Movement disorders can be divided into hypokinetic and hyperkinetic. Hypokinetic movement disorders refer primarily to disorders with decreased amplitude and/or speed of movement (parkinsonism), whereas hyperkinetic movement disorders are those displaying excess of movement (chorea, dystonia, myoclonus, tics, and tremor).
I. HYPOKINETIC MOVEMENT DISORDERS
A. Parkinson’s disease (PD) is the most common cause of degenerative parkinsonism. PD’s response to dopaminergic medications is robust, compared with the atypical parkinsonisms (previously referred to as Parkinson-plus syndromes) in which the response, if any, is partial and transient.
1. Nonpharmacologic management of PD includes education about the disease, support of patient and family, appropriate nutrition, and exercise. Exercise can improve symptoms and their response to treatment, in addition to reducing fatigue, enhancing sleep, and potentially yielding a disease-modifying effect in the long run. A well-balanced diet is essential for PD patients because of the increased risk of malnutrition and weight loss. Redistribution of dietary protein can be beneficial in the care of patients with advanced PD as protein interferes with absorption of levodopa in the gastrointestinal tract.
2. Pharmacologic therapy for PD.
a. Neuroprotection. None of the currently available therapies is firmly acknowledged as disease modifying. The selective monoamine oxidase B (MAO-B) inhibitors selegiline (deprenyl) and rasagiline (Azilect; Teva) have been tested in designs suggestive of such an effect, but their symptomatic benefit may be masking any putative neuroprotective effects. A study testing the potential disease-modifying effect of coenzyme Q10 at a dose of 2,400 mg per day was stopped at an interim analysis for failing to meet a prespecified milestone. Levodopa’s introduction in the 1970s dramatically reduced the mortality from this disease, but this effect is believed to result from a purely symptomatic effect.
b. Symptomatic management of PD. Many drugs are useful for improving parkinsonian symptoms. Initiation of PD treatment can be tailored to patients’ age, employment status, predominant PD symptoms, severity of illness, intercurrent medical problems, side-effect profile of previous medications, and cost.
(1) Selegiline (Eldepryl; Somerset) and Rasagiline (Azilect, TEVA) are irreversible, selective inhibitors of MAO-B at the recommended doses of up to 5 mg twice a day and 1 mg every day, respectively. Zelapar (Zydis selegiline; Valeant) is an orally disintegrating formulation of selegiline, taken at 2.5 mg per day.
Adverse effects of selegiline are relatively infrequent when the drug is used early in the disease. Occasional patients report insomnia related to its amphetamine-like metabolite. To minimize insomnia, selegiline should not be used late in the afternoon or evening. Rasagiline, on the other hand, does not produce amphetamine-like metabolites. Although a restricted low-tyramine diet has been recommended by the American Food and Drug Administration to minimize the “cheese effect,” such effect is not expected at the recommended doses, designed to maintain selectivity of MAO-B receptors.
Patients taking MAO-B inhibitors should not be given meperidine (Demerol; Sanofi) for pain control or dextromethorphan. Although serotonin syndrome has been reported in patients taking selegiline with selective serotonin reuptake inhibitors (SSRIs), many patients are on this combination without adverse effects.
(2) Amantadine (Symmetrel; Endo Pharmaceuticals) is used in the management of mild to moderate PD and is most helpful in addressing tremor and levodopa-induced dyskinesias. The various mechanisms of action include N-methyl-D-aspartate receptor antagonism, blockade of dopamine reuptake, stimulation of dopamine receptors, and promotion of dopamine release.
Amantadine is excreted unchanged in the urine. The usual dosage range is 100 mg two or three times a day (Table 41.1). Elderly patients and those sensitive to the effects of medications should probably start with 25 mg per day for a few days, using a syrup formulation.
Adverse effects of amantadine may be mild but some are intolerable. The most common are leg edema and livedo reticularis, a mottling discoloration of the lower limbs, as well as effects associated with its anticholinergic properties, such as disorientation and hallucinations, as well as dry mouth and blurry vision, especially in older patients.
(3) Anticholinergic drugs have been used for many years in the management of PD. Dopamine depletion in the striatum causes a relative “hypercholinergic” state that responds to the use of anticholinergic drugs. Many centrally acting anticholinergic drugs are available, but the two most commonly used in the United States are trihexyphenidyl (Artane; Lederle) and benztropine (Cogentin; Merck). Biperiden(Akineton; Knoll), orphenadrine (Norflex; 3M Pharmaceuticals), and procyclidine (Kemadrin; GlaxoSmithKline) are rarely used.
Anticholinergics may be used early in the course of PD. Tremor remains the only practical indication for their use given the poor side-effect profile (see below). Typically, trihexyphenidyl is started at doses of 1 mg per day and increased weekly up to 2 mg four times per day until symptomatic control is obtained or side effects develop. Benztropine usually is started at 0.5 mg per day and titrated up to 4 mg per day. If anticholinergics are to be discontinued, this should be done gradually to avoid withdrawal effects.
Adverse effects of anticholinergic medications include both peripheral antimuscarinic side effects (e.g., dry mouth, impaired visual accommodation, urinary retention, constipation, tachycardia, and impaired sweating) and central effects (e.g., sedation, dysphoria, memory difficulties, confusion, and hallucinations).
(4) Dopamine receptor agonists directly stimulate dopamine receptors. The currently commercially available dopamine agonists in the United States are pramipexole (Mirapex; Boehringer-Ingelheim), ropinirole (Requip; GlaxoSmithKline), apomorphine (Apokyn, Ipsen), and bromocriptine (Parlodel; Novartis). The long-acting ergot derivative bromocriptine and cabergoline are rarely used for the treatment of PD. Their use in North America has been largely restricted to the treatment of hyperprolactinemia. The transdermally delivered rotigotine (Neupro; UCB/Schwarz Pharma) is temporarily off the market due to the development of crystals in the patch system. Pergolide (Permax; Elan) was withdrawn from the US market due to increased risk of cardiac valvulopathy. Lisuride and piribedil are available in other countries.
