Edward F. Domino, MS, MD and Shannon C. Miller, MD, FASAM, DFAPA
CHAPTER OUTLINE
■ DEFINITION (DRUGS IN THIS CLASS)
■ SUBSTANCES INCLUDED IN THIS CLASS
■ FORMULATIONS, METHODS OF USE, AND ABUSE
■ HISTORICAL FEATURES
■ EPIDEMIOLOGY
■ PHARMACOKINETICS
■ PHARMACODYNAMICS
■ DRUG-DRUG INTERACTIONS
■ NEUROBIOLOGY
■ CONCLUSIONS AND FUTURE RESEARCH
DEFINITION (DRUGS IN THIS CLASS)
Several heterogeneous chemicals are antagonists of the N-methyl-D-aspartate (NMDA) receptor subtype of the major excitatory neurotransmitter, glutamic acid, in the brain. Termed dissociatives, these substances can be distinguished pharmacologically and clinically from true hallucinogens (see Chapter 14). A simplified view suggests that dissociatives and hallucinogens share common features; however, hallucinogens affect primarily 5HT2A receptors instead of NMDA receptors. Hallucinogens are associated with a different 5HT2A-associated clinical syndrome of intoxication (whereby dissociation or impaired reality testing is less typically involved and visual hallucinations are more commonly involved). Dissociatives include various arylcyclohexylamines (of which phencyclidine [PCP] and ketamine are best known), dizocilpine (MK-80l), dextromethorphan (DXM), and the gaseous anesthetic, nitrous oxide. Ketamine and nitrous oxide are used clinically in animals and humans as general anesthetics, usually in combination with other agents. DXM is used clinically as an antitussive in more than 100 over-the-counter cough preparations.
Most of the known NMDA antagonists are drugs of abuse when used in subanesthetic doses/concentrations. Subanesthetic doses of PCP and ketamine induce a psychotomimetic state that resembles many but not all of the signs and symptoms of schizophrenia (1). DXM in large doses is readily metabolized to dextrorphan (DXO), a significant NMDA antagonist pharmacodynamically akin to PCP and ketamine (2). Nitrous oxide or “laughing gas” is not typically classified as a dissociative (more commonly considered an inhalant); however, given its NMDA antagonist and dissociative-like clinical effect, it merits inclusion in this chapter.
Dissociatives share the clinical effect of causing a dissociative state of intoxication that is desired by the user. The pharmacology of hallucinogens and other drugs capable of producing psychosis is discussed in their respective chapters in this section. The specific treatment of dissociative intoxication and withdrawal states is discussed in Section 6 of this book.
SUBSTANCES INCLUDED IN THIS CLASS
The chemical structures of abused arylcyclohexylamines are shown in Figure 15-1. PCP and ketamine are the principal abused illicit compounds. DXM is the principal abused over-the-counter compound. Ketamine (Ketalar, Ketalin, Keta, Calypsol, etc.) is a racemic mixture of D- and L-isomers. The (S)-isomer is more potent and is claimed to have less dysphoric effects. Other members of this class that are less commonly abused include cyclohexamine (N-ethyl-1-phenylcyclohexylamine, CI, 400), 1-(1-2-thienylcyclohexyl) piperidine, 1-(1-phenylcyclohexyl) pyrrolidine, and 4-methyl pip PCP (1-(phenylcyclohexyl)-4-methylpiperidine). Dextromethorphan (DM, DXM, D-3-methoxy- N-methylmorphinan) is the D-isomer of a codeine analog, methorphan. In contrast to the L-isomer, which is an opioid analgesic, DXM is not. The PCP-derived designer drug N-(1-phenylcyclohexyl)-3-methoxypropanamine and the ketamine analog methoxetamine should also be included in this class.
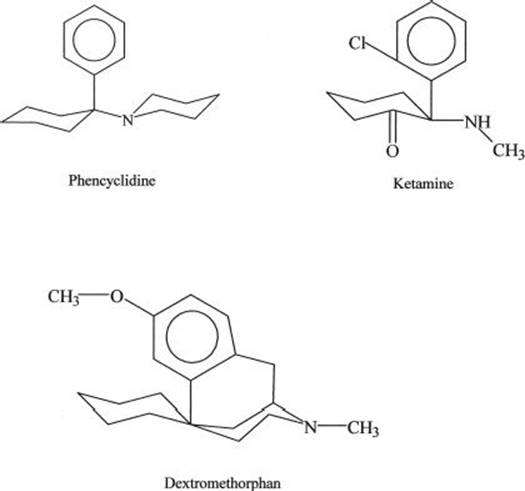
FIGURE 15-1 Chemical structures of major abused dissociative drugs.
FORMULATIONS, METHODS OF USE, AND ABUSE
U.S. Food and Drug Administration-approved Formulations
Ketamine (Ketalar) is available as a sterile solution for use in general anesthesia in both animals and humans as a U.S. Controlled Substances Act Schedule III substance. It has also been used for prehospital analgesia and anesthesia (3) and conscious sedation (4). It is used most often in children, who appear less susceptible than adults to emergent delirium (5). In medical settings, it is injected intravenously or intramuscularly in doses of 0.1 to 1.0 mg/kg, depending on its clinical use. However, it is also effective when insufflated, smoked, or taken orally. It is abused by various routes in different doses. Doses of ketamine as large as 900 to 1,000 mg given intravenously or intramuscularly are lethal.
PCP is no longer available as a medical commercial preparation approved by the U.S. Food and Drug Administration (FDA). It is available in many illicit preparations in various forms and is used in a wide range of doses. It has many of the pharmacologic effects of ketamine but is more potent, longer acting, and more likely to produce seizures. Doses of only 120 mg of PCP may cause death. It has been used as a general anesthetic in animals. It is prepared illegally in various forms: powder, tablets, and liquid (salt in water, base in ether). The latter are typically sprayed onto plant leaves such as ginger, marijuana, mint, oregano, or parsley and then smoked. PCP and some of its congeners are Schedule I substances.
Legal DXM preparations are administered orally. They are usually available as DXM hydrobromide and less commonly as DXM polistirex. Capsules, tablets, lozenges, or solutions of DXM hydrobromide are available alone or in combination with many other substances as cough, cold, and flu relief preparations. The usual antitussive dosage for adults is 10 to 20 mg every 4 hours or 30 mg every 6 to 8 hours, not to exceed 120 mg daily. Extended release forms are given as 60 mg twice daily. Larger doses of DXM are abused for their mental effects. DXM has a low toxicity and high therapeutic index (6). Death is unlikely even at very high doses but has been reported. The additional ingredients in over-the-counter preparations make for additional hazards: decongestant/pseudoephedrine (causing cardiac toxicity), antihistamine/chlorpheniramine (antihistamine toxicity), pain/acetaminophen (liver toxicity), and bromides (bromide toxicity). A serotonin syndrome has also been reported (2).
