Edited by Mark Little
OUTLINE
29.1 Approach to the poisoned patient 951
29.2 Cardiovascular drugs 958
29.3 Antipsychotic drugs 965
29.4 Antidepressant drugs 969
29.5 Lithium 974
29.6 Antihistamine and anticholinergic poisoning 976
29.7 Paracetamol 979
29.8 Salicylate 984
29.9 Antidiabetic drugs 987
29.10 Colchicine 990
29.11 Theophylline 993
29.12 Iron 995
29.13 Drugs of abuse 998
29.14 Methaemoglobinaemia 1005
29.15 Cyanide 1008
29.16 Corrosive ingestion 1011
29.17 Hydrofluoric acid 1014
29.18 Pesticides 1017
29.19 Ethanol and other ‘toxic’ alcohols 1024
29.20 Carbon monoxide 1031
29.1 Approach to the poisoned patient
Lindsay Murray
Essentials
1 Self-poisoning is a manifestation of an underlying psychiatric, drug and alcohol or social disorder.
2 A wide range of clinical manifestations of toxicity may be observed following drug overdose.
3 An accurate risk assessment predicts the likely clinical course and informs planning for subsequent investigation, management and disposition.
4 The mainstay of management is timely institution of an appropriate level of supportive care.
5 The role of gastrointestinal decontamination is controversial. Except in select cases, it is unlikely that these procedures have a significant impact on clinical outcome when performed more than 1 h following ingestion.
6 Specific antidotes and techniques of enhanced elimination are rarely indicated, but their timely use may be life saving in specific instances.
Introduction
Drug overdose in adults usually occurs in the context of self-poisoning, which may be either recreational or an act of deliberate self-harm.
Deliberate self-poisoning accounts for 1–5% of all public hospital admissions in Australia [1,2]. The bulk of the medical management of cases presenting to hospital is carried out in the emergency department (ED) and the emergency physician is expected to be expert in the field. Although the management must vary considerably according to the nature and severity of the poisoning, some general principles apply.
Above all, it must be remembered that the acute overdose presentation is only a discrete time-limited event in the course of the underlying condition, which is usually psychiatric or social in origin.
Pathophysiology and clinical features
The effects of ingestion of pharmaceuticals or illicit drugs range from the non-toxic to the life threatening and may involve any system. Poisoning is a dynamic presentation and the patient may present at varying points in the time course of the poisoning. Consequently, rapid clinical deterioration or improvement may be observed after the initial presentation and assessment.
Acute morbidity and mortality from poisoning is usually a consequence of the cardiovascular, respiratory or central nervous system (CNS) complications of the poisoning. Less commonly, hepatic, renal or metabolic effects are potentially life threatening.
The most frequent life-threatening respiratory complication of poisoning is ventilatory failure, which is usually a consequence of CNS depression. Less commonly, it is secondary to ventilatory muscle paralysis. The frequency and depth of respirations are reduced. Respiratory failure may also be caused by direct pulmonary toxicity or complications, such as pulmonary aspiration or non-cardiogenic pulmonary oedema (Table 29.1.1).
Table 29.1.1
Toxic causes of respiratory failure

ARDS: acute respiratory distress syndrome.
Cardiovascular manifestations of poisoning include tachycardia, bradycardia, hypertension, hypotension, conduction defects and arrhythmias (Table 29.1.2). Bradycardia is relatively rarely observed and is associated with a number of potentially life-threatening ingestions. Tachycardia is commonly observed and is usually benign. It may be due to intrinsic sympathomimetic or anticholinergic effects of a drug or a reflex response to hypotension or hypoxia. Hypotension is also commonly observed and may be due to a number of different causes (Table 29.1.2). Hypertension is unusual. Severe hypertension is usually associated with illicit drug use and is important because it may produce complications such as intracerebral haemorrhage.
Table 29.1.2
Cardiovascular effects of poisoning

CNS manifestations of poisoning include decreased level of consciousness, agitation or delirium, seizures and disordered temperature regulation. A decreased level of consciousness is a common presentation of poisoning and is associated with many drugs, some of which are listed in Table 29.1.1. Although usually a direct drug effect, CNS depression is occasionally secondary to hypoglycaemia, hypoxia or hypotension. Common causes of agitation or delirium following overdose are listed in Table 29.1.3. Toxic seizures are potentially life threatening and important causes are listed in Table 29.1.4.
Table 29.1.3
Toxic causes of agitation or delirium

Table 29.1.4
Toxic causes of seizures
Amphetamines
Bupropion
Carbamazepine
Chloroquine
Cocaine
Isoniazid
Mefanamic acid
Theophylline
Tramadol
Tricyclic antidepressants
Venlafaxine
Hypothermia is usually a complication of environmental exposure secondary to a decreased level of consciousness or altered behaviour. Hyperthermia is a direct toxic effect and causes are listed in Table 29.1.5. Severe hyperthermia is rapidly lethal if not corrected.
Table 29.1.5
Toxic causes of hyperthermia
Amphetamines
Anticholinergics
Cocaine
MAO inhibitors
Salicylates
Serotonin syndrome
Metabolic and other manifestations of poisoning include hyper- and hypoglycaemia, hyper- and hyponatraemia, acidosis and alkalosis and hepatic failure.
Acute poisoning is distinguished from many other forms of acute illness in that, given appropriate supportive care over a relatively short period, a full recovery can usually be expected. A small number of potentially fatal poisonings may demonstrate progressive toxicity despite full supportive care. These are the so-called cellular toxins and include colchicine, iron, salicylate, cyanide, paracetamol, theophylline and digoxin. In some of these cases, early aggressive gastrointestinal decontamination, timely administration of antidotes or the institution of techniques of enhanced elimination may be life saving.
Mortality or morbidity may also result from specific complications of a poisoning. These include trauma, pulmonary aspiration, adult respiratory distress syndrome, rhabdomyolysis, renal failure and hypoxic encephalopathy. These complications usually occur prior to arrival in the ED.
Pulmonary aspiration frequently complicates a period of decreased level of consciousness or a seizure. It is a leading cause of in-hospital morbidity and mortality following overdose. This complication is characterized by rapid onset of dyspnoea, cough, fever, wheeze and cyanosis.
Rhabdomyolysis occurs as a direct toxic effect (rare) or secondary to excessive muscular hyperactivity, seizures, hyperthermia or prolonged coma with direct muscle compression. The urine is dark and acute renal failure can develop secondary to tubular deposition of myoglobin.
Assessment
Risk assessment
A risk assessment should be made as soon as possible in the management of the poisoned patient. Only resuscitation is a greater priority (Table 29.1.6). Risk assessment is a distinct quantitative cognitive step through which the clinician attempts to predict the likely clinical course and potential complications for the individual patient at that particular presentation [3]. An accurate risk assessment allows informed decision making in regard to all subsequent management steps including duration and intensity of supportive care and monitoring, screening and specialized testing, decontamination, enhanced elimination, antidotes and disposition. Factors that are taken into account when formulating this risk assessment include: the agent(s), the dose, the time since ingestion, the clinical features present and patient factors (Table 29.1.6). Specialized testing may refine risk assessment. Access to specialized poisons information in the form of a poisons information centre or in-house databases is often necessary to formulate an accurate risk assessment.
Table 29.1.6
Risk assessment-based approach to poisoning

Reproduced from Murray L, Daly F, Little M, Cadogan M. Toxicology handbook, 2nd edn. Sydney: Elsevier; 2011.
History
Every effort should be made to obtain information as to the type and dose of drug ingested, the time of ingestion and the progression of symptoms since ingestion. History provided by the patient, if they are awake, is usually reliable and should not be dismissed.
Physical examination
The focused physical examination of the poisoned patient aims to:
![]() identify any immediate threats to life and the need for intervention
identify any immediate threats to life and the need for intervention
![]() establish a baseline clinical status
establish a baseline clinical status
![]() corroborate the history
corroborate the history
![]() identify intoxication syndromes
identify intoxication syndromes
![]() identify possible alternative diagnoses
identify possible alternative diagnoses
![]() identify any complications of the poisoning.
identify any complications of the poisoning.
The initial physical examination of the overdose patient in many ways parallels the primary survey of the trauma patient. The airway, breathing and circulation are assessed and stabilized as necessary. The level of consciousness should be assessed, the presence of seizure activity noted and the blood glucose and temperature measured.
A more complete examination is carried out when the patient is stable. This should include a full neurological examination, including assessment of the level of consciousness and mental status, pupil size, muscle tone and movements and the presence or absence of focal neurological signs. Poisoning normally causes global CNS depression and focal signs suggest an alternative diagnosis or a CNS complication, such as cerebral haemorrhage.
Other features that should be specifically sought are any evidence of associated trauma, the state of hydration, the condition of the skin, in particular the presence of pressure areas, the presence or absence of bowel sounds and the condition of the urine.
Several toxic autonomic syndromes, or ‘toxidromes’, have been described in relation to poisoning. The principal ones are listed in Table 29.1.7. Identification of these syndromes may narrow the differential diagnosis in cases of unknown poisoning.
Table 29.1.7
Toxic autonomic syndromes or ‘toxidromes’

NSSRI: non-selective serotonin re-uptake inhibitor; SSRI: selective serotonin re-uptake inhibitor.
Poisons information
Information on the clinical course and toxic doses of specific pharmaceutical and non-pharmaceutical poisons is available on a 24 h basis throughout Australia by telephoning 131126. The poison information centres are staffed by trained poisons information specialists and are also able to refer cases to clinical toxicologists for consultation.
Treatment
The management of poisoning should be approached in a systematic way. Following initial resuscitation, further treatment is informed by the risk assessment (see Table 29.1.6).
Resuscitation, supportive care and monitoring
Supportive care is the key element in the management of poisoning. The vast majority of poisonings result in temporary dysfunction of one or more of the body systems. If appropriate support of the system in question is instituted in a timely fashion and continued until the toxic substance is metabolized or excreted, a good outcome can be anticipated. In severe poisonings, supportive care may be very aggressive and possible interventions are listed in Table 29.1.8.
Table 29.1.8
Supportive care measures for the poisoned patient

The specific supportive management of a number of manifestations or complications of poisoning warrants further mention insofar as it may differ from the standard management of such conditions with other aetiologies.
Cardiopulmonary arrest from poisoning should be aggressively resuscitated. Direct current cardioversion is rarely successful in terminating toxic arrhythmias and should not take precedence over establishing adequate ventilation and oxygenation, cardiac compressions, correction of acidosis or hypovolaemia and the administration of specific antidotes. Resuscitative efforts should be continued beyond the usual time frame. In cardiac arrest due to drugs with direct cardiac toxicity, the use of cardiopulmonary bypass or extracorporeal membrane oxygenation (ECMO) until the drug is metabolized may be life saving.
In general, intravenous benzodiazepines are the drugs of choice for control of toxic seizures. Large doses may be required. Hypoxia and hypoglycaemia must be corrected if they are contributory factors. Patients with toxic seizures do not generally need long-term anticonvulsant therapy. Isoniazid-induced seizures are difficult to control without administration of an adequate dose of the specific antidote, pyridoxine.
The management of pulmonary aspiration is essentially supportive, with supplemental oxygenation and intubation and mechanical ventilation if necessary. Neither prophylactic antibiotics nor corticosteroids have been shown to be helpful in the management of this condition, which is essentially a chemical pneumonitis.
Toxic hypertension rarely requires specific therapy. Most cases are mild and simple observation is sufficient. Agitation or delirium is a feature of many intoxications associated with hypertension and adequate sedation with benzodiazepines usually lowers the blood pressure. Severe toxic hypertension is most likely in toxicity from cocaine or amphetamine-type drugs and treatment may be indicated to avoid complications, such as cardiac failure or intracerebral haemorrhage. The drug of choice in this situation is sodium nitroprusside by intravenous infusion. The extremely short duration of action of this vasodilator allows accurate control of hypertension during the toxic phase and avoids the development of hypotension once toxicity begins to wear off.
Management of rhabdomyolysis consists of treatment of the causative factors, fluid resuscitation and careful monitoring of fluids and electrolytes. The role of mannitol and urinary alkalinization in reducing the risk of renal failure is not clear. Established acute renal failure requires haemodialysis, often for up to 6 weeks.
Decontamination
The aim of decontamination of the gastrointestinal tract is to bind or remove ingested material before it is absorbed into the circulation and able to exert its toxic effects. This is a very attractive concept and has long been considered one of the fundamental interventions in management of the overdose patient.
However, gastrointestinal decontamination should not be regarded as a routine procedure in the management of the patient presenting to the ED following an overdose. The decision to perform gastrointestinal decontamination and the choice of method should be based on an assessment of the likely benefit, the likely risk and the resources required. Gastrointestinal decontamination should only be considered where there is likely to be a significant amount of a significantly toxic material remaining in the gut. It is never indicated when the risk assessment predicts a benign course. Efforts at decontamination technique should never take precedence over the institution of appropriate supportive care.
Three basic approaches to gastrointestinal decontamination are available: gastric emptying, administration of an adsorbent and catharsis.
Gastric emptying can be attempted by the administration of an emetic, most commonly syrup of ipecac, or by gastric lavage. In volunteer studies, both of these techniques removed highly variable amounts of marker substances from the stomach even if performed immediately after ingestion and the effect diminished rapidly with time to the point of being negligible after 1 hour [4,5]. Clinical outcome trials have failed to demonstrate improved outcome as a result of routine gastric emptying in addition to administration of activated charcoal, except, perhaps, in patients presenting unconscious within 1 h of ingestion [6–8].
The principal adsorbent available to clinicians is activated charcoal (AC), which effectively binds most pharmaceuticals and chemicals, and is currently the decontamination method of choice for most poisonings. Materials that do not bind well to charcoal are listed in Table 29.1.9.
Table 29.1.9
Materials that do not bind well to activated charcoal

Charcoal is ‘activated’ by treatment in acid and steam at high temperature. This process removes impurities and greatly increases the surface area available for binding. Activated charcoal (AC) is packaged as a 50 g dose premixed with water or sorbitol, which is likely to be sufficient for the majority of ingestions. Adult patients are usually able to drink AC slurry from a cup. If the level of consciousness is too impaired to allow this, they should be intubated first. Administration of AC is absolutely contraindicated unless the patient has an intact or protected airway.
Volunteer studies demonstrate that the effect of AC diminishes rapidly with time and that the greatest benefit occurs if it is administered within 1 h. There is as yet no evidence that AC improves clinical outcome [9].
There is no evidence to suggest that the addition of a cathartic, such as sorbitol, to AC improves clinical outcome [10].
Apart from rarely employed endoscopic and surgical techniques, whole-bowel irrigation (WBI) is the most aggressive form of gastrointestinal decontamination. Polyethylene glycol solution (Golytely is administered via a nasogastric tube at a rate of 2 L/h until a clear rectal effluent is produced. This usually takes about 6 h and requires one-to-one nursing. In volunteer studies, this technique reduced the absorption of slow-release pharmaceuticals and so may be of benefit in life-threatening overdoses of these agents. Again, clinical benefit has not yet been conclusively demonstrated [11]. The use of WBI has also been reported in the management of potentially toxic ingestions of iron, lead and packets of illicit drugs. Whole-bowel irrigation is contraindicated if there is evidence of ileus or bowel obstruction and in patients who have an unprotected airway or haemodynamic compromise.
Enhanced elimination
A number of techniques are available to enhance the elimination of toxins from the body. Their use is rarely indicated, as only a very few drugs capable of causing severe poisoning have pharmacokinetic parameters that render them amenable to these techniques (Table 29.1.10).
Table 29.1.10
Techniques of enhanced elimination

Multiple-dose AC (25–50 g every 3–4 h) may enhance drug elimination by interrupting the enterohepatic circulation or by ‘gastrointestinal dialysis’. Gastrointestinal dialysis is the movement of a toxin across the gastrointestinal wall from the circulation into the gut down a concentration gradient that is maintained by charcoal binding. For this technique to be effective, a drug must undergo considerable enterohepatic circulation or, in the case of ‘gastrointestinal dialysis’, have a small volume of distribution, small molecular weight, low protein binding, slow endogenous elimination and bind to charcoal [12]. The advantages of this technique are that it is non-invasive and simple to carry out.
Alkalinization of the urine enhances urinary excretion of drugs that are filtered at the glomerulus and are unable to be reabsorbed across the tubular epithelium when in an ionized form at alkaline pH. For elimination to be effectively enhanced by this method, the drug must be predominantly eliminated by the kidneys in the unchanged form, have a low pKa, be distributed mainly to the extracellular fluid compartment and be minimally protein bound.
Haemodialysis (HD) and haemoperfusion (HP) are both very invasive techniques and for that reason are reserved for potentially life-threatening intoxications. Only a small number of drugs that have small volumes of distribution, slow endogenous clearance rates, small molecular weights (HD) and bind to charcoal (HP) will have their rates of elimination significantly enhanced by these procedures.
Antidotes
Very few drugs have effective antidotes. Occasionally, however, timely use of an antidote may be life saving or substantially reduce morbidity, time in hospital or resource requirements. Antidotes that may be indicated in the ED setting are listed in Table 29.1.11. However, it must be remembered that antidotes are also drugs and are frequently associated with adverse effects of their own. An antidote should only be used where a specific indication exists and then only at the correct dose, by the correct route and with appropriate monitoring. Because many antidotes are so infrequently used, obtaining sufficient supplies when the need arises can be difficult. Every ED must review its stocking of antidotes and have a plan for obtaining further supplies should the need arise.
Table 29.1.11
Useful emergency antidotes
|
Poisoning |
Antidote |
|
Atropine |
Physostigmine |
|
Benzodiazepines |
Flumazenil |
|
Cyanide |
Dicobalt edetate, hydroxocobalamin |
|
Digoxin |
Digoxin-specific |
|
Insulin |
Dextrose |
|
Iron |
Desferoxamine |
|
Isoniazid |
Pyridoxine |
|
Methaemoglobinaemia |
Methylene blue |
|
Methanol and ethylene glycol |
Ethanol, fomepizole |
|
Organophosphates and carbamates |
Atropine, oximes |
|
Opioids |
Naloxone |
|
Paracetamol |
N-acetyl cysteine |
|
Sulphonylureas |
Dextrose, octreotide |
|
Tricyclic antidepressants |
Sodium bicarbonate |
|
Warfarin, brodifacoum |
Vitamin K |
Differential diagnosis
It is essential to exclude important non-toxic diagnoses in the patient presenting with coma or altered mental status presumed to be due to drug overdose. These diagnoses include head injury, intracerebral haemorrhage or infarction, CNS infection, hyponatraemia, hypoglycaemia, hypo- or hyperthermia, post-ictal states and psychiatric disorders.
Clinical investigations
Investigations should only be performed if they are likely to affect the management of the patient. They are employed as either screening tests or for specific purposes.
In poisoning, screening tests aim to identify occult toxic ingestions for which early specific treatment might improve outcome. The recommended screening tests for acute poisoning are the 12-lead ECG and the serum paracetamol level. The ECG is used to exclude conduction defects which may predict potentially life-threatening cardiotoxicity. The serum paracetamol is useful to ensure that paracetamol poisoning is diagnosed within the time available for effective antidotal treatment.
Other specific investigations may be indicated to exclude important differential diagnoses, confirm a specific poisoning for which significant complications might be anticipated, assess the severity of intoxication, assess response to treatment or assess the need for a specific antidote or enhanced elimination technique.
The patient with only minor manifestations of poisoning may require no other blood tests apart from a screening paracetamol level. Pregnancy should be excluded in women of childbearing age by serum or urine β-HCG if necessary. More seriously ill patients may require electrolyte, renal and liver function tests and a full blood count, creatine kinase and arterial blood gases. Urinalysis reveals myoglobinuria in significant rhabdomyolysis.
Routine qualitative drug screening of urine or blood in the overdose patient is rarely useful in planning management.
Measurement of serum drug concentrations is only useful if this provides important diagnostic or prognostic information or assists in planning management. Some drug levels that may be useful are listed in Table 29.1.12. For most cases, drug overdose management is guided by clinical findings and not by drug levels. Some drugs commonly taken in overdose for which serum concentrations are of no value in planning management are listed in Table 29.1.13.
Table 29.1.12
Drug levels that may be helpful in the management of selected cases of overdose
Carbamazepine
Digoxin
Dilantin
Lithium
Iron
Paracetamol
Phenobarbitone
Salicylate
Theophylline
Valproate
Table 29.1.13
Drug levels that are not helpful in the management of overdose
|
CNS drugs |
Cardiovascular drugs |
|
Antidepressants |
ACE inhibitors |
|
Benzodiazepines |
β-Blockers |
|
Benztropine |
Calcium channel blockers |
|
Cocaine |
Clonidine |
|
Newer antipsychotics |
|
|
Opiates |
|
|
Phenothiazines |
Radiology has a limited role in the management of overdose. A chest X-ray is indicated in any patient with a significantly decreased level of consciousness, seizures or hypoxia. It may show evidence of pulmonary aspiration. A computed tomography scan of the head may be indicated to exclude other intracranial pathology in the patient with an altered mental status. The abdominal X-ray is useful in evaluating overdose of radiopaque metals including iron, lithium, potassium, lead and arsenic.
Disposition
Both the medical and the psychiatric disposition of the overdose patient must be considered. A good risk assessment is essential to determining timely and safe disposition.
The majority of overdose patients who remain stable at 4–6 h after the ingestion do not need further close monitoring and may be admitted to a non-monitored bed until manifestations of toxicity completely resolve. An emergency observation ward is ideal for this purpose.
Any patient who develops clinical manifestations of intoxication severe enough to require the institution of specific supportive care measures requires admission to an intensive care environment. A few patients will require admission for prolonged monitoring based on the history of the ingestion. For example, anyone with a history of ingestion of colchicine, organophosphates, slow-release theophylline or slow-release calcium channel blockers requires admission because of the possibility of delayed onset of severe toxicity.
Psychiatric evaluation of deliberate self- poisoning cases is indicated as soon as the patient’s medical condition permits. All such patients must be continuously supervised until the psychiatric evaluation has taken place.
Controversies
![]() The role of, choice of method and indications for gastric decontamination remain controversial. These procedures are no longer regarded as routine, but there are likely to be subgroups of overdose patients who may derive clinical benefit from gastrointestinal decontamination. These groups have not yet been precisely identified.
The role of, choice of method and indications for gastric decontamination remain controversial. These procedures are no longer regarded as routine, but there are likely to be subgroups of overdose patients who may derive clinical benefit from gastrointestinal decontamination. These groups have not yet been precisely identified.
![]() The clinical and economic utility of establishing specialized toxicology treatment centres.
The clinical and economic utility of establishing specialized toxicology treatment centres.
References
1. McGrath J. A survey of deliberate self-poisoning. Med J Aust. 1989;150:317–322.
2. Pond SM. Prescription for poisoning. Med J Aust. 1995;162:174–175.
3. Murray L, Daly F, Little M, Cadogan M, eds. Toxicology handbook. 2nd ed. Sydney: Elsevier Australia; 2011.
4. American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position paper: ipecac syrup. J Toxicol Clin Toxicol. 2004;42:133–143.
5. American Academy of Clinical Toxicology, European Assoication of Poison Centres and Clinical Toxicologists. Position paper: gastric lavage. J Toxicol Clin Toxicol. 2004;42:933–943.
6. Kulig K, Bar-Or D, Kantrill SV, et al. Management of acutely poisoned patients without gastric emptying. Ann Emerg Med. 1990;14:562–567.
7. Merigian KS, Woodard M, Hedges JR, et al. Prospective evaluation of gastric emptying in the self-poisoned patient. Am J Emerg Med. 1990;8:479–483.
8. Pond SM, Lewis-Driver DJ, Williams G, et al. Gastric emptying in acute overdose: a prospective randomised controlled trial. Med J Aust. 1995;163:345–349.
9. American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position paper: single-dose activated charcoal. J Toxicol Clin Toxicol. 2005;43:61–87.
10. American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position paper: cathartics. J Toxicol Clinl Toxicol. 2004;42:243–253.
11. American Academy of Clinical Toxicology, European Association of Poisons Centres and Clinical Toxicologists. Position paper: whole bowel irrigation. J Toxicol Clin Toxicol. 2004;24:843–854.
12. Chyka PA. Multiple-dose activated charcoal and enhancement of systemic drug clearance: summary of studies in animals and human volunteers. Clin Toxicol. 1995;33:399–405.
29.2 Cardiovascular drugs
Betty Shuk Han Chan and Angela Chiew
Essentials
1 Calcium channel blockers, β-blockers, digoxin and sodium channel blocker poisonings are associated with potentially life-threatening toxicity.
2 The key to the management of calcium channel blocker and β-blocker toxicity rests with aggressive supportive care of the circulation including early use of hyperinsulinaemia euglycaemic therapy.
3 The onset of toxicity following overdose with slow-release formulations of calcium channel blockers may be delayed.
4 Early aggressive decontamination with whole-bowel irrigation is important in the management of slow-release calcium channel blocker overdose.
5 The key to management of sodium channel blockers is sodium bicarbonate therapy and hyperventilation.
6 Early identification of patients presenting with potentially severe digoxin toxicity and appropriate use of the specific Fab fragment antibody is life saving.
7 The management of clonidine poisoning is largely supportive.
8 Intravenous lipid emulsion therapy should be reserved for the treatment of severe local anaesthetic toxicity. It is not standard treatment in other overdoses.
Calcium channel blockers and β-blockers
Introduction
The calcium channel blockers (CCBs) and β-blockers are widely prescribed in the community. In overdose, they present with similar clinical pictures of potentially life-threatening impairment of cardiac function. The management of both types of overdose is similar and they are discussed together.
Pharmacokinetics
Standard CCB preparations are rapidly absorbed from the gastrointestinal tract, with onset of action occurring within 30 min. Pharmacokinetic parameters are shown in Table 29.2.1. Verapamil and diltiazem undergo significant first-pass hepatic clearance. Verapamil is metabolized to norverapamil, which possesses 15–20% of verapamil’s pharmacological activity and is renally excreted. Diltiazem is metabolized to deacetyldiltiazem, which has half the potency of the parent compound and undergoes biliary excretion. The elimination half-lives of all CCBs may be prolonged following massive overdose. Amlodipine has a longer plasma half-life (30–50 h) than other CCBs.
Table 29.2.1
Pharmacological profiles of the calcium channel blockers

NR: normal release; SR: slow release.
Adapted from Kerns W II, Kline J, Ford MD. β-Blocker and calcium channel blocker toxicity. Emerg Med Clin N Am 1994;12:365–89 with permission.
Importantly, slow-release preparations of both verapamil and diltiazem are widely prescribed and are associated with much longer times to peak plasma concentration and clinical effect.
Absorption of β-blockers is rapid, with peak clinical effects occurring within 1–4 h. Pharmacokinetic parameters of the principal β-blockers are detailed in Table 29.2.2. Agents with high lipid solubility, such as propranolol, penetrate the blood–brain barrier better than the water-soluble agents and hence cause greater central nervous system (CNS) toxicity.
Table 29.2.2
Pharmacological profiles of the β-blockers

Adapted from Kerns W II, Kline J, Ford MD. β-Blocker and calcium channel blocker toxicity. Emerg Med Clin N Am 1994;12:365–89 with permission.
Pathophysiology
CCBs antagonize the entry of extracellular calcium into cardiac and smooth muscle, but not skeletal muscle. Upon entry into cells, calcium participates in mechanical, electrical and biochemical reactions. It is involved in excitation–contraction of cardiac and smooth muscles, as well as phase O depolarization in the sinus and atrioventricular (AV) nodes by calcium influx through channels. CCBs affect myocardial contractility and slow conduction through the sinus and AV nodes. Contraction of smooth muscle is mediated by calcium influx, which is inhibited by CCBs. This results in vasodilatation and secondary reflex tachycardia from an increase in sympathetic activity.
The different classes of CCB have somewhat different pharmacological and toxic effects, as a consequence of their different binding characteristics to the dihydropyridine (DHP) receptors. Verapamil, a phenylalkylamine, produces more profound cardiac conduction defects and equal reductions in systemic vascular resistance when compared with other CCBs on a mg/kg basis. Verapamil is more likely to produce symptomatic decreases in blood pressure, heart rate and cardiac output than diltiazem, a benzothiazepine. The DHPs, which include amlodipine, felodipine, lercanidipine, nifedipine and nimodipine, preferentially bind to vascular smooth muscle and predominantly decrease systemic and coronary vascular resistance. With the exception of felodipine, they also produce a reflex tachycardia by the unloading of baroreceptors.
β-Blockers prevent the binding of catecholamines to β-receptors (β1, β2). β1-Receptors are located in the myocardium, kidney and eye and β2-receptors in adipose tissue, pancreas, liver and both smooth and skeletal muscle. β1-Stimulation produces increased chronotropy and inotropy in the heart, increased renin secretion in the kidney and increased aqueous humor production. β2-Stimulation relaxes smooth muscle in the blood vessels, bronchial tree, intestinal tract and uterus.
Blockade of β-receptors results in blunting of the metabolic, chronotropic and inotropic effects of catecholamines. Some β-blockers, especially propranolol, may also impede sodium entry via myocardial fast inward sodium channels, thus slowing phase 0 of the action potential. This results in a prolonged QRS duration on the electrocardiogram and produces cardiotoxicity in overdose similar to that of the tricyclic antidepressants.
The different β-blockers have slightly differing pharmacological properties, including selectivity for β-adrenoreceptors, intrinsic sympathomimetic activity and membrane-stabilizing activity. The relative affinity for β-adrenoreceptors may influence expression of toxicity. Atenolol, esmolol and metoprolol are β1-selective agents and therapeutic use of these drugs is less likely to produce the peripheral vasoconstriction, bronchospasm and disturbances in glucose homoeostasis that result from β2 inhibition. However, pharmacological specificity decreases with increasing dose. Several β-blockers have partial agonist activity such that, although they block the β-receptor to catecholamines, they also weakly stimulate the receptor. This partial agonist activity may have a protective effect in overdose.
Clinical features
Calcium channel blockers
The severity of toxicity is determined by a number of factors, including the amount and characteristics of the drug ingested, the underlying health of the patient, co-ingestants and delay until treatment. The majority of serious cases and deaths result from the ingestion of verapamil or diltiazem, the most toxic of the CCBs. Ingestion of as few as 10 tablets of the higher dose formulation of verapamil or diltiazem can cause severe toxicity. Elderly patients and those with congestive cardiac failure may develop toxicity with ingestions of two to three times their normal daily dose. The principal clinical features are shown in Table 29.2.3. Ingestion of toxic amounts of standard preparations typically produces symptoms within 2 h, although maximal toxicity may not occur for up to 6–8 h. The slow-release preparations can produce significant toxicity with onset of symptoms more than 6 h post-ingestion. The major threats to life are myocardial depression and hypotension. Overdose of DHPs often produces tachycardia with normal blood pressure during the first 30 min, followed later by hypotension and bradycardia in large ingestions (>10 mg/kg). Even though amlodipine has been reported to be less toxic than verapamil and diltiazem, it can cause severe shock in large overdoses. With verapamil and diltiazem poisoning, nausea, vomiting, hyperglycaemia and metabolic acidosis can develop. All CCBs can cause symptoms of cerebral hypoperfusion, such as syncope, lethargy, lightheadedness, dizziness, altered mental status, seizures and coma.
Table 29.2.3
Clinical features of CCB overdose

β-Blockers
In one large series of patients with β-blocker overdose, 30–40% of patients remained asymptomatic and only 20% developed severe toxicity. Most of the life-threatening presentations or deaths that have been reported in the literature are due to overdosage of propranolol or sototol. Significant toxicity is more likely to develop in patient ingestions with these β-blockers, in patients with pre-existing cardiac disease or where there is co-ingestion of other drugs with effects on the cardiovascular system, especially CCBs and cyclic antidepressants. Ingestion of more than 1.5 g propranolol is associated with severe toxicity. If β-blocker toxicity is to develop, it is usually observed within 6 h of ingestion.
Sinus node suppression, conduction abnormalities and decreased contractility are typical. First-degree AV block, AV dissociation, right bundle branch block and intraventricular conduction delay have been reported.
Propranolol in overdose, has sodium channel blocking effect, that is characterized by cardiotoxicity including prolongation of the QRS interval and ventricular arrhythmias that more closely resemble tricyclic antidepressant overdose. Sotalol has both β-blocker activity and class 3 antiarrhythmic properties. Class 3 drugs lengthen the duration of the QT interval owing to prolongation of the action potential in His–Purkinje tissue. Therefore, ventricular arrhythmias, such as torsades de pointes, are more common with sotalol.
Hypotension occurs as a result of negative inotropic effect. In addition, CNS effects, such as depressed conscious level and seizures, can occur, especially with the more lipid-soluble and membrane-depressant agents, such as propranolol. Hypoglycaemia is reported following atenolol overdose.
Clinical investigation
The ECG is essential in evaluating and monitoring toxic conduction defects. Serum drug levels are unhelpful in management. Patients with severe toxicity require monitoring of serum electrolytes and glucose. Serum calcium must be closely monitored if calcium salts are administered therapeutically.
Treatment
The primary aim in both β-blocker and CCB toxicity is to restore perfusion to vital organs by increasing cardiac output and the methods used are similar.
Supportive management may include airway and ventilatory support, intravenous fluid administration, early implemenation of hyperinsulinaemia euglycaemic therapy and administration of inotropes. Transcutaneous or transvenous pacing may be tried in cases with profound bradycardia, but often is of limited benefit. Severe cases may require studies on cardiac output and peripheral vascular resistance using either Swan–Ganz catheter or pulse contour cardiac output monitoring (PiCCO) and invasive blood pressure monitoring.
If safe to do so, oral-activated charcoal should be administered as soon as practicable to all those presenting after ingestion of slow-release preparations and may be considered for other β-blocker ingestions. More aggressive decontamination, with whole-bowel irrigation, is indicated following overdose with slow-release CCBs.
A number of drugs play a role in the management of significant CCB or β-blocker poisoning, although none is a completely effective antidote. Suggested doses are shown in Table 29.2.4.
Table 29.2.4
Useful drugs in the management of CCB and β-blocker toxicity
|
CCBs |
β-Blockers |
|
|
Calcium |
Calcium gluconate 10% 30 mL (child 0.6 mL/kg) IV over 10 min OR calcium chloride 10% 10 mL (child 0.2 mL/kg) IV over 10 min. Repeat every 5 min as required. Further administration guided by serum calcium concentrations |
|
|
Catecholamines |
Adrenaline (epinephrine) infusion started at 1 μg/kg/min and titrate to maintain organ perfusion |
Isoprenaline or adrenaline (epinephrine) infusion titrated to maintain organ perfusion |
|
Sodium bicarbonate |
A bolus dose of sodium bicarbonate 8.4% 1–2 mmol/kg, every 3–5 min, to correct severe metabolic acidosis to a pH greater than 7.3 |
A bolus dose of sodium bicarbonate 8.4% 1–2 mmol/kg, every 3–5 min, titrated to a narrowing of the QRS complex, resolution of arrhythmias |
|
Hyperinsulinaemia euglycaemia |
Actrapid 1 U/kg IV bolus followed by an infusion starting at 1 U/kg/h. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia |
Actrapid 1 U/kg IV bolus followed by an infusion commencing at 1 U/kg/h. Give with 50% dextrose 50 mL followed by infusion to maintain euglycaemia |
Calcium, an inotropic agent, is the initial drug of choice for CCB toxicity and has also been used successfully for β-blocker poisoning. Administration must be closely monitored, with ionized calcium measured 30 min after commencing the infusion and then second-hourly. Catecholamines are useful in attempting to restore adequate tissue perfusion.
Hyperinsulinaemic euglycaemia therapy (HIET) is increasingly advocated as therapy for hypotension unresponsive to fluids and calcium salts, with many toxicologists using HIET early in the management of these poisonings if inotropes are being considered. This therapy is supported by animal work and multiple human case reports, but a randomized controlled trial is lacking. Insulin administration switches cardiac cell metabolism from fatty acids to carbohydrates. It restores calcium fluxes and improves myocardial contractility. The recommended initial dose of actrapid is 1 U/kg IV followed by an infusion commencing at 1 U/kg/h. Although the optimal dose is still to be determined, there are some human case reports suggesting increasing doses up to 10 U/kg/h. This should be accompanied by an initial bolus dose of 50 mL 50% dextrose followed by an infusion of dextrose to maintain euglycaemia. Case reports often describe patients needing no more than 25 g/h of dextrose while poisoned (i.e. 50 mL/h of 50% dextrose).
The use of glucagon is supported only by case reports and some animal studies. There are no clinical trials supporting its efficacy in either calcium channel or β-blocker poisoning. Due to the significant doses often required, it is frequently difficult to source adequate stocks of glucagon for use as an inotropic agent. As such, its use in the treatment of calcium channel or β-blocker poisoning is not routinely recommended.
Severe propranolol toxicity is usually due to sodium channel blockade and treatment is similar to tricyclic antidepressant poisoning, including intubation, ventilation and sodium bicarbonate.
There are no clinically effective methods of enhancing the elimination of CCBs or β-blockers. When all else fails, extra-corporeal life support has been shown to allow organ perfusion until reversal of cardiac dysfunction and elimination of the drugs.
Disposition
Following overdose of β-blockers or standard CCBs, patients should be observed in a monitored environment for at least 6 h. Overdoses of slow-release CCBs require monitoring for at least 16 h from the time of ingestion. All symptomatic patients should be admitted to a monitored environment until toxicity resolves.
Digoxin
Introduction
Both acute and chronic digoxin toxicity are potentially life-threatening presentations to the emergency department (ED). Early recognition and administration of the specific Fab fragment antidote, if indicated, usually results in a good outcome.
Pharmacokinetics
Digoxin is moderately well absorbed following oral administration, with a bioavailability in the range of 50–80%. The initial volume of distribution is relatively small, but it is then slowly redistributed, predominantly to skeletal muscle, to give a relatively large volume of distribution of approximately 7 L/kg. Digoxin is excreted predominantly unchanged by the kidney, with an elimination half-life of about 36 h.
Pathophysiology
At a subcellular level, digoxin inhibits the function of Na–K ATPase, which leads to intracellular depletion of potassium and accumulation of sodium and calcium ions. Alteration of ionic fluxes affects cell membrane conduction. At toxic concentrations of digoxin, the effects on the cardiac conducting system produce decreased conduction velocity throughout the system, increased refractoriness at the AV node and enhanced automaticity of the Purkinje fibres. Vagal tone is also enhanced. In acute digoxin poisoning, the sudden loss of Na–K ATPase function produces hyperkalaemia.
Clinical features
Two distinct clinical presentations of digoxin toxicity are observed: acute and chronic. Both are characterized by cardiac arrhythmias and virtually all types of arrhythmia have been reported in the context of digoxin toxicity.
Acute digoxin overdose in adults is usually intentional. The therapeutic margin for digoxin is relatively narrow and any ingestion with suicidal intent is regarded as potentially life threatening.
The non-cardiac manifestations of toxicity are nausea and vomiting and hyperkalaemia. Nausea and vomiting occur early and may be the presenting complaint. The most common cardiac manifestations are sinus bradycardia, increased ventricular ectopy, sinoatrial node arrest and first-, second- or third-degree heart block. Ventricular tachycardia and fibrillation may occur. In significant acute overdose, progressive worsening of the conduction disturbance over a period of hours is usually observed.
Chronic digoxin toxicity may be precipitated by therapeutic errors, intercurrent illnesses that decrease renal elimination of digoxin or by drug interactions. Common drug interactions include those with quinidine, CCBs, amiodarone and indomethacin. The patient is commonly elderly. Reduced muscle mass and reduced renal function in the elderly mean that both the volume of distribution and rate of elimination of digoxin may be substantially reduced.
Nausea and vomiting are also common manifestations of chronic digoxin toxicity and are frequent presenting symptoms. Neurological manifestations are characteristic of chronic toxicity and include visual disturbances, weakness and fatigue. The most common cardiovascular manifestations of chronic digoxin toxicity are arrhythmias and these may be sinus bradycardia, atrial fibrillation with slowed ventricular response or a junctional escape rhythm, atrial tachycardia with block and ventricular tachycardia and fibrillation.
Death from digoxin toxicity results from pump failure, severe cardiac conduction impairment or ventricular arrhythmia.
Clinical investigations
The most important investigations are the ECG, serum electrolytes and creatinine and serum digoxin concentration.
The ECG is invaluable in documenting the type and severity of any cardiac conduction defect. Serial ECGs may demonstrate worsening of the cardiac conduction defects as toxicity progresses.
In acute poisoning, the serum potassium rises as Na–K ATPase function is progressively impaired. Hyperkalaemia denotes significant acute digoxin toxicity. Prior to the availability of a specific antidote for digoxin poisoning, a serum potassium concentration>5.5 mEq/L was associated with a high probability of lethal outcome. Hyperkalaemia seldom occurs in chronic digoxin poisoning, unless patient has acute renal failure. In fact, these patients are frequently hypokalaemic and hypomagnesaemic secondary to chronic diuretic use. Both these electrolyte disorders are important as they exacerbate digoxin toxicity.
Serum digoxin levels taken at 6 hours post ingestion are useful in assessing and confirming toxicity, but must be carefully interpreted in the context of the clinical presentation. They do not accurately correlate with clinical toxicity. Therapeutic concentrations are usually quoted as 0.6–1.0 nmol/L (0.5–0.8 mcg/L). Significant chronic toxicity may be associated with 0.6–1.0 nmol/L (0.5–0.8 mcg/L) elevations of the serum digoxin concentration. This is particularly the case in the presence of pre-existing cardiac disease, hypokalaemia or hypomagnesaemia. Following acute overdose, the serum digoxin concentration is relatively high compared to tissue concentrations, until distribution is completed by 6–12 h post-ingestion. However, early concentrations greater than 15 nmol/L indicate serious poisoning.
Treatment
The best outcome is associated with early recognition of digoxin toxicity.
For chronic toxicity with minimal symptoms, management may involve no more than observation, cessation of digoxin administration, correction of hypokalaemia and hypomagnesaemia and appropriate management of any factors that contributed to the development of toxicity. However, the presence of any cardiovascular system effects, particularly in elderly patients, is an indication for the administration of Fab fragments of digoxin-specific antibodies. Apart from brady-tachy arrhythmias that are associated with haemodynamic instability, patients who have increased automaticies with cardiac or gastrointestinal symptoms may be considered for digoxin specific antibody. This is especially for patients who have renal failure with a Creatinine clearance <30 ml/min. From an economic viewpoint, the potential reduction in length of stay of 1-2 days as a result of treatment with digoxin specific antibody needs to take into consieration of the increase in the cost of 1 vial of digoxin Fab to A$850 per 40 mg vial.
Following acute overdose, the patient should be initially managed in a monitored area with full resuscitative equipment available. Immediate attention to the airway, breathing and circulation may be required. Intravenous access should be established and blood sent for urgent electrolytes and serum digoxin concentration. Although digoxin is well bound by charcoal, administration is usually difficult because of repetitive vomiting and attempts should not detract from other interventions.
The specific antidote to digoxin poisoning is Fab fragments of digoxin-specific antibodies, which should be administered as soon as possible in any potentially life-threatening digoxin intoxication. Commonly accepted indications for the administration of Fab fragments are listed in Table 29.2.5.
Table 29.2.5
Indications for administration of fab fragments of digoxin-specific antibodies following acute overdose
Hyperkalaemia (K>5.5 mmol/L) associated with digoxin toxicity
History of ingestion of more than 10 mg of digoxin
Haemodynamically unstable cardiac arrhythmia
Cardiac arrest from digoxin toxicity
Serum digoxin concentration greater than 15 nmol/L
Fab fragments of digoxin-specific antibodies
These are derived from IgG antidigoxin antibodies produced in sheep. Removal of the Fc fragments of the antibodies greatly reduces the potential for hypersensitivity reactions and contributes to the remarkable safety profile of the product. Intravenously administered Fab fragments bind digoxin in the intravascular space on a mole-for-mole basis. As binding continues, digoxin moves down a concentration gradient from the tissue compartments to the intravascular compartment. Bound digoxin is inactive. A clinical response is usually observed within 20–30 min of administration. The Fab–digoxin complexes are excreted in the urine.
The extraordinary clinical efficacy of digoxin-specific fragments has been well documented in a few multicentre studies. These studies demonstrated the safety of the product, with the adverse reactions reported being hypokalaemia (4%), rapid atrial fibrillation or worsening of congestive cardiac failure (3%) and allergic reaction (0.8%).
The correct dose of Fab fragments may be calculated on the basis that 40 mg (one vial) will bind 0.5 mg of digoxin. If the dose ingested is unknown and/or a steady-state serum digoxin concentration is not available, dosing of Fab fragments must be empiric. A reasonable empiric dosing is to give 2 vials initially and then check for a clinical response. Further digoxin specific antibody may be given to neutralise half of the body burden once digoxin concentration is available. If the digoxin ingested dose or serum concentration is known, give half of the equimolar dose is adequate to stablise patient with severe digoxin toxiicty. In cardiac arrest, give a bolus dose (5 vials), and this dose can be repeated after 30 to 60 minutes if there has been no clinical response. Smaller doses (two vials) are usually sufficient to reverse the effects of chronic toxicity.
It is important that ED staff are aware of the amount and location of supplies of Fab fragments within their own institution and know the most rapid way to acquire further stocks should the need arise.
Serum digoxin concentrations will be extremely high following the administration of Fab fragments because most assays measure both bound and unbound digoxin.
Disposition
Patients with mild, chronic digoxin toxicity (gastrointestinal symptoms only) may be discharged after cessation of digoxin therapy provided there are no significant electrolyte disturbances, renal failure or other precipitating medical conditions. Following administration of Fab fragments, cases of chronic toxicity with conduction defects usually require medical admission for observation and treatment of intercurrent illness.
Acute overdoses require close observation for at least 12 h. Those that develop toxicity require admission and an appropriate level of monitoring. Following successful administration of digoxin-specific Fab fragments, patients must be carefully monitored for hypokalaemia and worsening of any underlying medical conditions for which digoxin may have been prescribed therapeutically. All intentional ingestions require psychiatric evaluation prior to medical discharge.
Clonidine
Introduction
Clonidine, an imidazoline derivative, is a central α2-adrenergic agonist. It was first developed in the 1960s as a nasal decongestant. It is currently used for the management of hypertension, attention deficit hyperactivity disorder (ADHD) as well as withdrawal symptoms from drug and alcohol addiction, tobacco withdrawal and Tourette’s syndrome. Clonidine toxicity often mimics that of opioids.
Pharmacokinetics
Clonidine is well absorbed with a bioavailability of almost 100%. The peak concentration in plasma and effect is observed within 1–3 h. The elimination half-life is 6–24 h with a mean half-life of 12 h. Half of the administered dose is excreted unchanged by the kidney.
Pathophysiology
Clonidine activates central α2-receptors. This results in a reduction in CNS sympathetic outflow at the vasomotor centre in the medulla oblongata. Clonidine is thought to reduce blood pressure through a reduction in cardiac output as well as its weak peripheral α-adrenergic antagonist properties. Clonidine also stimulates parasympathetic outflow and this may contribute to the slowing of heart rate as a consequence of increased vagal tone. Paradoxically, clonidine overdose can result in an initial hypertension from its partial α1-adrenergic agonist effect. It is suggested that clonidine’s inhibition of sympathetic outflow is mediated through endogenous opiate release.
Clinical features
Clonidine can cause transient hypertension from initial vasoconstriction with parenteral administration followed by hypotension. In addition to bradycardia and conduction defects, it can cause a central chlorpromazine-like effect with sedation. Other CNS symptoms include coma, seizure, miosis, reduced respiration and hypothermia. The median onset of symptoms following clonidine ingestion is 30 min and patients are usually symptomatic on arrival at the ED. Symptoms usually resolve by 24 h.
Investigations
The ECG is essential in evaluating and monitoring for bradycardia and conduction defects.
Treatment
The management of clonidine poisoning is primarily supportive. Hypotension usually responds to intravenous fluids. Atropine has been shown to abolish bradycardia in some case reports. Occasionally, inotropes may be required to maintain haemodynamic stability. Hypertension is usually short lived and rarely requires treatment. Patients are usually symptomatic on arrival and the benefits of administering activated charcoal are unlikely to outweigh the risk of aspiration.
Disposition
Patients should be observed in hospital until they are asymptomatic and bradycardia has resolved. They do not require ongoing cardiac monitoring for a stable sinus bradycardia.
Class 1c Antiarrhythmics
Introduction
Apart from the class one antiarrhythmics, many drugs in overdose cause sodium channel blockade. Drugs highly associated with sodium channel blockade and QRS widening are shown in Table 29.2.6. Overdose with class 1c antiarrhythmic drugs is among the most serious ingestions and is associated with a high morbidity and mortality. Overdoses with these agents are rare but they may be rapidly lethal with profound cardiovascular collapse. Drugs in this class include flecainide and propafenone; they are used for the treatment of SVT and ventricular arrhythmias. Class 1c antiarrhythmics block the fast inward sodium channel during phase 0 of the action potential. They have slow offset kinetics and cause complete blockade of sodium channel for a much longer duration than class 1a and 1b antiarrhythmics.
Table 29.2.6
Drugs highly associated with QRS widening and sodium channel blockade

*Antipsychotic drugs are not highly associated with QRS widening, but are commonly taken in overdose.
Reproduced with permission from Toxicology and Wilderness Expert Group. Drugs highly associated with QRS widening and sodium channel blockade (Table 17.1) [revised 2011 June]. In: Therapeutic guidelines eTG complete [Internet]. Melbourne: Therapeutic Guidelines Limited; 2012 Nov.
Pharmacokinetics
Flecainide has a high oral bioavailability and a rapid onset of action of 30 to 60 min. It has a long elimination half-life of 7–23 h. In adults, ingestions of 800 mg or more should be considered as life threatening. Similarly, propafenone has a long elimination half-life.
Clinical Features
In overdose, they have a rapid onset of clinical symptoms, typically with 30 min to 2 h. Overdose symptoms include nausea, vomiting, hypotension, bradycardia, varying degrees of atrioventricular block and tachyarrhythmia. In severe cases, coma and seizures may occur. Severe intoxication is frequently fatal because of the rapid onset of hypotension and ventricular arrhythmias.
Investigations
The ECG is essential in evaluating and monitoring the QRS duration, conduction defects and QT interval. The most common and important ECG change in overdose is QRS widening (more than 120 ms). Although QT prolongation occurs in flecainide overdose, torsades de pointes is rare.
Treatment
The mainstay of management of class 1c overdose is good supportive care including inotropic support, gastrointestinal decontamination and early and repeated doses of sodium bicarbonate to treat any broad complex arrhythmias and hypotension. Treatment is similar to other sodium channel blocking agents, such as the tricyclic antidepressants and includes plasma alkalinization to a pH of 7.5 with hyperventilation and repeated boluses of sodium bicarbonate. The use of antiarrhythmic drugs is problematic and are generally contraindicated and sufficient amounts of sodium bicarbonate should be used first.
Disposition
Asymptomatic patients with a normal ECG 4 h after ingestion of flecainide are unlikely to develop toxicity. Patients who are symptomatic should be admitted to a monitored area.
Cardiac arrest due to cardiovascularly active drugs
It is important for clinicians to be aware that cardiac arrest due to overdose of cardiovascularly active drugs may necessitate prolonged CPR and resuscitative manoeuvres and consideration of heroic measures, such as cardiopulmonary bypass (Fig. 29.2.1). Consultation with a clinical toxicologist is always recommended prior to cessation of resuscitative efforts in these cases.

