Derwood H. Pamphilon1 & Zbigniew M. Szczepiorkowski2
1Retired from NHS Blood and Transplant, Bristol, UK
2Transfusion Medicine Service, Cellular Therapy Center, Dartmouth-Hitchcock Medical Center and Geisel School of Medicine at Dartmouth, Hanover, New Hampshire, USA
Introduction
In recent years there have been considerable advances in cellular therapies. The most widely used type of cellular therapy has been haemopoietic stem cell transplantation (HSCT) from its inception in 1968. In many cases patients with haematological and nonhaematological diseases are cured after HSCT. There have also been advances in the immunotherapy of cancer and viral infections and in the use of cellular therapy for tissue regeneration and repair.
A number of different agencies and professional bodies are involved in the regulation and accreditation of cellular therapy both in the USA and Europe. The regulations and standards depend on the source of the cell to be transplanted, the way it is used and the nature of any manipulations carried out. As a result of this the last decade has seen a seemingly bewildering growth in regulatory and accreditation requirements and these have put pressure on both clinical and laboratory services.
The drivers for these are:
· traceability of products from donor to recipient;
· microbiological safety;
· enhanced product quality.
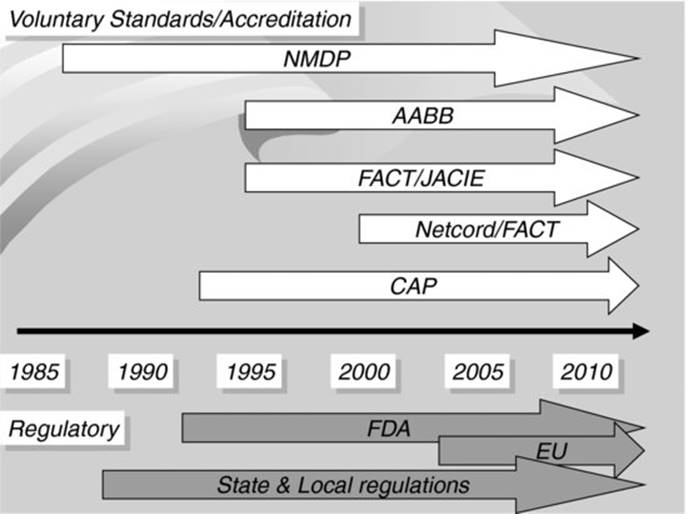
There are multiple organizations involved in the process of accreditation and standard setting of haemopoietic stem cell transplant programmes. Figure 38.1 illustrates the timeline of this involvement by different organizations.
Fig 38.1 Timeline of involvement of different organizations in the field of cellular therapy.
AABB (formerly the American Association of Blood Banks); CAP, College of American Pathologists; FACT, Foundation for the Accreditation of Cellular Therapy; EU, European Union; FDA, US Food and Drug Administration; JACIE, Joint Accreditation Committee (ISCT and EBMT); NMDP, National Marrow Donor Program.
Haemopoietic stem cell transplant activity
The numbers of transplants has increased considerably in recent years so that between 50 and 70 000 are performed each year. The majority are autografts and related (usually sibling) donor procedures, although World Marrow Donor Association (WMDA) data show that the number of transplants using unrelated donor stem cells increased from 3237 in 1997 to 10 981 in 2008 [1]. The overall number of unrelated donors available on international registries reported to the WMDA in its 10th Annual Report increased from 4.8 to 14.6 million during that time period. In the last 10–15 years there has been a switch to the use of peripheral blood progenitor cells (PBPC), which are now regarded as the source of choice in 98% of autografts and 74% of allografts in Europe as well as a great increase in the use of cord blood (CB) units so that, in patients under the age of 16, they now account for 30% of transplants and its use is increasing in adults as well.
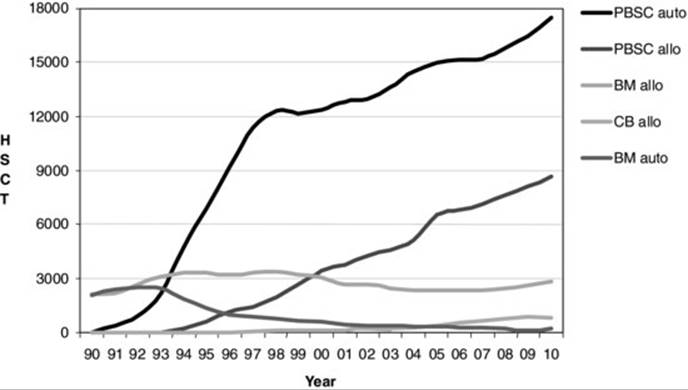
In 1990, 4200 HSCTs were reported to the European Blood and Marrow Transplant Group (EBMT), a number that had risen by 2008 to 26 810 (Figure 38.2). Autologous transplants comprised 65% of these and 35% were allografts; 46% of first-time allografts are now from identical siblings and 49% from unrelated HSCT has increased in all diseases reported to the EBMT and CIBMTR with the exception of chronic myeloid leukaemia (CML), where the advent of the tyrosine kinase inhibitor imatinib has reduced numbers (see Chapter 41).
