Elaine M. Kaptein
Thyrotoxicosis, particularly severe thyrotoxicosis, is associated with multiple alterations in cardiovascular function, renal hemodynamics, and renal tubular reabsorptive function. Renal function is altered due to a combination of direct and indirect effects of thyroid hormone excess on the kidney (Fig. 33.1). The indirect effects include changes in cardiovascular hemodynamics and the renin–angiotensin–aldosterone system. Reversible alterations in sodium and water metabolism, calcium, phosphate, vitamin D, and uric acid homeostasis also occur.
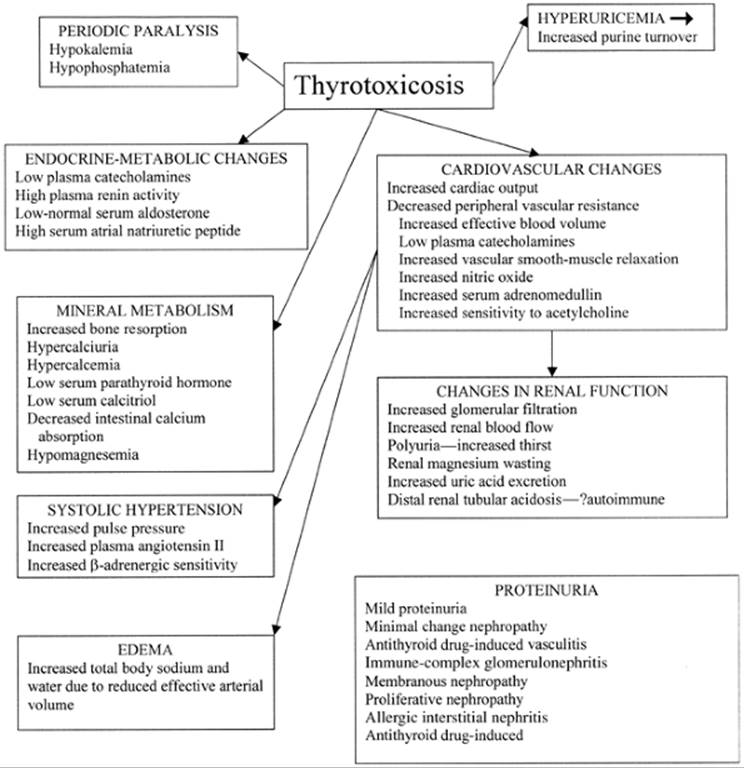
FIGURE 33.1. Diagram of the cardiovascular and other changes affecting renal function in patients with thyrotoxicosis.
HEMODYNAMIC CHANGES
Thyrotoxicosis results in a hyperkinetic circulation with increased heart rate and cardiac output, decreased peripheral vascular resistance, and increased blood flow to most organs, including the kidneys (see Chapter 31) (1). The decrease in peripheral vascular resistance may be due to relaxation of vascular smooth muscle cells, increased β-adrenergic receptor activity, and an increase in effective blood volume (2) (Fig. 33.1).
Nitric oxide is an important factor in regulating vascular tone, renal sodium excretion, and the renal pressure diuresis-natriuresis response, and therefore arterial blood pressure (3). Nitric oxide synthase activity is increased in the left ventricle, aorta and vena cava, and renal cortex and medulla of thyrotoxic rats (3). In thyrotoxic humans, decreased vascular resistance is largely due to increased endothelial production of nitric oxide and increased vascular sensitivity to acetylcholine, the endothelial-dependent vasodilator (4). Furthermore, the vasoconstrictor response to norepinephrine is increased, whereas plasma catecholamine concentrations are low (see Chapter 38) (4). These vascular abnormalities are corrected by correction of thyrotoxicosis (4). In addition, serum concentrations of the endothelial vasodilatory hormone adrenomedullin are high in patients with thyrotoxicosis (2). Thus, an increase in basal endothelial cell function and endothelial hyperresponsiveness to vasodilating stimuli provide an explanation for the decrease in peripheral vascular resistance and maintenance of a hyperkinetic state in patients with thyrotoxicosis (4). Activation of the renin–angiotensin–aldosterone system may play a causative role in development of cardiac hypertrophy in thyrotoxicosis (5,6).