FIG. 29.2.1 Flow chart management of cardiac arrest following poisonings. *Toxin with available antidote, e.g. natural toxin, digoxin, organophosphates; 4-AP: 4-aminopyridine; BB: β-blocker; CCB: calcium channel blocker; ECMO: extracorporeal membrane oxygenation; HIE: high-dose insulin euglycaemia; IABP: intra-aortic balloon pump; ILE: intravenous lipid emulsion; LA: local anaesthetic; NACB: sodium channel blocker; WCT: wide complex tachycardia. (From Gunja N, Graudins A. Management of cardiac arrest following poisoning. Emerg Med Australas 2011; 23:16–22, with permission.)
Controversies
![]() The advantages of glucagon over other inotropic agents in the management of CCB and β-blocker overdose are questionable. It is now rarely used as a first-line agent.
The advantages of glucagon over other inotropic agents in the management of CCB and β-blocker overdose are questionable. It is now rarely used as a first-line agent.
![]() The indications for initiation of hyperinsulinaemia euglycaemia therapy in CCB and β-blocker overdose are not well defined. This therapy is being advocated as first-line therapy for toxic hypotension.
The indications for initiation of hyperinsulinaemia euglycaemia therapy in CCB and β-blocker overdose are not well defined. This therapy is being advocated as first-line therapy for toxic hypotension.
![]() There are case reports of the successful use of cardiopulmonary bypass to maintain an adequate cardiac output following severe CCB, β-blocker and sodium channel blocker overdoses. Techniques such as this and extracorporeal membrane oxygenation may play a role in the management of otherwise fatal cases of cardiovascular collapse.
There are case reports of the successful use of cardiopulmonary bypass to maintain an adequate cardiac output following severe CCB, β-blocker and sodium channel blocker overdoses. Techniques such as this and extracorporeal membrane oxygenation may play a role in the management of otherwise fatal cases of cardiovascular collapse.
![]() The dosing regime of digoxin specific antibody is likely to be lower than the full equimolar dose that was previously advocated. It is proposed that giving half of the equimolar dose is adequate to stabilise patients with severe digoxin toxicity. The cost of the digoxin antibody should be weighed against the costs of additional in-hospital care that may be incurred if they are withheld.
The dosing regime of digoxin specific antibody is likely to be lower than the full equimolar dose that was previously advocated. It is proposed that giving half of the equimolar dose is adequate to stabilise patients with severe digoxin toxicity. The cost of the digoxin antibody should be weighed against the costs of additional in-hospital care that may be incurred if they are withheld.
![]() A clinical response to naloxone may occur in up to 31% of cases of clonidine toxicity but the clinical value of this intervention is doubtful.
A clinical response to naloxone may occur in up to 31% of cases of clonidine toxicity but the clinical value of this intervention is doubtful.
Further reading
1. Antman EM, Stone PH, Muller JE, et al. Calcium channel blocking agents in the treatment of cardiovascular diseases: Part E basic and clinical electrophysiological effects. Ann Intern Med. 1980;93:875–885.
2. Antman EM, Wenger FL, Butler VP, et al. Treatment of 150 cases of life threatening digitalis intoxication with digoxin specific Fab antibody fragments: final report of multicenter study. Circulation. 2004;81:1744–1752.
3. Auzinger GM, Scheinkestel CD. Successful extracorporeal life support in a case of severe flecanide intoxication. Crit Care Med. 2001;29:887–890.
4. Bateman DN. Digoxin-specific antibody fragments: how much and when? Toxicological reviews. 2004;3:135–143.
5. Baud FJ, et al. Clinical review: aggressive management and extracorporeal support for drug-induced cardiotoxicity. Crit Care. 2007;11:207.
6. Buckley N, Dawson AH, Howarth D, et al. Slow release verapamil poisoning Use of polyethylene glycol whole bowel lavage and high dose calcium. Med J Aust. 1993;158:202.
7. Engebretsen KM, Kaczmarek KM, Morgan J, Holger J. High-dose insulin therapy in beta-blocker and calcium channel blocker poisoning. Clintox. 2011;49:277–283.
8. Gunja N, Graudins A. Management of cardiac arrest following poisoning. Emerg Med Australas. 2011;23:16–22.
9. Holger JS, Engerbretsen KM, Fritzlar SJ, et al. Insulin versus vasopressin and epinephrine to treat β-blocker toxicity. Clin Toxicol. 2007;45:396–401.
10. Kolecki PF, Curry SC. Poisoning by sodium channel blocking agents. Crit Care Clin. 1997;13:829–848.
11. Love J, Howell JM, Litovitz TL, et al. Acute beta blocker overdose: factors associated with the development of cardiovascular morbidity. J Toxicol Clin Toxicol. 2000;38:275–281.
12. Seger D. Clonidine toxicity revisited. Clin Toxicol. 2002;40:145–155.
29.3 Antipsychotic drugs
Dino Druda and Shaun Greene
Essentials
1 Antipsychotics can cause numerous adverse effects at therapeutic doses, which may limit compliance and require changes to treatment regimens.
2 Extrapyramidal effects are less pronounced with the newer agents.
3 Clozapine is associated with agranulocytosis and myocarditis with therapeutic use and requires strict surveillance.
4 Following overdose, antipsychotics predominantly cause CNS depression and cardiovascular effects.
5 Amisulpride can cause significant QT prolongation which may result in torsades de pointes.
6 The mainstay of management of antipsychotic overdose is supportive.
7 Neuroleptic malignant syndrome is a rare, idiosyncratic adverse reaction which may be lethal without timely diagnosis and treatment.
Introduction
The antipsychotics form a heterogeneous group of medications that has evolved since chlorpromazine was first used to treat schizophrenia in the 1950s. The first-generation or so-called ‘typical’ antipsychotics caused many adverse effects, especially movement disorders such as extrapyramidal symptoms (EPS) and tardive dyskinesia (TD). They also have very little efficacy in treating the negative symptoms of schizophrenia (social withdrawal, anhedonia, poverty of speech, etc.). This led to the development and marketing of the second-generation, or atypical, antipsychotics in the late 1980s. In general, these drugs have fewer tendencies to cause movement disorders and have efficacy in managing the negative symptoms of schizophrenia, while maintaining efficacy in the management of acute psychosis. For this reason, they have largely replaced the older antipsychotic as first-line therapy in the treatment of schizophrenia and psychotic disorders.
Pharmacology
There are numerous ways of classifying the antipsychotic drugs; typical or atypical as described above (Table 29.3.1), by their chemical structure or according to neuroreceptor binding affinity.
Table 29.3.1
Typical and atypical antipsychotic drugs

All antipsychotic drugs produce their beneficial therapeutic effects by antagonizing the dopamine D2 receptors in the mesolimbic system. The first-generation antipsychotics were classified as high or low potency depending on their affinity for the D2 receptor. However, antagonism of the other D2 receptors leads to many of the adverse clinical effects. Antagonism of D2 receptors in the nigrostriatal pathway leads to movement disorders (EPS, akathisia, TD), antagonism of the D2 receptors in the mesocortical area can contribute to the negative symptoms and antagonism of D2 receptors in the anterior pituitary stimulates prolactin secretion which can lead to gynaecomastia and galactorrhoea. Blockade of D2 receptors in the anterior hypothalamus is associated with alterations in temperature regulation, which can lead to hypo- or hyperthermia and may be involved in the development of neuroleptic malignant syndrome, which is discussed later. The antiemetic effect of some of the antipsychotics is due to antagonism of the D2 receptors in the chemoreceptor trigger zone in the medulla.
The newer atypical antipsychotics also derive therapeutic efficacy from affinity and antagonism at various serotonin (5-HT) receptors. Antagonism at the 5-HT2A receptor is implicated both in increasing the efficacy of treating the negative symptoms of schizophrenia and also in reducing the incidence of EPS.
Agents with high antagonism of muscarinic M1 and M2 receptors (e.g. olanzapine, quetiapine) can cause an agitated delirium and peripheral features characteristic of anticholinergic toxicity (flushing, dry skin, tachycardia, urinary retention, etc.). Drugs that have a higher anticholinergic activity than dopaminergic tend to cause fewer extrapyramidal effects. High relative antagonism of histamine H1 receptors leads to sedation and, to a lesser extent, hypotension. Antagonism at the α1-adrenergic receptor can result in hypotension (e.g. quetiapine, clozapine) and clozapine also antagonizes the α2-receptor, although the clinical significance of this is uncertain.
Several first-generation antipsychotics can block voltage-gated fast sodium channels which, in overdose, can lead to slowing of cardiac conduction thus prolonging the QRS complex and impairing myocardial contractility. Blockade of the delayed rectifier potassium channel causes delayed repolarization and leads to prolongation of the QT interval.
Despite the heterogeneous nature of the antipsychotics, in general, they share similar pharmacokinetic properties. They are well absorbed after oral administration, with peak serum concentrations usually occurring within 2–6 hours of ingestion. This may be delayed following overdose of agents with significant anticholinergic properties. They are lipophilic, have large volumes of distribution and the majority of the agents are highly protein bound. They are extensively metabolized in the liver, with some having active metabolites.
Clinical effects
Adverse effects
Adverse effects at therapeutic doses may be dose related or idiosyncratic.
Extrapyramidal syndromes
These are a heterogeneous group of disorders characterized by abnormal neuromuscular activity. They can be particularly distressing to patients and may lead to difficulties with compliance and cessation of treatment. There are four well-recognized syndromes–acute dystonia, akathisia, parkinsonism and tardive dyskinesia. Of these, the first three are usually reversible, whereas tardive dyskinesia is irreversible, but occurs late, usually after months to years of treatment.
EPS are more common among the first-generation typical antipsychotics, especially those with high potency, such as haloperidol. Atypical antipsychotics are associated with a lower incidence of EPS, although it must be appreciated that EPS can occur with the use of any antipsychotic. Reactions are usually idiosyncratic, although can occur following overdose.
Acute dystonia is characterized by sustained involuntary muscle contraction, which commonly involves the face, head and neck, but can also involve the extremities. Rarely, the larynx can be involved, which may be life threatening. Risk factors for developing dystonia include male gender, young age and previous history of dystonic reaction. Onset is usually within a few hours of exposure, but may be delayed for several days.
Akathisia is characterized by an unpleasant sensation of restlessness or unease and, often, the patient is unable to remain still. It can be difficult to diagnose and can often be attributed to the underlying psychiatric condition rather than to the treatment.
Drug-induced parkinsonism is similar to idiopathic Parkinson’s disease, with rigidity and bradykinesia, although the characteristic tremor may be less pronounced. It is more common in older patients and patients on high potency agents.
Tardive dyskinesia is characterized by repetitive, involuntary, purposeless movements, classically involving the muscles of the face and mouth, although the limbs and trunk may be involved. It usually appears after months or years of therapy with antipsychotic medication and is usually resistant to treatment.
Cardiovascular effects
Cardiovascular effects of antipsychotics with therapeutic use include tachycardia, postural hypotension and ECG changes. Postural hypotension may be multifactorial, with α1-adrenergic blockade and direct myocardial depression playing a role. ECG changes may be diverse, with QRS prolongation, QT prolongation and non-specific ST-segment and T-wave changes being reported.
Seizures
All antipsychotics can lower the seizure threshold. However, seizures rarely complicate therapeutic use of antipsychotics, unless the patient has underlying risk factors, such as organic brain disease or epilepsy.
Metabolic syndromes
Chronic use of many of the antipsychotics is associated with the development of a metabolic syndrome, which can lead to weight gain, dyslipidaemia, hypertension and impaired glucose tolerance. These can be distressing and also contribute to the development of cardiovascular disease and type II diabetes. The development of these adverse effects can affect compliance with treatment. Metabolic affects are particularly associated with the use of olanzapine and clozapine.
Neuroleptic malignant syndrome (NMS)
NMS is a rare idiosyncratic adverse reaction, which can occur with any of the antipsychotic medications. Risk factors, diagnosis and management of NMS are described later in this chapter.
Clozapine
Clozapine is associated with a number of idiosyncratic effects that can occur with therapeutic use and requires more vigilant surveillance. These include agranulocytosis and myocarditis. These should be considered in the differential diagnosis if a patient on clozapine presents unwell.
Overdose
Following overdose of antipsychotics, the most common and significant manifestations involve the CNS and cardiovascular system.
Dose-dependent CNS depression occurs, ranging from lethargy and somnolence to coma and seizures. Airway protective reflexes may be impaired, requiring intensive care. Many of the agents can cause significant anticholinergic delirium with associated peripheral effects, such as urinary retention, flushed skin and reduction in sweat and saliva secretion.
The most common cardiovascular effects are tachycardia and hypotension. Tachycardia may be due to anticholinergic effects and also as a response to hypotension. Hypotension often occurs as a result of peripheral α1-receptor blockade, leading to vasodilatation.
ECG changes are often present after overdose and can include QRS prolongation and QT interval prolongation. Significant arrhythmias are uncommon, apart from following overdose with amisulpride, which can cause torsades des pointes.
Some of the specific clinical features following overdose of individual agents are described below.
Amisulpride
Overdose of amisulpride commonly causes QT prolongation, bradycardia and hypotension. Episodes of torsades de pointes have been reported and so amisulpride is considered to be particularly cardiotoxic. Onset of cardiotoxicity may be delayed for greater than 12 hours and QT prolongation can persist for many hours, with the potential to develop torsades de pointes abruptly. Ingestions greater than 4 g have been associated with development of prolonged QT and ingestions greater than 8 g can cause significant sedation and hypotension.
Chlorpromazine
Ingestions of greater than 15 mg/kg of chlorpromazine in children are associated with significant toxicity. Large overdoses>5 g, especially in drug-naïve patients, may lead to significant CNS depression, which may require intubation and intensive care due to loss of airway protective reflexes. Significant hypotension also occurs following overdose.
Clozapine
Acute overdose of clozapine leads to CNS depression, which is more pronounced in clozapine-naïve patients, who may require intubation. Seizures are reported in overdose. Despite the known anticholinergic properties of clozapine, hypersalivation is common. Miosis is classically described, but mydriasis can also occur. Agranulocytosis does not occur after a single overdose.
Haloperidol
Sedation and EPS are common following overdose of haloperidol. Haloperidol has also been associated with QT prolongation and arrhythmias following large ingestions or intravenous administration.
Olanzapine
Onset of clinical features following overdose is within 6 hours. Sedation and anticholinergic effects are the most common manifestations, leading to a combination of agitation and drowsiness, which may require intubation. Miosis may be noted. Tachycardia is common, but significant ECG abnormalities are rare.
Quetiapine
Quetiapine is available in immediate and extended release preparations. Overdose causes dose-related CNS depression and tachycardia. Hypotension and seizures are also reported after larger ingestions. Ingested doses of greater than 3 g are associated with increased length of stay and ICU admission. QTc prolongation is often reported, although the clinical significance of this is unclear, as there have been no reported cases of torsades de pointes. It may be that the prolongation of the QTc is as a result of overcorrection for tachycardia rather than intrinsic cardiotoxicity.
Risperidone
Risperidone is relatively benign following overdose, with tachycardia and dystonia being the most common effects. Onset of dystonia may be delayed and may recur after treatment.
Ziprasidone
Ziprasidone is associated with QT prolongation, both with therapeutic use and following overdose. Torsades de pointes has been reported after ziprasidone overdose with co-ingestants, but not in isolation.
Investigations
Antipsychotic toxicity is primarily diagnosed on history and examination for typical clinical features. Serum drug concentrations are not usually available in a clinically useful time frame or helpful in the management of acute overdose. In the case of patients on clozapine who present unwell to hospital, then a clozapine concentration can be measured. For these patients, a WCC is helpful when looking for agranulocytosis and troponin may be elevated in cases of clozapine-induced myocarditis. If this is suspected, then ECG and echocardiography may also be required.
Initial ECG evaluation should occur for all patients following antipsychotic overdose and any abnormality of the QRS or QT intervals warrants continuous cardiac monitoring. If initial ECG is normal, then it should be repeated after 6 hours in asymptomatic patients. More prolonged cardiac monitoring is required for patients following overdose with amisulpride or ziprasidone.
Treatment
The management of antipsychotic toxicity is primarily supportive. Attention to initial resuscitation should occur initially. Mild sedation requires no specific treatment. Patients with significantly decreased conscious state with loss of airway protective reflexes will require intubation, ventilation and intensive care.
Decontamination with activated charcoal can be considered if the presentation is within an hour and there is no clinical sign of CNS depression. Otherwise, administration of activated charcoal should be delayed until after the airway has been secured with intubation. There is no evidence for any benefit from enhanced elimination techniques, either with multidose activated charcoal or extracorporeal techniques and so they are not indicated.
Hypotension should initially be treated with an appropriate bolus of crystalloid solution. If there is no response to initial fluid bolus, then vasopressors may be required. There have been reports of worsening hypotension following the administration of adrenaline to patients who are hypotensive following quetiapine overdose, so noradrenaline is the preferred initial vasopressor. Patients with ventricular arrhythmias or with prolonged QRS should be managed with sodium bicarbonate in a similar fashion to patients with significant tricyclic antidepressant (TCA) toxicity. Intravenous bicarbonate 8.4% (1 mL=1 mmol) 1–2 mmol/kg boluses should be administered to obtain an arterial pH of 7.50–7.55. The pH may then be maintained in this range using hyperventilation in intubated patients. QT prolongation requires no specific management other than cardiac monitoring and correction of any potential contributing electrolyte abnormalities, such as hypokalaemia or hypomagnesaemia. Should torsades de pointes develop, it should be treated in the first instance with IV magnesium sulphate 50% 2–4 mL (1–2 g or 4–8 mmol) infusion over 10 minutes. Chemical or electrical overdrive pacing may also be required to avoid further instances.
Seizures are often self-limiting and require no treatment. However, should intervention be necessary, benzodiazepines should be used first line. Barbiturates and/or general anaesthesia are used for refractory seizures or status. There is no role for other anticonvulsants, such as phenytoin, in the management of drug-induced seizures.
Anticholinergic delirium should be managed initially with non-pharmacological measures, such as nursing in a quiet area and limiting stimulation. Urinary retention should be sought and relieved with a urinary catheter, as the distress caused by this may contribute to any agitation. Should medication be required, titrated doses of benzodiazepines (e.g. diazepam) should be used, although it must be appreciated that benzodiazepines may contribute to any CNS depressant effects of the ingested antipsychotic.
Acute dystonia is managed with an anticholinergic agent, such as benztropine (1–2 mg given IV or IM). Benzodiazepines may also be used. Repeat dosing may be required as the dystonia can recur.
Disposition
Patients should be observed for 6 hours after overdose of most antipsychotics. If they remain asymptomatic and have a normal ECG at this time, then they can be medically cleared for discharge. This observation period should be extended for 12 hours following significant ingestion of extended release preparations of quetiapine, ingestions of>4 g amisulpride and ingestions of ziprasidone.
Patients with significant symptoms should be admitted for observation until symptoms are resolving. Intensive care admission is required for patients requiring intubation and ventilation. In the event of any concerning ECG abnormalities, such as QT prolongation, then admission for cardiac monitoring is recommended until these changes are resolving.
Neuroleptic malignant syndrome
Neuroleptic malignant syndrome is a rare, idiosyncratic, potentially life-threatening adverse reaction that has been reported to occur with all the antipsychotics. There is a large variation in reported incidence, but recent pooled data suggest an incidence of 0.01–0.2%. The pathophysiology of NMS is not completely understood, but is thought to be due to central dopamine blockade, especially in the nigrostriatal and hypothalamic pathways. Risk factors for the development of NMS include male gender, young age, use of high potency antipsychotics, recent increase in dose, parenteral administration, dehydration and organic brain disease.
Clinical features
The onset of NMS occurs typically over 1–3 days. The typical clinical features are a combination of altered mental status, hyperthermia (temperature>38°C), autonomic dysfunction and muscular rigidity (‘lead pipe’ rigidity). The altered mental status ranges from delirium and confusion to stupor and coma. Autonomic dysfunction can manifest as tachycardia, cardiac arrhythmias, respiratory irregularities and hypo- or hypertension. The muscular rigidity is classically described as ‘lead-pipe’ rigidity, with increased tone and resistance to passive movement. There may be superimposed tremor leading to cogwheeling. Other neuromuscular abnormalities include bradykinesia, dystonia, mutism and dysarthria.
Differential diagnosis
There have been numerous diagnostic criteria proposed for NMS, but none are universally accepted. Alternate diagnoses must be considered and excluded, particularly CNS infection. Other conditions to be considered in the differential include heatstroke, thyrotoxicosis, serotonin toxicity, anticholinergic syndrome, malignant catatonia, non-convulsive status epilepticus, phaeochromocytoma and drug intoxication (MAOIs, sympathomimetics).
However, NMS must be considered in any patient on antipsychotic medication who is unwell, particularly when there is altered mental status, fever or muscle rigidity, especially if there has been a recent change in the antipsychotic regimen.
Investigations
There is no diagnostic test for NMS, although there are characteristic laboratory abnormalities in some cases. Increased muscle enzymes (CK, LDH, AST) are often present in cases of NMS. Leucocytosis is also frequently observed. There may be other electrolyte disturbance, metabolic acidosis and coagulation abnormalities.
Investigations to rule out alternate diagnoses need to be carried out, including CT scan of the brain and lumbar puncture to rule out CNS infection.
Treatment
Once NMS is diagnosed, it is essential to institute aggressive supportive care. The offending drug must be immediately withdrawn. Patients are often dehydrated, so fluid resuscitation should be commenced. Hyperthermia must be managed aggressively with passive and active cooling. If the temperature is>39.5°C, then intubation and neuromuscular paralysis should be considered. There is increased risk of thromboembolism, so prophylaxis should be commenced.
Benzodiazepines are often used early in the treatment of NMS and they may be effective in ameliorating symptoms in milder cases. Bromocriptine is a centrally acting dopamine agonist that can only be administered orally or via NG tube. The starting dose is 2.5 mg three times daily, increased to a daily maximum of 40 mg. It needs to be continued for 1–2 weeks, as premature discontinuation can lead to rebound symptoms. Potential adverse effects include vomiting and worsening of psychosis.
Dantrolene interferes with calcium release in skeletal muscle cells and therefore reduces skeletal muscle activity. It may be useful in cases of NMS with prominent hyperthermia and muscle rigidity. It can be given by IV infusion at 2–3 mg/kg/day. However, there are conflicting reports regarding the efficacy and outcome benefit of dantrolene in the management of NMS. Electroconvulsive therapy (ECT) has been advocated for cases of NMS refractory to pharmacological treatments, although its efficacy is unclear.
Controversies
![]() The clinical significance of QTc prolongation following quetiapine overdose is unclear. It is thought to be due to overcorrection of the QT for tachycardia and, as yet, there have been no reported cases of torsades de pointes.
The clinical significance of QTc prolongation following quetiapine overdose is unclear. It is thought to be due to overcorrection of the QT for tachycardia and, as yet, there have been no reported cases of torsades de pointes.
![]() The optimum time for cardiac monitoring following significant amisulpride or ziprasidone overdose is yet to be clearly defined.
The optimum time for cardiac monitoring following significant amisulpride or ziprasidone overdose is yet to be clearly defined.
![]() Many antipsychotics are highly lipophilic and there have been case reports of lipid rescue therapy being used to treat significant toxicity. However, evidence for definite outcome benefit is lacking.
Many antipsychotics are highly lipophilic and there have been case reports of lipid rescue therapy being used to treat significant toxicity. However, evidence for definite outcome benefit is lacking.
![]() The efficacy of specific therapies for NMS, such as dantrolene, bromocriptine and ECT, is controversial and evidence of outcome benefit is lacking.
The efficacy of specific therapies for NMS, such as dantrolene, bromocriptine and ECT, is controversial and evidence of outcome benefit is lacking.
Further reading
1. Balit CR, Isbister GK, Hackett LP, Whyte IM. Quetiapine poisoning: a case series. Ann Emerg Med. 2003;42:751–758.
2. Burns MJ. The pharmacology and toxicology of atypical antipsychotic agents. Clin Toxicol. 2001;39:1–14.
3. Hawkins DJ, Unwin P. Paradoxical and severe hypotension in response to adrenaline infusions in massive quetiapine overdose. Crit Care Resusc. 2008;10:320–322.
4. Isbister GK, Balit CR, Kilham HA. Antipsychotic poisoning in young children. Drug Safety. 2005;28:1029–1034.
5. Isbister GK, Balit CR, Macleod D, Duffull SB. Amisulpride overdose is frequently associated with QT prolongation and torsades de pointes. J Clin Psychopharmacol. 2010;30:391–395.
6. Juurlink DN. Antipsychotics. In: Nelson LS, Levin NA, Howland MA, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York: McGraw-Hill; 2010;1003–1015.
7. Levine M, Ruhn A-M. Overdose of atypical antipsychotics: clinical presentation, mechanisms of toxicity and management. CNS Drugs. 2012;26:601–611.
8. Minns AB, Clark RF. Toxicology and overdose of atypical antipsychotics. J Emerg Med. 2012;43:906–913.
9. Morgan M, Hackett LP, Isbister GK. Olanzapine overdose: a series of analytically confirmed cases. Internatl Clin Psychopharmacol. 2007;22:183–186.
10. Page CB, Calver LA, Isbiter GK. Risperidone overdose causes extrapyramidal effects but not cardiac toxicity. J Clinic Psychopharmacol. 2010;30:387–390.
11. Reulbach U, Dutsch C, Biermann T, et al. Managing an effective treatment of neuroleptic malignant syndrome. Crit Care. 2007;11:R4.
12. Strawn JR, Keck PE, Caroff SN. Neuroleptic malignant syndrome. Am J Psychiatr. 2007;164:870–876.
29.4 Antidepressant drugs
Shaun Greene and Dino Druda
Essentials
1 Tricyclic antidepressant (TCA) overdose is associated with severe cardiovascular toxicity, seizures, coma and death.
2 Sodium bicarbonate is the specific treatment of TCA cardiotoxicity.
3 Overdose of extended-release bupropion is associated with dose-related delayed onset of seizures.
4 Selective serotonin reuptake inhibitors generally produce relatively mild toxicity, however, citalopram and escitalopram are associated with QT prolongation and torsades des pointes.
5 Selective serotonin reuptake inhibitors are associated with serotonin toxicity in overdose and following interactions with other serotonergic drugs.
6 Selective noradrenaline reuptake inhibitors produce a sympathomimetic toxidrome and delayed onset of seizures.
7 Serotonin toxicity may be life threatening and can occur following administration of multiple serotonergic agents or serotonergic drug overdose. Treatment is supportive with cessation of serotonergic drugs and administration of a serotonin receptor antagonist in selected cases.
Introduction
Severity of clinical toxicity following overdose (OD) of antidepressant drugs available in Australia varies according to the class of drug. Toxicity is dose dependent and produces clinical manifestations affecting multiple organ systems. Cardiovascular and neurological features can be life threatening. Early risk assessment and aggressive supportive care are essential in ensuring a good outcome.
Tricyclic antidepressants
Although efficacious in treating depression, tricyclic antidepressants (TCAs) are relatively more toxic than other classes of antidepressants in OD. Significant toxicity including death is associated with ingested doses of more than 10 mg/kg in adults and 5 mg/kg in children. Of the tricyclic antidepressants available in Australia (Table 29.4.1), dothiepin is associated with the greatest toxicity. Cardiovascular system dysfunction and coma typically manifest rapidly following significant ingestion. Good outcome is dependent on aggressive airway management, utilization of sodium bicarbonate and provision of supportive care in a critical care environment.
Table 29.4.1
Tricyclic antidepressants available in Australia
Amitriptyline
Clomipramine
Dothiepin
Doxepin
Imipramine
Nortriptyline
Trimipramine
Pharmacology
The tertiary amine structure of TCAs non-selectively interacts with multiple receptors throughout the body, most of which are not implicated in positive antidepressant effects. Pharmacodynamic interactions include:
![]() Inhibition of central nervous system (CNS) serotonin and noradrenaline reuptake and modulation of genetic expression of serotonin, β-adrenergic and other CNS receptors contribute to antidepressant effects. This pharmacodynamic property does not contribute significantly to classical TCA toxicity, but is likely responsible for TCA-related serotonin toxicity (described later in this chapter).
Inhibition of central nervous system (CNS) serotonin and noradrenaline reuptake and modulation of genetic expression of serotonin, β-adrenergic and other CNS receptors contribute to antidepressant effects. This pharmacodynamic property does not contribute significantly to classical TCA toxicity, but is likely responsible for TCA-related serotonin toxicity (described later in this chapter).
![]() Binding to inactivated cardiac sodium channels producing rate-dependent inhibition of sodium conductance leading to membrane stabilizing effects, QRS prolongation and potentially lethal arrhythmias and impaired myocardial contractility.
Binding to inactivated cardiac sodium channels producing rate-dependent inhibition of sodium conductance leading to membrane stabilizing effects, QRS prolongation and potentially lethal arrhythmias and impaired myocardial contractility.
![]() Stimulation of central postsynaptic histamine receptors producing CNS depression, sedation and coma.
Stimulation of central postsynaptic histamine receptors producing CNS depression, sedation and coma.
![]() Antagonism of muscarinic acetylcholine receptors producing anticholinergic effects including tachycardia, agitation and urinary retention.
Antagonism of muscarinic acetylcholine receptors producing anticholinergic effects including tachycardia, agitation and urinary retention.
![]() Antagonism of peripheral α1-adrenergic receptors producing peripheral vasodilatation.
Antagonism of peripheral α1-adrenergic receptors producing peripheral vasodilatation.
![]() Varying degrees of antagonism at potassium, chloride and γ-aminobutyric acid (GABA) receptors.
Varying degrees of antagonism at potassium, chloride and γ-aminobutyric acid (GABA) receptors.
Tricyclic antidepressants are well absorbed following ingestion, undergo extensive first pass metabolism, are hepatically metabolized (often producing active metabolites), are highly protein bound and are lipophilic and therefore widely distributed throughout the body. Half-lives are relatively long (10–81 hours) and often observed to be longer following OD.
Clinical features
The most common clinical features of significant TCA overdose are CNS depression (varying from agitated delirium to coma) and sinus tachycardia; these manifest rapidly within 1–2 hours of exposure. Risk assessment based on dose ingested and associated anticholinergic, CNS and cardiovascular clinical effects are described in Table 29.4.2. Agitated delirium secondary to anticholinergic receptor agonism is not always evident in more severe cases as coma predominates. Sodium channel blockade and α-receptor mediated peripheral vasodilatation lead to supraventricular and ventricular arrhythmias, hypotension and asystole in a dose-dependent manner. Anticholinergic features may become more apparent during the recovery phase as histamine receptor-induced sedation resolves.
Table 29.4.2
Tricyclic antidepressants: dose-related risk assessment and clinical effects

