition of subcortical structures that normally suppress extrapyramidal motor activity. In many patients, excitatory movements are coincident with the early slow phase of the EEG, which corresponds to the beginning of deep anesthesia.83 It is possible that myoclonus could occur on awakening if the extrapyramidal system emerged more quickly than the cortex that inhibits it.118 The fact that etomidate-induced myoclonic activity may be associated with seizure activity on the EEG suggests caution in the use of this drug for the induction of anesthesia in patients with a history of seizure activity.83 Conversely, others have not documented seizure-like activity on the EEG in association with etomidate-induced myoclonus.117
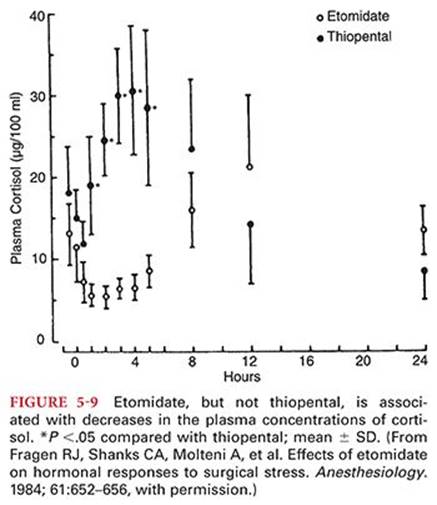
Adrenocortical Suppression
Etomidate causes adrenocortical suppression by producing a dose-dependent inhibition of the conversion of cholesterol to cortisol (Fig. 5-9).119,120 The specific enzyme inhibited by etomidate appears to be 11-β-hydroxylase as evidenced by the accumulation of 11-deoxycorticosterone.121 This enzyme inhibition lasts 4 to 8 hours after an induction dose of etomidate. Conceivably, patients experiencing sepsis or hemorrhage and who might require an intact cortisol response would be at a disadvantage should etomidate be administered.122 Conversely, suppression of adrenocortical function could be considered desirable from the standpoint of “stress-free” anesthesia. Nevertheless, in at least one report, it was not possible to demonstrate a difference in the plasma concentrations of cortisol, corticosterone, or adrenocorticotrophic hormone in patients receiving a single dose of etomidate or thiopental.123 In a retrospective study of more than 3,000 cardiac surgical patients who received etomidate for induction of anesthesia, there was no evidence to suggest that etomidate exposure was associated with severe hypotension, longer mechanical ventilation hours, longer length of hospital stay, or in-hospital mortality.124 In stark contrast, another large scale retrospective study demonstrated that anesthetic induction with etomidate, rather than propofol, was associated with increased 30-day mortality and cardiovascular morbidity after noncardiac surgery.125 The clinical benefit of minimizing cardiac suppression should be carefully weighed against the potential for worsened long-term outcomes when using propofol in high-risk patients.
Allergic Reactions
The incidence of allergic reactions following administration of etomidate is very low.126 When reactions have occurred, it is difficult to separate the role of etomidate from other concomitantly administered drugs (neuromuscular blocking drugs) that are more likely to evoke histamine release than etomidate.
Benzodiazepines
Benzodiazepines are drugs that exert, in slightly varying degrees, five principal pharmacologic effects: anxiolysis, sedation, anticonvulsant actions, spinal cord–mediated skeletal muscle relaxation, and anterograde amnesia (acquisition or encoding of new information).127 The amnestic potency of benzodiazepines is greater than their sedative effects resulting in a longer duration of amnesia than sedation. Stored information (retrograde amnesia) is not altered by benzodiazepines.128 Benzodiazepines do not produce adequate skeletal muscle relaxation for surgical procedures nor does their use influence the required dose of neuromuscular blocking drugs. The frequency of anxiety and insomnia in clinical practice combined with the efficacy of benzodiazepines has led to widespread use of these drugs. For example, it is estimated that 4% of the population uses “sleeping pills” sometime during a given year, and 0.4% of the population uses hypnotics for more than a year.129 Although benzodiazepines are effective for the treatment of acute insomnia, their use for management of chronic insomnia is decreasing. Compared with barbiturates, benzodiazepines have fewer tendencies to produce tolerance, less potential for abuse, a greater margin of safety, and elicit fewer and less serious drug interactions. Unlike barbiturates, benzodiazepines do not induce hepatic microsomal enzymes. Benzodiazepines are intrinsically far less addicting than opioids, cocaine, amphetamines, or barbiturates.
Midazolam is the most commonly used benzodiazepine in the perioperative period. Furthermore, the context-sensitive half-times for diazepam and lorazepam are prolonged; therefore, only midazolam is likely to be used for prolonged administration when prompt recovery is desired. However, the longer context-sensitive half-time of lorazepam makes this drug an attractive choice to facilitate sedation of patients in critical care environments. Unlike other drugs administered IV to produce CNS effects, benzodiazepines, as a class of drugs, are unique in the availability of a specific pharmacologic antagonist, flumazenil.
Structurally, benzodiazepines are similar and share many active metabolites.
Mechanism of Action
Benzodiazepines appear to produce all their pharmacologic effects by facilitating the actions of GABA.130 Benzodiazepines do not activate the GABAA receptors but rather enhance the affinity of the receptors for GABA (Fig. 5-10).131 As a result of increased affinity for GABA, there is more frequent channel openings, resulting in increased chloride conductance and hyperpolarization of the postsynaptic cell membrane. The postsynaptic neurons are thus rendered more resistant to excitation. This resistance to excitation is presumed to be the mechanism by which benzodiazepines produce anxiolysis, sedation, anterograde amnesia, alcohol potentiation, and anticonvulsant and skeletal muscle relaxant effects.

Benzodiazepines interact with a site located between the α and γ subunits of the GABAA receptor. The γ subunit is required for benzodiazepine binding. The α1- and α5-containing GABAA receptors are important for sedation, whereas anxiolytic activity is due to interaction with α2 and α5 subunit–containing receptors.132,133 The α1-containing GABAA receptors are the most abundant receptor subtypes accounting for approximately 60% of GABAAreceptors in the brain. α2 Subunits have more restricted expression, principally in the hippocampus and amygdala. The α5-containing GABAA receptors are principally extrasynaptic and are responsible for modulation of the resting membrane potential. This anatomic distribution of receptors is consistent with the minimal effects of these drugs outside the CNS (minimal circulatory effects). In the future, it may be possible to design benzodiazepines that selectively activate specific GABAA receptor types to produce anxiolysis without sedation.
The GABAA receptor is a large macromolecule that contains physically separate binding sites (principally α, β, and γ subunits) not only for GABA and the benzodiazepines but also barbiturates, etomidate, propofol, neurosteroids, and alcohol. Acting on a single receptor at different binding sites, the benzodiazepines, barbiturates, and alcohol can produce synergistic effects to increase GABAA receptor–mediated inhibition in the CNS. This property explains the pharmacologic synergy of these substances and, likewise, the risks of combined overdose, which can produce life-threatening CNS depression. This synergy is also the basis for pharmacologic cross-tolerance between these different classes of drugs and is consistent with the clinical use of benzodiazepines as the first-choice drugs for detoxication from alcohol. Conversely, benzodiazepines have a built-in ceiling effect that prevents them from exceeding the physiologic maximum of GABA inhibition. The low toxicity of the benzodiazepines and their corresponding clinical safety is attributed to this limitation of their effect on GABAergic neurotransmission.
Differences in the onset and duration of action among commonly administered benzodiazepines reflect differences in potency (receptor binding affinity), lipid solubility (ability to cross the blood–brain barrier and redistribute to peripheral tissues), and pharmacokinetics (uptake, distribution, metabolism, and elimination). All benzodiazepines are highly lipid soluble and are highly bound to plasma proteins, especially albumin. Hypoalbuminemia owing to hepatic cirrhosis or chronic renal failure may increase the unbound fraction of benzodiazepines, resulting in enhanced clinical effects produced by these drugs. Following oral administration, benzodiazepines are highly absorbed from the gastrointestinal tract and after IV injection, they rapidly enter the CNS and other highly perfused organs.
Nucleoside Transporter Systems
Benzodiazepines decrease adenosine degradation by inhibiting the nucleoside transporter, which is the principal mechanism whereby the effect of adenosine is terminated through reuptake into cells.134Adenosine is an important regulator of cardiac function (reduces cardiac oxygen demand by slowing heart rate and increases oxygen delivery by causing coronary vasodilation) and its physiologic effects convey cardioprotection during myocardial ischemia.
Electroencephalogram
The effects of benzodiazepines on the EEG resemble those of barbiturates in that α activity is decreased and low-voltage rapid β activity is increased. This shift from α to β activity occurs more in the frontal and rolandic areas with benzodiazepines, which, unlike the barbiturates, do not cause posterior spread. In common with barbiturates, however, tolerance to the effects of benzodiazepines on the EEG does not occur. Midazolam, in contrast to barbiturates and propofol, is unable to produce an isoelectric EEG.
Side Effects
Fatigue and drowsiness are the most common side effects in patients treated chronically with benzodiazepines. Sedation that could impair performance usually subsides within 2 weeks in patients chronically treated with benzodiazepines. Patients should be instructed to ingest benzodiazepines before meals and in the absence of antacids because meals and antacids may decrease absorption from the gastrointestinal tract. Chronic administration of benzodiazepines does not adversely affect systemic blood pressure, heart rate, or cardiac rhythm. Although effects on ventilation seem to be absent, it may be prudent to avoid these drugs in patients with chronic lung disease characterized by hypoventilation and/or decreased arterial oxygenation as they may interact with other medications to have adverse effects. Decreased motor coordination and impairment of cognitive function may occur, especially when benzodiazepines are used in combination with other CNS depressant drugs. Acute administration of benzodiazepines may produce transient anterograde amnesia, especially if there is concomitant ingestion of alcohol. For example, there have been reports of profound amnesia in travelers who have ingested triazolam combined with alcohol to facilitate sleep on airline flights across several time zones.135
Drug Interactions
Benzodiazepines exert synergistic sedative effects with other CNS depressants including alcohol, inhaled and injected anesthetics, opioids, and α2 agonists. Anesthetic requirements for inhaled and injected anesthetics are decreased by benzodiazepines. Although benzodiazepines, especially midazolam, potentiate the ventilatory depressant effects of opioids, the analgesic actions of opioids are reduced by benzodiazepines.136,137 Indeed, antagonism of benzodiazepine effects with flumazenil results in enhanced analgesic effects of opioids.
Hypothalamic-Pituitary-Adrenal Axis
Benzodiazepine-induced suppression of the hypothalamic-pituitary-adrenal axis is supported by evidence of suppression of cortisol levels in treated patients.138 In animals, alprazolam produces dose-dependent inhibition of adrenocorticotrophic hormone and cortisol secretion.139 This suppression is enhanced compared with other benzodiazepines and may contribute to the unique efficacy of alprazolam in the treatment of major depression.
Dependence
Even therapeutic doses of benzodiazepines may produce dependence as evidenced by the onset of physical or psychologic symptoms after the dosage is decreased or the drug is discontinued. Symptoms of dependence may occur after more than 6 months’ use of commonly prescribed low-potency benzodiazepines. It is misleading to consider dependence as evidence of addiction in the absence of inappropriate drug-seeking behaviors. Withdrawal symptoms (irritability, insomnia, tremulousness) have a time of onset that reflects the elimination half-time of the drug being discontinued. Typically, symptoms of withdrawal appear within 1 to 2 days for short-acting benzodiazepines and within 2 to 5 days for longer acting drugs.
Aging
Aging and liver disease affect glucuronidation less than oxidative metabolic pathways. Lorazepam, oxazepam, and temazepam are metabolized only by glucuronidation and have no active metabolites. For this reason, these benzodiazepines may be preferentially selected in elderly patients over benzodiazepines, such as diazepam, and that is metabolized by hepatic microsomal enzymes to form active metabolites. Elderly patients may also be intrinsically sensitive to benzodiazepines, suggesting that the enhanced response to these drugs that occurs with aging has pharmacodynamic as well as pharmacokinetic components. Long-term benzodiazepine administration may accelerate cognitive decline in elderly patients. Benzodiazepine withdrawal symptoms in the elderly include confusion. Postoperative confusion is more common in elderly long-term benzodiazepine users (daily use for >1 year) than in short-term users or nonusers of benzodiazepines.140
Platelet Aggregation
Benzodiazepines may inhibit platelet-activating factor–induced aggregation resulting in drug-induced inhibition of platelet aggregation. Midazolam-induced inhibition of platelet aggregation may reflect conformational changes in platelet membranes.141 Although benzodiazepines significantly inhibit platelet aggregation in vitro, they do not appear to affect the risk of hemorrhagic complications in patients with severe, chemotherapy-induced thrombocytopenia142; the clinical significance of benzodiazepine-induced inhibition of platelet aggregation in the surgical arena is unclear.
Midazolam
Midazolam is a water-soluble benzodiazepine with an imidazole ring in its structure that accounts for stability in aqueous solutions and rapid metabolism.143 This benzodiazepine has replaced diazepam for use in preoperative medication and conscious sedation. As with other benzodiazepines, the amnestic effects of midazolam are more potent than its sedative effects. Thus, patients may be awake following administration of midazolam but remain amnestic for events and conversations (postoperative instructions) for several hours.
Commercial Preparation
The pK of midazolam is 6.15, which permits the preparation of salts that are water soluble. The parenteral solution of midazolam used clinically is buffered to an acidic pH of 3.5. This is important because midazolam is characterized by a pH-dependent ring-opening phenomenon in which the ring remains open at pH values of less than 4, thus maintaining water solubility of the drug (Fig. 5-11). The ring closes at pH values of greater than 4, as when the drug is exposed to physiologic pH, thus converting midazolam to a highly lipid-soluble drug (see Fig. 5-11). The water solubility of midazolam obviates the need for a solubilizing preparation, such as propylene glycol required for other benzodiazepines that can produce venoirritation or interfere with absorption after intramuscular (IM) injection. Indeed, midazolam causes minimal to no discomfort during or after IV or IM injection. Midazolam is compatible with lactated Ringer solution and can be mixed with the acidic salts of other drugs, including opioids and anticholinergics.

Pharmacokinetics
Midazolam undergoes rapid absorption from the gastrointestinal tract and prompt passage across the blood–brain barrier. Despite this prompt passage into the brain, midazolam is considered to have a slow effect-site equilibration time (0.9 to 5.6 minutes) compared with other drugs such as propofol and thiopental. In this regard, IV doses of midazolam should be sufficiently spaced to permit the peak clinical effect to be appreciated before a repeat dose is considered. Only about 50% of an orally administered dose of midazolam reaches the systemic circulation, reflecting a substantial first-pass hepatic effect. As for most benzodiazepines, midazolam is extensively bound to plasma proteins; this binding is independent of the plasma concentration of midazolam (Table 5-2).143,144 The short duration of action of a single dose of midazolam is due to its lipid solubility, leading to rapid redistribution from the brain to inactive tissue sites as well as rapid hepatic clearance. The context-sensitive half-times for diazepam and lorazepam are prolonged compared with midazolam.
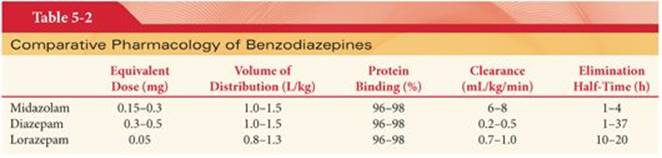
The elimination half-time of midazolam is 1 to 4 hours, which is much shorter than that of diazepam (see Table 5-2).143 The elimination half-time may be doubled in elderly patients, reflecting age-related decreases in hepatic blood flow and possibly enzyme activity. The volume of distribution (Vd) of midazolam and diazepam are similar, probably reflecting their similar lipid solubility and high degree of protein binding. Elderly and morbidly obese patients have an increased Vd of midazolam resulting from enhanced distribution of the drug into peripheral adipose tissues. The clearance of midazolam is more rapid than that of diazepam, as reflected by the context-sensitive half-time. As a result of these differences, the CNS effects of midazolam would be expected to be shorter than those of diazepam. Indeed, tests of mental function return to normal within 4 hours after the administration of midazolam in healthy young patients.
The institution of cardiopulmonary bypass is associated with a decrease in the plasma concentration of midazolam and an increase on termination of cardiopulmonary bypass.22 These changes are attributed to redistribution of priming fluid into body tissues. In addition, benzodiazepines are extensively bound to protein, and changes in protein concentrations and pH that accompany institution and termination of cardiopulmonary bypass may have significant effects on the unbound and pharmacologically active fractions of these drugs. The elimination half-time of midazolam is prolonged after cardiopulmonary bypass.
Metabolism
Midazolam is rapidly metabolized by hepatic and small intestine cytochrome P450 (CYP3A4) enzymes to active and inactive metabolites (Fig. 5-12).143 The principal metabolite of midazolam, 1-hydroxymidazolam, has approximately half the activity of the parent compound.145 This active metabolite is rapidly conjugated to 1-hydroxymidazolam glucuronide and is subsequently cleared by the kidneys. This glucuronide metabolite has substantial pharmacologic activity when present in high concentrations, as may occur in critically ill patients with renal insufficiency who are receiving continuous IV infusions of midazolam over prolonged periods of time. In these patients, the glucuronide metabolite may have synergistic sedative effects with the parent compound.146 The other pharmacologically active metabolite of midazolam, 4-hydroxymidazolam, is not present in detectable concentrations in the plasma following IV administration of midazolam.

