David S. Cooper
The ideal treatment of thyrotoxicosis would be directed at its cause. This is possible in only a few patients, for example, those with exogenous thyrotoxicosis or thyrotropin (TSH)-secreting pituitary adenomas. In patients with thyrotoxicosis due to its more common causes, especially Graves' disease, an autonomously functioning thyroid adenoma (toxic adenoma), and multinodular goiter, the fundamental causes are not known. Therapy is therefore directed at inhibiting thyroidal thyroxine (T4) and triiodothyronine (T3) synthesis and release or destroying thyroid tissue. Ancillary treatment involves ameliorating the effects of T4 and T3 on peripheral tissues. There are several means of accomplishing these goals, and their efficacy depends to some extent on the cause of the thyrotoxicosis. Because this is so, an attempt should be made to determine the cause; this usually can be achieved by history and physical examination, aided by selected tests such as measurement of thyroid radioiodine uptake and thyroid radionuclide imaging (see Chapter 12, and Chapters 23,24,25,26,27,28 and 29, in which the different causes of thyrotoxicosis are discussed).
This chapter considers the three forms of treatment of thyrotoxicosis—antithyroid drugs, radioactive iodine (radioiodine), and thyroidectomy—that are in wide use now. The emphasis is on treatment of the thyroid hyperactivity (hyperthyroidism) caused by Graves' disease, because it is the most common cause of thyrotoxicosis, and it is the disorder in which the relative merits of different treatments are most vigorously debated. When appropriate, the treatment of some of the other causes of thyrotoxicosis and of special patients (e.g., children, pregnant women, and patients with thyrotoxic storm) are mentioned. Information about treatment of some of the less-common causes of the disorder can be found in the chapters addressing those disorders.
THYROTOXICOSIS CAUSED BY GRAVES' DISEASE
Graves' disease is an autoimmune disease characterized by thyrotoxicosis caused by hypersecretion of the thyroid gland, thyroid hyperplasia, infiltrative ophthalmopathy, and localized myxedema. The thyroid hypersecretion and hyperplasia are caused by TSH receptor–stimulating antibodies (TSHR-Abs), which are antibodies against the TSH receptor on the cell membrane of thyroid follicular cells that mimic the effects of TSH (see section on pathogenesis of Graves' disease in Chapter 23) (1).
Autoimmune diseases tend to wax and wane over time, and Graves' disease is no exception. Although spontaneous remissions occur in patients who are not treated, Graves' disease—or rather the thyrotoxicosis that results from it—is virtually always treated because spontaneous remissions in untreated patients are uncommon and because the resulting thyrotoxicosis can have deleterious effects on multiple organ systems. In addition, there are several safe and effective therapies from which to choose, although each has certain drawbacks. Which therapy is best is a matter of debate, and opinions vary from country to country and from continent to continent (2,3). There is no “best” treatment, and the choice depends on several factors. Among the most important are the physician's experience and the patient's preferences. In some situations (e.g., in pregnant women and elderly patients), the therapeutic choices are more limited.
The chief therapeutic objective is to alleviate the patient's thyrotoxicosis. Antithyroid drugs act by decreasing thyroid hormone production. Whether the remissions that sometimes occur during or after antithyroid drug therapy are spontaneous, are due to amelioration of thyrotoxicosis, or are due to drug effects on the immune system is a matter of debate and is discussed later in detail. In contrast, surgery and radioiodine reduce the mass of thyroid tissue, but are not thought to alter the underlying Graves' disease, except possibly by removing intrathyroidal lymphocytes, a source of TSHR-Ab. Hypothyroidism usually follows the latter two treatments, but also may occur during or after drug therapy (4,5), possibly because of autoimmune destruction of the thyroid gland (6,7). Thus, the end result may be the same, regardless of the form of therapy.
DRUG THERAPY OF THYROTOXICOSIS CAUSED BY GRAVES' DISEASE
Antithyroid Drugs
Antithyroid drugs have been a mainstay of treatment of patients with Graves' thyrotoxicosis for almost 60 years (8,9). They can be given to patients with other forms of thyrotoxicosis (e.g., toxic nodular goiter), but they are not usually the primary mode of therapy for these conditions. These drugs inhibit the synthesis of T4 and T3, leading to gradual reduction in their serum concentrations. After several weeks or a few months, the dosage usually can be reduced; in some patients the drug can be discontinued, and the patient may remain euthyroid for months or years. A remission of Graves' disease, usually defined as being euthyroid for at least 1 year after treatment was stopped, occurs in about one half the patients. Thereafter, some patients have recurrent thyrotoxicosis (10), but others never do (11) (Fig. 45.1).
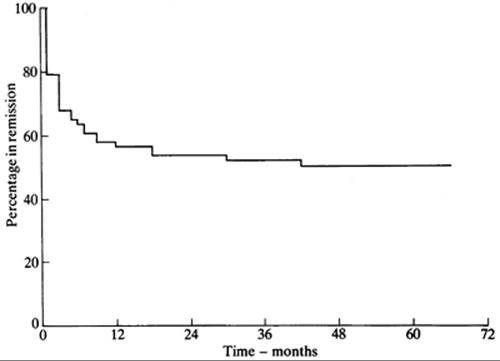
FIGURE 45.1. Kaplan-Meier plot showing the percentage of 72 patients with Graves' thyrotoxicosis remaining in remission after discontinuation of antithyroid drug therapy. Therapy was stopped at time 0. (From Young ET, Steel NR, Taylor JJ, et al. Prediction of remission after antithyroid drug treatment in Graves' disease. QJM 1988;250:175, with permission.)
The antithyroid drugs to be considered here are heterocyclic compounds known as thioamides that contain a thioureylene group (Fig. 45.2). Three drugs of this type are available: methimazole [1-methyl 2-mercaptoimidazole; MMI (Tapazole)], carbimazole (1-methyl-2-thio-3 carbethoxy-imidazole), and propylthiouracil (6-propyl-2-thiouracil; PTU). MMI and PTU are used in the United States and South America, MMI in Europe and Japan, and carbimazole mainly in the United Kingdom. Carbimazole is rapidly metabolized to MMI (12) and has no properties not shared by MMI; therefore, these two drugs can be considered as one.
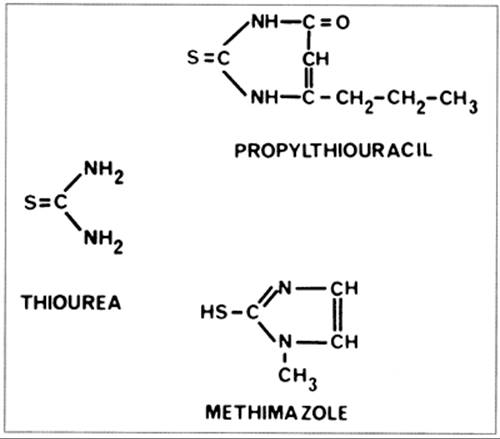
FIGURE 45.2. The structure of thiourea and two antithyroid drugs, propylthiouracil and methimazole, used clinically.
The origin of antithyroid drugs dates back to the early 1940s, with the serendipitous observations of two groups working independently at the Johns Hopkins Medical School. Richter and Clisby, who were studying taste preferences in laboratory animals, noted that the bitter substance phenylthiocarbamide caused goiter in rats (13). The MacKenzies, who were studying the gut flora of guinea pigs, recognized that the nonabsorbable antibiotic sulfaguanidine also caused goiter (14). They (15) and Astwood (16) subsequently determined that the cause of the goiter was stimulation of the thyroid by the pituitary gland, consequent to pharmacologic inhibition of thyroid hormone production. Within 18 months after the observations that sulfaguanidine and thiourea caused goiter, Astwood proposed that goitrogens could be used to treat thyrotoxicosis, screened many potentially useful compounds using a bioassay system, and conducted clinical studies with thiourea and thiouracil. Indeed, he coined the term “antithyroid drug” (17).
Mechanism of Action
The antithyroid drugs have intrathyroidal and extrathyroidal actions. The chief intrathyroidal actions are inhibition of iodine oxidation and organification, inhibition of iodotyrosine coupling, possible alteration of the structure of thyroglobulin, and possible inhibition of thyroglobulin biosynthesis. The main extrathyroidal action is inhibition of conversion of T4 to T3 (by PTU, but not MMI). The drugs' immunosuppressive actions, if they exist, could be at either or both sites.
Intrathyroidal Actions
Detailed descriptions of antithyroid drug pharmacology can be found in Chapter 4 and in the section on effects of drugs and other substances on thyroid hormone synthesis and metabolism in Chapter 11. Antithyroid drugs are actively transported into the thyroid gland (18,19,20), by a mechanism that is similar but probably not identical to the iodide transport system (21). They do not inhibit iodide transport or block the release of stored T4 and T3. Their most important actions are to interfere with thyroid peroxidase–mediated iodide oxidation, organification of iodine, and iodotyrosine coupling. With respect to the organification of iodine, the drugs compete with tyrosyl residues in thyroglobulin for oxidized iodine (22,23). As a result, the active iodine species is diverted away from tyrosyl residues in thyroglobulin, so that fewer are iodinated. The drugs themselves ultimately are oxidized and degraded. Antithyroid drugs also interfere with the peroxidase-catalyzed coupling process by which iodotyrosyl residues are coupled to form T4 and T3; the drug concentrations required to inhibit coupling are less than those required to inhibit iodine organification (24).
In addition to reducing iodine organification and iodotyrosyl coupling, the drugs may bind to thyroglobulin after they have been oxidized (25). Such binding could change the conformation of the thyroglobulin molecule, perhaps rendering it more resistant to subsequent iodination or hydrolysis. In addition, the drugs may inhibit the biosynthesis of thyroglobulin (26), although the concentrations required are probably higher (10-3 mol/L) than are achieved in vivo (27). They also may inhibit the growth of thyroid follicular cells (28,29).
Extrathyroidal Actions
Propylthiouracil, but not MMI, blocks the conversion of T4 to T3 in peripheral tissues (and the thyroid) by inhibiting the activity of type 1 T4-deiodinase (see Chapter 7.) The mechanism is uncertain but may involve competition between the drug and cofactors for the reaction, which include reduced sulfhydryl groups (30). In addition, PTU may bind covalently to the enzyme via a selenosulfide bond (31), thereby inactivating it (32). MMI is not an inhibitor of this reaction, but other imidazole derivatives do inhibit T4 conversion to T3 in vitro (33). The clinical importance of the ability of PTU to block this conversion is discussed later.
Effects on the Immune System
Methimazole and PTU may have immunosuppressive as well as antithyroid actions (34,35,36). Although for the purpose of classification these putative effects are extrathyroidal, they probably involve actions on intrathyroidal immune function as well. The central question is whether the effects are caused by the action of the drugs on the immune system, or whether the abatement of autoimmune phenomena is simply the result of the decline in thyroid secretion induced by the drugs. In studies in vitro, the observed activity can be directly ascribed to the drug, but in studies in vivo the distinction between direct and indirect effects is ambiguous.
Despite some negative results (37), numerous in vitro studies have documented an effect of antithyroid drugs on various arms of the immune system. The drugs inhibit lymphocyte transformation (38), and they may have other inhibitory (39,40,41) [as well as stimulatory (39,40,41)] effects on lymphocyte, monocyte (39), and neutrophil (39,42) function and on formation of soluble mediators such as interleukin-2 (IL-2) (43). The formation of free radicals, which may be important in T-cell responsiveness and in complement-mediated thyroid-cell injury, may be inhibited by MMI (44). In addition, the drugs may reduce expression of major histocompatibility complex (MHC) class II (HLA-DR) molecules on thyroid cells, which may be important for the initiation or maintenance of Graves' disease (45). Antithyroid drugs may reduce HLA-DR expression directly (46) or by inhibiting secretion of interferon-γ, which induces class II molecules (47). MMI reduces MHC class I messenger RNA concentrations in cultured thyroid cells (FRTL-5 cells) (48). Finally, antithyroid drugs may influence the immune system by inducing expression of Fas ligand (FasL) on thyroid cells, which could lead to activation of Fas on lymphocytes and consequently Fas-induced apoptosis of these cells (49).
There is also strong in vivo evidence, albeit circumstantial, for an immunologic effect of antithyroid drugs. The thyroid glands of patients with hyperthyroidism who were treated with an antithyroid drug before thyroidectomy were depleted of lymphocytes, as compared with patients who had received only the β-adrenergic receptor antagonist propranolol (50). In addition, the serum concentrations of TSHR-Ab, whether measured by bioassay or receptor assay, and other antithyroid antibodies decline during antithyroid drug therapy (51,52). The effects appear to be specific for thyroid-related antibodies, because the serum concentrations of antiparietal cell antibodies did not change in patients with coexisting autoimmune gastritis (53). Serum concentrations of the immunomodulator intercellular adhesion molecule-1 (ICAM-1) (54), and of some cytokines and soluble cytokine receptors, also decrease in response to antithyroid drug therapy, including those of IL-1β (55), soluble IL-2 receptors (55,56), and soluble IL-6 receptors (57). A study of thyroid aspirates from treated versus untreated patients suggested that thyrocyte HLA-DR expression is reduced by thioamide therapy (58). Furthermore, antithyroid drug treatment results in changes in cell-mediated immunity in patients with Graves' disease. For example, an increase (normalization) in suppressor T-cell number during treatment was found in several (59,60,61), but not all (62), studies, and helper T-cell (60) and natural killer cell activity decrease (63). Also, MMI decreases the number of activated T cells within the thyroid itself, as compared with the pretreatment number (60).
Despite the evidence for an immunomodulatory effect of antithyroid drugs, several caveats are necessary. With regard to the in vitro data, the effective doses have varied from 10-4 to 10-5 mol/L, whereas intrathyroidal concentrations in vivo are unlikely to exceed 5 × 10-5 mol/L (27,64), thus casting some doubt on the pharmacologic relevance of the observed effects. The changes in serum autoantibody concentrations and in T-cell subsets do not occur in all patients, and the changes that do occur are variable. The reasons for this are unclear, but they must relate to the question of whether remissions of Graves' disease are spontaneous, or whether they are induced by the antithyroid drug (see later discussion). Finally, any changes in immune response markers that may be induced by antithyroid drugs inevitably occur when thyroid secretion is declining and thyrotoxicosis is improving (34). Thus, if the thyrotoxic state were responsible for perpetuation of the altered immunity, then its correction should reduce the alterations. Therapy for Graves' thyrotoxicosis with potassium perchlorate leads to a decline in serum TSHR-Ab concentrations in a manner similar to that which occurs during antithyroid drug treatment (65), but since perchlorate also may have immunosuppressive effects (66), the matter remains unresolved.
Despite these reservations, additional in vivo data indicate an immunosuppressive effect of antithyroid drugs. First, administration of MMI causes serologic and histologic attenuation of experimental autoimmune thyroiditis in rats (67,68,69). Second, in one study of euthyroid patients with chronic autoimmune (Hashimoto's) thyroiditis, administration of carbimazole caused a decline in serum antithyroid peroxidase antibody concentrations (52), a result not confirmed, however, by two other studies (70,71). Finally, MMI, but not glucocorticoids, blocked the increase in serum TSHR-Ab concentrations that occurred in patients with Graves' disease treated with radioiodine (see later discussion), suggesting that an organ-specific effect, rather than generalized immunosuppression, is of primary importance (72). In another study, patients treated with either PTU or carbimazole had identical decrements in serum thyroid hormone concentrations, but the carbimazole-treated patients had greater decreases in serum TSHR-Ab concentrations and increases in the number of suppressor T cells, suggesting, indirectly, an effect on the immune system independent of thyroid function (73).
To summarize, antithyroid drugs can inhibit immune function in vitro, but the concentrations of drug required may be higher than are attained within the thyroid gland during treatment. Changes in serum concentrations of antithyroid antibodies and TSHR-Ab and in T-cell subsets occur in patients receiving chronic antithyroid drug therapy, but changes in thyroid function occur concomitantly, making it impossible to distinguish cause and effect satisfactorily.
CLINICAL PHARMACOLOGY OF THE ANTITHYROID DRUGS
Methimazole
MMI is almost completely absorbed from the gastrointestinal tract (74,75). Peak serum concentrations occur 1 to 2 hours after ingestion and are in the range of 300 ng/mL (2.6 mmol/L) after a 15-mg oral dose (Table 45.1) (75). The serum concentrations are dose related and correlate with effects on iodine organification (76). Carbimazole is rapidly converted to MMI in serum: 10 mg of carbimazole yields about 6 mg MMI (12). The serum half-life of MMI is 6 to 8 hours, but little is bound to serum proteins (75,77). The serum half-life is similar in patients with thyrotoxicosis (75,77), but it may be shorter in patients who do not respond to the drug (78). Drug clearance is unchanged in patients with renal disease (74) but is slowed in those with hepatic disease (77).
TABLE 45.1. SELECTED PHARMACOLOGIC FEATURES OF ANTITHYROID DRUGS
Propylthiouracil
Methimazole
Serum protein binding
~75%
Nil
Serum half-life
~1–2 h
~4–6 h
Volume of distribution
~20 L
~40 L
Metabolism of drug during illness
Severe liver disease
Normal
Decreased
Severe kidney disease
Normal
Normal
Transplacental passage
Low
Higher
Concentrations in breast milk
Low
Higher
From Cooper DS, Bode HH, Nath B, et al. Methimazole pharmacology in man: studies using a newly developed radioimmunoassay for methimazole. J Clin Endocrinol Metab 1984;58:473; Cooper DS, Saxe VC, Meskell M, et al. Acute effects of propylthiouracil (PTU) on thyroidal iodine organification and peripheral iodothyronine deiodination: correlation with serum PTU levels measured by radioimmunoassay. J Clin Endocrinol Metab 1982;54:101; Cooper DS, Steigerwalt S, Migdal S. Pharmacology of propylthiouracil in thyrotoxicosis and chronic renal failure. Arch Intern Med 1987;147:785; Kampmann JP, Hansen JEM. Serum protein binding of propylthiouracil. Br J Clin Pharmacol 1983;16:549; Zaton A, Martinez A, DeGandarias JM. The binding of thioureylene compounds to human serum albumin. Biochem Pharmacol1988;37:3127; and Kampmann JP, Hansen IM, Johansen K, et al. Propylthiouracil in human milk. Lancet 1980;2:736, with permission.
Intrathyroidal MMI concentrations are about 500 to 2000 ng/g (about 5 × 10-5 mol/L) (27,64). The intrathyroidal turnover of MMI is slow, the concentrations 17 to 20 hours after ingestion being similar to those 3 to 6 hours after ingestion (27), which may account for the longer duration of action of MMI as compared with PTU. The effects of MMI dissipated within 24 hours in one study (79), but other studies suggest a longer duration of action (80). Little MMI is excreted in the urine, and neither the products of metabolism nor their fate is known (81). Because it is not protein bound (77) and is lipid soluble, MMI freely crosses membranes [e.g., placenta (82) and breast epithelium (77)]. Given its relatively long serum (and intrathyroidal) half-life and its long duration of action, MMI is effective when given as a single daily dose (83,84,85,86).
Although the potency of MMI is commonly regarded as being about 10 times that of PTU, it is almost surely greater, and it may be up to 50 times more potent (76). Indeed, thyrotoxicosis can be controlled in most patients with doses of MMI that are less, for example, 10 to 15 mg daily, than those traditionally thought to be necessary (85,86). The difference in potency between MMI and PTU is probably not due to an actual difference at the biochemical level, but rather to differences in uptake into and metabolism within the thyroid gland, because in vitro MMI is not a significantly more potent inhibitor of thyroid peroxidase-catalyzed reactions (23). One study found an increase in prednisolone clearance in patients taking MMI, possibly related to hepatic enzyme induction (87). Therefore, patients requiring glucocorticoid therapy for Graves' ophthalmopathy may need higher doses if they are also taking MMI.
Propylthiouracil
Orally administered PTU is almost completely absorbed. Peak serum concentrations occur about 1 hour after ingestion and are dose dependent, with peak concentrations of about 3 mg/mL (18 mmol/L) after a 150-mg oral dose (88) (Table 45.1). Serum PTU concentrations correlate with the drug's effects on iodine oxidation and organification and with inhibition of T4-deiodinase activity (84). There is little information about intrathyroidal concentrations, which are most relevant to efficacy and duration of action (20). The serum half-life of PTU is in the range of 1 to 2 hours, and it is not altered in patients with thyrotoxicosis (85) or hepatic (89) or renal failure (90), in children (91) or in elderly patients (92). PTU is strongly (80% to 90) protein bound (93), largely to serum albumin (94), and is ionized at physiologic pH (95). This has implications for PTU therapy in pregnant and lactating women (see later discussion), because free (i.e., unbound) drug concentrations are low and ionized drug may not freely cross membranes. Most of an ingested dose of PTU is excreted in the urine, after conjugation with glucuronide in the liver (19).