Clinical investigations
A 12-lead ECG is the most valuable prognostic investigation following TCA overdose. A terminal 40 ms axis between 120 and 270 degrees is a sensitive indicator of TCA presence but this is difficult to measure at the bedside. Measurement of maximal limb lead QRS duration is a useful predictor of toxicity. Prolongation of greater than 100 ms is associated with an increased incidence of coma, need for intubation, seizures, hypotension and arrhythmias. One study demonstrated no seizures or arrhythmias in patients with a QRS duration that remained<100 ms. Ventricular arrhythmias were predicted in one study by a QRS duration>160 ms. The finding of a positive R wave of greater than 3 mm in amplitude in lead aVR or a ratio of>0.7 between the amplitude of R and S waves in aVR are sensitive markers for seizures and arrhythmias. A rightward frontal plane QRS vector (indicated by an S wave in lead I and an R wave in aVR) is associated with TCA toxicity. Although QT prolongation is observed in TCA therapy and toxicity, this finding is not predictive of clinical toxicity.
TCA blood concentrations can be measured, but are poorly correlated with degree of clinical toxicity.
Treatment
Patients with significant clinical toxicity or those with a recent (previous 3–4 hours) reported ingestion of a potentially toxic amount of a TCA receive aggressive supportive care in a resuscitation area. Early securing of the airway via endotracheal intubation is indicated when there is any decrease in conscious state. Poor respiratory function and secondary hypoxia potentially worsen TCA toxicity.
Administration of activated charcoal should be considered within 1 hour of ingestion, provided facilities exist to protect the airway if decreased consciousness or seizures occur. Activated charcoal may reduce TCA absorption if administered to intubated patients via a nasogastric tube up to 4 hours post-ingestion.
Early aggressive use of sodium bicarbonate in conjunction with hyperventilation is indicated where there is any cardiovascular dysfunction in conjunction with QRS prolongation (>100 ms), hypotension unresponsive to initial intravenous fluid (see further discussion below) or in the presence of any arrhythmia. Intravenous bicarbonate 8.4% (1 mL=1 mmol) 1–2 mmol/kg boluses should be administered to obtain an arterial pH of 7.50–7.55. pH should then be maintained in this range using hyperventilation. Sodium bicarbonate provides hypertonic sodium, competitively overcoming sodium channel blockade. Alkalization improves sodium channel function and reduces the free concentration of TCA available to produce toxicity. Acid–base manipulation using sodium bicarbonate is more effective than hyperventilation alone in TCA toxicity. Sodium bicarbonate may be prophylactically beneficial in cases where there is a significant history of TCA ingestion and a QRS duration of>100 ms.
Other therapies, including concentrated hypertonic saline (3% sodium chloride) and lignocaine, may be beneficial in treating resistant arrhythmias. Class 1a antiarrhythmics, including procainamide, quinidine and phenytoin, are contraindicated.
Hypotension is treated with intravenous crystalloid (up to 20–30 mL/kg). Administration of sodium bicarbonate is indicated if hypotension persists. Inotropes are indicated for resistant hypotension despite intravenous fluid administration and normalization of acid–base status.
Disposition
Patients who are well with no CNS depression and a normal ECG 6 hours post-reported TCA ingestion are safe for medical discharge. Those with any signs of significant toxicity require admission to a critical care environment.
Bupropion
Pharmacology
Bupropion is a drug with a unicyclic structure used as a smoking cessation aid in Australia and as an antidepressant in a number of other countries. It has a structure similar to amphetamine and inhibits reuptake of dopamine, with a lesser effect on noradrenaline and serotonin. Bupropion is licensed for use as an antidepressant in some countries. It is only available as an extended release preparation in Australia. Bupropion and its active metabolite hydroxyl-bupropion have half-lives of approximately 20 hours. Both are relatively highly protein bound (84% and 77%). Bupropion has a volume of distribution of 2000 L. Mild anticholinergic properties may be evident following OD. Bupropion OD is characterized by a high risk of seizures; studies suggest hydroxyl-bupropion is responsible, although the exact mechanism is unclear.
Clinical features
Bupropion is only available as a modified release preparation in Australia. In therapeutic doses, bupropion may cause mild hypertension, postural hypotension in sporadic cases, headache, agitation, seizures and gastrointestinal irritation. Cardiac conduction abnormalities are not reported in therapeutic dosing.
Tachycardia, hypertension, nausea, vomiting, tremor and hallucinations may be observed following bupropion OD; however, severe agitation and seizures are the most significant clinical features; seizures are typically delayed up to 6–8 hours (in some cases up 16 hours) post-exposure and are dose dependent. In a series of 59 patients presenting following OD of modified release bupropion, seizures occurred in 30% of those ingesting<4.5 g, 50% with 4.5–9 g ingested and 100% of individuals who ingested>9 g.
QT and QRS prolongation, arrhythmias and hypotension have been reported following ingestions of>9 g of bupropion. QT prolongation is likely to be related to coexisting tachycardia. A QT nomogram should be utilized in the risk assessment of any measured QT prolongation related to bupropion toxicity. Deaths have been reported following massive ingestions.
Treatment
Provision of meticulous supportive care is the mainstay of management following bupropion OD. Patients should be managed in a monitored area with resuscitation facilities available in case of seizures or cardiovascular deteriorations. Activated charcoal should be considered in patients who are cooperative and not agitated within 4 hours of ingestion. Intravenous benzodiazepines should be administered in incremental doses to control agitation at an early stage as they may increase the threshold for seizure activity. Resistant agitation may require sedation and intubation. Seizures are managed using benzodiazepines.
Hypotension is initially treated with intravenous crystalloid. QRS prolongation is treated with intravenous sodium bicarbonate (see tricyclic antidepressant section).
Disposition
Patients who are well with a normal ECG and no agitation or seizures 16 hours post-bupropion exposure are safe for medial discharge. Patients with significant agitation, ongoing seizures, or CVS instability require admission to a critical care environment.
Monoamine oxidase inhibitors
Pharmacology
Monoamine oxidase inhibitors (MAOIs) either reversibly or irreversibly inhibit function of the enzymes monoamine oxidase A and B (MAO-A, MAO-B), leading to increased CNS concentrations of adrenaline, noradrenaline, serotonin and dopamine. The pharmacodynamic effects of non-selective irreversible MAOIs (including phenelzine and tranylcypromine) are not overcome until MAO is resynthesized, resulting in dose-dependent toxicity that may last for days. Moclobemide, a selective reversible inhibitor of MAO-A is more benign in OD, but may cause severe serotonin toxicity when combined with other serotonergic agents.
The MAOIs are well absorbed orally, undergo extensive first-pass metabolism, readily cross the blood–brain barrier and have moderate volumes of distribution. Peak concentrations occur within 2 or 3 hours of ingestion. Metabolites are renally eliminated.
Clinical features
Overdose of irreversible MAOIs is characterized initially by peripheral sympathomimetic stimulation and central nervous system excitation. Symptoms do not usually manifest until 6–12 hours post-overdose. Initial symptoms include tachycardia, agitation, restlessness, hyperreflexia and voluntary movements. As toxicity progresses, there is progressive muscle rigidity, respiratory failure, decreasing conscious state, hyperthermia, rhabdomyolysis, coma and cardiovascular collapse. Clinical toxicity may last for days.
Lone overdose of the reversible selective MAOI moclobemide generally produces only mild symptoms including tachycardia, nausea and anxiety. Overdose of moclobemide with another serotonergic agent may lead to significant serotonin toxicity.
MAOI adverse effects in therapeutic doses include the development of serotonin toxicity when combined with other serotonergic agents. The tyramine reaction may occur following ingestion of tyramine-containing foods. Tyramine is an indirect acting sympathomimetic. MAOI inhibition of tyramine metabolism can lead to a hyperadrenergic crisis with severe hypertension, intracranial haemorrhage, renal failure, disseminated intravascular coagulopathy (DIC) and rhabdomyolysis.
Treatment
Management of MAOI toxicity is primarily supportive. Asymptomatic patients presenting within 2 hours of ingestion of tranylcypromine or phenelzine may benefit from administration of activated charcoal. Patients with evidence of severe toxicity are managed in a resuscitation area with particular attention to supporting organ function and limiting complications. Hypertension is initially treated using titrated doses of intravenous benzodiazepines. Refractory hypotension requires treatment with intravenous nitrates, sodium nitroprusside or phentolamine. Beta-adrenergic blockers are contraindicated; unopposed α-receptor stimulation may worsen toxicity. Hypothermia unresponsive to sedation with benzodiazepines must be treated aggressively along conventional lines. Serotonin toxicity requires specific care as outlined below. Moclobemide toxicity usually only requires basic supportive care.
Disposition
Patients who remain clinically well 6 hours post-ingestion of moclobemide or 12 hours post-ingestion of phenelzine or tranylcypromine are safe for medical discharge. Patients who are symptomatic following overdose of an irreversible MAOI require admission to an intensive care unit.
Selective serotonin reuptake inhibitors
Selective serotonin reuptake inhibitors (SRRIs) are associated with fewer adverse effects both in therapeutic use and OD compared to TCAs. They are utilized in treating depression, obsessive–compulsive disorder, panic/anxiety disorders, eating disorders and chronic pain syndromes. Table 29.4.3 lists SSRIs available in Australia.
Table 29.4.3
Serotonin reuptake inhibitors available in Australia
Selective serotonin reuptake inhibitors (SSRIs)
Citalopram, escitalopram, fluoxetine, fluvoxamine, paroxetine, sertraline
Combined selective serotonin and noradrenaline reuptake inhibitors (SNRIs)
Venlafaxine, desvenlafaxine, duloxetine, reboxetine
Serotonin reuptake inhibition with α-adrenergic antagonism
Mirtazepine
Pharmacology
SSRIs increase synaptic concentrations of serotonin in the CNS via interaction with G-protein coupled serotonin receptors (14 different receptors have been described currently). With extended use, downregulation of serotonin inhibitory autoreceptors occurs, leading to increased serotonin synthesis and decreased reuptake.
SSRIs are well absorbed post-ingestion. SSRIs and metabolites are substrates for and inhibitors of numerous hepatic microsomal enzymes, the most important being CYP2D6. Hepatic metabolism produces various metabolites; many are active and capable of prolonging therapeutic effect and also increasing the probability of adverse drug interactions.
Clinical features
Therapeutic use of SSRIs is associated with headache, insomnia, sexual dysfunction, gastrointestinal symptoms, dizziness and fatigue. Serotonin toxicity can occur in therapeutic use if exposure to another serotonergic agent occurs.
OD is rarely associated with severe adverse effects. Mild CNS depression may occur, but is not severe. Nausea, vomiting, tachycardia, diaphoresis and dizziness are reported, but are generally mild and self-limiting.
QT prolongation and torsades des pointes have been reported following citalopram and escitalopram OD. Fluoxetine, citalopram and escitalopram may cause mild bradycardia.
Treatment
Activated charcoal is only indicated following massive ingestion of an SSRI within the previous hour. There is evidence that cardiovascular toxicity may be limited if activated charcoal is administered to patients who have taken>300 mg escitalopram or>600 mg citalopram within the previous 4 hours.
Treatment is primarily supportive. Regular ECGs and cardiac monitoring are indicated following ingestion>1000 mg citalopram (or 600 mg if activated charcoal has not been given within 4 hours of ingestion) or>400 mg escitalopram (or 400 mg if activated charcoal has not been given within 4 hours of ingestion). Cardiac monitoring is continued for at least 13 hours post-exposure in these cases or until the QT interval has normalized.
Combined serotonin and noradrenaline reuptake inhibitors
Duloxetine, reboxetine, venlafaxine and desvenlafaxine are the combined selective serotonin and noradrenaline reuptake inhibitor antidepressants available in Australia.
Pharmacology
Selective serotonin and noradrenaline reuptake inhibitor (SNRIs) inhibit both serotonin and noradrenaline reuptake in the CNS. Rapid downregulation of β-adrenergic receptors may increase the speed of onset of therapeutic effect compared to SSRIs.
SNRIs are rapidly absorbed after oral administration and are hepatically metabolized. Desvenlafaxine and venlafaxine are only available in Australia in modified release formulations.
Clinical features
Overdose produces a noradrenergic mediated sympathomimetic toxidrome characterized by tachycardia, tremor, nausea, vomiting, dizziness and agitation. Venlafaxine is associated with seizures following ingestions of>5 g. Increasing agitation may herald onset of seizures. Hyperthermia and rhabdomyolysis may occur with large ingestions. Cardiovascular toxicity may occur following ingestions of>8 g of venlafaxine. QRS prolongation, QT prolongation, ventricular arrhythmias and hypotension have been reported. Desvenlafaxine is more potent than venlafaxine therapeutically (50 mg desvenlafaxine is equivalent to 75 mg venlafaxine) and therefore thresholds for toxic effects are likely to be lower with desvenlafaxine.
Treatment
Following large ingestions of SNRIs, patients should be managed in an area equipped with cardiac monitoring and facilities to manage seizures. Activated charcoal may be beneficial if administered within an hour of a large ingestion. Intubation and administration of activated charcoal within 4 hours of ingestion of>5 g of venlafaxine should be considered.
Agitation should be controlled using incremental doses of benzodiazepines. Seizures are normally self-limiting, but may require treatment with intravenous benzodiazepines.
Hypotension is treated initially with intravenous fluid. Arrhythmias associated with QRS prolongation should be treated with sodium bicarbonate (see TCA section).
Disposition
Patients should be observed for at least 6 hours post-ingestion of standard release preparations and 16 hours post-ingestion of modified release preparations or until clinically well. Cardiac monitoring is indicated following large ingestions (>5 g venlafaxine). Ingestion of>5 g of venlafaxine mandates 24 hours of observation due to the risk of delayed seizures.
Mirtazapine
Mirtazapine is a unique antidepressant; in addition to blocking the reuptake of serotonin, mirtazapine is a centrally acting α2-antagonist (increasing concentrations of serotonin and noradrenaline) and serotonin receptor (5-HT2 and 5-HT3) agonist.
Overdose of mirtazapine typically only causes minor effects including minor sedation. Tachycardia, more significant CNS sedation and seizures have been reported following large ingestions. Treatment is supportive.
Serotonin toxicity
Pharmacology
Serotonin toxicity (traditionally described as serotonin syndrome) is a consequence of excess serotonin acting at CNS and peripheral receptor sites. Table 29.4.4 lists drugs associated with serotonin toxicity. Although the exact pathophysiological mechanism is not fully elucidated, 5-HT1A and 5-HT2A receptors appear to be predominantly involved.
Table 29.4.4
Selected drugs associated with serotonin toxicity
Increased serotonin production and release
Tryptophan, lysergic acid diethylamide (LSD)
Increased release of stored serotonin
Amphetamines (including MDMA), cocaine, lithium, mirtazapine
Impaired reuptake of serotonin into presynaptic nerve
SSRIs (fluoxetine, citalopram, sertraline, fluvoxamine, paroxetine), bupropion, fentanyl, cocaine, tramadol, venlafaxine
Inhibition of serotonin metabolism
Monoamine oxidase inhibitors (moclobemide, phenelzine, tranylcypromine), methylene blue, linezolid
Although serotonin toxicity may occur following lone therapeutic or excess exposure to a serotonergic agent, it most commonly occurs in the context of combination use of serotonergic drugs or in instances where there has been an inadequate ‘wash-out’ time between substitution of one serotonergic agent for another.
Clinical features
Serotonin toxicity normally develops within hours of exposure to the offending agent or agents. A number of diagnostic criteria have been suggested, but none widely validated. Most commonly, the diagnosis has been defined as the presence of clinical findings affecting three distinct systems (in the context of exposure to a serotonergic agent):
![]() autonomic system: tachycardia, flushing, diaphoresis, mydriasis, hyperthermia, tachypnoea, diarrhoea
autonomic system: tachycardia, flushing, diaphoresis, mydriasis, hyperthermia, tachypnoea, diarrhoea
![]() central nervous system: altered conscious state, elevated mood, akathesia, insomnia, agitation, anxiety, confusion, seizures
central nervous system: altered conscious state, elevated mood, akathesia, insomnia, agitation, anxiety, confusion, seizures
![]() neuromuscular: hyperreflexia, clonus (inducible or spontaneous), hypertonia (lower limbs>upper limbs), myoclonus, tremor, rigors, rigidity, incoordination.
neuromuscular: hyperreflexia, clonus (inducible or spontaneous), hypertonia (lower limbs>upper limbs), myoclonus, tremor, rigors, rigidity, incoordination.
Toxicity is seen along a spectrum and varies from subclinical non-specific feelings of anxiety and apprehension, through to life-threatening hyperthermia, autonomic instability, cardiovascular dysfunction, multiorgan failure, seizures and rigidity. Most patients recover, but deaths have been reported. The variety and variable severity of symptoms may make the diagnosis challenging and it is not uncommon for the differential diagnosis to include neuromuscular malignant syndrome (NMS). Distinguishing features include the slower onset of NMS and the typical associated ‘bradykinetic’ clinical picture and severe muscle rigidity. Serotonergic toxicity is characterized by hyperexcitability with hyperreflexia and clonus. Other differential diagnoses include CNS infection, anticholinergic syndrome, sympathomimetic syndrome, acute dystonia, malignant hyperthermia and non-convulsive seizures. Blood concentrations of serotonergic drugs do not correlate with clinical toxicity.
Treatment
Cessation of the serotonergic agents implicated in toxicity and avoidance of further serotonergic agents are mandatory in all cases. Benzodiazepines are first-line treatment to reduce muscle rigidity or to treat seizures. Cases of severe hyperthermia or muscle rigidity should be treated aggressively: sedation with neuromuscular paralysis and admission to a critical care environment.
The use of a number of antidotes known to antagonize 5-HT1 and 5-HT2 receptors has been reported. No clinical trials have examined the safety or efficacy of these drugs. They include cyproheptadine, olanzapine, chlorpromazine and propranolol. Of these, cyproheptadine appears to be the most widely utilized. The dose is 12 mg orally (via a nasogastric tube in the intubated patient) as a single dose followed by 4–8 mg orally 8-hourly. Chlorpromazine may be useful in cases where additional sedation is required, but the possibility of dehydration may combine with the peripheral vasodilating effects of chlorpromazine to produce significant hypotension.
Controversies
![]() The role of sodium bicarbonate in preventing development of neurological and cardiovascular toxicity following TCA ingestion is not well defined.
The role of sodium bicarbonate in preventing development of neurological and cardiovascular toxicity following TCA ingestion is not well defined.
![]() Many TCAs are highly lipophilic and there have been case reports of lipid rescue therapy being used to treat significant toxicity. However, evidence for definite outcome benefit is lacking.
Many TCAs are highly lipophilic and there have been case reports of lipid rescue therapy being used to treat significant toxicity. However, evidence for definite outcome benefit is lacking.
![]() Diagnostic criteria for serotonin toxicity have not been widely validated.
Diagnostic criteria for serotonin toxicity have not been widely validated.
![]() The efficacy of serotonin receptor antagonists in treating serotonin toxicity has not been well studied.
The efficacy of serotonin receptor antagonists in treating serotonin toxicity has not been well studied.
![]() Indications for administration of serotonin antagonists in the treatment of serotonin toxicity are poorly defined.
Indications for administration of serotonin antagonists in the treatment of serotonin toxicity are poorly defined.
Further reading
1. Balit CR, Lynch CN, Isbister GK. Bupropion poisoning: a case series. Med J Aust. 2003;178:61–63.
2. Bateman ND. Tricyclic antidepressant poisoning: central nervous system effects and management. Toxicol Rev. 2005;24:181–186.
3. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med. 2005;352:1112–1120.
4. Bradberry SM, Thanacoody HKR, Watt BE, et al. Management of the cardiovascular complications of tricyclic antidepressant toxicity: role of sodium bicarbonate. Toxicol Rev. 2005;24:195–204.
5. Dunkley EJ, Isister GK, Sibbritt D, et al. The hunter serotonin toxicity criteria: simple and accurate diagnostic decision rules for serotonin toxicity. Q J Med. 2003;96:635–642.
6. Isbister GK. Electrocardiogram changes and arrhythmias in venlafaxine overdose. Br J Clin Pharmacol. 2009;67:572–576.
7. Isbister GK, Bowe SJ, Dawson A, et al. Relative toxicity of selective serotonin re-uptake inhibitors (SSRIs) in overdose. Clin Toxicol. 2004;42:277–285.
8. Isbister GK, Hackett LP, Dawson AH, et al. Moclobemide poisoning: toxicokinetics and occurrence of serotonin toxicity. Br J Clin Pharmacol. 2003;56:441–450.
9. Liebelt EL, Francis PD, Woolf AD. ECG lead aR versus QRS interval in predicting seizures and arrhythmias in acute tricyclic antidepressant toxicity. Ann Emerg Med. 1995;26:195–201.
10. Mills KC. Monoamine oxidase inhibitor toxicity. Emerg Med. 1993;15:58–71.
11. Thanacoody HKR, Thomas SHL. Tricyclic antidepressant poisoning–cardiovascular toxicity. Toxicol Rev. 2005;24:205–214.
12. Whyte IM, Dawson AH, Buckley NA. Relative toxicity of venlafaxine and selective serotonin reuptake inhibitors in overdose compared to tricyclic antidepressants. Q J Med. 2003;96:369–374.
29.5 Lithium
Mark Monaghan
Essentials
1 Chronic lithium toxicity is associated with significant morbidity and mortality, especially where diagnosis and treatment are delayed. Acute lithium overdose, unless massive, has a more benign course.
2 Chronic lithium poisoning presents with neurological dysfunction. Acute lithium overdose presents with gastrointestinal dysfunction.
3 Consider the diagnosis of lithium intoxication and check a serum lithium concentration in any patient on lithium therapy who presents unwell.
4 Chronic lithium intoxication usually develops because of impaired lithium excretion. The underlying factors must be identified and corrected.
5 Serum lithium levels correlate with central nervous system (CNS) levels and clinical severity in chronic but not acute intoxication.
6 Haemodialysis effectively enhances lithium elimination but is rarely required in patients with normal renal function. This intervention is more likely to be necessary in chronic intoxication than acute overdose.
Introduction
Lithium, the metal with the lowest molecular weight, is usually dispensed as the carbonate salt. It is widely used in the therapy of bipolar disorder and a number of other conditions. Both immediate-release and sustained-release preparations are available. This drug has a relatively narrow therapeutic index and chronic intoxication develops relatively frequently. Acute overdose is less common.
Pharmacokinetics
Standard lithium preparations are rapidly and completely absorbed after oral administration with peak serum levels occurring at 2–4 h. Absorption and time to peak level is delayed after administration of sustained-release preparations and following overdose. Once absorbed, lithium is slowly redistributed from the intravascular space to the total body water. Lithium is not metabolized and its elimination is almost exclusively renal. Lithium is freely filtered at the glomerulus but, under normal circumstances, approximately 80% of filtered ions are reabsorbed in the proximal tubule and only 20% are excreted in the urine. Under these circumstances, renal clearance of lithium is approximately 10–40 mL/min and its elimination half-life is 20–24 h. The renal elimination of lithium is greatly affected by sodium and water balance and by the presence of drugs that affect renal tubular reabsorption of sodium. In the early stages following acute overdose, renal elimination is much greater because lithium is relatively concentrated in the intravascular compartment and available for filtration at the glomerulus.
Clinical features
Acute lithium overdose
Patients who take a significant overdose of lithium carbonate as with any other metal salt, develop rapid onset of gastrointestinal toxicity characterized by nausea, vomiting, abdominal pain and diarrhoea. This gastrointestinal disturbance can be very severe and may result in significant fluid and electrolyte losses. It is usually observed where more than 25 g are ingested, but can occur following smaller doses. Gastrointestinal upset is not a prominent feature of chronic lithium toxicity.
Acute lithium overdose is much less likely to result in significant neurotoxicity than is chronic lithium toxicity. Neurotoxicity can develop following massive acute overdose if renal clearance is sufficiently impaired so as to allow redistribution of sufficient lithium from the intravascular compartment to tissue compartments before it could be excreted. This situation may develop if there is pre-existing renal failure or if inadequate fluid resuscitation leads to dehydration, sodium depletion or renal impairment as a consequence of the fluid losses from gastrointestinal toxicity.
Chronic lithium toxicity
Chronic lithium toxicity may develop in association with prolonged excessive dosing or, more commonly, as a result of impaired lithium excretion due to intercurrent illness or a drug interaction. Lithium excretion is impaired in renal failure and congestive cardiac failure because of reduced filtration at the glomerulus and also in water or sodium depletion states because of increased reabsorption of sodium (and lithium) in the proximal tubule. A number of drugs including non-steroidal anti-inflammatory drugs (NSAIDs), selective serotonin reuptake inhibitors (SSRIs), neuroleptics, angiotesin converting enzyme (ACE) inhibitors, thiazide diuretics and topiramate may either impair lithium excretion or exacerbate toxicity.
The clinical features of chronic lithium toxicity are almost exclusively neurological and the following severity grading system is widely used:
![]() Grade I (mild): nausea, vomiting, tremor, hyperreflexia, agitation, muscle weakness, ataxia
Grade I (mild): nausea, vomiting, tremor, hyperreflexia, agitation, muscle weakness, ataxia
![]() Grade II (serious): stupor, rigidity, hypotonia, hypotension
Grade II (serious): stupor, rigidity, hypotonia, hypotension
![]() Grade III (life threatening): coma, seizures, myoclonia, cardiovascular collapse.
Grade III (life threatening): coma, seizures, myoclonia, cardiovascular collapse.
The differential diagnosis for this presentation is broad and includes non-convulsive status epilepticus, serotonin and neuroleptic malignant syndromes, electrolyte abnormalities and CNS pathologies, such as sepsis.
While minor benign ECG changes may be observed, Lithium toxicity is generally not associated with significant cardiovascular effects, although delayed bradycardia has been reported.
Chronic lithium therapy is also associated with nephrogenic diabetes insipidus and thyroid dysfunction, which may complicate the clinical presentation of toxicity.
Clinical investigations
Essential laboratory investigations in the assessment of lithium toxicity are serum electrolytes, renal function and serum lithium concentration. Serial serum lithium concentrations are often required. Other investigations are performed as indicated to evaluate and manage intercurrent disease processes and to exclude important differential diagnoses.
Therapeutic serum lithium concentrations are generally quoted as 0.6–1.2 mEq/L, although clinical evidence of lithium toxicity can be observed at concentrations within this range, particularly in the elderly. More commonly in cases of chronic intoxication, mild toxicity is observed at lithium concentrations of 1.5–2.5 mEq/L, severe toxicity at concentrations of 2.5–3.5 mEq/L and life-threatening toxicity at concentrations>3.5 mEq/L. Following acute overdose, serum lithium concentrations do not correlate with clinical severity as they do not reflect CNS concentrations; however, when performed serially, they are useful in guiding management. Peak serum lithium concentrations>4.0 mEq/L are frequently observed following acute overdose in patients who do not go on to develop neurotoxicity. Seum levels in chronic toxicity are more equilibrated with and therefore more accurately reflect CNS levels.
Treatment
Acute lithium overdose
The vast majority of acute poisonings can be managed solely with good supportive care. Intravenous access should be established and infusion of normal saline commenced during the initial assessment. Administration should be sufficient to correct any sodium or water deficits arising as a result of the toxic gastroenteritis and to ensure a good urine output. Excessive administration of normal saline or attempts at forced diuresis do not further enhance lithium excretion. A serum lithium concentration, renal function and electrolytes should be performed as part of the initial assessment and repeated as necessary to guide further management. In particular, the serum lithium should be followed until falling and<2 mEq/L.
Activated charcoal does not bind lithium well and need not be administered unless there has been a significant co-ingestion. On the basis of a single volunteer study, whole-bowel irrigation has been recommended for overdose of extended-release preparations but the gastrointestinal upset renders this intervention technically difficult in patients with large ingestions. Sodium polystyrene sulphonate has been proposed as an effective alternative absorbent, which may also enhance elimination of lithium. At this stage, this agent is not widely used and repeated administration can cause hypokalaemia.
Haemodialysis is rarely indicated following acute overdose in the patient with normal renal function who receives good supportive care. It may be necessary in the presence of renal failure or in the patient post-massive ingestion who goes on to develop neurotoxicity in the presence of a slowly falling serum lithium concentration.
Chronic lithium toxicity
The diagnosis of lithium toxicity should be considered in any individual on lithium therapy who presents to the emergency department unwell, in particular with evidence of neurological dysfunction. The diagnosis should be confirmed or excluded by ordering a serum lithium concentration as part of the initial work-up. A precipitating illness that has resulted in impaired lithium excretion will usually be present and require assessment and treatment on its own merits.
Appropriate supportive care measures should be instituted on arrival. Once the diagnosis of chronic lithium toxicity is confirmed, further care is orientated towards management of the precipitating medical condition and enhancing lithium excretion by optimizing renal function and correcting any water or sodium deficits with intravenous normal saline. Therapy with lithium carbonate and any drugs contributing to lithium toxicity should be immediately discontinued.
Enhanced elimination of lithium by haemodialysis may be attempted in severe or worsening chronic lithium neurotoxicity. The aim of this intervention is to minimize the duration of neurological dysfunction and avoid permanent neurological sequelae. Lithium has physicochemical and pharmacokinetic properties that render it very suitable for enhancing elimination by haemodialysis: low molecular weight, high water solubility, small volume of distribution, no plasma protein binding and an endogenous renal clearance rate much lower than that achieved by haemodialysis. There is, however, no evidence that haemodialysis improves clinical outcome or survival rates.
The indications for haemodialysis are difficult to define. It should be considered in any patient with an elevated serum lithium concentration and severe or life-threatening neurotoxicity. It may be considered in the patient with less severe toxicity in whom adequate renal function and a falling lithium concentration are unable to be established with initial fluid resuscitation. Once instituted, haemodialysis should be continued until the serum lithium is<1 mEq/L. Some rebound in serum lithium may be noted after intermittent haemodialysis is discontinued, which may be avoided if prolonged intermittent renal replacement therapy is used or continuous arteriovenous (AV) or venovenous (VV) haemodiafiltration (HDF) is sustained for>16 h. The combination of haemodialysis followed by CVVHDF is recommended by some authors. The decision to dialyse can usually be made some 8–12 h after admission.
Disposition and prognosis
Patients with chronic lithium intoxication require admission for management of their fluid and electrolyte status, monitoring of renal function and serum lithium concentration and management of intercurrent illnesses. Ideally, admission should be to an institution with a capacity to perform haemodialysis where toxicity is moderate or severe. Following haemodialysis, neurological recovery may be delayed well beyond the removal of lithium and permanent neurological deficits are reported.
Acute lithium overdose usually has an excellent outcome with good supportive care and may be admitted to a non-monitored setting for intravenous fluids and monitoring of fluid and electrolytes and lithium concentrations. The asymptomatic patient with normal renal function and lithium level falling to below 2 mEq/L is fit for medical discharge. This usually occurs within 24 h. Psychiatric evaluation is mandatory and may take place while waiting for lithium levels to fall.
Controversies
![]() The indications for and preferred method of gastrointestinal decontamination following acute lithium overdose remain undefined.
The indications for and preferred method of gastrointestinal decontamination following acute lithium overdose remain undefined.
![]() Precise criteria for haemodialysis in chronic lithium intoxication remain undefined.
Precise criteria for haemodialysis in chronic lithium intoxication remain undefined.
![]() Continuous arterio- or venovenous haemofiltration have been proposed as alternatives to haemodialyis for enhancement of lithium elimination. Although lower clearances are achieved with these methods, they are often easier to institute and may minimize rapid transcellular fluid and electrolyte shifts. At the moment they can only be recommended where haemodialysis is not available.
Continuous arterio- or venovenous haemofiltration have been proposed as alternatives to haemodialyis for enhancement of lithium elimination. Although lower clearances are achieved with these methods, they are often easier to institute and may minimize rapid transcellular fluid and electrolyte shifts. At the moment they can only be recommended where haemodialysis is not available.
Further reading
1. Bailey AR, Sathianathan VJ, Chiew AL, et al. Comparison of intermittent haemodialysis, prolonged intermittent renal replacement therapy and continuous renal replacement haemofiltration for lithium toxicity: a case report. Crit Care Resusc. 2011;13:120–122.
2. Eyer F, Pfab R, Felgenhauer N, et al. Lithium poisoning: pharmacokinetics and clearance during different therapeutic measures. J Clin Psychopharmacol. 2006;26:325–330.
3. Hansen HE, Amdisen A. Lithium intoxication. Q J Med. 1978;47:123–144.
4. Jaeger A, Saunder P, Kopferschmidt J, et al. When should dialysis be performed in lithium poisoning? A kinetic study in 14 cases of lithium poisoning. Clin Toxicol. 1993;31:429–447.
5. Meertens JH, Jagernath DR, Eleveld DJ, et al. Haemodialysis followed by continuous veno-venous haemodiafiltration in lithium intoxication; a model and a case. Eur J Intern Med. 2009;20:e70–e73.
6. Netto I, Phutane VH. Reversible ithium neurotoxicity: review of the literature. Prim Care Companion CNS Disord. 2012;14:1.
7. Oakley PW, Whyte IM, Carter GL. Lithium toxicity: an iatrogenic problem in susceptible individuals. Aust NZ J Psychiatr. 2001;35:833–840.
8. Shou M. Long lasting neurological sequelae after lithium intoxication. Acta Psychiatr Scand. 1984;70:594.
9. Strayhorn JM, Nash JL. Severe neurotoxicity despite ‘therapeutic’ serum lithium levels. Dis Nerv Syst. 1977;38:107–111.
10. Waring WS. Management of lithium toxicity. Toxicol Rev. 2006;25:221–230.
11. Waring WS. Delayed cardiotoxicity in chronic lithium poisoning: discrepancy between serum lithium concentrations and clinical status. Basic Clin Pharmacol Toxicol. 2007;100:353–355.
29.6 Antihistamine and anticholinergic poisoning
Naren Gunja and Andis Graudins
Essentials
1 Anticholinergic toxicity is a relatively common and often unrecognized toxicological problem in the emergency department.
2 Many common psychotropic medications may cause antimuscarinic effects, such as neuroleptics and tricyclic antidepressants.
3 H1-receptor antagonists are readily accessible and a common cause of anticholinergic poisoning.
4 Significant central nervous system and cardiovascular toxicity may infrequently complicate large ingestions of first-generation H1-receptor antagonists.
5 H2-receptor antagonist overdose rarely produces any significant clinical effects.
Introduction
Anticholinergic toxicity is a common side effect of many pharmaceutical agents, natural remedies and plants, both in therapeutic dosing and in overdose (Table 29.6.1). Symptoms and signs may range from mild manifestations of the syndrome (e.g. dry mouth and blurred vision) to severe anticholinergic delirium with agitation, hallucinations and aggressive behaviour.
Table 29.6.1
Anticholinergic agents
Pharmaceuticals
Anticholinergic agents
Atropine
Benzhexol
Benztropine
Oxybutynin
Scopolamine
Antipsychotic agents
Clozapine
Olanzapine
Phenothiazines
Quetiapine
Risperidone
Cyclic antidepressants
Amitriptyline
Chlormipramine
Dothiepin
Doxepin
Imipramine
Nortriptyline
First-generation H1-receptor blockers
Chlorpheniramine
Cyproheptadine
Dexchlorpheniramine
Diphenhydramine
Doxylamine
Orphenadrine
Pheniramine
Promethazine
Others
Amantadine
Carbamazepine
Botanicals
Datura spp. (Jimson weed or thorn apple)
Brugmansia spp. (Angel’s trumpet)
Atropa belladona (deadly nightshade)
The antihistamine agents are a diverse group of drugs that can be broadly classified, based upon receptor specificity, into H1- and H2-receptor antagonists. The H1-receptor antagonists are widely used in the treatment of allergic conditions, nasal congestion, and as over-the-counter sleep aids. This group can be further divided into the ‘first-generation’ agents, which tend to be more lipophilic and are more sedating, and the ‘second-generation’ or non-sedating agents. The H2-receptor antagonists are primarily used in the treatment of peptic ulcer disease and gastro-oesophageal reflux, but may also be used in conjunction with H1-antagonists in the treatment of severe allergic reactions.
Antihistamine agents are relatively easy to obtain and frequently ingested in overdose or abused recreationally for their sedating and anticholinergic effects. The incidence of antihistamine poisoning and abuse in Australia is not well characterized. Other prescription drugs may also result in anticholinergic toxicity both in therapeutic dosing and in overdose. These may also be intentionally abused for their anticholinergic effects. Chinese and traditional herbal medicines may result in anticholinergic toxicity, either directly from the herbal agent ingested or as a result of contamination with anticholinergic agents, such as atropine or scopolamine. The intentional abuse of botanicals (e.g. Datura spp.) may also present with anticholinergic toxicity. In view of the easy availability of many of these pharmaceutical and herbal agents, the emergency physician should include a detailed drug history in the evaluation of any patient presenting with evidence of mental status change and anticholinergic symptoms or signs. In particular, polypharmacy and drug interactions between multiple agents with the potential for anticholinergic effects should be included in the differential diagnosis of elderly patients presenting with mental status changes.
Pharmacodynamics and pharmacokinetics
The H1-antagonists are a diverse group of agents that reversibly block the action of histamine at H1-receptors. High lipid solubility results in good central nervous system (CNS) penetration and sedation. The first-generation agents also block muscarinic, α-adrenergic and serotonergic receptors. Local anaesthetic effects due to sodium channel blockade may mimic the antiarrhythmic properties of class 1a antiarrhythmic agents. Diphenhydramine, dimenhydrinate and cyproheptadine, in particular, may prolong the cardiac muscle cell action potential duration by this mechanism. The second-generation H1-antagonists (fexofenadine, loratadine) have much less CNS penetration and are more histamine-receptor specific with little or no effect at other receptor subtypes.
All the H1-antagonists are well absorbed orally with peak serum concentrations occurring within 2–4 h. Absorption may be delayed in overdose due to anticholinergic effects seen with the first-generation agents. Bioavailability is limited by significant first-pass metabolism. Some agents may be converted to active metabolites (e.g. hydroxyzine). Volume of distribution and protein binding are generally high. Elimination half-lives for the first-generation agents are between 2 and 6 h. The second-generation agents generally have longer half-lives (e.g. loratadine 8.3 h).
The H2-antagonist agents are generally well tolerated with few side effects with therapeutic dosing. Cimetidine inhibits hepatic microsomal enzyme metabolism and reduces the metabolism of drugs eliminated by this pathway. This may result in increased serum concentrations and clinical effects of co-ingested medications.
All drugs with anticholinergic side effects have the potential to slow gastric emptying and produce gastrointestinal ileus when taken in overdose. As a result, absorption of these agents may be slowed and result in the potential for prolonged toxicity.
Clinical features
The anticholinergic toxidrome is usually manifest by a combination of peripheral and central muscarinic cholinergic receptor blockade. Peripheral effects may include sinus tachycardia, cutaneous vasodilatation and flushing, low-grade temperature, warm dry skin with an absence of axillary sweat, dry mucous membranes, gastrointestinal ileus and urinary retention. CNS effects include mydriasis with blurred vision due to the inhibition of visual accommodation, delirium, confusion, visual hallucinations, incoherent speech, agitation, combativeness, aggression and coma. Patients presenting with anticholinergic syndrome will often have an impaired perception of their environment which may result in behaviour that could injure the patient. Anticholinergic symptoms and signs may be prominent with ingestion of first-generation antihistamines. Over half of patients ingesting>1 g of promethazine are likely to develop delirium. Even therapeutic doses of some of the H1-antagonist agents may be sufficient to produce an anticholinergic delirium in susceptible individuals (especially the elderly and children). Topical use of these agents, particularly on broken skin surfaces, may also result in anticholinergic delirium.
In patients who present to hospital several hours following poisoning with an anticholinergic agent, the peripheral features of the toxidrome may be absent. This may also occur in elderly people with mild-to-moderate anticholinergic delirium resulting from the side effects of therapeutic drug administration.
Other manifestations of H1-antagonist toxicity may include CNS and cardiovascular effects and rhabdomyolysis. Overdose of first-generation agents commonly produces sedation, confusion, agitation and ataxia. Large ingestions may result in coma and seizures. Pheniramine, a commonly abused antihistamine in Australia, appears to be more proconvulsant than other agents following overdose, with a reported incidence of seizures of 30%. Seizures have been reported with other first-generation H1-antagonists, such as orphenadrine and diphenhydramine. Fatal doses of diphenhydramine in adults range from 20 to 40 mg/kg. Doxylamine poisoning may result in non-traumatic rhabdomyolysis. Hypotension, due to α-receptor blockade, can occur following large ingestions of first-generation agents. Conduction defects are infrequent following poisoning with first-generation H1-antagonists. Orphenadrine, diphenhydramine and dimenhydrinate poisoning can result in QRS-interval prolongation, broad-complex tachycardia and ventricular arrhythmias similar to that seen in cyclic antidepressant poisoning. Promethazine does not appear to cause seizures or arrhythmias.
Overdose with H2-antagonists, such as cimetidine, usually results in little or no evidence of toxicity. Doses of up to 15 g have failed to produce clinical toxicity.
Clinical investigations
A 12-lead electrocardiograph should be performed to check for the presence of sinus tachycardia, QRS and QT-interval duration. Bedside blood glucose testing is indicated in all patients with altered mental status. Blood for serum electrolytes and paracetamol level should be collected. In patients with mental status changes not easily explained by drug intoxication, other organic causes for cognitive impairment should be ruled out. Serum antihistamine levels are not readily available and do not influence patient management. Standard ‘drugs-of-abuse’ urine screens do not detect antihistamines or most other agents with anticholinergic toxicity. Bedside bladder scan should be performed to rule out urinary retention, a common finding in anticholinergic poisoning.
Treatment
The mainstay of therapy for poisoning with anticholinergic agents is supportive care in a safe environment. Comatose or hypoventilating patients should have appropriate airway intervention and ventilatory support. Hypotension should be treated initially with intravenous crystalloid boluses. Hypotension refractory to fluids may necessitate the use of pressor agents, such as noradrenaline. Agitation and seizures can be controlled using parenteral benzodiazepines in the first instance. Barbiturates (thiopentone, phenobarbitone) may be considered in refractory seizures. Bladder catheterization should be performed in cases of urinary retention.
Gastrointestinal decontamination, if indicated, should be performed with a single-dose of oral activated charcoal. Charcoal given within 2 h has been shown to reduce the risk of promethazine-induced delirium. The benefit of activated charcoal in patients presenting with minimal or no signs of toxicity more than 2 h following ingestion is doubtful. Methods of enhancing elimination of antihistamines are ineffective because of their large volumes of distribution and high protein binding.
The reversible acetylcholinesterase inhibitor physostigmine has been used in the diagnosis and management of anticholinergic agitation and delirium. Physostigmine rapidly reverses the effects of anticholinergic delirium and may prevent the need for escalating doses of benzodiazepines to control agitation. Physostigmine may decrease the need for other interventions, such as cerebral computed tomography scanning and lumbar puncture in patients with suspected anticholinergic delirium.
When using physostigmine, an initial test dose 0.5 mg IV is followed by 1.0–2.0 mg over the following 3–5 min in an adult. A partial response may necessitate further 0.5–1.0 mg boluses. Clinical effects may last from 30 to 120 min. Caution should be exercised in using physostigmine in patients with suspected antihistamine or cyclic antidepressant poisoning with ECG evidence of cardiac conduction delay because of the risk of precipitating cardiac asystole.
Broad-complex tachycardia resulting from severe poisoning with orphenadrine, diphenhydramine or dimenhydrinate should be treated with serum alkalinization with intravenous sodium bicarbonate boluses (1.0–2.0 mmol/kg) as for severe cyclic antidepressant poisoning. Symptomatic bradycardia and high degree atrioventricular block should be initially treated with atropine. Unresponsive cases may need cardiac pacing or inotropic support.
Disposition
Patients who present with minimal or no signs of toxicity require 4–6 h of observation and monitoring. They may be medically cleared if, at the end of this period, they are alert with a normal ECG and no signs of delirium or urinary retention. Patients with persistent mental status changes require further observation but, if the ECG is normal, do not require further cardiac monitoring. The duration of anticholinergic delirium may be from 12 h to several days depending on the agent and dose ingested. Severe toxicity, if it is to develop, will be evident within 2–3 h of ingestion. Those patients with poisoning complicated by coma, seizures or cardiovascular system toxicity require admission and observation in an intensive care or high-dependency setting.
All patients with intentional ingestions or suspicion of self-harm require psychiatric assessment prior to discharge.
Controversies
![]() The precise indications, contraindications and timing of physostigmine continue to be controversial in the management of anticholinergic delirium.
The precise indications, contraindications and timing of physostigmine continue to be controversial in the management of anticholinergic delirium.
Further reading
1. Beaver KM, Gavin TJ. Treatment of acute anticholinergic poisoning with physostigmine. Am J Emerg Med. 1998;16:505–507.
2. Burns MJ, Linden CH, Graudins A, et al. A comparison of physostigmine and benzodiazepines for the treatment of anticholinergic poisoning. Ann Emerg Med. 2000;35:374–381.
3. Clark RF, Vance MV. Massive diphenhydramine poisoning resulting in a wide-complex tachycardia: successful treatment with sodium bicarbonate. Ann Emerg Med. 1992;21:318–321.
4. Farrell M, Heinrichs M, Tilelli JA. Response of life threatening dimenhydrinate intoxication to sodium bicarbonate administration. J Toxicol Clin Toxicol. 1991;29:527–535.
5. Feinberg M. The problems of anticholinergic adverse effects in older patients. Drugs Aging. 1993;3:335–348.
6. Koppel C, Ibe K, Tenczer J. Clinical symptomatology of diphenhydramine overdose: an evaluation of 136 cases in 1982 to 1985. J Toxicol Clin Toxicol. 1987;25:53–70.
7. Koppel C, Tenczer J, Ibe K. Poisoning with over-the-counter doxylamine preparations: an evaluation of 109 cases. Hum Toxicol. 1987;6:355–359.
8. Page CB, Duffull SB, Whyte IM, et al. Promethazine overdose: clinical effects, predicting delirium and the effect of charcoal. Q J Med. 2009;102:123–131.
9. Pentel P, Peterson CD. Asystole complicating physostigmine treatment of tricyclic antidepressant overdose. Ann Emerg Med. 1980;9:588–590.
10. Rimmer SJ, Church MK. The pharmacology and mechanism of action of histamine H1 antagonists. Clin Exp Allerg. 1990;20:3–17.
11. Schneir AB, Offerman SR, Ly BT, et al. Complications of diagnostic physostigmine administration to emergency department patients. Ann Emerg Med. 2003;42:14–19.
12. Suchard JR. Assessing physostigmine’s contraindication in cyclic antidepressant ingestions. J Emerg Med. 2003;25:185–191.
29.7 Paracetamol
Andis Graudins
Essentials
1 Paracetamol poisoning is one of the most common toxicological presentations to Australasian emergency departments.
2 The decision to treat patients with antidotal therapy following acute single ingestions should be made using the paracetamol treatment nomogram.
3 N-acetylcysteine (NAC) prevents liver toxicity, however, this effect decreases with delay to treatment. Patients presenting more than 8 h post-ingestion should have NAC commenced while waiting for the return of serum paracetamol concentrations and liver function tests.
4 The paracetamol treatment nomogram cannot be used to assess the risk of hepatotoxicity following repeated supratherapeutic ingestions.
5 Paracetamol overdose should be excluded in all patients with suspected deliberate self-poisoning, especially when presenting with impaired conscious state and in anyone with evidence of unexplained hepatic impairment on liver function studies.
6 Extended-release formulations of paracetamol are available in Australia and should be sought when taking the drug history.
7 The routinely recommended dose of NAC infusion may not be sufficient to prevent development of hepatotoxicity following massive ingestion of paracetamol (>50 g). Clinical toxicologist advice is recommended.
8 In patients where timing of paracetamol ingestion or history of exposure cannot be reliably elicited to make a risk assessment, treatment with NAC should be commenced until the infusion is completed or the clinical scenario can be clarified and there is no biochemical evidence of liver toxicity.
Introduction
Poisoning with paracetamol is common in Australia, as well as other Western countries. In the USA, over 100 000 potential paracetamol poisonings are reported annually to the American Association of Poison Control Centers. In the UK, paracetamol poisoning accounts for up to 43% of poisoning exposures presenting to emergency departments.
Pharmacokinetics and pathophysiology
Paracetamol (N-acetyl para-aminophenol, acetaminophen) is rapidly absorbed from the gastrointestinal (GI) tract in therapeutic doses with peak plasma concentrations occurring within 30–60 min with tablet formulations and less than 30 min with liquid preparations. Bioavailability increases with size of the dose, ranging from 68% following 500 mg to 90% following 1–2 g orally. Time to peak plasma concentration may be delayed in the presence of co-ingestants that delay gastric emptying, such as opioids, antihistamines and anticholinergic agents. The volume of distribution for paracetamol is approximately 1 L/kg, with around 50% plasma protein binding. Metabolism occurs primarily in the liver with small amounts also metabolized renally. Metabolites are renally excreted with less than 4% excreted unchanged in the urine. Elimination half-life is approximately 1.5–2.5 h following therapeutic dosing. Paracetamol is metabolized by three mechanisms. With therapeutic dosing, approximately 60% is conjugated to glucuronide metabolites and 35% to sulphate metabolites. Less than 5% of paracetamol is metabolized by microsomal enzymes. CYP2E1 is the major isoenzyme but CYP2A and CYP1A2 are also significant. Microsomal metabolism produces a reactive intermediary metabolite, N-acetyl-para-benzoquinoneimine (NAPQI). This is rapidly conjugated with glutathione to produce non-toxic mercapturic acid and cysteine metabolites that are renally excreted. Elimination half-life is the same for adults, children and elderly patients but may be slightly elevated in neonates.
In overdose, glucuronidation and sulphation pathways are rapidly saturated, resulting in increased metabolism of paracetamol by the microsomal enzyme pathway. When glutathione stores are depleted by more than 70%, NAPQI accumulates in the liver and binds to hepatocytes, resulting in cell death and predominantly centrilobular hepatic necrosis.
Microsomal metabolism of paracetamol may be enhanced by barbiturates, carbamazepine, oral contraceptives, chronic alcohol ingestion or starvation. Inhibition of microsomal metabolism may occur in the presence of acute alcohol ingestion and with the administration of 4-methylpyrazole. Therapeutic doses of cimetidine do not decrease excretion of mercapturate metabolites of paracetamol following therapeutic doses in humans. There are no human studies to support the use of cimetidine in prevention of hepatotoxicity following paracetamol poisoning.
A sustained-release formulation of paracetamol (Panadol Osteo) has been on the market in Australia for the management of arthritis pain since 2002. This formulation contains 665 mg of paracetamol in a bilayer tablet with one-third being immediate-release and two-thirds sustained-release. It has been designed to release paracetamol slowly and maintain a therapeutic concentration for up to 8 h. Human volunteer data in simulated overdose suggests a delay to, and reduction in, peak paracetamol concentration. Comparison with immediate-release paracetamol at similar doses showed reduction in peak paracetamol concentration and area under the curve by more than 50% and delay to peak paracetamol concentration from 1 to 3 h. Pharmacokinetic data following deliberate self-poisoning with this formulation suggests that there may be a delay in peak serum concentration which may go undetected with a single 4-hour serum paracetamol estimation. Panadol Osteo overdose may also be associated prolonged absorption and detectable paracetamol concentrations beyond the duration of the standard 20 h N-acetylcysteine treatment protocol. Massive ingestions of immediate-release paracetamol (>50 g), particularly with co-ingestants that slow GI motility (opioids, antihistamines, anticholinergic agents), can also be associated with delayed peak and prolonged elevation of serum paracetamol concentrations.
An isolated small rise in INR has been observed in patients with paracetamol poisoning in the absence of hepatic impairment. Mild elevations in INR and reduced levels of functional Factor VII occurred in 66% of patients with an extrapolated 4-hour paracetamol concentration greater or equal to 1000 μmol/L (150 mg/L). This effect appears to be related to inhibition of vitamin K-dependent activation of coagulation factors.
Clinical features
The clinical features of early paracetamol poisoning are non-specific and do not permit diagnosis on clinical grounds. Classically, untreated poisoning progresses through four stages of toxicity. Stage 1 lasts about 24 h and is a subclinical period where the patient may exhibit only mild nausea, vomiting and malaise. During this period, paracetamol is being metabolized, glutathione stores are being depleted and hepatotoxicity is in its early stages. In severe poisoning, mild elevations of hepatic aminotranferases may be apparent as early as 16 h post-ingestion. In stage 2, nausea and vomiting resolve. Patients may develop right upper quadrant pain and hepatic tenderness 24–48 h post-ingestion. Liver function begins to deteriorate, with increasing aminotransferases, bilirubin and prothrombin time. Stage 3 is essentially a continuum of the above between 72 and 96 h post-ingestion. Hepatic function deteriorates and chemical hepatitis, jaundice and encephalopathy may develop. Peak aminotransferases are seen around 72 h post-ingestion. Stage 4 is either the stage of resolution with a fall in aminotransferase concentrations or, less commonly, the development of fulminant hepatic failure. Renal failure may also develop as a consequence of paracetamol toxicity. This may either be independent of hepatoxicity with direct renal toxicity from renal microsomal enzymatic metabolism of paracetamol to NAPQI or as a consequence of liver failure induced hepatorenal syndrome.
Other manifestations of acute paracetamol poisoning may include coma, lactic acidaemia and myocardial damage. Coma results from massive ingestion of paracetamol and is independent of any hepatic impairment. Serum paracetamol concentrations greater than 6500 μmol/L (1000 mg/L) may present with coma. Similarly, massive overdose may result in cardiac changes, such as ST–T-wave changes, bundle-branch block and sinus bradycardia.
In general, most patients recover from paracetamol toxicity. The overall untreated mortality is less than 1% and that of untreated patients with hepatotoxicity around 3.5%.
There are a number of ‘over-the-counter’ cough and cold preparations containing paracetamol in combination with other agents. These include sympathomimetics, such as pseudoephedrine, antihistamines, such as diphenhydramine, or cough suppressants, such as dextromethorphan. Patients may present with symptoms and signs of an acute toxidrome from one or more of these agents. Compound analgesics may also be ingested. These may result in the development of associated salicylate, opioid or caffeine (Panadol Extra) toxicity.
Assessment of risk of hepatotoxicity
The risk of hepatotoxicity following acute ingestion of paracetamol is dose dependent. In healthy adults, hepatotoxicity may result from ingestion of more than 200 mg/kg or 10 g, whichever is the least. In children less than 6 years old, ingestion of more than 200 mg/kg may result in toxic serum concentrations. The threshold for toxicity may be less in patients with underlying hepatic impairment (e.g. chronic alcoholic liver disease, chronic active hepatitis), severe malnutrition or in the presence of microsomal enzyme-inducing agents.
The paracetamol-treatment nomogram shows a clear relationship between the serum paracetamol concentration and the potential for subsequent hepatotoxicity following a single ingestion of immediate-release paracetamol. The nomogram begins at 4 h post-ingestion to allow for absorption and distribution of paracetamol. Serum concentrations taken less than 4 h post-ingestion may be unreliable in predicting the potential for hepatotoxicity.
The risk of hepatotoxicity from untreated acute paracetamol ingestion can be estimated from the nomogram. Patients with a serum concentration falling above a line from 1300 μmol/L (200 mg/L) at 4 h post-ingestion to 170 μmol/L (25 mg/L) at 16 h post-ingestion (the ‘probable toxicity’ line) will have a 60% chance of developing hepatotoxicity (AST>1000 IU/L) if left untreated. This risk increases to 87% in untreated patients with paracetamol concentrations above 2000 μmol/L (300 mg/L) 4 h post-ingestion.
The current ‘treatment line’ in Australasia is the ‘possible hepatotoxicity’ line, 1000 μmol/L (150 mg/L) at 4 h post-ingestion to 125 μmol/L (16 mg/L) at 16 h post-ingestion. This was adopted to allow for errors in calculation of the time of ingestion. The safety of treatment decisions to commence NAC based upon this 1000 μmol/L at 4 h nomogram line has been demonstrated in the USA in over 11 000 patients, where no patients treated with NAC within 15 h of ingestion died. In contrast, use of the higher line (1300 μmol/L at 4 h) has been associated with isolated reports of untreated patients who subsequently developed acute hepatic failure or suffered a fatal outcome with concentrations below this line.
With the recognition that there are numerous ‘at-risk’ groups that may have a lower threshold for hepatotoxicity, it has previously been recommended that the treatment line be dropped by 50% of the ‘probable toxicity line’. It must be noted that lowering of the treatment threshold is purely empiric in these cases and there have been no studies to confirm this approach.
As a result, Australasian treatment guidelines for paracetamol poisoning utilize a single-nomogram line approach to the management of paracetamol poisoning. The current nomogram (Fig. 29.7.1) starts at 1000 μmol/L (150 mg/L at 4 h) and parallels the treatment approach practised in North America. This provides an additional margin of safety for patients who may possess risk factors, provides a margin of error for estimation of time of ingestion and removes the need for potentially confusing additional lines.

FIG. 29.7.1 Paracetamol treatment nomogram. For use in the risk assessment of acute paracetamol ingestion at a single point in time.
Repeated supratherapeutic dosing with paracetamol is associated with a risk of hepatotoxicity, particularly in those with the hepatic risk factors. Liver failure has been reported in retrospective case series with chronic use of as little as 4 g a day in patients with underlying acute illnesses with associated decreased oral intake. However, prospective evaluation of the risk of liver failure with therapeutic doses of paracetamol in chronic alcoholics does not provide an indication that there is an increased susceptibility to liver failure in this subset of patients. It is important to note that the paracetamol-treatment nomogram is not useful in the assessment of hepatotoxic risk in these patients. In alcoholic patients, raised hepatic aminotransferases into the thousands are suggestive of a toxin-induced hepatitis as seen with paracetamol. Both alcoholic hepatitis and viral hepatitis rarely produce aminotransferase that rises above 1000 IU/L.
Antidotal therapy with N-acetylcysteine
N-acetylcysteine (NAC) is effective at preventing the development of hepatotoxicity.
(AST>1000 IU/L) following paracetamol poisoning. It is metabolized to cysteine in the liver and is a precursor to glutathione, necessary for the inactivation of the toxic metabolite NAPQI. Additionally, NAC may act as a substrate for hepatic sulphation, thus reducing the amount of paracetamol being shunted to the microsomal pathway of metabolism. In Australia, NAC is administered according to the 20-h intravenous protocol described by Prescott (150 mg/kg over 15 min, 50 mg/kg over 4 h, 100 mg/kg over 16 h). There is no need empirically to commence NAC therapy in patients presenting within 8 h of acute ingestion. The incidence of any hepatotoxicity following institution of therapy within 8 h of ingestion is very low (1–6%) and independent of the route of dosing (IV versus oral). The incidence of hepatotoxicity increases to 40% if NAC is delayed from 10 to 16 h following ingestion and may be as high as 87% if delayed from 16 to 24 h in patients treated with the 20 h intravenous protocol. However, NAC probably limits the degree of hepatic damage even in late presenting patients. In addition, the dose of NAC may need to be increased or prolonged beyond the standard 20 h regimen in cases of massive ingestion of immediate-release or extended-release paracetamol (>50 g) or where serum concentration of paracetamol remains persistently elevated. Clinical toxicologist advice is recommended in these settings.
Adverse reactions to intravenous NAC are either anaphylactoid, allergy-like, phenomena (urticaria, bronchospasm, hypotension) usually occurring during or soon after the administration of the intravenous loading dose or gastrointestinal reactions (nausea, vomiting) related to sulphydryl groups on the molecule. Adverse reactions may also be seen following administration of oral NAC. Anaphylactoid reactions are not IgE-mediated but related to direct histamine release from mast cells. They are dose dependent in nature and usually respond to slowing or cessation of the infusion for a short period. Occasionally, administration of antihistamines and/or adrenaline may be necessary. The incidence of anaphylactoid reactions may be as high as 20%. A prospective study varying the rate of infusion of the NAC loading dose found a small, non-significant difference in the incidence of anaphylactoid reactions between the standard 15 min loading-dose rate to a 1 h loading-dose rate (18 vs 14%, respectively). Many local dosing guidelines now recommend a 1-h infusion rate for the NAC loading dose. The history of a previous adverse reaction to NAC does not preclude its use in the event of subsequent presentations for paracetamol poisoning. Life-threatening reactions are rare but have uncommonly been reported in patients with pre-existing asthma.
Treatment
Management of paracetamol poisoning is tailored according to the specific clinical scenario.
Acute overdose presenting within 8 h of ingestion
GI decontamination with activated charcoal (AC) should be considered in cooperative patients presenting within 1–2 h of ingestion. Early administration of AC may reduce the risk of reaching a toxic 4-h paracetamol concentration and the need for subsequent antidotal therapy. Administration of AC more than 2 h post-ingestion is unlikely to affect serum paracetamol concentrations. Antidotal therapy with NAC is commenced if the serum paracetamol concentration falls above the paracetamol-nomogram line. Clinically well patients treated within 8 h of ingestion do not require blood tests at the end of their 20-h NAC infusion if they remain well (no nausea, vomiting, anorexia, abdominal pain or tenderness) and do not fall into one of the at risk groups that may require prolonged NAC therapy.
Pregnant patients are treated in a similar fashion to other patients. Paracetamol crosses the placenta and in overdose may result in an increased risk of spontaneous abortion. Cord blood samples taken from newborns of mothers being treated with NAC for paracetamol poisoning have shown that therapeutic serum NAC concentrations occur in the fetal circulation. There are reports of neonates being overdosed with paracetamol (after birth) being successfully treated with IV NAC.
Acute overdose presenting 8–24 h post-ingestion
In view of the increased incidence of hepatotoxicity with delayed antidote administration, NAC therapy should be commenced on presentation. Blood is then taken for serum paracetamol concentration and liver function tests (LFTs). Antidotal treatment may be ceased if the paracetamol level is non-toxic and liver function normal. Otherwise, a full 20-h course of NAC is administered. Prolonged NAC infusion, usually at the 16-h bag (100 mg/kg), is indicated if repeat LFTs indicate rising aminotransferases prior to the end of 20-h course.
Acute overdose presenting more than 24 h post-ingestion
Patients presenting more than 24 h following paracetamol ingestion may still benefit from antidotal therapy with NAC. Therapy should be commenced if the patient has a detectable serum paracetamol level, there is evidence of aminotransferase elevation suggesting paracetamol hepatic injury or there is clinical evidence of paracetamol hepatotoxicity (nausea, vomiting, right upper quadrant pain). Patients may benefit from prolonged duration of NAC therapy if serum aminotransferases and/or prothrombin-time/INR continue to rise after 24 h of therapy. NAC should be continued at a rate of 100 mg/kg/12 h until prothrombin-time/INR and liver function begins to normalize or the patient requires liver transplantation.
Acute overdose with unknown time of ingestion
The time of ingestion of a single overdose of paracetamol may be unknown. This may especially occur in patients with altered mental status from co-ingestants or other causes. In view of the relative safety of NAC as an antidote, it should be commenced empirically in these patients to avoid delayed therapy using the standard 20-h NAC protocol. Serum paracetamol, LFTs and prothrombin-time/INR are collected. A more accurate history of overdose may be elicited when the patient is awake. Treatment may be ceased if accurate history is subsequently elicited or if aminotransferase enzymes are normal at the end of 20 h of NAC therapy.
The staggered acute overdose
In patients presenting with a history of more than one paracetamol overdose over several hours, a worse case scenario can be adopted. An assumption is made that the total dose of paracetamol has been ingested as a single-dose at the earliest possible time and is greater than 200 mg/kg. The serum paracetamol concentration is plotted on the nomogram based on this time point. Treatment is initiated if it is above the nomogram line.
Repeated supratherapeutic ingestion
Current consensus guidelines suggest that in adults and children over 6 years of age with normal liver function, the risk of hepatic injury may be increased if more than 200 mg/kg or 10 g (whichever is the least) are ingested over 24 h, or more than 150 mg/kg or 6 g are ingested per 24 h for the preceding 48 h or, in patients with underlying liver impairment, more than 100 mg/kg or 4 g a day is ingested per 24 h (Table 29.7.1). In these groups, a biochemical risk assessment should be made. If serum paracetamol is less than 70 μmol/L (10 mg/L) and serum aminotransferases are less than 50 IU/L, no treatment is required. If either assay is elevated, NAC should be commenced and LFTs reassessed 8 h later. If these are not rising and the patient is well, NAC therapy may be ceased. Otherwise, the full 20-h course should be administered or continued further until aminotransferases begin to fall. Static aminotransferases suggest an alternative cause for hepatic pathology. The reason for analgesic overuse should also be sought.
Table 29.7.1
Paracetamol dosing associated with hepatic injury in adults and children over 6 years of age

Adapted from Dart RC, et al. Acetaminophen poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol 2006;44:1–18.
Paracetamol-induced hepatic failure
The development of hepatic failure is uncommon following paracetamol poisoning. The risk is greater in late presenting patients. Patients with evidence of developing fulminant hepatic failure following paracetamol poisoning exhibit clinical signs of encephalopathy and liver failure. Poorer prognosis is associated with an INR more than 2 at 24 h or more than 3 at 48 h, INR increasing between days 3 and 4, serum creatinine greater than 200 μmol/L, pH<7.3 despite fluid resuscitation, or rising serum lactate. Patients may benefit from prolonged NAC therapy along with supportive care in a specialized liver unit. A lower mortality is reported in patients with hepatic failure treated with NAC. Early consultation with a liver transplantation unit should be sought.
Sustained-release paracetamol ingestion
Delayed peak serum paracetamol concentration and delayed crossing of the nomogram line is possible following overdose with extended-release paracetamol (Panadol Osteo). The administration of NAC more than 20 h post-ingestion may be of benefit in view of the sustained-release nature of this product. To avoid delays in N-acetylcysteine treatment, this should be commenced if the reported ingested dose is greater than 200 mg/kg or 10 g (whichever is the least). Serum paracetamol concentration should be estimated 4 or more hours post-ingestion and a second estimation collected 4 h after the first. If both concentrations fall below the nomogram line, treatment may be discontinued, otherwise NAC should be administered for the full 20-h dose. Large ingestions (>30 g) of this formulation have the potential to result in prolonged paracetamol absorption. Serial paracetamol estimations are useful in this setting to ascertain when the concentration is falling to an insignificant level. If paracetamol concentration is detectable prior to completion of the 20-h infusion or if LFTs suggest developing hepatotoxicity, treatment with NAC should continue with the 100 mg/kg/16-h infusion.
Massive ingestions of paracetamol
This may be suspected in two situations: (1) in patients with history of large ingestions of paracetamol (>500 mg/kg or>50 g); (2) where serum paracetamol concentration is very high and suggests a massive ingestion (>3000 μmol/L or 450 mg/L). In these cases, the standard N-acetylcysteine dosing regimen may not be adequate to prevent hepatotoxicity developing. Clinical toxicologist advice is recommended. Consideration should be given to increasing the dose of NAC, particularly in the 16-h maintenance infusion. An empiric doubling of the 16-h infusion dose to 200 mg/kg/16 h has been suggested in these cases as it provides more NAC to mitigate hepatotoxicity. Serum paracetamol concentrations should be followed serially. If these remain detectable prior to cessation of the 20-h regimen, NAC treatment should be continued beyond this period, repeating the 16-h infusion dose, until paracetamol concentration is undetectable and there is no evidence of hepatotoxicity.
Controversies
![]() The optimal duration of and dose regimen for N-acetylcysteine treatment in atypical paracetamol poisoning presentations, such as supratherapeutic ingestions, massive overdoses and extended-release paracetamol poisoning.
The optimal duration of and dose regimen for N-acetylcysteine treatment in atypical paracetamol poisoning presentations, such as supratherapeutic ingestions, massive overdoses and extended-release paracetamol poisoning.
![]() The clinical significance of suggested ‘risk factors’ for the development of hepatotoxicity. Most of the suggested factors are theoretical and have never been validated. Current treatment guidelines should be able to be applied without taking these factors into account.
The clinical significance of suggested ‘risk factors’ for the development of hepatotoxicity. Most of the suggested factors are theoretical and have never been validated. Current treatment guidelines should be able to be applied without taking these factors into account.
![]() Variations in international recommendations on the threshold for treatment of acute paracetamol poisoning utilizing different nomogram cut-offs for toxicity.
Variations in international recommendations on the threshold for treatment of acute paracetamol poisoning utilizing different nomogram cut-offs for toxicity.
Further reading
1. Buckley NA, Whyte IM, O’Connell DL, Dawson AH. Activated charcoal reduces the need for N-acetylcysteine treatment after acetaminophen (paracetamol) overdose. J Toxicol Clin Toxicol. 1999;37:753–757.
2. Chiew A, Day P, Salonikas C, et al. The comparative pharmacokinetics of modified-release and immediate-release paracetamol in a simulated overdose model. Emerg Med Australas. 2010;22:548–555.
3. Daly FFS, Fountain J, Graudins A, et al. Consensus statement: new guidelines for the management of paracetamol (Acetaminophen) poisoning in Australia and New Zealand–explanation and elaboration. Med J Aust. 2008;188:296–301.
4. Flanagan RJ, Mant TG. Coma and metabolic acidosis early in severe acute paracetamol poisoning. Hum Toxicol. 1986;5:179–182.
5. Graudins A, Chiew A, Chan B. Overdose with modified-release paracetamol results in delayed and prolonged absorption of paracetamol. Intern Med J. 2010;40:72–76.
6. Green TJ, Sivilotti MLA, Langmann C, et al. When do the aminotransferases rise after acute acetaminophen overdose? Clin Toxicol (Phila). 2010;48:787–792.
7. Keays R, Harrison PM, Wendon JA, et al. Intravenous acetylcysteine in paracetamol induced fulminant hepatic failure: a prospective controlled trial. Br Med J. 1991;303:1026–1029.
8. Kerr F, Dawson AH, Whyte IM, et al. The Australasian clinical toxicology investigators collaboration randomized trial of different loading infusion rates of N-acetylcysteine. Ann Emerg Med. 2005;45:402–408.
9. Prescott LF, Illingworth RN, Critchley JA, et al. Intravenous N-acetylcysteine: the treatment of choice for paracetamol poisoning. Br Med J. 1979;2:1097–1100.
10. Sivilotti MLA, Green TJ, Langmann C, et al. Multiplying the serum aminotransferase by the acetaminophen concentration to predict toxicity following overdose. Clin Toxicol (Phila). 2010;48:793–799.
11. Waring WS, Stephen AF, Robinson OD, et al. Lower incidence of anaphylactoid reactions to N-acetylcysteine in patients with high acetaminophen concentrations after overdose. Clin Toxicol (Phila). 2008;46:496–500.
12. Whyte IM, Buckley NA, Reith DM, et al. Acetaminophen causes an increased international normalized ratio by reducing functional factor VII. Ther Drug Monitor. 2000;22:742–748.
29.8 Salicylate
Digby Green and Andis Graudins
Essentials
1 Salicylate pharmacokinetics become complex and alter markedly following overdose.
2 Therapeutic serum salicylate concentrations range from 1.1 to 2.2 mmol/L (15–30 mg/dL).
3 Treatment and disposition decisions cannot be made on the basis of a single serum salicylate concentration.
4 The Done nomogram is unreliable and should not be used in the management of salicylate poisoning.
5 Urinary alkalinization is an effective method of enhanced elimination of salicylate. Haemodialysis is rarely indicated.
6 Chronic salicylate poisoning is an insidious condition, mostly seen in the elderly, manifested by an unexplained metabolic acidosis that may be incorrectly attributed to another medical condition.
Introduction
Salicylate is a plant-derived compound, now pharmaceutically manufactured and widely used in a variety of pharmaceutical preparations and over-the-counter herbal products, cough and cold remedies, ointments and topical rubefacients. Despite this, salicylate poisoning is an infrequent presentation to Australian emergency departments, due largely to the preference of paracetamol as the over-the-counter analgesic of choice. Acute deliberate self-poisoning with salicylate results in a well-recognized constellation of symptoms and signs that are dose related. Emergency physicians have a number of treatment modalities available to manage this condition. Chronic intoxication, in contrast, occurs more commonly in the elderly with multiple co-morbidities and is more likely to require haemodialysis. Chronic intoxication is associated with significant morbidity and mortality. Children rarely ingest sufficient amounts to cause toxicity, but ingestion of small amounts (>5 mL) of topical agents containing methyl salicylate can result in severe toxicity in children under 5 years of age. Attention should be given to the quoted units of measurement, standard or SI, to avoid incorrect interpretation of serum drug concentration.
Pharmacology and pathophysiology
Aspirin (acetylsalicylic acid, ASA) is rapidly absorbed in the acid medium of the upper gastrointestinal (GI) tract and undergoes rapid hydrolysis to form salicylic acid. Peak serum salicylate concentrations are reached within 2 h of therapeutic dosing. Absorption is erratic and delayed following overdose partly due to pylorospasm and pharmacobezoar formation. Overdose with sustained-release or enteric-coated preparations may delay peak serum concentrations for up to 24 h.
Salicylic acid has a pKa of 3.0 and exists predominantly in the unionized form at a pH of 7.4. It is highly protein bound (85–90%) following therapeutic doses with a very small apparent volume of distribution (0.1–0.2 L/kg). Plasma protein binding becomes saturated and free salicylate concentration rises in overdose. As pH falls, a greater proportion of salicylate exists in the unionized form and movement into the extravascular compartments, including the central nervous system (CNS), is enhanced with resulting increases in the volume of distribution and tissue toxicity.
Salicylic acid is metabolized in the liver and kidney to form salicyluric acid, glycine, glucuronic, acyl and salicyl phenolic conjugates. These conjugates are excreted renally along with small amounts of free salicylate. The elimination half-life following therapeutic dosing is around 4 h. Salicylate metabolism is saturated when plasma salicylate concentration rises above the therapeutic range. Elimination kinetics change from first-order to zero-order, dramatically increasing the elimination half-life. Urinary excretion of unchanged salicylate is minimal when the urine pH is acidic. As urine pH increases, a greater proportion of filtered salicylate is in an ionized state and is unavailable for reabsorption in the proximal convoluted tubule. An increase in urine pH from 5.0 to 8.0 results in up to 1000-fold increase in ionized salicylate excretion.
At therapeutic doses, salicylate acts as an analgesic, antipyretic, antiplatelet and anti-inflammatory agent primarily by way of its inhibitory effects on prostaglandin synthesis mediated by irreversible inhibition of cyclo-oxygenase enzymes one and two (COX-1 and COX-2). Overdose results in toxic effects on the CNS, acid–base balance, cellular metabolism, coagulation, lungs and the GI tract. CNS effects include an initial direct stimulation of the medullary respiratory centre producing an increase in rate and depth of respiration and a corresponding primary respiratory alkalosis, tinnitus, deafness and confusion. In severe poisoning, where systemic acidaemia enhances cerebral penetration of unionized salicylate, coma, convulsions and cerebral oedema occur.
Metabolic effects include direct uncoupling of oxidative phosphorylation and inhibition of Krebs cycle enzymes leading to systemic acidaemia, hyperglycaemia, hyperthermia, derangement of carbohydrate, amino acid and lipid metabolism. Increased oxygen consumption and carbon dioxide production are also apparent. Dehydration results from increased insensible respiratory and cutaneous fluid losses, as well as from nausea and vomiting from GI irritation. Inhibition of platelet aggregation as well as vitamin-K-sensitive clotting factor function may produce a mild coagulopathy. Haemorrhage rarely occurs in humans or animals following severe salicylate poisoning. Salicylate-induced non-cardiogenic pulmonary oedema is also reported in association with severe poisoning.
Clinical features
The degree of toxicity following acute ingestion of salicylate is dose related (Table 29.8.1). The most useful features in risk assessment are the clinical signs and symptoms, the acid–base status and serum salicylate concentration and the reported dose ingested.
Table 29.8.1
Dose-related effect of aspirin toxicity