Metabolism of midazolam is slowed in the presence of drugs (cimetidine, erythromycin, calcium channel blockers, antifungal drugs) that inhibit cytochrome P450 enzymes resulting in unexpected CNS depression.147 Cytochrome P450 3A enzymes also influence the metabolism of fentanyl. In this regard, the hepatic clearance of midazolam is inhibited by fentanyl as administered during general anesthesia.148Overall, the hepatic clearance rate of midazolam is 5 times greater than that of lorazepam and 10 times greater than that of diazepam.
Renal Clearance
The elimination half-time, Vd, and clearance of midazolam are not altered by renal failure.149 This is consistent with the extensive hepatic metabolism of midazolam.
Effects on Organ Systems
Central Nervous System
Midazolam, like other benzodiazepines, produces decreases in CMRO2 and cerebral blood flow analogous to barbiturates and propofol. Midazolam causes dose-related changes in regional cerebral blood flow in brain regions associated with the normal functioning of arousal, attention, and memory.150 Cerebral vasomotor responsiveness to carbon dioxide is preserved during midazolam anesthesia.43 Patients with decreased intracranial compliance show little or no change in ICP when given midazolam doses of 0.15 to 0.27 mg/kg IV. Thus, midazolam is an acceptable alternative to barbiturates for induction of anesthesia in patients with intracranial pathology. There is some evidence, however, that patients with severe head trauma but ICP of less than 18 mm Hg may experience an undesirable increase in ICP when midazolam (0.15 mg/kg IV) is administered rapidly (Fig. 5-13).151 Similar to thiopental, induction of anesthesia with midazolam does not prevent increases in ICP associated with direct laryngoscopy for tracheal intubation.152 Although midazolam may improve neurologic outcome after incomplete ischemia, benzodiazepines have not been shown to possess neuroprotective activity in humans. Midazolam is a potent anticonvulsant effective in the treatment of status epilepticus. Prolonged sedation of infants in critical care units (4 to 11 days) with midazolam and fentanyl has been associated with encephalopathy on withdrawal of the benzodiazepine.153 Paradoxical excitement occurs in less than 1% of all patients receiving midazolam and is effectively treated with a specific benzodiazepine antagonist, flumazenil.154

Ventilation
Midazolam produces dose-dependent decreases in ventilation with 0.15 mg/kg IV producing effects similar to diazepam, 0.3 mg/kg IV.155 Patients with chronic obstructive pulmonary disease experience even greater midazolam-induced depression of ventilation.156 Transient apnea may occur after rapid injection of large doses of midazolam (>0.15 mg/kg IV), especially in the presence of preoperative medication that includes an opioid.157 In healthy volunteers, midazolam alone produced no ventilatory depressant effects, whereas the combination of midazolam, 0.05 mg/kg IV, and fentanyl, 2 µg/kg IV, resulted in arterial hypoxemia and/or hypoventilation.158 Midazolam, 0.05 or 0.075 mg/kg IV, was shown to depress resting ventilation in healthy volunteers, whereas spinal anesthesia (mean sensory level T6) stimulated resting ventilation, and the combination had a modest synergistic effect for depressing resting ventilation.159 Benzodiazepines also depress the swallowing reflex and decrease upper airway activity.
Cardiovascular System
Midazolam, 0.2 mg/kg IV, for induction of anesthesia produces a greater decrease in systemic blood pressure and increase in heart rate than does diazepam, 0.5 mg/kg IV.160 Conversely, these midazolam-induced hemodynamic changes are similar to the changes produced by thiopental, 3 to 4 mg/kg IV.161 Cardiac output is not altered by midazolam, suggesting that blood pressure changes are due to decreases in systemic vascular resistance. In this regard, benzodiazepines may be beneficial in improving cardiac output in the presence of congestive heart failure. In the presence of hypovolemia, administration of midazolam results in enhanced blood pressure–lowering effects similar to those produced by other IV induction drugs.162 Midazolam does not prevent blood pressure and heart rate responses evoked by intubation of the trachea. In fact, this mechanical stimulus may offset the blood pressure–lowering effects of large doses of midazolam administered IV. The effects of midazolam on systemic blood pressure are directly related to the plasma concentration of the benzodiazepine. However, a plateau plasma concentration appears to exist (ceiling effect) above which little further change in systemic blood pressure occurs.
Clinical Uses
Preoperative Medication
Midazolam is the most commonly used oral preoperative medication for children. Oral midazolam syrup (2 mg/mL) is effective for producing sedation and anxiolysis at a dose of 0.25 mg/kg with minimal effects on ventilation and oxygen saturation even when administered at doses as large as 1 mg/kg (maximum, 20 mg).163 Midazolam, 0.5 mg/kg administered orally 30 minutes before induction of anesthesia, provides reliable sedation and anxiolysis in children without producing delayed awakening (Fig. 5-14).164 Although it is recommended that oral midazolam be administered at least 20 minutes before surgery, there is evidence that significant anterograde amnesia is present when 0.5 mg/kg orally is administered 10 minutes before surgery.165 Midazolam crosses the placenta but the fetal to maternal ratio is significantly less than that for other benzodiazepines.

Intravenous Sedation
Midazolam in doses of 1.0 to 2.5 mg IV (onset within 30 to 60 seconds, time to peak effect 3 to 5 minutes, duration of sedation 15 to 80 minutes) is effective for sedation during regional anesthesia as well as for brief therapeutic procedures. The effect-site equilibrium time for midazolam must be considered in recognizing the likely time of peak clinical effect and the need for supplemental doses of midazolam.
The most significant side effect of midazolam when used for sedation is depression of ventilation caused by a decrease in the hypoxic drive, particularly in concert with other anesthetic drugs. Midazolam-induced depression of ventilation is exaggerated (synergistic effects) in the presence of opioids and other CNS depressant drugs.137 Patients with chronic obstructive pulmonary disease may also manifest exaggerated depression of ventilation following administration of benzodiazepines to produce sedation. It is important to appreciate that increasing age greatly increases pharmacodynamic variability and is associated with generally increased sensitivity to the hypnotic effects of midazolam.166
Induction of Anesthesia
Although seldom used for this purpose currently, anesthesia can be induced by administration of midazolam, 0.1 to 0.2 mg/kg IV, over 30 to 60 seconds. Nevertheless, thiopental usually produces induction of anesthesia 50% to 100% faster than midazolam (Fig. 5-15).167 Onset of unconsciousness (synergistic interaction) is facilitated when a small dose of opioid (fentanyl, 50 to 100 µg IV or its equivalent) precedes the injection of midazolam by 1 to 3 minutes. The dose of midazolam required for the IV induction of anesthesia is also less when preoperative medication includes a CNS depressant drug. In healthy patients receiving small doses of benzodiazepines, the cardiovascular depression associated with these drugs is minimal. When significant cardiovascular responses occur, it is most likely a reflection of benzodiazepine-induced peripheral vasodilation. As with depression of ventilation, cardiovascular changes produced by benzodiazepines may be exaggerated in the presence of other CNS depressant drugs such as propofol and thiopental.

Maintenance of Anesthesia
Midazolam may be administered to supplement opioids, propofol, and/or inhaled anesthetics during maintenance of anesthesia. The context-sensitive half-time for midazolam increases modestly with an increasing duration of administration of a continuous infusion of this benzodiazepine.15 Anesthetic requirements for volatile anesthetics are decreased in a dose-dependent manner by midazolam. Awakening after general anesthesia that includes induction of anesthesia with midazolam is 1.0 to 2.5 times longer than that observed when thiopental is used for the IV induction of anesthesia.168 Gradual awakening in patients who receive midazolam is rarely associated with nausea, vomiting, or emergence excitement.
Postoperative Sedation
Long-term IV administration of midazolam (loading dose 0.5 to 4 mg IV and maintenance dose 1 to 7 mg per hour IV) to produce sedation in intubated patients results in relative saturation of peripheral tissues with midazolam and clearance from the systemic circulation becomes less dependent on redistribution into peripheral tissues and more dependent on hepatic metabolism.169 In addition, pharmacologically active metabolites may accumulate with prolonged IV administration of the parent drug. Under these conditions, plasma concentrations of midazolam decrease more slowly (emergence delayed) after discontinuation of the IV infusion compared with single IV injections. Emergence time is also a function of the plasma concentrations of midazolam at the time the IV infusion is discontinued. Patients maintained at higher plasma concentrations of midazolam take longer to awaken than patients maintained at lower plasma concentrations for comparable periods of time. The concomitant administration of analgesic doses of opioids greatly decreases the needed dose of midazolam and results in a more rapid recovery from sedation following discontinuation of the IV infusion of midazolam.169Emergence time from midazolam infusion is increased in elderly patients, obese patients, and in the presence of severe liver disease.
Paradoxical Vocal Cord Motion
Paradoxical vocal cord motion is a cause of nonorganic upper airway obstruction and stridor that may manifest postoperatively. Midazolam 0.5 to 1 mg IV may be an effective treatment for paradoxical vocal cord motion.170
Diazepam
Diazepam is a highly lipid-soluble benzodiazepine with a more prolonged duration of action compared with midazolam. Because of the beneficial aspects of midazolam pharmacology, parenteral diazepam is seldom used as part of current anesthetic regimens.
Commercial Preparation
Diazepam is dissolved in organic solvents (propylene glycol, sodium benzoate) because it is insoluble in water. The solution is viscid, with a pH of 6.6 to 6.9. Dilution with water or saline causes cloudiness but does not alter the potency of the drug. Injection by either the IM or IV route may be painful. Diazepam is also available in a unique soybean formulation for IV injection. This formulation is associated with a lower incidence of pain on injection and thrombophlebitis.
Pharmacokinetics
Diazepam is rapidly absorbed from the gastrointestinal tract after oral administration, reaching peak concentrations in about 1 hour in adults but as quickly as 15 to 30 minutes in children. There is rapid uptake of diazepam into the brain, followed by redistribution to inactive tissue sites, especially fat, as this benzodiazepine is highly lipid soluble. The Vd of diazepam is large, reflecting extensive tissue uptake of this lipid-soluble drug (see Table 5-2). Women, with a greater body fat content, are likely to have a larger Vd for diazepam than men. Diazepam rapidly crosses the placenta, achieving fetal concentrations equal to and sometimes greater than those present in the maternal circulation.171 The duration of action of benzodiazepines is not linked to receptor events but rather is determined by the rate of metabolism and elimination.
Protein Binding
The protein binding of benzodiazepines parallels their lipid solubility. As such, highly lipid-soluble diazepam is extensively bound, presumably to albumin (see Table 5-2). Cirrhosis of the liver or renal insufficiency, with associated decreases in plasma concentrations of albumin, may manifest as decreased protein binding of diazepam and an increased incidence of drug-related side effects.172 The high degree of protein binding limits the efficacy of hemodialysis in the treatment of diazepam overdose.
Metabolism
Diazepam is principally metabolized by hepatic microsomal enzymes using an oxidative pathway of N-demethylation. The two principal metabolites of diazepam are desmethyldiazepam and oxazepam, with a lesser amount metabolized to temazepam (Fig. 5-16). Desmethyldiazepam is metabolized more slowly than oxazepam and is only slightly less potent than diazepam. Therefore, it is likely that this metabolite contributes to the return of drowsiness that manifests 6 to 8 hours after administration of diazepam, as well as to sustained effects usually attributed to the parent drug. Alternatively, enterohepatic recirculation may contribute to recurrence of sedation.173 The plasma concentration of diazepam at this time is clinically insignificant and probably reflects its rapid removal as a conjugate of glucuronic acid. Ultimately, desmethyldiazepam is excreted in urine in the form of oxidized and glucuronide conjugated metabolites. Unchanged, diazepam is not appreciably excreted in urine. Benzodiazepines do not produce enzyme induction.

Elimination Half-Time
The elimination half-time of diazepam is prolonged, ranging from 21 to 37 hours in healthy volunteers (see Table 5-2). Cirrhosis of the liver is accompanied by up to fivefold increases in the elimination half-time of diazepam.174Likewise, the elimination half-time of diazepam increases progressively with increasing age, which contributes to the increased sensitivity of these patients to the drug’s sedative effects.174Prolongation of the elimination half-time of diazepam in the presence of cirrhosis of the liver is due to decreased protein binding of the drug, leading to an increased Vd. In addition, hepatic clearance of diazepam is likely to be decreased, reflecting decreased hepatic blood flow characteristic of cirrhosis of the liver. Compared with lorazepam, diazepam has a longer elimination half-time but shorter duration of action because it dissociates more rapidly than lorazepam from GABAA receptors, permitting more rapid redistribution to inactive tissue sites.
Desmethyldiazepam, the principal metabolite of diazepam, has an elimination half-time of 48 to 96 hours. As such, the elimination half-time of the metabolite may exceed that of the parent drug. Plasma concentrations of diazepam often decline more rapidly than plasma concentrations of desmethyldiazepam. This pharmacologically active metabolite can accumulate in plasma and tissues during chronic use of diazepam. Prolonged somnolence associated with high doses of diazepam is likely to be caused by sequestration of the parent drug and its active metabolite, desmethyldiazepam, in tissues, presumably fat, for subsequent release back into the circulation. A week or more is often required for elimination of these compounds from plasma after discontinuation of chronic diazepam therapy.
Effects on Organ Systems
Diazepam, like other benzodiazepines, produces minimal effects on ventilation and the systemic circulation. Hepatic and renal functions are not altered appreciably. Diazepam does not increase the incidence of nausea and vomiting. There is no change in the circulating plasma concentrations of stress-responding hormones (catecholamines, arginine vasopressin, cortisol).
Ventilation
Diazepam produces minimal depressant effects on ventilation, with detectable increases in PaCO2 not occurring until 0.2 mg/kg IV is administered. This slight increase in PaCO2 is due primarily to a decrease in tidal volume. Nevertheless, rarely, small doses of diazepam (<10 mg IV) have produced apnea.175 Combination of diazepam with other CNS depressants (opioids, alcohol) or administration of this drug to patients with chronic obstructive airway disease may result in exaggerated or prolonged depression of ventilation.
The slope of the line depicting the ventilatory response to carbon dioxide is decreased nearly 50% within 3 minutes after the administration of diazepam, 0.4 mg/kg IV (Fig. 5-17).176 This depression of the slope persists for about 25 minutes and parallels the level of consciousness. Despite the decrease in slope, the carbon dioxide response curve is not shifted to the right as observed with depression of ventilation produced by opioids. These depressant effects on ventilation seem to be a CNS effect because the mechanics of respiratory muscles are unchanged.
Cardiovascular System
Diazepam administered in doses of 0.5 to 1 mg/kg IV for induction of anesthesia typically produces minimal decreases in systemic blood pressure, cardiac output, and systemic vascular resistance that are similar in magnitude to those observed during natural sleep (10% to 20% decreases) (Table 5-3).177 Because of its relative hemodynamic stability, high-dose diazepam was once used for cardiac surgery. There is a transient depression of baroreceptor-mediated heart rate responses that is less than the depression evoked by volatile anesthetics but that could, in hypovolemic patients, interfere with optimal compensatory changes.178 In patients with increased left ventricular end diastolic pressure, a small dose of diazepam significantly decreases this pressure. Diazepam appears to have no direct action on the sympathetic nervous system, and it does not cause orthostatic hypotension.

The incidence and magnitude of systemic blood pressure decreases produced by diazepam seem to be less than those associated with barbiturates administered IV for the induction of anesthesia.179Nevertheless, occasionally, a patient may unpredictably experience hypotension with even small doses of diazepam.180 The addition of nitrous oxide after induction of anesthesia with diazepam is not associated with adverse cardiac changes (see Table 5-3).177Therefore, nitrous oxide can be administered in the presence of diazepam to ensure absence of patient awareness during surgery. This contrasts with direct myocardial depression and decreases in systemic blood pressure that occur when nitrous oxide is administered in the presence of opioids. Likewise, prior administration of diazepam, 0.125 to 0.5 mg/kg IV, followed by injection of fentanyl, 50 µg/kg IV, is associated with decreases in systemic vascular resistance and systemic blood pressure that do not accompany administration of the opioid alone.
Skeletal Muscle
Skeletal muscle relaxant effects reflect actions of diazepam on spinal internuncial neurons and not actions at the neuromuscular junction.181 Presumably, diazepam diminishes the tonic facilitatory influence on spinal γ neurons, and, thus, skeletal muscle tone is decreased. Tolerance occurs to the skeletal muscle relaxant effects of benzodiazepines.
Overdose
CNS intoxication can be expected at diazepam plasma concentrations of greater than 1,000 ng/mL. Despite massive overdoses of diazepam, serious sequelae (coma) are unlikely to occur if cardiac and pulmonary functions are supported and other drugs such as alcohol are not present.
Clinical Uses
Diazepam remains a popular oral drug for preoperative medication of adults and is the benzodiazepine most likely to be selected for management of delirium tremens and treatment of local anesthetic–induced seizures. Production of skeletal muscle relaxation by diazepam is often used in the management of lumbar disc disease and may be of value in the rare patient who develops tetany. Midazolam has largely replaced diazepam for IV sedation and the preoperative medication of children.
Anticonvulsant Activity
The prior administration of diazepam, 0.25 mg/kg IV, to animals protects against the development of seizures due to local anesthetic toxicity. Evidence for this protection is an increased convulsant dose of lidocaine in benzodiazepine-pretreated animals (Fig. 5-18).182 Diazepam, 0.1 mg/kg IV, is effective in abolishing seizure activity produced by lidocaine, delirium tremens, and status epilepticus.

