Ilene Claudius
![]() Sickle cell anemia-related emergencies in children include vaso-occlusive crises, hematologic crises, and infections. All children with sickle cell anemia presenting with fever, pain, respiratory distress, or a change in neurologic function require a rapid and thorough emergency department (ED) evaluation.
Sickle cell anemia-related emergencies in children include vaso-occlusive crises, hematologic crises, and infections. All children with sickle cell anemia presenting with fever, pain, respiratory distress, or a change in neurologic function require a rapid and thorough emergency department (ED) evaluation.
EPIDEMIOLOGY
![]() In the United States about 8% of the African-American population carries the hemoglobin (HbS) gene and about 0.15% are homozygous (HbSS).
In the United States about 8% of the African-American population carries the hemoglobin (HbS) gene and about 0.15% are homozygous (HbSS).
![]() While vaso-occlusive crises are most common, infection is the leading cause of death.
While vaso-occlusive crises are most common, infection is the leading cause of death.
![]() Symptoms are rarely seen before 4 months of age due to the predominance of fetal hemoglobin.
Symptoms are rarely seen before 4 months of age due to the predominance of fetal hemoglobin.
PATHOPHYSIOLOGY
![]() When deoxygenated, HbS polymerizes abnormally, creating chains that distort the red blood cell membrane into the characteristic sickle shape, impeding blood flow. The resultant tissue hypoxia and acidosis encourages further sickling.
When deoxygenated, HbS polymerizes abnormally, creating chains that distort the red blood cell membrane into the characteristic sickle shape, impeding blood flow. The resultant tissue hypoxia and acidosis encourages further sickling.
![]() Decline in splenic function from infarction begins at 4 months of age and results in susceptibility to overwhelming infection with encapsulated organisms.
Decline in splenic function from infarction begins at 4 months of age and results in susceptibility to overwhelming infection with encapsulated organisms.
VASO-OCLUSIVE CRISES
![]() Vaso-occlusive episodes represent tissue ischemia and infarction from intravascular sickling. Bones, soft tissue, viscera, and the central nervous system can all be affected.
Vaso-occlusive episodes represent tissue ischemia and infarction from intravascular sickling. Bones, soft tissue, viscera, and the central nervous system can all be affected.
PAIN CRISES
CLINICAL FEATURES
![]() The classic pain crisis is usually typical (ie, similar to prior episodes) in body location, character, and severity for each individual patient. Long bones and the lower back are common sites of pain.
The classic pain crisis is usually typical (ie, similar to prior episodes) in body location, character, and severity for each individual patient. Long bones and the lower back are common sites of pain.
![]() Crises can be triggered by stress, cold, dehydration, hypoxia, anemia, or infection, but most often occur without a specific cause.
Crises can be triggered by stress, cold, dehydration, hypoxia, anemia, or infection, but most often occur without a specific cause.
![]() Often there are no physical findings, although pain, local tenderness, swelling, and warmth may occur.
Often there are no physical findings, although pain, local tenderness, swelling, and warmth may occur.
![]() Dactylitis (painful swelling of hands or feet) due to ischemia of the metatarsal/metacarpal nutrient vessels may be the initial presenting manifestation of sickle cell anemia in infants. A low-grade temperature elevation can accompany dactylitis.
Dactylitis (painful swelling of hands or feet) due to ischemia of the metatarsal/metacarpal nutrient vessels may be the initial presenting manifestation of sickle cell anemia in infants. A low-grade temperature elevation can accompany dactylitis.
![]() Abdominal pain crises from mesenteric, hepatic, or splenic ischemia commonly present as recurrent episodes of abrupt-onset poorly localized pain and tenderness without rebound or rigidity.
Abdominal pain crises from mesenteric, hepatic, or splenic ischemia commonly present as recurrent episodes of abrupt-onset poorly localized pain and tenderness without rebound or rigidity.
DIAGNOSIS AND DIFFERENTIAL
![]() Pain crises can be associated with low-grade fever and leukocytosis.
Pain crises can be associated with low-grade fever and leukocytosis.
