Shmuel Benenson, MD
Lisa Green, MD, MPH
Alan H. DeCherney, MD
Microbial infection has always been a grave threat to obstetric and gynecologic patients. Developments in antimicrobial therapy, however, have led to decreases in puerperal and postoperative morbidity and perinatal mortality. Indeed antibiotic development is one of the most important advances in medicine in the 20th century. Empiric antibiotics for bacterial infections improve clinical symptoms and outcome. As a consequence, clinicians prescribe antibiotics very frequently and sometimes even when it is not necessary. This approach has led to huge overuse of antibiotics, which in turn has led to the appearance of multidrug-resistant bacteria. Clinicians need to adopt an approach where effective antibiotic treatment is given to those who have bacterial infections, while at the same time antibiotic use is limited when not indicated.
SELECTION OF ANTIMICROBIAL DRUGS
Several considerations are pertinent to most infections encountered in obstetric and gynecologic practice. First, the majority of patients are generally healthy and free of debilitating illness, with the exception of some elderly and oncology patients. Second, the lower genital tract (vagina and cervix) contains a complex flora (eg, anaerobes, gram-positive and gramnegative aerobes, and Candida), whereas the upper genital tract (uterus, fallopian tubes, and ovaries) is sterile. Infections in the upper genital tract usually result from spread of the lower genital tract flora when the upper tract is anatomically disrupted (eg, by sexually transmitted disease, by surgery, or during delivery). For this reason, most infections, such as postpartum or postoperative infection and pelvic inflammatory disease, are polymicrobial. Third, cultures must be obtained when infection is suspected (eg, pelvic abscess, chorioamnionitis), and empiric antibiotic therapy targeted at the potential organisms is usually indicated before culture results are available. However, in some gynecologic infections, because of laboratory limitations, culture results may not be available in a timely fashion, or tests may not even be performed at all. In some cases, surgical intervention (“source control”) rather than antibiotic treatment (“antibiotic control”) is the main component of treatment. Fourth, when selecting antibiotic agents in a pregnant woman, the potential risk for the fetus should be taken into account.
To serve as a guide to antibiotic selection, 5 tables are provided. Tables 44–1 and 44–2 provide the classification and dosages of selected β-lactam antibiotics and antibiotics from other classes. Table 44–3presents the main or serious adverse events of antibiotics commonly used in obstetric-gynecologic practice and risk categories of antimicrobials in pregnancy. Table 44–4 shows recommended drugs and alternatives against selected bacteria encountered in obstetric-gynecologic practice. Finally, Table 44–5 shows suggested regimens for main clinical diagnoses.
Table 44–1. Antibiotic dosage of selected β-lactam agents.
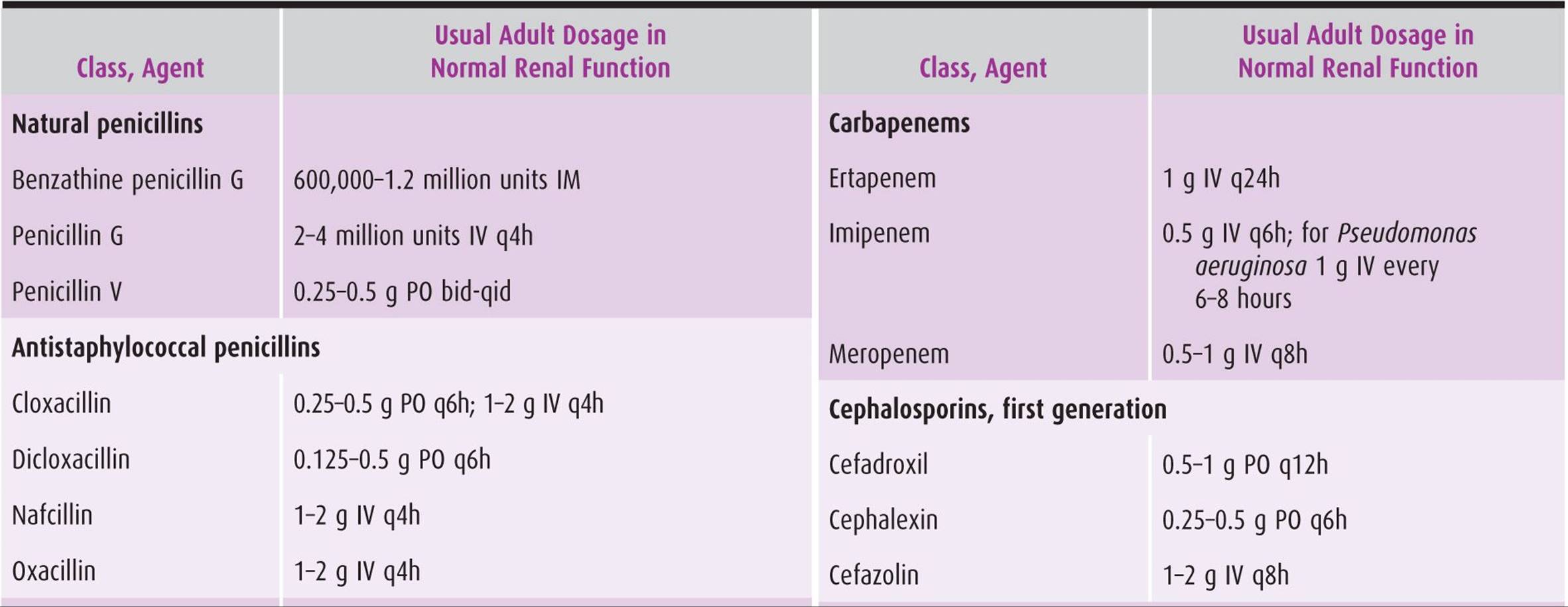
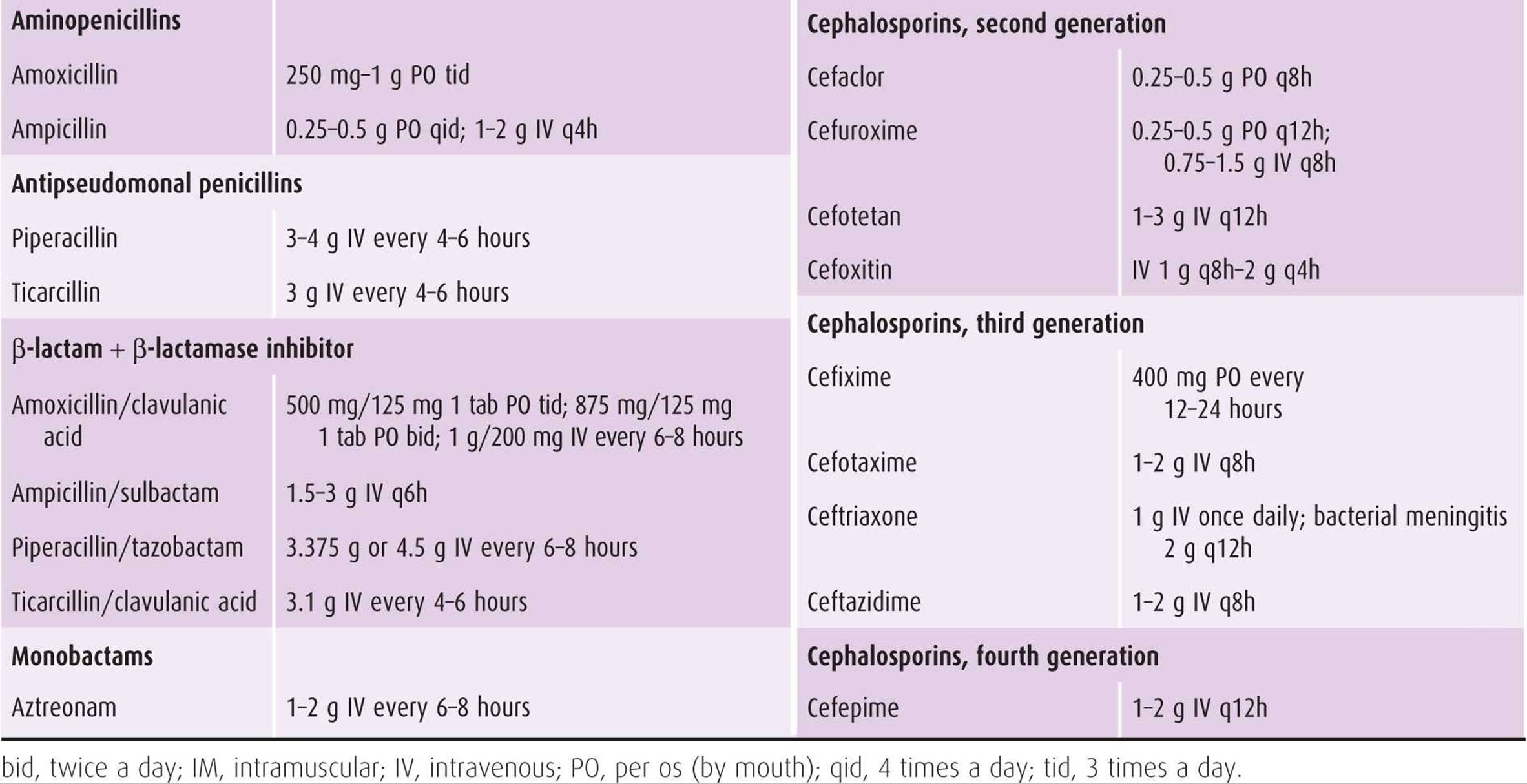
Table 44–2. Antibiotic dosage of selected antimicrobial agents other than β-lactams.
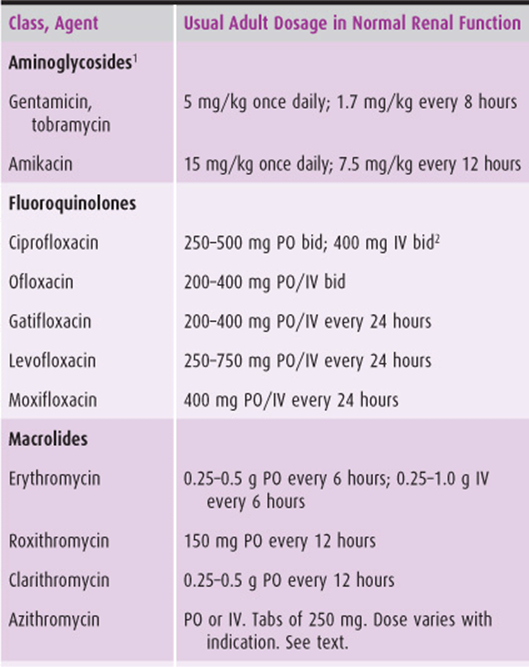
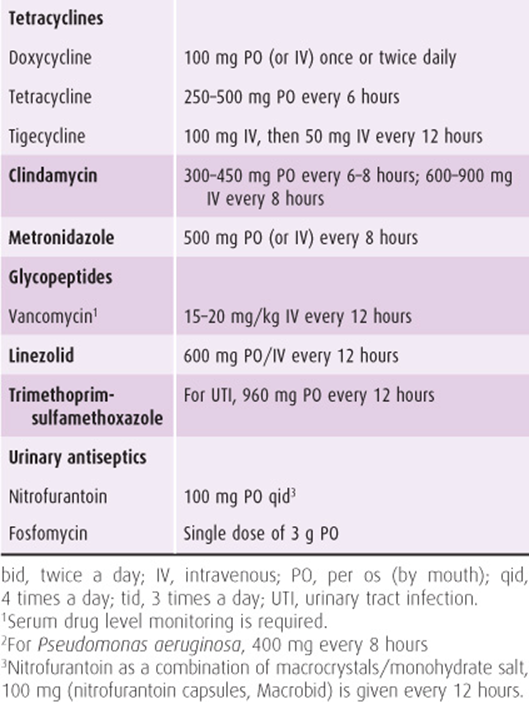
Table 44–3. Main or serious adverse events of antibiotics commonly used in obstetric-gynecologic practice and pregnancy risk categories.