HYPERTENSION
Systolic hypertension occurs in up to one third of patients with thyrotoxicosis (7). In one study, the mean systolic blood pressure was 10 mm Hg higher and diastolic blood pressure was 5 mm Hg lower during thyrotoxicosis than after euthyroidism was attained (1). Systolic blood pressure correlated with cardiac output, whereas diastolic blood pressure was related to systemic vascular resistance (1).
Thyrotoxicosis increases nitric oxide synthase activity, which as noted above regulates vascular tone, renal sodium excretion, and blood pressure (6). Thyrotoxicosis also results in activation of the renin–angiotensin system, the effects of which are blocked by angiotensin-converting enzyme inhibitors (6). In normal subjects, there is a functional balance between angiotensin II and nitric oxide production. In thyrotoxic rats, thyroxine (T4) administration increased blood pressure and plasma concentrations of angiotensin II and nitrates/nitrites (6). Simultaneous administration of T4 and subpressor doses of an inhibitor of nitric oxide synthesis caused a marked increase in blood pressure, which was attenuated by losartan, an angiotensin receptor–blocking drug (6). Plasma angiotensin II and nitric oxide concentrations were increased by T4, whereas both were reduced in the rats given T4 and the nitric oxide inhibitor (6). Impaired synthesis of nitric oxide results in increased sensitivity to the pressor effect of T4, and the latter is attenuated by losartan. Thus, increased nitric oxide synthase activity may play a protective homeostatic role in ameliorating the prohypertensive effects of T4 in thyrotoxicosis, as well as antagonizing the pressor actions of angiotensin II (6).
RENAL GLOMERULAR AND TUBULAR FUNCTION
In humans with thyrotoxicosis and animals treated with excess T4, renal plasma flow and glomerular filtration rate are increased, probably because of the increase in cardiac output and decrease in peripheral resistance (8,9). Intrarenal vasodilatation also occurs (8). The renal content of the endogenous vasoconstrictor substance endothelin is lower in thyrotoxic rats, which may contribute to renal vasodilatation (10).
The glomerular filtration rate, as measured by inulin clearance, and renal blood flow, as measured by paraaminohippurate clearance, are increased by 12% to 20% in patients with thyrotoxicosis and normalize during antithyroid treatment (9,11) (Fig. 33.1). Serum creatinine concentrations tend to be low in patients with thyrotoxicosis and increase during antithyroid treatment (11). Normal subjects given large doses of thyroid hormone have a decrease in serum creatinine concentrations before substantial muscle wasting occurs. Mean 24-hour urine creatinine excretion is significantly lower in patients with thyrotoxicosis due to loss of muscle mass, and it occurs despite an increase in the renal tubular secretion of creatinine (12,13,14).
Thyrotoxicosis induces both hypertrophy and hyperplasia of renal tubular epithelial cells. The increases are proportional to the increases in glomerular filtration rate and renal plasma flow. Intrarenal renin and angiotensin production are increased, as a result of increased renal renin messenger RNA expression, which leads to an increase in plasma renin activity and plasma angiotensin II concentrations and renal hypertrophy (15). The renal hypertrophy can be prevented by the angiotensin II antagonist losartan, indicating that it is dependent on intrarenal renin–angiotensin activation (15). Thyroid hormone also stimulates renal production of epidermal growth factor and renal growth in young animals (16), independent of protein intake and pituitary activity (8).
The functional and morphologic changes in renal tubules that occur in thyrotoxicosis are accompanied by an increase in renal tubular capacity for active transport. For example, transepithelial voltage and Na+,K+-adenosine triphosphatase (ATPase) activity are increased, with consequent increases in sodium transport. T4 also stimulates sodium-dependent phosphate transport in cultured renal tubular cells (17), without changing sodium-dependent transport of sulfate, glucose, or proline (18,19). In thyrotoxic rats, proximal tubular sodium-proton exchange is increased (19); in particular, the predominant isoform (NHE3) of the exchanger in proximal tubules is increased (20). These changes are not associated with clinically important increases in reabsorption of phosphate or bicarbonate.