![]() Temperatures >38.3°C (101°F) or a left shift in the leukocyte distribution likely indicate an infectious cause (eg, osteomyelitis, septic arthritis).
Temperatures >38.3°C (101°F) or a left shift in the leukocyte distribution likely indicate an infectious cause (eg, osteomyelitis, septic arthritis).
![]() Consider avascular necrosis of the femoral head in patients with hip pain (see Fig. 86-1).
Consider avascular necrosis of the femoral head in patients with hip pain (see Fig. 86-1).
![]() With abdominal pain, cholelithiasis, splenic sequestration, and non-sickle cell anemia-related causes should be considered as well.
With abdominal pain, cholelithiasis, splenic sequestration, and non-sickle cell anemia-related causes should be considered as well.
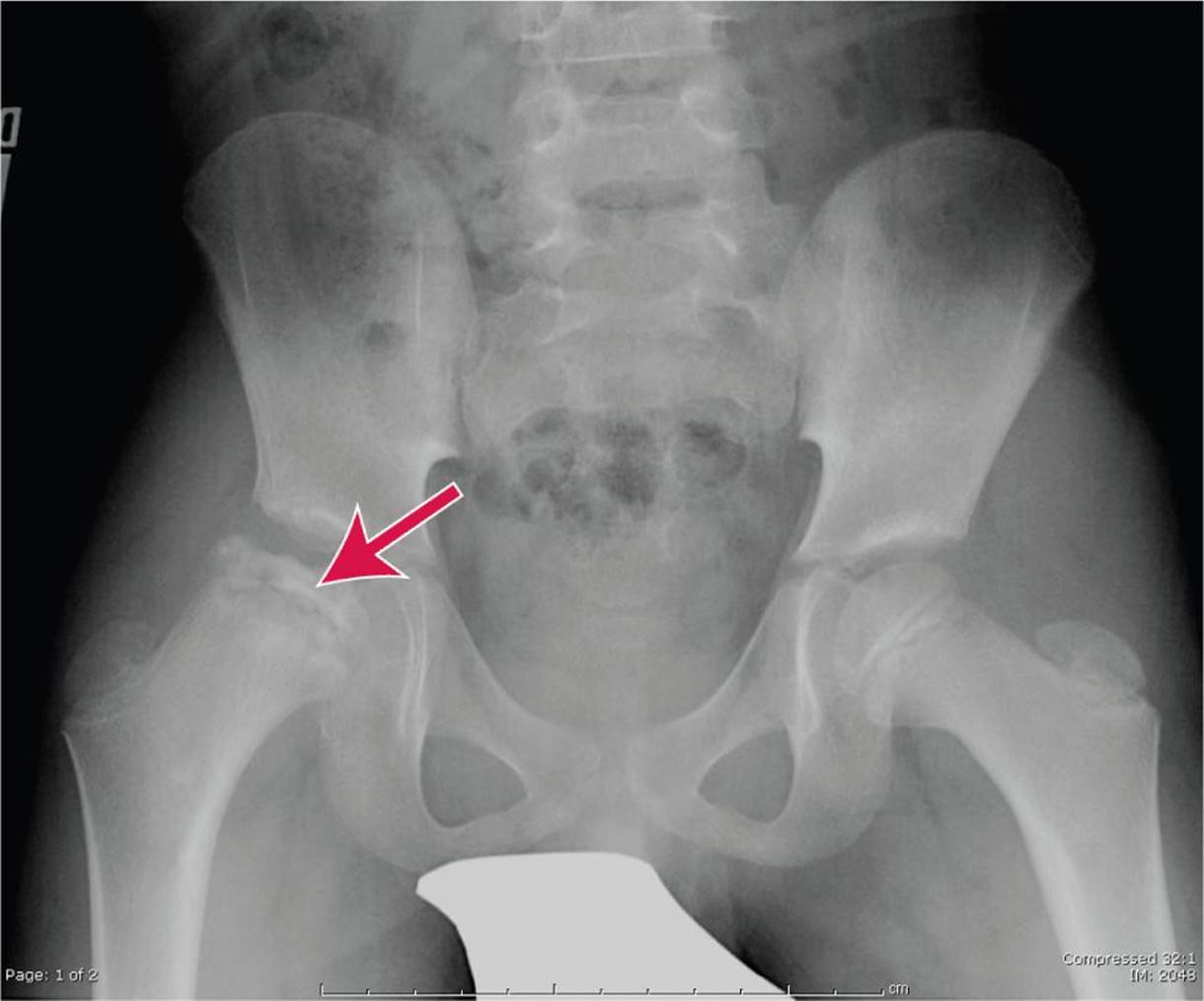
FIG. 86-1. Avascular necrosis of the femoral head. (Courtesy of Hollie Jackson, MD.)