The diagnosis of chronic salicylate poisoning is often missed. Recurrent dosing with aspirin, usually in the context of a viral illness or chronic pain, results in accumulation of plasma salicylate and prolongation of the elimination half-life. Patients may present with non-specific symptoms or signs suggesting inflammatory or infective aetiology, such as confusion, delirium, fever, dehydration or hyperglycaemia. The history of excessive salicylate ingestion may not be elicited and the clinical findings erroneously attributed to other conditions, such as septicaemia, cardiogenic pulmonary oedema, cerebrovascular accidents or diabetic ketoacidosis. The presence of an unexplained metabolic acidosis may be the vital clue leading to the diagnosis. Delay in the diagnosis of chronic salicylate poisoning is associated with an increased morbidity and mortality.
Clinical investigations
Salicylate intoxication should be suspected in any patient with clinical signs suggestive of poisoning, an unexplained respiratory alkalaemia or metabolic acidosis. Patients in whom the diagnosis is suspected should have blood drawn for serum electrolytes, urea, creatinine, blood glucose, prothrombin time and paracetamol and salicylate concentration. An arterial or venous blood gas is necessary to assess acid–base status and urine pH should be checked.
Patients with mild or early poisoning may present with a pure respiratory alkalosis due to respiratory centre stimulation and hypokalaemia. Urine pH may initially be alkaline as a response to hyperventilation. Adult patients with moderate-to-severe poisoning may present with a mixed acid–base disturbance of respiratory alkalosis and metabolic acidosis. Urine pH is commonly acidic in this setting due to increased excretion of hydrogen ions. A metabolic acidosis with normal or falling serum pH signifies development of potentially severe salicylate poisoning. Co-ingestion of sedatives may depress respiratory drive leading to loss of respiratory compensation for the metabolic acidosis and an earlier deterioration in acid–base status.
The Done nomogram was developed in 1960 to relate peak serum salicylate concentration to clinical severity of salicylate poisoning. This nomogram cannot be relied upon in the risk assessment of salicylate toxicity. The combined use of serial clinical observation, blood-gas estimations and serial salicylate measurements to monitor for ongoing absorption of aspirin will give the best indication of the degree of toxicity and response to treatment.
Treatment
Patients presenting following salicylate ingestion should have intravenous access established, blood drawn for serum salicylate estimation, electrolytes and blood sugar level. In moderate-to-severe poisoning these should be repeated every 3–4 h in view of the potential erratic salicylate absorption. Intravenous rehydration is often necessary in view of the increased insensible fluid losses due to hyperventilation and pyrexia and vomiting from GI irritation. Attention to fluid balance is particularly important in the very young, elderly or those with cardiac disease. Central venous and arterial pressure monitoring may be necessary in severe cases as well as urinary catheterization and hourly urine measurement.
Gastrointestinal decontamination with activated charcoal should be performed on presentation and in patients who present several hours following ingestion in view of the potential for delayed aspirin absorption. Whole-bowel irrigation with polyethylene glycol-electrolyte solution may be considered in patients with ingestion of large amounts of sustained-release formulation aspirin. Repeat-dose activated charcoal may be of benefit where there is ongoing absorption of salicylate evidenced by rising serial serum levels. Multiple-dose activated charcoal does not enhance salicylate elimination but may inhibit ongoing GI absorption from pharmacobezoars or aspirin concretions.
Pulmonary oedema should be treated with continuous positive pressure ventilation by mask or endotracheal intubation. Salicylate poisoning results in high minute volumes and respiratory alkalosis. Ventilation strategies are aimed at providing appropriate positive ventilation and preventing respiratory acidaemia. Seizures should be treated with parenteral benzodiazepines.
Urinary salicylate excretion can be enhanced by urinary alkalinization which may reduce salicylate elimination half-life from 20 to 5 h. The aim of urinary alkalinization is to increase urine pH above 7.5 to enhance the trapping of ionized salicylate in the urine. Indications include the presence of symptoms, acid–base abnormalities or serum salicylate levels greater than 2.2 mmol/L (30 mg/dL). Patients with clinical symptoms and signs of salicylate toxicity should have urinary alkalinization commenced while awaiting the results of drug assays and electrolyte concentrations. Urinary alkalinization is accomplished by an initial loading dose of intravenous sodium bicarbonate (0.5–1.0 mmol/kg) followed by an infusion of 100–150 mmol of sodium bicarbonate in 1 L of 5% dextrose solution at a rate of 100–250 mL/h adjusted to urine pH. Urine output should be maintained between 1 and 2 mL/kg/h. Serum potassium should be maintained within normal limits by the addition of supplemental potassium to the bicarbonate infusion (40 mmol per bag). In the presence of systemic hypokalaemia, the potassium ions are retained in the renal tubules in preference to hydrogen. This makes it extremely difficult to achieve urinary alkalinization. Serial serum electrolytes, salicylate concentrations and urinary pH should be measured every 3–4 h. The endpoint for therapy is a serum salicylate concentration with in the therapeutic range (1.1–2.2 mmol/L or 15–30 mg/dL), resolution of clinical signs of toxicity and normalization of acid–base status.
Extracorporeal removal of salicylate is infrequently required and the accepted clinical indications are listed in Table 29.8.2. Intermittent high-flow haemodialysis (HD) is the preferred option as it can rapidly normalize acid–base, fluid balance and electrolyte abnormalities as well as remove salicylate from the blood.
Table 29.8.2
Indications for haemodialysis in salicylate poisoning
Metabolic acidosis refractory to optimal supportive care and urinary alkalinization
Evidence of end-organ injury (i.e. cerebral oedema, seizures, rhabdomyolysis, pulmonary oedema)
Renal failure and fluid overload
Serum aspirin concentration>6.0 mmol/L or 100 mg/dL in acute poisoning
Serum aspirin concentration>4.0 mmol/L or 60 mg/dL in chronic poisoning
Note: in elderly patients, with chronic salicylate toxicity, the suggested serum threshold for haemodialysis is lower at 2.2–4.4 mmol/L (30–60 mg/dL).
Severe cases of salicylate intoxication may require endotracheal intubation and ventilation as a direct result of toxicity or co-ingestants. Importantly, minute volume should be large to maintain the hyperventilation. However, in such circumstances, urinary alkalinization, followed by haemodialysis is essential to prevent a worsening metabolic acidosis as ventilatory manipulation is often insufficient to maintain alkalaemia.
Disposition
In view of the potential for delayed and erratic salicylate absorption, patients require serial salicylate concentrations and observation for a minimum of 12 h. Salicylate estimations earlier than 6 h post-ingestion do not usually reflect peak serum concentrations following overdose.
Patients without clinical evidence of salicylate toxicity may be medically cleared in the presence of normal arterial or venous blood gas and two falling serum salicylate levels in the therapeutic range (1.1–2.2 mmol/L; 15–30 mg/dL) 3–4 h apart. Patients with evidence of acid–base abnormalities, end-organ dysfunction or requiring urinary alkalinization should be admitted to a high-dependency or intensive care unit. Transfer to a tertiary referral centre with facilities for haemodialysis should be considered if criteria for severe toxicity are present.
Controversies
![]() The threshold for initiating urinary alkalinization is not well defined. Many clinicians now prefer to alkalinize any symptomatic patient with a view to minimizing the duration of medical admission.
The threshold for initiating urinary alkalinization is not well defined. Many clinicians now prefer to alkalinize any symptomatic patient with a view to minimizing the duration of medical admission.
![]() Although there is minimal case-controlled evidence supporting the use of continuous arteriovenous or venovenous haemodialysis in severe salicylate poisoning, newer high-flow continuous venovenous haemodialysis units may be able to remove significant amounts of salicylate and provide an alternative to intermittent high-flow dialysis in selected cases.
Although there is minimal case-controlled evidence supporting the use of continuous arteriovenous or venovenous haemodialysis in severe salicylate poisoning, newer high-flow continuous venovenous haemodialysis units may be able to remove significant amounts of salicylate and provide an alternative to intermittent high-flow dialysis in selected cases.
Further reading
1. Chalasani N, Roman J, Jurado RL. Systemic inflammatory response syndrome caused by chronic salicylate intoxication. South Med J. 1996;89:479–482.
2. Dargan PI, Wallace CI, Jones AL. An evidence based flowchart to guide the management of acute salicylate (aspirin) overdose. Emerg Med J. 2002;19:206–209.
3. Dugandzic RM, Tierney MG, Dickinson GE, et al. Evaluation of the validity of the Done nomogram in the management of acute salicylate intoxication. Ann Emerg Med. 1989;18:1186–1190.
4. Herres J, Ryan D, Salzman M. Delayed salicylate toxicity with undetectable initial levels after large-dose aspirin ingestion. Am J Emerg Med. 2009;27(1173):e1–e3.
5. Jacobsen D, Wiik-Larsen E, Bredesen JE. Haemodialysis or haemoperfusion in severe salicylate poisoning? Hum Toxicol. 1998;7:161–163.
6. Mayer AL, Sitar DS, Tenenbein M. Multiple-dose charcoal and whole-bowel irrigation do not increase clearance of absorbed salicylate. Arch Intern Med. 1992;152:393–396.
7. Notarianni L. A reassessment of the treatment of salicylate poisoning. Drug Safe. 1992;7:292–303.
8. O’Malley GF. Emergency department management of the salicylate-poisoned patient. Emerg Med Clin N Am. 2007;25:333–346.
9. Pearlman BL, Gambhir R. Salicylate intoxication; a clinical review. Postgrad Med. 2009;121:162–168.
10. Thisted B, Krantz T, Strom J, et al. Acute salicylate self-poisoning in 177 consecutive patients treated in ICU. Acta Anaesthesiol Scand. 1987;31:312–316.
11. Wortzman DJ, Grunfeld A. Delayed absorption following enteric-coated aspirin overdose. Ann Emerg Med. 1987;16:434–436.
12. Wrathall G, Sinclair R, Moore A, Pogson D. Three case reports of the use of haemodiafiltration in the treatment of salicylate overdose. Hum Exp Toxicol. 2001;20:491–495.
29.9 Antidiabetic drugs
Jason Armstrong
Essentials
1 Deliberate self-poisoning with insulin or sulphonylureas may lead to life-threatening hypoglycaemia requiring prolonged observation and treatment over several days.
2 Octreotide blocks endogenous insulin secretion and is indicated in the management of symptomatic sulphonylurea toxicity.
3 Central venous access is often required following deliberate insulin overdose to facilitate treatment with concentrated glucose solutions.
4 Metformin is associated with life-threatening lactic acidosis. It does not cause significant hypoglycaemia in overdose.
Introduction
Diabetes mellitus (DM) is a chronic metabolic condition caused by an absolute (type I) or relative (type II) lack of insulin. In Australia, over one million people have diabetes and 100 000 people are diagnosed every year with the condition. Aboriginal and Maori populations in Australasia have some of the highest rates of type II diabetes in the world [1]. For these reasons, antidiabetic medications are readily available and frequently taken in overdose by both diabetic and non-diabetic individuals.
The three major groups of antidiabetic medications are insulin, sulphonylureas and biguanides, all of which have been used for over 50 years. Toxicity can result from intentional overdose, but also from decreased clearance of the medication at therapeutic dosing, due to underlying hepatic or renal disease.
A number of new agents for type II DM have been developed recently, including dipeptidyl peptidase-IV (DPP-IV) inhibitors, incretin mimetics, thiazolidinediones, alpha-glucosidase inhibitors and glinides. Overdose with these medications is less likely to cause significant clinical effects.
Insulin
Pharmacology and pathophysiology
Insulin is synthesized by the pancreatic β islet cells as a pro-hormone packaged inside secretory vesicles. It is secreted primarily in response to elevated serum glucose levels and becomes metabolically active when pro-insulin is cleaved by serum proteases to form insulin and C-peptide. Exogenous insulin, administered therapeutically in the management of type I and II DM, does not contain C-peptide.
Insulin is eliminated by hepatic metabolism (60%) and renal clearance (40%). A number of preparations are available and these have varying durations of action. However, following overdose, the usual pharmacokinetic properties of insulin may be altered because the injected dose forms a subcutaneous or intramuscular depot. Slow and erratic release of insulin from the depot can result in a markedly extended duration of action (up to several days) even with short-acting preparations [2].
Insulin promotes the intracellular movement of glucose, potassium, magnesium and phosphate, as well as decreasing ketone production from the breakdown of fatty acids. It inhibits the breakdown of fat and protein to release glucose (gluconeogenesis) and stimulates the synthesis of glycogen, protein and triglycerides.
In overdose, the principal effect of clinical significance is hypoglycaemia, which may be prolonged and profound following self-administration of large doses subcutaneously or intramuscularly. Hypoglycaemia tends to be more profound and prolonged in non-diabetic patients [3]. Insulin toxicity also causes electrolyte abnormalities, the most important of which is hypokalaemia, secondary to intracellular shift of potassium. Hypophosphataemia and hypomagnesaemia are also reported.
Clinical features
The clinical features of insulin toxicity are the neuropsychiatric and autonomic manifestations of hypoglycaemia. Autonomic symptoms and signs include diaphoresis, tremor, nausea, palpitations and tachycardia; neuropsychiatric features are confusion, agitation, seizures, coma and focal neurological deficits. These manifestations are usually evident within hours of self-administration of an insulin overdose and the patient frequently presents in coma. The suspicion of deliberate overdose is entertained when recurrent profound hypoglycaemia occurs following an initial response to dextrose administration. If the history of deliberate overdose is known at the time of presentation, then profound prolonged hypoglycaemia should be anticipated.
Prolonged severe hypoglycaemia can cause permanent neurological sequelae or death.
Clinical investigations
Serial measurement of blood glucose concentrations, usually at the bedside, allows titration of dextrose administration. Serial measurements of electrolytes are necessary to monitor hypokalaemia and potassium replacement. Serum magnesium and phosphate levels may also be affected.
If surreptitious or malicious administration is suspected, assays of insulin and C-peptide levels can be useful to provide objective evidence of the presence of exogenous insulin, as endogenous insulin levels should always be suppressed in the presence of hypoglycaemia unless an insulinoma is present.
Treatment
Management of insulin overdose is essentially supportive and requires administration of sufficient concentrated dextrose solution to maintain euglycaemia until all the insulin is absorbed from the depot site and its hypoglycaemic action terminated. After initial correction of hypoglycaemia with 50% dextrose, a 10% dextrose infusion should be commenced at 100 mL/h and blood sugar levels followed closely. Further boluses of dextrose and titration of the infusion rate are implemented as necessary. Very large dose of dextrose may be required, sometimes over days [2,3]. Frequently, it is necessary to administer a 50% dextrose infusion to maintain euglycaemia and this usually requires placement of a central venous line because concentrated dextrose solutions can cause sclerosing thrombophlebitis in peripheral veins.
Hypokalaemia due to intracellular shifts should be anticipated and supplemental K+ administered (e.g. 10–40 mmol/h IV in adults), guided by serial monitoring. Excessive K+ administration should be avoided. Hyponatraemia and volume overload are other complications of hypertonic dextrose therapy.
Disposition
Patients who report an overdose of insulin should be admitted and observed with bedside blood glucose assays for at least 8 h after self-administration. They are medically fit for discharge if they remain asymptomatic and euglycaemic at this stage. Those who develop hypoglycaemia requiring dextrose therapy should be admitted to a high-dependency or intensive care unit for ongoing dextrose infusion, potassium supplementation and close monitoring of blood sugar and electrolytes.
The duration of therapy required is variable. Dextrose therapy may be withdrawn by halving the rate of infusion every 2–4 h once hyperglycaemia develops, guided by regular bedside assessments of serum glucose. This minimizes the risk of precipitous hypoglycaemia. It may be particularly difficult to wean dextrose infusions in non-diabetic patients, as the large load of infused dextrose tends to stimulate endogenous insulin secretion after the effects of the initial overdose have worn off. In these cases, a slower weaning regimen may be required to prevent hypoglycaemia. It is sensible to avoid withdrawing dextrose infusions overnight when clinical features of hypoglycaemia are less easily recognized.
Patients are medically fit for discharge if they remain asymptomatic and euglycaemic 6 h after dextrose therapy is ceased. All intentional overdoses require psychiatric assessment once their medical condition has stabilized.
Sulphonylureas
Pharmacology and pathophysiology
Sulphonylureas are the most commonly prescribed oral hypoglycaemics in Australasia. Currently available agents include glibenclamide, gliclazide, glimepiride and glipizide. These agents bind to and block outgoing K+ channels on the pancreatic β cells leading to depolarization of the cell membrane, which opens voltage-gated Ca2+ channels and causes insulin release secondary to Ca2+ influx [4]. The result is a hyperinsulinaemic state. Although in therapeutic dose the duration of action is usually from 12 to 24 h, this can be markedly prolonged following overdose. Sulphonylureas undergo hepatic metabolism and have a combination of active and inactive metabolites, which are excreted renally. An exaggerated therapeutic effect can therefore occur when these agents accumulate in patients with coexistent hepatic or renal disease.
Clinical features
Sulphonylurea-induced hypoglycaemia may occur as a complication of therapy, inadvertent administration to a non-diabetic patient or as a consequence of deliberate self-poisoning. The hypoglycaemia after intentional ingestion is likely to be particularly profound and prolonged.
Treatment
Hypoglycaemia should be corrected immediately once identified with bedside blood glucose testing or suspected on clinical grounds. An initial bolus of 50 mL 50% dextrose followed by an infusion of 10% dextrose at 100 mL/h is appropriate for hypoglycaemia secondary to deliberate self-poisoning with sulphonylureas. However, hypoglycaemia is frequently refractory to dextrose supplementation in this setting and early use of octreotide is then indicated to maintain euglycaemia (see below).
Activated charcoal can be administered to patients who present within a few hours of intentional ingestion of sulphonylureas (especially with extended-release preparations) but does not take precedence over resuscitation and correction of hypoglycaemia.
Elderly patients with sulphonylurea-induced hypoglycaemia often have intercurrent medical illnesses that require treatment. Euglycaemia may be relatively easy to maintain with intravenous or oral dextrose supplementation, but octreotide may also have a role in therapeutic management (see below).
Octreotide
Octreotide is a synthetic octapeptide analogue of the naturally occurring foregut hormone somatostatin. It suppresses insulin release from pancreatic β cells by binding to Ca2+ channels on the cell membrane, inhibiting Ca2+ influx and subsequent insulin release.
Octreotide is now seen as first-line antidotal therapy in patients with hypoglycaemia secondary to sulphonylurea overdose. Early administration of octreotide may greatly reduce or abolish the dextrose requirement, obviate the need for central venous access and greatly simplify subsequent management and disposition. Therapy can be initiated with an initial bolus of 50 μg IV followed by an infusion of 25 μg/h. An alternative dosing regimen is 100 μg by intramuscular or subcutaneous injection every 6 h. Once initiated, octreotide therapy should be continued for at least 24 h before withdrawal is attempted. Octreotide is well tolerated, with nausea and vomiting only occasionally reported [4,5].
For the treatment of hypoglycaemia due to therapeutic accumulation of sulphonylurea agents, a single dose of octreotide 25–50 μg subcutaneously may be adequate to prevent recurrent hypoglycaemia during in-patient management.
Disposition
Patients with a history of sulphonylurea overdose should be admitted and observed with bedside blood glucose assays for at least 8 h after ingestion or up to 12 h if a slow-release preparation. They are medically fit for discharge if they remain asymptomatic and euglycaemic at this stage.
Patients who require treatment for hypoglycaemia need admission, usually for several days. Discharge can occur once they are tolerating a normal diet and their blood glucose level remains normal 6 h after cessation of glucose and/or octreotide therapy.
Patients who develop hypoglycaemia on therapeutic doses of sulphonylureas should be admitted for at least 24 h to monitor serum glucose and to review their medication regimen.
In all cases of sulphonylurea overdose or toxicity, discharge should not occur at night, due to the increased risk of occult hypoglycaemia.
Metformin
Pharmacology and pathophysiology
Metformin is the only biguanide currently available in Australasia. It is rapidly absorbed from the gastrointestinal tract, minimally metabolized and excreted almost entirely by the kidneys. The major antidiabetic effect is to inhibit gluconeogenesis, as well as to increase tissue sensitivity to insulin, thereby improving HbA1c control. It does not cause hypoglycaemia at therapeutic doses and, even following massive overdose, clinically significant hypoglycaemia is rarely observed. However, metformin is associated with life-threatening lactic acidosis because it blocks intracellular oxidative pathways leading to increased anaerobic metabolism. Metformin-associated lactic acidosis can occur during therapeutic dosing when impaired renal function leads to drug accumulation or when intercurrent illness leads to tissue hypoperfusion. It is also reported after massive overdose, even in non-diabetic patients. The threshold for this effect is not well established but is probably over 10 g [6]. The risk of toxicity from an acute ingestion will be exacerbated by other agents that cause hypotension or decreased renal perfusion.
Clinical features
The majority of metformin overdoses are associated with only minor symptoms. In particular, hypoglycaemia is not a feature of the presentation. Paediatric ingestions are unlikely to cause significant toxicity, provided that co-ingestion of sulphonylureas can confidently be excluded [7]. Where the clinical course is complicated by lactic acidosis, the insidious onset of non-specific symptoms, such as nausea, malaise and lethargy, may be observed. As lactate levels rise, the patient’s condition will deteriorate with progressive tachypnoea, cardiovascular instability and altered mental state [8]. Patients who develop progressive lactic acidosis while on therapeutic metformin may present severely unwell.
Metformin-associated lactic acidosis carries a significant mortality risk if it is not recognized and treated effectively [9].
Clinical investigation
Urgent electrolytes, renal function and lactate levels are indicated in any patient on metformin therapy who presents unwell or in any patient who becomes symptomatic while being observed following deliberate self-poisoning with metformin.
Treatment
Most cases of metformin overdose can be managed supportively. Maintenance of euvolaemia is imperative and IV crystalloid should be given to ensure effective renal perfusion and clearance. If lactate levels are elevated, serial estimations of pH and lactate must be performed until they return to the normal range. If lactate rises above 10 mmol/L or worsening acidosis, renal dysfunction and clinical deterioration occur, immediate treatment with lactate-free haemodialysis is indicated. This not only corrects the acid–base disturbance, but also rapidly removes metformin from the circulation. Either intermittent or continuous dialysis techniques can be used, as long as flow rates are adequate to ensure effective clearance [8].
Temporary improvement in acidosis can be achieved by infusion of NaHCO3 while organizing dialysis, but this does not address ongoing metformin toxicity and progressive deterioration is likely without definitive therapy.
Disposition
Patients can be discharged following metformin overdose if they remain clinically well with normal haemodynamic parameters and acid–base status. Those who develop significant lactic acidosis require intensive care admission and consideration for haemodialysis. All patients on therapeutic metformin who develop lactic acidosis require admission for close clinical and biochemical monitoring and consideration for haemodialysis.
Other agents
Dipeptidyl peptidase-IV (DPP-IV) inhibitors (‘gliptins’, e.g. sitagliptin) prevent hydrolysis of the endogenous foregut incretin hormones, glucagon-like peptide-1 (GLP-1) and glucose dependent insulinotropic polypeptide (GIP). This results in increased insulin release and a decrease in endogenous glucagon activity. There is a possibility of gliptin-induced hypoglycaemia as a consequence of therapy or overdose [10].
Exanetide, an incretin analogue, is a synthetic, long-acting polypeptide derived from the saliva of the Gila monster (Heloderma suspectum). It is administered by subcutaneous injection and adverse effects include significant vomiting. As with gliptin toxicity, drug-induced hypoglycaemia is likely to be mild, transient and readily responsive to supplemental dextrose [11].
Thiazolidinediones (‘glitazones’, e.g. pioglitazone) are used in type II DM. They improve insulin sensitivity in skeletal muscle and adipose tissue via the receptor peroxisome proliferator-activated receptor gamma (PPAR-γ) and also inhibit hepatic gluconeogenesis. They improve insulin resistance and thereby act to decrease circulating insulin levels. They do not stimulate insulin secretion and are not associated with hypoglycaemia
Alpha-glucosidase inhibitors (acarbose) are oligosaccharide agents that inhibit the activity of enzymes in the GI endoluminal brush border. Their action decreases the breakdown of complex sugars to monosaccharides, thereby decreasing the postprandial rise in blood glucose levels. They are not absorbed to any significant degree and do not cause hypoglycaemia or other systemic effects in overdose.
Glinides (e.g. repaglinide) are not commonly available in Australasia, but are prescribed more frequently in other countries. Their mode of action is to stimulate insulin secretion from the pancreas, but via a different part of the membrane receptor than sulphonylurea agents [12]. There are limited data available on overdose presentations, but there is potential for hypoglycaemia requiring therapy with IV dextrose. Because of the short half-life of glinides in comparison to sulphonylureas, prolonged toxicity is unlikely to result.
Controversies
![]() Glucagon is sometimes given pre-hospital for symptomatic hypoglycaemia. It can raise serum glucose due to enhanced breakdown of hepatic glycogen stores, but this effect is short lived and unreliable. It does not have a role in the in-patient management of deliberate self-poisoning with insulin or sulphonylureas.
Glucagon is sometimes given pre-hospital for symptomatic hypoglycaemia. It can raise serum glucose due to enhanced breakdown of hepatic glycogen stores, but this effect is short lived and unreliable. It does not have a role in the in-patient management of deliberate self-poisoning with insulin or sulphonylureas.
![]() Surgical excision of insulin depot stores has been attempted but is not indicated as medical management is effective in dealing with all clinical manifestations of insulin overdose.
Surgical excision of insulin depot stores has been attempted but is not indicated as medical management is effective in dealing with all clinical manifestations of insulin overdose.
![]() The optimal dose and route of administration of octreotide in sulphonylurea overdose is not well defined. Recommendations are empiric. Greater doses than those quoted above might be necessary following massive overdose of sulphonylureas in non-diabetic patients.
The optimal dose and route of administration of octreotide in sulphonylurea overdose is not well defined. Recommendations are empiric. Greater doses than those quoted above might be necessary following massive overdose of sulphonylureas in non-diabetic patients.
![]() The role of insulin assays in determining ongoing requirement for octreotide therapy in sulphonylurea toxicity could be explored. Currently, insulin levels are not routinely monitored in this setting.
The role of insulin assays in determining ongoing requirement for octreotide therapy in sulphonylurea toxicity could be explored. Currently, insulin levels are not routinely monitored in this setting.
References
1. AusDiab Report. International Diabetes Institute, Melbourne.<http://www.diabetes.com.au>[Accessed Feb. 2008].
2. Samuels MH, Eckel RH. Massive insulin overdose: detailed studies of free insulin levels and glucose requirements. Clin Toxicol. 1989;27:157–168.
3. Haskell RJ, Stapczynski JS. Duration of hypoglycaemia and need for intravenous glucose following intentional overdoses of insulin. Ann Emerg Med. 1984;13:505–511.
4. McLaughlin SA, Crandall CS, McKinney PE. Octreotide: an antidote for sulphonylurea induced hypoglycaemia. Ann Emerg Med. 2000;36:133–138.
5. Glatstein M, Scolnik D, Bentur Y. Octreotide for the treatment of sulfonylurea poisoning. Clin Toxicol. 2012;50:795–804.
6. Wills BK, Bryant SM, Bucley P, Seo B. Can acute overdose of metformin lead to lactic acidosis? Am J Emerg Med. 2010;28:857–861.
7. Spiller HA, Weber JA, Winter ML, et al. Multicenter case series of pediatric metformin ingestion. Ann Pharmacother. 2000;34:1385–1388.
8. Dell’Aglio DM, Perino LJ, Todino JD, et al. Metformin overdose with a resultant serum pH of 6.59: survival without sequelae. J Emerg Med. 2010;39:77–80.
9. Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579.
10. Karagiannis T, Paschos P, Paletas K, et al. Dipeptidyl peptidase-4 inhibitors for treatment of type 2 diabetes mellitus in the clinical setting: systematic review and meta-analysis. Br Med J. 2012;344:e1369.
11. Lovshin JA, Drucker DJ. Incretin-based therapies for type 2 diabetes mellitus. Nat Rev Endocrinol. 2009;5:262–269.
12. Frandsen KB, Tambascia MA. Repaglinide and prandial glucose regulation: the rational approach to therapy in type 2 diabetes? Arquiv Brasil Endocrinol Metabol. 1999;43:325–335.
29.10 Colchicine
Lindsay Murray
Essentials
1 All deliberate self-poisonings with colchicine should be regarded as potentially life threatening.
2 May present asymptomatically or with gastrointestinal symptoms only.
3 Consider the diagnosis in patients presenting with gastrointestinal symptoms followed by development of multiorgan failure, especially bone marrow failure.
4 The key points in management are early recognition of the potential severity of this intoxication, early gastrointestinal decontamination and aggressive supportive care.
Introduction
Colchicine is an alkaloid extracted from the plants, Colchicum autumnale (Autumn Crocus) or Gloriosa superba (Glory Lily). It has traditionally been widely used in the treatment of acute gout but has also been prescribed for conditions including familial Mediterranean fever, scleroderma, primary biliary cirrhosis and recurrent pericarditis.
Colchicine poisoning is relatively rare, most commonly occurring in the context of deliberate self-poisoning or therapeutic overdose. Severe toxicity from therapeutic administration of oral colchicine is unusual, but can occur in the elderly or patients with renal or hepatic disease. In this situation, the appearance of gastrointestinal symptoms usually acts as a safety mechanism and results in discontinuation of the drug before the appearance of more severe symptoms. Poisoning is also reported from ingestion of Colchicum autumnale itself.
It is important for the emergency physician to be familiar with the recognition and management of colchicine poisoning because it is associated with high mortality and the potential seriousness of the intoxication is often underestimated at initial presentation.
Toxicokinetics
Colchicine is rapidly absorbed following oral administration, with peak levels occurring from 0.5 to 2 h post-ingestion. Absorption is not significantly delayed following overdose. Bioavailability following oral administration ranges from 25 to 40% because of extensive first-pass hepatic metabolism. Following absorption, colchicine rapidly distributes from plasma to tissues, where it binds with high affinity to intracellular binding sites. The distribution half-life is from 45 to 90 min and the apparent volume of distribution is 21 L/kg in patients with toxicity. Terminal elimination half-lives in toxic patients range from 10.6 to 31.7 h, elimination being via renal excretion, hepatic metabolism via CYP3a4 and enterohepatic circulation. Drug clearance is significantly reduced in patients with renal and hepatic insufficiency.
Pathophysiology
Colchicine binds to tubulin and prevents its polymerization to form microtubules [1]. Microtubules are not only essential components of the cell cytoskeleton during mitosis, but are also integral to other cellular processes, such as endocytosis, exocytosis, phagocytosis, cell motility and protein assembly in the Golgi apparatus. In toxic doses, colchicine causes mitosis to arrest in metaphase with serious consequences for the rapidly dividing cells of the gut mucosa and bone marrow. As colchicine-induced microtubular disruption continues, it affects cell shape, intracellular transport and the secretion of hormones, enzymes and neurotransmitters, resulting in toxicity to virtually every cell in the body [2].
Clinical features
Severe colchicine poisoning presents as a relatively distinct clinical syndrome characterized by early onset of gastrointestinal symptoms followed by delayed onset of multiorgan toxicity and a high incidence of mortality.
In the largest reported series of colchicine poisoning (69 cases), ingestions estimated at<0.5 mg/kg were associated with gastrointestinal symptoms and coagulation disturbances only and a mortality of 0%. Ingestions of 0.5–0.8 mg/kg were associated with bone-marrow aplasia and a mortality of 10% and ingestions>0.8 mg/kg with cardiovascular collapse and 100% mortality at 72 h [3]. However, a number of fatalities have been reported following ingestions of doses<0.5 mg/kg, therefore, any overdose of colchicine should be regarded as potentially serious. The highest reported overdose that survived with aggressive supportive care is 1.38 mg/kg [4].
It is convenient to divide the clinical course of colchicine toxicity into three sequential (and usually overlapping) stages (Table 29.10.1). Less severe cases may not progress beyond the first stage. The most severe cases die during the second stage.
Table 29.10.1
Clinical stages of significant colchicine toxicity

