Jeffery N. Wilkins, MD, FASAM, DFAPA, Itai Danovitch, MD, MBA, and David A. Gorelick, MD, PhD
CHAPTER OUTLINE
■ STIMULANTS
■ HALLUCINOGENS
■ MARIJUANA
■ DISSOCIATIVE ANESTHETICS
■ INHALANTS
■ CLUB DRUGS
■ MDMA (“ECSTASY”)
■ GAMMA-HYDROXYBUTYRATE
■ HERBS OF ABUSE
■ FLUNITRAZEPAM
■ SEROTONIN SYNDROME
■ WITHDRAWAL FROM MULTIPLE DRUGS
■ POPULATION-SPECIFIC CONSIDERATIONS
This chapter reviews the treatment of acute intoxication and withdrawal states associated with the use of stimulants such as cocaine and methamphetamine (including their smokable forms “crack” and “ice”); hallucinogens such as lysergic acid diethylamide (LSD); marijuana; dissociative anesthetics such as phencyclidine (PCP), ketamine, and dextromethorphan (DXM); “club drugs” such as 3,4-methylenedioxymethamphetamine (MDMA or “ecstasy”) and gamma-hydroxybutyrate (GHB); and common herbal drugs of abuse. It also reviews the treatment of the serotonin syndrome and withdrawal from multiple drugs. Psychiatric and medical complications are considered separately because they often are treated with different modalities and in different settings (e.g., in psychiatric vs. medical emergency departments). Not all of the substances reviewed here have clinically distinct intoxication or withdrawal syndromes or are there pharmacologic treatments for all such syndromes.
Successful treatment of acute intoxication, overdose, or withdrawal can facilitate entry into addiction treatment by reducing uncomfortable withdrawal symptoms that negatively reinforce drug taking. Even when successful, these early stages of treatment often are followed by relapse to substance use, with patients potentially reentering a “revolving door” of repeated detoxification programs. Short-term treatment of acute intoxication or withdrawal does not obviate the need for long-term treatment of addiction.
Pharmacologic treatment of drug intoxication and overdose generally follows one of the three approaches: increased clearance of drug from the body, either by increasing catabolism or by increasing excretion, or both (1); blockade of the neuronal site to which the drug binds to exert its effect (as through the use of naloxone to block the muopioid receptor in the treatment of opiate overdose); and counteracting effects of the drug through alternative neuropharmacologic action.
Pharmacologic treatment of any drug withdrawal syndrome generally follows one of the two approaches: suppression by a cross-tolerant medication from the same pharmacologic class—usually a longer-acting one to provide a milder, controlled withdrawal (as in the use of the opioid methadone for opiate detoxification)—and/or reducing the signs and symptoms of withdrawal by targeting the neurochemical or receptor systems that mediate withdrawal (as in the use of the nonopiate clonidine to treat opiate withdrawal).
The application of these pharmacologic treatment approaches to the drugs reviewed in this chapter is limited. There may be no practical method for altering drug clearance (as with marijuana), or no specific drug receptor sites may have been identified. Even when a receptor site has been identified, there may not be a clinically useful antagonist. Finally, current understanding of the neuropharmacologic processes that mediate intoxication or withdrawal may be too limited to suggest appropriate pharmacologic interventions. Thus, clinical stabilization, supportive management, and palliation of symptoms often remain the mainstays of treatment.
STIMULANTS
Stimulant Intoxication
The acute psychological and medical effects of cocaine, amphetamines, and other stimulants are attributable principally to increases in catecholamine neurotransmitter activity. Enhanced catecholamine activity occurs through blockade of the presynaptic neurotransmitter reuptake pumps (as by cocaine) and by presynaptic release of catecholamines (as by amphetamines) (2). Resulting stimulation of the corticomesolimbic dopamine brain reward circuit mediates the desired (and addicting) psychological effects of stimulants. The resulting stimulation of the sympathetic nervous system leads to peripheral vasoconstriction (with organ ischemia), increased heart rate, and lowered seizure threshold, among other adverse effects. Table 46-1 lists acute medical complications of stimulant intoxication.
TABLE 46-1 ACUTE MEDICAL COMPLICATIONS OF STIMULANT INTOXICATION

aAll pulmonary complications except hyperventilation and pulmonary edema come primarily from the smoked route of administration.
Sources: Ghuran A, Nolan J. Recreational drug misuse: issues for the cardiologist. Heart 2000;83:627–633; Neiman J, Haapaniemi HM, Hillblom M. Neurological complications of drug abuse: pathophysiological mechanisms. Eur J Neurol2000;7:595–606; Schuckit MA. Drug and alcohol abuse. A clinical guide to diagnosis and treatment, 6th ed. New York: Springer, 2006; Tashkin DP. Airway effects of marijuana, cocaine, and other inhaled illicit agents. Curr Opin Pulm Med2001;7:43–61. Refs. (3–6).
Blockade of presynaptic catecholamine reuptake sites or postsynaptic receptors should, in principle, be an effective treatment for stimulant intoxication. Several medications have shown promise in attenuating the acute subjective effects of stimulants, such as bupropion (7), aripiprazole (8), risperidone (9), topiramate (10), and modafinil (11). Another method of attenuating the effects of stimulant intoxication might be to decrease drug availability in the central nervous system (CNS) by binding it peripherally with antidrug antibodies or by increasing its catabolism (1). The latter approach could be implemented with catalytic antibodies or with the endogenous cocaine-metabolizing enzyme butyrylcholinesterase (BChE, E.C. 3.1.1.8) or other esterases.
Table 46-2 gives an overview of treatment for the acute psychiatric and medical complications of stimulant intoxication.
TABLE 46-2 TREATMENT OF ACUTE STIMULANT INTOXICATION

Psychological and Behavioral Effects of Stimulant Intoxication
The initial effects of stimulant intoxication include increased energy, alertness, and sociability; elation; euphoria; and decreased fatigue, need for sleep, and appetite (12,13). At this stage, users do not seek or need treatment. With high-dose or repeated use, stimulant intoxication usually progresses to unwanted effects such as anxiety, irritability, interpersonal sensitivity, hypervigilance, suspiciousness, grandiosity, impaired judgment, stereotyped behavior, and psychotic symptoms such as paranoia and hallucinations. Up to three-quarters of stimulant users report paranoia or psychotic symptoms associated with their use, although the contribution of acute versus chronic stimulant exposure is often unclear (14–16). These results may also reflect selection bias among users who come to medical or research attention. Stimulant users typically remain alert and oriented, but the delusional state may impair judgment, cognition, and attention.
Patients with stimulant-induced psychoses may closely resemble those with acute schizophrenia and may be misdiagnosed as such (14,17). Cocaine-induced psychosis may differ from acute schizophrenic psychosis in having less thought disorder and bizarre delusions and fewer negative symptoms such as alogia and inattention (14). Stimulant-induced hallucinations may be auditory, visual, or somatosensory (14,18). Tactile hallucinations are especially typical of stimulant psychosis, such as the sensation of something crawling under the skin (formication). Specific genetic variations may account for some differences in individual vulnerability to stimulant-induced psychosis (19). Panic reactions are common and may evolve into a panic disorder (5). This may be exacerbated by anxiety elicited by the physiologic symptoms commonly associated with stimulant use, such as palpitations and hyperventilation.
Very severe stimulant intoxication may produce an excited delirium or organic brain syndrome that can be fatal (20,21). Patients should be evaluated promptly for an acute neurologic lesion (e.g., intracranial bleeding) or a preexisting neuropsychiatric condition and be treated aggressively (22).
Management of Psychological and Behavioral Effects of Stimulant Intoxication
The initial clinical evaluation should include a drug use history and drug toxicology to confirm stimulant intoxication. As the patient’s condition permits, further evaluation should rule out other potential medical (hyperthyroidism, hypoglycemia) or neuropsychiatric (panic or bipolar affective disorder) conditions (23). The initial treatment approach is nonpharmacologic (24,25). The patient should be observed in a quiet environment with minimal sensory stimulation to avoid exacerbating symptoms. Treatment staff should interact in a calm and confident manner, using the “ART” approach developed at the Haight-Ashbury Free Clinic in San Francisco: acceptance of the patient’s immediate needs (such as pain relief or use of the bathroom) and reassurance that the condition is due to the drug and likely will dissipate within a few hours and “talk down” to provide reality orientation and avoid hostility. All procedures should be explained to the patient before initiation. Physical restraints to control agitation or violent behavior should be avoided unless absolutely necessary. The use of restraints can increase the risk of hyperthermia and rhabdomyolysis, with resulting severe medical complications (26,27).
If medication is needed, most experts prefer benzodiazepines (such as diazepam [10 to 30 mg PO or 2 to 10 mg IM or IV] or lorazepam [2 to 4 mg PO or 1 to 2 IM or IV]) over antipsychotics to control severe agitation, anxiety, or psychotic symptoms (5,28,29), although there are very few controlled clinical trials (27). Benzodiazepines protect against the CNS and cardiovascular toxicities of stimulants, whereas antipsychotics may worsen the sympathomimetic and cardiovascular effects, lower the seizure threshold, increase the risk of hyperthermia, or precipitate extrapyramidal reactions (28). Parenteral benzodiazepine dosing may be repeated every 5 to 10 minutes until light sedation is achieved. If an antipsychotic is needed to control psychosis, a high-potency agent such as droperidol, haloperidol (5 to 10 mg PO, IM, or IV), or risperidone (2 to 4 mg PO) is preferred because of its minimal anticholinergic activity. Anticholinergic activity should be avoided because it may contribute to delirium and hyperthermia (by impairing heat dissipation from sweating). Chlorpromazine (30) and haloperidol (31) have been used safely in treating children with severe amphetamine poisoning. There is little evidence to guide treatment with second-generation antipsychotics, such as ziprasidone, risperidone, quetiapine, and olanzapine. Aripiprazole has been suggested as a promising medication because of its partial dopamine agonism (32).
A psychotic or agitated patient who has not responded to initial treatment should be hospitalized until the episode has resolved. This usually occurs within a few days if no more stimulants are ingested (5). Psychiatric symptoms that persist beyond a few days suggest an etiology other than stimulant use (24,33,34). Transient psychotic symptoms during periods of abstinence (“flashbacks”) have been reported among methamphetamine users (35).
Medical Effects of Stimulant Intoxication
Mild stimulant intoxication (the state desired by users) may be accompanied by one or more self-limiting physiologic effects such as restlessness, sinus tachycardia, hyperventilation, mydriasis, bruxism, headache, diaphoresis, or tremor. These do not usually bring the individual to medical attention or require treatment. Higher doses or repeated use is associated with more serious medical events, including nausea and vomiting, acute coronary syndrome (unstable angina or myocardial infarction, usually resulting in chest pain), cardiac tachyarrhythmia, hypertension, seizures, stroke, hyperthermia, or rhabdomyolysis (3,4,28,36–38). Acute medical complications associated with acute stimulant intoxication are summarized in Table 46-1.
Stimulant use should be high on the list of possible diagnoses for any younger patient presenting with one of these events, especially in the absence of other risk factors (39,40). Urine or blood samples for toxicologic analysis should be obtained to determine what drugs, if any, the patient has ingested recently. Even if an apparently adequate history has been obtained, the patient or collateral informants may not know the true content of any street drugs that have been used. A history of stimulant use within the preceding 96 hours or a positive toxicology test is highly suggestive. The actual blood cocaine concentration has little prognostic significance (41).
Nontraumatic chest pain is a common presenting complaint among stimulant users who seek acute medical care. The differential diagnosis includes acute coronary syndrome, acute aortic dissection, pneumothorax or pneumo-mediastinum (especially among drug smokers), endocarditis or pneumonia (especially among injection drug users), pulmonary embolus, myocarditis or cardiomyopathy, or musculoskeletal pain after a seizure (28,38). About 1% to 6% of patients with cocaine-associated chest pain and up to one-fourth of those with methamphetamine-associated chest pain will have an acute myocardial infarction (28,42). The risk for infarction is greatest during the first 1 to 3 hours after cocaine use and then declines rapidly (42). Concurrent use of multiple stimulants (e.g., cocaine and methamphetamine) may enhance cardiotoxicity (18), whereas concurrent use of opioids (such as “speedballing”) may mask the diagnosis (43).
The electrocardiogram is not always helpful diagnostically because of its low sensitivity and positive predictive value and the high frequency (more than one-third in some studies) of benign early repolarization among patients presenting with cocaine-associated chest pain (44,45). The best laboratory test for acute myocardial infarction is serial blood levels of cardiac troponin I (39,42). Its high specificity (around 95%) for acute myocardial infarction is not affected by recent cocaine use because it does not cross-react with skeletal muscle troponin. In contrast, myoglobin and creatine kinase (CK) levels may be elevated due to cocaine-associated rhabdomyolysis.
Patients who present with nontraumatic stimulant-associated chest pain usually should be observed for 9 to 12 hours while undergoing evaluation (46). Delayed complications are rare, so resolution of symptoms with a negative evaluation warrants discharge. Patients who have persistent chest pain despite standard treatment, hypotension, congestive heart failure, or cardiac arrhythmia require hospitalization for further evaluation and treatment. Even patients with confirmed acute myocardial infarction have a favorable prognosis, possibly because of their relatively young age and good underlying health (42).
Rhabdomyolysis may be due to a direct effect of the drug, hyperthermia, excessive muscle activity, or trauma (26). The usual symptoms of myalgia and muscle tenderness and swelling often are absent in rhabdomyolysis associated with stimulants. The diagnosis is suggested by a plasma CK level greater than five times normal (with other tissue sources ruled out) and a urine dipstick positive for heme but without red blood cells (indicating free myoglobin [or hemoglobin] in the urine).
Management of Medical Effects of Stimulant Intoxication
The first priority in the management of severe acute stimulant intoxication is maintenance of basic life support functions (29). Vital signs, hydration status, and neurologic status should be monitored closely. Activated charcoal or gastric lavage with isotonic saline may be helpful if a large amount of stimulant has been taken orally within the preceding hour (5,25). This can be done by oral intake or via a nasogastric tube in the awake, cooperative patient. Activated charcoal (50 to 100 g orally) may be just as effective as gastric lavage and minimizes the risk of aspiration. Ipecac-induced vomiting is not recommended.
Severe hypertension (e.g., diastolic blood pressure >120) that lasts more than 15 minutes should be treated promptly to avoid CNS hemorrhage (5). Rhabdomyolysis should be treated vigorously with intravenous fluid to maintain a urine output of greater than 2 mL/kg/h to avoid myoglobinuric renal failure (5,25). Maintenance of urine pH >5.6 with sodium bicarbonate (1 mmol/kg IV) helps to prevent the dissociation and precipitation of myoglobin.
