BACTERIAL INFECTIONS – MENINGITIS
ACUTE BACTERIAL MENINGITIS
In most cases the infection causing meningitis arises in the nasopharynx; intravascular invasion (bacteraemia) and penetration of the blood–brain barrier follow mucosal involvement with entry into the CSF. Bacteria may invade the subarachnoid space directly by spread from contiguous structures, e.g. sinuses and fractures. Specific characteristics of the capsule determine whether meninges are breached. Humoral defences against bacteria are absent in the CSF offering little resistance to infection.
Causative organisms
|
In neonates |
– Gram –ve bacilli, e.g. E. coli, Klebsiella. |
|
Haemophilus influenzae. |
|
|
In children |
– Haemophilus influenzae. Pneumococcus (Strep. pneumoniae). |
|
Meningococcus. (Neisseria meningitidis). |
|
|
In adults |
– Pneumococcus. Meningococcus. |
Other bacteria – Listeria monocytogenes, Streptococcus pyogenes and Staphylococcus aureus – are occasionally responsible.
Host factors (congenital or acquired immune deficiency, hyposplenism and alcoholism) predispose to infection, as do environmental factors (overcrowding and poverty).
Infections of mixed aetiology (two or more bacteria) may occur following head injury, mastoiditis or iatrogenically after lumbar puncture.
Pathology
The presence of the blood–brain barrier limits host defence mechanisms and enables multiplication of organisms.

A purulent exudate most evident in the basal cisterns extends throughout the subarachnoid space.
The underlying brain, although not invaded by bacteria, becomes congested, oedematous and ischaemic.
The integrity of the pia mater normally protects against brain abscess formation.
The cytokines, interleukin, tumour necrosis factor, and prostaglandin E2 are released as part of an acute inflammatory response. They increase vascular permeability, cause a loss of cerebrovascular autoregulation and exacerbate neuronal injury.
The inflammatory exudate may also affect vascular structures crossing the subarachnoid space producing an arteritis or venous thrombophlebitis with resultant infarction. Similarly, cranial nerves may suffer direct damage.
Hydrocephalus can result from CSF obstruction.
Clinical
The classical clinical triad is fever, headache and neck stiffness.
|
Prodromal features (variable) |
Meningitic symptoms |
|
A respiratory infection otitis media or pneumonia associated with muscle pain |
Severe frontal/occipital headache |
|
Stiff neck |
|
|
Photophobia. |
Systemic signs: – High fever. Transient purpuric or petechial skin rash in meningococcal meningitis.

Associated neurological signs
– Impaired conscious level
– Focal or generalised seizures are frequent.
– Cranial nerve signs occur in 15% of patients.
– Sensorineural deafness (not due to concurrent otitis media but to direct cochlear involvement) – 20%
– Focal neurological signs – hemiparesis, dysphasia, hemianopia, papilloedema – occur in 10%.
Non-neurological complications

Features specific to causative bacteria
|
Haemophilus meningitis |
Meningococcal meningitis |
Pneumococcal meningitis |
|
Generally occurs in small children. Preceding upper respiratory tract infection. Onset abrupt with a brief prodrome. |
Often occurs in epidemics where the organism is carried in the nasopharynx. Septicaemia can occur with arthralgia; purpuric skin rash. When overwhelming, confluent haemorrhages appear in the skin due to disseminated intravascular coagulation. |
Predominantly an adult disorder. Often associated with debilitation, e.g. alcoholism. May result from pneumonia, middle ear, sinus infection or follow splenectomy. Onset may be explosive, progressing to death within a few hours. |
|
Outcome |
||
|
Generally good Less than 5% mortality. |
Gradual onset – good prognosis. Sudden onset with septicaemia – poor outcome. Overall mortality − 10%. |
Mortality − 20%. Poor prognostic signs – coma, seizures, increased protein in CSF. |
Investigations
1. If patient has altered consciousness, focal signs, papilloedema, a recent seizure or is immunocompromised a CT brain should be done before LP. However, do not delay treatment – take blood cultures and commence antibiotics (see below) prior to scanning.
2. If above signs are absent or CT scan excludes a mass lesion → confirm diagnosis with a lumbar puncture and identify the organism.

– cell count is elevated, 100–10 000 cells/mm3(80–90% polymorphonuclear leucocytes).
– glucose is depressed.
– enzyme lactic dehydrogenase is elevated.
– culture CSF
Serological/immunological tests
The latex particle agglutination (LA) test, for the detection of bacteria antigen in CSF, has a sensitivity 80% for haemophilus and pneumococcus and 50% for meningococcus (100% specificity). The polymerase chain reaction (PCR), for the detection of bacteria nucleic acid in CSF, is available for all the suspected organisms. The specificity and sensitivity of PCR is unknown and the delay (3 to 5 days) to process results, makes the test less helpful than the combination of Gram’s stain, culture, and the LA test.
Blood cultures
– Organism isolated in 80% of cases of Haemophilus meningitis.
– Pneumococcus and meningococcus in less than 50% of patients.
3. Check serum electrolytes.
– important in view of the frequency of inappropriate antidiuretic hormone secretion.
4. Detect the source of infection.
|
– Chest X-ray – pneumonia – Sinus X-ray – sinusitis |
– Skull X-ray – fracture – Petrous views – mastoiditis |
Treatment
Once meningitis is suspected, treatment must commence immediately, often before identification of the causative organism. Antibiotics must penetrate CSF, be in appropriate bactericidal dosage and be sensitive to causal organism once identified.
Initial therapy (before organism identification)
|
Neonates (above 1 month) |
– ampicillin, + aminoglycoside and cephalosporin |
|
Children (under 5 years) |
– vancomycin + 3rd generation cephalosporin |
|
Adults |
– vancomycin + 3rd generation cephalosporin |
|
Immunocompromised patient |
– vancomycin + ampicillin + cephalosporin |
Steroids
A four-day regimen of dexamethasone, starting before or with the first dose of antibiotics, is now recommended in children with haemophilus and adults with bacterial meningitis likely to be pneumococcal. Meta-analysis found a risk reduction of neurological sequelae and mortality of about 30% in pneumococcal meningitis, with no clear difference with other organisms.
Therapy after organism identification
|
ORGANISM |
ANTIBIOTIC |
ALTERNATIVE THERAPY |
|
Haemophilus |
Ampicillin or 3rd generation cephalosporin according to sensitivities |
Chloramphenicol Fluoroquinolone Cefepime |
|
Pneumococcus |
Benzylpenicillin or 3rd generation cephalosporin according to sensitivities |
Chloramphenicol Fluoroquinolone Meropenem |
|
Meningococcus |
Benzylpenicillin or 3rd generation cephalosporin according to sensitivities |
Chloramphenicol Fluoroquinolone Meropenem |
|
E. coli |
3rd generation cephalosporin |
Aztreonam, fluoroquinolone, meropenem, ampicillin |
|
Listeria |
Ampicillin ± gentamicin |
Chloramphenicol Cotrimoxazole |
Duration
![]()
Pneumococcus – continue for 10–14 days after afebrile
Monitoring
In a deteriorating patient, CT scan will exclude the development of hydrocephalus, abscess or subdural empyema. In suspected sinus thrombosis MR venography may be required.
Remove any source of infection, e.g. mastoidectomy or sinus clearance.
In meningococcal meningitis the risk to household contacts is increased (500–800x) and chemoprophylaxis should be offered – rifampicin 600 mg b.d. for 48 hours. Vaccines are also available.
Meningitis/CSF shunts
Meningitis infection may follow CSF drainage operations for hydrocephalus. This may occur in the immediate postoperative period or be delayed for weeks or months. Clinical features of raised intracranial pressure may coexist due to shunt blockage. Bacteraemia is inevitable and blood cultures identify the responsible organism – usually Staphylococcus albus. The infection seldom resolves with antibiotic therapy alone and shunt removal is usually required.
BACTERIAL INFECTIONS – CNS TUBERCULOSIS
Tuberculosis is an infection caused in man by one of two mycobacteria – Mycobacterium tuberculosis and Mycobacterium bovis. The disease involves the nervous system in 10% of patients.
MENINGITIS
This is the commonest manifestation of tuberculous infection of the nervous system. In children, it usually results from bacteraemia following the initial phase of primary pulmonary tuberculosis.
In adults, it may occur many years after the primary infection.
Following bacteraemia, metastatic foci of infection lodge in:
1. Meninges
2. Cerebral or spinal tissue
3. Choroid plexus

Rupture of these encapsulated foci results in spread of infection into the subarachnoid space. In adults, reactivity of metastatic foci may occur spontaneously or result from impaired immunity (e.g. recent measles, alcohol abuse, administration of steroids).
The clinical features of tuberculous meningitis (TBM) result from:
|
– Infection. |
|
|
– Exudation |
– which may obstruct the basal cisterns and result in hydrocephalus. |
|
– Vasculitis |
– secondary to inflammation around vessels, resulting in infarction of brain and spinal cord. |
The basal meninges are generally most severely affected.
Clinical features
The majority of patients are adults; childhood TBM is now rare. Non-specific prodromal symptoms develop over 2–8 weeks.

Staging is useful for predicting outcome.
Seizures may occur at the onset. Involuntary movements (chorea, myoclonus) occur in 10%.
Atypically the illness may develop slowly over months presenting with dementia or rapidly like pyogenic (bacterial) meningitis. Occasionally cerebral features prevail rather than signs of meningitis.
Untreated, the illness may progress from phase 1 to death over a 3-week period.
Arachnoiditis inflammatory exudate may result in hydrocephalus/dementia/blindness.
TUBERCULOUS MENINGITIS
Investigations
• General: Anaemia, leucocytosis. Hyponatraemia (if inappropriate ADH secretion occurs).
• Cerebrospinal fluid
– Cell count, differential count, cytology (50–4000/mm3 – predominantly lymphocytes)
– Glucose, with a simultaneous blood sugar (<50% blood glucose)
– Protein (>1g/l)
– Acid-fast stain, Gram stain, appropriate bacteriologic culture and sensitivity, India ink (all causes of lymphocytic meningitis)
– Cryptococcal antigen, herpes antigen (other causes of lymphocytic meningitis)
– Culture for M. tuberculosis (50–80% positive)
– Polymerase chain reaction (PCR) to detect Mycobacterium DNA – specificity and sensitivity 100% and 70%.
• Tuberculin skin test: Positive in 50% of cases. (Negative if recent steroids or acquired primary infection.)
• Chest x-ray: – hilar lymphadenopathy/infiltrate/cavitations/effusion/scar.
• CT scan and MRI – hydrocephalus, basal meningeal thickening, infarcts, oedema, tuberculomas and obliteration of the subarachnoid space.