HISTORICAL FEATURES
The discovery of PCP has been well documented by those involved with its therapeutic development (7). The drug was developed as an intravenous anesthetic. The unique anesthesia it produced was complicated by a prolonged emergence delirium. This quickly led to its demise as a clinically useful agent. PCP is associated with symptoms that model both the positive (delusions, hallucinations) and negative (blunted affect, ambivalence, asociality, autistic-like effects) symptoms of schizophrenia, making for perhaps one of the more useful pharmacologic models of schizophrenia (8,9). Its trade name was Sernyl or Semylan. Years later, PCP was rediscovered by the drug abuse community and has also been known as “PCP,” “angel dust,” “hog,” and “crystal” (10).
The desirable anesthetic properties of PCP were retained in the short-acting arylcyclohexylamine derivative ketamine, which produced a briefer emergence delirium. The term dissociative anesthetic was coined to emphasize that the anesthetized patient was psychologically “disconnected” from his or her environment. Ketamine subsequently was discovered by the drug abuse community, where it is known as “K,” “super K,” “special K,” and “cat Valium,” among others. Ketamine has the reputation among users as being medically safe to use because it is made and packaged by pharmaceutical companies, most often for veterinary use (11).
Arylcyclohexylamine abuse occurs primarily in large metropolitan areas. Because the drugs are easy to synthesize, they are relatively inexpensive substitutes for many street drugs. The user may not realize that he or she has used an arylcyclohexylamine because the drugs frequently are misrepresented as LSD-25, amphetamine, or synthetic marijuana. Moreover, they may be added to marijuana by the user to enhance marijuana’s desired effects.
In contrast to the arylcyclohexylamines, MK-801 (dizocilpine) was developed as an anticonvulsant (12) and subsequently was used as a brain-protective agent; however, it was discarded because of its PCP-like effects (13). Clinical trials of MK-801 have been extremely limited, and the results are not publicly available. Very little is known of its properties in humans.
The history of DXM begins with the synthesis of race-methorphan (deoxydihydrothebaiodine) or methorphan (Dromoran) and its patenting by Hoffmann-LaRoche in 1954 as an opioid analgesic. After the D- and L -isomers were isolated, it was discovered that the D-isomer was antitussive and had less analgesic and narcotic-like properties. DXM is nearly equal to codeine as an antitussive. However, unlike codeine, DXM is fairly devoid of other opioid effects such as analgesia, central nervous system depression, and respiratory suppression. Although it is metabolized to DXO, an NMDA receptor antagonist, its binding sites in the brain include more than the NMDA receptor. DXM’s mechanism in low doses as an antitussive is unknown. In doses of 300 to 1,800 mg (20 to 120 times the recommended dose), DXM produces PCP-like mental effects (14,15). However, larger doses (237 times the recommended dose) are regularly abused (16). DXM abuse has been a concern since at least the 1960s. The over-the-counter tablet form of DXM, Romilar, was replaced by a cough syrup in 1973 to reduce its recreational use. In 1990, the FDA Drug Abuse Advisory Committee assessed DXM use by teenagers and recommended against placing the drug on the Controlled Drug Schedule but requested more study of the problem (17). Although abuse of DXM began originally with abuse of liquid cough syrup (known to hamper the abuse of large doses of DXM because of the distasteful nature of cough syrup), more convenient consumer products have since been developed, including both high-dose (30 mg) tablets as well as high-dose gel capsules, which are preferred by those who abuse DXM. Acid-base extraction techniques have been developed by users to “free base” the DXM alone (18). DXM has become popular, particularly among children and adolescents, owing to this population incorrectly perceiving DXM as a “SMART” drug to abuse; that is, they perceive DXM abuse as without stigma, not costing much money to procure, having easy access at local stores, being devoid of medical risks, and not included in routine employment or home-based drug testing (19). The FDA continues to express its concern over the abuse of DXM, in particular the purified powder form purchased via the Internet (20). Slang terms for the cough medicine preparations are “dex,” “robo,” “skittles” (owing to some tablets appearing similar to red Skittles candies), “tussin,” and “triple Cs.” Recreational use of DXM is described as “dexing,” “roboing,” and “robotripping” (referring to the popular DXM cough syrup Robitussin®).
Nitrous oxide has been known for more than 225 years. It is widely used today in anesthesia. In addition, its recreational use as “laughing gas” has been well described since it first was discovered. Ketamine and nitrous oxide still are medically used in humans as anesthetic agents. Ketamine is used in circumstances in which other anesthetic agents are relatively contraindicated. In contrast, nitrous oxide is widely used today as part of the mixture of anesthetics used to achieve “balanced anesthesia.”
The desirable “brain-protective” properties of NMDA antagonists, including DXM, have been pursued by the pharmaceutical industry in developing relatively weak derivatives of amantadine and other so-called sigma receptor agonists and antagonists (21). For example, amantadine is an antiviral agent used in the prophylaxis and therapy of influenza A and to treat parkinsonism. Patients with Parkinson disease who took amantadine reported that it improved their motor symptoms. The mechanism of action of amantadine is unclear, but may include dopamine release or blockade of its reuptake and possible muscarinic anti-cholinergic action. Amantadine and a related compound, memantine, are NMDA receptor antagonists (22), but are relatively weak and do not appear to be abused.
EPIDEMIOLOGY
PCP abuse may be more a problem in large cities like Washington, DC, Philadelphia, Miami, and Los Angeles than in the rest of the United States (23). Ketamine is typically used with other drugs (24); however, sole use of ketamine has been reported (11) and may be increasing, particularly in Asian countries. Although ketamine has often been self-administered by insufflation, there is an emerging problem in youth of injecting ketamine. Such youth are more likely to engage in multiple injections, shared bottles of ketamine, and use of syringes obtained from secondary sources-practices that increase risk for hepatitis C, HIV, and other infectious diseases (25).
DXM is considered one of the most commonly abused over-the-counter medications in the United States and was first included in the Monitoring the Future epidemiologic surveys in 2006. The proportion of US students who reported in 2006 having used DXM during the prior year for the expressed purpose of “getting high” was 4%, 5%, and 7% in grades 8, 10, and 12, respectively (26). Rates in 2011 were similar, at 3%, 6%, and 5%, respectively (27). Poison Control Center data from the first half of the 2000 to 2010 decade reflected increasing DXM abuse, particularly among adolescents (28). For example, cases of DXM abuse reported to the California Poison Control System (28) increased 10-fold in all age groups and 15-fold in adolescents between 1999 and 2004. Similar trends were seen in national databases. Approximately 75% of California cases were aged 9 to 17 years. The highest frequency of abuse was in 15- to 16-year-olds. The most commonly abused DXM product was Coricidin HBP Cough & Cold Tablets. The extent of DXM abuse is likely far greater than what has been reported, because only the most severe cases are reported to poison control databases, and nearly all routine drug screening kits still do not screen or test for DXM or DXO. Studies of DXM in blood samples of suspected impaired drivers in the state of Wisconsin between 1999 and 2005 also supported an increasing prevalence of DXM-positive drivers, with a mean concentration of 207 ng/mL—compared with an expected therapeutic concentration range of 0.5 to 5.9 ng/mL (the highest concentrations being in males aged 16 to 20 years) (29). Intentional abuse of Coricidin products reported to the Illinois poison center occurred primarily among adolescents and was associated with significant short-term clinical effects and $353,314 in hospital charges annually (2001–2006) (30). The national upward trend in DXM abuse in the first half of the decade (2000–2005) has since peaked at 17.6 calls per million population in 2006 and plateaued at 15.7 per million in 2010 (31).