Benzodiazepines in sedative doses are the initial treatment of choice for both acute cardiovascular and CNS toxicity from stimulants (39,44). Hypertension or tachycardia that does not respond to sedation alone may be treated with an alpha-adrenergic blocker such as phentolamine (2 to 10 mg IV over 10 minutes). Beta-adrenergic blockers such as propranolol or esmolol should be used with caution because of the risk of unopposed alpha-adrenergic stimulation by the stimulant, resulting in vasoconstriction and worsening hypertension (39,42,45,47). However, despite concerns about the use of beta blockers, a recent study has not found them to be associated with worse outcomes (48). The combined alpha-and beta-adrenergic blocker labetalol shows little alpha-adrenergic antagonism in clinical practice and also should be avoided. If alpha-adrenergic blockade is ineffective, direct vasodilation with sodium nitroprusside infusion (0.25 to 10 μg/kg/min) or nitroglycerin (5 to 100 μg IV) can be used. There is no evidence that rapid lowering of blood pressure compromises peripheral (including cerebral) circulation in an otherwise intact patient. Calcium channel blockers may reduce vasospasm, but their role remains unclear; calcium channel blockers enhance CNS toxicity in animal studies, have inconsistent effects in case series, and should be avoided in patients with heart block or heart failure. Dexmedetomidine, an α2-adrenergic receptor agonist, has shown promise in ameliorating the acute cardiovascular effects of severe cocaine intoxication, as well as providing sedation (49,50). It must be given by IV infusion (not bolus) to avoid hypertension and bradycardia.
Treatment of cocaine-induced cardiac tachyarrhythmias begins with correction of any exacerbating conditions such as myocardial ischemia, hypoxia, electrolyte abnormalities, or acid–base disturbance (42,46). Arrhythmias occurring several hours after cocaine use are usually secondary to myocardial ischemia. Standard arrhythmia management is usually appropriate, including use of lidocaine. Arrhythmias occurring immediately after cocaine use are usually from the sodium channel blocking action of cocaine. These may respond to sodium bicarbonate. Lidocaine (which also blocks sodium channels) should be used cautiously in this context because of animal studies suggesting it exacerbates cocaine-associated arrhythmias and seizures. Class IA antiarrhythmic medications (such as quinidine, procainamide, or disopyramide) should be avoided because of their potential additive effect on QRS and QT interval prolongation. There are no data on the use of amiodarone for cocaine-associated arrhythmias. Treatment of cardiac arrest is the same as for non–cocaine users; the outcome may be more favorable than for drug-free patients (51).
The treatment of stimulant-associated acute coronary syndrome largely resembles that for the non–drug-associated syndrome, with the exception of avoiding use of beta-adrenergic blockers and labetalol (46,47). Initial treatment includes oxygen, benzodiazepine for sedation, morphine for pain, sublingual nitroglycerin for vasodilation, and aspirin for antiplatelet action, while evaluation is continuing. Further treatment can include phentolamine or intravenous nitrates (10 μg/kg/min) to lower blood pressure and reverse coronary artery vasoconstriction. The role of calcium channel blockers is not well defined (39,42). They may be useful in patients who have not responded to benzodiazepines and nitroglycerin.
Both fibrinolytic therapy and percutaneous transluminal coronary angioplasty have a role in the treatment of confirmed myocardial infarction (39,42). Angioplasty may be preferable to fibrinolysis because of the increased risk of intracranial hemorrhage in cocaine users. Elevated body temperature (>102°F orally) is a marker for poor prognosis and should be managed aggressively to avert hyperthermic crisis (as by cold water sponging, cooling blankets, ice packs, ice water gastric lavage, or cold peritoneal lavage) (5,52). Untreated hyperthermia may result in rhabdomyolysis and renal failure.
Intravenous benzodiazepines (diazepam 5 to 10 mg or lorazepam 2 to 10 mg over 2 minutes, repeated as needed) are recommended to control seizures stemming from stimulant intoxication (49,52). Fosphenytoin (15 to 20 mg/kg at 100 to 150 mg/min) or phenobarbital (15 to 20 mg/kg over 20 minutes) also can be used. However, the latter may cause hypotension or prolonged sedation.
Excretion of amphetamine can be increased by acidifying the urine to pH <6.6 (as with 500 mg of oral ammonium chloride every 3 to 4 hours), which inhibits renal reabsorption of amphetamine (53). The actual clinical usefulness of this maneuver is uncertain (13). Acidification is contraindicated in the presence of myoglobinuria, if renal or hepatic function is abnormal, or in overdose situations, when plasma acidification may compromise cardiovascular function (33).
Stimulant Withdrawal
Abrupt cessation of stimulant use is associated with depression, anxiety, fatigue, difficulty concentrating, anergia, anhedonia, increased drug craving, increased appetite, hyper-somnolence, and increased dreaming (because of increased REM sleep) (54–56). The initial period of intense symptoms is commonly termed the “crash,” but most symptoms are mild and self-limited, resolving within 1 to 2 weeks without treatment.
Hospitalization for stimulant withdrawal is rarely indicated on medical grounds and has not been shown to improve the short-term outcome for stimulant addiction (57,58). Pharmacologic treatment has focused more on long-term treatment of addiction than on short-term treatment of acute withdrawal (59,60). Most clinical trials that used medication during the early withdrawal period have continued to use such medication for at least several weeks, with the additional goal of treating the addiction itself.
Medical Effects of Stimulant Withdrawal
The first week of stimulant withdrawal has been associated with myocardial ischemia (61), possibly because of coronary vasospasm. Other medical effects of stimulant withdrawal are relatively minor, including nonspecific musculoskeletal pain, tremors, chills, and involuntary motor movement (62). These rarely require specific medical treatment.
Management of Stimulant Withdrawal
The stimulant withdrawal syndrome has been hypothesized to be the result of decreased levels of brain dopamine activity resulting from chronic stimulant exposure. This so-called “dopamine deficiency” hypothesis of withdrawal has not been consistently supported by clinical studies (63–65), but has generated the use of dopamine agonists to treat cocaine withdrawal, most commonly bromocriptine and amantadine. However, no medication has been shown consistently effective in controlled clinical trials (66) or is any medication approved for the treatment of stimulant withdrawal by any national regulatory authority. Administration of a cross-tolerant or similarly acting stimulant has not been systematically evaluated as a short-term treatment for stimulant withdrawal (24). No controlled clinical trial has directly compared the benefits of medication versus a supportive milieu.
Symptoms of stimulant withdrawal are best treated supportively with rest, exercise, and a healthy diet (5,24). Short-acting benzodiazepines such as lorazepam may be helpful in selected patients who develop agitation or sleep disturbance. Severe (suicidal ideation) or persistent (>2 to 3 weeks) depression may require antidepressant treatment (5,66a) and psychiatric admission. The risk of relapse is high during the early withdrawal period, in part because drug craving is easily triggered by encounters with drug-associated stimuli. This issue is better addressed by psychosocial treatment, such as supportive therapy, cognitive–behavioral therapy, relapse prevention, and contingency management, than by medication.
HALLUCINOGENS
Hallucinogen Intoxication
Hallucinogens have in common the ability to change or distort sensory perceptions in a clear sensorium. Most hallucinogens fall into one of two chemical groups (see Chapter 14). Indolealkylamine hallucinogens (including LSD, psilocybin, or N,N-dimethyltryptamine) are structurally related to serotonin; phenylethylamine hallucinogens (including 3,4,5-tri-methoxyphenylethylamine [mescaline], 3,5- dimethoxy-4-methylamphetamine [DOM, STP]) are structurally related to norepinephrine. Both indolealkylamine and phenylethyl-amine hallucinogens generate psychedelic (LSD-like) experiences and thus are often categorized together (67,68). In contrast, 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”) has characteristics of both a hallucinogen and a stimulant and is considered separately (see also Chapter 14). PCP and its close analog ketamine are anesthetics that are used for their dissociative and euphoric effects. Both MDMA and PCP are considered in their own section below (see also Chapter 15).
Psychological and Behavioral Effects of Hallucinogen Intoxication
The acute psychological and behavioral effects of hallucinogen intoxication are summarized in Table 46-3. The subjective experience is influenced greatly by set and setting, that is, the expectations and personality of the user, coupled with the environmental and social conditions of use. Mood can vary from euphoria and feelings of spiritual insight to depression, anxiety, and terror. Perception usually is intensified and distorted, with alterations in the sense of time, space, and body boundaries. While illusions (visual and auditory distortions of perception) are common, true hallucinations (perceptions that do not have any basis in reality) are not. Synesthesia, a blending of the senses wherein colors are heard and sounds are seen, is a common perceptual distortion. Cognitive function may range from clarity to confusion and disorientation, although reality testing usually remains intact.
TABLE 46-3 ACUTE PSYCHOLOGICAL AND BEHAVIORAL EFFECTS OF INTOXICATION WITH LSD, MARIJUANA, PCP, OR MDMA
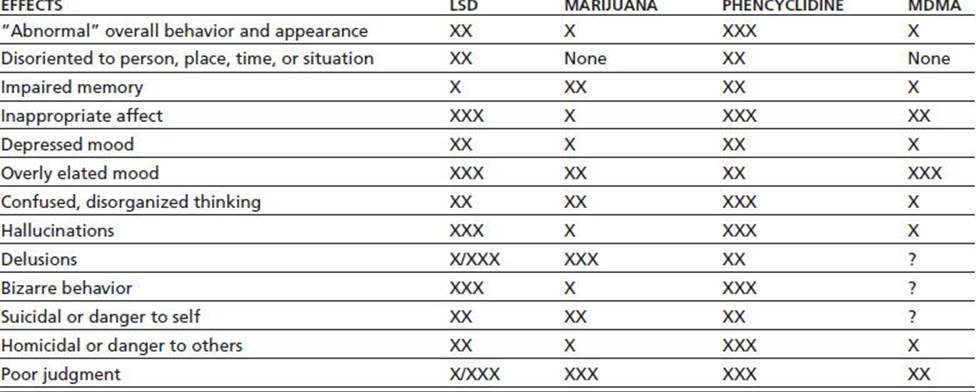
Relative weighting: X = mild; XX = moderate; XXX = marked; /= common/rare; ? = insufficient research.
MDMA, 3,4-methylenedioxymethamphetamine.
Sources: Brust JCM. Acute neurologic complications of drug and alcohol abuse. Neurol Clin N Am 1998;16:503–519; Frecska E, Luna LE. The adverse effects of hallucinogens from intramural perspective. Neuropsychopharmacol Hung2006;8:189–200; Abraham HD, Aldridge AM, Gogia P. The psychopharmacology of hallucinogens. Neuropsychopharmacology 1996;14:285–298. Refs. (69–71).
A “bad trip” (69) usually takes the form of an anxiety attack or panic reaction, with the user feeling out of control (70,72,73). An experience of depersonalization may precipitate the fear of losing one’s mind permanently. Panic reactions are more common in those who have limited experience with hallucinogens, but previous “positive” experiences provide no protection against an adverse reaction (74). While higher doses are associated with more intense experiences, adverse reactions are less a function of dose than of context and environment. Hallucinogens may trigger a transient psychosis even in psychologically normal users; however, a true psychotic episode is rare. Hallucinogen-induced psychosis may resemble acute paranoid schizophrenia (68,74). The two usually can be distinguished because patients with schizophrenia tend to have auditory (rather than visual) hallucinations and a history of prior mental illness. Hallucinogen users, unlike patients with schizophrenia, usually retain at least partial insight that their symptoms are drug related.
Hallucinogen ingestion may result in an acute toxic delirium that is characterized by delusions, hallucinations, agitation, confusion, paranoia, and inadvertent suicide attempts (e.g., attempts to fly or perform other impossible activities).
Medical Effects of Hallucinogen Intoxication
Acute medical complications of hallucinogen intoxication are summarized in Table 46-4. Sympathomimetic effects are common, particularly pupillary dilation, hyperreflexia, piloerection, tachycardia, and increases in blood pressure. Dizziness, paresthesias, headache, nausea, or tremor may occur. Body temperature should be monitored and any elevation treated promptly. Dry skin, increased muscle tone, agitation, and seizures are warning signs of a potential hyperthermic crisis. Patients may not respond to anticonvulsant medication until body temperature is lowered. Complications that require treatment are rare in the absence of overdose (68,69).
Table 46-4 A cute Medical Complications of Intoxication with LSD, MDMA, Marijuana, or PCP

MDMA, 3,4-methylenedioxymethamphetamine; HR, heart rate; BP, blood pressure.
Sources: Ghuran A, Nolan J. Recreational drug misuse: issues for the cardiologist. Heart 2000;83:627–633; Brust JCM. Acute neurologic complications of drug and alcohol abuse. Neurol Clin N Am 1998;16:503–519; Frecska E, Luna LE. The adverse effects of hallucinogens from intramural perspective. Neuropsychopharmacol Hung 2006;8:189–200; Kalant H. The pharmacology and toxicology of “ecstasy” (MDMA)and related drugs. Can Med Assoc J 2001;165(7):917–928; Schuckit MA. Drug and alcohol abuse. A clinical guide to diagnosis and treatment, 6th ed. New York: Springer, 2006; Selden BS, Clark RF, Curry SC. Marijuana. Emerg Med Clin North Am 1990;8:527–539. Refs. (75,76).
Oral LSD is rapidly absorbed, so that ipecac-induced vomiting or gastric lavage usually is not helpful and may exacerbate the patient’s psychological distress. There is no evidence that LSD binds to charcoal. Gastric lavage may be useful in psilocybin ingestion or when there is doubt as to the identity of the ingested mushrooms (70,72).
Management of Hallucinogen Intoxication
Initial treatment is supportive. The patient should be placed in a quiet environment with minimal sensory stimulation, but should be observed because of the risk of unintended self-injury (as the result of delusions or hallucinations) or of suicide (as the result of depression). The presence of a familiar person usually is comforting. Unless the patient presents in an acutely agitated or threatening state, physical restraints are contraindicated because they may exacerbate anxiety and increase the risk of rhabdomyolysis associated with muscle rigidity or spasms. The use of “gentle restraints” in combination with muscle massage and individualized counseling may be helpful (74).
The “talk down” or reassurance technique may be helpful. The clinician, in a concerned and nonjudgmental manner, discusses the patient’s anxiety reaction, stressing that the drug’s effects are temporary and that the patient will recover completely.
For patients who do not respond to reassurance alone, oral benzodiazepines such as lorazepam (1 to 2 mg) or diazepam (10 to 30 mg) are the drugs of choice (68,72). When oral medication is too slow, or the patient will not take oral medication, intramuscular lorazepam (2 mg, repeated hourly as needed) may be effective. If benzodiazepines are insufficient, a high-potency antipsychotic such as haloperidol (5 to 10 mg orally or 2 mg intramuscularly) may be needed. The role of second-generation antipsychotics in this situation remains unclear, but 5-HT2A receptor antagonism may be a useful property (68,72,73). Phenothiazines should be avoided because they are associated with poor outcomes (77) and may exacerbate unsuspected anticholinergic poisoning.