EMERGENCY DEPARTMENT CARE AND DISPOSITION
![]() Hydrate with oral or IV fluids (D5.45 normal saline) at a rate 1½ times maintenance. IV boluses of normal saline are indicated in the dehydrated or hypotensive patient (20 mL/kg), with careful attention to avoid fluid overload.
Hydrate with oral or IV fluids (D5.45 normal saline) at a rate 1½ times maintenance. IV boluses of normal saline are indicated in the dehydrated or hypotensive patient (20 mL/kg), with careful attention to avoid fluid overload.
![]() Mild to moderate pain can often be managed with oral hydration and analgesics, such as narcotics and nons-teroidal anti-inflammatory drugs (NSAIDs).
Mild to moderate pain can often be managed with oral hydration and analgesics, such as narcotics and nons-teroidal anti-inflammatory drugs (NSAIDs).
![]() Parenteral, long-acting narcotics (morphine 0.1–1.0 milligram/kg IV/IM or hydromorphone 0.015 milligram/kg IV/IM) and NSAIDs (ketorolac 0.5 milligram/kg) are indicated if oral regimens are inadequate.
Parenteral, long-acting narcotics (morphine 0.1–1.0 milligram/kg IV/IM or hydromorphone 0.015 milligram/kg IV/IM) and NSAIDs (ketorolac 0.5 milligram/kg) are indicated if oral regimens are inadequate.
![]() Oxygen is rarely useful in a patient without hypoxemia.
Oxygen is rarely useful in a patient without hypoxemia.
![]() Consider transfusion for acute drop in hemoglobin from baseline, or Hb <5 grams/dL.
Consider transfusion for acute drop in hemoglobin from baseline, or Hb <5 grams/dL.
![]() Admission is warranted for poor pain control or inadequate oral fluid intake.
Admission is warranted for poor pain control or inadequate oral fluid intake.
ACUTE CHEST SYNDROME
![]() Acute chest syndrome, due to ischemia and infarction, is frequently a complication of pneumonia in children.
Acute chest syndrome, due to ischemia and infarction, is frequently a complication of pneumonia in children.
CLINICAL FEATURES
![]() Acute chest syndrome should be considered in all patients with sickle cell disease (SCD) who present with complaints of chest pain. Signs and symptoms include pleuritic chest pain, cough, fever, dyspnea, hypoxia, tachypnea, rhonchi, or rales. Patients can deteriorate rapidly.
Acute chest syndrome should be considered in all patients with sickle cell disease (SCD) who present with complaints of chest pain. Signs and symptoms include pleuritic chest pain, cough, fever, dyspnea, hypoxia, tachypnea, rhonchi, or rales. Patients can deteriorate rapidly.
![]() Common concurrent infections include Chlamydia, Mycoplasma, viral, Streptococcus pneumoniae, Staphylococcus aureus, and Haemophilus influenzae.
Common concurrent infections include Chlamydia, Mycoplasma, viral, Streptococcus pneumoniae, Staphylococcus aureus, and Haemophilus influenzae.
DIAGNOSIS AND DIFFERENTIAL
![]() Chest radiographs should be obtained, but may be normal initially (see Fig. 86-2).
Chest radiographs should be obtained, but may be normal initially (see Fig. 86-2).
![]() A complete blood count (CBC) typically indicates a leukocytosis in pneumonia or acute chest crisis. Low Hb may indicate need for transfusion. Low platelets predict poor outcome.