WATER AND ELECTROLYTE METABOLISM
Patients with thyrotoxicosis rarely have discernible abnormalities in water metabolism. Serum electrolyte concentrations are usually normal. Occasional patients have polydipsia and polyuria, with 24-hour urine volumes as high as 3 or 4 L (21), associated at times with slightly hypotonic plasma. Some thyrotoxic patients have mild impairment in urinary concentrating ability (22), but this is not clinically important (23), and release of vasopressin in response to osmotic stimuli is normal (24). The polyuria is due to increased thirst, an explanation supported by observations in thyrotoxic animals (25,26). In patients with thyrotoxicosis, the sensation of thirst is initiated at a lower serum osmolality than when they are euthyroid (24). This increase in thirst may be secondary to the thirst-stimulating effect of high plasma angiotensin II concentrations (27). The increased thirst (polydipsia) and polyuria revert to normal after treatment of thyrotoxicosis.
Patients with thyrotoxicosis have high plasma renin activity and serum atrial natriuretic peptide concentrations (2,28,29,30). A higher cardiac preload in thyrotoxicosis may initiate secretion of atrial natriuretic peptide (2). The increase in plasma renin activity and serum atrial natriuretic peptide concentrations may also be due to a direct effect of T4 on gene expression (2). The high serum atrial natriuretic peptide concentrations may contribute to the decrease in peripheral vascular resistance in thyrotoxicosis (2).
Thyrotoxic patients have normal or low serum aldosterone concentrations, particularly in relation to their plasma renin activity (2,30). Urinary excretion of sodium and potassium is normal when patients consume usual amounts of those ions. Whereas total body exchangeable potassium is decreased in thyrotoxicosis, as a result of a decrease in total muscle mass (31), total exchangeable sodium is often increased. Thyrotoxicosis in rats reduces sodium retention (6), which may be caused by increased renal perfusion and perhaps high serum atrial natriuretic peptide concentrations.
EDEMA
Thyrotoxic patients may have pitting edema involving the ankles, legs, and sacrum (32). Periorbital edema is usually a manifestation of Graves' ophthalmopathy rather than of thyrotoxicosis. Edema in thyrotoxic patients results from renal salt and water retention in response to a reduction in effective arterial volume, and the sodium retention contributes to an increase in blood volume and venous pressure. The edema that develops under these circumstances does not imply the presence of congestive heart failure. In support of this concept, exercise testing in thyrotoxic patients with edema usually results in a substantial increase in cardiac output with little or no increase in right atrial or pulmonary artery pressure. If the additional circulatory load imposed by high cardiac output overwhelms myocardial reserve, or if myocardial function is impaired by organic heart disease or by the thyrotoxicosis itself, congestive failure ensues (see Chapter 31). Severe thyrotoxicosis also may be associated with protein-calorie malnutrition and hypoalbuminemia, an additional cause of plasma volume expansion and edema.
MINERAL METABOLISM
Thyrotoxicosis causes increased bone resorption, as evidenced by low bone density and an increase in markers of bone resorption, such as urinary excretion of hydroxyproline, pyridinoline and deoxypyridinoline cross-links, and N-telopeptide of type I collagen (33). There is a compensatory increase in bone formation, as evidenced by increased serum osteocalcin and bone-specific alkaline phosphatase concentrations (see Chapter 40) (33). The increase in bone resorption is greater than that of bone formation (33); the result is osteopenia, and in some patients osteoporosis. These changes in mineral metabolism result in an increase in serum calcium concentrations and occasionally in hypercalcemia. The increase in serum calcium concentrations inhibits parathyroid hormone secretion, resulting in hypercalciuria and decreased renal 1α-hydroxylation of 25-hydroxyvitamin D, and therefore decreased intestinal calcium absorption. Occasional patients have renal calculi, nephrocalcinosis, and reversible renal insufficiency (34).
All these changes, including osteoporosis, are reversible with treatment of thyrotoxicosis.