Fig 38.2 Trends in allogeneic and autologous BM and PB HSCT 1990–2010 (data from Helen Baldomero, EBMT).
The structure of SCT programmes
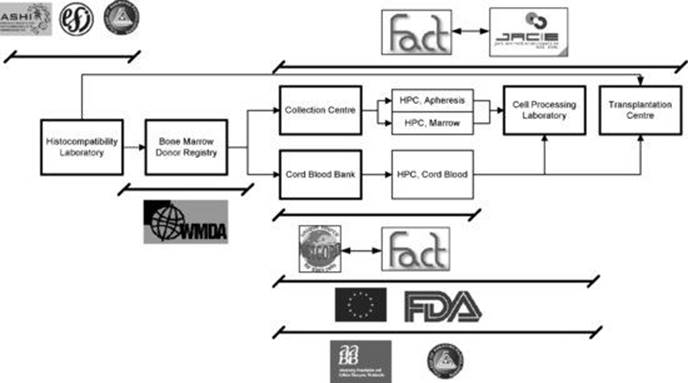
Figure 38.3 shows the journey of an allogeneic stem cell product from a registry or sibling donor or CB unit where a blood sample is typed in the histocompatibility and immunogenetics (H&I) laboratory to determine the HLA type, via the marrow, peripheral blood or CB collection facility and the cell processing laboratory to the clinical transplant unit. The various accreditation and regulatory bodies involved and their area of involvement are shown.
Fig 38.3 Regulatory environment for haemopoietic stem cell transplantation.
Note that this figure does not reflect all potential regulatory reporting requirements for transplantation centres.
European Union Directives and Legislation
Documents published in Europe in 1978 and 1994 stressed the need for international standardization of tissue and cell collection practices and the harmonization of legislation relating to the collection and transplantation of substances of human origin. It was recommended that there should be functional definitions of tissue banks and that such banks should be:
· nonprofit making;
· licensed by national health authorities;
· the cells collected should be tested for infectious disease markers (IDMs);
· appropriate records should be kept;
· there should be consent for removal or collection of cells and tissues.
In 2004 the European Union (EU) Directive on Tissues and Cells was published as Directive 2004/23/EC [2]. In 2006 two technical annexes supplying more detailed information were published as Commission Directives 2006/17/EC (donation, procurement and testing) and 2006/86/EC (coding, processing, preservation, storage and distribution) [3,4]. The Directives are legally binding with a requirement that they are transposed into European law.
In 2005 Competent Authorities (CA) were appointed or established in the UK and other European Member States and these subsequently became responsible for the licensing of facilities storing tissues or cells. In the UK the Human Tissue (Quality and Safety for Human Application) Regulations, which translate the EU Directive into UK law, were published in 2006 and in 2007; all three directives were fully implemented. Other EU countries have also made similar arrangements. The Directive states that it ‘lays down standards of quality and safety for human tissues and cells intended for human applications, in order to ensure a high level of protection of human health’ and that it was established to ‘help to reassure the public that human tissues and cells that are procured in another Member State, nonetheless carry the same guarantees as those in their own country’. Of relevance to HPC transplantation and immunotherapy, the following cells and tissues are included within the scope of the Directive:
· haemopoietic stem cells from peripheral blood, bone marrow and CB;
· donor leucocytes and other cellular therapies;
· adult and embryonic stem cells.
The various sections of the Directive describe:
i the requirements for the person in charge of a cellular therapy or tissue facility (the responsible person or designated individual);
ii the arrangements for the facility itself and its staffing;
iii the role of the CA and the need for 2-yearly inspections;
iv the requirements for consent;
v traceablilty with retention of key records for a period of 30 years;
vi the reporting of adverse events and reactions to the CA;
vii conditions to be met when stem cells are imported or exported.
The Human Tissue Act (HT Act) 2004
This key piece of legislation serves as a good example of how national legislation for cell and tissue collection and processing operates [5,6]. It was introduced in the UK in 2006, repealing and replacing the HT Act of 1961 as well as the Anatomy Act (1989) and Human Organ Transplants Act (1989). It established the Human Tissue Authority (HTA) as the CA for the UK. Its aim is to regulate the collection, storage, use and disposal of human bodies, body parts, organs and tissues.