Dopamine receptor agonists may relieve all of the cardinal manifestations of PD. Despite the theoretical advantages over levodopa by acting directly on striatal dopamine receptors while circumventing the degenerating dopaminergic neurons, dopamine agonists are less effective than levodopa, yielding a lower risk of dyskinesia and motor fluctuation compared with it, and have an extensive list of potential side effects. Agonists can be used both as monotherapy and as adjuncts to levodopa. To minimize side effects, the dosage of a dopamine agonist should be increased gradually until the desired effect is obtained. Table 41.1 shows common dosages for different antiparkinsonian drugs. Apomorphine can only be administered subcutaneously as a rescue treatment for intractable and disabling wearing off. An apomorphine challenge is required to determine the correct dose of the drug while the patient is pretreated with an antiemetic, such as domperidone or trimethobenzamide.
TABLE 41.1 Antiparkinsonian Medications
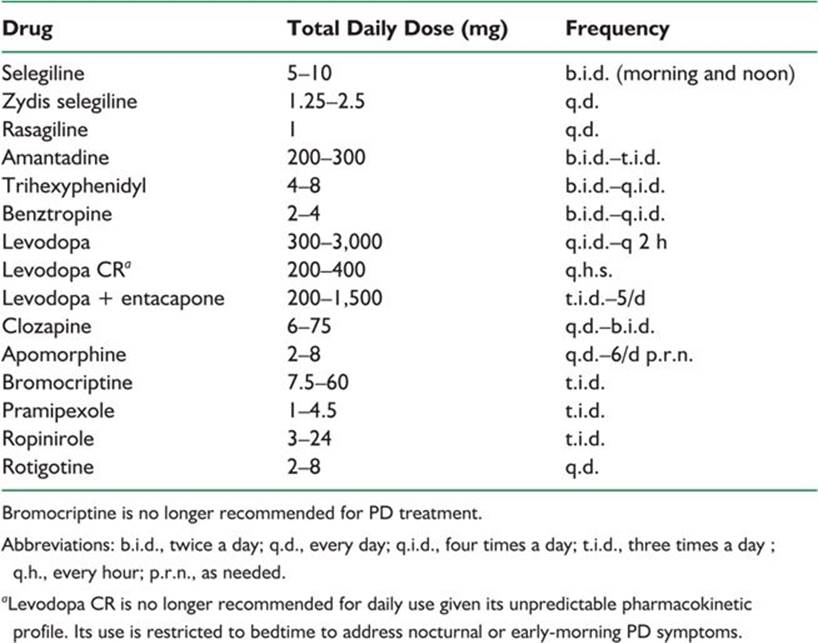
Dopaminergic adverse effects of dopamine agonists include nausea, vomiting, postural hypotension, and excessive daytime sleepiness and psychiatric manifestations including visual hallucinations and impulse-control disorders. Elderly and cognitively impaired patients are more prone to psychiatric side effects. Impulse control disorders (excessive shopping, compulsive gambling, and hypersexuality, among others) may develop 20 months after the onset of therapy, which demands regular monitoring. Also in the long term, dopamine agonists can cause leg edema and livedo reticularis. Older ergot-derived dopamine agonists such as bromocriptine and cabergoline in rare instances cause pulmonary and retroperitoneal fibrosis, cardiac valvulopathy, vasospasm, and erythromelalgia and can exacerbate angina and peptic ulcer disease.
(5) Levodopa is the most effective antiparkinsonian medication. It is mainly absorbed in the proximal small intestine by a carrier-mediated process for neutral amino acids and is similarly transported across the blood–brain barrier. Once in the brain, it is converted to dopamine by the enzyme aminoacid dopa decarboxylase. Levodopa is administered in combination with a peripheral dopa decarboxylase inhibitor (carbidopa in North America or benserazide in Europe). Inhibition of peripheral dopa decarboxylase markedly reduces the required total daily dose of levodopa and minimizes the gastrointestinal side effects and hypotension caused by peripheral conversion of levodopa to dopamine.
Available preparations of levodopa include immediate-release carbidopa–levodopa (Sinemet; Dupont), an orally disintegrating tablet (Parcopa; Azur Pharma), and a controlled-release preparation (Sinemet CR, Dupont). A minimum of 75 mg per day of carbidopa is required for appropriate peripheral decarboxylation. Carbidopa–levodopa preparations are available as 10 per 100, 25 per 100, and 25 per 250 tablets (carbidopa milligrams per levodopa milligrams) and as 25 per 100 and 50 per 200 tablets in the controlled-release preparation. Sustained-release preparations are 30% less bioavailable and substantially more erratic in its pharmacokinetics than the immediate-release forms. Because of the latter, Sinemet CR is no longer recommended for the control of daytime symptoms, and its use is reserved for reemergence of night-time or early-morning symptoms.
Levodopa generally relieves all of the cardinal signs of PD—bradykinesia, tremor, and rigidity. Delaying levodopa in any patient older than 70 years of age or in younger individuals insufficiently treated with dopamine agonists or MAO-B inhibitors is no longer recommended. Unlike the core deficits of tremor, bradykinesia, and rigidity, axial deficits, such as impaired postural reflexes, hypophonia, and dysphagia, are less reliably improved. A lack of response to levodopa may suggest a diagnosis of one of the atypical parkinsonisms, but an adequate trial with doses up to 1,500 mg of levodopa should be tried before considering anyone nonresponder. Treatment with carbidopa–levodopa usually is initiated using 25 per 100 immediate-release tablets titrating slowly upward to minimize acute side effect.