Thyrotoxicosis may result in magnesium deficiency, with decreased serum total and ionized magnesium concentrations, due to renal magnesium wasting (33). Low serum magnesium concentrations may contribute to the low serum parathyroid hormone concentrations in thyrotoxicosis.
URIC ACID METABOLISM
Patients with long-standing thyrotoxicosis may have high serum uric acid concentrations (11,35). In one study, 28% of patients with toxic nodular goiter and 39% of those with thyrotoxicosis caused by Graves' disease had hyperuricemia, as compared with 2% to 10% of normal subjects (11). Renal clearance of uric acid was increased in proportion to the increase in glomerular filtration rate, and therefore the fractional excretion of uric acid was normal (11). The occurrence of high serum uric acid concentrations in the presence of increased urinary clearance of uric acid indicates that production of uric acid is increased more than is renal uric acid clearance (11). The increase in uric acid production is most likely due to increased purine turnover. There are no reports of an increase in gout in thyrotoxic patients.
THYROTOXIC PERIODIC PARALYSIS
Thyrotoxic periodic paralysis is characterized by localized or generalized attacks of muscle weakness, even flaccid paralysis, that last for a few hours to several days. Most patients are men, with the male to female ratio being 20:1; Asian men are particularly susceptible to the disorder, but it has been described in many ethnic/racial groups (36,37,38,39). The episodes of muscle weakness are associated with a decrease in serum potassium concentrations, although the concentration is not always subnormal (40), and may be accompanied by hypophosphatemia and hypomagnesemia (41). Hypophosphatemia may be caused by cellular uptake of phosphate in conjunction with the cellular uptake of potassium. Total body potassium content is normal; the hypokalemia results from the shift of potassium into cells, and is associated with low urinary potassium excretion and a low transtubular potassium concentration gradient (41,42).
Thyrotoxicosis increases Na+, K+-ATPase activity in kidney, liver, and other tissues, and the activity of the enzyme increases in response to adrenergic stimulation (43,44,45). An increase in activity of this enzyme could explain the shift in potassium, but its periodic activation is unexplained. Na+,K+-ATPase-independent activation of potassium uptake may be more important (46). The epidemiology, clinical manifestations, and treatment of this unusual manifestation of thyrotoxicosis are discussed in more detail in Chapter 41.
RENAL TUBULAR ACIDOSIS
Occasional patients with thyrotoxicosis have renal tubular acidosis, which has been characterized as distal because there is failure to achieve maximal urinary acidification. Renal bicarbonate wasting (proximal renal tubular acidosis) has not been described; in fact, expression of the proximal tubular sodium proton exchanger is increased in patients with thyrotoxicosis (20,47). Hypokalemia, a common feature of distal renal tubular acidosis, may exacerbate a tendency to periodic paralysis. Renal tubular acidosis rarely results from hypercalcemia and hypercalciuria, which can cause nephrocalcinosis, tubular damage, and impairment of urinary acidification (34,48). Renal tubular acidosis also may occur in association with thyrotoxicosis caused by Graves' disease in the absence of nephrocalcinosis, and may persist after resolution of the thyrotoxicosis (49). The renal tubular acidosis in these patients may have an autoimmune basis; antibodies to renal tubular cells were demonstrated in the serum of one patient with Graves' thyrotoxicosis and renal tubular acidosis (50). Urine pH, serum bicarbonate concentrations, and urine calcium excretion should be measured in the occasional thyrotoxic patient who has nephrolithiasis (49). Some patients with autoimmune thyroid disease and renal tubular acidosis also have Sj□gren's syndrome, and this syndrome by itself may be associated with renal tubular acidosis (51).
PROTEINURIA AND IMMUNE COMPLEX GLOMERULONEPHRITIS
Some patients with autoimmune thyroid disease (Graves' disease or chronic autoimmune thyroiditis) have proteinuria, which is usually mild; it was found in 36% of patients in one study, most of whom were euthyroid (52,53) (Fig. 33.1). Administration of T4 induces proteinuria in rats, which is unaffected by partial blockade of nitric oxide synthase or losartan. Thus, proteinuria may be the result of a direct effect of T4 excess on glomerular permeability (6). Minimal change nephropathy has been reported in patients with autoimmune thyroid disease, including one patient who had multiple simultaneous relapses of Graves' thyrotoxicosis and the nephrotic syndrome (53,54).
