Opioids remain the mainstay of modern perioperative care and pain management. The modern word opium is derived from the Greek word opion (“poppy juice”); the opium poppy (Papaver somniferum) is the source of 20 distinct alkaloids. Written mention of the medicinal use of poppy juice dates back to at least 300 BC, although religious use likely goes back much further.1 Drugs derived from opium are referred to as opiates. Morphine, the best-known opiate, was isolated in 1803, followed by codeine in 1832, and papaverine in 1848. Morphine can be synthesized but it is more easily derived from opium. The term narcotic is derived from the Greek word for stupor and traditionally has been used to refer to potent morphine-like analgesics with the potential to produce physical dependence. The development of synthetic drugs with morphine-like properties has led to the use of the term opioid to refer to all exogenous substances, natural and synthetic, that bind specifically to any of several subpopulations of opioid receptors and produce at least some agonist (morphine-like) effects. Opioids are unique in producing analgesia without loss of touch, proprioception, or consciousness. A convenient classification of opioids includes opioid agonists, opioid agonist–antagonists, and opioid antagonists (Table 7-1).

Chemical Structure of Opium Alkaloids
The active components of opium can be divided into two distinct chemical classes: phenanthrenes and benzylisoquinolines. The principal phenanthrene alkaloids present in opium are morphine, codeine, and thebaine (Fig. 7-1). The principal benzylisoquinoline alkaloids present in opium, which lack analgesic activity, are papaverine and noscapine. The three rings of the phenanthrene core are composed of 14 carbon atoms. The fourth piperidine ring includes a tertiary amine nitrogen and is present in most opioid agonists. At pH 7.4, the tertiary amine nitrogen is highly ionized, making the molecule water soluble. These are chiral molecules, with levorotatory isomers being biologically active at opioid receptors.

Semisynthetic Opioids
Simple modification of the morphine molecule yields many derivative compounds with differing properties. For example, substitution of a methyl group for the hydroxyl group on carbon 3 results in methylmorphine (codeine). Substitution of acetyl groups on carbons 3 and 6 results in diacetylmorphine (heroin). Thebaine has insignificant analgesic activity but serves as the precursor for etorphine (analgesic potency >1,000 times morphine).
Synthetic Opioids
Synthetic opioids contain the phenanthrene nucleus of morphine but are manufactured by synthesis rather than chemical modification of morphine. Morphine derivatives (levorphanol), methadone derivatives, benzomorphan derivatives (pentazocine), and phenylpiperidine derivatives (meperidine, fentanyl) are examples of groups of synthetic opioids. There are similarities in the molecular weights (236 to 326) and pKs of phenylpiperidine derivatives and amide local anesthetics.
Fentanyl, sufentanil, alfentanil, and remifentanil (Fig. 7-2) are synthetic opioids that are widely used to supplement general anesthesia or as primary anesthetic drugs in very high doses. There are important clinical differences between these opioids.2–4 The major pharmacodynamic differences between these drugs are potency and rate of equilibration between the plasma and the site of drug effect (biophase).

Mechanism of Action
Opioids act as agonists at specific opioid receptors at presynaptic and postsynaptic sites in the central nervous system (CNS) (mainly the brainstem and spinal cord) as well as in the periphery.5–7 These same opioid receptors normally are activated by three endogenous peptide opioid receptor ligands known as enkephalins, endorphins, and dynorphins. Opioids mimic the actions of these endogenous ligands by binding to opioid receptors, resulting in activation of pain-modulating (antinociceptive) systems.
Existence of the opioid in the ionized state appears to be necessary for strong binding at the anionic opioid receptor site. Only levorotatory forms of the opioids exhibit agonist activity. Indeed, the naturally occurring form of morphine is the levorotatory isomer. The affinity of most opioid agonists for receptors correlates well with their analgesic potency.
The principal effect of opioid receptor activation is a decrease in neurotransmission.8 This decrease in neurotransmission occurs largely by presynaptic inhibition of neurotransmitter release (acetylcholine, dopamine, norepinephrine, substance P), although postsynaptic inhibition of evoked activity may also occur. The intracellular biochemical events initiated by occupation of opioid receptors with an opioid agonist are characterized by increased potassium conductance (leading to hyperpolarization), calcium channel inactivation, or both, which produce an immediate decrease in neurotransmitter release.
All opioid receptor classes couple to intracellular guanine (G) proteins. Upon binding of an opioid agonist to the extracellular domain of the receptor, the receptor changes shape, which activates the G protein bound to its intracellular domain. The G protein replaces its bound guanine diphosphate (GDP) with guanine triphosphate (GTP) and dissociates into two active subunits. Subsequent mechanisms include inhibition of adenylate cyclase, decrease the conductance of voltage-gated calcium channels, or opening of inward-flowing potassium channels. Any of these effects ultimately results in decreased neuronal activity. Opioid receptors also modulate the phosphoinositide-signaling cascade and phospholipase C. The prevention of calcium ion inflow results in suppression of neurotransmitter release (substance P) in many neuronal systems. Hyperpolarization results from actions at potassium channels, thus preventing excitation or propagation of action potentials. Opioid receptors may regulate the functions of other ion channels including excitatory postsynaptic currents evoked by N-methyl-D-aspartate (NMDA) receptors.
Opioid receptor–mediated inhibition of adenylate cyclase is not responsible for an immediate effect but may have a delayed effect, possibly via a reduction in cyclic adenosine monophosphate (cAMP)–responsive neuropeptide genes and reduction in neuropeptide messenger RNA concentrations. Depression of cholinergic transmission in the CNS as a result of opioid-induced inhibition of acetylcholine release from nerve endings may play a prominent role in the analgesic and other side effects of opioid agonists. Opioids do not alter responsiveness of afferent nerve endings to noxious stimulation nor do they block conduction of nerve impulses along peripheral nerves (as opposed to local anesthetics).
Opioid Receptors
Opioid receptors are classified as µ, δ, and κ receptors8,9 (Table 7-2). The names of the three subtypes developed from the ligands originally found to bind to them or their tissue of origin (mu—morphine, kappa—ketocyclazocine, delta—isolated from mouse vas deferens). These opioid receptors belong to a superfamily of seven transmembrane-segment G protein–coupled receptors that includes muscarinic, adrenergic, γ-aminobutyric acid, and somatostatin receptors. The opioid receptors have been cloned and their amino acid sequences defined.10,11 A single µ-receptor gene has been identified and six distinct µ receptors subtypes have been characterized.

In the brain, opioid receptors are primarily found in the periaqueductal gray, locus ceruleus, and the rostral ventral medulla. In the spinal cord, opioid receptors are found both on interneurons and primary afferent neurons in the dorsal horn. Consequently, direct application of opioid agonists to the spinal cord can produce intense analgesia.12 Outside the CNS, opioid receptors are found on sensory neurons and immune cells. Immune cells recruited to sites of inflammation also secrete opioid peptides to provide local analgesia.13 For example, intraarticular morphine is known to produce analgesia after knee surgery, presumably through action on peripheral nerves.14
The µ receptors are principally responsible for supraspinal and spinal analgesia. Theoretically, activation of a subpopulation of µ receptors (mu1) is speculated to produce analgesia, whereas mu2 receptors are responsible for hypoventilation, bradycardia, and physical dependence. Nevertheless, cloning of the µ receptors does not support the existence of separate mu1 and mu2 receptor subtypes.9 It is possible that such subtypes result from posttranslational modification of a common precursor protein. Whether β-endorphins or even morphine itself is the endogenous ligand for µ receptors is unclear.15 Endomorphins are peptides with high affinity and selectivity for µ receptors that are present in the brain.
Activation of κ receptors results in inhibition of neurotransmitter release via N-type calcium channels. Respiratory depression characteristic of µ receptor activation is less prominent with κ receptor activation, although dysphoria and diuresis may accompany activation of these receptors. κ receptor–mediated analgesia may be less effective for high-intensity painful stimulation than µ opioid–mediated. Opioid agonist–antagonists often act principally on κ receptors. δ receptors respond to the endogenous ligands known as enkephalins, and these opioid receptors may serve to modulate the activity of the µ receptors.
Functional and physical interactions between these receptor subtypes have been noted.16,17 Heteromerization between µ and δ opioid receptors leads to distinct receptor pharmacology in that doses of δ receptor ligands (agonists and antagonists) too low to trigger signaling can potentiate the binding and signaling of µ receptor agonists. Chronic, but not acute, morphine treatment results in an increase in µ-δ heteromers in key areas of the CNS that are implicated in pain processing.18
Endogenous Pain Modulating Mechanisms
The logical reason for the existence of opioid receptors and endogenous opioid agonists is to function as an endogenous pain suppression system. Once pain is consciously perceived, it has served its purpose and it is reasonable to posit that the ability to dampen this perception would have a survival benefit. Opioid receptors are located in areas of the brain (periaqueductal gray matter of the brainstem, amygdala, corpus striatum, and hypothalamus) and spinal cord (substantia gelatinosa) that are involved with pain perception, integration of pain impulses, and responses to pain (Fig. 7-3).19 It is speculated that endorphins inhibit the release of excitatory neurotransmitters from terminals of nerves carrying nociceptive impulses. As a result, neurons are hyperpolarized, which suppresses spontaneous discharges and evoked responses. Analgesia induced by electrical stimulation of specific sites in the brain or mechanical stimulation of peripheral areas (acupuncture) most likely reflects release of endorphins.20 Even the analgesic response to a placebo may also involve the release of endorphins. Sustained pain and stress induces the regional release of endogenous opioids interacting with µ opioid receptors in a number of cortical and subcortical brain regions. The activation of the µ opioid receptor system is associated with reductions in the sensory and affective ratings of the pain experience, with distinct neuroanatomic involvement.21,22

In addition, a recent study demonstrated that positive treatment expectancy substantially enhanced (doubled) the analgesic benefit of remifentanil, whereas negative treatment expectancy abolished remifentanil analgesia.23 These subjective effects were substantiated by significant changes in the neural activity in brain regions involved with the coding of pain intensity. The positive expectancy effects were associated with activity in the endogenous pain modulation system, and the negative expectancy effects with activity in the hippocampus.23 On the basis of subjective and objective evidence, we contend that an individual’s expectation of a drug’s effect critically influences its therapeutic efficacy and that regulatory brain mechanisms differ as a function of expectancy.
Common Opioid Side Effects
An ideal opioid agonist would have a high specificity for receptors, producing desirable responses (analgesia) and little or no specificity for receptors associated with side effects. To date, however, all opioids possess similar side effects that vary only in degree. Therefore, a focus on the effects of morphine provides a suitable starting point.
Cardiovascular System
Morphine, even in large doses, given to supine and normovolemic patients is unlikely to cause direct myocardial depression or hypotension. The same patients changing from a supine to a standing position, however, may manifest orthostatic hypotension and syncope, presumably reflecting morphine-induced impairment of compensatory sympathetic nervous system responses. For example, morphine decreases sympathetic nervous system tone to peripheral veins, resulting in venous pooling and subsequent decreases in venous return, cardiac output, and blood pressure.24
Morphine can also evoke decreases in systemic blood pressure due to drug-induced bradycardia or histamine release. Morphine-induced bradycardia results from increased activity of the vagal nerves, which probably reflects stimulation of the vagal nuclei in the medulla. Morphine may also exert a direct depressant effect on the sinoatrial node and acts to slow conduction of cardiac impulses through the atrioventricular node. These actions, may, in part, explain decreased vulnerability to ventricular fibrillation in the presence of morphine. Administration of opioids (morphine, fentanyl) in the preoperative medication or before the induction of anesthesia tends to slow heart rate during exposure to volatile anesthetics with or without surgical stimulation.25
Opioid-induced histamine release and associated hypotension are variable in both incidence and severity. The magnitude of morphine-induced histamine release and subsequent decrease in systemic blood pressure can be minimized by (a) limiting the rate of morphine infusion to 5 mg per minute intravenously (IV), (b) maintaining the patient in a supine to slightly head-down position, and (c) optimizing intravascular fluid volume. Conversely, administration of morphine, 1 mg/kg IV, over a 10-minute period produces substantial increases in the plasma concentrations of histamine that are paralleled by significant decreases in systemic blood pressure and systemic vascular resistance (Fig. 7-4).26 It is important to recognize, however, that not all patients respond to this rate of morphine infusion with the release of histamine, emphasizing the individual variability associated with the administration of this drug. In contrast to morphine, the infusion of fentanyl 50 µg/kg IV over a 10-minute period does not cause release of histamine in any patient (see Fig. 7-4). Pretreatment of patients with H1 and H2 receptor antagonists does not alter release of histamine evoked by morphine but does prevent changes in systemic blood pressure and systemic vascular resistance.27

Morphine does not sensitize the heart to catecholamines or otherwise predispose to cardiac dysrhythmias as long as hypercarbia or arterial hypoxemia does not result from ventilatory depression. Tachycardia and hypertension that occur during anesthesia with morphine are not pharmacologic effects of the opioid but rather are responses to painful surgical stimulation that are not suppressed by morphine. Both the sympathetic nervous system and the renin-angiotensin axis contribute to these cardiovascular responses. Large doses of morphine or other opioid agonists may decrease the likelihood that tachycardia and hypertension will occur in response to painful stimulation, but once this response has occurred, administration of additional opioid is unlikely to be effective.
During anesthesia, however, opioids are commonly administered with inhaled or IV anesthetics to ensure amnesia. The combination of an opioid agonist such as morphine or fentanyl with nitrous oxide results in cardiovascular depression (decreased cardiac output and systemic blood pressure plus increased cardiac filling pressures), which does not occur when either drug is administered alone.28 Likewise, decreases in systemic vascular resistance and systemic blood pressure may accompany the combination of an opioid and a benzodiazepine, whereas these effects do not accompany the administration of either drug alone (Fig. 7-5).29

Opioids have been increasingly recognized as playing a role in protecting the myocardium from ischemia. Through several mechanisms, most prominently though σ and κ receptors, opioids enhance the resistance of the myocardium to oxidative and ischemic stresses. Mitochondrial adenosine triphosphate (ATP)–regulated potassium channels (KATP) appear to be central to this signaling pathway.30
Ventilation
All opioid agonists produce dose-dependent and gender-specific depression of ventilation, primarily through an agonist effect at mu2 receptors leading to a direct depressant effect on brainstem ventilation centers.8 Because analgesic and ventilatory effects of opioids occur by similar mechanisms, it is assumed that equianalgesic doses of all opioids will produce some degree of ventilatory depression and reversal of ventilatory depression with an opioid antagonist always involves some reversal of analgesia. Opioid-induced depression of ventilation is characterized by decreased responsiveness of these ventilation centers to carbon dioxide as reflected by an increase in the resting PaCO2 and displacement of the carbon dioxide response curve to the right. Opioid agonists also interfere with pontine and medullary ventilatory centers that regulate the rhythm of breathing, leading to prolonged pauses between breaths and periodic breathing. It is possible that opioid agonists diminish sensitivity to carbon dioxide by decreasing the release of acetylcholine from neurons in the area of the medullary ventilatory center in response to hypercarbia. In this regard, physostigmine, which increases CNS levels of acetylcholine, may antagonize depression of ventilation but not analgesia produced by morphine.
Depression of ventilation produced by opioid agonists is rapid and persists for several hours, as demonstrated by decreased ventilatory responses to carbon dioxide. High doses of opioids may result in apnea, but the patient remains conscious and able to initiate a breath if asked to do so. Death from an opioid overdose is almost invariably due to depression of ventilation.
Clinically, depression of ventilation produced by opioids manifests as a decreased frequency of breathing that is often accompanied by a compensatory increase in tidal volume. The incompleteness of this compensatory increase in tidal volume is evidenced by predictable increases in the PaCO2. Many factors influence the magnitude and duration of depression of ventilation produced by opioid agonists. For example, advanced age and the occurrence of natural sleep increase the ventilatory depressant effects of opioids. Conversely, pain from surgical stimulation counteracts depression of ventilation produced by opioids. Likewise, the analgesic effect of opioids slows breathing that has been rapid and shallow due to pain.
Opioids produce dose-dependent depression of ciliary activity in the airways. Increases in airway resistance after administration of an opioid are probably due to a direct effect on bronchial smooth muscle and an indirect action due to release of histamine.
Cough Suppression
Opioids depress cough by effects on the medullary cough centers that are distinct from the effects of opioids on ventilation. The greatest cough suppression occurs with opioids that have bulky substitutions at the number 3 carbon position (codeine). One useful property of dextrorotatory isomers (such as dextromethorphan) is that they can suppress cough but do not produce analgesia or depression of ventilation. Thus, in some cases, opioids can be safely sold over-the-counter.
Central Nervous System
In the absence of hypoventilation, opioids decrease cerebral blood flow and possibly intracranial pressure (ICP). These drugs must be used with caution in patients with head injury because of their (a) associated effects on wakefulness, (b) production of miosis, and (c) depression of ventilation with associated increases in ICP if the PaCO2 becomes increased. Furthermore, head injury may impair the integrity of the blood–brain barrier, with resultant increased sensitivity to opioids.
The effect of morphine on the electroencephalogram (EEG) resembles changes associated with sleep. For example, there is replacement of rapid α waves by slower δ waves. Recording of the EEG fails to reveal any evidence of seizure activity after administration of large doses of opioids (see the section “Fentanyl”). Opioids do not alter the responses to neuromuscular blocking drugs. Skeletal muscle rigidity, especially of the thoracic and abdominal muscles, is common when large doses of opioid agonists are administered rapidly and intravenously.31 Clonic skeletal muscle activity (myoclonus) occurring during administration of opioids may resemble grand mal seizures, but the EEG does not reflect seizure activity. Skeletal muscle rigidity may be related to actions at opioid receptors and involve interactions with dopaminergic and γ-aminobutyric acid–responsive neurons.
Miosis is due to an excitatory action of opioids on the autonomic nervous system component of the Edinger-Westphal nucleus of the oculomotor nerve. Tolerance to the miotic effect of morphine is not prominent. Miosis can be antagonized by atropine, and profound arterial hypoxemia in the presence of morphine can still result in mydriasis.
Rigidity
Rapid IV administration of large doses of an opioid (particularly fentanyl and its derivatives as used in cardiac surgery) can lead to generalized skeletal muscle rigidity. This can be severe enough to interfere with manual ventilation. Although generally termed chest wall rigidity, evidence supports the conclusion that the majority of resistance to ventilation is due to laryngeal musculature contraction. Inhibition of striatal release of γ-aminobutyric acid and increased dopamine production are the likely explanations for opioid-induced increased skeletal muscle tone.32 The reported incidence of difficult ventilation after a moderate dose of sufentanil ranges from 84% to 100%.33Treatment is muscle relaxation with neuromuscular blocking drugs or opioid antagonism with naloxone.
Sedation
Postoperative titration of morphine frequently induces sedation that precedes the onset of analgesia.34 The usual recommendation for morphine titration includes a short interval between boluses (5 to 7 minutes) to allow evaluation of its clinical effect. Sedation occurs in up to 60% of patients during morphine titration and represents a common reason to discontinue morphine titration for postoperative analgesia. The assumption that sleep occurs when pain is relieved is not necessarily accurate and morphine-induced sedation should not be considered as an indicator of appropriate analgesia during IV morphine titration.
Biliary Tract
Opioids can cause spasm of biliary smooth muscle, resulting in increases in biliary pressure that may be associated with epigastric distress or biliary colic. This pain may be confused with angina pectoris. Naloxone will relieve pain caused by biliary spasm but not myocardial ischemia. Conversely, nitroglycerin will relieve pain due to either biliary spasm or myocardial ischemia. Equal analgesic doses of fentanyl, morphine, meperidine, and pentazocine increase common bile duct pressure 99%, 53%, 61%, and 15% above predrug levels, respectively.35 During surgery, opioid-induced spasm of the sphincter of Oddi may appear radiologically as a sharp constriction at the distal end of the common bile duct and be misinterpreted as a common bile duct stone. It may be necessary to reverse opioid-induced biliary smooth muscle spasm with naloxone so as to correctly interpret the cholangiogram. Glucagon, 2 mg IV, also reverses opioid-induced biliary smooth muscle spasm and, unlike naloxone, does not antagonize the analgesic effects of the opioid.36 However, biliary muscle spasm does not occur in most patients who receive opioids. Indeed, the incidence of spasm of the sphincter of Oddi is about 3% in patients receiving fentanyl as a supplement to inhaled anesthetics.37
Contraction of the smooth muscles of the pancreatic ducts is probably responsible for increases in plasma amylase and lipase concentrations that may be present after the administration of morphine. Such increases may confuse the diagnosis when acute pancreatitis is a possibility.
Gastrointestinal Tract
Commonly used opioids such as morphine, meperidine, and fentanyl can produce spasm of the gastrointestinal smooth muscles, resulting in a variety of side effects including constipation, biliary colic, and delayed gastric emptying.
Morphine decreases the propulsive peristaltic contractions of the small and large intestines and enhances the tone of the pyloric sphincter, ileocecal valve, and anal sphincter. The delayed passage of intestinal contents through the colon allows increased absorption of water. As a result, constipation often accompanies therapy with opioids and may become a debilitating problem in patients who require chronic opioid therapy, as little tolerance develops to this effect. Of interest, opium was used to treat diarrhea before its use as an analgesic was popularized.
Increased biliary pressure occurs when the gallbladder contracts against a closed or narrowed sphincter of Oddi. Passage of gastric contents into the proximal duodenum is delayed because there is increased tone at the gastroduodenal junction. In this regard, preoperative medication that includes an opioid could slow gastric emptying (potentially increase the risk of aspiration) or delay the absorption of orally administered drugs. All these effects may be reversed or prevented by a peripheral-acting opioid antagonist (see the section “Methylnaltrexone”).
Nausea and Vomiting
Opioid-induced nausea and vomiting are caused by direct stimulation of the chemoreceptor trigger zone in the floor of the fourth ventricle. This may reflect the role of opioid agonists as partial dopamine agonists at dopamine receptors in the chemoreceptor trigger zone. Indeed, apomorphine is a profound emetic and is also the most potent of the opioids at dopamine receptors. Stimulation of dopamine receptors as a mechanism for opioid-induced nausea and vomiting is consistent with the antiemetic efficacy of butyrophenones and phenothiazines. Morphine may also cause nausea and vomiting by increasing gastrointestinal secretions and delaying passage of intestinal contents toward the colon.
Morphine depresses the vomiting center in the medulla. As a result, IV administration of morphine produces less nausea and vomiting than the intramuscular (IM) administration of morphine, presumably because opioid administered IV reaches the vomiting center as rapidly as it reaches the chemoreceptor trigger zone. Nausea and vomiting are relatively uncommon in recumbent patients given morphine, suggesting that a vestibular component may contribute to opioid-induced nausea and vomiting.
Genitourinary System
Morphine can increase the tone and peristaltic activity of the ureter. In contrast to similar effects on biliary tract smooth muscle, the same opioid-induced effects on the ureter can be reversed by an anticholinergic drug such as atropine. Urinary urgency is produced by opioid-induced augmentation of detrusor muscle tone, but, at the same time, the tone of the urinary sphincter is enhanced, making voiding difficult.
Antidiuresis that accompanies administration of morphine to animals has been attributed to opioid-induced release of arginine vasopressin hormone (antidiuretic hormone). In humans, however, administration of morphine in the absence of painful surgical stimulation does not evoke the release of this hormone.38 Furthermore, when morphine is administered in the presence of an adequate intravascular fluid volume, there is no change in urine output.
Cutaneous Changes
Morphine causes cutaneous blood vessels to dilate. The skin of the face, neck, and upper chest frequently becomes flushed and warm. These changes in cutaneous circulation are in part caused by the release of histamine. Histamine release probably accounts for urticaria and erythema commonly seen at the morphine injection site. In addition, morphine-induced histamine release probably accounts for conjunctival erythema and pruritus. Localized cutaneous evidence of histamine release, especially along the vein into which morphine is injected, does not represent an allergic reaction.
Placental Transfer
Opioids are readily transported across the placenta. Therefore, depression of the neonate can occur as a consequence of administration of opioids to the mother during labor. In this regard, maternal administration of morphine may produce greater neonatal depression than meperidine does.39 This may reflect immaturity of the neonate’s blood–brain barrier. Chronic maternal use of an opioid can result in the development of physical dependence in the fetus. Subsequent administration of naloxone to the neonate can precipitate neonatal abstinence syndrome.
Drug Interactions
The ventilatory depressant effects of some opioids may be exaggerated by amphetamines, phenothiazines, monoamine oxidase inhibitors, and tricyclic antidepressants. For example, patients receiving monoamine oxidase inhibitors may experience exaggerated CNS depression and hyperpyrexia after administration of an opioid agonist, especially meperidine. This exaggerated response may reflect alterations in the rate or pathway of metabolism of the opioid. Sympathomimetic drugs appear to enhance analgesia produced by opioids. The cholinergic nervous system seems to be a positive modulator of opioid-induced analgesia in that physostigmine enhances and atropine antagonizes analgesia.
Hormonal Changes
Prolonged opioid therapy may influence the hypothalamic-pituitary-adrenal axis and the hypothalamic-pituitary-gonadal axis, leading to endocrine and immune effects.40,41 Morphine may cause a progressive decrease in plasma cortisol concentrations. The main effects of opioids on the hypothalamic-pituitary-gonadal axis involve modulation of hormone release including increased prolactin and decreased luteinizing hormone, follicle-stimulating hormone, testosterone, and estrogen concentrations.
Overdose
The principal manifestation of opioid overdose is depression of ventilation manifesting as a slow breathing frequency, which may progress to apnea. Pupils are symmetric and miotic unless severe arterial hypoxemia is present, which results in mydriasis. Skeletal muscles are flaccid, and upper airway obstruction may occur. Pulmonary edema commonly occurs, but the mechanism is not known. Hypotension and seizures develop if arterial hypoxemia persists. The triad of miosis, hypoventilation, and coma should suggest overdose with an opioid. Treatment of opioid overdose is mechanical ventilation of the patient’s lungs with oxygen and administration of an opioid antagonist such as naloxone. Administration of an opioid antagonist to treat opioid overdose may precipitate acute withdrawal in dependent patients.
Provocation of Coughing
Paradoxically, preinduction administration of fentanyl, sufentanil, or alfentanil may be associated with significant reflex coughing.42 The exact cause of opioid-induced cough is unclear but is thought to be due to imbalance between sympathetic and vagal innervation of the airways and/or stimulation of juxtacapillary irritant receptors.43 Morphine and hydromorphone do not appear to cause this reaction.
Pharmacodynamic Tolerance and Physical Dependence
Pharmacodynamic tolerance and physical dependence with repeated opioid administration are characteristics of all opioid agonists and are among the major limitations of their clinical use. Cross-tolerance develops between all the opioids. Tolerance can occur without physical dependence, but the reverse does not seem to occur.
Tolerance is the development of the requirement for increased doses of a drug (in this case, an opioid agonist) to achieve the same effect previously achieved with a lower dose. Such acquired tolerance usually takes 2 to 3 weeks to develop with analgesic doses of morphine, although acute tolerance can develop much more quickly with highly potent opioids.44 Tolerance develops to analgesic, euphoric, sedative, depression of ventilation, and emetic effects of opioids but not to their effects on miosis and bowel motility. The potential for physical dependence depends on the agonist effect of opioids. Indeed, physical dependence does not occur with opioid antagonists and is less likely with opioid agonist–antagonists. When opioid agonist actions predominate, there often develops, with repeated use, both psychological and physiologic need for the drug.
Physical dependence on morphine usually requires about 25 days to develop but may occur sooner in emotionally unstable persons. Some degree of physical dependence, however, occurs after only 48 hours of continuous medication. When physical dependence is established, discontinuation of the opioid agonist produces a typical withdrawal abstinence syndrome (Table 7-3).45 Initial symptoms of withdrawal include yawning, diaphoresis, lacrimation, or coryza. Insomnia and restlessness are prominent. Abdominal cramps, nausea, vomiting, and diarrhea reach their peak in 72 hours and then decline over the next 7 to 10 days. During withdrawal, tolerance to morphine is rapidly lost, and the syndrome can be terminated by a modest dose of opioid agonist. The longer the period of abstinence, the smaller the dose of opioid agonist that will be required.