The HT Act ensured that consent became the fundamental principle underpinning the lawful storage and use of organs and tissues. In addition, the Act also applies to the removal of transplantable material from the deceased. Consent is required when tissue is removed from the living or deceased for the purposes of:
· anatomical examination;
· determining the cause of death;
· obtaining scientific or medical information about a person relevant to another;
· public display;
· research in connection with disorders or functioning of the human body (unless the material is made anonymous and for specific, ethically approved research);
· transplantation.
The HT Act is supported by two governmental regulations (Statutory Instruments 2006 no. 1659 (37) and 2006 no. 1260 (38)), directions issued by the HTA to help explain and interpret the Act and also a number of Codes of Practice, of which three are particularly relevant to cell and tissue therapies [7]. These are:
i consent;
ii removal, collection, retention and disposal of organs and tissues;
iii donation of allogeneic bone marrow and peripheral blood stem cells for transplantation.
Obtaining legally valid consent is extremely important and the HTA states that it is a positive act, that it is voluntary and may be withdrawn at any time. Appropriate information should be provided and the person giving consent must have the capacity to do so. Children may consent if they are competent to do so. Consent prior to death is sufficient for organ and tissue donation and relatives have no legal right to overrule such consent.
The United States Food and Drug Administration (FDA)
The Food and Drug Administration (FDA) of the Department of Health and Human Services of the USA has been involved in the area of cellular therapy since early 1990s [8]. Through a series of public meetings and notices in the Federal Register the FDA recognized the need for regulatory oversight in the area of cell, gene and tissue therapies and products. Initial guidance documents were issued in 1993, 1996, 1997 and 1998 based on the Public Health Service Act, Section 361 (42 USC 264).
FDA Good Tissue Practice (GTP) Regulations for human cells, tissues and tissue-based products (HCT/Ps) require institutions shipping HPC, Cord Blood; HPC, Apheresis; and TC, Apheresis, but not HPC, Marrow, to be registered with FDA as manufacturers. There are specific requirements for donors who may be eligible/ineligible based on a suitability determination and defined in final guidance documents (see below). An annual update is required.
The regulatory approach implemented by the FDA based on the 1997 proposal for regulation included cellular therapy products (named HCT/P – human cells, tissues and tissue-based products) with gene therapy and tissues rather than with blood components. This different approach had significant implications for the field by defining minimal requirements for establishments involved in manufacturing of HCT/P. The FDA also introduced a concept of risk assessment which includes: (1) the relationship between the donor and the recipient (i.e. autologous, allogeneic related, allogeneic unrelated); (2) the amount of processing and manipulation (nonmanipulated, minimally manipulated and more than minimally manipulated); and (3) the purpose for which the tissues are used (homologous and nonhomologous use, where homologous use is defined as repair, replacement or supplementation of a recipient's cells or tissues with an HCT/P that performs the same basic functions in the recipients as in the donor).
The last of the aspects of risk assessment has been debated by the cellular therapy community as one that assigns potentially a different level of regulatory scrutiny based on the intended use despite equivalent risk in the first two areas, such as donor and the level of manipulation.
For very practical reasons it is common among cellular therapy practitioners in the USA to discuss products as ‘351’ and ‘361’ products. This nomenclature relates to two different sections of the Public Health Service Act. The 361 products are covered in 21 Code of Federal Regulations (CFR) 1271 A, B, C, D, E, F (i.e. Good Tissue Practice), while the 351 products are covered in multiple regulations including 21 CFR 1271 C, D; 21 CFR 207.20 (f); 21 CFR 210–211; 21 CFR 807.20 (d); 21 CFR 820.1 (a); 21 CFR 312 (investigational new drug regulations (IND)) and others. The 361 products are defined in 21 CFR 1271.10 as (1) minimally manipulated; (2) intended for homologous use; (3) do not involve combination with a drug or a device, except for a sterilizing, preserving or storage agent, if the agent does not raise new clinical safety concerns; and (4) does not have a systemic effect and is not dependent upon the metabolic activity of living cells for its primary function, or has a systemic effect and is for autologous use, or for allogeneic use in a first-degree and second-degree blood relative, or for reproductive use. All products that do not fulfil these requirements are considered 351 products.
Based on the assignment of 351 and 361 products there are different requirements for biological product deviation reporting.
It is important to note that there are tissues excluded from 21 CFR 1271 that include vascularized organs; whole blood and blood components; human milk and minimally manipulated bone marrow. Thus, HPC, Marrow (minimally manipulated), is regulated by different set of regulations, which are under the authority of the Health Resources and Services Administration (HRSA).
For a thorough discussion of FDA regulatory activities and current guidance documents the reader is referred to the agency website http://www.fda.gov/cber/gene.htm and http://www.fda.gov/cber/tiss.htm.