Immune complex glomerulonephritis has been described in a few patients with thyrotoxicosis, with immune complexes containing thyroglobulin and antithyroglobulin antibodies implicated as a cause of the renal disease (55,56,57). The most common renal lesion was membranous glomerulonephritis, although proliferative changes also have been described (55,56,57,58). Similar patterns of immune complex deposition have been reported in patients with chronic autoimmune thyroiditis and hypothyroidism (59). The relative rarity of this entity contrasts with the frequency of circulating immune complexes in thyroid disease, estimated to be as high as 17% in thyrotoxicosis and higher in chronic autoimmune thyroiditis (60).
Proteinuria and even the nephrotic syndrome have been reported in rare patients with Graves' thyrotoxicosis after treatment with radioiodine (52,61). Biopsy findings have included membranous nephropathy, and immunofluorescence microscopy in several patients revealed thyroglobulin and thyroid peroxidase in the glomerular basement membrane and the mesangium (61). These changes could occur as a result of autoimmunization caused by radiation-induced release of thyroid antigens, but more likely are simply related to the autoimmune thyroid disease.
RENAL COMPLICATIONS OF ANTITHYROID DRUG THERAPY
Renal complications of antithyroid drug therapy are rare. Proteinuria has been reported in patients taking propylthiouracil or, rarely, methimazole, usually in patients with a drug-induced vasculitis (62,63,64) or lupuslike reaction (65). The full syndrome of lupus erythematosus with diffuse proliferative lupus nephritis was reported in one patient treated with propylthiouracil (66). Antineutrophil cytoplasmic antibody (ANCA)–positive vasculitis is an uncommon but well-recognized complication of therapy with propylthiouracil (see Chapter 45). Both C-ANCA (cytoplasmic) and P-ANCA (perinuclear) related disease has been described. Nephritis with or without systemic involvement is present in about two thirds of ANCA-positive patients (67,68,69,70). The duration of propylthiouracil therapy in these patients before onset of nephritis varied from weeks to years, more often the latter. Cessation of therapy may result in remission of the renal disease, but because of persistent renal disease, some patients have been treated with a glucocorticoid or cyclophosphamide, and a few patients have had permanent renal disease (70).
Two patients with thyrotoxicosis who developed severe allergic interstitial nephritis causing acute renal failure soon after initiation of propylthiouracil therapy have been reported. Both patients had a generalized rash, eosinophilia, and fever; both required hemodialysis and improved with glucocorticoid therapy (71,72). Another patient with amiodarone-induced thyrotoxicosis developed acute renal failure due to acute interstitial nephritis and fatal Stevens-Johnson syndrome while receiving propylthiouracil (73).
REFERENCES
1. Marcisz C, Jonderko G, Kucharz E. Changes of arterial pressure in patients with hyperthyroidism during therapy. Med Sci Monit 2002;8:CR502.
2. Diekman MJ, Harms MP, Endert E, et al. Endocrine factors related to changes in total peripheral vascular resistance after treatment of thyrotoxic and hypothyroid patients. Eur J Endocrinol 2001;144:339.
3. Quesada A, Sainz J, Wangensteen R, et al. Nitric oxide synthase activity in hyperthyroid and hypothyroid rats. Eur J Endocrinol 2002;147:117.
4. Napoli R, Biondi B, Guardasole V, et al. Impact of hyperthyroidism and its correction on vascular reactivity in humans. Circulation 2001;104:3076.
5. Basset A, Blanc J, Messas E, et al. Renin-angiotensin system contribution to cardiac hypertrophy in experimental hyperthyroidism: an echocardiographic study. J Cardiovasc Pharmacol 2001;37:163.