The efficacy of diazepam as an anticonvulsant may reflect its ability to facilitate the actions of the inhibitory neurotransmitter GABA. In contrast to barbiturates, which inhibit seizures by nonselective depression of the CNS, diazepam selectively inhibits activity in the limbic system, particularly the hippocampus. If diazepam is administered to terminate seizures, a longer acting antiepileptic drug such as fosphenytoin is also administered.
Lorazepam
Lorazepam resembles oxazepam, differing only in the presence of an extra chloride atom on the ortho position of the 5-phenyl moiety. Lorazepam is a more potent sedative and amnesic than midazolam and diazepam, whereas its effects on ventilation, the cardiovascular system, and skeletal muscles resemble those of other benzodiazepines.
Pharmacokinetics
Lorazepam is conjugated with glucuronic acid in the liver to form pharmacologically inactive metabolites that are excreted by the kidneys. This contrasts with formation of pharmacologically active metabolites after the administration of midazolam and diazepam. The elimination half-time is 10 to 20 hours, with urinary excretion of lorazepam glucuronide accounting for greater than 80% of the injected dose (see Table 5-2). Compared with midazolam, lorazepam has a much slower metabolic clearance. This may be explained by the slower hepatic glucuronidation of lorazepam compared with more rapid oxidative hydroxylation of midazolam. Because formation of glucuronide metabolites of lorazepam is not entirely dependent on hepatic microsomal enzymes, the metabolism of lorazepam is less likely than that of diazepam to be influenced by alterations in hepatic function, increasing age, or drugs that inhibit P450 enzymes such as cimetidine. Indeed, the elimination half-time of lorazepam is not prolonged in elderly patients or in those treated with cimetidine. Lorazepam has a slower onset of action than midazolam or diazepam because of its lower lipid solubility and slower entrance into the CNS.
Clinical Uses
Lorazepam undergoes reliable absorption after oral and IM injection, which contrasts with diazepam. After oral administration, maximal plasma concentrations of lorazepam occur in 2 to 4 hours and persist at therapeutic levels for up to 24 to 48 hours. The recommended oral dose of lorazepam for preoperative medication is 50 µg/kg, not to exceed 4 mg.183 With this dose, maximal anterograde amnesia lasting up to 6 hours occurs, and sedation is not excessive. Larger oral doses produce additional sedation without increasing amnesia. The prolonged duration of action of lorazepam limits its usefulness for preoperative medication when rapid awakening at the end of surgery is desirable.
After a single IV dose (1 to 4 mg), the onset of effect occurs within 1 to 2 minutes, with a time-to-peak effect of 20 to 30 minutes, and a duration of sedative effects ranging from 6 to 10 hours.184 Infusions of lorazepam to produce postoperative sedation result in significant delays in emergence from sedation compared with midazolam.169 Obesity prolongs the sedative effects of lorazepam reflecting the larger volume of distribution and longer elimination half-time.
A slow onset limits the usefulness of lorazepam for (a) IV induction of anesthesia, (b) IV sedation during regional anesthesia, or (c) use as an anticonvulsant. Like diazepam, lorazepam is effective in limiting the incidence of emergence reactions after administration of ketamine. Although it is insoluble in water and thus requires use of solvents such as polyethylene glycol or propylene glycol, lorazepam is alleged to be less painful on injection and to produce less venous thrombosis than diazepam.
Lorazepam may be used as an economic alternative to midazolam for postoperative sedation of intubated patients. The risk of delayed emergence from sedation is increased when lorazepam is used for postoperative sedation and amnestic effects may last for several days. Delayed emergence from sedation may delay weaning from mechanical ventilation.
Oxazepam
Oxazepam, a pharmacologically active metabolite of diazepam, is commercially available (see Fig. 5-16). Its duration of action is slightly shorter than that of diazepam because oxazepam is converted to pharmacologically inactive metabolites by conjugation with glucuronic acid. The elimination half-time is 5 to 15 hours. Like lorazepam, the duration of action of oxazepam is unlikely to be influenced by hepatic dysfunction or administration of cimetidine.
Oral absorption of oxazepam is relatively slow. As a result, this drug may not be useful for the treatment of insomnia characterized by difficulty falling asleep. Conversely, oxazepam may be used for treatment of insomnia characterized by nightly awakenings or shortened total sleep time.
Alprazolam
Alprazolam has significant anxiety-reducing effects in patients with primary anxiety and panic attacks. Based on these effects, alprazolam may be an alternative to midazolam for preoperative medication.185Inhibition of adrenocorticotrophic hormone and cortisol secretion may be more prominent with alprazolam than with other benzodiazepines.
Clonazepam
Clonazepam is a highly lipid-soluble benzodiazepine that is well absorbed after oral administration. Clonazepam is metabolized to inactive conjugated and unconjugated metabolites that appear in the urine. The elimination half-time is 24 to 48 hours. Clonazepam is particularly effective in the control and prevention of seizures, especially myoclonic and infantile spasms.
Flurazepam
Flurazepam is chemically and pharmacologically similar to other benzodiazepines but is used exclusively to treat insomnia. After administration of 15 to 30 mg orally to adults, a hypnotic effect occurs in 15 to 25 minutes and lasts 7 to 8 hours. The period of rapid eye movement sleep is decreased by this drug. The principal metabolite of flurazepam is desalkylflurazepam. This metabolite is pharmacologically active and has a prolonged elimination half-time that may manifest as daytime sedation (hangover). Furthermore, repeated doses of flurazepam may result in accumulation of this metabolite, producing cumulative sedation.
Temazepam
Temazepam is an orally active benzodiazepine administered exclusively for the treatment of insomnia. Oral absorption is complete, but peak plasma concentrations do not reliably occur until about 2.5 hours after its administration. Metabolism in the liver results in weakly active to inactive metabolites that are conjugated with glucuronic acid. The elimination half-time is about 15 hours. Temazepam, 15 to 30 mg orally, does not alter the proportion of rapid eye movement sleep to total sleep in adults. Despite the relatively long elimination half-time, temazepam, as used to treat insomnia, is unlikely to be accompanied by residual drowsiness the following morning. Tolerance or signs of withdrawal do not occur, even after nightly administration for 30 consecutive days.
Triazolam
Triazolam is an orally absorbed benzodiazepine that is effective in the treatment of insomnia. Peak plasma concentrations after oral administration of 0.25 to 0.50 mg to adults occur in about 1 hour. The elimination half-time is 1.7 hours, rendering triazolam one of the shortest acting benzodiazepines. The two principal metabolites of triazolam have little if any hypnotic activity, and their elimination half-time is less than 4 hours. For these reasons, residual daytime effects or cumulative sedation effects with repeated doses of triazolam seem less likely than with other benzodiazepines.
Triazolam does not change the proportion of rapid eye movement to total sleep time. Rebound insomnia, however, may occur when this drug is discontinued. Marked anterograde amnesia has developed when this drug has been self-administered in attempts to facilitate sleep when traveling through several time zones.135 In otherwise healthy elderly patients, triazolam causes a greater degree of sedation or psychomotor impairment than in young persons.186These effects are due to decreased clearance and higher plasma concentrations rather than from an increased sensitivity to the drug. For these reasons, it is recommended that the dose of triazolam be decreased 50% in elderly persons.
Flumazenil
Flumazenil, a 1,4-imidazobenzodiazepine derivative, is a specific and exclusive benzodiazepine antagonist with a high affinity for benzodiazepine receptors, where it exerts minimal agonist activity.187,188 As a competitive antagonist, flumazenil prevents or reverses, in a dose-dependent manner, all the agonist effects of benzodiazepines. Flumazenil also effectively antagonizes the benzodiazepine component of ventilatory depression that is present during combined administration of a benzodiazepine and opioid.137 Metabolism of flumazenil is by hepatic microsomal enzymes to inactive metabolites.
Dose and Administration
The dose of flumazenil should be titrated individually to obtain the desired level of consciousness. The recommended initial dose is 0.2 mg IV (8 to 15 µg/kg IV), which typically reverses the CNS effects of benzodiazepine agonists within about 2 minutes. If required, further doses of 0.1 mg IV (to a total of 1 mg IV) may be administered at 60-second intervals. Generally, total doses of 0.3 to 0.6 mg IV have been adequate to decrease the degree of sedation to the required extent in patients sedated or anesthetized with benzodiazepines, whereas total doses of 0.5 to 1.0 mg IV are usually sufficient to completely abolish the effect of a therapeutic dose of a benzodiazepine. In patients who are unconscious due to an overdose with an unknown drug or drugs, failure to respond to IV doses of flumazenil of more than 5 mg probably indicates the involvement of intoxicants other than benzodiazepines or the presence of functional organic disorders. The duration of action of flumazenil is 30 to 60 minutes, and supplemental doses of the antagonist may be needed to maintain the desired level of consciousness. An alternative to repeated doses of flumazenil to maintain wakefulness is a continuous low-dose infusion of flumazenil, 0.1 to 0.4 mg per hour.187 The administration of flumazenil to patients being treated with antiepileptic drugs for control of seizure activity is not recommended as it could precipitate acute withdrawal seizures.189
Side Effects
Flumazenil-induced antagonism of excess benzodiazepine agonist effects is not followed by acute anxiety, hypertension, tachycardia, or neuroendocrine evidence of a stress response in postoperative patients.190,191 Reversal of benzodiazepine agonist effects with flumazenil is not associated with alterations in left ventricular systolic function or coronary hemodynamics in patients with coronary artery disease.192 The weak intrinsic agonist activity of flumazenil most likely attenuates evidence of abrupt reversal of agonist effects. Flumazenil does not alter anesthetic requirements (MAC) for volatile anesthetics, suggesting that these drugs do not exert any of their depressant effects on the CNS at benzodiazepine receptors.193 Flumazenil, administered at about 10 times the clinically recommended dose, has no agonist effects on resting ventilation or psychomotor performance in normal individuals.194
Short-Acting Nonbenzodiazepine Benzodiazepines
Benzodiazepine refers to a specific chemical structure consisting of a benzene ring and a diazepine ring, hence the name benzodiazepine. Unfortunately, the name has also come to refer to a pharmacologic class of drugs with a shared clinical activity and a shared molecular binding site on the GABAA receptor at the interface between the α and γ subunits, the benzodiazepine site. Eventually, drugs were found that bound to the same receptor, and exhibited the same pharmacology, but did not consist of a benzene ring bound to a diazepine ring. These drugs were given the cumbersome but vaguely amusing name: nonbenzodiazepine benzodiazepine. The agents that have been approved are zaleplon (Sonata), zolpidem (Ambien), and more recently, eszopiclone (Lunesta).
Zaleplon, zolpidem, and eszopiclone exert activity at the GABA receptor complex.195 These drugs seem to have more selectivity for certain subunits of GABA receptors, resulting in a clinical profile for treatment of sleeping disorders that is more efficacious with fewer side effects than occur with conventional benzodiazepines. Their use has steadily risen during the past decade, with 3% of Americans now reporting use of one or more of these agents during the prior month.196 Due to variations in binding to GABA receptor subunits, these drugs show differences in their effect on sleep stages. Zaleplon (10 mg orally) has a rapid elimination so there are few residual side effects after taking a single dose at bedtime. It may be particularly useful for patients with delayed onset of sleep. By comparison, zolpidem (10 mg orally) has a delayed elimination, prolonging drug effect. This may result in residual sedation and side effects but may be used for sustained treatment of insomnia with less waking during the night. All of these agents are slightly effective for insomnia, but their overall effects are of questionable clinical importance.197
Barbiturates
The introduction of thiopental in 1934 revolutionized the practice of anesthesia. This rapid-acting barbiturate made it possible to induce general anesthesia in seconds, avoiding a slow, often unpleasant, more dangerous induction with diethyl ether. The current edition of this text marks a turning point in anesthetic pharmacology: Thiopental and other barbiturate sedative-hypnotics were imported from manufacturers overseas, but these companies have now ceased exporting barbiturates to the United States in order to protest their use as a part of the lethal injection “cocktail” for capital punishment.198 We will still include a discussion of barbiturate pharmacology in this chapter, and this is done for several reasons: First, it is conceivable that shipments of these drugs may resume. Second, some anesthesiologists who practice outside of the United States use these drugs. Most importantly, the pharmacokinetics and pharmacodynamics of barbiturates are the prototypes and comparators for almost all of our clinically used IV anesthetics. To understand the literature on drugs like propofol, etomidate, and midazolam, it is critical to know the properties of barbiturates to which they were often compared, as these were the gold standard during their development.
Barbiturates’ Use in Anesthesia
The clinically used barbiturates are derived from barbituric acid. The substitutions on this molecule determine the physicochemical properties, pharmacokinetics, and the relative potency to produce various effects. Oxybarbiturates (pentobarbital, secobarbital) have oxygen at the second position. Replacement of the oxygen with a sulfur atom results in the corresponding thiobarbiturates (thiopental, thiamylal), which are much more lipid soluble and have greater hypnotic potency. A phenyl group at the fifth position (phenobarbital) increases the anticonvulsant, but not hypnotic, potency. On the other hand, a methyl group on the nitrogen (as with methohexital) increases hypnotic potency but lowers the seizure threshold and causes myoclonus during induction.
Mechanism of Action
Barbiturates are one of the earliest examples of CNS depressants that act in part by potentiating GABAA channel activity. At clinically used concentrations, they also act on glutamate, adenosine, and neuronal nicotinic acetylcholine receptors. Studies in knock-in mice have shown that GABAA receptors containing β3 subunits are responsible for the immobilizing activity of pentobarbital and partly responsible for the hypnotic activity.199 The interaction of barbiturates (as well as propofol and etomidate acting at different sites) functions allosterically to increase the affinity of GABA for its binding site, thereby increasing the duration of the GABAA-activated opening of chloride channels (see Fig. 5-10). Barbiturates can also mimic the action of GABA by directly activating GABAA receptors at higher doses.
Pharmacokinetics
Thiopental causes rapid onset and rapid awakening after a single IV dose due to rapid uptake then rapid redistribution out of the brain into inactive tissues (Fig. 5-19).200 As previously discussed, this is the basis for the short action of most other highly lipophilic drugs. Ultimately, elimination from the body depends almost entirely on metabolism because less than 1% of thiopental is recovered unchanged in urine.201 The time required for the plasma concentration of thiopental to decrease 50% after discontinuation of a prolonged infusion (context-sensitive half-time) is lengthy. The drug is sequestered in fat and skeletal muscle then it reenters the circulation and prevents the plasma concentration from dropping rapidly.15

Thiobarbiturates are metabolized in hepatocytes and, to a small extent, in extrahepatic sites such as the kidneys and possibly the CNS. Metabolites (particularly hydroxythiopental and the 5-carboxylic acid) are usually inactive and are always more water soluble than the parent compound, which facilitates renal excretion. Ultimately, metabolism of thiopental is almost complete (99%). Hepatic clearance of thiopental is characterized by a low hepatic extraction ratio and capacity-dependent elimination. This means factors affecting hepatic enzyme activity should change clearance. However, the reserve capacity of the liver to oxidize barbiturates is huge, so hepatic dysfunction must be extreme before a prolonged duration of action occurs.
In pediatric patients, the elimination half-time of thiopental is shorter than in adults.202 This is due to more rapid hepatic clearance of thiopental by pediatric patients. Therefore, recovery after large or repeated doses of thiopental may be more rapid for infants and children than for adults. Protein binding and Vd of thiopental are not different in pediatric and adult patients. Elimination half-time is prolonged during pregnancy because of the increased protein binding of thiopental.
Pharmacodynamics and Clinical Applications
Premedication
Oral and injectable barbiturates have been replaced by benzodiazepines for preanesthetic medication. Drowsiness may last for only a short time after a sedative-hypnotic dose of a barbiturate is administered orally, but residual CNS effects characterized as “hangover” may persist. The rapid onset of action of barbiturates renders these drugs useful for treatment of grand mal seizures, but, again, benzodiazepines are probably superior, providing a more specific site of action in the CNS. Rectal administration of barbiturates, especially methohexital, 20 to 30 mg/kg, has been used to induce anesthesia in uncooperative or young patients.203 Loss of consciousness after rectal administration of methohexital correlates with a plasma concentration greater than 2 µg/mL.204
Induction of Anesthesia
The relative potency of barbiturates used for IV induction of anesthesia assumes that thiopental is 1, thiamylal is 1.1, and methohexital is 2.5. At a blood pH of 7.4, methohexital is 76% nonionized compared with 61% for thiopental, which is consistent with the greater potency of methohexital. These drugs produce minimal to no direct effects on skeletal, cardiac, or smooth muscles. Induction dose requirements for thiopental vary with patient age, weight, and most importantly with cardiac output. The dose of thiopental required to induce anesthesia decreases with age, reflecting a slower passage of barbiturate from the central compartment to peripheral compartments (Fig. 5-20).205,206The dose of thiopental needed to produce anesthesia in early pregnancy (7 to 13 weeks’ gestation) is decreased about 18% compared with that for nonpregnant females (Fig. 5-21).207 Thiopental requirements, for unknown reasons, seem to be increased in children for more than 1 year after thermal injury.208 Despite a contrary clinical impression, thiopental dose requirements (with EEG suppression as the endpoint) are not different between nonalcoholics and alcoholics with abstinence of 9 to 17 days and 30 days (Fig. 5-22).209
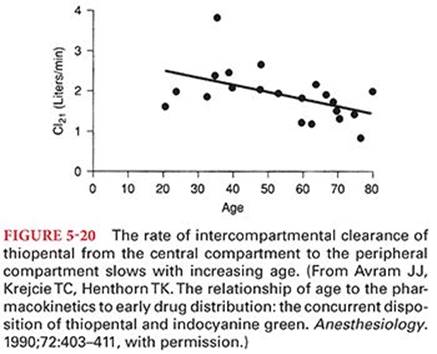


Methohexital is the only barbiturate with pharmacodynamic effects sufficiently different from thiopental and thiamylal to offer an alternative for IV induction. One advantage of methohexital is its effect to lower the seizure threshold. Methohexital, but not thiopental, is effective in inducing seizure activity in patients with psychomotor epilepsy undergoing temporal lobe resection of seizure-producing areas.210,211 The decreased anticonvulsant effect of methohexital is useful during electroconvulsive therapy because the therapeutic effect is related to the duration of the seizure. The principal disadvantage of methohexital is the incidence of excitatory phenomena, such as involuntary skeletal muscle movements (myoclonus) and other signs of excitatory activity including hiccoughs. These phenomena are dose dependent and may be decreased by pretreatment with opioids.
Occasionally, IV administration of a barbiturate is used as a supplement to inhaled anesthetics or as the sole anesthetic for brief and usually pain-free procedures such as cardioversion or electroconvulsive therapy. When high doses of methohexital are administered in a continuous infusion for neuroanesthesia, postoperative seizures occur in about one-third of patients.212 Thiopental infusion is seldom a satisfactory choice for maintenance of anesthesia because of its long context-sensitive half-time and prolonged recovery period.15
Even before the removal of barbiturates from the U.S. market, propofol had replaced them for induction of anesthesia in most cases. The time to awaken from a single induction dose of propofol was not that different, but it produced less nausea and generally patients met recovery milestones (voiding, walking) more rapidly, especially in those where rapid awakening is considered desirable.
Treatment of Increased Intracranial Pressure and Ischemic Injury
Barbiturates can be administered to decrease refractory ICP that remains increased despite other measures. Barbiturates decrease ICP by decreasing cerebral blood volume through drug-induced cerebral vascular vasoconstriction and an associated decrease in cerebral blood flow. The decrease in cerebral blood flow and increase in the perfusion-to-metabolism ratio made thiopental a useful drug for induction of anesthesia in patients with increased ICP (Fig. 5-23).213 The drug can be titrated to a level that produces EEG burst suppression, and an isoelectric EEG occurs with maximal (~55%) barbiturate-induced depression of CMRO2. However, this therapy produces significant hypotension, and improved outcome after head trauma has not been demonstrated in patients treated with barbiturates, despite the ability of these drugs to decrease and control ICP.214

Barbiturate therapy has also been used to improve brain survival after global cerebral ischemia due to cardiac arrest, but the efficacy for this indication remains unproven.215 There are data suggesting that neuropsychiatric complications after cardiopulmonary bypass (presumably due to embolism) clear more rapidly in patients treated prospectively with thiopental to maintain an isoelectric EEG.216 There is insufficient evidence, however, to support routine use of this therapy.
In contrast to global cerebral ischemia, animal studies consistently show improved outcome with barbiturate therapy of incomplete (focal) cerebral ischemia that permits drug-induced metabolic suppression.217 In this regard, barbiturate-induced decreases in CMRO2 exceed decreases in cerebral blood flow, which may provide protection to poorly perfused areas of the brain. The routine use of barbiturates during cardiac surgery or after stroke is not recommended because moderate degrees of hypothermia (33°C to 34°C) appear to provide superior neuroprotection without prolonging the recovery phase.
Side Effects
Side effects, especially on the cardiovascular system, inevitably accompany the clinical use of barbiturates. In normovolemic subjects, thiopental, 5 mg/kg IV, produces a transient 10- to 20-mm Hg decrease in blood pressure that is offset by a compensatory 15 to 20 beats per minute increase in heart rate (Fig. 5-24).218 The mild and transient decrease in systemic blood pressure that accompanies induction of anesthesia with barbiturates is principally due to peripheral vasodilation, reflecting depression of the medullary vasomotor center and decreased sympathetic nervous system outflow from the CNS. This dose of thiopental produces minimal to no evidence of direct myocardial depression.

