Multifactorial
Principles
• Polygenic disease
• Environmental trigger
• Susceptibility allele
• Protective allele
Major Phenotypic Features
• Age at onset: Childhood through adulthood
• Polyuria, polydipsia, polyphagia
• Hyperglycemia
• Ketosis
• Wasting
History and Physical Findings
F.C., a 45-year-old father with late-onset diabetes mellitus, was referred to the genetics clinic for counseling regarding his children's risk for diabetes. F.C. developed glucose intolerance (inability to maintain normal blood glucose levels after ingestion of sugar) at the age of 39 years and fasting hyperglycemia at 45 years. He did not have a history of other medical or surgical problems. The findings from his physical examination were normal except for moderate abdominal obesity; his body mass index [weight in kilograms/(height in meters)2] was 31.3, with the excess adiposity distributed preferentially around his waist. He had five children by two different partners; a child from each relationship had developed insulin-dependent (type 1) diabetes mellitus (IDDM) before 10 years of age. His sister developed IDDM as a child and died during adolescence from diabetic ketoacidosis. The geneticist explained that given his family history, F.C. might have a late-onset form of IDDM and that his current, non–insulin-dependent diabetes mellitus was probably an antecedent to the development of IDDM. After discussing the possible causes of and prognostic factors for the development of IDDM, F.C. elected to enroll himself and his children, who were all minors, in a research protocol studying the prevention of IDDM. As part of that study, he and his children were tested for anti-islet antibodies. Both he and an unaffected daughter had a high titer of anti-islet antibodies; the daughter also had an abnormal glucose tolerance test result but not fasting hyperglycemia. As part of the study protocol, F.C. and his daughter were prescribed low-dose insulin injections.
Background
Disease Etiology and Incidence
IDDM (now typically called type 1 diabetes, MIM 222100) is usually caused by autoimmune destruction of islet β cells in the pancreas; this autoimmune reaction is triggered by an unknown mechanism. The destruction of islet β cells causes insulin deficiency and thereby dysregulation of anabolism and catabolism, resulting in metabolic changes similar to those observed in starvation (Fig. C-26). Among North American white individuals, IDDM is the second most common chronic disease of childhood, increasing in prevalence from 1 in 2500 at 5 years of age to 1 in 300 at 18 years of age.
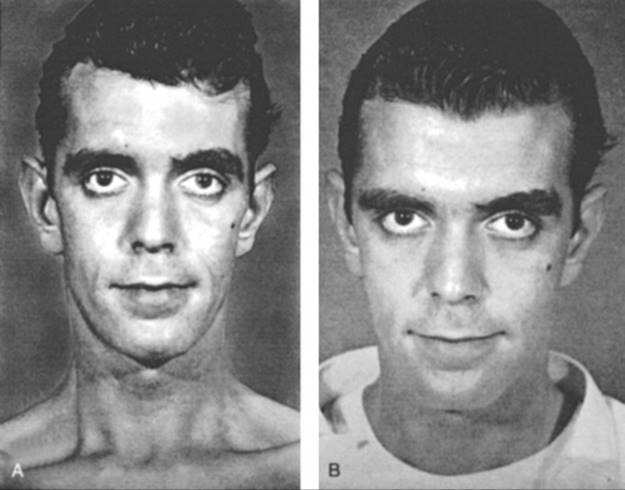
FIGURE C-26 A 28-year-old man with insulin-dependent diabetes mellitus. A, Photograph after 3 weeks of polydipsia and polyuria. B, Photograph after 5-kg weight gain with 10 days of insulin replacement. See Sources & Acknowledgments.
Pathogenesis
IDDM usually results from a genetic susceptibility and subsequent environmental insult (see Chapter 8) and only very rarely from an environmental insult or a genetic mutation alone. Although approximately 90% of IDDM occurs in patients without a family history of diabetes, observations supporting a genetic predisposition include differences in concordance between monozygotic (33% to 50%) and dizygotic twins (1% to 14%), familial clustering, and differences in prevalence among different populations (see Chapter 8). More than a dozen different genetic susceptibility loci have been reported in humans, although few have been identified consistently and reproducibly. One of the few confirmed loci is the HLA locus that may account for as much as 30% to 60% of the genetic susceptibility. Approximately 95% of white patients express a DR3 or DR4 allele, or both, compared with 50% of controls; this association apparently arises not because DR3 and DR4 are susceptibility alleles, but because of linkage disequilibrium between DR and DQ. The DQβ1*0201 allele, which segregates with DR3, and DQβ1*0302, which segregates with DR4, appear to be the primary susceptibility alleles. In contrast, DQβ1*602, which segregates with DR2, appears to be a protective allele; that is, it negates the effect of a susceptibility allele when both are present. Both of the DQβ1 susceptibility alleles have a neutral amino acid at position 57, a site within the putative antigen-binding cleft, whereas protective or neutral DQβ1 alleles have an aspartic acid at position 57. This substitution of an uncharged amino acid for aspartic acid is predicted to change the specificity of antigen binding to the DQ molecule.