PHARMACOKINETICS
The pharmacokinetics of PCP in humans have never been well studied with psychoactive doses using modern methods (6). Blood PCP concentrations from 7 to 240 ng/mL (mean, 75) were found in arrested persons intoxicated in public or driving under its influence. The blood/plasma concentration ratio is 1. The plasma half-life (t½) of PCP has been reported to vary from 7 to 46 hours, suggesting the influence of dose and/or multiphase elimination processes. A terminal elimination phase (gamma) t½ of 1 to 4 days has been reported in cases of severe PCP poisoning (6). PCP is biotransformed in the liver to several metabolites and excreted in the urine as both free and glucuronide conjugates. Acidification of the urine increases its renal clearance because PCP is a base. However, this maneuver is no longer recommended clinically because of the risk of increasing urinary myoglobin precipitation (32).
Ketamine’s greater lipophilicity than PCP’s accounts for its rapid onset, short anesthetic duration of action, and shorter period of emergence delirium. Plasma concentrations of ketamine vary widely depending on the dose, route, and time elapsed since administration. Anesthetic doses produce plasma or serum concentrations of 1 to 6.3 ?g/mL.
Nonanesthetic psychoactive blood concentrations of ketamine are in the low nanograms per milliliter range (100 to 400). Ketamine follows a three-phase plasma pharmacokinetic model when given intravenously (6,33). There is a brief initial (alpha) phase with t½ of about 7 minutes because of rapid redistribution, followed by a longer elimination (beta) phase with t½ of 3 to 4 hours. As used in general anesthesia, an intravenous dose of 2.0 mg/kg produces rapid induction. This dose produces an onset in 30 seconds, with the coma lasting for 8 to 10 minutes. The intramuscular injection of ketamine has a latency of 3 to 5 minutes and a duration of 10 to 20 minutes or more, depending on the dose administered.
DXM is readily absorbed from the gut. Peak serum levels are reached at 2 to 3 hours for immediate release and 6 hours for sustained release preparations. DXO levels peak at 1.6 to 7 hours (34). Humans have a genetic polymorphism for the biotransformation of DXM (6). Rapid metabolizers have a plasma elimination t½ of about 3.4 hours, and slow metabolizers may have t½ exceeding 24 hours. Slow metabolizers of DXM represent about 10% to 15% of the population. Both O- and N-demethylation of DXO occur, with the N-demethylated version also having antitussive effects. Subsequent biotransformation results in various less active compounds. Phenotypic “slow” metabolizers of DXM report fewer intoxication effects than normal subjects (35). Thus, clinically slow metabolizers might be at higher risk for developing DXM dependence/addiction (2).
PHARMACODYNAMICS
Depending on the dose and specific arylcyclohexylamine ingested, patients who have taken PCP or ketamine present with widely different neurologic and psychiatric signs and symptoms. These signs and symptoms can be generally subdivided into three major clinical pictures: (a) confusion, delirium, and psychosis; (b) semicoma and coma; and (c) coma with seizures. Patients may become progressively more obtunded and eventually comatose, or the reverse, with the patient emerging from coma and showing emergence delirium. Table 15-1 lists the various signs and symptoms of PCP intoxication at different intravenous doses. Figure 15-2 shows the correlation of the molecular target sites with doses, concentrations, and clinical effects (36). Most PCP abusers do not grossly overdose themselves to the point of semicoma and coma. Hence, most patients intoxicated with PCP show a clinical picture of confusion, delirium, and psychosis. Rats show marked behavioral sensitization to both PCP and MK-801, with asymmetric cross-sensitization (37–39). The significance of this phenomenon for humans is unknown. Whether individuals who abuse PCP or ketamine show enhanced psychotomimetic effects over time is also unknown. Tolerance occurs with PCP and to a greater degree with continuous dosing (40). Animal models show severe withdrawal after cessation of PCP exposure: vocalizations, bruxism, oculomotor hyperactivity, diarrhea, piloerection, somnolence, tremor, and seizures (41). However, human dissociative withdrawal is not formally recognized by the Diagnostic and Statistical Manual, and human evidence remains limited (42,43).
TABLE 15-1 DOSE-RELATED EFFECTS OF PCP IN HEALTHY SUBJECTS
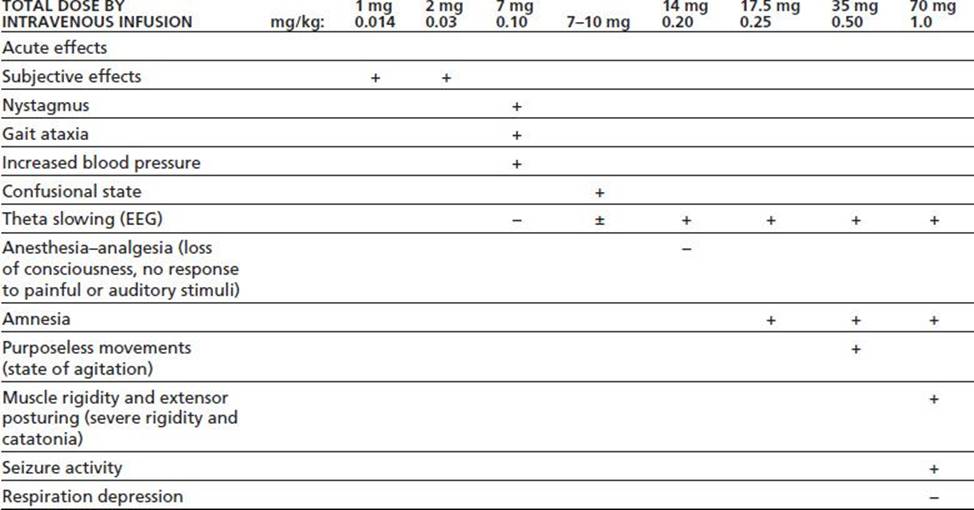
Data summarized by Burns & Lerner in Domino EF, ed. Phencyclidine: historical and current perspectives. Ann Arbor, MI: NPP Books, 1981:450.
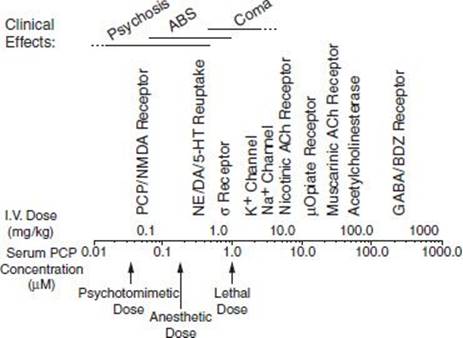
FIGURE 15-2 PCP doses, serum concentrations, molecular target sites, and clinical effects.