Pharmacodynamic tolerance has been related to neurologic changes that take place after long-term exposure to the opioid.45 The classic explanation for tolerance to a receptor agonist involved changes occurring at the level of the receptors and involve receptor desensitization. Opioid receptors on the cell membrane surfaces become gradually desensitized by reduced transcription and subsequent decreases in the absolute numbers of opioid receptors (downregulation). A second mechanism proposed to explain pharmacodynamic tolerance involves upregulation of the cAMP system. Acutely, opioids inhibit functional activity of cAMP pathways by blocking adenylate cyclase, the enzyme that catalyzes the synthesis of cAMP. Long-term opioid exposure is associated with gradual recovery of cAMP pathways and tolerance develops. Increased synthesis of cAMP may be responsible for physical dependence and physiologic changes associated with withdrawal. Upregulation of cAMP has been most clearly demonstrated in the locus ceruleus of the brain. Clonidine, a centrally acting α2-adrenergic agonist that diminishes transmission in sympathetic pathways in the CNS, is an effective drug in suppressing withdrawal signs in persons who are physically dependent on opioids. Tolerance is not due to enzyme induction, because no increase in the rate of metabolism of opioid agonists occurs.
Long-term pharmacodynamic tolerance characterized by opioid insensitivity may persist for months or years in some individuals and most likely represents persistent neural adaptation.45 In this regard, NMDA glutamate receptors are important in the development of opioid tolerance and increased pain sensitivity. Prolonged exposure to opioids activates NMDA receptors via second messenger mechanisms and also downregulates spinal glutamate transporters. The resultant high synaptic concentrations of glutamate and NMDA receptor activation contribute to opioid tolerance and abnormal pain sensitivity (pronociceptive or sensitization process). The observation that treatment with small doses of ketamine (an NMDA receptor antagonist) abolishes the acute opioid tolerance seen with remifentanil supports this hypothesis.46
Opioid Agonists
Opioid agonists include but are not limited to morphine, meperidine, fentanyl, sufentanil, alfentanil, and remifentanil (see Table 7-1).47 The most notable feature of the clinical use of opioids is the extraordinary variation in dose requirements for effective treatment of pain.48 This interindividual variation emphasizes that usual doses of opioids may produce inadequate or excessive opioid effects. Opioid rotation may be useful when dose escalation is not effective in treating pain.
Morphine
Isolated in 1806 and named after Morpheus, the Greek god of dreams, morphine is the prototype opioid agonist to which all other opioids are compared. In humans, morphine produces analgesia, euphoria, sedation, and a diminished ability to concentrate. Other sensations include nausea, a feeling of body warmth, heaviness of the extremities, dryness of the mouth, and pruritus, especially in the cutaneous areas around the nose. The cause of pain persists, but even low doses of morphine increase the threshold to pain and modify the perception of noxious stimulation such that it is no longer experienced as pain. Continuous, dull pain is relieved by morphine more effectively than is sharp, intermittent pain. In contrast to nonopioid analgesics, morphine is effective against pain arising from the viscera as well as from skeletal muscles, joints, and integumental structures. Analgesia is most prominent when morphine is administered before the painful stimulus occurs.49 In the absence of pain, however, morphine may produce dysphoria rather than euphoria.
Pharmacokinetics
Morphine is well absorbed after IM administration, with onset of effect in 15 to 30 minutes and a peak effect in 45 to 90 minutes. The clinical duration of action is about 4 hours. Morphine can be administered orally for treatment of chronic pain recognizing that absorption from the gastrointestinal is limited by significant first-pass metabolism in the liver, which limits the bioavailability of an orally administered dose to approximately 25% (1 mg of IV morphine ~4 mg of oral morphine). Morphine is usually administered IV in the perioperative period, thus eliminating the unpredictable influence of drug absorption. The peak effect (equilibration time between the blood and brain) after IV administration of morphine is delayed compared with opioids such as fentanyl and alfentanil, requiring about 15 to 30 minutes (Table 7-4). Morphine inhaled as an aerosol from a nebulizer may act on afferent nerve pathways in the airways to relieve dyspnea as associated with lung cancer and associated pleural effusion.50 However, profound depression of ventilation may follow aerosol administration of morphine.51 The onset and duration of the analgesic effects of morphine are similar after IV administration or inhalation via a pulmonary drug delivery system that produces a fine aerosol.52
Plasma morphine concentrations after rapid IV injections do not correlate closely with the drug’s pharmacologic activity, likely due to the delay in transit of morphine across the blood–brain barrier. Cerebrospinal fluid (CSF) concentrations of morphine peak 15 to 30 minutes after IV injection and decay more slowly than plasma concentrations (Fig. 7-6).53 As a result, the analgesic and ventilatory depressant effects of morphine may not be evident during the initial high plasma concentrations after IV administration of the opioid. Likewise, these same drug effects persist despite decreasing plasma concentrations of morphine. Moderate analgesia probably requires maintenance of plasma morphine concentrations of at least 0.05 µg/mL.54

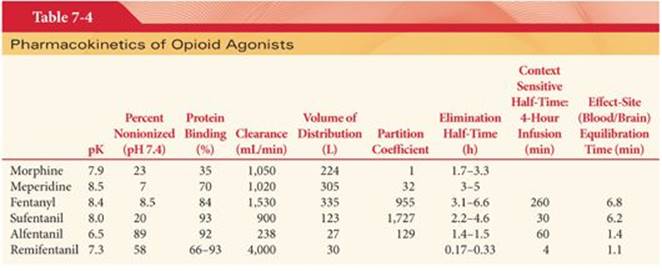
Only a small amount of administered morphine gains access to the CNS. For example, it is estimated that less than 0.1% of morphine that is administered IV has entered the CNS at the time of peak plasma concentrations. Reasons for poor penetration of morphine into the CNS include (a) relatively poor lipid solubility, (b) high degree of ionization at physiologic pH, (c) protein binding, and (d) rapid conjugation with glucuronic acid. Alkalinization of the blood, as produced by hyperventilation of the patient’s lungs, will increase the nonionized fraction of morphine and thus enhance its passage into the CNS. Nevertheless, respiratory acidosis, which decreases the nonionized fraction of morphine, results in higher plasma and brain concentrations of morphine than are present during normocarbia (Fig. 7-7).55 This suggests that carbon dioxide–induced increases in cerebral blood flow and enhanced delivery of morphine to the brain are more important than the fraction of drug that exists in either the ionized or nonionized fraction. In contrast to the CNS, morphine accumulates rapidly in the kidneys, liver, and skeletal muscles. Morphine, unlike fentanyl, does not undergo significant first-pass uptake into the lungs.56
Metabolism
Metabolism of morphine is primarily conjugation with glucuronic acid in hepatic and extrahepatic sites, especially the kidneys. About 75% to 85% of a dose of morphine appears as morphine-3-glucuronide, and 5% to 10% as morphine-6-glucuronide (a ratio of 9:1). Morphine-3-glucuronide is detectable in the plasma within 1 minute after IV injection, and its concentration exceeds that of unchanged drug by almost 10-fold within 90 minutes (Fig. 7-8).53An estimated 5% of morphine is demethylated to normorphine, and a small amount of codeine may also be formed. Metabolites of morphine are eliminated principally in the urine, with only 7% to 10% undergoing biliary excretion. Morphine-3-glucuronide is detectable in the urine for up to 72 hours after the administration of morphine. A small fraction (1% to 2%) of injected morphine is recovered unchanged in the urine.

Morphine-3-glucuronide is pharmacologically inactive, whereas morphine-6-glucuronide produces analgesia and depression of ventilation via its agonist actions at µ receptors.57 In fact, the ventilatory response to carbon dioxide is impacted similarly by morphine and morphine-6-glucuronide (Fig. 7-9).58 The duration of action of morphine-6-glucuronide is greater than that of morphine, and it is possible that the majority of analgesic activity attributed to morphine is actually due to morphine-6-glucuronide, especially with long-term administration of morphine.59 Morphine and morphine-6-glucuronide bind to µ opioid receptors with comparable affinity, whereas the analgesic potency of morphine-6-glucuronide is 650-fold higher than morphine.60

Renal metabolism makes a significant contribution to the total metabolism of morphine, which offers a possible explanation for the absence of any decrease in systemic clearance of morphine in patients with hepatic cirrhosis or during the anhepatic phase of orthotopic liver transplantation.61
Elimination of morphine glucuronides may be impaired in patients with renal failure, causing an accumulation of metabolites and unexpected ventilatory depressant effects of small doses of opioids (Fig. 7-10).62 Indeed, prolonged depression of ventilation (>7 days) has been observed in patients in renal failure after administration of morphine.63 Formation of glucuronide conjugates may be impaired by monoamine oxidase inhibitors, which is consistent with exaggerated effects of morphine when administered to patients being treated with these drugs.

Elimination Half-Time
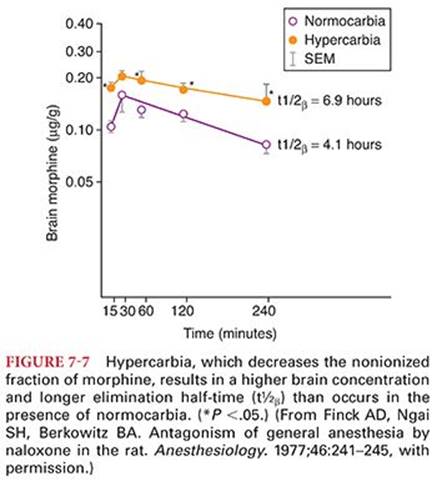
After IV administration of morphine, the elimination of morphine-3-glucuronide is somewhat longer than for morphine (see Table 7-4 and Fig. 7-8).53 The decrease in the plasma concentration of morphine after initial distribution of the drug is principally due to metabolism because only a small amount of unchanged opioid is excreted in the urine. Plasma morphine concentrations are higher in the elderly than in young adults (Fig. 7-11).54 In the first 4 days of life, the clearance of morphine is decreased and its elimination half-time is prolonged compared with that found in older infants.64 This is consistent with the observation that neonates are more sensitive than older children to the respiratory depressant effects of morphine. Patients with renal failure exhibit higher plasma and CSF concentrations of morphine and morphine metabolites than do normal patients, reflecting a smaller volume of distribution (Vd).65Possible accumulation of morphine-6-glucuronide suggests the need for caution when administering morphine to patients with renal dysfunction. Concentrations of morphine in the colostrum of parturients receiving patient-controlled analgesia with morphine are low and it is unlikely that significant amounts of drug will be transferred to the breast-fed neonate.66
Gender
Gender may affect opioid analgesia but the direction and magnitude of these differences depend on many interacting variables including the opioid used.67 Morphine exhibits greater analgesic potency and slower speed of offset in women than men.68 This observation is consistent with higher postoperative opioid consumption in men compared with women. Likewise, morphine decreases the slope of the ventilatory response to carbon dioxide in women, whereas in men, there was no significant effect.69 Morphine has no demonstrated effect on the apneic threshold in women but causes an increase in men. Hypoxic sensitivity is decreased by morphine in women but not men.
Side Effects
Side effects described for morphine are also characteristic of other opioid agonists, although the incidence and magnitude may vary.
Meperidine
First synthesized in 1939, meperidine (also referred to as pethidine) is a synthetic opioid agonist at µ and κ opioid receptors and is derived from phenylepiperidine (Fig. 7-2). There are several analogues of meperidine, including fentanyl, sufentanil, alfentanil, and remifentanil. Meperidine shares several structural features that are present in local anesthetics including a tertiary amine, an ester group, and a lipophilic phenyl group. Indeed, meperidine administered intrathecally blocks sodium channels to a degree comparable with lidocaine. Structurally, meperidine is similar to atropine, and it possesses a mild atropine-like antispasmodic effect on smooth muscle.
Pharmacokinetics
Meperidine is about one-tenth as potent as morphine. The duration of action of meperidine is 2 to 4 hours, making it a shorter acting opioid agonist than morphine. In equianalgesic doses, meperidine produces equivalent sedation, euphoria, nausea, vomiting, and depression of ventilation to morphine. Meperidine is absorbed from the gastrointestinal tract, but extensive first-pass hepatic metabolism (up to 80%) limits its oral usefulness.
Metabolism
Hepatic metabolism of meperidine is extensive, with about 90% of the drug initially undergoing demethylation to normeperidine and hydrolysis to meperidinic acid.70 Normeperidine subsequently undergoes hydrolysis to normeperidinic acid. Urinary excretion is the principal elimination route and is pH dependent. For example, if the urinary pH is <5, as much as 25% of meperidine is excreted unchanged. Indeed, acidification of the urine can be considered in an attempt to speed elimination of meperidine. Decreased renal function can predispose to accumulation of normeperidine.
Normeperidine has an elimination half-time of 15 hours (35 hours in patients in renal failure) and can be detected in urine for as long as 3 days after administration of meperidine. This metabolite is about one-half as active as meperidine as an analgesic. In addition, normeperidine produces CNS stimulation. Normeperidine toxicity manifesting as myoclonus and seizures is most likely during prolonged administration of meperidine as during patient-controlled analgesia, especially in the presence of impaired renal function.70 Normeperidine may also be important in meperidine-induced delirium (confusion, hallucinations), which has been observed in patients receiving the drug for longer than 3 days, corresponding to accumulation of this active metabolite.
Elimination Half-Time
The elimination half-time of meperidine is 3 to 5 hours (see Table 7-4). Because clearance of meperidine primarily depends on hepatic metabolism, it is possible that large doses of opioid would saturate enzyme systems and result in prolonged elimination half-times. Nevertheless, elimination half-time is not altered by doses of meperidine up to 5 mg/kg IV. About 60% of meperidine is bound to plasma proteins. Elderly patients manifest decreased plasma protein binding of meperidine, resulting in increased plasma concentrations of free drug and an apparent increased sensitivity to the opioid. The increased tolerance of alcoholics to meperidine and other opioids presumably reflects an increased volume of distribution, resulting in lower plasma concentrations of meperidine for a given dose.
Clinical Uses
The clinical use of meperidine has declined greatly in recent years. Meperidine is the only opioid considered adequate for surgery when administered intrathecally, owing to its ability to block sodium channels in a way similar to local anesthetics in addition to its µ-mediated opioid acitivity.71 An IM injection of meperidine for postoperative analgesia results in peak plasma concentrations that vary three- to fivefold as well as a time required to achieve peak concentrations that varies three- to sevenfold among patients.72 The minimum analgesic plasma concentration of meperidine is highly variable among patients; however, in the same patient, differences in concentrations as small as 0.05 µg/mL can represent a margin between no relief and complete analgesia. A plasma meperidine concentration of 0.7 µg/mL would be expected to provide postoperative analgesia in about 95% of patients.73 Normeperidine toxicity has been described in patients receiving meperidine for patient-controlled analgesia.70 Therefore, because there are other effective agents, patient-controlled analgesia with meperidine cannot be recommended.
Meperidine may be effective in suppressing postoperative shivering that may result in detrimental increases in metabolic oxygen consumption. The antishivering effects of meperidine may reflect stimulation of κ receptors (estimated to represent 10% of its activity) and a drug-induced decrease in the shivering threshold (not present with alfentanil, clonidine, propofol, or volatile anesthetics).74–76 In addition, meperidine is a potent agonist at α2receptors, which might contribute to antishivering effects.77 Indeed, clonidine is even more effective than meperidine in reducing postoperative shivering. Butorphanol (a κ receptor agonist-antagonist) stops shivering more effectively than opioids with a predominant µ opioid receptor agonist effect. Evidence for a role of κ receptors in the antishivering effects of meperidine and butorphanol is the failure of naloxone to completely inhibit this drug-induced effect.
Unlike morphine, meperidine is not useful for the treatment of diarrhea and is not an effective cough suppressant. During bronchoscopy, the relative lack of antitussive activity of meperidine makes this opioid less useful. Meperidine is not used in high doses because of significant negative cardiac inotropic effects plus histamine release in a substantial number of patients.78
Side Effects
The side effects of meperidine generally resemble those described for morphine. Meperidine, in contrast to morphine, rarely causes bradycardia but instead may increase heart rate, reflecting its modest atropine-like qualities. Large doses of meperidine result in decreases in myocardial contractility, which, among opioids, is unique for this drug. Delirium and seizures, when they occur, presumably reflect accumulation of normeperidine, which has stimulating effects on the CNS.
Serotonin syndrome (autonomic instability with hypertension, tachycardia, diaphoresis, hyperthermia, behavioral changes including confusion and agitation, and neuromuscular changes manifesting as hyperreflexia) occurs when drugs capable of increasing serotonin administration are administered. In severe cases, coma, seizures, coagulopathy, and metabolic acidosis may develop. Administration of meperidine to patients receiving antidepressant drugs (monoamine oxidase inhibitors, fluoxetine) may elicit this syndrome.79
Meperidine readily impairs ventilation and may be even more of a ventilatory depressant than morphine. This opioid promptly crosses the placenta, and concentrations of meperidine in umbilical cord blood at birth may exceed maternal plasma concentrations.39 Meperidine may produce less constipation and urinary retention than morphine. After equal analgesic doses, biliary tract spasm is less after meperidine injection than after morphine injection but greater than that caused by codeine.35 Meperidine does not cause miosis but rather tends to cause mydriasis, reflecting its modest atropine-like actions. A dry mouth and an increase in heart rate are further evidence of the atropine-like effects of meperidine. Transient neurologic symptoms have been described following the administration of intrathecal meperidine for surgical anesthesia.80
The pattern of withdrawal symptoms after abrupt discontinuation of meperidine differs from that of morphine in that there are few autonomic nervous system effects. In addition, symptoms of withdrawal develop more rapidly and are of a shorter duration compared with those of morphine.
Fentanyl
Fentanyl is a phenylpiperidine-derivative synthetic opioid agonist that is structurally related to meperidine (see Fig. 7-2). As an analgesic, fentanyl is 75 to 125 times more potent than morphine. It was first synthesized by Janssen Pharmaceutica in 1960 during an assay of meperidine derivatives and subsequently released as the citrate salt under the trade name Sublimaze.81
Pharmacokinetics
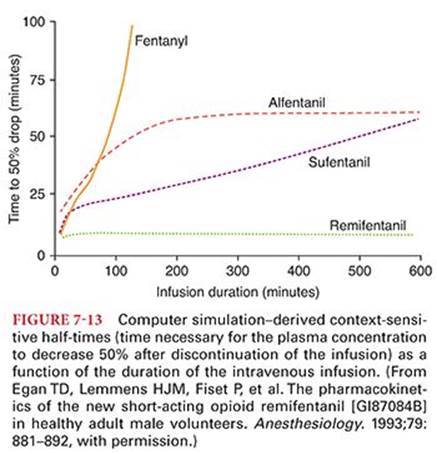
A single dose of fentanyl administered IV has a more rapid onset and shorter duration of action than morphine. Despite the clinical impression that fentanyl produces a rapid onset, there is a distinct time lag between the peak plasma fentanyl concentration and peak slowing on the EEG. This delay reflects the effect-site equilibration time between blood and the brain for fentanyl, which is 6.4 minutes. The greater potency and more rapid onset of action reflect the greater lipid solubility of fentanyl compared with that of morphine, which facilitates its passage across the blood–brain barrier. Consequently, plasma concentrations of fentanyl (unlike morphine) correlate well with CSF concentrations. Likewise, the short duration of action of a single dose of fentanyl reflects its rapid redistribution to inactive tissue sites such as fat and skeletal muscles, with an associated decrease in the plasma concentration of the drug (Fig. 7-12).82