Diagnosis
Diagnosis is based on the clinical presentation with characteristic CSF findings.
DIFFERENTIAL DIAGNOSIS – Viral meningoencephalitis – Subacute/chronic meningitis (see pages 517–8).
Treatment
If suspect, commence antituberculous treatment.
Recommended treatment programme:
Normal regime:

Drug resistance suspected due to previous antituberculous therapy, e.g.
– Developing countries
– History of previous infection.
→ Add a fourth drug – streptomycin (1 g daily) or ethambutal (25 mg/kg daily).
Isoniazid and pyrazinamide penetrate meninges well; other drugs penetrate less well especially when the inflammation begins to settle.
Side effects:
– Isoniazid may produce peripheral neuropathy – protect with pyridoxine 50 mg daily.
– Ethambutol may produce optic atrophy – check colour vision.
– Streptomycin may cause 8th cranial nerve damage (vertigo and deafness).
– Nausea, vomiting, abnormal liver function and skin rashes may occur with all antituberculous drugs.
Evidence concerning the duration of anti-tuberculous treatment is conflicting. Conventionally therapy is given for 6–9 months, although some still recommend it for 24 months.
Intrathecal therapy: Since CSF penetration, especially with streptomycin, is poor, some recommend intrathecal treatment. Streptomycin 50 mg may be given daily or more frequently in seriously ill patients.
When obstructive hydrocephalus occurs, combined intraventricular (through the shunt reservoir or drainage catheter) and lumbar intrathecal treatment injections may be administered.
Steroid therapy: A recent Cochrane review reported that adjunctive steroids reduce neurological sequelae, hearing loss and mortality in patients with TBM without HIV. Insufficient data are available to recommend the use of steroids in HIV positive TBM.
Hydrocephalus
Progressive dilatation of the ventricles impairing conscious level requires CSF drainage – either temporarily with a ventricular catheter (permitting intraventricular drug administration) or permanently with a ventriculoperitoneal/atrial shunt. Surgery may also be considered for co-existent tuberculomas and tuberculous abscesses though these often resolve with drug therapy.
The course of treated tuberculous meningitis
Outcome is influenced by the patient’s age, general state of health, timing of initiation of treatment and the development of arachnoiditis and vascular complications.
Treatment in early stages is associated with a 10% mortality, in later stages with a 50% mortality. Of those who survive, neurological sequelae persist in 30% – hemiplegia, hypothalamic/pituitary dysfunction, blindness, deafness, dementia and epilepsy.
With treatment, CSF sugar quickly returns to normal; the cellular reaction gradually diminishes over 3–4 months; the protein level may take a similar time to return to normal.
Tuberculous meningitis in AIDS
Atypical mycobacteria such as M. avium and fortuitum should be considered. Response to treatment is generally good. TBM tends to occur in the earlier phases of immunodeficiency with CD4 T cell count, at <400 per mm3.
OTHER FORMS OF CNS TUBERCULOUS INFECTION
TUBERCULOMAS OF THE BRAIN
Tuberculomata may occur in cerebral hemispheres, cerebellum or brain stem with or without tuberculous meningitis, and may produce a space-occupying effect. They consist of caseating granulomas made up of epitheloid cells and macrophages containing mycobacteria. Lesions may be single or multiple. CT and MRI demonstrate lesions but appearances are not pathognomonic. Most resolve over a few weeks with antituberculous therapy.

POTT’S DISEASE
Chronic epidural infection follows tuberculous osteomyelitis of the vertebral bodies. This arises in the lower thoracic region, can extend over several segments and may spread through the intervertebral foramen into pleura, peritoneum or psoas muscle (psoas abscess).
TUBERCULOUS MENINGOMYELITIS
Infection of the leptomeninges results in an exudate that encases the spinal cord and nerve roots. This produces back pain, paraesthesia, lower limb weakness and loss of bowel and bladder control. Imaging may be normal while CSF shows high protein, lymphocytes and rarely acid fast bacilli. This disorder is now more frequent in AIDS patients. Differential diagnosis includes cytomegalovirus, cryptococcus, syphilis and lymphoma. Laminectomy and meningeal biopsy may be required to establish diagnosis. When suspected, empirical theory with antituberculous drugs is appropriate.
Clinical features:

SPIROCHAETAL INFECTIONS OF THE NERVOUS SYSTEM
SYPHILIS
This infectious disease is caused by the spirochaete Treponema pallidum. Entry is by:
– inoculation through skin or mucous membrane (sexually transmitted) – acquired syphilis.
– transmission in utero – congenital syphilis.
In the last 30 years, there has been a steady decline in incidence regardless of race and ethnicity. Despite this, it still remains an important health problem in certain geographic areas.
Up to 10% of patients with HIV will test positive for syphilis. All patients with neurosyphilis should be tested for this.

The chancre or primary sore on skin or mucous membrane represents the local tissue response to inoculation and is the first clinical event in acquired syphilis.
The organism, although present in all lesions, is more easily demonstrated in the primary and secondary phases.
In congenital syphilis fetal involvement can occur even though many years may elapse between the mother’s primary infection and conception.
Widespread recognition and efficient treatment of the primary infection have greatly reduced the late or tertiary consequences.
Not all patients untreated in the secondary phase progress to the tertiary phase.
In HIV patients the neurological complications occur earlier and advance more quickly.
Investigations
Spirochaetes can be demonstrated microscopically by dark field examination in primary and secondary phase lesions.
Serological diagnosis depends on detection of antibodies.
1. Non-specific (Reagin) antibodies (IgG and IgM).
Reagin tests involve complement fixation.
The Venereal Disease Research Laboratory (VDRL) test is the commonest and when strongly positive indicates active disease (may be negative in HIV).
2. Specific treponemal antibodies (do not differentiate between past and present infection). Fluorescent treponemal antibody absorption (FTA) test and Treponema immobilisation (TPI) test.
3. Treponema pallidum DNA can be detected in the CSF of patients by PCR (sensitivity 60%).
SPIROCHAETAL INFECTION – NEUROSYPHILIS
The initial event in neurosyphilis is meningitis. Of all untreated patients 25% develop an acute symptomatic syphilitic meningitis within 2 years of the primary infection.
ACUTE SYPHILITIC MENINGITIS: Three clinical forms are recognised:

Late neurological complications occur in only 7% of untreated cases.
These forms are exceptionally rare and the clinical syndromes mentioned above seldom occur in a ‘pure’ form.
MENINGOVASCULAR SYPHILIS
‘Early’ late manifestation resulting in an obliterative endarteritis and periarteritis.
Presents as a ‘stroke’ in a young person – hemisphere, brain stem or spinal. Granulations around the base of the brain may produce cranial nerve palsies or even hydrocephalus.
CSF – lymphocytes 100/mm3, protein ↑, gammaglobulin ↑, positive serology. Penicillin arrests progression.
SPINAL SYPHILIS
Chronic meningitis with subpial damage to the spinal cord.
Presents as a progressive paraplegia, occasionally with radicular pain and wasting in upper limbs – ERB’s PARAPLEGIA. CSF – as meningovascular syphilis. Penicillin arrests progression.
OCULAR MANIFESTATIONS
Meningitis around optic nerve with subpial necrosis may be the only manifestation of late syphilis.
Presents as a constriction of the visual fields with a progressive pallor of the optic disc:
– if both eyes are affected, the vision is rarely saved.
– if only one eye is involved, treatment with penicillin will save the other.
Neuroretinitis, uveitis and chorioretinitis occur, especially in HIV patients.
GENERAL PARESIS
Characterised by dementia – with memory impairment, disordered judgement and disturbed affect – manic behaviour, delusions of grandeur (rare).
There are two phases:
1. Pre-paralytic – with progressive dementia.
2. Paralytic – when corticospinal and extrapyramidal symptoms and signs develop associated with involuntary movements (myoclonus).
Argyll Robertson pupils may be present (see page 146).
At autopsy, meningeal thickening, brain atrophy and perivascular infiltration with plasma cells and lymphocytes are evident; culture from the cortex may reveal an occasional treponema.
CSF – lymphocytes 50/mm3, protein ↑ 0.5–2 g/l, gammaglobulin ↑.
Reagin tests in CSF positive in the majority.
Treatment in the preparalytic phase will halt progression in 40%.
TABES DORSALIS
Posterior spinal root and posterior column dysfunction account for symptoms.

The CSF is more normal than in general paresis. The Reagin test may be negative in 30 per cent. Treatment may produce some improvement; it will not reverse joint destruction.
SYPHILITIC GUMMA presenting as an intracranial mass is extremely rare.
TREATMENT OF NEUROSYPHILIS
|
Penicillin G. |
2–4 megaunits i.v. |
(When patient sensitive to penicillin |
|
or |
4-hourly for 10 days. |
↓ |
|
Procaine Penicillin |
600 000 units i.m. daily for 15 days. |
erythromycin or tetracycline may be given orally over 30 days.) |
|
Benzathine Penicillin |
2–4 megaunits i.m. weekly × 3. |
To prevent congenital syphilis penicillin should be given to all neonates and infected mothers during the first 4 months of pregnancy.
The Jarisch-Herxheimer reaction – tachycardia/fever – occurs in one-third of patients within a few hours of commencing treatment; it is believed to be due to endotoxin release from killed organisms. Steroids should counter the reaction, especially in tertiary syphilis.
CSF follow up: CSF is checked initially and at 6 monthly intervals until normal.
Cell count and degree of positivity of VDRL are the best indicators of persistent infection.
Failure of treatment is common in HIV positive patients and more frequent retesting of blood and CSF is necessary.
SPIROCHAETAL INFECTION
LYME DISEASE (NEUROBORRELIOSIS)
Originally described in the community of Old Lyme, this is a disorder, caused by the spirochaete Borrelia burgdorferi, characterised by relapsing and remitting arthralgia associated with a characteristic skin rash (erythema chronicum migrans) and neurological features. The organism, related to the treponemes, is prevalent throughout Europe and North America and is carried by ixodes ticks.
Clinical features
Only a minority of persons bitten by an infected tick develop the disease. Spirochaetocidal activity in normal serum and the immune response normally provide protection. It rarely occurs in HIV patients.

Diagnosis

Treatment
Stage 1 – Oral antibiotics: penicillin, erythromycin or tetracycline.
Stage 2 – I.V. penicillin G. 20 million units for 10 days (or ceftriaxone).
Stage 3 – as stage 2.
If symptoms persist – wrong diagnosis with misleading titres, or – immune mediated damage.
Steroids can be used in late stages when symptoms have not responded to antibiotics.
LEPTOSPIROSIS
Leptospira interrogans is transmitted to man in the infected urine of wild and domestic animal carriers. Subclinical infection commonly occurs in high-risk occupations, e.g. sewer workers. Symptomatic illness is usually mild and only 10% of patients develop jaundice and haemorrhagic complications (Weil’s disease).
Clinical features

Diagnosis
A combination of abnormal liver and renal function with elevated creatine kinase suggest the diagnosis. Leptospirae can be isolated from blood and CSF (in the immune phase) but diagnosis is usually confirmed by demonstrating agglutinating antibodies (ELISA detected IgM).
Treatment
The disease is usually self limiting and therapy unnecessary. Early treatment in the leptospiraemic phase with Penicillin G 12 million units daily and tetracycline 500 mg four times per day may minimize the immune-mediated complications. Support of hepatic/renal failure and management of haemorrhagic complications may be life-saving.
PARASITIC INFECTIONS OF THE NERVOUS SYSTEM – PROTOZOA
TOXOPLASMOSIS
A world-wide parasitic infection affecting many species, including man.
Organism: An anaerobic intracellular protozoan, Toxoplasma gondii.
The majority of infections in man are asymptomatic (30% of the population have specific antibodies indicating previous exposure).
In the host