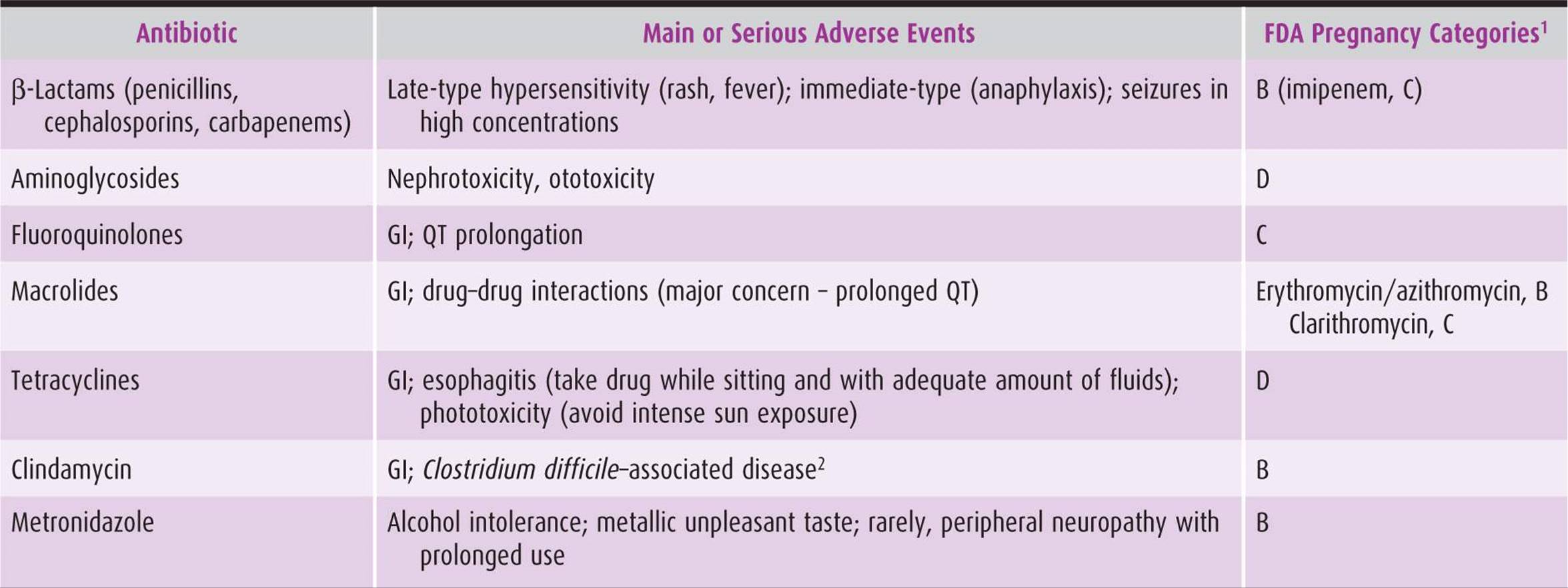

Table 44–4. Recommended and alternative antimicrobial drugs against selected bacteria encountered in obstetric-gynecologic practice.
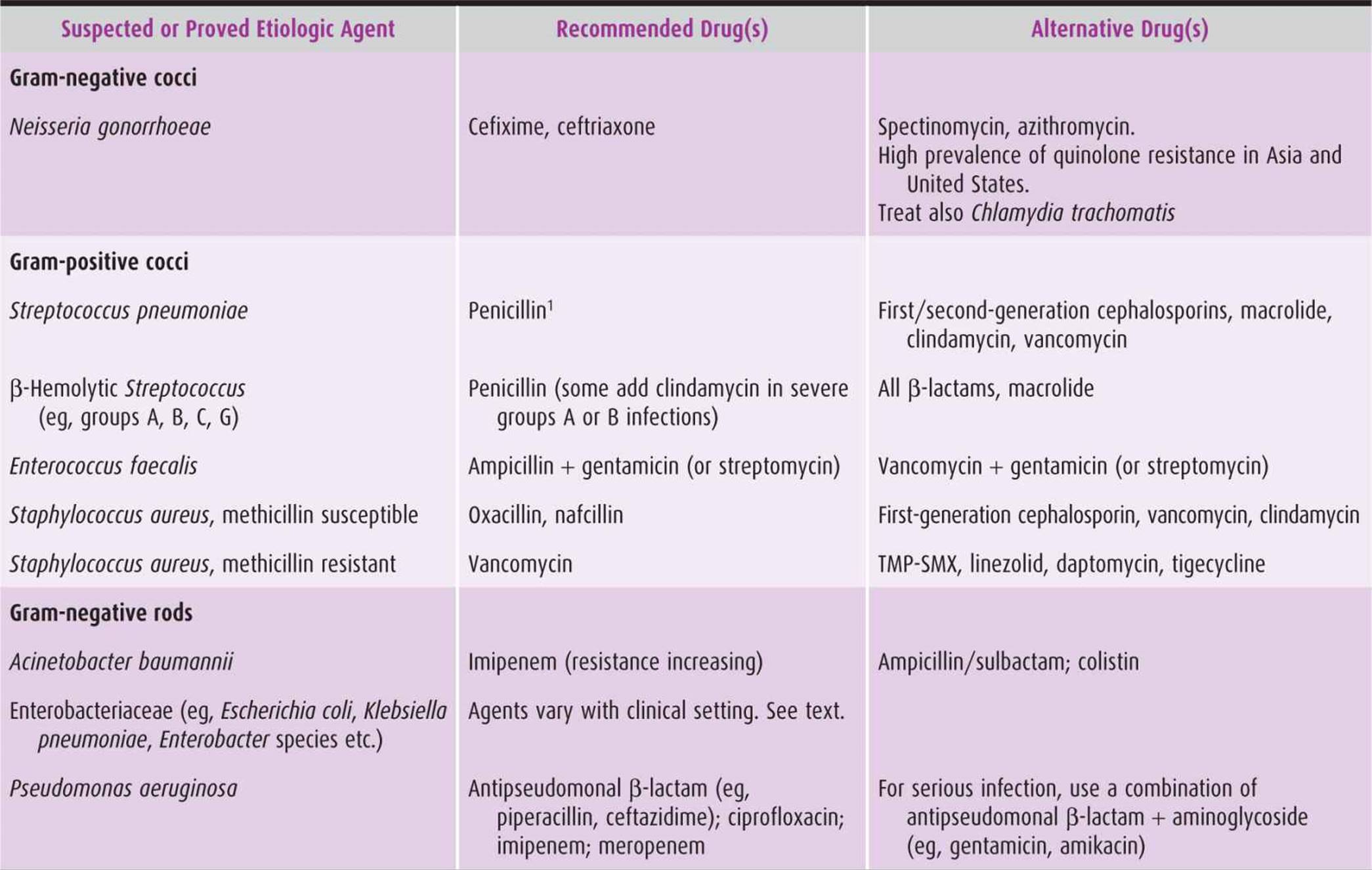
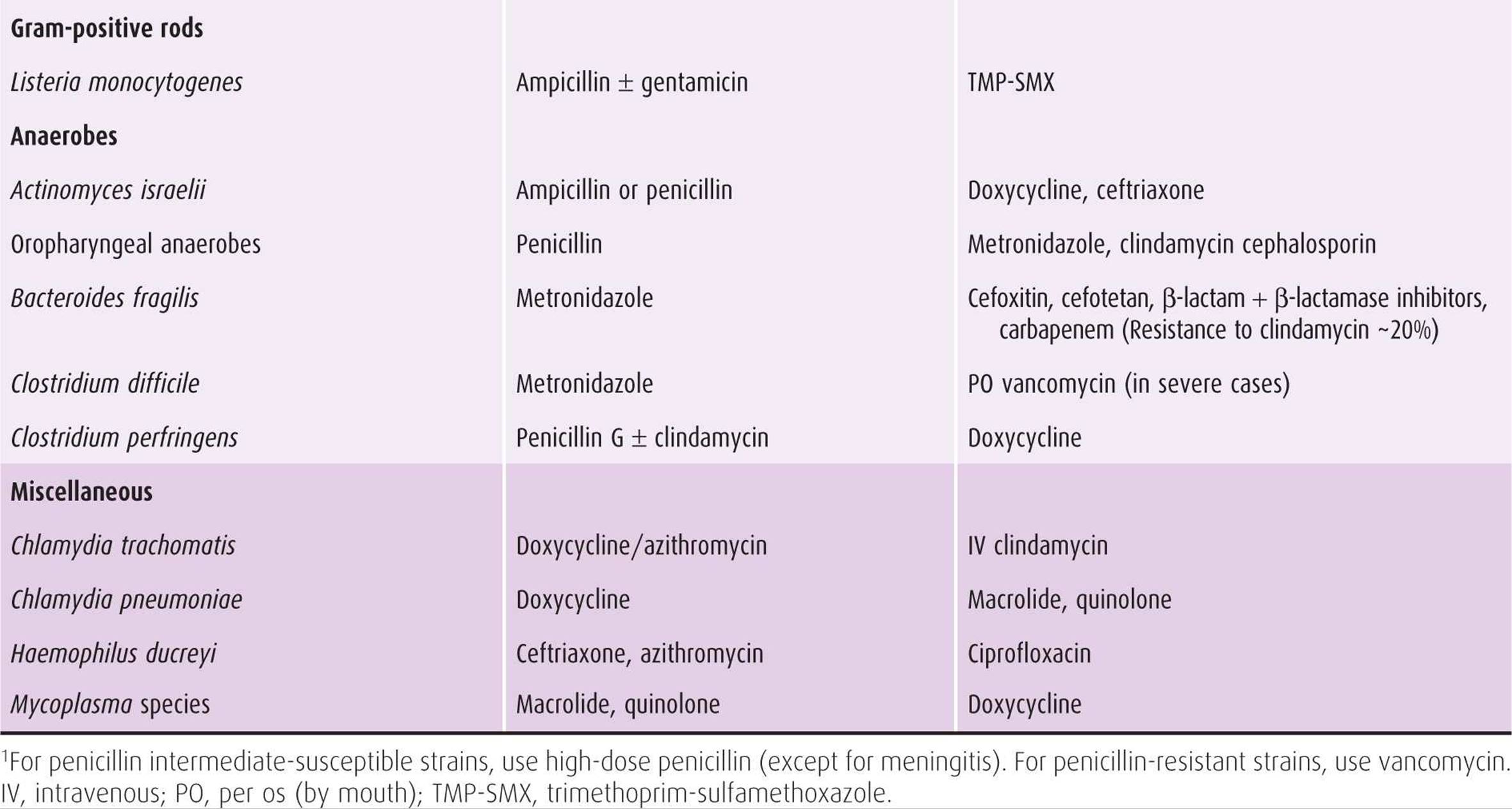
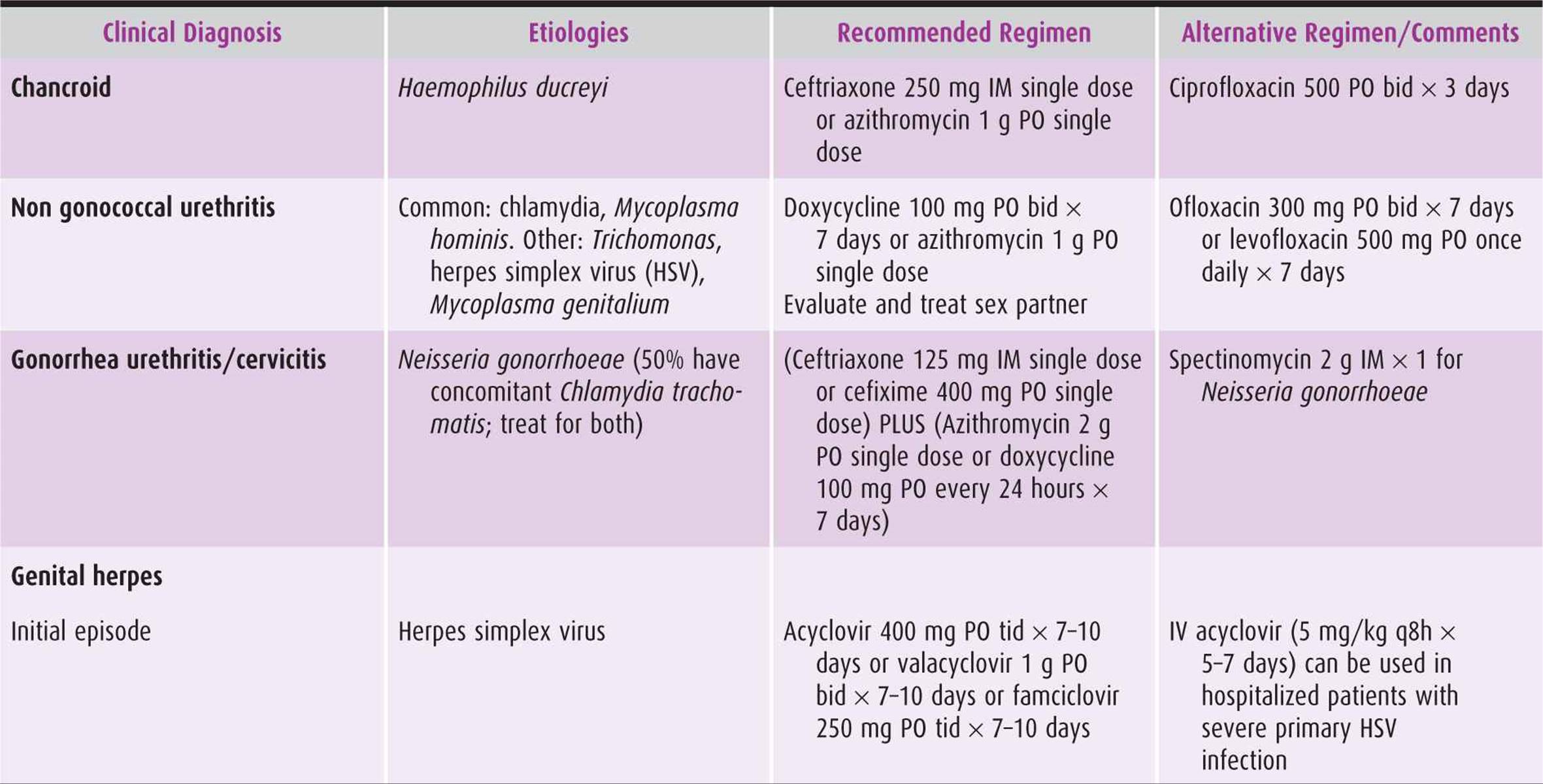
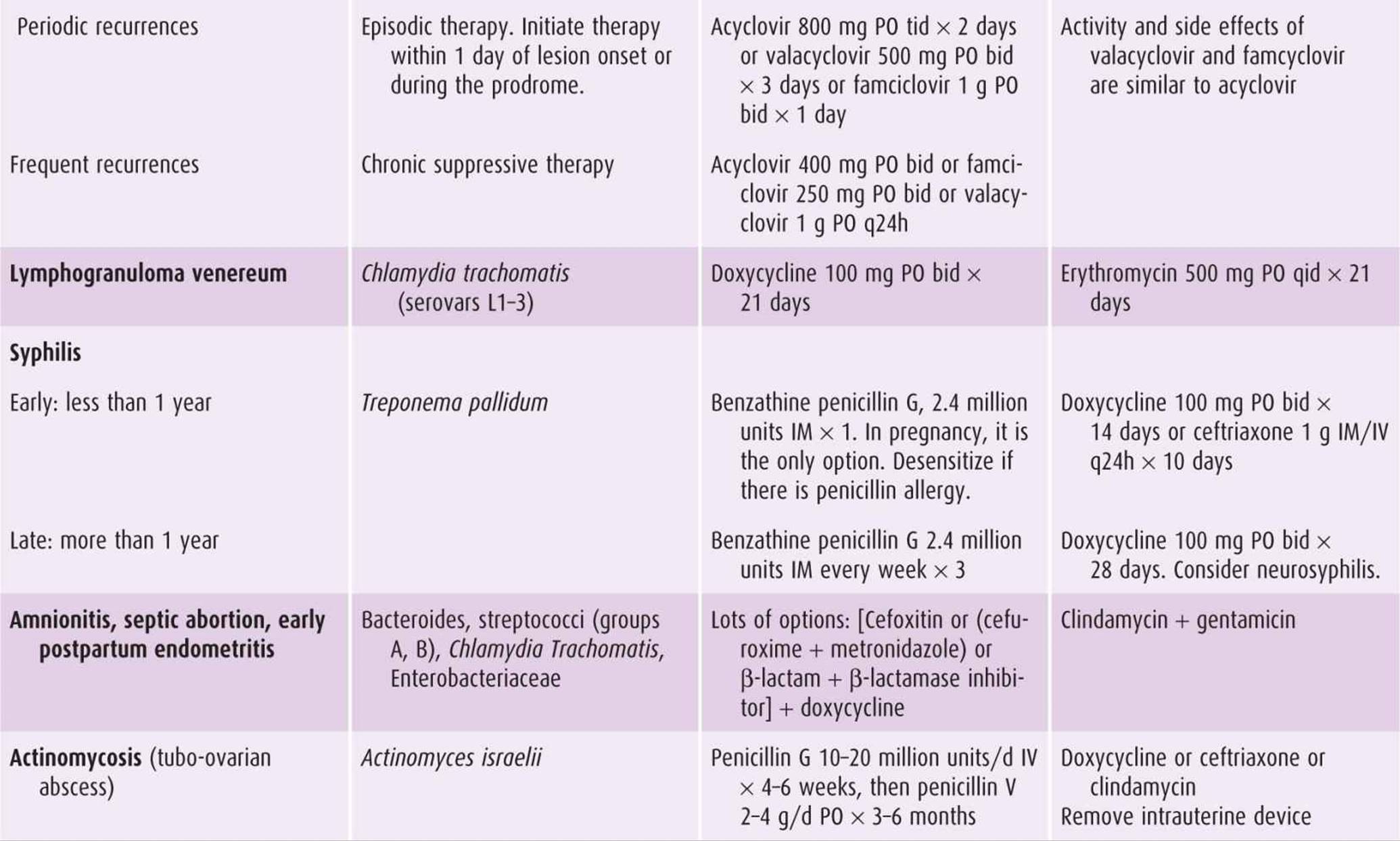
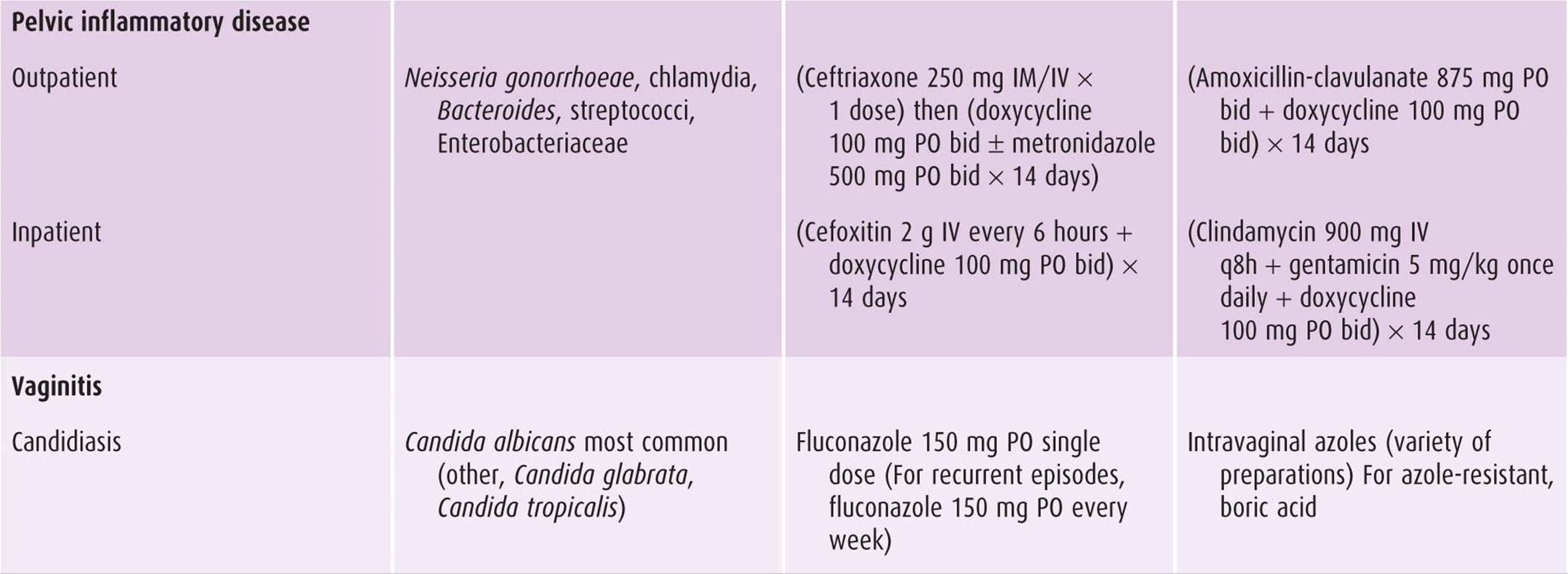
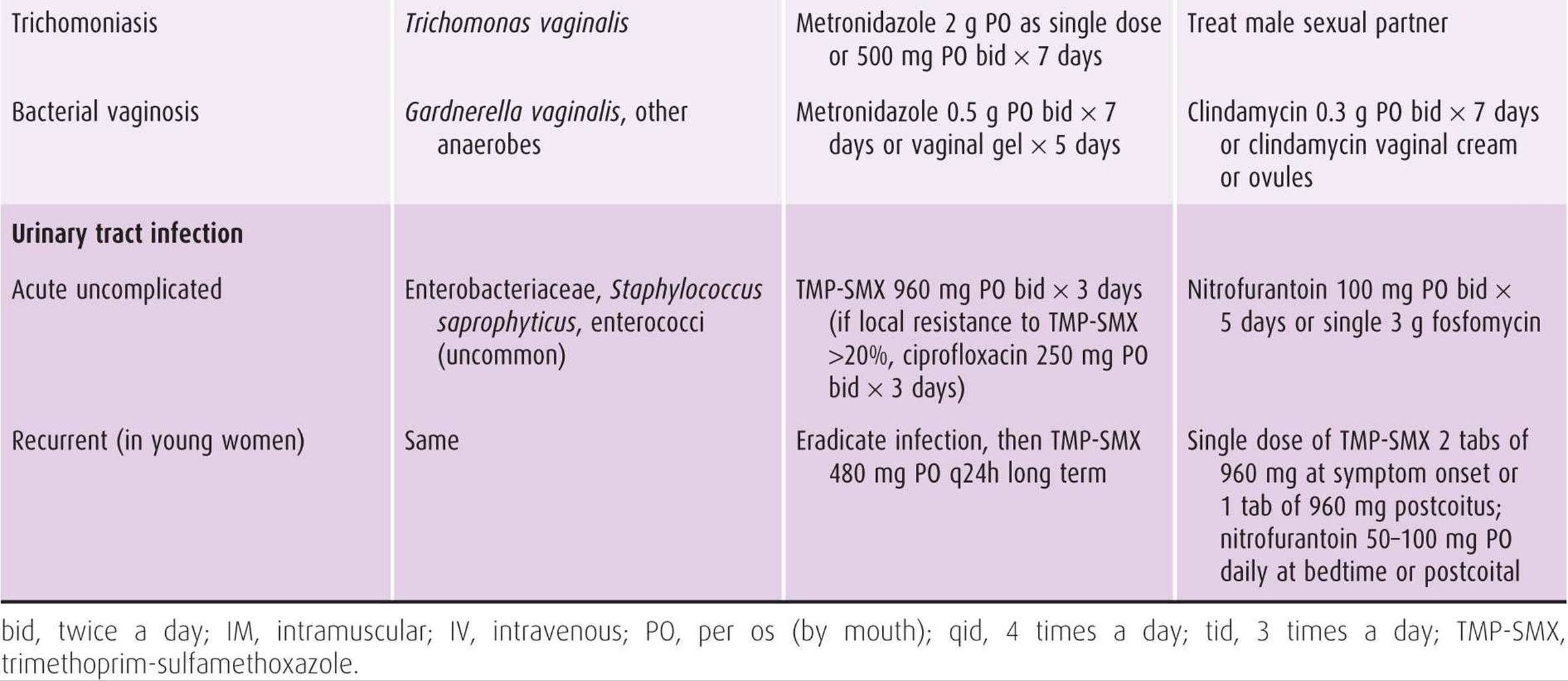
Table 44–5. Treatment regimens for selected infections in obstetric-gynecologic practice.
The following steps merit consideration in each patient.
A. Infectious Diagnosis?
The physician must attempt to decide on clinical grounds whether the patient has an infection or whether the symptoms and signs are caused by a noninfectious cause.
B. Diagnostic Microbiology
Before beginning antimicrobial drug treatment for a suspected infection, always attempt to obtain appropriate clinical specimens for culture in order to identify the causative infectious organism and its susceptibility to antimicrobial drugs. Gram stains of sterile body fluids or abscess fluid are one of the most useful tools available to direct an empiric antibiotic treatment. Cultures should be obtained from the suspected site of infection, and if infection is severe, blood cultures should be taken.
C. The Pathogen
The most likely pathogen (or pathogens) causing infection should be defined. This assessment is based on clinical information and epidemiology (eg, age, organ involved, community-acquired vs. hospital-acquired infection; Table 44–5).