As the disease progresses, patients may develop motor complications in the form of motor fluctuations with wearing off toward the end of a dose cycle (reemergence of parkinsonian deficits or appearance of end-of-dose or early-morning dystonia) or choreic or choreathetoid movements (dyskinesia). Wearing off can be improved by decreasing the interdose interval of levodopa, increasing the individual levodopa doses, adding a catechol-O-methyl- transferase (COMT) or MAO-B inhibitor, or considering apomorphine subcutaneous injections. Choreic movements of the upper body predominantly head and neck is often indicative of peak-dose dyskinesia and requires lowering the levodopa doses, increasing the interdose interval, or adding amantadine. Choreic movements predominantly of the lower body, especially legs, feet, and pelvis, may occur at the beginning of or at the end of a dose cycle of levodopa and are referred to as diphasic dyskinesia. Unlike peak-dose dyskinesias, diphasic dyskinesias are treated by increasing the dose of levodopa or decreasing the interdose interval. In general, motor complications, particularly dyskinesia, begin after 2 to 10 years of levodopa therapy. Younger patients are more prone to dyskinesia and motor fluctuations earlier in the course of the disease. When medication adjustments do not improve these motor complications, deep brain stimulation (DBS) of the subthalamic nucleus (STN) or internal pars of the globus pallidus (GPi) may be considered (see section 3, below).
The short half-life of immediate-release levodopa is believed to be a major factor in the development of motor complications. A gel formulation of levodopa continuously administered intrajejunally is under investigation and expected to become available in the United States in 2012. A similar preparation has been available in Europe for over a decade (Duodopa; Abbott).
Adverse effects of levodopa can be classified into acute and chronic. In the short term, nausea, vomiting, and hypotension-related lightheadedness can be addressed by adding extra carbidopa (Lodosyn; Bristol-Myers Squibb), 25 to 75 mg three times per day, to enhance the peripheral decarboxylation and minimize bioavailability of dopamine outside the brain, where is typically toxic. Another option is to use a peripheral dopamine receptor blocker such as domperidone in doses of 10 to 20 mg three times a day. Domperidone is not currently available in the United States but can be readily obtained from Canadian online pharmacies. In the long term, patients may develop changes in behavioral complications in the form of psychosis, paranoia, sexual preoccupation, impulse control disorder, mania, or agitation. Visual hallucinations can be quite vivid in the form of people or animals. Mental status changes usually are dose-dependent and typically lessen with medication reduction, although at the expense of deterioration of motor function. Quetiapine (Seroquel; Astra-Zeneca) or clozapine (Clozaril; Novartis) may be considered in these cases, as the only antipsychotics safe to use in PD. Reports of accelerated melanoma growth in PD patients taking levodopa have been published. However, melanoma seems to be more prevalent among PD patients regardless of their exposure to levodopa or other PD drugs.
(6) COMT inhibitors are used as adjuncts to levodopa. By blocking the peripheral conversion of levodopa to 3-O-methyldopa, COMT inhibitors increase the bioavailability of levodopa. Two COMT inhibitors are available—tolcapone (Tasmar; Valeant) and entacapone (Comtan; Novartis). A carbidopa–levodopa–entacapone preparation (Stalevo; Novartis) is also available. Tolcapone is used in doses of 100 to 200 mg three times a day, and entacapone 200 mg is given with each dose of levodopa up to 2,000 mg per day. If tolcapone is used, liver function tests should be monitored periodically at least for the first 6 months because of earlier reports of rare cases of tolcapone-induced liver failure, some fatal. Side effects of COMT inhibitors are related to increased bioavailability of levodopa. In addition, diarrheaand a brownish-orange discoloration of the urine may occur.
(7) Visual hallucinations and psychosis are adverse effects that can occur in association with any antiparkinsonian medication. Any potential triggering event, such as infections or metabolic derangements, should be actively sought and treated, if present. Otherwise, decreasing or discontinuing the dose of dopaminergic medications is warranted, in the following order if hallucinations persist: anticholinergics, amantadine, selegiline/rasagiline, and dopamine agonists. If worsening of motor symptoms make such a reduction impossible, the judicious use of one of the safe atypical antipsychotics quetiapine or clozapine, or an acetylcholinesterase inhibitor, such as rivastigmine, may be necessary. Clozapine is started at a dose of usually 6.25 mg at bedtime. Because 1% to 2% of patients taking clozapine may experience agranulocytosis, patients should be monitored with weekly blood counts. Quetiapine is effective in the management of dopaminergic-induced psychosis at doses of 12.5 mg to 300 at night without worsening parkinsonism.
(8) Constipation is a common problem among PD patients. Management should include dietary modifications, increasing fluid and fiber intake, exercise, and minimizing or eliminating the use of anticholinergic medications. Psyllium (Metamucil; Procter & Gamble) 1 tsp (5 ml) two to four times a day may be added. Osmotic agents such as sorbitol and lactulose are often helpful. Agents that stimulate intestinal motility such as bisacodyl(Dulcolax; Novartis) could be added. Some patients may need enemas.
(9) Other potential problems of patients with PD include nocturia, urinary urgency and frequency, erectile dysfunction, dysphagia, orthostatic hypotension, and sleep problems (see Chapters 7, 9, 18, and 31). Depression requires special mention as it affects about 50% of patients with PD and may respond to antidepressant medications such as SSRIs and SNRIs, given the involvement of serotonin but importantly of norepinephrine in PD.
3. Surgery. The stereotactic surgical options to treat PD patients include ablative procedures and DBS at different targets—ventral intermediate nucleus of the thalamus (Vim), GPi, and STN. The success of surgical treatment of patients with PD depends on a careful selection of the appropriate candidates. First, only patients with idiopathic PD should be considered. Patients with advanced disease, poor response to levodopa, dementia, uncontrolled depression, uncontrolled hallucinations, and unstable medical problems are unlikely to benefit from these procedures. Surgical candidates should undergo a presurgical neuropsychological evaluation to rule out substantial cognitive dysfunction.
a. Thalamotomy and thalamic DBS are procedures directed at managing contralateral medically intractable tremor regardless of etiology. Neither of these two procedures improves other features of PD. Tremor reduction occurs in approximately 80% of patients. Bilateral thalamotomy is no longer recommended due to the high risk of dysarthria. Bilateral thalamic DBS may interrupt bilateral thalamic output with fewer side effects than thalamotomy, allowing for adjustment of stimulation to maximize benefits and minimize side effects.
