Miriam Jacob and W. H. Wilson Tang
DEFINITION OF HEART FAILURE
There are multiple definitions of heart failure, but it is fundamentally a clinical syndrome. Similar to anemia or acute renal failure, heart failure is not a “standalone” diagnosis, but rather always possesses an etiology In some cases, however, the etiology cannot be determined. Virtually any form of heart disease can lead to heart failure. In general, heart failure is defined as a clinical syndrome characterized by shortness of breath and fatigue at rest or with exertion in the presence of underlying structural and/or functional heart disease. In advanced cases, salt and water retention are manifested by edema and organ dysfunction.
DEMOGRAPHICS OF HEART FAILURE
Heart failure is a common condition in the United States affecting more than 5 million people, with an overall self- reported prevalence of 2.4%. The incidence increases with age. At the age of 40, the lifetime risk of developing heart failure is one in five. In those older than 65, 10 per 1,000 people will have heart failure. The most common preceding diagnosis is hypertension (75%). Not only is it common in the general population, but it is also associated with high morbidity and mortality. There has been a 164% increasing hospitalization rate over the past 15 years, now accounting for almost 1 million hospitalizations and approximately 3.5 million outpatient visits, annually Heart failure accounts for about 7% of all deaths due to cardiovascular disease. Those who are diagnosed also have a shorter survival; 50% of those diagnosed with heart failure die in 5 years. In-hospital mortality is as high as 5% to 8% (Roger et al., 2011).
The morbidity associated with heart failure in an individual affects both ambulatory and inpatient care. The average patient takes six medications. Seventy-eight percent of patients have at least two hospitalizations per year. Up to 30% are readmitted within 90 days of hospitalization (Schrier and Gheorghiade, 2011). In the Medicare population, 18% have heart failure, which translates to 6 billion dollars of Medicare payments. They average one hospitalization per year, with an average stay of 7 days. These patients use outpatient services often, with about 10 physical office visits per year (Schneider et al., 2009). The annual cost attributed to heart failure is approximately $46 billion (Schrier and Gheorghiade, 2011).
THE INDEX EVENT
The event may be obvious, such as a sudden loss of a large mass of contractile tissue (i.e., an acute myocardial infarction), or it may be completely silent, such as the early expression of a mutant gene. In many cases, such as familial cardiomyopathy and the onset of valvular heart disease or hypertension, heart failure occurs after a lengthy latency period, or it may develop acutely, such as from acute aortic insufficiency due to bacterial endocarditis. The index event could take the form of acute lymphocytic myocarditis and manifest as heart failure only many months or years later. There are infinite genetic and environmental influences, which is why the natural history of heart failure and the pace at which it unfolds is so variable among individual patients. Uncertainty about the index event or etiology also makes the prognosis for any individual patient unclear. Given the heterogeneity in the presentation of heart failure, the description includes the stage of heart failure from being at risk (stage A), asymptomatic with structural heart disease (stage B), symptomatic heart failure (stage C), and those with refractory heart failure (stage D) (Fig. 15.1; Hunt et al., 2009).
Identifying the underlying etiology of the heart failure is an important part of the evaluation as it may directly impact long-term prognosis. Common etiologies include:
![]() Hypertension
Hypertension
![]() Myocardial ischemia/infarction
Myocardial ischemia/infarction
![]() Genetic (familial or hereditary)
Genetic (familial or hereditary)
![]() Diabetes mellitus
Diabetes mellitus
![]() Myocarditis (infectious, giant cell)
Myocarditis (infectious, giant cell)
![]() Infiltrative or restrictive (hemochromatosis, amyloidosis, sarcoidosis, Fabry disease)
Infiltrative or restrictive (hemochromatosis, amyloidosis, sarcoidosis, Fabry disease)
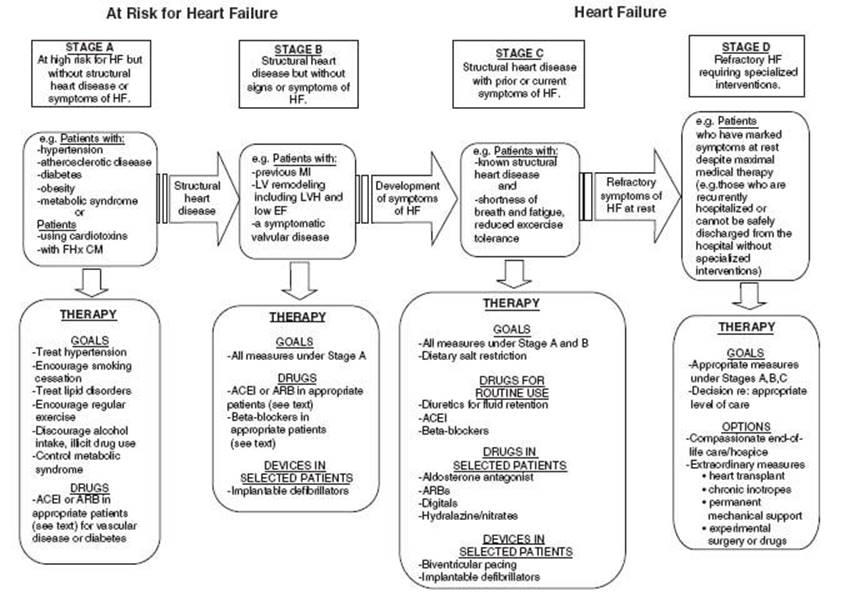
FIGURE 15.1 Stages in the development of heart failure/recommended therapy by stage. (Redrawn from Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration with the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53(15):e1-e90, with permission from Elsevier.)