Transmission: Eating uncooked meat or contact with faeces of an infected dog or cat (definitive hosts).
There are two forms of toxoplasmosis:
|
CONGENITAL – when a previously unaffected woman contracts infection during pregnancy (subclinical infection); transplacental spread results in fetal infection. Premature delivery occurs in 25%. Neurological complications: – hydrocephalus, – aqueduct stenosis, – microcephaly. Non-neurological featues: – skin rash, jaundice, hepatosplenomegaly, choroidoretinitis. Skull X-ray shows: – curvilinear calcification (basal ganglion and periventricular regions). Varying degrees of organ involvement may occur. The only manifestation may be choroidoretinitis in an otherwise healthy child. |
ACQUIRED – symptomatic infection is uncommon and may be associated with underlying systemic disease or immunosuppression (e.g. AIDS). Fever and fatigue with muscle weakness and lymphadenopathy result. Abnormal lymphocytes in peripheral blood leads to confusion with infectious mononucleosis. The neurological features are those of a meningoencephalitis with focal signs and depressed conscious level. Choroidoretinitis occasionally occurs.
|
Diagnosis:
Organisms are seldom identified.
IgG antibodies indicate previous exposure, positive IgM and high or rising IgG confirm active infection.
Serological tests may be negative in AIDS.
In acquired infection CT shows characteristic ring shaped contrast enhancement. MRI is even more sensitive. Brain biopsy is necessary for exclusion of CNS lymphoma and for definitive diagnosis.
N.B. Rubella, cytomegalovirus and herpes simplex can also spread transplacentally and cause jaundice and hepatosplenomegaly. Cytomegalovirus may also produce choroidoretinitis and intracranial calcification.
Treatment
Sulphadiazine and pyrimethamine (Dapaprim) with folinic acid for 6 weeks. In AIDS newer drugs, such as clarithromycin and azithromycin, have also been used with some success. In this patient group recurrence after discontinuation of therapy mandates life long treatment. Give steroids when choroidoretinitis is present.
MALARIA
Plasmodium falciparum, the agent of malignant tertiary malaria, is responsible for cerebral malaria. Infected red blood cells adhere to vascular endothelium and block the microcirculation. Endothelial damage produces cerebral oedema. Confusion, focal signs, convulsions and coma occur. Diagnosis depends on demonstrating parasites in peripheral blood. Parenteral anti malaria treatment (chloroquine), exchange transfusion and supportive therapy may be life saving. Overall mortality is 10%. Complete recovery without sequelae is expected in survivors.
VIRAL INFECTIONS
General principles
Invasion of the nervous system may occur as part of a generalised viral infection.
Occasionally nervous system involvement is disproportionately severe and symptoms of generalised infection are slight.
Viruses enter the body through the: respiratory tract, gastrointestinal tract, genitourinary tract or by inoculation through the skin.

After CNS penetration, the clinical picture depends upon the particular virus and the cells of the nervous system which show a specific susceptibility.

Some viruses cause a chronic, progressive infection, others remain dormant for many years within the nervous system before becoming symptomatic.
MENINGITIS
Meningitis is the commonest type of viral infection of the central nervous system. The term aseptic meningitis includes viral meningitis as well as other forms of meningitis where routine culture reveals no other organisms.
|
Common causal viruses – |
Rare causal viruses – |
|
ENTEROVIRUSES |
LYMPHOCYTIC CHORIOMENINGITIS |
|
MUMPS VIRUS |
HUMAN IMMUNODEFICIENCY VIRUS (HIV) |
|
HERPES SIMPLEX (subtype 2) |
WEST NILE VIRUS |
|
EPSTEIN-BARR VIRUS (EBV) |
VIRAL INFECTIONS – MENINGITIS
Clinical features of acute aseptic meningitis

Enterovirus infection e.g. Coxsackie or echo viruses – affects children/young adults and occurs seasonally in late summer.
Spread is by the faecal/oral route.
Mumps – affects children/young adults. Winter/spring incidence.
Herpes simplex (type 2) – accounts for 5% of viral meningitis. Develops in 25% of patients with primary genital infection (suspect in sexually active adults). Can cause a recurrent meningitis (Mollaret’s meningitis).
Lymphocytic choriomeningitis – affects any age and is a consequence of airborne spread from rodent droppings.
Human Immunodeficiency Virus (HIV) – suspect in high risk groups (page 515). HIV antibodies are often absent and develop 1–3 months later during convalescence.
Investigations
The CSF cell count is elevated (lymphocytes or monocytes) with a normal glucose and protein. PCR detection of viral DNA/RNA in CSF though diagnostic, is rarely thought necessary. Virus may be cultured from throat swabs or stool. Serological tests on serum in acute and convalescent phases are especially valuable in detecting mumps and herpes simplex (type 2).
Differential diagnosis
From other causes of an aseptic meningitis which are usually subacute or chronic in onset:
– Tuberculous or fungal meningitis
– Leptospirosis
– Sarcoidosis
– Carcinomatous meningitis
– Partially treated bacterial meningitis
– Parameningeal chronic infection which evokes a meningeal response, e.g. mastoiditis.
The self-limiting and mild nature of viral meningitis should not lead to confusion with these more serious disorders.
Prognosis is excellent and treatment symptomatic.
VIRAL INFECTIONS – PARENCHYMAL
Viruses may act:
directly → acute viral encephalitis or meningoencephalitis, or indirectly via the immune system→ allergic or postinfectious encephalomyelitis and postvaccinial encephalomyelitis.
Also, a ‘toxic’ encephalopathy may develop during the course of a viral illness in which inflammation is not a pathological feature – REYE’S SYDNROME.
ACUTE VIRAL ENCEPHALITIS
Viral infection causes neuronal and glial damage with associated inflammation and oedema.
Viral encephalitis is a worldwide disorder with the highest incidence in the tropics.
Common causal viruses:
|
World-wide: – Mumps – Herpes simplex – Varicella zoster – Epstein-Barr – Arboviruses |
Rare forms in specific areas: |
|
|
St Louis West Nile Russian spring/summer |
– Arthropod-borne – USA – Arthropod-borne – Africa/India – Arthropod-borne – eastern Europe |
Encephalitis following childhood infections – measles, varicella, rubella – is presumed to be postinfectious and not due to direct viral invasion, though the measles virus has occasionally been isolated from the brain.
Clinical features:
Signs and symptoms:
General: pyrexia, myalgia, etc.
Specific to causative virus, e.g. features of infectious mononucleosis (Epstein-Barr).
Meningeal involvement (slight) → neck stiffness, cellular response in CSF.
Signs and symptoms of parenchymal involvement – focal and/or diffuse.

In general, the illness lasts for some weeks.
Prognosis is uncertain and depends on the causal virus as do neurological sequelae.
Herpes Simplex (HSV) and Varicella-Zoster (VZV) commonly cause disease in humans.
HERPES SIMPLEX ENCEPHALITIS
HSV-1 is the commonest cause of sporadic encephalitis.
One third occur due to primary infection; two thirds have pre-existing antibodies (reactivation).
Clinical features
A world-wide disorder occurring during all seasons and affecting all ages.
Incidence: 1/250 000
General symptoms at onset – headache, fever – with evolution over several days to seizures and impaired conscious level.

Investigations
MR imaging: T2 weighted MRI showing temporal and orbitofrontal hyperintensities typical of herpes simplex encephalitis.
CSF examination reveals 5–500 lymphocytes. The protein is mildly elevated and the glucose is normal.
EEG examination shows generalised slowing with bursts of ‘periodic’ high voltage slow wave complexes over the involved temporal lobe.
Polymerase chain reaction (PCR) on CSF may be negative in the first 48 hours. The quantity of HSV DNA then increases and, if initially negative and the clinical course is suggestive, the examination should be repeated. Also paired sera and CSF should be sent for HSV antibody (CSF HSV-specific antibody can still be detected up to 30 days).
Brain biopsy seldom required in view of the above new diagnostic techniques.


Treatment: Acyclovir inhibits DNA synthesis, is relatively non-toxic and significantly reduces morbidity and mortality. When the diagnosis is considered, treatment must start without delay.
Varicella-Zoster Virus (VZV) encephalitis may complicate chicken pox, or a cutaneous zoster eruption. CSF shows a mild lymphocytosis (<100 cells/mm3), a slight increase in the protein and a normal glucose. PCR detects VZV DNA. The virus can be grown from CSF and antibodies detected. Treatment with acyclovir or famciclovir is effective. Vasculitis may complicate.
VIRAL INFECTIONS – REYE’S SYNDROME
REYE’S SYNDROME
This rare encephalopathy, associated with fatty changes in the liver and other viscera, is almost exclusively confined to children. It is due to aspirin useage in infection with Influenza A, Influenza B or varicella–zoster viruses.
Incidence
Since 1980 the incidence of this condition has dropped dramatically, in part due to avoidance of salicylates in children.
Pathology
Neurons and glial cells are swollen; the liver, heart and kidney show fatty infiltration.
Pathogenesis
Viral synergism with an environmental factor, e.g. salicylates, may be responsible.
Morphological changes in mitochondria indicate a central role.

Death results from raised intracranial pressure.
Investigations
|
– Raised liver enzymes (ALT & AST) – Elevated serum ammonia |
– Hypoglycaemia (in infants) – Prolonged prothrombin time |
– Increase in serum fatty acids Aminoaciduria |
CT/MRI show appearances of diffuse cerebral oedema
Differential diagnosis
Consider other causes of raised intracranial pressure in childhood, especially
– lead encephalopathy,
– lateral sinus thrombosis, e.g. following mastoiditis.
Treatment
Treatment aims at lowering intracranial pressure with the aid of intracranial pressure monitoring (see page 52). In addition, blood glucose must be maintained and any associated coagulopathy treated. Reduction of ammonia may be achieved by peritoneal dialysis or exchange transfusion.
Prognosis
Early diagnosis and supportive treatment has reduced the mortality from 80% to 30%.
When raised intracranial pressure is present, mortality increases to 50% and a high proprotion of survivors have cognitive disorders.
A condition similar to Reye’s syndrome occurs in some children with family history of ‘sudden infant death’. A deficiency of medium chain acetyl-CoA dehydrogenase (an enzyme essential for fatty acid metabolism) is found. Carnitine deficiency results as a consequence of ‘alternative pathway’ fatty acid metabolism. Siblings of children with Reye’s syndrome should be screened for this disorder.
VIRAL INFECTIONS – CHRONIC DISORDERS
In these disorders the infection results in a chronic progressive neurological condition.
The evidence of a viral etiology is:
Direct – finding of inclusion bodies, demonstration of viral particles or isolation of virus.
Indirect – relationship of onset of symptoms to a preceding viral illness, transmission of illness from one host to the next
N.B. Not all these features are present in any one illness
SUBACUTE SCLEROSING PANENCEPHALITIS (SSPE)
Caused by measles-like paramyxovirus – isolated from brain biopsy.
Less common with the availability of widespread primary measles vaccination.
Clinical features: A world-wide disorder. Incidence: 1 per million per year. Onset: between ages 7–10 years.
Stage 1: Behavioural problem, declining school performance, progression → dementia
Stage 2: Chorioretinitis, myoclonic jerks, seizures, ataxia, dystonia
Stage 3: Lapses into rigid comatose state