Ventilation
Barbiturates also produce dose-dependent depression of medullary and pontine ventilatory centers. Thiopental decreases the sensitivity of the medullary ventilatory center to stimulation of carbon dioxide, and apnea is especially likely in the presence of other depressant drugs. Resumption of spontaneous ventilation after a single IV induction dose of barbiturate is characterized by a slow frequency of breathing and decreased tidal volume. Laryngeal reflexes and the cough reflex are not depressed until large doses of barbiturates have been administered.
Somatosensory Evoked Responses
Thiopental produces dose-dependent changes in median nerve somatosensory evoked responses and brainstem auditory evoked responses. However, some response is always obtainable,219 so thiopental is an acceptable drug to administer when the ability to monitor somatosensory evoked potentials is desirable.
Other Effects
Enzyme Induction
Barbiturates, especially phenobarbital, stimulate an increase in liver microsomal protein content (enzyme induction) after 2 to 7 days of sustained drug administration. Altered drug responses and drug interactions may reflect barbiturate-induced enzyme induction, resulting in accelerated metabolism of (a) other drugs, such as oral anticoagulants, phenytoin, and tricyclic antidepressants; or (b) endogenous substances, including corticosteroids, bile salts, and vitamin K. The production of heme is accelerated, and this may exacerbate acute intermittent porphyria in susceptible patients.
Intraarterial Injection
Inadvertent intraarterial injection of thiopental usually results in immediate, intense vasoconstriction and excruciating pain that radiates along the distribution of the artery. Vasoconstriction may obscure distal arterial pulses, and blanching of the extremity is followed by cyanosis. Gangrene and permanent nerve damage may occur.
Treatment of accidental intraarterial injection of a barbiturate includes immediate attempts to dilute the drug, prevention of arterial spasm by injecting vasodilators such as lidocaine or papaverine, and general measures to sustain adequate blood flow.
Allergic Reactions
Allergic reactions in association with IV administration of barbiturates for induction of anesthesia most likely represent anaphylaxis (antigen–antibody interaction). Nevertheless, thiopental can also produce signs of an allergic reaction in the absence of prior exposure, suggesting an anaphylactoid response.220 Although true anaphylaxis can occur, some of these reactions appear to be anaphylactoid responses due to direct release of histamine from tissue mast cells.220–222 The incidence of allergic reactions to thiopental is estimated to be 1 per 30,000 patients.223 The majority of reported cases are in patients with a history of chronic atopy who often have received thiopental previously without adverse responses.
Non–γ-Aminobutyric Acid Sedatives and Hypnotics
Ketamine
Ketamine is a phencyclidine derivative that produces “dissociative anesthesia,” which is characterized by evidence on the EEG of dissociation between the thalamocortical and limbic systems.224,225Dissociative anesthesia resembles a cataleptic state in which the eyes remain open with a slow nystagmic gaze. The patient is noncommunicative, although wakefulness may appear to be present. Varying degrees of hypertonus and purposeful skeletal muscle movements often occur independently of surgical stimulation. The patient is amnesic, and analgesia is intense. Ketamine has advantages over propofol and etomidate in not requiring a lipid emulsion vehicle for dissolution and in producing profound analgesia at subanesthetic doses. However, the frequency of emergence delirium limits the clinical usefulness of ketamine as a sole agent. Ketamine is a drug with significant abuse potential, emphasizing the need to take appropriate precautions against unauthorized nonmedical use.
Structure–Activity Relationships
Ketamine is a water-soluble molecule that structurally resembles phencyclidine. The presence of an asymmetric carbon atom results in the existence of two optical isomers of ketamine.224 The left-handed optical isomer of ketamine is designated S(+)-ketamine and the right-handed optical isomer is designated R(−)-ketamine. The racemic form of ketamine has been the most frequently used preparation although S(+)-ketamine is clinically available. When studied separately, S(+)-ketamine produces (a) more intense analgesia, (b) more rapid metabolism and thus recovery, (c) less salivation, and (d) a lower incidence of emergence reactions than R(−)-ketamine.226,227 For example, the analgesic potency of S(+)-ketamine is approximately twice that of racemic ketamine and four times greater than R(−)-ketamine. Ketamine isomer induces less fatigue and cognitive impairment than equianalgesic small-dose racemic ketamine.228 Both isomers of ketamine appear to inhibit uptake of catecholamines back into postganglionic sympathetic nerve endings (cocaine-like effect). The fact that individual optical isomers of ketamine differ in their pharmacologic properties suggests that this drug interacts with specific receptors to induce these behaviors. The preservative used for ketamine is benzethonium chloride.
Mechanism of Action
The mechanism of action of ketamine-induced analgesia and dissociative anesthesia is unknown. Ketamine is known to interact with multiple CNS receptors but clear association between receptor interaction and specific behavior has not been established. Ketamine binds noncompetitively to the phencyclidine recognition site on N-methyl-D-aspartate (NMDA) receptors. In addition, ketamine exerts effects at other sites including opioid receptors, monoaminergic receptors, muscarinic receptors, and voltage-sensitive sodium and L-type calcium channels and neuronal nicotinic acetylcholine receptors.229–231 Unlike propofol and etomidate, ketamine has only weak actions at GABAA receptors. Inflammatory mediators produced locally by compression of nerve roots can activate neutrophils that then adhere to blood vessels and impair blood flow. Ketamine suppresses neutrophil production of inflammatory mediators and improves blood flow.232 Direct inhibition of cytokines in blood by ketamine may contribute to the analgesic effects of this drug.
N-Methyl-D-Aspartate Receptor Antagonism
NMDA receptors (members of the glutamate receptors family) are ligand-gated ion channels that are unique in that channel activation requires binding of the excitatory neurotransmitter, glutamate with glycine as an obligatory coagonist (Fig. 5-25).224 Ketamine inhibits activation of NMDA receptors by glutamate and decreases presynaptic release of glutamate. The interaction with phencyclidine binding sites appears to be stereoselective, with the S(+) isomer of ketamine having the greatest affinity.

Opioid Receptors
Ketamine has been reported to directly interact with µ, δ, and κ opioid receptors.233 In contrast, other studies have suggested ketamine may be an antagonist at µ receptors and an agonist at κ receptors. Ketamine also weakly interacts with σ receptors.
Monoaminergic Receptors
The antinociceptive action of ketamine may involve activation of descending inhibitory monoaminergic pain pathways.
Muscarinic Receptors
Ketamine anesthesia is partially antagonized by anticholinesterase drugs. The fact that ketamine produces anticholinergic symptoms (emergence delirium, bronchodilation, sympathomimetic action) suggests that an antagonist effect of ketamine at muscarinic receptors is more likely than an agonist effect.
Sodium Channels
Consistent with its mild local anesthetic–like properties, ketamine interacts with voltage-gated sodium channels, sharing a binding site with local anesthetics.230
Neuronal Nicotinic Acetylcholine Receptors
Ketamine interacts with both heteromeric and homomeric α7 nicotinic acetylcholine receptors.231 In α7-type nicotinic receptors, a single subunit has been identified as a binding site in the extracellular loop between transmembrane segments 2 and 3.234 Nicotinic inhibition by ketamine does not appear to affect sedation or immobility but may play a role in its analgesic effects.235
Pharmacokinetics
The pharmacokinetics of ketamine are similar to thiopental in rapid onset of action, relatively short duration of action, and high lipid solubility (see Table 5-1). Ketamine has a pK of 7.5 at physiologic pH. Peak plasma concentrations of ketamine occur within 1 minute after IV administration and within 5 minutes after IM injection. Ketamine is not significantly bound to plasma proteins and leaves the blood rapidly to be distributed into tissues. Initially, ketamine is distributed to highly perfused tissues such as the brain, where the peak concentration may be four or five times that present in plasma. The extreme lipid solubility of ketamine (5 to 10 times that of thiopental) ensures its rapid transfer across the blood–brain barrier. Furthermore, ketamine-induced increases in cerebral blood flow could facilitate delivery of drug and thus enhance rapid achievement of high brain concentrations. Subsequently, ketamine is redistributed from the brain and other highly perfused tissues to less well-perfused tissues, the release of which results in late psychodynamic effects after emergence. Ketamine has a high hepatic clearance rate (1 L per minute) and a large Vd (3 L/kg), resulting in an elimination half-time of 2 to 3 hours. The high hepatic extraction ratio suggests that alterations in hepatic blood flow could influence ketamine’s clearance rate.
Metabolism
Ketamine is metabolized extensively by hepatic microsomal enzymes. An important pathway of metabolism is demethylation of ketamine by cytochrome P450 enzymes to form norketamine (Fig. 5-26).236 In animals, norketamine is one-fifth to one-third as potent as ketamine. This active metabolite may contribute to prolonged effects of ketamine (analgesia), especially with repeated doses or a continuous IV infusion. Norketamine is eventually hydroxylated and then conjugated to form more water-soluble and inactive glucuronide metabolites that are excreted by the kidneys. After IV administration, less than 4% of a dose of ketamine can be recovered from urine as unchanged drug. Fecal excretion accounts for less than 5% of an injected dose of ketamine. Chronic administration of ketamine stimulates the activity of enzymes responsible for its metabolism. Accelerated metabolism of ketamine as a result of enzyme induction could explain, in part, the observation of tolerance to the analgesic effects of ketamine that occurs in patients receiving repeated doses of this drug. Indeed, tolerance may occur in burn patients receiving more than two short-interval exposures to ketamine.237 Development of tolerance is also consistent with reports of ketamine dependence.236

Clinical Uses
Ketamine is a unique drug evoking intense analgesia at subanesthetic doses and producing prompt induction of anesthesia when administered IV at higher doses. Inclusion of an antisialagogue in the preoperative medication is often recommended to decrease the likelihood of coughing and laryngospasm due to ketamine-induced salivary secretions. Glycopyrrolate may be preferable, as atropine or scopolamine can easily cross the blood–brain barrier and could theoretically increase the incidence of emergence delirium (see the section “Emergence Delirium”).
Analgesia
Intense analgesia can be achieved with subanesthetic doses of ketamine, 0.2 to 0.5 mg/kg IV.238 Plasma concentrations of ketamine that produce analgesia are lower after oral than IM administration, presumably reflecting a higher norketamine concentration due to hepatic first-pass metabolism that occurs after oral administration. Analgesia is thought to be greater for somatic than for visceral pain. The analgesic effects of ketamine are likely due to its activity in the thalamic and limbic systems, which are responsible for the interpretation of painful signals. Small doses of ketamine are also useful adjuvants to opioid analgesia.239
Spinal cord sensitization is responsible for pain associated with touching or moving an injured body part that would normally not be painful. Central to the development of spinal cord sensitization is activation of NMDA receptors, which are located in the spinal cord dorsal horn. NMDA receptors are excitatory amino acid receptors that are important in pain processing and the modulation of pain.240 Excitatory amino acids, particularly glutamate, acting at NMDA receptors play an important role in spinal nociceptive pathways. Inhibition of spinal NMDA receptors by drugs such as ketamine, magnesium, and dextromethorphan is useful in the management of postoperative pain including decreases in analgesic consumption. Analgesia can be produced during labor without associated depression of the neonate.241,242Neonatal neurobehavioral scores of infants born by vaginal delivery with ketamine analgesia are lower than those for infants born with epidural anesthesia but higher than the scores in infants delivered with thiopental–nitrous oxide anesthesia.243 Postoperative sedation and analgesia after pediatric cardiac surgery can be produced by continuous infusions of ketamine, 1 to 2 mg/kg/hour. Ketamine is useful as an analgesic adjuvant in patients with preexisting chronic pain syndromes who require surgery.
Neuraxial Analgesia
The efficacy of extradural ketamine is controversial. Although ketamine has been reported to interact with opioid receptors, the affinity for spinal opioid receptors may be 10,000-fold weaker than that of morphine.244 It seems likely that extradural effects of ketamine (30 mg) are due to both spinal and systemic effects and possibly interaction with local anesthetic binding sites on voltage-gated sodium ion channels. Overall, the epidural effects of ketamine are relatively small but in combination with other epidural analgesics (opioids, local anesthetics), an additive or synergistic effect may occur.245 Intrathecal administration of ketamine (5 to 50 mg in 3 mL of saline) produces variable and brief analgesia, unless the ketamine is also combined with epinephrine to slow systemic absorption. The neuraxial use of ketamine to produce analgesia appears to be of limited value and is not an approved indication.229
Induction of Anesthesia
Induction of anesthesia is produced by administration of ketamine, 1 to 2 mg/kg IV or 4 to 8 mg/kg IM. Injection of ketamine IV does not produce pain or venous irritation. The need for large IV doses reflects a significant first-pass hepatic effect for ketamine. Consciousness is lost in 30 to 60 seconds after IV administration and in 2 to 4 minutes after IM injection. Unconsciousness is associated with maintenance of normal or only slightly depressed pharyngeal and laryngeal reflexes. Return of consciousness usually occurs in 10 to 20 minutes after an injected induction dose of ketamine, but return to full orientation may require an additional 60 to 90 minutes. Emergence times are even longer after repeated IV injections or a continuous infusion of ketamine. Amnesia persists for about 60 to 90 minutes after recovery of consciousness, but ketamine does not produce retrograde amnesia.
Because of its rapid onset of action, ketamine has been used as an IM induction drug in children and difficult-to-manage mentally challenged patients regardless of age. Due to its intense analgesic activity, ketamine has been used extensively for burn dressing changes, débridements, and skin grafting procedures. The excellent analgesia and ability to maintain spontaneous ventilation in an airway that might otherwise be altered by burn scar contractures are important advantages of ketamine in these patients. Tolerance may develop, however, in burn patients receiving repeated, short-interval anesthesia with ketamine.237
Induction of anesthesia in acutely hypovolemic patients is often accomplished with ketamine, taking advantage of the drug’s cardiovascular-stimulating effects. In this regard, it is important to recognize that ketamine, like all injected anesthetics, may become a myocardial depressant if endogenous catecholamine stores are depleted and sympathetic nervous system compensatory responses are impaired.246
The administration of ketamine to patients with coronary artery disease is complicated by increased myocardial oxygen requirements that may accompany this drug’s sympathomimetic effects on the heart. Furthermore, the absence of cardioprotective effects (preconditioning) associated with racemic ketamine is a consideration when this drug is administered to patients with known coronary artery disease (see the section on preconditioning). Nevertheless, induction of anesthesia with administration of diazepam, 0.5 mg/kg IV, and ketamine, 0.5 mg/kg IV, followed by a continuous infusion of ketamine, 15 to 30 µg/kg/minute IV, has been used for anesthesia in patients with coronary artery disease historically.236 The combination of subanesthetic doses of ketamine with propofol for production of total IV anesthesia has been reported to produce more stable hemodynamics than propofol and fentanyl while avoiding the undesirable emergence reactions that may accompany administration of higher doses of ketamine.247
The beneficial effects of ketamine on airway resistance due to drug-induced bronchodilation make this a potentially useful drug for rapid IV induction of anesthesia in patients with asthma.248
Ketamine should be used cautiously or avoided in patients with systemic or pulmonary hypertension or increased ICP, although this recommendation may deserve reevaluation based on more recent data (see the sections “Central Nervous System” and “Cardiovascular System”). Nystagmus associated with administration of ketamine may be undesirable in operations or examinations of the eye performed under anesthesia.
Ketamine has been administered safely to patients with malignant hyperthermia and does not trigger the syndrome in susceptible swine.249 Extensive experience with ketamine for pediatric cardiac catheterization has shown the drug to be useful, but its possible cardiac-stimulating effects must be considered in the interpretation of catheterization data.
Reversal of Opioid Tolerance
Subanesthetic doses of ketamine are effective in preventing and reversing morphine-induced tolerance.250 Although the mechanism of opioid-induced tolerance is unknown, it is believed to involve interaction between NMDA receptors, the nitric oxide pathway, and µ-opioid receptors. Administration of subanesthetic doses of ketamine (0.3 mg/kg/hour) reduces the likelihood of opioid tolerance and improves analgesia.
Improvement of Psychiatric Disorders
NMDA receptors for glutamate are thought to be involved in the pathophysiology of mental depression and the mechanism of action of antidepressants. Ketamine in small doses improved the postoperative depressive state in patients with mental depression.251 Intermittent treatment with low-dose ketamine also results in long-term suppression of obsessions and compulsions in patients with obsessive compulsive disorder.252
Restless Leg Syndrome
A single case report describes symptomatic improvement in two patients with restless leg syndrome treated with oral ketamine.253 It is possible that ketamine inhibits neuroinflammation in the spinal cord or higher centers. Within the spinal cord, restless leg syndrome may reflect NMDA receptor activation and production of inflammatory mediators that impair spinal cord blood flow.
Side Effects
Ketamine is unique among injected anesthetics in its ability to stimulate the cardiovascular system and produce emergence delirium.225 Although generally considered contraindicated in patients with increased ICP, it must be recognized that many of the early studies of ketamine’s effects on ICP were conducted on spontaneously breathing subjects.225
Central Nervous System
Ketamine is traditionally considered to increase cerebral blood flow and CMRO2, although there is also evidence suggesting that this may not be a valid generalization.225
Intracranial Pressure
Ketamine is reported to be a potent cerebral vasodilator capable of increasing cerebral blood flow by 60% in the presence of normocapnia.254 As a result, patients with intracranial pathology are commonly considered vulnerable to sustained increases in ICP after administration of ketamine. Nevertheless, in mechanically ventilated animals with increased ICP, there was no further increase in ICP after administration of ketamine, 0.5 to 2.0 mg/kg IV.255Furthermore, anterior fontanelle pressure, an indirect monitor of ICP, decreases in mechanically ventilated preterm neonates after administration of ketamine, 2 mg/kg IV.256 In patients requiring craniotomy for brain tumor or cerebral aneurysm resection, administration of ketamine, 1 mg/kg IV, did not increase middle cerebral artery blood flow velocity, and ICP decreased modestly (Fig. 5-27).257 In patients with traumatic brain injury, the administration of ketamine, 1.5, 3.0, and 5.0 mg/kg IV, during mechanical ventilation of the lungs resulted in significant decreases in ICP regardless of the dose of ketamine.258 These results in patients suggest that ketamine can be administered to patients with mildly increased ICP if administered with mild hyperventilation without adversely altering cerebral hemodynamics. Prior administration of thiopental, diazepam, or midazolam has been shown to blunt ketamine-induced increases in cerebral blood flow.
Neuroprotective Effects
Activation of NMDA receptors has been implicated in cerebral ischemic damage.229 The antagonist effect of ketamine on NMDA receptors suggests a possible neuroprotective role for this drug although this remains an unproved hypothesis. Indeed, S(+) ketamine offers no greater neuroprotection than remifentanil.259
Electroencephalogram
Ketamine’s effects on the EEG are characterized by abolition of alpha rhythm and dominance of θ activity. Onset of δ activity coincides with loss of consciousness. At high doses, ketamine produces a burst suppression pattern. Ketamine-induced excitatory activity occurs in both the thalamus and limbic systems without evidence of subsequent spread of seizure activity to cortical areas.260 As such, ketamine would be unlikely to precipitate generalized convulsions in patients with seizure disorders. Indeed, ketamine does not alter the seizure threshold in epileptic patients.261 Although myoclonic- and seizure-like activity may occur in normal patients, EEG evidence of cortical epileptic activity is absent and ketamine is considered to possess anticonvulsant activity.262
Somatosensory Evoked Potentials
Ketamine increases the cortical amplitude of somatosensory evoked potentials.263 This ketamine-induced increase in amplitude is attenuated by nitrous oxide. Auditory and visual evoked responses are decreased by ketamine.
Cardiovascular System
Ketamine produces cardiovascular effects that resemble sympathetic nervous system stimulation. Indeed, a direct negative cardiac inotropic effect is usually overshadowed by central sympathetic stimulation.
Hemodynamic Effects
Systemic and pulmonary arterial blood pressure, heart rate, cardiac output, cardiac work, and myocardial oxygen requirements are increased after IV administration of ketamine (Table 5-4).264 The increase in systolic blood pressure in adults receiving clinical doses of ketamine is 20 to 40 mm Hg, with a slightly smaller increase in diastolic blood pressure. Typically, systemic blood pressure increases progressively during the first 3 to 5 minutes after IV injection of ketamine and then decreases to predrug levels over the next 10 to 20 minutes. The cardiovascular-stimulating effects on the systemic and pulmonary circulations are blunted or prevented by prior administration of benzodiazepines or concomitant administration of inhaled anesthetics, including nitrous oxide.225,265 Likewise, ketamine administered to mildly sedated infants fails to produce hemodynamic changes in either the systemic or pulmonary circulation.266