Following a significant acute oral overdose the patient may remain asymptomatic for between 2 and 24 h. The toxic patient then develops severe nausea, vomiting, diarrhoea and abdominal pain. This symptomatology corresponds to gastrointestinal mucosal damage and impairment of secretion of normal mucosal enzymes [5]. During this stage, fluid losses from vomiting and diarrhoea may be significant enough to result in hypovolaemic shock.
Multisystem organ failure is characteristic of the second stage, with onset from 24 to 72 h following ingestion. Respiratory, neurological, renal, haematological and cardiovascular involvement is typical. Acute adult respiratory distress syndrome may be a consequence of hypovolaemic shock or sepsis or occur as a result of direct damage to the pulmonary vasculature [6]. Bone marrow suppression is heralded by lymphopaenia, followed by granulocytopaenia, reticulocytopaenia and thrombocytopaenia, reaching a nadir at 4–8 days following ingestion.
Sepsis may complicate this stage of toxicity [3]. Disseminated intravascular coagulopathy was noted to be a frequent complication in one large series of patients with colchicine toxicity [3]. Fever occurs commonly and may be a direct drug effect or a sign of complicating infection. Shock, frequently observed during this phase, is cardiogenic and/or hypovolaemic in origin and is strongly associated with death [7]. Cardiac rhythm disturbances, including sinus bradycardia and sinus arrest, complete atrioventricular block and sudden cardiac arrest have been reported. Renal failure in acute colchicine toxicity is multifactorial and related to prolonged hypotension, hypoxia, sepsis and rhabdomyolysis. Metabolic derangements described include metabolic acidosis, hyperglycaemia, hypokalaemia, hypocalcaemia, hypophosphataemia and hypomagnesaemia [8]. Neurological disturbances include delirium, coma, seizures, transverse myelitis and ascending paralysis. Death is common during this period and usually occurs as a result of profound cardiogenic shock, sudden cardiac arrest or sepsis. Cardiac arrest has been observed as early as 36 h following acute colchicine ingestion.
In those who survive stage two, a rebound leucocytosis occurs at 7 or more days after initial symptoms and corresponds to the recovery of bone-marrow function. Alopecia commonly occurs at about this time. Complete recovery is the rule in patients surviving stage two.
Differential diagnosis
The diagnosis of colchicine poisoning is usually evident when the history and clinical features are taken into account. Difficulties and delayed diagnosis occur when the history of ingestion of colchicine or colchicine-containing plant material is not obtained. In the absence of such a history, colchicine poisoning has been misdiagnosed as gastroenteritis, sepsis or an acute abdomen requiring laparotomy. Colchicine poisoning should be considered whenever progressive multiple organ dysfunction, especially with bone marrow depression, develops following predominantly gastrointestinal symptoms.
Clinical investigations
Given the potential for severe multisystem organ failure as described above, extensive baseline laboratory studies should be performed upon presentation. These include electrolytes, full blood count, coagulation profile, renal function tests, liver function tests, electrocardiography and chest radiography. These studies need to be repeated during a hospital admission at intervals dictated by the patient’s clinical course. Although colchicine concentrations in biological fluids can be measured, they are not readily available and not useful in the management of colchicine poisoning.
Treatment
The key points in the management of acute colchicine toxicity are early recognition of the potential severity of this intoxication, early gastrointestinal decontamination and aggressive supportive care.
Decontamination of the gut by the administration of oral-activated charcoal is the management priority for the patient presenting in the first (asymptomatic) stage of colchicine intoxication; prevention of absorption of even small amounts may favourably affect the severity of the intoxication and the ultimate outcome. In patients who present later (during the second stage), resuscitative efforts take precedence over gastrointestinal decontamination.
Careful monitoring of vital signs and cardiac rhythm should be instituted upon arrival. An IV cannula should be placed and IV fluid therapy commenced in any symptomatic patient. In those patients who present with substantial delay, immediate resuscitative measures may be required. Baseline laboratory studies as outlined above should be performed.
All patients with colchicine overdose require admission to hospital for a minimum of 24 h observation. Careful monitoring, not only of vital signs and cardiac rhythm but also of fluid and electrolyte status and blood cell counts, is mandatory. Further supportive therapy is dictated by clinical status and may include intravenous crystalloid rehydration, plasma expansion, inotropes, artificial ventilation, correction of electrolyte and acid–base disturbances, correction of coagulation disorders and antibiotic treatment of infectious complications.
Because of colchicine’s large volume of distribution and high affinity to intracellular binding sites, attempts to enhance elimination by repeat-dose activated charcoal, haemodialysis or haemoperfusion are unlikely to be effective.
Disposition
All patients in whom colchicine toxicity is diagnosed or even suspected require admission. The asymptomatic patient should be observed for a minimum of 24 h. If no symptoms of intoxication (diarrhoea, vomiting or abdominal pain) are evident at the end of that period, colchicine toxicity may be confidently excluded and the patient discharged. The symptomatic patient should be admitted to an intensive care unit for careful monitoring and supportive care as outlined above.
Prognosis
As noted above, a relatively high mortality is associated with colchicine overdose. Prognosis is to a large extent determined by the dose ingested. Early resuscitation and provision of excellent supportive care improve prognosis. Patients who present late, in whom the diagnosis is delayed or where the potential seriousness of the presentation is underestimated initially do worse. In patients who survive stage two, a complete recovery can be anticipated. The alopecia observed during the recovery phase is not permanent, with hair growth commencing after the first month.
Controversies
![]() The bone-marrow suppression associated with colchicine toxicity has been reported to respond to the administration of granulocyte colony-stimulating factor [9]. However, it is unclear whether these reports represent a true therapeutic response or the natural course of recovery.
The bone-marrow suppression associated with colchicine toxicity has been reported to respond to the administration of granulocyte colony-stimulating factor [9]. However, it is unclear whether these reports represent a true therapeutic response or the natural course of recovery.
![]() Colchicine-specific Fab fragments have been produced in goats immunized with a conjugate of colchicine and serum albumin and effectively reverse colchicine toxicity in mice [10]. When administered to a patient with severe colchicine toxicity, rapid improvement in haemodynamic parameters and ultimate survival were observed [11]. Unfortunately, colchicine-specific Fab fragments are not yet commercially available.
Colchicine-specific Fab fragments have been produced in goats immunized with a conjugate of colchicine and serum albumin and effectively reverse colchicine toxicity in mice [10]. When administered to a patient with severe colchicine toxicity, rapid improvement in haemodynamic parameters and ultimate survival were observed [11]. Unfortunately, colchicine-specific Fab fragments are not yet commercially available.
References
1. Borizy GG, Taylor EW. The mechanism of action of colchicine: binding of colchicine-H3 to cellular protein. J Cell Biol. 1967;34:525–533.
2. Stapczynski JS, Rothstein RJ, Gaye WA, et al. Colchicine overdose: report of two cases and a review of the literature. Ann Emerg Med. 1981;10:364–369.
3. Bismuth C, Gautier M, Conso F. Aplasie médullaire après intoxication aiguë à la colchicine. Nouv Presse Med. 1977;6:1625–1629.
4. Iosfina I, Lan J, Chin C, et al. Massive colchicine overdose with recovery. Case Rep Nephrol Urol. 2012;2:20–24.
5. Stemmermann GN, Hayashi T. Colchicine intoxication A reappraisal of its pathology based on a study of three fatal cases. Hum Pathol. 1971;2:321–332.
6. Heaney D, Derghazarian CB, Pineo GF, et al. Massive colchicine overdose: report on the toxicity. Am J Med Sci. 1976;271:233–238.
7. Sauder P, Kopferschmitt J, Jaeger A, et al. Haemodynamic studies in eight cases of acute colchicines poisoning. Hum Toxicol. 1983;2:169–179.
8. Putterman C, Ben-Cherit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features and management. Semin Arthritis Rheum. 1991;3:143–155.
9. Harris R, Marx G, Gillett M, Kark A. Colchicine-induced bone marrow suppression: treatment with granulocyte colony-stimulating factor. J Emerg Med. 2000;18:435–440.
10. Sabouraud A, Urtizberea M, Grandgeorge M, et al. Dose-dependent reversal of acute murine colchicine poisoning by goat colchicine-specific Fab fragments. Toxicology. 1991;68:121–132.
11. Baud FJ, Sabouraud A, Vicaut E, et al. Brief report: treatment of severe colchicine overdose with colchicine-specific Fab fragments. N Engl J Med. 1995;332:642–645.
Further reading
1. Finkelstein Y, Aks SE, Hutson JE, et al. Colchicine poisoning: the dark side of an ancient drug. Clin Toxicol. 2010;48:407–414.
29.11 Theophylline
Lindsay Murray
Essentials
1 Theophylline toxicity is associated with life-threatening seizures and cardiac arrhythmias.
2 Serum theophylline levels are useful in assessing and managing acute theophylline toxicity.
3 Onset of maximal toxicity may be significantly delayed following overdose of sustained-release preparations.
4 Techniques of enhancing drug elimination play an important role in the management of severe theophylline toxicity.
5 Early identification of high-risk patients allows the institution of enhanced elimination techniques before life-threatening complications develop.
Introduction
Theophylline, a methylxanthine derivative related to caffeine, has long been used in the treatment of asthma and chronic airflow limitation. Although the use of the drug has declined, both acute and chronic theophylline toxicity continue to result in potentially life-threatening presentations to the emergency department (ED).
Therapeutic blood concentrations of theophylline are generally regarded as being between 55 and 110 μmol/L (10 and 20 mg/L). A single ingestion of more than 10 mg/kg of theophylline by an adult is capable of producing a blood concentration above this range.
Pharmacokinetics
Theophylline is well absorbed orally, with a bioavailability of almost 100%. The rate of absorption depends on the pharmaceutical formulation. The most commonly prescribed preparations are sustained-release and, following overdose of these preparations, peak absorption may be delayed up to 15 h.
Once absorbed, theophylline is rapidly distributed with a relatively small volume of distribution (0.3–0.7 L/kg). Theophylline is metabolized via the cytochrome P450 system to produce active and inactive metabolites. Only about 10% of absorbed theophylline is excreted unchanged in the urine. The rate of metabolism is extremely variable and decreases with time. Theophylline metabolism exhibits saturable kinetics. At higher doses of theophylline, relatively small increments in dose are associated with disproportionate increases in serum concentration. In cases of severe intoxication, endogenous elimination of theophylline is very slow.
Pathophysiology
The precise mechanisms of toxicity of theophylline are unknown. Proposed mechanisms include inhibition of phosphodiesterase leading to elevated concentrations of intracellular cAMP, augmented plasma catecholamine activity, competitive antagonism of adenosine and changes in intracellular calcium transport.
Clinical features
Two different clinical syndromes of theophylline poisoning are recognized: acute and chronic. Both are potentially life threatening, although the chronic form is associated with greater morbidity and mortality.
Chronic intoxication is the most common clinical presentation and occurs when excessive doses of theophylline are administered repeatedly or where intercurrent illness or drug interaction interferes with hepatic metabolism. Theophylline has a notoriously narrow therapeutic index and up to 15% of patients with a serum theophylline concentration in the therapeutic range have clinical manifestations of toxicity.
Acute intoxication is usually the result of deliberate overdose with suicidal intent, but is occasionally observed following inadvertent iatrogenic overdose. Toxicity is anticipated following a single acute ingestion of>10 mg/kg and life-threatening toxicity is anticipated with>50 mg/kg.
The clinical manifestations of theophylline intoxication are numerous and principally affect the gastrointestinal, cardiovascular, central nervous, musculoskeletal and metabolic systems.
The gastrointestinal tract is particularly sensitive to theophylline toxicity, the most prominent symptom being vomiting. This is usually severe and frequently refractory to treatment with antiemetics.
Sinus tachycardia is an almost universal manifestation of theophylline toxicity. However, severe intoxication is also associated with more unstable rhythms, including supraventricular tachycardia, atrial fibrillation, atrial flutter, multifocal atrial tachycardia and ventricular tachycardia [1]. Refractory hypotension may occur in severe toxicity as a result of β2-mediated peripheral vasodilatation.
Central nervous system manifestations most commonly consist of anxiety and insomnia. With more severe intoxication, tachypnoea from respiratory centre stimulation and seizures occur. Seizures can develop suddenly, may be repetitive, are difficult to treat and are associated with poor outcome.
Metabolic complications of theophylline poisoning include hypokalaemia, hypophosphataemia, hypomagnesaemia, hyperglycaemia and metabolic acidosis [2]. Hypokalaemia is frequent following acute overdose, occurs early and is a consequence of intracellular shift of potassium secondary to catecholamine excess [3,4]. Musculoskeletal manifestations include muscle aches, increased muscle tone and myoclonus.
Chronic intoxication usually occurs in elderly patients and is associated with vomiting and tachycardia. The metabolic abnormalities are less frequently observed. Seizures and cardiac arrhythmias occur more frequently and at much lower serum theophylline concentrations than in acute intoxication [5,6].
Following acute overdose, especially where sustained-release preparations are involved, the clinical manifestations of severe toxicity may be delayed up to 12 h. These patients usually present with severe vomiting before the onset of more severe toxicity, including seizures and arrhythmias.
Clinical investigations
The diagnosis of theophylline toxicity is suspected on history and clinical presentation and confirmed by documentation of a significant serum theophylline concentration. The serum theophylline concentration is also invaluable in the assessment of severity and ongoing management of theophylline poisoning. Although theophylline is readily measured, it is not detected on routine drug screens.
Patients with acute theophylline overdose generally exhibit signs of minor toxicity at serum concentrations from 110 to 220 μmol/L (20–40 mg/L), moderate toxicity with concentrations from 220 to 440 μmol/L (40–80 mg/L) and severe toxicity with concentrations greater than 440 μmol/L (80 mg/L). Serum theophylline concentrations of greater than 550 μmol/L (100 mg/L) are frequently fatal [7]. After an acute overdose, serum theophylline should be measured every 3 h or so until a falling concentration is documented.
In chronic theophylline poisoning, serious toxicity is observed at lower serum concentrations and the measured concentration is not predictive of the severity of poisoning [8]. Seizures, arrhythmias and fatalities can occur at concentrations as low as 220–330 μmol/L (20–30 mg/L). In these patients, the best predictor of poor outcome is age over 60 years [9].
Other useful laboratory studies include electrolytes and creatinine, glucose, liver function tests (LFTs) and electrocardiogram (ECG).
Treatment
The initial management of theophylline poisoning follows the principles of general supportive care. Specific attention may need to be directed towards control of the airway, hypotension, tachyarrhythmias and seizures.
Hypotension usually responds to intravenous fluid administration. A noradrenaline (norepinephrine) infusion may be necessary in resistant cases. Supraventricular arrhythmias can be treated with a β-blocker, such as propranolol or esmolol, intravenously but this may induce bronchospasm in susceptible individuals. Seizures must be treated aggressively with high-dose benzodiazepines. If this fails, phenobarbitone and even general anaesthesia may be required. Phenytoin is ineffective and contraindicated. Metabolic disturbances do not generally require specific therapy. Severe hypokalaemia should be corrected with potassium supplementation.
Following acute overdose, oral-activated charcoal should be administered, even if presentation is delayed. Antiemetics are usually required for successful administration.
The pharmacokinetic properties of theophylline, especially the small volume of distribution, lend themselves to methods of enhanced elimination. Theophylline is relatively efficiently removed by haemodialysis, charcoal haemoperfusion and administration of repeat-dose activated charcoal [10].
Theophylline clearance rates of 100 mL/min have been reported with multiple-dose activated charcoal [10]. Again, aggressive antiemetic therapy may be necessary if this non-invasive method of enhancing drug elimination is to be effective. Administration of a selective serotonin antagonist, such as ondansetron, has proved particularly effective in this setting.
Both charcoal haemoperfusion and haemodialysis greatly increase the elimination of theophylline and are highly effective in achieving a good clinical outcome [11]. Such invasive methods are only indicated in potentially life-threatening theophylline toxicity. Commonly accepted indications include acute intoxication, where the serum theophylline is greater than 550 μmol/L; chronic intoxication, where it is greater than 220–330 μmol/L or in any patient with intractable hypotension, ventricular ectopy or resistant seizures [5,7]. Ideally, patients at greatest risk of developing arrhythmias or seizures should be identified early and haemodialysis or haemoperfusion instituted before these complications develop. Continuous venovenous haemofiltration has been successfully used as an alternative to standard intermittent haemodialysis in the treatment of severe theophylline poisoning with a reduction in the elimination half-life to 5.87 h reported [12].
Disposition
All patients with symptomatic theophylline toxicity require admission to hospital. Patients with acute overdose of sustained-release preparations should be admitted for monitoring and serial serum theophylline concentrations. Patients with moderate-to-severe theophylline toxicity require admission to a monitored bed.
Controversies
![]() Although charcoal haemoperfusion has been recommended as the most effective way to enhance theophylline elimination, it has not yet been shown to be associated with any additional improvement in clinical outcome compared to haemodialysis.
Although charcoal haemoperfusion has been recommended as the most effective way to enhance theophylline elimination, it has not yet been shown to be associated with any additional improvement in clinical outcome compared to haemodialysis.
![]() Continuous renal replacement therapies offer a number of advantages over standard intermittent dialysis as a method of enhancing theophylline elimination. They are easily set up and run in most intensive care units and can be run 24 h a day. However, clearance rates are slower and these techniques are not currently recommended except where standard dialysis is not available or unfeasible because of haemodynamic instability.
Continuous renal replacement therapies offer a number of advantages over standard intermittent dialysis as a method of enhancing theophylline elimination. They are easily set up and run in most intensive care units and can be run 24 h a day. However, clearance rates are slower and these techniques are not currently recommended except where standard dialysis is not available or unfeasible because of haemodynamic instability.
References
1. Bender PR, Brent J, Kulig K. Cardiac arrhythmias during theophylline toxicity. Chest. 1991;100:884–886.
2. Hall KW, Dobson KE, Dalton JG, et al. Metabolic abnormalities associated with intentional theophylline overdose. Ann Intern Med. 1984;101:457–462.
3. Amitai Y, Lovejoy FH. Hypokalaemia in acute theophylline poisoning. Am J Emerg Med. 1988;6:214–218.
4. Shannon M, Lovejoy FH. Hypokalemia after theophylline intoxication The effects of acute vs chronic poisoning. Arch Intern Med. 1989;149:2725–2729.
5. Olson KR, Benowitz NL, Woo OF, Pond SM. Theophylline overdose: acute single ingestion versus chronic repeated overmedication. Am J Emerg Med. 1984;3:386–394.
6. Shannon M. Life-threatening events after theophylline overdose: a 10-year prospective analysis. Arch Intern Med. 1999;159:989–994.
7. Sessler C. Theophylline toxicity: clinical features of 116 consecutive cases. Am J Med. 1990;88:567–576.
8. Shannon M, Lovejoy F. Effect of acute versus chronic intoxication on clinical features of theophylline poisoning in children. J Pediatr. 1992;121:125.
9. Shannon M. Predictors of major toxicity after theophylline overdose. Ann Intern Med. 1993;119:1161–1167.
10. Kulig KW, Bar-Or D, Rumack BH. Intravenous theophylline poisoning and multiple-dose charcoal in an animal model. Ann Emerg Med. 1987;16:842.
11. Heath A, Knudsen K. Role of extracorporeal drug removal in acute theophylline poisoning–a review. Med Toxicol. 1987;2:294.
12. Henderson JH, McKenzie CA, Hilton PJ, Leach RM. Continuous venovenous haemofiltration for the treatment of theophylline toxicity. Thorax. 2001;56:242–243.
29.12 Iron
Zeff Koutsogiannis
Essentials
1 Acute iron poisoning is a potentially life-threatening condition.
2 The risk of severe toxicity is determined by the dose of elemental iron ingested not the weight of the iron salt.
3 Iron poisoning has both local (gastrointestinal) and systemic effects.
4 Early effective gastrointestinal decontamination with whole-bowel irrigation is important in the management of high-risk cases.
5 Chelation therapy with intravenous desferrioxamine is the definitive treatment for severe poisoning.
6 Generally, most patients recover, although presence of shock or coma indicates a poor prognosis.
7 Long-term sequelae are gastrointestinal scarring and obstruction but this is uncommon.
Introduction
The majority of exposures to iron occur in preschool children, but significant iron ingestions also occur in adults as a result of deliberate self-poisoning. It is also one of the most commonly ingested agents in self-poisoning during pregnancy as a result of its ready availability to obstetric patients. Iron supplements are often considered by patients and parents to be innocuous dietary supplements, leading to careless storage and handling and delays in seeking medical care following ingestions.
Due to education, different packaging, smaller dosages and toxicovigilance by poisons centres, iron toxicity has declined in the past decade, but significant poisonings still occur.
Pathophysiology
Iron is an essential element in red blood cell production, haemoglobin and myoglobin oxygenation and cytochrome function. The body cannot directly excrete iron so body stores are finely regulated by controlling absorption of iron from the gastrointestinal (GI) tract. After absorption across the GI mucosa in the ferrous form (Fe2+), iron is oxidized to the ferric state (Fe3+) and then stored bound to ferritin or transported across the cell membrane into the blood, where it binds to transferrin. Iron is extracted from transferrin in the bone marrow and used for haemoglobin synthesis. It is also removed from transferrin by the reticuloendothelial system and hepatocytes and stored as haemosiderin and ferritin. Total iron binding capacity (TIBC) is a measurement of the total amount of iron that transferrin can bind and normally exceeds serum iron by two- to threefold.
Ferritin is a large storage protein that reversibly binds to iron. When an iron deficit exists, iron is transported from ferritin and the GI tract. If the body’s iron requirements have been met, iron remains stored in the intestinal cell rather than bound to transferrin. Eventually, the intestinal cell dies and sloughs off into the lumen for elimination. This is the main mechanism limiting excessive iron absorption and the mechanism by which the body regulates iron balance.
Iron rarely exists as an unbound or ‘free’ element. It is free iron that is toxic to cellular processes. As a result, iron toxicity results from direct local (GI) effects and cellular toxicity (systemic effects).
Local effects
Iron preparations, like other metal salts, have a direct corrosive effect on the GI mucosa. In overdose, this can lead to irritation, ulceration, bleeding, ischaemia, infarction and perforation. Associated profound fluid losses can result in hypotension, shock and lactate formation leading to metabolic acidosis. The long-term sequelae of this corrosive action include GI scarring and obstruction. As the mucosal surface is disrupted, iron is absorbed passively down concentration gradients.
Systemic effects
When the absorbed iron exceeds the protein binding capacity, the free iron causes cellular dysfunction and death. Free iron is an intracellular toxin localizing to the mitochondria, forming free radicals and disrupting oxidative phosphorylation. The resultant mitochondrial dysfunction and destruction lead to cell death and can occur in any organ. Other systemic findings of iron poisoning include cardiovascular collapse, anion-gap metabolic acidosis, coagulopathy and encephalopathy. Metabolic acidosis persisting after correction of hypovolaemia and hypoperfusion is probably a result of mitochondrial toxicity. Coagulopathy developing early in iron poisoning results from inhibition of serum proteases while, in the later stages, it is due to hepatic dysfunction.
Toxic dose
In general, the risk of developing iron toxicity can be predicted from the dose of elemental iron ingested per kilogram body weight (Table 29.12.1). It is essential to calculate the dose of elemental iron rather than dose of iron salt. If the formulation of the iron salt is not known, then assume a worst-case scenario and calculate 105 mg of elemental iron per tablet.
Table 29.12.1
Risk assessment based on dose of elemental iron ingested
|
Risk assessment |
Dose ingested (mg/kg) |
|
Asymptomatic |
<20 |
|
Local (GI) symptoms only |
20–60 |
|
Risk of systemic toxicity |
60–120 |
|
Potentially lethal |
>120 |
Prevention
Iron poisoning is a major cause of unintentional poisoning death in young children making up almost one-third of all toxicological deaths in that age group in the 1980s to the 1990s. However, there has been a decrease in the incidence of non-intentional ingestion by young children and decreased mortality following the introduction of unit-dose packaging. This, together with education may further decrease the incidence of toxicity and late presentations.
Clinical features
The clinical course of iron poisoning is traditionally described as comprising five stages. Not all patients will experience all stages; they can die at any stage; can present at any stage and the time frames for each stage are imprecise and may overlap.
A more practical approach is to consider iron poisoning as comprising two clinical stages with a pathophysiological basis: GI toxicity and systemic toxicity.
Stage 1 (0–6 h)
This stage is dominated by symptoms and signs of GI injury particularly vomiting, but also abdominal pain, diarrhoea and GI bleeding. In severe cases, hypovolaemic shock secondary to GI losses can develop. The failure to develop any GI symptoms within 6 h of ingestion effectively excludes significant iron poisoning.
Stage 2 (2–24 h)
Also known as the ‘latent’ or ‘quiescent’ phase, this stage represents the period between resolution of GI symptoms and appearance of overt systemic toxicity. It is not a true quiescent phase as ongoing cellular toxicity occurs. Although clinicians should be wary of GI symptom resolution, most patients have in fact recovered and do not progress to Stage 3. Those with significant poisoning remain clinically ill with subtle signs (but should be easily identifiable) and progress to Stage 3.
Stage 3 (6–48 h)
This is the stage of systemic toxicity characterized by shock and multiorgan system failure. By definition, it represents severe toxicity. The shock is multifactorial arising from hypovolaemia, vasodilation and poor cardiac output with evidence of poor peripheral perfusion, worsening acidosis and acute renal failure. A coagulopathy may develop and lead to recurrent GI bleeding. Central nervous system effects include lethargy, coma and convulsions.
Stage 4 (2–5 days)
This is the hepatic phase of iron toxicity and is relatively uncommon. It is characterized by acute hepatic failure with jaundice, hepatic coma, hypoglycaemia, coagulopathy and elevated transaminase and ammonia levels. It has a high mortality.
Stage 5 (2–6 weeks)
This stage is relatively rare and represents the delayed sequelae from the corrosive effects of iron resulting in GI scarring. This results in gastric outlet (pyloric stricture) and small bowel obstructions.
Clinical investigations
Acute iron poisoning is a clinical diagnosis and all significantly symptomatic patients require treatment regardless of the iron level or results of other tests. However, serum iron levels, abdominal X-rays and other tests do play a role in determining management.
Serum iron concentration
Normal serum iron concentrations are between 10 and 30 μmol/L. Peak iron levels usually occur between 2 and 6 h after overdose, although they may sometimes be delayed. Frequent levels may need to be taken to determine the true peak. Repeat levels should be determined at 8–12 hours to rule out delayed absorption from sustained release preparations or bezoar formation. Although iron poisoning is a clinical diagnosis, iron levels have been used to determine toxicity and direct management. A serum iron concentration greater than 90 μmol/L at 4–6 h after an overdose is associated with a greater risk of subsequently developing systemic iron toxicity. However, it is intracellular and not serum iron that is responsible for systemic toxicity and, thus, during Stages 2 or 3, the iron level may be decreasing or even normal while the patient deteriorates. In the presence of desferrioxamine, the serum iron level is artificially lowered.
The TIBC is falsely elevated in the presence of high iron concentrations or desferrioxamine and is no longer regarded as useful in the assessment of iron poisoning.
Plain abdominal X-rays
Most iron preparations are radiopaque and an early abdominal X-ray is useful in confirming ingestion of iron and in subsequently guiding gastric decontamination. A negative X-ray does not exclude iron ingestion as the tablets may have disintegrated or not be radiopaque. Liquid preparations or chewable tablets are typically not radiopaque.
Other laboratory tests
Although leucocytosis and hyperglycaemia are frequently observed in iron poisoning, they are not useful in terms of diagnosis or management. The presence of an anion-gap metabolic acidosis is a useful marker of systemic iron poisoning and, as such, a low serum bicarbonate concentration is a good surrogate marker of systemic iron poisoning in places where serum iron levels are not readily available.
Other tests that are useful in managing patients with established iron poisoning include serum electrolytes, renal function, liver function, blood gases and clotting profile.
However, if the white cell count, blood glucose, radiographic findings are normal and there are no GI symptoms, serious toxicity is unlikely.
Differential diagnosis
Usually, the diagnosis is self-evident from the history of exposure, but iron poisoning needs consideration in the undifferentiated poisoning with an anion-gap metabolic acidosis and GI symptoms.
Treatment
The approach to management of a patient presenting following an iron overdose is determined by the initial risk assessment. This is based on the dose ingested and the presence or absence of GI and/or systemic features of iron poisoning. For most patients, a period of observation and good supportive care, often including intravenous fluids, will be sufficient. In those patients at risk of systemic poisoning or who present with established iron poisoning, aggressive decontamination measures and chelation therapy may be necessary to achieve a good outcome. The aim is to prevent the development of systemic toxicity.
Observation and supportive care
All patients demonstrating signs and symptoms consistent with clinical toxicity of Stages 1, 2 or 3 warrant further treatment. Aggressive fluid replacement with isotonic fluid is essential. An initial bolus of 20 mL/kg should be given, followed by boluses as needed to replace fluid losses and maintain urine output. Patients with established iron poisoning may require more advanced supportive care, including inotropic support, blood transfusions, correction of coagulopathy with fresh frozen plasma and correction of acidosis.
Any lethargic patient who is likely to deteriorate should be promptly intubated to facilitate safe decontamination.
Gastrointestinal decontamination
Iron is not well adsorbed to activated charcoal. Other modalities designed to reduce iron absorption from the GI tract, including oral bicarbonate, phosphate, magnesium hydroxide, oral calcium disodium EDTA and sodium polystyrene sulphonate, are equally ineffective. Thus, alternative methods of GI decontamination must be considered in patients who present following ingestion of more than 60 mg/kg of elemental iron, especially where unabsorbed iron is evident on abdominal X-ray.
Inducing emesis with syrup of ipecac is not recommended because it may mask the symptoms produced by iron, aggravate the GI irritation and can lead to an underestimation of the severity of the toxicity. Gastric lavage may be a useful option if performed early, but is often technically difficult in that the tablets tend to clump together, form pharmacobezoars and attach to the gastric mucosa. Endoscopy has been used to remove large iron loads, but this is also technically difficult. Surgical removal has been reported.
Whole-bowel irrigation (WBI) is widely advocated as the GI decontamination method of choice in the setting of iron poisoning, although there are no controlled trials. It should be initiated in any patient who has ingested more than 60 mg/kg of elemental iron and still has large number of iron tablets present in the GI tract on X-ray. The procedure is continued until there is a clear rectal effluent and no visible iron on X-ray. As iron has a direct corrosive effect on the GI mucosa, caution is therefore advised with the use of WBI in late presenters who may have sustained mucosal damage.
Chelation therapy
Desferrioxamine is the parenteral chelating agent of choice for iron poisoning. It binds Fe3+ to form ferrioxamine which is water soluble, red-to-orange in colour and renally excreted. Desferrioxamine binds free iron and iron in transit between transferrin and ferritin thus effecting a redistribution of iron from tissue sites back into plasma. It does not chelate iron bound to transferrin, haemoglobin, myoglobin or cytochrome enzymes.
Chelation therapy is indicated in any patient with established systemic iron toxicity or at risk of developing such toxicity. Thus, the indications are:
![]() systemic toxicity (shock, metabolic acidosis, altered mental status) irrespective of iron levels
systemic toxicity (shock, metabolic acidosis, altered mental status) irrespective of iron levels
![]() serum iron levels greater than 60 μmol/L and symptomatic
serum iron levels greater than 60 μmol/L and symptomatic
![]() serum iron levels greater than 90 μmol/L are generally regarded as being predictive of subsequent systemic toxicity and an indication to commence chelation therapy even if asymptomatic.
serum iron levels greater than 90 μmol/L are generally regarded as being predictive of subsequent systemic toxicity and an indication to commence chelation therapy even if asymptomatic.
Ferrioxamine’s red-to-orange colour is responsible for the classically described vin rose urine in patients given desferrioxamine but this colour change is an insensitive marker of the presence of free iron and the desferrioxamine intramuscular challenge test is no longer used.
Desferrioxamine is given as a continuous intravenous infusion starting slowly and aiming for a rate of 15 mg/kg/h. Administration rate may be limited by hypotension, the principal adverse effect. Intramuscular administration is not recommended as it is painful, requires multiple injections, has erratic absorption and higher side-effect profile. The precise endpoints for chelation therapy are unclear but therapy can be safely discontinued once the serum iron level is normal or low, the patient is clinically well and the anion-gap metabolic acidosis has resolved. Except under exceptional circumstances, desferrioxamine should not be continued for longer than 24 h or exceed 80 mg/kg/24 h because of the risk of pulmonary toxicity and acute respiratory distress syndrome (ARDS). Treatment duration of 6 h is usually sufficient.
Expert advice from a clinical toxicologist should be sought if using desferrioxamine therapy.
The approach to iron poisoning is not altered in the pregnant patient. Symptomatic iron overdose in pregnancy is associated with preterm labour, spontaneous abortion and maternal death. Desferrioxamine does not cause perinatal complications or fetal toxicity and is potentially life saving. It is therefore indicated in iron intoxication in pregnancy with clinical evidence of moderate to severe toxicity. The dose is based on the pre-pregnancy weight of the patient.
Enhanced elimination
Haemodialysis and haemoperfusion are not effective at removing iron but may be necessary to remove the ferrioxamine complex in patients with renal failure.
Exchange transfusion has been used in massive paediatric ingestions but it is of questionable value.
Disposition
Patients who have ingested less than 60 mg/kg of elemental iron and remain asymptomatic at 6 h may be medically discharged. Those with GI symptoms or requiring WBI because of large ingestion require admission for supportive care and ongoing observation and monitoring. Those with systemic toxicity and/or requiring chelation therapy require intensive care admission. All patients where deliberate self-poisoning is suspected require psychosocial assessment.
Prognosis
Most patients with iron overdose remain asymptomatic or develop minor GI toxicity only and do well with supportive care. Those with large ingestions should have an excellent outcome if recognized early and appropriate and timely decontamination and/or chelation therapy is instituted. Patients presenting late with established severe systemic toxicity have a poorer prognosis. Gastrointestinal stricture formation is a potential long-term sequela.
Controversies
![]() N-acetylcysteine may protect against iron-induced hepatotoxicity.
N-acetylcysteine may protect against iron-induced hepatotoxicity.
![]() New oral chelating agents, such as deferiprone (effective in patients with chronic iron overload states such as thalassaemia) and the hexadentate phenolic aminocarboxylate iron chelator sodium N, N″-bis(2-hydroxybenzyl) ethylenediamine-N, N″-diacetic acid (HBED) ligand have been shown to improve survival and enhance iron excretion in animal studies, but no human data are as yet available.
New oral chelating agents, such as deferiprone (effective in patients with chronic iron overload states such as thalassaemia) and the hexadentate phenolic aminocarboxylate iron chelator sodium N, N″-bis(2-hydroxybenzyl) ethylenediamine-N, N″-diacetic acid (HBED) ligand have been shown to improve survival and enhance iron excretion in animal studies, but no human data are as yet available.
![]() Modifications of desferrioxamine, such as conjugation with dextran or hydroxyethyl starch, have shown the potential for enhanced efficacy and improved patient tolerability. Additional research must be conducted to determine the role of these agents.
Modifications of desferrioxamine, such as conjugation with dextran or hydroxyethyl starch, have shown the potential for enhanced efficacy and improved patient tolerability. Additional research must be conducted to determine the role of these agents.
![]() Diazepam has been shown in animal studies to reduce mortality without chelation.
Diazepam has been shown in animal studies to reduce mortality without chelation.
Further reading
1. Banner W, Tong TG. Iron poisoning. Pediatr Clin N Am. 1986;33:393–409.
2. Chang T, Rangan C. Iron poisoning: a literature-based review of epidemiology, diagnosis and management. Pediatr Emerg Care. 2011;27:978–985.
3. Finch CA, Huebers H. Perspectives in iron metabolism. N Engl J Med. 1982;306:1520.
4. Henretig FM, Temple AR. Acute iron poisoning in children. Emerg Med Clin N Am. 1984;2:121.
5. Iron. In: WikiTox: open source clinical toxicology curriculum.<http://curriculum.toxicology.wikispaces.net/2.1.9.6+Iron>.
6. Jacobs J, Greene H. Acute iron intoxication. N Engl J Med. 1965;273:1124–1127.
7. Manoguerra AS, Booze LL, Scharman EJ, et al. Iron ingestion: an evidence-based consensus guideline for out of hospital management. Clin Toxicol. 2005;43:553–570.
8. Mills KC, Curry SC. Acute iron poisoning. Emerg Med Clin N Am. 1994;12:397–413.
9. Perrone J. Iron. In: Nelson LS, Levin NA, Howland MA, eds. Goldfrank’s toxicologic emergencies. 9th ed. New York: McGraw-Hill; 2010;629–638.
10. Tenenbein M. Position statement: whole bowel irrigation American academy of clinical toxicology; European association of poisons centres and clinical toxicologists. J Toxicol Clin Toxicol. 1993;35:753–762.
11. Tenenbein M. Benefits of parenteral deferoxamine for acute iron poisoning. J Toxicol Clin Toxicol. 1996;34:485–489.
12. Tenenbein M, Littman C, Stimpson RE, et al. Gastrointestinal pathology in adult iron overdose. J Toxicol Clin Toxicol. 1990;28:311–320.
29.13 Drugs of abuse
Kerry A Hoggett
Essentials
1 The diagnosis of intoxication with drugs of abuse is clinical. Good supportive care ensures optimal outcome in the majority of cases.
2 Predisposing factors for heroin overdose include co-ingestion of CNS depressant drugs, poor tolerance, high purity and reluctance to seek medical care. Naloxone is a useful adjunct in the management of airway and ventilation in opiate overdose.
3 Benzodiazepines are important in the management of the central nervous system and cardiovascular manifestations of amphetamine intoxication. Hyperthermia, decreased conscious state, headache, neurological signs or chest pain suggest life-threatening complications and warrant aggressive management and investigation.
4 Cocaine use is associated with both cardiac and non-cardiac toxicity and may be life threatening. Toxicity may be difficult to distinguish clinically from amphetamines, although it tends to be of shorter duration. Aggressive investigation and management is required for hyperthermia, seizures, chest pain and ventricular cardiac arrhythmias.
5 Gamma hydroxybutyric acid is a sedative-hypnotic drug causing CNS depression. Management is supportive.
6 Newer synthetic psychoactive agents (stimulants, hallucinogens and depressants) are becoming widely available, with evolving structural modifications and variable effects. Management is supportive.
7 Presentation to the emergency department following overdose provides an opportunity for intervention. Education to avoid future overdoses and referral to agencies specializing in detoxification and rehabilitation is appropriate.
Introduction
Australian data report 37.3% of people aged over 14 years have used illicit drugs at least once during their lifetime. In 2010, the most commonly used illicit drugs included ecstasy (10.3%), hallucinogens (8.8%), cocaine (7.3%) and methamphetamines (7.0%). Novel synthetic psychoactive drugs have become widely available from street vendors and Internet sources. The ongoing emergence of new ‘legal highs’ has been challenging for health professionals and law makers alike. For all agents, the complications of illicit drug abuse are classified into three groups: primary toxic effect, complications of intoxication and complications of administration/injection technique. Trauma must be considered in all intoxicated patients.
Opiates
Opiates are derivatives of the opium poppy, Papaver somniferum, which contains approximately 20 alkaloids, including morphine and codeine.
Aetiology, pharmacology and pathogenesis
Opiates are agonists, partial agonists or antagonists at μ (mu), δ (delta) and κ (kappa) receptors in the brain and spinal cord. The principal effects of opiates on the CNS are due to their action on μ receptors. Most opiates are well absorbed across mucous membranes and from subcutaneous and intramuscular sites. Oral opiates undergo extensive first-pass metabolism by glucuronidation (morphine), demethylation and oxidative metabolism. Metabolites are excreted in the urine and may accumulate in renal failure.
Morphine and heroin (diacetylmorphine) taken parenterally reach peak clinical effect within minutes and have a short half-life of 3 h. In contrast, methadone, slow-release oral morphine preparations and ingested transdermal patches have slow and erratic absorption, reach peak plasma concentrations after several hours and have long durations of effect. Transdermal fentanyl patches applied to the skin result in a subcutaneous depot of opiate from which absorption continues after patch removal.
Epidemiology
Opiate overdose is common among Australian heroin users. Factors that contribute to non-fatal and fatal opiate overdose include the co-ingestion of other central nervous system (CNS) depressant drugs, poor tolerance, high street purity and reluctance to seek medical care.
Clinical features
The diagnosis of opioid intoxication is clinical, based on history and examination. The clinical signs of opiate intoxication are related to their effect on the CNS and may be modified by co-ingested alcohol, drugs, trauma or medical complications. A Glasgow coma scale (GCS) less than 12 associated with respirations of 12 breaths/min or less, miotic pupils or circumstantial evidence of drug use is suggestive. ‘Track marks’ are not always evident. With increasing dose, analgesia, euphoria, miosis, sedation, coma, respiratory depression and apnoea occur. Hypoxia, hypercarbia and acidaemia lead to tachycardia, hypertension and variable pupillary responses, followed by bradycardia, hypotension and cardiac arrest as terminal events.
Respiratory depression may lead to hypoxic encephalopathy. Dependent areas suffer peripheral neuropathy, compartment syndrome or rhabdomyolysis which may combine with hypovolaemia and hypoxia to produce renal failure and hyperkalaemia. Hypothermia or hyperthermia can occur. Non-cardiogenic pulmonary oedema is an infrequent complication of opiate overdose, recognized in the context of resuscitation from apnoea, when hypoxia and other metabolic derangements are present. Symptoms usually develop within 4 h of presentation and only a minority require mechanical ventilation.
Complications of poor injection technique include cellulitis, thrombophlebitis, intra-arterial injection and embolization, mycotic aneurysm, endocarditis, anaerobic infection and blood-borne virus infection. A high index of suspicion is required. In addition, the purity of heroin varies and the drug is ‘cut’ with agents which have their own clinical effects.
Differential diagnosis
The differential diagnosis is that of any patient with an altered level of consciousness and includes toxicological and metabolic causes, sepsis, neurotrauma, stroke and post-ictal state. Clonidine ingestion may present with low GCS and miosis and may show partial response to naloxone. Toxicity from paracetamol or ibuprofen in co-formulation with opiates should be considered. The presence of fever without localizing symptoms and signs should raise the suspicion of bacteraemia secondary to parenteral drug abuse.
Clinical investigations
Thorough physical examination is usually adequate to exclude complications, such as aspiration, non-cardiogenic pulmonary oedema and compartment syndrome. Further investigations are directed at confirming or excluding alternative diagnoses and complications.
Treatment
Initial care is directed at the resuscitation and immediate life threats. All patients should have bedside blood glucose measurement. Patients with an altered level of consciousness should be closely monitored in a resuscitation area, positioned to minimize the probability of aspiration and moved frequently to prevent dependent injuries.
Naloxone is a short-acting opioid antagonist active at μ receptors, useful as an adjunct to support airway and ventilation. It may be given via the intravenous, intramuscular, subcutaneous or endotracheal routes. Naloxone is safe and rarely associated with serious complications. Bolus therapy (e.g. 0.4–2.0 mg intravenously or intramuscularly in an adult; 0.01 mg/kg in a child) reverses the respiratory depression of opioid intoxication, but may be complicated by rapid wakening, agitation and an acute withdrawal state in opioid-dependent patients. An alternative approach is the use of small intravenous doses (e.g. 0.04 mg) titrated to achieve airway control and ventilation while avoiding abrupt emergence. Intravenous doses of naloxone are effective within a few minutes. The duration of effect of naloxone (20–90 minutes) is less than that of most opioids, therefore, patients must be carefully monitored for resedation. Naloxone infusions may be useful in carefully selected patients who are intoxicated by long-acting opioids, to prevent airway compromise or need for intubation. However, absorption and elimination of opioids may be unpredictable and undulating CNS depression may persist despite continuous naloxone infusion. Intubation is a safe alternative to prolonged infusion for large ingestions.
The duration of observation in the emergency department (ED) and the need for admission depends on the opioid involved, route of administration, co-ingested drugs and the presence of co-morbidity or complications. Long-acting or slow-release opioids require observation for at least 12 hours. Following overdose with short-acting agents, patients can be safely discharged when they are ambulant and alert, have normal vital signs and oxygen saturation and at least 4–6 h have passed since naloxone administration. Discharge should not occur at night.
Other issues
Children and the elderly are more susceptible to opiate toxicity. In children, delayed onset of toxicity and prolonged effects are commonly seen. Co-morbid conditions and physiological changes in the elderly may result in prolonged intoxication. Prolonged observation may be required.
Heroin overdose indicates ongoing hazard and presentation to the ED provides an opportunity for intervention. Patients should be counselled regarding strategies to avoid future overdose and early activation of emergency medical services. If willing, the patient should be referred to agencies specializing in drug detoxification and rehabilitation.
Opioid withdrawal syndrome
The development of physiological dependence with repeated doses of opioid agonists leads to an abstinence or withdrawal syndrome when opioids are ceased. The symptoms represent the reverse of the central and peripheral effects of opioid administration, including anxiety, insomnia, apprehension, hyperventilation, mydriasis, nausea, vomiting, diarrhoea and abdominal pain. Symptoms usually occur 12 h after the last dose of morphine or heroin, peak at approximately 2 days and abate after 5 days. Seizures do not occur and the prognosis is good, even without medical intervention.
After exclusion of concomitant pathology, management should include treatment of dehydration, symptomatic care and referral to drug and alcohol services. Clonidine, an α2-receptor adrenergic agonist, is used to decrease autonomic symptoms. An initial dose of 1–2 μ/kg, 2–3 times per day, can be increased depending on side effects, such as orthostatic hypotension.
Amphetamines
Pharmacology and pathophysiology
Amphetamines refer to a broad group of related derivatives of β-phenylisopropylamine characterized clinically by CNS stimulatory and peripheral sympathomimetic responses. Amphetamines are structurally related to ephedrine and resemble the catecholamines. Substitutions on the basic structure of β-phenylisopropylamine include methamphetamine (‘ice’, ‘speed’), 3,4-methylenedioxymethamphetamine (MDMA, ‘ecstasy’, ‘Adam’ or ‘E’), 3,4-methylenedioxyethamphetamine (MDEA, ‘Eve’) and 3,4-methylenedioxyamphetamine (MDA, ‘love drug’) and para-methoxyamphetamine (PMA).
Amphetamines may be ingested, smoked, insufflated or injected. All are absorbed from the gastrointestinal (GI) system, with peak serum levels within 3 h. They are weak bases, 20% bound to plasma proteins and have large volumes of distribution. Half-lives vary from 8 to 30 h. Hepatic transformation is the major route of elimination, however, up to 30% of amphetamine and metamphetamine may be eliminated in the urine.
Amphetamines enhance the release of catecholamines and block their reuptake causing increased stimulation of central and peripheral adrenergic receptors leading to CNS excitation and a sympathomimetic syndrome. Higher doses lead to central serotonin release. Substitutions alter the hallucinogenic, behavioural and cardiovascular effects of the drugs at low doses. In overdose, it may be impossible to distinguish the exact amphetamine derivative involved as effects are more uniform.
Epidemiology
The use of amphetamine as a drug of abuse has been prevalent since its introduction in 1932. The 2010 National Drug Strategy Household Survey found 7% of Australians over 14 years reported using amphetamine or methamphetamine in their lifetime. 3,4-Methylenedioxymethamphetamine (MDMA, ecstasy) use was also common, with 10% of people over age 14 years reporting use in their lifetime.
Clinical features
The diagnosis of amphetamine intoxication is clinical. The predominant symptoms are those of CNS excitation and peripheral sympathomimetic response. Euphoria, apprehension and agitation are common. Tachypnoea, mydriasis, tremor, diaphoresis and hyperpyrexia may also be seen. After acute intoxication or binges, psychosis may occur with visual and tactile hallucinations, severe agitation and paranoia resembling paranoid schizophrenia. Myocardial infarction, aortic dissection, rhabdomyolysis, acidosis, acute cardiomyopathy, shock, renal failure and coagulopathy are documented. Symptoms may persist for several days. Death is secondary to hyperpyrexia, seizures, arrhythmia or intracerebral haemorrhage.
Hyponatraemia with cerebral oedema is reported following MDMA use and may be fatal, presenting initially as altered consciousness and seizures. Mechanisms contributing to hyponatraemia include psychogenic polydipsia and inappropriate secretion of antidiuretic hormone.
Complications of injection technique include cellulitis, thrombophlebitis, intra-arterial injection and embolization, mycotic aneurysm, staphylococcal pneumonia, endocarditis, anaerobic clostridial infection and blood-borne viral infections. In addition, chronic abuse of amphetamines may be complicated by a necrotizing vasculitis which may involve multiple organ systems and lead to renal failure, myocardial ischaemia and cerebrovascular disease.
Amphetamine dependence and withdrawal is recognized. Withdrawal is characterized by neurasthenic symptoms, including somnolence and intense cravings for amphetamines. Management is supportive.
Differential diagnosis
Intoxication by an amphetamine derivative may not be discernible clinically from cocaine, except for the increased psychotic features and longer duration of action. The differential diagnosis includes cocaine intoxication, anticholinergic delirium, serotonin syndrome, monoamine oxidase inhibitors, alcohol and benzodiazepine withdrawal, sepsis, hypoglycaemia, thyrotoxicosis and phaeochromocytoma.
Clinical investigations
Physical examination and investigations should be directed at excluding complications and alternative diagnoses. Seizures or coma should prompt investigation for complications, such as intracerebral haemorrhage or electrolyte disturbance.
Treatment
Initial attention must be directed at resuscitation and immediate threats to airway, breathing and circulation. All patients should receive oxygen and have a bedside blood glucose measurement. Close monitoring of the patient in a quiet area away from excessive stimulation may be advantageous. Patients exhibiting psychomotor acceleration or psychosis should be managed with an intravenous benzodiazepine titrated to achieve adequate sedation.
Hyperthermia and seizures should be managed aggressively. Hyperthermia is an important contributing factor to morbidity and mortality. Core temperature should be measured in all patients and continuous monitoring is recommended if the temperature is elevated. Mild-to-moderate hyperthermia (<39°C) usually responds to benzodiazepine sedation and fluid resuscitation. If this is unsuccessful, intubation and paralysis are indicated. There is no evidence that dantrolene has a role in the management of hyperthermia associated with psychostimulants. Benzodiazepines are first-line therapy for seizures, followed by barbiturates, then general anaesthesia and paralysis. Seizures in the context of acute hyponatraemia require initial rapid sodium replacement.
Following resolution of the acute intoxication phase, patients with persistent psychotic features may respond to an antipsychotic agent.
Disposition
The duration of observation in the ED and the need for admission will depend on the severity of intoxication, the influence of co-ingested drugs and the presence of co-morbidity or complications. Patients with mild intoxication (without hyperthermia or chest pain) may be observed in the ED and discharged when vital signs and mental status have returned to normal. The long half-lives of the amphetamines may dictate inpatient care if symptoms or abnormal vital signs do not resolve within a few hours.
Presentation to an ED provides an opportunity for preventative intervention. Patients should be counselled and offered strategies to avoid future toxicity or overdose or referred to drug detoxification services.
Cocaine
Pharmacology and pathophysiology
Coca leaves have been chewed by the natives of the South American Andes for approximately 1200 years and were first exported to Europe in 1580. Cocaine hydrochloride, or benzoylmethylecgonine hydrochloride, is a white powder prepared from the leaves of the Erythroxylon coca plant. ‘Freebase’ cocaine is an alkaloid prepared by mixing cocaine hydrochloride, water and baking soda and separating the precipitate. If the solvent is allowed to evaporate, pure cocaine crystals remain, known as ‘rock’ or ‘crack’. Cocaine reaches the cerebral circulation within seconds after smoking or insufflation. Gastrointestinal absorption may not peak for 90 min. Cocaine is hydrolysed by plasma and liver cholinesterase to an active metabolite. In animal models, cocaine is metabolized in the presence of ethanol to ethylecgonine. This is a myocardial depressant more potent than the sum of the depressant effects of cocaine and ethanol alone. Five to ten per cent of cocaine is excreted in the urine unchanged with a half-life of 60–90 min.
The pathophysiology of cocaine is complex. It is a CNS stimulant acting via enhanced release of noradrenaline, plus blockade of noradrenaline, dopamine and serotonin reuptake. With increasing doses, euphoria is followed by dysphoria, agitation, seizures and coma. Cocaine stimulates the medullary vasomotor centre resulting in hypertension and tachycardia. Small doses may produce transient bradycardia. At high levels, the medullary centre may be depressed, leading to respiratory depression.
Peripherally, cocaine inhibits the reuptake of adrenaline and noradrenaline and stimulates the presynaptic release of noradrenaline. This leads to a sympathomimetic response mediated through both α- and β-adrenoreceptors, leading to tachycardia, diaphoresis, vasoconstriction and hypertension. Increased psychomotor activity, vasoconstriction and direct hypothalamic toxicity, possibly mediated by dopamine receptors, contribute to hyperpyrexia.
Cocaine is an ester-type local anaesthetic that blocks fast sodium channels. In severe toxicity, hypotension may occur due to a direct toxic effect on the myocardium. Wide complex tachyarrhythmias are observed with cocaine toxicity. Sodium channel and potassium channel blockade occur, in addition to sympathomimetic, ischaemic and cardiomyopathic effects. Arrhythmias are related to multiple factors including dose, co-exposures, acid–base and electrolyte imbalance and genetic variability. Transient arrhythmias may account for syncope not attributable to seizures.
Epidemiology
In Australia, the prevalence of cocaine use is increasing, with 7.3% of the population aged over 14 years reporting cocaine use in their lifetime in 2010.
Clinical features
The predominant symptoms are those of CNS excitation and peripheral sympathomimetic response. CNS manifestations include agitation, altered mental state, seizures, dyspnoea, transient focal neurological signs, intracranial haemorrhage and coma. Cerebral infarction, transient ischaemic attacks, subarachnoid haemorrhage, cerebral vasculitis and migraine-like headache are described. Contributing mechanisms include hypertension, vasoconstriction, vasculitis, increased coagulability, altered cerebrovascular autoregulation and embolization of particulate matter.
Cardiovascular manifestations include tachycardia, hypertension, supraventricular or ventricular tachydysrrhythmias and syncope. Chest pain may be due to musculoskeletal, pulmonary or cardiovascular causes. Cocaine-induced myocardial ischaemia and infarction is multifactorial, due to increased myocardial oxygen demand, immediate or delayed coronary artery vasospasm, increased platelet aggregation, impaired thrombolysis, accelerated atherosclerosis and dilated cardiomyopathy. Aortic dissection associated with cocaine use is reported. A retrospective study of patients intoxicated with cocaine presenting with chest pain consistent with ischaemia found 6% suffered acute myocardial infarction.
Smoking cocaine may lead to respiratory complications including thermal airway injury, pneumothorax and pneumomediastinum, non-cardiac pulmonary oedema, interstitial pneumonitis and bronchiolitis obliterans. Tachypnoea, mydriasis, tremor, diaphoresis and hyperpyrexia may also be seen. Mesenteric vasoconstriction and vasculitis may lead to bowel ischaemia and infarction. Rhabdomyolysis complicated by renal failure and hyperkalaemia is reported.
Differential diagnosis
The differential diagnosis includes intoxication with other sympathomimetic agents, hallucinogenic agents, anticholinergic delirium, serotonin syndrome, monoamine oxidase inhibitors, alcohol and benzodiazepine withdrawal, sepsis, hypoglycaemia, thyrotoxicosis and phaeochromocytoma.
Clinical investigations
The diagnosis is clinical, based on history, sympathomimetic symptoms and signs and the exclusion of other conditions. Physical examination and investigations are directed at excluding complications and alternative diagnoses. All patients with altered vital signs should have a 12-lead ECG and continuous ECG monitoring.
Treatment
Initial care is directed towards the resuscitation and immediate threats to airway, breathing and circulation. Patients exhibiting CNS agitation or sympathomimetic cardiovascular effects should receive an intravenous benzodiazepine titrated to achieve sedation. This will control manifestations of cocaine toxicity in the majority of patients. Hyperthermia and seizures require aggressive resuscitation (as for amphetamines) to prevent a poor outcome. Seizures should be managed with benzodiazepines and barbiturates, escalating to general anaethesia if required.
Atrial tachycardia and hypertension usually respond to sedation with benzodiazepines. Atrial tachyarrhythmias are usually benign and rarely require specific treatment. Vasodilators, such as nitrates or phentolamine, are recommended for hypertension not responding to benzodiazepines. Ventricular arrhythmias should be treated according to advanced cardiac life support guidelines. In addition to defibrillation or cardioversion, immediate intravenous bolus bicarbonate (1–2 mEq/kg) and benzodiazepine sedation are used. Lignocaine may be an acceptable alternative. The use of β-adrenergic receptor blockers and class 1a and 1c antiarrhythmics in cocaine intoxication is contraindicated.
If myocardial ischaemia or infarction is suspected, investigation should be as for ischaemic chest pain of non-toxicological aetiology. Benzodiazepine sedation, nitrates and verapamil are recommended to decrease heart rate, reduce hypertension and reduce cocaine-induced coronary vasoconstriction. Primary angioplasty is considered the treatment of choice for cocaine-induced acute myocardial infarction. Thrombolytic therapy may be considered if angioplasty is not available, maximal medical management has failed, hypertension has been controlled and there is no evidence of intracranial haemorrhage. Ventricular arrhythmias occurring after the acute phase are presumed to be secondary to myocardial ischaemia and should therefore be treated in a standard manner. In patients with non-diagnostic electrocardiograms, short-term admission may be required to exclude myocardial infarction.
Disposition
The duration of observation in the ED and the need for admission depend on the severity of toxicity, co-ingested drugs and the presence of co-morbidity or complications. Patients with mild intoxication (without hyperthermia or ischaemic chest pain) may be observed in the ED and discharged when the patient is asymptomatic with normal vital signs and mental status.
Presentation to an ED provides an opportunity for preventative intervention. Patients should be counselled and offered strategies to avoid future toxicity or overdose or, if willing, referred to agencies specializing in drug detoxification and rehabilitation.
Gamma-hydroxybutyric acid
Pharmacology and pathophysiology
Gamma-hydroxybutyric acid (GHB) (4-hydroxybutanoate; sodium oxybate) is a sedative-hypnotic agent causing sedation and psychotropic effects. GHB is a short-chain fatty acid that acts as a neurotransmitter. It is one of the metabolites of gamma-aminobutyric acid (GABA). The mechanism by which GHB causes its effects is unclear, but is probably mediated by specific GHB and GABA-B receptors. It also has dopaminergic activity, increases acetylcholine and serotonin levels and may interact with endogenous opioids. GHB was developed as a short-acting anaesthetic agent, but lost favour due to poor analgesic properties and a propensity to cause seizure-like activity at the onset of coma.
GHB is ingested as a liquid and is rapidly absorbed, peaking within 15–45 min. It is metabolized by alcohol dehydrogenase to succinate, which enters the Krebs cycle. The average half-life is 20–50 min.
Epidemiology
GHB (‘grievous bodily harm’, ‘fantasy’, ‘scoop’, ‘liquid X’, ‘liquid E’) and its congeners gamma-butyrolactone (GBL) and 1,4-butanediol, have been advocated for body building, euphoria, sleep enhancement and sexual stimulation. Recreational use of GHB in Australia is uncommon, but increasing. The 2010 National Drug Strategy Household Survey reported that 0.8% of Australians over 14 years reported using GHB in their lifetime.
Clinical features
Most patients present to the ED following acute GHB intoxication. CNS effects with increasing dose include euphoria, then agitation followed rapidly by sedation and coma. Respiratory depression and apnoea may occur, but this is usually reported in the context of co-ingestants. Profound coma may occur but the patient may resist instrumentation of the airway or rouse when stimulated, only to relapse again when the stimulus is removed. Abrupt resolution of coma within 2–3 h of presentation is characteristic of GHB intoxication. However, co-ingested agents may cloud the clinical picture.
Agitation, myoclonus and generalized seizures are reported. ‘Seizures’ commonly represent myoclonic movements or may be generalized due to hypoxia or a co-ingested agent. Agitation is common on emergence from coma and patients may rapidly change from unresponsive to agitated and combative. Altered conscious state is associated with mild bradycardia and/or hypotension which rarely requires intervention. Chronic users may develop a withdrawal syndrome that mimics alcohol or sedative–hypnotic withdrawal.
Differential diagnosis
The differential diagnosis is that of any patient with an altered level of consciousness and includes toxicological and metabolic causes, sepsis, neurotrauma, stroke and post-ictal state. Persistent CNS depression beyond 6 h should prompt a search for alternative causes.
Investigations
The diagnosis of GHB intoxication is clinical. A thorough physical examination is usually adequate to exclude complications. Investigations are directed at excluding alternative diagnoses and complications.
Treatment
Management is supportive. Initial care is directed at resuscitation and immediate threats to airway, breathing and circulation. All patients should receive oxygen and have a bedside blood glucose measurement. Patients with an altered level of consciousness should be closely monitored in a resuscitation area, positioned to minimize the probability of aspiration and moved frequently to prevent dependent injuries. Intubation may required if the airway is threatened due to vomiting or prolonged coma due to co-ingested agents or complications. There is no specific antidote for GHB.
Disposition
The duration of observation will depend on the need for intubation, co-ingested agents or the presence of complications. Most patients recover within a few hours and may be safely discharged from the ED when they are ambulant and orientated.
Presentation to an ED also provides an opportunity for preventative intervention. Patients should be counselled regarding strategies to avoid future overdose. In addition, the patient may be referred to agencies specializing in drug detoxification and rehabilitation.
Emerging drugs of abuse–‘legal highs’
Introduction and epidemiology
Novel agents derived from synthetic chemicals, plants or fungal matter have recently come into use as alternatives to controlled drugs of abuse. These agents are structural analogues of endogenous neurotransmitters (especially serotonin, noradrenaline and dopamine) resulting in similar effects. There is a lack of data surrounding the pharmacology, toxicology, prevalence and effects of most of these agents. Most are sold as benign substances, such as ‘bath salts’ or ‘incense’ and labelled ‘not for human consumption’. Understanding of the effects of these agents is difficult given misleading packaging, unpredictable consistency and continual emergence of structural analogues. Caffeine is commonly added to the product, altering the observed clinical effects. There are three main groups of ‘legal highs’:
![]() stimulants (cathinones, synthetic cocaine, pipradols, piperazines)
stimulants (cathinones, synthetic cocaine, pipradols, piperazines)
![]() hallucinogens (cannabinoid receptor agonists, piperazines, methoxetamine)
hallucinogens (cannabinoid receptor agonists, piperazines, methoxetamine)
![]() depressants (GBL, novel opioids).
depressants (GBL, novel opioids).
Cathinones, piperazines, synthetic cannabinoid receptor agonists and methoxetamine are most well studied and will be considered here.
Stimulants: synthetic cathinones
Pharmacology and pathophysiology
Cathinone is a beta-ketone amphetamine analogue present in young leaves of the Catha edulis plant. Khat leaf chewing causes sympathomimetic effects similar to amphetamine, including tachycardia, hypertension, euphoria and increased alertness. Numerous synthetic phenylalkylamines have been derived, including mephedrone (‘meow-meow’, ‘MCAT’), methcathinone, methylone and methylenedioxypyrovalerone (MDPV; ‘bath salts’, ‘ivory wave’). Most cause sympathomimetic effects by increased release and reduced synaptic clearance of noradrenaline, dopamine and serotonin via monoamine uptake transporters. They are presented as a powder or pill which is insufflated, ingested or injected. The pharmacology of these agents in humans has not been well established. Reported onset of action after ingestion is 15–45 min with effects lasting 2–7 h.
Clinical features
Increased energy from ingestion of cathinones is reported by users to be better and longer lasting than cocaine. Twenty per cent report adverse symptoms including palpitations, GI upset and mental disturbance. Cardiac, neurological and psychiatric derangement is common, with agitation, sometimes severe and requiring restraint, being most common. A sympathomimetic toxidrome with chest pain, diaphoresis, tachycardia and hypertension may occur. Psychosis, anxiety, hallucinations and delusions are common, abnormal liver and renal function, rhabdomyolysis and hyponatraemia with seizures and death have been reported. Addiction potential and withdrawal symptoms are considered probable but are uncharacterized.
Clinical investigations
Diagnosis of intoxication is clinical, although cathinone levels can be obtained for forensic purposes.
Treatment
There are no data to suggest the optimal management of patients with cathinone toxicity. Routine decontamination and enhanced elimination are unlikely to be effective and there is no specific antidote. Management is supportive and has been extrapolated from that of other sympathomimetic agents, using benzodiazepine sedation as first line for agitation, seizures, psychosis, tachycardia and hypertension. Hyperthermia is treated aggressively with cooling measures and benzodiazepines, escalating to intubation and paralysis for fever with severe muscle rigidity. Treatment of hyponatraemia follows standard protocols depending on severity and clinical features.
Stimulants/hallucinogens: piperazines
Pharmacology and pathophysiology
Benzylpiperazine (BZP, ‘herbal party pills’, ‘BenzoFury’) is most well known among this group of agents, however, other agents, such as trifluoromethylphenylpiperazine (TFMPP), are commonly available. BZP is a dopamine reuptake inhibitor and increases dopamine release. BZP increases peripheral catecholamine release, causing sympathomimetic stimulation.
TFMPP is a direct serotonin agonist at 5HT-1 and 5HT-2 receptors and inhibits serotonin reuptake. Piperazines are metabolized by cytochrome P450 and may inhibit these enzymes, leading to drug interactions.
Clinical features
BZP and TFMPP are often combined to mimic closely the effects of MDMA. Effects depend on agent and dose administered, with stimulant effects predominant at lower doses and hallucinogenic effects (TFMPP) at higher doses; however, there is significant inter-individual variability. A sympathomimetic-type toxidrome is common with sinus tachycardia, hypertension, agitation, anxiety, confusion and gastrointestinal upset being reported. More severe effects include seizures, hyperthermia and related complications, movement disorders, chest pain/myocardial toxicity and hyponatraemia (rare). Effects are reported to last 6–8 h, however, lethargy, insomnia, anxiety, paranoia and mood disorders may persist for several days.
Clinical investigations
Diagnosis is clinical based on history and clinical features. While levels are available for forensic reasons, they do not correlate well with toxicity and are not clinically useful. Further investigation may be required to exclude complications or co-morbid conditions.
Treatment
Management is symptomatic and supportive. Routine gastrointestinal decontamination and enhanced elimination are unlikely to be of benefit. There is no specific antidote. Initial management is aimed at resuscitation and immediate life threats of airway, breathing, circulation and seizures along normal pathways. Intravenous benzodiazepine sedation may be required for seizures, agitation or generalized muscle rigidity and is adequate to control symptoms for most cases. Hyperthermia should be treated aggressively with cooling measures and titrated benzodiazepines with escalation to intubation and paralysis if necessary.
Hallucinogens: synthetic cannabinoid receptor agonists
Pharmacology and pathophysiology
Synthetic cannabinoid receptor agonists (‘kronic’, ‘spice’, ‘k2’, ‘chill out’, ‘chaos’) act at cannabinoid receptors CB1 and CB2, as well as NMDA receptors. There are seven structural groups of agents (JWH, CP and HU compounds) which are added to herbal mixes and smoked. These agents have a higher affinity for the cannabinoid receptors than tetrahydrocannabinol (THC). Pharmacokinetic properties have not been characterized.
Clinical features
Case series suggest psychiatric effects predominate including anxiety and paranoia, agitation and delusions. Tachycardia and diaphoresis are common. Seizures have been reported with some compounds. A withdrawal syndrome has been reported in habitual users.
Clinical investigations
Diagnosis is clinical based on history and examination findings. Further investigations are aimed at excluding complications and alternative diagnoses. Levels are not readily available within a clinically significant time frame, but can be obtained for occupational and forensic reasons.
Treatment
There is no specific antidote for cannabinoid receptor antagonists. Management is symptomatic and supportive. Benzodiazepine sedation may be required for agitation, hallucinations and seizures.
Hallucinogens: methoxetamine
Pharmacology and pathophysiology
Methoxetamine is a ketamine analogue/phencyclidine derivative which is an antagonist at NMDA receptors and inhibits dopamine reuptake from the synapse. It is absorbed by oral, nasal, rectal and intramuscular routes with rapid onset and short duration of symptoms.
Clinical features
Effects are similar to those of ketamine, with euphoria and perceptual disturbances. Severe nausea and vomiting, diarrhoea, anxiety and paranoia have been reported. Tachycardia and nystagmus are common. Respiratory depression may occur.
Treatment
Diagnosis is clinical. Management is symptomatic and supportive. Benzodiazepines, antiemetics and intravenous fluid are likely to be required. Respiratory support may be needed.
‘Body-packers’, ‘body-stuffers’
Epidemiology
Body-packers and body-stuffers conceal illicit drugs within body cavities. Patients may ingest many times a lethal dose but appear asymptomatic at presentation. A body-packer conceals a large quantity of an illicit drug inside a body cavity, usually the GI tract, in an attempt to smuggle it across international borders. Up to 1 kg of drugs may be carefully packaged in 50–150 packages and layered with wax to prevent leaking. A body-stuffer ingests smaller quantities of drugs before apprehension by the authorities. The package is usually poorly constructed and is more likely to leak. Time from ingestion to hospital arrival is usually short. The vagina and rectum are alternative sites for drug concealment in the body-stuffer. Heroin, cocaine, amphetamine derivatives, MDMA and cannabis are all reported in body-packers and -stuffers.
History may be misleading, especially if taken in the presence of law enforcement officers. Ideally, a detailed history should be obtained noting the type and amount of drug ingested, the method of packaging, symptoms of drug intoxication and any factors that may increase the likelihood of bowel obstruction or ileus. A thorough physical examination should include examination of the vagina and rectum and a search for any signs of drug intoxication.
Clinical investigations
There is a paucity of data regarding optimal imaging modality, decontamination and treatment. Imaging of the potential body-packer is controversial. Abdominal radiographs are often positive in body-packers, where multiple package–air interfaces may be seen. The sensitivity and specificity of plain abdominal films in large series is reported to be 85–90%. Importantly, negative plain radiographs do not exclude the diagnosis. Abdominal computed tomography (CT) scanning with oral contrast has been recommended, although sensitivity is not 100%. Qualitative urine drug screens do not change management and are not routinely indicated. In the presence of signs of intoxication, investigations for complications and co-morbidities may be appropriate.
Treatment
In view of the potential sudden lethality of both these practices, patients should receive a high triage priority and be managed in a resuscitation setting with secure IV access regardless of presenting symptoms or signs. Evidence of drug intoxication, either at presentation or during decontamination, represents a medical emergency and requires aggressive management. Initial care is directed at assessment and resuscitation of immediate threats to airway, breathing, circulation and the control of seizures as required.
Gastrointestinal decontamination of body- packers and body-stuffers is controversial and should be considered on a case-by-case basis. If cooperative, patients should receive activated charcoal to adsorb intraluminal drug. Whole bowel irrigation with polyethylene glycol at 2 L/h via nasogastric tube until all packages are passed has been recommended. However, a conservative approach to asymptomatic patients has recently been advocated with observation, laxatives and light diet until all packets are retrieved. The risk of late-onset drug intoxication, bowel obstruction, laparotomy or death is less than 5% with this approach. Surgical exploration is indicated if there is gastric outflow or bowel obstruction, concretion formation, ileus or perforation. If a cocaine or amphetamine body-packer exhibits toxicity, immediate surgical exploration to remove all packages has been advocated. Surgery is probably not necessary in the opiate body-packer as adequate resuscitation, supportive care and antidote therapy should ensure a favourable outcome. Body-stuffers with a single package located in the stomach may be amenable to endoscopic removal, especially if there is failure to progress.
All body-packers should remain in a closely monitored environment in the ED, observation unit or intensive care unit until all packages have been retrieved. Staff should be aware of potential signs of intoxication and be available to intervene in the event of complications. Patients are observed until all packages have been accounted for and there have been three normal package-free stools. Repeat radiology is performed to confirm clearance before discharge.
Controversies
![]() Optimal sedation regimen for cocaine and amphetamine-intoxicated patients.
Optimal sedation regimen for cocaine and amphetamine-intoxicated patients.
![]() Intubation vs expectant management for patients with GHB intoxication.
Intubation vs expectant management for patients with GHB intoxication.
![]() Optimal management of patients using synthetic agents or ‘legal highs’.
Optimal management of patients using synthetic agents or ‘legal highs’.
![]() Optimal imaging and gastrointestinal decontamination in body-packers.
Optimal imaging and gastrointestinal decontamination in body-packers.
Further reading
1. Fatovich DM, Bartu A, Davis G, et al. Morbidity associated with heroin overdose presentations to an emergency department: a 10 year record linkage study. Emerg Med Australas. 2010;22:240–245.
2. Glauser J, Queen JR. An overview of non-cardiac cocaine toxicity. J Emerg Med. 2007;32:181–186.
3. Greene S, Kerr F, Braitberg G. Review article: amphetamines and related drugs of abuse. Emerg Med Australas. 2008;20:391–402.
4. Hoffman RS. Treatment of patients with cocaine-induced arrhythmias: bringing the bench to the bedside. Br J Clin Pharmacol. 2010;69:448–457.
5. Lange RA, Hillis LD. Medical progress: cardiovascular complications of cocaine use. N Engl J Med. 2001;345:351–358.
6. Mason PE, Kerns WP. Gamma hydroxybutyric acid (GHB) intoxication. Acad Emerg Med. 2002;9:730–739.
7. Prosser JM, Nelson LS. The toxicology of bath salts: a review of synthetic cathinones. J Med Toxicol. 2012;8:33–42.
8. Rosenbaum CD, Carreiro SP, Babu KM. Here today, gone tomorrow…and back again? A review of herbal marijuana alternatives (K2, spice) Synthetic cathinones (bath salts), kratom, Salvia divinorum, methoxetamine and piperazines. J Med Toxicol. 2012;8:15–32.
9. Schep LJ, Slaughter RJ, Vale JA, et al. The clinical toxicology of the designer “party pills” benzylpiperazine and trifluoromethylphenylpiperazine. Clin Toxicol. 2011;49:131–141.
10. Sporer KA. Acute heroin overdose. Ann Intern Med. 1999;130:584–590.
11. Traub SJ, Hoffman RS, Nelson LS. Body packing–the internal concealment of drugs. N Engl J Med. 2003;349:2519–2526.
29.14 Methaemoglobinaemia
Robert Edwards
Essentials
1 Consider the diagnosis of methaemoglobinaemia in patients with cyanosis unresponsive to oxygen.
2 Multiple presentations can occur following incidents involving contamination of food or water. Early clinical recognition allows institution of treatment and prevention of further cases.
3 Pulse oximetry is misleading in methaemoglobinaemia. Readings do not usually fall below 85%.
4 Administer methylene blue to symptomatic patients with elevated methaemoglobin levels and to unstable patients with a history of exposure to an agent known to cause methaemoglobinaemia.
5 Methylene blue can cause haemolysis and methaemoglobinaemia if given to patients who do not have methaemoglobinaemia or who are G6PD deficient or if more than 5 mg/kg is used.
6 Failure to respond to methylene blue may result from too small or too large a dose, congenital enzyme or haemoglobin defects or an incorrect diagnosis.
Introduction
Although it is an uncommon presentation, the emergency physician must be able to diagnose methaemoglobinaemia because it is potentially fatal and can be readily treated with the antidote, methylene blue.
Aetiology and pathophysiology
Under normal conditions, methaemoglobin is continuously produced from haemoglobin by the oxidation of the iron molecule from the ferrous (Fe2+) to the ferric (Fe3+) state. In the normal physiological state, less than 1% of haemoglobin is methaemoglobin because it is continuously being reduced, predominantly by the enzyme NADH methaemoglobin reductase.
Excessive methaemoglobinaemia causes tissue hypoxia because methaemoglobin is incapable of carrying oxygen and causes a shift of the oxygen haemoglobin dissociation curve to the left.
Methaemoglobinaemia may be acquired or congenital. Congenital methaemoglobinaemia is due either to a deficiency of the enzyme NADH methaemoglobin reductase (a rare autosomal recessive condition) or the haemoglobinopathy, haemoglobin M (Milwaukee). The latter is transmitted with an autosomal inheritance. Homozygotes usually do not survive and heterozygotes live with a methaemoglobin level of around 15–30%. Patients with glucose-6-phosphate dehydrogenase (G6PD) deficiency are at increased risk of developing methaemoglobinaemia when exposed to an oxidizing agent, due to low levels of NADPH.
Acquired methaemoglobinaemia in adults arises as a consequence of accidental or intentional exposure to a therapeutic drug or other oxidizing agent. Oxidants that commonly result in excessive methaemoglobin production are listed in Table 29.14.1.
Table 29.14.1
Agents causing acquired methaemoglobinaemia