The duration of action of PTU is about 12 to 24 hours (80,96). This rate probably depends on several factors, including the rates at which the drug is concentrated and degraded within the thyroid. Clearly, the duration of action is longer than the serum half-life. Although PTU can sometimes be given satisfactorily as a single daily dose (97), it usually is given every 6 to 8 hours (98), at least when therapy is initiated. With time, the frequency and total daily dose often can be decreased (96).
Clinical Considerations in the Use of Antithyroid Drugs
The thioamide antithyroid drugs are chiefly used for the long-term treatment of patients with thyrotoxicosis caused by Graves' disease, with the expectation—or at least the hope—that a remission of Graves' disease will occur. In surveys of thyroidologists in the United States over a decade ago, radioiodine, not an antithyroid drug, was the preferred treatment for most patients, the exceptions being children, adolescents, and young adults (2,3). In contrast, an antithyroid drug is the treatment of choice in much of the rest of the world, including Europe, Japan, and South America (2).
The clinical factors that influence the choice of therapy and the likelihood of remission are discussed later. Antithyroid drugs rather than surgery are preferred for pregnant women, which is discussed separately, and for children and adolescents (also discussed later). They are often given before surgery and sometimes before radioiodine therapy, and they are standard therapy for neonates with Graves' disease, which is a transient condition.
Both MMI and PTU are very (at least 90) effective in controlling thyrotoxicosis due to Graves' disease, and to some extent the choice between the two drugs is a matter of personal preference. Given the advantages of MMI (see later in the chapter), it is hard to understand why PTU remains rather widely used. Only PTU inhibits extrathyroidal T4 conversion to T3, and although serum T3 concentrations do initially decline more rapidly after the initiation of PTU therapy, there is no evidence that the more rapid decline is clinically important, except possibly in patients with severe or life-threatening thyrotoxicosis (thyrotoxic storm) (99,100). In fact, MMI therapy results in more rapid normalization of serum T4 and T3 concentrations than does PTU therapy (101,102), probably related to the greater potency or the longer duration of action of MMI, as discussed earlier.
Generic MMI is available in either 5-, 10-, or 20 mg tablets (Tapazole¨ is only available as 5- and 10 mg tablets), and PTU in 50 mg tablets, a difference in formulation that means that fewer tablets of MMI need be given each day. The usual starting dose of MMI has been 20 to 30 mg daily, often in divided doses, but once-daily dosing—and lower doses—are adequate for most patients (103). In a prospective multicenter trial in Europe, 10 mg daily was nearly as effective as 40 mg daily (104); serum T4 and T3 concentrations were normal in 6 weeks in 85% of the patients given 10 mg daily and 92% of those given 40 mg daily. Patients living in areas of relative iodine deficiency had a more rapid response, an effect noted previously (105). There is little additional benefit of even higher doses (106). The usual starting dose of PTU is 100 mg three times daily.
One study found that baseline thyroid function is an important predictor of the required starting dosage. If the initial serum T4 concentration was above 20 µg/dl (260 nmol/L), a daily carbimazole dose of 20 mg (equal to about 15 mg MMI) was inadequate for many patients; in contrast, if the initial serum T4concentration was lower, a starting dose of 40 mg per day (equivalent to 30 mg MMI), caused hypothyroidism in a substantial number of patients (Fig. 45.3) (107).
Clearly, the dose should be increased if thyroid secretion does not decrease within 4 to 6 weeks. Doses of PTU as high as 2000 mg daily have been given to patients thought to be resistant to the drug, but in most instances the problem was poor compliance (108). In seriously ill patients who do not respond to high doses of antithyroid drugs, the addition of glucocorticoid therapy may provide additional benefit (109).
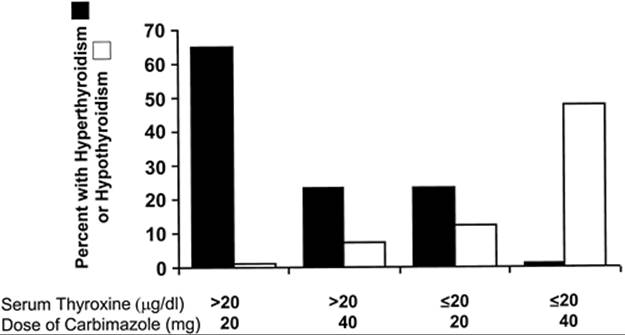
FIGURE 45.3. Percentages of patients with thyrotoxicosis who had persistent thyrotoxicosis or hypothyroidism 4 weeks after initiation of treatment with 20 mg/day or 40 mg/day of carbimazole, subdivided according to baseline serum thyroxine concentration ≤20 µg/dL or >21 µg/dL (260 nmol/L). Thirty-four patients were treated with 20 mg/day and 30 patients with 40 mg/day. (From Cooper DS. Antithyroid drugs in the management of patients with Graves' disease: an evidence-based approach to therapeutic controversies. J Clin Endocrinol Metab 2003; 88:3473, copyright 2003, The Endocrine Society, modified with permission; and from Page SR, Sheard CE, Herbert M, et al. A comparison of 20 or 40 mg per day of carbimazole in the initial treatment of hyperthyroidism. Clin Endocrinol (Oxf) 1996;45:511, with permission.)
As thyroid secretion decreases during the first several weeks or months after antithyroid therapy is initiated, the dose of drug should be decreased, for example, to 5 or even 2.5 mg MMI or 100 or 50 mg PTU daily, or hypothyroidism may supervene. Other factors that determine the speed of recovery include disease activity, the initial degree of thyroid hypersecretion, and the intrathyroidal stores of T4 and T3. The ability to reduce the dose without exacerbation of thyrotoxicosis reflects not only waning of disease activity, possibly reflected by a decline in TSHR-Ab production, but also because the goal of therapy changes, from relatively complete to partial inhibition of T4 and T3 synthesis.
If high doses of drug are required for control of thyrotoxicosis, remission is unlikely, and ablative therapy usually is selected. Some authors have argued that continuous high-dose antithyroid drug therapy is preferable to reducing the dose to maintain thyroid function within normal limits, because rates of remission may be higher as a result of greater putative immunosuppressive effects. However, high-dose therapy has not been widely used because this theory is unproven, it requires concomitant T4 therapy to prevent iatrogenic hypothyroidism, and because the frequency of serious side effects is higher with high doses of antithyroid drug (104,110,111).
The choice of antithyroid drug is an individual matter, based mainly on the physician's personal preferences and experience, but there are many reasons to prefer MMI (112). First, the likelihood of compliance is higher because MMI can be given once daily and fewer tablets per day are needed; once-daily dosing of PTU is less effective (97,98). Second, patients treated with MMI become euthyroid sooner (101,102,113,114). The costs of MMI and PTU are comparable when doses of MMI in the 10 to 20 mg/day range are given; at higher doses, MMI is more expensive than PTU. Finally, MMI may be safer than PTU (115), at least in terms of the most important side effect of these drugs, which is agranulocytosis (see later discussion). In some special circumstances—pregnancy and thyrotoxic storm—PTU may be preferable.
Side Effects of Antithyroid Drugs
Antithyroid drugs have multiple potential side effects (Table 45.2). Most are considered to be allergic reactions. Fever, urticaria or other rashes, and arthralgia occur in 1% to 5% of patients (115,116), usually within the first several weeks or months after initiation of therapy, and are more common in patients treated with higher doses (104,116,117,118). In one study, serum aminotransferase concentrations increased slightly in one third of patients within 2 months after starting PTU therapy (119). The results of baseline liver function tests, which are often abnormal in hyperthyroidism (120,121), were not predictive of this change in PTU-treated patients (119), and the high serum aminotransferase values resolved without discontinuation of therapy. Similar changes have not been reported for MMI. Serum alkaline phosphatase concentrations also may increase transiently during antithyroid drug therapy, not because of hepatobiliary dysfunction, but rather because of an increase in bone formation (121). Routine monitoring of liver function is not indicated.
TABLE 45.2. SIDE EFFECTS OF ANTITHYROID DRUGS
Minor
Common (1%–5%)
Urticaria or other rash
Arthralgia
Fever
Transient granulocytopenia
Uncommon (< 1%)
Gastrointestinal
Abnormalities of taste and smell
Arthritis
Major
Rare (0.2%–0.5%)
Agranulocytosis
Very rare (< 1%)
Aplastic anemia
Thrombocytopenia
Toxic hepatitis (PTU)
Cholestatic hepatitis (MMI)
Vasculitis, systemic lupus-like syndrome
Hypoprothrombinemia (PTU)
Hypoglycemia (due to anti-insulin antibodies) (MMI)
MMI, methimazole; PTU, propylthiouracil.
The more serious and rarer toxic reactions (major side effects) are agranulocytosis, aplastic anemia, hepatitis [with PTU (122)] and cholestasis [with MMI (122)], polyarthritis (123), and a lupus-like syndrome or vasculitis (124,125), all of which, with the possible exception of agranulocytosis, are more common in patients treated with PTU. Agranulocytosis, the most feared problem, probably occurs with equal frequency with both drugs (about 0.2% to 0.5%); the other severe reactions are less common. Fulminant PTU-induced hepatitis may be more common in children; deaths have been reported, and several other patients required liver transplantation (126). A few PTU-treated patients with isolated hypoprothrombinemia have been reported (127). MMI also can cause liver disease, usually cholestatic hepatitis; it may be severe (122,128,129). Patients should be warned about the potential for hepatotoxicity, and to discontinue the drug if they have malaise, jaundice, or dark urine. Patients with the lupus-like syndrome or vasculitis associated with PTU (and rarely MMI) may have skin involvement, glomerulonephritis, or pulmonary hemorrhage, and often have high serum concentrations of antineutrophil cytoplasmic antibodies (ANCA) (124,125,130). Reports of ANCA-related vasculitis, mostly with PTU, are predominantly from Asian countries; some patients were ANCA positive before treatment was started (131,132), and many develop ANCA but do not have vasculitis (132,133). In most cases, the antibody is myeloperoxidase-ANCA (MPO-ANCA) (131,133,134).
Like the minor side effects, these major side effects usually occur within the first several weeks or months after the initiation of therapy, when drug dosage is higher. However, they can occur during prolonged treatment (110) and may be more common when the drug is resumed than when it was first given (45).
The cause of MMI- or PTU-induced agranulocytosis is not known, but it may be an immunologic phenomenon. Some patients have evidence of lymphocyte sensitization (135), an association with certain HLA class II haplotypes has been reported (136), and some patients have antibodies to granulocytes and granulocytic progenitor cells (137).
Agranulocytosis, which is often defined as a granulocyte count less than 250 cells/mm3 (0.25 × 109/L), usually develops so suddenly that routine monitoring of the leukocyte count has been thought to be of little value. Elderly patients may be more susceptible to agranulocytosis (115,138). However, in one study granulocytopenia [granulocyte count less than 500/mm3 (0.5 × 109/L)] was detected by routine monitoring of leukocyte counts before agranulocytosis occurred, suggesting the onset may be gradual (138). In the patients who had granulocytopenia, prompt discontinuation of therapy led to an increase in leukocyte count. If this observation is confirmed, periodic leukocyte counts would be reasonable, at least during the first few months of therapy, when most reported cases of agranulocytosis have occurred. However, due to the low frequency of this side effect, the cost effectiveness of routine monitoring must be questioned.
Patients with agranulocytosis typically present with fever and evidence of infection, usually of the oropharynx. All patients should be warned of the possible symptoms, and given written instructions that the drug should be discontinued and a physician contacted immediately if they have any symptoms of infection. Agranulocytosis must be distinguished from the transient, mild granulocytopenia [granulocyte count < 1500/mm3 (1.5 × 109/L)] that occurs in up to 10% of antithyroid drug–treated patients, as well as that occasionally present in patients with thyrotoxicosis before therapy or in normal black subjects. As a practical matter, complete leukocyte counts should be obtained before initiation of therapy; if the baseline granulocyte count is normal but a subsequent count is < 1500/mm3 (1.5 × 109/L), the drug should be discontinued. If the drug is not discontinued, the leukocyte count should be repeated weekly until it is stable or increasing. It may be possible to distinguish those patients with granulocytopenia during therapy who will recover rapidly from those who are likely to have agranulocytosis. In one study, 25 of 28 patients (89%) with moderate granulocytopenia [granulocyte count 500 to 1000/mm3 [0.5 to 1.0 × 109/L)] and 4 of 6 patients (67%) with more severe granulocytopenia [granulocyte count 100 to 500/mm3[0.1 to 0.5 × 109/L)] had a normal granulocyte count 4 hours after a single injection of 75 mg of granulocyte colony stimulating factor (G-CSF) and subsequently recovered fully, whereas those patients who did not have a normal granulocyte count after G-CSF injection had progressive decreases in granulocyte counts (139). Thus, testing with G-CSF may allow recognition of those patients who will recover from those who will require additional care.
In addition to prompt discontinuation of the antithyroid drug, treatment of agranulocytosis typically involves the administration of broad-spectrum antibiotics and appropriate supportive measures; hospitalization should be avoided if possible, but is essential if the patient is febrile. The granulocyte count usually begins to increase within several days, but may not be normal for 10 to 14 days. G-CSF therapy has proven variably effective. Retrospective data suggest modest efficacy (140) in shortening the recovery time, but in a randomized trial of 24 patients, the combination of G-CSF (100 to 250 mg) and antibiotic therapy did not shorten the duration of agranulocytosis, as compared with antibiotic therapy alone (141). However, G-CSF may accelerate recovery in patients in whom the ratio of granulocytes to erythrocytes in the bone marrow is 0.5 or higher (142). Glucocorticoid therapy is probably ineffective (142).
In the case of minor drug-related side effects such as fever or rash, the side effect may subside in several days despite continuation of therapy, with or without a short course of antihistamine therapy. If the effect persists, the other antithyroid drug can be substituted, with reasonable probability that the side effect will subside. Substitution should not be attempted in the case of agranulocytosis or the other major side effects because cross-reactivity has been reported.
Patients with major side effects in whom antithyroid drug therapy is discontinued usually become thyrotoxic soon thereafter, if they were not thyrotoxic when the drug was discontinued. In them, antithyroid drug therapy is no longer an option. They should be treated with a β-adrenergic antagonist drug and inorganic iodine, an iodinated radiographic contrast agent, or lithium if the thyrotoxicosis is severe. If it is not, radioiodine therapy should be given as soon as practicable.
Other rare side effects of MMI are pancreatitis (143); hypoglycemia, caused by anti-insulin antibodies (the “insulin-autoimmune syndrome”), typically in Japanese patients (144,145); and myalgia and high serum creatine kinase concentrations (146). MMI can cause a decreased sense of taste (116), whereas PTU may cause a bitter or metallic taste.
Follow-up of Patients Taking an Antithyroid Drug for Graves' Disease
Once MMI or PTU therapy has been initiated, patients should be seen every 4 to 6 weeks until they are clinically and biochemically euthyroid. This usually occurs within 4 to 6 weeks with MMI, but it may take up to 12 weeks with PTU (101,113). As the thyrotoxicosis comes under control, the dose of antithyroid drug should be progressively reduced. Hypothyroidism, thyroid enlargement, or both may occur in patients if the dosage of drug is not decreased. Later, the frequency of follow-up visits can be decreased to every 2 to 3 months and then every 6 months.
The usual biochemical tests of thyroid function may be misleading early in the course of antithyroid drug therapy. TSH secretion is strongly inhibited by thyrotoxicosis, and therefore serum TSH concentrations may remain low for several months despite normalization of serum T4 and T3 concentrations. Some patients remain thyrotoxic despite having normal or even low serum T4 concentrations; they have persistently high serum T3 concentrations, indicating the need for an increase, rather than a decrease, in antithyroid drug dosage (147,148). This syndrome of so-called T3-predominant thyrotoxicosis is due to incomplete inhibition of thyroid hormone synthesis and may be associated with a low likelihood of remission (149). Although an enlarging thyroid gland may indicate that hypothyroidism has developed, it also may be indicative of persistent or increasing TSHR-Ab production and a low likelihood of remission.
Remissions and Antithyroid Drug Therapy
The primary goal of antithyroid drug therapy is to render the patient euthyroid. However, this form of treatment is usually chosen in anticipation that the patient will eventually have a remission of Graves' disease, and therefore will not need destructive therapy. Unfortunately, the ability to predict which patients are likely to have a remission is poor (150), and there have been only a few large studies of the possible clinical, biochemical, and pharmacologic features that correlate with remission or relapse (151,152,153). One way of organizing the often-conflicting information concerning the factors that might be related to remission is to distinguish the clinical or patient-related factors from the drug-related factors, for example, type or dose of drug or duration of therapy. The goal is to identify those patients in whom remission is unlikely, so that they are not given treatment destined to fail.
PRETREATMENT CLINICAL FACTORS RELATING TO REMISSION OF GRAVES' DISEASE
Certain pretreatment clinical characteristics seem to be associated with a low likelihood of long-term remission (i.e., the patient remains euthyroid for at least 1 year after antithyroid drug therapy is discontinued). They include a large goiter and more severe biochemical thyrotoxicosis, in both adults (Fig. 45.4) (151,152,153,154) and children (155). Another may be a high ratio of T3 to T4 in serum [T3 (ng/dL): T4 (µg/dL) >20) before (and during) therapy (149), but this is disputed (156). High baseline serum concentrations of TSHR-Ab may (152,157) or may not (151) be predictive of eventual relapse, but an undetectable serum TSHR-Ab concentration at the time of diagnosis is associated with a very high (90%) rate of remission (158). Patients with certain HLA haplotypes (especially HLA-DR3) may be less likely (159), whereas those who are HLA-DR4 positive may be more likely, to have a remission (160); others have not found HLA typing to be useful (154,155,156,157,158,159,160,161). Likewise, a negative family history of Graves' disease may (162) or may not (161) be associated with an increased likelihood of remission. Patients with allergic diseases (e.g., atopy, allergic rhinitis, asthma), and high serum IgE concentrations may be less likely to have a remission (163).
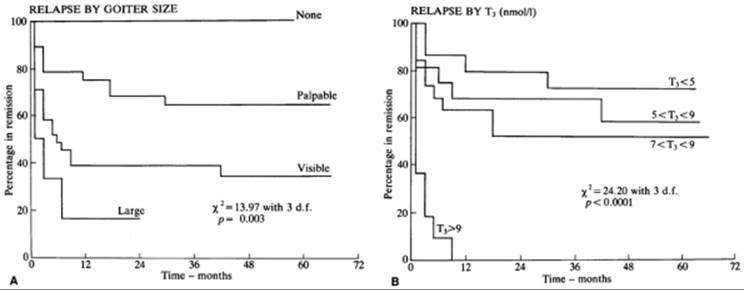
FIGURE 45.4. Likelihood of remission of Graves' thyrotoxicosis after discontinuation of antithyroid drug therapy in 72 patients as a function of (A) goiter size and (B) serum triiodothyronine (T3) concentration at the beginning of therapy. Therapy was discontinued at time 0. To convert serum T3 values to ng/dL, multiply by 65.1. (From Young ET, Steel NR, Taylor JJ, et al. Prediction of remission after antithyroid drug treatment in Graves' disease. QJM 1988;20:175, with permission.)
Factors not consistently related to the likelihood of remission are the sex of the patient (164), smoking (165,166), the presence of ophthalmopathy, or the duration of symptoms before the initiation of therapy. The remission rate may be higher in older patients, possibly due to milder thyrotoxicosis (167), but no relationship with age was found in another study (151). One study found that certain personality traits (e.g., hypochondriasis, depression, paranoia), as well as a higher prevalence of “daily hassles,” were more common in those who relapsed versus those who remained euthyroid (168). Unfortunately, the low predictive value of any clinical finding makes it difficult to know a priori which patients are likely to have a remission and which are not.
Therapy Factors Relating to Remission of Graves' Disease
Duration of Therapy
Although many patients are treated with an antithyroid drug for 1 to 2 years before it is discontinued (2), some patients have a remission within weeks or months after treatment is begun (169,170). Longer courses of therapy intuitively seem preferable, and there are convincing retrospective data (171), particularly in children (172), that the longer the drug is given, the more likely the patient is to have a remission. However, recent prospective randomized studies on this point are conflicting (Fig. 45.5). In a French study of 114 patients, 62% of those treated with MMI for 6 months relapsed, as compared with 42% of those treated for 18 months (173). However, in two other similarly designed studies, the relapse rates were similar among patients treated for 12 months and 24 months (174), and among patients treated for 18 and 42 months (175). Furthermore, among 100 patients given a “block-replace” regimen of carbimazole plus T4, the 1-year relapse rates were 41% in those treated for 6 months and 35% in those treated for 1 year (176). Given these results, treatment for 12 to 24 months seems reasonable, at which time treatment should be discontinued and the patient followed periodically.