The lungs also serve as a large inactive storage site, with an estimated 75% of the initial fentanyl dose undergoing first-pass pulmonary uptake.56 This nonrespiratory function of the lungs limits the initial amount of drug that reaches the systemic circulation and may play an important role in determining the pharmacokinetic profile of fentanyl. When multiple IV doses of fentanyl are administered or when there is continuous infusion of the drug, progressive saturation of these inactive tissue sites occurs. As a result, the plasma concentration of fentanyl does not decrease rapidly, and the duration of analgesia, as well as depression of ventilation, may be prolonged. Cardiopulmonary bypass causes clinically insignificant effects on the pharmacokinetics of fentanyl despite associated hemodilution, hypothermia, nonphysiologic blood flow and cardiopulmonary bypass–induced systemic inflammatory responses.83
Metabolism
Fentanyl is extensively metabolized by N-demethylation, producing norfentanyl, hydroxyproprionyl-fentanyl, and hydroxyproprionyl-norfentanyl. Norfentanyl is structurally similar to normeperidine and is the principal metabolite of fentanyl in humans. It is excreted by the kidneys and can be detected in the urine for 72 hours after a single IV dose of fentanyl. Less than 10% of fentanyl is excreted unchanged in the urine. The pharmacologic activity of fentanyl metabolites is believed to be minimal.84 Fentanyl is a substrate for hepatic P450 enzymes (CYP3A) and is susceptible to drug interactions that reflect interference with enzyme activity (less likely than with alfentanil).85
Elimination Half-Time
Despite the clinical impression that fentanyl has a short duration of action, its elimination half-time is longer than that for morphine (see Table 7-4). This longer elimination half-time reflects a larger Vd of fentanyl because clearance of both opioids is similar (see Table 7-4). The larger Vd of fentanyl is due to its greater lipid solubility and thus more rapid passage into tissues compared with the less lipid-soluble morphine. After an IV bolus, fentanyl distributes rapidly from the plasma to highly vascular tissues (brain, lungs, heart). More than 80% of the injected dose leaves the plasma in <5 minutes. The plasma concentrations of fentanyl are maintained by slow reuptake from inactive tissue sites, which accounts for persistent drug effects that parallel the prolonged elimination half-time. In animals, the elimination half-time, Vd, and clearance of fentanyl are independent of the dose of opioid between 6.4 and 640 µg/kg IV.86
A prolonged elimination half-time for fentanyl in elderly patients is due to decreased clearance of the opioid because Vd is not changed in comparison with younger adults.87 This change may reflect age-related decreases in hepatic blood flow, microsomal enzyme activity, or albumin production, as fentanyl is highly bound (79% to 87%) to protein. For these reasons, it is likely that a given dose of fentanyl will be effective for a longer period of time in elderly patients than in younger patients. A prolonged elimination half-time of fentanyl has also been observed in patients undergoing abdominal aortic surgery requiring infrarenal aortic cross-clamping.88 Somewhat surprising, however, is the failure of hepatic cirrhosis to prolong significantly the elimination half-time of fentanyl.89
Context-Sensitive Half-Time
As the duration of continuous infusion of fentanyl increases beyond about 2 hours, the context-sensitive half-time of this opioid becomes greater than sufentanil (Fig. 7-13).3,90 This reflects saturation of inactive tissue sites with fentanyl during prolonged infusions and return of the opioid from peripheral compartments to the plasma. This tissue reservoir of fentanyl replaces fentanyl eliminated by hepatic metabolism so as to slow the rate of decrease in the plasma concentration of fentanyl when the infusion is discontinued.
Cardiopulmonary Bypass
All opioids show a decrease in plasma concentration with initiation of cardiopulmonary bypass.91 The degree of this decrease is greater with fentanyl because a significant proportion of the drug adheres to the surface of the cardiopulmonary bypass circuit. The decrease is least with opioids that have a large Vd such that the addition of prime volume is less important. In this respect, sufentanil and alfentanil may provide more stable plasma concentrations during cardiopulmonary bypass. Elimination of fentanyl and alfentanil has been shown to be prolonged by cardiopulmonary bypass.
Clinical Uses
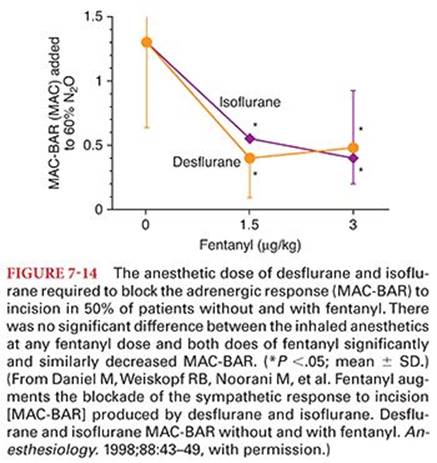
Fentanyl is administered clinically in a wide range of doses. For example, low doses of fentanyl, 1 to 2 µg/kg IV, are injected to provide analgesia. Fentanyl, 2 to 20 µg/kg IV, may be administered as an adjuvant to inhaled anesthetics in an attempt to blunt circulatory responses to (a) direct laryngoscopy for intubation of the trachea, or (b) sudden changes in the level of surgical stimulation. Timing of the IV injection of fentanyl to prevent or treat such responses should consider the effect-site equilibration time, which for fentanyl is prolonged compared with alfentanil and remifentanil. Injection of an opioid such as fentanyl before painful surgical stimulation may decrease the subsequent amount of opioid required in the postoperative period to provide analgesia.49 Administration of fentanyl 1.5 or 3 µg/kg IV 5 minutes before induction of anesthesia decreases the subsequent doses of isoflurane or desflurane with 60% nitrous oxide needed to block the sympathetic nervous system response to surgical stimulation (Fig. 7-14).92Large doses of fentanyl, 50 to 150 µg/kg IV, have been used alone to produce surgical anesthesia. Large doses of fentanyl as the sole anesthetic have the advantage of stable hemodynamics due principally to the (a) lack of direct myocardial depressant effects, (b) absence of histamine release, and (c) suppression of the stress responses to surgery. Disadvantages of using fentanyl as the sole anesthetic include (a) failure to prevent sympathetic nervous system responses to painful surgical stimulation at any dose, especially in patients with good left ventricular function; (b) unpredictable amnestic effects potentially leading to recall; and (c) postoperative depression of ventilation.93–95 Intrathecal fentanyl (maximal benefit achieved with 25 µg) can produce rapid, profound analgesia for early labor with minimal side effects.96
Fentanyl may be administered as a transmucosal preparation (oral transmucosal fentanyl) in a delivery device (several formulations are available, including a lozenge mounted on a handle or a film or rapid-dissolving preparation applied to the buccal mucosa) designed to deliver 5 to 20 µg/kg of fentanyl. The goal is to decrease preoperative anxiety and facilitate the induction of anesthesia, especially in children.97,98 In children 2 to 8 years of age, the preoperative administration of oral transmucosal fentanyl, 15 to 20 µg/kg 45 minutes before the induction of anesthesia, reliably induces preoperative sedation and facilitates induction of inhalation anesthesia.99 These same patients, however, are likely to experience decreases in breathing frequency and arterial oxygenation and an increased incidence of postoperative nausea and vomiting that is not influenced by prophylactic administration of droperidol. In children 6 years of age and younger, the preoperative administration of oral transmucosal fentanyl, 15 µg/kg, is associated with an unacceptably high incidence of preoperative vomiting.100 Conversely, another report did not observe an increased incidence of vomiting or arterial oxygen desaturation after premedication with oral transmucosal fentanyl.101 For treatment of postoperative pain after orthopedic surgery, 1 mg of oral transmucosal fentanyl is equivalent to 5 mg of IV morphine.102 Patients experiencing pain due to cancer may self-administer this opioid to the extent necessary to produce a desirable level of analgesia.
Transdermal fentanyl preparations delivering 75 to 100 µg per hour result in peak plasma fentanyl concentrations in about 18 hours that tend to remain stable during the presence of the patch, followed by a decreasing plasma concentration for several hours after removal of the delivery system, reflecting continued absorption from the cutaneous depot. These transdermal delivery systems were designed to produce stable, long-term fentanyl plasma concentrations in efforts to provide adequate, sustained analgesia for chronic, cancer-related pain. Each transdermal patch contains a depot of fentanyl that provides adequate drug to produce stable plasma fentanyl concentrations for 3 consecutive days. Transdermal fentanyl systems applied before the induction of anesthesia and left in place for 24 hours decrease the amount of parenteral opioid required for postoperative analgesia.103 Acute toxic delirium has been observed in patients with chronic pain due to cancer being treated with transdermal fentanyl for prolonged periods of time.104 It is possible that renal failure and accumulation of norfentanyl contributes to the possible toxic effects of prolonged use of transdermal fentanyl.
In dogs, maximal analgesic, ventilatory, and cardiovascular effects are present when the plasma concentration of fentanyl is approximately 30 ng/mL.105 Thus, the analgesic actions of fentanyl cannot be separated from its effects on ventilation and heart rate. The fact that all receptor-mediated effects are similar at the same plasma concentration of fentanyl suggests saturation of the opioid receptors. Evidence of opioid overdose has been observed when an upper body warming blanket was placed intraoperatively and came into contact with the fentanyl patch and has been linked to increased dermal blood flow and resultant drug uptake caused by the warming.106
Side Effects
The side effects of fentanyl resemble those described for morphine. Persistent or recurrent depression of ventilation due to fentanyl is a potential postoperative problem.107 Secondary peaks in plasma concentrations of fentanyl and morphine have been attributed to sequestration of fentanyl in acidic gastric fluid (ion trapping). Sequestered fentanyl could then be absorbed from the more alkaline small intestine back into the circulation to increase the plasma concentration of opioid and cause depression of ventilation to recur. This, however, may not be the mechanism for the secondary peak of fentanyl, because reabsorbed opioid from the gastrointestinal tract or skeletal muscles, as evoked by movement associated with transfer from the operating room, would be subject to first-pass hepatic metabolism. An alternative explanation for the secondary peak of fentanyl is washout of opioid from the lungs as ventilation to perfusion relationships are reestablished in the postoperative period.
Cardiovascular Effects
Unlike morphine, fentanyl, even in large doses (50 µg/kg IV), does not evoke the release of histamine (see Fig. 7-4).26 As a result, dilatation of venous capacitance vessels leading to hypotension is unlikely. Carotid sinus baroreceptor reflex control of heart rate is markedly depressed by fentanyl, 10 µg/kg IV, administered to neonates (Fig. 7-15).108 Therefore, changes in systemic blood pressure occurring during fentanyl anesthesia have to be carefully considered because cardiac output is principally rate dependent in neonates. Bradycardia is more prominent with fentanyl than morphine and may lead to occasional decreases in blood pressure and cardiac output.

Seizure Activity
Seizure-like activity has been described to follow rapid IV administration of fentanyl, sufentanil, and alfentanil.109 In the absence of EEG evidence of seizure activity, however, it is difficult to distinguish opioid-induced skeletal muscle rigidity or myoclonus from seizure activity. Indeed, recording of the EEG during periods of opioid-induced skeletal muscle rigidity fails to reveal evidence of seizure activity in the brain.110 Even plasma concentrations as high as 1,750 ng/mL after rapid administration of fentanyl, 150 µg/kg IV, do not produce EEG evidence of seizure activity.111 Conversely, opioids might produce a form of myoclonus secondary to depression of inhibitory neurons that would produce a clinical picture of seizure activity in the absence of EEG changes.
Somatosensory Evoked Potentials and Electroencephalogram
Fentanyl in doses exceeding 30 µg/kg IV produces changes in somatosensory evoked potentials that, although detectable, do not interfere with the use and interpretation of this monitor during anesthesia.112Opioids, including fentanyl, attenuate skeletal muscle movement at doses that have little effect on the EEG. This suggests that movement in response to surgical skin incision (used to measure minimum alveolar concentration [MAC]) primarily reflects the ability of a drug to obtund noxious reflexes and may not be the most appropriate measure for assessing consciousness or loss of consciousness.113 This opioid effect confounds the use of bispectral analysis as a measure of anesthetic adequacy when lack of movement with surgical skin incision is used to define efficacy.114
Intracranial Pressure
Administration of fentanyl and sufentanil to head injury patients has been associated with modest increases (6 to 9 mm Hg) in ICP despite maintenance of an unchanged PaCO2.115 These increases in ICP are typically accompanied by decreases in mean arterial pressure and cerebral perfusion pressure. In fact, increases in ICP do not accompany the administration of sufentanil when changes in mean arterial pressure are prevented (Fig. 7-16).116 This suggests that increases in ICP evoked by sufentanil (and presumably fentanyl) may have been due to autoregulatory decreases in cerebral vascular resistance due to decreases in systemic blood pressure resulting in vasodilation, increased blood volume, and increased ICP. Nevertheless, opioid-induced increases in ICP are similar in the presence of intact or impaired autoregulation, suggesting that mechanisms other than activation of the vasodilatory cascade need to be considered.117