Critically ill patients occasionally respond to ketamine with unexpected decreases in systemic blood pressure and cardiac output, which reflect depletion of endogenous catecholamine stores and exhaustion of sympathetic nervous system compensatory mechanisms, leading to an unmasking of ketamine’s direct myocardial depressant effects.246,267 Conversely, ketamine has been shown to decrease the need for inotropic support in septic patients, perhaps reflecting an inhibition of catecholamine reuptake.268,269
In shocked animals, ketamine is associated with an increased survival rate compared with animals anesthetized with halothane.270 Blood pressure may be better maintained in hemorrhaged animals anesthetized with ketamine. However, ketamine administration is associated with greater increases in arterial lactate concentrations than occur in animals with lower systemic blood pressures anesthetized with a volatile anesthetic.271 This suggests inadequate tissue perfusion despite maintenance of systemic blood pressure by ketamine. Presumably, ketamine-induced vasoconstriction maintains systemic blood pressure at the expense of tissue perfusion.
Cardiac Rhythm
The effect of ketamine on cardiac rhythm is inconclusive. There is evidence that ketamine enhances the dysrhythmogenicity of epinephrine.272 Conversely, ketamine may abolish epinephrine-induced cardiac dysrhythmias.
Mechanisms of Cardiovascular Effects
The mechanisms for ketamine-induced cardiovascular effects are complex. Direct stimulation of the CNS leading to increased sympathetic nervous system outflow seems to be the most important mechanism for cardiovascular stimulation.273 Evidence for this mechanism is the ability of inhaled anesthetics, ganglionic blockade, β blockade, cervical epidural anesthesia, and spinal cord transection to prevent ketamine-induced increases in systemic blood pressure and heart rate.274,275 Furthermore, increases in plasma concentrations of epinephrine and norepinephrine occur as early as 2 minutes after IV administration of ketamine and return to control levels 15 minutes later.276 In vitro, ketamine produces direct myocardial depression, emphasizing the importance of an intact sympathetic nervous system for the cardiac-stimulating effects of this drug.277 The role of ketamine-induced inhibition of norepinephrine uptake (reuptake) into postganglionic sympathetic nerve endings and associated increases of plasma catecholamine concentrations on the drug’s cardiac-stimulating effects are not known.272
Ventilation and Airway
Ketamine does not produce significant depression of ventilation. The ventilatory response to carbon dioxide is maintained during ketamine anesthesia and the PaCO2 is unlikely to increase more than 3 mm Hg.278 Breathing frequency typically decreases for 2 to 3 minutes after administration of ketamine. Apnea, however, can occur if the drug is administered rapidly IV or an opioid is included in the preoperative medication.
Upper airway skeletal muscle tone is well maintained, and upper airway reflexes remain relatively intact after administration of ketamine.279 Despite continued presence of upper airway reflexes, ketamine anesthesia does not negate the need for protection of the lungs against aspiration by placement of a cuffed tube in the patient’s trachea. Salivary and tracheobronchial mucous gland secretions are increased by IM or IV administration of ketamine, leading to the frequent recommendation that an antisialagogue be included in the preoperative medication when use of this drug is anticipated.
Bronchomotor Tone
Ketamine has bronchodilatory activity and is as effective as halothane or enflurane in preventing experimentally induced bronchospasm in dogs.248 Ketamine has been used in subanesthetic doses to treat bronchospasm in the operating room and ICU. Successful treatment of status asthmaticus with ketamine has been reported.280 In the presence of active bronchospasm, ketamine may be recommended as the IV induction drug of choice. The mechanism by which ketamine produces airway relaxation is unclear, although several mechanisms have been suggested, including increased circulating catecholamine concentrations, inhibition of catecholamine uptake, voltage-sensitive calcium channel block, and inhibition of postsynaptic nicotinic or muscarinic receptors.229
Hepatic or Renal Function
Ketamine does not significantly alter laboratory tests that reflect hepatic or renal function.
Allergic Reactions
Ketamine does not evoke the release of histamine and rarely, if ever, causes allergic reactions.281
Platelet Aggregation
Ketamine inhibits platelet aggregation possibly by suppressed formation of inositol 1,4,5-triphosphate and subsequent inhibition of cytosolic free calcium concentrations.282 Drug-induced effects on platelet aggregation are a consideration in patients with known bleeding disorders undergoing surgery.
Emergence Delirium (Psychedelic Effects)
Emergence from ketamine anesthesia in the postoperative period may be associated with visual, auditory, proprioceptive, and confusional illusions, which may progress to delirium. Cortical blindness may be transiently present. Dreams and hallucinations can occur up to 24 hours after administration of ketamine. The dreams frequently have a morbid content and are often experienced in vivid color. Dreams and hallucinations usually disappear within a few hours.
Mechanisms
Emergence delirium probably occurs secondary to ketamine-induced depression of the inferior colliculus and medial geniculate nucleus, leading to misinterpretation of auditory and visual stimuli.236Furthermore, the loss of skin and musculoskeletal sensations results in decreased ability to perceive gravity, thereby producing a sensation of bodily detachment or floating in space. Opioids that act as κ agonists produce similar psychedelic effects suggesting a potential role for ketamine interaction with κ receptors.
Incidence
The observed incidence of emergence delirium after ketamine ranges from 5% to 30% and is partially dose dependent.236 Factors associated with an increased incidence of emergence delirium include (a) age older than 15 years, (b) female gender, (c) doses of ketamine of greater than 2 mg/kg IV, and (d) a history of personality problems or frequent dreaming.236 In healthy volunteers, the incidence of psychedelic effects is related to the plasma concentration of ketamine (Fig. 5-28).283 It is possible that the incidence of dreaming is similar in children, but this age group is less able to communicate the dream’s occurrence. Indeed, there are reports of recurrent hallucinations in children as well as in adults receiving ketamine.284,285 Nevertheless, psychological changes in children after anesthesia with ketamine or inhaled drugs are not different.286 Likewise, no significant long-term personality differences are present in adults receiving ketamine compared with thiopental.287

Emergence delirium occurs less frequently when ketamine is used repeatedly. For example, it is rare for emergence delirium to occur after three or more anesthetics with ketamine. Finally, inhaled anesthetics can also produce auditory, visual, proprioceptive, and confusional illusions, but the incidence of such phenomena, especially unpleasant experiences, is indeed greater after anesthesia that includes administration of ketamine.
Prevention
A variety of drugs used in preoperative medication or as adjuvants during maintenance of anesthesia have been evaluated in attempts to prevent emergence delirium after administration of ketamine. Benzodiazepines have proved the most effective in prevention of this phenomenon, with midazolam being more effective than diazepam.288,289 A common approach is to administer the benzodiazepine IV about 5 minutes before induction of anesthesia with ketamine. Inclusion of thiopental or inhaled anesthetics may decrease the incidence of emergence delirium attributed to ketamine. Conversely, the inclusion of atropine in the preoperative medication may increase the incidence of emergence delirium.290
Despite contrary opinions, there is no evidence that permitting patients to awaken from ketamine anesthesia in quiet areas alters the incidence of emergence delirium.291 Prospective discussion with the patient of the common side effects of ketamine (dreams, floating sensations, blurred vision) is likely to decrease the incidence of emergence delirium and reduce concern if it occurs, as much as any other approach.236
Drug Interactions
The importance of an intact and normally functioning CNS in determining the cardiovascular effects of ketamine is emphasized by hemodynamic depression rather than stimulation that occurs when ketamine is administered in the presence of inhaled anesthetics. For example, depression by inhaled anesthetics of sympathetic nervous system outflow from the CNS prevents the typical increases in systemic blood pressure and heart rate that occur when ketamine is administered alone.274 Ketamine administered in the presence of volatile anesthetics may result in hypotension.292 Presumably, volatile anesthetics depress sympathetic nervous system outflow from the CNS, thus unmasking the direct cardiac depressant effects of ketamine. Diazepam, 0.3 to 0.5 mg/kg IV, or an equivalent dose of midazolam, is also effective in preventing the cardiac-stimulating effects of ketamine. In the presence of verapamil, the blood pressure–elevating effects of ketamine may be attenuated, whereas drug-induced increases in heart rate are enhanced.293 β Blockade reduces ketamine-induced increase in heart rate and blood pressure.
Ketamine-induced enhancement of nondepolarizing neuromuscular blocking drugs may reflect interference by ketamine with calcium ion binding or its transport.294 Alternatively, ketamine may decrease sensitivity of postjunctional membranes to neuromuscular blocking drugs. The duration of apnea after administration of succinylcholine is prolonged, possibly reflecting inhibition of plasma cholinesterase activity by ketamine.
Pharmacologic activation of adenosine triphosphate–regulated potassium (KATP) channels mimics ischemic preconditioning and decreases infarct size or improves functional recovery of ischemic-reperfused viable (stunned) myocardium. Conversely, pharmacologic blockade of (KATP) channels can antagonize the cardioprotective effects of ischemic preconditioning. In an animal model, ketamine blocked the cardioprotective effects of ischemic preconditioning and this effect was due to the R(−) isomer.295 Conversely, S(+)-ketamine does not block the cardioprotective effects of preconditioning or alter myocardial infarct size (Fig. 5-29).296 In patients at risk for myocardial infarction during the perioperative period, drugs known to block preconditioning should be used with caution, whereas drugs known to elicit early and late preconditioning (opioids, volatile anesthetics) may be beneficial.

Dextromethorphan
Dextromethorphan (d-isomer of levorphanol) is a low-affinity NMDA antagonist that is a common ingredient in over-the-counter cough suppressants. It also has activity at multiple other ligands including neuronal nicotinic receptors. It is equal in potency to codeine as an antitussive but lacks analgesic or physical dependence properties. Unlike codeine, this drug rarely produces sedation or gastrointestinal disturbances. Its euphoric effects lead to a significant abuse potential. Signs and symptoms of intentional excessive intake of dextromethorphan include systemic hypertension, tachycardia, somnolence, agitation, slurred speech, ataxia, diaphoresis, skeletal muscle rigidity, seizures, coma, and decreased core body temperature. Hepatotoxicity may be a consideration when dextromethorphan with acetaminophen is ingested in excessive amounts.
Dexmedetomidine
Dexmedetomidine is a highly selective, specific, and potent α2-adrenergic agonist (1,620:1 α2 to α1).297,298 One of the highest densities of α2 receptors is present in the pontine locus ceruleus, an important source of sympathetic nervous system innervation of the forebrain and a vital modulator of vigilance. The sedative effects evoked by dexmedetomidine most likely reflect inhibition of this nucleus.299
Dexmedetomidine is the dextroisomer and pharmacologically active component of medetomidine, which has been used for many years in veterinary practice for its hypnotic, sedative, and analgesic effects. Compared with clonidine, dexmedetomidine is 7 to 10 times more selective for α2 receptors and has a shorter duration of action than clonidine. In this regard, dexmedetomidine is considered a full agonist at the α2 receptor, whereas clonidine is a partial agonist (ratio of α2 to α1 activity for clonidine is 220:1).298 Atipamezole is a specific and selective α2 receptor antagonist that rapidly and effectively reverses the sedative and cardiovascular effects of IV dexmedetomidine.300
The quality of sedation produced by α2 agonists differs from sedation produced by drugs (midazolam, propofol) that act on GABA.301 For example, dexmedetomidine, acting on α2 receptors, produces sedation by decreasing sympathetic nervous system activity and the level of arousal. The result is a calm patient who can be easily aroused to full consciousness. Amnesia is not assured. Drugs that activate GABA receptors produce a clouding of consciousness and can cause paradoxical agitation as well as tolerance and dependence.
Pharmacokinetics
The elimination half-time of dexmedetomidine is 2 to 3 hours compared with 6 to 10 hours for clonidine. Dexmedetomidine is highly protein bound (>90%) and undergoes extensive hepatic metabolism. The resulting methyl and glucuronide conjugates are excreted by the kidneys. Dexmedetomidine has weak inhibiting effects on cytochrome P450 enzyme systems that might manifest as increased plasma concentrations of opioids as administered during anesthesia.302
Clinical Uses
As with clonidine, pretreatment with dexmedetomidine attenuates hemodynamic responses to tracheal intubation, decreases plasma catecholamine concentrations during anesthesia, decreases perioperative requirements for inhaled anesthetics and opioids, and increases the likelihood of hypotension.303,304 Dexmedetomidine decreases MAC for volatile anesthetics in animals by greater than 90% compared with a plateau effect between 25% to 40% for clonidine (Fig. 5-30).305 In patients, isoflurane MAC was decreased 35% and 48% by dexmedetomidine plasma concentrations of 0.3 ng/mL and 0.6 ng/mL, respectively.306 Despite marked dose-dependent analgesia and sedation produced by this drug, there is only mild depression of ventilation. Dexmedetomidine in high doses (loading dose of 1 µg/kg IV followed by 5 to 10 µg/kg/hour IV) produces total IV anesthesia without associated depression of ventilation.307 The preservation of breathing provides a potential anesthetic technique for patients with a difficult upper airway. As with clonidine, dexmedetomidine has been reported to be effective in attenuating the cardiostimulatory and postanesthetic delirium effects of ketamine.308 Addition of 0.5 µg/kg dexmedetomidine to lidocaine being administered to produce IV regional anesthesia improves the quality of anesthesia and postoperative analgesia without causing side effects.309 Dexmedetomidine markedly increases the range of temperatures not triggering thermoregulatory defenses. For this reason, dexmedetomidine, like clonidine, is likely to promote perioperative hypothermia and also prove to be an effective treatment for nonthermally induced shivering.310 Severe bradycardia may follow the administration of dexmedetomidine and cardiac arrest has been reported in a patient receiving a dexmedetomidine infusion as a supplement to general anesthesia.311