Ketamine exists as L-and D-isomers, also chemically designated as the (S)- and (R)-enantiomers, respectively. It is available in the United States only as the combination of enantiomers. (S)-ketamine in vitro has a lower inhibition constant for the NMDA receptor and a higher one for the sigma-binding site than does (R)-ketamine (21). (S)-ketamine is the preferred intravenous anesthetic, especially in Germany and other European countries. There is some evidence of its superior analgesic potency. From a practical clinical point of view, the separate enantiomers have properties that are grossly similar to those of the racemic mixture. Thus, the pharmacology of ketamine is that of the mixture.
About a third of patients in the early clinical trials of ketamine as a general anesthetic experienced an obvious emergence delirium (44). Why two-thirds did not remains unexplained, but suggests the importance of preoperative and postoperative medications, dosage, environmental, psychological, or genetic factors. People with schizophrenia are much more susceptible to a prolonged psychotic episode related to PCP than are other individuals (45). In addition, environmental and genetic factors influence PCP biotrans-formation in animals and humans.
During and after slow intravenous administration of 2.0 mg/kg ketamine, the following sequence of eye signs is observed: blinking, staring, closure of lids, nystagmus, strabismus, and loss of lid reflex. Initially, when the patient falls into a dissociative or cataleptic state, the eyelids are widely open and horizontal or vertical nystagmus is seen. Later, the eyeballs become centrally fixed in a gaze. During this stage, both somatosensory and visual stimulation elicit evoked potentials in the cortex. This finding supports the contention that the patient’s brain cannot interpret the afferent impulses because of the disruption of the normal connections of the sensory cortex with association areas. The persistence of open eyes distinguishes ketamine-induced anesthesia from that caused by other intravenous and inhalational anesthetics or coma-producing substances. This dissociation between eye signs and anesthetic or coma depth is one of the major disadvantages of ketamine as an anesthetic agent because eye signs cannot be used to gauge the depth of anesthesia.
Ketamine induces coma in a dose-dependent manner. A minimum of 0.5 mg/kg intravenously is necessary to induce coma for approximately 1.5 minutes. A dose of 1.0 mg/kg induces coma for approximately 5.8 minutes, whereas a dose of 2.0 mg/kg induces coma for approximately 10 minutes. Persons who abuse ketamine aim to achieve the low-dose mental state that is considered reinforcing by substance abusers. One exception is the giving of large doses surreptitiously to an unsuspecting person, such as in the illegal act of drug-facilitated sexual assault. The actual rate at which ketamine is used for this purpose is unknown.
The clinical effects of ketamine are akin to PCP and include analgesia, dissociation, hallucinations, and anesthesia. Agitation and cardiovascular and respiratory stimulation tend to be less than with PCP. Violence and unintended trauma may also result (46). Long-term chronic effects include dysphoria, impaired memory and cognition, apathy, and irritability (47), as well as distortion in the subjective experience of time (48). Chronic ketamine use has been associated with increased serum levels of brain-derived neurotrophic factor (49). Some anecdotal evidence supports the potential for tolerance and physical dependence with ketamine, but this needs further study.
About 13 years ago, it was reported that subanesthetic doses of ketamine produced antidepressant effects in depressed patients (50). This surprising finding was ignored by most researchers. There were subsequent isolated case reports that ketamine was an antidepressant (51,52). It was the controlled clinical trial of ketamine for treatment-resistant major depression by Zarate et al. (53) that first convincingly documented its therapeutic effectiveness. In the past 7 years, there have been many other reports of its effectiveness. Zarate et al. (54) recently replicated ketamine’s antidepressant efficacy in bipolar depressed patients. Seventy-nine percent improved and remained significantly better after 3 days. The most common side effects were dissociative symptoms at the end of the 40 minutes of ketamine infusion. The same National Institute of Mental Health (NIMH) group studied the subjects’ brain electrical activity using magnetoencephalography (MEG) (55). Spontaneous MEG activity postketamine was the same whether the patients were less depressed or not. However, those patients who had an antidepressant effect postketamine had increased MEG activity to finger stimulation, indicating greater cortisol neuronal excitability in the somatosensory cortex. In a separate NIMH study, blood concentrations of brain-derived neurotropic factor (BDNF) and electroencephalogram (EEG) slow-wave sleep increased in the responding subjects (56). Furthermore, blood levels of the less active metabolite of ketamine were higher in bipolar nonresponders, indicating the need to measure both ketamine and its metabolite levels in future research studies.
DXM has significant serotonergic properties, including increasing the synthesis and release of serotonin, as well as inhibiting the reuptake of serotonin from the synaptic cleft. DXM in clinical therapeutic doses produces relatively few side effects. These include body rash, itching, nausea, and vomiting and are most likely when DMX is combined with the other ingredients in cough preparations. Depending on dose, the drug can cause drowsiness, dizziness, altered vision, cardiovascular, and significant central nervous system effects that may resemble PCP intoxication. Euphoria and hallucinosis can occur within 15 to 30 minutes of ingestion of intoxicating doses, with peak effects experienced after roughly 2.5 hours. Clinical effects of DXM are summarized in Table 15-2 (2). The intoxication state can persist in varying degrees for about 3 to 6 hours (called a “plateau”). Serotonin syndrome can result, especially when taken with other serotonergic drugs (57). DXO is a stronger NMDA receptor antagonist than DXM. DXO is relatively inactive at the mu, kappa, and delta opioid receptor sites; thus, it is essentially devoid of the more conventional opiate properties, although respiratory depression has been reported with massive ingestion (58).
TABLE 15-2 CHARACTERISTICS OF THE DXM INTOXICATION SYNDROME
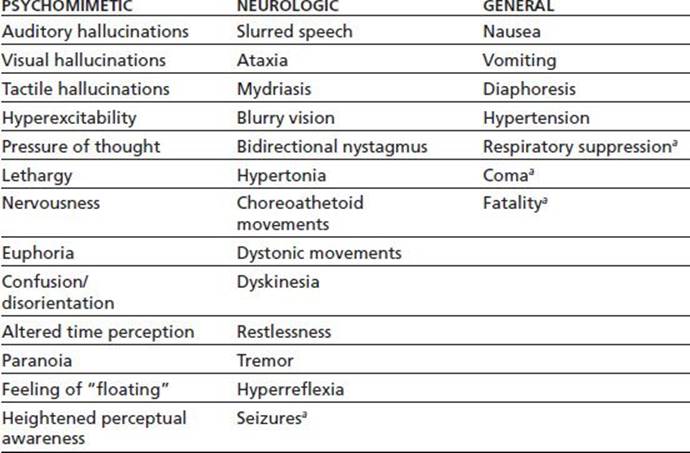
a Condition occurs only rarely.
From Bobo WV, Miller SC, Martin BD. The abuse liability of dextromethorphan among adolescents: a review. J Child Adolesc Subst Abuse 2005;14(4):55–75.