Drug Interactions
Analgesic concentrations of fentanyl greatly potentiate the effects of benzodiazepines and decrease the dose requirements of propofol. The opioid-benzodiazepine combination displays marked synergism with respect to hypnosis and depression of ventilation.118 In clinical practice, the advantage of synergy between opioids and benzodiazepines for the maintenance of patient comfort is carefully weighed against the disadvantages of the potentially adverse depressant effects of this combination.
Sufentanil
First synthesized in 1974, sufentanil is a thienyl analogue of fentanyl (see Fig. 7-2). The analgesic potency of sufentanil is 5 to 10 times that of fentanyl, which parallels the greater affinity of sufentanil for opioid receptors compared with that of fentanyl. Based on the plasma concentration necessary to cause 50% of the maximum slowing on the EEG (EC50), sufentanil is 12 times more potent than fentanyl.119An important distinction from fentanyl is the 1,000-fold difference between the analgesic dose of sufentanil and the dose that produces seizures in animals. This difference is 160-fold for fentanyl and may be important when large doses of opioid agonists are used to produce anesthesia. Transient skeletal muscle spasm has been described after the accidental intrathecal injection of a large dose of sufentanil (40 µg), suggesting an irritative effect produced by the opioid.120
Pharmacokinetics
The elimination half-time of sufentanil is intermediate between that of fentanyl and alfentanil (see Table 7-4).121 A single IV dose of sufentanil has a similar elimination half-time in patients with or without cirrhosis of the liver.122 A prolonged elimination half-time has been observed in elderly patients receiving sufentanil for abdominal aortic surgery.123 The Vd and elimination half-time of sufentanil is increased in obese patients, which most likely reflects the high lipid solubility of this opioid.124
A high tissue affinity is consistent with the lipophilic nature of sufentanil, which permits rapid penetration of the blood–brain barrier and onset of CNS effects (effect-site equilibration time of 6.2 minutes is similar to that of 6.8 minutes for fentanyl).119 A rapid redistribution to inactive tissue sites terminates the effect of small doses, but a cumulative drug effect can accompany large or repeated doses of sufentanil. Sufentanil undergoes significant first-pass pulmonary uptake (approximately 60%) after rapid IV injection.125 This pulmonary first-pass uptake is similar to fentanyl and greater than morphine (about 7%) and alfentanil (about 10%).
The extensive protein binding of sufentanil (92.5%) compared with that of fentanyl (79% to 87%) contributes to a smaller Vd, which is characteristic of sufentanil. Binding to α1-acid glycoprotein constitutes a principal proportion of the total plasma protein binding of sufentanil. Levels of α1-acid glycoprotein vary over a threefold range in healthy volunteers and are increased after surgery, which could result in a decrease in the plasma concentration of pharmacologically active unbound sufentanil. Lower concentrations of α1-acid glycoprotein in neonates and infants probably account for decreases in protein binding of sufentanil in these age groups compared with that in older children and adults.126 The resulting increased free fraction of sufentanil in the neonate might contribute to enhanced effects of this opioid. Indeed, fentanyl and its derivatives produce anesthesia and depression of ventilation at lower doses in neonates than in adults.127
Metabolism
Sufentanil is rapidly metabolized by N-dealkylation at the piperidine nitrogen and by O-demethylation. The products of N-dealkylation are pharmacologically inactive, whereas desmethyl sufentanil has about 10% of the activity of sufentanil. Less than 1% of an administered dose of sufentanil appears unchanged in urine. Indeed, the high lipid solubility of sufentanil results in maximal renal tubular reabsorption of free drug as well as its enhanced access to hepatic microsomal enzymes. Extensive hepatic extraction means that clearance of sufentanil will be sensitive to changes in hepatic blood flow but not to alterations in the drug-metabolizing capacity of the liver. Sufentanil metabolites are excreted almost equally in urine and feces, with about 30% appearing as conjugates. The production of a weakly active metabolite and the substantial amount of conjugated metabolite formation imply the possible importance of normal renal function for the clearance of sufentanil. Indeed, prolonged depression of ventilation in association with an abnormally increased plasma concentration of sufentanil has been observed in a patient with chronic renal failure.128
Context-Sensitive Half-Time
The context-sensitive half-time of sufentanil is actually less than that for alfentanil for continuous infusions of up to 8 hours in duration (see Fig. 7-13).3 This shorter context-sensitive half-time can be explained in part by the large Vd of sufentanil compared to alfentanil. After termination of a sufentanil infusion, the decrease in the plasma drug concentration is accelerated not only by metabolism but also by continued redistribution of sufentanil into peripheral tissue compartments. Compared with alfentanil, sufentanil may have a more favorable recovery profile when used over a longer period of time. Conversely, alfentanil has a pharmacokinetic advantage for the treatment of discrete and transient noxious stimuli because its short effect-site equilibration time allows rapid access of the drug to the brain and facilitates titration.
Clinical Uses
In volunteers, a single dose of sufentanil, 0.1 to 0.4 µg/kg IV, produces a longer period of analgesia and less depression of ventilation than does a comparable dose of fentanyl (1 to 4 µg/kg IV).129 Compared with large doses of morphine or fentanyl, sufentanil, 18.9 µg/kg IV, results in more rapid induction of anesthesia, earlier emergence from anesthesia, and earlier tracheal extubation.130 As observed with other opioids, sufentanil causes a decrease in cerebral metabolic oxygen requirements and cerebral blood flow is also decreased or unchanged.131 Bradycardia produced by sufentanil may be sufficient to decrease cardiac output. As observed with fentanyl, delayed depression of ventilation has also been described after the administration of sufentanil.132
Although large doses of sufentanil (10 to 30 µg/kg IV) or fentanyl (50 to 150 µg/kg IV) produce minimal hemodynamic effects in patients with good left ventricular function, the systemic blood pressure and hormonal (catecholamine) responses to painful stimulation such as median sternotomy are not predictably prevented.133 It seems unlikely that any clinically useful dose of sufentanil or fentanyl will abolish such responses in all patients. Use of large doses of opioids, including sufentanil or fentanyl, to produce IV induction of anesthesia may result in rigidity of chest and abdominal musculature. This skeletal muscle rigidity makes ventilation of the patient’s lungs with positive airway pressure difficult. Difficult ventilation during sufentanil-induced skeletal muscle rigidity may actually reflect obstruction at the level of the glottis or above, which can be overcome by tracheal intubation.134
Alfentanil
Alfentanil is an analogue of fentanyl that is less potent (one-fifth to one-tenth) and has one-third the duration of action of fentanyl (see Fig. 7-2). It was first synthesized in 1976. A unique advantage of alfentanil compared with fentanyl and sufentanil is the more rapid onset of action (rapid effect-site equilibration) after the IV administration of alfentanil. For example, the effect-site equilibration time for alfentanil is 1.4 minutes compared with 6.8 and 6.2 minutes for fentanyl and sufentanil, respectively (see Table 7-4).4,135
Pharmacokinetics
Alfentanil has a short elimination half-time compared with fentanyl and sufentanil (see Table 7-4). Cirrhosis of the liver, but not cholestatic disease, prolongs the elimination half-time of alfentanil.136 Renal failure does not alter the clearance or elimination half-time of alfentanil.137 The elimination half-time of alfentanil is shorter in children (4 to 8 years old) than adults, reflecting a smaller Vd in these younger patients.138
The rapid effect-site equilibration characteristic of alfentanil is a result of the low pKa of this opioid such that nearly 90% of the drug exists in the nonionized form at physiologic pH. It is the nonionized fraction that readily crosses the blood–brain barrier. The rapid peak effect of alfentanil at the brain is useful when an opioid is required to blunt the response to a single, brief stimulus such as tracheal intubation or performance of a retrobulbar block.
The Vd of alfentanil is four to six times smaller than that of fentanyl (see Table 7-4).139 This smaller Vd compared with that of fentanyl reflects lower lipid solubility and higher protein binding. Despite this lesser lipid solubility, penetration of the blood–brain barrier by alfentanil is rapid because of its large nonionized fraction at physiologic pH. Alfentanil is principally bound to α1-acid glycoprotein, a protein whose plasma concentration is not altered by liver disease. Because protein binding is similar, it is likely that a decreased percentage of adipose tissue in children is responsible for the short elimination half-time.
Metabolism
Alfentanil is metabolized predominantly by two independent pathways, piperidine N-dealkylation to noralfentanil and amide N-dealkylation to N-phenylpropionamide. Noralfentanil is the major metabolite recovered in urine, with <0.5% of an administered dose of alfentanil being excreted unchanged. The efficiency of hepatic metabolism is emphasized by clearance of about 96% of alfentanil from the plasma within 60 minutes of its administration.
Attempts to develop reliable infusion regimens to attain and maintain specific plasma concentrations of alfentanil have been confounded by the wide interindividual variability in alfentanil pharmacokinetics. The most significant factor responsible for unpredictable alfentanil disposition is the 10-fold interindividual variability in alfentanil systemic clearance, presumably reflecting variability in hepatic intrinsic clearance. In this regard, it is likely that population variability in P450 3A4 (CYP3A4) activity (most abundant P450 hepatic enzyme and the major isoform of P450 responsible for alfentanil metabolism and clearance) is the mechanistic explanation for the interindividual variability in alfentanil disposition.140 Alfentanil clearance is markedly influenced by CYP3A activity and alfentanil is a sensitive and validated probe for CYP3A activity.85 Alterations in P450 activity may be responsible for the ability of erythromycin to inhibit the metabolism of alfentanil and a resulting prolonged opioid effect.141
Context-Sensitive Half-Time
The context-sensitive half-time of alfentanil is actually longer than that of sufentanil for infusions up to 8 hours in duration (see Fig. 7-13).90 This phenomenon can be explained in part by the large Vd of sufentanil. After termination of a continuous infusion of sufentanil, the decrease in the plasma drug concentration is accelerated not only by metabolism but also by continued redistribution of sufentanil into peripheral compartments. Conversely, the Vd of alfentanil equilibrates rapidly; therefore, peripheral distribution of drug away from the plasma is not a significant contributor to the decrease in the plasma concentration after discontinuation of the alfentanil infusion. Thus, despite the short elimination half-time of alfentanil, it may not necessarily be a superior choice to sufentanil for ambulatory sedation techniques.
Clinical Uses
Alfentanil has a rapid onset and offset of intense analgesia reflecting its very prompt effect-site equilibration. This characteristic of alfentanil is used to provide analgesia when the noxious stimulation is acute but transient as associated with laryngoscopy and tracheal intubation and performance of a retrobulbar block. For example, administration of alfentanil, 15 µg/kg IV, about 90 seconds before beginning direct laryngoscopy is effective in blunting the systemic blood pressure and heart rate response to tracheal intubation.142 The catecholamine response to this noxious stimulation is also blunted by alfentanil, 30 µg/kg IV.142 Alfentanil in doses of 10 to 20 µg/kg IV blunts the circulatory but not the catecholamine release response to the sudden exposure to high inhaled concentrations of desflurane.143 Alfentanil, 150 to 300 µg/kg IV, administered rapidly, produces unconsciousness in about 45 seconds. After this induction, maintenance of anesthesia can be provided with a continuous infusion of alfentanil, 25 to 150 µg/kg/hour IV, combined with an inhaled anesthetic.144 Unlike other opioids, supplemental doses of alfentanil seem to be more likely to decrease systemic blood pressure that is increased after painful stimulation. Alfentanil increases biliary tract pressures similarly to fentanyl. Alfentanil, compared with equipotent doses of fentanyl and sufentanil, is associated with a lower incidence of postoperative nausea and vomiting in outpatients.145 Acute dystonia has been described after administration of alfentanil to a patient with untreated Parkinson’s disease.146 This may reflect an ability of opioids to decrease central dopaminergic transmission and suggests caution in administration of this opioid to patients with untreated Parkinson’s disease.
Remifentanil
Remifentanil is a selective µ opioid agonist with an analgesic potency similar to that of fentanyl (15 to 20 times as potent as alfentanil) and a blood–brain equilibration (effect-site equilibration) time similar to that of alfentanil (see Table 7-4).2,3,147–149 Although chemically related to the fentanyl family of short-acting phenylpiperidine derivatives, remifentanil is structurally unique because of its ester linkage (see Fig. 7-2). Remifentanil’s ester structure renders it susceptible to hydrolysis by nonspecific plasma and tissue esterases to inactive metabolites.3 This unique pathway of metabolism leads to (a) brief action, (b) precise and rapidly titratable effect due to its rapid onset and offset, (c) lack of accumulation, and (d) rapid recovery after discontinuation of its administration.
Ventilation
After administration of 0.5 µg/kg IV remifentanil, there is a decrease in the slope and downward shift of the carbon dioxide ventilatory response curve that reaches its nadir after about 150 seconds following injection (Fig. 7-17).150Recovery after this small dose of remifentanil was complete within about 15 minutes. The combination of remifentanil and propofol is synergistic resulting in severe depression of ventilation (Fig. 7-18).151


Pharmacokinetics
The pharmacokinetics of remifentanil are characterized by small Vd, rapid clearance, and low interindividual variability compared to other IV anesthetic drugs. The rapid metabolism of remifentanil and its small Vd mean that remifentanil will accumulate less than other opioids. Because of its rapid systemic clearance, remifentanil provides pharmacokinetic advantages in clinical situations requiring predictable termination of drug effect. Remifentanil’s pharmacokinetics is similar in obese and lean patients. Therefore, dosing regimens should be based on ideal (lean) body mass rather than total body weight.152
The most salient pharmacokinetic feature of remifentanil is the extraordinary clearance of nearly 3 L per minute, which is about eight times more rapid than that of alfentanil. Remifentanil has a smaller Vd than alfentanil. The combination of rapid clearance and small Vd produces a drug with a uniquely transient effect. In fact, the rate of decline (context-sensitive half-time) of the remifentanil plasma concentration will be nearly independent of the infusion duration (see Fig. 7-13).2,3 The rapid effect-site equilibration means that a remifentanil infusion rate will promptly approach steady state in the plasma and its effect site. It is estimated that remifentanil plasma concentrations will reach a steady state within 10 minutes of beginning an infusion. The relationship between infusion rate and opioid concentration will be less variable for remifentanil than for other opioids. Furthermore, the rapid clearance of remifentanil, combined with the rapid blood–brain equilibration, means changes in infusion rates will be paralleled by prompt changes in drug effect.
Based on analysis of the EEG response, it is estimated that remifentanil is about 19 times more potent than alfentanil (EC50 for EEG depression 20 ng/mL vs. 376 ng/mL).3 The effect-site equilibration time, however, is similar for both opioids, suggesting that remifentanil will have an alfentanil-like onset (see Table 7-4). For example, after a rapid IV injection, the peak effect-site concentration of remifentanil will be present within 1.1 minutes, compared with 1.4 minutes for alfentanil. The effect, however, will be more transient after administration of remifentanil than alfentanil.
Metabolism
Remifentanil is unique among the opioids in undergoing metabolism by nonspecific plasma and tissue esterases to inactive metabolites.3 The principal metabolite, remifentanil acid, is 300 to 4,600-fold less potent than remifentanil as a µ agonist and is excreted primarily by the kidneys. N-dealkylation of remifentanil is a minor metabolic pathway in humans. Remifentanil does not appear to be a substrate for butyrylcholinesterases (pseudocholinesterase), and thus its clearance should not be affected by cholinesterase deficiency or anticholinergics.2 Additionally, it is likely that remifentanil’s pharmacokinetics will be unchanged by renal or hepatic failure because esterase metabolism is usually preserved in these states.153 In this regard, the clearance of remifentanil is not altered during the anhepatic phase of liver transplantation. Hypothermic cardiopulmonary bypass decreases clearance of remifentanil by an average of 20%, presumably reflecting the effect of temperature on blood and tissue esterase activity. Esterase metabolism appears to be a very well-preserved metabolic system with little variability between individuals, which contributes to the predictability of drug effect associated with the infusion of remifentanil.
Elimination Half-Time
An estimated 99.8% of remifentanil is eliminated during the distribution (0.9 minute) and elimination (6.3 minutes) half-time. Clinically, remifentanil behaves like a drug with an elimination half-time of 6 minutes or less.
Context-Sensitive Half-Time
Context-sensitive half-time for remifentanil is independent of the duration of infusion and is estimated to be about 4 minutes.2,3,154 This drug’s rapid clearance is responsible for the lack of accumulation even during prolonged periods of infusion. In contrast, the context-sensitive half-times for sufentanil, alfentanil, and fentanyl are longer and depend significantly on the duration of the infusion (see Table 7-4 and Fig. 7-13).
Clinical Uses
The clinical uses of remifentanil reflect the unique pharmacokinetics of this drug, which allow rapid onset of drug effect, precise titration to the desired effect, the ability to maintain a sufficient effect-site concentration to suppress the stress response, and rapid recovery from the drug’s effects. In cases where a profound analgesic effect is desired transiently (performance of a retrobulbar block), remifentanil may be useful. Prompt onset and short duration of action make remifentanil a useful selection for suppression of the transient sympathetic nervous system response to direct laryngoscopy and tracheal intubation in at-risk patients.155 Intermittent remifentanil administered as patient-controlled analgesia is an effective and reliable analgesic during labor and delivery.156 One additional benefit during labor would be rapid clearance from the neonatal circulation as well, thus reducing the risk of neonatal depression.157 Conceivably, remifentanil could be used for long operations, when a quick recovery time is desired (neurologic assessment, wake-up test) but at a significantly higher cost than other opioids.
Anesthesia can be induced with remifentanil, 1 µg/kg IV administered over 60 to 90 seconds, or with a gradual initiation of the infusion at 0.5 to 1.0 µg/kg IV for about 10 minutes, before administration of a standard hypnotic prior to tracheal intubation.158 The dose of hypnotic drug may need to be decreased to compensate for the synergistic effect with remifentanil. Remifentanil can be used as the analgesic component of a general anesthetic (0.25 to 1.00 µg/kg IV or 0.05 to 2.00 µg/kg/minute IV) or sedation techniques with the ability to rapidly recover from undesirable effects such as opioid-induced depression of ventilation or excessive sedation. Remifentanil, 0.05 to 0.10 µg/kg/minute, in combination with midazolam, 2 mg IV, provides effective sedation and analgesia during monitored anesthesia care in otherwise healthy adult patients.159 Midazolam also produces a dose-dependent potentiation of remifentanil’s depressant effect on breathing rate. Changes in remifentanil drug effect predictably follow changes in the infusion rate, making it possible to more precisely titrate to the desired response than with other opioids. Before cessation of the remifentanil infusion, a longer acting opioid should be administered to ensure analgesia when the patient awakens. The spinal or epidural administration of remifentanil is not recommended, as the safety of the vehicle (glycine, which acts as an inhibitory neurotransmitter) or opioid have not been determined.2 Remifentanil, 100 µg IV, attenuates the acute hemodynamic responses to electroconvulsive therapy and does not alter the duration of electroconvulsive-induced seizure activity.160
Side Effects
The advantage of remifentanil possessing a short recovery period may be considered a disadvantage if the infusion is stopped suddenly, whether it be deliberate or accidental. It is important to administer a longer acting opioid for postoperative analgesia when remifentanil has been administered for this purpose intraoperatively. All fentanyl analogs, including remifentanil, have been reported to induce “seizure-like” activity.161
Nausea and vomiting, depression of ventilation, and mild decreases in systemic blood pressure and heart rate may accompany the administration of remifentanil. Depression of ventilation produced by remifentanil is not altered by renal or liver dysfunction. Histamine release does not accompany the administration of remifentanil. ICP and intraocular pressure are not changed by remifentanil.162,163 High-dose remifentanil decreases cerebral blood flow and cerebral metabolic oxygen requirements without impairing cerebrovascular carbon dioxide reactivity.164 Remifentanil delays drainage of dye from the gallbladder into the duodenum but the delay is shorter than with other opioids.165
Hyperalgesia
Postoperative analgesic requirements in patients receiving relatively large doses of remifentanil intraoperatively are often surprisingly high, suggesting remifentanil may be associated with acute opioid tolerance (Fig. 7-19).166 In addition, delayed hyperalgesia may be produced by acute exposure to large doses of opioids. Tolerance to opioids is pharmacodynamic and tolerance is dose-dependent. Possible mechanisms for tolerance include alterations of the NDMA receptors and its intracellular second messenger systems. In this regard, NMDA receptor antagonists such as ketamine and magnesium block opioid tolerance. Subanesthetic ketamine has been shown to decrease morphine requirements and the development of hyperalgesia after intraoperative remifentanil use (Fig. 7-20).46,167 It is important to know that patients on chronic pain medication preoperatively would require higher doses of opioids postoperatively compared to naive patients and small doses ketamine would be an alternative therapeutic approach. However, those patients might be mistaken to have developed hyperalgesia to opioids.