The illness may occur after measles vaccination or following clinical infection at an early age (under 2 years).
Accompanying features of infection, i.e. pyrexia, leucocytosis, are absent.
Investigations

EEG – shows periodic high voltage slow wave complexes on a low voltage background trace.
Pathology
Changes involve both white and grey matter, especially in the posterior hemispheres. Brain stem, cerebellum and spinal cord are also affected. Oligodendrocytes contain eosinophilic inclusion bodies. Marked gliosis occurs with perivascular lymphocyte and plasma cell cuffing.
Treatment: There is no effective treatment. Since the introduction of measles vaccination there has been a marked reduction of SSPE.
Subacute measles encephalitis may follow measles infection in children on immunosuppressive drug treatment or with hypogammaglobulinaemia. The clinical course is different however from SSPE and EEG and CSF findings are less specific.
PROGRESSIVE RUBELLA PANENCEPHALITIS
Similar to SSPE with a fatal outcome, caused by rubella virus.
|
Presents at a later age (10–15 years) |
CSF shows high γ globulin. |
|
Progressive dementia. |
Antibodies elevated in serum and CSF to rubella. |
|
Ataxia. Spasticity. Myoclonus. |
Biopsy does not show inclusion bodies. |
|
Treatment: No effective treatment |
PRION DISEASES
Fatal conditions characterised by the accumulation of a modified cell membrane protein – Prion protein or PrP (proteinaceous infectious particle) within the central nervous system.
Clinical features are dependent on site and rate of deposition of PrP. A similar disorder in cattle, bovine spongiform encephalopathy (BSE) may be a source of infection in man.
The Prion theory
Experimental and epidemiological evidence supports transmissibility. Physical properties of the infective agent – heat and radiation resistance and absence of nucleic acid – suggests it is comprised solely of protein. This infectious protein when innoculated modifies normal cell membrane protein which acts as a template for further conversion to abnormal protein. This host-encoded protein accumulates without any inflammatory or immune response. In familial cases a point mutation in the prion gene explains disease susceptibility.
Creutzfeldt-Jakob disease (CJD)
A worldwide disorder with incidence 1:1 000 000. Approximately 90% of cases are sporadic and 10% familial caused by mutations in the prion protein (PRNP) gene on chromosome 20. Age of onset 50–60 years. Non specific symptoms at onset (anxiety and depression) are rapidly followed by myoclonus, ataxia, akinetic rigid state, dementia. Death within 12 months is usual. Iatrogenic disease occurs following corneal or dural grafts, depth electrodes and cadaveric derived human growth hormone treatment.

Investigation
EEG – 1–2 Hz triphasic sharp waves with periodic complexes
CSF – increase in protein 14–3–3 (a protein kinase inhibitor)
[The combined EEG and CSF findings, where positive, have diagnostic sensitivity/specificity of 97%]
Pathology
– Neuronal degeneration occurs with marked astrocytic proliferation and amyloid plaque formation. Vacuolation of glial cells results in a characteristic spongiform appearance.

Treatment – supportive.

New Variant CJD (vCJD)
Generally affects younger age group. Psychiatric symptoms of depression, anxiety, or withdrawal are common early manifestations. Neurological symptoms appear approximately 6 months later, with paraesthesias an early feature. Eventually sufferers exhibit ataxia, progressive dementia and involuntary movements (myoclonus, chorea, or dystonia).
Only 50% of patients have protein 14–3–3 proteins in CSF. EEG reveals non-specific slowing (the periodic complexes of sporadic CJD are absent).
PRNP gene mutations are found with patients homozygous for methionine at the 129 codon. Neuropathological changes – ‘florid’ plaques in the cerebral and cerebellar cortex, severe thalamic gliosis, and spongiform change with diffuse accumulation of prion proteins.
Gerstmann Straussler syndrome (GSS)
A similar disorder condition to CJD. Cases are familial and characterised by specific pathology of spongiform changes associated with amyloid plaques containing PrP immunoreactive proteins. Clinical features are nonspecific – ataxia, Parkinsonism, dementia. Death occurs within 5 years of contact.
Kuru
An extensively studied disorder of Papua New Guinea. It is of interest in view of man to man spread from cannibalism.
VIRAL INFECTIONS – MYELITIS AND POLIOMYELITIS
MYELITIS
Acute viral transverse myelitis is rare. It can occur in association with measles, mumps, Epstein-Barr, herpes zoster/simplex, enterovirus infections HIV, HTLV-1 and 2 and smallpox. Fever, back and limb pain precede paralysis, sensory loss and bladder disturbance. Initially paralysed limbs are flaccid, but over 1–2 weeks spasticity and extensor plantar responses develop. Good recovery occurs in 30%. Death from respiratory failure is rare (5%).
Investigations
Myelography when performed is normal. MRI may demonstrate focal cord signal changes. CSF shows elevated protein with a neutrophil or lymphocytic response. Serological tests will occasionally identify the causal virus. Electrophysiology distinguishes from Guillain-Barré syndrome.
Treatment
Supportive; the place of steroids remains unproven.
It is not clear whether the pathological effects (perivenous demyelination) result from direct or delayed (immunological) reactions to the virus.
POLIOMYELITIS
An acute viral infection in which the anterior horn cells of the spinal cord and motor nuclei of the brain stem are selectively involved. A major cause of paralysis and death 30 yrs ago, now rare with the introduction of effective vaccines and improved sanitation.
Causative viruses:
The poliovirus is a picornavirus (RNA virus).
Three immunological distinct strains have been isolated. Immunity to one does not result in immunity to the other two.
Coxsackie and echoviruses (also picornaviruses), may produce a clinically identical disorder. West Nile virus can produce a polio-like flaccid paralysis.
Pathology
Initially – inflammatory meningeal changes, followed by – inflammatory cell infiltration (polymorphs and lymphocytes) around the brain stem nuclei and anterior horn cells. Neurons may undergo necrosis or central chromatolysis.
Microglial proliferation follows.

Mode of spread
Spread by faecal/oral route. Once ingested the virus multiplies in the nasopharynx and gastrointestinal tract.

Epidemiology
A highly communicable disease which may result in epidemics.
Seasonal incidence – late summer/autumn.
World-wide distribution, although more frequent in northern temperate climates.
Prophylactic vaccination has produced a dramatic reduction in incidence in the last 25 years. In developing countries without a vaccination programme, the disease remains a problem.
Clinical features
Infection may result in:
– Subclinical course + resultant immunity (majority)
– Mild non-specific symptoms of viraemia + resultant immunity
– Meningism without paralysis (PREPARALYTIC) + resultant immunity
– Meningism followed by paralysis (PARALYTIC) + resultant immunity.
VIRAL INFECTIONS – POLIOMYELITIS

Diagnosis
During the meningeal phase, consider other causes of acute meningitis.
Once the paralytic phase ensues, distinguish from the Guillain-Barré syndrome and transverse myelitis.
The clinical picture + CSF examination (polymorphs and lymphocytes increased; protein elevated with normal glucose) are sufficient to reach the diagnosis.
Poliovirus RNA can be detected in faeces or CSF by PCR.
Prognosis
In epidemics, a mortality of 25% results from respiratory paralysis. Improvement in muscle power usually commences one week after the onset of paralysis and continues for up to a year.
Only a proportion of muscles remain permanently paralysed; in these, fasciculation may persist. In affected limbs, bone growth becomes retarded with shortening as well as thinning.
Treatment
The patient is kept on bed rest and fluid balance carefully maintained.
Respiratory failure may require ventilation.
Avoid the development of deformities in affected limbs with physiotherapy and splinting.
Post-Polio Syndrome
A significant proportion of polio patients develop late sequelae often 30 yrs after initial illness – fatigue, myalgia and progressive muscle atrophy with weakness are characteristic.
Prophylaxis

VIRAL INFECTIONS – VARICELLA-ZOSTER INFECTION
Varicella (chickenpox) and herpes zoster (shingles) are different clinical manifestations of infection by the same virus – Varicella–Zoster, a DNA human herpes virus.
|
Conditions caused: |
– an acute encephalitis – viral meningitis – myelitis |
– postinfectious encephalomyelitis – postinfectious polyneuropathy (Guillain-Barré syndrome). |
SHINGLES
This occurs after virus reactivation, dormant after the primary infection (chickenpox).

Clinical features
Patients are usually over 50 years of age. Sexes are affected equally. Recurrence is rare. Often occurs in immunocompromised patients e.g. lymphoma. Also associated with spinal/nerve root trauma.
Initial feature:

Motor weakness occurs in 20% due to damage of the anterior horn cell. More widespread spinal (myelitis) or encephalic involvement occurs in the immunodeficient. In these patients extensive cutaneous lesions are common (disseminated zoster).
Cranial nerve ganglia involvement:
– Trigeminal: usually ophthalmic division with vesicles above the eye and associated corneal ulceration – HERPES ZOSTER OPHTHALMICUS.
– Geniculate: vesicles within the external auditory meatus and ear drum. Ipsilateral deafness and facial weakness result. – RAMSAY HUNT SYNDROME.
Diagnosis: Based on clinical features. Virus DNA can be detected in vesicular fluid by PCR.
Treatment
This depends on the severity and location of skin lesions. Mild disease requires symptomatic treatment only. Severe disease, involvement in immunocompromised patients, or ophthalmic vesicles require acyclovir either orally or intravenously.
POST HERPETIC NEURALGIA
This is a condition which occurs in 10% of all patients. The incidence rises with age. A chronic, uncomfortable, burning pain presents in the territory of the involved dermatome. The pathogenesis is unknown.
Treatment with antidepressants, anticonvulsants, e.g. carbamazepine, transcutaneous stimulation (TCS) or sympathetic ganglion block may help, but results are unpredictable.
Varicella and Herpes Zoster CNS involvement
Patients with AIDS and other immunocompromising disorders risk severe, life-threatening CNS involvement – encephalitis, cerebral vasculitis, myelitis or brain stem encephalitis. Herpes Zoster ophthalmicus can be associated, in the middle aged, with delayed major cerebral artery territory infarction. This presents 4–6 weeks after infection. Stroke also occurs as a remote complication of childhood varicella, usually within 12 weeks of clinical chickenpox. These are both due to virus-induced damage to cerebral arteries (vasculitis). The role of anti-viral drugs and steroids is uncertain.
OPPORTUNISTIC INFECTIONS
These infections occur in immunocompromised patients. Certain types of immunological deficiency tend to be associated with specific forms of infection.

CLINICAL SYNDROMES, DIAGNOSIS AND TREATMENT

ACQUIRED IMMUNODEFICIENCY SYNDROME (AIDS)
Human immunodeficiency virus (HIV-I) has lymphotropic (CD4 lymphocytes) and neurotropic (microglial) properties. Neurological features develop in 80% of infected individuals manifesting as either direct effects of the HIV virus or infections, tumours and associated vascular disorders due to immunodeficiency. AIDS is the end stage of chronic infection.
Prevalence of AIDS and HIV infection
Certain individuals are ‘at risk’ of infection:

The incidence of HIV infection in ‘at risk’ groups varies considerably.
Sex education, supply of clean needles to addicts, active drug-dependence programmes and specific precautions in the preparation of blood products are necessary to limit its spread.
Current prevalence of HIV – USA 450/100,000
CLINICAL COURSE OF HIV INFECTION

NEUROLOGICAL PRESENTATIONS OF HIV INFECTION

Treatment
Opportunistic infection – treatment of specific infection (see page 514).
With known HIV + ve patients, invasive procedures such as biopsy are often avoided and trials of therapy are administered, e.g. cerebral toxoplasmosis – trial of pyrimethamine and sulphadiazine, monitored with CT/MRI. If lesions do not resolve → biopsy (? lymphoma).

