KAREN L. ROOS, ALLAN R. TUNKEL, DIEDERIK VAN DE BEEK, AND W. MICHAEL SCHELD
The meningitis syndrome has been recognized for centuries. Hippocrates realized the important intracranial consequences of otitic infection, and clear clinical descriptions of meningitis have been found dating from the sixteenth century. However, the syndrome of epidemic meningitis with a purpuric rash was not identified until 1805, when Viesseux wrote about an epidemic of “malignant purpuric fever” surrounding Geneva, Switzerland, the first clinical description of meningococcemia with meningitis. The pathologic hallmark of the condition, inflammation within the subarachnoid space (SAS), was described in autopsy reports in the French literature the following year. Danielson and Mann (1) recorded the first observations of meningococcemia and meningitis in the United States in 1806. Many of these early descriptions were collated in a treatise by Elisha North of Connecticut in 1811 and summarized in references 1 and 2. Then, as now, the disease could present dramatically in a fulminant form. The epidemic nature of meningococcemia was frightening to physicians and lay persons alike. For example, Dr. Samuel Woodward, of Torrington, Connecticut, wrote the following in The American Mercury, Hartford, in 1807:
The violent symptoms were great lassitude, with universal pains in the muscles, chills; heats, if any, were of short duration; unusual prostration of strength; delirium, with severe pain in the head; vomiting, with indescribable anxiety of stomach; eyes red and watery, and rolled up, and the head drawn back with spasm; pulse quick, weak, and irregular; petechiae and vibices all over the body, and a cadaverous countenance and smell; death often closed the scene in ten or fifteen hours after the first attack . . . the body, near the fatal period, and soon after, became as spotted as an adder. . . .
Similarly, the following was written by the Reverend Festus Foster of Petersham, Massachusetts, as a letter to the editor of The Worcester Spy, dated March 6, 1810:
I hasten to give you a sketch of the spotted fever in this place. It made its first appearance about the beginning of January last; but the instances were few and distant from each other, until last week. Although it had proved fatal in most instances, seven only had died belonging to this town, previous to the 25th of February. Since that time the disorder has come upon us like a flood of mighty waters. We have buried eight persons within the last eight days. About twelve or fifteen new cases appeared on Thursday last; many of them very sudden and violent. This was the most melancholy and alarming day ever witnessed in this place. Seven or eight physicians were continually engaged in the neighborhood north of the meeting house, and I believe not one half hour passed in the forenoon without presenting a new case. Pale fear and extreme anxiety were visible in every countenance. . . .
It is inconceivable that this fulminant form of meningococcemia had been previously unrecognized, especially given the excellent clinical descriptions of rashes in the literature from the period. One must speculate that the virulence of meningococci for humans changed in the early nineteenth century.
Meningococci were first isolated in 1887 by Anton Weichselbaum in Vienna; they were obtained from the cerebrospinal fluid (CSF) of six patients with meningitis and were initially named Diplococcus intracellularis meningitidis. All three of the major meningeal pathogens (Neisseria meningitidis, Streptococcus pneumoniae, and Haemophilus influenzae) were isolated and described in the last two decades of the nineteenth century. Quincke introduced lumbar puncture (LP) in 1891, and the major CSF alterations associated with meningitis (pleocytosis, hypoglycorrhachia, and elevated protein concentration) were well recognized by the turn of the century.
The treatment of bacterial meningitis in the early years of this century was dominated by methods for removal of large volumes of CSF and/or direct instillation of substances (e.g., dyes, enzymes) into the SAS. After early leads from European investigators, the first truly significant therapeutic modality for this disorder on a large scale, the systemic and intrathecal administration of antimeningococcal antisera raised in horses, was documented by Simon Flexner in 1913. Though toxic, antisera therapy reduced the mortality of meningococcal meningitis (from approximately 80% to 30%) during World War I and for decades thereafter. The principles of serum therapy were applied by Dr. Hattie Alexander and others to meningitis caused by H. influenzae in the 1940s.
The approach to the patient with bacterial meningitis was profoundly altered by the advent of antimicrobial therapy. The first successful account of the therapy of meningococcal meningitis with an antimicrobial agent in this country was published by Schwentker et al. (3) in 1937; nine patients survived after receiving subcutaneous and intraspinal injections of sulfanilamide, and the sole death occurred after eradication of the organism from CSF. The introduction of penicillin and other antimicrobial agents (e.g., streptomycin and chloramphenicol) ushered in the modern antimicrobial era, likened to an industrial revolution (4). These developments led to the widespread belief that serious bacterial infections were “solved.” Despite the introduction of myriad new antimicrobial agents and the development of newer diagnostic techniques, the mortality from meningitis caused by the three major bacterial pathogens has not changed appreciably in the last four decades. However, the use of the third-generation cephalosporins during the 1980s for therapy of gram-negative aerobic bacillary meningitis has substantially reduced the mortality of this condition. Recent years have revealed an explosion of new knowledge on the pathogenesis and pathophysiology of bacterial meningitis (see later discussion), with attendant ramifications on the use of adjunctive therapy (e.g., corticosteroids, nonsteroidal antiinflammatory agents, and monoclonal antibodies) for this disease.
EPIDEMIOLOGY
During 2003 to 2007, approximately 4,100 cases of bacterial meningitis occurred annually in the United States (5), but this disease is much more common in developing countries (see later discussion). In addition, the relative frequency with which each of the various bacterial species causes meningitis is age related. Gram-negative bacilli (principally Escherichia coli K1), group B streptococci, other enteric bacilli, and much less commonly, Pseudomonasspecies are the major causative agents during the neonatal period. Meningitis in children and adults is primarily caused by meningococci and pneumococci, although disease caused by aerobic gram-negative bacilli is increasing in frequency, especially in the elderly. N. meningitidis is the only major cause of epidemics of bacterial meningitis.
The development of meningitis depends on a complex array of factors, including virulence properties of the organisms, the carrier state, and the host’s humoral immune response. These factors differ among the major pathogens, for which reason epidemiology, carrier state, and host immunity are considered separately for each of the three major etiologic agents in this section.
The classification of the pathogens, putative virulence factors, and the clinical settings associated with less prevalent agents are discussed later (see the section “Etiology,” later in this chapter).
Haemophilus influenzae
H. influenzae type b (Hib) was previously the leading cause of bacterial meningitis in the United States but now accounts for only approximately 7% of cases (5). Meningitis caused by Hib displays an interesting bimodal seasonal pattern in northern Europe and the northern United States, with peaks in June and September through November (6,7).
The overall annual incidence of serious Hib disease differs between geographic locales and among populations. Significant interannual variations in the incidence of meningitis caused by Hib have also been reported within a single geographic area over time (8). This is an important consideration in assessing the efficacy of Hib vaccines. Before the advent of conjugate vaccines (see later discussion), the overall rate of Hib meningitis in the United States was approximately 60 per 100,000 children younger than 5 years of age (9), greater than the figures from other countries in northern Europe. These incidence rates differ markedly among age-groups (see later discussion) and in children younger than 6 years. In a 3-year nationwide prospective study on pediatric meningitis in Israel, the incidence of Hib meningitis during the first year of life was 67.1 per 100,000, and in children younger than 5 years of age, it was 18.5 per 100,000 (10). Some studies report a higher incidence in nonwhites (7,11). For example, the incidence rate for Hib meningitis for the total population of Washington State was 2.2, 3.4, and 13.5 per 100,000 for whites, blacks, and Native Americans, respectively (11). In contrast, others have found no differences between rates for blacks and whites younger than 1 year of age (12).
Before the availability of the current Hib vaccines, 1 in every 200 children developed invasive Hib disease by 5 years of age. Meningitis caused by Hib in the first 2 months of life is rare, presumably because of placental transfer of protective concentrations of maternal bactericidal antibody. Most cases occur between 4 months and 2 years of age. The highest rate of illness occurs in children 6 to 17 months of age; children older than 2 years of age have a lower incidence (6,8,9,11–17). Approximately 80% of cases develop in unvaccinated children younger than 2 years of age in this country, but this proportion varies by geographic locale. The proportion of cases is approximately 20% lower in this age-group in northern European countries. These differences in age distribution may directly influence the efficacy of candidate Hib vaccines. Nontypeable strains of H. influenzae are commonly carried in the nasopharynx of asymptomatic individuals. Carriage of encapsulated strains (usually type b) is rare: rates are less than 5% in children and less than 1% in adults. However, the carriage rates among household contacts of an index case are much higher: 20% to 25% overall and more than 50% among children younger than 5 years of age. This varies with the clinical disease. For example, carriage rates among children 5 years old are 20% and 55% for household contacts of epiglottitis and meningitis cases, respectively. This prevalence is reflected in the increased risk for serious Hib disease among household contacts of the index case, which is age dependent: 4% for children 2 years of age, 2% for children 2 to 3 years old, and 0.1% for children 4 to 5 years of age. The risk for Hib infection among household contacts is approximately 600-fold greater than the age-adjusted risk for the population at large and is the basis for chemoprophylactic strategies. Carriage is usually asymptomatic and may occur despite the presence of circulating anticapsular antibodies or effective eradication of meningitis following antibiotic therapy. The Hib carrier state may persist for weeks to months.
The occurrence of Hib meningitis is inversely proportional to the age-related concentration of type-specific anticapsular antibodies (18). Finnish studies, measuring anti–polyribosylribitol phosphate (anti-PRP) antibodies by radioimmunoassay, confirm the age-related susceptibility to systemic Hib disease: 90% of children (3 to 12 months old) had concentrations of less than 150 ng/mL, whereas adults had higher concentrations (19). These anti-PRP antibodies, in concert with complement, are (a) opsonic and bactericidal against Hib in vitro and (b) protective in vivo. Antibodies to Hib outer membrane proteins (Omps) also appear protective, but only against the homologous subtype. The anti-PRP response to infection is age related, being poor in infants; older children and adults develop higher titers. It is also dependent on PRP concentrations and clearance rates. PRP antigenemia may persist for weeks in younger children with Hib meningitis, delaying the antibody response. Approximately 80% of children with Hib meningitis develop an antibody response within 3 months. The antibody response is blunted in children with agammaglobulinemia or immunoglobulin G2 (IgG2) subclass deficiency, as well as in all children younger than 24 months of age receiving the Hib PRP vaccine, because this polysaccharide is a poor immunogen in this age-group (9). The age-related acquisition of protective anticapsular antibodies is too rapid to be accounted for by the low incidence of carriage or disease caused by Hib alone. Cross-reacting antigens from E. coli and other bacteria within the gut are postulated to serve as the primary immunogen.
Acquisition of Hib (nasopharyngeal carriage) and the concentration of circulating anticapsular antibody are the two main factors that determine risk for disease in most patients. Some of these risk factors have been alluded to (e.g., immunoglobulin or complement deficiency, household contacts of an index case). Other conditions that also may be important in increasing susceptibility to invasive Hib infection include sickle cell anemia, postsplenectomy states, CSF fistulas, chronic pulmonary infections, alcoholism, and probably lower socioeconomic status (e.g., Eskimos and American Indians). Day care outside the home and the presence of young siblings increases the risk for invasive disease, whereas breast-feeding is protective. The risk is highest for children younger than 2 years of age in day care but is not apparent for older children, is equally high for those in a family day care setting or those in a professional day care center (mean group size, 4 and 12 children, respectively), and is significantly higher (p < .02) within the first month of attendance, especially among younger children (20). The risk ratio doubles with each additional sibling younger than 7 years of age and is higher in twins (20). New associations have also suggested that the child’s previous state of health, especially a history of otitis media and/or previous hospitalization, increases the risk for serious Hib disease. Otitis media remains significant, especially for younger children, even after controlling for confounding variables such as day care attendance (21). Pharyngitis and otitis media are associated with Hib meningitis in approximately one half and two thirds of the cases, respectively.
Because of the bimodal seasonal occurrence of Hib meningitis, at least in northern latitudes, it has been suggested that preceding viral upper respiratory tract infections predispose to the acquisition of Hib and subsequent disease, but this issue remains controversial (22). Recent studies comparing the attack rates of meningitis between two ethnic groups living together in one geographic area (Jews and Bedouins in the Negev region of Israel) suggested that community-acquired bacterial meningitis is associated more strongly with the type of morbidity most prevalent in the region at any given time (e.g., upper respiratory tract or gastrointestinal infections) rather than any specific type of infection (23).
There has been a profound reduction (from 76% to 90%) in the incidence of invasive infections caused by Hib in the United States, specifically in young children (24–27), attributed, in part, to the widespread use of conjugate vaccines against Hib that were licensed for routine use in all children beginning at 2 months of age. In a study of Hib disease rates in Los Angeles County, California, Hib disease was nearly eradicated in a fully immunized population, demonstrating the importance of promotion of widespread use of these conjugate vaccines (28). Similar results have been observed outside the United States. In Finland, there has been a marked decrease in the number of cases of Hib meningitis from a peak of 43 cases per 100,000 population in the late 1970s to no cases in 1991 in the greater Helsinki area (29). Similarly, during a prospective study of bacterial meningitis in the northeast Thames region of the United Kingdom, there was an 87% decline in the number of cases of Hib meningitis in 1993 (7 cases) compared with 1991 and 1992 (50 cases) (30). Widespread usage beginning at 2 months of age has nearly eliminated serious invasive Hib disease in children in North America, Western Europe, Japan, and in many areas of Latin America. Unfortunately, for reasons of cost, Hib conjugate vaccines are used sparingly in many resource-limited settings, and Hib meningitis remains common in children in these areas. For example, Hib still accounted for 32% of cases in children younger than 5 years of age in Bulgaria in the mid-1990s (31). In addition, there was a report of emergence of cases in Nottingham, United Kingdom of invasive Hib disease in those previously vaccinated against the disease (32). Several explanations were put forth to explain this increase, most notably that the vaccination schedule in the United Kingdom was at 2, 3, and 4 months of age, and no booster dose was given (33). In areas in the developing world, declining rates of Hib meningitis have been reported since introduction of Hib conjugate vaccines, with effectiveness ranging from 88% to 94% (34–37).
The occurrence of meningitis caused by Hib in individuals older than 6 years of age should prompt efforts to exclude common accompanying conditions, such as otitis media, sinusitis, epiglottitis, CSF leaks, an immunodeficiency state, splenectomy or asplenic states, other parameningeal foci of infection, diabetes mellitus, and alcoholism (38,39). Although the incidence in children has dramatically declined, the incidence of invasive H. influenzae disease in adults is more complex. In one population-based study of the epidemiology and outcome caused by typeable and nontypeable H. influenzae among adults in Utah during 1998 to 2008, there was an increase in incidence over the study period from 0.14 per 100,000 person-years in 1998 to 1.61 per 100,000 person-years in 2008 (40); patients older than 65 years of age accounted for 51% of the cases and 67% of the deaths.
Neisseria meningitidis
Meningococcal infections continue to pose serious problems on all continents. They are influenced by multiple factors, including geography, season, climate, meningococcal serogroup, and population demographics (2,41). Although worldwide in distribution, the incidence of epidemic meningococcal meningitis and/or meningococcemia exhibits high geographic variability. The meningitis belt of sub-Saharan Africa represents a classic endemic area (Fig. 24.1). Although meningococcal infections were not recorded in the area until the 1880s, large outbreaks still occur regularly. Although the precise effects of climatic conditions on the incidence of meningitis are unresolved, the belt lies within the 300- and 1,100-mm rainfall lines. At least 390,000 cases with 53,000 deaths occurred within the seven countries of the belt in the 10-year period 1951 through 1960. The average annual incidence since 1950 has been estimated as approximately 70 cases per 100,000 population by the World Health Organization (WHO) (41). In 1988, more than 57,000 cases of meningococcal disease were reported from the African continent; in 1989, the number of cases increased to more than 70,000, with more than 40,000 reported from Ethiopia (42). Because reporting is often delayed and may be incomplete, this figure likely underestimates the actual number of cases. More than 180,000 cases were reported by the WHO for the regions in 1996, the highest yearly total in more than 40 years. Similarly, within a 1-month period (October 1974), 4,865 patients with meningococcal meningitis were treated at the major infectious diseases hospital in Sao Paulo, Brazil. The overall mean annual incidence of meningitis caused by N. meningitidis reached 370 cases per 100,000 population in the greater Sao Paulo area in that year. The attack rate was 517 cases per 100,000 inhabitants during a group C epidemic in Upper Volta (now Burkina Faso) in 1979, and recent studies documented an attack rate of 400 to 450 per 100,000 children up to 8 years of age in the Faeroe Islands (43). In contrast, the mean annual attack rate in the United States (1975 to 1980) was approximately 1.2 per 100,000 persons but was, again, age dependent: 17.1 per 100,000 in children younger than 1 year of age, 5.2 per 100,000 in 1- to 4-year-old children, and 0.3 per 100,000 among adults. Approximately 2,500 to 3,000 cases of meningococcal infection were reported annually in the United States between 1984 and 2003. In a multistate surveillance project conducted between 1989 and 1991 in the United States, the average annual incidence of meningococcal disease was 1.1 per 100,000; 46% of cases occurred in children 2 years of age, and the highest age-specific incidence was in children younger than 4 months of age (44). A similar figure of approximately 2 per 100,000 population was reported from Finland from 1976 through 1980.

The peak incidence of meningococcal meningitis in industrialized nations occurs in winter through early spring in both epidemic and endemic periods. Similar seasonal trends may also occur in tropical areas. For example, both the group C and group A meningococcal epidemics in the Sao Paulo area from 1971 to 1974 began in May or June, the point of transition from the rainy to the dry season. African outbreaks occur during the dry season from December to June. Annual outbreaks in the sub-Saharan meningitis belt tend to peak in late April and early May, when the dry desert wind (harmattan) has ceased and temperatures are high throughout the day, and terminate abruptly with the onset of the rainy season (41). Low humidity may alter the pharyngeal mucosal barrier, thereby predisposing it to infection. Although the introduction of a new virulent strain into a susceptible population may contribute to the epidemics, many other factors—including crowding, the presence of other respiratory pathogens, poor hygiene, and poorly defined environmental features—contribute to the initiation of a meningococcal epidemic (45).
Although meningococcal meningitis may be more prevalent in men and boys, the reports are often skewed by the inclusion of military recruits and chronic alcoholics. Meningitis caused by N. meningitidis is primarily a disease of children and young adults: fewer than 10% of cases occur in patients older than 45 years of age. In the United States and Finland, children younger than 5 years of age account for approximately 55% of cases during nonepidemic conditions, whereas in Zaria, Nigeria, the peak incidence occurs in 5- to 9-year-olds (43). Major epidemics are heralded by a “shift to the right” toward older age-groups (i.e., adolescents instead of children), a predictive feature of epidemics in the meningitis belt identified by prospective surveillance. Although meningococcal meningitis is unusual in adults, 33% of sporadic meningococcal disease occurred in adults in a 5-year population-based study in Atlanta (46). Underlying conditions such as congestive heart failure, multiple myeloma, and infection with human immunodeficiency virus (HIV) were prevalent in adults older than 24 years of age with meningococcal infection but unusual in the 18- to 24-year-old group.
Large-scale epidemics caused by serogroup A meningococci have occurred at 20- to 30-year intervals throughout the world in the last and this century and continue at approximately 8- to 12-year intervals in the African meningitis belt, where approximately 1% of the population is affected. These strains infrequently cause disease in the United States, but serious outbreaks caused by serogroups A, B, or C continue in many areas (Table 24.1). A predominant serogroup circulating in the African meningitis belt since 2000 is W135, an unusual occurrence (see later discussion). Serogroups B and C now cause most focal outbreaks and endemic disease in many areas (see the section “Etiology” later in this chapter).

N. meningitidis disease is exclusive to humans. No intermediate host, reservoir, or animal-to-human transmission has been proved. The nasopharynx is the natural reservoir for meningococci; transmission is facilitated by airborne droplets or close contact. The definition of “close contact” has not been clearly elucidated but generally refers to persons who have had prolonged (8 hours or longer) contact while in close proximity (3 feet or less to the patient) or direct exposure to the patient’s oral secretions (through kissing, mouth-to-mouth resuscitation, endotracheal intubation, or endotracheal tube management) within 1 week before the onset of the patient’s symptoms until 24 hours after initiation of appropriate antimicrobial therapy (47). Meningococcal colonization may result in an asymptomatic carrier state (which is most common) or in endemic, hyperendemic (e.g., a meningitis belt between epidemics at 10 to 50 cases per 100,000 per year), or epidemic disease. Although there is no clear relationship between carriage rate and overt disease, the development of the carrier state and the host immune response are, as for Hib meningitis, important variables in the epidemiology of meningococcal infections.
Approximately 6% of the population develops nasopharyngeal colonization with N. meningitidis yearly. Nasopharyngeal carriage rates vary with age and the population under study. The carriage rate is markedly influenced by age: 0.5% to 1% in children 3 to 48 months old, approximately 5% in adolescents 14 to 17 years old, and 20% to 40% in young adults. Analogous to Hib, carriage rates are higher in close contacts of an index case. Carriage rates of meningococci of approximately 40% have been documented in close family contacts of meningococcal cases. In closed populations (e.g., military barracks during early training), carriage rates of 20% to 60% are commonplace and may reach 90% during epidemics of meningococcal disease. Nasopharyngeal carriage usually persists for weeks to months, similar to Hib carriage. Spread of meningococcal disease is usually carrier mediated (i.e., not spread by case-to-case contacts) and largely accounts for the increased risk for disease (500- to 1,000-fold above the background endemic rate) in household contacts of an index case. The organism is often introduced into the home environment by an adult family member, with subsequent transmission to others; infants are colonized last of all. Although uncharacterized host and environmental factors contribute to containment of infection to the nasopharynx (thereby preventing disseminated disease), host immunity also plays an important role. Nevertheless, those individuals most recently colonized with meningococci appear to be at the greatest risk for invasive disease. Secondary systemic meningococcal disease often develops within 5 days of recognition of the index case, with 70% to 80% of secondary cases occurring within 14 days of the primary case.
As with Hib disease, the age-specific incidence of meningococcal infection is inversely proportional to the presence of serum bactericidal antibodies against serogroups A, B, and C. More than 50% of infants possess bactericidal antibody at birth as a result of transplacental transfer. The specifics of the antibody response may be responsible for the occurrence of meningococcal meningitis during the neonatal period. The group B capsular polysaccharide is a polymer consisting of two to eight linked sialic acid residues but is immunologically identical to the oligosaccharides of several human glycoproteins, including brain gangliosides. Immunologic tolerance thus exists in this age-group; although IgM antibody can be induced, the usual switch to IgG antibody production does not occur (43). Because IgM does not cross the placenta, IgG antibody to the serogroup B polysaccharide is lacking in neonates, contributing to the occurrence of group B meningococcal disease in this patient population. In addition, group B meningococcal capsular antigen is identical to the capsular polysaccharides of E. coli K1 and certain types of group B streptococci, major causes of neonatal sepsis and meningitis. The prevalence of antimeningococcal capsular antibodies is lowest between 6 and 24 months of age, increasing to approximately 70% by early adulthood. The inverse link between occurrence of invasive disease and bactericidal antibody was first documented during an outbreak of serogroup C meningococcal meningitis among army recruits in 1968 (48). In this study, the sera of only 5.6% of the recruits who developed meningococcal disease had bactericidal activity before the onset of illness, compared with 82.2% of control sera. Notably, 5 (38.5%) of 13 recruits without bactericidal antibody developed systemic illness after colonization with the group C strain.
Although recovery from invasive meningococcal disease generally confers lifelong immunity against the homologous serogroup, this is not the major immunizing process. Nasopharyngeal colonization, particularly with serogroups B, C, or Y, may elicit the development of bactericidal activity, primarily directed against the colonizing strain but also against heterologous organisms within 5 to 12 days of acquisition. Colonization with nongroupable meningococci or Neisseria lactamica may elicit protective immunity, especially in young children. N. lactamica is virtually nonpathogenic, but nasopharyngeal carriage rates of this organism are highest (4% to 20%) in children between 3 months and 12 years of age, whereas the age-adjusted carriage rates for N. meningitidis are only 0.5% to 2%. As with Hib, the carriage rates of pathogenic meningococci are too low in children to account for antibody formation, and the importance of other cross-reacting organisms has also been proposed: Bacillus pumilus for group A polysaccharide and E. coli for group C organisms. Paradoxically, an exuberant IgA response to meningococci may actually enhance the development of systemic disease. When a large proportion of induced anticapsular antibodies are of the IgA class, complement-mediated immune bacteriolysis by IgM is blocked, thus enhancing susceptibility to invasive disease. This peculiar immunologic phenomenon is transient, lasting only a few days following asymptomatic nasopharyngeal acquisition of N. meningitidis or closely related organisms.
In addition to antibody, an intact complement system is also a component of host defense against invasive meningococcal disease. Studies of extreme phenotypes have identified genetic correlates of increased susceptibility in the complement system (49). Recurrent or chronic neisserial infections have been associated with rare isolated deficiencies of late complement components (C5, C6, C7, or C8, and perhaps C9), occasionally in concert with failure to produce antimeningococcal antibodies. Recurrent episodes of neisserial infections may occur in these patients without an increase in susceptibility to other pathogens (50), and screening for complement defects is useful in patients with these syndromes. Complement deficiency also appears to predispose to meningitis caused by nongroupable meningococci and Neisseria-related bacteria (i.e., Moraxella and Acinetobacter species) (51). In addition, complement deficiency or depletion of early components (C1, C3, or C4) because of an underlying disease such as nephrotic syndrome, hepatic failure, systemic lupus erythematosus, presence of C3 nephritic factor, or multiple myeloma may predispose to the first episode of invasive meningococcal disease. An association between homozygous C4b deficiency (present in approximately 3% of the population) and the development of childhood meningitis was demonstrated. Up to 30% of patients with invasive meningococcal syndromes display decreased complement function. Properdin deficiency, or dysfunction with normal concentrations, also predisposes to meningococcal infections; this defect is reversible by vaccination (52). The mortality associated with meningococcal meningitis in patients with complement component deficiencies is actually lower than the general population (3% versus 19%, respectively). However, since invasive meningococcal infection occurs in 39% of patients with deficiency of terminal complement components and about 6% of those with properdin deficiency, a screening test (e.g., CH50) has been suggested (53) for all individuals with serious meningococcal disease, with further consideration for determination of specific complement and/or properdin concentrations if documented. Asplenic states increase the risk for serious infections by encapsulated organisms, especially Hib or S. pneumoniae but also meningococci.
In genetic case–control studies, invasive meningococcal disease was associated with the IL1RA + 2018T → C polymorphism, with susceptibility increased in homozygous IL1RA + 2018C carriers (49). CFH, SFTPA2, CEACAM3, and CEACAM6 polymorphisms were significantly associated with an increased susceptibility to meningococcal disease, whereas other CEACAM6 and SFTPA2 polymorphisms showed a protective effect (49). A genome-wide association study identified variants in the CFH region associated with host susceptibility to meningococcal disease (54). Further studies are needed to confirm these associations before a definite conclusion can be drawn on their role because of the limited sample size and lack of correction for multiple testing.
Although all the aforementioned factors (particularly recent colonization with a pathogenic strain in a nonimmune host) undoubtedly contribute to the pathogenesis of meningococcal disease, the precise determinants contributing to overt clinical illness (as opposed to the usual outcome of asymptomatic carriage) are poorly defined. Even during epidemics, only 1 in 1,000 to 5,000 colonized patients develop disease (43). Various predisposing factors, including crowding, lower socioeconomic status, and poor general health, have been proposed to explain the increased incidence among blacks in the United States and among alcoholics in Finland or Alaska (41). However, the influence of such conditions (e.g., overcrowding) has not been supported by studies in Nigeria. An antecedent viral infection has been suggested as another predisposing factor, because approximately one third of meningococcal cases follow symptoms referable to the upper respiratory tract. An outbreak of meningococcal disease followed a large influenza epidemic in Texas in 1981, and simultaneous outbreaks of meningococcal and influenza A2 infections have been described in institutional settings. Although meningococcal pneumonia may complicate influenza (e.g., the 1918 to 1919 pandemic), the role of viral infections in the enhancement of meningococcal dissemination is unproved (22).
The time from nasopharyngeal acquisition to bloodstream invasion is short (usually approximately 10 days). The incubation period may also be short, because “secondary” cases commonly occur within 1 to 4 days of the index case. Once the organism is bloodborne, more than 90% of meningococcal disease is manifested as meningitis and/or meningococcemia.
Although a single case of meningococcemia or meningitis in a college student engenders alarm, invasive disease due to this organism is no more common in such students when compared with nonstudent age-matched 18- to 22-year-old controls but is increased threefold in freshmen living in dormitories, a target of some vaccine recommendations. Active or passive smoking and binge alcohol consumption also appear to increase the risk of meningococcal disease in this age-group. The first quadrivalent meningococcal conjugate vaccine against serogroups A, C, Y, and W135 was licensed in January 2005 in the United States for routine use, starting at age 11 to 12 years; recent recommendations are for one vaccine in this age-group, with a booster dose given at age 16 years (55,56). A two-dose primary series administered 2 months apart is recommended for those age 2 to 54 years with persistent complement component deficiency or functional or anatomic asplenia and for adolescents with HIV infection. A two-dose primary series is also recommended for children 9 to 23 months of age at increased risk for invasive meningococcal disease (57). Lack of a vaccine against serogroup B meningococcus is a significant issue, although recent investigations of a multicomponent serogroup B vaccine (4CMenB) demonstrated that this vaccine was immunogenic (58–61); 84% to 100% of infants who were administered the vaccine developed bactericidal antibodies.
Streptococcus pneumoniae
Although pneumococcal meningitis occurs in all age-groups, pneumococci remain the most common cause of bacterial meningitis in adults. The annual incidence of pneumococcal meningitis has remained relatively stable in the United States for the past three decades until recently with introduction of the heptavalent pneumococcal conjugate vaccine (see later discussion). In seven studies analyzing data from diverse geographic areas in this country from 1959 to 1978, the annual incidence was 0.3 to 2.3 per 100,000 population, with a mean of 1.3 per 100,000. An identical infection rate of 1.3 per 100,000 persons annually was recorded from the Oklahoma City area in 1984 and extrapolated from several states and Los Angeles County in a relatively recent (1995) national survey conducted by the Centers for Disease Control and Prevention (CDC) (62,63); higher rates of invasive pneumococcal disease were reported at the extremes of age (see later discussion), in men and boys, and among blacks and American Indians as compared with whites. Nearly identical incidence figures (1.2 to 1.4/100,000 population annually) were reported from the Göteborg, Sweden area from 1975 through 1980 (64) and from Örebro county in Sweden (1.0/100,000 population annually) from 1981 through 1992 (65). Pneumococcal meningitis was, again, more common in men and boys, and most cases occurred from December through May. However, higher incidence rates have been reported from other areas. For example, surveillance studies from 1980 to 1986 among the Alaskan native population in the Yukon-Kusko-Kurin delta region of southwestern Alaska documented an extremely high frequency of invasive pneumococcal disease; the annual rate for pneumococcal meningitis was 13.2 per 100,000 persons overall (66). Perhaps more importantly, the annual incidence rate was 216 per 100,000 children younger than 2 years—18 times higher than that reported from Sweden (64) and 36- to 37-fold greater than United States rates derived from both passive and active surveillance (7,62,66). These rates of bacteriologically confirmed invasive pneumococcal disease were the highest reported for any population worldwide. Although most cases of invasive pneumococcal disease occurred during the Arctic summer, pneumococcal meningitis cases clustered in the winter (66), as described in other reports (7).
The risk for pneumococcal meningitis is age dependent, with increased incidence rates occurring at the extremes of age. For example, the number of cases per 100,000 persons per year in the Göteborg, Sweden area for 1970 through 1980 were as follows: 12.0 for infants younger than 12 months old, 0.4 to 0.9 for children and adults 2 to 39 years old, 1.2 to 1.6 for persons 40 to 70 years old, and 2.2 for those older than 70 years of age (64). The dramatic incidence among Alaskan native children younger than 2 years of age (216/100,000 annually) are noted earlier in this chapter. Similarly, the annual incidence rates for all invasive pneumococcal infections (including bacteremic pneumonia) in the Oklahoma City area in 1984 were 97 per 100,000 for infants younger than 1 year of age and 87 per 100,000 for elderly adults older than 80 years of age.
As with disease caused by the other major meningeal pathogens, pneumococcal meningitis follows recent nasopharyngeal colonization by a virulent strain. The rate of asymptomatic carriage varies with age, environment, geographic locale, and the presence of an upper respiratory tract infection. Pneumococci have been isolated from the upper respiratory tract of 5% to 70% of normal adults; approximately 25% acquire a new strain annually. Carriage rates decline with age (30% to 35% for children ages 6 to 11 years and 18% to 19% in adults) and are higher in closed populations (e.g., 27% to 58% in schools and orphanages, 50% to 60% in closed military populations). The duration of pneumococcal carriage varies from weeks to months and is longer in children than in adults. Most carrier strains in the normal population are of higher numbered capsular types and only infrequently are associated with invasive disease (see later discussion). Carriage is prolonged in individuals with low serum antibody concentrations against the homologous capsular type before colonization. Spread of this organism within the family unit is influenced by crowding, the season (greater in fall and winter), and the presence of pneumococcal disease (particularly pneumonia and otitis media). The precise relationship between pneumococcal carriage and the development of protective immunity is poorly defined. More than 50% of children develop a rise in type-specific antibody concentrations following colonization; this is rarely observed in adults, perhaps because of the relatively high antibody concentrations already present in adults. Nevertheless, otitis media often occurs in colonized infants despite the presence of type-specific antibodies. Antibody concentrations generally decline with time despite persistent carriage of a given strain (67). In addition, antibody responses to different capsular types vary considerably and are generally poor in infants younger than 2 years of age. Specific antibody responses tend to be higher after intermittent periods of nasopharyngeal carriage than after continuous ones. The antibody response after pneumococcal colonization, its influence on subsequent disease, and the impact of other environmental antigens require further study.
Studies of extreme phenotypes among patients with invasive pneumococcal disease have identified genetic correlates of increased susceptibility in the complement system and the signalling cascade after toll-like receptor pathways (49). Case–control studies showed that invasive pneumococcal disease was associated with certain MBL and C3-variant genotypes (49,68).
Several factors predispose to pneumococcal meningitis (69,70):
■ Pneumonia (15% to 25% of patients)
■ Otitis media
■ Sinusitis
■ CSF fistulas, leak
■ Head injury
■ Cochlear implants with positioners
■ Alcoholism, cirrhosis
■ Sickle cell disease, thalassemia major
■ Other splenic disease
■ Wiskott-Aldrich syndrome
■ Multiple myeloma
Pneumonia coexists much more commonly with pneumococcal meningitis than with the other two major pathogens. Acute otitis media is seen in approximately 30% of patients with pneumococcal meningitis; acute sinusitis may also be an important antecedent event. Pneumococci are the most common cause of recurrent meningitis in the setting of CSF leaks. Recent or remote head trauma is found in about 10% of patients with pneumococcal meningitis. Alcoholism, cirrhosis, and spontaneous bacterial peritonitis are underlying disorders in approximately 20% to 35% of patients with this disease. Pneumococci are the most common cause of meningitis in children with sickle cell anemia and commonly cause meningitis in other asplenic states or in the setting of primary or acquired immunodeficiencies (71). S. pneumoniaecauses 87% of pyogenic meningitis cases in sickle cell disease; cases in the 2- to 3-year-old group occur at a rate of 12 per 1,000 patient-years. The risk for pneumococcal meningitis in this age-group of children with sickle cell anemia is increased 36-fold as compared with that observed in control groups of black children, and it is increased 314-fold over that observed in whites. Pneumococcal meningitis also occurs with increased frequency in persons with the Wiskott-Aldrich syndrome, thalassemia major, childhood nephrotic syndrome, multiple myeloma, and chronic lymphocytic leukemia. Defects in immunoglobulin concentration or function, as well as poor alternative complement pathway–mediated opsonization of pneumococci, are common in many of these disorders. Pneumococcal meningitis is approximately 450-fold more common in patients with acquired immunodeficiency syndrome (AIDS) when compared with the general population (71), often despite prophylaxis with trimethoprim-sulfamethoxazole (TMP-SMX) to prevent other opportunistic infections. In one study of 352 episodes of community-acquired pneumococcal meningitis in adults, 245 (70%) were associated with an underlying disorder and the overall in-hospital mortality rate was 30% (72); death in patients younger than 60 years of age was more often caused by neurologic complications; in patients 60 years or older, death was more likely secondary to systemic complications. In children with cochlear implants with positioners who are beyond 24 months after implantation, the incidence of bacterial meningitis was 450 cases per 100,000 person-years compared with 0 cases in children without positioners (73); the updated overall incidence was 189 cases per 100,000 person-years, with most cases caused by S. pneumoniae (74). Outbreaks of pneumococcal meningitis have also been described during African outbreaks of meningococcal meningitis (75). In children who develop second episodes of pneumococcal meningitis, screening for congenital immunoglobulin deficiencies should be performed (53).
The rates of pneumococcal meningitis have decreased in children and adults since introduction of the heptavalent pneumococcal conjugate vaccine (from 1.13 to 0.79 cases per 100,000 between 1998 to 1999 and 2004 to 2005), although the increase in disease caused by serotypes not in the vaccine (19A, 22F, and 35B) is concerning (76). The Advisory Committee on Immunization Practices now recommends use of the 13-valent pneumococcal conjugate vaccine (77), which not only offers protection against the serotypes in the heptavalent vaccine (4, 6B, 9V, 14, 18C, 19F, 23F) but also protection against additional serotypes (serotypes 1, 3, 5, 6A, 7F, 19A).
ETIOLOGY
In a review of 296 episodes of community-acquired meningitis in adults reported from the Massachusetts General Hospital from 1962 through 1988, the most common pathogens were S. pneumoniae (37%), N. meningitidis (13%), and L. monocytogenes (10%) (78).
The epidemiology of bacterial meningitis changed dramatically in areas that have embraced Hib conjugate vaccines starting in the late 1980s. The influence of these vaccines from the four largest and most recent surveys conducted in the United States (7,62,79) is shown in Table 24.2. Hib was responsible for 45% to 48% of cases from 1978 through 1986 but only 7% of cases in a surveillance study (62) conducted in 1995 in laboratories serving all acute-care hospitals in 22 counties from four states (>10 million population). The incidence of Hib meningitis declined from 2.9 to 0.2 per 100,000 population from 1986 to 1995, respectively. The median age of patients with bacterial meningitis rose from 15 months to 25 years during this interval, perhaps the most dramatic change in the epidemiology of any bacterial infection in recent decades. As a result, approximately 70% of bacterial meningitis in the United States is due to pneumococci or meningococci. In the early pivotal trial in California, invasive (e.g., bacteremia and meningitis) pneumococcal disease was virtually eliminated in infants vaccinated with the heptavalent conjugate vaccine at 2, 4, and 6 months of age. Although targeted to children, recent data support a “herd” immunity phenomenon, similar to that experienced with Hib conjugate vaccines, leading to a decline in pneumococcal bacteremia and/or meningitis in the population (including adults) following introduction of conjugate pneumococcal vaccine in infancy. In another surveillance study among residents in eight surveillance areas representing 17.4 million persons from 1998 to 2007, the impact of the heptavalent pneumococcal conjugate vaccine was appreciated in which the incidence of meningitis caused by vaccine serotypes decreased from 0.61 cases per 100,000 population in 1998 to 1999 to 0.05 cases per 100,000 population in 2006 to 2007, although the number of cases of bacterial meningitis caused by nonvaccine serotypes increased by 61% (5); the mean age of all patients with meningitis increased from 30.3 years in 1998 to 1999 to 41.9 years in 2006 to 2007. However, despite the declining incidence of bacterial meningitis in the United States, the overall case-fatality rates did not change significantly (15.7% in 1998 to 1999 compared with 14.3% in 2006 to 2007; p = .50).