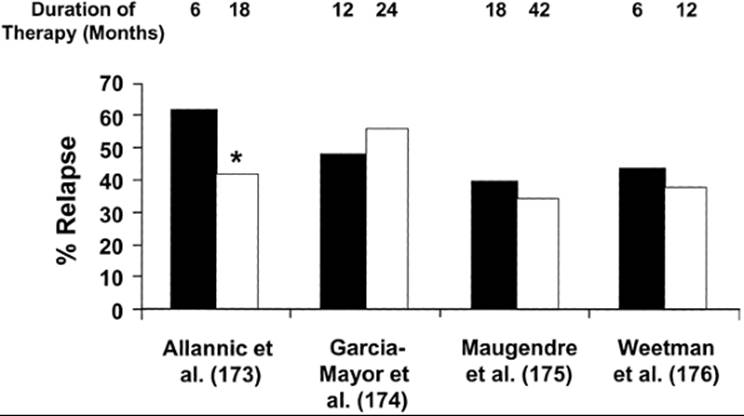
FIGURE 45.5. The rates of relapse of thyrotoxicosis caused by Graves' disease as a function of duration of antithyroid drug therapy in four prospective studies. The numbers overlying the bars are the number of months of in each treatment arm in each study. The rate of relapse was significantly lower in the patients treated longer in only one study (*p < 0.05, for the comparison between the two groups). (From Cooper DS. Antithyroid drugs in the management of patients with Graves' disease: an evidence-based approach to therapeutic controversies. J Clin Endocrinol Metab 2003;88:3473, copyright 2003, The Endocrine Society).
Drug Dose
The possibility that high-dose antithyroid drug therapy might increase the likelihood of remission is based on the suggestion that the drugs have immunosuppressive effects, as discussed earlier. One study did find that the remission rate was higher (75% vs. 42) with high-dose therapy (mean MMI dose 60 mg/day or PTU dose 700 mg/day), as compared with lower doses, but the study was confounded by the fact that the high-dose therapy group also received T3 (177). In a subsequent study by the same group in which the patients given both high and low doses of antithyroid drug were given T3, the rates of remission were similar (178), and they were similar in other prospective randomized studies in which high and low doses were compared (153,179,180,181).
Additional research casts doubt on the efficacy of high-dose regimens to improve the likelihood of remission. In one study there was no correlation between the MMI concentration in thyroid tissue obtained surgically and lymphocyte counts and numbers of activated T cells and antigen-presenting cells (182). In another, serum concentrations of β2-microglobulin, soluble HLA class I antigen, and soluble IL-2 receptor were high in patients with Graves' thyrotoxicosis initially and decreased similarly whether the patients received high- or low-dose antithyroid drug therapy (56). Given the lack of evidence that high-dose therapy is more likely to be followed by remission and the higher likelihood of side effects (104,177,179,181), high-dose therapy cannot be recommended.
Combination Antithyroid Drug and Thyroxine Therapy
The possibility that the combination of an antithyroid drug and T4 might improve the remission rate compared with an antithyroid drug alone is based on the hypothesis that T4, by maintaining lower serum TSH concentrations, would decrease the expression of antigens (e.g., the TSH receptor) that are responsible for perpetuating the production of TSHR-Ab. In a study of 109 Japanese patients, 97% of those treated with MMI and T4 for 18 months followed by T4 alone for 3 years remained in remission during the 3-year T4 treatment period, as compared with 62% in patients given MMI for 18 months (183). Multiple attempts to replicate these remarkable results have been unsuccessful. In one study in the United Kingdom, for example, in which patients were treated for 17 months with carbimazole or carbimazole plus T4, followed by T4 or no therapy for 18 months, the proportion who remained in remission was similar (184). Similar negative results have been reported from Canada and several other European countries (185,186,187,188,189,190). Differences in ethnicity or iodine intake may be partly responsible but are unlikely to be the sole explanations for the difference. Furthermore, another group of Japanese investigators found no difference in serum TSHR-Ab concentrations in patients treated with MMI alone as compared with patients treated with MMI and T4 (191).
Responses during Therapy Relating to Remission
Features during therapy that suggest that a patient may be entering remission include a decrease in goiter size (192), the ability to control the thyrotoxicosis with decreasing doses of drug, and normalization of the ratio of T4 to T3 in serum (148). Conversely, continuing thyroid enlargement, a requirement for a high dose of antithyroid drug, and persistence of high serum T3 concentrations are evidence of continuing Graves' disease. Numerous other tests have been proposed to determine whether a patient's Graves' disease may be in remission so that antithyroid drug therapy can be discontinued with a low likelihood of relapse, but none has the requisite sensitivity and specificity to be useful in individual patients (151,193). The best studied of these tests is measurement of serum TSHR-Ab (194). Serum TSHR-Ab concentrations tend to decrease during antithyroid drug therapy because amelioration of thyrotoxicosis, an immunosuppressive effect of the drug, spontaneous remission, or a combination of these factors. The failure of serum TSHR-Ab to become undetectable during antithyroid drug therapy signifies almost certain relapse after discontinuation of therapy (154,195). If the antibodies disappear, however, there is still a 30% to 50% chance of relapse (152,157,166,194). Thus, detectable serum TSHR-Ab activity, but not its absence, has predictive value.
Other tests proposed as predictors of relapse or remission in patients treated with an antithyroid drug include T3 suppression testing (196), thyrotropin-releasing hormone (TRH) testing (197), and measurements of serum thyroglobulin (198) and antithyroid peroxidase antibodies (199). These tests, if abnormal, indirectly indicate continuing production of TSHR-Ab, and therefore that remission has not occurred. Although each test has its proponents, in a large multicenter study of 451 patients (151) none of these tests had value in individual patients.
Discontinuation of Antithyroid Drug Therapy
In practice, it is most appropriate simply to reduce gradually and then discontinue the antithyroid drug after treatment for 12 to 24 months and follow the patient clinically and with serial measurements of serum TSH. Characteristically, as thyrotoxicosis recurs, serum TSH concentrations decrease, then serum T3concentrations increase, and then serum T4 concentrations increase. However, a decrease in serum TSH concentration does not always mean recurrent thyrotoxicosis is imminent; some patients have low serum TSH concentrations but normal serum T3 and T4 concentrations (subclinical thyrotoxicosis) for prolonged periods (see Chapter 79). These latter patients provide evidence that Graves' disease can be subclinical (e.g., a patient can have Graves' thyroid disease without being overtly thyrotoxic), and they have a higher rate of relapse compared with patients who have normal serum TSH concentrations at discontinuation of therapy (200).
Most relapses of thyrotoxicosis occur within 3 to 6 months after antithyroid drug therapy is discontinued (152) (Fig. 45.1); relapses within this interval probably reflect persistent Graves' disease, rather than remission and recurrence of the disease. Thereafter, the rate of relapse gradually declines to near zero. In the older literature, about 60% of patients were still in remission after 4 years (192), but this was before our current ability to assess thyroid function accurately. More recent studies suggest that the rate of recurrent thyrotoxicosis plateaus at about 50% at 5 years (10,11,154,201). Relapse may be particularly likely in the postpartum period; in one study, almost 50% of women who were in remission before becoming pregnant developed recurrent thyrotoxicosis after delivery (202). A controlled trial suggested that this high rate of relapse might be lowered by administration of T4 during pregnancy (203), but these data have not been confirmed. Because relapses can occur at any time in a patient's life, and hypothyroidism can occur many years after antithyroid drug therapy, lifelong follow-up is recommended for all patients with Graves' disease.
The physician should have in mind a treatment strategy that can be implemented if and when a relapse occurs. In children, a second course of antithyroid drug therapy usually is advised. In young adults, either a second course of antithyroid drug therapy or radioiodine therapy is acceptable, although the likelihood of remission during another course of antithyroid drug therapy may be low. For older adults, radioiodine therapy is usually recommended. Very long-term administration of an antithyroid drug is safe, and some patients may prefer to take a low daily dose of either MMI or PTU for decades rather than receive destructive therapy (204).
In summary, primary antithyroid drug therapy is a reasonable choice in children and younger adults who have mild to moderate thyrotoxicosis caused by Graves' disease and who are likely to be compliant with therapy, in patients who have a bias against radioiodine, and in those with severe ophthalmopathy (see later in the chapter). It is a less reasonable choice as primary therapy for patients with severe biochemical thyrotoxicosis [serum T3 concentration >600 ng/dL (9.2 nmol/L)] or a large goiter.
OTHER DRUGS USED IN THE TREATMENT OF THYROTOXICOSIS CAUSED BY GRAVES' DISEASE
Inorganic Iodide
The effects of iodide on thyroid function are complex and are discussed in detail in the section on the effect of excess iodide in Chapter 11. The major actions of iodide are to decrease T4 and T3 synthesis by inhibiting iodine oxidation and organification (the Wolff-Chaikoff effect), (205) and to block the release of T4and T3 from the thyroid by inhibiting thyroglobulin proteolysis; the latter is the more rapid and prominent action in thyrotoxic patients.
Patients treated with inorganic iodide alone improve quickly, and their serum T4 and T3 concentrations decrease substantially, but not usually to within the normal range, in 7 to 14 days (206,207). Subsequently, many patients escape from its inhibitory effects, and their symptoms worsen as their serum T4 and T3concentrations increase. However, in occasional patients with mild thyrotoxicosis, the disease can be controlled for prolonged periods with potassium iodide, given as Lugol's solution (8 mg iodide per drop) or as a saturated solution of potassium iodide (SSKI,35 to 50 mg iodide per drop) (208). Typical dosages for this and other indications are three to five drops of Lugol's solution three times a day or one drop of SSKI three times a day.
These dosages were empirically derived, and doses in the range of 5 to 10 mg/day would probably suffice.
The three major uses of iodide today are for preparation of patients for surgery, treatment of thyrotoxic storm, and after radioiodine therapy. Preoperative iodide therapy was introduced when iodide was the only available antithyroid drug; in addition to reducing thyroid secretion, it was thought to reduce the vascularity of the thyroid gland. It is primarily for that reason that it is given today (209), usually for 10 days before surgery in patients who have already received antithyroid drug therapy. However, in a controlled study, iodide was not more effective than placebo in reducing operative blood loss or making thyroid gland manipulation easier (210). In patients previously treated only with a β-adrenergic antagonist drug, iodide does reduce thyroid function and blood flow; patients treated with both may have fewer perioperative complications, as compared with patients treated with a β-adrenergic antagonist drug alone (211). Thyroid surgery as therapy for thyrotoxicosis is discussed in detail later.
Patients with severe thyrotoxicosis are sometimes treated with iodide because of its ability to block thyroid hormone release acutely (see Chapter 43). Finally, iodide has been given with mixed success after radioiodine therapy alone (212,213,214) and rarely in combination with an antithyroid drug (215) to reduce thyroid secretion quickly. However, the combination of iodide and MMI does not result in more rapid lowering of serum T4 and T3 concentrations than MMI alone (216). Potassium iodide should be started 1 week after administration of the radioiodine so as not to interfere with thyroid uptake of the radioiodine. Close follow-up is important because hypothyroidism can develop quickly (212), although another study found no effect of iodine versus no therapy after radioiodine treatment (214). Patients treated with radioiodine or surgery are less likely to escape from the inhibitory effects of iodide on iodine oxidation and organification (217), and therefore are more likely to have a sustained antithyroid response (see section on effect of excess iodide in Chapter 11). Thus, iodide can be given as a single agent to patients with recurrent thyrotoxicosis after surgery or after radioiodine therapy.
Iodide cannot be given with impunity. In patients with toxic nodular goiter it can increase serum T4 and T3 concentrations and worsen symptoms, especially if the patient's iodine intake was marginal. In addition, although rare, sensitivity to iodine can occur in the form of acneform eruptions (iodism), sialoadenitis, and vasculitis.
Iodinated Radiographic Contrast Agents
Orally administered iodinated radiographic contrast agents used for cholecystography [e.g., sodium iopanoate [Telepaque] and sodium ipodate (Oragrafin)] are iodinated triiodoaniline derivatives that inhibit T4-deiodinase activity, thereby acutely lowering serum T3 concentrations (218,219, and reviewed in 220 and 221). Additionally, the inorganic iodide formed as these compounds are deiodinated in vivo inhibits T4 and T3 release from the thyroid, and the iodide and perhaps also the compounds themselves inhibit synthesis of T4 and T3 (222). In normal subjects, serum T4 concentrations increase slightly, probably as a result of a decrease in T4 clearance and perhaps also a decrease in cellular T4 uptake (223) (see section on effect of drugs and other substances on thyroid hormone synthesis and metabolism in Chapter 11). Although early reports suggested that these agents might be useful as primary therapy for Graves' thyrotoxicosis (224), they have limited value in long-term therapy (225,226), because of escape from the inhibitory effect of iodide on T4 and T3 synthesis. In addition, their administration may make subsequent control with an antithyroid drug more difficult, presumably because of the large iodine load (226). Subsequent radioiodine therapy also has to be delayed, because radioiodine uptake remains low for several months (227).
Because it rapidly lowers serum T3 concentrations, ipodate has been used in conjunction with PTU (228) to decrease thyroid function in patients who required rapid control of thyrotoxicosis (e.g., in preparation for thyroidectomy) (229). In one study, MMI plus ipodate lowered serum T3 concentrations and pulse rate more rapidly than MMI alone or MMI plus SSKI (216). The results were similar in another study in which ipodate, PTU, and PTU and propranolol were compared (228). These drugs have also been given to lower serum T3 concentrations in patients with massive T4 overdose (poisoning) (230), thyrotoxicosis from subacute thyroiditis (231), and amiodarone-induced thyrotoxicosis (232), and in severe neonatal thyrotoxicosis (233). The usual dose is 1 g daily. These agents have few side effects (renal failure with high doses, one reported case of thrombocytopenia), and they have potential benefits in patients with thyrotoxic storm, T4 poisoning, and other conditions in which both rapid inhibition of thyroid secretion and inhibition of extrathyroidal T3 production might be beneficial. Iopanoate (Telepaque) and ipodate (Oragrafin) are no longer available in the United States.
The water-soluble radiographic contrast agents given intravenously for angiography, pyelography, and computed tomography also contain large amounts of iodine. They are deiodinated, and the iodide has its expected antithyroid action, but it is short-lived because the agents are very rapidly excreted. The contrast agents themselves have no effects on thyroid hormone secretion or metabolism in normal subjects.
Potassium Perchlorate
The perchlorate anion (CIO4-) is a competitive inhibitor of thyroid iodide transport (234). It was used in the past as therapy for thyrotoxicosis but was abandoned because of its side effects (aplastic anemia, gastric ulceration) and because of the advent of MMI and PTU. In doses of 400 to 600 mg/day, it has proven effective (65) and safe. It also has proven effective in combination with an antithyroid drug in patients with iodine-induced thyrotoxicosis. Blockade of iodine transport by perchlorate would seem to be a reasonable adjunct in these patients, who often are resistant to therapy with an antithyroid drug alone (235).
Lithium Carbonate
Lithium is well known to have antithyroid actions, but its mechanism(s) of action is still not understood. It is concentrated by the thyroid (236), probably by active transport. Its primary action is to inhibit T4 and T3 release, a process that is stimulated by TSH and mediated by cyclic adenosine monophosphate (237,238,239). It also may inhibit T4 and T3 synthesis (240,241). As with iodide, there is a tendency for the thyroid gland to escape from the inhibitory actions of lithium; therefore, lithium has limited value for long-term treatment of patients with thyrotoxicosis. Lithium has little advantage over MMI (241), and it has many side effects. In practice, it should not be given as primary treatment for thyrotoxicosis, but it is an option for patients with severe thyrotoxicosis who are allergic to iodide, and it may have an adjunctive role in patients treated with radioiodine (see later in the chapter) and in those with amiodarone-induced thyrotoxicosis (242). The dose of lithium is 300 to 450 mg orally every 8 hours, the goal being to maintain serum lithium concentrations in the range of 1 mEq/L.
Beta-Adrenergic Antagonist Drugs
Beta-adrenergic antagonist drugs are useful adjuncts in the treatment of patients with thyrotoxicosis (243). Many of the manifestations of thyrotoxicosis mimic a hyperadrenergic state (244), and blockade of adrenergic receptors provides patients with considerable relief from some symptoms of thyrotoxicosis, notably anxiety, palpitations, tremor, and heat intolerance. Although the clinical benefit is not due to changes in thyroid function, these drugs cause small, clinically unimportant decrements in serum T3 concentrations because of inhibition of extrathyroidal conversion of T4 to T3 (245).
Although these drugs improve the negative nitrogen balance (246) and decrease heart rate (247), cardiac output (248), and oxygen consumption (249) in patients with thyrotoxicosis, these measurements seldom become normal (250). Hence, these drugs are useful as primary therapy only in patients with transient thyrotoxicosis (e.g., the various forms of thyroiditis). On the other hand, they are useful as adjunctive therapy in alleviating symptoms during diagnostic evaluation or while awaiting the results of primary therapy.
Propranolol was the drug of this class originally given to patients with thyrotoxicosis, and it is still used widely, but some newer drugs of this class have a longer duration of action (long-acting propranolol, atenolol, metoprolol, and nadolol) or are more cardioselective (atenolol and metoprolol). The usual starting dose of propranolol is in the range of 80 mg/day; 50 mg/day of atenolol or metoprolol and 40 mg/day of nadolol have similar effects. High doses (e.g., 240 to 360 mg/day of propranolol) are occasionally needed to control symptoms and slow the heart rate (251). Propranolol and the cardioselective drug esmolol can be given intravenously to patients who are acutely ill.
In general, β-adrenergic antagonist drugs are well tolerated. The main side effects are nausea, headache, fatigue, insomnia, and depression; rare side effects are rash, fever, agranulocytosis, and thrombocytopenia. Complications related to the β-adrenergic antagonist actions are more common. Patients with a history of asthma should not be given these drugs, although a patient with mild asthma and, for example, marked tachycardia and palpitations could be given a cardioselective drug. Patients with a history of congestive heart failure also should not be given the drugs, except when the heart failure is clearly rate related or caused by atrial fibrillation (252). Even then, the drug should be given cautiously, preferably with digoxin. These drugs also are contraindicated in patients with bradyarrhythmias or Raynaud's phenomenon, and in patients being treated with a monoamine oxidase inhibitor.
Several studies have examined the potential usefulness of calcium channel–blocking drugs in thyrotoxicosis. In one study, diltiazem reduced resting heart rate by 17%, comparable with what can be achieved with a β-adrenergic antagonist drug (253). Calcium channel blockers have not been widely used in patients with thyrotoxicosis, but they should be considered for patients in whom β-adrenergic drugs are contraindicated.
RADIOACTIVE IODINE THERAPY FOR THYROTOXICOSIS
Since its introduction in the mid-1940s (254), radioiodine therapy has widely used for adults with thyrotoxicosis caused by Graves' disease (2). Although it is less rapidly effective than antithyroid drug therapy, it is in many other ways an ideal form of therapy; it is effective, safe, and relatively inexpensive. 131I is the isotope of choice; other isotopes of iodine (e.g., 125I-iodine) offer no clinical advantage. It is administered orally as a single dose in a capsule or in water, is rapidly and completely absorbed, and is quickly concentrated, oxidized, and organified by thyroid follicular cells. Although 131I emits both β and γ radiation, it is the ionizing effects of the β particles, which have a path length of 1 to 2 mm, that destroy thyroid follicular cells. Because this distance exceeds the diameter of a thyroid cell, adjacent cells are irradiated even if they do not concentrate iodine.
Initially, radioiodine causes cellular necrosis that provokes an inflammatory response (255). Indeed, some patients have mild thyroid tenderness a few days after treatment; others have transient worsening of their thyrotoxicosis, due to leakage of stored T4 and T3 from disrupted follicles into the circulation (256). Histologically, cellular necrosis and inflammation are seen, as are bizarre nuclear changes reminiscent of carcinoma (257); the latter can persist for years and can cause confusion in the interpretation of thyroid cytology. Therefore, if a patient has a nodule that warrants biopsy, this should be done before radioiodine is given (258). Over time, chronic inflammation and fibrosis result in a substantial decrease in the size of the thyroid gland, ultimately and perhaps inevitably resulting in hypothyroidism. Some patients have relatively normal thyroid function for years or even decades, but they are in the minority.
Practical Therapeutic Considerations
Although worldwide experience in the use of radioiodine is vast, no unanimity of opinion exists concerning the optimal radioiodine dose or the most satisfactory method of dose calculation. In general, a dose that will deliver about 50 to 150 Gy (5000 to 15,000 rad) to the thyroid reduces thyroid hormone secretion to normal or below normal in patients with Graves' disease, but higher doses are required in patients with toxic nodular goiter. To achieve doses in this range, various factors must be considered, including the size of the thyroid, its avidity for iodine (i.e., the 6- or 24-hour radioiodine uptake), the turnover of radioiodine within the gland, the physical half-life of the isotope (8 days in the case of 131I), and prior or planned antithyroid drug therapy, which may necessitate higher doses (see later discussion). When an antithyroid drug is given before radioiodine therapy, it should be discontinued for at least 3 days, lest it interfere with radioiodine organification or possibly act as a free radical scavenger (259), thereby diminishing the radiation effect.