D. Pathogen-Oriented Empiric Antibiotic Treatment
Selecting appropriate initial antimicrobial therapy is of high priority. Based on the probable pathogen causing the infection and the local susceptibility patterns, the physician should choose a drug (or drug combination) that is likely to be effective against the suspected microorganism.
E. Other Factors Influencing the Antibiotic Choice
1. Are there antibiotic allergies?
2. Pregnancy: Choose antibiotics in a pregnant woman in accordance with the US Food and Drug Administration’s (FDA) use-in-pregnancy drug rating system (Table 44–3).
3. Renal failure may affect not only the choice of antibiotic but also its dosages.
a. Dosages of antibiotics primarily excreted by the kidneys (eg, β-lactams, aminoglycosides, and fluoroquinolones) should be modified based on the creatinine clearance.
b. Renal function should be monitored in patients treated with antibiotics that are potentially nephrotoxic (eg, aminoglycosides).
c. Serum antibiotic levels should be monitored every 2 to 4 days when an aminoglycoside or vancomycin is used.
4. Severity of illness: In those who are seriously ill, the spectrum of the empiric antibiotic regimen needs to include coverage of potentially resistant pathogens.
F. Route of Administration
Intravenous (IV) antibiotics are preferred in serious infections. Oral therapy is effective for mild to moderately severe infections and for completion of therapy initially treated with IV antibiotics. For some antibiotics, absorption after oral administration depends on the proximity to other food intake (eg, amoxicillin-clavulanate and doxycycline should be taken immediately after eating food, whereas trimethoprim-sulfamethoxazole should be taken between meals).
G. Laboratory Results and Clinical Response
Based on the laboratory results (eg, culture and susceptibility) and on the patient clinical response, the desirability of changing the antimicrobial drug regimen should be considered. Conversion from a broad-spectrum to a narrow-spectrum drug should be carried out if possible. It is important to assess the clinical response of the patient before making changes based on culture results. Laboratory results should not automatically overrule clinical judgment.
H. Duration of Antimicrobial Therapy
In general, effective antimicrobial treatment results in marked clinical improvement within a few days. However, continued treatment for varying periods may be necessary to effect cure. The duration of therapy depends on clinical judgment. For most infections, the duration ranges from 5–7 to 10–14 days. For most postoperative and postpartum infections, IV antibiotics can be discontinued after the patient has been afebrile for 24–48 hours. Transfer from IV to oral can be made if the patient can eat and the absorption is not interrupted.
I. Adverse Reactions
The administration of antimicrobial drugs is occasionally associated with untoward reactions (Table 44–3). These reactions can be divided into 3 main groups.
1. Hypersensitivity—The most common reactions are fever and skin rashes. Hematologic or hepatic disorders and anaphylaxis are rare.
2. Direct toxicity—Most common are nausea, vomiting, and diarrhea. More serious toxic reactions are impairment of renal, hepatic, or hematopoietic function.
3. Suppression—Suppression of normal microbial flora and “superinfection” by drug-resistant microorganisms (such as Clostridium difficile).
J. Failure to Improve
When the patient is not improving despite adequate antibiotic treatment as determined by culture results, the following possibilities should be considered:
1. Presence of an undrained abscess, hematoma, or foreign body
2. Inappropriate antibiotic dose or route of administration
3. Low antibiotic concentration at the site of infection (eg, central nervous system [CNS])
4. Emergence of drug-resistant or -tolerant organism
5. Involvement of 2 or more microorganisms in the infectious process, of which only 1 was originally detected and used for drug selection
ANTIMICROBIAL DRUGS
1. Penicillins
The penicillins are among the most widely used antimicrobial drugs. The term penicillin is the generic term for a large group of antimicrobial substances, all of which share a common chemical nucleus consisting of a thiadolizine ring, the β-lactam ring, and a side chain; hence the synonym “β-lactams” for the whole group (ie, penicillins, cephalosporins, and carbapenems). The ring is essential for antibacterial activity, whereas the side chain determines the antibacterial spectrum and pharmacologic properties of a particular penicillin (Table 44–1). As a result of the common ring, the potential for allergic cross-sensitivity among penicillins is high. All β-lactam antibiotics inhibit formation of microbial cell wall. By binding to proteins in the cell wall (penicillin-binding proteins), they block the final transpeptidation reaction in the synthesis of cell wall peptidoglycan. This reaction results in bacterial cell death; thus, penicillin is bactericidal.
The most common mechanism of bacterial resistance against penicillin is the production of a β-lactamase enzyme, which destructs the β-lactam ring. This is the principal mechanism of penicillin resistance in Staphylococcus aureus, Bacteroides fragilis, and Enterobacteriaceae. When penicillin is combined with a β-lactamase inhibitor, the penicillin may escape breakdown.
Pharmacokinetics (Absorption, Distribution, & Excretion)
Acid-labile compounds are poorly absorbed and hence administered only parenterally (penicillin G, antipseudomonal penicillins). Acid-stable compounds vary in the proportion of absorption after oral administration (50% for cloxacillin, 60% for penicillin V, and 75% for amoxicillin). To minimize binding to foods, most oral penicillins should not be preceded or followed by food for at least 1 hour.
After absorption, penicillins are widely distributed in body fluids and tissues (lung, liver, kidney, muscle, bone, and placenta). The levels of penicillins in abscesses and peritoneal fluids are sufficient in the presence of inflammation. In many tissues, penicillin concentrations are equal to those in serum. Lower levels are found in the CNS; however, with active inflammation of the meninges, as in bacterial meningitis, penicillin levels in the cerebrospinal fluid exceed 1–10% of serum concentrations.
Most penicillins are rapidly excreted by the kidneys into the urine—90% by tubular secretion, which results in very high levels in the urine. Significant reduction in renal function must be taken into account in the administration of most penicillins.
Indications, Dosages, & Routes of Administration
A. Penicillin G, Penicillin V
In obstetric-gynecologic practice, penicillin is the drug of choice for treatment of infections caused by groups A and B streptococci, Treponema pallidum (causing syphilis), clostridia, and actinomycosis. Severe infections caused by enterococci should be treated by a synergistic combination of ampicillin (or penicillin G) and gentamicin. Penicillin is not recommended to treat Neisseria gonorrhoeae because of widespread resistance.
1. Aqueous penicillin G is given intravenously and produces high blood levels but is excreted rapidly. Therefore, for serious infections, it should be given every 4 hours (6 times daily). In adults with normal renal function, a dose of 18–24 million units a day is used in severe infections and 9–12 million units in other infections.
2. Benzathine penicillin G is an insoluble salt that is injected intramuscularly to establish a depot that yields very low drug levels for prolonged periods of time (ie, 3–4 weeks). An injection of 2.4 million units intramuscularly once per week for 1 or 3 weeks is the recommended treatment for early and late syphilis, respectively.
3. Penicillin V is made by a mild modification of the side chain of penicillin G, which allows it to resist gastric acid breakdown and thus can be given orally. Penicillin V is indicated in minor infections (eg, group A Streptococcuspharyngitis) in daily doses of 1–2 g (500 mg of penicillin V equals 800,000 units).
B. Amoxicillin, Ampicillin, Ticarcillin, Piperacillin
Several side-chain modifications of penicillin have been made to provide enhanced activity of these “broad-spectrum” penicillins.
1. Amoxicillin and ampicillin, in addition to being active against penicillin-susceptible organisms, are more active than penicillin against enterococci and Listeria monocytogenes. Due to widespread bacterial resistance among Enterobacteriaceae, these drugs can be given against Enterobacteriaceae (eg, to treat urinary tract infections) only after susceptibility studies are available. They are ineffective against Pseudomonas aeruginosa.
Amoxicillin is available only for oral use in daily doses of 1.0 g every 8 hours. Amoxicillin has replaced oral ampicillin. IV ampicillin is given in a dose of 1–2 g every 4 hours (ie, 6–12 g daily) depending on the organism involved and the severity and site of infection.
2. Ticarcillin and piperacillin were introduced for gramnegative bacteria. They are active against many Enterobacteriaceae and also P aeruginosa. Because resistance emerges rapidly, susceptibility testing is required. Their anti–gram-positive spectrum mimics that of ampicillin. For severe P aeruginosa infection, piperacillin is preferred (it is 4 times more active against P aeruginosa), and addition of an aminoglycoside is required in order to decrease the likelihood of emergence of resistance.
These drugs are administered IV only: piperacillin 4 g every 6 hours and ticarcillin 3 g every 4 hours.
C. β-Lactamase–Resistant Penicillins
These agents, which are resistant to destruction by β-lactamase, are used primarily to treat infections caused by βlactamase–producing S aureus. They are also active against other gram-positive aerobes (eg, group A Streptococcus, Streptococcus pneumoniae) but are inferior to penicillin for them. They are not active against the Enterobacteriaceae or enterococci.
1. Oral—Cloxacillin or dicloxacillin can be given in doses of 0.25–1.0 g every 6 hours in mild or localized staphylococcal infections. Food markedly interferes with absorption, and hence, they should not be preceded or followed by food for at least 1 hour.
2. IV—Nafcillin, oxacillin, and cloxacillin are the drugs of choice for the treatment of serious systemic methicillin-sensitive S aureus (MSSA) infections. The usual dose is 6–12 g/d (1–2 g every 4 hours), depending on the severity of the infection. Because these drugs are excreted primarily by hepatic mechanisms, dose reduction is not needed in renal failure (mild reduction of cloxacillin is needed in severe renal failure), but in hepatic failure, dose reduction is required.
D. Combinations of Penicillins plus β-Lactamase Inhibitors
Because the primary bacterial resistance mechanism against penicillin is through β-lactamase production, a β-lactamase inhibitor was combined with several penicillins. This inhibitor binds irreversibly to the β-lactamase, and thus allows the free penicillin to exert its antibacterial activity.
In addition to being active against the penicillin component–susceptible organisms (eg, streptococci, enterococci), these combinations are effective also against MSSA, B fragilis and many Enterobacteriaceae (some species of Enterobacteriaceae produce β-lactamases that are resistant to these inhibitors). They are not active against methicillin-resistant S aureus (MRSA) or vancomycin-resistant enterococci (VRE) because their mechanism of resistance is different.
Because of their wide spectrum of activity against bacteria involved in pelvic infections (eg, anaerobes, Enterobacteriaceae, enterococci), these combinations have been successful in many circumstances.
1. Oral—Amoxicillin-clavulanate is the only oral combination agent available. It should be administered at the start of the meal to decrease gastrointestinal side effects. The dose is 500 mg amoxicillin and 125 mg clavulanate given 3 times a day or 875 mg amoxicillin and 125 mg clavulanate given every 12 hours.
2. IV—Four parenteral combinations are available that are different in their anti–gramnegative spectrum of activity. Amoxicillin-clavulanate (amoxicillin 1.0 g, clavulanate 200 mg) and ampicillin-sulbactam (ampicillin 2.0 g, sulbactam 1.0 g) are administered 3 times daily and are active against many gramnegative organisms but not P aeruginosa. Ampicillin-sulbactam is sometimes active against resistant Acinetobacter baumannii. Ticarcillin-clavulanate (ticarcillin 3.0 g, clavulanate 0.1 g) given 4 to 6 times daily and piperacillin-tazobactam (piperacillin 4.0 g, tazobactam 0.5 g) given 3 to 4 times daily are active also against P aeruginosa. Piperacillin-tazobactam is the widest spectrum anti–gramnegative agent among these 4 agents.
Adverse Effects of Penicillins
Most of the serious side effects of the penicillins are due to hypersensitivity.
A. Allergy
Allergic reactions to penicillin occur in 1–10% of patients.
1. The most frequent (80–90%) are late reactions, which occur days after initiation of therapy. The common clinical signs are morbilliform rash and fever but also eosinophilia and interstitial nephritis (gastrointestinal symptoms are not a sign of allergy).
2. Immediate reactions (within 1 hour) occur as a result of histamine release from immunoglobulin (Ig) E–sensitized mast cells. These are the most dangerous reactions but fortunately are rare (0.05%). Immediate reactions may be associated with urticaria, angioedema, laryngeal edema, bronchospasm, and anaphylaxis.
3. Accelerated reactions occur in the first 1–72 hours and, except for laryngeal edema, are not life threatening.
The penicillin-allergic patient (any agent of the penicillin group; consider both trade and generic names) should be presumed to be allergic to other penicillins (unless skin tests prove otherwise). The risk of cross-allergy to a cephalosporin is estimated to be 10% and to a carbapenem 1%. In a patient with a history of delayed mild reaction to penicillin, cephalosporins can be used. When there is a history of immediate penicillin allergy, cephalosporins should not be administered (unless a cephalosporin skin test has been performed).