Highly active antiretroviral therapy (HAART) with effective treatment for infections and neoplastic complications has significantly improved outcome. Mean survival time for HIV-infected persons currently exceeds 10 years. The prolonged survival of HIV-infected persons increases their risk of developing PML or CNS lymphoma (these responding poorly to treatment).
HAART management comprises two nucleoside reverse transcriptase inhibitors (e.g. zidovudine and didanosine) and a protease inhibitor (e.g. ritonavir or indinavir), or a nonnucleoside reverse transcriptase inhibitor (nevirapine). This combination is given to HIV-infected individuals with detectable viral loads or immunologic dysfunction (less than 500 CD4+ cells/mm3). HAART results in immunological and neurocognitive improvement, even when HIV is advanced. Treatment aims at reducing the viral load to undetectable levels, PCR having a central role in monitoring therapy and identifying drug resistance.
SUBACUTE/CHRONIC MENINGITIS
This entity is characterised by symptoms and signs of meningeal irritation which persist and progress over weeks without improvement. Unlike acute meningitis, the onset is insidious; cranial nerve signs and focal deficits such as hemiparesis, dementia and gradual deterioration of conscious level may predominate. Predisposing factors include immunosuppression or immunocompromised host. The outcome depends upon aetiology and the early instigation of appropriate treatment.
Chronic meningitis is associated with certain CSF findings.
– Lymphocytosis + low glucose (relative to serum level)
– Lymphocytosis + normal glucose.
Diagnosis depends upon CSF examination.
Lumbar puncture should be performed in suspicious cases as soon as CT scan has ruled out a mass lesion.
SUBACUTE/CHRONIC MENINGITIS WITH A LOW CSF GLUCOSE
Western herbs shown to suppress or inhibit pro-inflammatory cytokine production


Despite extensive investigation, a group of patients with chronic meningitis exists in whom no cause is found.
DEMYELINATING DISEASE
Demyelinating disorders of the central nervous system affect myelin and/or oligodendroglia with relative sparing of axons.
The central nervous system is composed of neurons with neuroectodermal and mesodermal supporting cells.
The neuroectodermal cells comprise:
astrocytes
ependymal cells
oligodendrocytes.
The oligodendrocytes, like Schwann cells in the peripheral nervous system, are responsible for the formation of myelin around central nervous system axons.
One Schwann cell myelinates one axon but one oligodendrocyte may myelinate several contiguous axons, and the close proximity of cell to axon may not be obvious by light microscopy.

Oligodendrocytes are present in grey matter near neuronal cell bodies and in white matter near axons.
Myelin is composed of protein and lipids. Protein accounts for 20% of total content. The lipid fraction may be divided into:
cholesterol
glycophosphatides (lecithins)
sphingolipids (sphingomyelins).
The laying down of myelin in the central nervous system commences at the fourth month of fetal life in the median longitudinal bundle, then in frontal and parietal lobes at birth. Most of the cerebrum is myelinated by the end of the 2nd year. Myelination continues until the 10th year of life.
Myelin disorders may be classified as diseases in which:
1. Myelin is inherently abnormal or was never properly formed – these disorders generally present in infancy and early childhood and have a biochemical basis, e.g. leukodystrophy.
2. Myelin which was normal when formed breaks down as a consequence of pathological insult, e.g. multiple sclerosis.
MULTIPLE SCLEROSIS
Multiple sclerosis (MS) is a common demyelinating disease, normally characterised by focal disturbance of function and a relapsing and remitting course.
The disease occurs most commonly in temperate climates and prevalence differs at various latitudes:
|
Latitude (°N) |
Rate/100 000 (adjusted) |
|
|
Orkneys and Shetland……… |
60 |
309 |
|
England (Cornwall)………… |
51 |
63 |
|
Italy (Bari)…………… |
41 |
13 |
The disease usually occurs in young adults with a peak age incidence of 20–40 years. Slightly more females than males are affected. There is a 3% risk of disease if a sibling or parent is affected.
PATHOLOGY
Scattered lesions with a greyish colour, 1 mm to several cm in size, are present in the white matter of the brain and spinal cord and are referred to as plaques. The lesions lie in close relationship to veins (postcapillary venules) – perivenous distribution.

PATHOGENESIS
Immune deficiency has been suggested. This might explain the possible persistence of a latent virus and variations in immune status could be the basis of ‘relapses and remissions’. T lymphocytes and macrophages found in plaques may be sensitized to myelin antigens.
Hereditary/genetic factors appear significant. There is an increased familial incidence of multiple sclerosis. This has led to the study of histocompatibility antigens (HL-A). An association between A3, B7, B18 and DW2/DRW2 and multiple sclerosis has been demonstrated. Concordance rate in monozygotic twins is 30% and in dizygotes 5%. Affected women transmit MS to offsping more frequently than affected men suggesting that mitochondrial genes contribute to inheritance.
Viruses may be important in the development of multiple sclerosis, infection perhaps occurring in a genetically/immunologically susceptible host.
Elevated serum and CSF antibody titres have been found to:
– varicella zoster, measles, rubella and herpes simplex during relapse.
Biochemical: No biochemical effect has been demonstrated – myelin appears normal before breakdown and the proposed excess of dietary fats or malabsorption of unsaturated fatty acids is unproven.
In summary – the causation is probably multifactorial.

CLINICAL FEATURES
|
Peak age of onset |
– 20–25 years |
|
Childhood onset rare |
– 2% |
|
Patients presenting >50 years |
– 5% |
|
Patients presenting >60 years |
– 1% |
Multiple sclerosis is usually characterised by:
– Signs and symptoms of widespread white matter disease.
– A relapsing and remitting or progressive course.
Symptoms at onset
1. Vague symptoms: lack of energy, headache, depression, aches in limbs – may result in diagnosis of psychoneurosis. These symptoms are eventually associated with:
2.

Trigeminal neuralgia may be an early symptom of multiple sclerosis, and this should be considered in the young patient with paroxysmal facial pain.
Sensory symptoms
Numbness and paraesthesia are common and often minor and transient. Paraesthesia is more often due to posterior column demyelination than to spinothalamic tract involvement.

Posterior column lesions result in impaired vibration sensation and joint position sensation. In such cases a limb may be rendered ‘useless’ by the absence of positional awareness.
Lhermitte’s sign: with cervical posterior column involvement sudden neck flexion will evoke a ‘shock-like’ sensation in the limbs.
Spinothalamic lesions result in dysaesthesia – an unpleasant feeling of burning, coldness or warmth, with associated sensory loss to pain and temperature contralateral to the lesion.

Motor symptoms
Monoparesis and paraparesis are the most common motor symptoms. Hemiparesis and quadriparesis occur less commonly.
Paraparesis is the result of spinal demyelination, usually in the cervical region.
Signs: – Increased tone
– Hyperactive tendon reflexes, extensor plantar response and absent abdominal reflexes
– Pyramidal distribution weakness.

Loss of vision
Acute optic neuritis (Retrobulbar neuritis): Visual loss associated usually with a central scotoma and recovery over some weeks. This commonly occurs in young adults. The visual loss develops over several days and is often associated with pain on ocular movement (irritation of the dural membrane around the optic nerve). In milder forms, only colour vision is affected. Typically only one eye is affected, although occasionally both eyes simultaneously or consecutively are involved.

On examination: Disturbance of visual function ranges from a small central scotoma to complete loss. Fundal examination reveals swelling – papillitis – in up to 50% of patients, depending upon the proximity of the plaque to the optic nerve head.

‘Sheathing’ from an inflammatory exudate around peripheral retinal venules is common. Reduced visual acuity distinguishes papillitis from papilloedema.
Investigation: Visual evoked responses (VERs) show delay. High resolution CT or MRI of the optic nerve excludes tumour. MR confirms the presence of plaque.
Treatment: The optic neuritis study group showed that IV or oral steroids compared with placebo accelerated recovery though at 2 years there was no significant difference in eventual visual function. Oral steroids were associated with a higher risk of recurrent optic neuritis. Intravenous steroids appeared (within the next 2 years) to reduce the risk of subsequent MS.
Outcome: 90% of patients recover most vision, although symptoms may transiently return following a hot bath or physical exercise – Uhthoff’s phenomenon. Following recovery the optic disc develops an atrophic appearance with a pale ‘punched out’ temporal margin.
Subsequent course: The optic neuritis study group reported 12% of cases had developed clinically definite MS within 2 years (4% with a normal and 30% with an abnormal cranial MRI). Thereafter the risk is 5–6% per annum.
Acute bilateral optic neuritis: less common than unilateral disease and progression to MS not as likely. Occasionally followed by a transverse myelitis (Neuromyelitis optica, page 529). Examination of mitochondrial DNA distinguishes from Leber’s hereditary optic neuropathy (page 551).
Disturbance of ocular movement
Diplopia may result from demyelination affecting the brain stem pathway of the III, IV or VI cranial nerves. Abnormality of eye movements with or without diplopia occurs when supranuclear or internuclear connection are involved. The latter results from a lesion in the medial longitudinal fasciculus – internuclear ophthalmoplegia (I.N.O.) – and in young persons is pathognomonic of MS.

Nystagmus may be an incidental finding on neurological examination. It is unusual in multiple sclerosis when the eyes are in the primary position, and is commonly seen on lateral gaze. Pupillaryabnormalities may occur from:
– sympathetic involvement in the brain stem (Horner’s syndrome)
– III nerve involvement, or
– II nerve involvement.
The swinging light test is a sensitive test of impaired afferent conduction in the II nerve. Alternating the light from one eye to the other results eventually in ‘pupillary escape’ – the pupil dilates despite the presence of direct light.

OTHER FEATURES
Vestibular symptoms: Vertigo of central type may be a presenting problem or it may develop during the course of the illness. Hearing loss is rare.
Ataxia of gait and limb inco-ordination are frequently present. The gait ataxia may be cerebellar or sensory type (see Romberg’s test). Limb inco-ordination, intention tremor and dysarthria indicate cerebellar involvement.
Sphincter disturbance with urgency or precipitancy of micturition and eventual incontinence occurs. Conversely urinary retention in a young person may be the first symptom of disease. On direct questioning, impotence is frequently found.
Mental changes: Mood change – euphoria or depression occur. Cognitive impairment develops in advanced cases. Generalised fatigue is common.
Emotional lability: Uncontrolled outbursts of crying or laughing, result from involvement of pseudobulbar pathways.
Paroxysmal (symptoms occurring momentarily throughout any stage of the disease): paraesthesia, dysarthria, ataxia, pain, e.g. trigeminal neuralgia, photopsia (visual scintillations), epilepsy.
CLINICAL COURSE
The pattern of illness in individual sufferers cannot be predicted. Several different rates of disease activity and progression have been defined.