6. Rodriguez-Gomez I, Sainz J, Wangensteen R, et al. Increased pressor sensitivity to chronic nitric oxide deficiency in hyperthyroid rats. Hypertension 2003;42:220.
7. Marcisz C, Jonderko G, Kucharz EJ. Influence of short-time application of a low sodium diet on blood pressure in patients with hyperthyroidism or hypothyroidism during therapy. Am J Hypertens 2001;14:995.
8. Bradley SE, Stephan F, Coelho JB, et al. The thyroid and the kidney. Kidney Int 1974;6:346.
9. Hlad CJ, Bricker NS. Renal function and I131 clearance in hyperthyroidism and myxedema. J Clin Endocrinol Metab 1954;14: 1539.
10. Singh G, Sharma AC, Thompson EB, et al. Renal endothelin mechanism in altered thyroid states. Life Sci 1994;54:1901.
11. Sato A, Shirota T, Shinoda T, et al. Hyperuricemia in patients with hyperthyroidism due to Graves' disease. Metabolism 1995;44: 207.
12. Shirota T, Shinoda T, Yamada T, et al. Alteration of renal function in hyperthyroidism: increased tubular secretion of creatinine and decreased distal tubular delivery of chloride. Metabolism 1992;41:402.
13. Adlerberth A, Angeras U, Jagenburg R, et al. Urinary excretion of 3-methylhistidine and creatinine and serum concentrations of amino acids in hyperthyroid patients following preoperative treatment with antithyroid drug or β-blocking agents: results from a prospective, randomized study. Metabolism 1987;36: 637.
14. Ford HC, Lim WC, Chisnall WN, et al. Renal function and electrolyte levels in hyperthyroidism: urinary protein excretion and the plasma concentrations of urea, creatinine, urine acid, hydrogen ion and electrolytes. Clin Endocrinol (Oxf) 1989;30:293.
15. Kobori H, Ichihara A, Miyashita Y, et al. Mechanism of hyperthyroidism-induced renal hypertrophy in rats. J Endocrinol 1998;159:9.
16. Tang MJ, Lin YJ, Huang JJ. Thyroid hormone upregulates gene expression, synthesis and release of pro-epidermal growth factor in adult rat kidney. Life Sci 1995;57:1477.
17. Noronha-Blob L, Lowe V, Sacktor B. Stimulation by thyroid hormone of phosphate transport in primary cultured renal cells. J Cell Physiol 1988;137:95.
18. Beers KW, Dousa TP. Thyroid hormone stimulates the Na+-PO4 symporter but not the Na+-SO4 symporter in renal brush border. Am J Physiol 1993;265:F323.
19. Kinsella J, Sacktor B. Thyroid hormones increase Na+/K+ exchange activity in renal brush border membranes. Proc Natl Acad Sci U S A 1985;82:3606.
20. Azuma KK, Balkovetz DF, Magyar CE, et al. Renal Na+/H+ exchanger isoforms and their regulation by thyroid hormone. Am J Physiol 1996;270:C585.
21. Evered DC, Hayter CJ, Surveyor I. Primary polydipsia in thyrotoxicosis. Metabolism 1972;21:393.
22. Cutler RE, Glatte H, Dowling JT. Effect of hyperthyroidism on the renal concentrating mechanism in humans. J Clin Endocrinol 1967;27:453.
23. Katz AI, Emmanouel DS, Lindheimer MD. Thyroid hormone and the kidney. Nephron 1975;15:223.
24. Harvey JN, Nagi DK, Baylis PH, et al. Disturbance of osmoregulated thirst and vasopressin secretion in thyrotoxicosis. Clin Endocrinol (Oxf) 1991;35:29.
25. Thoday KL, Monney CT. Historical, clinical and laboratory features of 126 hyperthyroid cats. Vet Record 1992;131:257.
26. Hoey A, Page A, Brown L, et al. Cardiac changes in experimental hyperthyroidism in dogs. Aust Vet J 1991;68:352.
27. Phillips PA, Rolls BJ, Ledingham GG, et al. Angiotensin II–induced thirst and vasopressin release in man. Clin Sci 1985;68: 669.