![]() Substance use (alcohol, ephedra, cocaine)
Substance use (alcohol, ephedra, cocaine)
![]() Peripartum cardiomyopathy
Peripartum cardiomyopathy
![]() Connective tissue disease
Connective tissue disease
![]() Doxorubicin and other chemotherapy-induced cardiotoxicity
Doxorubicin and other chemotherapy-induced cardiotoxicity
![]() Infectious—Chagas, HIV Echovirus, Coxsackie
Infectious—Chagas, HIV Echovirus, Coxsackie
![]() Valvular heart disease
Valvular heart disease
![]() Other miscellaneous pathologies (e.g., pericardial disease)
Other miscellaneous pathologies (e.g., pericardial disease)
ADAPTIVE RESPONSES TO THE HEART FAILURE SYNDROME
The circulation adapts to a perceived disruption in homeostasis with both short-term and long-term adaptations. Shortterm adaptations include activation of the Frank-Starling mechanism and activation of the sympathetic nervous system (SNS). Long-term adaptations include heightened and alterations in the size and the shape of the heart (the so-called left ventricular [LV] remodeling). Although these adaptations may be somewhat protective in the short-term, over time they become counterproductive and contribute importantly to the pathogenesis of heart failure.
The Frank-Starling mechanism acts to increase the force of heart muscle contraction in response to an increase in end-diastolic volume (Fig. 15.2). In heart failure, however, this response is blunted, both at rest and during exercise. The force-frequency response is also attenuated in the failing heart, secondary to decreased norepinephrine (NE) stores and β-receptor density, which produces a decreased inotropic response to exercise so that less contractile force is generated in response to an increase in heart rate. Patients with heart failure can still call on the Frank-Starling mechanism, albeit at a reduced operational level. The inability to raise the stroke volume during exercise may be one of many reasons why patients have reduced exercise
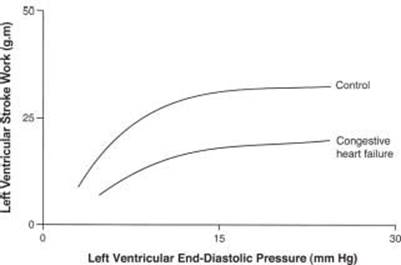
FIGURE 15.2 Heart failure is characterized by a diminished ability to increase the cardiac output or cardiac work in response to an increase in preload-Starlings law of the heart.
AUTONOMIC NERVOUS SYSTEM DYSFUNCTION
The SNS is activated early in the syndrome of heart failure, before overt signs and symptoms occur. Elevated plasma NE levels are observed and are an important marker of a poor prognosis. The mechanism that activates the SNS in heart failure is unknown. Increased local levels of synaptic NE in the heart increase the force of contraction and heart rate, offering early support for the failing heart. But this may also be the source of dysrhythmias and likely is responsible for the downregulation of β-adrenergic receptors. The failing heart tissue is itself also relatively depleted of NE, thus rendering the heart less responsive to sympathetic stimulation. There is less myocardial reserve in response to inotropic stimulation. The SNS also drives some of the increase in myocyte size, thus contributing to the LV remodeling process. Finally, the SNS activates the RAAS via β-receptors in the kidney, adding further to heightened peripheral resistance, salt, and water retention, and LV remodeling. In summary, early activation of the SNS in heart failure is “protective” by increasing heart rate, force of contraction, myocardial mass, and by protecting blood pressure, but there is a price to pay in the long run. Ultimately, excessive SNS activity is directly toxic to the heart and contributes importantly to the pathogenesis of heart failure.