In circumstances where penicillin is the clear drug of choice and where alternatives are likely to be less effective, an oral desensitization protocol may be used safely. One such indication is the treatment of a pregnant woman with syphilis.
B. Toxicity
High penicillin blood levels may lead to high spinal fluid levels. This may occur when high doses are given in the presence of renal failure and may lead to seizures. Large doses of penicillins given orally may lead to gastrointestinal upset, particularly nausea and diarrhea. These symptoms are most marked with oral amoxicillin or amoxicillin-clavulanate. Penicillins can cause pseudomembranous colitis.
2. Cephalosporins
The cephalosporins are bactericidal agents that inhibit bacterial cell wall synthesis, similar to the penicillins, and are part of the β-lactam family. They consist of a β-lactam ring attached to a dihydrothiazoline ring. Substitutions of chemical groups at various positions on the basic structure have resulted in a proliferation of drugs with varying pharmacologic properties and antimicrobial activities. In patients with impaired renal function, dosage adjustment is required for most cephalosporins (not including ceftriaxone).
Cephalosporins have been divided into 3 (or 4) major groups or “generations” based mainly on their antibacterial activity (Table 44–1):
1. First-generation cephalosporins have good activity against aerobic gram-positive organisms (streptococci, MSSA; excluding enterococci) and many community-acquired gramnegative organisms.
2. Second-generation drugs have a slightly extended spectrum against gramnegative bacteria (eg, cefuroxime) and some also against anaerobes (eg, cefoxitin).
3. Third- and fourth-generation cephalosporins can be divided into 2 subsets: one type (ie, ceftriaxone, cefotaxime) has significant anti–gram-positive activity and also anti–community-acquired gramnegative activity (not P aeruginosa); the other type (ie, ceftazidime) is less active against gram-positive organisms but has broad-spectrum anti–gramnegative bacteria activity, including P aeruginosa. The fourth-generation agent cefepime is active against both gram-positive and gramnegative organisms, including P aeruginosa.
Not all cephalosporins fit neatly into this grouping, and there are exceptions to the general characterization of the drugs in the individual generations. However, the generational classification of cephalosporins is useful for discussion purposes. In this part, only a limited number of agents that are particularly useful will be discussed.
The most common mechanisms for resistance are production of β-lactamases, alteration of the penicillin-binding proteins (PBPs), and changes in the outer membrane. Some of the β-lactamases are plasmid mediated, and others are inducible and chromosomal mediated (Enterobacter species, Citrobacter species, P aeruginosa), which lead to treatment failure and emergence of resistance in isolates initially susceptible. The resistance of S aureus to methicillin and cephalosporins is mediated by change of the PBP. Extended-spectrum β-lactamases (ESBLs) have been recognized in the early 1980s. They confer resistance to almost all cephalosporins and penicillins (even to those that seemed sensitive in vitro) but not to carbapenems. The clue for the presence of ESBL is resistance of an Enterobacteriaceae to 1 or all third-generation cephalosporins. Today, most laboratories are performing a specific test (double disc diffusion) in order to identify an ESBL producer.
Indications, Dosages, & Routes of Administration
A. First-Generation Cephalosporins
These drugs are active against gram-positive cocci, including S pneumoniae, viridans streptococci, groups A and B streptococci, and MSSA. They are also active against many community-acquired gramnegative bacteria. They are inactive against enterococci and MRSA (like all other cephalosporins) and P aeruginosa. Oral anaerobic bacteria are usually sensitive, but most bowel anaerobes (ie, B fragilis) are not.
1. Oral—In general, these agents are rapidly and thoroughly absorbed. They are useful for mild to moderate MSSA and group A streptococcal infections of the skin and soft tissue (eg, cellulitis).
1. Cephalexin and cephradine are given orally in doses of 0.25–0.5 g 4 times daily. When treating a significant S aureus infection, a dose of 1.0 g 4 times daily should be used initially.
2. Cefadroxil can be given in doses of 0.5–1 g twice daily.
2. IV—Cefazolin is the most commonly used agent.
1. It is among the drugs of choice for surgical prophylaxis in gynecologic operations and caesarean section.
2. It serves as an alternative to β-lactamase–resistant penicillins (eg, nafcillin, oxacillin) for known or suspected MSSA infections (eg, cellulitis).
3. Cefazolin is an alternative to penicillin for group B streptococcal prophylaxis in patients with a delayed mild allergic reaction to penicillin.
Cefazolin can be given in doses of 0.5–2 g every 8 hours (depends on the severity of the infection).
B. Second-Generation Cephalosporins
These drugs are active against organisms also covered by first-generation drugs, but they have an extended anti– gramnegative activity (community acquired). In addition, cefoxitin and cefotetan are active against bowel anaerobes, especially B fragilis. They have no activity against enterococci, MRSA, and P aeruginosa.
1. Oral—They are useful for respiratory tract infections and for simple cystitis. Cefuroxime axetil or cefprozil is given orally in doses of 0.25–0.5 g twice daily.
2. IV—Due to the spectrum of activity, these drugs can be used to treat obstetric and gynecologic infections. They have no advantage over first-generation cephalosporins for perioperative prophylaxis.
1. Cefuroxime is given in doses of 750 mg to 1.5 g 3 times daily. For intraabdominal polymicrobial infections, an antianaerobic drug (ie, metronidazole) should be added to cefuroxime.
2. Cefoxitin and cefotetan have enhanced anaerobic activity and hence can generally be used alone for intraabdominal infections. These drugs should not be relied on as monotherapy in patients with bacteremia due to B fragilisbecause they may not be active against 5–30% of these isolates. Cefoxitin is given in doses of 2 g every 6–8 hours, and cefotetan is given in a dose of 1–2 g twice daily.
C. Third-/Fourth-Generation Cephalosporins
Third-/fourth-generation cephalosporins have the broadest anti–gramnegative spectrum of all cephalosporins. In addition to anti–gramnegative activity, ceftriaxone, cefotaxime, and cefixime have significant anti– gram-positive activity (less than that of first and second generations), whereas ceftazidime (which is less active against gram-positives) is active also against P aeruginosa. The fourth-generation agent cefepime is active against both gram-positive and gramnegative organisms, including P aeruginosa. None of these agents has good activity against the bowel anaerobe B fragilis. Most of these drugs are active against N gonorrhoeae.
Ceftriaxone is eliminated primarily by biliary excretion, and no dosage adjustment is required in renal insufficiency. The other drugs in this generation are eliminated by the kidneys and thus require dosage adjustments in renal insufficiency.
1. Oral—Cefixime is used in a very limited fashion. In obstetric-gynecologic practice, this agent can be used to treat uncomplicated gonococcal infection by 400 mg given once (combined with doxycycline or azithromycin for cotreatment of chlamydia).
2. IV
1. Ceftriaxone is a widely used agent due to its broad spectrum of activity against community-acquired infections and its long half-life, allowing a once-daily regimen (except for CNS infections). One dose of ceftriaxone 250 mg intramuscularly is used for treating uncomplicated gonorrhea (combined with doxycycline or azithromycin for cotreatment of chlamydia). For non-CNS infections (eg, community-acquired pneumonia, abdominal/gynecologic infections, and urinary tract infections), a once-daily dose of 1 g is given.
2. Ceftazidime is, for practical purposes, exclusively an antiaerobic gramnegative agent. It has an excellent anti–P aeruginosa activity and is used to treat hospital-acquired infections where P aeruginosa is an option. Ceftazidime is given in doses of 1–2 g every 8 hours.
3. Cefepime is an extended-spectrum cephalosporin that has activity against gram-positive organisms comparable with ceftriaxone and anti–gramnegative activity comparable to ceftazidime, including an excellent activity against P aeruginosa. It is used to treat hospital-acquired infections where gram-positive and gramnegative organisms are a possibility. In many centers, it has replaced ceftazidime due to its wider spectrum. As with all cephalosporins, it is not active against MRSA, B fragilis, and enterococci. Cefepime is given in doses of 1–2 g every 12 hours.
Adverse Effects of Cephalosporins
A. Allergy
Cephalosporins, both oral and IV formulations, are very well tolerated. There is approximately a 1–3% rate of primary allergic reactions to cephalosporins (rashes, fever, and eosinophilia). Anaphylaxis is rare, occurring in less than 0.02% of recipients. The incidence of cross-allergy between cephalosporins and penicillins is estimated to be approximately 10%. In a patient with a history of delayed mild reaction to penicillin, cephalosporins are commonly used. With a history of an immediate reaction to penicillin (eg, bronchospasm, hypotension), cephalosporins should be avoided. The risk with third-generation cephalosporins in this setting is as low as 1%, similar to those without that history.
B. Toxicity
Positive Coombs reaction occurs in 3%. Other uncommon adverse effects include nephrotoxicity and hematologic effects (granulocytopenia and thrombocytopenia).
C. Superinfection
Pseudomembranous colitis caused by C difficile may occur more frequently in recipients of second- and third-generation cephalosporins.
3. Unique β-Lactam Antibiotics
Monobactams (Aztreonam)
Aztreonam is active against most gramnegative aerobes (including P aeruginosa) but not against gram-positive organisms or anaerobes. Aztreonam resembles aminoglycosides in activity without their nephrotoxicity. Because it is active against gramnegative aerobes only, combination therapy is necessary for suspected mixed infections with gram-positive organisms and/or anaerobes. The usual dose is 1–2 g IV every 6–8 hours. Although aztreonam has potentially less toxicity than gentamicin, gentamicin is much less expensive, and the majority of obstetric-gynecologic patients are at low risk for gentamicin toxicity. It is an alternative anti–gramnegative agent in patients with allergy to penicillins or cephalosporins because there is little risk of cross-sensitivity.
Carbapenems
Carbapenems have the broadest antibacterial spectrum of the β-lactam class, largely because they are so β-lactamase stable. Four carbapenems—ertapenem, doripenem, imipenem, and meropenem—are in clinical use. All have excellent activity against gram-positive cocci, excluding MRSA. Imipenem is active against Enterococcus faecalis but not the other carbapenems. The carbapenems have the broadest anti–gramnegative spectrum, including P aeruginosa (except for ertapenem), and are the drugs of choice for ESBL gramnegative producers. Carbapenems are highly active against most anaerobic species including the bowel anaerobes (eg, B fragilis). Generally speaking, doripenem, imipenem, and meropenem are therapeutically equivalent and interchangeable in most clinical situations; ertapenem is different in its lack of activity against P aeruginosa and A baumannii. During the past decade, carbapenem resistance in Enterobacteriaceae and A baumannii has emerged, mostly mediated by a β-lactamase that hydrolyses carbapenems.
All the carbapenems must be administered parenterally. They are well distributed to various body compartments and penetrate well into most tissues. The usual doses are ertapenem 1 g once daily; doripenem 500 mg every 8 hours; imipenem 500 mg every 6 hours; and meropenem 0.5–1 g every 8 hours. Dosage adjustment is required in renal insufficiency.
Because carbapenems have an unusual spectrum, they should be reserved for serious nosocomial infections for the treatment of highly resistant organisms. They should not be used as a first-line treatment for pelvic infections.
Carbapenems generally are well tolerated. The most common adverse effects of imipenem are nausea, vomiting, diarrhea, and skin rashes. All carbapenems, particularly imipenem, have been associated with seizures. The cross-allergy between carbapenems and penicillins is probably low (ie, ~1%).
4. Aminoglycosides
These agents are in most instances bactericidal. They penetrate the cell wall and membrane and inhibit protein synthesis by binding irreversibly to the 30S subunit of the bacterial ribosome. The most commonly used aminoglycosides are gentamicin, tobramycin, and amikacin.
Aminoglycosides are active against almost all gramnegative rods including P aeruginosa. Among resistant gramnegative bacilli, amikacin is the most frequently active, tobramycin is the next frequently active, and gentamicin is the least frequently active. Aminoglycosides have some activity against some gram-positive aerobes (eg, staphylococci, enterococci) but are never given alone for these bacteria but are administered together with a β-lactam agent. Gentamicin, together with penicillin or ampicillin, usually is bactericidal against enterococci, but the incidence of high-level resistance of enterococci to gentamicin is increasing. Anaerobes are not susceptible to aminoglycosides.