1. Relapsing and remitting Of all patients with MS, 70% pass through this stage. With each attack recovery is virtually complete. This phase of illness may persist for many years. The explanation of why relapses take place is unknown.
2. Secondary progressive and relapsing/remitting secondary progressive After a period of time, relapsing and remitting MS attacks are followed by incomplete recovery and cumulative loss of function and disability. At any one time, the chronic progressive stage accounts for 20% of all sufferers. Converting from relapsing and remitting to secondary progressive occurs on average 6–10 years after the initial symptoms.

3. Primary progressive This form is common in late onset MS (>45 yrs) and accounts for 15% of all patients. Symptoms and signs are usually spinal and relapses absent in the context of insidious progression.

4. Benign This is defined as low disability (EDSS <3) 10 years after onset. The true incidence of such cases is difficult to define and patients may still progress in time to major disability. Some support for a benign form comes from the occasional incidental autopsy finding of MS.

Recognition of different phases of MS is essential in selecting patients for new disease-modifying treatments. The degree of disability can be recorded using specific scales such as the Kurtzke score or the Extended Disability Status Score (EDSS).
[This is a 10 point non-linear scale where 1 = no symptoms or signs, 6 = a walking aid to achieve a short distance, 8 = restricted to bed/wheelchair and 10 = death due to MS.]
INVESTIGATIONS
The development of imaging and laboratory testing has advanced diagnostic accuracy.
Neurophysiological: may detect a second asymptomatic lesion (see page 54).

2. Somatosensory evoked response (SSEP) may detect central sensory pathway lesions.
3. Brain stem auditory evoked potential (BAEP) may detect brain stem lesions.
Cerebrospinal fluid examination by lumbar puncture
A mild pleocytosis (25 cells/mm3), mainly lymphocytes, is occasionally found. The total protein may be elevated, although this rarely exceeds 100 mg/l. An increase in gammaglobulin occurs in 50–60% of cases. Electrophoresis of CSF using agar or acrylamide shows discrete bands which are not present in serum.
These bands are found in up to 95% of patients with established disease and in 50-60% after the first attack. Oligoclonal bands (OCBs) are not specific to MS but persist indefinitely, unlike other inflammatory neurological diseases (see following page).

MRI
This has contributed enormously to the diagnosis and understanding of MS. Normal white matter appears dark with low signal intensity in T2 weighted images. Myelin breakdown produces a longer relaxation time and increased signal on T2. Gliosis produces similar changes. The presence of white matter abnormalities with a periventricular distribution is suggestive but not diagnostic of MS. Paramagnetic contrast (Gadolinium) will show active inflammation. A combination of MRI and CSF (oligoclonal band) will rule out MS if both are negative. MR may predict long term outcome – following a single episode of demyelination (e.g. optic neuritis or transverse myelitis). Those with cranial MR abnormalities will relapse sooner than those without. At present MRI does not correlate well with disability, but newer techniques may be more sensitive measures of disease progression.

Diagnosis requires two or more episodes of symptoms attributable to demyelination, at least 30 days apart at different sites in the central nervous system, and the exclusion of alternative pathologies. Research criteria have been developed for clinical studies which combine clinical features with investigation findings. The McDonald criteria (2001) allow for MRI scan evidence of the development of new lesions to lead to a diagnosis after a single clinical episode.
Primary progressive MS can be diagnosed after 1 year of progressive deficit, with brain or spine plaques along with CSF unmatched oligoclonal bands and exclusion of alternative diagnoses.
DIFFERENTIAL DIAGNOSIS
Many conditions mimic multiple sclerosis and unless strict diagnostic criteria are adhered to other treatable disorders will be missed.
Conditions with similar clinical presentations to MS
|
Inflammatory disorders – Systemic lupus erythematosus – Polyarteritis nodosa – Behçet’s disease |
Isolated cranial disorders – AVM – Meningioma |
|
Granulomatous disorders – Sarcoidosis – Wegener’s granulomatosis |
Miscellaneous disorders – Spinocerebellar degeneration – Mitochondrial disorders – Adrenoleukodystrophy – HTLV-1 myelopathy |
|
Isolated spinal cord/foramen magnum disorders – Extrinsic/intrinsic tumours – Vitamin B12 disease |
– Lyme disease – Acute disseminated encephalomyelitis |
Conditions with similar MRI appearances to MS
|
– Vasculitis – Sarcoidosis – Leukodystrophies – Acute disseminated encephalomyelitis |
– Small vessel vascular disease – Decompression sickness – Lyme disease – Chronic inflammatory demyelinating polyneuropathy |
Conditions with similar CSF profile to MS (presence of oligoclonal bands)
|
– HIV infection – Lyme disease – Syphilis |
– Chronic meningitis – Neurosarcoidosis – Subacute sclerosing panencephalitis (SSPE) |
SYMPTOMATIC
|
Spasticity |
Drugs: baclofen (GABA derivative); dantrolene (direct action on muscle); tizanidine (α2 adrenergic agonist); botulinum toxin |
|
Physiotherapy and splinting |
|
|
Intrathecal baclofen |
|
|
Urinary symptoms |
Detrusor instability – anticholinergics (oxybutinin, tolterodine); if severe intravesical botulinum toxin injections |
|
Nocturia – desmopressin spray |
|
|
Incomplete bladder emptying – intermittent self catheterisation |
|
|
Bowel symptoms |
Dietary manipulation; laxatives; suppositories/enemas |
|
Pain |
Analgesics; anticonvulsants; antidepressants; NSAIDs; transcutaneous electrical nerve stimulation |
|
Paroxysmal symptoms |
Seizures – anticonvulsants |
|
Fatigue |
Amantadine; modafinil |
|
Depression |
Clinical psychology; antidepressants, tricyclic or SSRI |
|
Tremor |
Betablockers; primidone; if severe deep brain stimulation |
|
Ataxia |
Walking aids; physiotherapy |
Symptom management will often require a coordinated multidisciplinary approach, particularly as the disease progresses.
ACUTE RELAPSE
Methylprednisone 3 g i.v. over 3 days. Check for infection beforehand, monitor blood glucose and consider an H2 blocker for ulcer prophylaxis. Methylprednisone can also be given orally.
MODIFY NATURAL HISTORY
Relapsing remitting MS
Betainterferon and glatiramer acetate reduce the relapse rate by about 30%. The evidence that this reduction translates into reduced disability is less clear. In the UK ambulant patients with two clinically significant relapses in the last 2 years would be eligible for treatment.
Natalizumab, a monoclonal antibody, reduces the relapse rate by over 60% with reduction in disability but is associated with a risk of developing progressive multifocal leucoencephalopathy (1 in 1000). As a result it is available only for patients with aggressive disease. Mitoxantrone is a chemotherapy agent which can be used in aggressive disease with risk of cardiotoxicity and leukemia.
A range of further agents are undergoing trials.
Primary progressive MS and secondary progressive MS
There are no effective disease modifying drugs currently available.
OTHER DEMYELINATING DISEASES
NEUROMYELITIS OPTICA (Devic’s disease)
A subacute disorder characterised by simultaneous or consecutive demyelination of the optic nerves and spinal cord. Whether a distinct entity or variant of MS is uncertain. Pathologically demyelination is associated with marked cavitation and necrosis (possibly due to severe oedema confined and compressed by the pia of the optic nerves and spinal cord). Systemic lupus erythematosus, Behçet’s disease and sarcoidosis produce a similar picture.
Clinical features
Visual loss is rapid, bilateral and occasionally total.
Spinal cord symptoms follow – hours, days or occasionally weeks later.
Back pain and girdle pain. Paraesthesia in lower limbs.
Paralysis may ascend to involve respiratory muscles.
Urinary retention is common.
Recovery is complete in 60–70% of patients. When recurrent attacks occur, this results in an aggressive course with a high fatality.
Examination

Investigations
Anti-aquaporin 4 antibody is positive in over 90% of patients.
Visual evoked responses are prolonged. The CSF shows an elevated protein with a lymphocytosis occasionally as high as 1000 cells per mm3. Gammaglobulin may be elevated and OCBs absent. MRI shows cord swelling with enhancement over several levels.
Treatment
Patients may respond to i.v. methylprednisone or plasma exchange. Supportive treatment is required to minimise complications (DVT/PE, decubiti, contractures). Ventilatory support is sometimes required and may be permanent. There appears to be a case for using immunosuppressive drugs (azathioprine, cyclophosphamide) to prevent relapses though evidence is as yet limited.
TRANSVERSE MYELITIS
This occasionally occurs as the first manifestation of MS but this also occurs with viral infection (e.g. herpes virus), vasculitis and atherosclerotic vascular disease. Only 4% of patients with normal cranial MRI progress to MS. Investigations should exclude other causes of acute spinal cord syndrome – spinal cord compression – by MRI.
ACUTE DISSEMINATED ENCEPHALOMYELITIS (ADEM) (postinfectious encephalomyelitis)
ADEM is an acute immune-mediated demyelinating disorder in which small foci of demyelination with a perivenous distribution are scattered throughout the brain and spinal cord. Lesions are 0.1–1.0 mm in diameter.

This disorder may follow upper respiratory and gastrointestinal infections (viral), viral exanthems (measles, chickenpox, rubella, etc.) or immunisation with live or killed virus vaccines (influenza, rabies).
Measles is the commonest cause occurring in 1 per 1000 primary infections; next Varicella zoster (chickenpox), 1 per 2000 primary infections.
Clinical features: Within days or weeks of resolution of the viral infection, fever, headache, nausea and vomiting develop. Meningeal symptoms (neck stiffness, photophobia) are then followed by drowsiness and multifocal neurological signs and symptoms – hemisphere brain stem/cerebellar/spinal cord and optic nerve involvement. Myoclonic movements are common.
Predominantly spinal, cerebral or cerebellar forms occur, though usually the picture is mixed. Optic nerve involvement takes the form of optic neuritis. Rarely the peripheral nervous system is involved.
Diagnosis: No diagnosis test. CSF – 20–200 mononuclear cells. Total protein and γ globulin raised. Peripheral blood may be normal or show neutrophilia, lymphocytosis or lymphopenia.

The electroencephalogram (EEG) shows diffuse slow wave activity. CT scan is normal. MRI shows small focal white matter changes, simultaneously enhancing with contrast indicating that all are of the same degree of acuteness (unlike MS).
Diagnosis is straightforward when there is an obvious preceding viral infection or immunisation. When viral infection immediately precedes, distinction from acute encephalitis is often impossible.
Separation from acute MS may be difficult. Fever, meningeal signs with elevated CSF protein above 100 mg/ml with cell count greater than 50 per mm3 suggest ADEM.
Pathology: demyelination is limited to perivascular areas and lesions do not approach the same size as in MS.
Outcome: The illness is typically monophasic.
The mortality rate is 20%.
Full recovery occurs in 50%.
Poor prognosis is associated with an abrupt onset and the degree of deficit.
Treatment: Steroids are used, although no controlled trials have been conducted. Large dosage is recommended during the acute phase. Cyclophosphamide may be used in refractory cases.
ACUTE HAEMORRHAGIC LEUKOENCEPHALITIS
This is a rare demyelinating disease. It is regarded as a very acute form of postinfectious/acute disseminated encephalomyelitis.
Clinical picture: Antecedent viral infection, depression of conscious level and multifocal signs and symptoms. Focal features may suggest a mass lesion or even herpes simplex encephalitis.
The diagnosis is only really possible at biopsy or autopsy, but elevated CSF pressure, lymphocytosis and erythrocytes in CSF and xanthochromic appearance of fluid are all suggestive.