Reflex control mechanisms are abnormal in heart failure (Fig. 15.3). Peripheral vascular resistance is increased, and there is defective parasympathetic control, an abnormal response to orthostasis, a blunted heart rate response to exercise and to pharmacologic vasodilation, impaired heart rate recovery from exercise, reduced heart-rate variability, and altered baroreceptor function. These abnormalities may improve following heart transplantation, suggesting that these are functional and not structural changes. They are rarely normalized. The precise cause of these abnormal reflex control mechanisms is not clear, but they may be the result of evolutionary forces that are acting to redistribute blood flow to more vital organs.
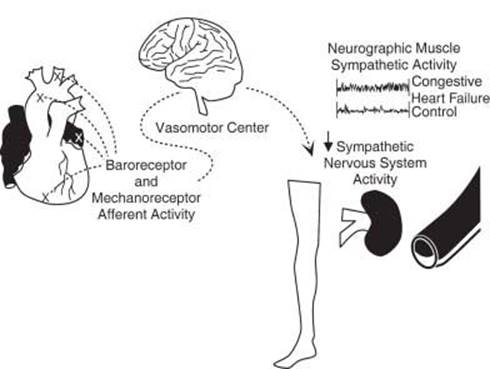
FIGURE 15.3 Baroreceptor and mechanoreceptor activation occurs when the heart is distended due to volume overload. This signal is processed by the brain and, in the setting of heart failure, fails to reduce sympathetic activity (the normal response). The result is enhanced sympathetic traffic to the periphery, vasoconstriction, and reduced renal blood
RENIN–ANGIOTENSIN–ALDOSTERONE SYSTEM
The RAAS is active in the circulation and in the tissue in heart failure. Probably 90% of the activity of the RAAS is embedded in the various tissues, including the heart, brain, and vasculature. This system, in conjunction with the SNS, plays a key role in the pathogenesis of the syndrome.
The RAAS is known to be activated by numerous mechanisms:
![]() Volume contraction
Volume contraction
![]() Low cardiac output
Low cardiac output
![]() Decreased renal blood flow
Decreased renal blood flow
![]() Hyponatremicperfusate to the macula densa
Hyponatremicperfusate to the macula densa
![]() β-Adrenergic stimulation to the kidney
β-Adrenergic stimulation to the kidney
![]() Diuretics
Diuretics
![]() Salt and water restriction
Salt and water restriction
Angiotensin-II (Ang II) is a small, potent peptide produced by the cleavage of Ang-I by angiotensin-converting enzyme (ACE). Ang-II has a vast array of biologic activities, most of which contribute importantly to the pathogenesis of heart failure:
![]() Vasoconstriction
Vasoconstriction
![]() Vascular and cardiac myocyte growth, hypertrophy
Vascular and cardiac myocyte growth, hypertrophy
![]() Activation of fibroblasts with increased collagen production
Activation of fibroblasts with increased collagen production
![]() Facilitation of NE release
Facilitation of NE release
![]() Stimulation of aldosterone release
Stimulation of aldosterone release
![]() Volume expansion
Volume expansion
![]() Thirst stimulation
Thirst stimulation
![]() Arginine vasopressin release
Arginine vasopressin release
![]() Proinflammatory activity
Proinflammatory activity
![]() Direct toxicity to the myocardium when present in excessive quantities
Direct toxicity to the myocardium when present in excessive quantities
![]() Mesangial hypertrophy in the kidney
Mesangial hypertrophy in the kidney
![]() Increased intraglomerular hydraulic pressure via postglomerular efferent arteriole vasoconstriction
Increased intraglomerular hydraulic pressure via postglomerular efferent arteriole vasoconstriction
COUNTERREGULATORY SYSTEMS (NATRIURETIC PEPTIDES)
B-type natriuretic peptide (BNP) is released from the myocardium during heart failure in response to increased myocardial wall tension. It circulates in quantities relative to the severity of heart failure, and is widely used as a marker for the diagnosis and severity of heart failure. The biologically active moiety, BNP, is a modest vasodilator with some diuretic and natriuretic properties. It also has antigrowth activity and reduces collagen synthesis in vitro. BNP also tends to offset activity of the SNS and the RAAS. This endogenous counterregulatory peptide is not able to stem the tide of forces that drive the progression of severe heart failure, as very high levels of plasma BNP and NT-pro-BNP are observed in patients with acute decompensation. It is possible, however, that the release of BNP in the early stages of heart failure may forestall the onset of more severe signs and symptoms particularly with its natriuretic effects and may be more counterregulatory toward the SNS and RAAS.