As shown in Table 24.1, the responsible organisms for community-acquired bacterial meningitis in adults older than 16 years of age are somewhat different, but S. pneumoniae, N. meningitidis, and L. monocytogenes predominate. Similar recent (e.g., after 2000) surveys for resource-limited settings are lacking and sorely needed to guide vaccine strategies in this age-group.
In the following sections, each of the major meningeal pathogens is considered, including relevant classification schemes. In addition, the potential virulence characteristics of each organism are briefly discussed.
Haemophilus influenzae
Haemophilus species are small, gram-negative, pleomorphic coccobacilli. They are facultative anaerobes that grow best anaerobically with 5% to 10% CO2 on blood-enriched media. H. influenzae requires both X factor (hematin) and V factor (nicotinamide adenine dinucleotide, nicotinamide adenine dinucleotide phosphate, or nicotinamide nucleoside) for growth under aerobic conditions and is nonhemolytic. Chocolate agar is most commonly employed for the initial isolation of H. influenzae.
H. influenzae strains are either encapsulated (typeable) or unencapsulated (nontypeable). Encapsulated strains are classified into six types, designated a through f, according to specific reactions with antisera directed against epitopes on capsular antigens. Methods for grouping include counterimmunoelectrophoresis, latex particle agglutination of culture supernatants, immunofluorescence, and the production of halos surrounding colonies on media containing antisera. Nearly all invasive H. influenzae infections are caused by serotype b (Hib). The capsule is a repeating polymer of PRP, an important virulence determinant of this organism (see later discussion). Hib also contains a lipooligosaccharide (here, designated lipopolysaccharide [LPS] for convenience) in the outer membrane, an additional virulence determinant. Hib strains have been further classified into subtypes based on electrophoretic mobility differences among Omps. Although the pathogenic role of these Omps is uncertain, subtype analysis is useful for epidemiologic studies. For example, in a survey of 256 invasive isolates from 22 states representing a variety of clinical settings, about 70% of cases were caused by strains of three subtypes (1H, 2L, and 3L) among the 21 Omp subtypes identified. In contrast, 84% of 80 invasive isolates studied in the Netherlands had the same Omp subtype pattern (type 1, identical to subtype 3L in the Granoff classification system), and no strains of subtype 1H, 1L, or 2H were found.
It has been recognized, largely on the basis of multilocus enzyme electrophoretic analysis by Musser et al. (80), that the natural populations of Hib from widely divergent geographic areas are clonal as a consequence of infrequent recombination of chromosomal genes. For example, 32 distinct multilocus enzyme genotypes, referred to as electrophoretic types (ETs), were apparent among 177 U.S. isolates by analysis of 16 metabolic enzymes, but 73% of invasive disease episodes were caused by strains belonging to only three ETs. In the largest and most comprehensive analysis (80), 2,209 encapsulated H. influenzae strains (including 1975 Hib) from 30 countries on six continents collected over a 40-year period were studied by multilocus electrophoresis of 17 chromosomally encoded metabolic enzymes, Omp subtyping, and the pattern of restriction fragment length polymorphism in the cap region (the chromosome region responsible for capsular expression). On the basis of allele profiles at the enzyme loci, 280 distinct ETs in two phylogenetic divisions were identified: the population structure is definitely clonal. Currently, nearly all invasive disease worldwide is caused by nine clones of Hib. One genetically distinct clone complex occurs with considerable frequency worldwide, but marked geographic variation occurs for other clones or clone families. Based on an extensive analysis, it appears that this distribution of clones on an intercontinental scale is largely accounted for by the patterns of racial/ethnic composition and historical demographic movements of the human host populations (80).
Neisseria meningitidis
Neisseria species are non–spore-forming, nonmotile, oxidase-positive, gram-negative cocci (measuring approximately 0.8 × 0.6 µm) that usually appear as biscuit- or kidney-shaped diplococci on smears of infected fluids. Because other organisms (e.g., Moraxella species) are similar morphologically, identification rests on biochemical or immunologic techniques. All Neisseria species are oxidase positive, and sugar fermentation reactions are usually sufficient for speciation within the genus. Gonococci ferment glucose (but not maltose or lactose) to acid, whereas meningococci ferment both glucose and maltose to acid. N. lactamica, a related organism occasionally present in throat cultures, ferments glucose, maltose, and lactose. Maltose-negative variants of meningococci have been isolated; these strains may be differentiated from gonococci by fluorescent antibody tests, coagglutination tests, or electrophoretic analysis of hexokinase isoenzymes. These strains usually acquire the ability to ferment maltose on subculture; true maltose-negative variants are rare, but this property is genetically linked to sulfadiazine resistance. Meningococci grow rapidly on blood, chocolate, gonococcal, or enriched Mueller-Hinton agar in a moist, 3% to 10% CO2 environment at 35°C to 37°C. A modified Thayer-Martin medium is employed for meningococcal isolation from contaminated sites, as in detection of the carrier state. The transoral approach for obtaining specimens is more practical and is at least as sensitive as older transnasal approaches for carrier detection. Because this organism is susceptible to drying and chilling, all specimens should be inoculated promptly.
As in other gram-negative bacteria, the ultrastructural characteristics of meningococci are complex. The surface components include capsular polysaccharide, fimbriae or pili, LPS, and Omp; several of these structures are important virulence determinants. Meningococci are classified by serogroups based on structural differences among capsular polysaccharides and agglutination reactions with specific antisera, and they are further defined by serotypes based on analysis of the Omp. Capsular polysaccharide detected by positive agglutination with reference antisera is uniformly present among invasive isolates, but 20% to 50% or more of carrier strains are unencapsulated (nongroupable). The serogroups have important epidemiologic and prevention-related implications. Thirteen serogroups are recognized (43), designated as follows: A, B, C, D, H, I, K, L, X, Y, Z, 29E, and W135. Most meningococcal disease is caused by organisms in serogroups A, B, C, and Y, although the proportion of cases caused by serogroup W135 is increasing. Although serious outbreaks of serogroup A, B, or C disease have occurred worldwide, most focal outbreaks and endemic disease in many countries are caused by serogroups B and C. For example, in the United States from 1975 to 1980, the distribution of serogroups among 12,980 cases was as follows: B, 56%; C, 19%; Y, 11%; W135, 10%; and A, 3%. Similar figures were reported for the period 1978 to 1981 (7). Serogroup W135 disease, especially among adults, has been increasing in the United States and apparently elsewhere (e.g., Senegal) since 1981. Group B organisms, especially B:15:P1.16, have emerged as important pathogens in northern Europe, causing serious local outbreaks peaking in the 10- to 20-year-old group. The continued high prevalence of serogroup B meningococcal disease has important implications because of the lack of an effective, widely available vaccine against this serogroup. An outbreak of serogroup C disease occurred in California in 1987 (81). Serious outbreaks caused by serogroup A continue in Nepal, Saudi Arabia, Chad, Sudan, Burkina Faso, and elsewhere. Approximately 40,000 cases of serogroup A disease occurred in Ethiopia in the spring of 1989 (Fig. 24.1).
There have been increases in the incidence of serogroup C disease in North America to equal or surpass the incidence of disease caused by serogroup B (42). From 1985 through 1992, a clonal serogroup C meningococcal strain (designated ET-15 and defined by multilocus enzyme electrophoresis) was associated with an increase in both the incidence and the mortality of meningococcal disease in Canada (82). This and four other closely related clonal strains were also implicated in a marked increase in outbreaks of serogroup C meningococcal disease in the United States (83). These studies suggested the emergence of a clonal group of virulent serogroup C meningococcal strains in North America, leading to an increase in the rate of meningococcal disease in some regions, an increased number of outbreaks, and a higher case-fatality rate (84).
In an analysis from the United States (85), part of an active and ongoing laboratory-based population-based surveillance for meningococcal infection from 1992 to 1996, the serogroup distribution for the three most common isolates was as follows: C, 35%; B, 32%; and Y, 26%. Similar results were noted in New York City through 2000 (86). In contrast, serogroup B accounted for 75% of meningococcal isolates in Italy (87). More than 98% of cases of invasive meningococcal disease are sporadic (88). From 1998 to 2007, a total of 2,262 cases of meningococcal disease were reported to the Active Bacterial Core surveillance sites, with an annual incidence of 0.53 cases per 100,000 population (89). The incidence decreased from 0.92 cases per 100,000 population in 1998 to 0.33 cases per 100,000 population in 2007; the incidence decreased to this historic low before the introduction of the quadrivalent meningococcal conjugate vaccine. Infants younger than 1 year of age had the highest incidence (5.38 cases per 100,000 population), although the distribution of serogroups is also different in this population (serogroup B, 3.08 per 100,000 population; serogroup C, 0.53 per 100,000 population; serogroup Y, 1.50 per 100,000 population). A recent outbreak of serogroup C disease was reported in New York City among men who have sex with men (90). During the outbreak of meningococcal disease coinciding with the Hajj pilgrimage in March 2000, the attack rate of W135 disease was 25 cases per 100,000 pilgrims (91); all outbreak-associated isolates of serogroup W135 were members of a single clone of the hypervirulent ET-37 complex, which occurred as the result of expansion of a clone that had been in circulation since 1970 (92). A high incidence of serogroup X cases was reported in Niger (93), representing 51% of 1,139 confirmed cases of meningococcal meningitis in 2006; serogroup X disease also emerged in Togo and Burkina Faso during 2006 to 2010 (94).
Serotypes within a serogroup of N. meningitidis are classified largely on analysis of Omp profiles in the cell envelope. At least 20 serotypes are recognized (95), resulting in a classification scheme that is useful in epidemiologic studies. Physicochemical characterization of the Omps, which might be candidate antigens for vaccines, has led to the designation of five major classes. Class 2 and 3 proteins are the major porins responsible for aqueous channels in the outer membrane. Class 5 proteins are surface exposed and may have a role in virulence, but the function of class 1 and 4 proteins is poorly defined. The serotype designation has important epidemiologic uses and may identify virulence characteristics among meningococci. For example, serotype 2 (2a, 2b) strains are responsible for approximately 50% of serogroup B disease (in which 15 serotypes have been described), followed by serotypes 15 and 9; in contrast, serotypes 4 and 6 have been isolated only from carriers. Serotype 2 is also responsible for about 80% of invasive serogroup C disease and is an important marker for serogroups Y and W135 pathogenic strains as well (95). All serogroup A meningococci, in contrast, are homogeneous with respect to Omp and show no homology to serotypes within other serogroups. Serotype analysis has also been linked to certain clinical characteristics (96). However, this technique is available only in research or reference laboratories. In addition, at least eight immunotypes of meningococci, classified by differences among LPS subtypes, are known to exist and may play a role in pathogenesis and disease expression. LPS immunotype analysis may also be useful in the further characterization of the epidemiology of meningococcal disease (41,43). The recent availability of monoclonal antibodies to detect variations in Omp and/or LPS will improve the resolution of these typing systems for epidemiologic analysis.
In addition to classification by serogroup, serotype, and LPS subtype, many reports have focused on multilocus enzyme electrophoresis for the characterization of the chromosomal genome of isolates and for estimation of the genetic relatedness among strains (97). These studies, similar to the analysis of Hib (80), have identified the clonal nature of N. meningitidis and have been useful for epidemiologic purposes. For example, electrophoretic variation (defined as <17% genetic distance between isolates) in seven metabolic enzymes and two Omp in 423 isolates of serogroup A strains recovered from 23 epidemics or outbreaks occurring in 38 countries on six continents over a 70-year period since 1915 identified 21 “clones” (designated A I-1 through A IV-4) containing 34 ETs (97). This technique has been useful for delineating similarities and differences among isolates from cases and carriers before, during, and after epidemics (98). It has also been useful for tracing movements of epidemic strains geographically over time (81). For example, following an epidemic in Nepal (1983 to 1984), a single clonal complex of serogroup A meningococci (III-1, representing 11 closely related ETs) was introduced into Saudi Arabia in 1987 by Muslim pilgrims traveling to Mecca for the annual hajj (98). The same strain was then introduced into sub-Saharan Africa following the hajj, causing an explosive outbreak in Chad in 1988. An analysis of 109 isolates of serogroup B meningococci in Norway has also revealed differences among carriers and cases (99). Although 78 ETs were identified, 91% of the cases of systemic disease in 1984 were caused by strains from the ET-5–ET-37 complex, whereas these isolates were recovered from only 7% and 9%, respectively, of healthy carriers. The most common clonal complex found among carriers was never isolated from patients with invasive disease, suggesting a low virulence potential for these clones. Two clones, ET-15 and ET-508, have been associated with outbreaks of meningococcal infection in North America (82,83,100). Clonal analysis will undoubtedly continue to contribute important information on the epidemiology of meningococcal infection, and it may prove useful in an analysis of virulence properties. As noted earlier, serogroup W135 has been circulating, particularly among pilgrims to Mecca during the hajj since 2000 in Africa, the Middle East, and worldwide (attack rate ≥25/100,000); all isolates were members of a single hypervirulent ET-37 complex (91,92). This occurred through the expansion of a clone that had been circulating since 1970. Vaccination with quadrivalent vaccine in hajj pilgrims from several areas (e.g., North America, Western Europe) appears to have reduced transmission of W135 meningococci to close contacts upon return (101,102).
The putative meningococcal virulence characteristics are as follows:
■ Capsular polysaccharide
■ Pili
■ IgA protease
■ LPS (endotoxin)
■ Omps
■ Outer membrane vesicles, or blebs
■ Metabolic pathways (e.g., iron)
As noted earlier, all isolates from invasive infections are encapsulated (serogroup positive), but 20% to 90% of isolates from carriers are unencapsulated (nontypeable). The capsular polysaccharide appears to be essential for meningococcal virulence, probably because of its antiphagocytic properties that allow the organism to escape host phagocytic clearance mechanisms within the bloodstream and/or CSF. Pili are protein surface appendages composed of identical pilin-repeating subunits. Pili from meningococci and gonococci are morphologically and chemically similar. Cross-reacting antibodies bind to a short peptide sequence (residues 69 to 94) of gonococcal pili that is essential for receptor-binding function. Fresh meningococcal isolates from carriers and cases contain 7 to 40 pili per diplococcus. Pili are important mediators of meningococcal adhesion to human nonciliated columnar nasopharyngeal epithelial cells, an important early step in the development of the carrier state. Extracellular proteases that cleave the IgA1 heavy chain in the hinge region are elaborated by pathogenic Neisseria species (i.e., meningococci and gonococci). Although the role of IgA proteases in the pathogenesis of disease is controversial, these enzymes are produced by only a few organisms (e.g., N. meningitidis, Neisseria gonorrhoeae, H. influenzae, S. pneumoniae, and Streptococcus sanguinis), many of which are important meningeal pathogens and may promote invasion at the pharyngeal portal of entry. Meningococcal LPS resembles H. influenzae LPS by a lack of the O-antigenic polysaccharide side chains found in enteric bacilli despite a smooth phenotype and proven virulence. LPS is clearly important in the genesis of an array of the clinical manifestations of meningococcemia and/or meningitis (see later discussion). Although the specific role for Omp in meningococcal virulence is unclear, these organisms release substantial amounts of cell surface material (in the form of outer membrane vesicles containing Omp and LPS) during growth in vitro and in vivo in the absence of cell lysis, a process exacerbated by antimicrobial agents. These outer membrane vesicles, or blebs, represent relevant vehicles for central nervous system (CNS) tissue damage during meningococcal infection (see later discussion). Tissue invasion may also be facilitated by the ability of meningococci to obtain iron from transferrin.
Streptococcus pneumoniae
Pneumococci are non–spore-forming, nonmotile, small (approximately 0.8 µm), gram-positive streptococci that typically appear as lancet-shaped diplococci with the tapered ends in juxtaposition in clinical specimens. They tend to associate in pairs rather than in short chains, although the latter morphology is facilitated by broth culture. Pneumococci are facultative anaerobes that flourish in a variety of supplemented artificial media. Optimal growth occurs in various media supplemented with serum or blood and glucose, at a pH level of 7.8 and a temperature of 37°C in an enriched CO2 environment. The organisms are catalase negative and relatively fastidious. Colonies on blood agar are initially dome shaped, but they become umbilicated with time as a result of the activity of autolytic enzymes (L-alaninemuramylamidase). Pneumococci are α-hemolytic (i.e., a greenish discoloration surrounds colonies on blood agar), although β-hemolysis occurs under anaerobic conditions. S. pneumoniae must be separated from other α-hemolytic streptococci in the laboratory; this is usually accomplished with a disk susceptibility test using the unique susceptibility of pneumococci to Optochin (ethylhydrocupreine hydrochloride). Pneumococci, but not other streptococci, are also sensitive to bile or bile salts (e.g., 10% deoxycholate), but the bile solubility test is now rarely performed in hospital laboratories.
Pneumococci are classified within serotypes on the basis of antigenic differences among capsular polysaccharides. These capsular substances are complex polysaccharides that form hydrophilic gels on the surface of the organism. At present, at least 90 serotypes have been identified, classified by two systems of nomenclature (American and Danish). Capsular polysaccharides are identified by agglutination; the Neufeld quellung reaction, characterized by capsular swelling and increased refraction in the presence of antisera, also useful for identifying pneumococci in clinical specimens (e.g., CSF); precipitation; and counterimmunoelectrophoresis. It is important to emphasize that cross reactions exist between individual pneumococcal serotypes, as well as with other bacteria (e.g., E. coli, Klebsiella pneumoniae, Hib, and S. sanguinis). For example, serotype 14 pneumococcal capsular polysaccharide cross-reacts with type III group B streptococci and with certain human ABO blood group isoantigens.
Capsular polysaccharide is essential for pneumococcal virulence, and a few serotypes are associated with most invasive infections. Encapsulated organisms (smooth colonies on agar) are virulent for humans and experimental animals, whereas unencapsulated (rough colonies on agar) strains are avirulent. Capsular polysaccharide enhances virulence through its antiphagocytic properties, as in Hib and meningococci.
Of the more than 90 known pneumococcal serotypes, only a few (usually the lower number types rather than the higher number types commonly found in the carrier state) account for most invasive pneumococcal infections. The predominant capsular types were types 1, 2, and 3 in the preantibiotic era. A different pattern has emerged in the past 40 years, and it differs between adults and children. A few capsular types cause the majority of bacteremic cases among adults: types 1, 3, 4, 7, 8, 9, 12, and 14, and less commonly types 6, 18, and 19 (these are not listed in rank order). Approximately 65% of cases are caused by eight serotypes, although no single serotype predominates among bacteremic adults. Capsular types 14, 6, 18, 19, 23, 1, 4, and 9 cause approximately 85% of serious infections in children, a pattern different from that observed in adults. Perhaps more important with respect to this discussion, there is a very strong correlation between serotypes causing pneumonia (or bacteremia) and those responsible for pneumococcal meningitis (69). For example, 76% to 86% of blood and CSF isolates were included among the 14 serotypes represented in the original pneumococcal vaccine. However, marked geographic variations exist among pneumococcal serotypes causing invasive disease, and the serotype distribution may change over time in a given locale. Furthermore, some serotypes may be more virulent than others and associated with less favorable outcomes, including death. Older studies of pneumococcal meningitis in the 1950s (summarized by Scheld [69]) noted an increased mortality with disease caused by serotypes 2, 8, and 12. Although type 12 caused only 2.5% of cases, it was responsible for 30.7% of the deaths. Similarly, serotype 6 was associated with four of eight deaths in a more recent survey.
The role of other cell surface components (such as C-polysaccharide antigens, cell wall antigens, M- or R-protein antigens) and putative toxins (such as hemolysin, neuraminidase, purpura-producing principle) in the pathogenesis and pathophysiology of pneumococcal meningitis remains poorly defined. Nevertheless, CSF inflammation is induced by pneumococcal cell wall and its constituents (particularly lipoteichoic acid) but not by purified pneumococcal capsular polysaccharide (see later discussion).
Gram-Negative Bacilli
Approximately 84% of cases of neonatal meningitis and sepsis attributable to E. coli are caused by strains bearing the K1 capsular polysaccharide antigen; this capsular type serves as a marker of neurovirulence. In addition to the K1 capsule, multiple other potential virulence factors for blood–brain barrier (BBB) traversal and meningitis have been documented in E. coli from phylogenetic groups A, D, and B2 (e.g., O45:K1 and O18:K1 strains) (103).
Aerobic gram-negative bacilli have become increasingly important in patients with bacterial meningitis (104). Beyond the neonatal period, aerobic gram-negative bacillary meningitis occurs in three major clinical settings: head trauma (approximately 30% of cases, especially in conjunction with CSF rhinorrhea or otorrhea); after neurosurgical procedures (approximately 50% of cases); and in association with other conditions (approximately 20% of cases), including strongyloidiasis, gram-negative bacteremia, ruptured brain abscess, or impairment of host defense mechanisms (e.g., steroids or AIDS). Many of these infections are nosocomial in origin, although community-acquired gram-negative bacillary meningitis is increasing in frequency, particularly in the elderly older than 71 years and in debilitated, alcoholic, or diabetic adults (105–107). The most common causes of gram-negative aerobic bacillary meningitis beyond the first month of life include Klebsiella species (about 40% of cases), E. coli (roughly 15% to 30%), and Pseudomonas aeruginosa (about 10% to 20%). In a recent study from Korea of 91 adult patients with nosocomial meningitis, Acinetobacter species accounted for 32.5% of cases (108).
Streptococcus agalactiae
Group B streptococci are the most common cause of invasive neonatal disease in the United States, accounting for approximately 11,000 cases of meningitis and/or bacteremia yearly. The incidence of group B streptococcal neonatal infections has remained relatively stable in this country until recently (109). The use of antimicrobial agents (e.g., ampicillin) in pregnant women with vaginal colonization with group B streptococci at the time of delivery has led to a decline in the incidence of neonatal invasive group B streptococcal disease, perhaps, as suggested by one study, with an increase in ampicillin-resistant E. coliearly-onset sepsis in low-birth-weight infants (110). However, it appears to be increasing in frequency in the developing world.
Group B streptococci are classified into six main serotypes (designated Ia, Ib/c, Ia/c, II, III, and IV) based on the expression of type-specific capsular polysaccharide antigens and various surface proteins as additional antigenic markers (111); additional candidate serotypes are under evaluation. Although all serotypes have been isolated from neonates with invasive disease, type III is responsible for the vast majority of meningeal infections, suggesting a high virulence potential and/or CNS tropism for this serotype. The chromosomal genetic diversity of S. agalactiae was studied and the clonal nature of the native bacterial populations was again demonstrated (112). A collection of 128 isolates representing all six serotypes, including 44 type III isolates from invasive episodes (18 recovered from CSF), were subjected to multilocus enzyme electrophoresis, an analysis based on electrophoretically demonstrable allelic profiles at 11 metabolic enzyme loci, all encoded at the chromosome level. Nineteen distinct ETs were identified in two primary phylogenetic divisions, each representing a multilocus clonal genotype. A single ET (ET-1), comprising 40 isolates of serotype III group B streptococci, formed the first phylogenetic division. These strains produced greater amounts of neuraminidase and were more virulent than the other type III isolates found in several of the 18 ETs in the second division. This newly evolved clone (ET-1) is responsible for most invasive disease episodes caused by group B streptococci type III in the United States (112). Overall, approximately 52% of group B streptococcal meningitis cases in the United States occur during the first month of life. In one recent review of 444 cases of neonatal bacterial meningitis over a 7-year period, group B streptococcus was the most common etiology in early-onset (occurring between day 0 to 4 of birth) and late-onset (occurring between day 5 and 28 of birth) disease, responsible for 77% and 50% of cases, respectively (113). In the United States, the overall mortality rate ranges from 7% to 27%. Survivors of group B streptococcal meningitis also have substantial long-term morbidity (114), indicating the need for ongoing developmental follow-up and the development of preventive strategies (see later discussion).
In addition to the common conditions of neonatal meningitis and postpartum fever and/or bacteremia in parturient women caused by group B streptococci, these organisms also cause serious infections in adults, including meningitis (115,116). Risk factors in adults include age older than 60 years, diabetes mellitus, parturition, cardiac disease, collagen-vascular diseases, malignancy, alcoholism, hepatic failure, renal failure, corticosteroid therapy, decubitus ulcers, neurogenic bladder, previous stroke, and AIDS. No underlying illnesses were found in 43% of patients in one review (116).
Listeria monocytogenes
L. monocytogenes remains an important cause of neonatal meningitis; the source is the genital tract or subclinical infection of the mother. Although L. monocytogenes may cause meningitis in normal adults, most patients are diabetic, alcoholic, elderly, or immunosuppressed. L. monocytogenes had been the major cause of bacterial meningitis among renal transplant recipients, but this is decreasing in frequency as a result of the use of TMP-SMX prophylaxis.
L. monocytogenes is widespread. Although clusters of nosocomial cases and focal outbreaks are reported, most cases of human listeriosis are sporadic. The incidence of L. monocytogenes infections is difficult to quantitate. Many countries in northern Europe have reported annual incidence figures of approximately 2 to 3 per million. After a large (142 cases) food-borne outbreak in Los Angeles County, California, in 1985, mandatory reporting of L. monocytogenes isolates by clinical laboratories was instituted. During the first year of active surveillance, 94 cases of listeriosis were reported (117), for an annual crude incidence of 11.7 per million persons, similar to figures (11.3 per million annually) reported from France in 1984. Listeriosis is undoubtedly underreported.
Approximately one third of cases in the United States are in neonates and/or their mothers (39% in the Los Angeles survey) (117). The proportion of perinatal infections in Europe is higher. Among the nonperinatal cases, various risk factors for listeriosis were identified, including immunosuppression as a documented history of steroid ingestion or chemotherapy (35% of cases, the single most important risk factor); age older than 75 years; renal disease; cancer; alcoholism and/or cirrhosis; and AIDS. Nevertheless, serious Listeria infections, including meningitis, remain uncommon in patients with AIDS (118,119), but diagnosis of Listeria meningitis in anyone younger than 50 years of age should prompt testing for HIV, if not done previously. Of the nonperinatal cases, only 2 of 57 had no definable underlying risk factor; 21 of 57 had meningitis (117). As stated earlier, L. monocytogenes remains a distinctly unusual cause of meningitis in developing countries. Listerial infection is most common in infants younger than 1 month of age (up to 10% of cases), adults older than 60 years of age, alcoholics, cancer patients, those receiving corticosteroid therapy, and immunosuppressed adults (e.g., renal transplant recipients) (120–122). In one recent study, patients with chronic lymphocytic leukemia had a greater than 1,000-fold risk of acquiring listeriosis (123). Other predisposing conditions include diabetes mellitus, liver disease, chronic renal disease, collagen-vascular diseases, pregnancy, and conditions associated with iron overload. Listeria meningitis has also been reported with use of anti–tumor necrosis factor-α (TNF-α) agents, such as infliximab in patients with Crohn disease (124) and ulcerative colitis (125), and etanercept in a patient with adult Still disease (126). Meningitis can also occur in immunocompetent children and adults (127,128). In nonperinatal cases, the route of transmission is often unknown. At least 1% of normal individuals excrete the organism in their stools, but contacts of symptomatic patients have much higher excretion rates (approximately 25%). Nevertheless, the true carriage rate, its duration, and its relationship to invasive disease are poorly defined. Although often considered a zoonosis, most patients do not report animal exposure. Reports have emphasized food-borne transmission of L. monocytogenes, a route of transmission that accounts for the overwhelming majority of sporadic cases. Many foods have been implicated, including coleslaw, Mexican-style cheese, raw vegetables, seafood, pasteurized milk, Swiss cheese, raw hot dogs, undercooked chicken, alfalfa sprouts, cantaloupe, diced celery, hog head cheese (a meat jelly made from hog heads and feet), and processed meats, thus pointing to the intestinal tract as the usual portal of entry (120,122,129–134). In one outbreak, 57 cases of listeriosis occurred in western Switzerland in association with the consumption of soft cheese (135); 40% of these cases were meningitis and 39% were meningoencephalitis. Some studies report a higher frequency of listeriosis in summer, opposite to the seasonal pattern seen with most other forms of bacterial meningitis. However, the incidence of invasive listeriosis has been decreasing, likely a result of a decrease in the prevalence of L. monocytogenes contamination of ready-to-eat food (136); this has been associated with a decrease in nonperinatal listeriosis-associated deaths (137).
L. monocytogenes is a gram-positive, non–spore-forming, catalase-positive, aerobic rod that may be difficult to culture on initial isolation but that, once grown, passes readily on a variety of laboratory media. The organisms may appear coccoid on Gram stains of clinical specimens, particularly CSF, and are often mistaken for pneumococci. More importantly, L. monocytogenes resembles diphtheroids and may thus be dismissed as a “contaminant,” a grave error. The presence of α-hemolysis and a characteristic tumbling motility at room temperature are used to separate L. monocytogenes from similar diphtheroid-like organisms. A hemolytic and cytolytic toxin (listeriolysin 0) of 52 kd appears essential for virulence; the toxin is expressed under conditions of low pH and low iron concentration and may facilitate phagolysosomal disruption and growth within mononuclear phagocytes. At least 11 serotypes are recognized, but more than 90% of invasive infections are caused by three serotypes: Ia, Ib, and IVb. The rate of unfavorable outcome among adults with Listeria meningitis was recently found to increase over a 14-year period from 27% to 61%, with the emerging L. monocytogenes serotype ST6 identified as the main factor leading to a poorer prognosis (138).
Staphylococcus epidermidis
Coagulase-negative staphylococci are very rare causes of bacterial meningitis in children and adults, except in the setting of an indwelling CSF shunt, where these organisms are the most prevalent pathogen. Therefore, this group is discussed elsewhere in this volume.
Staphylococcus aureus
Meningitis caused by Staphylococcus aureus is relatively unusual; this organism was responsible for 0.8% to 8.8% of cases in various surveys (139,140). S. aureus is the second most common cause of CSF shunt infections, accounting for 12% to 36% of cases. This organism is also frequently isolated from patients with nosocomial meningitis and is responsible for approximately 20% of such cases. Although secondary meningitis in the setting of infective endocarditis is an uncommon event, most of these infections are caused by S. aureus. Other important associated conditions have been recognized, including head trauma, neurosurgical procedures, various abscesses (cerebral, epidural, oral, abdominal), sinusitis, osteomyelitis, decubitus ulcers, pneumonia, cellulitis, injection drug use, malignancy, and infected intravascular grafts or shunts. Several other predisposing factors have been proposed. In a retrospective review of 28 cases of S. aureus meningitis seen from 1972 to 1982 at three North Carolina teaching hospitals, 22 occurred beyond the neonatal period (140). Among the adult patients (n = 20; mean age 52 years), 45% had an underlying condition (diabetes mellitus, malignancy, renal failure, immunosuppression), 35% had had head trauma or undergone neurosurgery (ventriculoperitoneal shunt, craniotomy), and about 20% developed meningitis in association with endocarditis or a paraspinal infection. Mortality was high (50% in adults), especially when S. aureus meningitis complicated a distant extracranial focus of infection (five of the six patients with purulent meningitis during active endocarditis died) (140). The prognosis for S. aureus infections of CSF shunts is more favorable. In a review of clinical and bacteriologic data from 61 postoperative and 43 hematogenous cases of S. aureusmeningitis from Denmark, postoperative cases had a lower mortality rate (18%) than cases resulting from hematogenous spread (56%); hematogenous S. aureus meningitis had a higher mortality rate related to age, presence of shock, and infections with strains of phage type 95 (141). Hospital-acquired cases are often caused by methicillin-resistant strains (142). In one series from 1999 to 2008 (143), S. aureus accounted for approximately 5% of cases of culture-proven bacterial meningitis in adults; since 2005, more than 75% of all cases were caused by methicillin-resistant Staphylococcus aureus (MRSA) and 52% (11 of 21 cases) of hematogenous cases were seen in injection drug users. In a multicenter review of 86 cases of MRSA meningitis in adults (144), the infection was nosocomial in 93% of cases; in those patients with postoperative meningitis, the most common predisposing conditions were the presence of CSF devices, neurosurgery, CSF leaks, and head trauma.
Anaerobic Bacteria
Meningitis caused by anaerobic bacteria is rare, accounting for fewer than 1% of pyogenic cases, except following the intraventricular rupture of a brain abscess. Anaerobic meningitis may be underrecognized because CSF is not routinely cultured anaerobically. Enriched media and proper transport of CSF to the laboratory, which are essential for isolation of anaerobes, are not uniformly performed. Only five cases caused by strict anaerobes were reported among 18,642 patients analyzed by the CDC in one study (7).
Anaerobic meningitis is associated with a variety of clinical conditions, including rupture of brain abscess or extension to the surface of the brain; chronic otitis, mastoiditis, or sinusitis; head trauma; neurosurgical procedures (e.g., craniotomy, laminectomy); congenital dural defects; abdominal trauma or surgery; gastrointestinal disease; head and neck cancer; suppurative pharyngitis; CSF shunts; and immunosuppression (particularly corticosteroid administration) (145,146). Most cases arise from spread of infection secondary to a contiguous focus of disease; anaerobic meningitis rarely complicates bacteremia from a distant extracranial focus.
A variety of bacterial species are responsible for anaerobic meningitis. Only nine cases of Bacteroides fragilis meningitis unaccompanied by a brain abscess had been reported in the modern era through 1987 (147). Seven occurred in premature infants or neonates (median age, 20 days), thereby complicating congenital defects or gastrointestinal disease such as necrotizing enterocolitis (148). Meningitis caused by Fusobacterium species, usually Fusobacterium necrophorum, generally occurs in older children (median age, about 5 years) or adults as a complication of acute or chronic otitis media (148). A variety of anaerobic gram-positive cocci have been isolated in a few cases, particularly peptostreptococci. Meningitis caused by Clostridium species almost always develops following head trauma or a neurosurgical procedure. For example, in a summary of 17 cases caused by Clostridium perfringens (149), only 3 were not associated with CNS trauma or surgery. Although the disease course is highly variable, some cases of clostridial meningitis are characterized by intracranial gas formation, visible on plain skull radiographs or computed tomography (CT); CSF white blood cell (WBC) concentrations exceeding 20,000/mm3; and death within hours of presentation. A few cases of meningitis caused by Actinomyces species (in the absence of brain abscess formation) and Propionibacterium acnes (usually subacute with a predominantly monocytic CSF pleocytosis) have also been reported. In approximately one eighth of patients, the infection is mixed, with anaerobic plus aerobic or microaerophilic organisms recovered from CSF.
Unusual Etiologic Agents
CNS infections caused by higher bacteria (e.g., Mycobacterium species, Nocardia species, Actinomyces species), spirochetes (e.g., Treponema pallidum, Borrelia burgdorferi, Leptospira species), Brucellaspecies, and so on, are discussed elsewhere in this volume. A plethora of bacteria have been documented as the cause of meningitis in isolated case reports or in small numbers of patients, including group A streptococci usually in association with pharyngitis, otitis media, and/or sinusitis (150); nonpneumococcal α-hemolytic streptococci such as Streptococcus mitis (151); enterococci (152,153); Streptococcus gallolyticus; diphtheroids (although P. acnes is an important etiologic agent in patients with CSF shunt and drain infections); N. gonorrhoeae (approximately 30 cases reported in the past 20 years); Neisseria subflava; Gardnerella vaginalis (one case report); many members of the Enterobacteriaceae in addition to E. coli and Salmonella species; Flavobacterium meningosepticum; Haemophilus species other than H. influenzae; and many others. Fewer than 0.5% of adult cases of bacterial meningitis are caused by group C streptococci but may occur in humans after contact with domestic animals (especially horses) or their unpasteurized products (154,155); however, mortality is high, perhaps because of the unpredictable susceptibility of this organism to β-lactam agents.
Despite the frequency with which the viridans streptococci cause bacteremia, they are unusual causes of meningitis (0.3% to 5% of culture-proven cases) (156). Streptococcus salivarius meningitis has been reported following spinal anesthesia (157,158) and myelogram procedures (159), supporting the importance of appropriate infection control practices (i.e., masks, proper aseptic technique, and safe injection practice) in those who perform spinal procedures. Streptococcus suis is the most frequent cause of bacterial meningitis in southern Vietnam and is associated with significant morbidity attributable to hearing loss (160); the pig is the natural reservoir of this microorganism and the main source of human infection. Risk factors for S. suis meningitis include eating “high-risk” dishes (such as undercooked pig blood or pig intestine) popular in parts of Asia, occupational exposure to pigs, and exposures to pig or pork in the presence of skin injuries (161).
Polymicrobial bacterial meningitis, with simultaneous recovery of two or more bacterial species from CSF, is unusual. Mixed infections account for about 1% of bacterial meningitis cases. In a review of 34 series encompassing 11,281 cases of bacterial meningitis, 116 cases (1%) were mixed (162). This condition appears to be evolving in the antibiotic era. Before 1950, nearly all cases occurred in children and were caused by combinations of bacteria commonly associated with meningitis (the three major meningeal pathogens). Since 1950, most cases have occurred in adults, with a wider spectrum of etiologic agents, particularly gram-negative aerobic bacilli. Approximately one third of cases were nosocomially acquired. Common predisposing conditions in the older population affected since 1950 include contiguous foci of infection, tumors in close proximity to the neuraxis (e.g., head and neck), rectal carcinoma, or fistulous communications with the CNS. The mortality rate is 63% for cases occurring after 1950. Several cases of meningitis caused by mixed bacterial and mycobacterial or fungal agents have also been reported.
Simultaneous isolation of viruses and bacteria from the CSF is rare; only seven well-documented cases were reported prior to 1988 (163). However, in a 1-year retrospective review from the Ohio State University published in 1986, 5 (2.8%) of 176 children with CSF enteroviral isolates also had bacterial meningitis (163). Conversely, CSF samples from 5 (4.8%) of 105 children with bacterial meningitis also grew an enterovirus. All the patients presented in late summer at the peak of the enterovirus season, and each case was caused by a different bacterial pathogen. Because the CSF formula was indistinguishable from that of patients with typical bacterial meningitis, and because the clinical course and response to therapy were similar to those of patients with typical bacterial meningitis, this condition may be underrecognized, as CSF viral cultures are rarely performed when bacterial meningitis is the likely diagnosis.
PATHOGENESIS AND PATHOPHYSIOLOGY
Despite the availability of effective antimicrobial therapy, bacterial meningitis continues to be a potentially fatal illness. Several investigators have examined the pathogenic and pathophysiologic mechanisms operating in meningitis, with the aim of improving the outcome of patients with this disorder. These pathogenetic and pathophysiologic mechanisms are discussed in detail in Chapter 23 and are detailed in a number of excellent reviews (164–174).
PATHOLOGY
Adams et al. (175) described the pathology of bacterial meningitis in 1948 based on examination of autopsy material from patients with H. influenzae meningitis. Experimental models of bacterial meningitis, knowledge of host defense mechanisms, and the pathophysiology of associated complications have subsequently allowed for a more complete understanding of the pathologic processes operating in this disorder.
Bacteria reach the meninges through one of the following pathways: (a) hematogenous dissemination from a distant site (e.g., nasopharynx, lung, skin, and genitourinary tract); (b) spread from an adjacent suppurative focus of infection (e.g., otitis media, sinusitis, and mastoiditis); and (c) a congenital or an acquired structural defect (176).
Once bacteria gain access to the SAS, an inflammatory process ensues (Fig. 24.2). Neutrophils migrate into the SAS, producing a purulent exudate. On gross examination, the exudate has a gray-yellow or yellow-green appearance (Fig. 24.3). It is most abundant in the cisterns at the base of the brain and over the convexities of the hemispheres in the rolandic and sylvian sulci (175) (Fig. 24.4). The tendency for exudate to accumulate in the cisterns at the base of the brain is explained by the anatomy of the SAS, which is deepest at the base of the brain. The various cisterns are expansions of the SAS, with the largest of these areas lying between the cerebellum and medulla and extending downward below the foramen magnum, the so-called cisterna magna or cerebellomedullary cistern (177). Purulent exudate accumulates in this cistern and extends into the other basal cisterns and onto the posterior surface of the spinal cord (Figs. 24.5 and 24.6). The exudate also extends into the arachnoidal sheaths of the cranial nerves and into the perivascular spaces of the cortex. A small amount of exudate may be found in the ventricular fluid and attached to the ventricular walls and choroid plexus; thus, the appearance of the ventricular fluid is usually cloudy by the end of the first week of the infection (175).