Evidence supporting an environmental component to the induction of IDDM in genetically susceptible individuals includes a concordance of less than 50% among monozygotic twins, a seasonal variation in incidence, and an increased incidence of diabetes among children with congenital rubella. Proposed environmental triggers include viral infections and early exposure to bovine albumin. Exposure to viruses and bovine albumin could cause autoimmune destruction of β cells by molecular mimicry, that is, sharing of antigenic determinants between β-cell proteins and the virus or bovine albumin. Approximately 80% to 90% of newly diagnosed patients with IDDM have anti-islet cell antibodies. These autoantibodies recognize cytoplasmic and cell surface determinants such as glutamic acid decarboxylase, carboxypeptidase H, ganglioside antigens, islet cell antigen 69 (ICA69), and a protein tyrosine phosphatase. Glutamic acid decarboxylase and ICA69, respectively, share epitopes with coxsackievirus B4 and bovine serum albumin.
Thus, IDDM appears to be an autoimmune disease, although the precise role of islet cell autoantibodies remains uncertain. Additional evidence for an autoimmune mechanism in IDDM includes an increased prevalence of other autoimmune diseases, mononuclear cell infiltrates of islets, and recurrent β-cell destruction after transplantation from a monozygotic twin. However, two lines of evidence suggest that progression to IDDM involves more than the development of autoantibodies. First, less than 1% of the general population develops diabetes although 10% have islet autoantibodies; and second, first-degree relatives and schoolchildren have remission rates of 10% to 78% for islet cell antibodies.
Phenotype and Natural History
Loss of insulin reserve occurs during a few to many years. The earliest sign of abnormality is the development of islet autoantibodies when blood glucose concentrations, glucose tolerance (ability to maintain normal blood glucose levels after ingestion of sugar), and insulin responses to glucose are normal. This period is followed by a phase of decreased glucose tolerance but normal fasting blood glucose concentration. With continued loss of β cells, fasting hyperglycemia eventually develops, but sufficient insulin is still produced to prevent ketosis; during this period, patients have non–insulin-dependent diabetes mellitus. Eventually, insulin production falls below a critical threshold, and patients become dependent on exogenous insulin supplements and have a propensity to ketoacidosis. Younger patients generally progress through these phases more rapidly than do older patients.
Although the acute complications of diabetes can be controlled by administration of exogenous insulin, the loss of endogenous insulin production causes many problems, including atherosclerosis, peripheral neuropathy, renal disease, cataracts, and retinopathy. Approximately 50% of patients eventually develop renal failure. The occurrence and severity of these complications are related to the genetic background and degree of metabolic control. Rigorous control of blood glucose levels reduces the risk for complications by 35% to 75%.
Management
Although pancreatic or islet transplantation can cure IDDM, the paucity of tissue for transplantation and complications of immunosuppression limit this therapy. Management of most patients emphasizes intensive control of blood glucose levels by injection of exogenous insulin.
The development of islet autoantibodies several years before the onset of IDDM has led to the development of studies to predict and prevent IDDM. The administration of insulin or nicotinamide appears to delay the development of IDDM in some patients.
Inheritance Risk
The risk for IDDM in the general population is approximately 1 in 300. With one affected sibling, the risk increases to 1 in 14 (1 in 6 if HLA identical, 1 in 20 if HLA haploidentical). The risk increases to 1 in 6 with a second affected first-degree relative in addition to an affected sibling and to 1 in 3 with an affected monozygotic twin. Children of an affected mother have a 1 in 50 to 1 in 33 risk for development of IDDM, whereas children of an affected father have a 1 in 25 to 1 in 16 risk. This paternity-related increased risk appears to be limited to fathers with an HLA DR4 allele.
Questions for Small Group Discussion
1. Discuss the difficulties of identifying the genetic components of polygenic diseases.
2. How might HLA susceptibility alleles affect susceptibility and protective alleles affect protection?
3. Discuss the underlying mechanisms for prevention of IDDM by exogenous insulin injections.
4. Compare risk counseling for fathers and mothers with IDDM. Discuss the teratogenic risks and mechanisms of maternal diabetes.
References
Alemzadeh R, Ali O. Diabetes mellitus. Kliegman RM, Stanton BF, St. Geme JW, et al. Nelson textbook of pediatrics. ed 19. WB Saunders: Philadelphia; 2011:1968–1997.
Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. 2010;464:1293–1300.
Chiang JL, Kirkman MS, Laffel LM, et al. Type 1 diabetes through the life span: a position statement of the American Diabetes Association. Diabetes Care. 2014;37:2034–2054.