BIOMARKERS IN HEART FAILURE
In search of insight into heart failure physiology at the bedside, there are many research-based and clinically available biomarkers that have been studied in heart failure including markers of inflammation, oxidative stress, myocardial injury, myocardial stress, extracellular matrix remodeling, and neurohormones. For example, natriuretic peptides have been found to correlate with changes in ventricular volume and pressure load, while at more advanced stages correlated with renal impairment and overall congestion. Both atrial natriuretic peptide (ANP) and BNP are produced from the ventricles in response to increase volume and/or pressure, which can predict both short-term and long-term outcomes in chronic heart failure patients, including rehospitalization and mortality. It has been shown to predict all-cause mortality in patients with no evidence of LV systolic dysfunction. With treatment, BNP decreases in acute heart failure. Both the biologically active BNP and the N-terminal fragment (NT-proBNP) can be measured in the blood (Wright and Struthers, 2006).
NE has also been shown to be a biomarker of heart failure outcomes although not as powerful a predictor as BNP and possessing much more heterogeneity. With activation of the SNS, NE is thought to be a marker as well as being directly toxic to myocytes. Over time, an increase in BNP or NE is associated with increased mortality (Anand et al. 2003).
Hyponatremia (serum sodium < 135 mEq/L) is associated with poor prognosis in many medical conditions. Worsening hyponatremia goes along with worsening systolic function, decreased glomerular filtration rate (GFR), and Arginine vasopressin (AVP) dysregulation. About 25% of patients with heart failure have hyponatremia, which is caused by neurohormonal dysregulation and diuretics (especially thiazides). Hyponatremia is associated with increased mortality at 30 days and at 1 year. Persistent hyponatremia is most morbid with increased heart failure hospitalizations and increased mortality as compared to treated hyponatremia or patients with normal serum sodium (Jao and Chiong, 2010).
As in acute coronary syndrome and some chronic diseases (like chronic renal failure), increased cardiac troponin is associated with worse prognosis. Cardiac troponin (either T or I) in various studies is elevated in 10% to 50% of chronic heart failure and 6% to 84% of acute heart failure patients. The possible mechanisms linking elevated troponin to worsening heart failure include increased wall stress, subendocardial ischemia, inflammatory cytokines, oxidative stress, altered calcium handling, and neurohormonal activation. Even with adjustment for BNP, increased troponin T or I is associated with worsening mortality with a hazard ratio from 2 to 5. In acute heart failure, as with hyponatremia, persistent troponin elevation is associated with a worse prognosis (Kociol et al., 2010).
INFLAMMATION AND OXIDATIVE STRESS
Inflammation has been implicated in the pathogenesis and progression of heart failure for many years. C-reactive protein (CRP) and proinflammatory cytokines (TNF-α, IL-1, IL-6, and IL-18) have been associated with worse outcomes. IL-6 has been associated with a hypertrophic response, while TNF-α (through activation of matrix metalloproteinases [MMPs]) is associated with ventricular dilation.
Oxidative stress results in an imbalance of reactive oxygen species and reactive nitrogen species, which exacerbates myocardial damage in heart failure. One must measure indirectly the effect of oxidative stress through oxidized low-density lipoprotein (LDL), myeloperoxidase (MPO), and urinary biopyrrins.
MPO is associated with endothelial dysfunction and seems to contribute to LV remodeling. Higher levels of MPO are seen in chronic heart failure and higher levels are seen with increasing severity of New York Heart Association (NYHA) class and predict increased mortality from heart failure. Several newer inflammatory markers have promising prognostic data, but all of these markers await validation of clinical studies and how to best link them to treatment strategies.