Edwards RJ, Ujma J. Extreme methaemoglobinaemia secondary to recreational use of amyl nitrite. Journal of Accident and Emergency Medicine 1995;12:138–42 with permission.
Nitrates, nitrites and local anaesthetics are the culprits most commonly reported in the medical literature. Recreational use of amyl, butyl or isobutyl nitrite can cause severe methaemoglobinaemia. Transdermal absorption of industrial nitrate solutions, ingestion of food and water contaminated with nitrates and the intravenous use or inhalation of nitrates may all cause methaemoglobinaemia. Sodium nitrite is commonly used commercially as a food preservative, colouring agent or corrosion inhibitor. In 2006, a total of five patients suffering methaemoglobinaemia (ranging from 21 to 57%) presented to a Sydney emergency department in two separate clusters due to Asian food being prepared with an additive containing 100% sodium nitrite bought in Asian grocery stores.
Therapeutic use of glyceryl trinitrate (GTN) has been reported to increase methaemoglobin levels up to 38%, but it is more likely to cause severe hypotension before methaemoglobinaemia develops. Prolonged use of high doses of GTN (>10 μg/kg/min) and the presence of renal or hepatic dysfunction make this complication more likely.
Local anaesthetics, prilocaine and benzocaine in particular, can cause methaemoglobinaemia even when applied topically and administered in standard doses.
Risk factors for the development of methaemoglobinaemia from topically applied local anaesthetics include excessive dosing, a break in the mucosal barrier and a partial deficiency of the enzyme NADH methaemoglobin reductase.
Dapsone therapy can cause both haemolytic anaemia and methaemoglobinaemia.
Aniline (aminobenzene) and its major metabolite, phenyl hydroxylamine, are potent methaemoglobin-forming agents, even after transdermal exposure. Aniline and related compounds, such as nitrobenzene, are used widely in industry, especially the chemical and rubber industries.
In more recent times, the widespread availability of drugs over the Internet has seen further reports of methaemoglobinaemia. This includes a report of two men who took a small amount of what they believed to be the recreational psychoactive drug 2C-E (a member of the phenethylamine family) produced in China which was later found to be pure aniline.
Clinical features
The symptoms and signs of methaemoglobinaemia are attributable to the effects of cellular hypoxia on the CNS and the heart. At levels between 25% and 40%, headache, weakness, anxiety, lethargy, syncope, tachycardia and dyspnoea are observed. Further elevations are associated with decreasing level of consciousness (45–55%) leading to coma, seizures, arrhythmias and cardiac conduction disturbances (55–70%). Levels above 70% are associated with mortality, but deaths can occur at lower levels.
With accidental exposures (either ingestion or cutaneous), multiple presentations of either family members or co-workers can be encountered after common exposure to the offending agent. It is not unusual in these circumstances for the relationship between the toxic substance and the presentations to be unclear initially.
The hallmark of methaemoglobinaemia is a deep cyanosis that is unresponsive to oxygen therapy. The cyanosis may be so deep that it is more brown than blue and has been termed chocolate cyanosis. A useful diagnostic clue is the classic chocolate brown appearance of the patient’s blood. This may be observed at methaemoglobin levels as low as 15–20%.
Clinical investigations
The diagnosis of methaemoglobinaemia is confirmed by spectrophotometric measurement of methaemoglobin. The result is expressed as a percentage of the total haemoglobin level. Analysis of the sample should be performed as soon as possible because methaemoglobin levels fall with time. The indications for spectrophotometry are:
![]() cyanosis unresponsive to oxygen
cyanosis unresponsive to oxygen
![]() tachypnoea or other features of hypoxia and history of exposure to methaemoglobin-inducing agents
tachypnoea or other features of hypoxia and history of exposure to methaemoglobin-inducing agents
![]() normal or raised PaO2 and low SpO2 on pulse oximetry
normal or raised PaO2 and low SpO2 on pulse oximetry
![]() chocolate brown appearance of arterial blood.
chocolate brown appearance of arterial blood.
Arterial blood gas analysis often demonstrates a metabolic acidosis with a normal oxygen tension. Other important investigations include a chest X-ray to exclude pulmonary pathology that might contribute to hypoxia and an ECG to assess cardiac rhythm and look for evidence of myocardial ischaemia or infarction. A full blood count to check haemoglobin (oxidizing agents, such as aniline and their metabolites, can cause a chemical-induced haemolytic anaemia) and electrolytes, urea, creatinine and liver function tests should also be performed. Consider a G6PD level if methylene blue fails to work.
Pulse oximetry
Methaemoglobin interferes with the accuracy of pulse oximetry. With increasing levels of methaemoglobin, pulse oximetry readings approach 85% (at around 30% methaemoglobin) and remain in the mid-eighties range. Methaemoglobin has a maximal light absorption at a wavelength similar to that of oxyhaemoglobin (660 nm) and is therefore not differentiated from oxyhaemoglobin.
Pulse co-oximeters which measure more than the standard two wavelengths of light, can distinguish not only methaemoglobin but also carboxyhaemoglobin and, if available, can confirm the diagnosis of methaemoglobin by direct transcutaneous measurement.
Treatment
Initial management includes assessment of the airway, breathing and circulation and institution of appropriate measures of care. Administration of oxygen therapy is often not associated with any clinical benefit but the presence of cyanosis not responsive to oxygen is a diagnostic clue.
Decontamination of the gastrointestinal (GI) tract or skin may be indicated.
Antidote
Methylene blue (tetramethyl thionine), 1–2 mg/kg IV, is a specific antidote for methaemoglobinaemia. Normally, 95% of methaemoglobin is reduced by the NADH methaemoglobin reductase system, a greater proportion is reduced by a second enzyme system, NADPH methaemoglobin reductase when methylene blue is present acting as a cofactor to methaemoglobin reductase.
NADPH is produced by the Embden–Myerhoff pathway and requires adequate G6PD activity. Thus, in states of G6PD deficiency, methylene blue may not be as effective.
Methylene blue is indicated for symptomatic patients with an elevated methaemoglobin level. Patients who are blue but asymptomatic do not require methylene blue. Symptoms can normally be expected in patients with levels greater than 15%, less if the patient is anaemic.
The dose of methylene blue is 1–2 mg/kg intravenously over 5 min. Unstable patients with cyanosis unresponsive to high flow oxygen and a history of oxidant exposure or ‘chocolate brown’ blood should be given methylene blue even if the methaemoglobin level is not available. A reduction in the methaemoglobin level and accompanying clinical improvement usually occur over 30–60 min. A further dose of 1 mg/kg can be given after 1 h if the methaemoglobin level remains elevated. Factors that may result in failure to respond are listed in Table 29.14.2.
Table 29.14.2
Reasons for failure of methaemoglobinaemia to respond to methylene blue (MB)

The side effects of methylene blue include dyspnoea, a feeling of pressure on the chest, restlessness, apprehension, tremor, nausea and vomiting. Paradoxically, methylene blue itself can oxidize haemoglobin to methaemoglobin if given in high doses (>5–7 mg/kg). Adverse effects are minimized if the correct dose is used. Methylene blue occasionally causes persistent blue discoloration of the patient or haemolytic anaemia. G6PD-deficient patients should not be given methylene blue as it may precipitate massive haemoloysis.
Continuous infusion of methylene blue has been used to treat prolonged methaemoglobinaemia formation associated with dapsone. Methylene blue has a short half-life, there may be delayed increase in methaemoglobin level up to 12 h after the administration of methylene blue. Therefore, serial monitoring of methaemoglobin level may be indicated. Aniline dye-induced methaemoglobinaemia may be resistant to treatment as its metabolite (phenylhydroxylamine) blocks uptake of methylene blue.
Other therapies
Exchange transfusion
Exchange transfusion is indicated for patients with G6PD deficiency or where there is failure to respond to methylene blue or where there is significant haemolysis and methylene blue relatively contraindicated.
N-acetylcysteine
N-acetylcysteine (NAC) has been proposed and used in case reports as a supplemental reducing substance in methaemoglobinaemia. It contains a reduced sulphydryl group. However, small in vitro and in vivostudies have not shown an improvement in methaemoglobin levels and its effectiveness has not been proven.
Controversies
![]() Treatment with hyperbaric oxygen has been recommended as an adjuvant treatment to methylene blue. Realistically, it should only be considered where there has been an inadequate response to methylene blue. The partial pressure of oxygen can be increased to such a degree so as to ensure adequate oxygen transport in the absence of functioning haemoglobin.
Treatment with hyperbaric oxygen has been recommended as an adjuvant treatment to methylene blue. Realistically, it should only be considered where there has been an inadequate response to methylene blue. The partial pressure of oxygen can be increased to such a degree so as to ensure adequate oxygen transport in the absence of functioning haemoglobin.
![]() Adjuvant treatment with ascorbic acid (vitamin C) has been recommended. It has a direct effect in reducing methaemoglobin, but this effect is too slow for it to be used as a primary treatment. The dose is 0.5–1.0 g 6-hourly, either orally or intravenously.
Adjuvant treatment with ascorbic acid (vitamin C) has been recommended. It has a direct effect in reducing methaemoglobin, but this effect is too slow for it to be used as a primary treatment. The dose is 0.5–1.0 g 6-hourly, either orally or intravenously.
Further reading
1. Berlin G, Brod AB, Hilden JO, et al. Acute dapsone intoxication: a case treated with continuous infusion of methylene blue, forced diuresis and plasma exchange. Clin Toxicol. 1984;22:537–548.
2. Center for Disease Control. Severe methemoglobinemia and hemolytic anemia from aniline purchased as 2C-E (4-ethyl2,5-dimethoxyphenethylamine), a recreational drug, on the Internet. MMWR. 2012;61:88–91.
3. Curry S. Methemoglobinemia. Ann Emerg Med. 1982;11:214–221.
4. Edwards RJ, Ujma J. Extreme methaemoglobinaemia secondary to recreational use of amyl nitrite. J Accid Emerg Med. 1995;12:134–137.
5. Goluboff N, Wheaton R. Methylene blue induced cyanosis and acute haemolytic anaemia complicating treatment of methaemoglobinaemia. J Paediatr. 1961;58:86–90.
6. Hunter L, Gordge L, Dargan PI. Methaemoglobinaemia associated with the use of cocaine and volatile nitrites as recreational drugs: a review. Br J Clin Pharmacol. 2011;7291:18–26.
7. Kellet PB, Copeland CS. Methemoglobinemia associated with benzocaine containing lubricant. Anesthesiology. 1983;59:463–464.
8. Maric P, Sayed SA, Heron LG, et al. Methaemoglobinaemia following ingestion of a commonly available food additive. Med J Am. 2008;188:156–158.
9. Reider HU, Frei FJ, Zbinden AM, Thomson DA. Pulse oximetry in methaemoglobinaemia Failure to detect low oxygen saturation. Anaesthesia. 1989;44:326–327.
10. Rosen PL, Johnson C, McGehee WG. Failure of methylene blue in toxic methaemoglobinaemia Association with glucose-6-phosphate dehydrogenase deficiency. Ann Intern Med. 1971;75:83–86.
11. Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis and management. South Med J. 2011;104:757–761.
12. Stucke AG, Riess ML, Connolly LA. Hemoglobin M (Milwaukee) affects arterial oxygen saturation and makes pulse oximetry unreliable. Anesthesiology. 2006;104:887–888.
29.15 Cyanide
George Braitberg
Essentials
1 Cyanide is a metabolic poison associated with a high mortality.
2 Cyanide toxicity is characterized by rapid onset of central nervous, respiratory and cardiovascular effects and by metabolic acidosis.
3 Cyanide exposure correlates well with serum lactate levels.
4 Prompt resuscitative efforts with high flow oxygen and administration of antidotes may be life saving; a number of alternative agents are available.
5 Cyanide poisoning from smoke inhalation is often overlooked and treatment is complicated by the potential coexistence of carboxy- and methaemoglobinaemia.
Introduction and epidemiology
Cyanide is used in a variety of commercial processes including metal extraction (especially gold) and recovery, metal hardening and in the production of agricultural and horticultural pest control compounds. Exposure can also occur to hydrogen cyanide (HCN) gas, produced when inorganic cyanide comes in contact with mineral acids as in electroplating, or accidentally when cyanide solutions are poured into acid waste containers. Cyanide off-gassing in house fires is well documented.
Death from cyanide poisoning is one of the most rapid and dramatic seen in medicine and antidotal therapy must be given early to alter outcome. Having said that, there is a number of case reports where patients with lethal cyanide blood levels have survived with good supportive care. A dose of 200 mg of ingested cyanide, or 3 min exposure to HCN gas, is potentially lethal.
Fortunately, serious acute cyanide poisoning is rare. However, the incidence of cyanide poisoning may be significantly underestimated. Blood cyanide concentrations greater than the toxic level of 40 μmol/L were found in 74% of victims found dead at the scenes of fires.
Cyanide is considered a likely agent of terrorism because it possesses attributes of the ideal chemical weapon. It is plentiful, available and, because of its use in industry and research laboratories, is widely distributed making it susceptible to theft, hijacking attempts and other terrorist acts. Cyanide does not require special knowledge to use and it is capable of causing mass incapacitation and casualties. It can be released in the atmosphere as a gaseous weapon or introduced into pharmaceuticals, the food supply and is considered a primary threat to water supplies. In Australia, cyanide has been used to adulterate medications.
Toxicokinetics and pathophysiology
The uptake of cyanide into cells is rapid and follows a first-order kinetic simple diffusion process. The half-life of cyanide is from 2 to 3 h.
While the precise in vivo action of cyanide is yet to be determined, it is thought that its major effect is due to binding with the ferric ion (Fe3+) of cytochrome oxidase, the last cytochrome in the respiratory chain. This results in inhibition of oxidative phosphorylation, halting electron transport, oxygen consumption and ATP formation. This leads to a net accumulation of hydrogen ions, a change in the NAD:NADH ratio and greatly increased lactic acid production. Other enzymatic processes, involving antioxidant enzymes, catalase, superoxide dismutase and glutathione, may contribute to toxicity. Cyanide is also a potent stimulator of neurotransmitter release in both the central and the peripheral nervous systems. Humans detoxify cyanide by transferring sulphane sulphur, R–Sx–SH, to cyanide to form thiocyanate (SCN). The availability of R–Sx–SH is the rate-limiting step.
Clinical features
Any acute cyanide exposure is potentially lethal. Onset of symptoms is usually rapid and can be within seconds to minutes for inhalation and within an hour for oral exposure. The ‘classical’ presentation is rapid onset of coma, seizures, shock and profound lactic acidosis.
Cyanide toxicity is characterized by effects on the central nervous system (CNS), respiratory and cardiovascular systems and by a marked metabolic acidosis.
CNS manifestations, in order of increasing severity of cyanide exposure, are headache, anxiety, disorientation, lethargy, seizures, respiratory depression, CNS depression and cerebral death. An initial tachypnoea gives way to respiratory depression as CNS depression develops.
Cardiovascular manifestations include hypertension followed by hypotension, tachycardia followed by bradycardia, arrhythmias, atrioventricular block and increased cardiac output followed by myocardial depression and cardiovascular collapse. Cyanide poisoning can shorten the QT interval to the point of ‘T on R’ phenomenon. The classic finding of bright red skin due to poor tissue oxygen (secondary to a decreased arteriovenous oxygen gradient) is not observed if significant myocardial, respiratory or CNS depression has already occurred, in which case the patient may appear cyanotic.
Clinical investigations
Arterial blood gas analysis and serum lactate measurements reveal metabolic acidosis with a raised lactate. Concentration decay curves suggest that serum lactate concentration is closely correlated to blood cyanide concentration. In smoke-inhalation victims without severe burns, plasma lactate concentrations above 10 mmol/L correlate with blood cyanide concentrations above 40 μmol/L, with a sensitivity of 87%, a specificity of 94% and a positive predictive value of 95%.
Cyanide is concentrated 10-fold by erythrocytes and whole-blood cyanide concentrations are used as the benchmark when comparing levels. A level of 40 μmol/L is considered toxic and a level of 100 μmol/L potentially lethal. Symptomatic intoxication starts at levels of about 20 μmol/L. Cyanide levels are usually not available in a clinically significant time frame.
A semiquantitative bedside test for cyanide in blood has been used but is not readily available.
Treatment
Resuscitation
Attention to airway, breathing, circulation and other resuscitative measures must be instituted immediately. Patients should be ventilated with 100% O2. Patients must be removed from enclosed or confined spaces with high airborne concentrations of cyanide. Rescuers, likewise, must not enter such areas without full protective clothing and proper respirators or self-contained breathing apparatus. Mouth-to-mouth breathing should never be done.
Decontamination
Exposed skin and eyes should be copiously flushed with water or normal saline in an attempt to decontaminate the patient. All clothes should be removed and bagged. Activated charcoal is given only after the airway is secured.
Antidote treatment principles
In a recent review for the Australian Resuscitation Council, Reade et al. concluded that:
Cardiorespiratory collapse, combined with either a high blood cyanide level or obvious evidence of cyanide poisoning, is a clear indication for use of an antidote. Under these circumstances, the use of antidotes with even narrow therapeutic indices would appear reasonable. However, the indication for use of a cyanide antidote is less clear when smaller quantities are ingested, in patients without cardiorespiratory collapse, or where the diagnosis is unclear. Under these circumstances, it is logical to avoid the more toxic antidotes and this is reflected in the TGA-approved product information for dicobaltedetate. Hydroxocobalamin and sodium thiosulphate have few adverse effects, justifying their use in lesser degrees of cyanide poisoning.
There is uncertainty about the prevalence and clinical significance of cyanide poisoning from smoke inhalation. Yeoh and Braitberg found 11 of 138 patients with fire-related deaths had potentially fatal blood cyanide levels. They suggested that in patients with severe burns, elevated lactate or carboxyhaemoglobin greater than 10%, the use of a safe antidote should be considered.
Therapeutic endpoints in treatment are improvement in conscious state, haemodynamic stability and improvement in metabolic acidosis.
Specific cyanide antidotes
Hydroxocobalamin (Cyanokit [package insert] Columbia, MD: Meridian Technologies, Inc., 2009)
A recent systematic review of cyanide poisoning management for the Australian Resuscitation Council has recommended this as the initial antidote for the management of adults with suspected severe cyanide poisoning.
Hydroxocobalamin (vitamin B12A) is the cyanide antidote most widely used in Europe. It complexes with cyanide, on a mole-for-mole ratio to form cyanocobalamin. Antidotal doses of hydroxocobalamin are approximately 5000 times the physiological dose.
Hydroxocobalamin and cyanocobalamin are excreted by the kidney. The half-life of hydroxocobalamin in cyanide-exposed patients is 26.2 h. As the half-life of cyanide in smoke inhalation victims is calculated to be between 1.2 and 3.0 h, it is suggested that hydroxocobalamin can be satisfactorily used as single-dose therapy. The amount of cyanocobalamin formed after a dose of 5 g hydroxocobalamin correlates linearly until a blood cyanide level of 40 μmol/L is reached. At higher blood cyanide concentrations, there is little further rise in plasma cyanocobalamin and it is suggested that the rate-limiting step in the formation of cyanocobalamin is the availability of antidote, not the absence of cyanide ions. In cases of ingestion of cyanide with suicidal intent (where blood cyanide levels may be>150 μmol/L or plasma lactate concentrations>20 μmol/L), the usual dose of 5–10 g may be insufficient.
Extensive research has demonstrated the safety of this drug. In healthy adult smokers, 5 g of IV hydroxocobalamin is associated with a transient reddish discoloration of the skin, mucous membranes and urine and a mean elevation in systolic blood pressure of 14%, with a concomitant 16% decrease in heart rate. No other clinical adverse effects are noted and allergic reactions are rare. There is substantial experimental evidence to support the efficacy of hydroxocobalamin at lower levels of toxicity Hydroxocobalamin has been shown to be safe and efficacious in mild-to-moderate cyanide poisonings with levels up to 150 μmol/L and has been given successfully to patients with severe cyanide toxicity.
There are no data comparing the efficacy of hydroxocobalamin with other antidotes, so it is not possible to make any definitive conclusion about which antidote is best. However, in the emergency situation hydroxocobalamin appears to offer a greater margin of safety.
Hydroxocobalamin is a strong red chromophore with absorption maxima at 274 nm and 351 nm and no absorption above 600 nm. Interference with co-oximetric and colorimetric laboratory measurements has been reported. Carlsson et al. found some clinically important result errors that might lead to misdiagnoses and incorrect treatment, including a falsely lowered carboxyhaemoglobin and a falsely elevated lactate. Depending on the pathology system used a positive bias on methaemaglobin measurements may occur.
Hydroxocobalamin has a reasonably long shelf life, but is expensive. Hydroxocobalamin has also been recommended as the treatment of choice for mass casualty chemical disasters where cyanide poisoning is suspected.
Cyanide antidote kit
Administration of sodium nitrite followed by sodium thiosulphate is a long-accepted antidote for cyanide poisoning. The Cyanide Antidote kit was originally introduced in 1970 (under the pharmaceutical name Eli Lilly Cyanide kit) and contains:
![]() amyl nitrite perles*
amyl nitrite perles*
![]() sodium nitrite 10 mL (30 mg/mL)
sodium nitrite 10 mL (30 mg/mL)
![]() sodium thiosulphate 50 mL (250 mg/mL).
sodium thiosulphate 50 mL (250 mg/mL).
The kit is based upon the premise that humans can tolerate up to 30% methaemoglobinaemia. Conversion of haemoglobin to methaemoglobin promotes the movement of cyanide out of the cytochrome system; 4 mg/kg of sodium nitrite takes 30 min to achieve 7–10.5% methaemoglobin. The formation of sodium thiocyanate allows for the reformation of Hb2+, restoring the oxygen-carrying capacity of haemoglobin. Cellular respiration can continue as normal with cyanide removed from the respiratory chain. The observation that dramatic improvements in symptoms have occurred well before methaemoglobin levels have peaked has led many authors to suggest different mechanisms of action, such as vasodilatation and extracellular redistribution of cyanide. In smoke inhalation victims with suspected combined carbon monoxide and cyanide poisoning, the addition of 10% methaemoglobin may have clinically significant synergistic detrimental effects on the oxyhaemoglobin dissociation curve; in this setting, methaemoglobin inducers should be avoided.
Amyl nitrite alone in the management of per-hospital mass casualty cyanide poisoning has been reviewed recently. Evidence for its use is limited to animal studies and its role in the management of human case reports relative to the other treatments administered (e.g. life support, sodium nitrite and sodium thiosulphate) is unclear. Amyl nitrite has significant adverse effects (hypotension, syncope, excessive methaemoglobinaemia and haemolysis in glucose-6-phosphate dehydrogenase [G6PD] deficient patients). On balance, its use is not recommended in the pre-hospital or hospital setting.
Thiosulphate on its own can function as a slow sulphur donor, converting cyanide to thiocyanate. Thiosulphate is relatively non-toxic, although nausea and vomiting may occur following treatment. When hydroxocobalamin is not available, given its relatively favourable adverse-effect profile, it should be given to all patients with suspected cyanide toxicity, including those with smoke inhalation. It has been effective in the treatment of toxicity due to sodium nitroprusside therapy.
Dicobalt edetate (Kelocyanor)
This inorganic cobalt salt was introduced as a cyanide antidote in the late 1950s. It complexes with cyanide to form cobalt cyanide, thus removing cyanide from the circulation and reducing toxicity. However, unless cyanide is forced into the extracellular fluid, tissue levels are minimally affected.
Adverse effects are considerable and may be life threatening. Severe hypotension, cardiac arrhythmias, convulsions and gross oedema are reported. These effects are exacerbated when the drug is administered to an individual who is not cyanide poisoned. The treating physician therefore faces a significant dilemma when presented with a critically ill patient in whom the history of exposure is unclear. The use of this antidote is only for confirmed cyanide poisoning cases where the patient has lost consciousness and safer antidotes are not available.
The recommended initial dose of dicobaltedetate is 300 mg IV. Further doses may be required.
Other therapies
Hyperbaric oxygen (HBO) has been proposed as a treatment in cyanide poisoning but remains controversial with conflicting animal data. In most published human reports, HBO is offered after a combination of modalities and it is not possible to determine the treatment effect specific to each.
Experimental antidotes, such as nano alpha-ketoglutarate and trimethoprim derivatives, are currently being studied.
Recommended antidotal regimen
The regimen below is recommended if available:
![]() 5–15 g of hydroxocobalamin IV over 30 min (but may be given as IV push if needed). Repeat if needed.
5–15 g of hydroxocobalamin IV over 30 min (but may be given as IV push if needed). Repeat if needed.
![]() Plus (as an adjunct to the treatment of severe cyanide poisoning following failure to respond to hydroxycobalamin).
Plus (as an adjunct to the treatment of severe cyanide poisoning following failure to respond to hydroxycobalamin).
![]() 12.5 g sodium thiosulphate (or 0.5 g/kg IV for paediatric patients up to the adult dose) as a bolus injection or infused over 10–30 min. One half of the initial dose can be administered 2 h later if toxicity reappears.
12.5 g sodium thiosulphate (or 0.5 g/kg IV for paediatric patients up to the adult dose) as a bolus injection or infused over 10–30 min. One half of the initial dose can be administered 2 h later if toxicity reappears.
Consultation with a toxicologist is recommended for all suspected cases of cyanide poisoning.
Controversies
![]() The effectiveness of any antidote to treat cyanide poisoning is based on case reports and animal data. Hydroxocobalamin is more commonly recommended as the choice of antidote in cyanide poisoning. Data on human exposure are limited. The current TGA approved product in Australia, dicobalt edetate, Kelocyanor, is not recommended because of its substantial risk profile. There is little indication for the use of methaemoglobin inducers (e.g. amyl nitrite) of the cyanide antidote kit in the pre-hospital or the hospital setting.
The effectiveness of any antidote to treat cyanide poisoning is based on case reports and animal data. Hydroxocobalamin is more commonly recommended as the choice of antidote in cyanide poisoning. Data on human exposure are limited. The current TGA approved product in Australia, dicobalt edetate, Kelocyanor, is not recommended because of its substantial risk profile. There is little indication for the use of methaemoglobin inducers (e.g. amyl nitrite) of the cyanide antidote kit in the pre-hospital or the hospital setting.
![]() Initiation of treatment for cyanide poisoning will be based upon clinical presentation and indirect laboratory results. There may be a greater importance of good resuscitation efforts without the use of antidotes in some cases.
Initiation of treatment for cyanide poisoning will be based upon clinical presentation and indirect laboratory results. There may be a greater importance of good resuscitation efforts without the use of antidotes in some cases.
Further reading
1. Baud FJ, Barriot P, Toffis V, et al. Elevated blood cyanide levels in victims of smoke inhalation. N Engl J Med. 1991;325:1761–1766.
2. Baud FJ, Borron SW, Bavoux E, et al. Relationship between plasma lactate and blood cyanide concentrations in acute poisoning. Br Med J. 1996;312:26–27.
3. Carlsson CJ, Hansen HE, Hilsted L, Malm J. An evaluation of the interference of hydroxycobalamin with chemistry and co-oximetry tests on nine commonly used instruments. Scand J Clin Lab Invest. 2011;71:378–386.
4. Eckstein M. Enhancing public health preparedness for a terrorist attack involving cyanide. J Emerg Med. 2008;35:59–65.
5. Forsyth JC, Mueller PD, Becker CE, et al. Hydroxocobalamin as a cyanide antidote: safety, efficacy and pharamacokinetics in heavily smoking normal volunteers. J Toxicol Clin Toxicol. 1993;31:277–294.
6. Hart GB, Strauss MB, Lennon PA, et al. Treatment of smoke inhalation by hyperbaric oxygen. J Emerg Med. 1985;3:111.
7. Houeto P, Hoffman JR, Imbert M, et al. Relation of blood cyanide to plasma cyanocobalamin concentration after a fixed dose of hydroxocobalamin in cyanide poisoning. Lancet. 1995;346:605–608.
8. Lavon O, Bentur Y. Does amyl nitrite have a role in the management of pre-hospital mass casualty cyanide poisoning? Clin Toxicol. 2010;48:477–484.
9. Marraffa JM, Howland MA. Antidotes for toxicological emergencies: a practical review. Am J Hlth Syst Pharm. 2012;89:199–212.
10. Reade MC, Davies SR, Morley PT, et al. Review article: management of cyanide poisoning. Emerg Med Australas. 2012;24:225–238.
11. Singh P, Kaur M. CN-scavenger: a leap towards development of a CN-antidote. Chem Commun. 2011;47:9122–9124.
12. Yeoh MJ, Braitberg G. Carbon monoxide and cyanide poisoning in fire related deaths in Victoria, Australia. J Toxicol Clin Toxicol. 2004;42:855–863.
29.16 Corrosive ingestion
Robert Dowsett
Essentials
1 Symptomatic patients may have burns to the airway or supraglottic tissues.
2 Decontamination has limited utility; care should be taken not to make patients vomit and no attempt should be made to neutralize corrosives.
3 Serious injuries to the oesophagus or stomach may occur in the absence of visible burns to the lips, mouth or throat.
4 Admit all symptomatic patients.
5 The major acute complications are perforation and necrosis.
6 The major long-term complication is oesophageal stricture.
Introduction
Strong corrosives capable of causing significant injury are those with a pH in solution of less than 2 or greater than 12 (Table 29.16.1). The pH of a solution is dependent on the concentration and dissociation constant (pKa) of the chemical. Strong acids have a pKa<0 and strong alkalis have a pKa>14 (Table 29.16.2). The extent of injury also depends on the volume ingested, contact time and viscosity.
Table 29.16.1
Approximate pH of some common solutions
|
Solution |
pH |
|
Battery acid (1% solution) |
1.4 |
|
Domestic toilet cleaner (1%) |
2.0 |
|
Bleach (1% solution) |
9.5–10.2 |
|
Automatic dishwasher detergents |
10.4–13 |
|
Laundry detergents |
11.6–12.6 |
|
Domestic ammonium cleaners |
11.9–12.4 |
|
Drain cleaner (containing NaOH, KOH) |
13.3–14 |
Table 29.16.2
pKa of some common corrosives
|
Chemical |
pKa |
Highly corrosive? |
|
Hydrochloric acid |
−3 |
Yes |
|
Bromic acid |
<1 |
Yes |
|
Nitric acid |
<1 |
Yes |
|
Sulphuric acid |
1.9 |
|
|
Arsenic acid |
2.3 |
|
|
Nitrous acid |
3.3 |
|
|
Hydrofluoric acid |
3.4 |
|
|
Ammonia |
9.3 |
|
|
Ammonium hydroxide |
9.3 |
|
|
Magnesium hydroxide |
10 |
|
|
Zinc hydroxide |
11 |
|
|
Calcium hydroxide |
11.6 |
|
|
Lithium hydroxide |
>14 |
Yes |
|
Potassium hydroxide |
>14 |
Yes |
|
Sodium hydroxide |
>14 |
Yes |
|
Calcium oxide |
>14 |
Yes |
|
Sodium carbonate |
>14 |
Yes |
|
Potassium carbonate |
>14 |
Yes |
|
Sodium hypochlorite |
>14 |
Yes |
Domestic bleaches, automatic dishwasher detergents, toilet bowel cleaners and drain cleaners are the commonest substances ingested, but severe injury generally does not occur unless large amounts are swallowed [1]. Deaths result mainly from the ingestion of acid-containing drain or toilet cleaners continuing a trend over the last 20 years with a decline in fatalities from alkali drain cleaners [2].
Pathophysiology
Acid–base reactions cause injury by disrupting organic macromolecules. Heat generation may cause thermal burns. Chemical reactions may also result in the production of other compounds that can cause additional injury to the gastrointestinal (GI) tract and lungs (Table 29.16.3).
Table 29.16.3
Chemical reactions resulting in the production of further toxic chemicals