Postoperative Sedation
Dexmedetomidine (0.2 to 0.7 µg/kg/hour IV) is useful for sedation of postoperative critical care patients in an ICU environment, particularly when mechanical ventilation via a tracheal tube is necessary. In comparison with remifentanil, dexmedetomidine infusions do not result in clinically significant depression of ventilation and sedation exhibits some similarity with natural sleep.312 Following tracheal extubation, dexmedetomidine-sedated patients breathe spontaneously and appear calm and relaxed.313 Both clonidine and dexmedetomidine are useful in the ICU to prevent drug withdrawal symptoms following long-term sedation with benzodiazepines. Because of its sympatholytic and vagomimetic actions, dexmedetomidine may be accompanied by systemic hypotension and bradycardia. The ability to specifically antagonize the sedative effects of dexmedetomidine with atipamezole may be useful.300
Scopolamine
Scopolamine is a naturally occurring anticholinergic alkaloid derived from the belladonna plant. Scopolamine, also known as hyoscine, is a lipid-soluble tertiary amine that readily crosses the blood–brain barrier, where it binds muscarinic cholinergic receptors.314 Although chiral, the naturally occurring and biologically active enantiomer is l-scopolamine.
Following IV administration, scopolamine undergoes a biphasic elimination, with an elimination half-life of approximately 4.5 hours.315 Scopolamine has a volume of distribution of approximately 100 L, hepatic clearance of 1 L per minute, and a renal clearance of just 70 mL per minute. Only 6% of an IV dose appears as unchanged drug in the urine. It is almost never given orally, as the bioavailability is unpredictable, ranging from 10% to 50%.315
Clinical Uses
Sedation
As shown in Table 5-5, compared to atropine and glycopyrrolate, the other commonly used anticholinergics, scopolamine is notable for more specificity for the central effects rather than peripheral effects. Scopolamine is the only anticholinergic drug used primarily for sedation. Scopolamine is approximately 100 times more potent than atropine in decreasing the activity of the reticular activating system. Scopolamine, in addition to depressing the cerebral cortex, also affects other areas of the brain, causing amnesia. Typical doses of scopolamine (0.3 to 0.5 mg IM or IV) usually cause sedation, whereas similar doses of atropine produce minimal CNS effects. Scopolamine also greatly enhances the sedative effects of concomitantly administered drugs, especially opioids and benzodiazepines. Indeed, the combination of IM morphine and scopolamine was once a very popular form of preoperative sedation, following introduction of morphine–scopolamine (1.2 mg) combinations for anesthesia in 1900.316
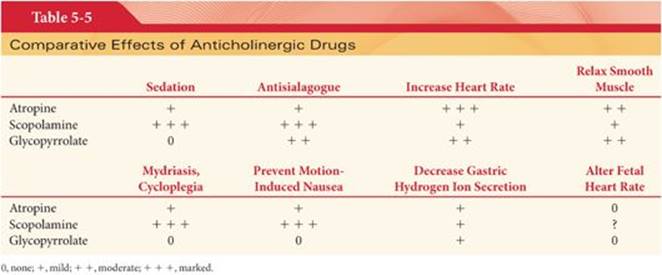
Occasionally, CNS effects of anticholinergic drugs, especially scopolamine, cause symptoms ranging from restlessness to somnolence. These symptoms are more likely to occur in elderly patients and should be considered as a possible explanation for delayed awakening from anesthesia or agitation in the early postoperative period. Inhaled anesthetics can potentiate the effects of anticholinergic drugs on the CNS, leading to an increased incidence of postoperative restlessness or somnolence. Physostigmine is effective in reversing restlessness or somnolence due to CNS effects of tertiary amine anticholinergic drugs. The typical dose of physostigmine for reversal of scopolamine sedation is 2 mg IV. Scopolamine has recently become a drug of abuse.
Antisialagogue Effect
Scopolamine is approximately three times more potent as an antisialagogue than atropine. For this reason, scopolamine is often selected when both an antisialagogue effect and sedation are desired results of preoperative medication. In equivalent antisialagogue doses, scopolamine, 0.3 to 0.5 mg IM, is less likely than atropine, 0.4 to 0.6 mg IM, to produce heart rate changes.
Antiemetic Effect
Scopolamine is commonly administered as a transcutaneous patch to prevent postoperative nausea and vomiting.
Side Effects
Mydriasis and Cycloplegia
Patients with glaucoma and parturients require special considerations in using anticholinergic drugs for preoperative medication. For example, the mydriatic effects of scopolamine are greater than those of atropine, suggesting caution in the administration of scopolamine to patients with glaucoma.317
Circular muscles of the iris that constrict the pupil are innervated by cholinergic fibers from the third cranial nerve, whereas fibers from the same nerve cause contraction of the ciliary muscles, allowing the lens to become more convex. Anticholinergic drugs placed topically on the cornea block the action of acetylcholine at both these sites, resulting in mydriasis and cycloplegia. Mydriasis produced by an anticholinergic drug is completely offset by topical placement on the cornea of an anticholinesterase drug such as pilocarpine.
Central Anticholinergic Syndrome
Scopolamine and, to a lesser extent, atropine can enter the CNS and produce symptoms characterized as the central anticholinergic syndrome. Symptoms range from restlessness and hallucinations to somnolence and unconsciousness. Presumably, these responses reflect blockade of muscarinic cholinergic receptors and competitive inhibition of the effects of acetylcholine in the CNS. Physostigmine, a lipid-soluble tertiary amine anticholinesterase drug administered in doses of 15 to 60 µg/kg IV, is a specific treatment for the central anticholinergic syndrome. Treatment may need to be repeated every 1 to 2 hours.
Overdose
Deliberate or accidental overdose with an anticholinergic drug produces a rapid onset of symptoms characteristic of muscarinic cholinergic receptor blockade. The mouth becomes dry, swallowing and talking is difficult, vision is blurred, photophobia is present, and tachycardia is prominent. The skin is dry and flushed, and a rash may appear especially over the face, neck, and upper chest (blush area). Even therapeutic doses of anticholinergic drugs sometimes may selectively dilate cutaneous vessels in the blush area. Body temperature is likely to be increased by anticholinergic drugs, especially when the environmental temperature is also increased. This increase in body temperature largely reflects inhibition of sweating by anticholinergic drugs, emphasizing that innervation of sweat glands is by sympathetic nervous system nerves that release acetylcholine as the neurotransmitter. Small children are particularly vulnerable to drug-induced increases in body temperature, with “atropine fever” occurring occasionally in this age group after administration of even a therapeutic dose of anticholinergic drug. Minute ventilation may be slightly increased due to CNS stimulation and the impact of an increased physiologic dead space due to bronchodilation. Arterial blood gases are usually unchanged. Skeletal muscle weakness and orthostatic hypotension, when present, reflect nicotinic cholinergic receptor blockade. Fatal events due to an overdose of an anticholinergic drug include seizures, coma, and medullary ventilatory center paralysis.
Small children and infants seem particularly vulnerable to developing life-threatening symptoms after an overdose with an anticholinergic drug. Physostigmine, administered in doses of 15 to 60 µg/kg IV, is the specific treatment for reversal of symptoms. Because physostigmine is metabolized rapidly, repeated doses of this anticholinesterase drug may be necessary to prevent the recurrence of symptoms.
References
1. Bryson HM, Fulton BR, Faulds D. Propofol: an update of its use in anaesthesia and conscious sedation. Drugs. 1995;50:513–559.
2. Fulton B, Sorkin EM. Propofol: an overview of its pharmacology and a review of its clinical efficacy in intensive care sedation. Drugs. 1995;50:636–657.
3. Smith I, White PF, Nathanson M, et al. Propofol: an update on its clinical use. Anesthesiology. 1994;81:1005–1043.
4. Ward DS, Norton JR, Guivarc’h PH, et al. Pharmacodynamics and pharmacokinetics of propofol in a medium-chain triglyceride emulsion. Anesthesiology. 2002;97:1401–1408.
5. Masaki Y, Tanaka M, Nishikawa T. Physicochemical compatibility of propofol-lidocaine mixture. Anesth Analg. 2003;97:1646–1651.
6. Song D, Hamza MA, White PF, et al. Comparison of a lower-lipid propofol emulsion with the standard emulsion for sedation during monitored anesthesia care. Anesthesiology. 2004;100:1072–1075.
7. Banaszczyk M, Carlo AT, Millan V, et al. Propofol phosphate, a water-soluble propofol prodrug: in vivo evaluation. Anesth Analg. 2002;95:1285–1292.
8. Fechner J, Ihmsen H, Hatterscheid D, et al. Pharmacokinetics and clinical pharmacodynamics of the new propofol prodrug GPI 15715 in volunteers. Anesthesiology. 2003;99:303–313.
9. Fechner J, Ihmsen H, Hatterscheid D, et al. Comparative pharmacokinetics and pharmacodynamics of the new propofol prodrug GPI 15715 and propofol emulsion. Anesthesiology. 2004;101:626–639.
10. Sneyd JR. Propofol and epilepsy. Br J Anaesth. 1999;82:168–169.
11. Yamakura T, Bertaccini E, Trudell JR, et al. Anesthetics and ion channels: molecular models and sites of action. Annu Rev Pharmacol Toxicol. 2001;41:23–51.
12. Kerz T, Hennes HJ, Feve A, et al. Effects of propofol on H-reflex in humans. Anesthesiology. 2001;94:32–37.
13. Court MH, Duan SX, Hesse LM, et al. Cytochrome P-450 2B6 is responsible for interindividual variability of propofol hydroxylation by human liver microsomes. Anesthesiology. 2001;94:110–119.
14. Takizawa D, Hiraoka H, Gota F, et al. Human kidneys play an important role in the elimination of propofol. Anesthesiology. 2005;102:327–330.
15. Hughes MA, Glass PSA, Jacobs JR. Context-sensitive half-time in multicompartment pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology. 1992;76:334–341.
16. Kuipers JA, Boer F, Olieman W, et al. First-pass lung uptake and pulmonary clearance of propofol: assessment with a recirculatory indocyanine green pharmacokinetic model. Anesthesiology. 1999;91:1780–1787.
17. Dawidowicz AL, Fornal E, Mardarowicz M, et al. The role of human lungs in the biotransformation of propofol. Anesthesiology. 2000;93:992–997.
18. He YL, Ueyama H, Tashiro C, et al. Pulmonary disposition of propofol in surgical patients. Anesthesiology. 2000;93:986–991.
19. Servin FS, Bourgeois B, Gomeni R, et al. Pharmacokinetics of propofol administered by target controlled infusion to alcoholic patients. Anesthesiology. 2003;99:576–585.
20. Masuda A, Asahi T, Sakamaki M, et al. Uric acid excretion increases during propofol anesthesia. Anesth Analg. 1997;85:144–148.
21. Dailland P, Cockshott ID, Lirzin JD, et al. Intravenous propofol during cesarean section: placental transfer, concentrations in breast milk, and neonatal effects. A preliminary study. Anesthesiology. 1989;71:827–834.
22. Gedney JA, Ghosh S. Pharmacokinetics of analgesics, sedatives and anaesthetic agents during cardiopulmonary bypass. Br J Anaesth. 1995;75:344–351.
23. Short TG, Lim TA, Tam YH. Prospective evaluation of pharmacokinetic model-controlled infusion of propofol in adult patients. Br J Anaesth. 1996;76:313–315.
24. Cheng SS, Yeh J, Flood P. Anesthesia matters: patients anesthetized with propofol have less postoperative pain than those anesthetized with isoflurane. Anesth Analg. 2008;106:264–269.
25. Pambianco DJ, Vargo JJ, Pruitt RE, et al. Computer-assisted personalized sedation for upper endoscopy and colonoscopy: a comparative, multicenter randomized study. Gastrointest Endosc. 2011;73:765–772.
26. Wahr JA, Plunkett JJ, Ramsay JG, et al. Cardiovascular responses during sedation after coronary revascularization: incidence of myocardial ischemia and hemodynamic episodes with propofol versus midazolam. Anesthesiology. 1996;84:1350–1360.
27. Parke TJ, Steven JE, Rice ASC, et al. Metabolic acidosis and fatal myocardial failure after propofol infusion in children: five case reports. BMJ. 1992;305:613–616.
28. Borgeat A, Wilder-Smith OHG, Suter PM. The nonhypnotic therapeutic applications of propofol. Anesthesiology. 1994;80:642–656.
29. Gan TJ, Glass PSA, Howell ST, et al. Determination of plasma concentrations of propofol associated with 50% reduction in postoperative nausea. Anesthesiology. 1997;87:779–784.
30. Gan TJ, Ginsberg B, Grant BS, et al. Double-blind, randomized comparison of ondansetron and intraoperative propofol to prevent postoperative nausea and vomiting. Anesthesiology. 1996;85:1036–1042.
31. Borgeat A. Subhypnotic doses of propofol do not possess antidopaminergic properties. Anesth Analg. 1997;84:196–198.
32. Chassard D, Lansiaux S, Duflo F, et al. Effects of subhypnotic doses of propofol on gastric emptying in volunteers. Anesthesiology. 2002;97:96–101.
33. Borgeat A, Wilder-Smith OHG, Saiah M, et al. Subhypnotic doses of propofol relieve pruritus induced by epidural and intrathecal morphine. Anesthesiology. 1992;76:510–512.
34. Avramov MN, Husain MM, White PF. The comparative effects of methohexital, propofol, and etomidate for electroconvulsive therapy. Anesth Analg. 1995;81:596–602.
35. Eames WO, Rooke GA, Sai-Chuen R, et al. Comparison of the effects of etomidate, propofol, and thiopental on respiratory resistance after tracheal intubation. Anesthesiology. 1996;84:1307–1311.
36. Brown RH, Greenberg RS, Wagner EM. Efficacy of propofol to prevent bronchoconstriction: effects of preservative. Anesthesiology. 2001;94:851–855.
37. Rieschke P, LaFleur BJ, Janicki PK. Effects of EDTA- and sulfite-containing formulations of propofol on respiratory system resistance after tracheal intubation in smokers. Anesthesiology. 2003;98:323–328.
38. Nishiyama T, Hanaoka K. Propofol-induced bronchoconstriction: two case reports. Anesth Analg. 2001;93:645–646.
39. Tibbs GR, Rowley TJ, Sanford RL, et al. HCN1 channels as targets for anesthetic and nonanesthetic propofol analogs in the amelioration of mechanical and thermal hyperalgesia in a mouse model of neuropathic pain. J Pharmacol Exp Ther. 2013;345:363–373.
40. Kaisti KK, Langsjo JW, Aalto S, et al. Effects of sevoflurane, propofol, and adjunct nitrous oxide on regional cerebral blood flow, oxygen consumption, and blood volume in humans. Anesthesiology. 2003;99:603–613.
41. Pinaud M, Leausque JN, Chetanneau A, et al. Effects of propofol on cerebral hemodynamics and metabolism in patients with brain trauma. Anesthesiology. 1990;73:404–409.
42. Girard F, Moumdjian R, Boudreault D, et al. The effect of propofol sedation on the intracranial pressure of patients with space-occupying lesion. Anesth Analg. 2004;99:573–577.
43. Strebel S, Kaufmann M, Guardiola PM, et al. Cerebral vasomotor responsiveness to carbon dioxide is preserved during propofol and midazolam anesthesia in humans. Anesth Analg. 1994;78:884–888.
44. Hewitt PB, Chu DKL, Polkey CE, et al. Effect of propofol on the electrocorticogram in epileptic patients undergoing cortical resection. Br J Anaesth. 1999;82:199–202.
45. Boisseau N, Madany M, Staccini P, et al. Comparison of the effects of sevoflurane and propofol on cortical somatosensory evoked potentials. Br J Anaesth. 2002;88:785–789.
46. Herrick IA, Craen RA, Gelb AW, et al. Propofol sedation during awake craniotomy for seizures: electrocorticographic and epileptogenic effects. Anesth Analg. 1997;84:1280–1284.
47. Veselis RA, Reinsel RA, Feshchenko VA, et al. The comparative amnestic effects of midazolam, propofol, thiopental, and fentanyl at equisedative concentrations. Anesthesiology. 1997;87:749–764.
48. Keidan I, Perel A, Shabtai EL, et al. Children undergoing repeated exposures for radiation therapy do not develop tolerance to propofol. Anesthesiology. 2004;100:251–254.
49. Rouby JJ, Andreev A, Leger P, et al. Peripheral vascular effects of thiopental and propofol in humans with artificial hearts. Anesthesiology. 1991;75:32–42.
50. Robinson BJ, Ebert TJ, O’Brien TJ, et al. Mechanisms whereby propofol mediates peripheral vasodilation in humans. Sympatho-inhibition or direct vascular relaxation? Anesthesiology. 1997;86:64–72.
51. Daniel M, Eger IE, Weiskopf RB, et al. Propofol fails to attenuate the cardiovascular response to rapid increases in desflurane concentration. Anesthesiology. 1996;84:75–80.
52. Lopatka CW, Muzi M, Eberft JT. Propofol, but not etomidate, reduces desflurane-mediated sympathetic activation in humans. Can J Anesth. 1999;46:342–347.
53. Kanaya N, Satoh H, Seki S, et al. Propofol anesthesia enhances the pressor response to intravenous ephedrine. Anesth Analg. 2002;94(5):1207–1211.
54. Deutschman CS, Harris AP, Fleisher LA. Changes in heart rate variability under propofol anesthesia: a possible explanation for propofol-induced bradycardia. Anesth Analg. 1994;79:373–377.
55. Lavoie J, Walsh EP, Burrows FA, et al. Effects of propofol or isoflurane anesthesia on cardiac conduction in children undergoing radiofrequency catheter ablation for tachydysrhythmias. Anesthesiology. 1995;82:884–887.
56. Sharpe MD, Dobkowski WB, Murkin JM, et al. Propofol has no direct effect on sinoatrial node function or on normal atrioventricular and accessory pathway conduction in Wolff-Parkinson-White syndrome during alfentanil/midazolam anesthesia. Anesthesiology. 1995;82:888–895.
57. Skei S, Ichimiya T, Hideaki T, et al. A case of normalization of Wolff-Parkinson-White syndrome conduction during propofol anesthesia. Anesthesiology. 1999;90:1779–1781.
58. Kleinsasser A, Kuenszberg E, Loeckinger A, et al. Sevoflurane, but not propofol, significantly prolongs the Q-T interval. Anesth Analg. 2000;90:25–27.
59. Egan TD, Brock UJG. Asystole after anesthesia induction with a fentanyl, propofol, and succinylcholine sequence. Anesth Analg. 1991;73:818–820.
60. Freysz M, Timourt Q, Betrix L, et al. Propofol and bradycardia. Can J Anaesth. 1991;28:137–138.
61. James MFM, Reyneke CJ, Whiffler K. Heart block following propofol: a case report. Br J Anaesth. 1989;62:213–215.
62. Tramer MR, Moore RA, McQuay HJ. Propofol and bradycardia: causation, frequency and severity. Br J Anaesth. 1997;78:642–651.
63. Tramer M, Moore A, McQuay H. Prevention of vomiting after paediatric strabismus surgery: a systematic review using the numbers-needed-to-treat method. Br J Anaesth. 1995;75:556–561.
64. Horiguchi T, Nishikawa T. Heart rate response to intravenous atropine during propofol anesthesia. Anesth Analg. 2002;95:389–392.
65. Bouillon T, Bruhn J, Radu-Radulescu L, et al. Mixed-effects modeling of the intrinsic ventilatory depressant potency of propofol in the non-steady state. Anesthesiology. 2004;100:240–250.
66. Nieuwenhuijs D, Sarton E, Teppema LJ, et al. Respirator sites of action of propofol: Absence of depression of peripheral chemoreflex loop by low-dose propofol. Anesthesiology. 2001;95:889–895.
67. Blouin RT, Seifert HA, Babenco HD, et al. Propofol decreases the hypoxic ventilatory response during conscious sedation and isohypercapnia. Anesthesiology. 1993;79:1177–1182.
68. Anand K, Ramsay MA, Crippin JS. Hepatocellular injury following the administration of propofol. Anesthesiology. 2001;95:1523–1524.
69. Mowafi HA, Al-Ghamdi A, Rushood A. Intraocular pressure changes during laparoscopy in patients anesthetized with propofol total intravenous anesthesia versus isoflurane inhaled anesthesia. Anesth Analg. 2003;97:471–474.