Pathology: Perivascular polymorph infiltration. Microscopic and macroscopic haemorrhage. Perivascular demyelination and necrotising changes in vessels.
Treatment: Steroids in high dosage should be used though evidence of value in this rare condition is scant.
PROGRESSIVE MULTIFOCAL LEUKOENCEPHALOPATHY
This is a demyelinative disease occurring in association with systemic illness in which cell-mediated and occasionally humoral immunity is depressed, e.g. AIDS (4% of cases), lymphoma, sarcoidosis, systemic lupus erythematosus. The disorder is due to reactivation of previous papovavirus (SV40 or JC virus) infection.
Clinical picture: Features of diffuse process – personality change, hemiparesis, cortical visual loss, seizures, etc. Duration of illness: 3–6 months. Non-remitting and fatal.
Pathology: Demyelination without inflammatory response, especially in subcortical white matter. Electron microscopy – papovavirus in oligodendroglia.
Diagnosis: CT scanning and MR reveal widespread multifocal white matter damage. Definitive diagnosis is made from brain biopsy. Virus can be isolated by inoculation on to glial tissue culture.
Treatment: reversal of any underlying immune deficit (for example highly active antiretroviral therapy (HAART) in patients with AIDS) may slow progression.
LEUKODYSTROPHIES
Inborn errors of metabolism may affect the normal development of myelin. These genetic disorders usually present in infancy or childhood but occasionally produce their first manifestations in adult life.
3 specific types are recognised
– Metachromatic leukodystrophy
– Globoid cell leukodystrophy
– Adrenomyeloneuropathy or adrenoleukodystrophy (ADL).
The last condition is sex linked, characterised by adrenal insufficiency and disordered myelin in brain, spinal cord and peripheral nerve. The clinical picture is highly variable and results from a defect in beta oxidation of very long chain fatty acids (VLCFA) which build up in blood and skin fibroblasts. Dietary treatments (Lorenzo’s oils) lower these and may slow progression of this fatal disorder. Heterozygote female carriers may become symptomatic with a late onset progressive myelopathy.
NEUROLOGICAL COMPLICATIONS OF DRUGS AND TOXINS
Introduction
Drugs and toxins commonly affect the nervous system. They cause a spectrum of disorders of which most are potentially reversible on withdrawal of the causal agent.
Diagnostic suspicion is especially dependent upon history:
– Availability of drugs.
– Occupational/industrial exposure to toxins.
Drug toxicity may result from:
– The chronic abuse of drugs, e.g. barbiturates, opiates.
– The side effects of drug therapy, e.g. anticonvulsants, steroids.
– The wilful overdosage of drugs, e.g. sedatives, antidepressants.
Toxin exposure may be:
– Accidental: industrial or household poisons, e.g. organophosphates, carbon monoxide, turpentine.
– Wilful: solvent abuse.
History and examination
When acute intoxication is suspected, the following clinical features are supportive.

Also: Note
– Puncture marks in narcotic addicts
– The presence of a snout area rash in solvent abusers
– Rashes in barbiturate poisoning
– Respiration rate in salicylate poisoning
– Skin colour in carbon monoxide poisoning.
Clinical features:
While the neurological picture is generally diffuse, certain pronounced symptoms occur with one drug or toxin and not with another. The following table should act as a guide to diagnosis and alert the clinician to the possible offending substance.
For treatment, the reader is advised to consult an appropriate pharmacology or general medical text.
DRUG-INDUCED NEUROLOGICAL SYNDROMES
HEADACHE
Vasodilators: antihistamines, sympathomimetics, calcium channel blockers, bronchodilators, ergotamine, cocaine
Dopamine agonists
Non-steroidal anti-inflammatories
SEIZURES
Antidepressants, antimicrobials: cycloserine, isoniazid, metronidazole, penicillin
Antineoplastics: vincristine, methotrexate.
Analgesics: pentazocine, fentanyl, opiates
Anaesthetics: ketamine, halothone, althesin
Bronchodilators. Sympathomimetics
Miscellaneous: amphetamine, baclofen, lithium, iodinated contrast media, insulin
CONFUSION/DELIRIUM
Anticholinergics. Anticonvulsants
Antimicrobials: iosiazid, rifampicin
Antineoplastics: vincristine
Dopamine agonists. Tranquillisers
Miscellaneous: cimetidine, ranitidine, lithium
PERIPHERAL NEUROPATHY
Antimicrobials: ethambutol, isoniazid, nitrofurantoin, metronidazole, dapsone
Antineoplastics: cytosine arabinoside, cisplatin, procarbazine, vincristine (and other vinca alkaloids)
Antirheumatics: colchicine, D-penicillamine, gold, indometacin
Miscellaneous: cimetidine, phenytoin
RETINOPATHY
Antimalarials: chloroquine, mepacrine
VIII NERVE DAMAGE
Aminoglycoside antibiotics: gentamicin, kanamycin, neomycin, streptomycin
Miscellanous: cisplatin, ethycrinic acid, quinine, salicylates
VISUAL DISTURBANCE
Phenothiazines Miscellaneous: ethambutol, indometacin, tamoxifen
OPTIC NEURITIS
Antimicrobials: chloramphenicol dapsone, isoniazid, streptomycin
Miscellaneous: chlorpropamide
MOVEMENT DISORDERS
Antiemetics: metoclopramide
Butyrophenones: haloperidol, droperidol
Dopamine agonists. Phenothiazines: chlorpromazine, thioridazine
Tricyclic antidepressants
ATAXIA
Anticonvulsants: carbamazepine, phenytoin, primidone
Antineoplastics: cytosine arabinoside, fluoracil
Phenothiazines: sedatives; barbiturates, chloral hydrate. Tranquillisers: diazepam
MUSCLE PAIN AND WEAKNESS
Antineoplastics: cytosine arabinoside, methotrexate, thiotepa
Statins
Miscellaneous: clofibrate, D-penicillamine, danazol, L-tryptophan.
Drug screen in suspected overdosage
Too often the clinician, when managing drug or toxin overdosage, requests a ‘drug screen’. The techniques used in detection, e.g. gas chromatography, thin-layer chromatography and immunological tests, are sophisticated and time-consuming and may require samples of serum, urine or both.
The clinician must ‘narrow down the field’ from the history and presenting symptoms/signs and discuss with the laboratory the class of drug or toxin he suspects. A knowledge of the blood level of some drugs, e.g. salicylates, barbiturates, is important in deciding the approach to treatment.
SPECIFIC SYNDROMES OF DRUGS AND TOXINS
NEUROLEPTIC MALIGNANT SYNDROME
A rare life-threatening disorder induced by initiation, increase or reintroduction of neuroleptic drugs (e.g. chlorpromazine, haloperidol). The condition appears to result from acute dopamine receptor blockade and is characterised by hyperpyrexia, bradykinesia, rigidity, autonomic disturbance, alteration of consciousness and high serum muscle enzymes (creatine kinase). The causal drug should be withdrawn and the patient cooled. Give dopamine agonists with dantrolene sodium to control bradykinesia and rigidity respectively. Death occurs in 15% from renal failure and/or cardiovascular collapse.
SEROTONIN SYNDROME
SSRIs can cause dystonia and occasionally low-grade fever, confusion, autonomic disturbance, restlessness and rigidity. Early recognition and drug withdrawal is vital for good outcome.
SOLVENT ABUSE
The abuse of volatile solvents is an increasing problem especially in children. The purpose of inhalation is to achieve a state of euphoria. Habituation develops. Commonly used substances are: aerosols, cleaning fluids, nail varnish remover, lighter fluids, ‘model’ glue. The ‘active’ components of these are simple carbon-based molecules, e.g. benzene, hexane and toluene.
|
Symptoms of acute intoxication: – Euphoria – Dysarthria, ataxia, diplopia – Delusions and hallucinations occur, followed by seizures if exposure has been prolonged. |
Symptoms of chronic abuse: – Behavioural disturbance. – Chronic ataxia. – Sensorimotor peripheral neuropathy. |
Treatment of acute intoxication is symptomatic; there are no specific antidotes.
Industrial exposure to hydrocarbons produces similar symptoms.
ORGANOPHOSPHATES
These are widely used as insecticides (sheep dip) and herbicides. They cause symptoms by phosphorylation of the enzyme acetyl cholinesterase. Acute intoxication produces seizures, autonomic disturbance and coma.
LEAD EXPOSURE
Lead has no biological function. It is present in normal diet as well as in the atmosphere from automobile fumes and in the water supply of old buildings containing lead tanks and piping. Occupation exposure occurs in plumbers, burners and smelters. Lead excess interferes with haem synthesis. This results in the accumulation of ‘blocked’ metabolites such as aminolevulinic acid (ALA) in serum and urine. It also inhibits oxidative enzymes (e.g. Superoxide dismutase). Anaemia occurs with a characteristic finding in the blood film (basophilic stippling). Both the peripheral and central nervous systems are affected.

Treatment
Chelating agents (e.g. calcium disodium edetate – EDTA – or D-penicillamine) and i.v. mannitol in acute encephalopathy with papilloedema.
In acute fulminating encephalopathy the mortality has been reduced to 5%, but neurological sequelae are common.
COMPLICATIONS OF RECREATIONAL DRUG ABUSE
The problems of drug abuse are of epidemic proportions. An increasing number of neurological syndromes are recognised.

All recreational drugs are associated with increased risk of cerebral or spinal infarction or intracerebral haemorrhage. (Mechanisms are varied – drug-induced hypertension, coagulopathies, foreign body (talc) embolisation and septic emboli from infective endocarditis.)
All intravenous drug abusers are at risk of HIV infection and its complications (page 515)
METABOLIC ENCEPHALOPATHIES
In general terms, the clinical features of metabolic encephalopathy are relatively stereotyped reflecting a generalised disturbance of function of both hemispheres.