Microscopic examination of the subarachnoid exudate in the early stages of infection demonstrates large numbers of neutrophils and bacteria (lying either free in the exudate or within neutrophils) (Fig. 24.7) (176). The role of the neutrophil in eradicating infection at this stage is unknown. The presence of free-living bacteria in the exudate suggests that phagocytosis by neutrophils is incomplete as a result of deficient opsonic activity in CSF; however, low CSF leukocyte concentrations in the presence of high CSF bacterial concentrations have been associated with a poor prognosis in both experimental and human meningitis (178). These observations suggest that the neutrophils have a beneficial role in partial control of the early stages of the infection. The presence of large numbers of neutrophils in the SAS and vessel walls may, however, also be detrimental to the host, as is discussed in the previous section.

Within the first 48 to 72 hours of infection, there is evidence of inflammation in the walls of the small and medium-sized subarachnoid arteries (Fig. 24.8). The endothelial cells swell and multiply, narrowing the lumen. The adventitia is infiltrated by neutrophils, and neutrophils and lymphocytes form a layer beneath the intima (Fig. 24.9). Subintimal arterial infiltration by neutrophils and lymphocytes is relatively unique to infection of the meninges, and it may be related to the anatomy of the meningeal arteries. It is only rarely observed in inflammatory processes in other organs (175). The adventitia of the subarachnoid vessels, as they enter the brain parenchyma, is formed by the arachnoid membrane. As arteries and veins enter the brain parenchyma, they carry with them a sleeve of arachnoid immediately surrounding the vessels and a sleeve of pia mater external to this. Between these two layers lies an extension of the SAS, known as the perivascular space or Virchow-Robin space, which is filled with CSF (177) (Fig. 24.10). Because the vessel wall is enveloped by the arachnoid membrane, it is affected early by any inflammatory process in the meninges. However, as shown in animal models of bacterial meningitis, the arachnoid membrane generally remains intact.



The meningeal veins become distended and develop mural inflammation during bacterial meningitis. There may be focal necrosis of the vessel wall, along with mural thrombus formation in the lumen of the vein or in the dural sinus (175) (Fig. 24.11). Hemorrhagic cortical infarction is the result of cortical venous and dural sinus thrombosis.

Toward the end of the first week of meningeal infection, there is a change in the cellular composition of the subarachnoid exudate. Neutrophils begin to degenerate and are removed by macrophages, which are derived from meningeal histiocytes. Lymphocytes and fibroblasts proliferate in the exudate. Microscopic changes in the brain parenchyma may also be present. The nuclei of neurons and glial cells become shrunken, pyknotic, and darkly staining (Fig. 24.12). Rod-shaped microglial cells and astrocytes increase in number in the cerebral and cerebellar cortex, brainstem, and spinal cord (Fig. 24.13). Astrocytic processes become swollen (Fig. 24.14). There is a loss of myelinated fibers in the subcortical white matter, cerebellum, and brainstem (175). Similar morphologic changes are seen in ischemic and hypoxic cortical injury, suggesting that ischemia and/or hypoxia may contribute to the pathologic changes from bacterial meningitis at this stage.



Also at the end of the first week of the infection, there is infiltration of the subependymal tissues and perivascular spaces by neutrophils and lymphocytes. The ependymal and subependymal tissues become edematous, and cells begin to die; desquamation of the ependymal lining also occurs. Rod-shaped microglial cells and swollen astrocytes proliferate and overgrow the remnants of the ependymal lining. An inflammatory infiltrate in the walls of the subependymal arteries may occlude the vessel, leading to tissue necrosis (175).
As the infection progresses, the subarachnoid exudate continues to accumulate. In some areas, it may become several millimeters thick (Fig. 24.15). Toward the end of the second week, the exudate separates into two layers. The outer layer, just beneath the arachnoid membrane, is composed of neutrophils and fibrin. The inner layer, which is contiguous with the pia, is composed of lymphocytes, plasma cells, and macrophages (175). As the subarachnoid exudate continues to accumulate, the flow of CSF may become obstructed.

The dynamics involved in obstruction of CSF flow are as follows: the bulk of CSF is formed by the choroid plexus in the lateral and third ventricles, and it flows through the cerebral aqueduct into the fourth ventricle. CSF leaves the fourth ventricle through the midline foramen of Magendie and the lateral foramina of Luschka to reach the SAS (177). When the foramina of Magendie and Luschka are blocked by exudate, the spinal fluid cannot circulate to the convexities of the brain, where it is normally absorbed. The flow of CSF is blocked at the level of the fourth ventricle, resulting in noncommunicating or obstructive hydrocephalus (Fig. 24.16).

From the fourth ventricle, CSF normally flows to the SAS at the base of the brain. From here, CSF flows up over the convexity of the hemispheres to be absorbed by the arachnoid villi in the intracranial venous sinuses (177). The presence of a fibrinopurulent exudate in the SAS interferes with the absorption of CSF by the arachnoid villi. This obstruction to CSF resorption resulting from inflammatory changes in the arachnoid granulations results in communicating hydrocephalus. When the subarachnoid exudate has been present for several weeks, there are (a) marked fibrosis of the arachnoid villi and (b) pockets of exudate walled off by adhesions between the arachnoid membrane and dura (175). These fibrotic changes produce further mechanical obstruction to the resorption of CSF by the arachnoid villi. The end results are (a) transependymal movement of CSF from the ventricular system into the brain parenchyma and (b) the development of interstitial cerebral edema.
The development of diffuse cerebral edema and increased ICP further complicates the pathologic changes already described. Cerebral edema is defined as an increase in the volume of the brain resulting from an increased water content (179) (Fig. 24.17). The cerebral edema in meningitis is a combination of vasogenic, cytotoxic, and interstitial edema (170). Vasogenic edema is a result of increased permeability of brain capillaries with the subsequent accumulation of water and protein molecules in the extracellular space, mainly in the subcortical white matter. Cytotoxic edema is caused by an accumulation of intracellular water and sodium with subsequent swelling of cells. Membrane polyunsaturated fatty acids and other toxic factors released from leukocytes contribute to the development of cytotoxic edema (69). Interstitial edema is a result of obstruction to CSF resorption, as discussed earlier in this chapter.
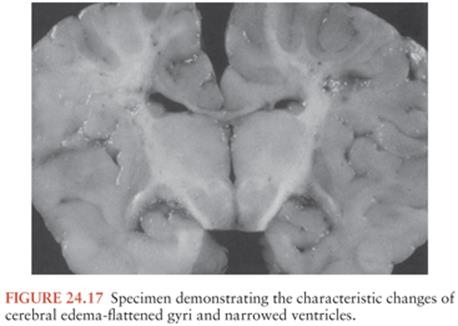
Cerebral edema leads to an increase in ICP, and increased ICP adversely affects cerebral perfusion pressure (CPP), defined as the difference between the systemic mean arterial pressure (MAP) and the ICP: CPP = MAP − ICP (180). Cerebral blood flow may be maintained at normal or near-normal levels in the presence of increased ICP, provided that the CPP is maintained at a range of at least 50 to 60 mm Hg. As ICP continues to rise, or if systemic arterial pressure decreases, cerebral ischemia and infarction may result.
Experimental evidence exists for a loss of autoregulation of cerebral blood flow in bacterial meningitis (181). This is another potential contributing factor to the development of cerebral ischemia in this infection. Cerebral blood flow is normally constant within a range of mean systemic arterial pressure from 50 to 150 mm Hg. When autoregulation is disturbed, systemic hypotension results in decreased cerebral blood flow and cerebral ischemia (182).
Cerebral edema may lead to herniation of brain tissue. Herniation may compress intracerebral arteries, leading to ischemia and infarction; it may also compress the surface of the brain against the dura, leading to necrosis of brain tissue. Tonsillar herniation, the downward displacement of the cerebellar tonsils through the foramen magnum, can result in apnea, hemodynamic instability, coma, and death (177,183).
The pathologic lesions described are typical of meningitis caused by bacteria, but some distinctions among lesions caused by H. influenzae, N. meningitidis, and S. pneumoniae infection have been observed. The subarachnoid exudate in H. influenzae meningitis is very thick and purulent, with loculated pockets of pus in the basilar cisterns and cerebral sulci. In contrast, the exudate in pneumococcal meningitis tends to be more extensive over the convexities of the hemispheres than in the basilar cisterns (Fig. 24.18). In meningococcal meningitis, the pathologic changes depend on the severity and duration of the infection. In acute fulminating meningococcemia, death may occur before pus can accumulate in the SAS. At autopsy, severe hyperemia and swelling of cerebral tissue are evident, with petechial hemorrhages in the gray and white matter (Fig. 24.19) and in the subependymal regions of the lateral ventricles. A hemorrhagic ependymitis is typical of severe lethal meningococcal infection (Fig. 24.20). The presence of pus in the SAS may be evident only by microscopic examination (183). The pathologic changes in meningococcal meningitis of longer duration are similar to those described for meningitis caused by pyogenic organisms in general.



Cranial and spinal nerve deficits, focal neurologic deficits, seizure disorders, and subdural effusions are well-recognized complications of meningitis. The cranial and spinal nerve deficits are usually transient and caused by exudate in the SAS surrounding the nerves. Focal neurologic deficits and seizure activity arise from cortical and subcortical ischemia and infarction (bland and hemorrhagic), which are the result of inflammation and thrombosis in arteries and veins. Subdural effusions are relatively common in the course of bacterial meningitis in children; they are the result of an increase in the permeability of the thin-walled capillaries and veins in the inner layer of the dura, with leakage of fluid into the subdural space.
Bacteremia also contributes to the pathologic features of this disease. Bacteremia is present in 30% to 90% of cases of bacterial meningitis. It can be either the primary event leading to development of meningitis or a secondary event arising from the clearance of bacteria from the SAS through the arachnoid villi to the bloodstream. Pneumococcal cell walls activate the alternative complement pathway, with the generation of chemotactic peptides in the systemic circulation and in CSF. The principal component of this activity is C5a, a highly chemotactic peptide that is a stimulus for an intense CSF accumulation of neutrophils. By this process, neutrophils also become sequestered in the pulmonary vascular bed, leading to cardiopulmonary dysfunction, neutrophil-mediated vascular damage, and the development of the acute respiratory distress syndrome and thereby further contributing to the morbidity and mortality of meningitis (69).
CLINICAL MANIFESTATIONS
Neonates
Clinical clues to the presence of meningitis in neonates are temperature instability (hypothermia or hyperthermia), listlessness, high-pitched crying, fretfulness, lethargy, refusal to feed, weak suck, irritability, jaundice, vomiting, diarrhea, or respiratory distress (184,185). Nuchal rigidity is not typically found in the neonate. A change in the child’s affect or state of alertness is one of the most important signs of meningitis. A bulging fontanelle (seen in one third of cases) usually occurs late during the course of illness; seizures are observed in 40% of neonates with bacterial meningitis.
Infants and Children
The symptoms and signs of acute bacterial meningitis in infants and children depend on the age of the child, duration of illness, and host response to infection (186); the clinical features can be subtle, variable, nonspecific, and even absent. The initial symptoms of bacterial meningitis in infants and children may be any of the following: fever, stiff neck, headache, lethargy, irritability, nausea, vomiting, and photophobia (Table 24.3). In children 1 to 4 years of age, fever (94%), vomiting (82%), and nuchal rigidity (77%) are the most common initial symptoms.

Although the symptoms are nonspecific, the combination of one or more of these symptoms with signs of meningeal irritation should suggest the diagnosis of meningitis. The classic signs of meningeal irritation are nuchal rigidity and Brudzinski and Kernig signs. Brudzinski actually described several signs of meningeal irritation, including the nape-of-the-neck sign, the identical contralateral reflex sign, and a reciprocal contralateral reflex sign, as well as others (187,188). The nape-of-the-neck sign is Brudzinski’s best-known sign and is universally recognized today as “Brudzinski sign.” The nape-of-the-neck sign is positive when passive flexion of the neck results in flexion of the hips and knees. The identical contralateral reflex sign is elicited with the patient in the supine position by passively flexing the hip and knee on one side. This sign is positive when the contralateral leg flexes with this maneuver. The reciprocal contralateral reflex sign is positive when the leg that has flexed in response to passive flexion of the other leg begins to extend spontaneously, resembling a “little kick.” The identical and reciprocal contralateral reflex signs are not elicited as often as the nape-of-the-neck sign (188). The manner in which Kernig sign is elicited and the interpretation of the results of the maneuver as it is done today are different from those originally described by Kernig (189). The maneuver as described by Kernig was performed with the patient in a seated position while the physician attempted to extend the knee passively. In the presence of meningitis, knee extension was resisted so that a “contracture of the extremities” was maintained (188,189). Today the sign is elicited with the patient in a supine position. The thigh is flexed on the abdomen, with the knee flexed. The leg is then passively extended. When meningeal inflammation is present, the patient resists leg extension (190). Nuchal rigidity and Brudzinski and Kernig signs are observed in fewer than 50% of children with bacterial meningitis.
The possibility of bacterial meningitis should be considered in every child with fever, vomiting, nuchal rigidity, and lethargy or an altered mental status. In a review of 110 cases of culture-proven bacterial meningitis in children, fever (≥38.5°C) was the most common symptom, being present in 94% of patients. The absence of fever, particularly hypothermia, was associated with a worse prognosis, perhaps related to the slower rate of bacterial replication in CSF when temperatures are elevated. Apart from fever, the most common symptoms were (a) vomiting (82%) and nuchal rigidity (77%) in 1- to 4-year-old children and (b) headache (92%) in children ages 5 to 12 years. Vomiting and nuchal rigidity were present in 80% of the children who were 12 months or older. Nuchal rigidity is a classic sign of meningitis but can be absent early in the course of this illness; therefore, the absence of nuchal rigidity should not exclude the diagnosis of bacterial meningitis (191).
In a review of 709 LPs done on children in an outpatient setting in which there was a concern for meningitis, the CSF was abnormal in 16% (192). There were 30 cases of bacterial meningitis, 70 of viral meningitis, and 12 of unknown etiology. Lethargy was more common in children with bacterial meningitis than in children with viral meningitis: 50% of the children with bacterial meningitis were lethargic, and 32% of the children with viral meningitis were lethargic (p >.14). Although vomiting is a symptom of meningeal irritation, it is a nonspecific symptom in children. Vomiting occurred in 336 children, 84 of whom had bacterial or viral meningitis. Fever was present in every child with meningitis. The temperature elevations were higher in bacterial meningitis than in viral meningitis; 80% of the children with bacterial meningitis had temperatures of 38.8°C or higher. In children with viral meningitis, 40% had temperatures of 38.8°C or greater (193). The possibility of meningitis in a child who does not or cannot complain of headache or stiff neck and who does not have meningeal signs should be suspected when fever accompanies changes in behavior, changes in mental status, or new onset of seizures.
In one recent review of children aged 2 months to 15 years who presented with suspected meningitis, the classic clinical signs had limited value in establishing the diagnosis (194). Clinical examination revealed nuchal rigidity in 65% of those with meningitis; Brudzinski and Kernig signs were elicited in 51% and 27% of those with meningitis, respectively. Therefore, physicians should have a low threshold for LP in patients at high risk for bacterial meningitis, given the serious nature of this disease.
In a review of 1,064 cases of bacterial meningitis in infants and children, there were no signs of meningeal irritation in 16 patients (1.5%). Eight patients were older than 2 years of age. LP was performed because of unexplained fever associated with an altered level of consciousness, behavioral changes, seizure activity, or petechial skin lesions. Meningitis was caused by N. meningitidis in seven patients, H. influenzae in six, S. pneumoniae in two, and Salmonella enteritidis in one. Most patients had a peripheral leukocytosis with a left shift. The peripheral WBC count was greater than 10,000 cells/mm3 in 12 patients and greater than 20,000 cells/mm3 in 7 patients (195). The results of this review suggest that although meningitis may occasionally occur without meningeal signs, there will usually be other signs or symptoms of intracranial infection and a peripheral leukocytosis.
Observational data that are useful in predicting the presence of serious illness (e.g., meningitis) in a febrile child include the following: (a) quality of cry, (b) reaction to parent stimulation, (c) level of consciousness, (d) color, (e) hydration, and (f) response to social stimulation. These six items were identified as significant and independent predictors of serious illness from a list of 14 observational items, scored by pediatricians, for 312 febrile children 24 months of age or younger (196). The quality of the cry in a child with a serious illness was weak, moaning, or high pitched. A healthy child was either not crying or had a strong cry with normal tone. Reaction to parental stimulation was judged based on the parent holding the child, talking to the child, or giving the child a bottle. The child with a serious illness did not stop crying or barely responded to stimulation by its parent. Consciousness was impaired in children with serious illnesses. They were lethargic, stuporous, or obtunded. Sick children were described as pale, cyanotic, or ashen. Signs of dehydration were present. The response to social stimulation was judged according to whether the child would smile when talked to or smiled at. Sick children did not respond to social stimulation. These six items, when used together, had a specificity of 88% and a sensitivity of 77% for the presence of serious illness. When combined with history and physical examination, the sensitivity of the six-item model increased to 92%. If all six of the observation items were normal in a child, the probability of that child having a serious illness was only 4.7% (196).
The possibility of meningitis in a febrile child may also be suggested by the tempo of the illness. The presentation of meningitis in children is that of either a subacute infection or an acute fulminant illness. Children with a subacute presentation have fever, lethargy, and nuchal rigidity that progresses over 1 to several days and is usually preceded by an upper respiratory tract infection or otitis media (197). Children with meningitis may also present with an illness that has been progressive over 24 to 72 hours or a fulminant illness that develops over several hours.
Children with a more rapidly progressive illness have signs and symptoms of meningeal irritation and increased ICP on initial presentation. CSF pressures exceeding 300 mm H2O are common in acute bacterial meningitis, and ICPs exceeding 500 to 600 mm H2O are not unusual (197,198). Increased ICP in bacterial meningitis in children is the consequence, in part, of vasogenic and cytotoxic cerebral edema, altered CSF resorption, and the inappropriate secretion of antidiuretic hormone (199) (see Chapter 23). The clinical manifestations of increased ICP include (a) an altered level of consciousness; (b) dilated, poorly reactive or nonreactive pupils; (c) abnormalities of ocular motility; (d) pathologically brisk lower extremity reflexes; and (e) bradycardia and hypertension, also known as Cushing reflex. The development of elevated ICP should be anticipated and monitored in a child with bacterial meningitis. The absence of papilledema does not exclude the presence of increased ICP. Papilledema is rarely observed early in the course of increased ICP and is usually not evident until increased ICP has been present for at least several hours (197,198). For this reason, the presence of papilledema at the time of the initial presentation should raise suspicion of a focal intracranial process such as a brain abscess or other localized mass lesion, and it is an indication for CT prior to LP.
Seizures occur in 30% to 40% of children with acute bacterial meningitis, usually during the first 3 days of illness (200). In one review of 52 cases of H. influenzae meningitis in children, seizures occurred in 44% (23 cases) (198). There has been a long-standing controversy about whether to do an LP in febrile children with new-onset seizures. The vast majority of children who present with a new-onset seizure associated with fever and who have a normal neurologic examination do not have meningitis. One series reviewed the results of LP performed on 328 children presenting with their first febrile convulsion. None of the children had meningeal signs. Meningitis was diagnosed by LP in four children (1.2%). Three of the children had viral meningitis, and one had H. influenzae meningitis. All four children were younger than 18 months of age. All the children in this series who were older than 18 months of age had unequivocal signs of meningitis (201). A similar observation was made in a review of LP performed on 304 children for evaluation of new-onset seizures associated with fever. There were 15 cases of meningitis, and in only one case were there no meningeal signs. In that case, the child had viral meningitis and recovered fully without treatment (202). These studies suggest that LP should not necessarily be routinely performed in children for evaluation of simple febrile convulsions in the absence of meningeal signs.
Convulsive seizure associated with fever is a problem unique to young children. A simple febrile seizure, as defined by the Consensus Development Meeting on Long-term Management of Febrile Seizures (1980), occurs between ages 3 months and 5 years in association with fever and is of brief duration (<15 minutes), nonfocal, nonrepetitive, and without associated neurologic deficits. If the seizure fits this definition and the child is awake and alert after the seizure, the yield of an LP is very low. If, however, there are clinical signs of meningitis, an LP is indicated. If the seizure has a focal onset or there is a focal neurologic deficit on examination, CT is indicated before LP is performed. All children with new-onset febrile convulsions in whom LP is not performed should be reexamined 1 to 4 hours after the initial examination to be sure that serious disease is not present (203).
The presence of a diffuse erythematous maculopapular rash may be an early manifestation of meningococcemia or may represent a viral illness. The presence of a purpuric or petechial rash on the trunk and lower extremities is suggestive of meningococcemia, although petechiae are sometimes seen in echovirus type 9 meningitis, acute staphylococcal endocarditis, and rarely pneumococcal or H. influenzaemeningitis (198,204). Petechiae are found in the skin, mucous membranes, or conjunctivae, but never in the nailbeds, of patients with meningococcemia; they usually fade in 3 or 4 days (205). Petechiae and/or purpura occurs in 50% to 75% of children with meningococcal meningitis. Children with fulminating meningococcal septicemia may have the Waterhouse-Friderichsen syndrome, characterized by the following: (a) sudden onset of a febrile illness, (b) large petechial hemorrhages in the skin and mucous membranes, (c) cardiovascular collapse, and (d) disseminated intravascular coagulation. Of all patients with a meningococcal infection, 10% to 20% have a fulminant meningococcal septicemia (206) (Color Figs. 24.21 to 24.26).






Focal neurologic signs, such as cranial nerve palsies with abnormalities of ocular motility, hemiparesis, visual field defects, and ataxia, may occur early or late in the course of bacterial meningitis in approximately 15% of children (197). Cranial nerve palsies likely develop as the nerve becomes enveloped by exudate in the arachnoidal sheath surrounding the nerve. Alternatively, cranial nerve palsies may be a sign of increased ICP. The presence of bilateral palsies of the sixth cranial nerve, manifested as weakness of lateral rectus muscles, is a well-recognized sign of increased ICP.
Hemiparesis may be caused by vasculitis and cerebral infarction or may be a sign of the presence of a large subdural effusion. Subdural effusions commonly develop in the course of bacterial meningitis in children and are not usually associated with clinical symptomatology. Subdural effusions develop when the infection in the adjacent SAS leads to an increase in the permeability of the thin-walled capillaries and veins in the inner layer of the dura. The result is leakage of albumin-rich fluid into the subdural space, usually a self-limited process. When the inflammatory process subsides, fluid formation usually ceases and the fluid in the subdural space is resorbed (197). Some subdural effusions are, however, clinically significant. The development of a hemiparesis or increased ICP may be the consequence of an enlarging subdural effusion causing mass effect. The presence of a prolonged fever in a child with a subdural effusion suggests that the effusion has become infected.
Ataxia may be the presenting sign of bacterial meningitis in a child. Ataxia is a sign of vestibular dysfunction, and in this clinical setting, it suggests the presence of labyrinthitis. In most children, it is a transient symptom; however, it has implications for prognosis, because auditory and vestibular disturbances usually occur together. As such, ataxia is associated with postmeningitic hearing loss (207).
Children with meningitis may develop hyponatremia and the syndrome of inappropriate secretion of antidiuretic hormone (SIADH) (208). The symptoms of hyponatremia and/or SIADH are lethargy, stupor, confusion, and/or seizure activity. When the following criteria are met, the diagnosis of SIADH should be considered: (a) serum sodium level less than 135 mEq/L, (b) serum osmolality less than 270 mOsm/L, (c) urine osmolality greater than two times the serum osmolality, (d) urine sodium greater than 30 mEq/L, and (e) no clinical evidence of hypovolemia or dehydration (209). SIADH is not the only cause of hyponatremia in children with bacterial meningitis. Hyponatremia may also develop when fluid therapy is too aggressive or as a result of the syndrome of cerebral salt casting. Regardless of the reason for hyponatremia, the serum sodium level should be monitored frequently in every child with bacterial meningitis. This is discussed in greater detail later in this chapter (see the section “Therapy”).
Ocular complications, including fundal abnormalities, cranial nerve palsies (see earlier discussion), pupillary dysfunction, and corneal or conjunctival lesions, are actually quite common during the course of bacterial meningitis in children, but only one case of transient cataract formation has been reported in association with meningococcal meningitis.
Bacterial meningitis is at least 30-fold more common in children with cochlear implants to address profound hearing loss than in controls (210). The major pathogen is S. pneumoniae and risk factors include use of a positioner to improve transmission of the electrical signal by pushing the electrode against the medial wall of the cochlea (voluntarily withdrawn from the market by the manufacturer in July 2002); and joint radiographic evidence of inner ear malformation and CSF leak. More cases occurred within 30 days of surgery, but sporadic cases were observed 24 months after implantation (210).
Adults
The typical presentation of bacterial meningitis in an adult is that of an upper respiratory tract infection during which a meningeal symptom, such as headache, nuchal rigidity, vomiting, or an altered level of consciousness, develops (211) (Table 24.3). The clinical signs and symptoms of bacterial meningitis in an adult are very similar to those in children, with a few exceptions. The most common bacterial cause of meningitis in adults is S. pneumoniae. The clinical presentation of meningitis caused by S. pneumoniae is somewhat different from that of meningitis caused by the meningococcus or H. influenzae. Adults with pneumococcal meningitis usually have an altered mental status on admission and rapidly become stuporous or comatose. In addition, recurrent seizure activity and focal neurologic deficits are more common in the early stages of pneumococcal meningitis than in meningococcal or H. influenzaemeningitis. These and other factors may contribute to the continued high mortality rate in pneumococcal meningitis. S. pneumoniae is associated with the highest mortality rate of the major meningeal pathogens. For example, a mortality of 36.8% was reported from one series of 55 cases of community-acquired pneumococcal meningitis (212). Death is more likely to occur in patients of advanced age, in the absence of meningismus, and in the presence of pneumonia, other extraneural complications, or a prolonged duration of illness prior to therapy (7 days). Nevertheless, it appears that most deaths occur later in the disease course as a consequence of cardiorespiratory insufficiency (213). They are not usually caused by early overwhelming CNS damage.
A classic description of the clinical presentation of bacterial meningitis in adults was presented by Carpenter and Petersdorf (211) in 1962. This review includes 209 cases of bacterial meningitis: 53 cases were caused by meningococci, 63 by pneumococci, 35 by H. influenzae, and 58 by other bacterial organisms. A reliable history of the onset of symptoms was obtained in 134 patients. Thirty-six patients (27%) had sudden onset of headache, confusion, lethargy, and loss of consciousness and sought hospitalization within the first 24 hours. Only 3 of these 36 patients had symptoms of respiratory tract infection. In contrast, 71 (53%) of the 134 patients had more slowly progressive symptoms of meningitis for 1 to 7 days before admission. Of these, 26 patients (37%) had respiratory symptoms. In 27 (20%) of the 134 patients, an infection in the respiratory tract developed 1 to 3 weeks before the first symptom of meningitis. The results of physical examination for meningeal signs were recorded in 199 cases. Either nuchal rigidity, Kernig signs, or Brudzinski signs were present in 161 patients (81%). Studies suggest that the latter two signs are less common in adults with meningitis than previously reported (214). Level of consciousness on admission was recorded in 191 patients (96%); only 9 (5%) patients were alert, 48 (24%) were lethargic, 44 (22%) were confused, and the remainder were obtunded or comatose (211). Patients with meningococcal meningitis were most often alert, and those with pneumococcal meningitis were more often obtunded.
Geiseler et al. (215) made observations of altered consciousness at presentation in bacterial meningitis, similar to those of Carpenter and Petersdorf (211). They recorded level of consciousness at the time of admission in 1,289 patients with community-acquired bacterial meningitis. Overall, 230 (17.8%) were alert, 672 (52.1%) were irritable or lethargic, 262 (20.3%) were stuporous or obtunded, and 125 (9.7%) were comatose and/or convulsing (215). In adults, as in children, lethargy or an altered mental status is the strongest indicator of bacterial meningitis.
Durand et al. (78) reviewed the charts of all cases of acute bacterial meningitis in individuals 16 years or older at the Massachusetts General Hospital from 1962 through 1988. The most common pathogen of community-acquired meningitis was S. pneumoniae. Ninety-five percent of patients had fever (temperature 37.7°C) on presentation. Neck stiffness was present in 88% of patients on initial physical examination. At the time of admission, 61 (22%) were alert, 142 (51%) were confused or lethargic, 61 (22%) were responsive only to pain, and 17 (6%) were unresponsive to all stimuli. On presentation or during the first 24 hours, 81 (29%) had focal seizures or focal neurologic findings. The most common predisposing factors for community-acquired meningitis were pneumonia, sinusitis, acute otitis media, altered immune state, alcoholism, and diabetes mellitus (78).
In another review of 696 episodes of community-acquired bacterial meningitis, the triad of fever, neck stiffness, and altered mental status was present in only 44% of episodes (216), although almost all patients (95%) presented with at least two of four symptoms (i.e., headache, fever, stiff neck, and altered mental status). Cerebrovascular complications may manifest at any time during the course of pneumococcal meningitis. In an observational study of 696 patients with community-acquired bacterial meningitis, cerebral infarction occurred in 174 (25%) episodes and was seen in 128 (36%) of 352 patients with pneumococcal meningitis (217).
Pneumonia is present on admission in 25% to 50% of adults with pneumococcal meningitis (211). Acute and chronic otitis media are also predisposing conditions for pneumococcal meningitis. In one series of 178 patients with pneumococcal meningitis, acute otitis media was present in 59 (33.1%) (215). When pneumonia or otitis media is not present, the possibility of a dural sinus fistula should be considered. S. pneumoniae is the most common causative agent in meningitis following head trauma (basilar skull fracture) or meningitis associated with a structural defect (either congenital or traumatic in origin) that creates a communication between the paranasal sinuses, nasopharynx, or middle ear and the SAS (218).
In adults aged 15 to 60 years, underlying host factors may increase the risk for meningitis while simultaneously blunting its presenting signs. Such predisposing factors include malignancy, alcoholism, sickle cell disease, diabetes, organ transplantation, splenectomy, high-dose steroid therapy, and long-term dialysis. In these clinical settings, the symptoms of meningitis may include altered sensorium, persistent headache, or new-onset seizures. Fever or nuchal rigidity may not develop (219).
The occurrence of H. influenzae meningitis in an adult should prompt consideration of the presence of (a) otitis media, (b) paranasal sinusitis, (c) other parameningeal foci of infection, (d) CSF leak from previous head trauma, or (e) a concurrent pneumonia, pharyngitis, or immunodeficiency disease. The clinical presentation of H. influenzae meningitis in adults is typical of bacterial meningitis and includes headache, fever, altered mental status, and nuchal rigidity (220).
Elderly
Meningitis should be suspected in every elderly patient who is febrile and either disoriented, stuporous, or comatose (Table 24.3). In a review of 54 cases of bacterial meningitis in the elderly, confusion was present in 92% (12 of 13) of the patients with pneumococcal meningitis and in 78% (7 of 9) of those with gram-negative meningitis on initial presentation. This review compared the clinical presentation of bacterial meningitis in the elderly (patients aged 50 years and older) with that in younger patients (aged 15 to 49 years). On initial presentation, the incidence of more severe abnormalities of mental status in the older age-group with bacterial meningitis was statistically different from that of the younger group, and concurrent pneumonia was present more often in the older patients than in the younger group (221). In another review of CNS infections in patients older than 65 years of age at the Mount Sinai Hospital in New York from 1970 through 1985, 28 cases of bacterial meningitis were identified (222). Although fever (often of low grade) was uniformly present, only 57% had meningismus and only 21% complained of headache. Pneumococci were the most common etiologic agent, and these cases were often (58%) accompanied by pneumonia, sinusitis, or otitis media; the overall mortality was approximately 40%.
Most elderly patients with meningitis have nuchal rigidity, that is, resistance to passive flexion of the neck. Resistance to passive movement of the neck is a common physical finding in elderly patients because of the presence of cervical spondylosis. It is important to be able to distinguish between the cervical rigidity of cervical spondylosis and nuchal rigidity resulting from meningitis. In nuchal rigidity, the neck resists flexion but can be passively rotated from side to side. In rigidity consequent to cervical spinal disease, lateral rotation, extension, and flexion of the neck are all associated with resistance. Similarly, hypertonicity of the neck muscles resulting from disease of basal ganglia, such as parkinsonism, can be distinguished from true nuchal rigidity.
Specific comments should be made about the presentation of nontraumatic, spontaneous gram-negative bacillary meningitis in the elderly or debilitated patient. In these patients, the classic signs and symptoms of meningitis may be subtle at initial presentation. They may have only low-grade fever and altered mental status without headache or nuchal rigidity; however, patients with spontaneous gram-negative bacillary meningitis tend to have a rapidly progressive fulminant course associated with bacteremia, shock, and coma (223). The elderly patient with gram-negative meningitis may rapidly become comatose after presenting with what at first appeared to be a minor illness. Once coma develops, nuchal rigidity may not be present, because this sign is lost during deep coma.
In a recent 30-year study of 185 patients 65 years of age and older, the diagnosis of community-acquired bacterial meningitis was more difficult because of the absence of characteristic meningeal signs (224); compared with adult patients younger than 65 years of age, the older patients showed greater neurologic severity with a high number presenting with coma on admission, seizures, and hemiparesis.
Posttraumatic Meningitis
Bacterial meningitis may develop following a traumatic head injury that produces a dural fistula between the SAS and the nasal cavity, paranasal sinuses, or middle ear. The infection may develop shortly after the injury or may not occur until months to years later (225). Traumatic head injury is the most common cause of recurrent meningitis in the adult (220). Conversely, congenital fistulous connections to the CNS, often via the middle ear in association with Mondini dysplasia, are the most common underlying process in children with recurrent bacterial meningitis (226). An immunodeficiency state may also be instrumental in the development of this syndrome.
A dural fistula develops when the force of the injury is sufficient to fracture bone and tear the dura. The most common site for dural fistula is in the anterior cranial fossa, in the area of the cribriform plate. Here, the bone is very thin and the dura is tightly adherent to the bone. A fracture in this area allows CSF to leak through torn arachnoid and dura into the nasal cavity, resulting in CSF rhinorrhea (225). There may also be loss of the sense of smell, or anosmia. CSF rhinorrhea can be distinguished from nasal secretions by testing the fluid for β2-transferrin.
Physical signs indicating a basilar skull fracture with the potential for development of a dural fistula and meningitis include periorbital ecchymoses, bruising behind the ear (Battle sign), hemotympanum, and/or blood in the external auditory canal (225).
In most patients, CSF rhinorrhea ceases spontaneously. Approximately one in four patients with CSF rhinorrhea develops meningitis (225); the reported frequency ranges from 9% to 36%. Surgery is indicated in patients who develop meningitis with persistent rhinorrhea. The management of meningitis occurring in this setting and the approach to demonstration of the location of the dural fistula is discussed later in this chapter.
Meningitis Following Neurosurgical Procedures
Meningitis complicating a neurosurgical procedure, such as a craniotomy, is usually insidious in onset and difficult to distinguish clinically from the neurologic abnormalities expected in the postoperative period. Although an altered level of consciousness and signs of meningeal irritation may be expected in the postoperative period, the presence of fever or prolonged obtundation should prompt an examination of the CSF.
Approximately 60% to 70% of all cases of meningitis complicating a neurosurgical procedure, with the exception of a shunting procedure, are caused by gram-negative bacilli (223). The remainder is caused by staphylococci, predominantly S. aureus. In the postneurosurgical patient, K. pneumoniae, Acinetobacter baumannii, and E. coli are the most common infecting gram-negative organisms. Craniotomy for trauma or for tumor represents the most common surgical procedure associated with postoperative gram-negative bacterial meningitis (227).
Although the surgical insertion and subsequent constant presence of an indwelling ventriculoperitoneal (VP) or ventriculoatrial (VA) shunt catheter for decompression of hydrocephalus allow bacteria to enter the CSF space, signs of meningitis usually do not accompany these infections in the early stages. The bacteria involved in early shunt infection gain entry to the lumen of the shunt from a contaminated wound or from the patient’s skin surface at the time of operation (228). The initial symptoms of shunt infection are nonspecific and include fever, nausea, vomiting, and lethargy. Fever is the most common manifestation of shunt infection. Virtually all patients have temperatures greater than 37.8°C, and most have temperatures of 38.8°C or more (229). Fever is often the sole manifestation of infection in patients with VA shunts, whereas patients with infected VP shunts are more likely to present with signs of shunt malfunction and/or signs of inflammation around the shunt reservoir or along the course of the tubing (229,230). Signs of shunt malfunction are secondary to progressive hydrocephalus and, in children, include enlarging cranial circumference, tense nonpulsatile fontanelle, and papilledema. Signs of shunt malfunction may be associated with signs of meningitis. Results of examination of CSF from the lumbar area may be negative; therefore, CSF should be obtained by aspiration from the infected shunt reservoir.
Infections of the CNS may also develop when subcutaneous CSF reservoirs, such as Ommaya and Rickham reservoirs, are placed for therapeutic purposes. These and other types of indwelling intraventricular catheters may lead to meningitis with coagulase-negative staphylococci, S. aureus, Corynebacterium species, or gram-negative bacilli. Infections usually occur within the first 3 months after insertion of the device and, as with infections of VP or VA shunts, are probably the consequence of contamination by skin flora during implantation or subsequent use for therapeutic purposes. In these patients, signs of meningitis are usually not present, but most will complain of fever, lethargy, headache, or nausea and vomiting (231).
Immunosuppressed Hosts
The risk for development of bacterial meningitis in an immunocompromised patient depends on a number of factors, such as the underlying disease and its treatment, the duration of immunosuppression, and the type of immune abnormality (232). There are four major types of host defense abnormalities in the immunocompromised patient: (a) defects in T-lymphocyte–macrophage function (cell-mediated immunity); (b) defects in humoral immunity; (c) defects in the number and function of neutrophils; and (d) loss of splenic function from surgery, disease, or radiotherapy, resulting in the inability to remove encapsulated bacteria. Knowledge of the type of host defense abnormality is often helpful in predicting the infecting organism (233,234).
Patients with defects in cell-mediated immunity include (a) those with lymphomas, particularly Hodgkin disease; (b) organ transplant recipients; (c) patients treated with daily corticosteroid therapy; and (d) patients with AIDS. These patients are most susceptible to CNS infection by microorganisms that are intracellular parasites, the eradication of which depends on an intact T-lymphocyte–macrophage system (233). L. monocytogenes is a causative organism of bacterial meningitis in patients with defective cell-mediated immunity due to hematologic malignancies, organ transplantation, pregnancy, chronic corticosteroid therapy, alcoholism, and advanced age (235). The incidence of L. monocytogenes meningitis in HIV-infected individuals is low due to pneumocystis prophylaxis with TMP-SMX. The clinical presentation of L. monocytogenes meningitis includes fever and headache, as well as an increased tendency for focal neurologic deficits and seizures during the initial presentation. Meningitis caused by this organism may also present with a clinical picture suggestive of an acute brainstem disorder or rhombencephalitis, with signs of ataxia, cranial nerve deficits, and nystagmus (236).
Patients with defective humoral immunity are unable to mount an antibody response to bacterial infection, and they are, therefore, unable to control infection caused by encapsulated bacteria. Patients with this type of host defense abnormality include those with chronic lymphocytic leukemia, multiple myeloma, or Hodgkin disease following chemotherapy and radiotherapy, among others. These patients are at particular risk for meningitis caused by S. pneumoniae, Hib, and less commonly N. meningitidis. The presentation of meningitis in these patients is often that of a fulminant illness resulting in death in several hours.
Patients with splenectomy may develop (a) overwhelming bacteremia and fulminant meningitis with the same organisms, resulting from loss of the filtering function of the splenic sinusoids in removing encapsulated bacteria from the bloodstream, and (b) a reduced ability to produce IgM opsonizing antibodies (233,234).
Patients with neutropenia are at particular risk for meningitis caused by P. aeruginosa and members of the Enterobacteriaceae family (234). The clinical presentation of bacterial meningitis in patients with neutropenia may be subtle, consisting of low-grade fever and lethargy or a change in headache pattern (233). Signs of meningeal irritation depend on the host’s ability to mount an inflammatory response; therefore, in the neutropenic patient they are often minimal.
DIFFERENTIAL DIAGNOSIS
Although the diagnosis of meningitis is usually made by examination of the CSF, the decision to perform spinal fluid analysis is based on the clinical presentation. When the signs and symptoms suggest meningitis, and the decision is made to examine the CSF, the next step is to be certain that a focal intracranial mass lesion does not exist that may predispose to brain herniation following LP. If the history and neurologic examination suggest a focal mass lesion, then LP should be delayed until a neuroimaging procedure, either a cranial CT scan, without and with contrast enhancement, or a cranial magnetic resonance imaging (MRI) scan is obtained.
LP is relatively contraindicated in the presence of a focal mass lesion because of the danger of brain herniation. However, it has become common practice to delay LP until a CT or MRI scan has been obtained despite the absence of focal neurologic deficits by history or examination. The time involved in waiting for a CT or MRI scan significantly delays treatment, and delay in treatment is the most critical factor in determining morbidity and mortality in bacterial meningitis. Therefore, if a CT or MRI scan is to be performed, antimicrobial therapy should be initiated promptly, pending results. In the absence of an altered level of consciousness, focal neurologic signs, and/or papilledema, an LP can be safely performed without first obtaining a CT or MRI scan. Although CT is commonly performed before LP in adults with suspected meningitis, the vast majority of scans are unnecessary and unlikely to reveal abnormalities, and clinical characteristics can be used rapidly to exclude patients that are unlikely to have abnormal findings on CT (237). Focal infectious lesions that have clinical presentations similar to those of meningitis and that can result in significant morbidity if LP is unknowingly performed include brain abscess, subdural empyema, and epidural abscess. The clinical presentation of each of these disorders has similarities and distinguishing features when compared with that of meningitis.
The most common symptom of a brain abscess is a hemicranial or generalized headache, generally seen in 70% to 75% of patients (238–242). A brain abscess presents as an expanding intracranial mass lesion rather than as an infectious process; as such, fever is present in only 45% to 50% of patients and usually is not prominent. More than 50% of patients have focal neurologic deficits, and one third of patients present with new-onset focal or generalized seizure activity. The findings on neurologic examination are related both to the site of the abscess and to the presence of raised ICP caused by an expanding mass lesion. Hemiparesis is the most common sign of a frontoparietal lobe abscess. A disturbance of language or behavior or an upper homonymous quadrantanopia is the sign of a temporal lobe abscess. Ataxia is the most common sign of a cerebellar abscess. Nuchal rigidity rarely occurs until the abscess has ruptured into the ventricle or until infection has spread to the SAS. Sudden worsening of the headache, accompanied by new-onset meningismus, may signify rupture of the abscess into the ventricular space (243).
Most patients with a subdural empyema initially complain of headache that is localized to the side of the subdural infection. The headache becomes increasingly more severe and generalized, and it is followed by an alteration in the level of consciousness. Fever, chills, and nuchal rigidity are present in most cases. Focal neurologic deficits are present in 80% to 90% of patients, and they include hemiparesis or hemiplegia, paralysis of horizontal gaze to the side opposite the lesion, and focal or generalized seizures (244). The diagnosis should be considered in patients with acute bacterial sinusitis in combination with severe intractable headache, varying degrees of altered level of consciousness, focal neurologic deficits, and/or signs of meningeal irritation (245,246). The presentation of a posterior fossa subdural empyema includes severe headache, vomiting, marked nuchal rigidity, cranial nerve deficits, and pupillary abnormalities (247); cerebellar signs were elicited in only 40% of patients in one study (248).
A typical presentation of an intracranial epidural abscess is an unrelenting hemicranial headache and fever that have developed during or after treatment for frontal sinusitis, mastoiditis, or otitis media. If the abscess is large, mild alterations of consciousness may occur; however, focal neurologic deficits, seizures, and signs of increased ICP do not develop until the infection has extended into the subdural space or a deeper intraparenchymal complication has occurred (244).
The decision to delay LP until CT or MRI scan is obtained may be made when the patient does not appear to be seriously ill or when there is uncertainty about the findings of neurologic examination. Patients with viral meningitis usually do not appear as ill as patients with bacterial meningitis and often have had symptoms for several days. When the history suggests a focal onset to the headache or a transient neurologic symptom, LP is best delayed until a CT or MRI scan has been obtained.
The initial symptoms of viral meningitis are fever, headache, lethargy, myalgias, and nuchal rigidity. There are several distinguishing clinical features of viral meningitis: (a) viral meningitis has a more insidious onset and a slower progression than bacterial meningitis; (b) patients with viral meningitis often complain of an incapacitating headache that is not relieved by analgesics, but they are otherwise awake and alert; (c) the fever is usually higher in bacterial meningitis than in viral meningitis; and (d) although generalized malaise may be present, stupor, obtundation, and coma do not occur in viral meningitis (219).
Altered level of consciousness, focal neurologic deficits, and new-onset seizure activity are symptoms of a viral encephalitis, meningoencephalitis, or bacterial meningitis. The presentation of herpes simplex virus (HSV) encephalitis is often subacute and on examination is characterized by (a) fever, confusion, or a change in behavior; (b) new-onset seizure activity; and/or (c) focal neurologic deficits. A history of hemicranial headache of several days’ duration, preceding the onset of the confusional state, is a classic presentation of this illness. HSV has a predilection for the temporal and orbitofrontal areas; therefore, a change in mentation or behavior is a common finding (249).
Signs and symptoms of meningitis represent the most common neurologic presentation of Lyme disease. Patients have headache, stiff neck, low-grade fever, a unilateral or bilateral (in 25% of cases) facial nerve palsy, or a radiculitis. The characteristic skin lesion of Lyme disease, erythema migrans (EM), precedes the symptoms of meningitis in approximately 80% to 90% of patients. Signs and symptoms of meningitis occur weeks to a few months after the initial infection, or they may be the first manifestation of the disease without antecedent EM (250).
The presence of a rash with meningitis suggests meningococcemia. As has been discussed, the classic lesions associated with fulminating meningococcal septicemia are large petechial hemorrhages in the skin and mucous membranes. Between 50% and 75% of children with meningococcal meningitis have a purpuric or petechial rash, principally on the trunk and lower extremities. Petechiae are found in the skin, mucous membranes, and conjunctivae, but not in the nailbeds, in meningococcemia. Petechiae are also sometimes seen on the trunk and extremities in echovirus type 9 meningitis, acute staphylococcal endocarditis, and rarely pneumococcal or H. influenzae meningitis except in asplenic patients (198,204). Petechiae may be found in the nailbeds in acute staphylococcal endocarditis. Petechial rashes should be promptly examined microscopically in the initial evaluation of meningococcemia after aspiration or after making a “touch preparation” on a glass slide; approximately 70% of these preparations will reveal the organisms, usually within vacuolated neutrophils. In fulminant meningococcemia, the organisms may be visualized in the peripheral blood smear. Although the sensitivity of this method is low, this simple test should always be performed in suspected meningococcemia.
Headache, fever, rash, and altered mental status are symptoms of rickettsial infections (see Chapter 27) and, as such, enter into the differential diagnosis of meningitis. A petechial rash is characteristic of Rocky Mountain spotted fever (RMSF), which is caused by Rickettsia rickettsii. The rash of typhus is a faint macular-papular pink rash (251). The rash of RMSF consists initially of 1- to 5-mm pink macules that are often noted first on the wrist and ankles and then spread centrally to the chest, face, and abdomen. The rash of RMSF usually does not involve the mucous membranes. Petechial lesions in the axillae and around the ankles, accompanied by lesions on the palms and soles of the feet, are characteristic of RMSF, but this classic pattern is often absent. The macules will initially blanch with pressure, but after a few days they become fixed and turn dark red or purple. Diagnosis can be made by biopsy of the lesions and staining of the specimen with fluorescent antibodies to R. rickettsii (252). A negative result does not exclude RMSF, because sensitivity of this test is only 70%.
The characteristic rash caused by an enterovirus consists of erythematous macules and papules on the face, neck, trunk, and to a lesser degree the extremities. Rarely, the rash associated with enteroviral infection may become petechial in nature.
The rash of Lyme disease, EM, begins as a red macule or papule at the site of the tick bite. It then expands centrifugally as an erythematous lesion with central clearing. This may be the only lesion, or the disease may disseminate to form multiple secondary ringlike lesions. Symptoms of meningitis may develop while the skin lesions are still present, or they may begin 1 to 6 months after the skin lesions have resolved (250).
Noninfectious neurologic disorders that have clinical presentations similar to those of meningitis are subarachnoid hemorrhage, neuroleptic malignant syndrome, and posterior fossa tumors. The classic presentation of a subarachnoid hemorrhage is the sudden onset of a severe, excruciating headache, or a sudden transient loss of consciousness followed by a severe headache. Most patients complain of vomiting. Syncope accompanies the explosive onset of headache in about 50% of cases. Nuchal rigidity develops within a few hours of the onset of a subarachnoid hemorrhage and is usually associated with a change in the level of consciousness. Low-grade fever may develop within several days. When an intracranial aneurysm ruptures into the brain parenchyma, a focal neurologic deficit is usually present. A unilateral palsy of the third nerve, with a dilated, nonreactive pupil, is suggestive of third nerve compression by an aneurysm at the junction of the posterior communicating artery and the internal carotid artery. The triad of headache, neck stiffness, and vomiting should raise suspicion of a warning leak from an aneurysm (252).
The symptoms of neuroleptic malignant syndrome (NMS) are fever, generalized “lead-pipe” rigidity (including cervical rigidity), fluctuating level of consciousness (ranging from agitation to stupor and coma), and autonomic instability. The latter is characterized by pallor, unstable blood pressure, diaphoresis, tachycardia, and arrhythmias. A leukocytosis of 15,000 to 30,000 cells/mm3, with a shift to the left, is common. Liver function abnormalities are usually seen, but the most specific laboratory abnormality in this disorder is marked elevation in the serum creatine kinase (CK) concentration, usually exceeding 10,000 IU/L (253).
Signs of a posterior fossa tumor are stiff neck, cranial nerve abnormalities, gait disturbance, vomiting, cerebellar deficits, and occasionally an altered level of consciousness.
A cranial CT or MRI scan and examination of the CSF will narrow the differential diagnosis. The possibility of the presence of increased ICP should be considered before LP. Increased ICP is an expected complication of bacterial meningitis and is not a contraindication to LP.
The clinical signs of increased ICP are (a) a dilated, nonreactive pupil; (b) drowsiness; (c) abnormalities of ocular motility, the most common of which are the consequence of unilateral or bilateral palsies of the sixth cranial nerve; and (d) bradycardia and hypertension, the Cushing reflex. Pupillary dilation is usually secondary to parenchymal midbrain distortion from either raised ICP or transtentorial herniation. Drowsiness or stupor is often the first sign of increasing ICP and is caused by interference with the reticular activating system in the brainstem.
If raised ICP appears likely and a focal intracranial mass lesion has been excluded by CT or MRI scan, LP can usually be safely performed. When the decision is made to delay LP until a CT or MRI scan has been obtained, blood cultures should be obtained and intravenous antibiotics and dexamethasone therapy begun while awaiting results of CT or MRI scan (see later discussion). Intravenous antibiotics usually do not sterilize the CSF in the time it takes to obtain a CT or MRI scan and spinal fluid. Blood cultures may identify the infecting organism in 50% to 80% of cases of bacterial meningitis (although this frequency varies with the causative organism), and they are more often positive in patients who have not received prior treatment with oral antibiotics (189).
LABORATORY DIAGNOSIS
Cerebrospinal Fluid
The typical CSF findings of bacterial and aseptic meningitis are compared in Table 24.4.