The ultimate goal of the FDA regulatory structure is to bring cellular therapy products to the licensed status. In October 2009, the FDA issued the guidance document regarding the biological licence application for HPC, Cord Blood. The regulations required that after October 2011 all CB units would be either licensed by the CB banks or issued based on the IND [9] (see the guidance document for more information). These requirements led to a significant effort by the CB banks to submit a biological licence application (BLA) to the FDA for approval. There are several CB banks that submitted BLAs and are in different stages of licence issuance. At the time of writing (November 2012) only three products, Hemacord (New York Blood Center); HPC Cord Blood (University of Colorado Medical School) and Duracord (Duke University School of Medicine) have been licensed.
The CB units that are not licensed are issued under IND protocol (e.g. NMDP is a holder of one of the INDs).
Nongovernmental (voluntary) accreditation
Many programmes elect to be accredited by one of the voluntary accrediting organizations in addition to observing governmental regulations. There are multiple reasons for voluntary accreditation ranging from recognition by healthcare insurance providers for reimbursement purposes through improved quality of care to fulfilling requirements by some of the local governmental regulations (e.g. Commonwealth of Massachusetts requires FACT accreditation from all Transplantation Centres).
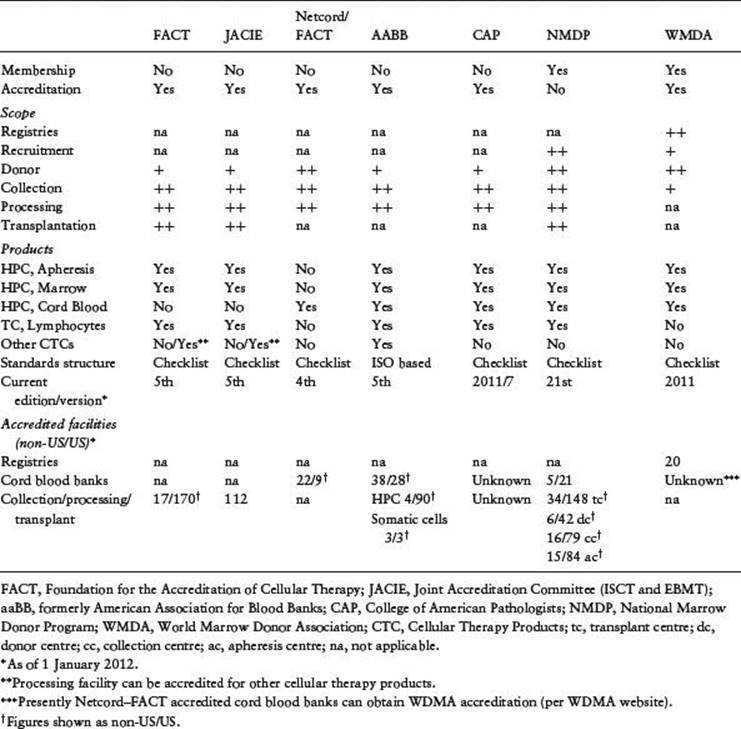
All accrediting organizations require adherence to local and governmental laws and regulations in addition to individual standards established by each of them. Each voluntary organization has a slightly different approach to the accreditation process, but generally an applicant facility needs to meet or exceed standards promulgated by the accrediting organization. The published standards, which are typically updated in defined time intervals, describe a minimum level of expectations. Table 38.1 summarizes major differences (and similarities) between different accrediting organizations.
Table 38.1 Overview of voluntary accreditation/registry organizations.
Standards are prepared by a group of experts within the organization, typically called ‘Standards Committee’, which establish minimum expectations for the facilities willing to participate in the accrediting programme. The draft standards are presented for public comments, which then may or may not be incorporated into the final standards. Some organizations, notably CAP, do not present the standards (in this particular case checklist questions) for comments. Once the final version of the standards is approved a tool is created, which is used during the inspection process.
The accreditation process generally consists of three phases: phase I – the application step, when the applicant facility submits necessary documentation to the accrediting body and certifies that it is compliant with the standards; phase II – the confirmation step, when the accrediting body using on-site inspection confirms that the applicant facility truly follows the standards; and phase III – the recognition step, when a certificate of accreditation is being issued based on the documentation submitted and the results of on-site inspection and, if necessary, satisfying responses to identified shortcomings in the applicant facility. The accreditation certificate has an expiration date and stipulates that if there are any significant changes in the programme structure and/or performance these will be promptly reported to the accrediting body. Each of the accrediting organizations may have additional requirements.
