Autosomal Recessive
Principles
• Ethnic variation in mutation frequency
• Variable expressivity
• Tissue-specific expression of mutations
• Genetic modifiers
• Environmental modifiers
Major Phenotypic Features
• Age at onset: Neonatal to adulthood
• Progressive pulmonary disease
• Exocrine pancreatic insufficiency
• Obstructive azoospermia
• Elevated sweat chloride concentration
• Growth failure
• Meconium ileus
History and Physical Findings
J.B., a 2-year-old boy, was referred to the pediatric clinic for evaluation of poor growth. During infancy, J.B. had diarrhea and colic that resolved when an elemental formula was substituted for his standard formula. As table foods were added to his diet, he developed malodorous stools containing undigested food particles. During his second year, J.B. grew poorly, developed a chronic cough, and had frequent upper respiratory infections. No one else in the family had poor growth, feeding disorders, or pulmonary illnesses. On physical examination, J.B.'s weight and height plotted less than the 3rd percentile and his head circumference at the 10th percentile. He had a severe diaper rash, diffuse rhonchi, and mild clubbing of his digits. The physical examination was otherwise normal. After briefly discussing a few possible causes of J.B.'s illness, the pediatrician requested several tests, including a test for sweat chloride concentration by pilocarpine iontophoresis; the sweat chloride level was 75 mmol/L (normal, <40 mmol/L; indeterminate, 40 to 60 mmol/L), a level consistent with cystic fibrosis. On the basis of this result and the clinical course, the pediatrician diagnosed J.B.'s condition as cystic fibrosis. J.B. and his parents were referred to the cystic fibrosis clinic for further counseling, mutation testing, and treatment. (Note: J.B. was born in 2008 prior to implementation of cystic fibrosis newborn screening in his home state.)
Background
Disease Etiology and Incidence
Cystic fibrosis (CF, MIM 219700) is an autosomal recessive disorder of epithelial ion transport caused by mutations in the CF transmembrane conductance regulator gene (CFTR) (see Chapter 12). Although CF has been observed in all races, it is predominantly a disease of those of northern European ancestry. The live birth incidence of CF ranges from 1 in 313 among the Hutterites of southern Alberta, Canada, to 1 in 90,000 among the Asian population of Hawaii. Among all U.S. whites, the incidence is 1 in 3200.
Pathogenesis
CFTR is an anion channel that conducts chloride and bicarbonate. It is regulated by ATP and by phosphorylation by cAMP-dependent protein kinase. CFTR facilitates the maintenance of hydration of airway secretions through the transport of chloride and inhibition of sodium uptake (see Chapter 12). Dysfunction of CFTR can affect many different organs, particularly those that secrete mucus, including the upper and lower respiratory tracts, pancreas, biliary system, male genitalia, intestine, and sweat glands.
The dehydrated and viscous secretions in the lungs of patients with CF interfere with mucociliary clearance, inhibit the function of naturally occurring antimicrobial peptides, provide a medium for growth of pathogenic organisms, and obstruct airflow. Within the first months of life, these secretions and the bacteria colonizing them initiate an inflammatory reaction. The release of inflammatory cytokines, host antibacterial enzymes, and bacterial enzymes damages the bronchioles. Recurrent cycles of infection, inflammation, and tissue destruction decrease the amount of functional lung tissue and eventually lead to respiratory failure (Fig. C-12).
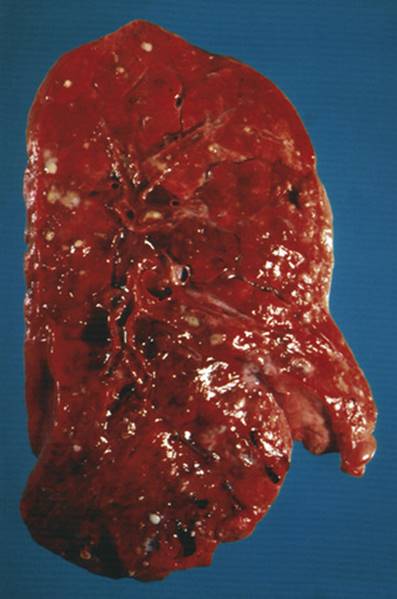
FIGURE C-12 A median cross section of a lung from a patient with cystic fibrosis (CF). Note the mucous plugs and purulent secretions within the airways. See Sources & Acknowledgments.
Loss of CFTR chloride transport into the pancreatic duct impairs the hydration of secretions and leads to the retention of exocrine enzymes in the pancreas. Damage from these retained enzymes eventually causes fibrosis of the pancreas.
CFTR also regulates the uptake of sodium and chloride from sweat as it moves through the sweat duct. In the absence of functional CFTR, the sweat has an increased sodium chloride content, and this is the basis of the historical “salty baby syndrome” and the diagnostic sweat chloride test.
Phenotype and Natural History
CF classically manifests in early childhood, although approximately 4% of patients are diagnosed in adulthood; 15% to 20% of patients present at birth with meconium ileus, and the remainder present with chronic respiratory complaints (rhinitis, sinusitis, obstructive lung disease) poor growth, or both later in life. The poor growth results from a combination of increased caloric expenditure because of chronic lung infections and malnutrition from pancreatic exocrine insufficiency. Five percent to 15% of patients with CF do not develop pancreatic insufficiency. More than 95% of male patients with CF are azoospermic because of congenital bilateral absence of the vas deferens. The progression of lung disease is the chief determinant of morbidity and mortality. Most patients die of respiratory failure and right ventricular failure secondary to the destruction of lung parenchyma and high pulmonary vascular resistance (cor pulmonale); the current average life expectancy is over 38 years in North America and many other regions of the world.