Patients usually recover sufficiently after several hours and may be released into the care of a responsible relative or friend. If psychosis does not resolve within 1 or 2 days, ingestion of a longer-acting drug such as PCP or DOM should be suspected (5). Symptoms that persist beyond a few days raise the strong likelihood of a preexisting or concurrent psychiatric or neurologic condition. Psychiatric problems that last more than a month probably are related to preexisting psychopathology.
Treatment for hallucinogen-induced delirium generally follows the guidelines for simple intoxication: Isolate the patient, and minimize sensory input until effects of the drug have worn off. Reassurance that the delirium will abate as the drug is metabolized also may be helpful. Pharmacologic treatment is not necessary in most cases and may confuse the clinical picture. If medication is needed, a drug with few anticholinergic properties is preferred for the reasons listed above; for example, diazepam may be given 15 to 30 mg orally, repeating 5 to 20 mg every 3 to 4 hours as needed.
Hallucinogen Withdrawal
Withdrawal symptoms, including fatigue, irritability, and anhedonia, are reported by about 10% of hallucinogen users (78). There is no evidence to suggest a clinically significant hallucinogen withdrawal syndrome (62,68), and such a syndrome is not recognized in the DSM-5 (79). The rapid development of tolerance (within 3 to 4 days) may explain in part why use of LSD-like drugs generally is intermittent. There is no role for medication in the treatment of hallucinogen withdrawal.
Some users of hallucinogens describe experiencing flashbacks, vivid memories, or brief recurrences of sensory distortions reminiscent of intoxication, during periods of sobriety. Flashbacks can occur spontaneously long after cessation of use and thus are not truly a withdrawal syndrome. In the DSM-5, they are diagnosed as “hallucinogen persisting perception disorder” (79). Initiation of selective serotonin reuptake inhibitors (SSRIs) or neuroleptics is associated with recurrences of flashbacks in at-risk individuals (80,81). Supportive measures, as well as the symptom-based pharmacologic interventions prescribed to manage hallucinogen intoxication, may be effective in managing such symptoms.
MARIJUANA
Marijuana Intoxication
The major psychological and physiologic effects of marijuana are mediated by the interaction of delta-9-tetrahydrocannabinol (THC) with specific cannabinoid (CB1) receptors on nerve cells (82,83), the regional distribution of which in the human brain is consistent with the known effects of marijuana (84). Other cannabinoids found in marijuana (e.g., cannabidiol, cannabinol) do not produce these typical marijuana effects (85). In animal and human studies, acute THC effects are reduced or blocked by CB1 receptor antagonists (86).
Psychological and Behavioral Effects of Marijuana Intoxication
The initial—usually desired—psychological effects of marijuana intoxication include relaxation, euphoria, slowed time perception, altered (often intensified) sensory perception, increased awareness of the environment, and increased appetite (87). Undesired effects may include impaired concentration, anterograde amnesia (88), and motor incoordination (89). As with hallucinogens, psychological set and social setting and prior experience with the drug can substantially influence the quality of the experience. Higher doses, repeated use, or a stressful setting is associated with adverse effects such as hypervigilance, anxiety, paranoia, derealization and depersonalization (commonly associated with altered time sense), acute panic (associated with anxiety), illusions or hallucinations (usually auditory or visual), psychosis, or delirium (68,90–92,92a). Acute marijuana- associated psychosis can be difficult to distinguish from schizophrenic psychosis other than by its transient time course (93). Marijuana-associated psychosis may be more likely to exhibit derealization/depersonalization experiences and visual, rather than auditory, hallucinations. Preexisting psychopathology increases the risk of adverse events such as panic attack or psychosis (94). Table 46-3 summarizes the acute adverse psychological effects of marijuana intoxication. Oral ingestion of marijuana can produce the same adverse reactions as does smoking, including psychosis (92a,95,96).
Medical Effects of Marijuana Intoxication
The acute physiologic effects of oral or smoked marijuana intoxication include conjunctival injection (“red eye”) due to vasodilation, tachycardia (sometimes with palpitations), orthostatic hypotension (sometimes resulting in syncope), and dry mouth (see Table 46-4). Neurologic signs include poor motor coordination, head jerks, and impairment of smooth pursuit eye movements (89). These generally are mild, are self-limiting, and do not require medical treatment (87). There are no well-established cases of human fatalities from exclusively marijuana overdose (91), although several cases of possible acute cardiovascular death have been reported (97) and marijuana smoking has been associated with atrial fibrillation and other tachyarrhythmias (98,99). Intravenous use of marijuana, although rare, can be associated with cardiovascular shock and renal failure (99).
Management of Marijuana Intoxication
Adverse effects of marijuana intoxication tend to be self-limited and often can be managed without medication. The patient should be kept in a quiet environment and offered supportive reassurance. If immediate pharmacologic intervention is needed to control severe agitation or anxiety, benzodiazepines are preferred to antipsychotics, although there are no controlled studies to confirm this. Psychosis usually responds to low doses of second-generation antipsychotics (92).
No medication is approved by any national regulatory authority for the treatment of marijuana intoxication. The selective cannabinoid CB1 receptor antagonist/inverse agonist rimonabant (developed for weight loss) blocked the acute psychological and cardiovascular effects of smoked marijuana in human laboratory studies (87). However, rimonabant and several similar medications were withdrawn from the market and from clinical development in 2008 because of psychiatric side effects (100). Should a future CB1 receptor antagonist prove safe and effective, it could be used to treat acute marijuana intoxication in the same way that naloxone acts on opiate intoxication.
Marijuana Withdrawal
Acute marijuana withdrawal is reported by up to one-third of heavy marijuana users in the community and more than half of those seeking treatment for marijuana dependence (101) and is a recognized clinical syndrome in the latest (fifth) edition of the American Psychiatric Association Diagnostic and Statistical Manual of Mental Disorders (DSM-5) (79). Symptoms are primarily psychological, including irritability, anxiety, depression, restlessness, anorexia, insomnia, and disturbed sleep (102). Much less common are physical symptoms such as gastrointestinal distress, diaphoresis, chills, nausea, shakiness, muscle twitches, and increased blood pressure (102). The syndrome is usually mild, comparable to tobacco withdrawal (103), and rarely needs medical attention, but may impair some normal activities of daily life (104). It may warrant clinical attention in the treatment of cannabis use disorders because withdrawal symptoms can serve as negative reinforcement for relapse among users trying to maintain abstinence (105).
Management of Marijuana Withdrawal
Marijuana withdrawal rarely requires treatment for intrinsic medical or psychiatric reasons, although treatment might be warranted in some cases to reduce the risk of relapse in persons trying to abstain while experiencing distressing withdrawal symptoms. In two controlled clinical trials involved treatment-seeking adults with cannabis dependence, both dronabinol (synthetic THC, 20 mg b.i.d.) and gabapentin (1,200 mg daily) significantly reduced marijuana withdrawal symptoms, although only gabapentin significantly reduced marijuana use (106).
DISSOCIATIVE ANESTHETICS
Phencyclidine, Ketamine, and Dextromethorphan Intoxication
PCP and its molecular analog ketamine are dissociative anesthetics (107,108); DXM is widely available as an antitussive in over-the-counter cough and cold medicines (109). The chemical agents in this class are relatively old, with PCP first synthesized just under 90 years ago (110) and both ketamine (111) and DXM approximately 50 years ago (112). Of the three, ketamine has received considerable attention in recent years because of its apparent ability to rapidly treat unipolar (113) and bipolar (114) depression and various pain syndromes (115). There is a rich literature for the abuse of these three dissociative agents. Both PCP and ketamine are considered controlled drugs in the United States; PCP is a Schedule II and ketamine is a Schedule III drug (116). The related drug DXM is not controlled and is widely available as an ingredient in over 100 different over-the-counter cough and cold medicines (117). At the recommended antitussive dose of 15 to 30 mg every 6 to 8 hours, adverse reactions are rare. However, about 5% to 10% of those of white European ethnicity are unable to demethylate DXM to dextrorphan (an active metabolite) because of a deficit in the liver cytochrome P450 CYP 2D6 isoenzyme. Thus, in the context of megadose use of DXM, this subset of individuals is at increased risk of toxicity from an acute excess in DXM levels (117).
The main effects of PCP and ketamine are mediated by their action as noncompetitive antagonists of the NMDA glutamate excitatory amino acid neurotransmitter receptor (111). In addition, direct effects on other neurotransmitter systems (such as dopamine) may occur at high doses (111,118) (see Chapter 15). In addition to NMDA antagonism, DXM has activity at the sigma receptor, which likely contributes to its therapeutic effects as a cough suppressant.
Psychological and Behavioral Effects of Dissociative Anesthetic Intoxication
Dissociative anesthetics produce a range of intoxicated states that can be grouped into three stages (118): Stage I, conscious, with psychological effects but (at most) mild physiologic effects; Stage II, stuporous or in a light coma, yet responsive to pain; and Stage III, comatose and unresponsive to pain. Table 46-3 summarizes the acute psychological and behavioral effects of PCP intoxication and overdose. The time course of psychological effects is highly variable and unpredictable, so that even a recovering patient should be kept under observation until all symptoms have resolved (typically at least 12 hours) (119). Patients may “emerge” from one stage of intoxication to the next; that is, a stuporous or comatose patient in Stage II or III may enter Stage I and become agitated and delirious (68,120). Similarly, a conscious patient in Stage I may suddenly become comatose (5). The entire clinical episode may require up to 6 weeks to resolve (5).
The psychiatric manifestations of Stage I intoxication can resemble a variety of psychiatric syndromes, making differential diagnosis difficult in the absence of toxicology results or a history of recent PCP, ketamine, or DXM intake. Common syndromes seen in treatment settings include delirium, psychosis without delirium, catatonia, hypomania with euphoria, and depression with lethargy. Agitated or bizarre behavior, with increased risk of violence, can occur with any psychiatric presentation (68,77,95,112). Because of the analgesic effect of PCP, patients may not report the existence of even serious injuries (which may be self-inflicted) (69).
Clinically significant psychological and behavioral effects of DXM begin to occur at approximately five times the therapeutic dose (117). These effects can be grouped into four dose-dependent plateaus (Table 46-5).
TABLE 46-5 PSYCHOLOGICAL AND BEHAVIORAL EFFECTS OF DXM INTOXICATION

Adapted from Schwartz RH. Adolescent abuse of dextromethorphan. Clin Pediatr 2005;44(7):565–568.
Medical Effects of Dissociative Anesthetic Intoxication
Intoxication at the mild Stage I desired by users is associated with few serious medical complications (see Table 46-4) (118). Common medical effects at this stage include nystagmus (especially horizontal), tachycardia, increased blood pressure, ataxia, dysarthria, numbness, increased salivation, and hyperreflexia (69). Higher stages are associated with severe medical effects, including hypertension, stroke, cardiac failure, seizures, rhabdomyolysis, acute renal failure, coma, and death (29).
The acute effects of ketamine tend to be less severe and of shorter duration than those of PCP, possibly due to its shorter half-life (111). Nystagmus occurs less often than with PCP (111).
Management of Psychological and Behavioral Effects of Dissociative Anesthetic Intoxication
Treatment of intoxication with dissociative anesthetics is largely supportive and aimed at controlling or reversing specific signs and symptoms (119). No clinically useful antagonist is yet available. The anticonvulsant lamotrigine (300 mg daily), which inhibits glutamate release, reduced the psychological and cognitive effects of ketamine in a small experimental trial (121). Mild Stage I intoxication is best treated without medication. The patient should be isolated in a quiet room with unobtrusive observation and minimal external stimuli. Frequent or intrusive contact or aggressive medical intervention may worsen the situation and should be avoided. Reassuring, reality-oriented communication (“talking down”) rarely works with such patients (69). Urine acidification and diuretics may increase renal clearance of PCP, but are of doubtful clinical utility at this level of intoxication and may exacerbate myoglobinuric renal failure (29,122). Benzodiazepines should be used if medication is needed to control severe anxiety, agitation, or psychotic behavior (29), although they may delay renal clearance of PCP at high doses (120).
If benzodiazepines are insufficient to control psychosis, high-potency first-generation antipsychotics, such as haloperidol or droperidol, or second-generation antipsychotics, such as risperidone or olanzapine, may be used (123,124). They are less likely than other antipsychotics to produce anticholinergic or cardiovascular side effects that may exacerbate PCP’s own anticholinergic and cardiovascular effects. No clinical trials have directly compared the efficacy and safety of first-versus second-generation antipsychotics or of benzodiazepines versus antipsychotics.
Management of Medical Effects of Dissociative Anesthetic Intoxication
The mild medical effects commonly associated with Stage I intoxication usually do not need specific medical treatment.
Tachycardia and hypertension can be treated with adrenergic blockers such as labetalol or calcium channel blockers such as verapamil, although there are no controlled trials to substantiate their efficacy. Severe hypertension can be treated with IV nitroprusside (29).
Stage II and III intoxication are medical emergencies that require treatment in a comprehensive medical setting to maintain life support functions until the drug has been eliminated from the body (118). Tables 46-6 and 46-7 summarize medical treatment for acute PCP intoxication. In this context, increasing the renal clearance of PCP with forced diuresis and urine acidification (pH <5) may be helpful (67), although this may exacerbate myoglobinuric renal failure (29). This can be done by administering ammonium chloride—2.75 mEq/kg in 60 mL of saline every 6 hours through a nasogastric tube and 2 g of IV ascorbic acid in 500 mL of IV fluid every 6 hours (125). IM ascorbic acid also has been used successfully (126). Caution should be exercised to avoid causing metabolic acidosis, especially in the presence of drugs such as barbiturates and salicylates, whose renal clearance is delayed by acidification. Activated charcoal may be helpful, but induced vomiting or gastric lavage is not (29,127,128). Dialysis is not helpful because these agents have a large volume of distribution.
TABLE 46-6 PROCEDURES FOR MANAGING ACUTE PCP INTOXICATION
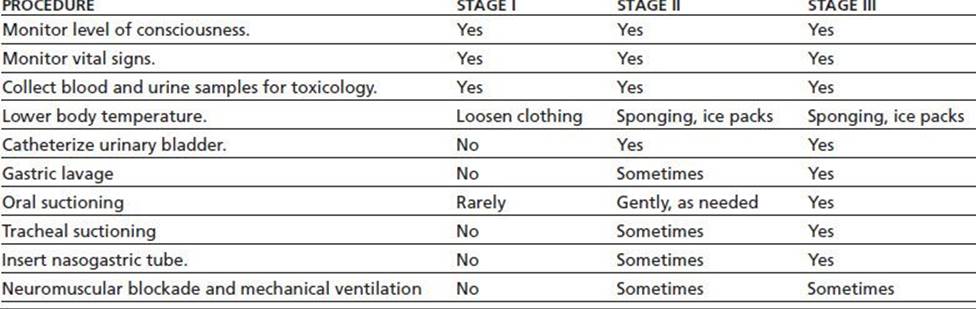
From Milhorn TH. Diagnosis and management of phencyclidine intoxication. Am Fam Phys 1991;43(4):1293–1302.