b. Pallidotomy and pallidal DBS, targeting the posteroventral GPi, are useful for PD patients with severe dyskinesia and motor fluctuations. Like bilateral thalamotomy, bilateral pallidotomy is no longer recommended due to the high incidence of dysarthria. Improvement in motor function and a marked anti-dyskinetic effect can be striking. PD symptoms that persist during the on state (e.g., freezing and dysarthria) do not respond well to pallidotomy. Improvement in the off-medication state is approximately 30% for pallidotomy and 40% for pallidal DBS.
c. STN DBS is currently the preferred surgical procedure for advanced PD with motor fluctuations and dyskinesia. The improvement is mainly during the off state with up to 60% improvement in motor scores reported. DBS may improve the quality of the on state by approximately 10%. STN stimulation allows for a greater reduction of antiparkinsonian medication compared with pallidal stimulation. GPi and STN DBS offer comparative benefits according to a recent head-to-head comparison study.
B. Atypical parkinsonisms are a group of rare degenerative conditions within the akinetic-rigid syndrome to which PD belongs.
1. Multiple system atrophy (MSA) is a progressive neurodegenerative disease characterized by a combination of parkinsonism, cerebellar dysfunction, and autonomic failure. The nomenclature MSA-P and MSA-C are used when parkinsonian or cerebellar features predominate, respectively. The parkinsonism tends to be tremorless. Clinical features that suggest the diagnosis are hyperreflexia or other corticospinal signs, severe early orthostatic hypotension and/or urinary incontinence, atypical levodopa-induced dyskinesias (affecting face [sardonic grin] and feet), Pisa syndrome (lateral truncal deviation), and inspiratory stridor.
a. Levodopa may provide transient improvement of parkinsonian sympoms albeit rarely sustained beyond 1 year. Other dopaminergic drugs are not indicated.
b. Orthostatic hypotension may be the greatest source of disability and can be worsened by levodopa. Nonpharmacologic measures include liberalizing salt and water intake, using waist- or thigh-high compressive leg stockings during the day, and raising the head of the bed 8 inches (20 cm) at night to minimize supine hypertension and excessive nocturnal diuresis. Patients should be careful rising from the sitting or supine positions and should avoid heavy meals. When pharmacotherapy is required, the peripheral α-1-adrenergic receptor agonist midodrine (ProAmatine; Roberts) may be used, starting at 2.5 mg three times a day, increasing by 2.5-mg weekly increments up to a maximum of 10 mg three times a day When midodrine is insufficient, treatment with the mineralocorticoid fludrocortisone (Florinef; Bristol-Myers Squibb) may be added at doses of 0.1 to 0.3 mg per day, as needed. The other potentially useful drugs are methylphenidate, pyridostigmine, erythropoietin, ergots, and desmopressin.
c. Urinary frequency or incontinence should be evaluated with the assistance of an urologist. Treatments such as oxybutynin (Ditropan; Alza), solifenacin (Vesicare; Astellas), tolterodine (Detrol; Pfizer), darifenacin(Enablex; Novartis), or trospium chloride (Sanctura; Esprit) for a spastic bladder or bethanechol (Urecholine; Odyssey) for a hypotonic bladder may provide relief. Some patients need intermittent or continuous catheterization. Sildefanil (Viagra; Pfizer), tadalafil (Cialis; Lilly), and vardenafil (Levitra; GlaxoSmithKline) may be useful for management of erectile dysfunction.
d. Dysarthria and dysphagia may benefit from evaluation by a speech therapist. Some patients with severe dysphagia need percutaneous gastrostomy. Gait difficulties and instability may necessitate use of supportive devices and physical therapy. Patients with MSA and inspiratory stridor should undergo a sleep study to determine whether concurrent obstructive sleep apnea requires treatment.
2. Progressive supranuclear palsy (PSP) is an atypical parkinsonism characterized by early postural instability and falls, disproportionate neck rigidity (sometimes with retrocollis), facial dystonia, supranuclear vertical gaze abnormalities, pseudobulbar affect, subcortical frontal dementia, and apathy.
a. Management of PSP is extremely limited. Antiparkinsonian medications are rarely helpful, although a trial of levodopa is prudent.
b. Symptomatic palliative therapies for PSP include management of dysarthria and dysphagia with the assistance of a speech therapist and may include the use of a gastrostomy tube among other measures. Because of the significantly decreased blinking rate, patients with PSP are at increased risk of keratitis and should use artificial tears. Blepharospasm and neck dystonia can be managed with botulinum toxin injections. Depression and emotional incontinence can be managed with antidepressants. Dystonia may benefit from amantadine. Gait instability can be managed with physical therapy and supportive devices.
II. HYPERKINETIC MOVEMENT DISORDERS
A. Chorea is an involuntary movement disorder characterized by irregular, dance-like jerky movements occurring within or between body parts in a random sequence. It can result from a variety of disorders of the basal ganglia.
1. Huntington’s disease (HD) is an autosomal-dominant degenerative brain disorder characterized by the insidious development of motor, cognitive, and psychiatric symptoms progressing toward death on average of about 20 years after onset of symptoms. The underlying genetic defect is the expansion of a CAG trinucleotide repeat of the HD gene, the product of which is a protein called huntingtin.Symptomatic treatment is directed at the major clinical features of the disease.
a. Choreiform movements can be reliably controlled with neuroleptics that have potent postsynaptic dopamine blocking effects such as haloperidol (Haldol; Ortho-McNeil). Benzodiazepines such as lorazepam and clonazepam may also decrease chorea. Tetrabenazine (Xenazine; Lundbeck), a reversible dopamine depleter and mild postsynaptic dopamine blocker, is effective in reducing chorea. Effective doses range from 12.5 to 50 mg three times a day.