28. Rolandi E, Santaniello B, Bagnasco M, et al. Thyroid hormones and atrial natriuretic hormone secretion: study in hyper- and hypothyroid patients. Acta Endocrinol (Copenh) 1992;127:23.
29. Tajiri J, Noguchi S, Naomi S, et al. Plasma atrial natriuretic peptide in patients with Graves' disease. Endocrinol Jpn 1990;37:665.
30. Shigimatsu S, Iwasaki T, Aizawa T, et al. Plasma atrial natriuretic peptide, plasma renin activity and aldosterone during treatment of hyperthyroidism due to Graves' disease. Horm Metab Res 1989;21:514.
31. Aikawa JK. Isotopic studies of the body potassium content in thyrotoxicosis. Proc Soc Exp Biol Med 1953;84:594.
32. Klatsky SA, Manson PN. Thyroid disorders masquerading as aging changes. Ann Plastic Surg 1992;28:420.
33. Pantazi H, Papapetrou PD. Changes in parameters of bone and mineral metabolism during therapy for hyperthyroidism. J Clin Endocrinol Metab 2000;85:1099.
34. Epstein FH, Freedman LR, Levitan H. Hypercalcemia, nephrocalcinosis and reversible renal insufficiency associated with hyperthyroidism. N Engl J Med 1958;258:782.
35. Giordano N, Santacroce C, Mattii G, et al. Hyperuricemia and gout in thyroid endocrine disorders. Clin Exp Rheumatol 2001; 19:661.
36. Ahlawat SK, Sachdev A. Hypokalaemic paralysis. Postgrad Med J 1999;75:193.
37. Shulkin D, Olson BR, Levey GS. Thyrotoxic periodic paralysis in a Latin-American taking acetazolamide. Am J Med Sci 1989; 297:337.
38. Mellgren G, Bleskestad IH, Aanderud S, et al. Thyrotoxicosis and paraparesis in a young woman: case report and review of the literature. Thyroid 2002;12:77.
39. Ober KP. Thyrotoxic periodic paralysis in the United States: report of 7 cases and review of the literature. Medicine 1992;71:109.
40. Yeo PP, O'Neill WC. Thyrotoxicosis and periodic paralysis. Med Grand Rounds 1984;3:10.
41. Lin SH, Davids MR, Halperin ML. Hypokalaemia and paralysis. Q J Med 2003;96:161.
42. Shizume K, Shishiba Y, Sakuma M, et al. Studies on electrolyte metabolism in idiopathic and thyrotoxic periodic paralysis. II: total exchangeable sodium and potassium. Metabolism 1966;15:145.
43. Kubota K, Ingbar SH. Influences of thyroid states and sympathoadrenal system on extrarenal potassium disposal. Am J Physiol 1990;258:E428.
44. Chan A, Shinde R, Chow CC, et al. In vivo and in vitro sodium pump activity in subjects with thyrotoxic periodic paralysis. BMJ 1991;303:1096.
45. Sterns RH, Spital A. Disorders of internal potassium balance. Semin Nephrol 1987;7:399.
46. Oh VMS, Taylor EA, Yeo SH, et al. Cation transport across lymphocytic plasma membranes in euthyroid and thyrotoxic men with and without hypokalaemic periodic paralysis. Clin Sci 1990;78:199.
47. Baum M, Dwarakanath V, Alpern RJ, et al. Effects of thyroid hormone on the neonatal renal cortical Na+/H+ antiporter. Kidney Int 1998;53:1254.
48. Dash SC, Jain S, Khanna KN, et al. Thyrotoxicosis, renal tubular acidosis and renal stone. J Assoc Phys India 1980;28:323.
49. Jaeger P, Portmann L, Wauters JP, et al. Report of 1 case without nephrocalcinosis. Am J Nephrol 1985;5:116.
50. Konishi K, Hayashi M, Saruta T. Renal tubular acidosis with autoantibody directed to renal collecting-duct cells. N Engl J Med 1994;331:1593.