Alkalis cause ‘liquefactive’ necrosis, a process that involves saponification of fats, dissolution of proteins and emulsification of lipid membranes. Acids cause ‘coagulative’ necrosis, a process that involves denaturation of protein.
In both settings, tissue injury can continue for several hours. Granulation tissue develops after 3–4 days, but collagen deposition may not begin until the second week, making the tissue extremely fragile during this period. Complete repair of the epithelium may take weeks. From the third week, newly deposited collagen begins to contract and may produce strictures of the oesophagus, stomach and affected bowel.
Hydrocarbon compounds can produce injury by dissolving lipids and coagulating proteins. Other chemicals can injure tissues by redox reactions and alkylation.
Narrowings in the GI tract are most at risk from corrosive ingestion: the cricopharyngeal area, the diaphragmatic oesophagus, antrum and pylorus [2]. Alkalis are more likely to produce oesophageal injury than are acids, which typically injure the stomach [2–4]. Solid corrosives are more likely to affect the mouth, pharynx and upper oesophagus and to cause deeper burns.
The main acute complications of corrosive ingestion are haemorrhage, perforation and fistula formation. These result from severe burns causing full-thickness necrosis. Tissue inflammation, necrosis and infection can result in hypovolaemia, acidosis and organ failure.
Full-thickness necrosis of the stomach may be associated with injury to the transverse colon, pancreas, spleen, small bowel, liver and kidneys. Perforation of the upper anterior oesophagus may lead to the formation of a tracheo-oesophageal fistula. Formation of a tracheo-oesophago-aortic fistula is a rapidly lethal complication.
Clinical features
Symptoms and signs associated with significant alkali ingestion include oropharyngeal pain, drooling, pain on swallowing, vomiting, abdominal pain and haematemesis [4]. If the larynx is involved, local oedema may produce respiratory distress, stridor and a hoarse voice [5].
Extensive tissue injury may be associated with fever, tachycardia, hypotension and tachypnoea.
Inspection of the oropharynx may reveal areas of mucosal burn. The absence of visible burns does not imply an absence of significant burns to the oesophagus [3,4,6].
Symptoms and signs associated with the life-threatening complications of oesophageal perforation and mediastinitis include chest pain, dyspnoea, fever, subcutaneous emphysema of the chest or neck and a pleural rub. Perforation of the abdominal oesophagus or stomach is associated with the clinical features of chemical peritonitis, including abdominal pain, fever and ileus [3].
The systemic effects of large acid ingestion include hypotension, metabolic acidosis, haemolysis, haemoglobinuria, nephrotoxicity, pulmonary oedema and hypotension. Systemic toxicity can result from the ingestion of arsenic, cyanide and other heavy metal salts, fluoride, ammonia, hydrazine, hydrochloric acid, nitrates, sulphuric acid and phosphoric acid. Ingestion of ammonia can cause coma, hypotension, acidosis, pulmonary oedema, liver dysfunction and coagulopathy. Systemic effects of phenol and related compounds include haemolysis and renal failure.
Long-term complications
The major late complication of corrosive ingestion is the development of an oesophageal stricture. All patients with full-thickness necrosis of the oesophageal wall develop strictures, as do 70% of those with deep ulceration [3]. Symptoms of oesophageal narrowing (principally dysphagia) may develop within 2 weeks; 80% occur within the first 2 months. Early onset of symptoms is associated with a more rapidly progressive and severe obstruction. Strictures do not develop in areas of superficial mucosal ulceration [7–9]. Strictures can also affect the mouth, pharynx and stomach. Only 40% of gastric outlet strictures become symptomatic [3]. A very late complication of alkali ingestion is the development of oesophageal carcinoma, reported to develop 22–81 years after exposure.
Clinical investigations
Initial investigations in symptomatic patients should include an ECG, arterial blood gas, blood count, type and cross-match, coagulation profile, serum electrolytes, blood glucose and liver and renal function.
Chest and upright abdominal X-rays should be assessed for evidence of mediastinal widening, pleural effusions and free air.
All patients who are symptomatic or have visible oropharyngeal burns should be considered for upper GI endoscopy within 24 hours. The entire upper GI tract may be safely examined with a small-diameter flexible endoscope, provided it is not retroflexed or forced through areas of narrowing [2,3]. It is not necessary to terminate the examination at the first circumferential or full-thickness lesion. The cricopharynx should be assessed initially to identify any laryngeal burns. If laryngeal oedema or ulceration is encountered, endotracheal intubation may be necessary before continuing with endoscopy.
A retrospective case series of 49 patients with caustic ingestion underwent computed tomography (CT) scanning and upper GI endoscopy to assess the degree of damage to the oesophagus and adjacent tissues [10]. In this small study, CT scanning was shown to be more sensitive and specific than endoscopy, although this was not statistically significant. Contrast oesophagography with a water-soluble contrast agent is useful for the detection of perforation, but is less sensitive than endoscopy in evaluating ulceration.
Oesophageal burns can be graded according to the depth of ulceration and the presence of necrosis (Table 29.16.4). Injuries can be divided into three main groups:
![]() Mucosal inflammation or superficial ulceration only. These injuries will heal completely and are not at risk of stricture formation.
Mucosal inflammation or superficial ulceration only. These injuries will heal completely and are not at risk of stricture formation.
![]() Areas of deep ulceration or discrete areas of necrosis or circumferential ulceration of any depth. Stricture formation may occur.
Areas of deep ulceration or discrete areas of necrosis or circumferential ulceration of any depth. Stricture formation may occur.
![]() Deep circumferential burns or extensive areas of necrosis. These patients are at high risk of perforation and stricture formation.
Deep circumferential burns or extensive areas of necrosis. These patients are at high risk of perforation and stricture formation.
Table 29.16.4
Classification of gastrointestinal corrosive burns
|
Grade I |
First-degree |
|
Mucosal inflammation |
Mucosal inflammation, oedema or superficial sloughing |
|
Grade IIA |
Second-degree |
|
Haemorrhages, erosions and superficial ulceration |
Damage extends to all layers of, but not through, the oesophagus |
|
Grade IIB |
|
|
Isolated discrete or circumferential superficial ulceration |
|
|
Grade IIIA |
Third-degree |
|
Small scattered areas of necrosis |
Ulceration through to perioesophageal tissues |
|
Grade IIIB |
|
|
Extensive necrosis involving the whole oesophagus |
Treatment
Patients should initially be assessed for the presence of any symptomatic airway burns or respiratory distress. The need for urgent intubation should be considered in any patient with stridor or hypoxia.
Efforts at decontamination must not induce vomiting, as this may exacerbate the oesophageal injury. The mouth should be rinsed thoroughly with water. Dilution of an ingested solid chemical by drinking 250 mL of water or milk is recommended. Patients should otherwise be given nothing by mouth. Neutralization, aspiration and administration of activated charcoal are all contraindicated.
Patients with persistent symptoms should be admitted for observation and undergo endoscopy 12–24 hours later. Further management is dictated by the findings at endoscopy.
Patients with endoscopic evidence of superficial injury can be managed on a general medical ward with supportive care only. Patients with deep discrete ulceration, circumferential ulceration or isolated areas of necrosis should be admitted to high-dependency or the intensive care unit and kept nil by mouth. Intravenous fluid replacement, accurate fluid and electrolyte balance and symptom control are the mainstays of therapy. These patients may require prolonged IV access and parenteral feeding and central venous access should be considered.
If perforation or penetration is suspected clinically or documented by endoscopy or contrast radiography, urgent laparotomy with or without thoracotomy must be considered. Early excision of areas with extensive full-thickness necrosis has been proposed, but this needs to be weighed against mortality rates of 40–50% for patients undergoing such emergency surgery.
Prophylactic broad-spectrum antibiotics are only indicated where there is evidence of GI tract perforation.
Strictures are dilated by endoscopy 3–4 weeks after ingestion. Reconstructive surgery may be required if the oesophageal lumen becomes completely obstructed or if perforation occurs.
Disposition
Asymptomatic patients can be discharged after observation and do not require investigation. They should be instructed to return if they develop pain, respiratory symptoms or difficulty swallowing. Symptomatic patients should be admitted for endoscopy with subsequent disposition dependent on the findings, as detailed above.
Controversies
![]() The value of administering oral fluids following ingestion of a liquid corrosive is controversial but probably of little value.
The value of administering oral fluids following ingestion of a liquid corrosive is controversial but probably of little value.
![]() The use of corticosteroids to prevent oesophageal strictures following corrosive ingestion is controversial. Clinical trials show contradictory results but the balance of available evidence and current opinion would suggest a lack of efficacy [7,11,12]. However, in all studies steroids were commenced after endoscopy. If they are to be effective steroids should be commenced on presentation. There is no role for continuing steroids in patients with Grade III injuries as they may increase the risk of perforation.
The use of corticosteroids to prevent oesophageal strictures following corrosive ingestion is controversial. Clinical trials show contradictory results but the balance of available evidence and current opinion would suggest a lack of efficacy [7,11,12]. However, in all studies steroids were commenced after endoscopy. If they are to be effective steroids should be commenced on presentation. There is no role for continuing steroids in patients with Grade III injuries as they may increase the risk of perforation.
![]() CT scanning may be a less invasive approach to the assessment of corrosive ingestions but further studies are needed before it can be recommended.
CT scanning may be a less invasive approach to the assessment of corrosive ingestions but further studies are needed before it can be recommended.
References
1. Bronstein AC, Spyker DA, Cantilena LR, et al. 2011 Annual Report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 29th Annual Report. Clin Toxicol. 2012;50:911–1164.
2. Sugawa C, Lucas CE. Caustic injury of the upper gastrointestinal tract in adults: a clinical and endoscopic study. Surgery. 1989;106:802–806.
3. Zargar SA, Kochhar R, Mehta S, et al. The role of fiberoptic endoscopy in the management of corrosive ingestion and modified endoscopic classification of burns. Gastrointest Endoscop. 1991;37:165–169.
4. Gorman RL, Khin-Maung-Gyi MT, Klein-Schwartz W, et al. Initial symptoms as predictors of esophageal injury in alkaline corrosive ingestions. Am J Emerg Med. 1992;10:189–194.
5. Moulin D, Bertrand JM, Buts JP, et al. Upper airway lesions in children after accidental ingestion of caustic substances. J Pediatr. 1985;106:408–410.
6. Crain EF, Gershel JC, Mezey AP. Caustic ingestions: symptoms as predictors of esophageal injury. Am J Dis Child. 1984;138:863–865.
7. Anderson KD, Rouse TM, Randolph JG. A controlled trial of corticosteroids in children with corrosive injury of the esophagus. N Engl J Med. 1990;323:637–640.
8. Webb WR, Koutras P, Eckker RR, et al. An evaluation of steroids and antibiotics in caustic burns of the esophagus. Ann Thorac Surg. 1970;9:95–102.
9. Cannon S, Chandler JR. Corrosive burns of the esophagus: analysis of 100 patients. Eye Ear Nose Throat Month. 1963;42:35–44.
10. Ryu HH, Jeung KW, Lee BK, et al. Caustic injury: can CT grading system enable prediction of esophageal stricture? Clin Toxicol. 2010;48:137–142.
11. Howell JM, Dalsey WC, Hartsell FW, et al. Steroids for the treatment of corrosive esophageal injury: a statistical analysis of past studies. Am J Emerg Med. 1992;10:421–425.
12. Fulton JA, Hoffman RS. Steroids in second degree caustic burns of the esophagus: a systematic pooled analysis of fifty years of human data: 1956–2006. Clin Toxicol. 2007;45:402–408.
29.17 Hydrofluoric acid
Sam Alfred and Andis Graudins
Essentials
1 Patients and medical staff are often unaware of the presence of hydrofluoric acid (HF) in household cleaning products.
2 Topical HF exposures may result in the gradual onset of severe local pain out of proportion to any clinical signs evident on presentation.
3 Patients may not relate their dermal symptoms to HF exposure due to the delay in onset of pain that occurs with domestic low concentration preparations.
4 Systemic toxicity may be life threatening and is expected following dermal burns caused by high concentration solutions or involving body surface areas of greater than 5% and following significant inhalations or ingestions.
5 Systemic toxicity is typically manifest as severe hypocalcaemia, hypomagnesaemia, hyperkalaemia and ventricular arrhythmias.
6 Ingestion and inhalation of HF may also result in significant gastrointestinal or respiratory injury.
Introduction
The inorganic acid of fluoride, hydrofluoric acid (HF), is a moderately corrosive chemical widely used in industry for the etching of glass, metal and stone and in the preparation of silicon computer chips. HF is also a common constituent of rust and scale removers, car wheel cleaners, brick cleaners and solder flux mixtures. These products may be for either commercial or home use and are often found in containers with inadequate labelling in regard to the potential toxicity. Concentrations of commercially available HF may vary from 50 to 100%. Products containing HF for domestic use generally have a concentration of less than 10%, but higher concentration products may be obtained illicitly for home use.
The most common route of accidental exposure to HF is topical [1–3]. This may occur when high-concentration HF leaks through damaged gloves in the industrial setting or when HF products are used in the home without gloves. Massive topical HF exposure and inhalational exposure to HF may also occur in the industrial setting. Finally, ingestion of HF products may occur accidentally in the home in the paediatric age group or as a result of deliberate self-harm in adults.
Pathophysiology
HF is a relatively weak acid with less corrosive effects than other stronger acids, such as hydrochloric or sulphuric. In particular, low concentrations of HF (<20%) may result in little or no perceptible corrosive injury to the skin immediately following exposure. This is due to the relatively low dissociation constant (pKa=3.8), which limits the concentration of free hydrogen ions on the skin surface [2–4]. As HF tends to remain in an undissociated neutral state, its ability to penetrate through the skin into deeper tissues is enhanced. Gradual dissociation of HF producing free fluoride ions in the tissues leads to local tissue injury characterized by liquefactive necrosis rather than the coagulative necrosis more commonly associated with acid burns [2,3]. As the concentration of HF increases so does the potential for corrosive injury [3,5]. Nevertheless, chemical burns may result from exposures to dilute (<5%) solutions of HF and fatal systemic poisoning has resulted from relatively small (<5% total body surface area [BSA]) burns caused by more concentrated solutions [6].
The primary mechanism of tissue damage resulting from exposure to HF is related to fluoride toxicity following on from dissociation of the acid in exposed tissues [2,3]. A number of pathological mechanisms may be involved in both local and systemic fluoride poisoning. Fluoride binds divalent cations, especially calcium and magnesium, to form insoluble fluoride salts. The resulting hypocalcaemia and hypomagnesaemia may have profound local and systemic effects on cellular and organ functions. Fluoride is a cellular poison. It inhibits both aerobic and anaerobic metabolic enzyme systems and interferes with cellular respiration[6]. Fluoride also interferes with Na+/K+ ATPase activity and opens calcium-dependent potassium channels in cell membranes, resulting in the leak of potassium into the extracellular space with the potential for systemic hyperkalaemia [1,7]. Precipitation of calcium may also interfere with calcium-dependent clotting factors, resulting in coagulopathy. Finally, exposure to HF may produce direct corrosive injury.
Fatal systemic fluoride poisonings have also been reported following inhalational and gastrointestinal (GI) exposures [6]. Once absorbed, fluoride ions are distributed to virtually every tissue and organ resulting in widespread disruption of organ function. Fluoride is slowly eliminated in the urine and elevations of urinary fluoride excretion can be detected following exposure to HF, although these do not correlate with clinical toxicity [3].
Clinical features
Exposure to HF in the industrial setting is usually recognized as such and patients will often have been decontaminated and had topical therapy applied prior to arrival at hospital. They are also likely to be in possession of appropriate information in the form of material safety data sheets. Acute dermal exposures in the domestic setting may present a more difficult diagnostic dilemma. Domestic product labels may be incomplete and offer no advice regarding the use of protective apparel such as gloves. Additionally, the onset of the signs and symptoms of HF injury may be delayed after exposure to low concentration domestic products and the patient may not recognize that the symptoms are related to chemical exposure [3].
Highly concentrated (>70%) HF contains enough free hydrogen ions to produce a burning sensation on the skin providing some degree of warning of an acute exposure with symptom onset within 1–2 h [1,5]. However, low concentrations of HF (<10%), found in products, such as over-the-counter rust removers, often produce no symptoms at the time of contact and patients can present with gradually increasing pain from 6 to 12 h following exposure [1].
The primary presenting complaint of acute topical HF injury is pain out of all proportion to any physical signs. HF exposure should always be considered in this situation. The pain is usually described as a tingling sensation that progresses to a burning pain and then to a typical deep, throbbing and severe pain [1,5,8]. This means there is often a delay of some hours to onset of significant symptoms and presentation.
Visible evidence of HF burns also follows a fairly common pattern. Initially, the burn site is erythematous and may be oedematous. As tissue injury progresses, the site becomes pale and blanched, progressing to a classical silvery grey appearance [2]. Local vesiculation and frank tissue necrosis may ensue. This process can progress over several days in untreated patients, resulting in the development of deep ulceration and extensive tissue loss.
Dermal exposure to HF commonly occurs on the hands or feet with relatively small areas of the skin being exposed. Systemic fluoride poisoning is rarely a problem under these circumstances. The risk of systemic toxicity increases with the percentage of BSA exposed to HF and the concentration of HF [3]. In general, if more than 3–5% BSA has been exposed to HF, there is a risk of hypocalcaemia [3]. Systemic fluoride toxicity is more likely following large dermal exposures, ingestion or inhalation of HF [3].
Systemic fluoride toxicity is manifest by various effects, the most lethal of which are the severe electrolyte abnormalities produced by direct interaction with fluoride or effects on cell membranes and cellular enzyme systems [3,6,7]. Hypocalcaemia is due to the complexing of calcium by fluoride ions. Hypomagnesaemia may also occur. However, the primary cause of the lethal arrhythmias (refractory ventricular tachycardia, ventricular fibrillation and pulseless idioventricular rhythm) is the development of hyperkalaemia [4,7]. Patients with systemic fluoride poisoning also develop a significant metabolic acidosis [3,4]. This is the result of fluoride interference with intracellular metabolism. The systemic manifestations of significant hypocalcaemia include carpopedal spasm, hyperreflexia, tetany and coagulopathy. Headache, paraesthesiae and visual complaints may be noted. In severe cases, coma, seizures, shock and dysrhythmias often precede death.
Fluoride inhalation is associated with pulmonary injury, including the development of non-cardiogenic pulmonary oedema, adult respiratory distress syndrome (ARDS) and the potential for systemic fluoride toxicity [3].
Clinical investigations
No investigations are necessary following dermal exposures to dilute domestic preparations involving less than 5% of BSA. Following significant dermal exposures to HF or any ingestion or inhalational exposure, serum electrolytes including magnesium and calcium, baseline coagulation studies and a 12-lead ECG (looking for evidence of hypocalcaemia or hyperkalaemia) are indicated. A chest radiograph should be performed in any patient with respiratory symptoms or severe systemic toxicity.
Treatment
All patients with significant dermal HF exposures (>5% BSA, exposure to concentrated industrial preparations) or any ingestion or inhalation exposures should have continuous cardiac monitoring and intravenous access established on arrival.
The initial management of an acute topical HF exposure is thorough skin decontamination with generous water irrigation. This ideally should be performed as a first-aid measure as soon as possible following the exposure as the delayed presentation of most patients makes it unlikely that significant amounts of HF still remain on the surface of the skin. Despite experimental evidence of enhanced decontamination with hexafluorine preparations, clinical experience suggests hexafluorine preparations offer no benefit in terms of local burn minimization or prevention of systemic toxicity when compared to water irrigation of exposed surfaces [9,10]. Other first-aid measures in the work place for known or suspected HF burns include topical treatments, such as calcium gluconate gel (2.5–10%) or soaks with quaternary ammonium salts, such as benzalkonium or benzethonium chloride. Topical therapies are intended to form insoluble complexes with any surface fluoride ion thus preventing tissue penetration and minimizing deeper injury. Topical therapy is probably of little value once fluoride ions penetrate to deeper tissues but should be initiated on presentation to the emergency department (ED). Calcium gluconate gel can be applied to the hand in a rubber glove. It may provide relief to some patients with low concentration HF exposures to the digits. In most cases, topical therapy is a temporizing measure until more invasive methods of calcium administration can be employed. If calcium gluconate gel is not readily available, a 4.8% preparation can be rapidly prepared by mixing one ampoule of calcium gluconate in 10 g of KY jelly.
The definitive treatment of dermal HF burns involves the administration of calcium gluconate into the tissues affected by the exposure [1–3,5,8]. This may be achieved by a number of methods: direct tissue infiltration, regional intravenous infusion using a Bier’s block technique and intra-arterial infusion [1–3,5,8]. The choice of method depends upon the site and concentration of HF involved.
Direct injection of approximately 0.5 mL/cm2 of 10% calcium gluconate solution at the burn site can be considered in areas with little skin tension, such as the trunk, forearms and legs. A small needle (25 gauge) should be used to minimize discomfort and care should be taken to infiltrate into, around and beneath the burn area as completely as possible. Only calcium gluconate should be used for local infiltration as calcium chloride produces direct injury when injected into tissues [3].
HF burns to the hands are relatively common. In view of the lack of loose tissues in the digits, direct dermal injection may be extremely painful and only small amounts of calcium gluconate may be injected. Additionally, the introduction of hyperosmolar calcium solutions to these limited tissue spaces may exacerbate oedema and result in vascular compromise [1,2]. HF may also penetrate beneath the fingernails. In the past, removal of fingernails was advocated to allow for injection of calcium gluconate into the nail bed. Fortunately the advent of focused, parenteral calcium administration techniques to the affected limb have meant that nail removal is less frequently required.
Two techniques are available for direct injection of calcium gluconate into digital HF burns. The first is intra-arterial infusion of calcium gluconate. This technique involves inserting an arterial cannula into the radial (for burns of the thumb, index and middle fingers) or brachial artery (for more extensive hand involvement) and slowly infusing a dilute solution of calcium gluconate utilizing an infusion pump. This allows the calcium to be delivered to the affected tissues through the vascular supply and avoids the pain and tissue distension associated with direct injection [8]. A typical dose is 10–20 mL of 10% calcium gluconate in 50–100 mL of 5% dextrose infused over 4 h and repeated as necessary. The endpoint for therapy is the absence of pain. The number of intra-arterial infusions required for pain relief may vary from one to four or five and depends on the concentration of HF to which exposure occurred. There is case report level evidence for the use of continuous infusions [11].
Regional intravenous calcium gluconate infusion using a Bier’s block technique has also been employed in the treatment of HF burns to the limbs [5]. Success has been observed for digital, hand and forearm exposures as well as for exposures to the leg [5]. The technique is similar to that described by Bier for regional limb anaesthesia and has the advantages of relative simplicity and of not requiring arterial cannulation. An intravenous cannula is inserted in the dorsum of the hand of the affected limb and the arm is raised to exsanguinate the superficial venous system. A pneumatic tourniquet is applied to the upper arm and inflated to a pressure 100 mmHg above systolic blood pressure. Ten to 15 mL of 10% calcium gluconate is diluted to a total volume of 50 mL with normal saline and injected via the cannula into the ischaemic arm. The tourniquet is sequentially released after 20 to 25 min [5]. Pain relief is usually apparent within 30 min of tourniquet release.
There have been no controlled studies comparing any of these techniques in the treatment of HF burns. However, intra-arterial calcium infusion appears to be a better technique for distal digital exposures, particularly in cases where exposure has been to high concentrations (>20%) or where multiple digits are involved [5]. Intra-arterial infusion of calcium has the advantages of more focal provision of calcium to the site of digital exposures and the potential for multiple infusions in patients with ongoing pain. If the intravenous route is selected as the primary therapy and is unsuccessful following one treatment, intra-arterial calcium infusion should then be used. The use of intra-arterial magnesium sulphate in place of calcium gluconate has resulted in tissue necrosis requiring surgical debridement in a small case series and cannot be recommended.
It is sometimes difficult to determine whether ongoing pain at the exposure site is due to continued tissue destruction from fluoride still present in the tissues or established tissue damage. This is particularly the case with patients who have had digital exposures to high concentrations of HF and received multiple infusions of intra-arterial calcium which do not seem to produce further pain relief. It also applies to patients who present more than 24 h post-exposure with ongoing pain despite calcium therapy. In both instances, failure to achieve pain relief with repeated infusions of calcium gluconate suggests that pain may be related to established tissue damage rather than ongoing tissue destruction.
Ocular HF exposures can result in serious consequences if left untreated. Patients should be treated as for other chemical exposures to the eye with copious saline irrigation and local anaesthetic drops for pain relief. Calcium gluconate (10–20 mL/L) may be added to saline irrigation fluid, although animal studies suggest that calcium gluconate eye drops are no better than copious irrigation with normal saline and may, in fact, result in delayed corneal healing [12]. In contrast, clinical case reports of calcium gluconate eye drop use suggest that this treatment is not harmful, but controlled studies are lacking.
Systemic fluoride poisoning resulting from HF ingestion, inhalation or significant dermal exposures is potentially life threatening. Patients with HF ingestion should receive rapid GI decontamination. Aspiration of HF through a small bore nasogastric tube may limit absorption if the patient presents within an hour of ingestion. Calcium or magnesium-containing antacids can complex intragastric HF and prevent some systemic absorption of fluoride ions, although any benefit is likely to be marginal at best. Endoscopy should be performed following HF ingestion as soon as the patient is clinically stable to assess the extent of any upper GI corrosive injury [1]. Nebulized calcium gluconate has been administered acutely to patients following HF inhalation. Serum calcium, magnesium and potassium levels should be closely monitored. Intravenous calcium and magnesium replacement should be commenced prior to any fall in serum Ca2+ or Mg2+ concentrations and replacement doses may be guided by the calculated dose of fluoride ingested. Large amounts of calcium (200–300 mmol) have been used in severe cases of systemic HF poisoning with hypocalcaemia [3,4]. Hyperkalaemia may be recognized on the 12-lead ECG, but close monitoring of serum potassium levels is warranted. Hyperkalaemia in systemic fluoride poisoning may be resistant to standard measures of potassium reduction, such as insulin, glucose and bicarbonate infusions. Ventricular arrhythmias associated with systemic fluoride poisoning may be refractory to cardioversion and defibrillation and may not respond to antiarrhythmic agents [7]. Haemodialysis is indicated for severe or refractory hypocalcaemia, hyperkalaemia or clinical toxicity (e.g. arrhythmias) and may be useful for the removal of fluoride ions [3]. Calcium and magnesium monitoring and replacement should continue during this procedure.
Disposition
Patients with minor dermal exposures in whom ED treatment produces complete resolution of symptoms may be discharged home with follow up arranged within 24 h or should pain return. Those patients in whom tissue damage is evident require referral to a plastic or hand surgeon.
Patients at risk of systemic fluoride poisoning (exposure to high concentration solutions, greater than 5% BSA burns, inhalations and ingestions) require admission to an intensive care unit for ongoing monitoring and management of the electrolyte disturbances and other complications of systemic toxicity.
All patients with eye exposures require early ophthalmological referral.
Controversies
![]() Relative value of intra-arterial versus regional intravenous calcium gluconate administration.
Relative value of intra-arterial versus regional intravenous calcium gluconate administration.
![]() Role of hexafluorine preparations in decontamination.
Role of hexafluorine preparations in decontamination.
References
1. Salzman M, O’Malley RN. Updates on the evaluation and management of caustic exposures. Emerg Med Clin N Am. 2007;25:459–476.
2. Burd A. Hydrofluoric acid-revisited. Burns. 2004;30:720–722.
3. Dunser MW, Ohlbauer M, Rieder J, et al. Critical care management of major hydrofluoric acid burns: a case report, review of the literature, and recommendations for therapy. Burns. 2004;30:391–398.
4. Chan BS, Duggin GG. Survival after a massive hydrofluoric acid ingestion. J Toxicol Clin Toxicol. 1997;35:307–309.
5. Graudins A, Burns MJ, Aaron CK. Regional intravenous infusion of calcium gluconate for hydrofluoric acid burns of the upper extremity. Ann Emerg Med. 1997;30:604–607.
6. Caravati EM. Acute hydrofluoric acid exposure. Am J Emerg Med. 1988;6:143–150.
7. Cummings CC, McIvor ME. Fluoride-induced hyperkalemia: the role of Ca++ dependent K+ channels. Am J Emerg Med. 1988;6:1.
8. Vance MV, Curry SC, Kunkel DB, et al. Digital hydrofluoric acid burns: treatment with intra arterial calcium infusion. Ann Emerg Med. 1986;15:890–896.
9. Hojer J, Personne M, Hulten P, Ludwigs U. Topical treatments for hydrofluoric acid burns: a blind controlled experimental study. J Toxicol Clini Toxicol. 2002;40:861–866.
10. Hulten P, Hojer J, Ludwigs U, Janson A. Hexafluorine vs standard decontamination to reduce systemic toxicity after dermal exposure to hydrofluoric acid. J Toxicol Clin Toxicol. 2004;42:355–361.
11. Lin TM, Tsai CC, Lin SD, Lai CS. Continuous intra-arterial infusion therapy in hydrofluoric acid burns. J Occupat Environ Med. 2000;42:892–897.
12. Beiran I, Miller B, Bentur Y. The efficacy of calcium gluconate in ocular hydrofluoric acid burns. Hum Exp Toxicol. 1997;16:223–228.
29.18 Pesticides
Darren M Roberts
Essentials
1 Acute pesticide poisoning is an important cause of morbidity and mortality worldwide.
2 Existing systems for classifying the toxicity of pesticides are imperfect. The toxicity of even ‘slightly hazardous’ pesticides is sometimes significant. Moderate-to-highly toxic pesticides may have a case-fatality rate of between 5 and 70% in patients with self-poisoning.
3 In addition to the pesticide constituent, other components of the formulation can also contribute to its toxicity. For example, concentrated formulations, inclusion of certain salts, solvents or surfactants can lead to worse outcomes.
4 Many pesticides have a delayed onset of poisoning. All patients with oral exposure should be monitored for a minimum of 6–12 h post-ingestion.
5 Resuscitation and supportive care are priorities in management of acute pesticide poisoning. Patients manifesting significant poisoning require prolonged admission, preferably in an intensive care unit.
6 The specific antidotes for anticholinesterase pesticide poisoning are atropine and, possibly, pralidoxime. These should be administered as soon as possible and titrated to effect.
7 The mortality from acute paraquat poisoning is high and due to multiorgan failure or progressive pulmonary fibrosis. Ingestion of as little as 20 mL of 20% w/v solution is sufficient to cause death. No satisfactory treatments have been confirmed to date.
8 The toxic component of glyphosate-containing herbicides appears to be the surfactant and potentially the isopropylamine salt. The mechanism of toxicity is not confirmed, but severe poisoning is associated with multiorgan toxicity and metabolic acidosis. Treatment is supportive.
Introduction
Pesticide poisoning occurs worldwide. Poisoning may occur due to either acute (intentional self-poisoning) or chronic (such as occupational) exposures. Acute poisoning is of more importance to the emergency physician and is the focus of this chapter.
A pesticide is any chemical used for the control of a plant or animal, which encompasses hundreds of chemicals. They can be subclassified in terms of their intended target, the most common being insecticides, herbicides (selective or non-selective), fungicides, rodenticides and nematocides. Other methods for classification that have been used include toxicity to animal species (LD50; dose that kills 50% of animal subjects), mechanism of action and chemical structure.
Worldwide, pesticides are the most common cause of death from acute self-poisoning. As with pharmaceutical poisoning, the toxicity of pesticides varies between individual compounds but, in general, pesticides are intrinsically more toxic than pharmaceuticals. However, not all pesticide exposures lead to clinically significant poisoning. In Australasia, most acute pesticide exposures are accidental and the majority of patients do not require admission to hospital.
An accurate risk assessment is necessary in each patient with acute pesticide poisoning. This considers the dose ingested, time since ingestion, clinical features, patient factors and available medical facilities. An understanding of the clinical toxicology of a pesticide in the context of the exposure allows for the likely complications to be anticipated and, with appropriate treatment, these may be prevented. If a patient presents to a facility that is unable to provide sufficient medical and nursing care or does not have ready access to necessary antidotes, then arrangements should be made to transport the patient rapidly and safely to a healthcare facility where this is available.
Due to the low incidence, pesticide poisoning is not always considered in the differential diagnosis. A number of case reports from Australia have described a delay in the diagnosis of significant pesticide poisoning because it was not considered initially. These delays did not appear to affect adversely patient outcomes, but they highlight the importance for clinicians to be familiar with the clinical features of pesticide poisoning.
This chapter primarily focuses on agrochemicals used in Australasia, in particular insecticides (organophosphorus pesticides [OPs] and carbamates) and herbicides (glyphosate and paraquat).
Aetiology, pathogenesis and pathology
Acute intentional self-poisoning with pesticides commonly requires admission to hospital and ongoing care. However, significant poisoning may also occur with accidental (e.g. storage of a pesticide in a milk carton) or criminal exposures.
The pathophysiology of acute pesticide poisoning and, therefore, the clinical manifestations vary widely between individual compounds. Many pesticides induce multisystem toxicity due to interactions with a number of physiological systems. Where known, the mechanism of toxicity in humans is discussed for each pesticide; often this bears little relation to the mechanism of action in the target pest. For many pesticides, the mechanism of toxicity is poorly described so less information is available to guide management of these exposures.
It should be noted that proprietary pesticide products contain co-formulants, in particular hydrocarbon-based solvents. Herbicide products also contain surfactants to enhance herbicide penetration into the plant. These co-formulants can contribute to the toxicity of a pesticide product and, in some cases, are more toxic than the active pesiticide constituent.
Epidemiology
Acute pesticide poisoning is a major issue in developing countries of the Asia-Pacific region and OPs are considered the most important cause of death from acute poisoning worldwide. In developed countries, however, the incidence of severe pesticide poisoning is relatively low. In rural areas, the incidence of severe pesticide poisoning may be higher compared to urban regions due to ready access, including concentrated formulations.
Prevention
Primary exposures
Regulatory restrictions to the availability of the more toxic pesticides may contribute to a decrease in mortality from pesticide self-poisoning. In Australia, for example, pesticides with a high case fatality, such as paraquat, organochlorines and parathion, are heavily regulated so poison exposures are increasingly rare. Proper storage, handling and use of pesticides can prevent accidental exposures and associated health consequences.
Secondary exposures or nosocomial poisoning
Secondary exposure refers to staff and family members being exposed to patients with acute pesticide poisoning, predisposing them to nosocomial poisoning. Few cases of nosocomial poisoning, if any, have been confirmed by abnormal cholinesterase activities. While mild symptoms, such as nausea, dizziness, weakness and headache, have been reported, these resolved after exposure to fresh air and were probably due to inhalation of the hydrocarbon solvent. Universal precautions including nitrile gloves are most likely to provide sufficient protection for staff members. Dermal decontamination is performed by washing spilt pesticide off the patient with soap and water and removing and discarding contaminated clothes.
Anticholinesterase pesticides
Anticholinesterase pesticides are among the most widely used pesticides and include organophosphorus (organophosphate, OP, OGP) and carbamate compounds. In Australasia, the most commonly encountered anticholinesterase compounds are chlorpyrifos, dimethoate, fenthion, malathion (maldison), diazinon and propoxur (carbamate).
The relationship between exposure and clinical toxicity is poorly defined and therefore all exposures should be treated as significant. Deliberate self-poisoning by ingestion is the scenario most likely to result in severe toxicity, reflecting the larger exposure. Carbamates appear to be less toxic and induce poisoning of a shorter duration than OPs, although notable exceptions include carbofuran and carbosulfan which are associated with severe toxicity and death.
Mechanism of toxicity
The effects of anticholinesterase compounds on human physiology are multiple, complex and incompletely described. Inhibition of acetylcholinesterase (AChE), thus preventing the hydrolysis of acetylcholine, is considered the most important mechanism. Accumulation of acetylcholine at cholinergic synapses causes excessive stimulation of postsynaptic receptors. This interferes with systemic nervous function, producing a range of clinical manifestations which are known as the acute cholinergic crisis (Table 29.18.1).
Table 29.18.1
Clinical manifestations and treatment of acute anticholinesterase poisoning
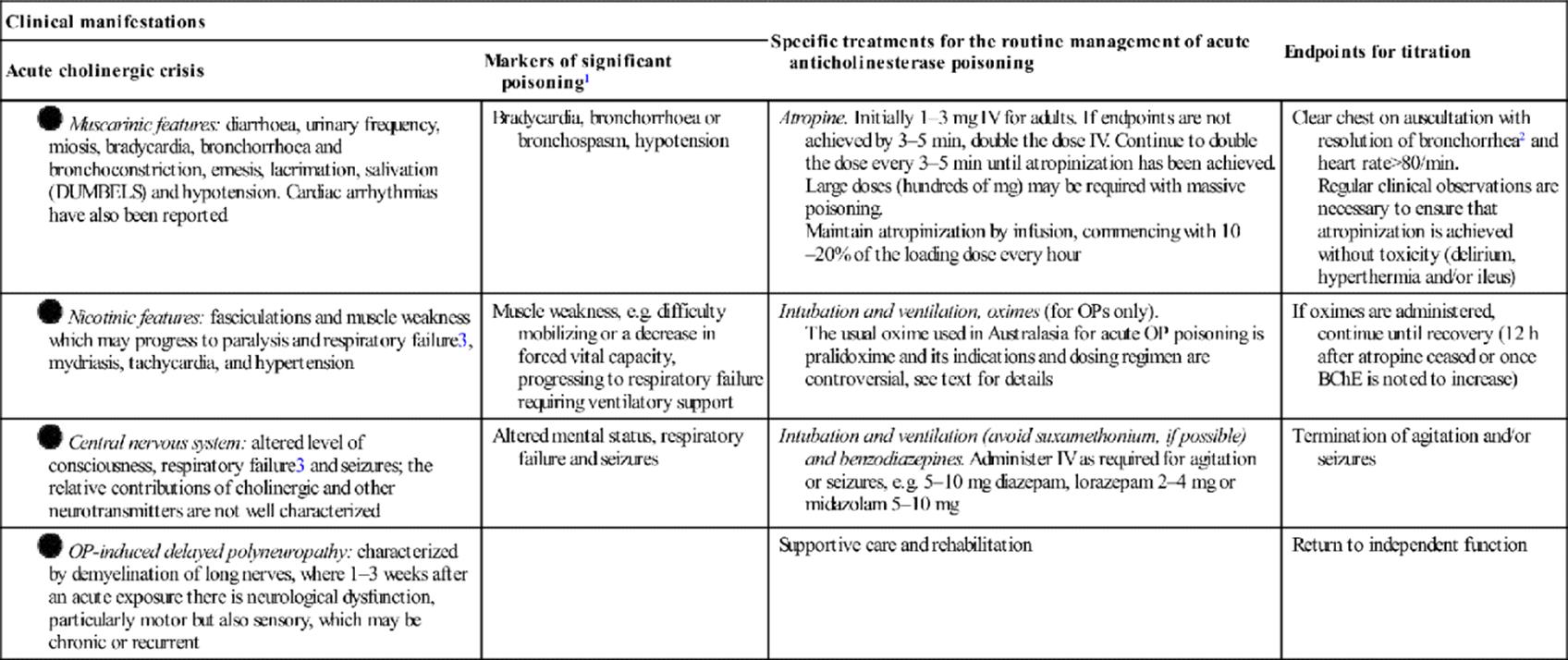
IV: intravenously; BchE: butyrylcholinesterase; OP: organophosphorus pesticides.
1A guide for identifying patients with a significant exposure and to guide clinical management in terms of who requires close observation and specific treatments, rather than for prognostication.
2Focal crepitations and/or wheeze may be noted when there has been pulmonary aspiration.
3Respiratory failure occurs due to centrally- and/or peripherally-mediated mechanisms. It may manifest either during the acute cholinergic crisis (type I paralysis) or suddenly during an apparent recovery phase (intermediate syndrome, or type II paralysis). Weakness of neck flexors is an early sign of significant muscle weakness. Intermediate syndrome is noted in approximately 5% of patients in various series.
Inhibition of other esterases also contributes to the clinical manifestations of acute poisoning. Inhibition of neuropathy target esterase leads to organophosphorus-induced delayed polyneuropathy (see Table 29.18.1).
Enzyme inhibition by an anticholinesterase compound is potentially reversible using an oxime, particularly when treatment is initiated early post-exposure. In the case of OP-inhibited AChE, in the absence of reactivation, a large proportion of inhibited AChE undergoes irreversible inhibition (‘ageing’) and enzyme resynthesis is required for restoration of nervous function. The rate of these competing reactions varies between individual OPs which also influences the clinical manifestations and response to oximes. Carbamates are structurally different to OPs such that there is spontaneous reactivation of carbamate-inhibited AChE and ageing does not occur.
Marked differences in the clinical manifestations of acute anticholinergic poisoning from different compounds are observed. This may reflect the variability in potency of enzyme inhibition, physiological adaptations following prolonged stimulation, pharmacokinetic factors, additional mechanisms of toxicity such as oxidative stress, inter-patient differences and/or other unknown factors.
Clinical features
The initial and prominent manifestation of acute anticholinesterase poisoning is the acute cholinergic crisis (see Table 29.18.1). The duration and manifestations of the acute cholinergic crisis vary between individual anticholinesterase compounds, as mentioned above.
Gastrointestinal symptoms are most prevalent following oral exposures, probably a result of high pesticide concentrations in the gut prior to absorption and the hydrocarbon solvent. Tachycardia does not consistently correlate with hypotension or pneumonitis, but can be secondary to catecholamine release from the adrenal medulla under nicotinic stimulation.
Differential diagnosis
In situations where the history is not forthcoming, the differential diagnosis is broad and includes other toxins (clonidine, opioids, dopamine antagonists, such as chlorpromazine or haloperidol), funnel web spider envenoming and pontine haemorrhage.
Clinical investigations
The diagnosis and management of acute anticholinesterase poisoning is primarily clinical but measurement of cholinesterase activity can assist. The reference ranges are wide due to the large inter-individual variability in baseline AChE and butyrylcholinesterase (BChE, plasma cholinesterase, pseudocholinesterase) activities. Cholinesterase inhibition is generally noted prior to clinical effects. AChE and BChE are generally depressed within 6 h, although enzyme inhibition may progress until 12–24 h post-ingestion. AChE or BChE activity that is less than 80% of the lower reference range is consistent with significant anticholinesterase exposure. Patients in whom cholinesterase activity is higher than this might still have been exposed, but to a minimal degree only.
Erythrocyte AChE is structurally similar to synaptic AChE and their activities decrease in a similar manner following exposure to anticholinesterase compounds. The degree of AChE inhibition is considered the most useful biomarker of severity because it appears to correlate with severity of OP poisoning. In severe poisoning due to an anticholinesterase agent, the erythrocyte AChE activity is less than 20% of normal. Serial measurements of erythrocyte AChE activity can be useful for confirming reactivation of the enzyme by an oxime. For example, if AChE activity normalizes following initiation of oxime therapy, then this suggests that ageing has not occurred and that the dose of oximes is sufficient.
BChE inhibition is a sensitive biomarker of anticholinesterase exposure but has no relation to the severity of poisoning because the affinity of anticholinesterase compounds for BChE is highly variable and differs to that of AChE. Serial measurements of BChE cannot be used for measuring the effect of oximes because there is rapid ageing. However, these may be useful for confirming systemic elimination of the anticholinesterase compounds. Here, once BChE activity starts to increase (the rate of this depends on hepatic function) this suggests that the plasma concentration of the anticholinesterase compound is negligible.
Cholinesterase mixing tests have been used in the management of patients with acute OP poisoning to titrate the oxime regimen. In one method, the patient’s plasma is mixed with an equal volume of non-poisoned donor (control) plasma. If BChE activity in the mixed sample is less than the mean of the samples from the patient and control, it suggests that free anticholinesterase compounds are present. It has been suggested that a decrease in cholinesterase activity in mixing studies is an indication to increase the dose of oximes, although the AChE response to oximes is probably a more useful parameter to guide oxime dosing. Despite being widely used, neither of these approaches to monitor therapy has been confirmed to improve outcome.
Blood gases and routine blood laboratory analyses are recommended for measuring metabolic and respiratory derangements; hypokalaemia secondary to vomiting and diarrhoea is not uncommon.
Criteria for diagnosis
Acute anticholinesterase poisoning is diagnosed on the basis of a history of exposure and development of characteristic clinical features (see Table 29.18.1). Therefore, a high index of clinical suspicion is necessary. Since the correlation between intent, dose and severity of toxicity appears to be poor and the clinical manifestations between individual compounds differ, each patient with an anticholinesterase exposure requires a thorough review.
The onset of cholinergic toxicity is variable, however, the majority of patients who develop severe poisoning are symptomatic within 6 h. Patients remaining asymptomatic for 12 h post-ingestion are unlikely to develop significant clinical toxicity. A possible exception is highly lipophilic compounds, such as fenthion. These may produce only subtle cholinergic features initially but then go on to cause progressive muscle weakness over a number of days, including respiratory failure, requiring ventilatory support.
Where there is doubt regarding the diagnosis or significance of an OP exposure, quantification of BChE or AChE activity is helpful (if available). BChE is particularly useful because it is more widely available and a sensitive marker of exposure.
Treatment
Resuscitation and early considerations
As with all acute poisonings, initial management begins with immediate assessment and management of disturbances in airway, breathing and circulation. Because suxamethonium is metabolized by BChE, this agent should not be used for intubation of patients with acute anticholinesterase poisoning because the duration of paralysis will be prolonged by many hours. Continuous clinical monitoring, including pulse oximetry, cardiac monitoring and blood pressure are required. Hypotension not responding to intravenous fluid loading may be due in part to OP-induced decrease in systemic vascular resistance which requires intravenous vasopressors.
Although the volume ingested as per history appears to be a poor predictor of the amount absorbed, all patients with intentional poisoning who are symptomatic should be managed in a centre with access to intensive care facilities due to the potential for severe poisoning to develop. Gastrointestinal decontamination with oral activated charcoal can be given to patients presenting within 1–2 h of ingestion if the airway is protected, although this was not shown to be useful in a randomized controlled trial.
During the immediate assessment and resuscitation of the patient, all patients should undergo some degree of dermal decontamination (discussed above).
Subsequent interventions depend on changes to the clinical observations while the patient is monitored. Antidotal therapy should be administered rapidly, as outlined in Table 29.18.1. Muscarinic features of the acute cholinergic syndrome should be reversible with adequate doses of atropine. Oximes, such as pralidoxime, are claimed to reverse muscle weakness if administered promptly, although clinical studies confirming this are lacking (discussed later). Established OP-induced delayed polyneuropathy (OPIDP) does not respond to antidotes; instead supportive care is the priority.
Mild poisoning and dermal exposures
Patients who present with a history of accidental poisoning who are asymptomatic or minimally symptomatic (limited to mild gastrointestinal symptoms) often do not require hospital admission. Management priorities for these patients are rapid triage, a detailed risk assessment and consideration of forensic implications. If the exposure is trivial, the patient does not need medical review and can be observed at home or in the workplace. Other patients should be decontaminated and monitored clinically for a minimum of 6–12 h. If available, cholinesterase activity should be measured to exclude a significant exposure. A normal cholinesterase activity at 6 h post-exposure may be sufficient to exclude a significant oral exposure, although more research is required to confirm this observation.
Patients with a single acute dermal exposure rarely develop significant clinical effects and probably do not require medical assessment. Volunteer studies document that the risk of poisoning from a dermal exposure is far below that of an oral exposure. Although the rate of anticholinesterase absorption across the skin is slower than across the gut, patients who are asymptomatic at 12 h are unlikely to develop significant poisoning. Such patients should be given instructions to present for medical review if there is a significant worsening of symptoms. If there is significant concern regarding a dermal exposure, testing for changes in cholinesterase activity is recommended.
Moderate-to-severe poisoning
Patients with moderate-to-severe anticholinesterase poisoning experience prolonged and complicated hospital admissions. Close observation is required to monitor for a rapid clinical deterioration, even if there is an apparent recovery from the acute cholinergic crisis. Therefore, following resuscitation, these patients require ongoing management in an intensive care unit (ICU). Priorities post-admission to ICU include careful titration of antidotes and supportive care, including ventilation and inotropes/vasopressors.
Antidotes
The three most widely used antidotes for anticholinesterase poisoning are muscarinic antagonists (usually atropine), oximes (usually pralidoxime in Australasia) and benzodiazepines. The indications and dosing regimen of these specific antidotes are described in Table 29.18.1.
Antimuscarinic agents
Atropine is the most widely used antimuscarinic agent. It is carefully titrated to reverse muscarinic effects and has no effect on the neuromuscular features.
Oximes
Oximes are used to reverse neuromuscular blockade by reactivating the inhibited AChE before ageing occurs and should therefore be administered as early as possible. In general, oximes are more effective in poisoning due to diethyl OPs (e.g. chlorpyrifos, diazinon) than dimethyl OPs (e.g. dimethoate, fenthion, malathion), due in part to slower ageing of inhibited AChE by diethyl OPs.
Evidence supporting the indications for oxime therapy, their efficacy and the optimal dosing regimen is lacking and controversial. The earlier clinical studies reported either no effect or harm from oximes. However, these conclusions were limited by inadequacies in study design and also because the commonly used oxime dosing regimen was 1 g every 6 h which was less than that advocated by the WHO at the time. This prompted further research.
Two subsequent randomized controlled trials (RCTs) are worthy of mention. One RCT (n=200 patients in India) concluded efficacy from higher doses of pralidoxime iodide: pralidoxime iodide 2 g loading dose in all patients, followed by either 24 g/day for 48 h, then 1 g every 4 h until recovery (higher dose) or the 1 g every 4 h (lower dose) until recovery. AChE activity was not measured in this study. The other RCT (n=235 patients in Sri Lanka) reported no benefit from pralidoxime dosed according to a WHO recommended regimen: pralidoxime chloride 2 g loading dose, followed by a constant infusion of 0.5 g/h for up to 7 days; this was compared to saline. AChE was measured at multiple points in this study, confirming severe poisoning on arrival and an appropriate response to pralidoxime therapy. A concern in the latter study was the higher mortality in the intervention group, although this was not statistically significant. However, because the study was terminated early due to slow recruitment, the importance of this observation cannot be determined. Regarding dose equivalents, 1 g of pralidoxime iodide is equivalent to 650 mg of pralidoxime chloride. Both of these RCTs acknowledged limitations in their study designs and/or patient cohort and recommended further studies.
Because carbamate-inhibited AChE does not undergo ageing, the role for oximes appears limited. However, it remains controversial given that data have been presented to suggest that oximes may increase the reactivation of carbamate-inhibited AChE, although not consistently. Oximes appear to increase carbaryl toxicity for reasons that are not understood.
In summary, the efficacy of oximes in anticholinesterase poisoning is not confirmed. Due to limitations in the existing literature, studies exploring differing dosing regimens, types of oximes (obidoxime may be more effective) and selection criteria are required. Until that time, it is reasonable to conclude that oximes are not a standard of care in the treatment of anticholinesterase poisoning. However, it is not unreasonable to administer obidoxime or lower doses of pralidoxime (e.g. 1 g pralidoxime chloride every 6 h) to patients with significant anticholinesterase poisoning, in particular by diethyl OPs, and monitor for reactivation of AChE.
Benzodiazepines
Benzodiazepines are recommended for use in patients with agitation or seizures. It is proposed that early use of benzodiazepines may prevent cognitive deficits or improve the central control of respiration preventing the need for intubation, however, this has not yet been sufficiently studied.
Prognosis
The mortality in patients with anticholinesterase poisoning is variable, which may reflect differences in degree of exposure, reporting, resources, genetics or the types of compounds encountered. However, the mortality can exceed 10%, compared to a mortality of less than 0.5% for pharmaceuticals.
Various tools are proposed to classify the severity of OP poisoning, but few have been widely adopted or validated. Generalized approaches to prognostication in OP poisoning are difficult given that individual compounds vary markedly in the onset, severity and manifestations of clinical toxicity. Further, they are not often useful for guiding management.
In the case of dermal exposures, prognosis appears favourable.
Paraquat (bipyridyl herbicides)
Paraquat is a non-selective contact herbicide and is considered one of the most toxic pesticides available. The mortality from acute poisoning is high, varying between 50 and 90%. Fortunately, cases of acute paraquat poisoning are increasingly rare in a number of countries due to restrictions in availability. However, paraquat continues to be an important cause of death in a number of countries in Asia, where it is widely used in subsistence farming.
Diquat is another bipyridyl herbicide that is more widely available in Australasia.
Mechanism of toxicity
Oral exposures to paraquat are most likely to lead to poisoning. Because paraquat formulations are highly irritating (and potentially corrosive), gastrointestinal toxicity occurs with all oral exposures.
Paraquat is rapidly absorbed (although the bioavailability is low) and distributed to all tissues. Free oxygen radicals are generated and non-specifically damage the lipid membrane of cells, inducing cellular toxicity and death. The free oxygen radicals produced by paraquat require oxygen for generation so supplemental inspired oxygen may exacerbate pulmonary injury.
The extent of dysfunction depends on the concentration of paraquat at the cellular level and the efficiency of protective mechanisms, such as intracellular glutathione, which is a free radical scavenger. Following an exposure of only 10–20 mL of the 20% w/v solution, these protective mechanisms can be overwhelmed leading to multisystem toxicity and death within 24–48 h.
Paraquat displays specific toxicity in the lung and kidney due to active uptake in type II pneumocytes and renal tubular cells. Because paraquat concentrates in these cells, they are more susceptible to injury than other cells. Therefore, in the event of a smaller exposure where multisystem toxicity does not occur, kidney and lung injury may occur. Kidney injury decreases the excretion of paraquat which increases systemic exposure. Pneumonitis can be followed by progressive pulmonary fibrosis and these are associated with impaired oxygenation.
Diquat does not concentrate in the pneumocytes as readily as paraquat. Therefore, if the patient survives the acute phase of multiorgan dysfunction, delayed pulmonary fibrosis is less likely to occur.
Clinical features
Severe gastrointestinal toxicity including vomiting and diarrhoea is the initial manifestation of acute paraquat poisoning. Necrosis of the oral mucosa is often noted about 12 h post-ingestion; it has been reported in patients who drink the paraquat solution without swallowing, despite the brief contact time. Oesophageal perforation with extensive subcutaneous emphysema may also occur due to the corrosive effects of the formulation.
Patients ingesting more than 20 mL are likely to develop severe poisoning with multisystem toxicity. This manifests as pneumonitis, hypotension, hepatitis, acute kidney injury and severe diarrhoea. Death within 48 h of ingestion is expected.
Patients ingesting less than 20 mL are still at risk of death, but this is more likely to be delayed by weeks or months post-ingestion. The primary mechanism of death is respiratory failure attributed to pulmonary fibrosis following increasing dyspnoea and hypoxaemia. This is associated with various degrees of hepatic and kidney impairment; kidney injury recovers within approximately 2–3 weeks in survivors.
Differential diagnosis
Acute paraquat poisoning may resemble sepsis or poisoning with another cellular poison, such as phosphine (aluminium or zinc phosphides), colchicine or iron. Oropharyngeal necrosis is more marked in paraquat poisoning.
Clinical investigations
A range of investigations has been proposed and tested in patients with acute paraquat poisoning. Because outcomes from paraquat poisoning are generally poor, their principal role is to define prognosis.
It is useful to confirm that an exposure to paraquat is significant because this will guide subsequent management. The easiest method to do this is by the dithionite urine test which involves the addition of 1 g of sodium dithionite solution and 1 g of sodium bicarbonate (or 1–2 mL of 1% sodium dithionite in 1–2 M sodium hydroxide) to 10 mL of urine. A blue colour change indicates paraquat ingestion and green colour change indicates diquat ingestion. The darker the colour change, the higher the concentration. If the test is negative on urine passed 6 h after ingestion, a significant exposure is unlikely.
The concentration of paraquat can be quantified in plasma to estimate prognosis, as discussed later. Unfortunately, it is often difficult to locate laboratories that are able to measure paraquat concentrations in a clinically useful time frame, thereby limiting its use.
Other investigations are useful to determine the evolution of toxicity to other organ systems, in particular serial measurements of serum electrolytes, kidney and liver function. A raised admission creatinine concentration is associated with worse outcomes. If the rate of increase in plasma creatinine concentration exceeds 4.3 μmol/L/h over 12 h then death is more likely. Serial blood gas measurements and chest X-rays demonstrate progression of pulmonary injury.
Criteria for diagnosis
The diagnosis of paraquat poisoning is made on the basis of a history of exposure and the clinical symptoms, so a high index of clinical suspicion is required. The urinary dithionite test is a simple and quick method for confirming (or hopefully excluding) paraquat poisoning.
Treatment
Because death is reported following ingestion of as little as 10–20 mL, all exposures should be treated as significant and observed in hospital for 6–12 h post-ingestion. This allows for the dithionite urinary test to be conducted and to evaluate for changes in plasma creatinine concentration. Patients should be treated symptomatically, including intravenous fluids and analgesia according to usual guidelines.
Systemic exposure to paraquat may be decreased by either reducing absorption or increasing clearance. Both Fuller’s earth and activated charcoal have been advocated to decrease absorption, but Fuller’s earth is of limited availability and activated charcoal has not been demonstrated to improve outcomes.
Adequate hydration is required for optimal renal function which promotes paraquat clearance. Extracorporeal methods for increasing paraquat clearance have been largely disappointing. Haemoperfusion reduces lethality in dogs if commenced within a few hours of ingestion. Increasing data published in non-English language journals suggest clinical benefits from early haemoperfusion (possibly up to 12 hours post-ingestion). So, despite limitations in the reporting and design of these studies, an effect of haemoperfusion is not currently ruled out.
Antioxidants (e.g. vitamin C, vitamin E, acetylcysteine) and anti-inflammatories (corticosteroids, salicylates) are proposed therapies but are inadequately studied. Enthusiasm for immunosuppression with cyclophosphamide and corticosteroids followed positive findings in a couple of small studies, but limitations in study design rendered these findings inconclusive. A larger randomized controlled trial was recently completed and reported no mortality benefit from immunosuppression with cyclophosphamide, methylprednisolone and dexamethasone. The role of other forms of immunosuppression, anti-inflammatories and antioxidants in the treatment of acute paraquat poisoning requires more research.
Despite these gaps in the data, it is obvious that, in the absence of treatment, the majority of patients will die. This has led some clinicians to treat patients with a number of treatments concurrently (e.g. activated charcoal, acetylcysteine, vitamins C and E, immunosuppression and either haemodialysis or haemoperfusion) in the hope that there will be a favourable outcome. The choice of whether to commence this treatment is largely a personal one and requires discussion with the patient and family. Such a treatment regimen is probably reasonable in patients with a faintly positive dithionite urinary test. However, it is unlikely to be of assistance to patients in whom this test is strongly positive or those with evolving multiorgan dysfunction. Instead, palliation should be the priority, including oxygen for hypoxia and morphine for dyspnoea and oropharyngeal or abdominal pain.
Prognosis
Mortality is higher in patients who experience a peripheral burning sensation than those who do not.
Much research has focused on the evaluation of markers of prognosis in patients with acute paraquat poisoning. Two predictors of death are well established: the dose ingested or the plasma concentration of paraquat, relative to the time of poisoning. When the plasma concentration of paraquat is available within a timely manner, this can be used with the time since ingestion to predict prognosis using either a nomogram or the calculated severity index of paraquat poisoning (SIPP). A number of nomograms have been developed and they perform similarly. The SIPP is calculated by multiplying the paraquat plasma concentration (mg/L) by the time since ingestion (hours): SIPP<10 predicts survival, SIPP 10–50 predicts death from lung fibrosis, and SIPP>50 predicts death from circulatory failure.
Direct markers of paraquat-induced organ toxicity that allow earlier prognostication might assist with determining which patients should be treated, palliated or discharged with confidence that harm will not occur.
Alternative markers of prognosis have also been explored, although few have been validated. These include measurement of the respiratory index, temporal changes in haematological or biochemical measures, such as creatinine (discussed above), or lactic acidosis and pulmonary surfactants.
Glyphosate
Glyphosate is a non-selective herbicide that acts by inhibiting the enzymatic synthesis of aromatic amino acids in plants. This target enzyme is not present in humans. Both ready-to-use (≈1–5%) and concentrated (≈30–50%) formulations requiring dilution are available.
Glyphosate is absorbed from the gastrointestinal tract and does not penetrate the skin to a significant extent. Respiratory, ocular and dermal symptoms may occur following occupational use but are usually of minor severity. Ingestion is the most significant route of exposure in clinical toxicology.
Mechanism of toxicity
The mechanism of toxicity of glyphosate-containing herbicides in humans is inadequately described. Experimentally, there appears to be minimal (if any) mammalian toxicity from glyphosate itself. Toxicity has been largely attributed to surfactant co-formulants and potentially the type of glyphosate salt. Polyoxyethyleneamine (POEA; tallow amine) is the most common surfactant formulated in these products.
Poisoning is more severe following ingestion of concentrated formulations. This may reflect either the total dose ingested or direct effects of the highly irritating/corrosive compounds present in these products.
Patients with severe poisoning manifest multisystem effects. This suggests that glyphosate-containing herbicides may be non-specific in their action or that they interfere with physiological functions that are common to a number of systems. Proposed mechanisms include disruption of cellular membranes and uncoupling of oxidative phosphorylation, although these may be inter-related.
Clinical features
Abdominal pain with nausea, vomiting and/or diarrhoea are the most common manifestations of acute poisoning. These may be mild and self-resolving but, in severe poisoning (particularly due to the concentrated solutions), there may be inflammation, ulceration, haemorrhage or infarction of the gastrointestinal tract. Severe diarrhoea may also occur and vomiting may be recurrent, leading to dehydration.
Multiorgan dysfunction is noted with severe poisoning, including hypotension, kidney or liver dysfunction, pulmonary oedema or pneumonitis, altered level of consciousness and/or metabolic acidosis. It is not understood which of these clinical features reflect primary or secondary toxic effects of the glyphosate-containing herbicides. These effects may be transient or severe, progressing over 12–72 h to shock and death. Some patients who subsequently died demonstrated only mild symptoms at the time of admission.
Differential diagnosis
The differential diagnoses are wide, including any poisoning or medical condition associated with gastrointestinal symptomatology and progressive multisystem toxicity.
Clinical investigations
There are no specific clinical investigations to guide management.
Targeted laboratory and radiological investigations should be conducted in patients demonstrating anything more than mild gastrointestinal symptoms. Serial blood gases may be useful for detection of metabolic disequilibria. Hyperkalaemia is reported with products where glyphosate is formulated as the potassium salt.
Although glyphosate plasma concentrations appear to correlate with outcomes, this assay is not available for clinical use.
Endoscopy can identify erosions or ulceration of the gastrointestinal tract, but this investigation is associated with a risk of viscus rupture.
Criteria for diagnosis
The principal criterion for diagnosis of acute poisoning with a glyphosate-containing herbicide is a history of exposure. Therefore, a high index of suspicion is necessary for diagnosis. A number of clinical criteria for the classification of severity have been suggested, but none has been validated.
Treatment
All patients presenting with a history of acute ingestion should be observed for a minimum of 6 h. Patients reporting a history of intentional ingestion and gastrointestinal symptoms should be observed for at least 24 h given that the severity of poisoning may progress.
Treatment of acute poisoning with glyphosate-containing herbicides is empiric. All patients should receive prompt resuscitation and supportive care. Activated charcoal may be given orally if the patient presents within 1–2 h of ingestion, although a randomized controlled trial did not support its efficacy for pesticides in general. Intravenous fluids should be administered to replace gastrointestinal losses. Because the aetiology of hypotension may be multifactorial, including fluid losses or a decrease in cardiac output and/or peripheral resistance, haemodynamic monitoring is recommended to guide treatment of hypotension not responding to routine volumes of intravenous fluids. Biochemical and acid–base abnormalities should be corrected where possible.
No specific antidote has been proposed or tested for the treatment of acute poisoning with glyphosate-containing herbicides. This relates largely to the unknown mechanism of toxicity of these products.
The literature is conflicting regarding the role of haemodialysis outside of the more common indications (resistant acidaemia, hyperkalaemia or fluid overload in the context of kidney injury). If it is to be used, it is anticipated that early initiation would optimize outcomes.
Prognosis
All intentional exposures should be considered significant. A correlation between increasing dose and glyphosate plasma concentration, increasing age and delayed presentation to hospital with severe poisoning and death has been suggested.
Mortality from acute poisoning with glyphosate-containing herbicides varies between studies (reflecting various biases) but may be as high as 30%. The mortality was 3.2% in a prospective multicentre study in rural hospitals in Sri Lanka with limited medical resources.
Tools for estimating prognosis in acute poisoning with glyphosate-containing herbicides have not been described in detail. Patients developing marked multiorgan dysfunction, including kidney injury, hypotension, pulmonary oedema, sedation, arrhythmias) are more likely to die. Patients with more extensive erosions of the upper GI tract developed more severe systemic poisoning and required prolonged admission.
Controversies
![]() Anticholinesterase pesticides:
Anticholinesterase pesticides:
![]() significance of other potential mechanisms of toxicity
significance of other potential mechanisms of toxicity
![]() role of oximes including dose and indications and the relative efficacy of individual oximes
role of oximes including dose and indications and the relative efficacy of individual oximes
![]() role of other proposed antidotes and treatments including α2-adrenergic receptor agonists (e.g. clonidine), BChE replacement therapy, gastric lavage, extracorporeal blood purification, magnesium sulphate, organophosphorus hydrolases and blood alkalinization with sodium bicarbonate
role of other proposed antidotes and treatments including α2-adrenergic receptor agonists (e.g. clonidine), BChE replacement therapy, gastric lavage, extracorporeal blood purification, magnesium sulphate, organophosphorus hydrolases and blood alkalinization with sodium bicarbonate
![]() importance of regulatory restrictions on ‘highly toxic’ anticholinesterase agents in decreasing mortality.
importance of regulatory restrictions on ‘highly toxic’ anticholinesterase agents in decreasing mortality.
![]() Paraquat:
Paraquat:
![]() efficacy of anti-inflammatories, antioxidants and enhanced elimination on clinical outcomes.
efficacy of anti-inflammatories, antioxidants and enhanced elimination on clinical outcomes.
![]() Glyphosate:
Glyphosate:
![]() the relative importance of glyphosate, its salt and other co-formulants on the severity of poisoning
the relative importance of glyphosate, its salt and other co-formulants on the severity of poisoning
![]() clinical and analytical predictors of the development of significant poisoning.
clinical and analytical predictors of the development of significant poisoning.
Further reading
1. Bradberry SM, Proudfoot AT, Vale JA. Glyphosate poisoning. Toxicol Rev. 2004;23:159–167.
2. Buckley NA, Eddleston M, Li Y, Bevan M, Robertson J. Oximes for acute organophosphate pesticide poisoning. Cochrane Database Syst Rev. 2011;2:CD005085.
3. Eddleston M, Buckley NA, Eyer P, Dawson AH. Management of acute organophosphorus pesticide poisoning. Lancet. 2008;371:597–607.
4. Eddleston M, Eyer P, Worek F, et al. Differences between organophosphorus insecticides in human self-poisoning: a prospective cohort study. Lancet. 2005;366:1452–1459.
5. Eddleston M, Eyer P, Worek F, et al. Pralidoxime in acute organophosphorus insecticide poisoning–a randomised controlled trial. PLoS Med. 2009;6:e1000104.
6. Eddleston M, Juszczak E, Buckley NA, et al. Randomised controlled trial multiple dose activated charcoal in acute self-poisoning. Lancet. 2008;371:579–587.
7. Eddleston M, Phillips MR. Self poisoning with pesticides. Br Med J. 2004;328:42–44.
8. Gawarammana IB, Buckley NA. Medical management of paraquat ingestion. Br J Clin Pharmacol. 2011;72:745–757.
9. Li LR, Sydenham E, Chaudhary B, You C. Glucocorticoid with cyclophosphamide for paraquat-induced lung fibrosis. Cochrane Database Syst Rev. 2012;7:CD008084.
10. Pawar KS, Bhoite RR, Pillay CP, et al. Continuous pralidoxime infusion versus repeated bolus injection to treat organophosphorus pesticide poisoning: a randomised controlled trial. Lancet. 2006;368:2136–2141.
11. Roberts DM, Aaron CK. Management of acute organophosphorus pesticide poisoning. Br Med J. 2007;334:629–634.
12. Roberts DM, Buckley NA, Mohamed F, et al. A prospective observational study of the clinical toxicology of glyphosate-containing herbicides in adults with acute self-poisoning. Clin Toxicol. 2010;48:129–136.
13. Senarathna L, Eddleston M, Wilks MF, et al. Prediction of outcome after paraquat poisoning by measurement of the plasma paraquat concentration. Q J Med. 2009;102:251–259.
29.19 Ethanol and other ‘toxic’ alcohols
David McCoubrie
Essentials
1 Ethanol is a major cause of morbidity, mortality and emergency department (ED) presentation in most Western societies. Presentations may result from acute intoxication, withdrawal or medical complications of chronic ethanol ingestion.
2 Ethanol causes central nervous system (CNS) depression that can be synergistic with other CNS depressants and can be life threatening without supportive care.
3 Ethanol withdrawal is encountered commonly in the emergency department and has a mortality of up to 5% without medical therapy.
4 Wernicke’s encephalopathy (W/E) is under diagnosed. The raised likelihood of W/E in patients with altered mental status and a suspected history of prolonged alcohol abuse should prompt early treatment with parenteral thiamine.
5 Methanol and ethylene glycol, the toxic alcohols, are potentially lethal when ingested even in relatively small volumes.
6 They exert toxic effects through the production of organic acid metabolites and dialysis is the recommended treatment.
7 An elevated anion-gap metabolic acidosis and a raised osmolar gap are associated with increased mortality in toxic alcohol ingestions.
8 Osmolar gap has a poor sensitivity and is incapable of excluding toxic alcohol ingestion.
Introduction
Alcohols are hydrocarbons that contain a hydroxyl (OH) group. Ethanol, a two-carbon primary alcohol, is the most commonly used recreational drug in Australasia and elsewhere in the Western world. Ethanol misuse is a major cause of mortality and morbidity both directly and indirectly and emergency departments (EDs) deal with the results on a daily basis. It was estimated that, in 1997, 3290 Australians died from injury due to high-risk drinking and there were 72 302 hospitalizations. In excess of 30% of all ED presentations are deemed to be ethanol related.
The complications of chronic alcohol consumption contribute to the development of a number of medical and surgical emergencies, many of which are dealt with elsewhere in this text. This chapter confines its discussion to acute ethanol intoxication, ethanol withdrawal and two other important ethanol specific emergency presentations–Wernicke’s encephalopathy and alcoholic ketoacidosis.
A number of other alcohols, although far less frequently implicated in ED presentation than ethanol, are metabolized to form toxic organic acids and produce life-threatening clinical syndromes. These alcohols include methanol and ethylene glycol and are termed ‘toxic alcohols’. Early recognition and intervention can prevent significant morbidity and mortality.
Ethanol
Pharmacology
Ethanol is a small molecule that is rapidly and almost completely absorbed from the stomach and small intestine. Ethanol is both water and lipid soluble and rapidly crosses lipid membranes to distribute uniformly throughout the total body water. Ethanol is principally eliminated by hepatic metabolism with smaller amounts (5–10%) excreted unchanged by the kidneys, lungs and in sweat.
Ethanol is oxidized by cytosolic and microsomal cytochrome P450 (2EI and 1A2) alcohol dehydrogenases (ADH) to acetaldehyde which, in turn, is metabolized by aldehyde dehydrogenase to acetate. Acetate is converted to acetyl-CoA and enters the Krebs cycle to be finally metabolized to carbon dioxide and water. Entry of acetyl-CoA in the Krebs cycle is dependent on adequate thiamine stores. Importantly, the ADH system is saturated at relatively low blood ethanol concentrations, which results in blood ethanol elimination moving from first-order to zero-order kinetics. The rate of ethanol metabolism in non-tolerant adults is approximately 10 g/h and blood ethanol levels fall by about 0.02 g/dL/h. An alternative pathway for ethanol metabolism is via the microsomal ethanol oxidizing system, the activity of which increases in response to chronic alcohol exposure. Metabolism by this route is relatively important at very high blood ethanol concentrations and in chronic alcoholics.
The mechanism of action of ethanol is poorly understood. However, ethanol acts as a central nervous system (CNS) depressant, at least partially by enhancing the effect of γ-aminobutyric acid (GABA) at GABAA receptors. Tolerance to the CNS depressant effect develops with chronic exposure.
Clinical presentation
Acute ethanol intoxication
The clinical features associated with acute ethanol intoxication predominantly relate to the CNS and progress with increasing blood alcohol level, although there is remarkable inter-individual variation, most commonly as a function of tolerance. Initial features include a sense of well-being, increased self-confidence and disinhibition. With increasing blood concentrations, impaired judgement, impaired coordination and emotional lability develop. At very high concentrations, ethanol can cause coma, respiratory depression, loss of airway protective reflexes and even death.
Presentation to the ED is usually as a result of the social and behavioural consequences of the alteration in higher CNS functions. Ethanol is frequently implicated in trauma, drowning, violence, self-harm, domestic and sexual abuse and other acute social and psychiatric emergencies. Ethanol is a common co-ingestant in deliberate self-poisoning.
Many other important medical and surgical conditions that cause altered mental status may be incorrectly ascribed to ethanol intoxication or coexist with ethanol intoxication. Table 29.19.1 lists an example differential diagnosis.
Table 29.19.1
Differential diagnosis of acute ethanol intoxication