DRUG-DRUG INTERACTIONS
Many centrally acting drugs can produce an additive pharmacodynamic interaction with all of the agents described herein. Therapeutic combinations of ketamine with benzodiazepines reduce its emergence delirium, depending on the pharmacokinetics of the drug involved. Clonidine and related alpha-adrenergic agonists such as dexmedetomidine have been given clinically with ketamine to reduce its dissociative effects.
DXM can induce a serotonin syndrome when taken with monoamine oxide inhibitors, selective serotonin reuptake inhibitors, or other serotonergically active substances. Genetic polymorphism in the biotransformation of DXM via CYP2D6 may enhance the toxicity of the former by inhibitors of the latter. These include many drugs such as chlorpromazine, delavirdine, fluoxetine, miconazole, paroxetine, pergolide, quinidine, quinine, ritonavir, and ropinirole, as well as acute ingestion of large amounts of alcohol.
NEUROBIOLOGY
The action of arylcyclohexylamines on the NMDA type of glutamate receptor was first described by Anis et al. (59). Other investigators suggested different mechanisms of action involving biogenic amines and sigma-binding sites (60–62). However, noncompetitive blockade of NMDA receptors is a primary mechanism of action of low concentrations of these agents (1). This important conclusion stimulated interest in the role of glutamic acid in schizophrenia and related psychotic disorders (63–65). Krystal et al. (66–70) have been especially active in studying ketamine and possible antagonists in human volunteers. Moreover, understanding the role of glycine and other agonists in modulating NMDA receptor function has led to possible novel therapeutic approaches to schizophrenia (65).
Recent imaging data show that ketamine-induced antagonism of the NMDA receptor is directly correlated with negative symptoms of schizophrenia (assessed with the Brief Psychiatric Rating Scale [BPRS] negative subscale), suggesting that dissociatives may induce negative symptoms via NMDA antagonism (71). It has also been hypothesized that dissociatives induce positive symptoms via enhancing glutamate release. As discussed by Deakin et al. (72), Olney and Farber (73) previously suggested that NMDA antagonists block excitation of gamma aminobutyric acid (GABA) inter-neurons (74), resulting in removal of GABAergic inhibition of cholinergic, serotonergic, and glutamatergic afferents to the posterior retrosplenial cingulate cortex. This suggests a mechanism for triple excitoxicity and the subsequent posterior cingulate pyramidal cell neurodegeneration observed after PCP administration. Subsequent studies using in vivo microdialysis confirmed that the administration of NMDA antagonists increased glutamate release in the frontal cortex. Alternatively, mGluR2 (metabotropic glutamate 2/3 receptor) agonists, acting presynaptically to decrease the release of glutamate, reverse the behavioral effects of PCP in rats (75).
Ketamine administration induces a rapid, focal decrease in ventromedial frontal cortex regional blood oxygenation level-dependent (BOLD) fMRI signals that strongly correlate with its dissociative effects. This results in significantly increased BOLD activity in the mid-posterior cingulate, thalamus, and temporal cortical regions-increases correlated with BPRS psychosis scores (72). Pretreatment with lamotrigine (a sodium channel blocker that decreases glutamate release) prevented many of the BOLD changes and increases in BPRS psychosis scores. Thus, dissociatives may induce positive symptoms via enhancing glutamate release (72). There may be other mechanisms at play that relate to the association of positive and negative symptomatology with dissociative exposure.
Because ketamine is classified as a general anesthetic, it has typically been administered in experimental antidepressant trials by an anesthesiologist infusing a single subanesthetic intravenous dose, followed by at least 24 hours of inpatient observation (76,77). Depression response rates at 24 hours postinfusion range from 25% to 85% and fall to 14% to 70% at 72 hours postinfusion. Most adverse effects have been mild, without significant changes in blood pressure, pulse, or respirations noted. While ketamine and dissociatives remain a promising area of research for depression, limitations remain (very few randomized controlled trials exist, an active placebo is typically lacking, long-term data are scant, and risks remain uncertain). Thus, this approach remains experimental. Proposed mechanisms for the rapid antidepressant action of ketamine include ketamine-mediated blockade of NMDA receptors at rest, resulting in release of BDNF via desuppression of its translation. Previous studies suggest increased BDNF function as one possible mechanism of action for traditional antidepressants (77).
The action of nitrous oxide as an NMDA antagonist is another major advance in our knowledge (78–80) that may have clinical applications (78–80). Nitrous oxide is thought to stimulate the neuronal release of endogenous opioid peptide or dynorphins; the molecular aspects of this process are as yet unknown (81). Moreover, nitrous oxide may have an excitatory action on neurons via GABA-A receptor- mediated disinhibition (82).
Addiction Liability
Why these substances are reinforcing is difficult to understand (as they are not reported by users as eliciting a state of pure euphoria), except in the context of individuals who wish to experience the feelings of floating in space, dissociation, sensory isolation, mental distortions, and so forth that dissociatives provide. Dissociatives are self-administered by animals. Rhesus monkeys self-administer PCP, and social stimulation among monkeys in adjoining cages enhances reinforcing strength of PCP (83). Changes in dopaminergic or cAMP signal cascades induced by single or repeated PCP doses in mice likely play a role in the development of PCP-induced rewarding effects (84). Rodent and primate animal studies of DXM support reinforcement by DXO, akin to PCP. Stimulus discrimination from DXO and stimulus generalization to PCP are reported for DXM. Moreover, DXM is also strongly self-administered (85).
Very little work has been done to develop medications to treat dissociative abuse (86). Reformulation of DXM preparations could reduce abuse liability (87). An anti-PCP monoclonal antibody for PCP addiction is under development (88).
Toxicity/Adverse Effects
Since the 1970s, when Olney described the neurotoxic effects of glutamic acid, reducing its excess has been a target for brain-protective agents. NMDA antagonists have remarkable effects on brain neurons (89), including toxicity (90), that can be reduced or prevented (91). These agents induce significant vesicular changes (termed Olney lesions) in rat brain posterior cingulate retrosplenial neurons. Not all species of animals evidence these changes. The relationship of such neurotoxicity to humans who use or abuse of NMDA antagonists remains unclear. Such neurotoxic changes are reduced by pretreatment with benzo-diazepines, further supporting the mechanism of NMDA antagonists blocking GABA interneuron activity, resulting in disinhibition of cholinergic, serotonergic, and glutamatergic afferents-resulting in excitotoxicity. Such vesicular Olney lesions are not evident in DXM toxicity in rats (92). However, repeated high-dose administration of DXM during adolescence in rats may induce permanent deficits in cognitive function; increased expression of NMDA receptor AR 1 subunits in the prefrontal cortex and hippocampus may play a role in these DXM-induced memory deficits (93). This has troublesome implications in the setting of the increasing prevalence of DXM abuse in adolescents coincident with a remarkable period of brain growth during this age period. For example, young children iatrogenically exposed to dissociative anesthetics might have later onset of learning disorders (94). Human studies show impairments in working and episodic memory, among other cognitive problems, correlating with ketamine exposure levels (95). Human chronic ketamine users, compared to controls, show less bilateral dorsal prefrontal gray matter, with duration of use negatively correlating with gray matter volume and estimated total lifetime consumption of ketamine negatively correlating with gray matter volume in the left superior frontal gyrus (96). This same research group found white matter abnormalities in bilateral frontal and left temporoparietal cortices, with anisotropy values negatively correlating with the total lifetime ketamine consumption (indicating pathology of the white matter/axons in these brain regions (97)). Studies using dissociative drug intoxication as a model of cognitive dysfunction in schizophrenia to develop new pharmacotherapies may also reveal solutions for the cognitive consequences of chronic dissociative use (98,99).