RENAL RETENTION OF SALT AND WATER
A hallmark of advanced heart failure is retention of salt and water. This leads to the well-recognized signs and symptoms of tissue congestion such as pulmonary edema, ascites, and leg edema. The mechanism of salt and water retention early in the natural history of heart failure is still not well understood and may be a result of underlying defect at the level of tubular function or response to salt and volume load. In the later stages, reduction in renal blood flow undoubtedly contributes to the problem. The kidney somehow perceives a reduction in effective circulating volume, and unleashes a host of mechanisms, including activation of the RAAS, to conserve and expand circulating volume. GFR is protected early in heart failure by vasoconstriction of the efferent glo- merular arterioles. This is due to Ang-II, which also stimulates the release of aldosterone from the adrenal cortex and contributes to sodium reabsorption and water retention. Eventually, this adaptation wanes, intraglomerular hydraulic pressure falls, and GFR is reduced. The development of renal insufficiency heralds the onset of a dwindling prognosis. Salt and water retention are further aggravated by intense activation of the SNS, causing edema and congestion. Increased release of arginine vasopressin diminishes free water clearance, leading to hyponatremia and more vasoconstriction. Eventually, the “goal” of volume expansion is met, but at the expense of circulatory and tissue congestion. Circulatory homeostasis is not achieved.
LEFT VENTRICULAR REMODELING
Another hallmark of heart failure is that the heart gradually changes size and shape as the syndrome progresses. LV mass increases, cells drop out, myocytes slip away from each other, collagen increases, the heart becomes more stiff, the myocytes become larger and elongate, the chamber dimension increases, wall tension increases adding to reduced performance (the law of Laplace), and the heart simply becomes less efficient over time. The failing heart is exquisitely sensitive to higher afterload (Fig. 15.4), consistent with the notion that the dilated heart performs more poorly. In a sense, these changes define heart failure at the organ level. The remodeling process is due to a confluence of forces, including perverse loading conditions and unrelenting activity of various neurohormones. As the heart hypertrophies, capillary density is reduced, leading to a form of “energy starvation” from oxygen deprivation. High-energy phosphate use is altered. Eventually, myocardial contractility is reduced. The following processes, currently under intense study, contribute to the remodeling of the heart:
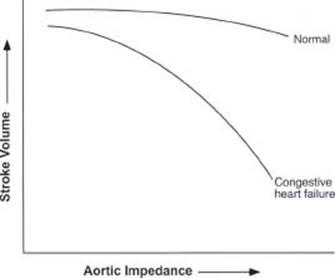
FIGURE 15.4 The failing heart is exquisitely sensitive to afterload. As impedance to ejection increases (increased vascular resistance, increased wall tension, etc.), the performance of the left ventricle diminishes proportionately. On the contrary, acute vasodilation with nitroprusside leads to a marked increase in cardiac output
![]() Increased myocardial mass (hypertrophy)
Increased myocardial mass (hypertrophy)
![]() Increased myocyte size (elongation and increased width)
Increased myocyte size (elongation and increased width)
![]() Cellular necrosis and apoptosis (cell dropout)
Cellular necrosis and apoptosis (cell dropout)
![]() Collagen deposition (reactive and replacement)
Collagen deposition (reactive and replacement)
![]() Myocyte slippage (increased MMPs, decreased tissue inhibitors of MMPs or tissue inhibitors of metalloproteinases [TIMPs])
Myocyte slippage (increased MMPs, decreased tissue inhibitors of MMPs or tissue inhibitors of metalloproteinases [TIMPs])
![]() Chamber enlargement
Chamber enlargement
![]() Increased wall tension
Increased wall tension
![]() Decreased myocardial performance
Decreased myocardial performance
![]() Impaired filling due to increased muscle and chamber stiffness
Impaired filling due to increased muscle and chamber stiffness
![]() Reversion to the “fetal genetic program” (enhanced BNP synthesis, progrowth)
Reversion to the “fetal genetic program” (enhanced BNP synthesis, progrowth)
![]() Activation of neurohormones
Activation of neurohormones
![]() Increased synthesis and release of counterregulatory hormones (i.e., BNP)
Increased synthesis and release of counterregulatory hormones (i.e., BNP)
ABNORMAL CELLULAR MECHANISMS
Important changes occur at the cellular level in the setting of heart failure. These abnormalities undoubtedly contribute to reduced contractility, as they are imbedded in the contractile units, that is, the sarcomere and the myocyte itself. Many of these abnormalities have been observed in vitro, in the laboratory setting only, but some have been derived from failing human hearts extirpated at the time of heart transplantation. It is not entirely clear whether these molecular abnormalities are primary features that contribute quantitatively to the failing heart or whether they are secondary or the so-called epiphenomena that occur as a consequence or a result of the heart failure syndrome. Nevertheless, it is important to consider them:
![]() Decreased β-receptor density in the heart
Decreased β-receptor density in the heart
![]() Increased Gi coupling protein in the heart
Increased Gi coupling protein in the heart
![]() α- to β-myosin heavy-chain transition (decreased myosin ATPase enzyme velocity) in the myocytes
α- to β-myosin heavy-chain transition (decreased myosin ATPase enzyme velocity) in the myocytes
![]() Defect in sarcolemma calcium uptake
Defect in sarcolemma calcium uptake
![]() Defect in calcium-ATPase (SERCA) and phospholamban (Ca2+ exchange)
Defect in calcium-ATPase (SERCA) and phospholamban (Ca2+ exchange)
![]() Abnormal contractile proteins
Abnormal contractile proteins
These changes, observed at the molecular level, likely contribute to reduced inotropy and may serve as a substrate for rhythm disturbances. They may be a vestige of evolutionary forces that initially allow the heart to operate in a more economical manner in the face of excessive inotropic stimulation. Over time, these “adaptations” contribute to impaired organ function.