b. Depression affects at least 30% to 50% of patients with HD. In HD patients, the suicide rate is four to eight times greater than in the general population. Depression can be managed with all standard agents used for the management of major depression. SSRIs typically are the drugs of choice for HD. Mirtazapine (Remeron; Organon) can be helpful in the care of HD patients with cachexia, anxiety, and insomnia because it can increase body weight and assist in sleep induction.
c. Irritability and aggressive behavior are common psychiatric manifestations in HD. Propranolol, valproic acid, and carbamazepine are potentially useful to treat aggressive behavior related to frustration and impatience.
d. Mania and hypomania can occur in HD. Approximately 10% of HD patients may exhibit hypomanic behavior. Mania in HD responds better to carbamazepine and oxcarbazepine than to lithium. Other therapeutic alternatives are valproic acid and clonazepam.
e. Psychosis has an estimated frequency of 3% to 25% among patients with HD and is more common among patients with early-onset disease. Atypical antipsychotic agents such as clozapine, quetiapine, olanzapine, and risperidone are effective in controlling psychotic symptoms. These drugs are associated with increased risk of hyperglycemia and diabetes.
2. Other causes of chorea include neurodegenerative disorders (e.g., chorea-acanthocytosis, Wilson’s disease (WD), and dentatorubropallidoluysian atrophy), Sydenham’s chorea, systemic lupus erythematosus, hyperthyroidism, and drug-induced chorea (e.g., phenytoin, oral contraceptives, stimulants, or antiparkinsonian drugs). Regardless of the underlying cause, the movements can be improved with the use of neuroleptics. However, disease-specific treatments should be pursued as appropriate (e.g., warfarin in antiphospholipid antibody syndrome, penicillin in Sydenham’s chorea). The risk of tardive dyskinesia (TD) is uncertain when neuroleptics are used in the treatment of chorea.
3. Hemiballismus is a severe form of chorea, with violent, flailing movements of the proximal aspect of the limbs on one side of the body. It is classically caused by lesions in the contralateral STN, but lesions outside the STN are more common. Treatment includes supportive care, prevention of self-injury, and pharmacologic agents such as benzodiazepines, neuroleptics, and catecholamine-depleting agents (reserpine or tetrabenazine). Valproic acid and other gamma-aminobutyric acid (GABA)-ergic drugs may be alternative therapeutic options. Surgical alternatives exist for patients who do not appropriately respond to medical therapy.
B. Tics are common movement disorders, affecting as many as 20% of children. They are brief, rapid, purposeless, repetitive movements involving one or more muscular groups. They are differentiated from other paroxysmal movement disorders by their partial voluntary control with suppressability when performing complex tasks, premonitory “urge,” and stereotypic appearance.
1. Tourette’s syndrome (TS) is a childhood-onset neuropsychiatric disorder characterized by motor and phonic tics. Tics wax and wane and tend to improve considerably during adulthood. Obsessive–compulsive behavior and attention deficit disorder (ADD) are comorbid conditions frequently associated with TS and may be more disabling than tics themselves.
a. Tics do not require treatment unless they are troublesome to the patient. The first step in treatment is education and reassurance. If further intervention is needed, clonidine (Catapres; Boehringer-Ingelheim) starting at 0.05 mg at bedtime and increased 0.05 mg every few days can be considered. The efficacy of clonidine for tic control is modest, however. Guanfacine starting at 0.5 to 1 mg at bedtime is another option that may be less sedating than clonidine.
Neuroleptics are the most efficacious agents for tic suppression. Haloperidol (Haldol, McNeil Laboratories) is probably the most commonly used neuroleptic for tics. Pimozide (Orap; Gate) was developed specifically for use in TS and may cause less sedation than haloperidol does. Pimozide may prolong the QT interval. Other neuroleptics such as trifluoperazine (Stelazine; GlaxoSmithKline) and thiothixene(Navane; Pfizer) can also be helpful. Atypical antipsychotics such as risperidone (Risperdal; Janssen), ziprasidone (Geodon; Pfizer), aripripazole (Abilify; Otsuka), and olanzapine (Zyprexa; Lilly) are being used with increasing frequency, though data from controlled trials are lacking. The necessary dosage can vary widely among patients and at different times for a given patient, given the fluctuating severity of the natural history of tics. Sedation and depression may be troublesome side effects. Although the risk of TD appears to be low among patients with TS, this potential long-term adverse effect must be discussed with patients and documented in the medical record. Clonazepam(Klonopin; Roche) and baclofen may be helpful to some patients. Botulinum toxin injections may be helpful for some tics.
b. Management of obsessive–compulsive behavior associated with TS is identical to that of the purely psychiatric condition. SSRIs and clomipramine (Anafranil; Novartis, may be used in this regard. The major adverse effects of clomipramine are sedation and anticholinergic effects.
c. ADD and other behavioral disorders of children may be difficult to control. Clonidine, tricyclic antidepressants, or selegiline may be effective. Use of CNS stimulants such as methylphenidate (Ritalin; Novartis) may ease ADD. Modafinil (Provigil; Cephalon) may be useful as well. The diverse behavioral abnormalities sometimes exhibited by children with TS not infrequently necessitate family counseling and other nonpharmacologic approaches.
C. Myoclonus is a shock-like, brief, involuntary movement caused by muscular contraction (positive myoclonus) or muscular inhibition (negative myoclonus). Myoclonus can originate from the cortex, subcortical areas, brainstem, or spinal cord. Common causes of myoclonus include metabolic derangements, such as renal and hepatic failure, and epileptiform disorders.
1. Diagnosis should include a thorough history and physical examination plus blood glucose and electrolytes, drug and toxin screen, renal and hepatic function tests, brain imaging, and EEG. A search for inborn errors of metabolism and paraneoplastic antibodies may be indicated in some cases.