Although estimations of gland size by physical examination are unreliable, the use of measurements of gland volume by ultrasonography for dose calculation does not improve outcome (260). Calculation of the biologic half-life of radioiodine is laborious but may be useful (261). Several less quantitative methods for determining radioiodine dosage have been proposed, including giving all patients the same fixed dosage (262,263). One common approach (264) is to use the following formula, in which the administered dose is (in millicuries):

Using this formula, typical doses are in the range of 5 to 15 mCi (185 to 555 MBq), yielding a radiation dose of 50 to 100 Gy (5000 to 10,000 rad). The choice of microcuries per gram of thyroid tissue is empiric; higher doses should be given to patients with a relatively low 24-hour radioiodine uptake (< 50), a large goiter, those with severe thyrotoxicosis (associated with more rapid intrathyroidal iodine turnover), and those with a toxic nodular goiter. The dose should also be increased by 25% in patients treated with an antithyroid drug before and those who will be treated with one after radioiodine administration (265) (see later in the chapter). In a recent retrospective analysis the patients who failed to respond to therapy were young and had a large thyroid gland, more severe thyrotoxicosis, prior exposure to an antithyroid drug, or a higher radioiodine uptake value, as compared with those in whom treatment was successful (266). The authors recommended a dose that would deliver 11 mCi (407 MBq) of radioiodine for such patients, compared with a dose of 8 mCi (296 MBq) for the majority of patients. It also seems sensible to give higher doses to patients who require a second dose of radioiodine and to those patients in whom the risk of persistent disease should be minimized, such as the elderly or those with cardiac disease.
Some have suggested that large doses should be given to most patients and that hypothyroidism should be accepted as a desired consequence, rather than as a side effect, of therapy (267,268). Most patients become euthyroid, regardless of how the dose is determined (269), and ultimately develop hypothyroidism; lowering the likelihood of hypothyroidism by lowering the dose simply results in delay or failure to cure the thyrotoxicosis (270), necessitates additional therapy, and only delays hypothyroidism (271,272). Giving a fixed dose of radioiodine may simplify and reduce the cost of therapy, because radioiodine uptake need not be measured, and in randomized trials the outcome was similar in patients given calculated doses and those given fixed doses (262,263).
Thyroid secretion declines gradually within weeks to months after radioiodine treatment. Symptoms can be controlled during this interval with a β-adrenergic antagonist drug, if necessary; occasional patients may benefit from antithyroid drug or potassium iodide therapy, as discussed earlier. If either is given, it should be discontinued after several months to determine the efficacy of the radioiodine. The rate at which improvement occurs depends on factors that are poorly understood, but almost certainly include the initial level of thyroid function, the size of the thyroid, and the rate of intrathyroidal radioiodine turnover, as well as the dose of radioiodine. Although no relationship between serum TSHR-Ab values and outcome was found in several studies (273,274), an inverse correlation between serum TSHR-Ab concentrations at baseline and the response to radioiodine was found in other studies (275,276). Serum TSHR-Ab concentrations tend to increase during the first year after radioiodine therapy, and then decline (273). This may be the cause of the post-radioiodine exacerbations that occasionally occur several months after therapy (277).
In general, 50% to 75% of patients have normal thyroid function and some shrinkage of goiter within 6 to 8 weeks after radioiodine therapy (278). Overall, over 80% to 90% of patients become euthyroid or hypothyroid after one dose of radioiodine (given according to the formula discussed earlier), 10% to 20% require a second dose, and a rare patient needs a third dose (278). The figures vary when other treatment philosophies are used. Because radioiodine sometimes acts slowly, an additional dose should not be given for 6 to 12 months.
Complications and Potential Risks of Radioiodine Therapy
Hypothyroidism
Hypothyroidism is an inevitable consequence of radioiodine therapy (279). In the past several decades its frequency has increased, and it has appeared sooner after radioiodine administration (280,281,282), probably because of the use of higher doses as well as the increased ease of detection of hypothyroidism using serum TSH determinations. Hypothyroidism develops in as many as 90% of patients within the first year after therapy (280), with a continuing rate of 2% to 3% per year thereafter. The rapidity with which hypothyroidism develops may relate to not only the dose of radioiodine but also immunologic factors; for example, hypothyroidism is particularly common in patients who have high serum antithyroid peroxidase antibody concentrations (283). Therapy with an antithyroid drug, given before or soon after radioiodine treatment, may lessen the rate of hypothyroidism and increase the risk of persistent thyrotoxicosis (284,285); one retrospective study suggested that these changes in the efficacy of radioiodine therapy were more frequent with PTU than MMI (286). In contrast, two prospective randomized trials found no effect of MMI pretreatment on the outcome of radioiodine therapy (287,288). In another prospective study the frequency of hypothyroidism 1 year later was higher in patients given MMI 4 days after radioiodine therapy as compared with patients treated with radioiodine alone (289).
In addition to permanent hypothyroidism, some patients have transient hypothyroidism (290,291,292,293), possibly due to transient thyroid injury, persistent TSH suppression, or both. Transient hypothyroidism usually occurs about 2 months after therapy and lasts for 1 to 4 months; serum TSH concentrations are often low, indicative of persistent suppression of TSH secretion. If hypothyroidism develops in the first 2 months after radioiodine therapy, particularly if there is persistence of goiter (293), therapy with T4 may be withheld for 1 to 2 months unless the patient is unacceptably symptomatic.
Thyroid and Other Tumors
Despite the advantages of radioiodine therapy, it continues to be a controversial form of treatment, particularly for children and young adults (294). The major concern has been the possible carcinogenic effects of ionizing radiation, particularly late effects that might not be detected for decades. It is clear that external head and neck irradiation is associated with an increased rate of thyroid carcinoma (see section on pathogenesis of thyroid carcinoma in Chapter 70). However, notwithstanding a few case reports (295), there is little evidence that radioiodine therapy for thyrotoxicosis is a risk factor for thyroid carcinoma (296,297,298,299,300,301,302). With the exception of small bowel cancer in one study (303), there is no evidence for increased mortality from any other form of cancer (298,299,300,301), including leukemia (301,304).
Long-term follow-up data on radioiodine therapy in children and adolescents are sparse. In one study (296), thyroid adenomas appeared to be more frequent in patients who received radioiodine therapy as children or adolescents. In the largest studies dealing exclusively with children (305,306,307), the longest of which had a 14-year follow-up, the incidence of thyroid carcinoma, leukemia, or other cancers was not increased, nor was there evidence of abnormal reproductive histories in women. In a smaller study, 3 of 18 children treated with radioiodine developed thyroid nodules, one of which was a low-grade follicular carcinoma (308). Thus, because extensive long-term follow-up data are not available for children, most are treated with an antithyroid drug. Radioiodine has gained increasing acceptability as a first-choice therapy in adolescents (294). A more complete discussion of the treatment of thyrotoxicosis in children can be found at the end of this section and in Chapter 76.
Teratogenicity and Chromosomal Damage
Pregnancy, or the possibility of pregnancy, is an absolute contraindication to radioiodine therapy. Thus, a history of recent menses or a pregnancy test must be obtained in all sexually active women before the administration of radioiodine. In those rare women given radioiodine inadvertently before the 10th week of pregnancy (before the fetal thyroid can concentrate iodine), the outcome was normal (309).
If the radioiodine is given later, the fetal thyroid may be damaged, with consequent fetal hypothyroidism. Candid discussion with the parents is required in this unfortunate circumstance. Management options include fetal blood sampling for measurement of serum TSH, maternal or intraamniotic T4 therapy, and careful follow-up with immediate evaluation at the time of birth. Among them the latter is most appropriate, given that infants with spontaneously occurring congenital hypothyroidism are normal at birth (see Chapter 75).
A more far-reaching question is that of possible genetic damage from radioiodine, with consequent deleterious health effects on the offspring of treated patients. Several studies have documented leukocyte chromosomal abnormalities in patients who received therapeutic doses of radioiodine (310,311), and minor abnormalities in those who received as little as 20 mCi (740 MBq) (312), but the clinical importance of these findings is uncertain. The whole body is exposed to radiation after radioiodine therapy, with gonadal radiation of particular concern because of γ irradiation from radioiodine in the urinary bladder. The estimates of the gonadal (ovarian) radiation dose after radioiodine therapy have varied by more than tenfold (313). A rough estimate of the dose is about 0.2 rad/mCi (0.054 Gy/Bq) of administered radioiodine (313), so that the dose to the ovaries would be 1 to 3 rad (0.01 to 0.03 Gy) for a woman receiving a usual therapeutic dose of 10 mCi (370 MBq). This dose is similar to that from several commonly performed radiologic procedures (e.g., barium enema and intravenous pyelography). The genetic risk of radioiodine therapy is less than the spontaneous rate of genetic abnormalities (0.003% or less vs. 0.8), estimates that are borne out by the negative clinical data that are available (305,306,314). Thus, Safa and colleagues (306) found no increase in congenital abnormalities in 86 children of 43 women who received radioiodine therapy as children. In a related study, none of 33 children of women who received radioiodine had cytogenetic abnormalities (315). In summary, there is no evidence that radioiodine therapy for thyrotoxicosis has adverse effects on the health of the offspring of treated patients. It seems prudent, however, to advise that pregnancy be avoided for at least 6 months after radioiodine therapy and until thyroid function is normal (316).
Despite these reassuring data, unnecessary exposure to radioiodine by family members, particularly children, should be minimized (317). It is almost impossible to avoid minor exposure as a result of environmental contamination (318). Because most of the radioactivity is excreted in the urine, patients should be instructed to use appropriate hygienic measures. Small amounts of radioiodine appear in the saliva, so that recently treated patients should not share food or drink with others and should avoid kissing them for several days after therapy. Because the thyroid is a source of γ radiation, parents are usually instructed not to hug their children for several days, and close contact with pregnant women should be avoided for a similar time period (318). Intimate physical contact should be avoided for several days. Lactation is also an absolute contraindication to radioiodine therapy because iodine is secreted into milk.
Miscellaneous Side Effects
There are no allergic reactions to radioiodine, even among people who are sensitive to iodide or iodinated contrast agents, because the mass of iodine in a typical radioiodine dose is only 1 mg (279). Persons who are allergic to radiographic contrast agents and seafood (allergic to proteins in the meat, not iodine) can safely be treated with radioiodine. Occasional patients note nausea, possibly caused by radiation gastritis. Mild anterior neck pain, caused by radiation thyroiditis, occurs rarely and is easily treated with a nonsteroidal antiinflammatory drug. Other rare associations with radioiodine therapy, not necessarily complications, are hypoparathyroidism, hyperparathyroidism, and vocal cord palsy (279).
Postradioiodine Worsening of Thyroid Function
Occasional transient exacerbation of thyrotoxicosis (256), possibly more frequent in patients with a toxic multinodular goiter (319), and very rare instances of thyrotoxic storm (320,321) have been reported as a result of release of T4 and T3 from the damaged thyroid follicles, but the transient increase in serum hormone concentrations usually is clinically silent, particularly in patients treated with a β-adrenergic antagonist drug.
Because MMI and PTU block the synthesis but not the release of T4 and T3, they deplete hormonal stores within the thyroid. In patients with severe thyrotoxicosis, elderly patients, and patients with cardiac disease, pretreatment with one of these drugs is prudent, not only because thyroid secretion is normalized sooner, but also because pretreatment may minimize the risk of aggravation of thyrotoxicosis after radioiodine therapy. Pretreatment is not necessary in most young patients with thyrotoxicosis. While pretreatment may not prevent worsening of thyroid function after radioiodine, the occasional biochemical exacerbations are less severe because the post-therapy level of thyroid function is reduced due to the pretreatment (322,323). Thyroid function may worsen in the few days between discontinuation of an antithyroid drug and administration of radioiodine, but the change is small and of little clinical importance (323,324). However, if that is a concern clinically, lithium given on the day of antithyroid drug withdrawal prevents any deterioration of thyroid function caused by antithyroid drug withdrawal (325).
Lithium therapy after radioiodine treatment may also prevent post-therapy worsening of thyroid function (326).
Radioiodine Therapy and Graves' Ophthalmopathy
The relationship between radioiodine therapy and the subsequent development or worsening of Graves' ophthalmopathy is an area of continuing controversy (327,328). Two large retrospective studies failed to document a relationship between the type of therapy for Graves' thyrotoxicosis (an antithyroid drug, radioiodine, or surgery) and subsequent changes in the eyes (329,330). Some other studies found that radioiodine therapy may lead to the development or worsening of ophthalmopathy (331,332,333,334), possibly because of the release of thyroid antigens and an increase in serum TSHR-Ab concentrations after therapy (273,335,336).
This question was examined in three prospective controlled studies of different antithyroid treatments. In the first, of 114 patients randomly assigned to treatment with MMI, surgery, or radioiodine, ophthalmopathy developed or worsened in 10%, 16%, and 33% of patients, respectively, during a 2-year follow-up period (333). The patients who received radioiodine became hypothyroid for a few weeks before T4 therapy was initiated, whereas the drug and surgery groups were not hypothyroid at any time. Since other data suggest that post-therapy hypothyroidism may be related to worsening ophthalmopathy (337), there is the possibility that it was the posttherapy hypothyroidism rather than the radioiodine therapy per se that was responsible for the increased risk of ophthalmopathy in this study.
In the second prospective study, 40 patients with Graves' disease were randomly assigned to be treated with radioiodine or surgery. The likelihood of worsening after therapy was similar in the two groups (45% vs. 35) (338). In the third study, 3% of 148 patients treated with MMI had the onset or worsening of eye disease, as compared with 15% of 150 patients treated with radioiodine, during a 1-year follow-up period (Fig. 45.6) (334). Among the radioiodine-treated patients, ophthalmopathy developed or worsened in 6% of nonsmokers as compared with 23% of smokers, suggesting that smoking may be an additional risk factor for radioiodine-related worsening of eye disease (339). The post-radioiodine therapy development or worsening of ophthalmopathy may be preventable by glucocorticoid therapy (0.4 to 0.5 mg/kg prednisone/day) for 3 months beginning immediately after radioiodine administration (331,334).
FIGURE 45.6. Kaplan-Meier plot showing the percentages of patients with Graves' thyrotoxicosis treated with radioiodine, radioiodine plus prednisone, or methimazole (MMI) who developed or had progression of ophthalmopathy during or after therapy. At 12 months there were 145 (of 145) patients in the radioiodine-prednisone group, 144 (of 148) patients in the MMI group, and 127 (of 150) patients in the radioiodine group. (From Bartalena L, Marcocci C, Bogazzi F, et al. Relation between therapy for hyperthyroidism and the course of Graves' ophthalmopathy. N Engl J Med 1998;338:73, with permission.)
Based on these results, patients with Graves' thyrotoxicosis should be counseled that eye disease is more likely to occur or worsen after radioiodine therapy than antithyroid drug (or surgical) therapy. They should also be counseled about the risks and benefits of adjunctive glucocorticoid therapy.
Indications for Radioiodine Therapy
The indications for radioiodine therapy remain somewhat controversial. Although children and adolescents are being treated with radioiodine with increasing frequency (294), an antithyroid drug is usually preferred in these age groups (2). Others believe that all patients with thyrotoxicosis caused by Graves' disease should be given a trial of antithyroid drug therapy, in the hope that a remission will occur. However, remissions are uncommon in patients with moderate or severe thyrotoxicosis. For them, therefore, radioiodine is the most appropriate and perhaps cost-effective therapy, because it permanently ameliorates thyrotoxicosis quickly and safely.
SURGICAL THERAPY FOR THYROTOXICOSIS
Subtotal thyroidectomy is the oldest form of therapy for thyrotoxicosis. Although surgery was the only available therapy for many decades, it is presently performed in the United States only under special circumstances: in children, adolescents, and pregnant women who are allergic to or noncompliant with antithyroid drugs; in patients with large goiters or severe ophthalmopathy (340); and in patients who prefer destructive therapy but are apprehensive about radioiodine therapy (341).
Subtotal thyroidectomy usually is defined as removal of most of the thyroid gland, leaving a few grams of the posterior portion of each lobe (see Chapter 17) (342). Many surgeons now recommend total or near total thyroidectomy rather than subtotal thyroidectomy because of the possibility of recurrent thyrotoxicosis due to thyroid regrowth with the latter procedure (343). Although the mortality of subtotal or total thyroidectomy now is close to zero (342,343,344,345), two worrisome complications of surgery can occur, albeit rarely (1% to 2), even in the most expert hands: recurrent laryngeal nerve damage and hypoparathyroidism. Other complications are transient hypocalcemia, postoperative bleeding, wound infection, and formation of keloids or otherwise unsightly scars. Although the skill of the surgeon is of paramount importance in avoiding perioperative morbidity (346), the number of surgeons experienced in performing thyroidectomy has decreased as other therapies increasingly dominate the treatment of thyrotoxicosis.
In a recent meta-analysis of thyroid surgery for Graves' disease, 100% of patients who underwent a total thyroidectomy were hypothyroid and remained so after follow- up periods ranging from 4 to 12 years (343). In contrast, after subtotal thyroidectomy, hypothyroidism occurs in the first postoperative year in 12% to 80% of patients (341,347,348), with late-onset hypothyroidism in an additional 1% to 3% per year (347), possibly reflecting the natural history of Graves' disease. The development of hypothyroidism depends on several factors, most importantly the size of the thyroid remnant, but also the presence of antithyroid antibodies, perhaps reflecting autoimmune destruction of the remnant (349), and the duration of follow-up. In addition, 5% to 15% of patients have recurrent thyrotoxicosis (342,343,344,350,351). It is more common in patients who before treatment have high serum TSHR-Ab concentrations (352) and severe ophthalmopathy (353); these patients should have a total rather than a subtotal thyroidectomy. The recurrences may develop many years after surgery; in one study, 43% developed more than 5 years after surgery (354). Radioiodine is the treatment of choice for patients who have recurrent thyrotoxicosis after subtotal thyroidectomy.
Preoperative treatment has changed in recent years. Although an antithyroid drug in combination with inorganic iodide was standard therapy for several decades, many now recommend a β-adrenergic antagonist drug, with or without iodide (211,348,355). The latter regimen allows surgery to be performed sooner rather than later, because it has been customary to give the antithyroid drug until the patient is euthyroid, which may take 4 to 6 weeks, and then give iodide for 10 to 14 more days before surgery. The propranolol (or similar drug) regimens consist of treatment for several weeks with doses sufficient to lower the resting pulse rate to < 80 beats/min, with or without potassium iodide, for 10 to 14 days before surgery. Patients treated with propranolol are not clinically or biochemically euthyroid when they undergo surgery, even if they are also treated with iodide (356), and may still have high serum T4concentrations and require continued propranolol therapy postoperatively. Furthermore, patients treated in this way have more postoperative problems (e.g., fever and tachycardia), especially those with severe thyrotoxicosis (355,356,357). Therefore, unless surgery must be performed quickly for some reason, it seems wiser to treat patients who are to undergo surgery in the traditional manner with an antithyroid drug and then iodide. If surgery must be performed urgently, preparation for several days with a β-adrenergic antagonist drug (propranolol 40 mg every 6 hours), high doses of a glucocorticoid and dexamethasone should be safe and effective (229).
The addition of iodide (one to three drops of SSKI daily) to the traditional antithyroid drug regimen 10 days before surgery is controversial because of the lack of convincing evidence that it decreases blood loss during surgery (210), despite studies demonstrating a decrease in thyroid blood flow (209,358). Nevertheless, it is a commonly recommended practice (342). Even if it does not affect blood loss, its antithyroid action is helpful.
SPECIAL CONSIDERATIONS IN THE TREATMENT OF THYROTOXICOSIS
Treatment of Children and Adolescents
Children and adolescents can be treated with radioiodine (294), but an antithyroid drug is preferable, if only for the reason that most of the patients and their parents prefer to avoid destructive therapy and what it entails (see Chapter 76) (2,3). MMI is the drug of choice because of the ease of once-a-day administration. If antithyroid drugs are unsatisfactory or not tolerated, either radioiodine therapy or surgery is appropriate. Although radioiodine has obvious advantages over surgery, not the least being cost, surgery is still as acceptable alternative because of lingering doubts about radioiodine-induced carcinogenesis and genetic damage (359).
Children and adolescents with Graves' thyrotoxicosis are usually treated for many months or even many years; therefore, data on remission rates as a function of duration of treatment are available (172,360). These data suggest that longer periods of therapy are associated with more frequent remission; in one study (172), >75% of patients were in remission after treatment for about 11 years. On the other hand, some children and adolescents ultimately require radioiodine because of drug side effects (which may be more common in children and adolescents), poor compliance with drug therapy, or failure to achieve long-term remission (361,362,363).