Opening Pressure
The first step in examination of the CSF is measurement of the opening pressure with an air-water manometer. This step is often neglected, but knowledge of the presence of raised ICP is important in management of the patient. Normal CSF pressure, with the patient in the lateral recumbent position, is usually defined as less than 180 mm H2O (254). However, normal opening pressure can be as high as 250 mm H2O in obese patients (255). CSF pressure should not be measured with the patient in a seated position. If the spinal needle is inserted with the patient seated, the patient should then be moved to a lateral recumbent position and the opening pressure recorded. Elevated CSF pressure in the range of 200 to 500 mm H2O is common in bacterial meningitis.
Appearance
Normal CSF is clear. The presence of more than 200 WBCs/mm3, more than 400 red blood cells (RBCs)/mm3, bacteria (>105 CFU/mL), or an elevated protein concentration makes the fluid appear cloudy or turbid. When the LP is traumatic and the initial CSF sample appears bloody, the fluid should clear as flow continues. Xanthochromia refers to a yellow or yellow-orange color in the supernatant of centrifuged spinal fluid; it may be used to distinguish CSF that is bloody secondary to subarachnoid hemorrhage from CSF that is bloody as a result of a traumatic LP. When CSF is bloody secondary to traumatic LP, the supernatant of the centrifuged fluid is clear. In subarachnoid hemorrhage, the supernatant is xanthochromic within 2 hours after the hemorrhage. Elevated CSF protein concentrations (>150 mg/dL) also cause xanthochromia (254,256) and are the usual reason for xanthochromia in bacterial meningitis.
Glucose
The normal CSF glucose concentration is greater than 45 mg/dL. A glucose concentration of less than 40 mg/dL occurs in approximately 58% of patients with bacterial meningitis (257). However, the CSF glucose may be falsely low in the presence of hypoglycemia, or it may be erroneously interpreted as normal in the presence of CNS infection when the serum glucose is elevated. An accurate interpretation of the CSF glucose concentration is done by determining the CSF-to-serum glucose ratio. A normal CSF-to-serum glucose ratio is about 0.6 (257). Values less than 0.31 are an indication of low CSF glucose, and they are observed in approximately 70% of patients with bacterial meningitis (257). A decreased CSF-to-serum glucose ratio is also consistent with fungal or tuberculous meningitis, carcinomatous meningitis, mumps encephalitis, subarachnoid hemorrhage in 15% to 20% of patients, and several other conditions (258).
Protein
Any process that disrupts the BBB results in an elevated CSF protein concentration. Values greater than 50 mg/dL in CSF obtained from the lumbar SAS, as well as ventricular CSF protein concentrations greater than 15 mg/dL, are considered abnormal. When the LP is traumatic and there is blood in the CSF, the true protein concentration is corrected by subtracting 1 mg of protein per deciliter for every 1,000 RBCs in CSF (258).
White Blood Cell Count
The CSF abnormalities characteristic of bacterial meningitis include a polymorphonuclear (PMN) pleocytosis, a low glucose concentration, and an elevated protein concentration. The CSF should be examined promptly after it is obtained, because WBCs in the CSF begin to disintegrate after about 90 minutes. The normal WBC count in the CSF of adults and children is 0 to 5 mononuclear cells (lymphocytes and monocytes)/mm3; a WBC count of greater than 10 cells indicates a pathologic process such as infection. Normal CSF does not contain PMN leukocytes; however, following centrifugation, an occasional PMN leukocyte may be seen. It has been stressed that for the CSF to be considered normal, no more than a single PMN leukocyte should be seen in the differential count, accompanied by a total WBC count of less than 5 cells/mm3 (254,256). However, most CSF differential counts are now performed on cytocentrifuged specimens in hospital laboratories. In these preparations, a few PMN leukocytes may be seen even in the absence of disease (i.e., when minimal blood contamination is present) or in association with a high peripheral leukocyte concentration (258).
A traumatic puncture or an intracerebral or subarachnoid hemorrhage introduces RBCs and WBCs into the CSF. The correction factor for the WBC count in the presence of blood in the CSF is as follows: (a) If the peripheral RBC and WBC counts are normal, then 1 WBC/700 RBCs can be subtracted from the total WBC count in CSF; and (b) in the presence of an abnormal peripheral WBC or RBC count, the following formula can be used (258), although valid studies on the diagnostic accuracy in meningitis are lacking:
True WBC (CSF) = Actual WBC (CSF) − WBC (Blood) × RBC (CSF)/RBC (Blood)
Generalized seizures may induce a transient CSF pleocytosis consisting predominantly of PMN leukocytes. However, to attribute a CSF pleocytosis to seizure activity, the following criteria should be met: (a) The fluid should be clear and colorless, (b) the opening pressure should be normal, (c) the CSF glucose concentration should be normal, (d) the WBC count should not exceed 80 cells/mm3, (e) there should be no meningeal signs or other evidence of infection, and (f) Gram stain results should be negative (259). Even if these conditions are met, patients should usually be treated with antibiotics until the results of bacterial cultures are known. There also remains the possibility that a viral encephalitis, with a predominance of PMN leukocytes in CSF, is the cause of the seizure activity.
In large reported series of bacterial meningitis, in 90% of cases, there are greater than 100 WBCs/mm3 in the CSF, and in 65% to 70% of cases, there are greater than 1,000 WBCs/mm3 (211,215,260,261). The differential count usually shows a predominance of PMN leukocytes. In about 10% of cases of bacterial meningitis, there may be a predominance of lymphocytes early in the infection, especially if the total WBC concentration is less than 1,000/mm3. In one series, 32% (13 of 41) of patients with bacterial meningitis with a CSF WBC concentration of 1,000 cells/mm3 or less had a predominance of lymphocytes (262). In addition, in about 20% to 75% of patients with viral meningitides, the CSF may initially have a predominance of PMN leukocytes, with an eventual shift (over the course of several hours) to a monocytic predominance. This has led to controversy about the necessity for repeated LP and the time period in which a repeated LP should be obtained to demonstrate a shift in cell type. It is our feeling that a repeated LP is usually not necessary unless there is further clinical deterioration. In the presence of a lymphocytic pleocytosis, the results of CSF chemistries, Gram stain, and other tests (see later discussion) suggest the diagnosis. If bacterial meningitis is suspected, even though there is a predominance of lymphocytes, the patient should be treated with antibiotics until the results of bacterial cultures are known.
Gram Stain and Culture
Examination of CSF by Gram stain allows for rapid, accurate identification of the infecting organism. If the CSF is cloudy, smears should be obtained from fresh, uncentrifuged fluid for Gram stain. If the CSF is clear, smears should be obtained from the centrifuged sediment. The Gram stain is positive in identifying the organism in 60% to 90% of cases of bacterial meningitis (257,263). However, the probability of detecting bacteria on a Gram-stained specimen depends on the number of organisms present. Most smears will be positive when the CSF bacterial concentration is greater than 105 CFU/mL. Only 25% of smears are positive when the bacterial concentration is 103 CFU/mL or less (197).
Cerebrospinal Fluid Lactate
The lactic acid concentration in CSF has been reported to be useful in differentiating between bacterial and viral meningitis, especially in those patients who have been partially treated with antibiotics prior to examination of the CSF, as well as in those patients with low CSF WBC concentrations. In a European study, the lactic acid in CSF was measured in 50 patients with acute bacterial meningitis. In 46 patients (92%), the CSF lactate concentration was 3.5 mmol/L or greater. The investigators in this study concluded that CSF lactate was useful in the diagnosis of acute bacterial meningitis if it was 3.5 mmol/L or more (264). Other studies have demonstrated that in most cases of acute bacterial meningitis, the CSF lactate concentration is 3.8 mmol/L or more (254). Although the sensitivity of the CSF lactate level is high for bacterial meningitis, its specificity is low.
In a review of the lactic acid concentrations in 493 samples of CSF from 434 adults with various CNS conditions, the lactate concentration was greater than 35 mg/dL in 50 cases. Only 19 of the 50 cases of infective meningitis were caused by either bacterial or viral pathogens. Although the lactic acid concentration was elevated in most cases of bacterial meningitis in this study, the CSF samples with elevated lactic acid concentrations had cell counts and chemistries suggestive of bacterial meningitis; therefore, the elevated lactate concentration provided little additional information (265). In this review, as in others, an elevated CSF lactic acid concentration was nonspecific. Other causes of elevated CSF lactate concentrations include recurrent seizure activity, cerebral ischemia, hypocapnia, closed head injury, neoplasms, and craniotomy (254,265). Although the source of the CSF lactate is debated, cerebral hypoxia/ischemia, anaerobic glycolysis, vascular compromise, and metabolism of the CSF leukocytes are all potentially important factors (266).
Additional studies have examined whether elevated CSF lactate concentrations are useful in differentiating bacterial from nonbacterial meningitis in patients who have not received prior antimicrobial therapy (267). Two metaanalyses, one including 25 studies with 1,692 patients (adults and children) (268) and the other including 31 studies with 1,885 patients (269), concluded that the CSF lactate concentration is useful in the differentiation of bacterial from aseptic meningitis. In another study of adult patients, the CSF lactate concentration had a negative predictive value 99% and positive predictive value 82% for bacterial meningitis when it was 3.8 mmol/L or higher (270). However, in patients who received antimicrobial therapy prior to LP, CSF lactate concentrations had a substantially lower sensitivity compared to those who had not been treated with antimicrobial agents (269), such that the usefulness of CSF lactate in patients pretreated with antimicrobial therapy is probably limited.
Other Tests
The CSF concentration of several other substances also increases in the presence of bacterial meningitis, including various enzymes (e.g., lactate dehydrogenase and creatine phosphokinase) and fibrin-degradation products; however, the elevations are nonspecific. Tests for these compounds are rarely, if ever, performed in hospital laboratories.
Combinations of Cerebrospinal Fluid Tests
Although many of the aforementioned tests commonly performed on CSF may suggest the diagnosis of bacterial meningitis, none is irrefutable evidence of this disease, except a positive culture and/or positive stains. Combinations of test results with clinical parameters may permit a more accurate assessment of the probability of bacterial versus viral meningitis. This approach appears to have merit, as emphasized in a retrospective review of 422 patients with acute meningitis at Duke University (271). The following CSF parameters were individual predictors of bacterial meningitis with greater than 99% certainty: glucose level of less than 34 mg/dL; CSF-to-blood glucose ratio less than 0.23; leukocyte counts more than 2,000/mm3 or neutrophil counts more than 1,180/mm3. Although any one of these results predicted bacterial meningitis with high probability, none could rule it out. A multiple regression model using four parameters (age, month of onset, CSF-to-blood glucose ratio, and CSF neutrophil concentration) proved highly reliable in separating bacterial from viral meningitis. Although the model requires further validation, a nomogram is included in the article and should be consulted for more precise analysis of gram-negative cases (271).
A number of other studies have examined combinations of clinical features, with or without test results, to develop models in an attempt to accurately predict the likelihood of bacterial meningitis compared to other potential etiologies (most often viruses) (272). In several retrospective studies of immunocompetent patients older than 1 month of age with acute bacterial or viral meningitis, a CSF glucose concentration less than 34 mg/dL, a CSF-to-blood glucose ratio less than 0.23, a CSF protein concentration greater than 220 mg/dL, more than 200 leukocytes per cubic millimeter of CSF, and more than 1,180 neutrophils per cubic millimeter of CSF were found to be individual predictors of bacterial rather than viral meningitis, with 99% certainty or better. Many other prediction models have been developed. In a recently published metaanalysis of bacterial meningitis score validation studies in which 5,312 patients were identified from eight studies, 4,896 (92%) had sufficient clinical data to calculate the Bacterial Meningitis Score, which identifies children with CSF pleocytosis who are at very low risk of bacterial meningitis (low-risk features were negative CSF Gram stain, CSF absolute neutrophil count <1,000 cells/mm3, CSF protein <80 mg/dL, and peripheral absolute neutrophil count <10,000 cells/mm3) (273). The combined sensitivity was 99.3%, specificity 62.1%, and negative predictive value 99.7%, indicating that this scoring system could be used to assist clinical decision making for the management of children with CSF pleocytosis. If used, these models should be limited to the age cohort in which they were developed. Despite the positive results of this metaanalysis and other studies, clinical judgment should continue to be used in decisions about the need for administration of empirical therapy in patients with suspected bacterial meningitis (272). Prediction models may be most useful in doubtful cases, when they can be used to suggest a reconsideration of the diagnosis.
Partially Treated Meningitis
The effect of oral antibiotic therapy on CSF analysis was studied in two prospective studies of 281 children with Hib meningitis. Ninety-four (33%) children had been treated with more than one dose of antibiotics within 1 week before admission. Compared with results in untreated children, the results of CSF analysis in children pretreated with antibiotics showed significant decreases in the percentage of neutrophils (p < .03), protein concentration (p < .001), and percentage with a positive Gram stain or culture (p < .05). Differences in total WBC count, glucose concentration, CSF-to-serum glucose ratio, and blood culture results were not statistically significant. When adjustment was made for duration of illness before admission, only the difference in CSF protein concentration remained statistically significant (p< .01) between children who were pretreated as compared with untreated children. The duration of illness preceding admission was significantly longer in children who had been treated with antibiotics compared with that in untreated children. These observations suggested that the natural progression of illness in the pretreated group was less rapid than that in the untreated group and possibly accounted for the differences in numbers of WBCs and bacteria in the pretreated group, in whom infection was less fulminant (274).
Intravenous antibiotic therapy, even for as long as several days prior to initial LP, does not markedly alter the chemical or morphologic characteristics of the CSF in cases of bacterial meningitis (197). CSF was examined in 68 children with acute bacterial meningitis on admission and 44 to 68 hours after intravenous antibiotic therapy. Initial antibiotic therapy in all cases consisted of ampicillin (200 mg/kg daily in six divided doses) in combination with chloramphenicol (100 mg/kg daily in four divided doses). In those cases in which meningococci, pneumococci, or group A streptococci were isolated from CSF culture, aqueous penicillin G (400,000 U/kg daily in six divided doses) was substituted. In 65 children with meningitis caused by Hib, pneumococci, group A streptococci, and meningococci, intravenous antibiotic therapy did not significantly alter the CSF protein, glucose, or WBC concentrations. However, bacteria were not evident on smear and did not grow in culture from CSF obtained after intravenous antibiotic therapy of this duration (275).
In general, bacteria should not be seen on Gram stain or grow in culture from CSF examined 24 hours after treatment has begun with appropriate antibiotic therapy. The CSF glucose concentration approaches normality by the third day of antibiotic therapy in 80% of patients, but it may remain low for as long as 10 days. The CSF protein concentration remains elevated for at least 10 days. The WBC count in CSF remains elevated in more than 50% of cases after a standard 7- to 10-day course of antibiotic therapy, but it typically decreases when compared with the value obtained prior to therapy or early in the course of bacterial meningitis (256).
Rapid Diagnostic Tests
Several techniques have been developed for the rapid detection of bacterial antigens in the CSF, including the latex agglutination test, the staphylococcal or other coagglutination tests, and counterimmunoelectrophoresis, among many others (276). These techniques use serum containing bacterial antibodies or commercially available antisera directed against the capsular polysaccharide to detect the presence of bacterial antigens in CSF. Counterimmunoelectrophoresis requires specialized equipment and expertise and is rarely performed in hospital laboratories. Due to unacceptably poor sensitivity of latex agglutination for the diagnosis of bacterial meningitis in CSF samples with a negative Gram stain (277), this test has been abandoned in most hospital laboratories and cannot be routinely recommended, although might be considered for patients who have been pretreated with antimicrobial therapy and when CSF Gram stain and culture results are negative, where available (278).
The limulus amebocyte lysate test can detect minute quantities of endotoxin (e.g., 10 ng/mL) in the CSF. It is reported to have a sensitivity of 77% to 100%, with some studies reporting sensitivities of 97% to 99% for detecting gram-negative endotoxin. It has been recommended as a useful method for the detection of gram-negative bacterial meningitis (279). It is occasionally employed in the setting of an abnormal CSF following neurosurgery or head trauma. Nevertheless, the results of the test rarely change patient management because physicians should employ antimicrobials with activity against gram-negative aerobic bacilli in this clinical setting, even if the limulus lysate test result is negative.
C-Reactive Protein and Procalcitonin
The C-reactive protein (CRP) is an acute-phase reactant that, when present in concentrations greater than 100 ng/mL in CSF, is quite sensitive for differentiating bacterial from viral meningitis. The CRP response is minimal in viral meningitis. CRP concentrations in CSF may be elevated in other CNS inflammatory or necrotic conditions and thus are not specific for bacterial meningitis; however, when cell counts and chemistries suggest meningitis, the CRP concentration is useful in distinguishing between bacterial meningitis and viral meningitis (280,281). In this circumstance, a negative CSF CRP result excludes bacterial meningitis with 99% certainty. Because CRP is produced in the liver, serum CRP may be useful in differentiating bacterial from viral meningitis as well. A normal serum CRP has a negative predictive value of about 99% for acute bacterial meningitis. Thus, in patients where the CSF Gram stain is negative and the differential diagnosis is between acute bacterial (or partially treated bacterial) and viral meningitis, a normal serum CRP concentration excludes bacterial meningitis with about 99% certainty and these patients may be safely observed in the absence of antibacterial therapy.
A normal serum procalcitonin (another acute-phase reactant) concentration has nearly identical predictive value to the CRP (282–284). In one study, a serum procalcitonin concentration of more than 0.2 ng/mL had a sensitivity and specificity of up to 100% in the diagnosis of bacterial meningitis (285), although false-negative results have been reported (286). In another study, serum procalcitonin, at a cutoff of 0.28 ng/mL, had a sensitivity of 95%, specificity of 100%, negative predictive value of 100%, and positive predictive value of 97% in the diagnosis of bacterial meningitis (270). In patients with meningitis in whom the CSF Gram stain is negative and analysis of other parameters is inconclusive, serum concentrations of CRP or procalcitonin that are normal or below the limit of detection have a high negative predictive value in the diagnosis of bacterial meningitis, so that these patients (i.e., with a presumptive diagnosis of viral meningitis) can be carefully observed without initiation of antimicrobial therapy (278,284).
Other Diagnostic Markers
Other markers that have been studied as markers for acute bacterial meningitis in children and adults include CSF concentrations of cortisol, heparin-binding protein, soluble triggering receptor expressed on myeloid cells 1, interleukin-6, interleukin-12, interleukin-1β, tumor necrosis factor-α, complement component B, and complement component 3 (272). Most of these studies included low numbers of patients, limiting their generalizability. In one study, heparin-binding protein had a sensitivity of 100% and specificity of 99.2% in the differentiation of bacterial from aseptic meningitis (287).
An immunochromatographic test for detection of S. pneumoniae in CSF was found to be 100% sensitive and specific for diagnosing pyogenic pneumococcal meningitis (288), although more studies are needed to demonstrate the usefulness of this test in the diagnosis of pneumococcal meningitis; the overall sensitivity of the test is 95% to 100% (289).
Polymerase Chain Reaction
Polymerase chain reaction (PCR) assays that use specific bacterial primers to detect the nucleic acid of S. pneumoniae, N. meningitidis, H. influenzae, E. coli, S. agalactiae, and L. monocytogenes in CSF are available. These assays are reported to be highly sensitive (290–295). In clinical practice, culture results are often reported before the results of the PCR assay are known limiting the usefulness of this assay. A broad-range bacterial PCR that can be performed in 2 hours and that can detect small numbers of viable and nonviable organisms in CSF has been developed. This could be useful as a screening test for bacterial meningitis and in patients who have been treated with antimicrobial therapy in whom CSF culture is often negative (296); in this study, the test characteristics for broad-based bacterial PCR demonstrated a sensitivity of 100%, a specificity of 98.2%, a positive predictive value of 98.2%, and a negative predictive value of 100%. In another study with use of a multiplex PCR assay for detection of N. meningitidis, S. pneumoniae, and Hib, the overall specificity and positive predictive value were 100% and the negative predictive value was 99.1% to 99.5% (297). Multiplex assays for detecting genes of meningeal pathogens were 100% specific for detecting its target organisms or serogroups, and the lower limit of detection was similar to that for the singleplex assays (298). In another study, the sensitivity of broad-range PCR was higher than that of culture (59% versus 43%), whereas the specificity was 97% for both methods of diagnosis (299). Therefore, broad-based bacterial PCR can be used to detect the most common microorganisms in only one test and has adequate sensitivity and excellent specificity (272). The broad-based bacterial PCR can be done within 2 hours in most industrialized countries, although they are scarce in resource-poor countries. PCR may be particularly useful in patients with bacterial meningitis who have received prior antimicrobial therapy and are more likely to have negative CSF cultures (300). The sensitivity and specificity of PCR in CSF for the diagnosis of pneumococcal meningitis are 92% to 100% and 100%, respectively (289). Real-time PCR has also been used for the diagnosis of L. monocytogenes meningoencephalitis (301). Problems with false-positive results arise when using PCR, although further refinements in this technique may lead to its usefulness in the diagnosis of bacterial meningitis, particularly when CSF Gram stain and cultures are negative. Another potential application of PCR is rapid detection of the in vitro susceptibility of meningeal pathogens to specific antimicrobial agents. In one report, a novel real-time PCR-hybridization assay was developed for the rapid detection of penicillin susceptibility in S. pneumoniae; when applied to 24 pneumococcal DNA-positive CSF extracts, penicillin-sensitive S. pneumoniae was detected in all instances (302). Further studies may establish the usefulness of this rapid technique in allowing clinicians to decide on the use of specific antimicrobial therapy in patients with bacterial meningitis (see later discussion).
Neuroimaging
In the acute stage of bacterial meningitis, the CT scan may be normal or it may demonstrate enhancement of the meninges and ependyma with widening of the cisterns at the base of the brain and the cortical sulci, a result of the accumulation of purulent exudate in the basal cisterns and over the convexities of the hemispheres (303). However, the presence of these abnormalities on CT scan contributes very little to the diagnosis of meningitis. The diagnosis is made by the clinical presentation and analysis of the CSF. The extent of meningeal enhancement on CT also does not influence management or prognosis. The value of CT in suspected bacterial meningitis is in the exclusion of other CNS pathologic processes and in the investigation of the complications of this infection, including (a) prolonged fever for several days after the initiation of antibiotic therapy, (b) fever that develops after an afebrile period during therapy (secondary fever), (c) prolonged obtundation or coma, (d) new or recurrent seizure activity, (e) signs of increased ICP, and (f) focal neurologic deficits.
The most common causes of prolonged fever in patients with bacterial meningitis are subdural effusions, drug fever, and concomitant arteritis or pneumonia. In published series, 9% to 13% of patients with Hib or with pneumococcal or meningococcal meningitis had fever for 10 days or longer after the initiation of appropriate antibiotic therapy. In approximately 25% of these patients, the fever was attributed to the presence of a subdural effusion. The most common causes of secondary fever are nosocomial infections and subdural effusions (304). Although the intracranial complications of meningitis are demonstrated well by CT scan, the results of the CT scan rarely influence the management of children with meningitis and prolonged fever in the absence of other clinical features suggesting CNS complications (305). In one review of 107 children with bacterial meningitis who underwent CT scan, one or more abnormalities were found in 52% of cases (306). However, the majority of findings did not require specific intervention.
Subdural effusions are a relatively common complication of bacterial meningitis, being reported in 20% to 50% of infants and children with meningitis. Only a small percentage is clinically significant (307–310). In most cases, the fluid in the subdural space is sterile and is resorbed when the inflammatory process subsides; however, when a subdural effusion is demonstrated by CT in a patient with prolonged fever, the possibility of the development of a subdural empyema is raised. Subdural effusions are typically low-density collections of fluid adjacent to the inner border of the skull that are hypodense to brain and nearly isodense to spinal fluid (Fig. 24.27). They are often bilateral and may flatten and displace the frontal horns posteriorly. When a subdural effusion becomes purulent, its density on CT scan appears higher than that of CSF. After the administration of an intravenous contrast agent, there is significant enhancement, when the effusion is an empyema, at the border between the extraaxial fluid collection and the underlying cortex (Fig. 24.28). Sterile subdural effusions do not typically demonstrate contrast enhancement of the medial border (303) (Fig. 24.29).



The possibility of raised ICP secondary to diffuse cerebral edema or obstructive or communicating hydrocephalus should be considered in patients with a progressive or prolonged alteration of consciousness. The CT abnormalities consistent with diffuse cerebral edema include (a) loss of differentiation between gray matter and white matter; (b) compression of the ventricles, giving the frontal horns a slitlike appearance; (c) loss of sulcal markings; and (d) lack of visualization of the perimesencephalic, suprasellar, or quadrigeminal cisterns (303) (Fig. 24.30). The CT appearance of communicating hydrocephalus is an enlargement of the entire ventricular system, including the fourth ventricle, with periventricular lucencies surrounding the frontal horns (Fig. 24.31). The latter abnormality represents transependymal movement of CSF from the ventricular system into the brain parenchyma as a result of blockage in the normal CSF resorption pathways (303). The development of an obstructive hydrocephalus secondary to blockage of CSF flow by exudate at the foramina of Magendie and Luschka has the CT appearance of dilated lateral and third ventricles, with nonvisualization of the fourth ventricle.


The development of seizure activity and/or focal neurologic symptoms and signs during the course of meningitis are clear-cut indications for neuroimaging. The cause of these abnormalities may be cerebritis, brain abscess, cortical infarction, enlarging subdural effusions, or empyema. Areas of cerebritis can easily be missed by CT scan. When they are visualized by CT, they appear as low-density lesions on the noncontrasted scan; after contrast administration, they are surrounded by an inhomogeneous “halo.” There may also be diffusion of contrast medium into the low-density center of an area of cerebritis. As the abscess matures and a capsule is formed, it becomes a low-density lesion with a sharply demarcated dense ring of contrast enhancement, surrounded by a variable hypodense region of edema (311) (see Chapter 25).
Cortical infarctions complicating bacterial meningitis are the result of vasculitis. The CT appearance of a cortical infarction is that of a hypodense lesion that conforms to a vascular territory. Following the administration of contrast, cortical infarctions have a gyriform, nodular, or ring pattern of enhancement (306) (Fig. 24.32). Hemorrhagic infarctions are characteristically associated with hyperdense areas on noncontrasted scans (303).