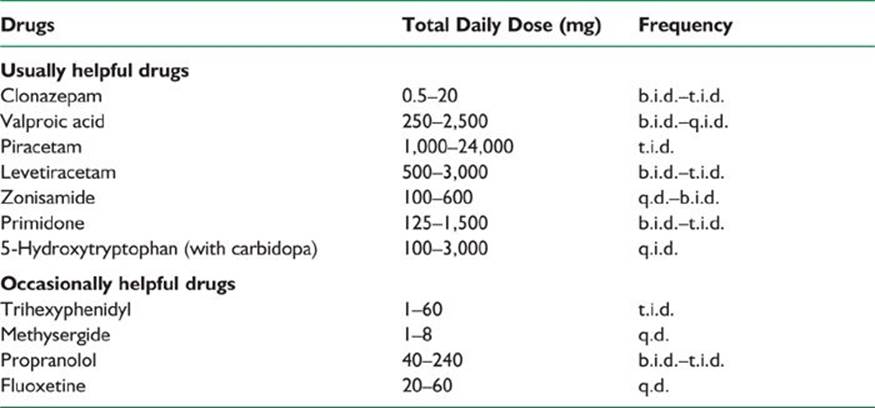
2. The ideal therapy for myoclonus is to manage the underlying condition. However, symptomatic treatment should be used if treatment is likely to make a significant functional impact (Table 41.2). Clonazepam at 2 to 6 mg per day, valproic acid 250 to 1,500 mg per day, levetiracetam (Keppra; UCB) 500 to 4,000 mg per day, and piracetam up to 24 g per day (not available in the United States) are first-line drugs for the management of myoclonus. Acetazolamide, zonisamide, primidone, 5-hydroxytryptophan, and tetrabenazine can also be helpful. Many patients need polytherapy to control myoclonus.
D. TD is a generic term used to describe persistent involuntary movements that occur as a consequence of long-term treatment with dopamine receptor antagonists (neuroleptics). Antipsychotics are the main causative group of neuroleptics, but antiemetics such as metoclopramide, prochlorperazine, and promethazine may also behave as TD-causing neuroleptics. The risk factors for development of classic TD include old age, female gender, mood disorder, and “organic” brain dysfunction. Classic TD usually consists of oral–buccal–lingual dyskinesia that may be associated with a variety of repetitive limb or trunk stereotyped movements.
TABLE 41.2 Medications Used in the Management of Myoclonus
1. The pathophysiologic mechanism of TD is not completely understood, but it is thought to be related to an increased number and affinity of postsynaptic D2 dopamine receptors in the striatum. The patient’s condition may initially improve after the neuroleptic agent is restarted or after the dosage is increased. Unfortunately, this is likely to perpetuate the problem.
2. The ideal management of TD would be prevention of this condition by avoiding unnecessary use of neuroleptics and using the minimally effective dose. Anticholinergic medications can worsen classic TD.
a. Dopamine depleters such as reserpine or tetrabenazine have been among the most useful medications to treat TD. Dosages of reserpine usually are started at 0.10 to 0.25 mg three times a day and may be gradually increased to 3 to 5 mg per day. Reserpine may cause parkinsonism, depression, orthostatic hypotension, and peptic ulcer disease. Tetrabenazine can be started at 25 mg per day and gradually increased up to 150 mg per day in divided doses. The most common limiting side effects are sedation, depression, and parkinsonism.
b. Benzodiazepines may prove useful for patients with mild symptoms. Long-acting agents such as clonazepam (usually 1.5 to 3 mg per day) provide the most consistent relief of symptoms.
c. Branched-chain amino acids (Tarvil; SHS North America) have been shown to significantly decrease TD symptoms in males. It is used at a dose of 222 mg per kg three times a day.
d. Neuroleptics, used in the lowest possible dosages, may be necessary if symptoms markedly interfere with activities of daily living.
e. Tardive dystonia is a subtype of TD that typically affects younger people. It usually involves the neck and trunk muscles. Management of tardive dystonia differs from that of classic TD in that anticholinergics are potentially beneficial, and botulinum toxin can be used in focal or segmental forms. Dopamine depleters are also useful.
f. For many patients, combination therapy is most effective. The use of a benzodiazepine with either a dopamine depleter or a low dosage of an atypical neuroleptic may be necessary. Vitamin E might prevent further deterioration of TD, but it is not really clear if it can improve TD symptoms. The eventual rate of remission is approximately 60% and improvement is slow, taking as long as 2 years.
E. Dystonia is a syndrome of sustained muscle contraction causing abnormal repetitive movements, twisting, or abnormal postures. Up to 21 different types of dystonia so far can be differentiated genetically and are designated as DYT1 to 21. Idiopathic dystonia can be generalized or restricted to a particular muscle group. With the exception of DYT3 (X-linked Lubag disease), patients with primary idiopathic dystonia have no gross or microscopic abnormalities. Secondary dystonia includes several inherited inborn errors of metabolism such as dopa-responsive dystonia (DRD or DYT5) and WD. Trauma, vascular disease, space-occupying lesions, drugs, and toxins are other causes of secondary dystonia.
1. DRD or DYT5 is an autosomal dominant disorder caused by a mutation in the GTP cyclohydrolase I gene. It usually becomes apparent during childhood with a gait disorder, foot cramping, or toe walking. It can involve the trunk and arms and can be misdiagnosed as cerebral palsy. Patients with this form of dystonia have few symptoms after first awakening but the symptoms progress throughout the day. This disorder is exquisitely sensitive to small doses of levodopa (50 to 200 mg). A brief trial of levodopa for childhood-onset dystonia is frequently recommended to exclude DRD.
2. Medical management of dystonia of any cause can be attempted with the medications listed in Table 41.3. None of these medications provides complete relief of symptoms. Combinations of medications can be beneficial. Extremely high doses of anticholinergic drugs such as trihexyphenidyl have been reported to benefit more than 50% of patients in some trials (sometimes at doses greater than 100 mg per day). Therapy usually is started with 1 mg per day and increased 1 to 2 mg per week divided on a three times a day schedule until control of symptoms is achieved or intolerable adverse effects appear. The combination of a dopamine depleter such as reserpine, an anticholinergic, and a postsynaptic dopamine blocker may be beneficial to patients with severe dystonia. GPi DBS can be offered to patients to with pharmacologically intractable and disabling dystonia.