Treatment of Elderly Patients
Elderly patients respond well to antithyroid drug therapy, and they are probably more likely to have a remission than are younger patients (167). Nevertheless, in the United States most are treated with radioiodine (2). They are more often pretreated with an antithyroid drug to reduce the risk of exacerbation of thyrotoxicosis after radioiodine therapy, which seems prudent (364). Antithyroid drug therapy may be resumed after radioiodine administration to ensure continuing euthyroidism while the radioiodine is taking effect (365). In patients with mild thyrotoxicosis, a β-adrenergic antagonist drug can be given as sole adjunctive therapy, both before and after radioiodine, with the usual caveats about these drugs in patients with underlying cardiac or pulmonary disease. With regard to the radioiodine dose, it should be high enough to ensure prompt resolution of the thyrotoxicosis, with a minimum chance for recurrence.
Most elderly patients in the United States with thyrotoxicosis probably have Graves' disease (366), but the frequency of solitary toxic adenoma and toxic multinodular goiter as the cause increases sharply with age (see later discussion). Patients with thyroid nodular disease usually require higher doses of radioiodine, because the goiters may be large and the radioiodine uptake relatively low (367). Hospitalization is required is some states in the United States if more than 30 mCi (1110 MBq) of radioiodine is given, and in some countries hospitalization is required even if very low doses are to be given. Patients with a toxic multinodular goiter should not be given iodide after radioiodine therapy or in preparation for surgery, for fear of exacerbating the thyrotoxicosis (368).
Thyrotoxicosis in Pregnant Women and during Lactation
Pregnancy
A full discussion of this topic is beyond the scope of this chapter; additional information can be found in Chapter 80 and in recent reviews (369). An antithyroid drug is the treatment of choice for pregnant women with thyrotoxicosis. In North America, PTU has usually been preferred because it has been thought that its fractional transfer to the fetus was less than that of MMI (82). However, this may not be the case (370). MMI is an acceptable alternative to PTU (371,372), and in fact it is the first-line drug in pregnant women in many parts of the world. A minor birth defect (aplasia cutis) and more severe birth defects (MMI “embryopathy”) have been rarely associated with MMI, and the latter anomaly even more rarely with PTU (373,374,375). This embryopathy includes choanal atresia and/or esophageal atresia.
Except for the very rare congenital abnormalities described above, antithyroid drugs are not teratogenic (376), but neonatal thyroid function can be affected by their transplacental passage. Fetal wastage and maternal morbidity are important problems in pregnant women with untreated or inadequately treated thyrotoxicosis (377), but to minimize fetal exposure to the antithyroid drug the dose should be just adequate, keeping the serum free T4 value in the upper part of the normal range or slightly high (371,378). Fortunately, thyrotoxicosis, which in pregnant women is nearly always caused by Graves' disease, often spontaneously improves in the later months of pregnancy, permitting the dose of drug to be lowered or even discontinued (378). Doses of PTU < 150 mg/day are seldom associated with fetal thyroid dysfunction (379). If affected, neonatal thyroid function usually is only slightly depressed (380), it recovers quickly after delivery, and children exposed to an antithyroid drug in utero have no developmental or intellectual deficits (381,382). Combined antithyroid drug and T4therapy is not recommended because it does not prevent neonatal hypothyroidism and because it may result in the administration of higher doses of the antithyroid drug than are necessary. After delivery, there often is an exacerbation of mild thyrotoxicosis in the mother (202).
Other drugs may be given to pregnant women with thyrotoxicosis. A β-adrenergic antagonist drug can be given to alleviate bothersome symptoms and usually is considered to be safe in pregnancy (369). However, long-term treatment was associated with spontaneous abortion in one report (383). One of these drugs also can be given for preoperative therapy, should that become necessary because the patient is allergic to or noncompliant with antithyroid drug therapy. Potassium iodide also could be given preoperatively. Long-term iodide therapy was thought to be contraindicated in pregnant women because of the risk of development of fetal goiter, but in a Japanese study of pregnant women with mild thyrotoxicosis treated with 6 to 40 mg/day of potassium iodide alone, starting as early as 11 weeks of gestation, no fetal or neonatal abnormalities were noted (384).
Lactation
Antithyroid drug therapy is probably safe in nursing mothers, based not only on measurements of drug in milk but also clinical studies of breastfed infants. Although the concentration of MMI in milk is higher than that of PTU, neither drug has deleterious effects on thyroid function in newborn infants (95,385,386,387,388). It seems reasonable to permit breast feeding in women who desire to do so. The American Academy of Pediatrics has judged both PTU and MMI to be compatible with safe breast feeding (389).
THYROTOXIC STORM
Thyrotoxic storm is a rare condition characterized by uncompensated thyrotoxicosis, with fever, tachycardia or tachyarrhythmia, and altered mental status (99). It is almost always precipitated by an event such as infection, surgery, or trauma in a previously untreated or poorly controlled patient. Thyrotoxic storm is considered in detail in Chapter 43 and elsewhere in this book.
THYROTOXICOSIS DUE TO TOXIC ADENOMA AND MULTINODULAR GOITER
Toxic adenoma and multinodular goiter are unusual causes of thyrotoxicosis in young adults, but are more common among older adults (390,391). Treatment is rather straightforward; because the problem is intrinsic thyroid autonomy rather than external stimulation, spontaneous remissions do not occur. Therefore, an antithyroid drug is rarely the treatment of choice, except as preparation for therapy with radioiodine or surgery (392,393,394,395). In most patients with a toxic adenoma, the hyperthyroidism can be cured with radioiodine; posttreatment hypothyroidism is less common than in Graves' disease because the radioiodine is not concentrated by the suppressed, normal paranodular tissue (396,397,398). Even after a euthyroid state has been achieved however, the nodule may not disappear (399).
Theoretical arguments have been presented suggesting that the paranodular tissue receives potentially carcinogenic doses of γ radiation from the large doses of radioiodine sometimes required to treat these patients (400), but carcinoma is so rare that it is likely coincidental (295). Large doses of radioiodine may be needed to treat patients with a toxic adenoma, but 90% of patients were cured with a mean radioiodine dose of 10 mCi (370 MBq) in one study (396). On the other hand, surgery would be reasonable in a young patient with a large (>5 cm) adenoma. Ethanol injections have been used successfully for treatment of thyroid adenomas in Europe (401), but multiple injections that are often painful are needed, and therefore the procedure has not gained wide acceptance.
Most patients with toxic multinodular goiters are over 50 years old, and they often have other illnesses. Particular care must be taken to eliminate thyrotoxicosis in these patients. They may require large and often multiple doses of radioiodine (402), although a study from the United Kingdom found a higher cure rate at 6 months in patients with toxic nodular goiter than in patients with Graves' disease (403). Most patients should be treated initially with an antithyroid drug to reduce thyroid function quickly and to minimize the risk of exacerbation after radioiodine administration. Even if the thyrotoxicosis is cured, there may be little change in thyroid size because the goiters often contain many poorly or nonfunctioning nodules as well as the hyperfunctioning ones. Thus, surgery may be a better option in patients with a very large goiter, especially if there is evidence of tracheal narrowing or substernal extension; functional airway compromise from a goiter, substernal or otherwise, is an indication for surgery.
REFERENCES
1. McIver B, Morris JC. The pathogenesis of Graves' disease. Endocrinol Metab Clin North Am 1998;27:73.
2. Solomon B, Glinoer D, Lagasse R, et al. Current trends in the management of Graves' disease. J Clin Endocrinol Metab 1990; 70:1518.
3. Wartofsky L, Glinoer D, Solomon B, et al. Differences and similarities in the diagnosis and treatment of Graves' disease in Europe, Japan, and the United States. Thyroid 1991;1:129.
4. Hirota Y, Tamai H, Hayashi Y, et al. Thyroid function and histology in forty-five patients with hyperthyroid Graves' disease in clinical remission more than ten years after thionamide drug treatment. J Clin Endocrinol Metab 1986;62:165.
5. Wood LC, Ingbar SH. Hypothyroidism as a late sequela in patients with Graves' disease treated with antithyroid agents. J Clin Invest 1979;64:1429.
6. Tamai H, Hirota Y, Kasagi K, et al. The mechanism of spontaneous hypothyroidism in patients with Graves' disease after antithyroid drug treatment. J Clin Endocrinol Metab 1987;64: 718.
7. Tamai H, Kasagi K, Takaichi Y, et al. Development of spontaneous hypothyroidism in patients with Graves' disease treated with antithyroidal drugs: clinical, immunological, and histological findings in 26 patients. J Clin Endocrinol Metab 1989;69: 49.
8. Astwood EB. Treatment of hyperthyroidism with thiourea and thiouracil. JAMA 1943;122:78.
9. Astwood EB. Chemotherapy of hyperthyroidism. Harvey Lect 1944;40:195.
10. Sugrue D, McEvoy M, Feely J, et al. Hyperthyroidism in the land of Graves: results of treatment by surgery, radio-iodine and carbimazole in 837 cases. QJM 1980;49:51.
11. Hedley AJ, Young RE, Jones SJ, et al. Antithyroid drugs in the treatment of hyperthyroidism of Graves' disease: long-term follow-up of 434 patients. Clin Endocrinol (Oxf) 1989;31:209.
12. Jansson R, Dahlberg PA, Lindstrom B. Comparative bioavailability of carbimazole and methimazole. Int J Clin Pharm Ther Toxicol 1983;21:505.
13. Richter CR, Clisby KH. Toxic effects of the bitter tasting phenylthiocarbamide. Arch Pathol 1942;33:46.
14. MacKenzie JB, MacKenzie CG, McCollum EV. The effect of sulfanilylguanidine on the thyroid of the rat. Science 1941;94: 518.
15. MacKenzie CG, MacKenzie JB. Effect of sulfonamides and thioureas on the thyroid gland and basal metabolism. Endocrinology 1943;32:185.
16. Astwood EB, Sullivan J, Bissell A, et al. Action of certain sulfonamides and of thiourea upon the function of the thyroid gland of the rat. Endocrinology 1943;32:210.
17. Vanderlaan WP. Antithyroid drugs in hyperthyroidism. In: Van Middlesworth L, ed. The thyroid gland. Chicago: Year Book Medical Publishers, 1986:333.
18. Lazarus JH, Marchant B, Alexander WD, et al. 35S-antithyroid drug concentration and organic binding of iodine in the human thyroid. Clin Endocrinol (Oxf) 1975;4:609.
19. Marchant B, Alexander WD. The thyroid accumulation, oxidation and metabolic rate of 35S-methimazole in the rat. Endocrinology 1972;91:747.
20. Marchant B, Alexander WD, Robertson JWK, et al. Concentration of 35S-propylthiouracil by the thyroid gland and its relationship to anion trapping mechanism. Metabolism 1971; 20:989.
21. Connell JMC, Ferguson MM, Chang DSC, Alexander WD. Influence of sodium perchlorate on thioureylene antithyroid drug accumulation in mice. J Endocrinol 1983;98:183.
22. Davidson B, Soodak M, Neary JT, et al. The irreversible inactivation of thyroid peroxidase by methylmercaptoimidazole, thiouracil, and propylthiouracil in vitro and its relationship to in vivo findings. Endocrinology 1978;103:871.
23. Taurog A, Dorris M. A reexamination of the proposed inactivation of thyroid peroxidase in the rat thyroid by propylthiouracil. Endocrinology 1989;124:3038.
24. Engler H, Taurog A, Dorris ML. Preferential inhibition of thyroxine and 3,5,3′-triiodothyronine formation by propylthiouracil and methylmercaptoimidazole in thyroid peroxidase-catalyzed iodination of thyroglobulin. Endocrinology 1982;110:190.
25. Papapetrou PD, Mothon S, Alexander WD. Binding of the 35-S of 35S-propylthiouracil by follicular thyroglobulin in vivo and in vitro. Acta Endocrinol 1975;79:248.
26. Monaco F, Santolamazza C, DeRos I, et al. Effects of propylthiouracil and methylmercaptoimidazole on thyroglobulin synthesis. Acta Endocrinol 1980;93:32.
27. Jansson R, Dahlberg PA, Johansson H, et al. Intrathyroidal concentrations of methimazole in patients with Graves' disease. J Clin Endocrinol Metab 1983;57:129.
28. Taniguchi S, Yoshida A, Mashiba H. Direct effect of methimazole on rat thyroidal cell growth induced by thyrotropin and insulin-like growth factor I. Endocrinology 1989;124:2046.
29. Korytkowski M, Cooper D. Antithyroid drug effects on function and growth of FRTL-5 cells. Thyroid 1992;2:345.
30. Leonard JL, Rosenberg IN. Thyroxine 5′-deiodinase activity of rat kidney: observations on activation by thiols and inhibition by propylthiouracil. Endocrinology 1978;103:2137.
31. Berry MJ, Kieffer JD, Harney JW, et al. Selenocysteine confers the biochemical properties characteristic of the type I iodothyronine deiodinase. J Biol Chem 1991;266:14155.
32. Visser TJ, Overmeeren EV. Binding of radioiodinated propylthiouracil to rat liver microsomal fractions. Biochem J 1979; 183:167.
33. Visser TJ, Overmeeren E, Fekkes D, et al. Inhibition of iodothyronine 5′-deiodinase by thioureylenes: structure-activity relationship. FEBS Lett 1979;103:314.
34. Volpe R. Evidence that the immunosuppressive effects of antithyroid drug are mediated through actions on the thyroid cell, modulating thyrocyte-immunocyte signaling. Thyroid 1994;4: 217.
35. Wartofsky L. Has the use of antithyroid drugs of Graves' disease become obsolete? Thyroid 1993;3:335.
36. Weetman AP, McGregor AM, Hall R. Evidence for an effect of antithyroid drugs on the natural history of Graves' disease. Clin Endocrinol (Oxf) 1984;24:163.
37. Bagnasco M, Venuti D, Ciprandi G, et al. The effect of methimazole on the immune system is unlikely to operate directly on T lymphocytes. J Endocrinol Invest 1990;13:493.
38. Wall JR, Manwar GL, Greenwood DM, et al. The in vitro suppression of lectin induced 3H-thymidine incorporation into DNA of peripheral blood lymphocytes after the addition of propylthiouracil. J Clin Endocrinol Metab 1976;43: 1406.
39. Balazs C, Kiss E, Leovey A, et al. The immunosuppressive effect of methimazole on cell-mediated immunity is mediated by its capacity to inhibit peroxidase and to scavenge free oxygen radicals. Clin Endocrinol (Oxf) 1986;25:7.
40. Hallengren B, Forsgren A, Melander A. Effects of antithyroid drugs on lymphocyte function in vitro. J Clin Endocrinol Metab 1980;51:298.
41. Weiss I, Davies TF. Inhibition of immunoglobulin secreting cells by antithyroid drugs. J Clin Endocrinol Metab 1981;53: 1223.
42. Imamura M, Aoki N, Saito T, et al. Inhibitory effects of antithyroid drugs on oxygen radical formation in human neutrophils. Acta Endocrinol 1986;112:210.
43. Weetman AP. Effect of the anti-thyroid drug methimazole on interleukin-1 and interleukin-2 levels in vitro. Clin Endocrinol (Oxf) 1986;25:133.
44. Weetman AP, Tandon N, Morgan BP. Antithyroid drugs and release of inflammatory mediators by complement-attacked thyroid cells. Lancet 1992;340:633.
45. Volpe R. Graves' disease. In: Burrow GN, Oppenheimer JH, Volpe R, eds. Thyroid function and disease. Philadelphia: WB Saunders, 1989:214.
46. Bodolay E, Suranyi P, Juhasz F, et al. Methimazole blocks Graves' IgG but not interferon-HLA-DR expression by thyroid cells. Immunol Lett 1988;18:167.
47. Davies TF, Yang C, Platzer M. The influence of antithyroid drugs and iodine on thyroid cell MHC class II antigen expression. Clin Endocrinol (Oxf) 1989;31:125.
48. Saji M, Moriarty J, Ban T, et al. Major histocompatibility complex class I gene expression in rat thyroid cell is regulated by hormones, methimazole, and iodide as well as interferon. J Clin Endocrinol Metab 1992;75:871.
49. Mitsiades N, Poulaki V, Tseleni-Balafouta S, et al. Fas ligand expression in thyroid follicular cells from patients with thionamide-treated Graves' disease. Thyroid. 2000;10:527.
50. Beck JS, Young RJ, Simpson JG, et al. Lymphoid tissue in the thyroid gland and thymus of patients with primary thyrotoxicosis. Br J Surg 1973;60:769.
51. Hardisty CA, Fowles A, Munro DS. The effect of radioiodine and thyroid drugs on serum long acting thyroid stimulator protector (LATS-P): a three year prospective study. Clin Endocrinol (Oxf) 1984;20:547.
52. McGregor AM, Petersen MM, McLachlan SM, et al. Carbimazole and the autoimmune response in Graves' disease. N Engl J Med 1980;303:302.
53. McGregor AM, Rees-Smith B, Hall R, et al. Specificity of the immunosuppressive action of carbimazole in Graves' disease. BMJ 1982;284:1750.
54. Sonnet E, Massart C, Gibassier J, et al. Longitudinal study of soluble intercellular adhesion molecule-1 (ICAM-1) in sera of patients with Graves' disease. J Endocrinol Invest 1999;22: 430.
55. Tsatsoulis A, Vlachoyiannopoulos PG, Dalekos GN, et al. HM. Increased serum interleukin-1 beta during treatment of hyperthyroidism with antithyroid drugs. Eur J Clin Invest 1995; 25:654.
56. Escobar-Morreale H, Serrano-Gotarredona J, Villar L, et al. Methimazole has no dose-related effect on the serum concentrations of soluble class I major histocompatibility complex antigens, soluble interleukin-2 receptor, and beta 2-microglobulin in patients with Graves' disease. Thyroid 1996;6:29.
57. Salvi M, Girasole G, Pedrazzoni M, et al. Increased serum concentrations of interleukin-6 (IL-6) and soluble IL-6 receptor in patients with Graves' disease. J Clin Endocrinol Metab 1996; 81:2976.
58. Zantut-Wittmann DE, Tambascia MA, da Silva Trevisan MA, et al. Antithyroid drugs inhibit in vivo HLA-DR expression in thyroid follicular cells in Graves' disease. Thyroid 2001;11:575.
59. Madec AM, Allannic H, Genetet N, et al. T lymphocyte subsets at various stages of hyperthyroid Graves' disease: effect of carbimazole treatment and relationship with thyroid-stimulating antibody levels or HLA status. J Clin Endocrinol Metab 1986;62:117.
60. Totterman TH, Karlsson FA, Bengtsson M, et al. I. Induction of circulating activated suppressor-like T cells by methimazole therapy for Graves' disease. N Engl J Med 1987;316:15.
61. Ohashi H, Okugawa T, Itoh M. Circulating active T cell subsets in autoimmune thyroid diseases: differences between untreated and treated patients. Acta Endocrinol 1991;125:502.
62. Charreire J, Karsenty G, Bouchard P, et al. Effect of carbimazole treatment on specific and nonspecific immunological parameters in patients with Graves' disease. Clin Exp Immunol 1984;57:633.
63. Wang PW, Luo SF, Huang BY, et al. Depressed natural killer in Graves' disease and during antithyroid medication. Clin Endocrinol (Oxf) 1988;28:205.
64. Okuno A, Yano K, Inyaku F, et al. Pharmakokinetics of methimazole in children and adolescents with Graves' disease. Acta Endocrinol 1987;115:112.
65. Wenzel KW, Lente JR. Similar effects of antithyroid drugs and perchlorate on thyroid stimulating immunoglobulins in Graves' disease: evidence against an immunosuppressive effect of antithyroid drugs. J Clin Endocrinol Metab 1984;58:62.
66. Weetman AP, Gunn C, Hall R, et al. Immunosuppression by perchlorate. Lancet 1984;1:906.
67. Davies TF, Weiss I, Gerber M. Influence of methimazole on murine thyroiditis. J Clin Invest 1984;73:397.
68. Rennie DP, McGregor AM, Keast D, et al. The influence of methimazole on thyroglobulin-induced autoimmune thyroiditis in the rat. Endocrinology 1983;112:326.
69. Reinhardt W, Appel MC, Alex S, et al. The inhibitory effect of large doses of methimazole on iodine induced lymphocytic thyroiditis and serum anti-thyroglobulin antibody titers in BB/ Wor rats. J Endocrinol Invest 1989;12:559.
70. Jansson R, Karlsson A, Dahlberg PA. Thyroxine, methimazole, and thyroid microsomal autoantibody titres in hypothyroid Hashimoto's thyroiditis. BMJ 1985;290:11.