TABLE 46-7 MEDICATIONS FOR TREATING ACUTE PCP INTOXICATION

From Milhorn TH. Diagnosis and management of phencyclidine intoxication. Am Fam Phys 1991;43(4):1293–1302.
Another pharmacokinetic approach currently undergoing animal testing is administration of anti-PCP monoclonal antibody–binding fragments (1). These antibody fragments bind to PCP molecules in the body and prevent them from entering the brain, thereby reducing the acute effects of a PCP dose. Further research is needed to establish the safety and efficacy of this antibody approach in humans.
DXM toxicity may result from the other ingredients found in cough or cold preparations (e.g., acetaminophen, pseudoephedrine, phenylephrine, guaifenesin, antihistamines) (117). The evaluation and treatment of patients with suspected DXM overdose must attend to the possibility of acetaminophen or other concomitant toxicity.
Dissociative Anesthetic Withdrawal
Although a dissociative anesthetic withdrawal syndrome is not recognized in the DSM-5 (79) or DSM-IV-TR (129), about one-fourth of heavy PCP users report withdrawal symptoms (78), including depression, anxiety, irritability, hypersomnolence, diaphoresis, and tremor (78). It is not clear to what extent these represent a true withdrawal syndrome. DXM withdrawal has been associated with craving, dysphoria, and insomnia (117). Tricyclic antidepressants such as desipramine may reduce the psychological symptoms associated with discontinuation of PCP use, but there is no evidence that such treatment improves the outcome of PCP addiction (66a,130,131). The efficacy of SSRIs, which would be safer in this context, is unknown.
Prolonged Psychiatric Sequelae
Hallucinogens and dissociative anesthetics (PCP and ketamine) have the potential to trigger psychiatric sequelae that last beyond the period of acute intoxication, including prolonged states of anxiety, depression, psychosis, and cognitive dysfunction. The risk of a prolonged psychiatric reaction appears to depend on several factors: the patient’s premorbid psychopathology, the number of prior exposures to the drug, and a history of polydrug use (67,68,78,80). Prolonged reactions occasionally are reported in apparently well-adjusted individuals with no obvious risk factors. Prolonged psychotic reactions to PCP are almost always associated with premorbid psychopathology (124).
Treatment of prolonged anxiety or depression usually is psychosocial, but may involve medication if symptoms become sufficiently severe. Treatment of prolonged psychosis essentially follows guidelines for treatment of chronic functional psychosis. Patients may present with wide-ranging symptomatology: apathy, insomnia, hypomania, dissociative states, formal thought disorder, hallucinations, delusions, and paranoia. An observation period of at least several days with no or minimal medication (such as sedatives) is helpful to ensure an accurate diagnosis.
The term “flashback” (hallucinogen persisting perception disorder in the DSM-5 and DSM-IV-TR) has been given to brief episodes (often lasting a few seconds) in which perceptual aspects of a previous hallucinogenic drug experience are unexpectedly reexperienced after acute intoxication has resolved. Flashbacks are associated principally with LSD, although they can occur after use of other hallucinogens, MDMA, PCP, and, occasionally, marijuana (80). Flashbacks can precipitate considerable anxiety, particularly if the original drug experience had negative overtones. Reexperience of perceptual effects may be accompanied by somatic and emotional components of the original experience. Flashbacks may occur spontaneously or be triggered by stress, exercise, another drug (such as marijuana), or a situation reminiscent of the original drug experience.
Flashbacks usually are brief and self-limiting. Treatment may involve no more than alleviating anxiety with supportive reassurance. Over time, flashbacks tend to decrease in frequency, duration, and intensity, as long as no further hallucinogens are taken (80).
There have been no clinical trials of pharmacologic treatment for flashbacks (80). Benzodiazepines are helpful in treating secondary anxiety. Small case series suggest that clonazepam, clonidine, and haloperidol may be helpful, whereas case reports suggest that phenothiazines, risperidone, and SSRIs may worsen the condition.
Prolonged psychiatric sequelae also include dissociative drug-induced cognitive deficits and depressed mood. Over the past 10 years, a number of studies consistently demonstrated ketamine-induced cognitive dysfunction (132–136); depressed mood has also been identified in active ketamine users (134,135). Conversely, one study found no compelling evidence of long-term ketamine-induced changes in cognitive function (137), while another (138) proposed that a lower level of education in ketamine users contributed to the apparent ketamine-induced cognitive impairment. A recent study of 100 current and former ketamine users in Hong Kong (136), which controlled for level of education, demonstrated deficits in mental and motor speed, visual and verbal memory, and executive function in current users (N = 49), but not in former users (no use for past 30 days, N = 51) (136). Significant increases in depression (Beck Depression Inventory) were found in 72% of the current ketamine users (136). The above studies collectively suggest that repeated ketamine use produces cognitive deficits as well as depressed mood in the majority of active ketamine users, though not in former ketamine users (139); the issue of reversibility remains unclear. Recent neuroimaging studies of chronic ketamine users found reduced frontal gray matter volume (140) and bilateral frontal and left temporo-parietal white matter abnormalities (141).
INHALANTS
Inhalant Intoxication
Inhalants are a chemically heterogeneous group of volatile hydrocarbons (found in glue, fuel, paint, aerosol propellant, and other products) that can be inhaled for psychoactive effect (142,143). Inhalant intoxication produces initial euphoria or “rush,” followed by lightheadedness, excitability, and perceptual changes (142,143). Significant mood changes or cognitive impairment is rare. Higher doses or more prolonged exposure may cause dizziness, slurred speech, and motor incoordination, followed by drowsiness and headache. Intoxicated users rarely seek medical attention, in part because exposure tends to be self-limited and the duration of effect from a single exposure is usually only a few minutes.
Even a single episode of inhalant use can result in sudden death (144,145). Inhalant-induced brain neurotoxicity (146,147), especially to the white matter (148), as well as kidney, heart, and nerve damage (149,150), may complicate the clinical presentation of acute inhalant intoxication.
There is no specific treatment for inhalant intoxication (151). The patient should be assessed, stabilized, and monitored (especially cardiopulmonary status and hydration) in accordance with their clinical condition. Inhalants may sensitize the myocardium, so pressor medications and bronchodilators are relatively contraindicated.
Inhalant Withdrawal
Inhalant withdrawal is not a recognized clinical syndrome in the DSM-IV-TR (129) or DSM-5 (79), yet a growing literature describes an inhalant-induced withdrawal process. One study found that over 11% of patients evaluated for inhalant abuse reported withdrawal-like symptoms (152). Presumed inhalant withdrawal symptoms include depressed mood, fatigue, anxiety, difficulty concentrating, tachycardia, diaphoresis, muscle trembling or twitching, increased tearing and nasal secretions, headache, nausea and vomiting, and craving for inhalants (142,152,153). Some users report further use of inhalants to avoid experiencing these symptoms, suggesting that symptoms served as negative reinforcement for continued use (152). There is no specific treatment for inhalant withdrawal (151).
CLUB DRUGS
“Club drugs” are a pharmacologically heterogeneous group of drugs associated with a youth subculture that revolves around late-night dance parties known as “raves” or “trances” (108,154). The illicit use of these substances was popularized in this setting because of their perceived ability to enhance the sensory experience and allow for long periods of dancing to repetitive music. Common club drugs include MDMA (“ecstasy”), an amphetamine analog with stimulant and hallucinogenic properties, as well as GHB and flunitrazepam (Rohypnol), both of which are CNS depressants. Pharmacologic interactions from the concurrent use of multiple club drugs substantially increase the risk of toxicity (155).
MDMA (“ECSTASY”)
“Ecstasy” is the common street name for MDMA (see Chapter 14). Related amphetamine analogs such as 3,4-ethylenedioxy-ethylamphetamine (“eve”); 3,4-methylenedioxyamphetamine; and N-methyl-1-(3,4-methylenedioxyphenyl)-2-butanamine may also be present in street preparations. The effects of MDMA are those of a stimulant combined with a mild hallucinogen. “Herbal Ecstasy” often refers to preparations containing the stimulant ephedrine. “Liquid Ecstasy” is a street name for GHB (see the following section).
MDMA often is taken concurrently with other drugs, such as LSD (in a combination called “candyflipping”), for enhanced effect. DXM (available in over-the-counter cough medicines) is a frequent concomitant drug and may be substituted for MDMA in street preparations (117,156). “Stacking” is the practice of taking multiple MDMA doses over a short period, often alternating with other drugs to enhance the experience. For example, amphetamine or cocaine may be used initially to augment the experience, followed later by a CNS depressant, such as alcohol, marijuana, or GHB, to temper “coming down” (107). Menthol, camphor, or ephedrine may be applied to the nasal mucosa or chest wall to enhance the drug experience (154).
MDMA has good oral bioavailability and readily crosses the blood–brain barrier (75,155). The onset of action is within 30 minutes; peak plasma concentrations are achieved in 1 to 3 hours (155,157). The elimination half-life is 7 to 8 hours. Because MDMA is a weak acid, this is delayed to 16 to 31 hours with alkaline urine. MDMA is metabolized by several hepatic microsomal enzymes, chiefly CYP2D6.
Individuals who are genetically deficient in CYP2D6 (up to 10% of whites) are theoretically at increased risk of developing MDMA toxicity (158), though some studies suggest this risk is minimal (159).
MDMA appears to have nonlinear kinetics because the higher affinity enzymes become saturated at relatively low drug concentrations (75). This results in disproportionately large increases in drug concentrations in response to small increases in dose (158) and may account for the poor correlation between plasma concentration and toxicity (160). A major MDMA metabolite is methylenedioxyamphetamine (MDA), which also is pharmacologically active and has a longer elimination half-life of 16 to 38 hours (161).
MDMA Intoxication
The diagnosis of MDMA intoxication is made by history of drug intake and/or analysis of unused drug. Most signs and symptoms are not specific to MDMA, but resemble those of stimulants or hallucinogens. MDMA is not detected by routine urine or blood drug screens, which may be positive for amphetamines (products of MDMA metabolism) (154).
Gastric lavage with activated charcoal may be helpful within the first hour after ingestion, especially if other drugs also have been taken. Induced emesis is not recommended because of the risk of CNS depression (154). Acidification of urine would quicken MDMA elimination, but usually is contraindicated because it increases the risk of metabolic acidosis, thereby exacerbating renal toxicity from rhabdomyolysis (154).
Psychological and Behavioral Effects of MDMA Intoxication
Low to moderate oral doses of MDMA (50 to 150 mg) typically produce an intense initial effect (known as “coming on” or “rush”), especially if taken on an empty stomach, that may last 30 to 45 minutes (108). Desired effects include increased wakefulness and energy, euphoria, increased sexual desire and satisfaction, heightened sensory perception, sociability, and increased empathy and sense of closeness to others (75,154,162,163). The initial phase is followed by several hours of less intense experience (“plateau”), during which repetitive dancing is common. Users start to “come down” 3 to 6 hours after ingestion (108).
Undesired effects may occur with repeated use or at higher doses (164). These include hyperactivity, fatigue, insomnia, anxiety, agitation, impaired decision-making, flight of ideas, hallucinations, depersonalization, derealization, and bizarre or reckless behavior. Some users develop panic attacks, brief psychotic episodes, or delirium, which usually resolve rapidly as the drug effect wears off (165). Initial treatment is the same as for hallucinogen intoxication: placement in a quiet, reassuring environment, with observation to reduce the risk of unintended self-injury. Physical restraints are contraindicated because they may exacerbate anxiety and increase the risk of rhabdomyolysis. If severe or persisting symptoms require medication, benzodiazepines are preferred. Antipsychotics should be avoided as much as possible because they increase the risk of hyperthermia and seizures. A high-potency antipsychotic such as haloperidol should be used if necessary. The role of second-generation antipsychotics remains unclear. A few users may develop persisting depression or recurrent psychotic symptoms or panic attacks, which require psychiatric treatment.
Medical Effects of MDMA Intoxication
The acute physical effects of MDMA at low to moderate doses resemble those of a stimulant: increased muscle tension, jaw clenching, tooth grinding (bruxism), restlessness, insomnia, ataxia, headache, nausea, decreased appetite, dry mouth, dilated pupils, and increased heart rate and blood pressure (75,154,162,163). Doses greater than 200 mg are associated with life-threatening toxicities that can be grouped into four major syndromes (166). Most dangerous is hyperthermia, which results from a combination of increased physical activity (as through vigorous dancing), warm environment (as in a crowded, poorly ventilated dance club), and disruption of thermoregulation by the drug, often exacerbated by dehydration (167). The syndrome may resemble that of severe heatstroke. The high body temperature causes rhabdomyolysis (with resulting myoglobinuria and renal failure), liver damage, or disseminated intravascular coagulation (resulting in hemorrhage). Treatment is based on early recognition, close monitoring of serum creatinine kinase levels (to detect rhabdomyolysis), and reversal of the hyperthermia. Core body temperatures greater than 102°F call for urgent measures such as ice water sponging, gastric or bladder lavage with cool liquids, and intravenous infusion of chilled saline. Muscle paralysis with intubation may be required for refractory, ongoing rhabdomyolysis. Rhabdomyolysis treatment includes vigorous hydration and alkalinization of the urine to minimize myoglobin precipitation in the renal tubules.
Benzodiazepines help control both the hyperthermia and agitation. Antipsychotics should be avoided because they interfere with heat dissipation and lower the seizure threshold. Recent case series suggest that dantrolene (1 mg/kg IV) may be helpful. Because of similarities between MDMA toxicity and the serotonin syndrome (see the following section entitled Serotonin Syndrome), serotonin antagonists such as methysergide and cyproheptadine have been used successfully.
Acute hepatic toxicity from MDMA may be related to metabolism into reactive intermediaries that deplete hepatic glutathione, resulting in cell death (75,161). The clinical picture can vary from a mild hepatitis (marked by enlarged, tender liver and elevated serum liver enzymes) that resolves spontaneously over several weeks to fulminate liver failure requiring transplantation. Liver toxicity may be exacerbated by hyperthermia.