A recent study in rodents has shown that morphine-induced hyperalgesia is associated with glial activation (indicating that this is not an acute phenomenon) and involved anion dysequilibrium potential between microglia and neuron pathway.168
Codeine
Codeine is the result of the substitution of a methyl group for the hydroxyl group on the number 3 carbon of morphine (see Fig. 7-1). The presence of this methyl group limits first-pass hepatic metabolism and accounts for the efficacy of codeine when administered orally. The elimination half-time of codeine after oral or IM administration is 3.0 to 3.5 hours. About 10% of administered codeine is demethylated in the liver to morphine, which may be responsible for the analgesic effect of codeine, although codeine-6-glucuronide may also exert an analgesic effect.169 Consequently, interindividual variability in metabolism may lead to variable analgesic effect from codeine-containing drugs; some patients who claim that codeine is an ineffective drug for them may in fact poorly metabolize it to active forms. Any remaining codeine is demethylated to inactive norcodeine, which is conjugated or excreted unchanged by the kidneys.
Codeine is effective at suppressing cough at oral doses of 15 mg. Maximal analgesia, equivalent to that produced by 650 mg of aspirin, occurs with 60 mg of codeine. When administered IM, 120 mg of codeine is equivalent in analgesic effect to 10 mg of morphine. Most often, codeine is included in medications as an antitussive or is combined with nonopioid analgesics for the treatment of mild to moderate pain. The risk of physical dependence on codeine appears to be less than that of morphine and occurs only rarely after oral analgesic use. Codeine produces minimal sedation, nausea, vomiting, and constipation. Dizziness may occur in ambulatory patients. Even in large doses, codeine is unlikely to produce apnea. Administration of codeine IV is not recommended, because histamine-induced hypotension is likely.
Hydromorphone
First introduced in 1926, hydromorphone is a derivative of morphine that is about five times as potent as morphine but has a slightly shorter duration of action. It is less hydrophilic than morphine, leading to faster onset of analgesia. This opioid produces somewhat more sedation and evokes less euphoria than morphine. Because of rapid elimination and redistribution, oral dosing every 4 hours is needed to maintain adequate plasma concentrations for analgesia. Hydromorphone is an effective alternative to morphine in the treatment of opioid-responsive moderate to severe pain.170 The uses and side effects of hydromorphone are the same as those of morphine, although histamine release is less prominent with hydromorphone. Similar to other opioids, large doses of hydromorphone have been reported to cause agitation and myoclonus.171
Oxymorphone
Oxymorphone is the result of the addition of a hydroxyl group to hydromorphone. It is about 10 times as potent as morphine and seems to cause more nausea and vomiting. The potential for physical dependence is great. An oral preparation of oxymorphone (immediate release) produces maximum plasma concentrations in 30 minutes with associated rapid onset of analgesia.172
Oxycodone
Oxycodone is commonly used orally for treating acute pain associated with illness or injury. This agent is about twice as potent as oral morphine and has a similar duration of analgesic action. Sustained-release oral oxycodone preparations provide stable plasma concentrations for the treatment of moderate to severe pain. Abuse potential is great including tampering (crushing and powdering) for IV or intranasal injection to obtain a rapid and powerful opioid effect. New, abuse-resistant formulations that are not easily solubilized for IV injection are now widely marketed.
Hydrocodone
Like oxycodone, hydrocodone is a commonly used oral opioid for treating acute pain associated with illness or injury. This agent is similar in potency to oral morphine and has a similar duration of analgesic action. A derivative of codeine, hydrocodone is a useful oral opioid, commonly combined with acetaminophen. The approval by the U.S. Food and Drug Administration of one extended release formulation (Zohydro) provoked much opposition due to its abuse potential and lack of abuse-deterrent features.173 Originally classified as a schedule III drug (moderate abuse potential) in the United States, hydrocodone has subsequently been reclassified as schedule II (high abuse potential) due to increased reports of diversion and abuse.
Methadone
Methadone is a synthetic opioid agonist that produces analgesia in the setting of chronic pain syndromes and is highly effective by the oral route. The efficient oral absorption, prompt onset of action, and prolonged duration of action of methadone render this an attractive drug for suppression of withdrawal symptoms in physically dependent persons such as heroin addicts. Use of this agent is difficult, owing to the marked individual variation in both pharmacokinetic and pharmacodynamic effects. The long terminal elimination half-life of (mean of 26 hours) makes rapid titration impossible; indeed, altering the dose any more frequently than every 5 to 7 days is unwise in the chronic setting, as excess drug accumulation can lead to slow, delayed overdose, often appearing many days after the most recent dose change.
Opioid Withdrawal
Methadone can substitute for morphine at about one-fourth the dosage. Controlled withdrawal from opioids using methadone is milder and less acute than that from morphine. Methadone, 20 mg IV, produces postoperative analgesia lasting >24 hours, reflecting its prolonged (35 hours) elimination half-time. This drug is metabolized in the liver to inactive substances that are excreted in the urine and bile with small amounts of unchanged drug.
The side effects of methadone (depression of ventilation, miosis, constipation, biliary tract spasm) resemble those of morphine. Its sedative and euphoric actions are less than those produced by morphine. Methadone-induced miosis is less prominent than that caused by morphine, and complete tolerance to this action can develop.
Treatment of Chronic Pain
Methadone has been proposed as an alternative to slow-release formulations for treatment of chronic pain because of its low abuse potential. In addition, NMDA receptor antagonist activity may be useful in treatment of neuropathic pain and minimize the potential for development of tolerance. The principal disadvantage for use of methadone to treat chronic pain is its prolonged and unpredictable half-time. When methadone is administered more than once daily, as is common in treatment of chronic pain syndromes, the drug may accumulate and result in high plasma concentrations and associated depression of ventilation.174 For this reason, slow-release formulations (such as oxycodone) may be preferable to methadone for the treatment of outpatient postoperative pain.
Propoxyphene
Propoxyphene is structurally similar to methadone. Oral doses of 90 to 120 mg of propoxyphene produce analgesia and CNS effects similar to those produced by 60 mg of codeine and 650 mg of aspirin. The only clinical use of propoxyphene is treatment of mild to moderate pain that is not adequately relieved by aspirin. Propoxyphene does not possess antipyretic or antiinflammatory effects, and antitussive activity is not significant.
Propoxyphene is completely absorbed after oral administration, but because of extensive first-pass hepatic metabolism (demethylation to norpropoxyphene), the systemic availability is limited. The elimination half-time after oral administration is about 14.5 hours. The most common side effects of propoxyphene are vertigo, sedation, nausea, and vomiting. Overdose is characterized by seizures, cardiac arrhythmias, and depression of ventilation.
Abrupt discontinuation of chronically administered propoxyphene results in a mild withdrawal syndrome. The incidence of abuse of propoxyphene is similar to that of codeine. Administration of this drug IV produces severe damage to veins and limits abuse by this route. Administration of propoxyphene in combination with alcohol and other CNS depressants may result in excessive drug-induced depression of ventilation.
Due to the unfavorable risk-benefit profile (and the availability of better alternatives), propoxyphene was voluntarily withdrawn from the United States market in 2010.
Tramadol
Tramadol is a centrally acting analgesic that has moderate affinity for µ receptors and weak κ and δ opioid receptor affinity but is 5 to 10 times less potent than morphine as an analgesic.175 In addition to µ opioid agonist effects, tramadol enhances the function of the spinal descending inhibitory pathways by inhibition of neuronal reuptake of norepinephrine and 5-hydroxytryptamine (serotonin) as well as presynaptic stimulation of 5-hydroxytryptamine release. In volunteers, naloxone antagonized only an estimated 30% of the effect of tramadol.176
Tramadol is a racemic mixture of two enantiomers, one of which is responsible for inhibition of norepinephrine uptake, whereas the other is responsible for inhibition of 5-hydroxytryptamine reuptake and facilitation of its release, plus the actions of this drug at µ receptors. In this regard, tramadol may be an exception to the argument that chiral mixtures should be avoided when technology exists to prepare a single, pure isomer.177 For example, the production of analgesia by tramadol with the absence of depression of ventilation and a low potential for the development of tolerance, dependence, and abuse may be a result of the complementary and synergistic antinociceptive interaction of the two enantiomers. Tramadol is metabolized by hepatic P450 enzyme systems to the major metabolite is O-desmethyltramadol, which also exerts modest stereoselective analgesic effects.
Tramadol 3 mg/kg administered orally, IM, or IV is effective for the treatment of moderate to severe pain. A marked decrease in postoperative shivering has been noted in treated patients and the minimal depressant effects on breathing are useful.178 Tramadol slows gastric emptying although the effect is small compared with other opioids.179 Tramadol is useful for the treatment of chronic pain because it is believed to be less likely to generate addiction and is not associated with major organ toxicity or significant sedative effects at standard doses. Toxicity from overdose can manifest as hypotension, bradycardia, seizures, coma, and rhabdomyolysis. Interestingly, IV lipid emulsion has also shown promise in reversing tramadol toxicity in a rabbit model.180 Disadvantages of tramadol include its interaction with Coumadin anticoagulants (not all reports confirm this interaction) and the occurrence of drug-related seizures (avoid in patients with epilepsy or those being treated with drugs that lower the seizure threshold such as antidepressants).181 A further drawback to the perioperative use of this drug as an analgesic is a high incidence of associated nausea and vomiting. Ondansetron may interfere with the analgesic component of tramadol that is due to effects on the reuptake and release of 5-hydroxytryptamine.
Heroin
Heroin (diacetylmorphine) is a synthetic opioid produced by acetylation of morphine. It was developed in 1898 and was originally claimed to have no addictive potential. When administered parenterally, heroin acts in a markedly different way than morphine. For example, there is rapid penetration of heroin into the brain, where it is hydrolyzed to the active metabolites monoacetylmorphine and morphine. The unique rapid entrance into the CNS is most likely caused by the lipid solubility and chemical structure of heroin. Compared with morphine, parenteral heroin has a (a) more rapid onset, (b) less opioid-induced nausea, and (c) greater potential for physical dependency. This greater liability for physical dependence is the reason that heroin is not available legally for clinical use in the United States.182,183
Opioid Agonist–Antagonists
Opioid agonist–antagonists include, but are not limited to, pentazocine, butorphanol, nalbuphine, buprenorphine, nalorphine, bremazocine, and dezocine (Fig. 7-21). These drugs bind to µ receptors, where they produce limited responses (partial agonists) or no effect (competitive antagonists). In addition, these drugs often exert partial agonist actions at other receptors, including κ and δ receptors. Antagonist properties of these drugs can attenuate the efficacy of subsequently administered opioid agonists. The side effects are similar to those of opioid agonists, and, in addition, these drugs may cause dysphoric reactions. The advantages of opioid agonist–antagonists are the ability to produce analgesia with limited depression of ventilation and a low potential to produce physical dependence. Furthermore, these drugs have a ceiling effect such that increasing doses do not produce additional responses. This ceiling effect on depression of ventilation, however, is often accompanied by an equally modest ability to decrease anesthetic requirements. In general, agonist-antagonist drugs should be reserved for patients who are unable to tolerate a pure agonist.

Pentazocine
Pentazocine is a benzomorphan derivative that possesses opioid agonist actions as well as weak antagonist actions. It is presumed to exert its agonist effects at δ and κ receptors. Concomitant opioid antagonist activity is weak, being only about one-fifth as potent as nalorphine. Nevertheless, antagonist effects of pentazocine are sufficient to precipitate withdrawal symptoms when administered to patients who have been receiving opioids on a regular basis. The agonist effects of pentazocine are antagonized by naloxone. Indeed, physical dependence to pentazocine can be demonstrated by abrupt withdrawal precipitated by naloxone.
Pharmacokinetics
Pentazocine is well absorbed after oral or parenteral administration. First-pass hepatic metabolism is extensive, with only about 20% of an oral dose entering the circulation. Metabolism of pentazocine occurs by oxidation of terminal methyl groups, and resulting inactive glucuronide conjugates are excreted in the urine. An estimated 5% to 25% of an administered dose of pentazocine is excreted unchanged in the urine, and <2% undergoes biliary excretion. The elimination half-time is 2 to 3 hours.
Clinical Uses
Pentazocine, 10 to 30 mg IV or 50 mg orally, is used most often for the relief of moderate pain. An oral dose of 50 mg is equivalent in analgesic potency to 60 mg of codeine. Pentazocine is useful for treatment of chronic pain when there is a high risk of physical dependence. Placement in the epidural space produces a rapid onset of analgesia that is of shorter duration than that produced by morphine.
Side Effects
The most common side effect of pentazocine is sedation, followed by diaphoresis and dizziness. Sedation is prominent after epidural placement of pentazocine, presumably reflecting activation of κ receptors. Nausea and vomiting are less common than with morphine. Dysphoria, including fear of impending death, is associated with high doses of pentazocine. This tendency to dysphoria limits the physical dependence liability of pentazocine. Pentazocine produces an increase in the plasma concentrations of catecholamines, which may account for increases in heart rate, systemic blood pressure, pulmonary artery blood pressure, and left ventricular end-diastolic pressure that accompany administration of this drug. Pentazocine, 20 to 30 mg IM, produces analgesia, sedation, and depression of ventilation similar to 10 mg of morphine. Increasing the IM dose above 30 mg does not produce proportionate increases in these responses. The increase in biliary tract pressure is less than that produced by equal analgesic doses of morphine, meperidine, or fentanyl.35 Pentazocine crosses the placenta and may cause fetal depression. In contrast to morphine, miosis does not occur after administration of pentazocine.
Butorphanol
Butorphanol is an agonist-antagonist opioid that resembles pentazocine. Compared with pentazocine, its agonist effects are about 20 times greater, whereas its antagonist actions are 10 to 30 times greater. It is speculated that butorphanol has a (a) low affinity for µ receptors to produce antagonism, (b) moderate affinity for κ receptors to produce analgesia and antishivering effects, and (c) minimal affinity for σ receptors, so the incidence of dysphoria is low.
Butorphanol is rapidly and almost completely absorbed after IM injection. In postoperative patients, 2 to 3 mg IM produces analgesia and depression of ventilation similar to 10 mg of morphine. Intranasal butorphanol has been used for the treatment of postoperative pain and migraine pain. The intraoperative use of butorphanol, like pentazocine, seems to be limited. The elimination half-time of butorphanol is 2.5 to 3.5 hours. Metabolism is principally to inactive hydroxybutorphanol, which is eliminated largely in the bile and to a lesser extent in the urine.
Side Effects
Common side effects of butorphanol include sedation, nausea, and diaphoresis. Dysphoria, reported frequently with other opioid agonist–antagonists, is infrequent after administration of butorphanol. Depression of ventilation is similar to that produced by similar doses of morphine. Like pentazocine, analgesic doses of butorphanol increase systemic blood pressure, pulmonary artery blood pressure, and cardiac output. Also, similar to pentazocine, the effects of butorphanol on the biliary and gastrointestinal tract seem to be milder than those produced by morphine. It may be difficult to use an opioid agonist effectively as an analgesic in the presence of butorphanol. This must be remembered when considering the use of butorphanol or any other opioid-agonist for preoperative medication. Withdrawal symptoms do occur after acute discontinuation of chronic therapy with butorphanol, but symptoms are mild.
Nalbuphine
Nalbuphine is an agonist-antagonist opioid that is related chemically to oxymorphone and naloxone. It is equal in potency as an analgesic to morphine and is about one-fourth as potent as nalorphine as an antagonist. Nalbuphine is metabolized in the liver and has an elimination half-time of 3 to 6 hours. Naloxone reverses the agonist effects of nalbuphine. Nalbuphine, 10 mg IM, produces analgesia with an onset of effect and duration of action similar to those of morphine. Depression of ventilation is similar to that of morphine until 30 mg IM of nalbuphine is exceeded, after which no further depression of ventilation occurs (ceiling effect).184 Sedation is the most common side effect, occurring in about one-third of patients treated with nalbuphine. The incidence of dysphoria is less than that with pentazocine or butorphanol but is qualitatively similar and increases in frequency as the dose of nalbuphine is increased. In contrast to pentazocine and butorphanol, nalbuphine does not increase systemic blood pressure, pulmonary artery blood pressure, heart rate, or atrial filling pressures. For this reason, nalbuphine may be useful to provide sedation and analgesia in patients with heart disease, as during cardiac catheterization. Abrupt withdrawal of nalbuphine after chronic administration produces withdrawal symptoms that are milder than those of morphine and more severe than those of pentazocine. The abuse potential of nalbuphine is low.
The antagonist effects of nalbuphine are speculated to occur at µ receptors. As a result, the subsequent use of morphine-like drugs for anesthesia or postoperative analgesia after preoperative medication with nalbuphine may not provide adequate analgesia. Conversely, the antagonist effects of nalbuphine at µ receptors could be an advantage in the postoperative period to reverse lingering ventilatory depressant effects of opioid agonists while still maintaining analgesia. Nalbuphine, 10 to 20 mg IV, reverses postoperative depression of ventilation caused by fentanyl but maintains analgesia.185 Evidence of recurrent hypoventilation often occurs 2 to 3 hours after administration of nalbuphine to antagonize the effects of fentanyl.
Buprenorphine
Buprenorphine is an agonist-antagonist opioid derived from the opium alkaloid thebaine. Its analgesic potency is great, with 0.3 mg IM being equivalent to 10 mg of morphine. After IM administration, the onset of buprenorphine effect occurs in about 30 minutes, and the duration of action is at least 8 hours. It is estimated that the affinity of buprenorphine for µ receptors is 50 times greater than that of morphine, and subsequent slow dissociation from these receptors accounts for its prolonged duration of action and resistance to antagonism with naloxone. After IM administration, nearly two-thirds of the drug appears unchanged in the bile and the remainder is excreted in urine as inactive metabolites.
Buprenorphine is effective in relieving moderate to severe pain such as that present in the postoperative period and that associated with cancer, renal colic, and myocardial infarction. Placed in the epidural space, the high lipid solubility (five times that of morphine) and affinity for opioid receptors limits cephalad spread and the likelihood of delayed depression of ventilation.186 The antagonist effects of buprenorphine reflect the ability of this drug to displace opioid agonists from µ receptors.
Side Effects
The side effects of buprenorphine include drowsiness, nausea, vomiting, and depression of ventilation. These are similar in magnitude to the side effects of morphine but may be prolonged and resistant to antagonism with naloxone. Pulmonary edema has been observed after administration of buprenorphine.187 In contrast to other opioid agonist-antagonists, dysphoria is unlikely to occur in association with administration of this drug. Because of its antagonist properties, buprenorphine can precipitate withdrawal in patients who are physically dependent on morphine. Conversely, withdrawal symptoms in patients who are physically dependent on buprenorphine develop slowly and are of lesser intensity than those associated with morphine. In this respect, withdrawal from buprenorphine resembles that from other opioid agonist–antagonists, and the risk of abuse is low.
Nalorphine
Nalorphine is equally potent with morphine as an analgesic but is not clinically useful because of a high incidence of dysphoria. The high incidence of dysphoria may reflect activity of this drug at σ receptors. The antagonist actions of nalorphine reflect its ability to displace opioid agonists from µ receptors.
Bremazocine
Bremazocine is a benzomorphan derivative that is twice as potent as morphine as an analgesic but, in animals, does not produce depression of ventilation or evidence of physical dependence. It is speculated that bremazocine interacts selectively with κ receptors. Failure of naloxone to reverse sedation produced by bremazocine is further evidence that this drug is acting at sites other than µ receptors.
Dezocine
Dezocine, 0.15 mg/kg IM, is an opioid agonist-antagonist with the analgesic potency, onset, and duration of action in the relief of postoperative pain comparable to morphine. Absorption of dezocine, 10 to 15 mg, after IM administration is rapid and complete, with analgesia occurring after about 30 minutes. After IV administration of dezocine, 5 to 10 mg, the onset of analgesia occurs in about 15 minutes. Elimination of dezocine is principally in the urine as a glucuronide conjugate. Like other opioid agonist–antagonists, dezocine exhibits a ceiling effect for depression of ventilation that parallels its analgesic activity.188 Large doses of dezocine administered IV to humans do not produce significant changes in systemic blood pressure, pulmonary artery pressure, or cardiac output.
Dezocine has a high affinity for µ receptors and a moderate affinity for δ receptors. The interaction at δ receptors serves to facilitate the effect of agonist activity at µ receptors. The incidence of dysphoria is minimal after administration of dezocine, presumably reflecting the low affinity of this drug for σ receptors.
Meptazinol
Meptazinol is a partial opioid agonist with relative selectivity at mu1 receptors. As a result, depression of ventilation does not occur with analgesic doses of meptazinol (100 mg IM is equivalent to morphine, 8 mg IM). The onset of analgesia is rapid, but the duration of action is <2 hours. Bioavailability after oral administration is <10%. Metabolism is to inactive glucuronide conjugates that are excreted by the kidneys. Protein binding is 20% to 25%, and the elimination half-time is about 2 hours. Physical dependence does not occur, miosis is slight, and constipation is absent. Nausea and vomiting are common side effects. Meptazinol cannot be substituted for an opioid agonist in patients physically dependent on opioids.
Opioid Antagonists
Minor changes in the structure of an opioid agonist can convert the drug into an opioid antagonist at one or more of the opioid receptor sites (Fig. 7-22).189 The most common change is substitution of an alkyl group for a methyl group on an opioid agonist. For example, naloxone is the N-alkyl derivative of oxymorphone (see Fig. 7-21).

Naloxone, naltrexone, and nalmefene are pure µ opioid receptor antagonists with no agonist activity. The high affinity for opioid receptors characteristic of pure opioid antagonists results in displacement of the opioid agonist from µ receptors. After this displacement, the binding of the pure antagonist does not activate µ receptors and antagonism occurs.
Naloxone
Naloxone is a nonselective antagonist at all three opioid receptors. Naloxone is selective when used to (a) treat opioid-induced depression of ventilation as may be present in the postoperative period, (b) treat opioid-induced depression of ventilation in the neonate due to maternal administration of an opioid, (c) facilitate treatment of deliberate opioid overdose, and (d) detect suspected physical dependence. Naloxone, 1 to 4 µg/kg IV, promptly reverses opioid-induced analgesia and depression of ventilation. The short duration of action of naloxone (30 to 45 minutes) is presumed to be due to its rapid removal from the brain. This emphasizes that supplemental doses of naloxone will likely be necessary for sustained antagonism of opioid agonists. In this regard, a continuous infusion of naloxone, 5 µg/kg/hour, prevents depression of ventilation without altering analgesia produced by neuraxial opioids.190
Naloxone is metabolized primarily in the liver by conjugation with glucuronic acid to form naloxone-3-glucuronide. The elimination half-time is 60 to 90 minutes. Naloxone is absorbed orally, but metabolism during its first pass through the liver renders it only one-fifth as potent as when administered parenterally.
Side Effects
Antagonism of opioid-induced depression of ventilation is accompanied by an inevitable reversal of analgesia. It may be possible, however, to titrate the dose of naloxone such that depression of ventilation is partially but acceptably antagonized to also maintain partial analgesia.
Nausea and vomiting appear to be closely related to the dose and speed of injection of naloxone. Administration of naloxone slowly over 2 to 3 minutes rather than as a bolus seems to decrease the incidence of nausea and vomiting. Awakening occurs either before or simultaneously with vomiting, which ensures that the patient’s protective upper airway reflexes have returned and the likelihood of pulmonary aspiration is minimized.
Cardiovascular stimulation after administration of naloxone manifests as increased sympathetic nervous system activity, presumably reflecting the abrupt reversal of analgesia and the sudden perception of pain. This increased sympathetic nervous system activity may manifest as tachycardia, hypertension, pulmonary edema, and cardiac dysrhythmias.191 Even ventricular fibrillation has occurred after the IV administration of naloxone and the associated sudden increase in sympathetic nervous system activity.192
Naloxone can easily cross the placenta. For this reason, administration of naloxone to an opioid-dependent parturient may produce acute withdrawal in the neonate.
Role in Treatment of Shock
Naloxone produces dose-related improvement in myocardial contractility and survival in animals subjected to hypovolemic shock and, to a lesser extent, in those subjected to septic shock.193 The beneficial effects of naloxone in the treatment of shock occur only with doses >1 mg/kg IV, suggesting that the beneficial effects of this drug are not opioid receptor–mediated or, alternatively, are mediated by opioid receptors other than µ receptors—possibly δ and κ receptors.
Antagonism of General Anesthesia
The occasional observation that high doses of naloxone seem to antagonize the depressant effect of inhaled anesthetics may represent drug-induced activation of the cholinergic arousal system in the brain, independent of any interaction with opioid receptors.194 A role of endorphins in the production of general anesthesia is not supported by data demonstrating a failure of naloxone to alter anesthetic requirements (MAC) in animals.
Naltrexone
Naltrexone, in contrast to naloxone, is highly effective orally, producing sustained antagonism of the effects of opioid agonists for as long as 24 hours. It has found a role in the treatment of alcoholism, possibly by reducing the pleasure associated with ethanol intoxication.195
Nalmefene
Nalmefene is a pure opioid antagonist that is a 6-methylene analogue of naltrexone (see Fig. 7-22).196 Nalmefene is equipotent to naloxone. The recommended dose is 15 to 25 µg IV administered every 2 to 5 minutes until the desired effect is achieved, with the total dose not exceeding 1 µg/kg. Prophylactic administration of nalmefene significantly decreases the need for antiemetics and antipruritic medications in patients receiving IV patient-controlled analgesia with morphine.197 The primary advantage of nalmefene over naloxone is its longer duration of action, which might provide a greater degree of protection from delayed depression of ventilation due to residual effects of the opioid as the antagonist is cleared. Compared with the brief elimination half-time of naloxone, the half-time of nalmefene is about 10.8 hours. This longer duration of action is likely due to the slower clearance of nalmefene compared with naloxone. Nalmefene is metabolized by hepatic conjugation, with <5% excreted unchanged in the urine. As with naloxone, acute pulmonary edema has occurred after the IV administration of nalmefene.198
Methylnaltrexone
Methylnaltrexone is a quaternary opioid receptor antagonist. The highly ionized quaternary methyl group limits the transfer of methylnaltrexone across the blood–brain barrier. As a result, methylnaltrexone is active at peripheral rather than central opioid receptors as demonstrated by its failure to penetrate the CNS sufficiently to promote withdrawal in morphine-dependent animals.
In humans, methylnaltrexone attenuates morphine-induced changes in the rate of gastric emptying and also decreases the incidence of nausea.199 The attenuation of morphine-induced nausea may be due to antagonism of morphine at the chemoreceptor trigger zone (located outside the blood–brain barrier) or through limitation of the delay in gastric emptying, which, in itself, may cause nausea. Presumably, methylnaltrexone could prevent the undesirable effects of opioids on gastric emptying and possibly vomiting without altering centrally mediated analgesia.
Alvimopan
Alvimopan (Fig. 7-23) is a newer µ-selective oral peripheral opioid antagonist. Its oral bioavailability is approximately 6% and its metabolism also relies on gut flora.200 It was approved by the U.S. Food and Drug Administration for treatment of postoperative ileus and has shown mixed results in subsequent clinical trials for treating ileus or opioid-induced constipation. Although likely safe in the acute setting, concern about a potential increase in cardiovascular events with long-term use has dampened enthusiasm for this drug.201