Intoxication and Overdose
Intoxication is discussed in greater detail in Section 6 of this book. Although a preliminary diagnosis of arylcyclo-hexylamine intoxication can be made on the basis of history, clinical signs, and symptoms, only a drug-positive blood or urine specimen will unequivocally establish it. Most clinically used drug screening panels include PCP but not the other agents discussed herein; thus, a request may be required for specialized testing. A large variety of different chemical assays is available, but gas chromatography-mass spectrometry is often ideal for confirmation (100).
The brain wave changes induced by arylcyclohexyl-amines are unusual, and an EEG can be helpful if the patient is uncooperative or comatose. Serum skeletal creatinine phosphokinase levels are increased, and the urine can contain myoglobin because of rhabdomyolysis. End-organ kidney damage may result.
The first step in the differential diagnosis of arylcyclo-hexylamine intoxication is identifying whether the patient is in coma with or without seizures, emerging from coma, descending into coma, or in a psychotic state. The patient in coma-with or without seizures-has a differential diagnosis that includes all other causes of coma and seizures. The history and laboratory analysis are crucial. This is further discussed in Section 6 of this book.
Psychotic manifestations of arylcyclohexylamine poisoning can be confused with catatonic schizophrenia, an acute toxic psychosis induced by hallucinogens, and various acute organic brain syndromes. Arylcyclohexylamine intoxication can induce an organic brain syndrome, as well as cardiovascular and renal complications that are seldom, if ever, seen with other psychiatric syndromes. Lower urinary tract symptoms are common (101). Body image loss (especially numbness of the entire body), feelings of being in outer space, and (less commonly) visual hallucinations suggest arylcyclohexylamine abuse, as opposed to classic hallucinogens such as LSD-25 or related agents. DXM is associated with psychosis at doses greater than 300 to 600 mg or in fast metabolizers of DXO (16). Psychosis may occur at lower doses when DXM is combined with other drugs such as alcohol. Folate deficiency may also be associated with DXM abuse (102). DXM abuse may result in brain damage, seizure, loss of consciousness, irregular heart beat (20), and death. Respiratory depression from DXM may be reversed with naloxone (103). An evidence-based consensus guideline for out-of-hospital management of DXM toxicity is available (104). Although DXM has obvious toxicity, it may hold promise for antidepressant effects (105).
CONCLUSIONS AND FUTURE RESEARCH
Dissociatives include an array of compounds sharing antagonist activity at the NMDA receptor (among other actions on the human brain) and resulting in a clinical syndrome involving dissociation or disconnection of the brain from its external and internal environments. Such a disconnection is described by users as the desired end state when abusing these compounds; however, it is not uncommon for users to exceed the dosing required for these effects, resulting in untoward psychiatric and medical effects. It is often only then that such patients present for medical assistance. Antidotes or other effective treatments do not yet exist for these compounds, making supportive care the basic treatment modality. The addiction liability of these drugs requires further exploration. The evaluation of this drug class for its neuroprotective qualities may prove increasingly fruitful as the world population increases in average life span, and the need for such agents increases as well. The rapid antidepressant qualities of ketamine infusion are a recent discovery already resulting in increased research (54), with interest in extending this research to DXM (105). Possible future expanded clinical use of ketamine as an antidepressant raises concern for future ketamine/dissociative abuse from a novel group of users-depressed individuals. Finally, the increasing prevalence and significance of DXM abuse is alarming, particularly for the concentrated involvement of young people who appear unaware of its potential toxicities. Preliminary data suggest neuronal toxicity, and resultant neuropsychologic impairment may result from DXM abuse, particularly in the adolescent, developing the brain. Public policy may be increasingly directed towards controlled access to DXM versus its current over-the-counter (or behind the counter) availability.
REFERENCES
1.Krystal JH, et al. Comparative and interactive human psychopharmacologic effects of ketamine and amphetamine: implications for glutamatergic and dopaminergic model psychoses and cognitive function. Arch Gen Psychiatry 2005;62(9):985–994.
2.Bobo WV, Miller SC, Martin BD. The abuse liability of dextromethorphan among adolescents: a review. J Child Adolesc Subst Abuse 2005;14(4):55–75.
3.Svenson JE, Abernathy MK. Ketamine for prehospital use: new look at an old drug. Am J Emerg Med 2007;25(8):977–980.
4.Mikhael MS, Wray S, Robb ND. Intravenous conscious sedation in children for outpatient dentistry. Br Dent J 2007;203(6):323–331.
5.Bhutta AT. Ketamine: a controversial drug for neonates. Semin Perinatol 2007;31(5):303–308.
6.Baselt RC. Disposition of toxic drugs and chemicals in man. Seal Beach, CA: Biomedical Publications, 2011.
7.Domino EF. PCP (phencyclidine), historical and current perspectives. Ann Arbor:NPP Books, 1981.
8.Ernst A, et al. Molecular validation of the acute phencyclidine rat model for schizophrenia: identification of translational changes in energy metabolism and neurotransmission. J Proteome Res2012;11(7):3704–3714.
9.Domino EF, Luby ED. Phencyclidine/schizophrenia: one view toward the past, the other to the future. Schizophr Bull 2012;38(5): 914–919.
10.Domino E. From Sernyl to angel dust: the return of PCP. Univ Mich Med Cent J 1981;XLVII:1–5.
11.Bobo WV, Miller SC. Ketamine as a preferred substance of abuse. Am J Addict 2002;11(4):332–334.
12.Troupin AS, Mendius JR, Cheng F, et al. MK-801. In: Meldrum BS, Porter RJ, eds. New anticonvulsant drugs. London, UK: Libbey, 1986:191–201.
13.Piercey M, Hoffmann W, Kaczkofsky P. Functional evidence for PCP-like effects of the anti-stroke candidate MK-801. Psychopharmacology (Berl) 1988;96(4):561–562.
14.McFee RB, Mofenson HC, Caraccio TR. Dextromethorphan: another “ecstasy”? Arch Fam Med 2000;9(2):123.
15.Nordt S. “DXM”: a new drug of abuse? Ann Emerg Med 1998;31(6):794.
16.Miller S. Dextromethorphan psychosis, dependence and physical withdrawal. Addict Biol 2005;10(4):325–327.
17.U.S. Food and Drug Administration. Drug Abuse Advisory Committee, Open Session, Vol. 1. Washington, DC: Public Health Service, 1990.
18.Hendrickson RG, Cloutier RL. “Crystal Dex:” free-base dextromethorphan. J Emerg Med 2007;32(4):393–396.