A complete blood count (CBC) typically indicates a leukocytosis in pneumonia or acute chest crisis. Low Hb may indicate need for transfusion. Low platelets predict poor outcome.
![]() Asthma, pulmonary hypertension, and fat embolus (from bony infarct) are other complications of sickle cell anemia to consider in the patient with respiratory distress.
Asthma, pulmonary hypertension, and fat embolus (from bony infarct) are other complications of sickle cell anemia to consider in the patient with respiratory distress.
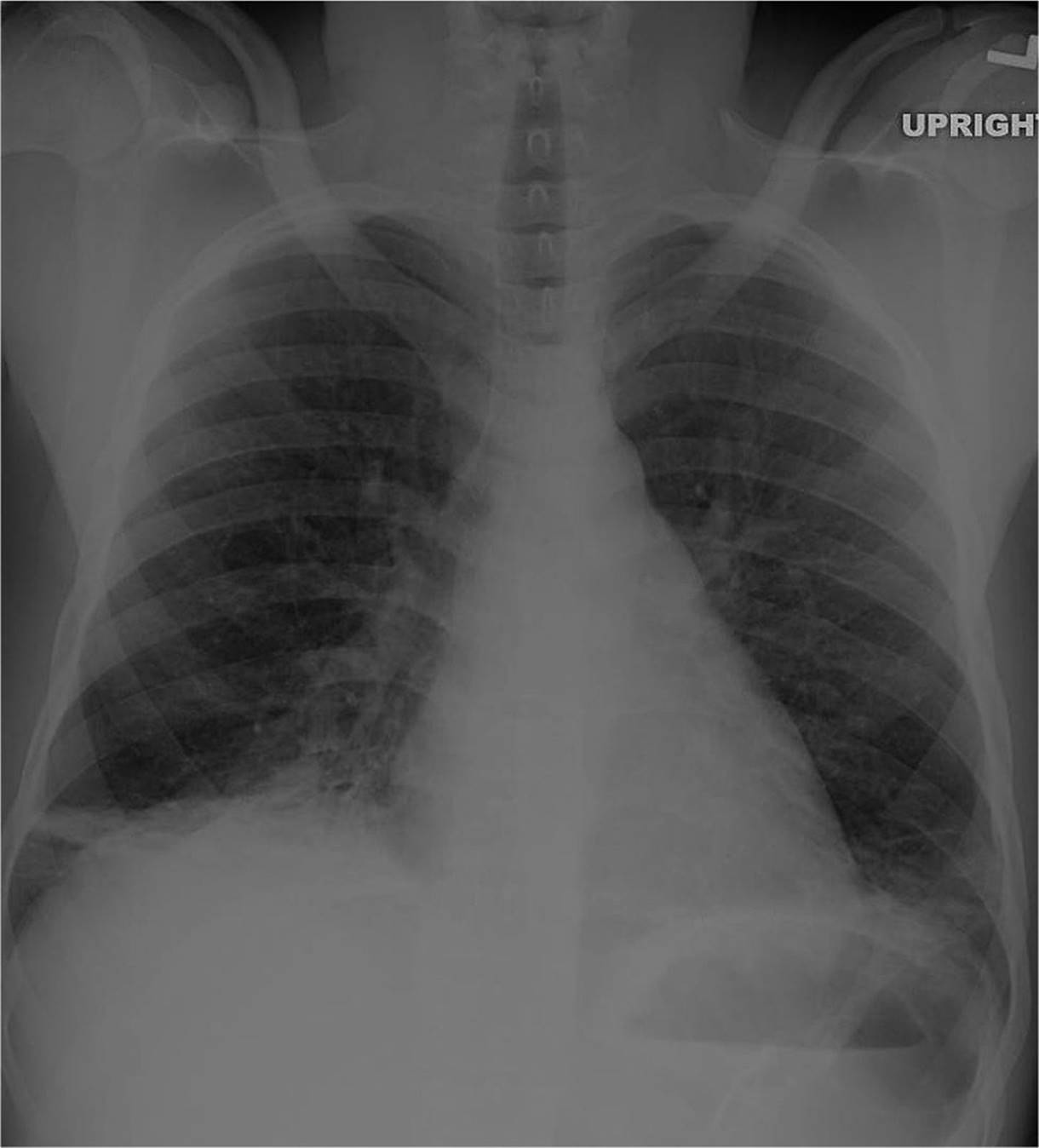
FIG. 86-2. Acute chest crisis. (Courtesy of Hollie Jackson, MD.)