(i) FACT and JACIE
In 1994 the Foundation for Accreditation of Cell Therapy (FACT) in the USA initiated a voluntary inspection and accreditation scheme for cell therapy facilities. Five years later its European counterpart, the Joint Accreditation Committee of the ISCT (International Society for Cell Therapy) and EBMT (JACIE), was founded. FACT–JACIE is a voluntary system that accredits clinical transplant programmes as well as the cell collection, processing and banking elements that are covered by current EU legislation [10]. Whilst FACT–JACIE accreditation is not compulsory, there are pressures for the clinical, collection and laboratory parts of HSCT programmes to comply with their requirements in some countries. These include purchasing agreements with healthcare funders. The primary aim of FACT and JACIE is to improve the quality of HSCT in North America, Europe and elsewhere by providing a means whereby transplant centres, HPC collection facilities and processing facilities can demonstrate high quality practice. This is achieved through external inspection of facilities to ensure compliance with the FACT–JACIE standards. A further aim is to ensure consistency between the standards and other national and international standards, including the EU Tissues and Cells Directive (Directive 2004/23/EC) and the related Commission Directives 2006/17/EC and 2006/86/EC (see above).
FACT–JACIE accreditation is voluntary, but provides a means whereby transplant facilities can demonstrate that they are working within a quality system that covers all aspects of the transplantation process and that they can show compliance with the requirements of insurance companies or national and/or international regulatory authorities. Accreditation of HPC transplant facilities is through on-line submission of documentation and Centres may apply for accreditation as complete programmes comprising a clinical programme, collection facility and processing laboratory or, for example, as a single collection or processing facility that may serve a number of clinical programmes.
The FACT–JACIE standards
The 5th edition of the standards was published in 2012 and covers all aspects of clinical transplant programmes, bone marrow and peripheral blood stem cell collection facilities, as well as processing laboratories. The standards also apply to the use of therapeutic cells (TC) derived from the peripheral blood or bone marrow, including donor lymphocytes and mesenchymal stem cells. The standards cover the clinical use of HPC(CB) by clinical programmes but not the collection or banking of CB, which is covered by the related Netcord–FACT standards and inspected and accredited by FACT–Netcord (see below). The standards are available on the FACT and JACIE websites and are structured as shown in Table 38.2, and contain essential principles that apply throughout:
Table 38.2 Structure of the 5th edition of the FACT–JACIE standards.
|
Clinical (B) |
Collection (C+Cm) |
Processing (D) |
|
General |
General |
General |
|
Clinical Unit |
Collection Facility |
Processing Facility |
|
Personnel |
Personnel |
Personnel |
|
Ouality Management |
Ouality Management |
Ouality Management |
|
Policies and Procedures |
Policies and Procedures |
Policies and Procedures |
|
Donor Selection, Evaluation, and Management |
Donor Evaluation and Management |
Process Controls |
|
Therapy Administration |
Coding and Labeling |
Coding and Labeling |
|
Clinical Research |
Process Controls |
Distribution |
|
Data Management |
Cellular Therapy Product Storage |
Storage |
|
Cellular Therapy Product Transportation and Shipping |
Transportation, Shipping, and Receipt |
|
|
Records |
Records |
Disposal |
|
Direct Distribution to Clinical Program |
Records |
· Establishment and maintenance of a Quality Management Programme (QMP).
· Requirement for documentation of policies, procedures, actions, requests, which extends to all aspects of transplant activity. For example, the initial diagnosis of a patient must be documented in the clinical notes using source material or reports. A request from the clinical unit to the laboratory for issue of cells must be made in writing. A potential donor must not only be properly evaluated for eligibility but the programme must have clear written criteria for what constitutes an eligible donor and must clearly document whether the donor meets these criteria.
· Personnel must not only be appropriately qualified, they must be trained in the procedures they regularly perform and their competency to perform the task after training must be assessed and documented.
· Validation of all equipment and procedures. Validation is a term used to describe the activity required to prove that any procedure, process, equipment, material, activity or system actually leads to the expected results. For example, a new apheresis machine must be shown to produce the expected results in terms of cell yields.
Also important is the requirement for close cooperation and interaction between the different parts of the programme, especially important where a clinical programme may use an offsite collection and/or processing facility.
Quality management (QM)
An active quality management programme (QMP) is essential to the FACT–JACIE standards. A QMP is a mechanism to ensure that procedures are being carried out by all staff members in line with agreed standards. In a transplant programme, this ensures that the clinical, collection and laboratory units are all working together to achieve good communication, effective common work practices and increased guarantees for patients. It is a means of rapidly identifying errors or accidents and resolving them so that the possibility of repetition is minimized. It assists in training and clearly identifies the roles and responsibilities of all staff. Once the required level of quality has been achieved, the remaining challenge is to maintain this standard of practice. With a working quality management system in place and adequate resources, the fundamental elements necessary to sustain the programme are continued staff commitment and vigilance. The culture and systems for quality management are well established in laboratories but are relatively new in clinical units and many programmes have experienced difficulty setting up a QMP to cover the clinical programme and collection facility. It is recommended that HSCT programmes have dedicated quality managers.