In the absence of a clear history, the diagnosis of ethanol intoxication is only confirmed upon determination of a breath or blood ethanol concentration. Because ethanol consumption is so ubiquitous, a positive reading does not exclude coexisting pathology.
Ethanol withdrawal syndrome
A withdrawal syndrome usually develops within 6–24 h of cessation or reduction in ethanol consumption in dependent individuals. Symptoms can begin any time after the blood ethanol concentration begins to fall and blood ethanol is frequently still measurable in withdrawing patients. The duration of the syndrome may be from 2 to 7 days. Although the pathophysiology is not well understood, the syndrome presents as unopposed sympathetic and CNS stimulation. It is associated with a mortality of 5% and early clinical recognition of this syndrome is important.
Patients may present to the ED already in withdrawal after deliberately abstaining from alcohol or after stopping drinking due to intercurrent illness or lack of funds to buy alcohol. Alternatively, ethanol-dependent patients may begin to withdraw while being treated in the ED, particularly where their stay is prolonged.
Clinical features of mild ethanol withdrawal are those of mild autonomic hyperactivity and include nausea, anorexia, coarse tremor, tachycardia, hypertension, hyperreflexia, insomnia and anxiety. In more severe cases, the patient goes on to develop more pronounced anxiety, insomnia, irritability, tremor, tachycardia, hyperreflexia, hypertension, fever, visual hallucinations, seizures and delirium. Symptoms usually peak by 50 h. Delirium tremens represents the extreme end of the spectrum of ethanol withdrawal. It is an uncommon but potentially lethal complication.
Wernicke’s encephalopathy
This is an acute neuropsychiatric syndrome that develops in certain alcohol-dependent individuals as a result of thiamine deficiency. It is a spectrum disorder that is classically described as a triad of:
![]() oculomotor disturbance (usually nystagmus and ocular palsies)
oculomotor disturbance (usually nystagmus and ocular palsies)
![]() abnormal mentation (usually confusion)
abnormal mentation (usually confusion)
![]() ataxia.
ataxia.
In up to 20% of cases, the signs and symptoms of the classic triad are not evident at presentation. Less common presentations include stupor, hypothermia, cardiovascular instability, seizures, visual disturbances, hallucinations and alterations in behaviour. In extremis, the condition may present with hyperthermia, hypertonia, spastic paresis, dyskinesias and coma.
Wernicke’s encephalopathy is a clinical diagnosis and constitutes a medical emergency with significant morbidity and a mortality of 10–20 % if left untreated. For this reason, the emergency physician must maintain a high index of suspicion in any patient with altered mental status and a suspicion of prolonged heavy ethanol intake.
Alcoholic ketoacidosis
Alcoholic ketoacidosis (AKA), also termed alcoholic acidosis, is an often unrecognized potentially life-threatening medical condition that develops in the alcoholic patient in response to starvation. The normal response to starvation is increased gluconeogenesis from pyruvate. In the alcoholic patient, pyruvate is preferentially converted to lactate. In response, fatty-acid metabolism is increased as an alternative source of energy, resulting in the production of acetyl-CoA and acetoacetate which, in turn, is reduced to β-hydroxybutyrate (BOHB), producing the ketoacidotic state.
Patients with AKA usually present with a history of prolonged heavy alcohol misuse preceding a bout of particularly excessive intake, which has been terminated several days earlier by nausea, severe vomiting and abdominal pain. There may be a history of previous episodes requiring brief admissions with labels of ‘query pancreatitis’ or ‘alcoholic gastritis’. Examination usually reveals tachypnoea, tachycardia, hypotension and diffuse epigastric tenderness on palpation. In contrast to patients with diabetic ketoacidosis, mental status is usually normal. The presence of an altered mental state should prompt consideration of other causes, especially hypoglycaemia and acute ethanol intoxication.
Toxic alcohol poisoning is an important differential diagnosis. Toxic alcohol acidosis does not produce ketosis and, in contrast, does cause significant alteration to conscious state, visual symptoms (methanol) and renal failure/crystalluria (ethylene glycol).
Clinical investigations
The excretion of ethanol by the lungs, although relatively unimportant in terms of ethanol elimination, obeys Henry’s law, i.e. the ratio between the concentration of ethanol in the alveolar air and blood is constant. This allows breath sampling of ethanol to estimate reliably blood ethanol concentration.
Most non-tolerant adults would be expected to develop some impairment of higher functions at blood ethanol concentrations in the range of 0.025–0.05 mg/dL (5–11 mmol/L) and to develop significant CNS depression in the range of 0.25–0.4 mg/L (55–88 mmol/L).
In a patient presenting with acute intoxication, no investigations may be necessary; however, blood or breath ethanol levels (BAL) are frequently useful to confirm the diagnosis. A BAL of zero is highly significant in a patient with an altered level of consciousness, as ethanol intoxication is excluded and other diagnoses need to be considered. A positive blood ethanol level does not exclude alternative diagnoses.
Other investigations should be performed as clinically indicated in an effort to exclude coexisting pathologies and alternative diagnoses as detailed above.
In the patient with AKA, bedside investigations reveal a low/normal glucose, low or absent breath ethanol and urinary ketones (these may be low or absent due to the inability of bedside assays to detect all ketone moieties, especially BOHB). Laboratory investigation will reveal an anion-gap (AG) acidosis (this may be severe with AG>30) and mild hyperlactaemia insufficient to account for the AG.
Treatment
Acute ethanol intoxication
Severe ethanol intoxication with CNS depression is life threatening but a good outcome is assured by timely institution of supportive care. In particular, attention may need to be given to the airway and ventilation. Hypotension generally responds to intravenous crystalloid infusion. The blood sugar level must be checked and normoglycaemia maintained. Intravenous thiamine should be administered, particularly to those with chronic ethanol abuse. There is no specific antidote to ethanol intoxication.
Less severe ethanol intoxication presents a management challenge to the emergency physician when it results in a combative or violent patient threatening harm to self or staff or threatening to discharge against medical advice. Such patients frequently require chemical sedation with titrated doses of intravenous benzodiazepines or butyrophenones in order to facilitate assessment and observation, ensure safety for patient and staff and prevent unsafe discharge.
Ethanol withdrawal
The key to management of this condition is early recognition and institution of adequate dosing of benzodiazepines. Large doses of benzodiazepines may be required to control symptoms. The risk and likely severity of ethanol withdrawal can usually be anticipated if an accurate history of alcohol intake and previous withdrawals is obtained. Coexisting conditions should be managed on their own merits. It is important to exclude hypoglycaemia and correct if present. Thiamine 200 mg (preferably intravenously) should be immediately given to any chronic alcoholic patient who presents with or develops an altered mental status (see Wernicke’s encephalopathy below). Benzodiazepines are first-line therapy for seizures resulting from ethanol withdrawal. Phenytoin is not effective in treating or preventing withdrawal seizures.
The management of ethanol withdrawal in the ED or observation ward is greatly facilitated by the use of ethanol withdrawal charts. These charts facilitate recognition of the first signs of ethanol withdrawal and timely administration of benzodiazepines in adequate doses. An example of such a chart is shown in Figure 29.19.1. Benzodiazepine, usually diazepam, administration is titrated to the clinical features of withdrawal. The total dose required to manage withdrawal is highly variable. Benzodiazepines are usually given orally but can be administered intravenously to the uncooperative or severely withdrawing patient. With extreme withdrawal, refractory to benzodiazepines, small aliquots of ethanol may be effective in controlling severe symptoms.

FIG. 29.19.1 An example of an alcohol withdrawal chart.
Wernicke’s encephalopathy
As Wernicke’s encephalopathy is a clinical diagnosis with high mortality if untreated, any known or suspected alcoholic patient who presents with altered mental status should receive thiamine 200 mg IV during the initial assessment. Recommendations for thiamine dosing in patients with suspected Wernicke’s vary between 200 and 500 mg tds, although there is less evidence supporting the 500 mg dose. These doses ought to be continued until conscious state clears or alternative diagnosis is determined. Parenteral administration is vital as thiamine is variably absorbed orally with low bioavailablility. An example of an emergency department thiamine administration guideline is found in Figure 29.19.2. If dextrose administration is required, it must follow thiamine replacement as it may acutely worsen the neurological status of the thiamine-deficient patient. Magnesium is a co-factor for thiamine-dependent transketolase and so any magnesium deficiency should be corrected.

FIG. 29.19.2 Guideline for thiamine administration in the emergency department. (Prepared by Dr Kerry Hoggett, ED Consultant and Clinical Toxicology Fellow, November 2011. On behalf of the Thiamine Working Party, RPH and FH, Government of Western Australia Department for Health.)
Alcoholic ketoacidosis
Initial resuscitation should include administration of adequate volumes of crystalloids to treat hypovolaemia followed by parenteral thiamine and infusion of dextrose-containing fluids. Potassium and magnesium supplementation should be given according to serum electrolyte results. Administration of dextrose, usually an infusion of 5% dextrose, is essential as it stimulates insulin release, inhibits glucagon release and so inhibits fatty-acid oxidation. Thiamine facilitates entry of pyruvate into the Krebs cycle. Administration of insulin or bicarbonate is not necessary.
Fluid, electrolyte and acid–base status should be closely monitored and further therapy tailored to the clinical response. Careful evaluation and treatment of the coexisting medical disorders is essential.
Disposition
The disposition of many ethanol-intoxicated patients presenting to the ED is determined by the associated medical, surgical, psychiatric or social issues. Ethanol-intoxicated patients should only be discharged from the ED when their subsequent safety can be ensured. Discharge into the care of a competent relative or friend is sometimes appropriate. Other patients, particularly if aggressive or neurologically impaired, require admission to a safe environment until such time as the intoxication resolves and they can be reassessed. An observation ward attached to the ED may be the most appropriate place if available. More severely intoxicated patients requiring airway control and support of ventilation should be admitted to the intensive care unit.
Patients in ethanol withdrawal may require admission for management of the precipitating medical or surgical illness. For those patients who wish to complete withdrawal with a view to abstinence, the remainder of the withdrawal may be managed in a general medical ward, specialized medical or non-medical detoxification centre or at home. Medical detoxification is mandatory where a severe withdrawal syndrome is anticipated. In any case, ongoing psychosocial support will be required and it is important for EDs to have a good knowledge of the locally available drug and alcohol services to ensure appropriate referral.
Patients with Wernicke’s encephalopathy should be admitted for ongoing care and thiamine and magnesium supplementation. The ophthalmoplegia and nystagmus usually have a good response to thiamine within hours to days. Ataxia and mental changes improve more slowly if at all and have a poorer prognosis. Up to 50% of cases will show no response despite thiamine therapy.
Patients with ethanol-induced ketoacidosis also require admission for ongoing dextrose and thiamine, monitoring of fluids and electrolytes and management of the precipitating medical condition. Mortality from ethanol-induced ketoacidosis per se is rare with early recognition and treatment, but death may occur as a result of the underlying medical condition, particularly if unrecognized.
Ideally, any patient with an ethanol-related presentation should be offered referral to drug and alcohol rehabilitation services for counselling.
Toxic alcohols
Epidemiology
Both methanol and ethylene glycol poisoning are extremely rare in Australasia. This is primarily due to their limited availability.
Methanol is found in model aeroplane fuel and laboratory solvents. There is no methanol in ‘methylated spirits’ sold in Australia (this is, in fact, pure ethanol with bittering agents to minimize palatability). Methanol is more freely available in other countries where it is found in household cleaning agents and windshield de-icer. Incorrect distillation of ethanol for human consumption, usually from home-made stills, has resulted in mass poisoning incidents with severe toxicity and fatalities.
Ethylene glycol is most commonly encountered as a constituent of radiator antifreeze or coolant. It is also found in hydraulic fluids and solvent preparations. Significant poisoning in Australasia almost always occurs following deliberate ingestion.
Toxicology
Methanol and ethylene glycol are both small molecules that are rapidly absorbed from the gastrointestinal (GI) tract with a volume of distribution that approximates total body water (0.6 L/kg). Toxic alcohols are oxidized initially by hepatic cytosolic and microsomal alcohol dehydrogenases (ADH) and then further metabolized by aldehyde dehydrogenase into acidic moieties. Methanol is metabolized initially to form formaldehyde and then to formic acid. Ethylene glycol is metabolized to glycoaldehyde and then to glycolate, glyoxylate and oxylate. The plasma half-lives of the toxic alcohols are appreciably increased in the presence of ethanol because ethanol has a much higher affinity for ADH: four times that of methanol and eight times that of ethylene glycol. As a result, the presence of ethanol greatly delays the onset of clinical and biochemical features of toxicity.
Methanol toxicity is mediated through the formation of formic acid. Formic acid binds to cytochrome oxidase resulting in impairment of cellular respiration. Its half-life is prolonged (up to 20 h) and its metabolism is dependent on the presence of tetrahydrofolate. The presence of systemic acidosis enhances the movement of formic acid intracellularly. The initial acidosis is secondary to formic acid, however, as cellular respiration is disturbed and toxicity progresses, a concurrent lactic acidosis is usually evident. Accumulation of formic acid manifests as increasing AG acidosis, gastrointestinal and neurological toxicities.
Ethylene glycol itself is a direct irritant to the GI tract and has CNS depressant effects similar to those of ethanol. The major toxicity is mediated through the acid metabolites, glycolate and oxylate. Oxalate complexes with calcium, leading to crystal deposition chiefly in the renal tubules and the CNS. Myocardium and lungs can also be affected. In addition, these acids appear to be inherently toxic. Complexing with calcium produces systemic hypocalcaemia and may manifest with prolongation of the QT interval. A profound AG acidosis develops and is principally attributed to glycolic acid accumulation although a concurrent lactic acidosis (type B) also contributes.
Predictors of toxicity
Toxic dose
The lethal dose of methanol is conservatively estimated as 0.5–1.0 mL/kg of a 100% solution. Clinical toxicity and visual sequelae may be seen with smaller doses, perhaps as little as 0.25 mL/kg.
The lethal dose of ethylene glycol is thought to be in the order of 1.0 mL/kg of a 100% solution.
Biochemical markers
Biochemical predictors of mortality in toxic alcohol ingestions are an elevated anion-gap metabolic acidosis and a raised osmolar gap.
When available, serum level of methanol and ethylene glycol greater than 50 mg/dL are associated with severe toxicity.
Clinical features
Methanol
Initially, mild CNS depression typical of ethanol intoxication is evident. A latent period (6–24 h) is classically observed during which time the patient may appear asymptomatic. Progressive ophthalmic, GI and CNS symptoms may then develop. Hyperpnoea is usually observed secondary to the metabolic acidosis. Progressive obtundation leading to coma and seizures heralds the onset of cerebral oedema and signifies poorer prognosis. Those who recover from serious CNS toxicity can display extrapyramidal movement disorders. Retinal toxicity may be irreversible in up to one-third of cases.
Ethylene glycol
The progression of clinical features following ingestion of ethylene glycol is described in three stages: neurological, cardiopulmonary and renal. These stages are artificial and toxicity may progress in a rapid manner with concurrent toxicities being observed. Initially, an intoxication syndrome analogous to ethanol occurs along with nausea and vomiting due to mucosal irritation. A progressively severe AG acidosis with renal failure and hypocalcaemia is characteristic. Crystalluria may be observed. With severe poisoning, renal failure progresses rapidly. Central nervous system depression is observed with severe manifestations including seizures, coma and cerebral oedema. Hyperpnoea occurs secondary to the metabolic acidosis.
Clinical investigations
Direct assay of methanol or ethylene glycol concentrations in serum is rarely readily available. In the absence of direct assays, the ability to exclude a potentially lethal toxic alcohol ingestion at presentation is limited. The combination of an osmolar gap (OG) and a wide AG acidosis is highly suggestive of either methanol or ethylene glycol intoxication. However, a normal OG does not exclude toxic alcohol ingestion. In the presence of a profound acidotic state, it is possible that a toxic alcohol has been largely metabolized and thus no longer sufficiently present to raise the OG. Additionally, baseline OGs may vary from−14 to+10 between individuals and so a ‘normal’ OG may mask a large occult increase representing a potentially lethal ingestion. Similarly, a normal AG at presentation is not sufficient to exclude toxic alcohol ingestion. Early in the clinical course an AG may be normal, only to develop rapidly as metabolism progresses. This is particularly so in the presence of ethanol where the onset of an AG acidosis will be delayed until the ethanol itself has been preferentially metabolized.
Falls in serum bicarbonate and arterial pH correlate well with levels of toxic organic acid metabolites in the circulation and, in the absence of direct assays, are their chief surrogate markers. In this context, it is common practice to exclude toxic ingestion where there is a normal venous bicarbonate (>20) 8 h after the serum or breath ethanol has been documented as undetectable.
When available in a clinically useful time frame, direct assays may shorten hospital assessment times especially with accidental exposures. The interpretation of serum methanol and ethylene glycol concentrations requires consideration of time since ingestion, ethanol co-ingestion and acid–base status.
Treatment
The definitive care for methanol and ethylene glycol ingestions is dialysis with concurrent ADH blockade therapy. All cases of deliberate self-poisonings with a toxic alcohol need to be managed in a facility with easy access to dialysis if clinical intoxication becomes apparent. ADH blockade therapy can impede the progression of clinical toxicity and permit safe transfer to an appropriate facility.
Alcohol dehydrogenase blockade
Blockade of ADH can be achieved by the administration of either ethanol or the specific ADH antagonist fomepizole (not currently available in Australasia). These agents prevent metabolism of toxic alcohols and the accumulation of their organic acid metabolites. ADH blockade significantly increases the half-life of parent toxic alcohols. In the presence of ethanol, half-life may increase to about 50 h for methanol and for ethylene glycol up to 20 h. ADH blockade with ethanol, although an essential element of care, is not definitive in most circumstances owing to the practical difficulty maintaining prolonged elevated blood ethanol concentration and obtaining serial toxic alcohol levels. However, where serial levels are available, there is evidence that fomepizole can be used in isolation to treat toxic alcohol ingestions.
Ethanol therapy can be initiated with a loading dose of 8 mL/kg of 10% ethanol intravenously or 1.8 mL/kg of 43% ethanol orally (equivalent to 3×40 mL shots of vodka in a 70 kg adult). Maintenance therapy requires an infusion of 1–2 mL/h of 10% ethanol or 0.2–0.4 mL/h of 43% ethanol orally (equivalent to one 40 mL shot of vodka each hour in a 70 kg adult). The ethanol concentration should be maintained in the range of 100–150 mg/dL (22–33 mmol/L) by careful titration of maintenance administration guided by frequent blood ethanol concentrations.
Haemodialysis
Haemodialysis represents definitive care for confirmed toxic alcohol ingestions. It effectively removes parent toxic alcohols and their acidic metabolites. Lactate free and bicarbonate buffered dialysates may assist the correction of acidaemia. Commonly accepted indications for haemodialysis are listed in Table 29.19.2. Endpoints for haemodialysis are listed in Table 29.19.3. Ethanol is also rapidly cleared by dialysis and ethanol infusion rates need to be increased (usually doubled) during haemodialysis.
Table 29.19.2
Indications for haemodialysis in toxic alcohol poisoning
Severe metabolic acidosis (pH<7.25)
Renal failure (ethylene glycol)
History of a large toxic alcohol ingestion and osmolar gap>10 mmol/L
Visual symptoms (methanol)
Ethylene glycol or methanol levels>50 mg/dL (if available)
Table 29.19.3
Endpoints for haemodialysis in toxic alcohol poisoning
Correction of acidosis
Osmolar gap<10 mmol/L
Ethylene glycol or methanol level<20 mg/dL (if available)
Supportive care and co-factor therapy
Folinic or folic acid administration is recommended in methanol poisoning (folinic acid 2 mg/kg IV qid) to aid in endogenous metabolism of formic acid. Pyridoxine and thiamine supplementation is recommended in ethylene glycol poisoning when the patient is thought to be deplete (e.g. alcoholics), again to aid endogenous metabolism of the pathogenic acids.
In methanol poisoning, systemic acidaemia enhances the movement of formic acid into the intracellular compartment. Correction with intravenous bicarbonate if pH<7.3 is recommended.
Calcium replacement in ethylene glycol poisoning is contentious given that it may promote calcium oxalate crystal formation. Consequentially, calcium should only be replaced if there is symptomatic hypocalcaemia (including prolongation of the QT interval) or intractable seizures.
Prognosis
Prompt ADH blockade therapy and dialysis ensures an excellent outcome in toxic ingestions who present before the development of established end-organ toxicity. Delayed diagnosis and treatment is associated with death and permanent neurological and renal sequelae, including blindness in the case of methanol poisoning.
Controversies
![]() It has been suggested that EDs could play a pivotal role in reducing ethanol-related morbidity by adopting procedures to detect and refer individuals who misuse ethanol. A number of centres have successfully done trial screening and brief intervention strategies for hazardous ethanol consumption.
It has been suggested that EDs could play a pivotal role in reducing ethanol-related morbidity by adopting procedures to detect and refer individuals who misuse ethanol. A number of centres have successfully done trial screening and brief intervention strategies for hazardous ethanol consumption.
![]() It is unclear whether fomezipole provides sufficient advantages over ethanol as an ADH blocker in toxic alcohol poisoning so as to justify the expense of importing and stocking it in Australasia.
It is unclear whether fomezipole provides sufficient advantages over ethanol as an ADH blocker in toxic alcohol poisoning so as to justify the expense of importing and stocking it in Australasia.
![]() Co-factor therapy in toxic alcohol poisoning is of unproven efficacy, however, there are few contraindications to their administration.
Co-factor therapy in toxic alcohol poisoning is of unproven efficacy, however, there are few contraindications to their administration.
Further reading
1. Barceloux DG, Bond GR, Krenzelok EP, et al. American academy of clinical toxicology ad hoc committee on the treatment guidelines for methanol poisoning American Academy of Clinical Toxicology practice guidelines on the treatment of methanol poisoning. J Toxicol Clin Toxicol. 2002;40:415–446.
2. Barceloux DG, Krenzelok EK, Olson K, et al. American Academy of Clinical Toxicology practice guidelines on the treatment of ethylene glycol poisoning. J Toxicol Clin Toxicol. 1999;37:537–560.
3. Coulter CV, et al. Methanol and ethylene glycol acute poisonings–predictors of mortality. Clin Toxicol. 2011;49:900–906.
4. Fulop M. Alcoholic ketoacidosis. Endocrinol Metabol Clin N Am. 1993;22:209–219.
5. Galvin R, et al. EFNS guidelines for diagnosis, therapy, and prevention of Wernicke encephalopathy. Eur J Neurol 2010;1408–1418.
6. Hoffman RS, Smilkstein MJ, Howland MA, et al. Osmol gaps revisited: normal values and limitations. J Toxicol Clin Toxicol. 1993;31:81–93.
7. Hungerford DW, Pollock DA, Todd KT. Acceptability of emergency department-based screening and brief intervention for alcohol problems. Acad Emerg Med. 2007;7:1383–1392.
8. McGuire LC, Cruickshank AM, Munro PT. Alcoholic ketoacidosis. Emerg Med J. 2006;23:417–420.
9. Mégarbane B, Borron SW, Baud FJ. Current recommendations for treatment of severe toxic alcohol poisonings. Intens Care Med. 2005;31:189–195.
10. Sechi G, Serra A. Wernicke’s encephalopathy: new clinical settings and recent advances in diagnosis and management. Lancet Neurol. 2007;6:442–455.
11. Turner RC, Lichstein PR, Peden JG, et al. Alcohol withdrawal syndromes: a review of pathophysiology, clinical presentation and treatment. J Gen Intern Med. 1989;4:432–444.
12. Zuburan C, Fernandes JG, Rodnight R, et al. Wernicke-Korsakoff syndrome. Postgrad Med J. 1997;73:27.
29.20 Carbon monoxide
Nicholas Buckley
Essentials
1 Carbon monoxide is the commonest agent used in completed suicides by poisoning in Australia and the UK.
2 Carbon monoxide is produced by incomplete combustion and is found in car exhaust, faulty heaters, fires and in industrial settings.
3 Carbon monoxide poisoning may result in significant long-term neuropsychological sequelae.
4 Oxygen increases the elimination of carbon monoxide–and the extent of increase is proportional to the inspired oxygen pressure.
5 The optimal mode of oxygen delivery to improve clinical outcomes remains controversial.
Introduction
Carbon monoxide (CO) poisoning is an important cause of mortality and morbidity from poisoning. Immediate resuscitation including 100% oxygen therapy is essential and the long-term results of most patients will be good with this simple intervention. It is unclear whether any additional intervention will reduce the low but important risk of serious long-term neurological damage.
Aetiology, pathophysiology and pathology
Carbon monoxide is a colourless, odourless, tasteless and non-irritant gas, produced by incomplete combustion of hydrocarbons [1]. Small amounts are also produced endogenously by normal metabolic processes. The most common sources of significant exposure are car exhausts, cigarette smoke, fires and faulty home heaters and barbecues. Catalytic converters reduce the production of carbon monoxide and are in all cars manufactured in the last decade or two. Carboxyhaemoglobin (COHb) concentrations in cigarette smokers range as high as 10%.
The pathophysiology of CO exposure is complex and incompletely understood. Upon exposure, CO binds to haemoglobin with an affinity 210 times that of oxygen, thereby decreasing the oxygen-carrying capacity of blood. CO can also produce injury by several other mechanisms, including direct disruption of cellular oxidative processes, binding to myoglobin and cytochrome oxidases and causing peroxidation of brain lipids [1]. However, the end result is tissue hypoxia, leading to varying degrees of end-organ damage and eventually death. The severity of poisoning is a function of the duration of exposure, the ambient concentration of CO and the underlying health status of the exposed individual. Although useful for diagnosis when detected, the initial COHb level correlates poorly with outcome [2].
Epidemiology
Poisoning with CO is an important cause of unintentional and intentional injury worldwide. In the USA alone, an estimated 1000–2000 accidental deaths due to CO exposure occur each year, resulting from an estimated 40 000 exposures [3]. In Australia and the UK, it is the most common agent in completed suicide by poisoning.
Prevention
Prevention of environmental or occupational exposure is possible by use of CO air monitors. COHb concentrations are increased for any given inspired CO concentration if the person is exercising or at high altitudes (increased breathing rate and pulmonary blood flow). These considerations are relevant to acceptable levels of exposure.
The introduction of catalytic converters has reduced CO production in vehicle exhaust and this, in turn, appears to be leading to a reduction in fatal suicidal poisoning in some countries [4,5].
Clinical features
The signs and symptoms of acute carbon monoxide poisoning are shown in Table 29.20.1[6] and severity broadly correlates with maximum COHb concentration. Initial symptoms are non-specific and probably predominantly due to compensatory mechanisms to maintain tissue oxygen delivery to vital organs (e.g. tachycardia, headache, dizziness, gastrointestinal symptoms). Signs with more severe toxicity directly reflect tissue hypoxia with central nervous and cardiovascular toxicity being the most critical manifestations. Death results rapidly when impaired oxygenation of the heart prevents the compensatory increase in cardiac output. The skin is classically cherry pink, although severely ill patients are often pale or cyanosed. Pre-existing cerebral or cardiovascular disease, anaemia and volume depletion or cardiac failure increase toxicity (for a given COHb). These people all have a reduced ability to compensate by increasing cardiac output or redistributing blood supply to vital organs. Cardiac toxicity is common in moderate to severe poisoning. Screening with cardiac enzymes with further testing with echocardiography or SPECT have been suggested [7,8]. Abnormal troponin has also been linked to greater long-term mortality [9].
Table 29.20.1
Typical clinical symptoms and signs relative to COHb (normal=0.5%) [6]
|
COHb (%) |
Symptoms and signs |
|
<10 |
Nil (commonly found in smokers) |
|
10–20 |
Nil or vague non-descript symptoms |
|
30–40 |
Headache, tachycardia, confusion, weakness, nausea, vomiting, collapse |
|
50–60 |
Coma, convulsions, Cheyne–Stokes breathing, arrhythmias, ECG changes |
|
70–80 |
Circulatory and ventilatory failure, cardiac arrest, death |
Buckley NA, Dawson AH, Whyte IM. Hypertox. Assessment and treatment of poisoning. www.hypertox.com www.wikitox.com; 2012 with permission.
Due to low oxygen pressures, the high affinity of fetal haemoglobin for CO and the much longer half-life of CO in the fetal circulation, the fetus is particularly susceptible to CO poisoning. The outcome of significant CO poisoning in the mother is often fetal death or neurological damage.
Delayed or persistent neuropsychiatric sequelae occur, largely confined to those who have prolonged loss of consciousness at some stage [10]. Long-term follow up is necessary as more subtle defects can develop or become apparent over a few weeks to months. The most common problems encountered are depressed mood (even in those accidentally exposed) and difficulty with higher intellectual functions (especially short-term memory and concentration) [11,12]. More severe problems include parkinsonism and speech problems. Neuropsychological testing may detect subtle defects not apparent on crude mini-mental state testing. The incidence of sequelae depends on the definition used–major deficits are relatively uncommon but neuropsychiatric complaints related to memory or concentration may occur in as many as 25–50% of patients with a loss of consciousness [11,12].
Differential diagnosis
In suicide attempts, the diagnosis of CO poisoning is generally apparent from the circumstances when the person is found. The major diagnostic issue is whether there is some other deliberate self-poisoning as this is extremely common. In unconscious patients, the ECG, paracetamol concentration and electrolytes should be reviewed with this possibility in mind [6].
A large proportion of victims of smoke inhalation also have cyanide poisoning. This rarely leads to a change in management (due to problems with administering the cyanide antidotes in this setting) but should be suspected when CNS effects are out of proportion with COHb concentrations and if there is a marked lactic acidosis.
Clinical investigations
Blood gases and oximetry
Most pulse oximeters do not attempt to measure COHb but merely the ratio of oxyHb to deoxyHb. Even those that have co-oximeters are not sufficiently accurate to use for either screening or quantification in a hospital setting [13]. Blood gases with a co-oximeter are required to quantify COHb. COHb concentrations (plus or minus a back calculation based on estimated half-life since removal) provide a rough guide to the extent of exposure. However, it is difficult to estimate accurately the oxygen dose received pre-hospital and therefore the half-life. There is also substantial variability between individuals in the extent they are able to compensate for high COHb. Therefore, the correlation with acute and long-term clinical effects is not good. COHb may confirm (or possibly exclude) the diagnosis but should not be used to estimate long-term prognosis.
ECG
Patients should have a baseline ECG (electrocardiogram), repeated 6 h later and ECG monitoring for at least 24 h and cardiac enzymes if the initial ECG is abnormal. The most important signs seen are those of cardiac ischaemia and these are identical to those seen in coronary artery disease.
Biochemistry
Cardiac enzymes should be measured when there is severe clinical toxicity or ECG changes. Metabolic acidosis, predominantly due to lactate will provide an indication of tissue hypoxia. Electrolytes (sodium, potassium, magnesium) should be measured as low concentrations of any of these may exacerbate cardiac toxicity. S100B concentrations (an astroglial structural protein), indicating acute neurological injury, have the potential to be useful in estimating neurological damage as early elevation correlates with long-term morbidity [14–16].
Criteria for diagnosis
A high COHb (>15%) with typical symptoms or signs confirms the diagnosis of acute CO poisoning.
In some parts of the world, it is common to attribute many non-specific presentations to chronic carbon monoxide exposure, often despite COHb concentrations that are normal or within the range of those seen in ‘healthy’ smokers. There are no agreed on criteria for making a diagnosis of chronic carbon monoxide poisoning, but the diagnosis should not be seriously entertained without confirmation of high ambient CO concentrations in the proposed environmental source.
Treatment
Initial management is directed towards securing the airway and stabilizing respiration and circulation. If there is impaired consciousness, ensure the airway is maintained with intubation if necessary. The comatose patient should be placed on a cardiac monitor, a 12-lead ECG performed, an intravenous line inserted and blood drawn for full blood count, electrolytes, lactate, COHb, blood sugar and cardiac enzymes. If awake, the patient should be reassured and discouraged from activity, for muscle activity will increase oxygen demand [6].
Metabolic acidosis should not be treated directly unless the acidosis itself contributes to toxicity (pH<7.0). It should respond to improved oxygenation and ventilation and the net effect of acidosis on oxygen delivery is probably beneficial.
Oxygen
This decreases the biological half-life substantially from 4 h in ambient air to approximately 40 min in a 100% oxygen atmosphere (Fig. 29.20.1) [6]. One hundred per cent oxygen should be administered with mechanically assisted ventilation if necessary. In patients able to tolerate it, continuous positive airway pressure by mask may allow 100% oxygen delivery without intubation. Four to 6 h of 100% normobaric oxygen will remove over 90% of the carbon monoxide. If the only available oxygen delivery device is a Hudson mask, it should be remembered that at a flow rate of 15 L/min no more than 60% oxygen is delivered. At these concentrations the half-life of CO is still around 90 min and a longer period of oxygen may be required for severe poisonings (Fig. 29.20.1). Oxygen toxicity is unlikely with less than 24 h treatment but the risk increases with increasing exposure.

FIG. 29.20.1 Approximate decline in COHb from 50% according to the inspired oxygen concentration and pressure.
When immediately available, hyperbaric oxygen (HBO) should be considered for patients with serious CO poisoning. Oxygen at 2–3 atmospheres will further reduce the half-life of COHb to about 20 min (see Fig. 29.20.1) but, more importantly, it causes very rapid reversal of tissue hypoxia due to oxygenation of tissue from oxygen dissolved in the plasma.
Controversy exists on the benefits, risks and indications for HBO (see Controversies below). Indications for HBO commonly used by hyperbaric facilities are simply those factors that indicate a higher risk of long-term neuropsychiatric sequelae [11,12]. These include:
![]() loss of consciousness at anytime during or following exposure
loss of consciousness at anytime during or following exposure
![]() abnormal neuropsychiatric testing or neurological signs
abnormal neuropsychiatric testing or neurological signs
![]() pregnancy.
pregnancy.
Complications of HBO therapy [11,12] include:
![]() decompression sickness
decompression sickness
![]() rupture of tympanic membranes
rupture of tympanic membranes
![]() damaged sinuses
damaged sinuses
![]() oxygen toxicity
oxygen toxicity
![]() problems due to lack of monitoring.
problems due to lack of monitoring.
Follow up
As well as psychiatric follow up for all patients who have been poisoned with CO due to self-harm, patients should have a neuropsychiatric follow up at 1–2 months to evaluate any long-term neuropsychiatric injury.
Other treatments
Animal models suggest possible benefits from use of allopurinol and N-acetylcysteine to protect against oxidative damage during hypoxic/reperfusion injury [17]. Numerous experimental (i.e. never used in humans) agents have also been suggested. Their use cannot be recommended outside of clinical trials; however, they may be a more logical treatment to prevent neurological damage from reactive oxygen species than hyperbaric oxygen.
Controversies
![]() The major controversy is about the benefits, risks and indications for HBO–and resolving this ‘clinical uncertainty’ with further trials will likely be frustrated by some extremely certain HBO clinicians [18]. There have been eight HBO randomized clinical trials (RCTs) reporting very conflicting outcomes. Some have concluded that HBO is harmful [19,20] and others that it is beneficial [21]. Systematic reviews have found no evidence for benefit from combined analysis of the trials [11,12]. They also find empiric evidence of multiple biases that operated to inflate the benefit of HBO in two positive trials. In contrast, the interpretation of negative trials was hampered by low rates of follow up, unusual interventions for control patients and inclusion of less severely poisoned patients.
The major controversy is about the benefits, risks and indications for HBO–and resolving this ‘clinical uncertainty’ with further trials will likely be frustrated by some extremely certain HBO clinicians [18]. There have been eight HBO randomized clinical trials (RCTs) reporting very conflicting outcomes. Some have concluded that HBO is harmful [19,20] and others that it is beneficial [21]. Systematic reviews have found no evidence for benefit from combined analysis of the trials [11,12]. They also find empiric evidence of multiple biases that operated to inflate the benefit of HBO in two positive trials. In contrast, the interpretation of negative trials was hampered by low rates of follow up, unusual interventions for control patients and inclusion of less severely poisoned patients.
![]() In centres with a chamber, the use of HBO, when it can be given rapidly and safely, may be justifiable based on the biological rationale that it is the most efficient means of rapidly increasing oxygen delivery and removing carbon monoxide. However, transferring patients between hospitals for delayed use of HBO, particularly over long distances, is not justifiable on current evidence from RCTs, animal studies [22,23] or the known pathophysiology of CO.
In centres with a chamber, the use of HBO, when it can be given rapidly and safely, may be justifiable based on the biological rationale that it is the most efficient means of rapidly increasing oxygen delivery and removing carbon monoxide. However, transferring patients between hospitals for delayed use of HBO, particularly over long distances, is not justifiable on current evidence from RCTs, animal studies [22,23] or the known pathophysiology of CO.
![]() The use of measuring S110B protein in the assessment of prognosis of these patients.
The use of measuring S110B protein in the assessment of prognosis of these patients.
References
1. Weaver LK. Carbon monoxide poisoning. Crit Care Clin. 1999;15:297–317.
2. Seger D, Welch L. Carbon monoxide controversies: neuropsychologic testing, mechanism of toxicity, and hyperbaric oxygen. Ann Emerg Med. 1994;24:242–248.
3. Hampson NB. Emergency department visits for carbon monoxide poisoning in the Pacific Northwest. J Emerg Med. 1998;16:695–698.
4. Amos T, Appleby L, Kiernan K. Changes in rates of suicide by car exhaust asphyxiation in England and Wales. Psychol Med. 2001;31:935–939.
5. Mott JA, Wolfe MI, Alverson CJ, et al. National vehicle emissions policies and practices and declining US carbon monoxide-related mortality. J Am Med Assoc. 2002;288:988–995.
6. Buckley, NA, Dawson AH, Whyte IM. Hypertox. Assessment and treatment of poisoning.<www.hypertox.com www.wikitox.com>; 2012. [Accessed Aug. 2012].
7. Lippi G, Rastelli G, Meschi T, et al. Pathophysiology, clinics, diagnosis and treatment of heart involvement in carbon monoxide poisoning. Clin Biochem. 2012;45:1278–1285.
8. Ahn KT, Park JH, Kim MS, et al. Prevalence and clinical outcomes of left ventricular systolic dysfunction after carbon monoxide exposure. Int J Cardiol. 2011;153:108–110.
9. Henry CR, Satran D, Lindgren B, et al. Myocardial injury and long-term mortality following moderate to severe carbon monoxide poisoning. J Am Med Assoc. 2006;295:398–402.
10. Pepe G, Castelli M, Nazerian P, et al. Delayed neuropsychological sequelae after carbon monoxide poisoning: predictive risk factors in the Emergency Department A retrospective study. Scand J Trauma Resusc Emerg Med. 2011;19:16.
11. Buckley NA, Isbister GK, Stokes B, Juurlink DN. Hyperbaric oxygen for carbon monoxide poisoning: a systematic review and critical analysis of the evidence. Toxicol Rev. 2005;24:75–92.
12. Buckley NA, Juurlink DN, Isbister G, et al. Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev 2011;CD002041.
13. Touger M, Birnbaum A, Wang J, et al. Performance of the RAD-57 pulse co-oximeter compared with standard laboratory carboxyhemoglobin measurement. Ann Emerg Med. 2010;56:382–388.
14. Park E, Ahn J, Min YG, et al. The usefulness of the serum s100b protein for predicting delayed neurological sequelae in acute carbon monoxide poisoning. Clin Toxicol (Phila). 2012;50:183–188.
15. Ide T, Kamijo Y, Ide A, et al. Elevated S100B level in cerebrospinal fluid could predict poor outcome of carbon monoxide poisoning. Am J Emerg Med. 2012;30:222–225.
16. Brvar M, Mozina H, Osredkar J, et al. S100B protein in carbon monoxide poisoning: a pilot study. Resuscitation. 2004;61:357–360.
17. Omaye ST. Metabolic modulation of carbon monoxide toxicity. Toxicology. 2002;180:139–150.
18. Buckley NA, Isbister GK, Juurlink DN. Hyperbaric oxygen for carbon monoxide poisoning: evidence versus opinion. Toxicol Rev. 2005;24:159–160.
19. Scheinkestel CD, Bailey M, Myles PS, et al. Hyperbaric or normobaric oxygen for acute carbon monoxide poisoning: a randomised controlled clinical trial. Med J Aust. 1999;170:203–210.
20. Annane D, Chadda K, Gajdos P, et al. Hyperbaric oxygen therapy for acute domestic carbon monoxide poisoning: two randomized controlled trials. Intens Care Med. 2011;37:486–492.
21. Weaver LK, Hopkins RO, Chan KJ, et al. Hyperbaric oxygen for acute carbon monoxide poisoning. N Engl J Med. 2002;347:1057–1067.
22. Bunc M, Luzar B, Finderle Z, et al. Immediate oxygen therapy prevents brain cell injury in carbon monoxide poisoned rats without loss of consciousness. Toxicology. 2006;225:138–141.
23. Brvar M, Finderle Z, Suput D, Bunc M. S100B protein in conscious carbon monoxide-poisoned rats treated with normobaric or hyperbaric oxygen. Crit Care Med. 2006;34:2228–2230.
*Commercial kits such as Nithiodote are now available that omit amyl nitrite (Nithiodote [sodium nitrite injection and sodium thiosulphate injection] package insert. Scottsdale, AZ: Hope Pharmaceuticals; 2011).