Acute cardiovascular toxicity from MDMA is the result of increased catecholamine activity (75,168). This may cause hypertension, with risk of blood vessel rupture and hemorrhage, or tachycardia and cardiac arrhythmia. The preferred treatment is an adrenergic antagonist with both alpha-and beta-blocking activities, combined with a vasodilator such as nitroglycerin or nitroprusside if needed to control blood pressure. A pure beta-adrenergic blocker should be avoided because of the remaining unopposed alpha-adrenergic stimulation, resulting in vasoconstriction and worsening hypertension. Hypertensive crisis unresponsive to mixed adrenergic blockers and vasodilators should be treated with an alpha-adrenergic antagonist such as phentolamine (3). Cardiac ischemia or arrhythmia should be treated by standard clinical protocols. Agitation should be controlled with a short-acting benzodiazepine such as lorazepam.
In addition to direct MDMA-mediated neurotoxicity (169), acute toxicity can result from hyponatremia (“water intoxication”), which may cause seizures and intra-cranial fluid shifts that compress the brain stem into the foramen magnum (75). The hyponatremia is caused by loss of sodium in sweat (as during vigorous dancing in a warm environment) and hemodilution from drinking large amounts of water and the antidiuretic effect of MDMA. The conservative initial treatment is fluid restriction. Profound hyponatremia has been treated with hypertonic saline solution (170). Intravenous benzodiazepines should be used to control seizures.
MDMA Withdrawal
Symptoms during the first few days after MDMA use may resemble a mild form of stimulant withdrawal or “crash,” with depression, anxiety, fatigue, and difficulty concentrating (75,108). These usually resolve without treatment.
Medical Effects of MDMA Withdrawal
There is no evidence of a physically prominent or distinctive withdrawal syndrome associated with MDMA that would require specific pharmacologic treatment. Users may complain of muscle pain and stiffness in the jaw, neck, lower back, and limbs for the first 2 to 3 days after use (75), which may be the result of MDMA-induced muscle tension and the vigorous dancing often associated with MDMA use. There is some evidence of increased variability of heart rate and blood pressure for several days after MDMA use.
GAMMA-HYDROXYBUTYRATE
GHB (sometimes termed “liquid ecstasy”) is a naturally occurring metabolite of the neurotransmitter gamma-aminobutyric acid (GABA) that may itself function as an agonist at both the GABA-B receptor and a putative GHB receptor (171–174). It is approved for the treatment of narcolepsy (sodium oxybate, Xyrem, Schedule III controlled substance) (107,118), but is also used recreationally (154,175). GHB became popular in the late 1980s, when it was marketed and sold in health food stores as a supplement for body building and other putative health effects. Use of GHB increased well beyond the supplement market, in part because of its reputed euphoric, aphrodisiac, disinhibitory, and amnestic effects (155). GHB’s short duration of action, minimal “hangover” effects, and nondetectability by standard drug screens contributed to its popularity. The legal precursors gamma-butyrolactone (GBL, an industrial solvent found in floor strippers and some household products) and 1,4-butanediol (1,4-BD), which are readily metabolized to GBH in the body, are also used recreationally (176), as are structural analogs such as β-phenyl-GABA (phenibut), developed in the Soviet Union as a sedative/anxiolytic but now readily available in the United States and Europe as a nutritional supplement (174).
GHB is taken orally as a liquid or in a powder mixed into drinks. A typical dose is 1 to 3 teaspoons or capfuls, though variations in concentration make it challenging to determine the actual dosage of GHB in recreational preparations. GHB is rapidly absorbed from the gastrointestinal tract and readily crosses the blood–brain barrier. Effects begin within 15 minutes of ingestion and last 2 to 4 hours (177). The blood elimination half-life is about 30 minutes, largely because of rapid redistribution into other tissues.
GHB Intoxication
The diagnosis of GHB intoxication is based on clinical suspicion, a history of drug ingestion, or analysis of unused drug. The signs and symptoms of GHB intoxication are not specific and are difficult to differentiate from other CNS depressants. GHB is not detected by routine drug toxicology assays. Definitive detection requires fluid analysis utilizing gas chromatography/mass spectrometry (178), which commonly takes 7 to 14 days.
There is no proven antidote for GHB intoxication. Physostigmine, naloxone, and flumazenil have reversed some GHB effects in small case series or animal studies (179), but should be considered experimental. Gastric lavage usually is not helpful because of rapid gastrointestinal absorption, but activated charcoal may be.
Psychological and Behavioral Effects of GHB Intoxication
The desired acute effects of GHB at low oral doses (<20 mg/kg) include relaxation, euphoria, sedation, disinhibition, sociability, and anterograde amnesia that has been linked to sexual assault (107,180– 183). Higher doses produce somnolence, confusion, and hallucinations (181). Unintended overdose may occur because of GHB’s very steep dose–response curve, narrow therapeutic index, and the great variability in potency of street preparations. First-time users often underestimate the potency of GHB. The effects are prolonged and intensified when taken with other CNS depressants, such as alcohol. Patients recovering from acute GHB intoxication may wake up abruptly with a clear sensorium or may go through a brief period of agitation and combativeness.
Medical Effects of GHB Intoxication
Low to moderate oral doses of GHB may cause headache, dizziness, ataxia, hypotonia, and vomiting (108,154,175– 177). Higher doses (>30 mg/kg) may cause incontinence, myoclonic movements, bradycardia, hypotension, hypothermia, generalized tonic–clonic seizures, and coma (29,184). Concurrent ingestion of other drugs, including alcohol, substantially increases the severity of GHB intoxication (185). Most patients with pure GHB intoxication recover completely within several hours with supportive care and do not require intubation (186). However, death may result from respiratory depression, so that intubation and mechanical ventilation may be indicated in severe cases. Seizures should be controlled with benzodiazepines, symptomatic bradycardia with atropine, and symptomatic hypotension with intravenous saline (29). Similar adverse effects occur with GBL and 1,4-BD (187).
GHB Withdrawal
Cessation of chronic GHB or GBL use leads to a discrete withdrawal syndrome resembling that of sedative–hypnotic withdrawal, presumably mediated by unopposed excitation in the neurotransmitter systems ordinarily inhibited by GABA-B (and GHB) receptors (75). Anxiety, restlessness, insomnia, tremor, nystagmus, tachycardia, and hypertension usually appear 2 to 12 hours after the last dose (107,188– 193). Mild symptoms usually resolve gradually over 1 to 2 weeks. More severe withdrawal may cause delirium with hallucinations, psychosis, agitation, and autonomic instability (185) and may present similarly to delirium tremens (190). GHB withdrawal seizures are rare but have been reported (185). Physical dependence may develop within 1 week of repeated daily dosing.
Most cases of GHB withdrawal can be managed with a long-acting benzodiazepine, tapering the dose after the symptoms are controlled (as for sedative–hypnotic withdrawal) (107,191). Severe cases may require high doses (several 100 mg) or parenteral administration. Patients unresponsive to benzodiazepines may benefit from barbiturates, a mood stabilizer such as gabapentin, or low-dose antipsychotics (108). Because of the unpredictability of GHB withdrawal and vulnerability to severe complications such as delirium and potential lethality (194), detoxification is best undertaken in a hospital setting (185). Mild withdrawal syndromes may be managed in an outpatient setting with close supervision (188).
HERBS OF ABUSE
Herbs are plants used for medicinal, culinary, or spiritual purposes. Many herbs contain psychoactive compounds with stimulant, anxiogenic, anxiolytic, hallucinogenic, euphoric, or dissociative effects (195,196). These properties have long been recognized in many indigenous cultures.
The psychoactive profile of herbs, combined with the fact that production, sale, and purchase of most herbs are largely unregulated, has contributed to a growing market for their recreational use (197). Internet distribution of herbs makes them widely available to minors (198). The perception that herbs are safer than illicit drugs, coupled with the absence of clearly established dosing parameters, contributes to their misuse. Routine toxicology screens do not detect many of these substances, so that identifying specific intoxication syndromes may be challenging. Accurate diagnosis may rest on collateral information from family, friends, and first responders, in addition to a thorough clinical examination.
Intoxication
Herbs of abuse often contain multiple psychoactive compounds, so that intoxication syndromes may not fit neatly into distinctive classifications. For clarity, herbs of abuse may be categorized as predominantly hallucinogenic or stimulating. Table 46-8 describes basic characteristics of some of the commonest herbs of abuse.
Hallucinogenic herbs achieve their psychotomimetic effects principally through activity at serotonergic or cholinergic receptors. Stimulating herbs generally augment the activity of norepinephrine or dopamine. Thus, the manifestations and management of intoxication syndromes for this varied group of substances generally follow that for hallucinogen or stimulant intoxication.
Table 46-8 COMMON HERBAL DRUGS OF ABUSE

DMT, N,N-dimethyltryptamine; GABA, γ-aminobutyric acid; LSA, lysergic acid hydroxyethylamide; MA, methamphetamine; MAO, monoamine oxidase.
Sources: Richardson WH, Slone CM, Michels JE. Herbal drugs of abuse: an emerging problem. Emerg Med Clin N Am 2007;254:35–57; Halpern JH. Hallucinogens and dissociative agents naturally growing in the United States. Pharmacol Ther 2004;102:131–138.
Management of Psychological, Behavioral, and Medical Effects
Management of intoxication with hallucinogenic herbs is largely supportive because most symptoms, including psychosis, are self-limited. The goal is to maintain safety, preventing patients from physically harming themselves or others. A quiet environment, with calm counseling and guidance, often avoids the need for pharmacologic interventions. Medications with anticholinergic properties are best avoided to minimize exacerbating substance-induced delirium. Physical restraints should be avoided because they increase psychological distress and may contribute to rhabdomyolysis. Patients who are agitated, in severe panic, or having distressing psychotic symptoms may be relieved by benzo-diazepines (e.g., lorazepam 2 mg PO/IM every 1 to 2 hours, titrated to mild sedation). In cases where predisposing factors or heavy chronic use contribute to prolonged psychotic symptoms, antipsychotic agents may be useful.
Management of intoxication with stimulant herbs is similar to that with hallucinogenic herbs, except that the former are more likely to generate hyperexcitable, agitated, and psychotic states. Patients with unstable vital signs should be closely monitored, including cardiac function, blood pressure, and body temperature. Beta-adrenergic blockers are generally avoided due to concern about unopposed alpha-adrenergic activity.
With one exception, there are no specific antidotes to intoxication with psychoactive herbs. Intoxication with herbs having anticholinergic activity (e.g., jimsonweed) has been successfully treated with physostigmine, a short-acting acetylcholinesterase inhibitor (195). Severe intoxication with betel nut, which has cholinergic activity, can be treated with atropine, a cholinergic antagonist.
Withdrawal
Most users of psychoactive herbs do not consume large-enough amounts for long-enough periods to develop physical dependence or a withdrawal syndrome. Some users of khat and betel nuts do experience a withdrawal syndrome, often including irritability, fatigue, and rhinorrhea (195). Protracted withdrawal symptoms (e.g., psychosis, depression, anxiety) should be treated symptomatically while the patient is evaluated for an underlying psychiatric disorder.
FLUNITRAZEPAM
Flunitrazepam (Rohypnol, also known as “roofies” or the “date rape pill”) is a potent, fast-acting benzodiazepine that frequently causes anterograde amnesia (121,176). It is legally manufactured and marketed in Europe and Latin America, but is illegal in the United States because of its association with “date rape,” although the epidemiologic evidence for this is limited (183). Flunitrazepam is difficult to detect with routine toxicology screens because of its low concentration.
Flunitrazepam Intoxication
Flunitrazepam intoxication resembles intoxication with other benzodiazepines and features sedation, disinhibition, anterograde amnesia, confusion, ataxia, bradycardia, hypotension, and respiratory depression (49,160). Overdose, alone and/or particularly concurrently with alcohol ingestion, can be lethal (49,160). When respiratory depression or circulatory compromise is severe, the benzodiazepine antagonist flumazenil (Romazicon) may be used, albeit cautiously. Flumazenil precipitates acute withdrawal in patients who are physically dependent on benzodiazepines and lowers the seizure threshold, thus increasing the risk of withdrawal seizures. Flumazenil is effective for about 20 minutes, so that repeated dosing is necessary to avoid resedation by flunitrazepam.
Flunitrazepam Withdrawal
A typical sedative–hypnotic withdrawal syndrome can develop after cessation of chronic flunitrazepam use (199). Withdrawal symptoms can develop up to 36 hours after the last dose and include anxiety, restlessness, tremors, headache, insomnia, and paresthesias. Treatment of withdrawal involves supportive measures and substitution with cross-tolerant medications such as lorazepam or clonazepam, followed by gradual tapering.
SEROTONIN SYNDROME
The serotonin syndrome is a potentially lethal condition associated with increased serotonergic activity in the CNS. Substances that increase serotonin activity, directly (MDMA), indirectly (SSRIs), or in combination, can trigger this syndrome (200). The serotonin syndrome is a triad of signs and symptoms, consisting of mental status changes (e.g., anxiety, confusion, agitation, lethargy, delirium, coma), autonomic hyperactivity (e.g., low-grade fever, tachycardia, diaphoresis, nausea, vomiting, diarrhea, dilated pupils, abdominal pain, hypertension, tachypnea), and neuromuscular abnormalities (e.g., myoclonus or clonus, nystagmus, hyperreflexia, rigidity, trismus, tremor) (201,202). The clinical presentation is highly variable, making diagnosis difficult, but neuromuscular signs are usually prominent, particularly in the lower extremities (202–204). The Hunter Toxicity Criteria Decision Rules are 84% sensitive and 97% specific for serotonin syndrome when compared with the gold standard of diagnosis by a medical toxicologist (203). The Hunter Criteria include recent ingestion of a serotonergic agent and at least one of the following:
1. Spontaneous clonus
2. Inducible clonus plus agitation or diaphoresis
3. Ocular clonus plus agitation or diaphoresis
4. Tremor plus hyperreflexia
5. Hypertonia plus temperature above 38°C plus ocular clonus or inducible clonus
The differential diagnosis includes neuroleptic malignant syndrome (with which it is most commonly confused), sepsis, heat stroke, delirium tremens, and sympathomimetic or anticholinergic poisoning. Patients with neuroleptic malignant syndrome differ from those with serotonin syndrome in that they are more likely to present with extrapyramidal signs and autonomic instability and rarely present with the neuromuscular changes common in serotonin syndrome.
The serotonin syndrome is the result of excessive stimulation of 5-HT2A, possibly with some contribution from 5-HT1A, receptors. This occurs through several different pathways: activation of serotonin receptors by agonists, enhanced release of serotonin (by MDMA or amphetamines), decreased presynaptic serotonin reuptake (by cocaine or SSRI antidepressants), decreased serotonin metabolism (by amphetamines or monoamine oxidase inhibitors), and increased serotonin synthesis. The serotonin syndrome is most commonly seen after ingestion of two or more drugs with such actions, but also may occur with a single drug.