Experience of centres implementing FACT and JACIE
It was anticipated that implementation of the FACT–JACIE standards would pose some difficulties for applicant centres, particularly in relation to establishing a quality management system (QMS). It was also anticipated that there would be resource implications in terms of staff time because of the amount of detailed documentation that is required to demonstrate compliance with the standards. To assess this in Europe a survey was designed to assess the difficulties experienced by centres in preparing for JACIE accreditation and the results showed that the most difficult part of preparation was implementing the quality management (QM) system, adverse event reporting system and other documentation. Lack of a culture of QM was cited as an important problem. The extra resources most frequently required were a quality manager and a data manager. Only 19% of centres needed to improve their physical facilities. There is clearly an important need for training of clinical staff (doctors and nurses) in QM. It is also important for centres to have a designated quality manager who has appropriate experience in QMS.
Improvements clearly depend on the level of existing services, so that failure to demonstrate improvement in, for example, facilities or data management may reflect good pre-existing resources. In other areas, e.g. adverse event reporting, the systems for monitoring performance were only set up as part of implementing JACIE, so it is difficult to monitor improvements without an established baseline for comparison. Indeed, implementation of JACIE may have the paradoxical effect of seeming to increase adverse events because these were not previously adequately reported. All centres felt that accreditation was worth the effort invested. In addition, with the implementation of the EU Directive on Safety of Tissues and Cells (2004/23/EC) it is likely that collection and processing facilities will increasingly view compliance with JACIE standards as important in providing evidence that they are complying with the requirements of the Directive.
A detailed analysis of transplant outcome in 107 000 European patients transplanted between 1999 and 2007 showed that acquisition of JACIE accreditation was associated with improvement in overall patient survival [11].
(ii) Netcord–FACT
In the same way that FACT–JACIE cooperate to produce a globally agreed set of standards and a guidance manual for accreditation of HSCT programmes, FACT collaborates with Netcord, which is an international organization for CB banking [12]. Their combined, international standards, first issued in 2000, are the gold standard for CB banks worldwide. The most recent standards (4th edition) were published in 2010. They comprise five sections that cover CB bank quality management, operations, donor management and collection, processing, donor selection and release. It should be noted that clinical transplantation is dealt with in the FACT–JACIE standards. The next edition is expected in 2013.
(iii) AABB
Established in 1947, AABB is an international, not-for-profit association dedicated to the advancement of science and the practice of transfusion medicine and related biological therapies. AABB membership consists of approximately 1800 institutions and over 8000 individuals, including physicians, scientists, administrators, medical technologists, nurses, researchers, blood donor recruiters and public relations personnel. Members are located in all 50 states and 80 foreign countries. In 1958, the Standards for a Blood Transfusion Service was published, and an independent accreditation programme was established. The AABB approach to the field of cellular therapies has aimed to balance flexibility in an outcome-based approach with the need for rigorous evidence-based standards. The standards are written using an ISO-based template. The ten chapter headings are based on the AABB Quality System Essentials (QSEs), published in 1997 as AABB Association Bulletin 97-4. The 10 QSEs correlate directly with ISO. The AABB Standards for Cellular Therapy Services (5th edition published in 2011) [13], which are revised and updated every 18 months, cover all cellular therapy product and cell sources including autologous, allogeneic and cadaveric donors. AABB also coordinates the production of a cell therapy product Circular of Information (COI) [14].
Under a QM system approach, each chapter progresses from general policies to specific procedures. The chapters are:
· Organization
· Resources
· Equipment
· Agreements
· Process Control
· Documents and Records
· Deviations and Nonconforming Products or Services
· Internal and External Assessments
· Process Improvement
· Safety and Facilities
The chapters open with broad statements (a part of the template for all AABB standards, which are amended, if necessary, to fit a particular field), which are followed by more specific standards and, finally, end with reference standards, which are most prescriptive. The reference standards are generally presented as tables or lists of activities/requirements.
The AABB accreditation is valid for 2 years and each accredited institution is assessed every 24 months. Recently, the AABB, following other accrediting organizations, introduced unannounced assessments. These occur on any day within 90 days of the accreditation expiration date.
(iv) College of American Pathologists
The College of American Pathologists (CAP) (www.cap.org) is a medical society serving nearly 16 000 physician members and the laboratory community throughout the world [15]. It is the world's largest association composed exclusively of pathologists and is widely considered the leader in laboratory quality assurance. The nearly 16 000 pathologist members of the CAP represent board-certified pathologists and pathologists in training worldwide. More than 6000 laboratories are accredited by the CAP and approximately 23 000 laboratories are enrolled in the College's proficiency testing programmes. There are two proficiency tests currently offered for the cellular therapy products, SCP (Stem Cell Test) and CBT (Cord Blood Test).