MRI scanning, like CT, is useful for evaluating the complications of bacterial meningitis. Subdural empyemas, cortical infarctions, and areas of cerebritis are more readily imaged by MRI than by CT, but in a sick patient an MRI scan is more difficult to obtain than a CT scan. It is considerably more difficult to manage a critically ill patient in an isolated MRI scanner suite than in the CT scanner.
The extent and degree of leptomeningeal enhancement from bacterial meningitis are well demonstrated by MRI scan after the intravenous administration of the paramagnetic contrast agent gadolinium (Fig. 24.33). Paramagnetic contrast agents produce local alterations in magnetic environments that directly affect the MRI signal obtained from protons. The image that is obtained after the administration of the contrast agent visualizes this effect on proton relaxation. The contrast agent itself is not visualized. Areas of active breakdown in the BBB are enhanced when scans are obtained after the administration of gadolinium (312). Pathologic examination of animals with experimental bacterial meningitis demonstrated that areas of contrast enhancement on both CT and MRI scans correlated with inflammatory cell infiltration, and gadolinium-enhanced T1-weighted MRI scans revealed inflammatory meningeal and ependymal lesions more effectively than did contrast-enhanced CT. Unenhanced T1- and T2-weighted MRI scans did not detect meningeal inflammation (313).

Subdural effusions can sometimes be distinguished from subdural empyemas by their MRI appearance. Subdural effusions are low-protein collections; therefore, they appear isointense to spinal fluid on MRI (Fig. 24.34). Subdural empyemas are more proteinaceous and therefore appear to have higher signal intensity than CSF on T2-weighted MRI scans (314).

MRI is superior to CT scan in visualizing a cortical infarction. Ischemia and/or infarction are common causes of focal neurologic deficits in bacterial meningitis. On T2-weighted MRI images, areas of infarction appear as areas of abnormal, increased signal intensity. An acute infarction is often not visualized on CT scan within the first 24 hours unless the infarction is large or associated with edema and mass effect. MRI is the most sensitive modality for these complications, particularly with regard to infarction, especially when seen on diffusion-weighted imaging, and ventriculitis (315). MR angiography and perfusion-weighted imaging may show vascular complications, including focal stenosis and irregularity of major intracranial arteries. CT angiography is more sensitive than MR angiography for demonstrating focal stenosis of small cerebral arteries.
INITIAL MANAGEMENT
The initial management of a patient with presumed bacterial meningitis is to obtain blood cultures, initiate antimicrobial and dexamethasone therapy if indicated, and obtain spinal fluid analysis to determine whether the CSF formula is consistent with that diagnosis (see earlier discussion) (278,316). Empirical antimicrobial therapy should be initiated based on the patient’s age and underlying disease status (Table 24.5). Although no prospective data are available on the timing of administration of antimicrobial therapy in patients with bacterial meningitis, a retrospective cohort study in patients with community-acquired bacterial meningitis demonstrated that a delay in initiation of antimicrobial therapy after patient arrival in the emergency department was associated with an adverse clinical outcome when the patient’s condition advanced to a high stage of prognostic severity (317), thus supporting the assumption that treatment of bacterial meningitis before it advances to a high level of clinical severity improves clinical outcome. This concept has also been supported by two retrospective studies: one demonstrated a reduction in mortality with early administration of antimicrobial therapy (318), and the other showed a benefit in terms of neurologic outcome and survival in patients who received antimicrobial therapy before the patient’s level of consciousness deteriorated to a score lower than 10 on the Glasgow Coma Scale (319). In another retrospective case study, delay in administration of antimicrobial therapy was associated with death; in the multivariate analysis, a delay of longer than 6 hours in antimicrobial administration after presentation conferred an 8.4-fold greater risk of death (320). An additional retrospective cohort study of 286 patients with community-acquired bacterial meningitis confirmed these results, in which early and adequate administration of antimicrobial therapy related to onset of overt signs of meningitis was independently associated with favorable outcome (odds ratio [OR] = 11.19) (321).

Some patients should have a noncontrast CT scan of the head performed before LP to rule out the presence of brain shift (as a result of an intracranial mass lesion or generalized brain edema) because of the potential risk of herniation (278). However, the time involved in waiting for a CT scan significantly delays the initiation of antimicrobial therapy, with the potential for increased morbidity and mortality in patients with bacterial meningitis. Therefore, emergency empirical antimicrobial therapy and adjunctive dexamethasone therapy if indicated, after obtaining blood cultures, should be initiated before sending the patient to the CT scanner. Although CSF cultures may be sterile after the initiation of antimicrobial therapy, pretreatment blood cultures and the CSF formula or Gram stain will provide evidence for or against a diagnosis of bacterial meningitis. In one retrospective review of 177 patients (39 of whom had received prior antimicrobial therapy) with CSF culture–proven bacterial meningitis (322), the combination of blood culture and CSF Gram stain, with or without latex agglutination, identified the causative bacterium in 92% of patients. Although some clinicians routinely order CT scans of the head before performance of an LP in adults with suspected bacterial meningitis, this is not necessary in most patients. In a study of 301 patients with bacterial meningitis (237), the clinical features at baseline that were associated with an abnormal finding on CT scan of the head were an age of at least 60 years, immunocompromised status, a history of CNS disease, a history of seizure within 1 week before presentation, and neurologic abnormalities (an abnormal level of consciousness, an inability to answer two consecutive questions correctly or to follow two consecutive commands, gaze palsy, abnormal visual fields, facial palsy, arm drift, leg drift, and abnormal language). It is reasonable to proceed with LP without CT scan of the head if the patient does not meet any of the following criteria: new-onset seizures, an immunocompromised state, signs that are suspicious for space-occupying lesions (papilledema or focal neurologic signs, not including cranial nerve palsy), or moderate to severe impairment of consciousness (278,323). Although the decision to perform a CT before LP must be individualized, these guidelines are useful in determining the patient groups that are more likely to have abnormal findings on neuroimaging studies.
Once the infecting meningeal pathogen is isolated and susceptibility testing known, antimicrobial therapy can be modified for optimal treatment (Tables 24.6 and 24.7) (278,316). Recommended dosages of antimicrobial agents for children with infections of the CNS are shown in Table 24.8, and those for adults are presented in Table 24.9. In addition, certain patients should receive adjunctive dexamethasone therapy when presenting with suspected or proven bacterial meningitis (185,186,278,316). This is discussed in more detail in the section “Adjunctive Therapy.”




TREATMENT
General Principles of Therapy
Bacteriologic cure of meningitis is defined as the eradication of bacteria from CSF. Effective antimicrobial therapy of bacterial meningitis depends on attaining adequate bactericidal activity in the CSF. Several factors, largely elucidated in experimental animal models of meningitis, determine whether bactericidal activity is achieved, including (a) the ability of an antibiotic to penetrate the BBB, (b) the activity of the antibiotic within purulent CSF, and (c) the rate of metabolism of an antibiotic and its rate of clearance from CSF (324–328).
The BBB poses physiologic restrictions, allowing only highly lipid-soluble substances or substances transported by carrier-mediated facilitated diffusion to traverse it under normal conditions (324,329). The ability of an antibiotic to penetrate the BBB depends on several factors: (a) degree of lipid solubility, (b) degree of ionization at physiologic pH, (c) protein binding in serum, (d) molecular size and structure of the antibiotic, and (e) status of the BBB. The BBB acts physiologically like a lipid bilayer. In general, the greater the lipid solubility of an antibiotic, the better its penetration into CSF. For example, chloramphenicol is a highly lipophilic substance that easily penetrates the BBB. The β-lactam antibiotics have poor lipid solubility, which limits their entry into CSF under normal conditions (324). The un-ionized form of a drug possesses greater lipid solubility than the ionized form. Thus, a lesser degree of ionization at the pH of serum and CSF increases entry of antibiotics into CSF by increasing their lipid solubility. Penicillin G has a high degree of ionization at the pH of plasma and CSF. This, combined with its low lipid solubility, may explain the poor penetration of penicillin G across intact meninges. The normal plasma-to-CSF pH gradient is approximately 0.1 pH unit (330). The plasma-to-CSF pH gradient is altered, however, by purulent meningitis. The accumulation of lactate in CSF during bacterial meningitis decreases the pH of CSF, increases the gradient, and enhances the penetration of some antibiotics into CSF. Conversely, as metabolic acidosis develops, the pH gradient is reversed and the penetration of antibiotics into CSF is reduced (324).
Protein binding and molecular size limit the ability of an antibiotic to enter the CSF. Only the free non–protein-bound portion of an antibiotic in serum can enter the CSF; therefore, highly protein-bound antibiotics have lower CSF concentrations than antibiotics with a lower degree of protein binding, other factors being equal. Increased binding to plasma proteins reduces the amount of antibiotic penetration into CSF; however, it is the concentration of free antibiotic in CSF relative to its minimum bactericidal concentration (MBC) that determines its therapeutic effectiveness (329).
Although the pharmacokinetics of an antibiotic greatly influence its ability to penetrate the BBB, the most important factor appears to be the presence of meningeal inflammation. A moderate degree of meningeal inflammation results in a marked increase in the penetrability of most antibiotics. In many instances, altered BBB permeability is essential for an antibiotic to be effective in bacterial meningitis. The morphologic alterations of the BBB observed in an adult rat model of experimental meningitis consist of an early and sustained increase in pinocytotic vesicles with a progressive separation of intercellular tight junctions in the cerebral microvasculature (331). These features may contribute to antimicrobial entry into purulent CSF.
Once the antibiotic penetrates into CSF, several factors influence its ability to eradicate the infection: (a) sufficient concentrations of free active drug must be achieved in CSF, because this form of the antibiotic is necessary for bactericidal effect. The high protein concentrations in purulent CSF limit the concentration of free, unbound antibiotic; (b) an antibiotic must achieve concentrations in CSF in vivo exceeding the MBC of the infecting organism by 10- to 20-fold for optimal efficacy (173); (c) the bactericidal activity of an antibiotic may be diminished by the coadministration of a bacteriostatic agent. For example, chloramphenicol inhibits the bactericidal effect of aminoglycosides against gram-negative aerobic bacilli within the CSF (332). Conversely, antimicrobial combinations may exert an enhanced, synergistic improvement in the rate of bactericidal activity within the CSF in vivo (e.g., ampicillin plus gentamicin versus L. monocytogenes or S. agalactiae); (d) early in the course of bacterial meningitis, there may be very large numbers of bacteria in CSF (i.e., >108 CFU/mL). Some antibiotics—in particular the β-lactam antibiotics—demonstrate an inoculum effect in vitro, such that the minimum inhibitory concentration (MIC) increases dramatically as the inoculum of the test strain is increased from 105 to 107 CFU/mL under standardized in vitro conditions (324). The inoculum effect may explain the failure of certain antibiotics in vivo, as the in vitro activity of an antibiotic is routinely determined in standard growth media using a bacterial concentration of 105 CFU/mL (325); (e) an antibiotic must remain physically stable in the presence of bacterial inactivating enzymes, such as β-lactamase and chloramphenicol acetyltransferase (329).
The effectiveness of an antibiotic in eradicating bacterial meningitis is also determined by its rate of metabolism and the activity of its metabolites. For example, cephalothin is metabolized in vivo to desacetylcephalothin, which is less active in vitro than the parent compound (324). In contrast, the metabolite of cefotaxime (desacetylcefotaxime) is as active in vitro as the parent compound. Antibiotics are removed from CSF either by simple resorption through arachnoid villi or by an energy-dependent active transport process that removes the antibiotic from the CSF to the intravascular compartment across the epithelium of the choroid plexus. This “exit pump” is inhibited by weak organic acids, such as salicylates and probenecid, and to some extent by meningitis itself. β-Lactam antibiotics are cleared from the CSF by this process. The third-generation cephalosporins (e.g., ceftriaxone and cefotaxime) possess decreased affinity (as compared with penicillin G) for the choroid plexus “exit pump,” so that they remain in CSF for a longer time (329).
There have also been investigations to determine whether continuous infusion of antimicrobial therapy improves outcome in patients with bacterial meningitis. In one study of 723 African children with bacterial meningitis randomly assigned to receive bolus or continuous infusion of cefotaxime for the first 24 hours of therapy, 272 children died, but the mode of administration did not significantly affect the proportion of children who died or were severely disabled at the time of hospital discharge (333); children with pneumococcal meningitis given continuous cefotaxime infusion were significantly less likely to die or have sequelae, however.
Antimicrobial Therapy for Specific Organisms
Neisseria meningitidis
Penicillin G and ampicillin are the preferred antibiotics for the treatment of meningitis caused by N. meningitidis (197,219,334). A 7-day course of therapy is adequate for most cases of uncomplicated meningococcal meningitis. There are reports of strains of N. meningitidis resistant to penicillin (335). However, β-lactamase–producing isolates are still rare (336). Penicillin-resistant strains that do not produce β-lactamase appear, instead, to have a reduced affinity for penicillin-binding proteins (e.g., PBP-2 and PBP-3) and have been reported from Spain, the United Kingdom, and other countries (335,337–339). In Spain, the number of relatively penicillin-resistant meningococcal isolates reached 20% in 1989 (340). However, in the United States in 1991, MICs of penicillin of 0.125 µg/mL were noted for only 3 of 100 isolates submitted to the CDC (341). Routine susceptibility testing of meningococcal isolates is recommended. Meningococcal meningitis caused by the relatively penicillin-resistant strains has, however, been successfully managed with penicillin therapy, and thus the clinical significance of this partial resistance is unclear at present (173). Nevertheless, this situation must be carefully monitored, because meningococci showing relative resistance to penicillin (i.e., MICs in the range of 0.1 to 1.0 µg/mL) are increasing in incidence worldwide. In Ontario, Canada, the prevalence of invasive meningococcal disease caused by strains with decreased in vitro susceptibility to penicillin was much higher (21.7%) in 2006 (342), although it did not change in frequency between 2000 and 2006. Cefotaxime or ceftriaxone should be used when relatively penicillin-resistant strains of meningococci are isolated and when a patient is allergic to penicillin (343). Because of potent in vitro activity and ease of administration (e.g., every 12 hours and perhaps effective at every 24 hours), ceftriaxone may well be the drug of choice for serious meningococcal infection, including meningitis. Chloramphenicol is also generally effective and widely used in developing countries.
Streptococcus pneumoniae
Initial therapy of pneumococcal meningitis includes a combination of a third- or fourth-generation cephalosporin (either ceftriaxone, cefotaxime, or cefepime) plus vancomycin until the results of antimicrobial susceptibility testing are known. The Clinical and Laboratory Standards Institute has recently redefined the in vitro susceptibility breakpoints for pneumococcal isolates from patients with meningitis as either susceptible or resistant, with intravenous penicillin breakpoints of 0.06 µg/mL or lower and 0.12 µg/mL or greater, respectively (344). A pneumococcal isolate with an MIC for cefotaxime or ceftriaxone of less than 0.5 µg/mL is considered susceptible, 1.0 µg/mL intermediate, and more than 2.0 µg/mL resistant (345). In the United States, approximately 34% of pneumococcal isolates are penicillin nonsusceptible (MICs in the intermediate and resistant ranges) and approximately 14% are resistant to ceftriaxone (346). The mechanisms by which S. pneumoniae develop resistance to β-lactam antibiotics (penicillin and extended-spectrum cephalosporins) is through alterations of one or more penicillin-binding proteins (347). Alterations in the penicillin-binding proteins lead to a decrease in their affinity for β-lactam antibiotics and thus a decreased susceptibility to the antibiotic (346). In Brazil, penicillin resistance was mainly detected in isolates of serotypes 14 (61%), 23F (16%), 6B (10%), and 19F (3%) (348). Results of recent surveillance studies in the United States show that the prevalence of penicillin-nonsusceptible S. pneumoniae ranges from 25% to more than 50% (349); rates are as high as 60% in some parts of Latin America and as high as 80% in some countries in Asia. Factors reported to predispose to resistance include the patient’s age (younger than 10 or older than 50 years); immunosuppression; prolonged hospital stay; children in day care settings; infection by serotypes 14 and 23; and frequent, prolonged, or prophylactic use of antimicrobial therapy. However, penicillin nonsusceptible strains have been isolated even when no risk factors or comorbidities are identified (350).
In view of the increasing number of strains resistant to penicillin, all CSF isolates of S. pneumoniae should be tested for sensitivity to penicillin and the third-generation cephalosporins by in vitro susceptibility testing. A third- or fourth-generation cephalosporin (i.e., cefotaxime or ceftriaxone or cefepime) is recommended for strains of pneumococci resistant to penicillin (MIC ≥0.12 µg/mL) but sensitive to the third-generation cephalosporins (MIC <1 µg/mL). For strains resistant to penicillin and the cephalosporins, vancomycin plus a third- or fourth-generation cephalosporin is the antimicrobial regimen of choice (351,352). The addition of rifampin or a ceftriaxone-rifampin regimen has also been recommended by some authorities, but rifampin may demonstrate indifference or slight antagonism when combined with β-lactam agents in standardized in vitro assays (353), although rifampin, without bacteriolytic activity, may protect against neuronal damage (354). Although concerns have been raised about use of vancomycin in patients with pneumococcal meningitis who are also receiving adjunctive dexamethasone, appropriate CSF concentrations of vancomycin may be attained as long as appropriate dosages of vancomycin are used. In a study of 14 patients, administration of intravenous vancomycin (at a continuous infusion of 60 mg/kg per day, after a 15 mg/kg loading dose) led to mean serum and CSF vancomycin concentrations of 25.5 µg/mL and 7.2 µg/mL, respectively (355). These data indicate that appropriate CSF concentrations can be attained when appropriate doses are used. Trough serum concentrations of 15 to 20 µg/mL are recommended (356). Intrathecal or intraventricular vancomycin is a reasonable consideration in patients not responding to parenteral therapy.
Vancomycin-resistant strains of pneumococci have not been seen, but strains of S. pneumoniae tolerant to vancomycin have been reported. Tolerance is the ability of a bacteria to survive in the presence of an antibiotic, neither growing nor being eradicated by the antibiotic. Tolerance may be a precursor for the development of antimicrobial resistance because it creates survivors of antibiotic therapy (357–359).
Imipenem has been utilized in the therapy of penicillin-resistant pneumococcal meningitis, although its proconvulsant activity may limit its usefulness; meropenem, a carbapenem with less seizure proclivity than imipenem, may be an effective alternative (360). However, in a study of 20 cefotaxime-resistant S. pneumoniae isolates (361), 4 were of intermediate susceptibility and 13 were resistant to meropenem, suggesting that meropenem may not be a useful alternative agent for the treatment of pneumococcal isolates that are highly resistant to penicillin and cephalosporins. Chloramphenicol is one agent that has been studied for the treatment of pneumococcal meningitis. However, clinical failures with chloramphenicol have been reported in patients with penicillin-resistant isolates, probably because of the poor bactericidal activity of chloramphenicol against these strains; 20 of 25 children had an unsatisfactory outcome (i.e., death, serious neurologic deficit, poor clinical response) in one study (362). Chloramphenicol resistance was also found in 27% of pneumococcal isolates in Malawi during 2004 to 2006 (363) and in 43% of isolates in Papua New Guinea (364).
Fluoroquinolones have shown efficacy in some small series or randomized trials in patients with gram-negative meningitis. In recent years, in response to drug-resistant pneumococci in particular, newer fluoroquinolones with improved activity against gram-positive cocci have been introduced. These agents (e.g., gatifloxacin, moxifloxacin, gemifloxacin, and garenoxacin) penetrate well into CSF and have produced excellent results in experimental models of multidrug-resistant pneumococcal meningitis, including vancomycin-tolerant strains (365–370). Pharmacodynamic analysis suggests that a Cmax CSF-to-MBC ratio of at least 5 and CSF concentrations above the MBC for the test strain for the entire dosing interval are necessary for optimal bactericidal activity. Furthermore, newer antipneumococcal fluoroquinolones demonstrate synergistic activity in vitro and in vivo in experimental models of pneumococcal meningitis and combination therapy appears to prevent quinolone resistance among pneumococci (370). A β-lactam (e.g., ceftriaxone)–potent antipneumococcal quinolone regimen is very promising (371,372) and may well supplant the currently favored ceftriaxone (or cefotaxime or cefepime) (373)—vancomycin in the future, pending evaluation in randomized controlled trials.
Gram-Negative Bacilli
The results of clinical trials in patients with gram-negative bacillary meningitis favor the use of a third- (or fourth-) generation cephalosporin over conventional aminoglycoside-containing regimens (219,223,374). Cefotaxime, ceftizoxime, ceftriaxone, and ceftazidime penetrate well into inflamed CSF and are highly active against gram-negative enteric bacilli (223,375). Cure rates of 78% to 94% have been achieved with the cephalosporins, compared with previous mortality rates of 40% to 90% with predominantly aminoglycoside-containing regimens (173).
However, given the emergence of strains of gram-negative bacilli that are resistant to the third-generation cephalosporins (376), the use of other intravenous agents, with or without intraventricular antimicrobials, may need to be considered and several have been used in patients with meningitis caused by aerobic gram-negative bacilli (278). In general, the aforementioned third-generation cephalosporins appear to be equally efficacious for the treatment of gram-negative bacillary meningitis, with the exception of meningitis caused by P. aeruginosa. Ceftazidime or cefepime is recommended when P. aeruginosa is suspected (377). Clinical trials suggest the efficacy of ceftazidime alone for Pseudomonas meningitis, but a combination of ceftazidime and an aminoglycoside may be used if response is delayed (173).
Although clinical experience is scant, the fluoroquinolones have demonstrated efficacy in animal models of gram-negative bacillary meningitis. Intravenous pefloxacin has shown good efficacy with bacteriologic eradication from the CSF in nine of ten patients with gram-negative aerobic bacillary meningitis failing conventional therapy in one study (378). However, the quinolones should be considered only for gram-negative bacillary meningitis caused by multiresistant strains or in patients unresponsive to standard therapies (379).
Although extended-spectrum penicillins (e.g., ticarcillin-clavulanate, temocillin) and aztreonam have proved effective in the therapy of experimental models of gram-negative bacillary meningitis in animals, their use is not recommended because the clinical experience with third-generation cephalosporins in humans is far more extensive. Meropenem has been successfully used in patients with gram-negative meningitis (including P. aeruginosa), and further investigations may confirm its efficacy in the therapy of bacterial meningitis (380). The worldwide database on the use of meropenem in the therapy of bacterial meningitis is quite extensive and encouraging, including cases of gram-negative bacillary meningitis failing third-generation cephalosporin therapy (e.g., Enterobacter species).
For empirical treatment of Acinetobacter meningitis, intravenous meropenem with or without an aminoglycoside administered by the intraventricular or intrathecal route has been recommended (381); if the organism is later found to be resistant to carbapenems, colistin (usually formulated as colistimethate sodium) or polymyxin B should be substituted for meropenem and may also need to be administered by the intraventricular or intrathecal route (382). Intravenous colistin (5 mg/kg per day) was successfully used to treat a patient with meningitis caused by a multidrug-resistant A. baumannii (383); intrathecal colistin was also efficacious in other cases of meningitis caused by this same multidrug-resistant organism (384,385) and intrathecal polymyxin E has also been used in a patient with Acinetobacter meningitis (386). In a summary of treatment of multidrug-resistant A. baumannii, a total of 14 patients were treated for CNS infection (ventriculitis or meningitis) with colistin given intravenously and/or either intrathecally or intraventricularly (387); sterilization was achieved in all cases and cure in 13 of 14 cases. In the presence of meningitis, CSF concentrations of colistin were shown to be 0.5 µg/mL (34% to 67% of serum concentrations) (388). Two cases of A. baumannii meningitis were also successfully treated with tigecycline (389).
Haemophilus influenzae type b
A third-generation cephalosporin, either cefotaxime or ceftriaxone, is recommended for the initial therapy of H. influenzae meningitis (334). There are few differences between cefotaxime and ceftriaxone for therapy of bacterial meningitis. Both are generally very active against the major meningeal pathogens, rapidly sterilize CSF cultures, and are safe and effective (390). The long half-life of ceftriaxone allows for twice-daily (or even once-daily) administration of this antibiotic. Several studies have documented that once-daily administration of ceftriaxone is safe and efficacious for the treatment of bacterial meningitis (336). However, this is not yet recommended as standard therapy for adults; a twice-daily dose is preferred (173). Ceftriaxone has shown promise as once-daily therapy for completion of the therapeutic course in the home setting in stable children with meningitis following an uncomplicated hospital stay. A 7- to 10-day course of antibiotics is generally recommended for Hib meningitis.
Despite initial enthusiasm, cefuroxime, a second-generation cephalosporin, is not recommended for the treatment of Hib meningitis. The in vitro bactericidal activity of this drug has been shown to be inferior to that of the third-generation cephalosporins, and there have been reports of an unusually high incidence of positive Gram stain and cultures in CSF obtained several days into treatment. In a prospective, multicenter study, 106 children with acute bacterial meningitis were randomly assigned to receive either ceftriaxone or cefuroxime (391). Delayed sterilization of CSF was more common among six patients given cefuroxime than in one patient given ceftriaxone (p = .112). When all children with positive CSF cultures (Hib, N. meningitidis, S. pneumoniae, S. agalactiae) were included in the analysis, ceftriaxone therapy, as compared with cefuroxime therapy, resulted in (a) more rapid sterilization of the CSF at follow-up LP at approximately 24 hours (2% versus 12% positive cultures; p = .11); (b) less moderate to profound sensorineural hearing loss at the 2-month follow-up examination (4% versus 17%; p < .05); and (c) reversible biliary pseudolithiasis on serial abdominal ultrasonography (16 of 35 versus 0 of 5; p < .001) (391). In another comparative trial, ceftriaxone also led to a more rapid clinical response as compared with cefuroxime (392). The third-generation cephalosporins, cefotaxime and ceftriaxone, are clearly preferable to cefuroxime in the treatment of Hib meningitis. Resistance of Hib to the third-generation cephalosporins and fluoroquinolones in vitro has not been described (393). A combination of chloramphenicol and ampicillin was at one time the recommended therapy for Hib meningitis. The use of a third-generation cephalosporin, either cefotaxime or ceftriaxone, has the following advantages over therapy with a combination of ampicillin plus chloramphenicol: (a) the need to monitor serum chloramphenicol concentrations is eliminated; (b) the potential toxicities of chloramphenicol are avoided; (c) the number of daily doses of antibiotics is decreased (199); (d) approximately 29% of Hib strains causing meningitis in the United States are resistant to ampicillin, through the production of β-lactamase, although a smaller number of strains are resistant to ampicillin because of reduced affinity for penicillin-binding proteins. An increasing number of Hib strains are resistant to chloramphenicol, through the production of chloramphenicol acetyltransferase (199). More than 50% of ampicillin-resistant H. influenzae CSF isolates from Spain are also chloramphenicol resistant (394). Thus, in some countries, Hib isolates resistant to both ampicillin and chloramphenicol are common (199). DNA coding for both the β-lactamase enzyme and the chloramphenicol acetyltransferase enzyme can reside on plasmids, although chromosomally mediated resistance, as, for example, to trimethoprim, has been described (395). Therefore, any β-lactamase–positive Hib isolate should be tested for susceptibility to chloramphenicol. In addition, therapy with the third-generation cephalosporins may result in a more rapid sterilization of the CSF as compared with therapy with ampicillin plus chloramphenicol.
The pharmacokinetics of chloramphenicol are highly variable among individuals; therefore, serum concentrations of this antibiotic must be monitored to ensure therapeutic concentrations while avoiding potential toxic concentrations, especially in infants. Therapeutic serum concentrations are in the range of 15 to 25 µg/mL, obtained 60 to 120 minutes after the completion of an intravenous or oral dose. Concentrations in excess of 30 µg/mL are associated with an increased incidence of bone marrow suppression, and levels exceeding 50 to 80 µg/mL may depress myocardial contractility (396). The pharmacology of chloramphenicol is altered in patients in shock or with liver disease. In either clinical situation, excessive serum concentrations of chloramphenicol could potentially decrease cardiac contractility. Chloramphenicol should, therefore, be avoided in patients with these conditions (396).
When chloramphenicol is used in combination with phenobarbital and phenytoin, serum concentrations of all three drugs must be monitored. Chloramphenicol inhibits hepatic microsomal enzymes and therefore prolongs the half-life of phenytoin in serum, resulting in toxic concentrations of phenytoin. Phenytoin, conversely, interferes with hepatic metabolism of chloramphenicol, resulting in toxic serum concentrations. Phenobarbital induces hepatic microsomal enzymes, increases chloramphenicol metabolism, and decreases serum chloramphenicol concentrations. These drug interactions interfere with the eradication of the infection as well as with the management of seizure activity (263,397).
As discussed, fluoroquinolone-resistant H. influenzae have not emerged, and these agents penetrate well into the CSF. Trovafloxacin was compared with ceftriaxone (± vancomycin) in a multicenter, randomized comparative trial conducted in 11 countries and enrolling children 3 months to 12 years of age (398). The major pathogens were Hib, 39%; N. meningitidis, 32%; and S. pneumoniae, 21%. The overall efficacy was similar in both groups (prompt CSF sterilization in 94% to 96%); fluoroquinolones may be an excellent alternative for Hib meningitis in patients with β-lactam allergy.
Streptococcus agalactiae
Penicillin G or ampicillin or a third-generation cephalosporin has been standard therapy for neonatal meningitis caused by group B streptococci (GBS) and is the recommended therapy for treatment of S. agalactiae meningitis in adults (278). Additionally, the number of penicillin-resistant strains of GBS appears to be increasing (399). Infection with strains of S. agalactiae resistant to tetracycline, erythromycin, lincomycin, and clindamycin has been reported; therefore, the use of these penicillin substitutes in patients with GBS infections is not recommended (399).
The therapy of GBS meningitis in patients with life-threatening penicillin allergy presents a problem. If third-generation cephalosporins must be avoided, then vancomycin or teicoplanin may be tried, but clinical experience is almost nonexistent.
Listeria monocytogenes
Ampicillin is the drug of choice (often combined with gentamicin during the initial phase of treatment) for meningitis caused by L. monocytogenes. In addition, in a recent retrospective review of patients with listeriosis (58% with primary bacteremia and 42% with meningitis), differences in mortality were not seen in those treated with ampicillin or with the combination of ampicillin and gentamicin. An alternative agent in a penicillin-allergic patient is TMP-SMX, which is bactericidal against Listeria in vitro. In one retrospective series, therapy with TMP-SMX plus ampicillin was associated with a lower failure rate and fewer neurologic sequelae than the combination of ampicillin plus an aminoglycoside (122), although more data are needed before this combination can be recommended. Oral therapy with TMP-SMX has been used in some patients with Listeria meningitis and may be considered in patients who demonstrate a rapid clinical response to intravenous therapy and in whom good adherence is expected (400). The third-generation cephalosporins are inactive against this organism (173). Intravenous vancomycin is not efficacious, although intraventricular vancomycin was successful in one case of recurrent L. monocytogenes meningitis (401). Meropenem may be a useful alternative as it is highly active against listeriae.
Staphylococci
Meningitis caused by S. aureus (MSSA) is treated with nafcillin or oxacillin (139,140,402,403). Vancomycin is the drug of choice for methicillin-resistant staphylococci and for patients allergic to penicillin. The CSF should be monitored during therapy, and if the spinal fluid continues to yield viable organisms after 48 hours of intravenous treatment, then either intrathecal or intraventricular vancomycin, 20 mg once daily (in adults), can be added (402–404). The role of adjunctive rifampin therapy is unclear, although the addition of rifampin or TMP-SMX should be considered in patients not responding to therapy and if the organism is susceptible (144). Linezolid has been used successfully in some patients with MRSA CNS infections (405,406). Daptomycin has been shown to have similar antibacterial activity to vancomycin in an experimental model of MRSA meningitis (407), and daptomycin plus rifampin has been successfully used in patients with MRSA meningitis (408–410).
Anaerobes
A combination of chloramphenicol and penicillin G has been recommended for meningitis caused by anaerobes. Penicillin G has excellent activity against most anaerobes, with the exception of Bacteroides fragilis. Chloramphenicol is active against most B. fragilis isolates. Analogous to the experience in patients with brain abscess, we prefer metronidazole for the therapy of the rare cases of anaerobic meningitis. Penicillin G should be used in addition pending culture results. Metronidazole is bactericidal against virtually all strict anaerobic organisms and penetrates into the CSF and brain well.
Empirical Antimicrobial Therapy by Age-Group and Underlying Condition
Neonates
Enteric gram-negative bacilli, streptococci (in particular S. agalactiae), and L. monocytogenes are the most common causative organisms of bacterial meningitis in neonates. A third-generation cephalosporin plus ampicillin is recommended as initial therapy in this age-group.
Children
A combination of cefotaxime or ceftriaxone or cefepime and vancomycin has become the antibiotic of choice for the initial treatment of acute meningitis in children in whom the etiologic agent has not been identified. The empirical therapy of bacterial meningitis in children should include coverage for S. pneumoniae, Hib, and N. meningitidis, which is provided by the third-generation cephalosporins; vancomycin is added for pneumococcal meningitis pending in vitro susceptibility testing. The recommended doses of these antibiotics are listed in Table 24.8.
Adults (Ages 15 to 50 Years)
S. pneumoniae and N. meningitidis are the causative organisms of approximately 85% of cases of bacterial meningitis in otherwise healthy adults (411). Empirical therapy of meningitis in adults should, therefore, be directed toward these organisms. Ceftriaxone (4 g per day in divided doses every 12 hours) or cefotaxime (up to 8 to 12 g per day in divided doses every 4 to 6 hours) or cefepime (6 g per day in divided doses every 8 hours) is effective therapy for meningitis caused by either of these organisms (412); vancomycin should be added until the results of antimicrobial susceptibility testing are known. The recommended doses of these antibiotics are listed in Table 24.9. All CSF isolates of pneumococci and meningococci should be tested for penicillin or cephalosporin resistance.
Older Adults
The most common organisms causing meningitis in adults older than 50 years are S. pneumoniae and enteric gram-negative bacilli; however, meningitis caused by Listeria and H. influenzae is increasingly recognized. For initial therapy of meningitis in elderly patients, either ceftriaxone or cefotaxime or cefepime plus vancomycin, in combination with ampicillin, is recommended (219,221,263). Meropenem may be an attractive candidate for monotherapy in this age-group in the future.
Duration of Therapy
The standards for the duration of therapy of bacterial meningitis have been derived from clinical experience rather than rigid scientific analysis (413). They are basically empirical. Although shorter courses of therapy may be equally efficacious, we recommend the following duration of treatment as general guidelines, not rigid standards, when the etiologic agent is known: N. meningitidis, 5 to 7 days; H. influenzae, 7 to 10 days; S. pneumoniae, 10 to 14 days; GBS, 14 to 21 days; and gram-negative aerobic bacilli and L. monocytogenes, 3 to 4 weeks. Nevertheless, it must be stressed that the patient’s response, as assessed by clinical and laboratory parameters, is the most important criterion in the decision to terminate therapy within this discretionary range. In a double-blind randomized trial of 5 or 10 days of therapy with ceftriaxone for bacterial meningitis in children beyond the neonatal period, it was determined that ceftriaxone could be discontinued in those patients who were stable after 5 days of treatment (414), although the uncertainties around organism-specific data (especially for S. pneumoniae) and the need for clinical judgment at day 5 should lead to caution in reducing treatment duration (415).
Meningitis Following Trauma
S. pneumoniae is the most common cause of meningitis following traumatic head injury in association with the formation of a dural sinus fistula (218). H. influenzae is a less common, but also important, pathogen in this setting. A third- or fourth-generation cephalosporin plus vancomycin is recommended for empirical treatment of meningitis in patients with closed head injury. The regimen can subsequently be modified based on the results of CSF cultures.
Meningitis Following Neurosurgical Procedures
The most common organisms causing meningitis in the patient who has undergone a neurosurgical procedure, with the exception of a shunting procedure, are gram-negative bacilli and staphylococci (223). Initial therapy of meningitis in the postneurosurgical patient should be directed against gram-negative bacilli, but also against P. aeruginosa and S. aureus (219). A third- or fourth-generation cephalosporin is recommended for the treatment of gram-negative bacillary meningitis (223,374). Ceftazidime or cefepime should be used. Cefepime is a fourth-generation cephalosporin that is also active against pseudomonads. Vancomycin should be added until infection with staphylococci is excluded.
Coagulase-negative staphylococci and S. aureus are the most common pathogens causing CSF shunt infections. Unless the organism is clearly susceptible to methicillin, vancomycin is recommended for shunt infections caused by staphylococci (139,402,403). Therapy of methicillin-resistant staphylococcal shunt infections should include a combination of intravenous vancomycin and either oral rifampin or intrashunt or intraventricular vancomycin (403,416). Although cefuroxime may enter ventricular fluid in the presence of an infected CSF shunt, the concentrations are quite variable (417). This agent is not, therefore, recommended for shunt infections.
Immunosuppressed Hosts
As has been discussed, the risk for development of bacterial meningitis in an immunocompromised patient depends on a number of factors, such as (a) the underlying disease and its treatment, (b) the duration of immunosuppression, and (c) the type of immune abnormality. Knowledge of the latter helps predict the infecting organism (233,234). Patients with defects in cell-mediated immunity are most susceptible to CNS infections by microorganisms that are intracellular parasites, the eradication of which depends on an intact T-lymphocyte–macrophage system. L. monocytogenes is the most common cause of bacterial meningitis in patients with defective cell-mediated immunity (234). Patients with defective humoral immunity are unable to mount an antibody response to a bacterial infection, and they are therefore unable to control infection caused by encapsulated bacteria. These patients are at particular risk for meningitis caused by S. pneumoniae, Hib, and, less commonly, N. meningitidis. Patients with neutropenia are at particular risk for meningitis caused by P. aeruginosa and members of the Enterobacteriaceae family (233). The choice of antibiotic for empirical treatment of bacterial meningitis in the immunosuppressed patient should be made based on the type of immune abnormality.
Adjunctive Therapy
As is discussed in Chapter 23, the generation of bacterial cell wall components in CSF during treatment of meningitis with antibiotics contributes to increased inflammation in the SAS (18). Bacterial cell wall components stimulate the release of inflammatory cytokines in the CNS, such as TNF, IL-1, and prostaglandins (173). It may be possible to reduce the inflammatory response in the SAS and thus improve the outcome of this infection by administering antiinflammatory agents in conjunction with antibiotics (172,418).
TNF is a macrophage-secreted hormone that is released in response to bacterial endotoxin. The injection of small doses of purified endotoxin into healthy volunteers causes the appearance of elevated serum concentrations of TNF within 90 minutes after the infusion, accompanied by symptoms of headache, fever, rigors, and myalgia (419). Endogenous TNF release has been observed in patients with sepsis and in those with meningococcemia. Damas et al. (420) detected very high serum concentrations of TNF (mean, 701 ± 339 pg/mL; normal, 75 ± 15 pg/mL) in patients in septic shock. Waage et al. (421) found elevated serum TNF concentrations in patients with meningococcal disease. The patients with the highest concentrations (>0.1 ng/mL) died. Ming et al. (422) found elevated concentrations of TNF in CSF during bacterial meningitis in both mice and humans. None of the CSF samples from patients with viral (echovirus, coxsackievirus, or mumps virus) meningitis or other neurologic diseases (e.g., multiple sclerosis) in this study contained measurable concentrations of TNF (422). This suggests that the presence of TNF in CSF may be specific for bacterial meningitis (173).
TNF induces IL-1 release from endothelial cells and macrophages (423). IL-1 represents a family of polypeptides that are both beneficial and detrimental to the host. The primary sources of IL-1 are monocytes and macrophages, but IL-1 is also produced by brain astrocytes and microglia. IL-1 is a potent chemoattractant for neutrophils, monocytes, B cells, and T cells; it has an important role in B-cell proliferation and antibody production, as well as in T-cell activation (424). IL-1 may, however, also be detrimental to the host. IL-1 released into tissue induces a proliferative response. IL-1 released by astrocytes into brain tissue may contribute to brain gliosis and scar formation (424). IL-1 increases the concentration of metabolites of arachidonic acid—most notably PGE2 and leukotriene B4, which are potent mediators of inflammation (423).
Possible therapeutic approaches to decrease the harmful effects of TNF and/or IL-1 might include (a) drugs or procedures to decrease their production, block their biologic activity, or enhance removal from the circulation, (b) passive immunization with antibodies against TNF and IL-1, and (c) drugs that interfere with IL-1–induced arachidonic acid metabolites. Corticosteroids are highly effective in reducing IL-1 production in vitro and in vivo. Many of the biologic activities of IL-1 are inflammatory; aspirin, acetaminophen, and nonsteroidal antiinflammatory agents can reduce fever, muscle PGE2 production, leukocyte chemotaxis, and so on. Therapeutic concentrations of nonsteroidal antiinflammatory agents and antipyretic blood levels of aspirin do not, however, reduce IL-1 production, IL-1–mediated lymphocyte activation, or IL-1 synthesis of acute-phase proteins (424).
Passive immunization with monoclonal antibodies directed against TNF and IL-1 may be a future therapeutic option. Beutler et al. (425) passively immunized mice with antiserum to murine TNF and protected them from the lethal effects of gram-negative bacteremia. A major limitation, however, of monoclonal antibody therapy for meningitis is the BBB. Even during active inflammation, the BBB is an effective barrier of antibody penetration into the CSF. To achieve sufficient antibody concentrations within the CSF, it would be necessary to produce serum concentrations of antibodies at least 20- to 100-fold higher than the expected protective concentration in serum or to administer the antibody by intrathecal injection (426).
Despite aggressive supportive care and the administration of appropriate antimicrobial agents, the outcome for patients with fulminant meningococcemia is often poor. Serum TNF concentrations correlate directly with outcome in this condition. Activated protein C (drotrecogin alfa activated) reduces mortality in patients with severe sepsis (427) but has been withdrawn from the worldwide market. Plasmapheresis has been attempted, although on an extremely limited scale, in meningococcemia and may lead to a rapid decrease in serum TNF concentrations and/or improved mortality and morbidity (428). Despite the lack of a controlled clinical trial, this approach definitely deserves further study.
Clinical trials suggest a beneficial effect from dexamethasone in the treatment of bacterial meningitis in children and adults. In a prospective, randomized trial, 429 patients with bacterial meningitis were treated with either (a) dexamethasone, ampicillin, and chloramphenicol or (b) ampicillin and chloramphenicol only. Dexamethasone was administered intramuscularly with the first dose of antibiotic, at a dose of 8 mg to children younger than 12 years and 12 mg to adults every 12 hours for 3 days. There were 56 cases of Hib meningitis, 106 cases of pneumococcal meningitis, and 267 cases of meningococcal meningitis. The case-fatality rate was significantly lowered in patients with pneumococcal meningitis receiving dexamethasone; only 7 of 52 patients died, compared with 22 of 54 patients not receiving dexamethasone (p < .01). Dexamethasone therapy also significantly reduced the incidence of hearing loss in patients with pneumococcal meningitis. None of the 45 surviving patients in the dexamethasone-treated group developed hearing loss, whereas 4 of 32 patients treated with antibiotics alone became deaf (p < .05) (429). However, there were no significant differences between groups in time to afebrility or improvement in CSF parameters, there was no documentation of possible adverse effects, an extraordinarily high percentage of patients presented in a comatose state, most patients (370 of 429) received inadequate therapy for 3 to 5 days before hospitalization, the antibiotics were administered intramuscularly, and no differences in mortality were noted in patients with meningococcal or Hib meningitis.
The results of a double-blind placebo-controlled trial of 200 infants and children with bacterial meningitis demonstrated a beneficial effect of dexamethasone therapy in reducing the incidence of sensorineural hearing loss. Patients were treated with ceftriaxone or cefuroxime, with either dexamethasone (0.15 mg/kg every 6 hours for 4 days) or placebo. Of 84 patients in the placebo-treated group, 13 (15.5%) had moderate or more severe bilateral hearing loss as compared with 3 (3.3%) of 92 of the dexamethasone-treated children (p < .01) (430). The beneficial effects of dexamethasone were observed only in the children receiving concurrent cefuroxime (which may be suboptimal therapy) and not in those treated with ceftriaxone, thus rendering interpretation difficult. Dexamethasone appeared to be of particular benefit in children with milder cases of Hib meningitis (430).
Similar trends suggesting a beneficial effect of dexamethasone were observed in a third randomized, placebo-controlled trial by the Dallas group for children receiving cefuroxime (431). Once again, patients receiving dexamethasone became afebrile sooner, and the CSF glucose concentration rose more rapidly during the first day of therapy. Although the small sample size precluded a significant result from an analysis of hearing loss, the data combined with the data of the previous study (430) continued to reveal an advantage for corticosteroid therapy (432). The use of cefuroxime, a suboptimal agent (see earlier discussion) (338), in approximately 160 of 260 patients in these trials had led some investigators to question the routine use of dexamethasone as adjunctive therapy based on the results of these clinical trials (433). A metaanalysis of 11 randomized clinical trials (as of 1988) of dexamethasone for adjunctive therapy confirmed benefit for H. influenzae type b meningitis, especially for hearing outcomes, and suggested benefit for pneumococcal meningitis in children if begun with or before parenteral antibiotics (434).
Another trial, from Costa Rica, randomized infants and children with bacterial meningitis to receive cefotaxime with either dexamethasone or placebo (435). In this study, the dexamethasone or placebo was administered 15 to 20 minutes before the first dose of cefotaxime in an attempt to attenuate the SAS inflammatory response maximally. When patients were monitored for a mean of 15 months, those who had received adjunctive dexamethasone had a significantly decreased incidence of one or more neurologic sequelae, although there was only a trend in reduction of audiologic impairment.
A review of the medical records of 97 infants and children with pneumococcal meningitis (treated from 1984 to 1990) demonstrated a beneficial effect of dexamethasone therapy in infants and children with fulminant meningeal infection, as defined by laboratory studies, altered level of consciousness, and the presence of septic shock and cerebrovascular instability. They accounted for two thirds of the deaths and had a significantly increased incidence of seizures and permanent bilateral moderate or greater hearing loss. Of the survivors, 1 of 8 steroid-treated patients, as compared with 7 of 13 nonsteroid-treated patients, had moderate or severe bilateral hearing loss (436). However, this was a retrospective review and there were no data on differences in outcome with regard to specific antibiotic used.
In a prospective, placebo-controlled double-blind trial of dexamethasone (given at a dosage of 0.4 mg/kg every 12 hours for 2 days) in 115 children with acute bacterial meningitis, Hib was the infecting organism in 30 (55%) of 55 patients in the placebo group, and in 37 (62%) of 60 patients in the dexamethasone-treated group. N. meningitidis was the infecting organism in 12 (22%) of 55 patients in the placebo group and in 16 (27%) of 60 patients in the dexamethasone-treated group. At follow-up examination 3, 9, and 15 months after discharge, 3 (5%) of 60 dexamethasone-treated patients had one or more neurologic or audiologic sequelae, compared with 9 (16%) of 55 placebo recipients (p = .065) (437).
Other studies questioned the routine use of adjunctive dexamethasone in infants and children with bacterial meningitis. In one trial, there were no significant reductions in audiologic and neurologic sequelae with adjunctive dexamethasone therapy, although dexamethasone was given within 24 hours of antimicrobial therapy (median, 11 hours) and the study was stopped prematurely because the standard of care became early administration of dexamethasone (438). Similarly, in the second trial, adjunctive dexamethasone was not associated with significant improvements in neurologic sequelae, developmental outcome, or unilateral or bilateral deafness (439). Dexamethasone was given within 4 hours of the first antimicrobial, and there was a lack of follow-up for 13% of the study population. In a very large study from Malawi, dexamethasone again failed to improve outcome in children with bacterial meningitis, but the evaluation of hearing loss was suboptimal and still suggested a steroid benefit (440).
The American Academy of Pediatrics recommends consideration of dexamethasone therapy in infants and children 2 months and older with proven or suspected bacterial meningitis. A daily dose of 10 to 12 mg/m2 (0.6 mg/kg) in four divided doses is recommended for 3 to 4 days (441). Therapy for 2 days may also be efficacious (437,442). Children should, however, be carefully monitored for potential complications of corticosteroid use, specifically gastrointestinal hemorrhage and hyperglycemia. The concomitant use of an intravenous H2 receptor antagonist is recommended to prevent gastrointestinal tract bleeding. If corticosteroids are used, they should definitely be administered early, that is, before or simultaneously with the first dose(s) of parenteral antimicrobial agents. This is particularly important, because administration of currently available bacteriolytic agents (e.g., ceftriaxone) leads to rapid release of free endotoxin from gram-negative organisms into CSF, with an attendant exaggeration of the host’s inflammatory response (443).
The results of a prospective, randomized, double-blind trial of adjunctive dexamethasone therapy for bacterial meningitis in 301 adults in five European countries over 9 years demonstrated that dexamethasone improves the outcome in adults with acute bacterial meningitis. The benefits were most striking in the patients with pneumococcal meningitis (444). In another clinical trial, patients with pneumococcal meningitis who were treated with dexamethasone had a lower fatality rate than those that were not treated with dexamethasone (429). There has been concern that dexamethasone would decrease the penetration of vancomycin into the CSF. In a prospective study of 11 adults with community-acquired pneumococcal meningitis that were treated with a combination of dexamethasone and vancomycin at a dose of 15 mg/kg every 8 hours or 7.5 mg/kg every 6 hours, there were four therapeutic failures (445). The dose of vancomycin was well below the recommended dose of 60 mg/kg per day. In a prospective randomized clinical trial of the bactericidal activity of vancomycin against cephalosporin-resistant pneumococci in CSF of children with acute bacterial meningitis, vancomycin in a dose of 60 mg/kg per day penetrated reliably into the CSF when the children were treated concomitantly with dexamethasone (0.6 mg/kg per day divided into four doses for 4 days) (446). The recommended dosage of dexamethasone for adults is 8 to 10 mg intravenously every 8 hours for 2 to 4 days. Dexamethasone therapy should not adversely affect the outcome of viral meningitis (447).
Many other clinical trials were undertaken to determine the effects of adjunctive dexamethasone on outcome in patients with bacterial meningitis (173,278). On the basis of previous data, and the apparent absence of serious adverse outcomes in adult patients who received dexamethasone, the routine use of adjunctive dexamethasone (given concomitant with or just prior to the first dose of an antimicrobial agent for maximal attenuation of the SAS inflammatory response) is warranted in most adults with pneumococcal meningitis (448). A recent study demonstrated a favorable trend toward reduced rates for death and hearing loss and no evidence that dexamethasone was harmful in patients with meningococcal meningitis (449). Adjunctive dexamethasone should not be used in patients who have already received antimicrobial therapy for several hours. Despite these positive benefits in terms of morbidity and mortality, there were some concerns regarding cognitive long-term outcome in patients treated with dexamethasone. However, a follow-up study of 87 eligible patients in which 46 were treated with adjunctive dexamethasone and 41 with placebo, neuropsychologic evaluation showed no significant differences between patients treated with dexamethasone or placebo (450). In an evaluation of 357 episodes of pneumococcal meningitis from 2006 to 2009 in the Netherlands since implementation of adjunctive dexamethasone on a large scale basis, the prognosis has improved with mortality rates decreasing from 30% to 20% (451).
Despite these positive benefits, the routine use of adjunctive dexamethasone in patients with bacterial meningitis in the developing world has been controversial. In one randomized, double-blind, placebo-controlled study in adolescents and adults in Vietnam with confirmed bacterial meningitis (452), patients who received adjunctive dexamethasone experienced a significant reduction in the risk of death at 1 month (relative risk [RR], 0.43) and the risk of death or disability at 6 months (RR, 0.56); the highest proportion of cases in this study were caused by S. suis, followed by S. pneumoniae. In contrast, in a randomized, double-blind, placebo-controlled study from Malawi, there were no significant differences in mortality at 40 days in the intention-to-treat analysis (56% in the dexamethasone group versus 53% in the placebo group) or when the analysis was restricted to patients with proven pneumococcal meningitis (53% in the dexamethasone group versus 50% in the placebo group) (453). However, in this trial, almost 90% of the patients were infected with HIV and most likely had advanced disease; delayed presentation was also associated with a poorer outcome, although adjusting for this factor in the analysis had no effect. These data suggest that adjunctive dexamethasone is not beneficial in resource-poor countries where a substantial number of patients are infected with HIV (454). In a Cochrane metaanalysis of 24 studies involving 4,041 participants, adjunctive dexamethasone did not reduce overall mortality, but there was a trend to lower mortality in adults; corticosteroids were associated with lower rates of severe hearing loss, any hearing loss, and neurologic sequelae, although these benefits were only seen in studies from high-income countries (455). In a subgroup analysis based on causative microorganism, corticosteroids reduced severe hearing loss in patients with H. influenzae meningitis and mortality in patients with S. pneumoniae meningitis.
The use of adjunctive dexamethasone is of particular concern in patients with pneumococcal meningitis caused by penicillin- and cephalosporin-resistant strains, in which case patients may require antimicrobial therapy with vancomycin (173,278). A diminished CSF inflammatory response after dexamethasone administration might significantly reduce vancomycin penetration into CSF and delay CSF sterilization, as shown in an experimental rabbit model of penicillin-and cephalosporin-resistant pneumococcal meningitis. This result was confirmed in another rabbit model of pneumococcal meningitis in which significantly lower CSF vancomycin concentrations and differences in bacterial killing were found in the dexamethasone-treated rabbits. However, CSF vancomycin penetration was not reduced by dexamethasone in a study in children (446), and in another study in which a continuous infusion of vancomycin was used (60 mg/kg per day), adequate CSF concentrations (7.2 µg/mL) were achieved despite the concomitant administration of adjunctive dexamethasone (355). CSF concentrations of ceftriaxone are not significantly altered in animals or patients treated with adjunctive dexamethasone (456,457). In contrast, in an experimental rabbit model of cephalosporin-resistant pneumococcal meningitis (458), concomitant use of dexamethasone with ceftriaxone resulted in higher CSF bacterial counts and a higher number of therapeutic failures. For any patient receiving adjunctive dexamethasone who is not improving as expected or who has a pneumococcal isolate for which the cefotaxime or ceftriaxone minimal inhibitory concentration (MIC) is 2.0 µg/mL or greater, a repeat LP 36 to 48 hours after initiation of antimicrobial therapy is recommended to document the sterility of CSF (278). In the study cited earlier, only 78 (72%) of 108 CSF cultures that were positive for S. pneumoniae were submitted for in vitro susceptibility testing, and all were susceptible to penicillin (444), a finding that is unusual in many areas of the world. In patients with pneumococcal meningitis caused by strains that are highly resistant to penicillin or cephalosporins, careful observation and follow-up are critical to determine whether use of adjunctive dexamethasone is associated with adverse clinical outcome in these patients (278,448).
In addition to corticosteroids, several other adjunctive approaches to the therapy of bacterial meningitis may be useful (418,459). These include (a) bactericidal but nonbacteriolytic antibiotics to reduce endotoxin and other injurious substance (e.g., outer membrane vesicle) release into CSF (a theoretical but as yet impractical method); (b) nonsteroidal antiinflammatory agents; (c) other prostaglandin inhibitors; (d) anti–endotoxin-binding agents; (e) monoclonal antibodies directed against complement factor 5 (460), endotoxin, cytokines, or leukocyte–endothelium adhesion molecules; (f) pentoxifylline; (g) cytokine antagonists; (h) nitric oxide synthase (NOS) inhibitors (i.e., aminoguanidine); (i) thalidomide, by blocking TNF release from microglia (461); (j) osmotic dehydrating agents (e.g., mannitol, glycerol); (k) scavengers of peroxynitrite; and (l) inhibitors of matrix metalloproteinases (MMPs).
Treatment of Complications
Raised Intracranial Pressure
ICP is usually increased in bacterial meningitis; therefore, this complication should be anticipated and treated promptly. The clinical signs of increased ICP are (a) an altered level of consciousness ranging from drowsiness to coma; (b) a dilated, poorly reactive, or nonreactive pupil; (c) abnormalities of ocular motility; and (d) bradycardia and hypertension—the Cushing reflex. Increasing ICP may be associated with only one or a combination of these clinical signs. Papilledema does not develop until increased ICP has been present for several hours; therefore, the absence of papilledema should not be used to exclude the presence of increased ICP. Increased ICP may lead to herniation. Signs of impending herniation include (a) midposition, nonreactive pupils; (b) unequal or dilated, nonreactive pupils; (c) skew deviation or dysconjugate eye movements; (d) decorticate or decerebrate posturing; and (e) bradycardia and abnormal respiratory patterns.
Patients who are awake and alert can be watched clinically for signs of advancing increased ICP. Patients who are stuporous or comatose may benefit from an ICP monitoring device.
ICP exceeding 20 mm Hg is abnormal and should be treated; however, outcome may be improved if pressures greater than 15 mm Hg are treated. The rationale for treating the smaller elevations in pressure is to avoid large elevations, or so-called “plateau waves,” that can lead to herniation and irreversible brainstem injury (180,263). Plateau waves are sustained elevations in ICP that may occur spontaneously or as the result of small increases in cerebral blood volume from hypoxia, fever, or intratracheal suctioning. When ICP is already high, plateau waves may be reached quickly and lead to brain death (182,263). The treatment of increased ICP is outlined in Table 24.10.