Tamper- or Abuse-Resistant Opioids
In recent years, prescribing of opioids for chronic noncancer pain has gained greater social acceptance. Correspondingly, the aggregate opioid consumption in the United States increased from 97 mg of morphine equivalents per person in 1997 to 710 mg in 2010.202 Along with this increase in prescription use, there has been a dramatic rise in drug diversion and abuse, both of which have a significant human and economic cost.203
In an attempt to provide effective oral opioid analgesia with less potential for abuse, oral opioid formulations have been developed to reduce the ability to rapidly ingest the active ingredient (crushing, snorting, injecting) with subsequent euphoric effects.204 Examples of this strategy include Suboxone (buprenorphine plus naloxone), Embeda (extended release morphine plus naltrexone), and OxyNal (oxycodone plus naltrexone). Convincing evidence of their efficacy is currently lacking, but they may prove to fill a useful role in chronic oral analgesic use by preventing opioid overdose due to crushing or injecting the extended-release opioid formulation.
Opioid Allergy
Although many patients claim “allergies” to opioids, true opioid allergy is rare. More often, predictable side effects of opioids such as localized histamine release, orthostatic hypotension, nausea, and vomiting are misinterpreted as an allergic reaction. Morphine contains a tertiary amine group, which causes nonimmune release of histamine. True allergic reactions to morphine are much rarer. Fentanyl is chemically dissimilar to morphine and, as such, does not cross-react with morphine derivatives.205 To date, there have been only four reported cases of fentanyl-induced anaphylaxis 206–209, one of which was subsequently retracted due to an undiagnosed latex allergy.210 In each of these cases, the reaction presented as hypotension and urticaria.
Opioid Immune Modulation
Opioid therapy may alter immunity through neuroendocrine effects or via direct effects on the immune system.40,211 Opioid receptors are present on immune cells, including T and B lymphocytes, dendritic cells, neutrophils, macrophages, and microglia.41 Prolonged exposure to opioids appears more likely than short-term exposure to produce immunosuppression especially in susceptible persons and abrupt withdrawal may also induce immunosuppression.
Of recent interest is the possible link between opioid-induced immunosuppression and cancer recurrence after resection.212–215 Opioids alter the development, differentiation, and function of immune cells, and particularly seem to depress natural killer (NK) cell activity (Fig. 7-24).216–218 NK cells are lymphoid lineage cells that induce apoptosis in target cells via mechanisms distinct from T lymphocytes and appear to be a major factor in tumor surveillance. Although immunosuppressant effects of opioids raise concerns, it is equally important to recognize that pain itself can impair immune function.

Anesthetic Requirements
The contribution of opioids to total anesthetic requirements can be estimated by determining the decrease in MAC of a volatile anesthetic in the presence of opioids. In animals, morphine decreases the MAC of volatile anesthetics in a dose-dependent manner, but there appears to be a ceiling effect to the anesthetic-sparing ability of morphine, with a plateau at 65% MAC.219 A single dose of fentanyl, 3 µg/kg IV 25 to 30 minutes before surgical incision, decreases isoflurane or desflurane MAC by about 50%.220 In animals, sufentanil decreases enflurane MAC by 70% to 90%.221 In patients, a sufentanil plasma concentration of 0.145 ng/mL produced a 50% decrease in isoflurane MAC, whereas plasma sufentanil concentrations of >0.5 ng/mL exhibited a ceiling effect.222,223 As with other opioids, alfentanil administered to animals decreases MAC in a dose-dependent manner until a plateau is reached at about a 70% decrease in MAC. The decrease in MAC produced by remifentanil is similar to that produced by other opioids and ranges from 50% to 91%, depending on the plasma concentration of remifentanil.224 These data cast serious doubt on the ability of opioid agonists to provide reliable amnesia during surgical procedures, even at extremely high doses.
Opioid agonist–antagonists are less effective than opioid agonists in decreasing MAC. For example, butorphanol, nalbuphine, and pentazocine maximally decrease MAC 11%, 8%, and 20%, respectively, even when the dose of these drugs is increased 40-fold.225 The ceiling effect for MAC parallels the ceiling effect for depression of ventilation and is consistent with the clinical impression that even large doses of opioid agonist–antagonists do not produce unconsciousness or even prevent patient movement in response to painful stimulation. Thus, the intraoperative role for these drugs is minimal.
Patient-Controlled Analgesia
As an alternative to intermittent bolus dosing of medication, patients may be provided with a mechanism to address their own analgesic requirements. Termed patient-controlled analgesia (PCA), this most typically involves a programmable electronic pump, which delivers a prescribed dose of medication upon patient demand. The rationale of this technique is that, by using frequent, small doses of opioid, patients will have better control of their pain by keeping effect-site concentrations in the therapeutic range for a larger proportion of the time. Rather than wide swings between inadequate analgesia and oversedation, the PCA regimen is designed to allow patients to self-titrate their dosing to optimize their pain management (Fig. 7-25).

Proposed advantages of this technique include decreased health care provider workload, increased patient satisfaction, lower opioid consumption, and the inherent safety of needing a conscious patient to self-administer a dose of opioid.226 Despite multiple studies, the most recent review by the Cochrane Collaboration suggests that PCA only provides marginally improved analgesia over conventional opioid therapy. Patient satisfaction, however, is higher with PCA.227
Typically, IV PCA opioids are used. Morphine, hydromorphone, and fentanyl are the most common choices. Suggested initial dosing regimens are listed in Table 7-5. Although PCA is usually used with IV opioids, there are a number of new technologies being evaluated, including transdermal fentanyl, sublingual sufentanil, oral opioids, intranasal opioids, and even inhaled opioids.226 None are currently used clinically.

Owing to the unique pharmacokinetic profile of remifentanil, it has found a role in PCA for labor and delivery. Because remifentanil undergoes nonenzymatic hydrolysis in both the maternal and fetal circulations, it has minimal effects on the neonate. In cases where epidural analgesia is contraindicated, remifentanil PCA has shown to provide good analgesia during the first stage of labor.228
Neuraxial Opioids
Placement of opioids in the epidural or subarachnoid space to manage acute or chronic pain is based on the knowledge that opioid receptors (principally µ receptors) are present in the substantia gelatinosa of the spinal cord.229Analgesia produced by neuraxial opioids, in contrast to regional anesthesia with local anesthetics, is not associated with sympathectomy, sensory block, or weakness. Analgesia is dose related (epidural dose is 5 to 10 times the subarachnoid dose) and effective for visceral pain. Neuraxial morphine may decrease the MAC for volatile anesthetics, although not all investigators have demonstrated this effect.230–232
Analgesia that follows epidural placement of opioids reflects diffusion of the drug across the dura to gain access to µ opioid receptors in the spinal cord as well as systemic absorption to produce effects similar to those that would follow IV administration of the opioid. For example, the mechanism of postoperative analgesia produced by epidural administration of highly lipophilic opioids (fentanyl, sufentanil) is primarily a reflection of systemic absorption. In fact, it has been proposed that epidural administration of lipophilic opioids may offer no clinical advantages over IV administration.233 Poorly lipid-soluble opioids such as morphine result in a slower onset of analgesia but a longer duration of action than lipid-soluble opioids when administered via the neuraxial route.
Pharmacokinetics
Opioids placed in the epidural space may undergo uptake into epidural fat, systemic absorption, or diffusion across the dura into the CSF.234 Epidural administration of opioids produces considerable CSF concentrations of drug. Penetration of the dura is highly influenced by lipid solubility, but molecular weight may also be important. Fentanyl and sufentanil are, respectively, approximately 800 and 1,600 times as lipid soluble as morphine. After epidural administration, CSF concentrations of fentanyl peak in about 20 minutes and sufentanil in about 6 minutes. In contrast, CSF concentrations of morphine, after epidural administration, peak in 1 to 4 hours. Furthermore, only about 3% of the dose of morphine administered epidurally crosses the dura to enter the CSF.235
The epidural space contains an extensive venous plexus, and vascular absorption of opioids from the epidural space is extensive. After epidural administration, fentanyl blood concentrations peak in 5 to 10 minutes, whereas blood concentrations of the more lipid-soluble sufentanil peak even sooner.236 In contrast, blood concentrations of morphine after epidural administration peak after 10 to 15 minutes. Epidural administration of morphine, fentanyl, and sufentanil produces opioid blood concentrations that are similar to those produced by an IM injection of an equivalent dose.234 The addition of epinephrine to the solution placed into the epidural space decreases systemic absorption of the opioid but does not influence the diffusion of morphine across the dura into the CSF. The addition of epinephrine to intrathecal morphine solutions enhances postoperative analgesia compared with intrathecal morphine alone.237 Vascular absorption after intrathecal administration of opioids is clinically insignificant.
Cephalad movement of opioids in the CSF principally depends on lipid solubility. For example, lipid-soluble opioids such as fentanyl and sufentanil are limited in their cephalad migration by uptake into the spinal cord, whereas less lipid-soluble morphine remains in the CSF for transfer to more cephalad locations. After lumbar intrathecal morphine administration, appreciable cervical CSF concentrations occur 1 to 5 hours after injection, whereas cervical CSF concentrations of highly lipid-soluble opioids are minimal after their epidural administration. The underlying cause of ascension of morphine is bulk flow of CSF. CSF ascends in a cephalad direction from the lumbar region, reaching the cisterna magna in 1 to 2 hours and the fourth and lateral ventricles by 3 to 6 hours.234 Coughing or straining, but not body position, can affect movement of CSF. The elimination half-time of morphine in CSF is similar to that in plasma.238
Side Effects
Side effects of neuraxial opioids are caused by the presence of drug in either the CSF or systemic circulation.234 In general, most side effects are dose dependent. Some side effects are mediated via interaction with specific opioid receptors, whereas others are nonspecific. Side effects are less common in patients chronically exposed to opioids. The four classic side effects of neuraxial opioids are pruritus, nausea and vomiting, urinary retention, and depression of ventilation.
Pruritus
Pruritus is the most common side effect with neuraxial opioids. It may be generalized but is more likely to be localized to the face, neck, or upper thorax. The incidence of pruritus varies widely and is often elicited only after direct questioning. Severe pruritus is rare, occurring in about 1% of patients. Pruritus is more likely to occur in obstetric patients, perhaps due to the interaction of estrogen with opioid receptors. The incidence may or may not be dose related. Pruritus usually occurs within a few hours of injection and may precede the onset of analgesia.
Although opioids may liberate the release of histamine from mast cells, this does not appear to be the mechanism for pruritus. Instead, pruritus induced by neuraxial opioids is likely due to cephalad migration of the opioid in CSF and subsequent interaction with opioid receptors in the trigeminal nucleus. An opioid antagonist such as naloxone is effective in relieving opioid-induced pruritus. Antihistamines may be an effective treatment for pruritus, but this is likely secondary to their sedative effect. Gabapentin, an anticonvulsant often used for treatment of neuropathic pain, has also shown some promise in the treatment of opioid-induced pruritus.239,240
Urinary Retention
The incidence of urinary retention varies widely and is most common in young males. Urinary retention with neuraxial opioids is more common than after IV or IM administration of equivalent doses of the opioid. The incidence of this side effect is not dose dependent or related to systemic absorption of the opioid. Urinary retention is most likely due to interaction of the opioid with opioid receptors located in the sacral spinal cord. This interaction promotes inhibition of sacral parasympathetic nervous system outflow, which causes detrusor muscle relaxation and an increase in maximum bladder capacity, leading to urinary retention. In humans, epidural morphine causes marked detrusor muscle relaxation within 15 minutes of injection that persists for up to 16 hours; it is readily reversed with naloxone.190
Depression of Ventilation
The most serious side effect of neuraxial opioids is depression of ventilation, which may occur within minutes of administration or may be delayed for hours. The incidence of ventilatory depression requiring intervention after conventional doses of neuraxial opioids is about 1%, which is the same as that after conventional doses of IV or IM opioids.234
Early depression of ventilation occurs within 2 hours of neuraxial injection of the opioid. Most reports of clinically important depression of ventilation involve epidural administration of fentanyl or sufentanil. This depression of ventilation most likely results from systemic absorption of the lipid-soluble opioid, although cephalad migration of opioid in the CSF may also be responsible. Clinically significant early depression of ventilation after intrathecal injection of morphine is unlikely.
Delayed depression of ventilation occurs more than 2 hours after neuraxial opioid administration and reflects cephalad migration of the opioid in the CSF and subsequent interaction with opioid receptors located in the ventral medulla. All reports of clinically significant delayed depression of ventilation involve morphine.234 Delayed depression of ventilation characteristically occurs 6 to 12 hours after epidural or intrathecal administration of morphine. Clinically important depression of ventilation has not been described more than 24 hours after the epidural or intrathecal injection of morphine.
Factors that increase the risk of delayed depression of ventilation, especially concomitant use of any IV opioid or sedative, must be considered in determining the dose of neuraxial opioid (see Table 7-3).234Coughing may affect the movement of CSF and increase the likelihood of depression of ventilation. Obstetric patients appear to be at less risk for ventilatory depression, perhaps because of the increased stimulation to ventilation provided by progesterone.
Detection of depression of ventilation induced by neuraxial opioids may be difficult. Arterial hypoxemia and hypercarbia may develop despite a normal breathing rate (Fig. 7-26).241 Pulse oximetry reliably detects opioid-induced arterial hypoxemia, and supplemental oxygen (2 L/min) is an effective treatment. The most reliable clinical sign of depression of ventilation, however, appears to be a depressed level of consciousness, possibly caused by hypercarbia.234 In patients receiving supplemental oxygen, arterial hypoxemia is a very late sign of hypoventilation; thus, pulse oximetry is of limited value in detection of opioid-induced respiratory depression in these patients. Prophylactic infusions of naloxone are of variable efficacy in protecting against depression of ventilation.190,242 Naloxone (0.25 µg/kg/hour IV) is effective in attenuating the side effects (nausea and vomiting, pruritus) associated with morphine-induced analgesia delivered by a patient-controlled IV delivery system.243