The CAP primarily accredits laboratories in clinical and anatomical pathology, but the accreditation process also includes other entities such as cellular therapy laboratories, HLA laboratories and reproductive laboratories.
The accreditation process, called the Laboratory Accreditation Program, is based on fulfilling the CAP checklist (self-assessment and on-site inspection), which consists of three major parts: the discipline specific checklist(s), the laboratory general checklist and the all common checklist. Each checklist component consists of subject header, declarative statement and evidence of compliance. These elements are designed to help both the inspectee and inspector in fulfilling the intent of the requirement. Furthermore, the inspectors are trained to use the ROAD (Read, Observe, Ask, Discover) inspection technique.
The questions on tissue banking were added to the transfusion medicine checklist in 1993. There were five questions covering the following: (1) documentation defining the authority, responsibility and accountability of the programme; (2) records documenting the type of processing and infectious disease testing for each tissue stored; (3) procedures defining storage conditions of the different tissues handled and retention of records; (4) records showing proper storage conditions; and (5) records allowing for identification of the donor and recipient for each tissue handled. In 2004 the CAP expanded the tissues section of the checklist and significantly expanded the haemopoietic progenitor cells section of the checklist. In general, the CAP checklists undergo frequent review by the CAP's scientific resource committees and the Commission on Laboratory Accreditation. New editions are released once a year.
The Reproductive Laboratory Accreditation checklist was created in 1993 and contains requirements for gametes and embryos. The September 2007 edition was revised to address or clarify requirements to prepare CAP-accredited laboratories better for their FDA inspections.
The CAP inspection is performed every other year and the inspection team consists of professionals from a CAP accredited facility that is led by a team leader who is an appropriately qualified Fellow of the College. The inspections generally last two days. The accredited facilities are also required to perform self-evaluation in the year when there is no on-site inspection. Any identified shortcomings need to be documented and addressed.
(v) The World Marrow Donor Association
The WMDA is an international organization that publishes standards to which HSCT donor registries wishing to achieve accreditation for their activities must adhere [16]. These standards are available on the WMDA website (www.worldmarrow.org) and include benchmark standards with which all registries must comply in order to be accredited for the first time, the rest being optional. For subsequent accreditation, the registry must comply with all of the standards. Important areas described by the standards include general organization of the donor registry, donor recruitment, assessment, counselling, histocompatibility and immunogenetic characterization of donors, other testing including an infectious disease marker, IT requirements, donor searches, collection and transport of cells. At the present time accreditation is given after a detailed review of documentation submitted by the registry by independent reviewers but site visits are not done, although a pilot scheme to introduce them is under way. Accreditation is valid for 5 years.
(vi) Histocompatibility Accreditation [17,18]
The American Society for Histocompatibility and Immunogenetics (ASHI) and its European counterpart – the European Federation of Immunogenetics (EFI) – accredit H&I laboratories after reviewing documentation and conducting a site visit. The College of American Pathologists also accredits histocompatibility laboratories. The accreditation by CAP fulfils requirements of the National Marrow Donor Program 21st edition of standards (October 2011), but at the time of writing has not been accepted by the FACT–JACIE standards. This creates an unfortunate situation where the transplant programmes cannot utilize CAP accredited histocompatibility laboratories for HLA testing.
Conclusions: How do HSCT programmes respond to the challenge?
The requirements of regulatory and accreditation bodies place huge demands on transplant programmes. In some cases they may need to construct new and improved facilities for HSC collection, processing and storage. A key feature is the need to develop robust quality management programmes as described above, which will include detailed policies and procedures to cover all their activities. Initial staff training and ensuring ongoing competency are crucial. HSCT programmes should remember that deficiencies commonly found at inspection involve the QMP, policies and procedures, donor assessment and testing and the labelling of cell therapy products. The interaction between the different component parts of programmes should work seamlessly and where, for example, cell processing or laboratory testing is performed outside the programme by external agencies, then service level agreements will need to be in place. Most units that achieve compliance with regulatory and accreditation standards feel that the exercise has been worthwhile and that the quality of the services that they offer has been improved.
Key points
1. There have been considerable advances in cellular therapy in the last 20 years and newer developments include the use of cell therapy products for regenerative medicine and immunotherapy.
2. The accreditation and regulatory environment has become increasingly complex and its aim is to enhance product quality and safety.
3. The development of robust quality systems is central to achieving compliance with these new requirements.
4. Some regulations are mandatory, e.g. the EU Directive and FDA requirements, whilst others, such as FACT–JACIE or AABB accreditation, are voluntary.
5. Increased resource is required to successfully implement the changes needed to achieve compliance.
References
1. World Marrow Donor Association Annual Report. Available at: http://www.worldmarrow.org/.
2. Directive 2004/23/EC of the European Parliament and Council on setting standards of quality and safety for the donation, procurement, testing, processing, preservation, storage and distribution of human tissues and cells. Available at: www.transfusionguidelines.org.uk.