PERIPHERAL VASCULAR AND SKELETAL MUSCLE ADAPTATIONS IN HEART FAILURE
Profound changes occur in the periphery in the setting of heart failure, and these likely are responsible for the impaired exercise tolerance and fatigue that commonly plagues patients. In addition to exercise intolerance, sleep disturbances occur, often in the form of obstructive and central sleep apnea. Blood flow is redistributed to the brain and skeletal muscles, away from the kidneys and the splanchnic beds. Abnormalities in reflex control underlie these changes. Skeletal muscles begin to atrophy, which contributes to fatigue. The causes of exercise intolerance are multiple and complex. On the cellular level, there is endothelial dysfunction with decreased nitric oxide in the periphery and decreased β-adrenergic myocardial receptor density. The skeletal myocyte switched from slow- to fast-twitch fiber with reduced mitochondrial size and enzymes and muscle atrophy. From the standpoint of cardiac physiology, there is chronotropic incompetence, reduced lung compliance, inability to increase stroke volume in response to exercise, and overall deconditioning.
PROGNOSIS
It is important to identify where an individual patient is situated in the natural history of heart failure, providing needed optimism for those in the early stages while allowing for advanced directives for patients in the end stages. However, data regarding prognosis are nearly always derived retrospectively from large databases and represent group data that may not apply to an individual patient. For example, a low “ejection fraction (EF)” is not a powerful risk factor in a group of patients with advanced disease, since the degree of compensation may influence the pace of disease progression. Physicians need to keep this in mind when interacting with patients and their families, who frequently ask about prognosis. Clearly, the overall prognosis has improved for heart failure over the past decades with many new and effective treatments. Nonetheless, heart failure is usually not “cured,” but it can be managed as a chronic condition. Many prognostic factors associate with a poor prognosis, including those shown in Table 15.1.
TABLE
15.1 Prognostic Factors Associated with a Poor Prognosis of Congestive Heart Failure

Several risk scores have been developed to stratify those at risk for heart failure and the survival with known heart failure. The Heart Failure Survival Score (HFSS) studied patients with severe heart failure and/or those being evaluated for cardiac transplant. It sought to identify those with poor event-free survival including more than just the peak maximum oxygen capacity (peak VO2). The score predicts event-free survival at 1 year—low risk approximately 93% and high risk 43%. The noninvasive HFSS model includes ischemic etiology, resting heart rate, EF, QRS ≥ 0.12 second (due to any cause), resting mean blood pressure, peak VO2, and serum sodium. The invasive model includes the pulmonary capillary wedge pressure. More recently, the Seattle Heart Failure Model was developed to estimate 1-, 2-, and 5-year survival with heart failure. The model was derived and validated in patients with various grades of HF (NYHA class I to IV). This model adds the effect of medications and devices. The independent predictors are NYHA class, ischemic etiology, diuretic dose, EF, systolic blood pressure, serum sodium, hemoglobin, percent lymphocytes, uric acid, and cholesterol (Table 15.2). This complex model is available for free online.