71. Romaldini JH, Werner MC, Rodriques HF, et al. Graves' disease and Hashimoto's thyroiditis: effects of high doses of antithyroid drugs on thyroid autoantibody levels. J Endocrinol Invest 1986;9:233.
72. Gamstedt A, Wadman B, Karlsson A. Methimazole, but not betamethasone, prevents 131I treatment-induced rises in thyrotropin receptor autoantibodies in hyperthyroid Graves' disease. J Clin Endocrinol Metab 1986;62:773.
73. Wilson R, McKillop JH, Pearson C, et al. Differential immunosuppressive action of carbimazole and propylthiouracil. Clin Exp Immunol 1988;73:312.
74. Jansson R, Lindstrom B, Dahlberg PA. Pharmacokinetic properties and bioavailability of methimazole. Clin Pharmacokinet 1985;10:443.
75. Okamura Y, Shigemasa C, Tatsulhara T. Pharmacokinetics of methimazole in normal subjects and hyperthyroid patients. Endocrinol Jpn 1986;33:605.
76. Low LCK, McCruden DC, Alexander WD. Intrathyroidal iodide binding rates and plasma methimazole concentrations in hyperthyroid patients on small doses of carbimazole. Br J Clin Pharmacol 1981;12:315.
77. Cooper DS, Bode HH, Nath B, et al. Methimazole pharmacology in man: studies using a newly developed radioimmunoassay for methimazole. J Clin Endocrinol Metab 1984;58:473.
78. Syrenicz A, Gawronska-Szklarz B, Wojcicki J, et al. Pharmacokinetic parameters of thiamazole in hyperthyroid patients responding rapidly and slowly to the treatment. Pol J Pharmacol Pharm 1991;43:207.
79. McCruden DC, Hilditch TE, Connell JMC, et al. Duration of antithyroid action of methimazole estimated with an intravenous perchlorate discharge test. Clin Endocrinol (Oxf) 1987;26:33.
80. Wartofsky L, Ingbar SH. A method for assessing the latency, potency and duration of action of antithyroid agents in man. In: Fellinger K, Hofer R, eds. Further advances in thyroid research. Wien: Verlag der Wiener Medizinischen Akademia, 1971;121.
81. Taurog A, Dorris ML. Propylthiouracil and methimazole display contrasting pathways of peripheral metabolism in both rat and human. Endocrinology 1988;122:592.
82. Marchant B, Brownlie BEW, Hart DM, et al. The placental transfer of propylthiouracil, methimazole and carbimazole. J Clin Endocrinol Metab 1977;45:1187.
83. MacFarlane IA, Davies D, Longson D, et al. Single daily dose short term carbimazole therapy for hyperthyroid Graves' disease. Clin Endocrinol (Oxf) 1983;18:557.
84. Messina M, Milani P, Gentile L, et al. Initial treatment of thyrotoxic Graves' disease with methimazole: a randomized trial comparing different dosages. J Endocrinol Invest 1987;10:291.
85. Roti E, Gardini E, Minelli R, et al. Methimazole and serum thyroid hormone concentrations in hyperthyroid patients: effects of single and multiple daily doses. Ann Intern Med 1989;111:181.
86. Shiroozu A, Okamura K, Ikenoue H, et al. Treatment of hyperthyroidism with a small single daily dose of methimazole. J Clin Endocrinol Metab 1986;63:125.
87. Legler UF. Impairment of prednisolone disposition in patients with Graves' disease taking methimazole. J Clin Endocrinol Metab 1988;66:221.
88. Cooper DS, Saxe VC, Meskell M, et al. Acute effects of propylthiouracil (PTU) on thyroidal iodine organification and peripheral iodothyronine deiodination: correlation with serum PTU levels measured by radioimmunoassay. J Clin Endocrinol Metab 1982;54:101.
89. Giles HG, Roberts EA, Orrego H, et al. Determination of free propylthiouracil clearance and single sample prediction of steady state. J Pharm Pharmacol 1982;34:62.
90. Cooper DS, Steigerwalt S, Migdal S. Pharmacology of propylthiouracil in thyrotoxicosis and chronic renal failure. Arch Intern Med 1987;147:785.
91. Hoffman WH, Miceli JN. Pharmacokinetics of propylthiouracil in children and adolescents with Graves' disease in the hyperthyroid and euthyroid states. Dev Pharmacol Ther 1988; 11:73.
92. Kampmann JP, Mortenson HB, Back D, et al. Kinetics of propylthiouracil in the elderly. Acta Med Scand 1979;624:93.
93. Kampmann JP, Hansen JEM. Serum protein binding of propylthiouracil. Br J Clin Pharmacol 1983;16:549.
94. Zaton A, Martinez A, DeGandarias JM. The binding of thioureylene compounds to human serum albumin. Biochem Pharmacol 1988;37:3127.
95. Kampmann JP, Hansen IM, Johansen K, et al. Propylthiouracil in human milk. Lancet 1980;2:736.
96. Barnes HV, Bledsoe T. A simple test for selecting the thioamide schedule in thyrotoxicosis. J Clin Endocrinol Metab 1972; 35: 250.
97. Greer MA, Meihoff WC, Studer H. Treatment of hyperthyroidism with a single daily dose of propylthiouracil. N Engl J Med 1965;272:888.
98. Gwinup G. Prospective randomized comparison of propylthiouracil. JAMA 78;239:2457.
99. Burch HB, Wartofsky L. Life-threatening thyrotoxicosis. Endocrinol Metab Clin North Am 1993;22:263.
100. Nicoloff JT. Thyroid storm and myxedema coma. Med Clin North Am 1985;69:1005.
101. Okamura K, Ikenoue H, Shiroozu A, et al. Reevaluation of the effects of methylmercaptoimidazole and propylthiouracil in patients with Graves' hyperthyroidism. J Clin Endocrinol Metab 1987;65:719.
102. Nicholas WC, Fischer RG, Stevenson RA, et al. Single daily dose of methimazole compared to every 8 hours propylthiouracil in the treatment of hyperthyroidism. South Med J 1995;88:973.
103. Mashio Y, Beniko M, Ikota A, et al. Treatment of hyperthyroidism with a small single daily dose of methimazole. Acta Endocrinol 1988;119:139.
104. Reinwein D, Benker G, Lazarus JH, et al. A prospective randomized trial of antithyroid drug dose in Graves' disease therapy. J Clin Endocrinol Metab 1993;76:1516.
105. Azizi F. Environmental iodine intake affects the response to methimazole in patients with diffuse toxic goiter. J Clin Endocrinol Metab 1985;61:374.
106. O'Malley BP, Rosenthal FD, Northover BJ, et al. Higher than conventional doses of carbimazole in the treatment of thyrotoxicosis. Clin Endocrinol (Oxf) 1988;29:281.
107. Page SR, Sheard CE, Herbert M, et al. A comparison of 20 or 40 mg per day of carbimazole in the initial treatment of hyperthyroidism. Clin Endocrinol (Oxf) 1996;45:511.
108. Cooper DS. Propylthiouracil levels in hyperthyroid patients unresponsive to large doses. Ann Intern Med 1985;102:328.
109. Jude EB, Dale J, Kumar S, et al. Treatment of thyrotoxicosis resistant to carbimazole with corticosteroids. Postgrad Med J 1996;72:439.
110. Cooper DS. Antithyroid drugs. N Engl J Med 1984;311:1353.
111. Werner MC, Romaldini JH, Bromberg N, et al. Adverse effects related to thionamide drugs and their dose regimen. Am J Med Sci 1989;297:216.
112. Cooper DS. Which antithyroid drug? Am J Med 1986;80:1165.
113. Kallner G, Vitols S, Ljunggren JG. Comparison of standardized initial doses of two antithyroid drugs in the treatment of Graves' disease. J Intern Med 1996;239:525.
114. Homsanit M, Sriussadaporn S, Vannasaeng S, et al. Efficacy of single daily dosage of methimazole vs. propylthiouracil in the induction of euthyroidism. Clin Endocrinol (Oxf) 2000;54:385.
115. Cooper DS, Goldminz D, Levin AA, et al. Agranulocytosis associated with antithyroid drugs. Ann Intern Med 1983;98:26.
116. Cooper DS. The side effects of antithyroid drugs. The Endocrinologist 1999;9:457.
117. Ducornet B, Duprey J. Effects secondaires des antithyroidiens de synthese. Ann Med Interne 1988;139:410.
118. Meyer-Gessner M, Benker G, Olbricht T, et al. Nebenwirkungen der antithyreoidalen therapie der hyperthyreose. Dtsch Med Wochenschr 1989;114:166.
119. Liaw Y-F, Huang M-J, Fan K-D, et al. Hepatic injury during propylthiouracil therapy in patients with hyperthyroidism. Ann Intern Med 1993;118:424.
120. Gurlek A, Cobaukara V, Bayraktar M. Liver tests in hyperthyroidism: effect of antithyroid therapy. J Clin Gastroenterol 1997;24:180.
121. Huang MJ, Li KL, Wei JS, et al. Sequential liver and bone biochemical changes in hyperthyroidism: prospective controlled follow-up study. Am J Gastroenterol 1994;89:1071.
122. Vitug AC, Goldman JM. Hepatotoxicity from antithyroid drugs. Horm Res 1985;21:229.
123. Bajaj S, Bell MJ, Shumak S, et al. Antithyroid arthritis. J Rheumatol 1998;25:1235.
124. Dolman KM, Gans RO, Vervaat TJ, et al. Vasculitis and antineutrophil cytoplasmic autoantibodies associated with propylthiouracil therapy. Lancet 1993;342:651.
125. Gunton JE, Stiel J, Caterson RJ, et al. Anti-thyroid drugs and antineutrophil cytoplasmic antibody positive vasculitis: a case report and review of the literature. J Clin Endocrinol Metab 1999;84:13.
126. Williams K, Nayak SU, Becker D, et al. Fifty years of experience with propylthiouracil-associated hepatotoxicity: what have we learned? J Clin Endocrinol Metab 1997;82:1727.
127. Gotta A, Sullivan CA, Seaman J, et al. Prolonged intraoperative bleeding caused by propylthiouracil-induced hypoprothrombinemia. Anesthesiology 1972;37:562.
128. Woeber KA. Methimazole-induced hepatotoxicity. Endocr Pract 2002;8:222.
129. Arab DM, Malatjalian DA, Rittmaster RS. Severe cholestatic jaundice in uncomplicated hyperthyroidism treated with methimazole. J Clin Endocrinol Metab 1995;80:1083.
130. Kitahara T, Hiromura K, Maezawa A, et al. Case of propylthiouracil-induced vasculitis associated with anti-neutrophil cytoplasmic antibody (ANCA): review of literature. Clin Nephrol 1997;47:336.
131. Sato H, Hattori M, Fujieda M, et al. High prevalence of antineutrophil cytoplasmic antibody positivity in childhood onset Graves' disease treated with propylthiouracil. J Clin Endocrinol Metab 2000;85:4270.
132. Guma M, Salinas I, Reverter JL, et al. Frequency of antineutrophil cytoplasmic antibody in Graves' disease patients treated with methimazole. J Clin Endocrinol Metab 2003;88: 2141.
133. Sera N, Ashizawa K, Ando T, et al. Treatment with propylthiouracil is associated with appearance of antineutrophil cytoplasmic antibodies in some patients with Graves' disease. Thyroid 2000;10:595.
134. Noh JY, Asari T, Hamada N, et al. Frequency of appearance of myeloperoxidase-antineutrophil cytoplasmic antibody (MPO-ANCA) in Graves' disease patients treated with propylthiouracil and the relationship between MPO-ANCA and clinical manifestations. Clin Endocrinol (Oxf) 2001;54:651.
135. Wall JR, Fang SL, Kuroki T, et al. In vitro immunoreactivity to propylthiouracil, methimazole, and carbimazole in patients with Graves' disease: a possible cause of antithyroid drug-induced agranulocytosis. J Clin Endocrinol Metab 1984;58:868.
136. Tamai H, Sudo T, Kimura A, et al. Association between the DRB1*08032 histocompatibility antigen and methimazole-induced agranulocytosis in Japanese patients with Graves' disease. Ann Intern Med 1996;124:490.
137. Fibbe WE, Claas FHJ, Star-Dijkstra WVD, et al. Agranulocytosis induced by propylthiouracil: evidence of a drug dependent antibody reacting with granulocytes, monocytes and haematopoietic progenitor cells. Br J Haematol 1986;64:363.
138. Tajiri J, Noguehi S, Murakami T, et al. Antithyroid drug-induced agranulocytosis. Arch Intern Med 1990;150:621.
139. Tajiri J, Noguchi S, Murakami N. Usefulness of granulocyte count measurement four hours after injection of granulocyte colony-stimulating factor for detecting recovery from antithyroid drug-induced granulocytopenia. Thyroid 1997;7:575.
140. Andres E, Kurtz JE, Perrin AE, et al. Haematopoietic growth factor in antithyroid-drug-induced agranulocytosis. QJM 2001;94:423.
141. Fukata S, Kuma K, Sugawara M. Granulocyte colony-stimulating factor (G-CSF) does not improve recovery from antithyroid drug-induced agranulocytosis: a prospective study. Thyroid 1999; 9:29.
142. Tamai H, Mukuta T, Matsubayashi S, et al. Treatment of methimazole-induced agranulocytosis using recombinant human granulocyte colony stimulating factor (rhG-CSF). J Clin Endocrinol Metab 193;77:1356.
143. Taguchi M, Yokota M, Koyano H, et al. Acute pancreatitis and parotitis induced by methimazole in a patient with Graves' disease. Clin Endocrinol (Oxf) 1999;51:667.
144. Hakamata M, Itoh M, Sudo Y, et al. Insulin autoimmune syndrome after third therapy with methimazole. Intern Med 1995;34:410.
145. Uchigata Y, Hirata Y. Insulin autoimmune syndrome (IAS, Hirata disease). Ann Med Interne (Paris) 1999;150:245.
146. Suzuki S, Ichikawa K, Nagai M, et al. Elevation of serum creatine kinase during treatment with antithyroid drugs in patients with hyperthyroidism due to Graves' disease. Arch Intern Med 1997;157:693.
147. Hegedüs L, Hansen JM, Bech K, et al. Thyroid stimulating immunoglobulins in Graves' disease with goitre growth, low thyroxine and increasing triiodothyronine during PTU treatment. Acta Endocrinol 1984;107:482.
148. Wenzel KW, Lente JR. Syndrome of persisting thyroid stimulating immunoglobulins and growth promotion of goiter combined with low thyroxine and high triiodothyronine serum levels in drug treated Graves' disease. J Endocrinol Invest 1983;6:389.
149. Takamatsu J, Sugawara M, Kuma K, et al. Ratio of serum triiodothyronine to thyroxine and the prognosis of triiodothyronine-predominant Graves' disease. Ann Intern Med 1984;100: 372.
150. Cooper DS. Antithyroid drugs for the treatment of hyperthyroidism caused by Graves' disease. Endocrinol Metab Clin North Am 1998;27:225.
151. Schleusener H, Schwander J, Fischer C, et al. Prospective multicentre study on the prediction of relapse after antithyroid drug treatment in patients with Graves' disease. Acta Endocrinol (Copenh) 1989;120:689.
152. Vitti P, Rago T, Chiovato L, et al. Clinical features of patients with Graves' disease undergoing remission after antithyroid drug treatment. Thyroid 1997;7:369.
153. Benker G, Rienwein D, Kahaly G, et al. Is there a methimazole dose effect on remission rate in Graves' disease? Results from a long-term prospective study. Clin Endocrinol (Oxf) 1998;49: 451.
154. Young ET, Steel NR, Talor JJ, et al. Prediction of remission after antithyroid drug treatment in Graves' disease. QJM 1988; 250:175.
155. Glaser NS, Styne DM. Predictors of early remission of hyperthyroidism in children. J Clin Endocrinol Metab 1997;82: 1719.
156. Torring O, Tallstedt L, Wallin G, et al. Graves' hyperthyroidism: treatment with antithyroid drugs, surgery, or radioiodine—a prospective randomized study. J Clin Endocrinol Metab 1996;81:2986.
157. Michelangeli V, Poon C, Taft J, et al. The prognostic value of thyrotropin receptor antibody measurement in the early stages of treatment of Graves' disease with antithyroid drugs. Thyroid 1998;8:119.
158. Kawai K, Tamai H, Matsubayashi S, et al. A study of untreated Graves' patients with undetectable TSH binding inhibitor immunoglobulins and the effect of antithyroid drugs. Clin Endocrinol(Oxf) 1995;43:551.
159. McGregor AM, Smith BR, Hall R, et al. Prediction of relapse in hyperthyroid Graves' disease. Lancet 1980;1:1101.
160. DeBruin TWA, Bolk JH, Bussemaker JK, et al. Graves' disease: immunological and immunogenetic indicators of relapse. BMJ 1988;296:1292.
161. Allannic H, Fauchet R, Lorcy Y, et al. A prospective study of the relationship between relapse of hyperthyroid Graves' disease after antithyroid drugs and HLA haplotype. J Clin Endocrinol Metab 1983;57:719.
162. Eshoj O, Kvetny J, Mogensen EF, et al. Prediction of the course of Graves' disease after medical antithyroid treatment. Acta Med Scand 1985;217:225.
163. Komiya I, Yamada T, Sato A, Kouki T, Nishimori T, Takasu N. Remission and recurrence of hyperthyroid Graves' disease during and after methimazole treatment when assessed by IgE and interleukin 13. J Clin Endocrinol Metab 2001;86:3540.
164. Allahabadia A, Daykin J, Holder RL, et al. Age and gender predict the outcome of treatment for Graves' hyperthyroidism. J Clin Endocrinol Metab 2000;85:1038.
165. Kimball LE, Kulinskaya E, Brown B, et al. Does smoking increase relapse rates in Graves' disease? J Endocrinol Invest 2002; 25:152.
166. Nedrebo BG, Holm PI, Uhlving S, et al. Predictors of outcome and comparison of different drug regimens for the prevention of relapse in patients with Graves' disease. Eur J Endocrinol 2002;147:583.
167. Yamada T, Aizawa T, Koizumi Y, et al. Age-related therapeutic response to antithyroid drug in patients with hyperthyroid Graves' disease. J Am Geriatr Soc 1994;42:513.
168. Fukao A, Takamatsu J, Murakami Y, et al. The relationship of psychological factors to the prognosis of hyperthyroidism in antithyroid drug-treated patients with Graves' disease. Clin Endocrinol (Oxf) 2003;58:550.
169. Bing RF, Rosenthal FD. Early remission in thyrotoxicosis produced by short courses of treatment. Acta Endocrinol (Copenh) 1982; 100:221.
170. Greer MA, Kammer H, Bouma DJ. Short-term antithyroid drug therapy for the thyrotoxicosis of Graves' disease. N Engl J Med 1977;297:173.
171. Tamai H, Nakagawa T, Fukino O, et al. Thionamide therapy in Graves' disease: relation of relapse rate to duration of therapy. Ann Intern Med 1980;92:488.
172. Lippe BM, Landaw EM, Kaplan SA. Hyperthyroidism in children treated with long term medical therapy: twenty-five percent remission every two years. J Clin Endocrinol Metab 1987; 64:1241.
173. Allannic H, Fauchet R, Orgiazzi J, et al. Antithyroid drugs and Graves' disease: a prospective randomized evaluation of the efficacy of treatment duration. J Clin Endocrinol Metab 1990; 70:675.
174. Garcia-Mayor RVG, Paramo C, Luna-Cano R, et al. Antithyroid drug and Graves' hyperthyroidism. Significance of treatment duration and TRAb determination on lasting remission. J Endocrinol Invest 1992;15:815.
175. Maugendre D, Gatel A, Campoin L, et al. Antithyroid drugs and Graves' disease—prospective randomized assessment of long-term teratment. Clin Endocrinol (Oxf) 1999;50:127.
176. Weetman AP, Pickerill AP, Watson P, et al. Treatment of Graves' disease with the block-replace regimen of antithyroid drugs: the effect of treatment duration and immunogenetic susceptibility on relapse. QJM 1994;87:337.
177. Romaldini JH, Bromberg N, Werner MC, et al. Comparison of effects of high and low dosage regimens of antithyroid drugs in the management of Graves' disease. J Clin Endocrinol Metab 1983;57:563.
178. Werner R, Romaldini J, Farah C, et al. Serum thyroid stimulating antibody, thyroglobulin levels, and thyroid suppressibility measurement as predictors of the outcome of combined methimazole and triiodothyronine therapy in Graves' disease. Thyroid 1991;1:293.
179. Jorde R, Ytre-Arne K, Stormer J, et al. Short-term treatment of Graves' disease with methimazole in high versus low doses. J Intern Med 1995;238:161.
180. Wilson R, Buchanan L, Fraser W, et al. Do higher doses of carbimazole improve remission in Graves' disease? QJM 1996; 89:381.