Nonetheless, in one study of 15 patients with bacterial meningitis in whom intracranial pressure was measured (462), intracranial pressure was successfully lowered in most patients by a broad range of measures, which consisted of sedation, steroids, normal fluid and electrolyte homeostasis, blood transfusion, albumin infusion, decrease of MAP, treatment with a prostacyclin analog, and eventually thiopental, ventriculostomy, and dihydroergotamine. In nonsurvivors, mean intracranial pressure was significantly higher and CPP was markedly lower than in survivors despite treatment; however, this was not a comparative study and the results should be interpreted with caution.
Elevating the head of the bed 30 degrees reduces the ICP. Turning the head to the side (particularly to the left) or hyperextending the neck may trigger an increase in ICP. Intratracheal suctioning or endotracheal intubation may increase ICP (182,263,463).
Hyperosmolar agents, such as mannitol, decrease ICP by decreasing cerebral edema. Mannitol remains almost entirely in the extracellular intravascular space, making this compartment hyperosmolar to brain tissue. The result is movement of water from brain tissue into the intravascular space. Mannitol can be given either as a bolus intravenous injection of 1 g/kg over 10 to 15 minutes or in small frequent doses of 0.25 g/kg every 2 to 3 hours. A bolus injection can be repeated at 3- to 4-hour intervals to maintain the serum osmolality between 315 and 320 mOsm/L (180,182,263,463).
Dexamethasone appears to be beneficial in reducing ICP and cerebral edema in animal models of bacterial meningitis (464,465). However, its efficacy for reducing cerebral edema in patients with bacterial meningitis has not been established. Steroids are known to be beneficial in reducing cerebral edema surrounding tumors. In this situation, cerebral edema is largely vasogenic in origin. Steroids reduce vasogenic edema by reducing the permeability of cerebral capillary endothelial cells (466). There is experimental evidence to suggest that steroids reduce interstitial edema in meningitis (467). Cerebral edema in meningitis is a combination of vasogenic, cytotoxic, and interstitial edema (170). The evidence that steroids decrease vasogenic and interstitial edema suggests a role for corticosteroids in the management of this complication, which may contribute to raised ICP.
The use of steroids to reduce cerebral edema in meningitis also has disadvantages. Steroids decrease inflammation in the meninges. As has been discussed, a moderate degree of inflammation in the meninges is required for the CSF penetration of many antibiotics. By reducing meningeal inflammation, the concentration of antibiotics in CSF is reduced. This may be most important several days into treatment, when meningeal inflammation has been reduced substantially by antibiotic treatment. Steroids should, therefore, be discontinued within approximately 4 days of treatment (441).
High-dose barbiturate therapy is useful when other modalities have failed to control ICP. Barbiturates decrease the cerebral metabolic demand for oxygen and thus decrease cerebral blood flow. The result is a decrease in ICP. Pentobarbital is administered in an initial dose of 5 to 10 mg/kg at a rate of 1 mg/kg per minute, followed by a dose of 1 to 3 mg/kg per hour. This therapy requires an intracranial monitoring device or an electroencephalogram (EEG) to monitor cerebral activity, because the clinical examination is severely limited by the depressive effects of barbiturate. Pentobarbital is administered until the ICP is reduced below 20 mm Hg or until the EEG demonstrates a suppression-burst pattern. Recommended serum concentrations of pentobarbital to reduce ICP are 20 to 40 µg/dL. A Swan-Ganz catheter should be in place to monitor cardiac output. High-dose barbiturates are associated with significant cardiac toxicity, including decreased cardiac output, decreased contractile force, arrhythmias, and hypotension. Pentobarbital is the recommended barbiturate when barbiturate coma is desired, because this drug has a relatively short half-life. The half-life of pentobarbital is 24 hours, compared with the longer half-life of phenobarbital (5 days). The use of pentobarbital allows for a more rapid reversal of barbiturate coma than does the use of phenobarbital. Pentobarbital coma is maintained until the ICP has been below 20 mm Hg for 24 hours. The dosage of barbiturate is then slowly decreased to prevent a rebound increase in ICP (263,463,468).
Seizures
Seizures occur in 30% to 40% of children with acute bacterial meningitis (200). They occur in more than 30% of adults with pneumococcal meningitis in the first few days of illness (218). In a nationwide prospective study on adults with community-acquired bacterial meningitis, seizures occurred in 17% of patients and were associated with severe CNS and systemic inflammation, structural CNS lesions, pneumococcal meningitis, and predisposing conditions (469). The high associated mortality rate warrants a low threshold for starting anticonvulsant therapy in those with clinical suspicion of a seizure. If not managed quickly and aggressively, status epilepticus may develop. Severe or prolonged seizure activity can produce permanent damage resulting from anoxic ischemic changes in areas of the temporal lobe, cerebellum, and thalamus (263,468). The increased energy requirements of discharging neurons cannot be met by cerebral blood flow during sustained seizure activity. The result is ischemic necrosis and loss of cortical neurons (441). Status epilepticus that is continuous for 90 minutes or longer can cause permanent neurologic sequelae.
For early termination of seizure activity, a short-acting anticonvulsant with a rapid onset of action (such as lorazepam or diazepam) is recommended. Lorazepam is administered intravenously in 1- to 4-mg doses in adults and in an initial dose of 0.05 mg/kg in children. Lorazepam has a duration of action three to four times longer than that of diazepam in adults (441); 4 mg of lorazepam is therapeutically equivalent to 10 mg of diazepam (470). Diazepam is administered in a dose of 0.25 to 0.4 mg/kg (maximum, 10 mg) at a rate of 1 to 2 mg per minute. The 10-mg dose may be repeated up to three times at intervals of 15 to 20 minutes (471). Diazepam has a half-life of 15 minutes; therefore, the blood level decreases rapidly. A long-acting anticonvulsant should be administered immediately after lorazepam or diazepam. The long-acting anticonvulsant of choice in children and adults is phenytoin or fosphenytoin. Phenytoin is administered in a dose of 18 to 20 mg/kg at a rate no faster than 50 mg per minute. Phenytoin can prolong the QT interval or lead to hypotension. If either of these side effects is observed, the rate of administration is decreased. Fosphenytoin is a water-soluble prodrug of phenytoin that is converted to phenytoin by nonspecific phosphatases. Doses of fosphenytoin are expressed as phenytoin equivalents. Infusion side effects are less common with fosphenytoin than with phenytoin (472). Fosphenytoin is administered in a dose of 18 to 20 mg/kg at a rate no faster than 150 mg per minute. Phenytoin is very effective in controlling convulsions without depressing consciousness or respiration (263). Intravenous phenytoin reaches peak brain and blood concentrations within 15 minutes (473). The half-life of phenytoin following a loading dose is approximately 36 hours (470). Serum concentrations greater than 25 µg/mL are usually necessary to terminate status epilepticus. If an 18- to 20-mg/kg dose of phenytoin fails to control seizure activity, an additional 500 mg of phenytoin can be given. A maintenance dose of 100 mg every 6 hours (in adults) should be started after the loading dose.
If fosphenytoin fails to control seizure activity, the patient can be treated with intravenous levetiracetam or valproic acid or intubated, mechanically ventilated, and treated with phenobarbital. For adults, phenobarbital is administered intravenously at a rate of 100 mg per minute until seizure activity stops, or to a loading dose of 20 mg/kg (263). The loading dose of phenobarbital in children is 20 mg/kg, administered intravenously at a rate of 30 mg per minute (474). The most common adverse effects of phenobarbital loading are hypotension and respiratory depression. If these complications are managed and seizure activity continues, an additional 10 mg/kg can be given (474). The primary reason for failure to control seizures is that anticonvulsants are administered in subtherapeutic doses or that the rate of administration is too slow.
The combination of phenytoin and phenobarbital controls seizure activity in the vast majority of patients. When they fail to do so, general anesthesia with pentobarbital can be tried. The dose of pentobarbital is the same for children and adults: a loading dose of 3 to 5 mg/kg and a maintenance dose of 1 to 2 mg/kg per hour (263,474). In the past, paraldehyde or a continuous intravenous diazepam drip was used to treat status epilepticus; however, paraldehyde is no longer available, and a diazepam drip is no longer recommended.
Fluid Management
Most children with bacterial meningitis are hyponatremic (serum sodium concentration <135 mEq/L) early in the course of their illness (441). Fifty percent of children have evidence of SIADH on admission to the hospital (208). Restriction of fluids to correct serum sodium is potentially important, because the degree and duration of hyponatremia correlate with the development of neurologic sequelae. However, a rigid adherence to fluid restriction, a time-honored practice in the treatment of hyponatremia in children with bacterial meningitis, is no longer recommended because of the adverse effects of hypovolemia on cerebral perfusion pressure. A Cochrane review on fluid therapy for acute bacterial meningitis concluded that there is some evidence that supports maintaining intravenous fluids rather than restricting them in the first 48 hours, in settings with high mortality rates and where patients present late. However, where children present early and mortality rates are lower, there is insufficient evidence to guide practice.
The initial rate of intravenous fluid administration should be approximately three fourths of normal maintenance requirements, or about 1,000 to 1,200 mL/m2 daily. A 5% dextrose solution with one-fourth to one-half normal saline and 20 to 40 mEq/L potassium is recommended. The serum sodium concentration and urine specific gravity should be measured every 6 to 12 hours (441). The mean duration of hyponatremia in children with Hib meningitis in one study was 20 hours (range, 0 to 240 hours) (475). The volume of fluids administered can be gradually increased when the serum sodium concentration rises above 135 mEq/L. In most cases, maintenance rates (1,500 to 1,700 mL/m2 daily) will be reached by 36 to 48 hours after admission. These recommendations do not apply to the child who is admitted in shock or who is severely dehydrated (441).
Subdural Effusion
Most subdural effusions do not need intervention and are associated with no permanent deficits (199). Routine subdural paracentesis should be avoided. Only the rare effusion becomes an empyema or is large enough to have a mass effect. In either instance, serial imaging of the fluid collection by CT or MRI scan will allow for early detection of these complications. Although rare, subdural empyema must be considered in patients with community-acquired bacterial meningitis and otitis or sinusitis, focal neurologic deficits, or epileptic seizures. S. pneumoniae is the predominant causative organism and neurosurgical intervention should be regarded as first-choice therapy in patients with empyema causing midline shift and focal neurologic abnormalities or a decreased level of consciousness (476).
PREVENTION
The chemoprophylaxis and immunoprophylaxis of bacterial infections of the CNS are considered in detail in Chapter 51.
References
1. Danielson L, Mann EA. The first American account of cerebrospinal meningitis. [Reprinted from Adams D, ed. Medical and agricultural register for the years 1806 and 1807, vol 1, no 5. Boston: Manning & Loring.] Rev Infect Dis. 1983;5:969–972.
2. North E. Concerning the epidemic of spotted fever in New England [reprinted excerpts]. Rev Infect Dis. 1980;2:811–816.
3. Schwentker FF, Gelman S, Long PH. The treatment of meningococcic meningitis with sulfanilamide. JAMA. 1937;108:1407–1408.
4. Scheld WM, Mandell GL. Landmark perspective. Sulfonamides and meningitis. JAMA. 1984;251:791–794.
5. Thigpen MC, Whitney CG, Messonnier NE, et al. Bacterial meningitis in the United States, 1998–2007. N Engl J Med. 2011;364:2016–2025.
6. Takala AK, Eskola J, Peltola H, et al. Epidemiology of invasive Haemophilus influenzae type b disease among children in Finland before vaccination with Haemophilus influenzae type b conjugate vaccine. Pediatr Infect Dis J. 1989;8:297–302.
7. Schlech WF III, Ward JI, Band JD, et al. Bacterial meningitis in the United States, 1978 through 1981. The National Bacterial Meningitis Surveillance Study. JAMA. 1985;253:1749–1754.
8. Sherry B, Emanuel I, Kronmal RA, et al. Interannual variation of the incidence of Haemophilus influenzae type b meningitis. JAMA. 1989;261:1924–1929.
9. Cochi SL, Broome CV, Hightower AW. Immunization of US children with Hemophilus influenzae type b polysaccharide vaccine. A cost-effectiveness model of strategy assessment. JAMA. 1985;253:521–529.
10. Dagan R, Isaachson M, Lang R, et al. Epidemiology of pediatric meningitis caused by Haemophilus influenzae type b, Streptococcus pneumoniae, and Neisseria meningitidis in Israel: a 3-year nationwide prospective study. Israeli Pediatric Bacteremia and Meningitis Group. J Infect Dis. 1994;169:912–916.
11. Ostroy PR. Bacterial meningitis in Washington state. West J Med. 1979;131:339–343.
12. Santosham M, Kallman CH, Neff JM, et al. Absence of increasing incidence of meningitis caused by Haemophilus influenza type b. J Infect Dis. 1979;140:1009–1012.
13. Fraser DW, Darby CP, Koehler RE, et al. Risk factors in bacterial meningitis: Charleston County, South Carolina. J Infect Dis. 1973;127:271–277.
14. Granoff DM, Basden M. Haemophilus influenzae infections in Fresno County, California: a prospective study of the effects of age, race, and contact with a case on incidence of disease. J Infect Dis. 1980;141:40–46.
15. Peltola H, Virtanen M. Systemic Haemophilus influenzae infection in Finland. Clin Pediatr (Phila). 1984;23:275–280.
16. Takala AK. Epidemiologic characteristics and risk factors for invasive Haemophilus influenzae type b disease in a population with high vaccine efficacy. Pediatr Infect Dis J. 1989;8:343–346.
17. Ward JI, Lum MK, Hall DB, et al. Invasive Haemophilus influenzae type b disease in Alaska: background epidemiology for a vaccine efficacy trial. J Infect Dis. 1986;153:17–26.
18. Fothergill LD, Wright J. Influenzal meningitis: relation of age incidence to bactericidal power of blood against causal organism. J Immunol. 1933;24:273–284.
19. Peltola H, Kayhty H, Virtanen M, et al. Prevention of Haemophilus influenzae type b bacteremic infections with the capsular polysaccharide vaccine. N Engl J Med. 1984;310:1561–1565.
20. Takala AK, Eskola J, Palmgren J, et al. Risk factors of invasive Haemophilus influenzae type b disease among children in Finland. J Pediatr. 1989;115:694–701.
21. Cochi SL, Fleming DW, Hightower AW, et al. Primary invasive Haemophilus influenzae type b disease: a population-based assessment of risk factors. J Pediatr. 1986;108:887–896.
22. Krasinski K, Nelson JD, Butler S, et al. Possible association of mycoplasma and viral respiratory infections with bacterial meningitis. Am J Epidemiol. 1987;125:499–508.
23. Rosenthal J, Dagan R, Press J, et al. Differences in the epidemiology of childhood community-acquired bacterial meningitis between two ethnic populations cohabiting in one geographic area. Pediatr Infect Dis J. 1988;7:630–633.
24. Michaels RH, Ali O. A decline in Haemophilus influenzae type b meningitis. J Pediatr. 1993;122:407–409.
25. Adams WG, Deaver KA, Cochi SL, et al. Decline of childhood Haemophilus influenzae type b (Hib) disease in the Hib vaccine era. JAMA. 1993;269:221–226.
26. Murphy TV, White KE, Pastor P, et al. Declining incidence of Haemophilus influenzae type b disease since introduction of vaccination. JAMA. 1993;269:246–248.
27. Broadhurst LE, Erickson RL, Kelley PW. Decreases in invasive Haemophilus influenzae diseases in US Army children, 1984 through 1991. JAMA. 1993;269:227–231.
28. Vadheim CM, Greenberg DP, Eriksen E, et al. Eradication of Haemophilus influenzae type b disease in southern California. Kaiser-UCLA Vaccine Study Group. Arch Pediatr Adolesc Med. 1994;148:51–56.
29. Peltola H, Kilpi T, Anttila M. Rapid disappearance of Haemophilus influenzae type b meningitis after routine childhood immunisation with conjugate vaccines. Lancet. 1992;340:592–594.
30. Urwin G, Yuan MF, Feldman RA. Prospective study of bacterial meningitis in North East Thames region, 1991–3, during introduction of Haemophilus influenzae vaccine. BMJ. 1994;309:1412–1414.
31. Kojouharova M, Gatcheva N, Setchanova L, et al. Childhood bacterial meningitis in Bulgaria: a population-based retrospective study in six regions during 1992–96. Int J Infect Dis. 2003;7:109–112.
32. Garner D, Weston V. Effectiveness of vaccination for Haemophilus influenzae type b. Lancet. 2003;361:395–396.
33. Steinhoff M, Goldblatt D. Conjugate Hib vaccines. Lancet. 2003;361: 360–361.
34. Gessner BD, Sutanto A, Linehan M, et al. Incidences of vaccine-preventable Haemophilus influenzae type b pneumonia and meningitis in Indonesian children: hamlet-randomised vaccine-probe trial. Lancet. 2005;365:43–52.
35. Cowgill KD, Ndiritu M, Nyiro J, et al. Effectiveness of Haemophilus influenzae type b conjugate vaccine introduction into routine childhood immunization in Kenya. JAMA. 2006;296:671–678.
36. Daza P, Banda R, Misoya K, et al. The impact of routine infant immunization with Haemophilus influenzae type b conjugate vaccine in Malawi, a country with high human immunodeficiency virus prevalence. Vaccine. 2006;24:6232–6239.
37. Adegbola RA, Secka O, Lahai G, et al. Elimination of Haemophilus influenzae type b (Hib) disease from The Gambia after the introduction of routine immunisation with a Hib conjugate vaccine: a prospective study. Lancet. 2005;366:144–150.
38. Takala AK, Eskola J, van Alphen L. Spectrum of invasive Haemophilus influenzae type b disease in adults. Arch Intern Med. 1990;150:2573–2576.
39. Farley MM, Stephens DS, Brachman PS Jr, et al. Invasive Haemophilus influenzae disease in adults. A prospective, population-based surveillance. CDC Meningitis Surveillance Group. Ann Intern Med. 1992;116: 806–812.
40. Rubach MP, Bender JM, Mottice S, et al. Increasing incidence of invasive Haemophilus influenzae disease in adults, Utah, USA. Emerg Infect Dis. 2011;17:1645–1650.
41. Peltola H. Meningococcal disease: still with us. Rev Infect Dis. 1983;5: 71–91.
42. Reido FX, Plikaytis BD, Broome CV. Epidemiology and prevention of meningococcal disease. Pediatr Infect Dis J. 1995;14:643–657.
43. Scheld WM. Meningococcal diseases. In: Warren KS, Mahmoud AAF, eds. Tropical and Geographical Medicine. 2nd ed. New York: McGraw-Hill; 1990:798–814.
44. Jackson LA, Wenger JD. Laboratory-based surveillance for meningococcal disease in selected areas, United States, 1989–1991. MMWR CDC Surveill Summ. 1993;42:21–30.
45. Schwartz B, Moore PS, Broome CV. The global epidemiology of meningococcal disease. Clin Microbiol Rev. 1989;2:S118–S124.
46. Stephens DS, Hajjeh RA, Baughman WS, et al. Sporadic meningococcal disease in adults: results of a 5-year population-based study. Ann Intern Med. 1995;123:937–940.
47. Gardner P. Clinical practice. Prevention of meningococcal disease. N Engl J Med. 2006;355:1466–1473.
48. Goldschneider I, Gotschlich EC, Artenstein MS. Human immunity to the meningococcus. I. The role of humoral antibodies. J Exp Med. 1969;129:1307–1326.
49. Brouwer MC, de Gans J, Heckenberg SG, et al. Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta-analysis. Lancet Infect Dis. 2009;9:31–44.
50. Ross SC, Densen P. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine (Baltimore). 1984;63:243–273.
51. Fijen CA, Kuijper EJ, Tjia HG, et al. Complement deficiency predisposes for meningitis due to nongroupable meningococci and Neisseria-related bacteria. Clin Infect Dis. 1994;18:780–784.
52. Sjoholm AG, Kuijper EJ, Tijssen CC, et al. Dysfunctional properdin in a Dutch family with meningococcal disease. N Engl J Med. 1988;319:33–37.
53. Overturf GD. Indications for the immunological evaluation of patients with meningitis. Clin Infect Dis. 2003;36:189–194.
54. Davila S, Wright VJ, Khor CC, et al. Genome-wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet. 2010;42:772–776.
55. Centers for Disease Control and Prevention Advisory Committee on Immunization Practices. Revised recommendations of the Advisory Committee on Immunization Practices to vaccinate all persons aged 11–18 years with meningococcal conjugate vaccine. MMWR Morb Mortal Wkly Rep. 2007;56:794–795.
56. Centers for Disease Control and Prevention. Updated recommendations for use of meningococcal conjugate vaccines—Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Morb Mortal Wkly Rep. 2011;60:72–76.
57. Centers for Disease Control and Prevention. Recommendation of the Advisory Committee on Immunization Practices (ACIP) for use of quadrivalent meningococcal conjugate vaccine (MenACWY-D) among children aged 9 through 23 months at increased risk for invasive meningococcal disease. MMWR Morb Mortal Wkly Rep. 2011;60:1391–1392.
58. Vesikari T, Esposito S, Prymula R, et al. Immunogenicity and safety of an investigational multicomponent, recombinant, meningococcal serogroup B vaccine (4CMenB) administered concomitantly with routine infant and child vaccinations: results of two randomised trials. Lancet. 2013;381: 825–835.
59. Snape MD, Pollard AJ. The beginning of the end for serogroup B meningococcus? Lancet. 2013;381:785–787.
60. Santolaya ME, O’Ryan ML, Valenzuela MT, et al. Immunogenicity and tolerability of a multicomponent meningococcal serogroup B (4CMenB) vaccine in healthy adolescents in Chile: a phase 2b/3 randomised, observer-blind, placebo-controlled study. Lancet. 2012;379:617–624.
61. Stephens DS. Prevention of serogroup B meningococcal disease. Lancet. 2012;379:592–594.
62. Schuchat A, Robinson K, Wenger JD, et al. Bacterial meningitis in the United States in 1995. Active Surveillance Team. N Engl J Med. 1997;337: 970–976.
63. Istre GR, Tarpay M, Anderson M, et al. Invasive disease due to Streptococcus pneumoniae in an area with a high rate of relative penicillin resistance. J Infect Dis. 1987;156:732–735.
64. Burman LA, Norrby R, Trollfors B. Invasive pneumococcal infections: incidence, predisposing factors, and prognosis. Rev Infect Dis. 1985;7: 133–142.
65. Kragsbjerg P, Källman J, Oleen P. Pneumococcal meningitis in adults. Scand J Infect Dis. 1994:659–666.
66. Davidson M, Schraer CD, Parkinson AJ, et al. Invasive pneumococcal disease in an Alaska native population, 1980 through 1986. JAMA. 1989;261:715–718.
67. Gray BM, Dillon HC Jr. Epidemiological studies of Streptococcus pneumoniae in infants: antibody to types 3, 6, 14, and 23 in the first two years of life. J Infect Dis. 1988;158:948–955.
68. Adriani KS, Brouwer MC, Geldhoff M, et al. Common polymorphisms in the complement system and susceptiblity to bacterial meningitis. J Infect. 2013;66:255–262.
69. Scheld WM. Bacterial meningitis in the patient at risk: intrinsic risk factors and host defense mechanisms. Am J Med. 1984;76:193–207.
70. Musher DM. Infections caused by Streptococcus pneumoniae: clinical spectrum, pathogenesis, immunity, and treatment. Clin Infect Dis. 1992;14:801–807.
71. Godeau B, Bachir D, Schaeffer A, et al. Severe pneumococcal sepsis and meningitis in human immunodeficiency virus-infected adults with sickle cell disease. Clin Infect Dis. 1992;15:327–329.
72. Weisfelt M, van de Beek D, Spanjaard L, et al. Clinical features, complications, and outcome in adults with pneumococcal meningitis: a prospective case series. Lancet Neurol. 2006;5:123–129.
73. Biernath KR, Reefhuis J, Whitney CG, et al. Bacterial meningitis among children with cochlear implants beyond 24 months after implantation. Pediatrics. 2006;117:284–289.
74. Wei BP, Robins-Browne RM, Shepherd RK, et al. Can we prevent cochlear implant recipients from developing pneumococcal meningitis? Clin Infect Dis. 2008;46:e1–e7.
75. Greenwood B. Pneumococcal meningitis epidemics in Africa. Clin Infect Dis. 2006;43:701–703.
76. Hsu HE, Shutt KA, Moore MR, et al. Effect of pneumococcal conjugate vaccine on pneumococcal meningitis. N Engl J Med. 2009;360:244–256.
77. Nuorti JP, Whitney CG; Centers for Disease Control and Prevention. Prevention of pneumococcal disease among infants and children—use of 13-valent pneumococcal conjugate vaccine and 23-valent pneumococcal polysaccharide vaccine—recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2010;59:1–18.
78. Durand ML, Calderwood SB, Weber DJ, et al. Acute bacterial meningitis in adults. A review of 493 episodes. N Engl J Med. 1993;328:21–28.
79. Wenger JD, Hightower AW, Facklam RR, et al. Bacterial meningitis in the United States, 1986: report of a multistate surveillance study. The Bacterial Meningitis Study Group. J Infect Dis. 1990;162:1316–1323.
80. Musser JM, Kroll JS, Granoff DM, et al. Global genetic structure and molecular epidemiology of encapsulated Haemophilus influenzae. Rev Infect Dis. 1990;12:75–111.
81. Moore PS, Reeves MW, Schwartz B, et al. Intercontinental spread of an epidemic group A Neisseria meningitidis strain. Lancet. 1989;2: 260–263.
82. Whalen CM, Hockin JC, Ryan A, et al. The changing epidemiology of invasive meningococcal disease in Canada, 1985 through 1992. Emergence of a virulent clone of Neisseria meningitidis. JAMA. 1995;273:390–394.
83. Jackson LA, Schuchat A, Reeves MW, et al. Serogroup C meningococcal outbreaks in the United States. An emerging threat. JAMA. 1995;273: 383–389.
84. Harrison LH. The worldwide prevention of meningococcal infection. Still an elusive goal. JAMA. 1995;273:419–421.
85. Rosenstein NE, Perkins BA, Stephens DS, et al. The changing epidemiology of meningococcal disease in the United States, 1992–1996. J Infect Dis. 1999;180:1894–1901.
86. Moura AS, Pablos-Mendez A, Layton M, et al. Epidemiology of meningococcal disease, New York City, 1989–2000. Emerg Infect Dis. 2003;9: 355–361.
87. Mastrantonio P, Stefanelli P, Fazio C, et al. Serotype distribution, antibiotic susceptibility, and genetic relatedness of Neisseria meningitidis strains recently isolated in Italy. Clin Infect Dis. 2003;36:422–428.
88. Bilukha OO, Rosenstein N; National Center for Infectious Diseases, Centers for Disease Control and Prevention. Prevention and control of meningococcal disease. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Recomm Rep. 2005;54:1–21.
89. Cohn AC, MacNeil JR, Harrison LH, et al. Changes in Neisseria meningitidis disease epidemiology in the United States, 1998–2007: implications for prevention of meningococcal disease. Clin Infect Dis. 2010;50:184–191.
90. Centers for Disease Control and Prevention. Notes from the field: serogroup C invasive meningococcal disease among men who have sex with men—New York City, 2010–2012. MMWR Morb Mortal Wkly Rep. 2013;61:1048.
91. Wilder-Smith A, Goh KT, Barkham T, et al. Hajj-associated outbreak strain of Neisseria meningitidis serogroup W135: estimates of the attack rate in a defined population and the risk of invasive disease developing in carriers. Clin Infect Dis. 2003;36:679–683.
92. Mayer LW, Reeves MW, Al-Hamdan N, et al. Outbreak of W135 meningococcal disease in 2000: not emergence of a new W135 strain but clonal expansion within the electophoretic type-37 complex. J Infect Dis. 2002;185:1596–1605.
93. Boisier P, Nicolas P, Djibo S, et al. Meningococcal meningitis: unprecedented incidence of serogroup X-related cases in 2006 in Niger. Clin Infect Dis. 2007;44:657–663.
94. Delrieu I, Yaro S, Tamekloe TA, et al. Emergence of epidemic Neisseria meningitidis serogroup X meningitis in Togo and Burkina Faso. PLoS One. 2011;6:e19513.
95. Frasch CE, Zollinger WD, Poolman JT. Serotype antigens of Neisseria meningitidis and a proposed scheme for designation of serotypes. Rev Infect Dis. 1985;7:504–510.
96. Spanjaard L, Bol P, de Marie S, et al. Association of meningococcal serotypes with the course of disease: serotypes 2a and 2b in the Netherlands, 1959–1981. J Infect Dis. 1987;155:277–282.
97. Olyhoek T, Crowe BA, Achtman M. Clonal population structure of Neisseria meningitidis serogroup A isolated from epidemics and pandemics between 1915 and 1983. Rev Infect Dis. 1987;9:665–692.
98. Crowe BA, Wall RA, Kusecek B, et al. Clonal and variable properties of Neisseria meningitidis isolated from cases and carriers during and after an epidemic in the Gambia, West Africa. J Infect Dis. 1989;159:686–700.
99. Caugant DA, Kristiansen BE, Froholm LO, et al. Clonal diversity of Neisseria meningitidis from a population of asymptomatic carriers. Infect Immun. 1988;56:2060–2068.
100. Kellerman SE, McCombs K, Ray M, et al. Genotype-specific carriage of Neisseria meningitidis in Georgia counties with hyper- and hyposporadic rates of meningococcal disease. J Infect Dis. 2002;186:40–48.
101. Lingappa JR, Al-Rabeah AM, Hajjeh R, et al. Serogroup W-135 meningococcal disease during the Hajj, 2000. Emerg Infect Dis. 2003;9:665–671.
102. Hahne S, Handford S, Ramsay M. W135 meningococcal carriage in Hajj pilgrims. Lancet. 2002;360:2089–2090.
103. Bonacorsi S, Clermont O, Houdouin V, et al. Molecular analysis and experimental virulence of French and North American Escherichia coli neonatal meningitis isolates: identification of a new virulent clone. J Infect Dis. 2003;187:1895–1906.
104. Unhanand M, Mustafa MM, McCracken GH Jr, et al. Gram-negative enteric bacillary meningitis: a twenty-one-year experience. J Pediatr. 1993;122:15–21.
105. Leonard MK, Murrow JR, Jurado R, et al. Salmonella meningitis in adults infected with HIV: case report and review of the literature. Am J Med Sci. 2002;323:266–268.
106. Chang WN, Tsai YC, Chien CC, et al. Frequent association with neurosurgical conditions in adult Proteus mirabilis meningitis: report of five cases. Clin Neurol Neurosurg. 2002;104:121–124.