EMERGENCY DEPARTMENT CARE AND DISPOSITION
![]() Monitor pulse oximetry and work of breathing.
Monitor pulse oximetry and work of breathing.
![]() Supplemental oxygen should be provided if respiratory distress is present or if oxygen saturation is persistently less than or equal to 94%.
Supplemental oxygen should be provided if respiratory distress is present or if oxygen saturation is persistently less than or equal to 94%.
![]() Adequate analgesia for chest pain should be provided (see above), as well as IV hydration with D5.45NS at 1 to 1½ times maintenance.
Adequate analgesia for chest pain should be provided (see above), as well as IV hydration with D5.45NS at 1 to 1½ times maintenance.
![]() Treat underlying bacterial pneumonia with empiric antibiotic therapy such as ceftriaxone 50 milligrams/kg and a macrolide.
Treat underlying bacterial pneumonia with empiric antibiotic therapy such as ceftriaxone 50 milligrams/kg and a macrolide.
![]() Consider transfusion, in consultation with a pediatric hematologist, for persistent Pao2 < 70 mm Hg or oxygen saturation drop of 10% from baseline. Packed red blood cell (PRBC) transfusions are indicated for Hb levels <10 grams/dL and exchange transfusions are indicated for patients with Hb levels >10 grams/dL in these circumstances.
Consider transfusion, in consultation with a pediatric hematologist, for persistent Pao2 < 70 mm Hg or oxygen saturation drop of 10% from baseline. Packed red blood cell (PRBC) transfusions are indicated for Hb levels <10 grams/dL and exchange transfusions are indicated for patients with Hb levels >10 grams/dL in these circumstances.
![]() All children with suspected acute chest syndrome warrant hospital admission.
All children with suspected acute chest syndrome warrant hospital admission.
ACUTE CENTRAL NERVOUS SYSTEM EVENTS
CLINICAL FEATURES
![]() Ischemic strokes are not uncommon in children with sickle cell anemia and can present with transient or persistent ischemic symptoms, seizure, altered mental status, or coma. Sickle cell anemia patients are also at increased risk of aneurysmal subarachnoid hemorrhage, although this rarely presents before adolescence.
Ischemic strokes are not uncommon in children with sickle cell anemia and can present with transient or persistent ischemic symptoms, seizure, altered mental status, or coma. Sickle cell anemia patients are also at increased risk of aneurysmal subarachnoid hemorrhage, although this rarely presents before adolescence.
DIAGNOSIS AND DIFFERENTIAL
![]() Computed tomography (CT) scan should be obtained to assess for acute hemorrhage.
Computed tomography (CT) scan should be obtained to assess for acute hemorrhage.
![]() Magnetic resonance imaging (MRI) scan of the brain may be useful to confirm the diagnosis of ischemic stroke, although should not delay treatment.
Magnetic resonance imaging (MRI) scan of the brain may be useful to confirm the diagnosis of ischemic stroke, although should not delay treatment.
![]() A lumbar puncture may be necessary to exclude sub-arachnoid hemorrhage or meningitis if symptoms are suggestive.
A lumbar puncture may be necessary to exclude sub-arachnoid hemorrhage or meningitis if symptoms are suggestive.
![]() A CBC, reticulocyte count, and type and screen should be obtained.
A CBC, reticulocyte count, and type and screen should be obtained.
EMERGENCY DEPARTMENT CARE AND DISPOSITION
![]() Transfuse or exchange transfuse to reduce the percentage of HbS <30%.
Transfuse or exchange transfuse to reduce the percentage of HbS <30%.
![]() Minimize secondary injury by correcting hypoglyc-emia, hypovolemia, and hyperpyrexia.
Minimize secondary injury by correcting hypoglyc-emia, hypovolemia, and hyperpyrexia.