The onset of the serotonin syndrome is within minutes to hours of medication initiation, increase in dose, or overdose. Laboratory abnormalities are nonspecific; no one test confirms the diagnosis. Elevated creatine phosphokinase, liver transaminases, white blood cell count, serum bicarbonate, and evidence of disseminated intravascular coagulation occur in severe cases (190). In severe cases, or in the absence of appropriate diagnosis and treatment, there may be progression to rhabdomyolysis, hyperthermia, renal failure, disseminated intravascular coagulation, and death.
Effective treatment of the serotonin syndrome requires early identification, immediate discontinuation of all serotonergic medications, close monitoring, and supportive care, usually including intravenous hydration (202,204). Such treatment usually results in a benign, self-limited course; many cases resolve within 24 hours. Muscle rigidity and spasm should be controlled with benzodiazepines to prevent rhabdomyolysis. Management of hyperthermia is vital to mitigate complications such as seizures, disseminated intravascular coagulation, and metabolic acidosis (202). Severe forms of the syndrome require more aggressive measures, including neuromuscular blocking agents, mechanical ventilation, and external cooling. Antipyretics are generally unhelpful because the source of hyperthermia is muscular activity, not alterations in hypothalamic temperature set point (201,202). Treatment with a 5-HT2A receptor antagonist (e.g., cyproheptadine, olanzapine, chlorpromazine) has been effective in case series, but has not yet been evaluated in controlled clinical trials.
WITHDRAWAL FROM MULTIPLE DRUGS
Multiple Sedative–Hypnotics
Withdrawal from dependence on multiple sedative– hypnotic agents, including alcohol, is best managed in the same way as withdrawal from a single such drug: by using tapering dosages of a single, longer-acting sedative–hypnotic (5,205). It usually is safest to focus on managing withdrawal of the longer-acting drug. The time course of withdrawal from multiple sedative–hypnotics is more unpredictable than from single drugs; for example, there may be a bimodal time course of symptomatology if one drug is short acting and the other is longer acting. The rate at which the dose is tapered usually should not exceed 10% per day. Successful withdrawal is facilitated by use of an anticonvulsant such as carbamazepine (206), although such use has not been evaluated in multiple drug withdrawal.
Sedative–Hypnotics with Other Drugs
In the pharmacologic management of patients withdrawing from both sedative–hypnotics and CNS stimulants, it is preferable to treat the sedative–hypnotic withdrawal first because this poses the greatest difficulty and medical risk. For concurrent addiction to sedative–hypnotics and opiates, concurrent pharmacologic treatment is recommended (5,205). The patient may be stabilized on an opiate (preferably oral methadone, although codeine can be used if methadone is not available) at the same time that the sedative–hypnotic dose is tapered by 10% per day. After the sedative–hypnotic withdrawal is completed, opiate withdrawal can begin. Clonidine has been suggested as adjunctive treatment for such mixed sedative–hypnotic and opiate withdrawal, because it can alleviate withdrawal symptoms from both drug classes, but this has not been evaluated systematically.
POPULATION-SPECIFIC CONSIDERATIONS
Neonates
Neonatal drug exposure is a substantial public health problem. Many drugs of abuse are readily transferred from the maternal circulation across the placenta to the fetus. Thus, perinatal drug use by the mother raises the possibility of drug intoxication or withdrawal in the newborn (207–209). Obtaining an accurate maternal drug use history for the period preceding delivery is essential. Meconium is the most accurate substrate for neonatal toxicology through the 3rd to 4th day of life, but such testing is not widely available.
Neonatal signs and symptoms of drug intoxication or withdrawal often are nonspecific, including sedation, irritability, restlessness, hypertonia, hyperreflexia, tremors, poor feeding, abnormal sleep patterns, respiratory difficulty, and seizures. Stimulants (such as cocaine), marijuana, LSD, and PCP all have been associated with a neonatal withdrawal syndrome, although one that usually is less intense than the opiate withdrawal syndrome (210).
Perinatal use of stimulants by the mother is associated with either bradycardia or tachycardia in the newborn (211). The additive cardiovascular effects of the stimulant and the normal catecholamine surge during labor may cause fetal distress and retard delivery (198). These cardiac effects usually resolve as the drug is eliminated from the body. Neonatal stimulant intoxication is associated with irritability, tremors, hyperactivity, abnormal movements, excessive sucking, and high-pitched and excessive crying for 1 to 2 days, followed by a period of lethargy and hypo-reactivity (212–214).
Treatment of drug-exposed newborns is largely supportive, with avoidance of overstimulation. Pharmacologic treatment should be used cautiously because it has its own potential for morbidity. Phenobarbital is the preferred medication for newborns with nonopiate drug withdrawal who do require pharmacologic treatment, as when seizures are a factor. A loading dose of 5 mg/kg/d is given until withdrawal is controlled, with adjustments of 10% to 20% every 2 to 3 days based on the response. Phenobarbital has a long half-life, so plasma concentrations should be checked periodically to avoid drug accumulation and overtreatment.
Older Adults
Rates of illicit drug use by the elderly are low but may be increasing (215). There are little published data on the treatment of drug intoxication or withdrawal in this age group. The elderly may be more susceptible to confusion and disorientation during withdrawal and to medication-induced delirium. The recommended dosing approach is “start low and go slow”; that is, start medication at a lower dose, and increase the dose in smaller increments than would be used in younger individuals.
Adolescents
Adolescence is the common age of onset for illegal drug use (216), and the developing adolescent brain may be especially vulnerable to the neurobiologic effects of drugs (217). Adolescents experience symptoms of drug withdrawal similar to those in adults, including physical symptoms (218). There are few published data on the treatment of drug intoxication or withdrawal in adolescents (219,220).
Women
Women often differ from men in their response to psychoactive drugs and to drug abuse treatment (221), but there has been little systematic study of gender differences in the treatment of drug intoxication and withdrawal. Limited anecdotal evidence suggests that pharmacologic treatment for women is similar to that for men, taking into account possible gender differences in medication pharmacokinetics. Two topics requiring further research are the influence of the menstrual cycle on intoxication and withdrawal and their treatment and the effects of intoxication and withdrawal and their treatment on pregnancy and the fetus.
ACKNOWLEDGMENTS
Dr. Gorelick is supported by the Intramural Research Program, National Institutes of Health, and National Institute on Drug Abuse. Dr. Wilkins is supported by the Cedars-Sinai Medical Center Lincy-Heyward/Moynihan Endowed Chair in Addiction Medicine and the U.S. Army Medical Department.
REFERENCES
1.Gorelick DA. Pharmacokinetic strategies for treatment of drug overdose and addiction. Future Med Chem 2012;4(2):227–243.
2.Howell LL, Kimmel HL. Monoamine transporters and psychostimulant addiction. Biochem Pharmacol 2008;75(1):196–217.
3.Ghuran A, Nolan J. Recreational drug misuse: issues for the cardiologist. Heart 2000;83:627–633.
4.Neiman J, Haapaniemi HM, Hillblom M. Neurological complications of drug abuse: pathophysiological mechanisms. Eur J Neurol 2000;7:595–606.
5.Schuckit MA. Drug and alcohol abuse. A clinical guide to diagnosis and treatment, 6th ed. New York: Springer, 2006.
6.Tashkin DP. Airway effects of marijuana, cocaine, and other inhaled illicit agents. Curr Opin Pulm Med 2001;7:43–61.
7.Newton TF, Roache JD, De La Garza R II, et al. Bupropion reduces methamphetamine-induced subjective effects and cue induced craving. Neuropsychopharmacology 2006;31(7):1537–1544.
8.Lile JA, Stoops WW, Vansickel AR, et al. Aripiprazole attenuates the discriminative-stimulus and subject-rated effects of D-amphetamine in humans. Neuropsychopharmacology2005;30(11):2103–2114.
9.Rush CR, Stoops WW, Hays LR, et al. Risperidone attenuates the discriminative-stimulus effects of d-amphetamine in humans.J Pharmacol Exp Ther 2003;306(1):195–204.
10.Johnson BA, Roache JD, Ait-Daoud N, et al. Effects of acute topiramate dosing on methamphetamine-induced subjective mood. Int J Neuropsychopharmacol 2007;10(1):85–98.
11.McGregor C, Srisurapanont M, Mitchell A, et al. Symptoms and sleep patterns during inpatient treatment of methamphetamine withdrawal: a comparison of mirtazapine and modafinil with treatment as usual. J Subst Abuse Treat 2008;35(3):334–342.
12.Angrist B. Clinical effects of central nervous system stimulants: a selective update. In: Engel J, Oreland L, Ingvar DH, et al, eds. Brain reward systems and abuse. New York: Raven Press, 1987:109–127.
13.Romanelli F, Smith KM. Clinical effects and management of methamphetamine abuse. Pharmacotherapy 2006;35(3): 334–342.
14.Fiorentini A, Volonteri LS, Dragogna F et al. Substance-induced psychoses: a critical review of the literature. Curr Drug Abuse Rev 2011;4(4):228–240.
15.Roncero C, Ros-Cucurull E, Daigre C, et al. Prevalence and risk factors of psychotic symptoms in cocaine-dependent patients. Actas Esp Psiquiatr 2012;40(4):187–197.
16.Smith MJ, Thirthalli J, Abdallah AB, et al. Prevalence of psychotic symptoms in substance users: a comparison across substances. Compr Psychiatry 2009;50(3):245–250.
17.Harris D, Batki SL. Stimulant psychosis: symptom profile and acute clinical course. Am J Addict 2000;9(1):28–37.
18.Darke S, Kaye S, McKetin R, et al. Major physical and psychological harms of methamphetamine use. Drug Alcohol Rev 2008;27(3):253–262.
19.Matsuzawa D, Hashimoto K, Miyatake R, et al. Identification of functional polymorphisms in the promoter region of the human PICK1 gene and their association with methamphetamine psychosis. Am J Psychiatry 2007;164:1105–1114.
20.Karch SB, Stephens BG. Drug abusers who die during arrest or in custody. J R Soc Med 1999;92:110–113.
21.Ruttenber AJ, McAnally HB, Wetli CV. Cocaine-associated rhabdomyolysis and excited delirium: different stages of the same syndrome. Am J Forensic Med Pathol 1999;20(2):120–127.
22.Miyashita T, Hayashi T, Ishida Y, et al. A fatal case of pontine hemorrhage related to methamphetamine abuse. J Forensic Leg Med 2007;14(7):444–447.
23.Brady WJ Jr, Duncan CW. Hypoglycemia masquerading as acute psychosis and acute cocaine intoxication. Am J Emerg Med 1999;17(3):318–319.
24.Ling W, Rawson R, Shoptaw S, et al. Management of methamphetamine abuse and dependence. Curr Psychiatry Rep 2006;8(5):345–354.
25.Roth BA, Benowitz NL, Olson KR. Emergency management of drug abuse-related disorders. In: Karch SB, ed. Drug abuse handbook. Boca Raton, FL: CRC Press, 1998:567–639.
26.Mohr WK, Petti TA, Mohr BD. Adverse effects associated with physical restraint. Can J Psychiatry 2003;48(5):330–337.
27.Srisurapanont M, Kittirattanapaiboon P, Jarusuraisin N. Treatment for amphetamine psychosis. Cochrane Database Syst Rev 2001;(1): CD003026.
28.Schep LJ, Slaughter RJ, Beasley DM. The clinical toxicology of metamfetamine. Clin Toxicol (Phila) 2010;48(7):675–694.
29.Mokhlesi B, Garimella PS, Joffe A, et al. Street drug abuse leading to critical illness. Intensive Care Med 2004;30:1526–1536.
30.Callaway CW, Clark RF. Hyperthermia in psychostimulant overdose. Ann Emerg Med 1994;24:68–75.
31.Ruha AM, Yarema MC. Pharmacologic treatment of acute pediatric methamphetamine toxicity. Pediatr Emerg Care 2006;22(12): 782–785.
32.Stoops WW, Lile JA, Lofwall MR, et al. The safety, tolerability, and subject-rated effects of acute intranasal cocaine administration during aripiprazole maintenance. Am J Drug Alcohol Abuse 2007; 33:769–776.
33.Hurlbut KM. Drug-induced psychoses. Emerg Med Clin North Am 1991;9(1):31–52.
34.Satel SL, Seibyl JP, Charney DS. Prolonged cocaine psychosis implies underlying major psychopathology. J Clin Psychiatry 1991;52(8): 349–350.
35.Yui K, Goto K, Ikemoto S. The role of noradrenergic and dopaminergic hyperactivity in the development of spontaneous recurrence of methamphetamine psychosis and susceptibility to episode recurrence. Ann N Y Acad Sci 2004;1025:296–306.
36.Nzerue CM, Hewan-Lowe K, Riley Jr LJ. Cocaine and the kidney: a synthesis of pathophysiologic and clinical perspectives. Am J Kidney Dis 2000;35(5):783–795.
37.Richards JR, Johnson EB, Stark RW, et al. Methamphetamine abuse and rhabdomyolysis in the ED: a 5-year study. Am J Emerg Med 1999; 17:681–685.
38.Phillips K, Luk A, Soor GS, et al. Cocaine cardiotoxicity: a review of the pathophysiology, pathology, and treatment options. Am J Cardiovasc Drugs 2009;9(3):177–196.
39.Schwartz BG, Rezkalla S, Kloner RA. Cardiovascular effects of cocaine. Circulation 2010;122(24):2558–2569.
40.Bhargava S, Arora RR. Cocaine and cardiovascular complications. Am J Ther 2011;18(4):e95–e100.
41.Blaho K, Logan B, Winbery S, et al. Blood cocaine and metabolite concentrations, clinical findings, and outcome of patients presenting to an ED. Am J Emerg Med 2000;18(5):593–598.
42.McCord J, Jneid H, Hollander JE, et al. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation 2008;117:1897–1907.
43.Attaran R, Ragavan D, Probst A. Cocaine-related myocardial infarction: concomitant heroin use can cloud the picture. Eur J Emerg Med 2005;12:199–201.
44.Mohamad T, Niraj A, Farah J et al. Spectrum of electrocardiographic and angiographic coronary artery disease findings in patients with cocaine-associated myocardial infarction. Coron Artery Dis2009;20(5):332–336.
45.Ramirez FD, Femenia F, Simpson CS, et al. Electrocardiographic findings associated with cocaine use in humans: a systematic review. Expert Rev Cardiovasc Ther 2012;10(1):105–127.
46.Hollander JE, Henry TO. Evaluation and management of the patient who has cocaine-associated chest pain. Cardiol Clin 2006;24: 103–114.
47.Damodaran S. Cocaine and beta-blockers: the paradigm. Eur J Intern Med 2010;21(2):84–86.
48.Rangel C, Shu RG, Lazar LD, et al. Beta-blockers for chest pain associated with recent cocaine use. Arch Intern Med 2010;170(10): 874–879.
49.Demaria S Jr, Weinkauf JL. Cocaine and the club drugs. Int Anesthesiol Clin 2011;49(1):79–101.