TABLE
15.2 Heart Failure Risk Scores
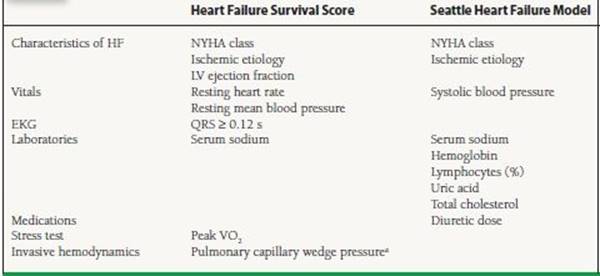
NYHA, New York Heart Association; LV left ventricular;VO2, oxygen consumption. “Only in the invasive model of HFSS.
SUMMARY
Heart failure is a complex syndrome, and its pathophysiology is inherently complex. Nevertheless, there are some unifying features. There are signs and symptoms of dyspnea, fatigue, and sometimes tissue congestion. There are underlying structural and/or functional abnormalities of the heart. The heart “adapts” to an index event to maintain circulatory homeostasis in the short term, but over the long term, there is further progression of heart failure, in part driven by these “adaptive” mechanisms. The heart enlarges, becomes more globular, and more inefficient. Excitation contraction becomes abnormal. Rhythm disturbances occur. LV performance diminishes, and often mitral and tricuspid insufficiency occurs. Exercise tolerance is diminished, salt and volume are retained, renal function deteriorates, and signs and symptoms worsen. We now have a much better understanding of how these events unfold, and treatment has improved markedly. However, the natural history of heart failure is highly variable in individual patients, making prognosis difficult to determine.
SUGGESTED READINGS
Aaronson KD, Schwartz JS, Chen TM, et al. Development and prospective validation of a clinical index to predict survival in ambulatory patients referred for cardiac transplant evaluation. Circulation. 1997;95(12):2660-2667.
Anand IS, Chandrashekhar Y, Ferrari R, et al. Pathogenesis of congestive state in chronic obstructive pulmonary disease. Circulation. 1992;86:1992:12-21.
Anand IS, Chandrashekhar Y, Ferrari R, et al. Pathogenesis of edema in chronic severe anemia: studies of hemodynamic variables, and plasma hormones. Br Heart J. 1993;70:357-362.
Anand IS, Fisher LD, Chiang Y, et al. Changes in brain natriuretic peptide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-HeFT). Circulation. 2003;107(9):1278-1283.
Braunwald E. Biomarkers in heart failure. N Engl J Med. 2008;358(20):2148-2159.
Cohn JN. From hypertension to heart failure. Eur Heart JSuppl. 2000;2(suppl A):A2-A5.
Harris P. Evolution and the cardiac patient. Cardiovasc Res. 1983;17:1-22.
Harris P. Congestive cardiac failure: central role of the arterial blood pressure. Br Heart J. 1987;58:190-203.
Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American nean Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53(15):e1-e90.
Jao GT, Chiong JR. Hyponatremia in acute decompensated heart failure: mechanisms, prognosis, and treatment options. Clin Cardiol. 2010;33(11):666-671.
Kociol RD, Pang PS, Gheorghiade M, et al. Troponin elevation in heart failure prevalence, mechanisms, and clinical implications. J Am Coll Cardiol. 2010;56(14):1071-1078.
Levy WC, Mozaffarian D, Linker DT, et al. The Seattle Heart Failure Model: prediction of survival in heart failure. Circulation. 2006;113(11):1424-1433.
Mann DL. Mechanisms and models in heart failure. Circulation. 1999;100:999-1008.
Packer M. The neurohormonal hypothesis: a theory to explain the mechanism of disease progression in heart failure. J Am Coll Cardiol. 1992;20:248-254.
Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics—2011 update: a report from the American Heart Association. Circulation. 2011;123(4):e18-e209.
Schneider KM, O’Donnell BE, Dean D. Prevalence of multiple chronic conditions in the United States' Medicare population. Health Qual Life Outcomes. 2009;7:82-93.
Schrier RW, Gheorghiade M. Challenge of rehospitalizations for heart failure: potential of natriuretic doses of mineralocorticoid receptor antagonists. Am Heart J. 2011;161(2):221-223.
Tang WHW, Francis GS. Natural history of heart failure. In: Kukin ML, Fuster V, eds. Oxidative Stress and Cardiac Failure. Armonk: Futura; 2003:3-47.
Wright GA, Struthers AD. Natriuretic peptides as a prognostic marker and therapeutic target in heart failure. Heart. 2006;92(2):149-151.
QUESTIONS AND ANSWERS
Questions
1. Which of the following statements about heart failure is true?
a. It is a clinical syndrome.
b. It can be caused by any form of heart disease.
c. It is diagnosed primarily by history and physical exam.
d. All of the statements are true.