181. Grebe SKG, Feek CM, Ford HC, et al. A randomized trial of short-term treatment of Graves' disease with high-dose carbimazole plus thyroxine versus low-dose carbimazole. Clin Endocrinol (Oxf) 1998;48:585.
182. Paschke R, Vogg M, Kristoferitsch R, et al. Methimazole has no dose-related effect on the intensity of the intrathyroidal autoimmune process in relapsing Graves' disease. J Clin Endocrinol Metab 1995;80:2470.
183. Hashizume K, Ichikawa I, Sakurai A, et al. Administration of thyroxine in treated Graves' disease. N Engl J Med 1991;324: 947.
184. McIver B, Rae P, Beckett G, et al. Lack of effect of thyroxine in patients with Graves' hyperthyroidism who are treated with an antithyroid drug. N Engl J Med 1996;334:220.
185. Rittmaster RS, Zwicker H, Abbott EC, et al. Effect of methimazole with or without exogenous l-thyroxine on serum concentrations of thyrotropin (TSH) receptor antibodies in patients with Graves' disease. J Clin Endocrinol Metab 1996;81:3283.
186. Pfeilschifter J, Zeigler R. Suppression of serum thyrotropin with thyroxine in patients with Graves' disease: effects on recurrence hyperthyroidism and thyroid volume. Eur J Endocrinol 1997;136:81.
187. Lucas A, Salinas I, Rius F, et al. Medical therapy of Graves' disease: does thyroxine prevent recurrence of hyperthyroidism? J Clin Endocrinol Metab 1997;82:2410.
188. Pujol P, Osman A, Grabar S, et al. TSH suppression combined with carbimazole for Graves' disease: effect on remission and relapse rates. Clin Endocrinol (Oxf) 1998;48:635.
189. Raber W, Kmen E, Waldhausl W, et al. Medical therapy of Graves' disease: effect on remission rates of methimazole alone and in combination with triiodothyronine. Eur J Endocrinol 2000;142:117.
190. Glinoer D, de Nayer P, Bex M. Effects of l-thyroxine administration, TSH-receptor antibodies and smoking on the risk of recurrence in Graves' hyperthyroidism treated with antithyroid drugs: a double-blind prospective randomized study. Eur J Endocrinol 2001;144:475.
191. Tamai H, Hayaki I, Kawai K, et al. Lack of effect of thyroxine administration on elevated thyroid stimulating receptor antibody levels in treated Graves' disease patients. J Clin Endocrinol Metab 1995;80:1481.
192. Hershman JM, Givens JR, Cassidy CE, et al. Long-term outcome of hyperthyroidism treated with antithyroid drugs. J Clin Endocrinol Metab 1966;26:803.
193. Benker G, Esser J, Kahaly G, et al. New therapeutic approaches in thyroidal autoimmune diseases. Klin Wochenschr 1990;68: 44.
194. Feldt-Rasmussen U, Schleusener H, Carayon P. Meta-analysis evaluation of the impact of thyrotropin receptor antibodies on long-term remission after medical therapy of Graves' disease. J Clin Endocrinol Metab 1994;78:98.
195. Teng CS, Yeung RTT. Changes in thyroid-stimulating antibody activity in Graves' disease treated with antithyroid drug and its relationship to relapse: a prospective study. J Clin Endocrinol Metab 1980;50:144.
196. Yamada T, Koizumi Y, Sato A, et al. Reappraisal of the 3,5,3′-triiodothyronine suppression test in the prediction of long-term outcome of antithyroid drug therapy in patients with hyperthyroid Graves' disease. J Clin Endocrinol Metab 1984;58:676.
197. Dahlberg PA, Karlsson FA, Jansson R, et al. Thyrotropin-releasing hormone testing during antithyroid drug treatment of Graves' disease as an indicator of remission. J Clin Endocrinol Metab 1985;61:1100.
198. Aizawa T, Ishiham M, Koizumi Y, et al. Serum thyroglobulin concentration as an indicator for assessing thyroid stimulation in patients with Graves' disease during antithyroid drug therapy. Am J Med 1990;89:175.
199. Hamada N, Ito K, Mimura T, et al. Retrospective evaluation of the significance of thyroid microsomal antibody in the treatment of Graves' disease. Acta Endocrinol (Copenh) 1987;114:328.
200. Cho BY, Shong MH, Yi KH, et al. Evaluation of serum basal thyrotrophin levels and thyrotrophin receptor antibody activities as prognostic markers for discontinuation of antithyroid drug treatment in patients with Graves' disease. Clin Endocrinol (Oxf) 1992;36:585.
201. Berglund J, Christensen SB, Dymling JF, et al. The incidence of recurrence and hypothyroidism following treatment with antithyroid drugs, surgery or radioiodine in all patients with thyrotoxicosis in Malmš during the period 1970–1974. J Intern Med 1991;229:435.
202. Amino N, Tanizawa O, Mori H, et al. Aggravation of thyrotoxicosis in early pregnancy and after delivery in Graves' disease. J Clin Endocrinol Metab 1982;55:108.
203. Hashizume K, Ichikawa K, Nishii Y, et al. Effect of administration of thyroxine on the risk of postpartum recurrence of hyperthyroid Graves' disease. J Clin Endocrinol Metab 1992;75:6.
204. Slingerland DW, Burrows BA. Long-term antithyroid treatment in hyperthyroidism. JAMA 1979;242:2408.
205. Burman KD, Wartofsky L. Iodine effects on the thyroid gland: biochemical and clinical aspects. Rev Endocr Metab Disord 2000;1:19.
206. Emerson CH, Anderson AJ, Howard WJ, et al. Serum thyroxine and triiodothyronine concentrations during iodide treatment of hyperthyroidism. J Clin Endocrinol Metab 1975;40:33.
207. Philippou G, Koutras DA, Piperingos G, et al. The effect of iodide on serum thyroid hormone levels in normal persons, in hyperthyroid patients, and in hypothyroid patients on thyroxine replacement. Clin Endocrinol (Oxf) 1992;36:573.
208. Thompson WO, Thompson PK, Brailey AG, et al. Prolonged treatment of exophthalmic goiter by iodine alone. Arch Intern Med 1930;45:481.
209. Chang DCS, Wheeler MH, Woodcock JP, et al. The effect of preoperative Lugol's iodine on thyroid blood flow in patients with Graves' hyperthyroidism. Surgery 1987;102:1055.
210. Coyle PJ, Mitchell JE. Thyroidectomy: is Lugol's iodine necessary? Ann R Coll Surg Engl 1982;64:334.
211. Peden NR, Gunn A, Browning MCK, et al. Nadolol and potassium iodide in the surgical treatment of thyrotoxicosis. Br J Surg 1982;69:638.
212. Ross DS, Daniels GH, DeStefano P, et al. Use of adjunctive potassium iodide after radioactive iodine (131I) treatment of Graves' hyperthyroidism. J Clin Endocrinol Metab 1983;57: 250.
213. Schimmel M, Utiger RD. Acute effect of inorganic iodide after 131I therapy for hyperthyroidism. Clin Endocrinol (Oxf) 1977; 6:329.
214. Bazzi MN, Bagchi N. Adjunctive treatment with propylthiouracil or iodine following radioiodine therapy for Graves' disease. Thyroid 1993;3:269.
215. Kasai K, Suzuki H, Shimoda SI. Effects of propylthiouracil and relatively small doses of iodide on early phase treatment of hyperthyroidism. Acta Endocrinol (Copenh) 1980;93:315.
216. Roti E, Robuschi G, Gardini E, et al. Comparison of methimazole, methimazole and sodium ipodate, and methimazole and saturated solution of potassium iodide in the early treatment of hyperthyroid Graves' disease. Clin Endocrinol (Oxf) 1988;28: 305.
217. Braverman LE, Woeber KA, Ingbar SH. Induction of myxedema by iodide in patients euthyroid after radioiodine or surgical treatment of diffuse toxic goiter. N Engl J Med 1969;281:816.
218. Burgi H, Wimpfheimer C, Burger A, et al. Changes of circulating thyroxine, triiodothyronine and reverse triiodothyronine after radiographic contrast agents. J Clin Endocrinol Metab 1976;43:1203.
219. Wu SY, Chopra IJ, Solomon DH, et al. Changes in circulating iodothyronines in euthyroid and hyperthyroid subjects given ipodate (Oragrafin), an agent for oral cholecystography. J Clin Endocrinol Metab 1978;46:691.
220. Braga M, Cooper DS. Oral cholecystographic agents and the thyroid. J Clin Endocrinol Metab 2001;86:1853.
221. Fontanilla JC, Schneider AB, Sarne DH. The use of oral radiographic contrast agents in the management of hyperthyroidism. Thyroid 2001;11:561.
222. Laurberg P. Multisite inhibition by ipodate of iodothyronine secretion from perfused dog thyroid lobes. Endocrinology 1985; 117:1639.
223. Fellicetta JV, Green WL, Nelp WB. Inhibition of hepatic binding of thyroxine by cholecystographic agents. J Clin Endocrinol Metab 1980;65:1032.
224. Wu SY, Shyh TP, Chopra IJ, et al. Comparison of sodium ipodate (Oragrafin) and propylthiouracil in early treatment of hyperthyroidism. J Clin Endocrinol Metab 1982;54:630.
225. Martino E, Balzano S, Bartalena L, et al. Therapy of Graves' disease with sodium ipodate is associated with a high recurrence rate of hyperthyroidism. J Endocrinol Invest 1991;14: 847.
226. Roti E, Gardini E, Minelli R, et al. Sodium ipodate and methimazole in the long-term treatment of hyperthyroid Graves' disease. Metabolism 1993;42:403.
227. Shen DC, Wu SY, Chopra IJ, et al. Long term treatment of Graves' hyperthyroidism with sodium ipodate. J Clin Endocrinol Metab 1985;61:723.
228. Sharp B, Reed AW, Tamagna EI, et al. Treatment of hyperthyroidism with sodium ipodate (Oragrafin) in addition to propylthiouracil and propranolol. J Clin Endocrinol Metab 1981; 53:622.
229. Baeza A, Aguayo M, Barria M, et al. Rapid preoperative preparation in hyperthyroidism. Clin Endocrinol (Oxf) 1991;35:439.
230. Brown RS, Cohen JH 3rd, Braverman LE. Successful treatment of massive acute thyroid hormone poisoning with iopanoic acid. J Pediatr 1998;132:903.
231. Chopra IJ, van Herle AJ, Korenman SG, et al. Use of sodium ipodate in management of hyperthyroidism in subacute thyroiditis. J Clin Endocrinol Metab 1995;80:2178.
232. Chopra IJ, Baber K. Use of oral cholecystographic agents in the treatment of amiodarone-induced hyperthyroidism. J Clin Endocrinol Metab 2001;86:4707.
233. Transue D, Chan J, Kaplan M. Management of neonatal Graves disease with iopanoic acid. J Pediatr 1992;121:472.
234. Soldin OP, Braverman LE, Lamm SH. Perchlorate clinical pharmacology and human health: a review. Ther Drug Monit 2001;23:316.
235. Martino E, Lombardi-Aghini F, Mariotti S, et al. Treatment of amiodarone associated thyrotoxicosis by simultaneous administration of potassium perchlorate and methimazole. J Endocrinol Invest 1986;9:201.
236. Berens SC, Wolff J, Murphy DL. Lithium concentration by the thyroid. Endocrinology 1970;87:1085.
237. Berens SC, Bernstein RS, Robbins J, et al. Antithyroid effects of lithium. J Clin Invest 1970;49:1357.
238. Burrow G, Burke WR, Himmelhoch JM, et al. Effect of lithium on thyroid function. J Clin Endocrinol Metab 1971;32: 647.
239. Williams JA, Berens SC, Wolff J. Thyroid secretion in vitro: inhibition of TSH and dibutyryl cyclic-AMP stimulated 131I release by Li. Endocrinology 1971;88:1385.
240. Kristenson O, Andersen HH, Pallisgaard G. Lithium carbonate in the treatment of thyrotoxicosis. Lancet 1976;1:603.
241. Turner JG, Brownlie BEW, Sadler WA, et al. An evaluation of lithium as an adjunct to carbimazole treatment in acute thyrotoxicosis. Acta Endocrinol 1976;83:86.
242. Dickstein G, Shechner C, Adawi F, et al. Lithium treatment in amiodarone-induced thyrotoxicosis. Am J Med 1997;102:454.
243. Geffner DL, Hershman JM. Beta-adrenergic blockade for the treatment of hyperthyroidism. Am J Med 1992;93:61.
244. Ginsberg AM, Clutter WE, Shah SD, et al. Triiodothyronine-induced thyrotoxicosis increases mononuclear leukocyte beta-adrenergic receptor density in man. J Clin Invest 1981;67: 1785.
245. Cooper DS, Daniels GH, Ladenson PW, et al. Hyperthyroxinemia in patients treated with high-dose propranolol. Am J Med 1982;73:867.
246. Georges LP, Santangelo RP, Mackin JF, et al. Metabolic effects of propranolol in thyrotoxicosis. I. Nitrogen, calcium and hydroxyproline. Metabolism 1975;24:11.
247. Valcavi R, Menozzi C, Roti E, et al. Sinus node function in hyperthyroid patients. J Clin Endocrinol Metab 1992;75:239.
248. Grossman W, Robin NI, Johnson LW, et al. Effects of beta blockade on the peripheral manifestations of thyrotoxicosis. Ann Intern Med 1971;74:875.
249. Saunders J, Hall SEH, Crowther A, et al. The effect of propranolol on thyroid hormones and oxygen consumption in thyrotoxicosis. Clin Endocrinol (Oxf) 1978;9:67.
250. O'Malley BP, Abbott RJ, Barnett DB, et al. Propranolol versus carbimazole as the sole treatment for thyrotoxicosis. A consideration of circulating thyroid hormone levels and tissue thyroid function. Clin Endocrinol (Oxf) 1982;16:545.
251. Feely J, Stevenson IH, Crooks J. Increased clearance of propranolol in thyrotoxicosis. Ann Intern Med 1981;94:472.
252. Klein I, Ojamaa K. Thyrotoxicosis and the heart. Endocrinol Clin North Am 1998;27:51.
253. Roti E, Montermini M, Roti S, et al. The effect of diltiazem, a calcium channel-blocking drug, on cardiac rate and rhythm in hyperthyroid patients. Arch Intern Med 1988;148:1919.
254. Becker DV, Sawin CT. Radioiodine and thyroid disease: the beginning. Semin Nucl Med 1996;26:155.
255. Jones BM, Kwok CC, Kung AW. Effect of radioactive iodine therapy on cytokine production in Graves' disease: transient increases in interleukin-4 (IL-4), IL-6, IL-10, and tumor necrosis factor-alpha, with longer term increases in interferon-gamma production. J Clin Endocrinol Metab 1999;84:4106.
256. Tamagna E, Levine GA, Hershman JM. Thyroid hormone concentrations after radioiodine therapy for hyperthyroidism. J Nucl Med 1979;20:387.
257. Dobyns BM, Vickery AL, Maloof F, et al. Functional and histologic effects of therapeutic doses of radioactive iodine on the thyroid of man. J Clin Endocrinol Metab 1953;13:548.
258. Centeno BA, Szyfelbein WM, Daniels GH, et al. Fine needle aspiration biopsy of the thyroid gland in patients with prior Graves' disease treated with radioactive iodine. Morphologic findings and practical pitfalls. Acta Cytol 1996;40:1189.
259. Sabri O, Zimny M, Schulz G, et al. Success rate of radioiodine therapy in Graves' disease: the influence of thyrostatic medication. J Clin Endocrinol Metab 1999;84:1229.
260. Tsuruta M, Nagayama Y, Yokoyama N, et al. Long-term follow-up studies on iodine-131 treatment of hyperthyroid Graves' disease based on the measurement of thyroid gland volume by ultrasonography. Ann Nucl Med 1993;7:193.
261. Berg G, Michanek A, Holmberg E, et al. Clinical outcome of radioiodine treatment of hyperthyroidism: a follow-up study. J Intern Med 1996;239:165.
262. Peters H, Fischer C, Bogner U, et al. Radioiodine therapy of Graves' hyperthyroidism: standard vs. calculated 131I activity. Results from a prospective, randomized, multicentre study. Eur J Clin Invest 1995;25:186.
263. Leslie WD, Ward L, Salamon EA, et al. A randomized comparison of radioiodine doses in Graves' hyperthyroidism. J Clin Endocrinol Metab 2003;88:978.
264. Beierwaltes WH. The treatment of hyperthyroidism with iodine-131. Semin Nucl Med 1978;8:95.
265. Crooks J, Buchanan WW, Wayne EJ, et al. Effect of pretreatment with methylthiouracil on results of 131-I therapy. BMJ 1960;1:151.
266. Alexander EK, Larsen PR. High dose of (131)I therapy for the treatment of hyperthyroidism caused by Graves' disease. J Clin Endocrinol Metab 2002;87:1073.
267. Erikson E, Erikson K, Wahlberg P. Treatment of hyperthyroidism with standard doses of radioiodine aiming at ablation. Acta Med Scand 1985;214:55.
268. Safa AM, Skillern PG. Treatment of hyperthyroidism with a large initial dose of sodium iodide I131. Arch Intern Med 1975; 135:673.
269. Franklyn JA, Daykin J, Droic Z, et al. Long-term follow-up of treatment of thyrotoxicosis by three different methods. Clin Endocrinol (Oxf) 1991;34:71.
270. Nordyke RA, Gilbert FI. Optimal iodine-131 dose for eliminating hyperthyroidism in Graves' disease. J Nucl Med 1991; 32:411.
271. Cevallos JL, Hagen GA, Maloof F, et al. Low-dose 131I therapy of thyrotoxicosis (diffuse goiter). N Engl J Med 1974;290:141.
272. Sridama V, McCormick M, Kaplan EL, et al. Long-term follow-up study of compensated low-dose 131I therapy for Graves' disease. N Engl J Med 1984;311:426.
273. Teng CS, Yeung RTT, Khoo RKK, et al. A prospective study of the changes in thyrotropin binding inhibitory immunoglobulins in Graves' disease treated by subtotal thyroidectomy or radioactive iodine. J Clin Endocrinol Metab 1980;50:1005.
274. Davies TF, Platzer M, Farid NR. Prediction of therapeutic response to radioiodine in Graves' disease using TSH-receptor antibodies and HLA-status. Clin Endocrinol (Oxf) 1982;16: 183.
275. Murakami Y, Takamatsu J, Sakane S, et al. Changes in thyroid volume in response to radioactive iodine for Graves' hyperthyroidism correlated with activity of thyroid-stimulating antibody and treatment outcome. J Clin Endocrinol Metab 1996; 81:3257.
276. Chiovato L, Fiore E, Vitti P, et al. Outcome of thyroid function in Graves' patients treated with radioiodine: role of thyroid-stimulating and thyrotropin-blocking antibodies and of radioiodine-induced thyroid damage. J Clin Endocrinol Metab 1998;83:40.
277. Stensvold AD, Jorde R, Sundsfjord J. Late and transient increases in free T4 after radioiodine treatment for Graves' disease. J Endocrinol Invest 1997;20:580.
278. Holm LE, Lundell G, Dahlqvist I, et al. Cure rate after 131I therapy for hyperthyroidism. Acta Radiol 1981;20:161.
279. Graham GD, Burman KD. Radioiodine treatment of Graves' disease. Ann Intern Med 1986;105:900.
280. Cunnien AJ, Hay ID, Gorman CA, et al. Radioiodine-induced hypothyroidism in Graves' disease: factors associated with the increasing incidence. J Nucl Med 1982;23:978.
281. Holm LE. Changing annual incidence of hypothyroidism after iodine-131 therapy for hyperthyroidism, 1951–1975. J Nucl Med 1982;23:108.
282. Peden NR, Hart IR. The early development of transient and permanent hypothyroidism following radioiodine therapy for hyperthyroid Graves' disease. Can Med Assoc J 1984;130:1141.
283. Ahmad AM, Ahmad M, Young ET. Objective estimates of the probability of developing hypothyroidism following radioactive iodine treatment of thyrotoxicosis. Eur J Endocrinol. 2002; 146:767.
284. Marcocci C, Gianchecchi D, Masini I, et al. A reappraisal of the role of methimazole and other factors on the efficacy and outcome of radioiodine therapy of Graves' hyperthyroidism. J Endocrinol Invest 1990;13:513.
285. Tuttle RM, Patience T, Budd S. Treatment with propylthiouracil before radioactive iodine therapy is associated with a higher treatment failure rate than radioiodine therapy alone in Graves' disease. Thyroid 1995;5:243.
286. Imseis RE, VanMiddlesworth L, Massie JD, et al. Pretreatment with propylthiouracil but not methimazole reduces the therapeutic efficacy of iodine-131 in hyperthyroidism. J Clin Endocrinol Metab 1998;83:685.