General Properties of Aminoglycosides
A. Pharmacokinetics
Aminoglycosides are not absorbed from the gut. The IV route is preferred, although the intramuscular route can be used if thrombocytopenia is absent. They are distributed widely in tissues and achieve reasonable concentrations in bone, synovial fluid, and peritoneal fluid. Urinary concentrations are high and exceed serum concentrations by 100 times. They have limited penetration into the CNS and probably intraabdominal abscesses. There is a variation in the volume of distribution and rate of excretion of amino-glycosides in individual patients with normal renal function. Thus, it is important to monitor serum levels in all patients every 3–4 days. Aminoglycoside excretion is entirely renal, mandating dose adjustment for renal failure. The serum half-life is 2–3 hours.
B. Dosing & Serum Level Monitoring
Aminoglycosides demonstrate a postantibiotic effect (ie, persistent suppression of bacterial growth after short antibiotic exposure) against aerobic gramnegative bacilli. Hence aminoglycosides can be administered as a single daily dose (SDD) or with the conventional multiple daily dosing regimen. The SDD approach appears to be comparable in efficacy to the traditional multiple daily dosing, is simpler to administer, and is less toxic. Some clinicians prefer to continue to use multiple daily dosing in the pregnant patient.
Creatinine clearance should be calculated before aminoglycoside administration, and doses should be corrected accordingly. In normal renal function, once-daily dose of gentamicin or tobramycin is 5 mg/kg and amikacin 15 mg/kg, given over 60 minutes. In conventional dosing, 1.7 mg/kg of gentamicin or tobramycin is given every 8 hours, and 7.5 mg/kg of amikacin is given every 12 hours.
In patients treated by SDD, trough level should be obtained before the next dose every 3–4 days. In patients treated by multiple daily dosing, peak levels (30 minutes after completion of the infusion, to ensure that enough quantity of drug was administered) and trough levels (before the next dose) should be monitored. If trough levels are elevated, the next dose needs to be reduced or the interval prolonged.
C. Clinical Use in Obstetrics & Gynecology
1. Aminoglycosides are used to treat complicated urinary tract infections (ie, pyelonephritis). Gentamicin is a primary agent unless the patient has recently received gentamicin and is possibly infected with a relatively resistant gramnegative bacterium; in this situation, amikacin should be used while awaiting culture results.
2. Aminoglycosides are used to treat intraabdominal infections (combined with antianaerobic agent).
3. Gentamicin can be used to treat P aeruginosa infections in combination with piperacillin.
4. Amikacin is used empirically to treat gramnegative bacteria in hospital-acquired infections especially in specialized areas (eg, intensive care units).
5. Aminoglycoside effectiveness is not optimal for sterilization of abscesses and in lower respiratory tract infections because of poor activity in the presence of low pH.
D. Adverse Effects
1. Hypersensitivity reactions are uncommon.
2. All aminoglycosides can cause varying degrees of nephrotoxicity, expressed by a rising blood urea nitrogen and creatinine. Changes are usually reversible with discontinuation, and rational use can prevent it. Risk factors for nephrotoxicity (and ototoxicity) include concomitant liver disease, concomitant use of other potentially nephrotoxic drugs (eg, vancomycin, nonsteroidal anti-inflammatory drugs), and prior renal disease. Prevention includes the following: use aminoglycosides for the shortest appropriate course, correct hypovolemia, avoid in the presence of risk factors, use SDD regimen, and monitor levels and serum creatinine. Serum creatinine levels should be obtained every 2–4 days.
3. Ototoxicity (eg, hearing loss, vertigo and loss of balance, or nystagmus) is frequently irreversible. Older patients are at greater risk.
5. Fluoroquinolones
All fluoroquinolones (FQs) inhibit bacterial DNA gyrase, a bacterial enzyme essential for DNA replication. They promote gyrase-mediated DNA breakage at specific sites, which leads to cell death. Quinolones are categorized based on spectrum of antimicrobial activity:
1. First-generation FQs (eg, nalidixic acid) are no longer used.
2. The second-generation agents, ciprofloxacin and ofloxacin, are very active against gramnegative organisms. Ciprofloxacin is the most potent FQ against P aeruginosa. They lack consistent activity against gram-positive cocci and anaerobes. There is increasing resistance of N gonorrhoeae against ciprofloxacin, once considered the drug of choice, and thus, FQs are no longer recommended in the United States for this indication.
3. Third- and fourth-generation FQs (gatifloxacin, levofloxacin, and moxifloxacin) have, in addition to anti–gramnegative activity (modest activity against P aeruginosa), enhanced anti–gram-positive activity, but limited activity against anaerobes. All FQs lack activity against MRSA and multidrug nosocomial gramnegative organisms.
All FQs are active against atypical respiratory pathogens (ie, Chlamydia pneumoniae, Mycoplasma pneumoniae, and Legionella pneumophila).
Their wide spectrums of activity, along with their excellent bioavailability, good tissue penetration, and safety, have made the FQs very attractive. The challenge is to limit their use to the appropriate setting where their enhanced spectrum is required, in order to reduce the risk of resistance selection and to lengthen their useful life.
Pharmacokinetics
Most FQs can be administered both orally and IV. Due to their excellent bioavailability, oral regimens provide similar serum levels as IV formulations, and thus, the oral route should be used preferentially whenever possible (ie, the patient is allowed to eat and does not have active nausea or vomiting). FQs have good tissue penetration. They are excreted mainly by the kidneys and require dose reduction in renal dysfunction (except moxifloxacin).
Clinical Use
1. Urinary tract infections (UTIs): Use of quinolones for uncomplicated community-acquired UTI should be reserved for infection due to organisms resistant to first-line treatment with trimethoprim-sulfamethoxazole (TMP-SMX). Ciprofloxacin is preferred when P aeruginosa is suspected. FQs are very useful in complicated UTI (eg, pyelonephritis).
2. Pelvic inflammatory disease as part of the antibiotic regimen. When N gonorrhoeae is a possibility, ceftriaxone is preferred over FQs due to increased resistance. In this setting, azithromycin or doxycycline should be added to treat the possibility of chlamydia.
3. intraabdominal infections: Ciprofloxacin plus metronidazole is a reasonable option (although usually, anti–P aeruginosa treatment is unnecessary) because this combination provides good anti–gramnegative and anaerobic activity.
4. Community-acquired pneumonia (CAP): The advanced-generation FQs (eg, gatifloxacin, levofloxacin, and moxifloxacin) are active against the common pathogens causing CAP (ie, S pneumoniae, Haemophilus influenzae, Moraxella catarrhalis, Mycoplasma and Chlamydia species, and L pneumophila). Consequently, they are included as a therapeutic option (“respiratory quinolones”) in the guidelines of CAP. However, in order to avoid the development of bacterial resistance, macrolides are preferred.
Dosing in Normal Renal Function
See Table 44–2.
Adverse Effects
FQs are well tolerated. The most common adverse effects are gastrointestinal symptoms, primarily nausea (5%), and CNS symptoms (1–4%) mainly in the elderly (eg, headache, dizziness, sleep disturbance, alteration of mood). The third-and fourth-generation FQs can cause QT prolongation and arrhythmia. Quinolone use may be associated with tendon rupture (eg, Achilles, shoulder, or hand) and hence should be discontinued with the first sign of tendon pain.
Quinolones should be avoided in pregnancy and nursing mothers because of the potential effect on developing cartilage.
Resistance
With widespread use of the FQs (partially due to their oral administration), the emergence of quinolone-resistant bacteria resulted (eg, P aeruginosa, nosocomial Enterobacteriaceae, N gonorrhoeae, Salmonella species).
6. Macrolides
The macrolides inhibit protein synthesis by reversibly binding to the 50S ribosomal subunit. Although generally bacteriostatic, they may be bactericidal under certain conditions or against certain microorganisms. Because their structure is different from that of β-lactams, macrolides are useful for β-lactam–allergic patients. Dose reduction is not necessary in mild to moderate renal failure (for azithromycin, no dose adjustment is needed even in severe renal failure). Resistance to one macrolide implies cross-resistance to other macrolides.
1. Erythromycin, the first macrolide, is active against many gram-positive bacteria (streptococci, MSSA, but not enterococci). Campylobacter jejuni, Bordetella pertussis, Haemophilus ducreyi, Mycoplasma, Chlamydia, and Legionella are also susceptible. It is not active against Enterobacteriaceae. It can be administered orally (250–500 mg every 6 hours) or IV (250 mg–1 g every 6 hours). Because of its common gastrointestinal side effects, the newer macrolides are often preferred.
2. Roxithromycin has a similar spectrum of activity as erythromycin. It is given solely by the oral route (150 mg twice daily) with fewer gastrointestinal side effects. It is not available in the United States.
3. The spectrum of activity of clarithromycin is similar to that of erythromycin except for enhanced activity against respiratory gramnegative bacteria (eg, H influenzae) and atypical mycobacteria. Only oral preparations are available (250–500 mg twice daily). Clarithromycin is relatively nontoxic.
4. Azithromycin is a macrolide compound with unique properties—high and sustained tissue antibiotic levels (which are much greater than the serum antibiotic levels) and prolonged tissue half-life (between 2 and 4 days), which decrease the duration of therapy needed. Its spectrum of activity is similar to that of clarithromycin. Both oral (250-mg tablet commonly distributed in a pack containing 6 tablets) and IV formulations are available. The dose and duration of treatment vary with the indication (eg, CAP: 500 mg on first day, then 250 mg once daily; chlamydia cervicitis: 1 g orally as a single dose).
Clinical Use
1. Community acquired pneumonia (CAP)
2. Sexually transmitted diseases:
a. Azithromycin (1 g orally once) is as effective as doxycycline (100 mg twice daily for 7 days) in treating chlamydia urethritis and cervicitis; cotreatment with single-dose ceftriaxone (250 mg intramuscularly) is necessary.
b. H ducreyi (chancroid) genital ulcer disease has been treated with a single dose of azithromycin 1 g orally.
c. Azithromycin may be used to treat Chlamydia trachomatis in pelvic inflammatory disease.
3. Macrolides are an alternative, in β-lactam–allergic patients, for the treatment of infections caused by groups A and B streptococci and MSSA.
4. Clarithromycin is active against Helicobacter pylori in duodenal/gastric ulcer as part of a combination therapy.
Adverse Effects
Gastrointestinal side effects (eg, nausea, vomiting, diarrhea) may occur; mostly with erythromycin. Allergic reactions are uncommon and are generally mild. Drug interactions are important (eg, elevation of warfarin blood levels) and have been reviewed elsewhere. Azithromycin has the least drug interactions of all the macrolides.
7. Tetracycline Group
The tetracyclines are bacteriostatic and act by interfering with protein synthesis at the ribosomal level (ie, 30S subunit). All have common basic chemical structures and antimicrobial activity. Doxycycline is the tetracycline of choice due to its superior pharmacokinetic properties, enhanced compliance, and lesser toxicity. Recently, a new generation (ie, glycylcyclines) that is also active against resistant bacteria has been developed. The clinical candidate of this class is tigecycline.
Antimicrobial Activity
Tetracyclines are active against a wide variety of organisms including the following:
1. Community-acquired respiratory tract pathogens (ie, S pneumoniae, H influenzae, M catarrhalis, and Mycoplasma, Legionella, and Chlamydia species)
2. C trachomatis
3. Treponema pallidum (syphilis)
4. The drug of choice for the treatment of Rickettsia and Brucella (with gentamicin)
5. They are not reliably active against N gonorrhoeae. 6. Tigecycline is also active against a variety of gram-positive and gramnegative organisms (including VRE, MRSA, and N gonorrhoeae, but not P aeruginosa).
Pharmacokinetics
The oral route is most commonly used, except for treatment of severe pelvic inflammatory disease (PID). Absorption is improved if the antibiotic is taken 1 hour before or 2 hours after meals. Doxycycline’s long half-life permits dosing every 12 to 24 hours, thus improving compliance. Tetracyclines are excreted via the urine, except for doxycycline, which is excreted in the feces (therefore, there is no need for doxycycline dose adjustment in renal failure). Oral contraceptive efficacy may be decreased with simultaneous use of tetracyclines.
Clinical Use in Obstetrics & Gynecology
Doxycycline is the drug of choice for genital infections caused by C trachomatis (eg, PID, cervicitis, lymphogranuloma venereum) in nonpregnant women.
1. In mucopurulent cervicitis, doxycycline 100 mg twice daily orally is given for 7 days (or single-dose azithromycin 1 g) in addition to a single dose of intramuscular ceftriaxone 250 mg (to treat N gonorrhoeae).
2. For PID, doxycycline 100 mg twice daily orally is given for 14 days (in combination with ceftriaxone and metronidazole). It can be administered IV with the same dosing regimen, but the oral route is preferred whenever possible.