3. Injection of botulinum toxin is the first line of treatment of many patients with focal and segmental dystonia. There are seven botulinum toxin serotypes, but only types A and B are available in the United States—botulinum toxin type A (onabotulinumtoxinA [Botox; Allergan], abobotulinumtoxinA [Dysport; Ipsen], and incobotulinumtoxinA [Xeomin, Merz]) and botulinum toxin type B(rimabotulinumtoxinB [MyoBloc; Solstice]). The toxin is injected directly into the affected muscle, producing reversible pharmacologic denervation. Injections usually are repeated at an average interval of 12 to 16 weeks when toxin type A or B are used. Potential side effects include excessive transient weakness of the injected and adjacent muscles, dry mouth, and local hematoma.
F. Tremor is probably the most common movement disorder. It is an involuntary, rhythmic oscillation of a body part.
1. Essential tremor (ET) typically includes both postural and kinetic tremor. A positive family history of tremor is common among ET patients. Tremor typically may improve with small amounts of alcohol. Generally, it is not possible to completely eliminate the tremor, and the goal of therapy should be to normalize activities of daily living. One-half to two-thirds of patients with ET benefit from pharmacologic therapy (Table 41.4). In some cases, the tremor may be quite refractory, and surgical treatment should be considered.
TABLE 41.3 Medications Used in the Management of Dystonia
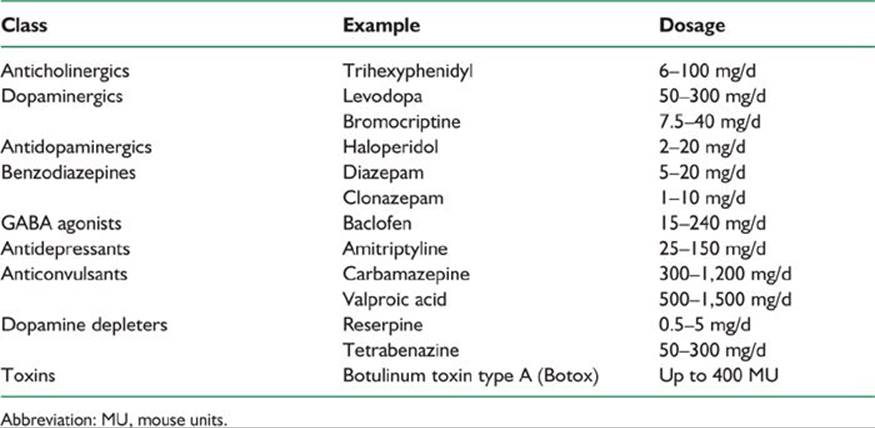
TABLE 41.4 Medications Used in the Management of Tremor
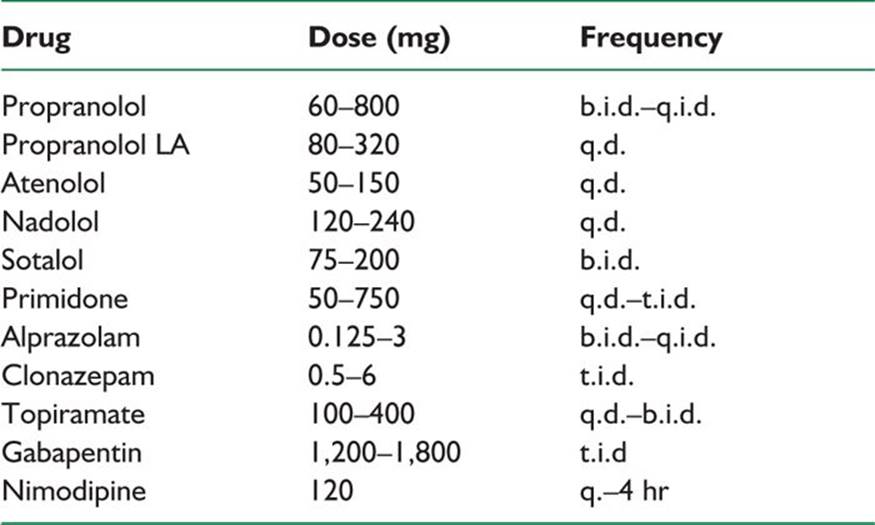
a. ß-Adrenergic receptor antagonists are used most extensively to manage ET. The clinical response to ß-blockers is variable and usually incomplete. These drugs reduce tremor amplitude but not tremor frequency and appear to be less effective in managing voice and head tremor. Nonselective ß-blockers such as propranolol (Inderal; Wyeth-Ayerst) are preferred. Propranolol should be started at small doses (e.g., 10 mg three times a day) and titrated upward as needed. Doses larger than 320 mg per day usually do not confer additional benefit. Potential side effects of ß-blockers include congestive heart failure, second- or third-degree atrioventricular block, worsening of obstructive lung disease, and masking of signs of hypoglycemia. The ß-blockers can also cause fatigue, nausea, diarrhea, rash, erectile dysfunction, and depression. Nadolol (Corgard; Bristol-Myers Squibb) is an option if propranolol causes CNS side effects as this drug does not readily cross the blood–brain barrier. Atenolol (Tenormin; Astra Zeneca), sotalol (Betapace; Bayer Healthcare), metoprolol (Lopressor; Novartis), and timolol (Blocadren; Merck) are potentially useful in the treatment of ET.
b. Primidone (Mysoline; Xcel) may improve ET. The mechanism of action of primidone for management of tremor is unknown. Primidone decreases the amplitude of tremor but does not alter its frequency. Treatment usually is started at 25 mg at bedtime, and a response may begin at doses between 50 and 350 mg per day. Doses up to 750 mg per day divided three times a day may be required for benefits to appear. Side effects include vertigo, nausea, unsteadiness, and drowsiness.