51. Mason A, Golding PL. Renal tubular acidosis and autoimmune thyroid disease. Lancet 1970;2:1104.
52. Weetman AP, Tomlinson K, Amos N, et al. Proteinuria in autoimmune thyroid disease. Acta Endocrinol (Copenh) 1985;109:341.
53. Holt S, Kingdon E, Morganstein D, et al. Simultaneous relapse of Graves' disease and minimal change glomerular disease. Nephrol Dial Transplant 2002;17:666.
54. Tanwani LK, Lohano V, Broadstone VL, et al. Minimal change nephropathy and Graves' disease: report of a case and review of the literature. Endocr Pract 2002;8:40.
55. Jordan SC, Buckingham B, Sakai R, et al. Studies of immune complex glomerulonephritis mediated by human thyroglobulin. N Engl J Med 1981;304:121.
56. O'Regan S, Fong JS, Kaplan BS, et al. Thyroid antigen-antibody nephritis. Clin Immunol Immunopathol 1976;6:341.
57. Matsuura M, Kikkawa Y, Akashi K, et al. Thyroid antigen-antibody nephritis: possible involvement of fucosyl-GMI as the antigen. Endocrinol Jpn 1987;34:587.
58. Horvath F, Teague P, Gaffney EF, et al. Thyroid antigen associated immune complex glomerulonephritis in Graves' disease. Am J Med 1979;67:901.
59. Jordan SC, Johnston WH, Bergstein JM. Immune complex glomerulonephritis mediated by thyroid antigens. Arch Pathol Lab Med 1978;102:530.
60. Calder EA, Penhale WJ, Barnes EW, et al. Evidence for circulating immune complexes in thyroid disease. BMJ 1974;2:30.
61. Becker BA, Fenves AZ, Breslau NA. Membranous glomerulonephritis associated with Graves' disease. Am J Kidney Dis 1999;33:369.
62. Griswold WR, Mendoza SA, Johnston W, et al. Vasculitis associated with propylthiouracil: evidence for immune complex pathogenesis and response to therapy. West J Med 1978;128:543.
63. McCormick RV. Periarteritis occurring during propylthiouracil therapy. JAMA 1950;144:1453.
64. Cassorla FG, Finegold DN, Parks JS, et al. Vasculitis, pulmonary cavitation, and anemia during antithyroid drug therapy. Am J Dis Child 1983;137:118.
65. Amrhein JA, Kenny FM, Ross D. Granulocytopenia, lupus-like syndrome, and other complications of propylthiouracil therapy. J Pediatr 1970;76:54.
66. Prasad GV, Bastacky S, Johnson JP. Propylthiouracil-induced diffuse proliferative lupus nephritis: review of immunological complications. J Am Soc Nephrol 1997;8:1205.
67. D'Cruz D, Chesser AM, Lightowler C, et al. Antineutrophil cytoplasmic antibody-positive crescentic glomerulonephritis associated with anti-thyroid drug treatment. Br J Rheumatol 1995; 34:1090.
68. Dolman KM, Gans RO, Vervaat TJ, et al. Vasculitis and antineutrophil cytoplasmic autoantibodies associated with propylthiouracil therapy. Lancet 1993;342:651.
69. Reynolds LR, Bhathena D. Nephrotic syndrome associated with methimazole therapy. Arch Intern Med 1979;139:236.
70. Winters MJ, Hurley RM, Lirenman DS. ANCA-positive glomerulonephritis and IgA nephropathy in a patient on propylthiouracil. Pediatr Nephrol 2002;17:257.
71. Reinhart SC, Moses AM, Cleary L, et al. Acute interstitial nephritis with renal failure associated with propylthiouracil therapy. Am J Kidney Dis 1994;24:575.
72. Fang JT, Huang CC. Propylthiouracil-induced acute interstitial nephritis with acute renal failure requiring haemodialysis: successful therapy with steroids. Nephrol Dial Transplant 1998;13:757
73. Dysseleer A, Buysschaert M, Fonck C, et al. Acute interstitial nephritis and fatal Stevens-Johnson syndrome after propylthiouracil therapy. Thyroid 2000;10:713.