3. Commission Directive 2006/17/EC implementing Directive 2004/23/EC of the European Parliament and Council as regards certain technical requirements for the donation, procurement and testing of human tissues and cells. Available at: www.transfusionguidelines.org.uk.
4. Commission Directive 2006/86/EC implementing Directive 2004/23/EC of the European Parliament and Council as regards traceability requirements, notification of severe adverse reactions and events and certain technical requirements for the coding, processing, preservation, storage and distribution of human tissues and cells. Available at: www.transfusionguidelines.org.uk.
5. The Human Tissue Act 2004 (except Scotland), ISBN 0 10 543004 8. Available at: www.hta.gov.uk.
6. The Human Tissue Act 2006 (Scotland), ISBN 0-10-590094-X. Available at: www.show.scot.nhs.uk.
7. Human Tissue Authority (HTA): Codes of Practice for Consent (Code 1), for Donation of Organs, Tissues and Cells (Code 2), for Removal, Storage and Disposal of Human Organs and Tissues (Code 5), for Donation of Allogeneic Bone Marrow and Peripheral Blood Stem Cells for Transplantation (Code 6) and for Import and Export of Human Bodies, Body Parts and Tissue (Code 8). Available at: http://www.hta.gov.uk.
8. FDA documents. Available at: www.fda.gov/cber/tiss.htm and www.fda.gov/cber/gene.htm.
9. Guidance for Industry Minimally Manipulated, Unrelated Allogeneic Placental/Umbilical CB Intended for Hematopoietic Reconstitution for Specified Indications; October 2009. Available at: http://www.fda.gov/cber/guidelines.htm.
10. Standards for Haematopoietic Progenitor Cell Collection, Processing and Transplantation, fifth edn, from the Foundation for the Accreditation of Cell Therapy (FACT) and the Joint Accreditation Committee of ISCT–Europe and EBMT (JACIE); 2012. Available at: www.jacie.org.
11. NETCORD–FACT International Standards for CB Collection, Processing, Testing, Banking, Selection and Release, 4th edn; 2010. Available from www.factwebsite.org.
12. AABB Standards for Cellular Therapy Services, 5th edition; 2011. Available from: www.aabb.org.
13. Circular of Information for the Use of Cellular Therapy Products, version 2009. Available from: www.aabb.org and www.factwebsite.org.
14. CAP Checklist. Available at www.cap.org under Accreditation and Laboratory Improvement.
15. World Marrow Donor Association (WMDA) Standards, March 2011. Available at www.worldmarrow.org.
16. Standards for Histocompatibility Testing: the American Society for Histocompatibility and Immunogenetics (ASHI) standards; 2011. Available at http://www.ashi-hla.org/images/uploads/2011ASHIStandards_Guidance.pdf.
17. Standards for Histocompatibility Testing: the European Federation for Immunogenetics (EFI) standards; 2010. Available at www.efiweb.org.
18. Hurley CK. (1999) Histocompatibility Testing Guidelines for Haematopoietic Stem Cell Transplantation using Volunteer Donors: Report from the World Marrow Donor Association. Quality Assurance and Donor Registries Working Groups of the World Marrow Donor Association. Bone Marrow Transplantation 1999; 24(2): 119–121.
Further reading
Cornish JM. JACIE accreditation in paediatric haemopoietic SCT. Bone Marrow Transplant 2008, October; 42 (Suppl. 2): S82–86. Review.
Gratwohl A, Brand R, Niederwieser D, Baldomero H, Chabannon C, Cornelissen J, de Witte T, Ljungman P, McDonald F, McGrath E, Passweg J, Peters C, Rocha V, Slaper-Cortenbach I, Sureda A, Tichelli A & Apperley J. Introduction of a quality management system and outcome after hematopoietic stem-cell transplantation. J Clin Oncol 2011, 20 May; 29(15): 1980–1986. Epub 11 April 2011.
Hurley CK, Foeken L, Horowitz M, Lindberg B, McGregor M & Sacchi N; WMDA Accreditation and Regulatory Committees. Standards, regulations and accreditation for registries involved in the worldwide exchange of hematopoietic stem cell donors and products. Bone Marrow Transplant 2010, May; 45(5): 819–824. Epub 22 February 2010.
Pamphilon D, Apperley JF, Samson D, Slaper-Cortenbach I & McGrath E. JACIE accreditation in 2008: demonstrating excellence in stem cell transplantation. Hematol Oncol Stem Cell Ther 2009; 2(2): 311–319.
Wall DA. Regulatory issues in cord blood banking and transplantation. Best Pract Res Clin Haematol 2010, June; 23(2): 171–177. Review. PMID: 20837328 [PubMed – indexed for MEDLINE].