![]() Thrombolysis is not recommended for acute ischemic stroke in this setting.
Thrombolysis is not recommended for acute ischemic stroke in this setting.
![]() There is no consensus on blood pressure management.
There is no consensus on blood pressure management.
![]() Coordinate care with pediatric hematology and neurology or neurosurgery, depending on type of stroke. All children with diagnosed or suspected CNS event should be admitted to the pediatric intensive care unit for close monitoring and further care.
Coordinate care with pediatric hematology and neurology or neurosurgery, depending on type of stroke. All children with diagnosed or suspected CNS event should be admitted to the pediatric intensive care unit for close monitoring and further care.
PRIAPISM
![]() Priapism, a painful sustained erection in the absence of sexual stimulation, occurs when sickled cells accumulate in the corpora cavernosa. It can affect all males with SCD regardless of age, and severe prolonged attacks can cause impotence.
Priapism, a painful sustained erection in the absence of sexual stimulation, occurs when sickled cells accumulate in the corpora cavernosa. It can affect all males with SCD regardless of age, and severe prolonged attacks can cause impotence.
![]() Patients with priapism should receive IV hydration with D5.45NS at VA 1½ times maintenance, appropriate analgesia, and bladder catheterization if the patient is unable to void spontaneously.
Patients with priapism should receive IV hydration with D5.45NS at VA 1½ times maintenance, appropriate analgesia, and bladder catheterization if the patient is unable to void spontaneously.
![]() Treatment options include oral α-adrenergic agonists (eg, terbutaline), needle aspiration of the corpora cavernosa, or intrapenile injection of dilute epinephrine.
Treatment options include oral α-adrenergic agonists (eg, terbutaline), needle aspiration of the corpora cavernosa, or intrapenile injection of dilute epinephrine.
![]() Transfusion or exchange transfusion to decrease the percentage of HbS may be necessary.
Transfusion or exchange transfusion to decrease the percentage of HbS may be necessary.
![]() Management and admission decisions should be made promptly in consultation with a urologist and pediatric hematologist.
Management and admission decisions should be made promptly in consultation with a urologist and pediatric hematologist.
HEMATOLOGICAL CRISES
ACUTE SPLENIC SEQUESTRATION CRISES
CLINICAL FEATURES
![]() The spleen of a young child with sickle cell anemia can enlarge, trapping much of the circulating blood volume. This condition can quickly progress to hypotension, shock, and death.
The spleen of a young child with sickle cell anemia can enlarge, trapping much of the circulating blood volume. This condition can quickly progress to hypotension, shock, and death.
![]() Classically, children present with sudden-onset left upper quadrant pain, pallor, and lethargy and have a markedly enlarged, tender, and firm spleen on abdominal examination.
Classically, children present with sudden-onset left upper quadrant pain, pallor, and lethargy and have a markedly enlarged, tender, and firm spleen on abdominal examination.
![]() Minor episodes can occur with insidious onset of abdominal pain, slowly progressive splenomegaly, and a more minor fall in hemoglobin level (Hb >6 grams/dL).
Minor episodes can occur with insidious onset of abdominal pain, slowly progressive splenomegaly, and a more minor fall in hemoglobin level (Hb >6 grams/dL).
![]() Because of splenic infarction, this rarely occurs after age 5 years in children with HbSS, but can occur later in patients with HbSC or HbS/β-thalassemia.
Because of splenic infarction, this rarely occurs after age 5 years in children with HbSS, but can occur later in patients with HbSC or HbS/β-thalassemia.
![]() Less commonly, sequestration can occur in the liver. Clinical features include an enlarged and tender liver with associated hyperbilirubinemia, severe anemia, and elevated reticulocyte count. Cardiovascular collapse is rare in this condition.
Less commonly, sequestration can occur in the liver. Clinical features include an enlarged and tender liver with associated hyperbilirubinemia, severe anemia, and elevated reticulocyte count. Cardiovascular collapse is rare in this condition.
DIAGNOSIS AND DIFFERENTIAL
![]() A CBC reveals a profound anemia with normal to elevated reticulocyte count.