2. The principal features of heart failure include all of the following except:
a. Activation of the renin-angiotensin-aldosterone system (RAAS) and sympathetic nervous system (SNS)
b. Left ventricular (LV) remodeling
c. The ability to mount a reflex tachycardia
d. Downregulation of β-adrenergic receptors
3. The inability to exercise properly in heart failure is due to all of the following except:
a. Reduced ejection fraction (EF)
b. Skeletal muscle atrophy
c. Endothelial dysfunction in peripheral vessels
d. Inability to increase stroke volume and heart rate
4. Which of the following statements about the prognosis for heart failure is true? (Select the best answer.)
a. It is fairly easy to predict in individual patients.
b. It is commonly assessed by measuring peak VO2 during exercise.
c. It is closely coupled to EF in individual patients.
d. It is commonly estimated by measuring neurohormones.
5. Which of the following characterizes heart failure?
a. Downregulation of β1- and β.-receptors
b. Downregulation primarily of β-receptors, with little change in β-receptors
c. Downregulation of G proteins and β1- and β2- receptors
d. Increase in myocardial norepinephrine (NE) stores
e. Intact baroreceptor function
6. All of the following neurohormones are associated with vasoconstriction, cell growth, hypertrophy, and sodium retention except:
a. Angiotensin-II (Ang-II)
b. Norepinephrine
c. Brain natriuretic peptide
d. Endothelin
e. Arginine vasopressin
Answers
1. Answer D: Heart failure, like renal failure or anemia, is a clinical syndrome with a constellation of signs and symptoms. It has many possible etiologies, since virtually any form of heart disease can lead to heart failure. Patients must have signs and symptoms (i.e., a low EF does not equal heart failure) that usually consist of dyspnea and fatigue at rest or with exertion. There must be underlying cardiac structural and/or functional abnormalities. There is no laboratory test for heart failure (i.e., a history and physical exam are necessary), though a plasma BNP level may help facilitate the diagnosis in certain settings.
2. Answer C: Patients with heart failure have well-documented disturbances of the autonomic nervous system and are unable to mount a reflex tachycardia in response to upright tilt, orthostasis, intense vasodilation, or other volume-depleting stimuli. In fact, the extent of this blunted sympathetic response is coupled to the severity of heart failure and is predictive of a poor prognosis. Similarly, patients with heart failure do not fully activate the parasympathetic arm of the autonomic nervous system in response to systemic pressor activity with phenylephrine (there is less vagal-induced slowing of the heart rate). Heart-rate variability is also blunted in patients with heart failure, and is also associated with a poor prognosis.
3. Answer A: There has been a very reproducible and consistently poor relationship noted between resting EF and exercise capacity (r = 0.20-25) in patients with chronic heart failure. This is likely because exercise capacity is limited in patients with chronic heart failure, not by abnormal central hemodynamics but by peripheral factors such as deconditioning and atrophy of skeletal muscles, changes in skeletal muscle oxidative enzymes, redistribution of blood flow away from skeletal muscles to more vital organs, and endothelial dysfunction in the peripheral vasculature due to a relative deficiency of local nitric oxide synthesis in blood vessels.
4. Answer B: There are almost as many “prognostic factors” in heart failure as there are stars in the clear night sky. Many of them are related to each other, and their independent contributions to prognosis are difficult to measure. Determining how much exercise the patient can do is perhaps the closest “factor” we have to a true “gold standard” for estimating prognosis. For example, the VO2max should be < 14 mL/kg/min for a patient to be considered for heart transplantation. Preserved exercise tolerance is a very powerful predictor of a better prognosis in patients with chronic heart failure.
5. Answer B: In chronic heart failure, it is primarily the β1-receptor that is downregulated. The density of cardiac β2-receptors is much less than that of the β1- receptors, and the β2-receptors may be less important in modulating positive inotropy. In addition to relatively selective β1-receptor downregulation that occurs in chronic heart failure, there is important uncoupling of the G-stimulating protein from the b-receptors, leading to a reduction in positive inotropic state.
6. Answer C: Natriuretic peptides modulate sodium and water (volume) regulation, vasodilation, natriuresis, antifibroblast proliferation, and anticollagen deposition, and have antiremodeling activity. Their biologic activities are nearly opposite to those of Ang-II, norepinephrine, endothelin, and arginine vasopressin