In addition to CF, mutations within CFTR have been associated with a spectrum of diseases, including obstructive azoospermia, idiopathic pancreatitis, disseminated bronchiectasis, allergic bronchopulmonary aspergillosis, atypical sinopulmonary disease, and asthma. Some of these disorders are associated with mutations within a single CFTR allele; others, like CF, are observed only when mutations are present in both CFTR alleles. A direct causative role for mutant CFTR alleles has been established for some but not all of these disorders.
A correlation between particular CFTR mutant alleles and disease severity exists only for pancreatic insufficiency. Secondary mutations or polymorphisms within a CFTR allele may alter the efficiency of splicing or protein maturation and thereby extend the spectrum of disease associated with some mutations. In addition, some mutations in CFTR cause disease manifestations only in certain tissues; for example, some mutations affecting the efficiency of splicing have a greater effect on wolffian duct derivatives than in other tissues because of a tissue-specific need for full-length transcript and protein. Environmental factors, such as exposure to cigarette smoke, markedly worsen the severity of lung disease among patients with CF.
Management
Because nearly 2000 different mutations and variants have been described across the CFTR gene, the diagnosis of CF is usually based on clinical criteria and sweat chloride concentration. Sweat chloride concentrations are normal in 1% to 2% of patients with CF; in these patients, however, an abnormal nasal transepithelial potential difference measurement is usually diagnostic of CF.
Currently there are no curative treatments of CF, although improved symptomatic management has increased the average longevity from early childhood to between 30 and 40 years. The objectives of medical therapy for CF are clearance of pulmonary secretions, control of pulmonary infection, pancreatic enzyme replacement, adequate nutrition, and prevention of intestinal obstruction. Although medical therapy slows the progression of pulmonary disease, the only effective treatment of respiratory failure in CF is lung transplantation. Pancreatic enzyme replacement and supplementation of fat-soluble vitamins treat the malabsorption effectively; because of increased caloric needs and anorexia, however, many patients also require caloric supplements. New drugs are being developed that either correct or enhance the function of CFTR proteins carrying certain mutations. Most patients also require extensive counseling to deal with the psychological effects of having a chronic fatal disease.
Newborn screening for CF has been implemented in all 50 U.S. states and in most Canadian provinces because detection in the newborn period, prevents the malnutrition seen in clinically undiagnosed pancreatic-insufficient patients. Long-term effects on survival and pulmonary disease progression are unclear.
Inheritance Risk
A couple's empirical risk for having a child affected with CF varies greatly, depending on the frequency of CF in their ethnic groups. For North Americans who do not have a family history of CF and are of northern European ancestry, the empirical risk for each to be a carrier is approximately 1 in 29, and such a couple's risk for having an affected child is therefore 1 in 3200. For couples who already have a child affected with CF, the risk for future children to have CF is 1 in 4. In 1997, a National Institutes of Health consensus conference recommended offering CF carrier testing to all pregnant women and couples considering a pregnancy in the United States. The American College of Obstetrics and Gynecology adopted those recommendations.
Prenatal diagnosis is based on identification of proven disease-causing CFTR mutations in DNA from fetal tissue, such as chorionic villi or amniocytes. Effective identification of affected fetuses usually requires that the mutations responsible for CF in a family have already been identified.
Questions for Small Group Discussion
1. Newborn screening for CF can be performed by testing immunoreactive trypsinogen (IRT) alone or by IRT followed by mutation screening. Discuss the risks and benefits of adding CFTR mutation screening to a newborn screening panel.
2. The most common CF mutation is ΔF508; it accounts for approximately 70% of all mutant CFTR alleles worldwide. For a couple of northern European origin, what is their risk for having an affected child if each tests negative for ΔF508? If one tests positive and the other tests negative for ΔF508?
3. What constitutes disease—a mutation in a gene or the phenotype caused by that mutation? Does detection of a mutation in the CFTR gene of patients with congenital bilateral absence of the vas deferens mean they have CF?
References
Barrett PM, Alagely A, Topol EJ. Cystic fibrosis in an era of genomically guided therapy. Hum Mol Genet. 2012;21(R1):R66–R71.
Boyle MP, de Boeck K. A new era in the treatment of cystic fibrosis: correction of the underlying CFTR defect. Lancet Respir Med. 2013;1:158–163.
Cystic Fibrosis Mutation Database. Available at: http://www.genet.sickkids.on.ca/cftr/.
Ferec C, Cutting GR. Assessing the disease-liability of mutations in CFTR. Cold Spring Harb Perspect Med. 2012;2:a009480.
Milla CE. Cystic fibrosis in the era of genomic medicine. Curr Opin Pediatr. 2013;25:323–328.
Tsui LC, Dorfman R. The cystic fibrosis gene: a molecular genetic perspective. Cold Spring Harb Perspect Med. 2013;3:a009472.