287. Andrade VA, Gross JL, Maia AL. Effect of methimazole pretreatment on the efficacy of radioactive iodine therapy in Graves' hyperthyroidism: one-year follow-up of a prospective randomized study. J Clin Endocrinol Metab 2001;86:3488.
288. Braga M, Walpert N, Burch HB, et al. The effect of methimazole on cure rates after radioiodine treatment for Graves' hyperthyroidism: a randomized clinical trial. Thyroid 2002;12:135.
289. Kung AW, Yau CC, Cheng AC. The action of methimazole and L-thyroxine in radioiodine therapy: a prospective study on the incidence of hypothyroidism. Thyroid 1995;5:7.
290. Uy HL, Reasner CA, Samuels MH. Pattern of recovery of the hypothalamic–pituitary–thyroid axis following radioactive iodine therapy in patients with Graves' disease. Am J Med 1995;99:173.
291. Gomez N, Gomez JM, Ortiz A, et al. Transient hypothyroidism after iodine-131 therapy for Graves' disease. J Nucl Med 1995;36:1539.
292. Aizawa Y, Yoshida K, Kaise N, et al. The development of transient hypothyroidism after iodine-131 treatment in hyperthyroid patients with Graves' disease: prevalence, mechanism, and prognosis. Clin Endocrinol (Oxf) 1997;46:1.
293. Sawers JSA, Toft AD, Irvine WJ, et al. Transient hypothyroidism after iodine-131 treatment of thyrotoxicosis. J Clin Endocrinol Metab 1980;50:226.
294. Rivkees SA, Sklar C, Freemark M. The management of Graves' disease in children, with special emphasis on radioiodine treatment. J Clin Endocrinol Metab 1998;83:3767.
295. Wiener JD, Thijs LG, Meijer S. Thyroid carcinoma after 131I treatment for hyperthyroidism. Acta Med Scand 1975;198:329.
296. Dobyns BM, Sheline GE, Workman JB, et al. Malignant and benign neoplasms of the thyroid in patients treated for hyperthyroidism: a report of the cooperative thyrotoxicosis therapy follow-up study. J Clin Endocrinol Metab 1974;38:976.
297. Holm LE, Dahlqvist I, Israelsson A, et al. Malignant thyroid tumors after iodine-131 therapy. N Engl J Med 1980;303:188.
298. Hoffman DA, McConahey WM, Diamond EL, et al. Mortality in women treated with hyperthyroidism. Am J Epidemiol 1982; 115:243.
299. Goldman MB, Maloof F, Monson RR, et al. Radioactive iodine therapy and breast cancer. Am J Epidemiol 1988;127:969.
300. Hall P, Lundell G, Holm LE. Mortality in patients treated for hyperthyroidism with iodine-131. Acta Endocrinol 1993;128:230.
301. Ron E, Doody MM, Becker DV, et al. Cancer mortality following treatment for adult hyperthyroidism. JAMA 1998;280:347.
302. Angusti T, Codegone A, Pellerito R, et al. Thyroid cancer prevalence after radioiodine treatment of hyperthyroidism. J Nucl Med 2000;41:1006.
303. Franklyn JA, Maisonneuve P, Sheppard M, et al. Cancer incidence and mortality after radioiodine treatment for hyperthyroidism: a population-based cohort study. Lancet 1999;353: 2111.
304. Hall P, Boice JD Jr, Berg G, et al. Leukaemia incidence after iodine-131 exposure. Lancet 1992;340:1.
305. Hayek A, Chapman EM, Crawford JD. Long-term results of treatment of thyrotoxicosis in children and adolescents with radioactive iodine. N Engl J Med 1970;283:949.
306. Safa AM, Schumacher OP, Rodriguez-Antunez A. Long-term follow-up results in children and adolescents treated with radioactive iodine (131I) for hyperthyroidism. N Engl J Med 1975;292:167.
307. Freitas JE, Swanson DP, Gross MD, et al. Iodine-131: optimal therapy for hyperthyroidism in children and adolescents? J Nucl Med 1979;20:847.
308. Sheline GE, Lindsay S, Bell HG. Occurrence of thyroid nodules in children following therapy with radioiodine for hyperthyroidism. J Clin Endocrinol Metab 1959;19:127.
309. Stoffer SS, Hamburger JI. Inadvertent 131I therapy for hyperthyroidism in the first trimester of pregnancy. J Nucl Med 1976;17:146.
310. Cantolino SJ, Schmickel RD, Ba UM, et al. Persistent chromosomal aberrations following radioiodine therapy for thyrotoxicosis. N Engl J Med 1966;275:739.
311. Nofal MM, Beierwaltes WH. Persistent chromosomal aberrations following radioiodine therapy. J Nucl Med 1964;5:840.
312. Vormittag W, Ring F, Kunze-Muhl E, et al. Structural chromosomal aberrations before and after administration of 20 mCi iodine-131. Mutat Res 1982;105:333.
313. Robertson JS, Gorman CA. Gonadal radiation dose and its genetic significance in radioiodine therapy of hyperthyroidism. J Nucl Med 1976;17:826.
314. Sarkar SD, Beierwaltes WH, Gill SP, et al. Subsequent fertility and birth histories of children and adolescents treated with 131I for thyroid cancer. J Nucl Med 1976;17:460.
315. Einhorn J, Hulten M, Lindsten J, et al. Clinical and cytogenetic investigation in children of parents treated with radioiodine. Acta Radiol 1972:11:193.
316. Gorman CA. Radioiodine and pregnancy. Thyroid 1999;9:721.
317. Kaplan MM, Meier DA, Dworkin HJ. Treatment of hyperthyroidism with radioactive iodine. Endocrinol Metab Clin North Am 1998;27:205.
318. Culver C, Dworkin HJ. Radiation safety considerations for post–iodine-131 hyperthyroid therapy. J Nucl Med 1991;32: 169.
319. Koornstra JJ, Kerstens MN, Hoving J, et al. Clinical and biochemical changes following 131I therapy for hyperthyroidism in patients not pretreated with antithyroid drugs. Neth J Med 1999;55:215.
320. McDermott MT, Kidd GS, Dodson LE, et al. Radioiodine-induced thyroid storm. Am J Med 1983;75:353.
321. Kadmon PM, Noto RB, Boney CM, et al. Thyroid storm in a child following radioactive iodine (RAI) therapy: a consequence of RAI versus withdrawal of antithyroid medication. J Clin Endocrinol Metab 2001;86:1865.
322. Andrade VA, Gross JL, Maia AL. Effect of methimazole pretreatment on serum thyroid hormone levels after radioiodine treatment in Graves' hyperthyroidism. J Clin Endocrinol Metab 1999;84:4012.
323. Burch HB, Solomon BL, Cooper DS, et al. The effect of antithyroid drug pretreatment on acute changes in thyroid hormone levels after (131)I ablation for Graves' disease. J Clin Endocrinol Metab 2001;86:3016.
324. Cooper DS. Antithyroid drugs and radioiodine therapy: a grain of (iodized) salt. Ann Intern Med 1994;121:612.
325. Bogazzi F, Bartalena L, Campomori A, et al. Treatment with lithium prevents serum thyroid hormone increase after thionamide withdrawal and radioiodine therapy in patients with Graves' disease. J Clin Endocrinol Metab 2002;87:4490.
326. Bogazzi F, Bartalena L, Brogioni S, et al. Comparison of radioiodine with radioiodine plus lithium in the treatment of Graves' hyperthyroidism. J Clin Endocrinol Metab 1999;84: 499.
327. Marcocci C, Bartalena L, Bogazzi F, et al. Relationship between Graves' ophthalmopathy and type of treatment of Graves' hyperthyroidism. Thyroid 1992;2:171.
328. Tallstedt L, Lundell G. Radioiodine treatment, ablation, and ophthalmopathy: a balanced perspective. Thyroid 1997;7:241.
329. Gwinup G, Elias AN, Ascher MS. Effect on exophthalmos of various methods of treatment of Graves' disease. JAMA 1982; 247:2135.
330. Sridama V, DeGroot LJ. Treatment of Graves' disease and the course of ophthalmopathy. Am J Med 1989;87:70.
331. Bartalena L, Marcocci C, Bogazzi F, et al. Use of corticosteroids to prevent progression of Graves' ophthalmopathy after radioiodine therapy for hyperthyroidism. N Engl J Med 1989;321:1349.
332. Vestergaard H, Laurberg P. Radioiodine and aggravation of Graves' ophthalmopathy. Lancet 1989;1:47.
333. Tallstedt L, Lundell G, Torring O, et al. Occurrence of ophthalmopathy after treatment for Graves' hyperthyroidism. N Engl J Med 1992;326:1733.
334. Bartalena L, Marcocci C, Bogazzi F, et al. Relation between therapy for hyperthyroidism and the course of Graves' ophthalmopathy. N Engl J Med 1998;338:73.
335. Atkinson S, McGregor AM, Kendall-Taylor P, et al. Effect of radioiodine on stimulatory activity of Graves' immunoglobulins. Clin Endocrinol (Oxf) 1982;16:537.
336. Kung WC, Yau CC, Cheng A. The incidence of ophthalmopathy after radioiodine therapy for Graves' disease: prognostic factors and the role of methimazole. J Clin Endocrinol Metab 1994;79:542.
337. Tallstedt L, Lundell G, Blomgren H, et al. Does early administration of thyroxine reduce the development of Graves' ophthalmopathy after radioiodine treatment? Eur J Endocrinol 1994;130:494.
338. Vazquez-Chavez C, Nishimura Meguro E, Espinosa Said L, et al. Effect of the treatment of hyperthyroidism on the course of exophthalmos. Rev Invest Clin 1992;44:241.
339. Bartalena L, Marcocci C, Tanda ML, et al. Cigarette smoking and treatment outcomes in Graves ophthalmopathy. Ann Intern Med 1998;129:632.
340. Winsa B, Rastad J, Larsson E, et al. Total thyroidectomy in therapy-resistant Graves' disease. Surgery 1994;116:1068.
341. Parwardhan NA, Moront M, Rao S, et al. Surgery still has a role in Graves' hyperthyroidism. Surgery 1993;114:1108.
342. Klementschitsch P, Shen K, Kaplan EL. Reemergence of thyroidectomy as treatment for Graves' disease. Surg Clin North Am 1979;59:35.
343. Palit TK, Miller CC III, Miltenburg DM. The efficacy of thyroidectomy for Graves' disease: a meta-analysis. J Surg Res 2000;90:161.
344. Maier WP, Derrick BM, Marks AD, et al. Long-term follow-up of patients with Graves' disease treated by subtotal thyroidectomy. Am J Surg 1984;147:267.
345. Sugino K, Mimura T, Ozaki O, et al. Management of recurrent hyperthyroidism in patients with Graves' disease treated by subtotal thyroidectomy. J Endocrinol Invest 1995;18:415.
346. Sosa JA, Bowman HM, Tielsch JM, et al. The importance of surgeon experience for clinical and economic outcomes from thyroidectomy. Ann Surg 1998;228:320.
347. Hedley AJ, Bewsher PD, Jones SJ, et al. Late onset hypothyroidism after subtotal thyroidectomy for hyperthyroidism: implications for long term follow-up. Br J Surg 1983;70:740.
348. Lee TC, Coffey RJ, Currier BM, et al. Propranolol and thyroidectomy in the treatment of thyrotoxicosis. Ann Surg 1982; 195:766.
349. Reid DJ. Hyperthyroidism and hypothyroidism complicating the treatment of thyrotoxicosis. Br J Surg 1987;74:1060.
350. Sugrue D, Drury MI, McEvoy M, et al. Long term follow-up of hyperthyroid patients treated by subtotal thyroidectomy. Br J Surg 1983;70:408.
351. Harada T, Shimaoka K, Arita S, et al. Follow up evaluation of thyroid function after thyroidectomy for thyrotoxicosis. World J Surg 1984;8:444.
352. Sugino K, Mimura T, Ozaki O, et al. Early recurrence of hyperthyroidism in patients with Graves' disease treated by subtotal thyroidectomy. World J Surg 1995;19:648.
353. Winsa B, Rastad J, Akerstrom G, et al. Retrospective evaluation of subtotal and total thyroidectomy in Graves' disease with and without endocrine ophthalmopathy. Eur J Endocrinol 1995; 132:406.
354. Kalk WJ, Durbach D, Kantor S, et al. Postthyroidectomy thyrotoxicosis. Lancet 1978;1:2911.
355. Lennquist S, Jortso E, Anderberg BO, et al. Beta blockers compared with antithyroid drugs as preoperative treatment in hyperthyroidism: drug tolerance, complications, and postoperative thyroid function. Surgery 1985;98:1141.
356. Peden NR, Browning MCK, Feely J, et al. The clinical and metabolic responses to early surgical treatment for hyperthyroid Graves' disease: a comparison of three preoperative treatment regimens. QJM 1985;221:579.
357. Feely J, Crooks J, Forrest AL, et al. Propranolol in the surgical treatment of hyperthyroidism, including severely thyrotoxic patients. Br J Surg 1981;68:865.
358. Marigold JH, Morgan AK, Earle DJ, et al. Lugol's iodine: its effect on thyroid blood flow in patients with thyrotoxicosis. Br J Surg 1985;72:45.
359. Rudberg C, Johansson H, Akerstrom G, et al. Graves' disease in children and adolescents. Late results of surgical treatment. Eur J Endocrinol 1996;134:710.
360. Buckingham BA, Costin G, Roe TF, et al. Hyperthyroidism in children. Am J Dis Child 1981;135:112.
361. Hamburger JI. Management of hyperthyroidism in children and adolescents. J Clin Endocrinol Metab 1985;60:1019.
362. Glaser NS, Styne DM. Predictors of early remission of hyperthyroidism in children. J Clin Endocrinol Metab 1997;82: 1719.
363. Zimmerman D, Lteif AN. Thyrotoxicosis in children. Endocrinol Metab Clin North Am 1998;27:109.
364. Cooper DS.Antithyroid drugs in the management of patients with Graves' disease: an evidence-based approach to therapeutic controversies. J Clin Endocrinol Metab 2003;88:3474.
365. Aro A, Huttunen JK, Lamberg B-A, et al. Comparison of propranolol and carbimazole as adjuncts to iodine-131 therapy of hyperthyroidism. Acta Endocrinol 1981;96:321.
366. Tibaldi JM, Barzel US, Albin J, et al. Thyrotoxicosis in the very old. Am J Med 1986;81:619.
367. Hamburger JI, Paul S. When and how to use higher 131I doses for hyperthyroidism. N Engl J Med 1968;279:1361.
368. Roti E, Uberti ED. Iodine excess and hyperthyroidism. Thyroid 2001;11:493.
369. Masiukiewicz US, Burrow GN. Hyperthyroidism in pregnancy: diagnosis and treatment. Thyroid 1999;9:647.
370. Mortimer RH, Cannell GR, Addison RS, et al. Methimazole and propylthiouracil equally cross the perfused human term placental lobule. J Clin Endocrinol Metab 1997;82:3099.
371. Momotani N, Noh JY, Ishikawa N, et al. Effects of propylthiouracil and methimazole on fetal thyroid status in mothers with Graves' hyperthyroidism. J Clin Endocrinol Metab 1997;82: 3633.
372. Wing DA, Millar LK, Koonings PP, et al. A comparison of propylthiouracil and methimazole in the treatment of hyperthyroidism in pregnancy. Am J Obstet Gynecol 1994;170:90.
373. Di Gianantonio E, Schaefer C, Mastroiacovo PP, et al. Adverse effects of prenatal methimazole exposure. Teratology 2001;64: 262.
374. Mandel SJ, Brent GA, Larsen PR. Review of antithyroid drug use during pregnancy and report of a case of aplasia cutis. Thyroid 1994;4:129.
375. Mandel SJ, Cooper DS. The use of antithyroid drugs in pregnancy and lactation. J Clin Endocrinol Metab 2001;86:2354.
376. Momotani N, Ito K, Hamada N, et al. Maternal hyperthyroidism and congenital malformation in the offspring. Clin Endocrinol (Oxf) 1984;20:695.
377. Davis LE, Lucas MJ, Hankins GDV, et al. Thyrotoxicosis complicating pregnancy. Am J Obstet Gynecol 1989;160:63.
378. Momotani N, Noh J, Oyanagi H, et al. Antithyroid drug therapy for Graves' disease during pregnancy. N Engl J Med 1986; 315:24.
379. Gardner DF, Cruikshank DP, Hays PM, et al. Pharmacology of propylthiouracil (PTU) in pregnant hyperthyroid women: correlation of maternal PTU concentrations with cord serum thyroid function tests. J Clin Endocrinol Metab 1986;62:277.
380. Cheron RG, Kaplan MM, Larsen PR, et al. Neonatal thyroid function after propylthiouracil therapy for maternal Graves' disease. N Engl J Med 1981;304:525.
381. Messer PM, Hauffa BP, Olbricht T, et al. Antithyroid drug treatment of Graves' disease in pregnancy: long-term effects on somatic growth, intellectual development and thyroid function of the offspring. Acta Endocrinol 1990;123:311.
382. Eisenstein Z, Weiss M, Katz Y, et al. Intellectual capacity of subjects exposed to methimazole or propylthiouracil in utero. Eur J Pediatr 1992;151:558.
383. Sherif IH, Oyan WT, Bosairi S, et al. Treatment of hyperthyroidism in pregnancy. Acta Obstet Gynecol Scand 1991;70: 461.
384. Momotani N, Hisaokat, Noh J, et al. Effects of iodine on thyroid status of fetus versus mother in treatment of Graves' disease complicated by pregnancy. J Clin Endocrinol Metab 1992; 75:738.
385. Lamberg BA, Ikonen E, Osterlung K, et al. Antithyroid treatment of maternal hyperthyroidism during lactation. Clin Endocrinol (Oxf) 1984;21:81.
386. Cooper DS. Antithyroid drugs: to breast-feed or not to breast-feed. Am J Obstet Gynecol 1987;157:234.
387. Momotani N, Yamashita R, Makino F, et al. Thyroid function in wholly breast-feeding infants whose mothers take high doses of propylthiouracil. Clin Endocrinol (Oxf) 2000;53:177.
388. Azizi F, Hedayati M. Thyroid function in breast-fed infants whose mothers take high doses of methimazole. J Endocrinol Invest 2002;25:493.
389. American Academy of Pediatrics, Committee on Drugs. Transfer of drugs and other chemicals into human milk. Pediatrics 2001;108:776.
390. Siegel RD, Lee SL. Toxic nodular goiter. Endocrinol Metab Clin North Am 1998;27:151.
391. Diez JJ. Hyperthyroidism in patients older than 55 years: an analysis of the etiology and management. Gerontology 2003; 49:316.
392. Cooke ST, Ratcliffe G, Fogelman I, et al. Prevalence of inappropriate drug treatment in patients with hyperthyroidism. BMJ 1985;291:1491.
393. Van Soestbergen MJM, Van der Vijver JCM, Graafland AD. Recurrence of hyperthyroidism in multinodular goiter after long-term drug therapy: a comparison with Graves' disease. J Endocrinol Invest 1992;15:797.
394. Franklyn JA. The management of hyperthyroidism. N Engl J Med 1994;330:1731.
395. Ferrari C, Reschini E, Paracchi A. Treatment of the autonomous thyroid nodule: a review. Eur J Endocrinol 1996;135:383.
396. Ross DS, Ridgway EC, Daniels GH. Successful treatment of solitary toxic thyroid nodules with relatively low-dose iodine-131, with low prevalence of hypothyroidism. Ann Intern Med 1984;101:488.
397. Huysmans DA, Corstens FH, Kloppenborg PW. Long-term follow-up in toxic solitary autonomous thyroid nodules treated with radioactive iodine. J Nucl Med 1991;32:27.
398. Burch HB, Shakir F, Fitzsimmons TR, et al. Diagnosis and management of the autonomously functioning thyroid nodule: the Walter Reed Army Medical Center experience, 1975–1996. Thyroid 1998; 8:871.
399. Goldstein R, Hart IA. Follow-up of solitary autonomous thyroid nodules treated with 131I. N Engl J Med 1983;309:1473.
400. Gorman CA, Robertson JS. Radiation dose in the selection of 131I or surgical treatment for toxic thyroid adenoma. Ann Intern Med 1978;89:85.
401. Zingrillo M, Torlontano M, Ghiggi MR, et al. Radioiodine and percutaneous ethanol injection in the treatment of large toxic thyroid nodule: a long-term study. Thyroid 2000;10:985.
402. Hamburger JI, Hamburger SW. Diagnosis and management of large toxic multinodular goiters. J Nucl Med 1985;26:888.
403. Franklyn JA, Daykin J, Holder R, et al.Radioiodine therapy compared in patients with toxic nodular or Graves' hyperthyroidism. QJM 1995;88:175.