50.Wong GT, Irwin MG. Poisoning with illicit substances: toxicology for the anaesthetist. Anaesthesia 2013;68(Suppl 1):117–124.
51.Hsue PY, McManus D, Selby V, et al. Cardiac arrest in patients who smoke crack cocaine. Am J Cardiol 2007;99:822–824.
52.Meehan TJ, Bryant SM, Aks SE. Drugs of abuse: the highs and lows of altered mental states in the emergency department. Emerg Med Clin North Am 2010;28(3):663–682.
53.Jenkins AJ, Cone EJ. Pharmacokinetics: drug absorption, distribution, and elimination. In: Karch SB, ed. Drug abuse handbook. Boca Raton, FL: CRC Press, 1998:151–201.
54.D’Souza MS, Markou A. Neural substrates of psychostimulant withdrawal-induced anhedonia. Curr Top Behav Neurosci 2010;3:119–178
55.McGregor C, Srisurapanont M, Jittiwutikarn J, et al. The nature, time course and severity of methamphetamine withdrawal. Addiction 2005;100:1320–1329.
56.Matuskey D, Pittman B, Forselius E, et al. A multistudy analysis of the effects of early cocaine abstinence on sleep. Drug Alcohol Depend 2011;115(1–2):62–66.
57.Mulvaney FD, Alterman AI, Boardman CR, et al. Cocaine abstinence symptomatology and treatment attrition. J Subst Abuse Treat 1999;16(2):129–135.
58.Rosenblum A, Foote J, Magura S, et al. Follow-up of inpatient cocaine withdrawal for cocaine-using methadone patients. J Subst Abuse Treat 1996;13(6):467–470.
59.Karila L, Gorelick D, Weinstein A, et al. New treatments for cocaine dependence: a focused review. Int J Neuropsychopharmacol 2008;11:425–438.
60.Vocci FJ, Appel NM. Approaches to the development of medications for the treatment of methamphetamine dependence. Addiction 2007;102(Suppl 1):96–106.
61.Nademanee K, Gorelick DA, Josephson MA, et al. Myocardial ischemia during cocaine withdrawal. Ann Intern Med 1989;111:376–380.
62.Khantzian EJ, McKenna GJ. Acute toxic and withdrawal reactions associated with drug use and abuse. Ann Intern Med 1979;90:361–372.
63.Satel SL, Price LH, Palumbo JM, et al. Clinical phenomenology and neurobiology of cocaine abstinence: a prospective inpatient study. Am J Psychiatry 1991;148:1712–1716.
64.Gill K, Gillespie HK, Hollister LE. Dopamine depletion hypothesis of cocaine dependence: a test. Hum Psychopharmacol 1991;6:25–29.
65.Volkow ND, Fowler JS, Wolf AP. Effects of chronic cocaine abuse on postsynaptic dopamine receptors. Am J Psychiatry 1992;147: 719–724.
66.Shoptaw SJ, Kao U, Heinzerling K, et al. Treatment for amphetamine withdrawal. Cochrane Database Syst Rev 2009;(2):CD003021.
66a.Giannini AJ, Malone DA, Giannini MC, et al. Treatment of depression in chronic cocaine and phencyclidine abuse with desipramine. J Clin Pharmacol 1986;26:211–214.
67.Halpern JH. Hallucinogens: an update. Curr Psychiatry Rep 2003;5: 347–354.
68.Pechnick, RN, Glasner-Edwards, S, Wilkins JN, et al. The Hallucinogens, Marijuana, and club drugs in Gabbard’s treatments of psychiatric disorders, 4th ed. Washington, DC: American Psychiatric Publishing, Inc, 2007:239–246.
69.Brust JCM. Acute neurologic complications of drug and alcohol abuse. Neurol Clin North Am 1998;16:503–519.
70.Frecska E, Luna LE. The adverse effects of hallucinogens from intramural perspective. Neuropsychopharmacol Hung 2006;8: 189–200.
71.Abraham HD, Aldridge AM, Gogia P. The psychopharmacology of hallucinogens. Neuropsychopharmacology 1996;14:285–298.
72.First MB, Tasman A, eds. Emergency treatment of hallucinogen intoxication. In: Clinical guide to the diagnosis and treatment of mental disorders. West Sussex, United Kingdom: John Wiley & Sons, Inc. 2010:195–201.
73.Van Amsterdam J, Opperhuizen A, van den Brink W. Harm potential of magic mushroom use: a review. Regul Toxicol Pharmacol 2011;59:423–429
74.Pechnick RN, Ungerleider JT. Hallucinogens. In: Lowinson JH, Ruiz P, Millman RB, et al., eds. Substance abuse: a comprehensive textbook, 3rd ed. Baltimore, MD: Williams & Wilkins, 1997:230–238.
75.Kalant H. The pharmacology and toxicology of “ecstasy” (MDMA) and related drugs. Can Med Assoc J 2001;165(7):917–928.
76.Selden BS, Clark RF, Curry SC. Marijuana. Emerg Med Clin North Am 1990;8:527–539.
77.Nielen RJ, van der Heijden FM, Tuinier S, et al. Khat and mushrooms associated with psychosis. World J Biol Psychiatry 2004;5:49–53.
78.Cottler LB, Schuckit MA, Helzer JE, et al. The DSM-IV field trial for substance use disorders: major results. Drug Alcohol Depend 1995; 38:59–69.
79.American Psychiatric Association. Diagnostic and statistical manual of mental disorders, 5th ed. Arlington, VA: American Psychiatric Publishing, 2013.
80.Halpern JH, Pope HG. Hallucinogen persisting perception disorder: what do we know after 50 years. Drug Alcohol Depend 2003;69:109–119.
81.Lerner AG, Shufman E, Kodesh A, et al. Risperidone-associated, benign transient visual disturbances in schizophrenic patients with a past history of LSD abuse. Isr J Psychiatry Relat Sci 2002;39(1):57–60.
82.Gomez-Ruiz M, Hernandez M, De MR, et al. An overview on the biochemistry of the cannabinoid system. Mol Neurobiol 2007;36:3–14.
83.Gonzalez R. Acute and non-acute effects of cannabis on brain functioning and neuropsychological performance. Neuropsychol Rev 2007;17:347–361.
84.Mackie K. Distribution of cannabinoid receptors in the central and peripheral nervous system. Handb Exp Pharmacol 2005;(168): 299–325.
85.Ilan AB, Gevins A, Coleman M, et al. Neurophysiological and subjective profile of marijuana with varying concentrations of cannabinoids. Behav Pharmacol 2005;16:487–496.
86.Huestis MA, Boyd SJ, Heishman SJ, et al. Single and multiple doses of rimonabant antagonize acute effects of smoked cannabis in male cannabis users. Psychopharmacology (Berl)2007;194:505–515.
87.Grotenhermen F. The toxicology of cannabis and cannabis prohibition. Chem Biodivers 2007;4:1744–1769.
88.Ranganathan M, D’Souza DC. The acute effects of cannabinoids on memory in humans: a review. Psychopharmacology (Berl) 2006;188(4):425–444.
89.Papafotiou K, Carter JD, Stough C. The relationship between performance on the standardised field sobriety tests, driving performance and the level of Delta9-tetrahydrocannabinol (THC) in blood. Forensic Sci Int 2005;155:172–178.
90.Johns A. Psychiatric effects of cannabis. Br J Psychiatry 2001;178: 116–122.
91.Kalant H. Adverse effects of cannabis on health: an update of the literature since 1996. Prog Neuropsychopharmacol Biol Psychiatry 2004;28:849–863.
92.Leweke FM, Gerth CW, Klosterkotter J. Cannabis-associated psychosis: current status of research. CNS Drugs 2004;18:895–910.
92a.Andre C, Jaber-Filho JA, Bento RM, et al. Delirium following ingestion of marijuana present in chocolate cookies. CNS Spectr 2006;11:262–264.
93.Nunez LA, Gurpegui M. Cannabis-induced psychosis: a cross-sectional comparison with acute schizophrenia. Acta Psychiatr Scand 2002;105:173–178.
94.Szuster RR, Pontius EB, Campos PE. Marijuana sensitivity and panic anxiety. J Clin Psychiatry 1988;49:427–429.
95.Favrat B, Menetrey A, Augsburger M, et al. Two cases of “cannabis acute psychosis” following the administration of oral cannabis. BMC Psychiatry 2005;5:17.
96.Bachs L, Morland H. Acute cardiovascular fatalities following cannabis use. Forensic Sci Int 2001;124:200–203.
97.Aryana A, Williams MA. Marijuana as a trigger of cardiovascular events: speculation or scientific certainty? Int J Cardiol 2007;118: 141–144.
98.Korantzopoulos P, Liu T, Papaioannides D, et al. Atrial fibrillation and marijuana smoking. Int J Clin Pract 2008;62:308–313.
99.Perez JA, Jr. Allergic reaction associated with intravenous marijuana use. J Emerg Med 2000;18:260–261.
100.Le Foll B, Gorelick DA, Goldberg SR. The future of endocannabinoid-oriented clinical research after CB1 antagonists. Psychopharmacology (Berl) 2009;205(1):171–174.
101.Budney AJ, Hughes JR. The cannabis withdrawal syndrome. Curr Opin Psychiatry 2006;19:233–238.
102.Vandrey R, Smith MT, McCann UD, et al. Sleep disturbance and the effects of extended-release zolpidem during cannabis withdrawal. Drug Alcohol Depend 2011;117(1):38–44.
103.Vandrey RG, Budney AJ, Hughes JR, et al. A within-subject comparison of withdrawal symptoms during abstinence from cannabis, tobacco, and both substances. Drug Alcohol Depend2008:92, 48–54.
104.Allsop DJ, Norberg MM, Copeland J, et al. The Cannabis Withdrawal Scale development: patterns and predictors of cannabis withdrawal and distress. Drug Alcohol Depend2011;119(1–2):123–129.
105.Levin KH, Copersino ML, Heishman SJ, et al. Cannabis withdrawal symptoms in non-treatment-seeking adult cannabis smokers. Drug Alcohol Depend 2010;111(1–2):120–127.
106.Mason BJ, Crean R, Goodell V, et al. A proof-of-concept randomized controlled study of gabapentin: effects on cannabis use, withdrawal and executive function deficits in cannabis-dependent adults. Neuropsychopharmacology 2012;37(7):1689–1698.
107.Britt GC, McCance-Katz EF. A brief overview of the clinical pharmacology of “club drugs.” Subst Use Misuse 2005;40:1189–1201.
108.Rome ES. It’s a rave new world: rave culture and illicit drug use in the young. Cleve Clin J Med 2001;68(6):541–550.
109.Williams JF, Kokotailo PK. Abuse of proprietary (over-the-counter) drugs. Adolesc Med Clin 2006;7:733–750.
110.Development of PCP, Center for Substance Abuse Research, University of Maryland, College Park, 2006. http://www.cesar.umd.edu/cesar/drugs/pcp.pdf. Retrieved June 4, 2013.
111.Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings). CNS Neurosci Ther 2013;19:370–380.
112.Expert Committee on Drug Dependence, World Health Organization. Dextromethorphan Pre-Review Report, 2012. http://www.who.int/medicines/areas/quality_safety/5.1Dextromethorphan_pre-review.pdf. Retrieved June 4, 2013.
113.aan het Rot M, Collins KA, Murrough JW, et al. Safety and efficacy of repeated dose intravenous ketamine for treatment-resistant depression. Biol Psychiatry 2010;67:139–145.
114.Diazgranados N, Ibrahim L, Brutsche NE, et al. A randomized add-on trial of an N-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry2010;67:793–802.
115.Persson J. Ketamine in pain management. CNS Neurosci Ther 2013;19:396–402.
116.DEA Controlled Substances. May 28, 2013. http://www.deadiversion.usdoj.gov/schedules/orangebook/c_cs_alpha.pdf. Retrieved June 4, 2013.
117.World Health Organization Expert Committee on Drug Dependence. Dextromethorphan: pre-review report. Thirty-fifth Meeting Hammamet, Tunisia, 4–8 June 2012. http://www.who.int/medicines/areas/quality_safety/5.1Dextromethorphan_pre-review.pdf. Retrieved June 19, 2013.
118.Gorelick DA, Balster RL. Phencyclidine (PCP). In: Bloom FE, Kupfer DJ, eds. Psychopharmacology: the fourth generation of progress. New York: Raven Press, 1995:1767–1776.
119.Baldridge EB, Bessen HA. Phencyclidine. Emerg Med Clin North Am 1990;8:541–550.
120.Milhorn TH. Diagnosis and management of phencyclidine intoxication. Am Fam Physician 1991;43(4):1293–1302.
121.Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine. Arch Gen Psychiatry 2000;57:270–276.
122.Gorelick D, Wilkins J, Wong C. Diagnosis and treatment of chronic phencyclidine abuse. NIDA Research Monograph 64. Rockville, MD: National Institute on Drug Abuse, 1986:218–228.
123.Giannini AJ, Underwood NA, Condon M. Acute ketamine intoxication treated by haloperidol: a preliminary study. Am J Ther 2000;7:389–391.
124.Carls KA, Ruehter VL. An evaluation of phencyclidine (PCP) psychosis: a retrospective analysis at a state facility. Am J Drug Alcohol Abuse 2006;32:673–678.
125.Weiss RD, Greenfield SF, Mirin SM. Intoxication and withdrawal syndromes. In: Hyman SE, ed. Manual of psychiatric emergencies. Boston, MA: Little, Brown & Co., 1994:279–293.
126.Giannini AJ, Loiselle RH, DiMarzio LR, et al. Augmentation of haloperidol by ascorbic acid in phencyclidine intoxication. Am J Psychiatry 1987;144:1207–1209.
127.Thirthalli J, Benegal V. Psychosis among substance users. Curr Opin Psychiatry 2006;19:239–245.
128.Chyka PA, Erdman AR, Manoguerra AS, et al. Dextromethorphan poisoning: an evidence-based consensus guideline for out-of-hospital management. Clin Toxicol (Phila)2007;45(6):662–677.
129.American Psychiatric Association. Diagnostic and statistical manual of mental disorders (4th ed., Text Revision). Washington, DC: Author, 2000.
130.Tennant FS, Rawson RA, McCann M. Withdrawal from chronic phencyclidine dependence with desipramine. Am J Psychiatry 1981;138:845–847.
131.Giannini AJ, Loiselle RH, Graham BH. Behavioral response to buspirone in cocaine and phencyclidine withdrawal. J Subst Abuse Treat 1993;10:523–527.
132.Morgan CJA, Monaghan L, Curran HV. Beyond the Khole: a 3-year longitudinal investigation of the cognitive and subjective effects of ketamine in recreational users who have substantially reduced their use of the drug. Addiction 2004;99:1450–1461.
133.Morgan CJA, Curran HV, Valerie C. Acute and chronic effects of ketamine upon human memory: a review. Psychopharmacology (Berl) 2006;188:408–424.