Sedation
Sedation after administration of neuraxial opioids appears to be dose related and occurs with all opioids but is most commonly associated with the use of sufentanil. When sedation occurs with neuraxial opioids, depression of ventilation must be considered. Mental status changes other than sedation may also occur with neuraxial opioids. Naloxone-reversible psychosis, catatonia, and hallucinations have been described.234
Central Nervous System Excitation
Tonic skeletal muscle rigidity resembling seizure activity is a well-known side effect of large IV doses of opioids, but this response is rarely observed after neuraxial administration. Myoclonic activity has been observed after neuraxial opioids and, in one report, progressed to a grand mal seizure.244 Although large doses of opioids reliably produce seizures in animals, clinically relevant doses of IV or neuraxial opioids are unlikely to be associated with generalized cortical seizure activity in humans.234 Cephalad migration of the opioid in CSF and subsequent interaction with nonopioid receptors in the brainstem or basal ganglia is the most likely explanation for opioid-induced CNS excitation. In this regard, opioids may block glycine or γ-aminobutyric acid–mediated inhibition.
Viral Reactivation
A link exists between the use of epidural morphine in obstetric patients and reactivation of herpes simplex labialis virus. Reactivation of the herpes virus occurs 2 to 5 days after epidural administration of the opioid.245 Manifestation of symptoms of herpes labialis (cold sores) characteristically occurs in the same sensory innervation as the primary infection, which are usually facial areas innervated by the trigeminal nerve. The underlying mechanism causing herpes virus reactivation likely involves cephalad migration of opioid in CSF and subsequent interaction with the trigeminal nucleus.
Neonatal Morbidity
Systemic absorption after epidural administration of an opioid results in predictable blood levels of the drug in the neonate immediately after birth. Clinically important depression of ventilation has been observed in the newborns of mothers receiving epidural opioids.234 The progress of labor in general does not seem to be adversely affected by neuraxial opioids.246 After administration of epidural fentanyl or sufentanil to parturients, the concentration of opioid in breast milk is negligible. As a general rule, opioid analgesics should not be withheld from women recovering from cesarean delivery due to concerns of neonatal drug exposure in breast milk.
Miscellaneous Side Effects
Epidural morphine has been associated with sustained erection (priapism) and inability to ejaculate in male volunteers. Naloxone-reversible miosis, nystagmus, and vertigo may occur after neuraxial opioids, most commonly morphine. Neuraxial opioids may delay gastric emptying, most likely reflecting an interaction of the opioid with a spinal cord opioid receptor.247 Neuraxial opioids, by inhibiting shivering, may cause decreased body temperature. Oliguria and water retention leading to peripheral edema have been reported after neuraxial opioid administration. Water retention is likely caused by release of vasopressin, stimulated by cephalad migration of the opioid in the CSF. Neuraxial opioids have been implicated as possible causes of spinal cord damage, especially following accidental use of opioids containing toxic preservatives.234 Clinical manifestations in these patients include sensory and motor neurologic dysfunction, myoclonic spasms, paresis, and paralysis. On the other hand, neuraxial opioids have been administered chronically without adverse sequelae.
References
1. Brownstein MJ. A brief history of opiates, opioid peptides, and opioid receptors. Proc Natl Acad Sci U S A. 1993;90(12):5391–5393.
2. Burkle H, Dunbar S, Van Aken H. Remifentanil: a novel, short-acting, mu-opioid. Anesth Analg. 1996;83(3):646–651.
3. Egan TD, Lemmens HJ, Fiset P, et al. The pharmacokinetics of the new short-acting opioid remifentanil (GI87084B) in healthy adult male volunteers. Anesthesiology. 1993;79(5):881–892.
4. Shafer SL, Varvel JR. Pharmacokinetics, pharmacodynamics, and rational opioid selection. Anesthesiology. 1991;74(1):53–63.
5. Pleuvry BJ. Opioid receptors and their relevance to anaesthesia. Br J Anaesth. 1993;71(1):119–126.
6. Stein C. The control of pain in peripheral tissue by opioids. N Engl J Med. 1995;332(25):1685–1690.
7. Stein C. Peripheral mechanisms of opioid analgesia. Anesth Analg. 1993;76(1):182–191.
8. Atcheson R, Lambert DG. Update on opioid receptors. Br J Anaesth. 1994;73(2):132–134.
9. Lambert DG. Recent advances in opioid pharmacology. Br J Anaesth. 1998;81(1):1–2.
10. Li S, Zhu J, Chen C, et al. Molecular cloning and expression of a rat kappa opioid receptor. Biochem J. 1993;295(pt 3):629–633.
11. Chen Y, Mestek A, Liu J, et al. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol Pharmacol. 1993;44(1):8–12.
12. Yaksh TL. Pharmacology and mechanisms of opioid analgesic activity. Acta Anaesthesiol Scand. 1997;41(1, pt 2):94–111.
13. Rittner HL, Brack A, Machelska H, et al. Opioid peptide-expressing leukocytes: identification, recruitment, and simultaneously increasing inhibition of inflammatory pain. Anesthesiology. 2001;95(2):500–508.
14. Heine MF, Tillet ED, Tsueda K, et al. Intra-articular morphine after arthroscopic knee operation. Br J Anaesth. 1994;73(3):413–415.
15. Kosterlitz HW. Biosynthesis of morphine in the animal kingdom. Nature. 1987;330(6149):606.
16. Prinster SC, Hague C, Hall RA. Heterodimerization of g protein-coupled receptors: specificity and functional significance. Pharmacol Rev. 2005;57(3):289–298.
17. Milligan G. G protein-coupled receptor dimerisation: molecular basis and relevance to function. Biochim Biophys Acta. 2007;1768(4):825–835.
18. Gupta A, Mulder J, Gomes I, et al. Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci Signal. 2010;3(131):ra54.
19. Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5(7):565–575.
20. Pomeranz B, Chiu D. Naloxone blockade of acupuncture analgesia: endorphin implicated. Life Sci. 1976;19(11):1757–1762.
21. Bie B, Brown DL, Naguib M. Synaptic plasticity and pain aversion. Eur J Pharmacol. 2011;667(1–3):26–31.
22. Zubieta JK, Smith YR, Bueller JA, et al. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293(5528):311–315.
23. Bingel U, Wanigasekera V, Wiech K, et al. The effect of treatment expectation on drug efficacy: imaging the analgesic benefit of the opioid remifentanil. Sci Transl Med. 2011;3(70):70ra14.
24. Lowenstein E, Whiting RB, Bittar DA, et al. Local and neurally mediated effects of morphine on skeletal muscle vascular resistance. J Pharmacol Exp Ther. 1972;180(2):359–367.
25. Cahalan MK, Lurz FW, Eger EI II, et al. Narcotics decrease heart rate during inhalational anesthesia. Anesth Analg. 1987;66(2):166–170.
26. Rosow CE, Moss J, Philbin DM, et al. Histamine release during morphine and fentanyl anesthesia. Anesthesiology. 1982;56(2):93–96.
27. Philbin DM, Moss J, Akins CW, et al. The use of H1 and H2 histamine antagonists with morphine anesthesia: a double-blind study. Anesthesiology. 1981;55(3):292–296.
28. Stoelting RK, Gibbs PS. Hemodynamic effects of morphine and morphine-nitrous oxide in valvular heart disease and coronary-artery disease. Anesthesiology. 1973;38(1):45–52.
29. Tomicheck RC, Rosow CE, Philbin DM, et al. Diazepam-fentanyl interaction—hemodynamic and hormonal effects in coronary artery surgery. Anesth Analg. 1983;62(10):881–884.
30. McPherson BC, Yao Z. Signal transduction of opioid-induced cardioprotection in ischemia-reperfusion. Anesthesiology. 2001;94(6):1082–1088.
31. Bowdle TA, Rooke GA. Postoperative myoclonus and rigidity after anesthesia with opioids. Anesth Analg. 1994;78(4):783–786.
32. Costall B, Fortune DH, Naylor RJ. Involvement of mesolimbic and extrapyramidal nuclei in the motor depressant action of narcotic drugs. J Pharm Pharmacol. 1978;30(9):566–572.
33. Bennett JA, Abrams JT, Van Riper DF, et al. Difficult or impossible ventilation after sufentanil-induced anesthesia is caused primarily by vocal cord closure. Anesthesiology. 1997;87(5):1070–1074.
34. Paqueron X, Lumbroso A, Mergoni P, et al. Is morphine-induced sedation synonymous with analgesia during intravenous morphine titration? Br J Anaesth. 2002;89(5):697–701.
35. Radnay PA, Brodman E, Mankikar D, et al. The effect of equi-analgesic doses of fentanyl, morphine, meperidine and pentazocine on common bile duct pressure. Anaesthesist. 1980;29(1):26–29.
36. Jones RM, Fiddian-Green R, Knight PR. Narcotic-induced choledochoduodenal sphincter spasm reversed by glucagon. Anesth Analg. 1980;59(12):946–947.
37. Jones RM, Detmer M, Hill AB, et al. Incidence of choledochoduodenal sphincter spasm during fentanyl-supplemented anesthesia. Anesth Analg. 1981;60(9):638–640.
38. Philbin DM, Wilson NE, Sokoloski J, et al. Radioimmunoassay of antidiuretic hormone during morphine anaesthesia. Can Anaesth Soc J. 1976;23(3):290–295.
39. Way WL, Costley EC, Leongway E. Respiratory sensitivity of the newborn infant to meperidine and morphine. Clin Pharmacol Ther. 1965;6:454–461.
40. Ballantyne JC, Mao J. Opioid therapy for chronic pain. N Engl J Med. 2003;349(20):1943–1953.
41. Ninkovic J, Roy S. Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids. 2013;45(1):9–24.
42. Tweed WA, Dakin D. Explosive coughing after bolus fentanyl injection. Anesth Analg. 2001;92(6):1442–1443.
43. Cho HB, Kwak HJ, Park SY, et al. Comparison of the incidence and severity of cough after alfentanil and remifentanil injection. Acta Anaesthesiol Scand. 2010;54(6):717–720.
44. Duttaroy A, Yoburn BC. The effect of intrinsic efficacy on opioid tolerance. Anesthesiology. 1995;82(5):1226–1236.
45. Mitra S, Sinatra RS. Perioperative management of acute pain in the opioid-dependent patient. Anesthesiology. 2004;101(1):212–227.
46. Joly V, Richebe P, Guignard B, et al. Remifentanil-induced postoperative hyperalgesia and its prevention with small-dose ketamine. Anesthesiology. 2005;103(1):147–155.
47. Cherny NI. Opioid analgesics: comparative features and prescribing guidelines. Drugs. 1996;51(5):713–737.
48. Aubrun F, Monsel S, Langeron O, et al. Postoperative titration of intravenous morphine in the elderly patient. Anesthesiology. 2002;96(1):17–23.
49. Woolf CJ, Wall PD. Morphine-sensitive and morphine-insensitive actions of C-fibre input on the rat spinal cord. Neurosci Lett. 1986;64(2):221–225.
50. Tooms A, McKenzie A, Grey H. Nebulised morphine. Lancet. 1993;342(8879):1123–1124.
51. Lang E, Jedeikin R. Acute respiratory depression as a complication of nebulised morphine. Can J Anaesth. 1998;45(1):60–62.
52. Thipphawong JB, Babul N, Morishige RJ, et al. Analgesic efficacy of inhaled morphine in patients after bunionectomy surgery. Anesthesiology. 2003;99(3):693–700; discussion 6A.
53. Murphy MR, Hug CC Jr. Pharmacokinetics of intravenous morphine in patients anesthetized with enflurane-nitrous oxide. Anesthesiology. 1981;54(3):187–192.
54. Berkowitz BA, Ngai SH, Yang JC, et al. The diposition of morphine in surgical patients. Clin Pharmacol Ther. 1975;17(6):629–635.
55. Finck AD, Berkowitz BA, Hempstead J, et al. Pharmacokinetics of morphine: effects of hypercarbia on serum and brain morphine concentrations in the dog. Anesthesiology. 1977;47(5):407–410.
56. Roerig DL, Kotrly KJ, Vucins EJ, et al. First pass uptake of fentanyl, meperidine, and morphine in the human lung. Anesthesiology. 1987;67(4):466–472.
57. Vaughan CW, Connor M. In search of a role for the morphine metabolite morphine-3-glucuronide. Anesth Analg. 2003;97(2):311–312.
58. Romberg R, Olofsen E, Sarton E, et al. Pharmacodynamic effect of morphine-6-glucuronide versus morphine on hypoxic and hypercapnic breathing in healthy volunteers. Anesthesiology. 2003;99(4):788–798.
59. Hanna MH, Peat SJ, Woodham M, et al. Analgesic efficacy and CSF pharmacokinetics of intrathecal morphine-6-glucuronide: comparison with morphine. Br J Anaesth. 1990;64(5):547–550.
60. Paul D, Standifer KM, Inturrisi CE, et al. Pharmacological characterization of morphine-6 beta-glucuronide, a very potent morphine metabolite. J Pharmacol Exp Ther. 1989;251(2):477–483.
61. Sear JW. Drug biotransformation by the kidney: how important is it, and how much do we really know? Br J Anaesth. 1991;67(4):369–372.
62. Chauvin M, Sandouk P, Scherrmann JM, et al. Morphine pharmacokinetics in renal failure. Anesthesiology. 1987;66(3):327–331.
63. Don HF, Dieppa RA, Taylor P. Narcotic analgesics in anuric patients. Anesthesiology. 1975;42(6):745–747.
64. Lynn AM, Slattery JT. Morphine pharmacokinetics in early infancy. Anesthesiology. 1987;66(2):136–139.
65. Hanna MH, D’Costa F, Peat SJ, et al. Morphine-6-glucuronide disposition in renal impairment. Br J Anaesth. 1993;70(5):511–514.
66. Baka NE, Bayoumeu F, Boutroy MJ, et al. Colostrum morphine concentrations during postcesarean intravenous patient-controlled analgesia. Anesth Analg. 2002;94(1):184–187, table of contents.
67. Kest B, Sarton E, Dahan A. Gender differences in opioid-mediated analgesia: animal and human studies. Anesthesiology. 2000;93(2):539–547.
68. Sarton E, Olofsen E, Romberg R, et al. Sex differences in morphine analgesia: an experimental study in healthy volunteers. Anesthesiology. 2000;93(5):1245–1254; discussion 6A.
69. Dahan A, Sarton E, Teppema L, et al. Sex-related differences in the influence of morphine on ventilatory control in humans. Anesthesiology. 1998;88(4):903–913.
70. Stone PA, Macintyre PE, Jarvis DA. Norpethidine toxicity and patient controlled analgesia. Br J Anaesth. 1993;71(5):738–740.
71. Cozian A, Pinaud M, Lepage JY, et al. Effects of meperidine spinal anesthesia on hemodynamics, plasma catecholamines, angiotensin I, aldosterone, and histamine concentrations in elderly men. Anesthesiology. 1986;64(6):815–819.
72. Austin KL, Stapleton JV, Mather LE. Multiple intramuscular injections: a major source of variability in analgesic response to meperidine. Pain. 1980;8(1):47–62.
73. Austin KL, Stapleton JV, Mather LE. Relationship between blood meperidine concentrations and analgesic response: a preliminary report. Anesthesiology. 1980;53(6):460–466.
74. Alfonsi P, Sessler DI, Du Manoir B, et al. The effects of meperidine and sufentanil on the shivering threshold in postoperative patients. Anesthesiology. 1998;89(1):43–48.
75. Ikeda T, Sessler DI, Tayefeh F, et al. Meperidine and alfentanil do not reduce the gain or maximum intensity of shivering. Anesthesiology. 1998;88(4):858–865.
76. Kurz A, Ikeda T, Sessler DI, et al. Meperidine decreases the shivering threshold twice as much as the vasoconstriction threshold. Anesthesiology. 1997;86(5):1046–1054.
77. Takada K, Clark DJ, Davies MF, et al. Meperidine exerts agonist activity at the alpha(2B)-adrenoceptor subtype. Anesthesiology. 2002;96(6):1420–1426.
78. Flacke JW, Flacke WE, Bloor BC, et al. Histamine release by four narcotics: a double-blind study in humans. Anesth Analg. 1987;66(8):723–730.
79. Tissot TA. Probable meperidine-induced serotonin syndrome in a patient with a history of fluoxetine use. Anesthesiology. 2003;98(6):1511–1512.
80. Lewis WR, Perrino AC Jr. Transient neurological symptoms after subarachnoid meperidine. Anesth Analg. 2002;94(1):213–214, table of contents.
81. Stanley TH. The history and development of the fentanyl series. J Pain Symptom Manage. 1992;7(3)(suppl):S3–S7.
82. Hug CC Jr, Murphy MR. Tissue redistribution of fentanyl and termination of its effects in rats. Anesthesiology. 1981;55(4):369–375.
83. Hudson RJ, Thomson IR, Jassal R, et al. Cardiopulmonary bypass has minimal effects on the pharmacokinetics of fentanyl in adults. Anesthesiology. 2003;99(4):847–854.
84. Peng PW, Sandler AN. A review of the use of fentanyl analgesia in the management of acute pain in adults. Anesthesiology. 1999;90(2):576–599.
85. Ibrahim AE, Feldman J, Karim A, et al. Simultaneous assessment of drug interactions with low- and high-extraction opioids: application to parecoxib effects on the pharmacokinetics and pharmacodynamics of fentanyl and alfentanil. Anesthesiology. 2003;98(4):853–861.
86. Murphy MR, Hug CC Jr, McClain DA. Dose-independent pharmacokinetics of fentanyl. Anesthesiology. 1983;59(6):537–540.
87. Bentley JB, Borel JD, Nenad RE Jr, et al. Age and fentanyl pharmacokinetics. Anesth Analg. 1982;61(12):968–971.
88. Hudson RJ, Thomson IR, Cannon JE, et al. Pharmacokinetics of fentanyl in patients undergoing abdominal aortic surgery. Anesthesiology. 1986;64(3):334–338.
89. Haberer JP, Schoeffler P, Couderc E, et al. Fentanyl pharmacokinetics in anaesthetized patients with cirrhosis. Br J Anaesth. 1982;54(12):1267–1270.
90. Hughes MA, Glass PS, Jacobs JR. Context-sensitive half-time in multicompartment pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology. 1992;76(3):334–341.
91. Gedney JA, Ghosh S. Pharmacokinetics of analgesics, sedatives and anaesthetic agents during cardiopulmonary bypass. Br J Anaesth. 1995;75(3):344–351.
92. Daniel M, Weiskopf RB, Noorani M, Eger EI II. Fentanyl augments the blockade of the sympathetic response to incision (MAC-BAR) produced by desflurane and isoflurane: desflurane and isoflurane MAC-BAR without and with fentanyl. Anesthesiology. 1998;88(1):43–49.
93. Hilgenberg JC. Intraoperative awareness during high-dose fentanyl—oxygen anesthesia. Anesthesiology. 1981;54(4):341–343.
94. Sprigge JS, Wynands JE, Whalley DG, et al. Fentanyl infusion anesthesia for aortocoronary bypass surgery: plasma levels and hemodynamic response. Anesth Analg. 1982;61(12):972–978.
95. Wynands JE, Townsend GE, Wong P, et al. Blood pressure response and plasma fentanyl concentrations during high- and very high-dose fentanyl anesthesia for coronary artery surgery. Anesth Analg. 1983;62(7):661–665.
96. Palmer CM, Cork RC, Hays R, et al. The dose-response relation of intrathecal fentanyl for labor analgesia. Anesthesiology. 1998;88(2):355–361.
97. Macaluso AD, Connelly AM, Hayes WB, et al. Oral transmucosal fentanyl citrate for premedication in adults. Anesth Analg. 1996;82(1):158–161.
98. Stanley TH, Leiman BC, Rawal N, et al. The effects of oral transmucosal fentanyl citrate premedication on preoperative behavioral responses and gastric volume and acidity in children. Anesth Analg. 1989;69(3):328–335.
99. Friesen RH, Lockhart CH. Oral transmucosal fentanyl citrate for preanesthetic medication of pediatric day surgery patients with and without droperidol as a prophylactic anti-emetic. Anesthesiology. 1992;76(1):46–51.
100. Epstein RH, Mendel HG, Witkowski TA, et al. The safety and efficacy of oral transmucosal fentanyl citrate for preoperative sedation in young children. Anesth Analg. 1996;83(6):1200–1225.
101. Dsida RM, Wheeler M, Birmingham PK, et al. Premedication of pediatric tonsillectomy patients with oral transmucosal fentanyl citrate. Anesth Analg. 1998;86(1):66–70.
102. Ashburn MA, Lind GH, Gillie MH, et al. Oral transmucosal fentanyl citrate (OTFC) for the treatment of postoperative pain. Anesth Analg. 1993;76(2):377–381.
103. Caplan RA, Ready LB, Oden RV, et al. Transdermal fentanyl for postoperative pain management. A double-blind placebo study. JAMA. 1989;261(7):1036–1039.
104. Kuzma PJ, Kline MD, Stamatos JM, et al. Acute toxic delirium: an uncommon reaction to transdermal fentanyl. Anesthesiology. 1995;83(4):869–871.
105. Arndt JO, Mikat M, Parasher C. Fentanyl’s analgesic, respiratory, and cardiovascular actions in relation to dose and plasma concentration in unanesthetized dogs. Anesthesiology. 1984;61(4):355–361.
106. Frolich MA, Giannotti A, Modell JH. Opioid overdose in a patient using a fentanyl patch during treatment with a warming blanket. Anesth Analg. 2001;93(3):647–648.
107. Becker LD, Paulson BA, Miller RD, et al. Biphasic respiratory depression after fentanyldroperidol or fentanyl alone used to supplement nitrous oxide anesthesia. Anesthesiology. 1976;44(4):291–296.
108. Murat I, Levron JC, Berg A, et al. Effects of fentanyl on baroreceptor reflex control of heart rate in newborn infants. Anesthesiology. 1988;68(5):717–722.
109. Manninen PH. Opioids and seizures. Can J Anaesth. 1997;44(5, pt 1):463–466.
110. Smith NT, Benthuysen JL, Bickford RG, et al. Seizures during opioid anesthetic induction—are they opioid-induced rigidity? Anesthesiology. 1989;71(6):852–862.
111. Murkin JM, Moldenhauer CC, Hug CC Jr, et al. Absence of seizures during induction of anesthesia with high-dose fentanyl. Anesth Analg. 1984;63(5):489–494.
112. Schubert A, Drummond JC, Peterson DO, et al. The effect of high-dose fentanyl on human median nerve somatosensory-evoked responses. Can J Anaesth. 1987;34(1):35–40.
113. Glass PS, Bloom M, Kearse L, et al. Bispectral analysis measures sedation and memory effects of propofol, midazolam, isoflurane, and alfentanil in healthy volunteers. Anesthesiology. 1997;86(4):836–847.
114. Sebel PS, Lang E, Rampil IJ, et al. A multicenter study of bispectral electroencephalogram analysis for monitoring anesthetic effect. Anesth Analg. 1997;84(4):891–899.