70. Fourcade O, Simon MF, Litt L, et al. Propofol inhibits human platelet aggregation induced by proinflammatory lipid mediators. Anesth Analg. 2004;99:393–398.
71. de Leon-Casasola, Weiss A, Lema MJ. Anaphylaxis due to propofol. Anesthesiology. 1992;77:384–386.
72. Laxenaire MC, Mata-Bremejo E, Moneret-Vautrin DA, et al. Life-threatening anaphylactoid reactions to propofol (Diprivan). Anesthesiology. 1992;77:275–280.
73. Badr AE, Mychaskiw GH, Eichhorn JH. Metabolic acidosis associated with a new formulation of propofol. Anesthesiology. 2001;94:536–538.
74. Cremer OL, Moons KG, Bouman EA, et al. Long-term propofol infusion and cardiac failure in adult head-injured patients. Lancet. 2001;357(9250):117–118.
75. Bray RJ. Fatal myocardial failure associated with a propofol infusion in a child. Anaesthesia. 1995;50:94.
76. Dearlove O, Dobson A. Does propofol cause death in children? Anaesthesia. 1995;50:916.
77. Burow BK, Johnson ME, Packer DL. Metabolic acidosis associated with propofol in the absence of other causative factors. Anesthesiology. 2004;101:239–241.
78. Salengros J-C, Velghe-Lenelle CE, Bollens R, et al. Lactic acidosis during propofol-remifentanil anesthesia in an adult. Anesthesiology. 2004;101:241–243.
79. Funston JS, Prough DS. Two reports of propofol anesthesia associated with metabolic acidosis in adults. Anesthesiology. 2004;101:6–8.
80. Culp KE, Augoustides JG, Ochroch AE, et al. Clinical management of cardiogenic shock associated with prolonged propofol infusion. Anesth Analg. 2004;99:221–226.
81. Wolf A, Weir P, Setage P, et al. Impaired fatty acid oxidation in propofol infusion syndrome. Lancet. 2001;357:606–607.
82. Hughes NJ, Lyons JB. Prolonged myoclonus and meningismus following propofol. Can J Anaesth. 1995;42:744–746.
83. Reddy RV, Moorthy SS, Dierdorf SF, et al. Excitatory effects and electroencephalographic correlation of etomidate, thiopental, methohexital, and propofol. Anesth Analg. 1993;77:1008–1011.
84. Follette JW, Farley WJ. Anesthesiologist addicted to propofol. Anesthesiology. 1992;77:817–818.
85. Earley PH, Finver T. Addiction to propofol: a study of 22 treatment cases. J Addict Med. 2013;7:169–176.
86. Monroe T, Hamza H, Stocks G, et al. The misuse and abuse of propofol. Subst Use Misuse. 2011;46:1199–1205.
87. Crowther J, Hrazdil J, Jolly DT, et al. Growth of microorganisms in propofol, thiopental, and a 1:1 mixture of propofol and thiopental. Anesth Analg. 1996;82:475–478.
88. Kuehnert MJ, Webb RM, Jochimsen EM, et al. Staphylococcus aureus bloodstream infections among patients undergoing electroconvulsive therapy traced to breaks in infection control and possible extrinsic contamination by propofol. Anesth Analg. 1997;85:420–425.
89. Nichols RL, Smith JW. Bacterial contamination of an anesthetic agent. N Engl J Med. 1995;333:184–185.
90. Warwick JP, Bladke D. Drawing up propofol [letter]. Anaesthesia. 1994;49:172.
91. Daskalopoulos R, Korcok J, Farhangkhgoee P, et al. Propofol protection of sodium-hydrogen activity sustains glutamate uptake during oxidative stress. Anesth Analg. 2001;93:1199–1204.
92. Peters CE, Korcok J, Gelb AW, et al. Anesthetic concentrations of propofol protect against oxidative stress in primary astrocyte cultures: comparison with hypothermia. Anesthesiology. 2001;94:313–321.
93. Krumholz W, Endrass J, Hempelmann G. Propofol inhibits phagocytosis and killing of Staphylococcus aureus and Escherichia coli by polymorphonuclear leukocytes in vitro. Can J Anaesth. 1994;41:446–449.
94. Kooy NW, Royall JA, Ye YZ, et al. Evidence for in vivo peroxynitrite production in human acute lung injury. Am J Respir Crit Care Med. 1995;151:1250–1254.
95. Sayin MM, Ozatamer O, Tasoz R, et al. Propofol attenuates myocardial lipid peroxidation during coronary artery bypass grafting surgery Br J Anaesth. 2002;89:242–246.
96. Doenicke AW, Roizen MF, Rau J, et al. Pharmacokinetics and pharmacodynamics of propofol in a new solvent. Anesth Analg. 1997;85:1399–1403.
97. Holley HS, Cuthrell L. Intraarterial injection of propofol. Anesthesiology. 1990;73:183–184.
98. MacPherson RD, Rasiah RL, McLeod LJ. Intraarterial propofol is not directly toxic to vascular endothelium. Anesthesiology. 1992;76:967–971.
99. Sundman E, Witt HR, Sandin R, et al. Pharyngeal function and airway protection during subhypnotic concentrations of propofol, isoflurane, and sevoflurane: volunteers examined by pharyngeal videoradiography and simultaneous manometry. Anesthesiology. 2001;95:1125–1132.
100. Kasraie N, Cousins TB. Propofol and the patient with hereditary coproporphyria. Anesth Analg. 1993;77:862–863.
101. Raff M, Harrison GG. The screening of propofol in MHS swine. Anesth Analg. 1989;68:750–751.
102. Sebel PS, Lowdon JD. Propofol: a new intravenous anesthetic. Anesthesiology. 1989;71:260–277.
103. Krauss JK, Akeyson EW, Giam P, et al. Propofol-induced dyskinesias in Parkinson’s disease. Anesth Analg. 1996;83:420–422.
104. Tomlin SL, Jenkins A, Lieb WR, et al. Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A receptors and animals. Anesthesiology. 1998;88(3):708–717.
105. Streisand JB, Jaarsma RL, Jay MA, et al. Oral transmucosal etomidate in volunteers. Anesthesiology. 1998;88:89–95.
106. Tomlin SL, Jenkins A, Lieb WR, et al. Stereoselective effects of etomidate optical isomers on gamma-aminobutyric acid type A receptors and animals. Anesthesiology. 1998;88:708–717.
107. Iibeigi MS, Davidson ML, Yarmush JM. An unexpected arousal effect of etomidate in a patient on high-dose steroids. Anesthesiology. 1998;89:1587–1589.
108. Holdcroft A, Morgan M, Whitwam JG, et al. Effect of dose and premedication on induction complications with etomidate. Br J Anaesth. 1976;48(3):199–205.
109. St Pierre M, Dunkel M, Rutherford A, et al. Does etomidate increase postoperative nausea? A double-blind controlled comparison of etomidate in lipid emulsion with propofol for balanced anaesthesia. Eur J Anaesthesiol. 2000;17:634–641.
110. Milde LN, Milde JH, Michenfelder JD. Cerebral functional, metabolic, and hemodynamic effects of etomidate in dogs. Anesthesiology. 1985;63:371–377.
111. Ebrahim ZY, DeBoer GE, Luders H, et al. Effect of etomidate on the electroencephalogram of patients with epilepsy. Anesth Analg. 1986;65:1004–1006.
112. Sloan TB, Ronai AK, Toleikis R, et al. Improvement of intraoperative somatosensory evoked potentials by etomidate. Anesth Analg. 1988;67:582–585.
113. Craido A, Maseda J, Navarro E, et al. Induction of anaesthesia with etomidate: haemodynamic study of 36 patients. Br J Anaesth. 1980;52:803–809.
114. Karliczek GF, Brenken U, Schokkenbrock R, et al. Etomidate-analgesic combinations for the induction of anesthesia in cardiac patients. Anaesthesist. 1982;31:213–220.
115. Sprung J, Ogletree-Hughes ML, Moravec CS. The effects of etomidate on the contractility of failing and nonfailing human heart muscle. Anesth Analg. 2000;91:68–75.
116. Choi SD, Spulding BC, Gross JB, et al. Comparison of the ventilatory effects of etomidate and methohexital. Anesthesiology. 1985;62:442–447.
117. Doenicke AW, Roizen MF, Kugler J, et al. Reducing myoclonus after etomidate. Anesthesiology. 1999;90:113–119.
118. Laughlin TP, Newberg LA. Prolonged myoclonus after etomidate anesthesia. Anesth Analg. 1985;64:80–82.
119. Fragen RJ, Shanks CA, Molteni A, et al. Effects of etomidate on hormonal responses to surgical stress. Anesthesiology. 1984;61:652–656.
120. Wagner RL, White PF, Kan PB, et al. Inhibition of adrenal steroidogenesis by the anesthetic etomidate. N Engl J Med. 1984;310:1415–1421.
121. Owen H, Spence AA. Etomidate. Br J Anaesth. 1984;56:555–557.
122. Longnecker DE. Stress free: to be or not to be? Anesthesiology. 1984;61:643–644.
123. Duthie DJR, Fraser R, Nimmo WS. Effect of induction of anaesthesia with etomidate on corticosteroid synthesis in man. Br J Anaesth. 1985;57:156–159.
124. Wagner CE, Bick JS, Johnson D, et al. Etomidate use and postoperative outcomes among cardiac surgery patients. Anesthesiology. 2014;120:579–589.
125. Komatsu R, You J, Mascha EJ, et al. Anesthetic induction with etomidate, rather than propofol, is associated with increased 30-day mortality and cardiovascular morbidity after noncardiac surgery. Anesth Analg. 2013;117:1329–1337.
126. Watkins JA. Etomidate: an “immunologically safe” anaesthetic agent. Anaesthesia. 1983;34:208–210.
127. Ashton A. Guidelines for the rational use of benzodiazepines: when and what to use. Drugs. 1994;48:25–40.
128. Ghoneim MM, Mewaldt SP. Benzodiazepines and human memory: a review. Anesthesiology. 1990;72:926–938.
129. Nowell PD, Mazumdar S, Buysse DJ, et al. Benzodiazepines and zolpidem for chronic insomnia: a meta-analysis of treatment efficacy. JAMA. 1997;278:2170–2177.
130. Goodchild CS. GABA receptors and benzodiazepines. Br J Anaesth. 1993;71:127–133.
131. Mohler H, Richards JG. The benzodiazepine receptor: a pharmacological control element of brain function. Eur J Anaesthesiol Suppl. 1988;2:15–24.
132. Low K, Crestani F, Keist R, et al. Molecular and neuronal substrate for the selective attenuation of anxiety. Science. 2000;290:131–134.
133. McKernan RM, Rosahl TW, Reynolds DS, et al. Sedative but not anxiolytic properties of benzodiazepines are mediated by GABAA receptor alpha1 subtype. Nat Neurosci. 2000;3:587–592.
134. Seubert CN, Morey TE, Martynuk AE, et al. Midazolam selectively potentiates the A2a- but not the A1-receptor–mediated effects of adenosine. Anesthesiology. 2000;92:567–577.
135. Morris HH, Estes ML. Traveler’s amnesia. Transient global amnesia secondary to triazolam. JAMA. 1987;258:945–946.
136. Gear RW, Miaskowski C, Heller PH, et al. Benzodiazepine mediated antagonism of opioid analgesia. Pain. 1997;71:25–29.
137. Gross JB, Blouin RT, Zandsberg S, et al. Effect of flumazenil on ventilatory drive during sedation with midazolam and alfentanil. Anesthesiology. 1996;85:713–720.
138. Petraglia F, Bakalakis S, Facchinetti F, et al. Effects of sodium valproate and diazepam on beta-endorphin, beta-lipotropin and cortisol secretion induced by hypoglycemic stress in humans. Neuroendocrinology. 1986;44:320–325.
139. Kalogeras KT, Calogero AE, Kuribayiashi T, et al. In vitro and in vivo effects of the triazolobenzodiazepine alprazolam on hypothalamic-pituitary adrenal function: pharmacological and clinical implications. J Clin Endocrinol Metab. 1990;70:1462–1471.
140. Kudoh A, Takase H, Takahira Y, et al. Postoperative confusion increases in elderly long-term benzodiazepine users. Anesth Analg. 2004;99:1674–1678.
141. Sheu JR, Hsiao G, Luk HN, et al. Mechanisms involved in the antiplatelet activity of midazolam in human platelets. Anesthesiology. 2002;96:651–658.
142. Rysler C, Stoffel N, Buser A, et al. Effect of beta-blockers, Ca2+ antagonists, and benzodiazepines on bleeding incidence in patients with chemotherapy induced thrombocytopenia. Platelets. 2010;21:77–83.
143. Reves JG, Fragen RJ, Vinik HR, et al. Midazolam: pharmacology and uses. Anesthesiology. 1985;62:310–324.
144. Greenblatt DJ, Abernathy DR, Locniskar A, et al. Effect of age, gender, and obesity on midazolam kinetics. Anesthesiology. 1984;61:27–35.
145. Johnson TN, Rostami-Hodjegan A, Goddard JM, et al. Contribution of midazolam and its 1-hydroxy metabolite to preoperative sedation in children: a pharmacokinetic-pharmacodynamic analysis. Br J Anaesth. 2002;89:428–437.
146. Bauer TM, Ritz R, Haberthur C, et al. Prolonged sedation due to accumulation of conjugated metabolites of midazolam. Lancet. 1995;346:145–150.
147. Hiller A, Olkkola KT, Isohanni P, et al. Unconsciousness associated with midazolam and erythromycin. Br J Anaesth. 1990;65:826–828.
148. Hase I, Oda Y, Tanaka K, et al. I.V. fentanyl decreases the clearance of midazolam. Br J Anaesth. 1997;79:740–743.
149. Vinik HR, Reves JG, Greenblatt DJ, et al. The pharmacokinetics of midazolam in chronic renal failure patients. Anesthesiology. 1983;59:390–394.
150. Veselis RA, Reinsel RA, Beattie BJ, et al. Midazolam changes regional cerebral blood flow in discrete brain regions: an H215O positron tomography study. Anesthesiology. 1997;87:1106–1117.
151. Papazian L, Albanese J, Thirion X, et al. Effect of bolus doses of midazolam on intracranial pressure and cerebral perfusion pressure in patients with severe head injury. Br J Anaesth. 1993;71:267–271.
152. Giffin JP, Cottrell JE, Shwiry B, et al. Intracranial pressure, mean arterial pressure, and heart rate following midazolam or thiopental in humans with brain tumors. Anesthesiology. 1984;60:491–494.
153. Bergman I, Steeves M, Burckart G, et al. Reversible neurologic abnormalities associated with prolonged intravenous midazolam and fentanyl administration. J Pediatr. 1991;119:644–649.
154. Thurston TA, Williams CGS, Foshee SL. Reversal of a paradoxical reaction to midazolam with flumazenil. Anesth Analg. 1996;83:192.
155. Forster A, Gardaz JP, Suter PM, et al. Respiratory depression of midazolam and diazepam. Anesthesiology. 1980;53:494–499.
156. Gross JB, Zebroski ME, Carel WD, et al. Time course of ventilatory depression after thiopental and midazolam in normal subjects and in patients with chronic obstructive pulmonary disease. Anesthesiology. 1983;58:540–544.
157. Kanto J, Sjovall S, Buori A. Effect of different kinds of premedication of the induction properties of midazolam. Br J Anaesth. 1982;54:507–511.
158. Bailey PL, Pace NL, Ashburn MA, et al. Frequent hypoxemia and apnea after sedation with midazolam and fentanyl. Anesthesiology. 1990;73:826–830.
159. Gauthier RA, Dyck B, Chung F, et al. Respiratory interaction after spinal anesthesia and sedation with midazolam. Anesthesiology. 1992;77:909–914.
160. Samuelson PN, Reves JG, Kouchoukos NT, et al. Hemodynamic responses to anesthetic induction with midazolam or diazepam in patients with ischemic heart disease. Anesth Analg. 1981;60:802–809.
161. Lebowitz PW, Cote ME, Daniels AL, et al. Comparative cardiovascular effects of midazolam and thiopental in healthy patients. Anesth Analg. 1982;61:661–665.
162. Adams P, Gelman S, Reves JG, et al. Midazolam pharmacodynamics and pharmacokinetics during acute hypovolemia. Anesthesiology. 1985;63:140–146.
163. Cote CJ, Cohen IT, Suresh S, et al. A comparison of three doses of a commercially prepared oral midazolam syrup in children. Anesth Analg. 2002;94:37–43.
164. McMillan CO, Spahr-Schopfer IA, Sikich N, et al. Premedication of children with oral midazolam. Can J Anaesth. 1992;39:545–550.
165. Kain ZN, Hofstadter MB, Mayes LC et al. Midazolam. Effects on amnesia and anxiety in children. Anesthesiology. 2000;93:676–684.
166. Jacobs JR, Reves JG, Marty J, et al. Aging increases pharmacodynamic sensitivity to the hypnotic effects of midazolam. Anesth Analg. 1995;80:143–148.
167. Sarnquist FH, Mathers WD, Brock-Utne J, et al. A bioassay of a water-soluble benzodiazepine against sodium thiopental. Anesthesiology. 1980;52:149–153.
168. Jensen S, Schou-Olesen A, Huttel MS. Use of midazolam as an induction agent: comparison with thiopental. Br J Anaesth. 1982;54:605–607.
169. Barr J, Zomorodi K, Bertaccini E, et al. A double-blind, randomized comparison of IV lorazepam vs. midazolam for sedation of ICU patients via a pharmacologic model. Anesthesiology. 2001;95:286–291.
170. Roberts KW, Crnkovic A, Steiniger JR. Post-anesthesia paradoxical vocal cord motion successfully treated with midazolam. Anesthesiology. 1998;89:517–519.
171. Dawes GS. The distribution and action of drugs on the fetus in utero. Br J Anaesth. 1973;45:766–769.
172. Greenblatt DJ, Koch-Weser J. Clinical toxicity of chlordiazepoxide and diazepam in relation to serum albumin concentration: a report from the Boston Collaborative Drug Surveillance Program. Eur J Clin Pharmacol. 1974;7:259–262.
173. Eustace PW, Hailey DM, Cox AG, et al. Biliary excretion of diazepam in man. Br J Anaesth. 1975;47:983–985.
174. Klotz U, Avant GR, Hoyumpa A, et al. The effects of age and liver disease on the disposition and elimination of diazepam in adult man. J Clin Invest. 1975;55:347–359.
175. Braunstein MC. Apnea with maintenance of consciousness following intravenous diazepam. Anesth Analg. 1979;58:52–53.
176. Gross JB, Smith L, Smith TC. Time course of ventilatory response to carbon dioxide after intravenous diazepam. Anesthesiology. 1982;57;18–21.
177. McCammon RL, Hilgenberg JC, Stoelting RK. Hemodynamic effects of diazepam-nitrous oxide in patients with coronary artery disease. Anesth Analg. 1980;59:438–441.
178. Marty J, Gauzit R, Lefevre P, et al. Effects of diazepam and midazolam on baroreflex control of heart rate and on sympathetic activity in humans. Anesth Analg. 1986;65:113–119.
179. Knapp RB, Dubow H. Comparison of diazepam with thiopental as an induction agent in cardiopulmonary disease. Anesth Analg. 1970;49:722–726.
180. Falk RB, Denlinger JK, Nahrwold ML, et al. Acute vasodilation following induction of anesthesia with intravenous diazepam and nitrous oxide. Anesthesiology. 1978;49:149–150.
181. Dretchen K, Ghoneim MM, Long JP. The interaction of diazepam with myoneural blocking agents. Anesthesiology. 1971;34:463–468.
182. De Jong RH, Heavner JE. Diazepam prevents and aborts lidocaine convulsions in monkeys. Anesthesiology. 1974;41:226–230.
183. Fragen RJ, Caldwell N. Lorazepam premedication: lack of recall and relief of anxiety. Anesth Analg. 1976;55:792–796.
184. Greenblatt DJ, Ehrenberg BL, Gunderman J, et al. Kinetic and dynamic study of intravenous lorazepam: comparison with intravenous diazepam. J Pharmacol Exp Ther. 1989;250:134–139.
185. Witte JL, Alegret C, Sessler DI, et al. Preoperative alprazolam reduces anxiety in ambulatory surgery patients: a comparison with oral midazolam. Anesth Analg. 2002;95:1601–1606.
186. Greenblatt DJ, Harmatz JS, Shapiro L, et al. Sensitivity to triazolam in the elderly. N Engl J Med. 1991;324:1691–1698.
187. Brogden RN, Goa KL. Flumazenil: a reappraisal of its pharmacological properties and therapeutic efficacy as a benzodiazepine antagonist. Drugs. 1991;42:1061–1089.
188. Ghoneim MM, Block RI, Ping Sum ST, et al. The interactions of midazolam and flumazenil on human memory and cognition. Anesthesiology. 1993;79:1183–1192.
189. Spivey WH. Flumazenil and seizures: analysis of 43 cases. Clin Ther. 1992;14:292–297.
190. White PF, Shafer A, Boyle WA, et al. Benzodiazepine antagonism does not provoke a stress response. Anesthesiology. 1989;70:636–639.
191. Kaukinen S, Kataja J, Kaukinen L. Antagonism of benzodiazepine-fentanyl anesthesia with flumazenil. Can J Anaesth. 1990;37:40–45.
192. Marty J, Nitenberg A, Philip I, et al. Coronary and left ventricular hemodynamic responses following reversal of flunitrazepam-induced sedation with flumazenil in patients with coronary artery disease. Anesthesiology. 1991;74:71–76.
193. Schwieger IM, Szlam F, Hug CC. Absence of agonistic or antagonistic effect of flumazenil (Ro 15-7088) in dogs anesthetized with enflurane, isoflurane, or fentanyl-enflurane. Anesthesiology. 1989;70:477–480.
194. Forster A, Crettenand G, Klopfenstein CE, et al. Absence of agonist effects of high-dose flumazenil on ventilation and psychometric performance in human volunteers. Anesth Analg. 1993;77:980–984.