107. Reichert MC, Medeiros EA, Ferraz FA. Hospital-acquired meningitis in patients undergoing craniotomy: incidence, evolution, and risk factors. Am J Infect Control. 2002;30:158–164.
108. Kim HI, Kim SW, Park GY, et al. The causes and treatment outcomes of 91 patients with adult nosocomial meningitis. Korean J Intern Med. 2012;27:171–179.
109. Dillon HC Jr, Khare S, Gray BM. Group B streptococcal carriage and disease: a 6-year prospective study. J Pediatr. 1987;110:31–36.
110. Stoll BJ, Hansen N, Fanaroff AA, et al. Changes in pathogens causing early-onset sepsis in very-low-birth-weight infants. N Engl J Med. 2002;347:240–247.
111. Henrichsen J, Ferrieri P, Jelinkova J, et al. Nomenclature of antigens of group B streptococci. Int J Syst Bacteriol. 1984;34:500.
112. Musser JM, Mattingly SJ, Quentin R, et al. Identification of a high-virulence clone of type III Streptococcus agalactiae (group B Streptococcus) causing invasive neonatal disease. Proc Natl Acad Sci U S A. 1989;86:4731–4735.
113. Gaschignard J, Levy C, Romain O, et al. Neonatal bacterial meningitis: 444 cases in 7 years. Pediatr Infect Dis J. 2011;30:212–217.
114. Libster R, Edwards KM, Levent F, et al. Long-term outcomes of group B streptococcal meningitis. Pediatrics. 2012;130:e8–e15.
115. Farley MM, Harvey RC, Stull T, et al. A population-based assessment of invasive disease due to group B Streptococcus in nonpregnant adults. N Engl J Med. 1993;328:1807–1811.
116. Dunne DW, Quagliarello V. Group B streptococcal meningitis in adults. Medicine (Baltimore). 1993;72:1–10.
117. Mascola L, Sorvillo F, Neal J, et al. Surveillance of listeriosis in Los Angeles County, 1985–1986. A first year’s report. Arch Intern Med. 1989;149:1569–1572.
118. Decker CF, Simon GL, DiGioia RA, et al. Listeria monocytogenes infections in patients with AIDS: report of five cases and review. Rev Infect Dis. 1991;13:413–417.
119. Berenguer J, Solera J, Diaz MD, et al. Listeriosis in patients infected with human immunodeficiency virus. Rev Infect Dis. 1991;13:115–119.
120. Lorber B. Listeriosis. Clin Infect Dis. 1997;24:1–9; quiz 10–11.
121. Mylonakis E, Hohmann EL, Calderwood SB. Central nervous system infection with Listeria monocytogenes. 33 years’ experience at a general hospital and review of 776 episodes from the literature. Medicine (Baltimore). 1998;77:313–336.
122. Clauss HE, Lorber B. Central nervous system infection with Listeria monocytogenes. Curr Infect Dis Rep. 2008;10:300–306.
123. Goulet V, Hebert M, Hedberg C, et al. Incidence of listeriosis and related mortality among groups at risk of acquiring listeriosis. Clin Infect Dis. 2012;54:652–660.
124. Kamath BM, Mamula P, Baldassano RN, et al. Listeria meningitis after treatment with infliximab. J Pediatr Gastroenterol Nutr. 2002;34:410–412.
125. Chuang MH, Singh J, Ashouri N, et al. Listeria meningitis after infliximab treatment of ulcerative colitis. J Pediatr Gastroenterol Nutr. 2010;50:337–339.
126. La Montagna G, Valentini G. Listeria monocytogenes meningitis in a patient receiving etanercept for Still’s disease. Clin Exp Rheumatol. 2005;23:121.
127. Zuniga M, Aguado JM, Vada J. Listeria monocytogenes meningitis in previously healthy adults: long-term follow-up. Q J Med. 1992;85:911–915.
128. Ben Shimol S, Einhorn M, Greenberg D. Listeria meningitis and ventriculitis in an immunocompetent child: case report and literature review. Infection. 2012;40:207–211.
129. Schuchat A, Deaver KA, Wenger JD, et al. Role of foods in sporadic listeriosis. I. Case-control study of dietary risk factors. The Listeria Study Group. JAMA. 1992;267:2041–2045.
130. Pinner RW, Schuchat A, Swaminathan B, et al. Role of foods in sporadic listeriosis. II. Microbiologic and epidemiologic investigation. The Listeria Study Group. JAMA. 1992;267:2046–2050.
131. Gottlieb SL, Newbern EC, Griffin PM, et al. Multistate outbreak of listeriosis linked to turkey deli meat and subsequent changes in US regulatory policy. Clin Infect Dis. 2006;42:29–36.
132. Centers for Disease Control and Prevention. Outbreak of invasive listeriosis associated with the consumption of hog head cheese—Louisiana, 2010. MMWR Morb Mortal Wkly Rep. 2011;60:401–405.
133. Centers for Disease Control and Prevention. Multistate outbreak of listeriosis associated with Jensen Farms cantaloupe—United States, August–September 2011. MMWR Morb Mortal Wkly Rep. 2011;60: 1357–1358.
134. Gaul LK, Farag NH, Shim T, et al. Hospital-acquired listeriosis outbreak caused by contaminated diced celery—Texas, 2010. Clin Infect Dis. 2013;56:20–26.
135. Bula CJ, Bille J, Glauser MP. An epidemic of food-borne listeriosis in western Switzerland: description of 57 cases involving adults. Clin Infect Dis. 1995;20:66–72.
136. Voetsch AC, Angulo FJ, Jones TF, et al. Reduction in the incidence of invasive listeriosis in foodborne diseases active surveillance network sites, 1996–2003. Clin Infect Dis. 2007;44:513–520.
137. Bennion JR, Sorvillo F, Wise ME, et al. Decreasing listeriosis mortality in the United States, 1990–2005. Clin Infect Dis. 2008;47:867–874.
138. Koopmans MM, Brouwer MC, Bijlsma MW, et al. Listeria monocytogenes sequence type 6 and increased rate of unfavorable outcome in meningitis: epidemiologic cohort study. Clin Infect Dis. 2013;57:247–253.
139. Schlesinger LS, Ross SC, Schaberg DR. Staphylococcus aureus meningitis: a broad-based epidemiologic study. Medicine (Baltimore). 1987;66:148–156.
140. Kim JH, van der Horst C, Mulrow CD, et al. Staphylococcus aureus meningitis: review of 28 cases. Rev Infect Dis. 1989;11:698–706.
141. Jensen AG, Espersen F, Skinhoj P, et al. Staphylococcus aureus meningitis. A review of 104 nationwide, consecutive cases. Arch Intern Med. 1993;153:1902–1908.
142. Arda B, Yamazhan T, Sipahi OR, et al. Meningitis due to methicillin-resistant Staphylococcus aureus (MRSA): review of 10 cases. Int J Antimicrob Agents. 2005;25:414–418.
143. Aguilar J, Urday-Cornejo V, Donabedian S, et al. Staphylococcus aureus meningitis: case series and literature review. Medicine (Baltimore). 2010;89:117–125.
144. Pintado V, Pazos R, Jimenez-Mejias ME, et al. Methicillin-resistant Staphylococcus aureus meningitis in adults: a multicenter study of 86 cases. Medicine (Baltimore). 2012;91:10–17.
145. Heerema MS, Ein ME, Musher DM, et al. Anaerobic bacterial meningitis. Am J Med. 1979;67:219–227.
146. Law DA, Aronoff SC. Anaerobic meningitis in children: case report and review of the literature. Pediatr Infect Dis J. 1992;11:968–971.
147. Feder HM Jr. Bacteroides fragilis meningitis. Rev Infect Dis. 1987;9: 783–786.
148. Tarnvik A. Anaerobic meningitis in children. Eur J Clin Microbiol. 1986;5:271–274.
149. Long JG, Preblud SR, Keyserling HL. Clostridium perfringens meningitis in an infant: case report and literature review. Pediatr Infect Dis J. 1987;6:752–754.
150. van de Beek D, de Gans J, Spanjaard L, et al. Group a streptococcal meningitis in adults: report of 41 cases and a review of the literature. Clin Infect Dis. 2002;34:e32–e36.
151. Bignardi GE, Isaacs D. Neonatal meningitis due to Streptococcus mitis. Rev Infect Dis. 1989;11:86–88.
152. Stevenson KB, Murray EW, Sarubbi FA. Enterococcal meningitis: report of four cases and review. Clin Infect Dis. 1994;18:233–239.
153. Knoll BM, Hellmann M, Kotton CN. Vancomycin-resistant Enterococcus faecium meningitis in adults: case series and review of the literature. Scand J Infect Dis. 2013;45:131–139.
154. Rajasekhar A, Clancy CJ. Meningitis due to group C Streptococcus: a case report and review of the literature. Scand J Infect Dis. 2010;42:571–578.
155. Minces LR, Brown PJ, Veldkamp PJ. Human meningitis from Streptococcus equi subsp. zooepidemicus acquired as zoonoses. Epidemiol Infect. 2011;139:406–410.
156. Lu CH, Chang WN, Chang HW. Adults with meningitis caused by viridans streptococci. Infection. 2001;29:305–309.
157. Centers for Disease Control and Prevention. Bacterial meningitis after intrapartum spinal anesthesia—New York and Ohio, 2008–2009. MMWR Morb Mortal Wkly Rep. 2010;59:65–69.
158. Rubin L, Sprecher H, Kabaha A, et al. Meningitis following spinal anesthesia: 6 cases in 5 years. Infect Control Hosp Epidemiol. 2007;28:1187–1190.
159. Hsu J, Jensen B, Arduino M, et al. Streptococcal meningitis following myelogram procedures. Infect Control Hosp Epidemiol. 2007;28:614–617.
160. Mai NT, Hoa NT, Nga TV, et al. Streptococcus suis meningitis in adults in Vietnam. Clin Infect Dis. 2008;46:659–667.
161. Nghia HD, Tu le TP, Wolbers M, et al. Risk factors of Streptococcus suis infection in Vietnam. A case-control study. PLoS One. 2011;6:e17604.
162. Downs NJ, Hodges GR, Taylor SA. Mixed bacterial meningitis. Rev Infect Dis. 1987;9:693–703.
163. Sferra TJ, Pacini DL. Simultaneous recovery of bacterial and viral pathogens from cerebrospinal fluid. Pediatr Infect Dis J. 1988;7:552–556.
164. Gerber J, Nau R. Mechanisms of injury in bacterial meningitis. Curr Opin Neurol. 2010;23:312–318.
165. Kim KS. Current concepts on the pathogenesis of Escherichia coli meningitis: implications for therapy and prevention. Curr Opin Infect Dis. 2012;25:273–278.
166. Koedel U, Klein M, Pfister HW. New understandings on the pathophysiology of bacterial meningitis. Curr Opin Infect Dis. 2010;23:217–223.
167. Koedel U, Scheld WM, Pfister HW. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. 2002;2:721–736.
168. Mook-Kanamori BB, Geldhoff M, van der Poll T, et al. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. 2011;24:557–591.
169. Quagliarello V, Scheld WM. Bacterial meningitis: pathogenesis, pathophysiology, and progress. N Engl J Med. 1992;327:864–872.
170. Scheld WM, Koedel U, Nathan B, et al. Pathophysiology of bacterial meningitis: mechanism(s) of neuronal injury. J Infect Dis. 2002; 186(suppl 2):S225–S233.
171. Tunkel AR. Bacterial Meningitis. Philadelphia: Lippincott Williams & Wilkins; 2001.
172. Tunkel AR, Scheld WM. Pathogenesis and pathophysiology of bacterial meningitis. Clin Microbiol Rev. 1993;6:118–136.
173. Tunkel AR, Wispelwey B, Scheld WM. Bacterial meningitis: recent advances in pathophysiology and treatment. Ann Intern Med. 1990;112: 610–623.
174. van der Flier M, Geelen SP, Kimpen JL, et al. Reprogramming the host response in bacterial meningitis: how best to improve outcome? Clin Microbiol Rev. 2003;16:415–429.
175. Adams RD, Kubik CS, Bonner FJ. The clinical and pathological aspects of B influenzal meningitis. Arch Pediatr. 1948;65:408–441.
176. Escourolle R, Poirier J. Pathology of infectious diseases. In: Escourolle R, Poirier J, eds. Manual of Basic Neuropathology. Philadelphia: WB Saunders; 1978:105–120.
177. Walton J. The cerebrospinal fluid (CSF): anatomy and physiology. In: Walton J, ed. Brain’s Disease of the Nervous System. Vol 1. 9th ed. Oxford: Oxford University Press; 1985:64–71.
178. Scheld WM. Pathogenesis and pathophysiology of pneumococcal meningitis. In: Sande MA, Smith AL, Root RK, eds. Bacterial Meningitis. London: Churchill Livingstone; 1985:37–69.
179. Miller JD, Adams JH. The pathophysiology of raised intracranial pressure. In: Adams JH, Corsellis JAN, Duchen LW, eds. Greenfield’s Neuropathology. 4th ed. New York: Wiley Medical; 1984:53–84.
180. Dacey RG. Monitoring and treating increased intracranial pressure. Pediatr Infect Dis J. 1987;6:1161–1163.
181. Sande MA, Scheld WM, McCracken GH. Summary of a workshop on the pathophysiology of bacterial meningitis: implications for new management strategies. Pediatr Infect Dis J. 1987;6:1167–1171.
182. Ropper AH. Raised intracranial pressure in neurologic disease. Semin Neurol. 1984;4:397–407.
183. Rorke LB, Pitts FW. Purulent meningitis: the pathologic basis of clinical manifestations. Clin Pediatr. 1963;2:64–71.
184. Bell WE. Bacterial meningitis in children: selected aspects. Pediatr Clin North Amer. 1992;39:651–658.
185. Saez-Llorens X, McCracken GH Jr. Bacterial meningitis in children. Lancet. 2003;361:2139–2148.
186. Kim KS. Acute bacterial meningitis in infants and children. Lancet Infect Dis. 2010;10:32–42.
187. Brudzinski J. Un signe nouveau sur les membres inférieurs dans les méningites chez les infants (signe de la nuque). Arch Med Enfants. 1909;12:745–752.
188. Verghese A, Gallemore G. Kernig’s and Brudzinski’s signs revisited. Rev Infect Dis. 1987;9:1187–1192.
189. Kernig VM. Über ein krankheits symptom der acuten meningitis. St Petersburg Med Wochenschr. 1882;7:398.
190. DeMyer W. Examination of the patient who has a disorder of consciousness. In: DeMyer W, ed. Technique of the Neurologic Examination. 3rd ed. New York: McGraw-Hill; 1980:359–404.
191. Valmari P, Peltola H, Ruuskanen O, et al. Childhood bacterial meningitis: initial symptoms and signs related to age, and reasons for consulting a physician. Eur J Pediatr. 1987;146:515–518.
192. Fontana A, Kristensen F, Dubs R, et al. Production of prostaglandin E and an interleukin-1 like factor by cultured astrocytes and C6 glioma cells. J Immunol. 1982;129:2413–2419.
193. Gururaj VJ, Russo RM, Allen JE, et al. To tap or not to tap. What are the best indicators for performing a lumbar puncture in an outpatient child? Clin Pediatr (Phila). 1973;12:488–493.
194. Amarilyo G, Alper A, Ben-Tov A, et al. Diagnostic accuracy of clinical symptoms and signs in children with meningitis. Pediatr Emerg Care. 2011;27:196–199.
195. Geiseler PJ, Nelson KE. Bacterial meningitis without clinical signs of meningeal irritation. South Med J. 1982;75:448–450.
196. McCarthy PL, Sharpe MR, Spiesel SZ, et al. Observation scales to identify serious illness in febrile children. Pediatrics. 1982;70:802–809.
197. Klein JO, Feigin RD, McCracken GH Jr. Report of the Task Force on Diagnosis and Management of Meningitis. Pediatrics. 1986;78:959–982.
198. Dodge PR, Swartz MN. Bacterial meningitis—a review of selected aspects. II. Special neurologic problems, postmeningitic complications and clinicopathological correlations. N Engl J Med. 1965;272:1003–1010.
199. Kaplan SL, Fishman MA. Update on bacterial meningitis. J Child Neurol. 1988;3:82–93.
200. Dunn EW. Neurologic complications of bacterial meningitis. In: Dunn DW, Epstein LG, eds. Decision Making in Child Neurology. Toronto: BC Decker; 1987:162–163.
201. Rutter N, Smales OR. Role of routine investigations in children presenting with their first febrile convulsion. Arch Dis Child. 1977;52:188–191.
202. Lorber J, Sunderland R. Lumbar puncture in children with convulsions associated with fever. Lancet. 1980;1:785–786.
203. Nelson KB, Ellenberg JH. Febrile seizures. In: Dreifuss FE, ed. Pediatric Epileptology. Boston: John Wright; 1983:173–198.
204. Tunkel AR, Scheld WM. Acute meningitis. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 4th ed. New York: Churchill Livingstone; 1995:831–865.
205. Harter DH, Jubelt B. Bacterial infections. In: Rowland LP, ed. Merritt’s Textbook of Neurology. Philadelphia: Lea & Febiger; 1984:53–72.
206. Frederiks JAM. Meningococcal meningitis. In: Vinken PJ, Bruyn GW, Klawans HL, et al, eds. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1988:21–40.
207. Kaplan SL, Feigin RD. Clinical presentations, prognostic factors and diagnosis of bacterial meningitis. In: Sande MA, Smith AL, Root RK, eds. Bacterial Meningitis. New York: Churchill Livingstone; 1985:83–94.
208. Kaplan SL, Feigin RD. The syndrome of inappropriate secretion of antidiuretic hormone in children with bacterial meningitis. J Pediatr. 1978;92:758–761.
209. Kanakriyeh M, Carvajal HF, Vallone AM. Initial fluid therapy for children with meningitis with consideration of the syndrome of inappropriate anti-diuretic hormone. Clin Pediatr (Phila). 1987;26:126–130.
210. Reefhuis J, Honein MA, Whitney CG, et al. Risk of bacterial meningitis in children with cochlear implants. N Engl J Med. 2003;349:435–445.
211. Carpenter RR, Petersdorf RG. Risk of bacterial meningitis in children with cochlear implants. Am J Med. 1962;33:262–275.
212. Stanek RJ, Mufson MA. A 20-year epidemiological study of pneumococcal meningitis. Clin Infect Dis. 1999;28:1265–1272.
213. Bruyn GA, Kremer HP, de Marie S, et al. Clinical evaluation of pneumococcal meningitis in adults over a twelve-year period. Eur J Clin Microbiol Infect Dis. 1989;8:695–700.
214. Thomas KE, Hasbun R, Jekel J, et al. The diagnostic accuracy of Kernig’s sign, Brudzinski’s sign, and nuchal rigidity in adults with suspected meningitis. Clin Infect Dis. 2002;35:46–52.
215. Geiseler PJ, Nelson KE, Levin S, et al. Community-acquired purulent meningitis: a review of 1,316 cases during the antibiotic era, 1954–1976. Rev Infect Dis. 1980;2:725–745.
216. van de Beek D, de Gans J, Spanjaard L, et al. Clinical features and prognostic factors in adults with bacterial meningitis. N Engl J Med. 2004;351:1849–1859.
217. Schut ES, Lucas MJ, Brouwer MC, et al. Cerebral infarction in adults with bacterial meningitis. Neurocrit Care. 2012;16:421–427.
218. Kaufman BA, Tunkel AR, Pryor JC, et al. Meningitis in the neurosurgical patient. Infect Dis Clin North Am. 1990;4:677–701.
219. Keroack MA. The patient with suspected meningitis. Emerg Med Clin North Am. 1987;5:807–826.
220. Spagnuolo PJ, Ellner JJ, Lerner PI, et al. Haemophilus influenzae meningitis: the spectrum of disease in adults. Medicine (Baltimore). 1982;61:74–85.
221. Gorse GJ, Thrupp LD, Nudleman KL, et al. Bacterial meningitis in the elderly. Arch Intern Med. 1984;144:1603–1607.
222. Behrman RE, Meyers BR, Mendelson MH, et al. Central nervous system infections in the elderly. Arch Intern Med. 1989;149:1596–1599.
223. Lefrock JL, Smith BR. Gram-negative bacillary meningitis in adults. In: Vinken PJ, Bruyn GW, Klawans HL, eds. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1988:103–115.
224. Cabellos C, Verdaguer R, Olmo M, et al. Community-acquired bacterial meningitis in elderly patients: experience over 30 years. Medicine (Baltimore). 2009;88:115–119.
225. Tunkel AR, Scheld WM. Acute infectious complications of head injury. In: Braackman R, ed. Handbook of Clinical Neurology—Head Injury. Amsterdam: Elsevier; 1990:317–326.
226. Kline MW. Review of recurrent bacterial meningitis. Pediatr Infect Dis J. 1989;8:630–634.
227. Berk SL, McCabe WR. Meningitis caused by gram-negative bacilli. Ann Intern Med. 1980;93:253–260.
228. Bayston R, Lari J. A study of the sources of infection in colonised shunts. Dev Med Child Neurol. 1974;16:16–22.
229. Schoenbaum SC, Gardner P, Shillito J. Infections of cerebrospinal fluid shunts: epidemiology, clinical manifestations, and therapy. J Infect Dis. 1975;131:543–552.
230. Forward KR, Fewer HD, Stiver HG. Cerebrospinal fluid shunt infections. A review of 35 infections in 32 patients. J Neurosurg. 1983;59:389–394.
231. Browne MJ, Dinndorf PA, Perek D, et al. Infectious complications of intraventricular reservoirs in cancer patients. Pediatr Infect Dis J. 1987;6:182–189.
232. Tunkel AR, Scheld WM. Central nervous system infection in the compromised host. In: Rubin RH, Young LS, eds. Clinical Approach to Infection in the Compromised Host. 3rd ed. New York: Plenum Press; 1994: 163–210.
233. Rubin RH, Hooper DC. Central nervous system infections in the compromised host. Med Clin North Am. 1985;69:281–293.
234. Armstrong D, Wong B. Central nervous system infections in immunocompromised hosts. Ann Rev Med. 1982;33:293–308.
235. Hussein AS, Shafran SD. Acute bacterial meningitis in adults. A 12-year review. Medicine (Baltimore). 2000;79:360–368.
236. John JF. Listeria monocytogenes. In: Vinken PJ, Bruyn GW, Klawans HL, eds. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1988: 89–101.
237. Hasbun R, Abrahams J, Jekel J, et al. Computed tomography of the head before lumbar puncture in adults with suspected meningitis. N Engl J Med. 2001;345:1727–1733.
238. Kaplan K. Brain abscess. Med Clin North Am. 1985;69:345–360.
239. Wispelwey B, Scheld WM. Brain abscess. Clin Neuropharmacol. 1987;10:483–510.
240. Hakan T, Ceran N, Erdem I, et al. Bacterial brain abscesses: an evaluation of 96 cases. J Infect. 2006;52:359–366.
241. Tonon E, Scotton PG, Gallucci M, et al. Brain abscess: clinical aspects of 100 patients. Int J Infect Dis. 2006;10:103–109.
242. Carpenter J, Stapleton S, Holliman R. Retrospective analysis of 49 cases of brain abscess and review of the literature. Eur J Clin Microbiol Infect Dis. 2007;26:1–11.
243. Lee TH, Chang WN, Su TM, et al. Clinical features and predictive factors of intraventricular rupture in patients who have bacterial brain abscesses. J Neurol Neurosurg Psychiatry. 2007;78:303–309.
244. Silverberg AL, DiNubile MJ. Subdural empyema and cranial epidural abscess. Med Clin North Am. 1985;69:361–374.
245. Hicks CW, Weber JG, Reid JR, et al. Identifying and managing intracranial complications of sinusitis in children: a retrospective series. Pediatr Infect Dis J. 2011;30:222–226.
246. DeMuri GP, Wald ER. Complications of acute bacterial sinusitis in children. Pediatr Infect Dis J. 2011;30:701–702.
247. Morgan DW, Williams B. Posterior fossa subdural empyema. Brain. 1985;108(pt 4):983–992.
248. Madhugiri VS, Sastri BV, Bhagavatula ID, et al. Posterior fossa subdural empyema in children—management and outcome. Childs Nerv Syst. 2011; 27:137–144.
249. Barnes DW, Whitley RJ. CNS diseases associated with varicella zoster virus and herpes simplex virus infection. Pathogenesis and current therapy. Neurol Clin. 1986;4:265–283.
250. Reik L. Lyme disease. In: Scheld WM, Whitley RJ, Durack DT, eds. Infections of the Central Nervous System. 2nd ed. New York: Lippincott-Raven; 1996:685–718.
251. Hornick RB. The typhus group. In: Wyngaarden JB, Smith LH, eds. Cecil Textbook of Medicine. Philadelphia: WB Saunders; 1988:1738–1742.
252. Mohr JP, Kistler JP, Zabramski JM, et al. Intracranial aneurysms. In: Barnett HJM, Mohr JP, Stein BM, et al, eds. Stroke: Pathophysiology, Diagnosis, and Management. New York: Churchill Livingstone; 1986: 643–677.
253. Guze BH, Baxter LR Jr. Current concepts. Neuroleptic malignant syndrome. N Engl J Med. 1985;313:163–166.
254. Dougherty JM, Roth RM. Cerebral spinal fluid. Emerg Med Clin North Am. 1986;4:281–297.
255. Corbett JJ, Mehta MP. Cerebrospinal fluid pressure in normal obese subjects and patients with pseudotumor cerebri. Neurology. 1983;33: 1386–1388.
256. Conly JM, Ronald AR. Cerebrospinal fluid as a diagnostic body fluid. Am J Med. 1983;75:102–108.
257. Marton KI, Gean AD. The spinal tap: a new look at an old test. Ann Intern Med. 1986;104:840–848.
258. Hayward RA, Oye RK. Are polymorphonuclear leukocytes an abnormal finding in cerebrospinal fluid? Results from 225 normal cerebrospinal fluid specimens. Arch Intern Med. 1988;148:1623–1624.
259. Schmidley JW, Simon RP. Postictal pleocytosis. Ann Neurol. 1981;9: 81–84.
260. Karandanis D, Shulman JA. Recent survey of infectious meningitis in adults: review of laboratory findings in bacterial, tuberculous, and aseptic meningitis. South Med J. 1976;69:449–457.
261. Swartz MN, Dodge PR. Bacterial meningitis—a review of selected aspects. I. General clinical features, special problems and unusual meningeal reactions mimicking bacterial meningitis. N Engl J Med. 1965;272:898–902.
262. Powers WJ. Cerebrospinal fluid lymphocytosis in acute bacterial meningitis. Am J Med. 1985;79:216–220.
263. Roos KL, Scheld WM. The management of fulminant meningitis in the intensive care unit. Crit Care Clin. 1988;4:375–392.
264. Lindquist L, Linne T, Hansson LO, et al. Value of cerebrospinal fluid analysis in the differential diagnosis of meningitis: a study in 710 patients with suspected central nervous system infection. Eur J Clin Microbiol Infect Dis. 1988;7:374–380.
265. Lannigan R, MacDonald MA, Marrie TJ, et al. Evaluation of cerebrospinal fluid lactic acid levels as an aid in differential diagnosis of bacterial and viral meningitis in adults. J Clin Microbiol. 1980;11:324–327.
266. Kolmel HW, von Maravic M. Correlation of lactic acid level, cell count and cytology in cerebrospinal fluid of patients with bacterial and non-bacterial meningitis. Acta Neurol Scand. 1988;78:6–9.
267. Genton B, Berger JP. Cerebrospinal fluid lactate in 78 cases of adult meningitis. Intensive Care Med. 1990;16:196–200.
268. Huy NT, Thao NT, Diep DT, et al. Cerebrospinal fluid lactate concentration to distinguish bacterial from aseptic meningitis: a systemic review and meta-analysis. Crit Care. 2010;14:R240.
269. Sakushima K, Hayashino Y, Kawaguchi T, et al. Diagnostic accuracy of cerebrospinal fluid lactate for differentiating bacterial meningitis from aseptic meningitis: a meta-analysis. J Infect. 2011;62:255–262.
270. Viallon A, Desseigne N, Marjollet O, et al. Meningitis in adult patients with a negative direct cerebrospinal fluid examination: value of cytochemical markers for differential diagnosis. Crit Care. 2011;15:R136.
271. Spanos A, Harrell FE Jr, Durack DT. Differential diagnosis of acute meningitis. An analysis of the predictive value of initial observations. JAMA. 1989;262:2700–2707.
272. Brouwer MC, Thwaites GE, Tunkel AR, et al. Dilemmas in the diagnosis of acute community-acquired bacterial meningitis. Lancet. 2012;380: 1684–1692.
273. Nigrovic LE, Malley R, Kuppermann N. Meta-analysis of bacterial meningitis score validation studies. Arch Dis Child. 2012;97:799–805.
274. Kaplan SL, Smith EO, Wills C, et al. Association between preadmission oral antibiotic therapy and cerebrospinal fluid findings and sequelae caused by Haemophilus influenzae type b meningitis. Pediatr Infect Dis. 1986;5:626–632.
275. Blazer S, Berant M, Alon U. Bacterial meningitis. Effect of antibiotic treatment on cerebrospinal fluid. Am J Clin Pathol. 1983;80:386–387.
276. Gray LD, Fedorko DP. Laboratory diagnosis of bacterial meningitis. Clin Microbiol Rev. 1992;5:130–145.
277. Tarafdar K, Rao S, Recco RA, et al. Lack of sensitivity of the latex agglutination test to detect bacterial antigen in the cerebrospinal fluid of patients with culture-negative meningitis. Clin Infect Dis. 2001;33:406–408.
278. Tunkel AR, Hartman BJ, Kaplan SL, et al. Practice guidelines for the management of bacterial meningitis. Clin Infect Dis. 2004;39:1267–1284.
279. Dwelle TL, Dunkle LM, Blair L. Correlation of cerebrospinal fluid endotoxinlike activity with clinical and laboratory variables in gram-negative bacterial meningitis in children. J Clin Microbiol. 1987;25:856–858.
280. Gray BM, Simmons DR, Mason H, et al. Quantitative levels of C-reactive protein in cerebrospinal fluid in patients with bacterial meningitis and other conditions. J Pediatr. 1986;108:665–670.
281. Abramson JS, Hampton KD, Babu S, et al. The use of C-reactive protein from cerebrospinal fluid for differentiating meningitis from other central nervous system diseases. J Infect Dis. 1985;151:854–858.
282. Sormunen P, Kallio MJ, Kilpi T, et al. C-reactive protein is useful in distinguishing Gram stain-negative bacterial meningitis from viral meningitis in children. J Pediatr. 1999;134:725–729.
283. Shimetani N, Shimetani K, Mori M. Levels of three inflammation markers, C-reactive protein, serum amyloid A protein and procalcitonin, in the serum and cerebrospinal fluid of patients with meningitis. Scand J Clin Lab Invest. 2001;61:567–574.
284. Nathan BR, Scheld WM. The potential roles of C-reactive protein and procalcitonin concentrations in the serum and cerebrospinal fluid in the diagnosis of bacterial meningitis. Curr Clin Top Infect Dis. 2002;22: 155–165.
285. Viallon A, Zeni F, Lambert C, et al. High sensitivity and specificity of serum procalcitonin levels in adults with bacterial meningitis. Clin Infect Dis. 1999;28:1313–1316.
286. Schwarz S, Bertram M, Schwab S, et al. Serum procalcitonin levels in bacterial and abacterial meningitis. Crit Care Med. 2000;28: 1828–1832.
287. Saha SK, Darmstadt GL, Yamanaka N, et al. Rapid diagnosis of pneumococcal meningitis: implications for treatment and measuring disease burden. Pediatr Infect Dis J. 2005;24:1093–1098.
288. Werno AM, Murdoch DR. Medical microbiology: laboratory diagnosis of invasive pneumococcal disease. Clin Infect Dis. 2008;46:926–932.
289. Moisi JC, Saha SK, Falade AG, et al. Enhanced diagnosis of pneumococcal meningitis with use of the Binax NOW immunochromatographic test of Streptococcus pneumoniae antigen: a multisite study. Clin Infect Dis. 2009;48(suppl 2):S49–S56.
290. Cherian T, Lalitha MK, Manoharan A, et al. PCR-enzyme immunoassay for detection of Streptococcus pneumoniae DNA in cerebrospinal fluid samples from patients with culture-negative meningitis. J Clin Microbiol. 1998;36:3605–3608.
291. Backman A, Lantz P, Radstrom P, et al. Evaluation of an extended diagnostic PCR assay for detection and verification of the common causes of bacterial meningitis in CSF and other biological samples. Mol Cell Probes. 1999;13:49–60.
292. Kristiansen BE, Ask E, Jenkins A, et al. Rapid diagnosis of meningococcal meningitis by polymerase chain reaction. Lancet. 1991;337:1568–1569.
293. Ni H, Knight AI, Cartwright K, et al. Polymerase chain reaction for diagnosis of meningococcal meningitis. Lancet. 1992;340:1432–1434.
294. Jaton K, Sahli R, Bille J. Development of polymerase chain reaction assays for detection of Listeria monocytogenes in clinical cerebrospinal fluid samples. J Clin Microbiol. 1992;30:1931–1936.
295. Radstrom P, Backman A, Qian N, et al. Detection of bacterial DNA in cerebrospinal fluid by an assay for simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and streptococci using a seminested PCR strategy. J Clin Microbiol. 1994;32:2738–2744.
296. Saravolatz LD, Manzor O, VanderVelde N, et al. Broad-range bacterial polymerase chain reaction for early detection of bacterial meningitis. Clin Infect Dis. 2003;36:40–45.
297. Tzanakaki G, Tsopanomichalou M, Kesanopoulos K, et al. Simultaneous single-tube PCR assay for the detection of Neisseria meningitidis, Haemophilus influenzae type b and Streptococcus pneumoniae. Clin Microbiol Infect. 2005;11:386–390.
298. Wang X, Theodore MJ, Mair R, et al. Clinical validation of multiplex real-time PCR assays for detection of bacterial meningitis pathogens. J Clin Microbiol. 2012;50:702–708.
299. Welinder-Olsson C, Dotevall L, Hogevik H, et al. Comparison of broad-range bacterial PCR and culture of cerebrospinal fluid for diagnosis of community-acquired bacterial meningitis. Clin Microbiol Infect. 2007;13:879–886.
300. Singhi SC, Mohankumar D, Singhi PD, et al. Evaluation of polymerase chain reaction (PCR) for diagnosing Haemophilus influenzae b meningitis. Ann Trop Paediatr. 2002;22:347–353.
301. Le Monnier A, Abachin E, Beretti JL, et al. Diagnosis of Listeria monocytogenes meningoencephalitis by real-time PCR for the hly gene. J Clin Microbiol. 2011;49:3917–3923.