115. Albanese J, Durbec O, Viviand X, et al. Sufentanil increases intracranial pressure in patients with head trauma. Anesthesiology. 1993;79(3):493–497.
116. Werner C, Kochs E, Bause H, et al. Effects of sufentanil on cerebral hemodynamics and intracranial pressure in patients with brain injury. Anesthesiology. 1995;83(4):721–726.
117. de Nadal M, Munar F, Poca MA, et al. Cerebral hemodynamic effects of morphine and fentanyl in patients with severe head injury: absence of correlation to cerebral autoregulation. Anesthesiology. 2000;92(1):11–19.
118. Bailey PL, Pace NL, Ashburn MA, et al. Frequent hypoxemia and apnea after sedation with midazolam and fentanyl. Anesthesiology. 1990;73(5):826–830.
119. Scott JC, Cooke JE, Stanski DR. Electroencephalographic quantitation of opioid effect: comparative pharmacodynamics of fentanyl and sufentanil. Anesthesiology. 1991;74(1):34–42.
120. Malinovsky JM, Lepage JY, Cozian A, et al. Transient muscular spasm after a large dose of intrathecal sufentanil. Anesthesiology. 1996;84(6):1513–1515.
121. Bovill JG, Sebel PS, Blackburn CL, et al. The pharmacokinetics of sufentanil in surgical patients. Anesthesiology. 1984;61(5):502–506.
122. Chauvin M, Ferrier C, Haberer JP, et al. Sufentanil pharmacokinetics in patients with cirrhosis. Anesth Analg. 1989;68(1):1–4.
123. Hudson RJ, Bergstrom RG, Thomson IR, et al. Pharmacokinetics of sufentanil in patients undergoing abdominal aortic surgery. Anesthesiology. 1989;70(3):426–431.
124. Schwartz AE, Matteo RS, Ornstein E, et al. Pharmacokinetics of sufentanil in obese patients. Anesth Analg. 1991;73(6):790–793.
125. Boer F, Hoeft A, Scholz M, et al. Pulmonary distribution of alfentanil and sufentanil studied with system dynamics analysis. J Pharmacokinet Biopharm. 1996;24(2):197–218.
126. Meistelman C, Benhamou D, Barre J, et al. Effects of age on plasma protein binding of sufentanil. Anesthesiology. 1990;72(3):470–473.
127. Yaster M. The dose response of fentanyl in neonatal anesthesia. Anesthesiology. 1987;66(3):433–435.
128. Wiggum DC, Cork RC, Weldon ST, et al. Postoperative respiratory depression and elevated sufentanil levels in a patient with chronic renal failure. Anesthesiology. 1985;63(6):708–710.
129. Bailey PL, Streisand JB, East KA, et al. Differences in magnitude and duration of opioid-induced respiratory depression and analgesia with fentanyl and sufentanil. Anesth Analg. 1990;70(1):8–15.
130. Sanford TJ Jr, Smith NT, Dec-Silver H, et al. A comparison of morphine, fentanyl, and sufentanil anesthesia for cardiac surgery: induction, emergence, and extubation. Anesth Analg. 1986;65(3):259–266.
131. Mayer N, Weinstabl C, Podreka I, et al. Sufentanil does not increase cerebral blood flow in healthy human volunteers. Anesthesiology. 1990;73(2):240–243.
132. Chang J, Fish KJ. Acute respiratory arrest and rigidity after anesthesia with sufentanil: a case report. Anesthesiology. 1985;63(6):710–711.
133. Philbin DM, Rosow CE, Schneider RC, et al. Fentanyl and sufentanil anesthesia revisited: how much is enough? Anesthesiology. 1990;73(1):5–11.
134. Abrams JT, Horrow JC, Bennett JA, et al. Upper airway closure: a primary source of difficult ventilation with sufentanil induction of anesthesia. Anesth Analg. 1996;83(3):629–632.
135. Scott JC, Stanski DR. Decreased fentanyl and alfentanil dose requirements with age. A simultaneous pharmacokinetic and pharmacodynamic evaluation. J Pharmacol Exp Ther. 1987;240(1):159–166.
136. Davis PJ, Stiller RL, Cook DR, et al. Effects of cholestatic hepatic disease and chronic renal failure on alfentanil pharmacokinetics in children. Anesth Analg. 1989;68(5):579–583.
137. Chauvin M, Lebrault C, Levron JC, et al. Pharmacokinetics of alfentanil in chronic renal failure. Anesth Analg. 1987;66(1):53–56.
138. Meistelman C, Saint-Maurice C, Lepaul M, et al. A comparison of alfentanil pharmacokinetics in children and adults. Anesthesiology. 1987;66(1):13–16.
139. Camu F, Gepts E, Rucquoi M, et al. Pharmacokinetics of alfentanil in man. Anesth Analg. 1982;61(8):657–661.
140. Kharasch ED, Russell M, Mautz D, et al. The role of cytochrome P450 3A4 in alfentanil clearance. Implications for interindividual variability in disposition and perioperative drug interactions. Anesthesiology. 1997;87(1):36–50.
141. Bartkowski RR, McDonnell TE. Prolonged alfentanil effect following erythromycin administration. Anesthesiology. 1990;73(3):566–568.
142. Miller DR, Martineau RJ, O’Brien H, et al. Effects of alfentanil on the hemodynamic and catecholamine response to tracheal intubation. Anesth Analg. 1993;76(5):1040–1046.
143. Yonker-Sell AE, Muzi M, Hope WG, et al. Alfentanil modifies the neurocirculatory responses to desflurane. Anesth Analg. 1996;82(1):162–166.
144. Ausems ME, Hug CC Jr, de Lange S. Variable rate infusion of alfentanil as a supplement to nitrous oxide anesthesia for general surgery. Anesth Analg. 1983;62(11):982–986.
145. Langevin S, Lessard MR, Trepanier CA, et al. Alfentanil causes less postoperative nausea and vomiting than equipotent doses of fentanyl or sufentanil in outpatients. Anesthesiology. 1999;91(6):1666–1673.
146. Mets B. Acute dystonia after alfentanil in untreated Parkinson’s disease. Anesth Analg. 1991;72(4):557–558.
147. Jhaveri R, Joshi P, Batenhorst R, et al. Dose comparison of remifentanil and alfentanil for loss of consciousness. Anesthesiology. 1997;87(2):253–259.
148. Rosow C. Remifentanil: a unique opioid analgesic. Anesthesiology. 1993;79(5):875–876.
149. Thompson JP, Rowbotham DJ. Remifentanil—an opioid for the 21st century. Br J Anaesth. 1996;76(3):341–343.
150. Babenco HD, Conard PF, Gross JB. The pharmacodynamic effect of a remifentanil bolus on ventilatory control. Anesthesiology. 2000;92(2):393–398.
151. Nieuwenhuijs DJ, Olofsen E, Romberg RR, et al. Response surface modeling of remifentanil-propofol interaction on cardiorespiratory control and bispectral index. Anesthesiology. 2003;98(2):312–322.
152. Egan TD, Huizinga B, Gupta SK, et al. Remifentanil pharmacokinetics in obese versus lean patients. Anesthesiology. 1998;89(3):562–573.
153. Hoke JF, Shlugman D, Dershwitz M, et al. Pharmacokinetics and pharmacodynamics of remifentanil in persons with renal failure compared with healthy volunteers. Anesthesiology. 1997;87(3):533–541.
154. Kapila A, Glass PS, Jacobs JR, et al. Measured context-sensitive half-times of remifentanil and alfentanil. Anesthesiology. 1995;83(5):968–975.
155. Thompson JP, Hall AP, Russell J, et al. Effect of remifentanil on the haemodynamic response to orotracheal intubation. Br J Anaesth. 1998;80(4):467–469.
156. Evron S, Glezerman M, Sadan O, et al. Remifentanil: a novel systemic analgesic for labor pain. Anesth Analg. 2005;100(1):233–238.
157. Kan RE, Hughes SC, Rosen MA, et al. Intravenous remifentanil: placental transfer, maternal and neonatal effects. Anesthesiology. 1998;88(6):1467–1474.
158. Hogue CW Jr, Bowdle TA, O’Leary C, et al. A multicenter evaluation of total intravenous anesthesia with remifentanil and propofol for elective inpatient surgery. Anesth Analg. 1996;83(2):279–285.
159. Avramov MN, Smith I, White PF. Interactions between midazolam and remifentanil during monitored anesthesia care. Anesthesiology. 1996;85(6):1283–1289.
160. Recart A, Rawal S, White PF, et al. The effect of remifentanil on seizure duration and acute hemodynamic responses to electroconvulsive therapy. Anesth Analg. 2003;96(4):1047–1450.
161. Haber GW, Litman RS. Generalized tonic-clonic activity after remifentanil administration. Anesth Analg. 2001;93(6):1532–1533.
162. Guy J, Hindman BJ, Baker KZ, et al. Comparison of remifentanil and fentanyl in patients undergoing craniotomy for supratentorial space-occupying lesions. Anesthesiology. 1997;86(3):514–524.
163. Warner DS, Hindman BJ, Todd MM, et al. Intracranial pressure and hemodynamic effects of remifentanil versus alfentanil in patients undergoing supratentorial craniotomy. Anesth Analg. 1996;83(2):348–353.
164. Klimscha W, Ullrich R, Nasel C, et al. High-dose remifentanil does not impair cerebrovascular carbon dioxide reactivity in healthy male volunteers. Anesthesiology. 2003;99(4):834–840.
165. Fragen RJ, Vilich F, Spies SM, et al. The effect of remifentanil on biliary tract drainage into the duodenum. Anesth Analg. 1999;89(6):1561–1564.
166. Guignard B, Bossard AE, Coste C, et al. Acute opioid tolerance: intraoperative remifentanil increases postoperative pain and morphine requirement. Anesthesiology. 2000;93(2):409–417.
167. Guignard B, Coste C, Costes H, et al. Supplementing desflurane-remifentanil anesthesia with small-dose ketamine reduces perioperative opioid analgesic requirements. Anesth Analg. 2002;95(1):103–108.
168. Ferrini F, Trang T, Mattioli TA, et al. Morphine hyperalgesia gated through microglia-mediated disruption of neuronal Cl(-) homeostasis. Nat Neurosci. 2013;16(2):183–192.
169. Vree TB, van Dongen RT, Koopman-Kimenai PM. Codeine analgesia is due to codeine-6-glucuronide, not morphine. Int J Clin Pract. 2000;54(6):395–398.
170. Angst MS, Drover DR, Lotsch J, et al. Pharmacodynamics of orally administered sustained-release hydromorphone in humans. Anesthesiology. 2001;94(1):63–73.
171. Thwaites D, McCann S, Broderick P. Hydromorphone neuroexcitation. J Palliat Med. 2004;7(4):545–550.
172. Gimbel J, Ahdieh H. The efficacy and safety of oral immediate-release oxymorphone for postsurgical pain. Anesth Analg. 2004;99(5):1472–1477.
173. Gershman JA, Fass AD. Hydrocodone rescheduling amendment and pipeline products on the horizon. P T. 2012;37(7):399–404.
174. Fishman SM, Wilsey B, Mahajan G, et al. Methadone reincarnated: novel clinical applications with related concerns. Pain Med. 2002;3(4):339–348.
175. Budd K, Langford R. Tramadol revisited. Br J Anaesth. 1999;82(4):493–495.
176. Collart L, Luthy C, Favario-Constantin C, et al. [Duality of the analgesic effect of tramadol in humans]. Schweiz Med Wochenschr. 1993;123(47):2241–2243.
177. Calvey TN. Chirality in anaesthesia. Anaesthesia. 1992;47(2):93–94.
178. Nieuwenhuijs D, Bruce J, Drummond GB, et al. Influence of oral tramadol on the dynamic ventilatory response to carbon dioxide in healthy volunteers. Br J Anaesth. 2001;87(6):860–865.
179. Crighton IM, Martin PH, Hobbs GJ, et al. A comparison of the effects of intravenous tramadol, codeine, and morphine on gastric emptying in human volunteers. Anesth Analg. 1998;87(2):445–449.
180. Vahabzadeh M, Moshiri M, Mohammadpour AH, et al. Promising effects of intravenous lipid emulsion as an antidote in acute tramadol poisoning. Reg Anesth Pain Med. 2013;38(5):425–430.
181. Kahn LH, Alderfer RJ, Graham DJ. Seizures reported with tramadol. JAMA. 1997;278(20):1661.
182. Angell M. Should heroin be legalized for the treatment of pain? N Engl J Med. 1984;311(8):529–530.
183. Mondzac AM. In defense of the reintroduction of heroin into American medical practice and H.R. 5290—the Compassionate Pain Relief Act. N Engl J Med. 1984;311(8):532–535.
184. Gal TJ, DiFazio CA, Moscicki J. Analgesic and respiratory depressant activity of nalbuphine: a comparison with morphine. Anesthesiology. 1982;57(5):367–374.
185. Bailey PL, Clark NJ, Pace NL, et al. Antagonism of postoperative opioid-induced respiratory depression: nalbuphine versus naloxone. Anesth Analg. 1987;66(11):1109–1114.
186. Lanz E, Simko G, Theiss D, et al. Epidural buprenorphine—a double-blind study of postoperative analgesia and side effects. Anesth Analg. 1984;63(6):593–598.
187. Gould DB. Buprenorphine causes pulmonary edema just like all other mu-opioid narcotics. Upper airway obstruction, negative alveolar pressure. Chest. 1995;107(5):1478–1479.
188. Gal TJ, DiFazio CA. Ventilatory and analgesic effects of dezocine in humans. Anesthesiology. 1984;61(6):716–722.
189. Glass PS, Jhaveri RM, Smith LR. Comparison of potency and duration of action of nalmefene and naloxone. Anesth Analg. 1994;78(3):536–541.
190. Rawal N, Schott U, Dahlstrom B, et al. Influence of naloxone infusion on analgesia and respiratory depression following epidural morphine. Anesthesiology. 1986;64(2):194–201.
191. Partridge BL, Ward CF. Pulmonary edema following low-dose naloxone administration. Anesthesiology. 1986;65(6):709–710.
192. Andree RA. Sudden death following naloxone administration. Anesth Analg. 1980;59(10):782–784.
193. Faden AI. Opiate antagonists and thyrotropin-releasing hormone. I. Potential role in the treatment of shock. JAMA. 1984;252(9):1177–1180.
194. Kraynack BJ, Gintautas JG. Naloxone: analeptic action unrelated to opiate receptor antagonism? Anesthesiology. 1982;56(4):251–253.
195. Srisurapanont M, Jarusuraisin N. Naltrexone for the treatment of alcoholism: a meta-analysis of randomized controlled trials. Int J Neuropsychopharmacol. 2005;8(2):267–280.
196. Dougherty TB, Porche VH, Thall PF. Maximum tolerated dose of nalmefene in patients receiving epidural fentanyl and dilute bupivacaine for postoperative analgesia. Anesthesiology. 2000;92(4):1010–1016.
197. Joshi GP, Duffy L, Chehade J, et al. Effects of prophylactic nalmefene on the incidence of morphine-related side effects in patients receiving intravenous patient-controlled analgesia. Anesthesiology. 1999;90(4):1007–1011.
198. Henderson CA, Reynolds JE. Acute pulmonary edema in a young male after intravenous nalmefene. Anesth Analg. 1997;84(1):218–219.
199. Foss JF, O’Connor MF, Yuan CS, et al. Safety and tolerance of methylnaltrexone in healthy humans: a randomized, placebo-controlled, intravenous, ascending-dose, pharmacokinetic study. J Clin Pharmacol. 1997;37(1):25–30.
200. Holzer P. Opioid antagonists for prevention and treatment of opioid-induced gastrointestinal effects. Curr Opin Anaesthesiol. 2010;23(5):616–622.
201. Bream-Rouwenhorst HR, Cantrell MA. Alvimopan for postoperative ileus. Am J Health Syst Pharm. 2009;66(14):1267–1277.
202. Manchikanti L, Helm S II, Fellows B, et al. Opioid epidemic in the United States. Pain Physician. 2012;15(3)(suppl):ES9–ES38.
203. Strassels SA. Economic burden of prescription opioid misuse and abuse. J Manag Care Pharm. 2009;15(7):556–562.
204. Lourenco LM, Matthews M, Jamison RN. Abuse-deterrent and tamper-resistant opioids: how valuable are novel formulations in thwarting non-medical use? [Review]. Exp Opin Drug Deliv. 2013;10(2):229–240.
205. Hepner DL, Castells MC. Anaphylaxis during the perioperative period. Anesth Analg. 2003;97:1381–1395.
206. Bennett MJ, Anderson LK, McMillan JC, et al. Anaphylactic reaction during anaesthesia associated with positive intradermal skin test to fentanyl. Can J Anaesth. 1986;33:75–78.
207. Fukuda T, Dohi S. Anaphylactic reaction to fentanyl or preservative. Can J Anaesth. 1986;33:826–827.
208. Zucker-Pinchoff B, Ramanathan S. Anaphylactic reaction to epidural fentanyl. Anesthesiology. 1989;71:599–601.
209. Cummings KC III, Arnaut KA. Case report: Fentanyl-associated intraoperative anaphylaxis with pulmonary edema. Can J Anaesth. 2007;54(4):301–306.
210. Zucker-Pinchoff B, Chandler MJ. Latex anaphylaxis masquerading as fentanyl anaphylaxis: Retraction of a case report. Anesthesiology. 1993;79:1153–1154.
211. Webster NR. Opioids and the immune system. Br J Anaesth. 1998;81(6):835–836.
212. Biki B, Mascha E, Moriarty DC, et al. Anesthetic technique for radical prostatectomy surgery affects cancer recurrence: a retrospective analysis. Anesthesiology. 2008;109(2):180–187.
213. Buggy DJ, Smith G. Epidural anaesthesia and analgesia: better outcome after major surgery? Growing evidence suggests so. BMJ. 1999;319(7209):530–531.
214. Exadaktylos AK, Buggy DJ, Moriarty DC, et al. Can anesthetic technique for primary breast cancer surgery affect recurrence or metastasis? Anesthesiology. 2006;4:660–664.
215. Cummings KC III, Xu F, Cummings LC, et al. A comparison of epidural analgesia and traditional pain management effects on survival and cancer recurrence after colectomy: a population-based study. Anesthesiology. 2012;116(4):797–806.
216. Beilin B, Shavit Y, Hart J, et al. Effects of anesthesia based on large versus small doses of fentanyl on natural killer cell cytotoxicity in the perioperative period. Anesth Analg. 1996;82(3):492–497.
217. Shavit Y, Ben-Eliyahu S, Zeidel A, et al. Effects of fentanyl on natural killer cell activity and on resistance to tumor metastasis in rats. Dose and timing study. Neuroimmunomodulation. 2004;11(4):255–260.
218. Yeager MP, Colacchio TA, Yu CT, et al. Morphine inhibits spontaneous and cytokine-enhanced natural killer cell cytotoxicity in volunteers. Anesthesiology. 1995;83(3):500–508.
219. Steffey EP, Eisele JH, Baggot JD, et al. Influence of inhaled anesthetics on the pharmacokinetics and pharmacodynamics of morphine. Anesth Analg. 1993;77(2):346–351.
220. Sebel PS, Glass PS, Fletcher JE, et al. Reduction of the MAC of desflurane with fentanyl. Anesthesiology. 1992;76(1):52–59.
221. Hall RI, Murphy MR, Hug CC Jr. The enflurane sparing effect of sufentanil in dogs. Anesthesiology. 1987;67(4):518–525.
222. Schwartz JG, Garriott JC, Somerset JS, et al. Measurements of fentanyl and sufentanil in blood and urine after surgical application. Implication in detection of abuse. Am J Forensic Med Pathol. 1994;15(3):236–241.
223. Brunner MD, Braithwaite P, Jhaveri R, et al. MAC reduction of isoflurane by sufentanil. Br J Anaesth. 1994;72(1):42–46.
224. Lang E, Kapila A, Shlugman D, et al. Reduction of isoflurane minimal alveolar concentration by remifentanil. Anesthesiology. 1996;85(4):721–728.
225. Murphy MR, Hug CC Jr. The enflurane sparing effect of morphine, butorphanol, and nalbuphine. Anesthesiology. 1982;57(6):489–492.
226. Palmer PP, Miller RD. Current and developing methods of patient-controlled analgesia. Anesthesiol Clin. 2010;28(4):587–599.
227. Hudcova J, McNicol E, Quah C, et al. Patient controlled opioid analgesia versus conventional opioid analgesia for postoperative pain. Cochrane Database Syst Rev. 2006;4:CD003348.
228. Hinova A, Fernando R. Systemic remifentanil for labor analgesia. Anesth Analg. 2009;109(6):1925–1929.
229. Cousins MJ, Mather LE. Intrathecal and epidural administration of opioids. Anesthesiology. 1984;61(3):276–310.
230. Drasner K, Bernards CM, Ozanne GM. Intrathecal morphine reduces the minimum alveolar concentration of halothane in humans. Anesthesiology. 1988;69(3):310–312.
231. Licina MG, Schubert A, Tobin JE, et al. Intrathecal morphine does not reduce minimum alveolar concentration of halothane in humans: results of a double-blind study. Anesthesiology. 1991;74(4):660–663.
232. Schwieger IM, Klopfenstein CE, Forster A. Epidural morphine reduces halothane MAC in humans. Can J Anaesth. 1992;39(9):911–914.
233. de Leon-Casasola OA, Lema MJ. Postoperative epidural opioid analgesia: what are the choices? Anesth Analg. 1996;83(4):867–875.
234. Chaney MA. Side effects of intrathecal and epidural opioids. Can J Anaesth. 1995;42(10):891–903.
235. Ionescu TI, Taverne RH, Drost RH, et al. Epidural morphine anesthesia for abdominal aortic surgery—pharmacokinetics. Reg Anesth. 1989;14(3):107–114.
236. Ionescu TI, Taverne RH, Houweling PL, et al. Pharmacokinetic study of extradural and intrathecal sufentanil anaesthesia for major surgery. Br J Anaesth. 1991;66(4):458–464.
237. Goyagi T, Nishikawa T. The addition of epinephrine enhances postoperative analgesia by intrathecal morphine. Anesth Analg. 1995;81(3):508–513.
238. Sjostrom S, Tamsen A, Persson MP, et al. Pharmacokinetics of intrathecal morphine and meperidine in humans. Anesthesiology. 1987;67(6):889–895.
239. Chiravanich W, Oofuvong M, Kovitwanawong N. Single dose of gabapentin for prophylaxis intrathecal morphine-induced pruritus in orthopedic surgery: a randomized controlled trial. J Med Assoc Thai. 2012;95(2):186–190.
240. Sheen MJ, Ho ST, Lee CH, et al. Preoperative gabapentin prevents intrathecal morphine-induced pruritus after orthopedic surgery. Anesth Analg. 2008;106(6):1868–1872.
241. Bailey PL, Rhondeau S, Schafer PG, et al. Dose-response pharmacology of intrathecal morphine in human volunteers. Anesthesiology. 1993;79(1):49–59; discussion 25A.
242. Morgan M. The rational use of intrathecal and extradural opioids. Br J Anaesth. 1989;63(2):165–188.
243. Gan TJ, Ginsberg B, Glass PS, et al. Opioid-sparing effects of a low-dose infusion of naloxone in patient-administered morphine sulfate. Anesthesiology. 1997;87(5):1075–1081.
244. Rozan JP, Kahn CH, Warfield CA. Epidural and intravenous opioid-induced neuroexcitation. Anesthesiology. 1995;83(4):860–863.
245. Crone LA, Conly JM, Storgard C, et al. Herpes labialis in parturients receiving epidural morphine following cesarean section. Anesthesiology. 1990;73(2):208–213.
246. Wong CA, Scavone BM, Peaceman AM, et al. The risk of cesarean delivery with neuraxial analgesia given early versus late in labor. N Engl J Med. 2005;352(7):655–665.
247. Kelly MC, Carabine UA, Hill DA, et al. A comparison of the effect of intrathecal and extradural fentanyl on gastric emptying in laboring women. Anesth Analg. 1997;85(4):834–838.