19.Miller SC. Coricidin® HBP cough and cold addiction. J Am Acad Child Adolesc Psychiatry 2005;44(6):509–510.
20.FDA. FDA Talk Paper. FDA warns against abuse of dextromethorphan (DXM), 2005.
21.Skuza G. Pharmacology of sigma receptor ligands from a behavioral perspective. Curr Pharm Des 2012;18(7):863–874.
22.Stoof JC, Booij J, Drukarch B. Amantadine as N-methyl-D-aspartic acid receptor antagonist: new possibilities for therapeutic applications? Clin Neurol Neurosurg 1992;94(Suppl):S4-S6.
23.Proceedings of the Community Epidemiology Working Group, National Institute of Drug Abuse. Epidemiologic trends in drug abuse: highlights and executive summary. Bethesda, MD: National Institutes of Health, Volume 1, 2011.
24.Lankenau SE, Clatts MC. Patterns of polydrug use among ketamine injectors in New York City. Subst Use Misuse 2005;40(9–10):1381–1397.
25.Lankenau SE, Clatts MC. Ketamine injection among high risk youth: preliminary findings from New York City. J Drug Issues 2002;32(3):893–905.
26.Johnston LD, O'Malley P, Bachman JG, et al. Teen drug use continues down in 2006, particularly among older teens; but use of prescription-type drugs remains high. [On-line]. Ann Arbor, MI: University of Michigan News and Information Services www.monitoringthefuture.org Accessed August 17, 2007
27.Johnston LD, O'Malley PM, Bachman JG, et al. Monitoring the future national survey results on drug use, 1975–2011, Volume 1, secondary school students. Ann Arbor: Institue for Social Research, The University of Michigan. Retrieved February 12, 2013 from http://www.monitoringthefuture.org/pubs.html
28.Bryner JK, et al. Dextromethorphan abuse in adolescence: an increasing trend: 1999–2004. Arch Pediatr Adolesc Med 2006;160(12):1217.
29.Cochems A, Harding P, Liddicoat L. Dextromethorphan in Wisconsin drivers. J Anal Toxicol 2007;31(4):227–232.
30.Yin S, Wahl M. Intentional Coricidin product exposures among Illinois adolescents. Am J Drug Alcohol Abuse 2011;37(6):509–514.
31.Wilson MD, et al. Monitoring trends in dextromethorphan abuse using the National Poison Data System: 2000–2010. Clin Toxicol 2011;49(5):409–415.
32.Stone CK, Humphries RL. Current diagnosis and treatment emergency medicine, 6th ed. available at: http://www.accessmedicine.com/content.aspx?aID=3099123. webcite: Access Medicine; 2011
33.Domino EF, et al. Plasma levels of ketamine and two of its metabolites in surgical patients using a gas chromatographic mass fragmentographic assay. Anesth Analg 1982;61(2):87–92.
34.Silvasti M, et al. Pharmacokinetics of dextromethorphan and dextrorphan: a single dose comparison of three preparations in human volunteers. Int J Clin Pharmacol Ther Toxicol1987;25(9):493.
35.Zawertailo LA, et al. Psychotropic effects of dextromethorphan are altered by the CYP2D6 polymorphism: a pilot study. J Clin Psychopharmacol 1998;18(4):332–337.
36.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 1991;148(10):1301–1308.
37.Xu X, Domino EF. Phencyclidine-induced behavioral sensitization. Pharmacol Biochem Behav 1994;47(3):603–608.
38.Xu X, Domino EF. Asymmetric cross-sensitization to the locomotor stimulant effects of phencyclidine and MK-801. Neurochem Int 1994;25(2):155–159.
39.Xu X, Domino EF. A further study on asymmetric cross-sensitization between MK-801 and phencyclidine-induced ambulatory activity. Pharmacol Biochem Behav 1999;63(3):413–416.
40.Balster RL. Clinical implications of behavioral pharmacology research on phencyclidine. NIDA Research Monograph. 64:148–62.
41.Balster RL, Woolverton WL. Continuous-access phencyclidine self-administration by rhesus monkeys leading to physical dependence. Psychopharmacology (Berl) 1980;70(1):5–10.
42.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 4th ed. Text Revision., Washington, DC: American Psychiatric Association, 2000.
43.Rawson RA, Tennant FS Jr, McCann MA. Characteristics of 68 chronic phencyclidine abusers who sought treatment. Drug Alcohol Depend 1981;8(3):223–227.
44.Domino EF. Taming the ketamine tiger. Anesthesiology 2010;113(3):678–684.
45.Luby ED, et al. Study of a new schizophrenomimetic drug: Sernyl. AMA Arch Neurol Psychiatry 1959;81:363–369.
46.White JM, Ryan CF. Pharmacological properties of ketamine. Drug Alcohol Rev 1996;15(2):145–155.
47.Soyka M, Krupinski G, Volki G. Phenomenology of ketamine induced psychosis. Sucht [Germ J Addiction Res Pract] 1993;5:327–331.
48.Coull JT, et al. Ketamine perturbs perception of the flow of time in healthy volunteers. Psychopharmacology (Berl) 2011;218(3):543–556.
49.Ricci V, et al. Chronic ketamine use increases serum levels of brain-derived neurotrophic factor. Psychopharmacology (Berl) 2011;215(1):143–148.
50.Berman RM, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 2000;47(4):351–354.
51.Kudoh A, Katagai H, Takazawa T. Antidepressant treatment for chronic depressed patients should not be discontinued prior to anesthesia. Can J Anesth 2002;49(2):132–136.
52.Correll GE, Futter GE. Two case studies of patients with major depressive disorder given low?dose (subanesthetic) ketamine infusions. Pain Med 2006;7(1):92–95.
53.Zarate CA Jr, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 2006;63(8):856–864.
54.Zarate CA Jr, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry 2012;71(11):939–946.
55.Cornwell BR, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry 2012;72(7):555–561.
56.Duncan WC, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacol 2013;16:301–311.
57.Monte AA, Chuang R, Bodmer M. Dextromethorphan, chlorphenamine and serotonin toxicity: case report and systematic literature review. Br J Clin Pharmacol 2010;70(6):794–798.
58.Paterson JW, Lulich KM. Antiallergic drugs and antitussives. In: Dukes NG, ed. Meyler’s side effects of drugs: an encyclopedia of adverse reactions and interactions. Amsterdam, The Netherlands: Elsevier, 1996.
59.Anis NA, et al. The dissociative anaesthetics, ketamine and phencyclidine, selectively reduce excitation of central mammalian neurones by N-methyl-aspartate. Br J Pharmacol1983;79(2):565–75.
60.Rao TS, et al. Differential effects of phencyclidine (PCP) and ketamine on mesocortical and mesostriatal dopamine release invivo. Life Sci 1989;45(12):1065–1072.
61.Rao TS, et al. Interactions of phencyclidine receptor agonist MK?801 with dopaminergic system: regional studies in the rat. J Neurochem 1990;54(4):1157–1162.