302. Kearns AM, Graham C, Burdess D, et al. Rapid real-time PCR for determination of penicillin susceptibility in pneumococcal meningitis, including culture-negative cases. J Clin Microbiol. 2002;40:682–684.
303. Enzmann DR. Acute meningitis. In: Enzmann DR, ed. Imaging of Infections and Inflammations of the Central Nervous System: Computed Tomography, Ultrasound, and Nuclear Magnetic Resonance. New York: Raven Press; 1984:188–211.
304. Lin TY, Nelson JD, McCracken GH Jr. Fever during treatment for bacterial meningitis. Pediatr Infect Dis J. 1984;3:319–322.
305. Kline MW, Kaplan SL. Computed tomography in bacterial meningitis of childhood. Pediatr Infect Dis J. 1988;7:855–857.
306. Friedland IR, Paris MM, Rinderknecht S, et al. Cranial computed tomographic scans have little impact on management of bacterial meningitis. Am J Dis Child. 1992;146:1484–1487.
307. Cabral DA, Flodmark O, Farrell K, et al. Prospective study of computed tomography in acute bacterial meningitis. J Pediatr. 1987;111:201–205.
308. Benson P, Nyhan WL, Shimizu H. The prognosis of subdural effusions complicating pyogenic meningitis. J Pediatr. 1960;58:670–682.
309. Syrogiannopoulos GA, Nelson JD, McCracken GH Jr. Subdural collections of fluid in acute bacterial meningitis: a review of 136 cases. Pediatr Infect Dis. 1986;5:343–352.
310. Goodman JM, Mealey J Jr. Postmeningitic subdural effusions: the syndrome and its management. J Neurosurg. 1969;30:658–663.
311. Britt RH, Enzmann DR, Yeager AS. Neuropathological and computerized tomographic findings in experimental brain abscess. J Neurosurg. 1981;55:590–603.
312. McNamara MT. Paramagnetic contrast media for magnetic resonance imaging of the central nervous system. In: Brant-Zawadzki MB, Normal D, eds. Magnetic Resonance Imaging of the Central Nervous System. New York: Raven Press; 1987:97–105.
313. Mathews VP, Kuharik MA, Edwards MK, et al. Dyke award. Gd-DTPA-enhanced MR imaging of experimental bacterial meningitis: evaluation and comparison with CT. AJR Am J Roentgenol. 1989;152:131–136.
314. Sze G, Zimmerman RD. The magnetic resonance imaging of infections and inflammatory diseases. Radiol Clin North Am. 1988;26:839–859.
315. Nickerson JP, Richner B, Santy K, et al. Neuroimaging of pediatric intracranial infection—part 1: techniques and bacterial infections. J Neuroimaging. 2012;22:e42–e51.
316. van de Beek D, Brouwer MC, Thwaites GE, et al. Advances in treatment of bacterial meningitis. Lancet. 2012;380:1693–1702.
317. Aronin SI, Peduzzi P, Quagliarello VJ. Community-acquired bacterial meningitis: risk stratification for adverse clinical outcome and effect of antibiotic timing. Ann Intern Med. 1998;129:862–869.
318. Miner JR, Heegaard W, Mapes A, et al. Presentation, time to antibiotics, and mortality of patients with bacterial meningitis at an urban county medical center. J Emerg Med. 2001;21:387–392.
319. Lu CH, Huang CR, Chang WN, et al. Community-acquired bacterial meningitis in adults: the epidemiology, timing of appropriate antimicrobial therapy, and prognostic factors. Clin Neurol Neurosurg. 2002;104:352–358.
320. Proulx N, Frechette D, Toye B, et al. Delays in the administration of antibiotics are associated with mortality from adult acute bacterial meningitis. QJM. 2005;98:291–298.
321. Lepur D, Barsic B. Community-acquired bacterial meningitis in adults: antibiotic timing in disease course and outcome. Infection. 2007;35:225–231.
322. Coant PN, Kornberg AE, Duffy LC, et al. Blood culture results as determinants in the organism identification of bacterial meningitis. Pediatr Emerg Care. 1992;8:200–205.
323. van de Beek D, de Gans J, Tunkel AR, et al. Community-acquired bacterial meningitis in adults. N Engl J Med. 2006;354:44–53.
324. Scheld WM. Theoretical and practical considerations of antibiotic therapy for bacterial meningitis. Pediatr Infect Dis. 1985;4:74–83.
325. Papastamelos AG, Tunkel AR. Antibacterial agents in infections of the central nervous system and eye. Infect Dis Clin North Am. 1995;9:615–637.
326. Tunkel AR, Scheld WM. Therapy of bacterial meningitis: principles and practice. Infect Control Hosp Epidemiol. 1989;10:565–569.
327. Sinner SW, Tunkel AR. Antimicrobial agents in the treatment of bacterial meningitis. Infect Dis Clin North Am. 2004;18:581–602, ix.
328. Nau R, Sorgel F, Eiffert H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev. 2010;23:858–883.
329. Reed MD. Current concepts in clinical therapeutics: bacterial meningitis in infants and children. Clin Pharm. 1986;5:798–809.
330. Barlow CF. Clinical aspects of the blood–brain barrier. Ann Rev Med. 1964;15:187–205.
331. Quagliarello VJ, Long WJ, Scheld WM. Morphologic alterations of the blood-brain barrier with experimental meningitis in the rat. Temporal sequence and role of encapsulation. J Clin Invest. 1986;77:1084–1095.
332. Cherubin CE, Marr JS, Sierra MF, et al. Listeria and gram-negative bacillary meningitis in New York City, 1972–1979. Frequent causes of meningitis in adults. Am J Med. 1981;71:199–209.
333. Pelkonen T, Roine I, Cruzeiro ML, et al. Slow initial beta-lactam infusion and oral paracetamol to treat childhood bacterial meningitis: a randomised, controlled trial. Lancet Infect Dis. 2011;11:613–621.
334. McCracken GH Jr, Nelson JD, Kaplan SL, et al. Consensus report: antimicrobial therapy for bacterial meningitis in infants and children. Pediatr Infect Dis J. 1987;6:501–505.
335. Van Esso D, Fontanals D, Uriz S, et al. Neisseria meningitidis strains with decreased susceptibility to penicillin. Pediatr Infect Dis J. 1987;6:438–439.
336. Cabellos C, Viladrich PF, Verdaguer R, et al. A single daily dose of ceftriaxone for bacterial meningitis in adults: experience with 84 patients and review of the literature. Clin Infect Dis. 1995;20:1164–1168.
337. Dillon JR, Pauze M, Yeung KH. Spread of penicillinase-producing and transfer plasmids from the gonococcus to Neisseria meningitidis. Lancet. 1983;1:779–781.
338. Sutcliffe EM, Jones DM, el-Sheikh S, et al. Penicillin-insensitive meningococci in the UK. Lancet. 1988;1:657–658.
339. Woods CR, Smith AL, Wasilauskas BL, et al. Invasive disease caused by Neisseria meningitidis relatively resistant to penicillin in North Carolina. J Infect Dis. 1994;170:453–456.
340. Saez-Nieto JA, Lujan R, Berron S, et al. Epidemiology and molecular basis of penicillin-resistant Neisseria meningitidis in Spain: a 5-year history (1985–1989). Clin Infect Dis. 1992;14:394–402.
341. Jackson LA, Tenover FC, Baker C, et al. Prevalence of Neisseria meningitidis relatively resistant to penicillin in the United States, 1991. Meningococcal Disease Study Group. J Infect Dis. 1994;169:438–441.
342. Brown EM, Fisman DN, Drews SJ, et al. Epidemiology of invasive meningococcal disease with decreased susceptibility to penicillin in Ontario, Canada, 2000 to 2006. Antimicrob Agents Chemother. 2010; 54:1016–1021.
343. Perez Trallero E, Garcia Arenzana JM, Ayestaran I, et al. Comparative activity in vitro of 16 antimicrobial agents against penicillin-susceptible meningococci and meningococci with diminished susceptibility to penicillin. Antimicrob Agents Chemother. 1989;33:1622–1623.
344. Centers for Disease Control and Prevention. Effects of new penicillin susceptibility breakpoints for Streptococcus pneumoniae—United States, 2006–2007. MMWR Morb Mortal Wkly Rep. 2008;57:1353–1355.
345. National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Susceptibility Testing. Tenth Informational Supplement (Aerobic Dilution). Wayne, PA: National Committee for Clinical Laboratory Standards; 2000.
346. Kaplan SL, Mason EO Jr. Mechanisms of pneumococcal antibiotic resistance and treatment of pneumococcal infections in 2002. Pediatr Ann. 2002;31:250–260.
347. Jacobs MR. Drug-resistant Streptococcus pneumoniae: rational antibiotic choices. Am J Med. 1999;106:19S–25S; discussion 48S–52S.
348. Barroso DE, Godoy D, Castineiras TM, et al. Beta-lactam resistance, serotype distribution, and genotypes of meningitis-causing Streptococcus pneumoniae, Rio de Janeiro, Brazil. Pediatr Infect Dis J. 2012;31:30–36.
349. Appelbaum PC. Resistance among Streptococcus pneumoniae: implications for drug selection. Clin Infect Dis. 2002;34:1613–1620.
350. Meyer CN, Samuelsson IS, Galle M, et al. Adult bacterial meningitis: aetiology, penicillin susceptibility, risk factors, prognostic factors and guidelines for empirical antibiotic treatment. Clin Microbiol Infect. 2004;10:709–717.
351. Bradley JS, Kaplan SL, Klugman KP, et al. Consensus: management of infections in children caused by Streptococcus pneumoniae with decreased susceptibility to penicillin. Pediatr Infect Dis J. 1995;14:1037–1041.
352. Tunkel AR, Scheld WM. Acute bacterial meningitis. Lancet. 1995;346: 1675–1680.
353. Giron KP, Gross ME, Musher DM, et al. In vitro antimicrobial effect against Streptococcus pneumoniae of adding rifampin to penicillin, ceftriaxone, or 1-ofloxacin. Antimicrob Agents Chemother. 1995;39:2798–2800.
354. Gerber J, Pohl K, Sander V, et al. Rifampin followed by ceftriaxone for experimental meningitis decreases lipoteichoic acid concentrations in cerebrospinal fluid and reduces neuronal damage in comparison to ceftriaxone alone. Antimicrob Agents Chemother. 2003;47:1313–1317.
355. Ricard JD, Wolff M, Lacherade JC, et al. Levels of vancomycin in cerebrospinal fluid of adult patients receiving adjunctive corticosteroids to treat pneumococcal meningitis: a prospective multicenter observational study. Clin Infect Dis. 2007;44:250–255.
356. Rybak M, Lomaestro B, Rotschafer JC, et al. Therapeutic monitoring of vancomycin in adult patients: a consensus review of the American Society of Health-System Pharmacists, the Infectious Diseases Society of America, and the Society of Infectious Diseases Pharmacists. Am J Health Syst Pharm. 2009;66:82–98.
357. McCullers JA, English BK, Novak R. Isolation and characterization of vancomycin-tolerant Streptococcus pneumoniae from the cerebrospinal fluid of a patient who developed recrudescent meningitis. J Infect Dis. 2000;181:369–373.
358. Novak R, Henriques B, Charpentier E, et al. Emergence of vancomycin tolerance in Streptococcus pneumoniae. Nature. 1999;399:590–593.
359. Liu HH, Tomasz A. Penicillin tolerance in multiply drug-resistant natural isolates of Streptococcus pneumoniae. J Infect Dis. 1985;152:365–372.
360. Bradley JS, Scheld WM. The challenge of penicillin-resistant Streptococcus pneumoniae meningitis: current antibiotic therapy in the 1990s. Clin Infect Dis. 1997;24(suppl 2):S213–S221.
361. Buckingham SC, Davis Y, English BK. Pneumococcal susceptibility to meropenem in a mid-south children’s hospital. South Med J. 2002;95: 1293–1296.
362. Friedland IR, Klugman KP. Failure of chloramphenicol therapy in penicillin-resistant pneumococcal meningitis. Lancet. 1992;339:405–408.
363. Cornick JE, Everett DB, Broughton C, et al. Invasive Streptococcus pneumoniae in children, Malawi, 2004–2006. Emerg Infect Dis. 2011; 17:1107–1109.
364. Manning L, Laman M, Greenhill AR, et al. Increasing chloramphenicol resistance in Streptococcus pneumoniae isolates from Papua New Guinean children with acute bacterial meningitis. Antimicrob Agents Chemother. 2011;55:4454–4456.
365. Cottagnoud P, Acosta F, Cottagnoud M, et al. Efficacies of BMS 284756 against penicillin-sensitive, penicillin-resistant, and quinolone-resistant pneumococci in experimental meningitis. Antimicrob Agents Chemother. 2002;46:184–187.
366. Cottagnoud P, Acosta F, Cottagnoud M, et al. Gemifloxacin is efficacious against penicillin-resistant and quinolone-resistant pneumococci in experimental meningitis. Antimicrob Agents Chemother. 2002;46:1607–1609.
367. Takahata M, Yamada H, Morita T, et al. Evaluation of T-3811ME (BMS-284756), a new des-F(6)-quinolone, for treatment of meningitis caused by penicillin-resistant Streptococcus pneumoniae in rabbits. Antimicrob Agents Chemother. 2002;46:1760–1765.
368. Rodriguez-Cerrato V, McCoig CC, Saavedra J, et al. Garenoxacin (BMS-284756) and moxifloxacin in experimental meningitis caused by vancomycin-tolerant pneumococci. Antimicrob Agents Chemother. 2003;47:211–215.
369. Pfister M, Zhang L, Hammarlund-Udenaes M, et al. Modeling of transfer kinetics at the serum-cerebrospinal fluid barrier in rabbits with experimental meningitis: application to grepafloxacin. Antimicrob Agents Chemother. 2003;47:138–143.
370. Kuhn F, Cottagnoud M, Acosta F, et al. Cefotaxime acts synergistically with levofloxacin in experimental meningitis due to penicillin-resistant pneumococci and prevents selection of levofloxacin-resistant mutants in vitro. Antimicrob Agents Chemother. 2003;47:2487–2491.
371. Tunkel AR, Scheld WM. Treatment of bacterial meningitis. Curr Infect Dis Rep. 2002;4:7–16.
372. Cottagnoud P, Tauber MG. Fluoroquinolones in the treatment of meningitis. Curr Infect Dis Rep. 2003;5:329–336.
373. Saez-Llorens X, O’Ryan M. Cefepime in the empiric treatment of meningitis in children. Pediatr Infect Dis J. 2001;20:356–361.
374. Landesman SH, Corrado ML, Shah PM, et al. Past and current roles for cephalosporin antibiotics in treatment of meningitis. Emphasis on use in gram-negative bacillary meningitis. Am J Med. 1981;71:693–703.
375. Cherubin CE, Eng RH, Norrby R, et al. Penetration of newer cephalosporins into cerebrospinal fluid. Rev Infect Dis. 1989;11:526–548.
376. O’Neill E, Humphreys H, Phillips J, et al. Third-generation cephalosporin resistance among Gram-negative bacilli causing meningitis in neurosurgical patients: significant challenges in ensuring effective antibiotic therapy. J Antimicrob Chemother. 2006;57:356–359.
377. Norrby SR. Role of cephalosporins in the treatment of bacterial meningitis in adults. Overview with special emphasis on ceftazidime. Am J Med. 1985;79:56–61.
378. Segev S, Barzilai A, Rosen N, et al. Pefloxacin treatment of meningitis caused by gram-negative bacteria. Arch Intern Med. 1989;149: 1314–1316.
379. Tunkel AR, Scheld WM. Treatment of bacterial meningitis. In: Wolfson JS, Hooper DC, eds. Quinolone Antimicrobial Agents. Washington, DC: American Society for Microbiology; 1993:381–395.
380. Klugman KP, Dagan R. Carbapenem treatment of meningitis. Scand J Infect Dis Suppl. 1995;96:45–48.
381. Kim BN, Peleg AY, Lodise TP, et al. Management of meningitis due to antibiotic-resistant Acinetobacter species. Lancet Infect Dis. 2009;9: 245–255.
382. van de Beek D, Drake JM, Tunkel AR. Nosocomial bacterial meningitis. N Engl J Med. 2010;362:146–154.
383. Jimenez-Mejias ME, Pichardo-Guerrero C, Marquez-Rivas FJ, et al. Cerebrospinal fluid penetration and pharmacokinetic/pharmacodynamic parameters of intravenously administered colistin in a case of multidrug-resistant Acinetobacter baumannii meningitis. Eur J Clin Microbiol Infect Dis. 2002;21:212–214.
384. Vasan W, Desmery P, Ilutovich S, et al. Intrathecal use of colistin. J Clin Microbiol. 2000;38:3523.
385. Cascio A, Conti A, Sinardi L, et al. Post-neurosurgical multidrug-resistant Acinetobacter baumannii meningitis successfully treated with intrathecal colistin. A new case and a systematic review of the literature. Int J Infect Dis. 2010;14:e572–e579.
386. Benifla M, Zucker G, Cohen A, et al. Successful treatment of Acinetobacter meningitis with intrathecal polymyxin E. J Antimicrob Chemother. 2004;54:290–292.
387. Katragkou A, Roilides E. Successful treatment of multidrug-resistant Acinetobacter baumannii central nervous system infections with colistin. J Clin Microbiol. 2005;43:4916–4917.
388. Antachopoulos C, Karvanen M, Iosifidis E, et al. Serum and cerebrospinal fluid levels of colistin in pediatric patients. Antimicrob Agents Chemother. 2010;54:3985–3987.
389. Tutuncu EE, Kuscu F, Gurbuz Y, et al. Tigecycline use in two cases with multidrug-resistant Acinetobacter baumannii meningitis. Int J Infect Dis. 2010;14(suppl 3):e224–e226.
390. McCracken GH Jr. Current management of bacterial meningitis in infants and children. Pediatr Infect Dis J. 1992;11:169–174.
391. Schaad UB, Suter S, Gianella-Borradori A, et al. A comparison of ceftriaxone and cefuroxime for the treatment of bacterial meningitis in children. N Engl J Med. 1990;322:141–147.
392. Lebel MH, Hoyt MJ, McCracken GH Jr. Comparative efficacy of ceftriaxone and cefuroxime for treatment of bacterial meningitis. J Pediatr. 1989;114:1049–1054.
393. Jorgensen JH. Update on mechanisms and prevalence of antimicrobial resistance in Haemophilus influenzae. Clin Infect Dis. 1992;14:1119–1123.
394. Campos J, Garcia-Tornel S, Gairi JM, et al. Multiply resistant Haemophilus influenzae type b causing meningitis: comparative clinical and laboratory study. J Pediatr. 1986;108:897–902.
395. Campos J, Chanyangam M, deGroot R, et al. Genetic relatedness of antibiotic resistance determinants in multiply resistant Hemophilus influenzae. J Infect Dis. 1989;160:810–817.
396. Kaplan SL. Haemophilus influenzae meningitis. In: Vinken PJ, Bruyn GW, Klawans HL, eds. Handbook of Clinical Neurology. Amsterdam: Elsevier; 1988:59–70.
397. Krasinski K, Kusmiesz H, Nelson JD. Pharmacologic interactions among chloramphenicol, phenytoin and phenobarbital. Pediatr Infect Dis. 1982;1:232–235.
398. Saez-Llorens X, McCoig C, Feris JM, et al. Quinolone treatment for pediatric bacterial meningitis: a comparative study of trovafloxacin and ceftriaxone with or without vancomycin. Pediatr Infect Dis J. 2002;21:14–22.
399. Lerner PI, Gopalakrishna KV, Wolinsky E, et al. Group B Streptococcus (S. agalactiae) bacteremia in adults: analysis of 32 cases and review of the literature. Medicine (Baltimore). 1977;56:457–473.
400. Gunther G, Philipson A. Oral trimethoprim as follow-up treatment of meningitis caused by Listeria monocytogenes. Rev Infect Dis. 1988;10:53–55.
401. Richards SJ, Lambert CM, Scott AC. Recurrent Listeria monocytogenes meningitis treated with intraventricular vancomycin. J Antimicrob Chemother. 1992;29:351–353.
402. Roos KL, Scheld WM. Staphylococcal infections of the central nervous system. In: Archer GL, Crossley K, eds. Staphylococci and Staphylococcal Diseases. New York: Churchill Livingstone; 2013.
403. Everett ED, Strausbaugh LJ. Antimicrobial agents and the central nervous system. Neurosurgery. 1980;6:691–714.
404. Gump DW. Vancomycin for treatment of bacterial meningitis. Rev Infect Dis. 1981;3(suppl):S289–S292.
405. Higa T, Tasaka T, Kubo Y, et al. Successful treatment of meningoencephalitis caused by methicillin-resistant Staphylococcus aureus with intravenous linezolid in an allogeneic cord blood stem cell transplant recipient. Scand J Infect Dis. 2008;40:990–992.
406. Sipahi OR, Bardak S, Turhan T, et al. Linezolid in the treatment of methicillin-resistant staphylococcal post-neurosurgical meningitis: a series of 17 cases. Scand J Infect Dis. 2011;43:757–764.
407. Bardak-Ozcem S, Turhan T, Sipahi OR, et al. Daptomycin versus vancomycin in treatment of methicillin-resistant Staphylococcus aureus meningitis in an experimental rabbit model. Antimicrob Agents Chemother. 2013;57:1556–1558.
408. Lee DH, Palermo B, Chowdhury M. Successful treatment of methicillin-resistant staphylococcus aureus meningitis with daptomycin. Clin Infect Dis. 2008;47:588–590.
409. Wallace MR, Sander AW, Licitra C, et al. Methicillin-resistant Staphylococcus aureus meningitis with daptomycin. Infect Dis Clin Pract. 2009;17:69–70.
410. Kelesidis T, Humphries R, Ward K, et al. Combination therapy with daptomycin, linezolid, and rifampin as treatment option for MRSA meningitis and bacteremia. Diagn Microbiol Infect Dis. 2011;71:286–290.
411. McCabe WR. Empiric therapy for bacterial meningitis. Rev Infect Dis. 1983;5(suppl 1):S74–S83.
412. Saez-Llorens X, Castano E, Garcia R, et al. Prospective randomized comparison of cefepime and cefotaxime for treatment of bacterial meningitis in infants and children. Antimicrob Agents Chemother. 1995;39: 937–940.
413. Radetsky M. Duration of treatment in bacterial meningitis: a historical inquiry. Pediatr Infect Dis J. 1990;9:2–9.
414. Molyneux E, Nizami SQ, Saha S, et al. 5 versus 10 days of treatment with ceftriaxone for bacterial meningitis in children: a double-blind randomised equivalence study. Lancet. 2011;377:1837–1845.
415. Snape MD, Kelly DF. Fine with five? Shorter antibiotic courses for childhood meningitis. Lancet. 2011;377:1809–1810.
416. Gardner P, Leipzig T, Phillips P. Infections of central nervous system shunts. Med Clin North Am. 1985;69:297–314.
417. Edwards MS, Baker CJ, Butler KM, et al. Penetration of cefuroxime into ventricular fluid in cerebrospinal fluid shunt infections. Antimicrob Agents Chemother. 1989;33:1108–1110.
418. Tunkel AR. Bacterial meningitis: non-antibiotic modes of therapy. Curr Opin Infect Dis. 1993;6:638–643.
419. Saez-Llorens X, Ramilo O, Mustafa MM, et al. Molecular pathophysiology of bacterial meningitis: current concepts and therapeutic implications. J Pediatr. 1990;116:671–684.
420. Damas P, Reuter A, Gysen P, et al. Tumor necrosis factor and interleukin-1 serum levels during severe sepsis in humans. Crit Care Med. 1989;17: 975–978.
421. Waage A, Halstensen A, Espevik T. Association between tumour necrosis factor in serum and fatal outcome in patients with meningococcal disease. Lancet. 1987;1:355–357.
422. Ming WJ, Bersani L, Mantovani A. Tumor necrosis factor is chemotactic for monocytes and polymorphonuclear leukocytes. J Immunol. 1987;138:1469–1474.
423. Tracey KJ, Lowry SF, Cerami A. Cachectin: a hormone that triggers acute shock and chronic cachexia. J Infect Dis. 1988;157:413–420.
424. Dinarello CA. An update on human interleukin-1: from molecular biology to clinical relevance. J Clin Immunol. 1985;5:287–297.
425. Beutler B, Milsark IW, Cerami AC. Passive immunization against cachectin/ tumor necrosis factor protects mice from lethal effect of endotoxin. Science. 1985;229:869–871.
426. Gigliotti F, Lee D, Insel RA, et al. IgG penetration into the cerebrospinal fluid in a rabbit model of meningitis. J Infect Dis. 1987;156:394–398.
427. Bernard GR, Vincent JL, Laterre PF, et al. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001; 344:699–709.
428. Drapkin MS, Wisch JS, Gelfand JA, et al. Plasmapheresis for fulminant meningococcemia. Pediatr Infect Dis J. 1989;8:399–400.
429. Girgis NI, Farid Z, Mikhail IA, et al. Dexamethasone treatment for bacterial meningitis in children and adults. Pediatr Infect Dis J. 1989;8: 848–851.
430. Lebel MH, Freij BJ, Syrogiannopoulos GA, et al. Dexamethasone therapy for bacterial meningitis. Results of two double-blind, placebo-controlled trials. N Engl J Med. 1988;319:964–971.
431. Lebel MH, Hoyt MJ, Waagner DC, et al. Magnetic resonance imaging and dexamethasone therapy for bacterial meningitis. Am J Dis Child. 1989;143:301–306.
432. McCracken GH Jr, Lebel MH. Dexamethasone therapy for bacterial meningitis in infants and children. Am J Dis Child. 1989;143:287–289.
433. Kaplan SL. Dexamethasone for children with bacterial meningitis. Should it be routine therapy? Am J Dis Child. 1989;143:290–292.
434. McIntyre PB, Berkey CS, King SM, et al. Dexamethasone as adjunctive therapy in bacterial meningitis. A meta-analysis of randomized clinical trials since 1988. JAMA. 1997;278:925–931.
435. Odio CM, Faingezicht I, Paris M, et al. The beneficial effects of early dexamethasone administration in infants and children with bacterial meningitis. N Engl J Med. 1991;324:1525–1531.
436. Kennedy WA, Hoyt MJ, McCracken GH Jr. The role of corticosteroid therapy in children with pneumococcal meningitis. Am J Dis Child. 1991;145:1374–1378.
437. Schaad UB, Lips U, Gnehm HE, et al. Dexamethasone therapy for bacterial meningitis in children. Swiss Meningitis Study Group. Lancet. 1993;342:457–461.
438. King SM, Law B, Langley JM, et al. Dexamethasone therapy for bacterial meningitis: better never than late? Can J Infect Dis. 1994;5:210–215.
439. Wald ER, Kaplan SL, Mason EO Jr, et al. Dexamethasone therapy for children with bacterial meningitis. Meningitis Study Group. Pediatrics. 1995;95:21–28.
440. Molyneux EM, Walsh AL, Forsyth H, et al. Dexamethasone treatment in childhood bacterial meningitis in Malawi: a randomised controlled trial. Lancet. 2002;360:211–218.
441. Kaplan SL, Fishman MA. Supportive therapy for bacterial meningitis. Pediatr Infect Dis J. 1987;6:670–677.
442. Syrogiannopoulos GA, Lourida AN, Theodoridou MC, et al. Dexamethasone therapy for bacterial meningitis in children: 2- versus 4-day regimen. J Infect Dis. 1994;169:853–858.
443. Arditi M, Ables L, Yogev R. Cerebrospinal fluid endotoxin levels in children with H. influenzae meningitis before and after administration of intravenous ceftriaxone. J Infect Dis. 1989;160:1005–1011.
444. de Gans J, van de Beek D; European Dexamethasone in Adulthood Bacterial Meningitis Study Investigators. Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347:1549–1556.
445. Viladrich PF, Gudiol F, Linares J, et al. Evaluation of vancomycin for therapy of adult pneumococcal meningitis. Antimicrob Agents Chemother. 1991;35:2467–2472.
446. Klugman KP, Friedland IR, Bradley JS. Bactericidal activity against cephalosporin-resistant Streptococcus pneumoniae in cerebrospinal fluid of children with acute bacterial meningitis. Antimicrob Agents Chemother. 1995;39:1988–1992.
447. Waagner DC, Kennedy WA, Hoyt MJ, et al. Lack of adverse effects of dexamethasone therapy in aseptic meningitis. Pediatr Infect Dis J. 1990;9:922–923.
448. Tunkel AR, Scheld WM. Corticosteroids for everyone with meningitis? N Engl J Med. 2002;347:1613–1615.
449. Heckenberg SG, Brouwer MC, van der Ende A, et al. Adjunctive dexamethasone in adults with meningococcal meningitis. Neurology. 2012;79:1563–1569.
450. Weisfelt M, Hoogman M, van de Beek D, et al. Dexamethasone and long-term outcome in adults with bacterial meningitis. Ann Neurol. 2006;60:456–468.
451. Brouwer MC, Heckenberg SG, de Gans J, et al. Nationwide implementation of adjunctive dexamethasone therapy for pneumococcal meningitis. Neurology. 2010;75:1533–1539.
452. Nguyen TH, Tran TH, Thwaites G, et al. Dexamethasone in Vietnamese adolescents and adults with bacterial meningitis. N Engl J Med. 2007;357:2431–2440.
453. Scarborough M, Gordon SB, Whitty CJ, et al. Corticosteroids for bacterial meningitis in adults in sub-Saharan Africa. N Engl J Med. 2007;357:2441–2450.
454. Greenwood BM. Corticosteroids for acute bacterial meningitis. N Engl J Med. 2007;357:2507–2509.
455. Brouwer MC, McIntyre P, Prasad K, et al. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2013;6:CD004405.
456. Gaillard JL, Abadie V, Cheron G, et al. Concentrations of ceftriaxone in cerebrospinal fluid of children with meningitis receiving dexamethasone therapy. Antimicrob Agents Chemother. 1994;38:1209–1210.
457. Buke AC, Cavusoglu C, Karasulu E, et al. Does dexamethasone affect ceftriaxone [corrected] penetration into cerebrospinal fluid in adult bacterial meningitis. Int J Antimicrob Agents. 2003;21:452–456.
458. Cabellos C, Martinez-Lacasa J, Tubau F, et al. Evaluation of combined ceftriaxone and dexamethasone therapy in experimental cephalosporin-resistant pneumococcal meningitis. J Antimicrob Chemother. 2000;45:315–320.
459. Tuomanen E. Partner drugs: a new outlook for bacterial meningitis. Ann Intern Med. 1988;109:690–692.
460. Woehrl B, Brouwer MC, Murr C, et al. Complement component 5 contributes to poor disease outcome in humans and mice with pneumococcal meningitis. J Clin Invest. 2011;121:3943–3953.
461. Peterson PK, Hu S, Sheng WS, et al. Thalidomide inhibits tumor necrosis factor-alpha production by lipopolysaccharide- and lipoarabinomannan-stimulated human microglial cells. J Infect Dis. 1995;172:1137–1140.
462. Lindvall P, Ahlm C, Ericsson M, et al. Reducing intracranial pressure may increase survival among patients with bacterial meningitis. Clin Infect Dis. 2004;38:384–390.
463. Marshall LF, Marshall SB. Medical management of intracranial pressure. In: Cooper PR, ed. Head Injury. 2nd ed. Baltimore: Lippincott Williams & Wilkins; 1987:177–196.
464. Tauber MG, Khayam-Bashi H, Sande MA. Effects of ampicillin and corticosteroids on brain water content, cerebrospinal fluid pressure, and cerebrospinal fluid lactate levels in experimental pneumococcal meningitis. J Infect Dis. 1985;151:528–534.
465. Syrogiannopoulos GA, Olsen KD, Reisch JS, et al. Dexamethasone in the treatment of experimental Haemophilus influenzae type b meningitis. J Infect Dis. 1987;155:213–219.
466. Fishman RA. Brain edema and disorders of intracranial pressure. In: Rowland LP, ed. Merritt’s Textbook of Neurology. 8th ed. Philadelphia: Lea & Febiger; 1989:262–266.
467. Scheld WM, Dacey RG, Winn HR, et al. Cerebrospinal fluid outflow resistance in rabbits with experimental meningitis. Alterations with penicillin and methylprednisolone. J Clin Invest. 1980;66:243–253.
468. Meldrum BS, Vigouroux RA, Brierley JB. Systemic factors and epileptic brain damage. Prolonged seizures in paralyzed, artificially ventilated baboons. Arch Neurol. 1973;29:82–87.
469. Zoons E, Weisfelt M, de Gans J, et al. Seizures in adults with bacterial meningitis. Neurology. 2008;70:2109–2115.
470. Leppik I. Status epilepticus. Paper presented at: the American Academy of Neurology Meeting; April 1983; San Diego, CA.
471. Dreifuss FE. Status epilepticus. In: Dreifuss FE, ed. Pediatric Epileptology: Classification and Management of Seizures in the Child. Boston: John Wright; 1983:221–230.
472. Lowenstein DH, Alldredge BK. Status epilepticus. N Engl J Med. 1998;338:970–976.
473. Treiman DM, Delgado-Escueta AV. Status epilepticus. In: Thompson RA, Green JR, eds. Critical Care of Neurological and Neurosurgical Emergencies. New York: Raven Press; 1980:53–99.
474. Dunn DW. Status epilepticus. In: Dunn DW, Epstein LG, eds. Decision Making in Child Neurology. Toronto: BC Decker; 1987:124.
475. Kaplan SL, Mason EO Jr, Mason SK, et al. Prospective comparative trial of moxalactam versus ampicillin or chloramphenicol for treatment of Haemophilus influenzae type b meningitis in children. J Pediatr. 1984;104:447–453.
476. Jim KK, Brouwer MC, van der Ende A, et al. Subdural empyema in bacterial meningitis. Neurology. 2012;79:2133–2139.
477. Sigurdardottir B, Bjornsson OM, Jonsdottir KE, et al. Acute bacterial meningitis in adults. A 20-year overview. Arch Intern Med. 1997;157:425–430.
478. McMillan DA, Lin CY, Aronin SI, et al. Community-acquired bacterial meningitis in adults: categorization of causes and timing of death. Clin Infect Dis. 2001;33:969–975.
479. Tauber MG, Burroughs M, Niemoller UM, et al. Differences of pathophysiology in experimental meningitis caused by three strains of Streptococcus pneumoniae. J Infect Dis. 1991;163:806–811.