3. Doxycycline is an alternative to penicillin for the treatment of syphilis (not in pregnant woman or in neurosyphilis).
The equivalent dose of tetracycline is 250–500 mg (orally only) every 6 hours.
Adverse Effects & Contraindications
1. Hypersensitivity reactions with fever or skin rashes are uncommon.
2. Toxic photosensitivity reactions consisting of a red rash on areas exposed to sunlight can occur. Patients should avoid intense sun exposure during doxycycline treatment.
3. Esophageal ulceration can occur with doxycycline. Patients should be told to take the drug while sitting and with adequate amount of fluids.
4. Thrombophlebitis can occur with IV doxycycline use.
5. Superinfection with Candida of the anogenital region can occur.
6. Tetracyclines should be avoided by pregnant women due to a possible hepatotoxicity to the mother and dental deformities in the child (also during lactation).
8. Clindamycin
Clindamycin inhibits protein synthesis at the ribosomal level (ie, 50S subunit) and is generally bacteriostatic. Its spectrum of activity includes gram-positive cocci (groups A and B streptococci, MSSA) and anaerobes, although there is increasing resistance of Bacteroides species to clindamycin. It is not active against gramnegative aerobes (eg, Enterobacteriaceae) or enterococci.
Pharmacokinetics
Clindamycin is well absorbed from the gastrointestinal tract, and food does not decrease its absorption. Therapeutic blood levels can be achieved by the oral or parenteral routes of administration. It penetrates most body tissues well, but not the cerebrospinal fluid. It is metabolized primarily by the liver; therefore, doses of clindamycin should be reduced in severe hepatic insufficiency. Dose adjustment is notrequired in renal failure. The oral dose is 300–450 mg every 6–8 hours. The IV dose is 600–900 mg every 8 hours.
Clinical Use in Obstetrics & Gynecology
1. In intraabdominal/pelvic infections (eg, tubo-ovarian abscess), clindamycin (which is active against gram-positive cocci and anaerobes) must be combined with an agent active against gramnegative organisms (eg, gentamicin, ciprofloxacin). Because B fragilis plays an important role in these infections and there is increasing resistance of B fragilis to clindamycin, many experts turned to metronidazole.
2. Clindamycin is an option for mixed aerobic and anaerobic perineal infection (ie, soft tissue infection) in combination with an agent active against gramnegative organisms.
3. Clindamycin is an alternative drug for the treatment of groups A and B streptococci and MSSA in patients allergic to both penicillin and cephalosporins.
4. Clindamycin is the drug of choice (in combination with high-dose penicillin) for the treatment of necrotizing fasciitis caused by group A Streptococcus.
5. Clindamycin 300 mg orally twice a day or 2% vaginal cream 5 g intravaginally at bedtime (both for 7 days) or clindamycin ovules 100 mg intravaginally at bed time for 3 days is an alternative to metronidazole for treatment of bacterial vaginosis.
Adverse Effects
1. Diarrhea is the most significant side effect of clindamycin: Antibiotic-associated diarrhea occurs in up to 20% of patients; C difficile–associated diarrhea (CDAD) occurs less frequently. When CDAD occurs, clindamycin should be discontinued if possible and oral (or IV) metronidazole or (in severe cases) oral vancomycin started.
2. Allergic reactions (eg, rash and fever) can occur.
3. Minor reversible elevations of hepatocellular enzymes are frequent.
9. Metronidazole
Metronidazole is mainly an agent for the treatment of anaerobic infections including B fragilis and Gardnerella vaginalis. It also has antiparasitic activity, including Entamoeba histolytica, Giardia lamblia, and Trichomonas vaginalis. Metronidazole inhibits DNA synthesis and is rapidly bactericidal.
Pharmacokinetics
The drug is absorbed very well and can be taken with food. Serum levels are similar after equivalent oral and IV doses. It has excellent penetration into almost all tissues. The drug is metabolized in the liver and excreted in the kidney. The usual daily dose is 500 mg orally or IV every 8 hours. Dose reduction is required in severe renal failure and significant hepatic impairment.
Clinical Use in Obstetrics & Gynecology
1. Metronidazole is the drug of choice for Bacteroides species, C difficile-associated disease, G vaginalis, E histolytica, and G lamblia.
2. It is indicated (in combination) for mixed aerobic/anaerobic infections (eg, intraabdominal and pelvic infections).
3. For Trichomonas vaginitis, the recommended dose is 2 g orally as a single dose or 500 mg twice daily for 7 days. Both sexual partners should be treated.
4. For bacterial vaginosis, the recommended regimens are 500 mg orally twice daily for 7 days or vaginal gel (1 applicator intravaginally) for 5 days. Treatment of sexual partners is not recommended unless balanitis is present. Topical treatment of bacterial vaginosis is not recommended in pregnancy.
Adverse Effects
In general, metronidazole is well tolerated.
1. Alcoholic beverages should not be consumed while taking metronidazole because of a disulfiram-like effect (ie, nausea, vomiting, and headaches).
2. Patients complain of a metallic unpleasant taste while on oral therapy.
3. With prolonged use, peripheral neuropathy may develop.
10. Vancomycin
Vancomycin is the first glycopeptides antibiotic. The primary effect of glycopeptides is inhibition of cell wall synthesis. Vancomycin is active only against gram-positive organisms. Except for enterococci, vancomycin is bactericidal; however, the addition of gentamicin increases the bactericidal activity against enterococci. The excessive use of vancomycin has contributed to the emergence of VRE and also S aureus with intermediate resistance or complete resistance to vancomycin. Prudent use of vancomycin is essential to preserve the effectiveness of this important antibiotic.
Pharmacokinetics & Dosage
Vancomycin is poorly absorbed. Very high stool concentrations after oral administration make it active against C difficile-associated disease (CDAD). The route of administration is mostly IV with diverse tissue penetration. Initial doses are based on actual weight, 15–20 mg/kg IV every 12 hours (over 1–2 hours to avoid the “red man syndrome”). Subsequent doses are based on measured trough serum levels. Target trough level in serious infections should be 15–20 μg/mL. Because vancomycin is excreted primarily by the kidneys, in renal failure, the dose must be reduced or the interval between doses increased.
Clinical Use in Obstetrics & Gynecology
Vancomycin is less active than β-lactam antibiotics (eg, nafcillin, oxacillin) for MSSA, and thus, β-lactam antibiotics should be preferred in this setting.
1. Vancomycin is the treatment of choice for MRSA infections.
2. It is an alternative for the treatment of MSSA and groups A and B Streptococcus in patients with a history of anaphylaxis to β-lactam antibiotics.
3. For severe cases of C difficile diarrhea, vancomycin 125 mg every 6 hours for 10–14 days is indicated instead of metronidazole. Discontinue other antibiotic agents if possible.
Adverse Effects
1. If vancomycin is infused too rapidly, it may cause flushing of the face, neck, or torso (“red man syndrome”), pruritus, and hypotension. To avoid this, vancomycin should be infused no more rapidly than 500 mg/h. Because this reaction is not immunologically mediated, it does not preclude further use.
2. Allergy (rashes other than red man syndrome) occurs in 5% of patients.
3. Nephrotoxicity and ototoxicity are very uncommon (occur mostly when vancomycin is used in combination with another nephrotoxic or ototoxic drug such as an aminoglycoside).
11. Linezolid
Linezolid belongs to the oxazolidinones; it interferes with bacterial protein synthesis by binding at the 50S ribosomal subunit. It is bacteriostatic and active only against gram-positive bacteria including MRSA and VRE, which are the main indications for use (although MRSA and VRE resistance to linezolid was described). Oral drug is 100% bio-available. The oral and IV doses are 600 mg every 12 hours; no dose modification is required for renal or hepatic failure. Reversible myelosuppression (ie, thrombocytopenia, anemia, and neutropenia) has been reported, most often after more than 2 weeks of therapy. Its use in obstetric and gynecologic practice currently is limited.
12. Spectinomycin
Spectinomycin structure is similar (but not identical) to the aminoglycosides. It inhibits ribosomal protein synthesis and is bactericidal. Spectinomycin is used only as an alternative agent for the treatment of N gonorrhoeae when the drugs of choice (eg, cephalosporin or azithromycin) cannot be used or in resistant strains. One injection of 2 g intramuscularly is given. No dose adjustment is required in renal failure. There are no known serious adverse reactions.
13. Trimethoprim-Sulfamethoxazole
The combination of trimethoprim and sulfamethoxazole (TMP-SMX) inhibits 2 sequential steps in the synthesis of folic acid by bacteria. Alone, each agent is bacteriostatic, but together, they are synergistic and bactericidal. The combination is available in oral and IV forms of 80 mg trimethoprim and 400 mg sulfamethoxazole. There are also double-strength (DS) tablets (160 mg trimethoprim and 800 mg sulfamethoxazole).
Antimicrobial Activity
1. TMP-SMX has a wide spectrum of activity against gram-positive cocci (including some strains of MRSA) and gramnegative bacteria.
2. It is not active against enterococci, P aeruginosa, or anaerobes.
3. It is active against L monocytogenes, Nocardia species, and Pneumocystis (carinii) jiroveci.
Pharmacokinetics
Because oral TMP-SMX is well absorbed, the oral route is usually preferred. Each component is distributed widely to most tissues. It is excreted mainly by the kidneys; therefore, dose reduction is required in renal failure. The long half-life allows twice-daily dosing.
Clinical Use in Obstetrics and Gynecology
1. TMP-SMX is used to treat acute uncomplicated UTI (cystitis) in females (in the presence of <20% resistance of local Escherichia coli). The dose is 960 mg twice a day for 3 days.
2. It is useful for susceptible pathogens in the treatment of pyelonephritis.
3. In a young woman with recurrent episodes of uncomplicated UTI (3 or more episodes per year), TMP-SMX 1 single-strength tablet orally every 24 hours long term, after eradication of infection, is a possibility. An alternative is self-administered single-dose treatment (TMP-SMX DS, 2 tablets, 320/1600 mg) at symptom onset or 1 DS tablet after coitus.
4. TMP-SMX is an alternative in L monocytogenes infection (eg, meningitis or amnionitis) in a penicillin-allergic patient.
5. It is considered the drug of choice for the treatment and prevention of P jiroveci (carinii) pneumonia.
Toxicity & Side Effects
Although skin rashes are common, this combination usually is well tolerated.
1. Mild gastrointestinal symptoms occur in 3%.
2. Skin rashes occur in 3–5%; most are benign; however, severe skin rashes (eg, exfoliative dermatitis, Stevens-Johnson syndrome) may occur. Rashes are more common in HIV patients.
3. Thrombocytopenia and neutropenia can occur. Complete blood count should be performed weekly.
4. It should be avoided in patients with glucose-6-phosphate dehydrogenase deficiency because hemolysis can be precipitated.
5. If TMP-SMX is used in patients receiving warfarin, it enhances the response to warfarin; thus, the risk of bleeding is increased.
14. Urinary Antiseptics (Nitrofurantoin, Fosfomycin)
Urinary antiseptics are agents that concentrate in the urine but do not produce therapeutic levels in the serum. Therefore, they are useful only for the treatment of lower UTIs. This feature has advantages, including reduced suppression of normal flora.
Nitrofurantoin
Nitrofurantoin is active for both gramnegative (eg, Enterobacteriaceae) and gram-positive (eg, enterococci, coagulase-negative staphylococci) bacteria causing UTI. Pseudomonas species are resistant. It is well absorbed (preferably taken with food) but reaches therapeutic concentrations only in urine. The drug is contraindicated in creatinine clearance of <40 mL/min (because the excretion to urine is reduced and the drug accumulates in the serum) and in hepatic insufficiency. The urine should not be alkalinized because the drug effect is reduced in alkaline urine.
For uncomplicated UTI, nitrofurantoin is taken for 3–7 days at 100 mg twice daily. For the prophylaxis of recurrent uncomplicated UTI, it can be given daily at bedtime or postcoital as a single 50- or 100-mg dose, but pulmonary toxicity is a risk with long-term use. Nitrofurantoin may be administered as a combination of macrocrystals/monohydrate salt (nitrofurantoin capsules, Macrobid). The usual dose of this combination is 100 mg every 12 hours.
Skin rashes occur in 1–5% of patients and are reversible. Major adverse effects include pneumonitis and polyneuropathies. Because these are more common in the elderly, Macrodantin should be administered with caution to people 60 years of age or older.