134.Morgan CJA, Muetzelfeldt L, Curran HV. Ketamine use, cognition and psychological wellbeing: a comparison of frequent, infrequent and ex-users with polydrug and non-using controls. Addiction 2009; 104:77–87.
135.Morgan CJA, Muetzelfeldt L, Curran HV. Consequences of chronic ketamine self-administration upon neurocognitive function and psychological wellbeing: a 1-year longitudinal study. Addiction2010;105:121–133.
136.Tang WK, Liang HJ, Lau CG, et al. Relationship between cognitive impairment and depressive symptoms in current ketamine users.J Stud Alcohol Drugs 2013;74:460–468.
137.Wolff K, Winstock AR. Ketamine: from medicine to misuse. CNS Drugs 2006;20:199–218.
138.Latvala A, Castaneda AE, Perälä J, et al. Cognitive functioning in substance abuse and dependence: a population-based study of young adults. Addiction 2009;104:1558–1568.
139.Morgan CJA, Curran HV, The Independent Scientific Committee on Drugs (ISCD). Ketamine use: a review. Addiction 2012; 107:27–38.
140.Liao Y, Tang J, Corlett PR. Reduced dorsal prefrontal gray matter after chronic ketamine use. Biol Psychiatry 2011;69:42–48.
141.Liao Y, Tang J, Ma M. Frontal white matter abnormalities following chronic ketamine use: A diffusion tensor imaging study. Brain 2010;133:2115–2122.
142.Howard MO, Bowen SE, Garland EL, et al. Inhalant use and inhalant use disorders in the United States. Addict Sci Clin Pract 2011;6(1):18–31.
143.Konghom S, Verachai V, Srisurapanont M, et al. Treatment for inhalant dependence and abuse. Cochrane Database Syst Rev 2010;(12):CD007537.
144.Pfeiffer H, Khaddam A, Brinkmann B, et al. Sudden death after isobutane sniffing: a report of two forensic cases. Int J Legal Med 2006;120:168–173.
145.Alper AT, Hasdemir H, Nurkalem Z, et al. Glue (toluene) abuse: increased QT dispersion and relation with unexplained syncope. Inhal Toxicol 2008;20:37–41.
146.Finch CK, Lobo BL. Acute inhalant-induced neurotoxicity with delayed recovery. Ann Pharmacother 2005;39:169–172.
147.Lubman DI, Yucel M, Lawrence AJ. Inhalant abuse among adolescents: neurobiological considerations. Br J Pharmacol 2008; 154:316–326.
148.Takagi M, Lubman DI, Yucel M. Solvent-induced leukoencephalopathy: a disorder of adolescence? Subst Use Misuse 2011;46:95–98.
149.Gautschi OP, Cadosch D, Zellweger R. Postural tremor induced by paint sniffing. Neurol India 2007;55:393–395.
150.Weintraub E, Gandhi D, Robinson C. Medical complications due to mothball abuse. South Med J 2000;93:427–429.
151.Williams JF, Storck M. Inhalant abuse. Pediatrics 2007;119: 1009–1017.
152.Ridenour TA, Bray BC, Cottler LB. Reliability of use, abuse, and dependence of four types of inhalants in adolescents and young adults. Drug Alcohol Depend 2007;91:40–49.
153.Perron BE, Howard MO, Vaughn MG, et al. Inhalant withdrawal as a clinically significant feature of inhalant dependence disorder. Med Hypotheses 2009;73:935–937.
154.Doyon S. The many faces of ecstasy. Curr Opin Pediatr 2001; 13:170–178.
155.Wu LT, Schlenger WE, Galvin DM. Concurrent use of methamphetamine, MDMA, LSD, ketamine, GHB, and flunitrazepam among American youths. Drug Alcohol Depend2006;84(1):102–113.
156.Baggott M, Heifets B, Jones R, et al. Chemical analysis of ecstasy pills. JAMA 2000;284(17):2190.
157.Oesterheld JR, Armstrong SC, Cozza KL. Ecstasy: pharmacodynamic and pharmacokinetic interactions. Psychosomatics 2004;45(1):84–87.
158.de la Torre R, Farre M, Ortuno J, et al. Non-linear pharmacokinetics of MDMA (’Ecstasy’) in humans. Br J Clin Pharmacol 2000;49:104–109.
159.Yang J, Jamei M, Heydari A, et al. Implications of mechanism-based inhibition of CYP2D6 for the pharmacokinetics and toxicity of MDMA. J Psychopharmacol 2006;20(6):842–849.
160.Henry JA, Jeffreys CJ, Dawling S. Toxicity and deaths from 3,4-methylenedioxymethamphetamine (“Ecstasy”). Lancet 1992;340: 384–387.
161.Monks TJ, Jones DC, Bai F, et al. The role of metabolism in 3,4-(+)-methylenedioxyamphetamine and 3,4-(+)-methylenedioxymethamphetamine (ecstasy) toxicity. Ther Drug Monit2004;26(2):132–136.
162.Burgess C, O’Donohoe A, Gill M. Agony and ecstasy: a review of MDMA effects and toxicity. Eur Psychiatry 2000;15:287–294.
163.El-Mallakh RS, Abraham HD. MDMA (Ecstasy). Ann Clin Psychiatry 2007;19:45–52.
164.Baylen CA, Rosenberg H. A review of the acute subjective effects of MDMA/ecstasy. Addiction 2006;101(7):933–947.
165.Vecellio M, Schopper C, Modestin J. Neuropsychiatric consequences (atypical psychosis and complex-partial seizures) of ecstasy use: possible evidence for toxicity-vulnerability predictors and implications for preventative and clinical care. J Psychopharmacol 2003;17(3):342–345.
166.Schifano F. A bitter pill. Overview of ecstasy (MDMA, MDA) related fatalities. Psychopharmacology (Berl) 2004;173(3–4): 242–248.
167.Green AR, O’shea E, Colado MI. A review of the mechanisms involved in the acute MDMA (ecstasy)-induced hyperthermic response. Eur J Pharmacol 2004;500(1–3):3–13.
168.Lai TI, Hwang JJ, Fang CC, et al. Methylene 3,4-dioxymethamphetamine-induced acute myocardial infarction. Ann Emerg Med 2003;42(6):759–762.
169.Cadet JL, Krasnova IN, Jayanthi S, et al. Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res 2007;11(3–4):183–202.
170.Halachanova V, Sansone RA, McDonald S. Delayed rhabdomyolysis after ecstasy use. Mayo Clin Proc 2001;76(1):112–113.
171.Crunelli V, Emri Z, Leresche N. Unravelling the brain targets of gamma-hydroxybutyric acid. Curr Opin Pharmacol 2006;6(1):44–52.
172.Snead OC III, Gibson KM. Gamma-hydroxybutyric acid. N Engl J Med 2005;352(26):2721–2732.
173.Karila L, Reynaud M. GHB and synthetic cathinones: clinical effects and potential consequences. Drug Test Anal 2011; 3(9):552–559.
174.Samokhvalov AV, Paton-Gay CL, Balchand K, et al. Phenibut dependence. BMJ Case Rep 2013;2013.
175.Shannon M, Quang LS. Gamma-hydroxybutyrate, gammabutyrolactone, and 1,4-butanediol: a case report and review of the literature. Pediatr Emerg Care 2000;16(6):435–440.
176.Ricaurte GA, McCann UD. Recognition and management of complications of new recreational drug use. Lancet 2005;365:2137–2145.
177.Abanades S. Gamma-hydroxybutyrate (GHB) in humans: pharmacodynamics and pharmacokinetics. Ann N Y Acad Sci 2006;1074:599.
178.Elliott SP. Gamma hydroxybutyric acid (GHB) concentrations in humans and factors affecting endogenous production. Forensic Sci Int 2003;133(1–2):9–16.
179.Caldicott D, Kuhn M. Gamma-hydroxybutyrate overdose and physostigmine: teaching new tricks to an old drug. Ann Emerg Med 2001;37(1):99–102.
180.Sumnall HR, Woolfall K, Edwards S, et al. Use, function, and subjective experiences of gamma-hydroxybutyrate (GHB). Drug Alcohol Depend 2008;92:286–290.
181.Miotto K, Darakjian J, Basch J, et al. Gamma-hydroxybutyric acid: patterns of use, effects and withdrawal. Am J Addict 2001;10(3):232–241.
182.Oliveto A, Gentry WB, Pruzinsky R, et al. Behavioral effects of gamma-hydroxybutyrate in humans. Behav Pharmacol 2010;21(4):332–342.
183.Beynon CM, McVeigh C, McVeigh J, et al. The involvement of drugs and alcohol in drug-facilitated sexual assault: a systematic review of the evidence. Trauma Violence Abuse2008;9(3):178–188.
184.Gonzalez A, Nutt DJ. Gamma hydroxybutyrate abuse and dependency. J Psychopharmacol 2005;19(2):195–204.
185.Galicia M, Nogue S, Miro O. Liquid ecstasy intoxication: clinical features of 505 consecutive emergency department patients. Emerg Med J 2011;28(6):462–466.
186.Mason PE, Kerns WP II. Gamma hydroxybutyric acid (GHB) intoxication. Acad Emerg Med 2002;9(7):730–739.
187.Zvosec D, Smith S, McCutcheon JR, et al. Adverse events, including death, associated with the use of 1,4-butanediol. N Engl J Med 2001;344(2):87–94.
188.Tarabar AF, Nelson LS. The gamma-hydroxybutyrate withdrawal syndrome. Toxicol Rev 2004;23(1):45–49.
189.Schep LJ. The clinical toxicology of γ-hydroxybutyrate, γ-butyrolactone and 1,4-butanediol. Clin Toxicol (Phila) 2012;50(6):458–470.
190.Evans R Gammabutyrolactone: withdrawal syndrome resembling delirium tremens. Jol Subst Use 2012;17(4):384–387.
191.McDonough M, Kennedy N, Glasper A, et al. Clinical features and management of gamma-hydroxybutyrate (GHB) withdrawal: a review. Drug Alcohol Depend 2004;75(1):3–9.
192.Wojtowicz JM. Withdrawal from gamma-hydroxybutyrate, 1,4-butanediol and gamma-butyrolactone: a case report and systematic review. CJEM 2008;10(1):69–74.
193.Bell J. Gamma-butyrolactone (GBL) dependence and withdrawal. Addiction 2011;106(2):442–447.
194.Zvosec DL. Case series of 226 γ-hydroxybutyrate-associated deaths: lethal toxicity and trauma. Am J Emerg Med 2011;29(3):319–332.
195.Richardson WH, Slone CM, Michels JE. Herbal drugs of abuse: an emerging problem. Emerg Med Clin North Am 2007;254:35–57.
196.Halpern JH. Hallucinogens and dissociative agents naturally growing in the United States. Pharmacol Ther 2004;102:131–138.
197.Dennehy CE, Tsourounis C, Miller AE. Evaluation of herbal dietary supplements marketed on the internet for recreational use. Ann Pharmacother 2005;39(10):1634–1639.
198.Yussman SM, Wilson KM, Klein JD. Herbal products and their association with substance use in adolescents. J Adolesc Health 2006;38(4):395–400.
199.Druid H, et al. Flunitrazepam: an evaluation of use, abuse and toxicity. Forensic Sci Int 2001;122(2–3):136–141.
200.Silins E, Copeland J, Dillon P. Qualitative review of serotonin syndrome, ecstasy (MDMA) and the use of other serotonergic substances: hierarchy of risk. Aust N Z J Psychiatry2007;41(8): 649–655.
201.Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005;352(11):1112–1120.
202.Christensen RC. Identifying serotonin syndrome in the emergency department. Am J Emerg Med 2005;23(3):406–408.
203.Dunkley EJ, Isbister GK, Sibbritt D, et al. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM 2003;96:635–642.
204.Perry PJ. Serotonin syndrome vs neuroleptic malignant syndrome: a contrast of causes, diagnoses, and management. Ann Clin Psychiatry 2012;24(2):155–162.
205.Gorelick DA, Wilkins JN. Special aspects of human alcohol withdrawal. In: Galanter M, ed. Recent developments in alcoholism, vol. 4. New York: Plenum Press, 1986:283–305.
206.Zullino DF, Khazaal Y, Hattenschwiler J, et al. Anticonvulsant drugs in the treatment of substance withdrawal. Drugs Today (Barc) 2004;40:603–619.
207.Kuschel C. Managing drug withdrawal in the newborn infant. Semin Fetal Neonatal Med 2007;12:127–133.
208.Oei J, Lui K. Management of the newborn infant affected by maternal opiates and other drugs of dependency. J Paediatr Child Health 2007;43:9–18.
209.Rayburn WF. Maternal and fetal effects from substance use. Clin Perinatol 2007;34:559–571.
210.Greene CM, Goodman MH. Neonatal abstinence syndrome: strategies for care of the drug-exposed infant. Neonatal Netw 2003;22(4):15–25.
211.Smith LM, Lagasse LL, Derauf C, et al. Prenatal methamphetamine use and neonatal neurobehavioral outcome. Neurotoxicol Teratol 2008;30(1):20–28.
212.Wagner CL, Katkaneni LD, Cox TH, et al. The impact of prenatal drug exposure on the neonate. Obstet Gynecol Clin North Am 1998;25(1):169–194.
213.Kandall SR. Treatment strategies for drug-exposed neonates. Clin Perinatol 1999;26(1):231–243.
214.Chomchai C, Na Manorom N, Watanarungsan P, et al. Methamphetamine abuse during pregnancy and its health impact on neonates born at Siriraj Hospital, Bangkok, Thailand. Southeast Asian J Trop Med Public Health 2004;35(1):228–231.
215.Simoni-Wastila L, Yang HK. Psychoactive drug abuse in older adults. Am J Geriatr Pharmacother 2006;4(4):380–394.
216.Kessler RC, Amminger GP, Gguilar-Gaxiola S, et al. Age of onset of mental disorders: a review of recent literature. Curr Opin Psychiatry 2007;20:359–364.
217.Schepis TS, Adinoff B, Rao U. Neurobiological processes in adolescent addictive disorders. Am J Addict 2008;17:6–23.
218.Stewart DG, Brown SA. Withdrawal and dependency symptoms among adolescent alcohol and drug abusers. Addiction 1995;90:627–635.
219.Bukstein OG, Bernet W, Arnold, V, et al. Practice parameter for the assessment and treatment of children and adolescents with substance use disorders. J Am Acad Child Adolesc Psychiatry 2005;44:609–621.
220.Fournier ME, Levy S. Recent trends in adolescent substance use, primary care screening, and updates in treatment options. Curr Opin Pediatr 2006;18:352–358.
221.Becker JB, Hu M. Sex differences in drug abuse. Front Neuroendocrinol 2008;29:36–47.