62.Rabin RA, Doat M, Winter JC. Role of serotonergic 5-HT2A receptors in the psychotomimetic actions of phencyclidine. Int J Neuropsychopharmacol 2000;3(04):333–338.
63.Abi-Saab W, et al. The NMDA antagonist model for schizophrenia: promise and pitfalls. Pharmacopsychiatry 1998;31(2):104–109.
64.Loh M, Rolls ET, Deco G. A dynamical systems hypothesis of schizophrenia. PLoS Comput Biol 2007;3(11):e228.
65.Javitt DC. Glycine transport inhibitors and the treatment of schizophrenia. Biol Psychiatry 2008;63(1):6–8.
66.Krystal JH, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry1994;51(3):199.
67.Krystal JH, et al. Interactive effects of subanesthetic ketamine and subhypnotic lorazepam in humans. Psychopharmacology (Berl) 1998;135(3):213–229.
68.Krystal JH, et al. Dose-related ethanol-like effects of the NMDA antagonist, ketamine, in recently detoxified alcoholics. Arch Gen Psychiatry 1998;55(4):354.
69.Krystal JH, et al. Interactive effects of subanesthetic ketamine and haloperidol in healthy humans. Psychopharmacology (Berl) 1999;145(2):193–204.
70.Krystal JH, et al. Dissociation of ketamine effects on rule acquisition and rule implementation: possible relevance to NMDA receptor contributions to executive cognitive functions. Biol Psychiatry2000;47(2):137–143.
71.Stone JM, et al. Relationship between ketamine-induced psychotic symptoms and NMDA receptor occupancy-a [123 I] CNS-1261 SPET study. Psychopharmacology (Berl)2008;197(3):401–408.
72.Deakin J, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry 2008;65(2):154.
73.Olney JW, Farber NB. Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 1995;52:998–1007.
74.Drejer J, Honore T. Phencyclidine analogues inhibit NMDA-stimulated [3H] GABA release from cultured cortex neurons. Eur J Pharmacol 1987;143(2):287–290.
75.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science 1998;281(5381):1349–1352.
76.Aan Het Rot M, et al. Ketamine for depression: where do we go from here? Biol Psychiatry 2012;72:537–547.
77.Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry 2012;169(11):1150–1156.
78.De Lima J, Hatch D, Torsney C. Nitrous oxide analgesia-a ‘sting in the tail’. Anaesthesia 2000;55(9):932–933.
79.Franks NP, Lieb WR. A serious target for laughing gas. Nat Med 1998;4(4):383–384.
80.Maze M, Fujinaga M. Recent advances in understanding the actions and toxicity of nitrous oxide. Anaesthesia 2001;55(4): 311–314.
81.Emmanouil DE, Quock RM. Advances in understanding the actions of nitrous oxide. Anesth Prog 2007;54(1):9–18.
82.Nagashima K, Zorumski CF, Izumi Y. Nitrous oxide (laughing gas) facilitates excitability in rat hippocampal slices through gamma-aminobutyric acid A receptor-mediated disinhibition. Anesthesiology2005;102(1):230–234.
83.Newman JL, Perry JL, Carroll ME. Social stimuli enhance phencyclidine (PCP) self-administration in rhesus monkeys. Pharmacol Biochem Behav 2007;87(2):280–288.
84.Noda Y, Nabeshima T. Involvement of signal transduction cascade via dopamine?D1 receptors in phencyclidine dependence. Ann N Y Acad Sci 2006;1025(1):62–68.
85.Nicholson KL, Hayes BA, Balster RL. Evaluation of the reinforcing properties and phencyclidine-like discriminative stimulus effects of dextromethorphan and dextrorphan in rats and rhesus monkeys. Psychopharmacology (Berl) 1999;146(1):49–59.
86.Miller SC. Treatment of dextromethorphan dependence with naltrexone. Addict Disord Their Treat 2005;4(4):145–148.
87.Miller SC. Dextromethorphan to dextrorphan: a pathway towards abuse liability. Hum Psychopharmacol 2011;26(1):89–90.
88.Lacy HM, et al. Engineering and characterization of a mouse/ human chimeric anti-phencyclidine monoclonal antibody. Int Immunopharmacol 2008;8(1):1–11.
89.David Bleakman, Andrew Alt, David Lodge, Daniel T. Monaghan, David E. Jane and Eric S. Nisenbaum. Ionotropic Glutamate Receptors. Chapter 10. in Handbook of Contemporary Neuropharmacology, David R. Sibley, Israel Hanin, Michael Kuhar, et. al, eds, John Wiley & Sons, Inc. 2007
90.Allen H, Iversen L. Phencyclidine, dizocilpine, and cerebrocortical neurons. Science 1990;247:221.
91.Olney J, et al. NMDA antagonist neurotoxicity: mechanism and prevention. Science 1991;254(5037):1515.
92.Carliss R, et al. Oral administration of dextromethorphan does not produce neuronal vacuolation in the rat brain. Neurotoxicology 2007;28(4):813–818.
93.Zhang TY, et al. Impairments in water maze learning of aged rats that received dextromethorphan repeatedly during adolescent period. Psychopharmacology (Berl) 2007;191(1):171–179.
94.Turner C, et al. Strategies to defeat ketamine-induced neonatal brain injury. Neuroscience 2012;210:384–392.
95.Morgan CJA, Muetzelfeldt L, Curran HV. Ketamine use, cognition and psychological wellbeing: a comparison of frequent, infrequent and ex-users with polydrug and non-using controls. Addiction2008;104(1):77–87.
96.Liao Y, et al. Reduced dorsal prefrontal gray matter after chronic ketamine use. Biol Psychiatry 2011;69(1):42–8.
97.Liao Y, et al. Frontal white matter abnormalities following chronic ketamine use: a diffusion tensor imaging study. Brain 2010;133(7):2115–2122.
98.Dawson N, et al. Modafinil reverses phencyclidine-induced deficits in cognitive flexibility, cerebral metabolism, and functional brain connectivity. Schizophr Bull 2012;38(3):457–474.
99.D'Souza DC, et al. Nicotine fails to attenuate ketamine-induced cognitive deficits and negative and positive symptoms in humans: implications for schizophrenia. Biol Psychiatry2012;72(9):785–794.
100.Lau SS, Domino EF. Gas chromatography mass spectrometry assay for ketamine and its metabolites in plasma. Biol Mass Spectrom 1977;4(5):317–321.
101.Winstock AR, et al. The prevalence and natural history of urinary symptoms among recreational ketamine users. BJU Int 2012;110(11):1762–1766.
102.Au W, et al. Cough mixture abuse as a novel cause of megaloblastic anaemia and peripheral neuropathy. Br J Haematol 2003;123(5):956–958.
103.Schneider SM, Michelson EA, Boucek CD. Dextromethorphan narcosis reversed by naloxone. Vet Hum Tox 1986;31:376.
104.Chyka PA, et al. Dextromethorphan poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol 2007;45(6):662–677.
105.Lauterbach EC. Dextromethorphan as a potential rapid-acting antidepressant. Med Hypotheses 2011;76(5):717–719.