c. Benzodiazepines may be used if the above drugs do not provide sufficient control of symptoms. Long-acting agents such as clonazepam can be used, but some patients may respond better to the use of a shorter-acting agent such as alprazolam (Xanax; Pfizer). Clonazepam at 1 to 3 mg per day can be very effective in orthostatic tremor. Potential adverse effects include sedation, ataxia, tolerance, and potential for abuse.
d. Botulinum toxin injections of limb tremor in ET may offer modest improvement of tremor that might be more cosmetic than functional as no improvement has been seen in functional scales. Transient weakness is the most common side effect.
e. Other drugs that may be considered for the management of ET are gabapentin (Neurontin; Pfizer), topiramate (Topamax; Ortho-McNeil), and zonisamide (Zonegran; Eisai). Clozapine) may also improve ET; however, the potential risk of idiosyncratic agranulocytosis makes this option unappealing.
f. Surgery can be used in selected cases when activities of daily living are severely affected despite medical management. Stereotactic thalamotomy or Vim DBS improve contralateral tremor.
G. WD is an autosomal recessive disorder of copper accumulation caused by a defect in copper excretion into the bile. Low-plasma levels of ceruloplasmin characterize WD. Copper deposits typically occur in the liver, iris, and basal ganglia. Other organs may be affected as well. A variety of movement disorders can accompany WD, including tremor, dystonia, chorea, dysphagia, dysarthria, and parkinsonism. Symptomatic management of the movement disorder is as discussed in other sections of this chapter.
1. Recommended screening methods for patients with a neurologic signs and symptoms of WD are as follows:
a. Serum ceruloplasmin and slit-lamp examination for Kayser–Fleischer’s (KF) rings. Approximately 90% of WD patients who have neurologic symptoms have a low-ceruloplasmin level. KF rings are present in 99.9% of WD patients who have neurologic symptoms.
b. 24-hour urine copper. In patients with neurologic WD, the 24-hour urine copper level is always more than 100 µg before chelation treatment. This value may be falsely elevated in patients with long-standing liver disease.
2. Copper-rich foods such as shellfish, chocolate, liver, nuts, and soy products should be avoided. However, this is not sufficient to avoid further accumulation, and zinc acetate is used to block mucosal absorption of copper. Zinc acetate is taken at a dose of 50 mg three times a day between meals. The toxicity of zinc is negligible, although it can cause abdominal discomfort. Zinc is the drug of choice for maintenance therapy after chelation and in presymptomatic or pregnant patients.
3. Penicillamine (Cuprimine; Aton) acts by means of reductive chelation of copper. It mobilizes large amounts of copper, mainly from the liver. The standard dose after a titration phase is 250 mg four times a day, each dose separated from food. Doses up to 1,500 mg per day can be used. Several potentially serious adverse effects are associated with penicillamine. Approximately 50% of patients treated with penicillamine have marked neurologic deterioration, and half of these patients do not recover to the pre-penicillamine level of function. Approximately one third of patients who start taking penicillamine have an acute hypersensitivity reaction. Other subacute potential toxicities include bone marrow suppression, membranous glomerulopathy, myasthenia gravis, reduced immune response, hepatitis, pemphigus, and a lupus erythematosus-like syndrome with a positive antinuclear antibody. A CBC with platelets and urinalysis are recommended every 2 weeks for the first 6 months of therapy and monthly thereafter.
4. Trientine (Syprine; Aton) is a chelating agent that induces urinary excretion of copper and has a more favorable side-effect profile than penicillamine, with a lower risk of neurological deterioration (25%), which makes it more favorable for consideration as initial WD treatment. Dosage and administration are identical to those of penicillamine. Although trientine promotes less copper excretion than does penicillamine, it does not cause a hypersensitivity reaction. The other toxicities are somewhat similar to those of penicillamine but less frequent.
5. Tetrathiomolybdate is an experimental drug that prevents absorption of copper from the intestine and is absorbed into the blood, where it binds to copper to form nontoxic complexes. It has been used successfully at a dose of 120 mg per day to manage acute WD with neurologic manifestations.
6. Liver transplantation is curative of WD.
![]()
Recommended Readings
Axer H, Axer M, Sauer H, Witte OW, Hagemann G. Falls and gait disorders in geriatric neurology. Clin Neurol Neurosurg. 2010;112(4):265–274.
Espay AJ. Management of motor complications in Parkinson disease: current and emerging therapies. Neurol Clin. 2010;28(4):913–925.
Factor SA, Molho ES. Emergency department presentations of patients with Parkinson’s disease. Am J Emerg Med. 2000;18(2):209–215.
Klein C, Schneider SA, Lang AE. Hereditary parkinsonism: Parkinson disease look-alikes—an algorithm for clinicians to “PARK” genes and beyond. Mov Disord. 2009;24(14):2042–2058.
Kleiner-Fisman G, Herzog J, Fisman DN, et al. Subthalamic nucleus deep brain stimulation: summary and metaanalysis of outcomes. Mov Disord. 2006;21(suppl 14):S290–S304.
Lang AE. When and how should treatment be started in Parkinson disease? Neurology. 2009;72(7, suppl):S39–S43.
Louis ED. Essential tremors: a family of neurodegenerative disorders? Arch Neurol. 2009;66(10):1202–1208.
Moro E, Lang AE, et al. Criteria for deep-brain stimulation in Parkinson’s disease: review and analysis. Expert Rev Neurother. 2006;6(11):1695–1705.
Ozelius LJ, Bressman SB. Genetic and clinical features of primary torsion dystonia. Neurobiol Dis. 2011;42(2):127–135.
Pahwa R, Factor SA, Lyons KE, et al. Practice parameter: treatment of Parkinson disease with motor fluctuations and dyskinesia (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2006;66(7):983–995
Poston KL, Frucht SJ. Movement disorder emergencies. J Neurol. 2008;255(suppl 4):2–13.
Postuma RB, Lang AE. Hemiballism: Revisiting a classic disorder. Lancet Neurol 2003;2(11):661-8.