Fosfomycin
Fosfomycin is used as a single oral dose of 3 g for treatment of uncomplicated UTI in women. The drug acts on the bacterial cell wall. It is active against the common pathogens of community-acquired UTI (eg, Enterobacteriaceae, Staphylococcus saprophyticus, and enterococci). P aeruginosa are usually resistant.
It is absorbed rapidly and excreted unchanged in the urine. Bactericidal concentrations persist for 24–48 hours in the urine. It is generally well tolerated, with diarrhea occurring in 9%. It may play a useful role in settings were compliance is a problem. Its high cost compared to the 3-day effective course of TMP-SMX is a limitation.
15. Antifungal Drugs
There are 3 main groups of antifungal agents: polyenes (eg, amphotericin B), azoles (eg, fluconazole), and echinocandins (eg, caspofungin). The major agents will be discussed briefly.
Amphotericin B
Amphotericin B is active and bactericidal against most invasive fungal pathogens (eg, Candida species and molds). It is poorly absorbed; hence, systemic infections must be treated IV. The dose for seriously ill patients is 0.7–1.0 mg/kg/d, typically infused over 4 hours. Thrice-weekly regimens may be used to minimize side effects. Elimination occurs via the biliary tract. Although amphotericin B is nephrotoxic, it does not accumulate in renal failure, and dose adjustments are made to minimize toxicity.
Fever and chills during infusion are common and usually diminish after the first week of treatment. Pretreatment by acetaminophen or ibuprofen is often used to reduce these symptoms. Nephrotoxicity is the main serious drug effect that limits its use. The incidence of renal failure may be reduced by saline infusion before each dose. Renal failure is usually reversible. The FDA pregnancy risk category is B.
To overcome the adverse effect–related dosage limitation, a liposomal delivery system was developed (eg, liposomal amphotericin [AmBisome]), allowing treatment with higher doses without increased systemic toxicity.
Triazoles (Fluconazole, Voriconazole)
These fungistatic agents are effective and less toxic alternatives to amphotericin B for the treatment of many systemic fungal infections. There are also some topical preparations and vaginal suppositories (eg, clotrimazole). They act by inhibition of the biosynthesis of ergosterol in the fungal cell membrane and, as a result, inhibition of cell growth.
Fluconazole
Fluconazole is available in both IV and oral preparations. Because absorption is excellent, the daily doses for oral and IV therapy are the same. It is excreted mainly in the kidneys; hence, dose adjustment is required in renal failure. It is active against yeasts (eg, Candida species, Cryptococcus species) but not against molds. The usual daily dose is 200–400 mg orally or IV once daily. In gynecology, it is often used to treat Candida vaginitis with a single oral dose of 150 mg. Alternatively, for this indication, there are many preparations of intravaginal azoles (eg, clotrimazole, miconazole) that are administered from one dose to 7–14 days. Fluconazole is well tolerated; side effects include rash (in <5%) and nausea and vomiting.
Voriconazole
Voriconazole is active against yeasts but also against molds. It is the drug of choice for the treatment of invasive aspergillosis. Voriconazole is available in both IV and oral preparations with excellent bioavailability. A loading dose of 6 mg/kg IV every 12 hours is followed by a maintenance dose of 4 mg/kg every 12 hours. With creatinine clearance of less than 50 mL/min, switch to oral therapy (due to accumulation of IV vehicle). The oral dose is 200 mg twice daily for patients who weigh more than 40 kg. Dose adjustment is necessary in hepatic insufficiency. The most frequent side effects include reversible visual disturbances and rash, both in 20%.
Echinocandins
Caspofungin is the first echinocandin. Its mechanism of action is inhibition of glucan (ie, an integral component of the fungal cell wall) synthesis. It is used to treat invasive candidiasis and invasive aspergillosis (an alternative to voriconazole). It is only available as IV formulation. Dose adjustment is necessary for patients with liver failure but not in renal failure. Caspofungin 70 mg IV on day 1 is followed by 50 mg IV once daily for maintenance. It is remarkably not toxic. There are also new agents in this class like anidulafungin. The FDA pregnancy risk category of the echinocandins is C.
16. Antiviral Drugs (Acyclovir, Valacyclovir, Famciclovir)
Only antiherpesvirus drugs will be discussed and not antiretrovirals or anticytomegalovirus agents.
Acyclovir acts by inhibition of viral DNA polymerase and thus blocks viral DNA synthesis. It is activated by the viral thymidine kinase. The antiviral spectrum of acyclovir is limited to herpesviruses (eg, herpes simplex virus [HSV] and varicella-zoster virus [VZV]). Valacyclovir is a prodrug of acyclovir. Famciclovir is a prodrug of penciclovir with similar spectrum of activity as acyclovir. The bioavailability of oral acyclovir is low (15–20%). Valacyclovir and famciclovir are readily absorbed after oral administration (bioavailability of 54–70% and 77%, respectively) and rapidly converted to their active form (acyclovir and penciclovir, respectively). Dose adjustment is required for these drugs in renal failure.
Clinical Use in Obstetrics & Gynecology
Acyclovir is the agent of choice for HSV and VZV and can be administered both orally and IV. Valacyclovir and famciclovir are comparably effective as oral alternatives and offer more convenient dosing regimen.
1. Acyclovir is effective in primary genital HSV infections. In outpatients, oral acyclovir (400 mg every 8 hours for 7–10 days) is used. Alternatively, oral valacyclovir or famciclovir (1,000 mg twice a day and 250 mg 3 times a day, respectively, both for 7–10 days) may be used. Topical acyclovir is less effective, and its use is discouraged. IV acyclovir (5 mg/kg every 8 hours for 5–7 days) can be used in hospitalized patients with severe primary HSV infections.
2. In recurrent genital HSV infections, patient-initiated treatment during the prodrome or at the first lesion appearance is associated with 2-day reduction in duration of symptoms. The options are acyclovir 800 mg 3 times a day for 2 days; valacyclovir 500 mg twice a day for 3 days; and famciclovir 1000 mg twice a day for 1 day.
3. Suppressive therapy reduces the frequency of genital HSV recurrences by 70–80% among patients who suffer frequent recurrences (ie, >6 per year). Effective regimens include acyclovir 400 mg twice a day, famciclovir 250 mg twice a day, and valacyclovir 1 g once daily.
4. Acyclovir treatment late in pregnancy reduces the frequency of caesarean sections among women who have recurrent genital herpes by diminishing the frequency of recurrences at term; the effect of antiviral therapy late in pregnancy on the incidence of neonatal herpes is not known. No data support the use of antiviral therapy among HSV-seropositive women without a history of genital herpes.
5. High-dose acyclovir is effective treatment for chickenpox and VZV in older adults.
6. For cytomegalovirus (CMV) infections, these agents are ineffective.
Toxicity & Side Effects
Oral agents (ie, acyclovir, valacyclovir, and famciclovir) are generally well tolerated. IV acyclovir may cause phlebitis and, in 5%, nephrotoxicity. Adequate prehydration may prevent it.
Available data do not indicate an increased risk for major birth defects in women treated with acyclovir during the first trimester, compared with the general population. However, data regarding prenatal exposure to valacyclovir and famciclovir are too limited to provide useful information on pregnancy outcomes.
Babinchak T, Ellis-Grosse E, Dartois N, Rose GM, Loh E; Tigecycline 301 Study Group; Tigecycline 306 Study Group. The efficacy and safety of tigecycline for the treatment of complicated intraabdominal infections: analysis of pooled clinical trial data. Clin Infect Dis 2005;41(Suppl 5):S354–S367. PMID: 16080073.
Betts RF, Chapman SW, Penn RL, eds. A Practical Approach to Infectious Diseases. 5th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
Centers for Disease Control and Prevention. Sexually transmitted diseases treatment guidelines. MMWR Recomm Rep 2010;59(RR-12): 1–110. PMID: 21160495.
Centers for Disease Control and Prevention. Update to CDC’s sexually transmitted diseases treatment guidelines, 2006: fluoroquinolones no longer recommended for treatment of gonococcal infections. MMWR Morb Mortal Wkly Rep 2007;56:332–336. PMID: 17431378.
Gilbert DN, Mollering RC, Eliopolus GM, Chambers HF, Saag MS. The Sanford Guide to Antimicrobial Therapy. 40th ed. Sperryville, VA: Antimicrobial Therapy, Inc.; 2010.
Mandell GL, Bennett JE, Dolin R, eds. Principle and Practice of Infectious Diseases. 7th ed. New York, NY: Churchill Livingstone; 2010.
Romano A, Viola M, Guéant-Rodriguez RM, Gaeta F, Pettinato R, Guéant JL. Imipenem in patients with immediate hypersensitivity to penicillins. N Engl J Med 2006;354:2835–2837. PMID: 16807429.
Society of Obstetricians and Gynaecologists of Canada. Screening and management of bacterial vaginosis in pregnancy. Available at: http://www.sogc.org/guidelines/documents/gui211CPG0808.pdf. Accessed August 2008.
ANTIMICROBIAL PROPHYLAXIS IN SURGERY
Indications
Antimicrobial prophylaxis reduces the incidence of wound infection after certain operations. It should be used only for procedures with high infection rates such as clean contaminated procedures (ie, involving mucosal surfaces such as the vagina), operations involving implantation of prosthetic material (eg, general surgeries where meshes are used, artificial joints), and procedures where the consequences of infection are serious.
Organisms Involved
For most gynecologic surgical site infections, the source of pathogens is the endogenous flora of the patient’s skin or vagina. Gynecologic surgical procedures, such as laparotomies or laparoscopies, do not breach surfaces colonized with bacteria from the vagina, and infections after these procedures more commonly result from skin bacteria only. The major pathogen in these clean surgeries is S aureus. Procedures breaching the endocervix, such as hysterosalpingogram, intrauterine device (IUD) insertion, endometrial biopsy, chromotubation, and dilation and curettage, may seed the endometrium and the fallopian tubes with microorganisms found in the upper vagina and endocervix (eg, Enterobacteriaceae, group B Streptococcus).
Timing
To be optimally effective, antibiotics must be given so that good tissue levels are present at the time of incision and for the duration of the operation. To achieve that goal, antibiotics should be started preferably less than 1 hour before incision for most agents (except for vancomycin and quinolones because they are infused over an hour). A convenient time to administer antibiotic prophylaxis is just before induction of anesthesia.
However, timing of antibiotic prophylaxis has historically been different with regard to caesarean delivery; the prophylactic antibiotic was given only after clamping of the umbilical cord. The rationale for this was to avoid exposure of the neonate to the antibiotics and to prevent any masking of newborn culture results in cases of a suspected newborn infection. In 2007, Sullivan and colleagues reported results from a randomized, prospective, double-blind trial comparing the administration of cefazolin an hour prior to skin incision with administration after cord clamping. The group receiving the earlier antibiotics had less overall infectious morbidity and less endometritis, with no associated increase in neonatal adverse effects. Other similar studies have found comparable results. In September 2010, this evidence prompted the American College of Obstetricians and Gynecologists (ACOG) to issue a Committee Opinion advocating that all patients undergoing caesarean delivery be given appropriate prophylactic antibiotics prior to skin incision.
Duration
In most instances, a single dose is sufficient. For procedures lasting more than 2 half-lives of prophylactic agent, intraoperative supplementary dose(s) may be required. In most cases, prophylaxis is not extended beyond 24 hours.
Antibiotic Prophylaxis Choices in Selected Procedures
1. Hysterectomy, either vaginal or abdominal: IV cefazolin 1–2 g 30 minutes before incision.
2. Cesarean section: IV cefazolin 1–2 g 30 minutes before incision.
3. Surgical abortion: first trimester, doxycycline 100 mg before procedure and 200 mg after; second trimester, IV cefazolin 1–2 g before procedure.
4. No antibiotic prophylaxis is indicated during IUD insertion, hysteroscopy, and endometrial biopsy.
An alternative in patients with immediate hypersensitivity to penicillins is clindamycin plus gentamicin. MRSA should be considered when it is prevalent in the community or in institutions where the rate of postoperative MRSA infection is high; in these circumstances, vancomycin should be considered.
ACOG Committee Opinion No. 465: antimicrobial prophylaxis for cesarean delivery: timing of administration. Obstet Gynecol 2010;116:791–792. PMID: 20733474.
ACOG Practice Bulletin No. 104: antibiotic prophylaxis for gynecologic procedures. Obstet Gynecol 2009;113:1180–1189. PMID: 19384919.
Camann W, Tuomala R. Antibiotic prophylaxis for cesarean delivery: always before skin incision! Int J Obstet Anesth 2011;20:1–2. PMID: 21126866.