195. Drover DR. Comparative pharmacokinetics and pharmacodynamics of short-acting hypnosedatives: zaleplon, zolpidem and zopiclone. Clin Pharmacokinet. 2004;43:227–238.
196. Bertisch SM, Herzig SJ, Winkelman JW, et al. National use of prescription medications for insomnia: NHANES 1999–2010. Sleep. 2014;37:343–349.
197. Huedo-Medina TB, Kirsch I, Middlemass J, et al. Effectiveness of non-benzodiazepine hypnotics in treatment of adult insomnia: meta-analysis of data submitted to the Food and Drug Administration. BMJ. 2012;345:e8343.
198. Woolston C. Death row incurs drug penalty. Nature. 2013;502:417–418.
199. Zeller A, Arras M, Jurd R, et al. Identification of a molecular target mediating the general anesthetic actions of pentobarbital. Mol Pharmacol. 2007;71:852–859.
200. Saidman LJ. Uptake, distribution, and elimination of barbiturates. In: Eger EI, ed. Anesthetic uptake and action. Baltimore, MD: Lippincott Williams & Wilkins; 1974.
201. Saidman LJ, Eger EI. The effect of thiopental metabolism on duration of anesthesia. Anesthesiology. 1966;27:118–126.
202. Sorbo S, Hudson RJ, Loomis JC. The pharmacokinetics of thiopental in pediatric surgical patients. Anesthesiology. 1984;61:666–670.
203. Manuli MA, Davies L. Rectal methohexital for sedation of children during imaging procedures. AJR Am J Roentgenol. 1993;160:577–580.
204. Liu LMP, Gaudreault P, Friedman PA, et al. Methohexital plasma concentrations in children following rectal administration. Anesthesiology. 1985;62:567–570.
205. Avram MJ, Krejcie TC, Henthorn TK. The relationship of age to the pharmacokinetics of early drug distribution: the concurrent disposition of thiopental and indocyanine green. Anesthesiology. 1990;72:403–411.
206. Stanski DR, Maitre PO. Population pharmacokinetics and pharmacodynamics of thiopental: the effect of age revisited. Anesthesiology. 1990;72:412–422.
207. Gin T, Mainland P, Chan MT, et al. Decreased thiopental requirements in early pregnancy. Anesthesiology. 1997;86:73–78.
208. Cote CJ, Petkau AJ. Thiopental requirements may be increased in children reanesthetized at least one year after recovery from extensive thermal injury. Anesth Analg. 1985;64:1156–1160.
209. Swerdlow BN, Holley FO, Maitre PO, et al. Chronic alcohol intake does not change thiopental anesthetic requirements, pharmacokinetics, or pharmacodynamics. Anesthesiology. 1990;72:455–461.
210. Ford FV, Morrell F, Whisler WW. Methohexital anesthesia in the surgical treatment of uncontrollable epilepsy. Anesth Analg. 1982;61:997–1001.
211. Rockoff MA, Goudsouzian NG. Seizures induced by methohexital. Anesthesiology. 1981;54:333–335.
212. Todd MM, Drummond JC, Sang H. The hemodynamic consequences of high-dose methohexital anesthesia in humans. Anesthesiology. 1984;61:495–501.
213. Bedford RF, Persing JA, Pobereskin L, et al. Lidocaine or thiopental for rapid control of intracranial hypertension. Anesth Analg. 1980;59:435–437.
214. Ward JD, Becker DP, Miller DJ, et al. Failure of prophylactic barbiturate coma in the treatment of severe head trauma. J Neurosurg. 1985;62:383–388.
215. Brain Resuscitation Clinical Trial I Study Group. Randomized clinical study of thiopental loading in comatose survivors of cardiac arrest. N Engl J Med. 1986;314:397–403.
216. Nussmeier NA, Arlund C, Slogoff S. Neuropsychiatric complications after cardiopulmonary bypass: cerebral protection by a barbiturate. Anesthesiology. 1986;64:165–170.
217. Todd MM, Chadwick HS, Shapiro HM, et al. The neurologic effects of thiopental therapy following experimental cardiac arrest in cats. Anesthesiology. 1982;57:76–86.
218. Filner BF, Karliner JS. Alterations of normal left ventricular performance by general anesthesia. Anesthesiology. 1976;45:610–620.
219. Drummond JC, Todd MM, U HS. The effect of high dose sodium thiopental on brain stem auditory and median nerve somatosensory evoked responses in humans. Anesthesiology. 1985;63;249–254.
220. Hirshman CA, Krieger W, Littlejohn G, et al. Ketamine-aminophylline-induced decrease in seizure threshold. Anesthesiology. 1982;56:464–467.
221. Etter MS, Helrich M, Mackenzie CF. Immunoglobulin E fluctuation in thiopental anaphylaxis. Anesthesiology. 1980;52:181–183.
222. Lilly JK, Hoy RH. Thiopental anaphylaxis and reagin involvement. Anesthesiology. 1980;53:335–337.
223. Clarke RSJ. Adverse effects of intravenously administered drugs in anaesthetic practice. Drugs. 1981;22:26–41.
224. Kohrs R, Durieux ME. Ketamine: teaching an old drug new tricks. Anesth Analg. 1998;87;1186–1193.
225. Reich DL, Silvay G. Ketamine: an update on the first twenty-five years of clinical experience. Can J Anaesth. 1989;36:186–197.
226. Kienbaum P, Heuter T, Paviakovic G, et al. S(+)-ketamine increases muscle sympathetic activity and maintains the neural response to hypotensive challenges in humans. Anesthesiology. 2001;94:252–258.
227. White PF, Ham J, Way WL, et al. Pharmacology of ketamine isomers in surgical patients. Anesthesiology. 1980;52:231–239.
228. Pfenninger EG, Durieux ME, Himmelseher S. Cognitive impairment after small-dose ketamine isomers in comparison to equianalgesic racemic ketamine in human volunteers. Anesthesiology. 2002;96:357–366.
229. Hirota K, Lambert DG. Ketamine: its mechanism(s) of action and unusual clinical uses. Br J Anaesth. 1996;77:441–444.
230. Wagner LE, Gingrich KJ, Kulli JC, et al. Ketamine blockade of voltage-gated sodium channels: evidence for a shared receptor site with local anesthetics. Anesthesiology. 2001;95:1406–1413.
231. Coates KM, Flood P. Ketamine and its preservative, benzethonium chloride, both inhibit human recombinant alpha7 and alpha4beta2 neuronal nicotinic acetylcholine receptors in Xenopus oocytes. Br J Pharmacol. 2001;134:871–879.
232. Weigand MA, Schmidt H, Zhao Q, et al. Ketamine modulates the stimulated adhesion molecule expression on human neutrophils in vitro. Anesth Analg. 2000;90:206–212.
233. Hurstveit O, Maurset A, Oye I. Interaction of the chiral forms of ketamine with opioid, phencyclidine, and muscarinic receptors. Pharmacol Toxicol. 1995;77:355–359.
234. Ho KK, Flood P. Single amino acid residue in the extracellular portion of transmembrane segment 2 in the nicotinic alpha7 acetylcholine receptor modulates sensitivity to ketamine. Anesthesiology. 2004;100:657–662.
235. Udesky JO, Spence NZ, Achiel R, et al. The role of nicotinic inhibition in ketamine-induced behavior. Anesth Analg. 2005;101:407–411.
236. White PF, Way WL, Trevor AJ. Ketamine: its pharmacology and therapeutic uses. Anesthesiology. 1982;56:119–136.
237. Demling RH, Ellerbee S, Jarrett F. Ketamine anesthesia for tangential excision of burn eschar: a burn unit procedure. J Trauma. 1978;18:269–270.
238. Himmelseher S, Durieux ME. Ketamine for perioperative pain management. Anesthesiology. 2005;102:211–220.
239. Subramaniam K, Balachundar S, Steinbrook RA. Ketamine as adjuvant analgesic to opioids: a quantitative and qualitative systematic review. Anesth Analg. 2004;99:482–495.
240. Liu H-T, Hollmann MW, Liu W-H, et al. Modulation of NMDA receptor function by ketamine and magnesium: part I. Anesth Analg. 2001;92:1173–1181.
241. Akamatsu TJ, Bonica JJ, Rhemet R. Experiences with the use of ketamine for parturition. I: primary anesthetic for vaginal delivery. Anesth Analg. 1974;53:284–287.
242. Janeczko GF, El-Etr AA, Youngest S. Low-dose ketamine anesthesia for obstetrical delivery. Anesth Analg. 1974;53:828–831.
243. Hodgkinson K, Marx GF, Kim SS, et al. Neonatal neurobehavioral tests following vaginal delivery under ketamine, thiopental, and extradural anesthesia. Anesth Analg. 1977;56:548–553.
244. Salt TE, Wilson DG, Prasad SK. Antagonism of N-methylaspartate and synaptic responses of neurones in the rat ventrobasal thalamus by ketamine and MK-801. Br J Pharmacol. 1988;94:443–448.
245. Sandler AN, Schmid R, Katz J. Epidural ketamine for postoperative analgesia. Can J Anaesth. 1998;45:99–102.
246. Waxman K, Shoemaker WC, Lippmann M. Cardiovascular effects of anesthetic induction with ketamine. Anesth Analg. 1980;59(5):355–358.
247. Guit JBM, Koning HM, Niemeijer RPE, et al. Ketamine as an analgesic for total intravenous anaesthesia with propofol. Anaesthesia. 1991;46:24–31.
248. Hirshman CA, Downes H, Farbood A, et al. Ketamine block of bronchospasm in experimental canine asthma. Br J Anaesth. 1979;51:713–718.
249. Dershwitz M, Sreter FA, Ryan JF. Ketamine does not trigger malignant hyperthermia in susceptible swine. Anesth Analg. 1989;69:501–503.
250. Eilers H, Philip LA, Bickler PE, et al. The reversal of fentanyl-induced tolerance by administration of “small-dose” ketamine. Anesth Analg. 2001;93:213–214.
251. Kudoh A, Takahira Y, Katagai H, et al. Small-dose ketamine improves the postoperative state of depressed patients. Anesth Analg. 2002;95:114–118.
252. Rodriguez CI, Kegeles L, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof of concept. Neuropsychopharmacology. 2013;38:2475–2483.
253. Kapur N, Friedman R. Oral ketamine: a promising treatment for restless legs syndrome. Anesth Analg. 2002;94:1558–1559.
254. Takeshita H, Okuda Y, Sari A. The effects of ketamine on cerebral circulation and metabolism in man. Anesthesiology. 1972;36:69–75.
255. Pfenninger E, Dick W, Ahnefeld FW. The influence of ketamine on both normal and raised intracranial pressure of artificially ventilated animals. Eur J Anaesthesiol. 1985;2:297–307.
256. Friesen RH, Thieme RE, Honda AT, et al. Changes in anterior fontanel pressure in preterm neonates receiving isoflurane, halothane, fentanyl, or ketamine. Anesth Analg. 1987;66:431–434.
257. Mayberg TS, Lam AM, Matta BF, et al. Ketamine does not increase cerebral blood flow velocity or intracranial pressure during isoflurane/nitrous oxide anesthesia in patients undergoing craniotomy. Anesth Analg. 1995;81:84–89.
258. Albanese J, Arnaud S, Rey M, et al. Ketamine decreases intracranial pressure and electroencephalographic activity in traumatic brain injury patients during propofol sedation. Anesthesiology. 1997;87:1328–1334.
259. Nagels W, Demeyere R, Van Hemelrijck J, et al. Evaluation of the neuroprotective effects of S(+)-ketamine during open-heart surgery. Anesth Analg. 2004;98:1595–1603.
260. Ferrer-Allado T, Brechner VL, Diamond A, et al. Ketamine-induced electroconvulsive phenomena in the human limbic and thalamic regions. Anesthesiology. 1973;38:333–344.
261. Celesia GG, Chen RC, Bamforth BJ. Effects of ketamine in epilepsy. Neurology. 1975;25:169–172.
262. Modica PA, Tempelhoff R, White PF. Pro- and anticonvulsant effects of anesthetics (part II). Anesth Analg. 1990;70:433–444.
263. Schubert A, Licine MG, Lineberry PJ. The effect of ketamine on human somatosensory evoked potentials and its modification by nitrous oxide. Anesthesiology. 1990;72:33–39.
264. Tweed WA, Minuck MS, Mymin D. Circulatory response to ketamine anesthesia. Anesthesiology. 1972;37:613–619.
265. Balfors E, Haggmark S, Nyhman H, et al. Droperidol inhibits the effects of intravenous ketamine on central hemodynamics and myocardial O2 consumption in patients with generalized atherosclerotic disease. Anesth Analg. 1983;62:193–197.
266. Hickey PR, Hansen DD, Cramoline GM, et al. Pulmonary and systemic hemodynamic responses to ketamine in infants with normal and elevated pulmonary vascular resistance. Anesthesiology. 1985;62:287–293.
267. Hoffman WE, Pelligrino D, Werner C, et al. Ketamine decreases plasma catecholamines and improves outcome from incomplete cerebral ischemia in rats. Anesthesiology. 1992;76:755–762.
268. Lundy PM, Lockwood PA, Thompson G, et al. Differential effects of ketamine isomers on neuronal and extraneuronal catecholamine uptake mechanisms. Anesthesiology. 1986;64:359–363.
269. Yli-Hankala A, Kirvela M, Randell T, et al. Ketamine anaesthesia in a patient with septic shock. Acta Anaesthesiol Scand. 1992;36:483–485.
270. Longnecker DE, Sturgill BC. Influence of anesthetic agents on survival following hemorrhage. Anesthesiology. 1976;45:516–521.
271. Weiskopf RB, Townley MI, Riordan KK, et al. Comparison of cardiopulmonary responses to graded hemorrhage during enflurane, halothane, isoflurane and ketamine anesthesia. Anesth Analg. 1981;60:481–492.
272. Koehntop DE, Liao JC, Van Bergen FH. Effects of pharmacologic alterations of adrenergic mechanisms by cocaine, tropolone, aminophylline and ketamine on epinephrine-induced arrhythmias during halothane nitrous oxide anesthesia. Anesthesiology. 1977;46:83–93.
273. Wong DHW, Jenkins LC. An experimental study of the mechanism of action of ketamine on the central nervous system. Can Anaesth Soc J. 1974;21:57–67.
274. Stanley TH. Blood pressure and pulse rate responses to ketamine during general anesthesia. Anesthesiology. 1973;39:648–649.
275. Traber DL, Wilson RD, Priano LL. Blockade of the hypertensive response to ketamine. Anesth Analg. 1970;49:420–426.
276. Baraka A, Harrison T, Kachachi T. Catecholamine levels after ketamine anesthesia in man. Anesth Analg. 1973;52:198–200.
277. Schwartz DA, Horwitz LD. Effects of ketamine on left ventricular performance. J Pharmacol Exp Ther. 1975;194:410–414.
278. Soliman MG, Brinale GF, Kuster G. Response to hypercapnia under ketamine anesthesia. Can Anaesth Soc J. 1975;22:486–494.
279. Taylor PA, Towey RM. Depression of laryngeal reflexes during ketamine anesthesia. Br Med J. 1971;2:688–689.
280. Sarma VJ. Use of ketamine in acute severe asthma. Acta Anaesthesiol Scand. 1992;36:106–107.
281. Laxenaire MC, Moneret-Vautrin D, Vervloet D. The French experience of anaphylactoid reactions. Int Anesthesiol Clin. 1985;23:145–160.
282. Nakagawa T, Hirakata H, Sato M et al. Ketamine suppresses platelet aggregation possibly by suppressed inositol triphosphate formation and subsequent suppression of cytosolic calcium increase. Anesthesiology. 2002;96:1147–1152.
283. Bowdle TA, Radant AD, Cowley DS, et al. Psychedelic effects of ketamine in healthy volunteers: relationship to steady-state plasma concentrations. Anesthesiology. 1998;88:82–88.
284. Fine J, Finestone SC. Sensory disturbances following ketamine anesthesia: recurrent hallucinations. Anesth Analg. 1973;52:428–430.
285. Meyers EF, Charles P. Prolonged adverse reactions to ketamine in children. Anesthesiology. 1978;49:39–40.
286. Modvig KM, Nielsen SF. Psychological changes in children after anesthesia: a comparison between halothane and ketamine. Acta Anaesthesiol Scand. 1977;21:541–544.
287. Moretti RJ, Hassan SZ, Goodman LI, et al. Comparison of ketamine and thiopental in healthy volunteers: effects on mental status, mood, and personality. Anesth Analg. 1984;63:1087–1096.
288. Cartwright PD, Pingel SM. Midazolam and diazepam in ketamine anaesthesia. Anaesthesia. 1984;59:439–442.
289. Toft P, Romer U. Comparison of midazolam and diazepam to supplement total intravenous anaesthesia with ketamine for endoscopy. Can J Anaesth. 1987;34:466–469.
290. Erbguth PH, Reiman B, Klein RL. The influence of chlorpromazine, diazepam and droperidol on emergence from ketamine. Anesth Analg. 1972;51:693–700.
291. Hejja P, Galloon S. A consideration of ketamine dreams. Can Anaesth Soc J. 1975;22:100–105.
292. Bidwai AV, Stanley HT, Graves CL, et al. The effects of ketamine on cardiovascular dynamics during halothane and enflurane anesthesia. Anesth Analg. 1975;54(5):588–592.
293. Fragen RJ, Avram MJ. Comparative pharmacology of drugs used for the induction of anesthesia. In: Stoelting RK, Barash PG, Gallagher TJ, eds. Advances in anesthesia. Chicago, IL: Year Book Medical Publishers; 1986:103–132.
294. Johnston RR, Miller RD, Way WL. The interaction of ketamine with d-tubocurarine, pancuronium, and succinylcholine in man. Anesth Analg. 1974;53:496–501.
295. Molojavyi A, Preckel B, Cofmere T, et al. Effects of ketamine and its isomers on ischemic preconditioning in the isolated rat heart. Anesthesiology. 2001;94:623–628.
296. Mullenheim J, Rulands R, Wietschorke T, et al. Late preconditioning is blocked by racemic ketamine, but not by S(+)-ketamine. Anesth Analg. 2001;93:265–270.
297. Bloor BC, Ward DS, Belleville JP, et al. Effects of intravenous dexmedetomidine in humans. II. Hemodynamic changes. Anesthesiology. 1992;77:1134–1142.
298. Sandler AN. The role of clonidine and alpha2-agonists for postoperative analgesia. Can J Anaesth. 1996;43:1191–1194.
299. Nelson LE, Lu J, Guo T, et al. The alpha2 adrenoreceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology. 2003;98:428–436.
300. Scheinin H, Aantaa R, Anttila M, et al. Reversal of the sedative and sympatholytic effects of dexmedetomidine with a specific alpha2 adrenoceptor antagonist atipamezole. A pharmacodynamic and kinetic study in healthy volunteers. Anesthesiology. 1998;89:574–584.
301. Shelly MP. Dexmedetomidine: a real innovation or more of the same? Br J Anaesth. 2001;87:677–678.
302. Buhrer M, Mappes A, Lauber R, et al. Dexmedetomidine decreases thiopental dose requirement and alters distribution pharmacokinetics. Anesthesiology. 1994;80:1216–1221.
303. Jalonen J, Hynynen M, Kuitunen A, et al. Dexmedetomidine as an anesthetic adjunct in coronary artery bypass grafting. Anesthesiology. 1997;86:331–345.
304. Kamibayashi T, Maze M. Clinical uses of alpha2-adrenergic agonists. Anesthesiology. 2000;93:1345–1349.
305. Segal IS, Vickery RG, Walton JK, et al. Dexmedetomidine diminishes halothane anesthetic requirements in rats through a postsynaptic alpha 2 adrenergic receptor. Anesthesiology. 1988;69:818–823.
306. Aantaa R, Maakola ML, Kallio A, et al. Reduction of the minimum alveolar concentration of isoflurane by dexmedetomidine. Anesthesiology. 1997;86:1055–1060.
307. Ramsay MAE, Luterman DL. Dexmedetomidine as a total intravenous anesthetic agent. Anesthesiology. 2004;101:787–790.
308. Levanen J, Makela ML, Scheinin H. Dexmedetomidine premedication attenuates ketamine-induced cardiostimulatory effects and postanesthetic delirium. Anesthesiology. 1995;82:1117–1125.
309. Memiş D, Turan A, Karamanlioğlu B, et al. Adding dexmedetomidine to lidocaine for intravenous regional anesthesia. Anesth Analg. 2004;98(3):835–840.
310. Talke P, Tayefeh F, Sessler DI, et al. Dexmedetomidine does not alter the sweating threshold, but comparably and linearly decreases the vasoconstriction and shivering thresholds. Anesthesiology. 1997;87:835–841.
311. Ingersoll-Weng E, Manecke GR, Thistlethwaite PA. Dexmedetomidine and cardiac arrest. Anesthesiology. 2004;100:738–739.
312. Hsu Y-W, Cortinez LI, Robertson KM, et al. Dexmedetomidine pharmacodynamics: part I. Crossover comparison of the respiratory effects of dexmedetomidine and remifentanil in healthy volunteers. Anesthesiology. 2004;101:1066–1076.
313. Venn RM, Bradshaw CJ, Spencer R, et al. Preliminary UK experience of dexmedetomidine, a novel agent for postoperative sedation in the intensive care unit. Anaesthesia. 1999;54:1136–1142.
314. Cortés R, Palacios JM. Muscarinic cholinergic receptor subtypes in the rat brain. I. Quantitative autoradiographic studies. Brain Res. 1986;362:227–238.
315. Putcha L, Cintrón NM, Tsui J, et al. Pharmacokinetics and oral bioavailability of scopolamine in normal subjects. Pharm Res. 1989;6:481–485.
316. Reis E. Scopolamine-morphine anesthesia. Cal State J Med. 1906;4:109–110.
317. Garde JF, Aston R, Endler GC, et al. Racial mydriatic response to belladonna premedication. Anesth Analg. 1978;57:572–576.