A CBC reveals a profound anemia with normal to elevated reticulocyte count.
EMERGENCY DEPARTMENT CARE AND DISPOSITION
![]() Transfusion with PRBCs is often required. Profoundly anemic children require more gentle transfusions, with the following amounts given over 3 to 4 hours: 3 mL/kg for pre-transfusion Hb <4 milligrams/dL, 5 mL/kg for pre-transfusion Hb 4 to 6 milligrams/dL, 10 mL/kg for pre-transfusion Hb >6 milligrams/dL.
Transfusion with PRBCs is often required. Profoundly anemic children require more gentle transfusions, with the following amounts given over 3 to 4 hours: 3 mL/kg for pre-transfusion Hb <4 milligrams/dL, 5 mL/kg for pre-transfusion Hb 4 to 6 milligrams/dL, 10 mL/kg for pre-transfusion Hb >6 milligrams/dL.
APLASTIC EPISODES
![]() Potentially life threatening, aplastic episodes are precipitated primarily by viral infections (classically, parvovirus B19), but can also be caused by bacterial infections, folic acid deficiency, or bone-marrow-suppressive drugs.
Potentially life threatening, aplastic episodes are precipitated primarily by viral infections (classically, parvovirus B19), but can also be caused by bacterial infections, folic acid deficiency, or bone-marrow-suppressive drugs.
![]() Patients present with gradual onset of pallor without pain or jaundice.
Patients present with gradual onset of pallor without pain or jaundice.
![]() A CBC reveals unusually low hemoglobin with decreased or absent reticulocytosis.
A CBC reveals unusually low hemoglobin with decreased or absent reticulocytosis.
![]() Blood transfusion may be required for severe anemia or symptoms (see guideline below). Hb <6 grams/dL or a drop of 3 grams/dL or symptoms are considered criteria for transfusion.
Blood transfusion may be required for severe anemia or symptoms (see guideline below). Hb <6 grams/dL or a drop of 3 grams/dL or symptoms are considered criteria for transfusion.
HEMOLYTIC CRISES
![]() Bacterial and viral infections in children with SCD can also precipitate an increasing degree of active hemolysis.
Bacterial and viral infections in children with SCD can also precipitate an increasing degree of active hemolysis.
![]() Onset is usually sudden. A CBC reveals a hemoglobin level decreased from baseline, with markedly increased reticulocytosis. Increased jaundice and pallor are noticed on physical examination.
Onset is usually sudden. A CBC reveals a hemoglobin level decreased from baseline, with markedly increased reticulocytosis. Increased jaundice and pallor are noticed on physical examination.
![]() Specific therapy is rarely required. Hematologic values return to normal as the infectious process resolves. Care should be directed toward treating the underlying infection. Close follow-up to monitor hemoglobin and reticulocyte count should be arranged at discharge.
Specific therapy is rarely required. Hematologic values return to normal as the infectious process resolves. Care should be directed toward treating the underlying infection. Close follow-up to monitor hemoglobin and reticulocyte count should be arranged at discharge.
INFECTIONS
CLINICAL FEATURES
![]() Due to functional asplenia, deficient antibody production, and impaired phagocytosis, bacterial infections, especially with encapsulated organisms, pose a serious and potentially fatal threat to young children with SCD and require aggressive management.
Due to functional asplenia, deficient antibody production, and impaired phagocytosis, bacterial infections, especially with encapsulated organisms, pose a serious and potentially fatal threat to young children with SCD and require aggressive management.
![]() Sickle cell anemia patients should receive penicillin prophylaxis until 5 years of age as well as routine vaccinations, including pneumococcus.
Sickle cell anemia patients should receive penicillin prophylaxis until 5 years of age as well as routine vaccinations, including pneumococcus.
DIAGNOSIS AND DIFFERENTIAL
![]() A CBC, reticulocyte count, and blood cultures should be obtained for all children with SCD and fever or history of fever. Clinical signs and symptoms should direct the remainder of the workup, including lumbar puncture as indicated.
A CBC, reticulocyte count, and blood cultures should be obtained for all children with SCD and fever or history of fever. Clinical signs and symptoms should direct the remainder of the workup, including lumbar puncture as indicated.
EMERGENCY DEPARTMENT CARE AND DISPOSITION
![]() Ill-appearing children or those <1 year of age should be treated empirically with an antibiotic active against S. pneumoniae and H. influenzae (eg, ceftriaxone 50 milligrams/kg).
Ill-appearing children or those <1 year of age should be treated empirically with an antibiotic active against S. pneumoniae and H. influenzae (eg, ceftriaxone 50 milligrams/kg).
![]() Well-appearing children over 1 year of age with temperature <40°C, WBC between 5,000 and 30,000 cells/mm3, platelets >100,000/mcL, Hb>5 grams/dL, no infiltrate on CXR, and no history of pneumococcal sepsis can be discharged after a single dose of ceftriaxone and a 4-hour period of observation with follow-up in 24 hours. Children not meeting these criteria should be admitted for parenteral antibiotics and observation.
Well-appearing children over 1 year of age with temperature <40°C, WBC between 5,000 and 30,000 cells/mm3, platelets >100,000/mcL, Hb>5 grams/dL, no infiltrate on CXR, and no history of pneumococcal sepsis can be discharged after a single dose of ceftriaxone and a 4-hour period of observation with follow-up in 24 hours. Children not meeting these criteria should be admitted for parenteral antibiotics and observation.
DISPOSITION GUIDELINES
![]() Hospital admission is warranted for the following:
Hospital admission is warranted for the following:
![]() Febrile children not meeting discharge criteria listed earlier
Febrile children not meeting discharge criteria listed earlier
![]() Acute chest crisis
Acute chest crisis
![]() Splenic sequestration
Splenic sequestration
![]() Any new CNS findings or presentations
Any new CNS findings or presentations
![]() Prolonged priapism
Prolonged priapism
![]() Any vaso-occlusive crises not responsive to analgesia and hydration (usually after 4–6 hours of therapy)
Any vaso-occlusive crises not responsive to analgesia and hydration (usually after 4–6 hours of therapy)
![]() Inability to maintain adequate hydration
Inability to maintain adequate hydration
![]() Inadequate assurance of follow-up
Inadequate assurance of follow-up
VARIANTS OF SICKLE CELL DISEASE
![]() Sickle cell trait is the carrier state of SCD and the most common variant. These patients are typically asymptomatic. They have vaso-occlusive complications only in the presence of extreme hypoxia or high altitude. They have minimal complications, the most common being hematuria (1%), most likely due to papillary necrosis in the renal medullary tissue.
Sickle cell trait is the carrier state of SCD and the most common variant. These patients are typically asymptomatic. They have vaso-occlusive complications only in the presence of extreme hypoxia or high altitude. They have minimal complications, the most common being hematuria (1%), most likely due to papillary necrosis in the renal medullary tissue.
![]() Sickle cell-Hemoglobin C disease is a heterozygous condition characterized by mild to moderate anemia and mild reticulocytosis. The smear reveals abundant target cells and few sickled cells. Many adults have splenomegaly, and these patients are at risk for pain crisis and organ infarcts. Most patients, however, have few clinical complications.
Sickle cell-Hemoglobin C disease is a heterozygous condition characterized by mild to moderate anemia and mild reticulocytosis. The smear reveals abundant target cells and few sickled cells. Many adults have splenomegaly, and these patients are at risk for pain crisis and organ infarcts. Most patients, however, have few clinical complications.
![]() Sickle cell-β-thalassemia disease is a heterozygous condition with varying degrees of severity of symptoms, dependent on the amount of normal β-hemoglobin chains that are produced. The severity can range from mild symptoms to a syndrome similar to SCD.
Sickle cell-β-thalassemia disease is a heterozygous condition with varying degrees of severity of symptoms, dependent on the amount of normal β-hemoglobin chains that are produced. The severity can range from mild symptoms to a syndrome similar to SCD.
For further reading in Tintinalli’s Emergency Medicine: A Comprehensive Study Guide, 7th ed., see Chapter 135, “Sickle Cell Disease,” by Ilene Claudius.