These features are characteristic but exceptions occur in specific encephalopathies –

Beware of the possibility of multiple pathology, e.g. an alcoholic patient with a chronic subdural haematoma may also have liver failure and thiamine deficiency.
CLASSIFICATION AND BIOCHEMICAL EVALUATION
Many metabolic disturbances cause an acquired encephalopathy in adults.
The most frequently encountered are:
|
– Hypoxic – Hypercapnoeic – Hypoglycaemic – Hyperglycaemic – Hepatic – Uraemic |
Less commonly: – Hyponatraemia. Hypernatraemia. – Hypokalaemia. Hyperkalaemia. – Hypocalcaemia. Hypercalcaemia. – Hypothyroidism. Lactic acidosis. – Addison’s disease. |
Drugs and toxins producing encephalopathy are dealt with separately (page 533).
Laboratory assessment of suspected metabolic encephalopathy
All patients should have a basic biochemical screen:
– Serum urea and electrolytes.
– Liver function (albumin, globulin, bilirubin, alkaline phosphatase and enzymes) and random blood glucose.
– Blood gases (pH, PO2 PCO2).
– Serum ammonia.
– Electroencephalography – slow wave activity (theta or delta) supports the diagnosis of a diffuse dysfunction: hepatic encephalopathy shows a specific triphasic slow wave configuration.
– CT scan – if the above tests are normal or coexisting structural brain disease is suspected.
Calculation of the anion gap may be helpful in the diagnosis of encephalopathies, especially lactic acidosis. The sum of the anions (Cl − and HCO −3) normally equals the sum of the cations (Na+ and K+). An increase in the gap in the absence of ketones, salicylates and uraemia suggests lactic acidosis or ethylene glycol poisoning.
HYPOXIC ENCEPHALOPATHY
Impaired brain oxygenation results from:
– Reduced arterial oxygen pressure – lung disease.
– Reduced haemoglobin to carry oxygen – anaemia or blood loss.
– Reduced flow of blood containing oxygen (ischaemic hypoxia) – due to reduced cardiac output (with reduced cerebral blood flow).
– Biochemical block of cerebral utilisation of oxygen – rare (e.g. cyanide poisoning).
When cerebral arterial PO2 falls below 35 mmHg (4.5 kPa), anaerobic metabolism takes over; this is not efficient and a further drop in PO2 will result in neurological dysfunction. The extent of hypoxic damage depends not only upon the duration of hypoxia but also on other factors, e.g. body temperature – hypothermia protects against damage. The irreversibility of hypoxic damage is explained by the ‘no flow phenomenon’ – after 3–5 minutes the endothelial lining of small vessels swells – even with reversal of hypoxia, flow through these vessels is no longer possible.
SPECIFIC ENCEPHALOPATHIES
Pathology
As a consequence of high metabolic demand, some areas are more susceptible than others.

Microscopic changes depend upon the delay between the hypoxic event and death.
|
Immediate: |
At 48 hours: |
At several days/weeks: |
|
Scattered petechial haemorrhages. |
Cerebral oedema associated with petechial haemorrhage. |
Necrosis in cortical grey matter and globus pallidus with associated astrocytic proliferation. The cerebellum and brain stem may also be affected. |
Clinical features:
e.g. Severe hypoxia from circulatory arrest

Delayed hypoxic encephalopathy refers to the rare occurrence of a full clinical recovery followed after some weeks by a progressive picture → deterioration of conscious level → death. Widespread subcortical demyelination is found at autopsy.
HYPERCAPNIC ENCEPHALOPATHY: the consequence of an elevated arterial carbon dioxide level.
Clinical features:
Headache, confusion, disorientation, involuntary movements.
Papilloedema, depressed limb reflexes, extensor plantar responses.
Diagnosis: A PCO2 greater than 50 mmHg (6 kPa) with a reduced PO2 is found on arterial blood sampling.
The presence of headache, confusion and papilloedema may suggest intracranial tumour. If hypercapnia has not been diagnosed, such patients inevitably are referred for CT brain scan.
HYPOGLYCAEMIA ENCEPHALOPATHY: the consequence of insufficient glucose reaching the brain and may result from – overdosage of diabetic treatment
– insulin secreting tumour – insulinoma
– hepatic disease with reduction of liver glycogen.
Serum glucose levels of 1.5 mmol/l are associated with the onset of encephalopathy. Levels at 0.5 mmol/l are associated with coma.
Pathology: Changes occur in the cerebral cortex – focal necrosis surrounded by neuronal degeneration. Subcortical grey matter (caudate nucleus) and cerebellum are vulnerable.
Clinical features: These, as with hypoxia, depend upon the duration and severity of hypoglycaemia.

Repeated mild to moderate episodes may result in a chronic cerebellar ataxia.
Repeated severe attacks may result in a mixed myelopathy/peripheral neuropathy which is distinguished from motor neuron disease by the presence of sensory signs.
HYPERGLYCAEMIC ENCEPHALOPATHY
Two types of encephalopathy develop as a consequence of hyperglycaemia:
|
Diabetic ketoacidotic coma |
Diabetic hyperosmolar non-ketotic coma |
|
Accumulation of acetone and ketone bodies in blood results in acidosis. Hyperventilation ensues with a reduction inPCO2 and HCO−3 Osmotic diuresis due to hyperglycaemia results in dehydration. |
This results from the hyperosmolar effect of severe hyperglycaemia. Reduction of the intracellular compartment results. Involuntary movements, seizures and hemiparesis may occur. Vascular thrombosis is not uncommon. Ketoacidosis is mild or does not occur. |
|
The neurological presentation is that of confusion, progression to coma and, if untreated, death. |
HEPATIC ENCEPHALOPATHY
Neurological signs and symptoms secondary to hepatic dysfunction may arise in:
– acute liver failure.
– chronic liver failure complicated by infection or gastrointestinal haemorrhage.
– chronic liver failure producing characteristic hepatocerebral degeneration.
Clinical features:

The encephalopathy is progressive.
Pathology
Neuronal loss with gliosis is noted in the cerebral cortex as well as basal ganglia, cerebellum and brain stem. Astrocytes with irregular and enlarged nuclei are characteristic.
Hepatocerebral degeneration (Wilson’s disease) (see page 373) is an abnormality of copper metabolism and leads to a deposition of copper in the brain, particularly the basal ganglia, and the liver. This leads to a slowly progressive neurological disorder. This can vary but the predominant features include dementia, dysarthria and ataxia, primitive reflexes, choreoathetosis, myoclonus, tremor and pyramidal signs.
Consciousness is not impaired.
URAEMIC ENCEPHALOPATHY
Clinical features:

Pathology:
Uraemia may produce non-specific pathological findings in the nervous system. Peripheral nervous system involvement occurs in chronic renal failure.
Dialysis encephalopathy is encountered in persons on renal dialysis exposed to high aluminium levels in the dialysate. The features are those of dementia, behavioural changes, seizures and myoclonus. The condition progresses unless aluminium levels are controlled.
Specific investigations and treatment of individual metabolic encephalopathies do not come within the scope of this book.
NUTRITIONAL DISORDERS
INTRODUCTION
Nutritional deficiency presents a massive problem in the developing world. In Western countries, alcoholism is the major cause of the neurological syndromes resulting from dietary deficiency with faddism and malabsorption disorders accounting for only a small number.
Vitamins appear important nutrients and certain disorders such as Wernicke Korsakoff syndrome (thiamine) or subacute combined degeneration (vit. B12) are attributed to a single deficiency. Others such as polyneuropathy result from multiple deficiency.
Vitamin deficiency in itself does not always produce symptoms; a dietary excess of carbohydrate seems essential for the development of the neurological features of thiamine deficiency.
As a rule, nutritional disorders of the nervous system present clinically in a symmetrical manner.
WERNICKE KORSAKOFF SYNDROME
This syndrome is comprised of an acute and a chronic phase:

Patients often demonstrate additional features of nutritional deficiency – peripheral neuropathy, trophic skin changes and autonomic dysfunction (arrhythmias, postural hypotension and hypothermia). Features of acute alcohol withdrawal often co-exist.

N.B. Korsakoff’s psychosis may also be caused by head injury, anoxia, epilepsy, encephalitis and vascular diseases.
WERNICKE’S SYNDOME
Diagnosed in 0.1–0.4% of hospital admissions.
Pathology:
Neuronal, axonal and myelin damage occur symmetrically in the mamillary bodies, the walls of the third ventricle, thalamus and periaqueductal grey matter. Secondary vascular proliferation and haemorrhages occur within these lesions.
WERNICKE KORSAKOFF SYNDROME

Investigation
• Serum B1 levels may be low.
• Pyruvate is elevated.
• Transketolase activity is decreased (enzyme in hexose monophosphate shunt).
• MRI may show mamillary body atrophy.
Treatment
Intravenous (i.v.) Pabrinex containing thiamine (B1), riboflavin (B2), pyridoxine (B6), is the only available treatment. Oral treatment is ineffective. Treatment must be given immediately to persons with a suggestive clinical picture and evidence of chronic alcohol use. Those with a history of alcohol abuse requiring IV glucose should be treated prophylactically (thiamine is critical for the metabolism of carbohydrate and levels can be exhausted by a sudden load).
With treatment – Eyes improve – in days, though nystagmus may persist for months.
– Ataxia improves – in weeks.
Overall mortality: 15% → coma → death.
Failure to recognise and promptly treat can result in Korsakoff’s syndrome or psychosis.
KORSAKOFF’S PSYCHOSIS
Sometimes encountered in traumatic or infective brain disorders, though normally overlaps with Wernicke’s syndrome.
Pathology
Lesions are identical in distribution to those of Wernicke’s syndrome without haemorrhages.
Clinical features
There is a disturbance of memory in which information cannot be stored. In addition the normal temporal sequence of established memories is disrupted, resulting in a semifictionalised account of the circumstances which the patient may find him/herself in (confabulation). This memory disturbance can only be tested for when the confusion of Wernicke’s disease has cleared.
Treatment
Acute treatment is with vitamin replacement as for Wernicke’s – there is no other specific treatment. The loss of short term memory causes significant disability and patients will often require closely supervised care.
B12 DEFICIENCY – SUBACUTE COMBINED DEGENERATION OF THE SPINAL CORD
B12 deficiency produces the specific neurological syndrome of subacute degeneration of the spinal cord (SADC). Two cobalamin-dependent enzymatic reactions occur in humans. The first reaction converts methylmalonyl-coenzyme A (CoA) to succinyl-CoA. The second involves the synthesis of methionine from homocysteine. Deficiency in B12 therefore results in an accumulation of homocysteine. Despite the importance of methionine to myelin sheath phospholipid methylation, the basis of neurological damage remains uncertain.
Causes
• Inadequate diet (e.g. strict vegans)
• Increased need – pregnancy
• Defective absorption – Pernicious anaemia
– decreased intrinsic factor (necessary for absorption)
– malabsorption (pancreatitis, coeliac disease, gastric surgery, tapeworm infestation etc.)
Pathology
Spinal cord demyelination with eventual axon loss – affects:

B12 deficiency resulting in neurological damage is usually associated with a macrocytosis, though a normal peripheral blood film may be found.
Clinical features
Onset is subacute, though can be acutely precipitated by exposure to nitrous oxide anaesthesia.
Paraesthesia of extremities is the presenting symptom.

Examination
– Gait is ataxic with positive Romberg’s test (sensory ataxia).
– Motor power is diminished distally.
– Plantar responses are extensor.
– Sensory loss: loss of vibration and joint position sensation in the lower limbs. Stocking/glove sensory loss is found when peripheral nerves are involved.
– Reflex findings are variable and depend on the predominance of peripheral nerve or corticospinal tract involvement.
Mini mental status examination (MMSE) test may suggest dementia.
Optic pallor and centrocaecal scotoma can be demonstrated.
Diagnosis
Suspect in paraparesis with combined upper and lower motor neuron signs with ‘stocking/glove’ sensory loss.
Differentiate from other causes of acute myelopathy, e.g. cord compression, multiple sclerosis.
Investigation
Peripheral blood film – may show a megaloblastic anaemia
Serum B12/Folate – low B12. (If folate low – investigate causes: diet/drugs/malabsorption) If serum B12 low (normal > 190 ng/l)
|
– measure intrinsic factor antibody |
|
|
– Schilling test (measure of capacity to absorb) |
– if normal – dietary |
|
– if low – repeat with intrinsic factor (normal = pernicious anaemia, abnormal = malabsorption and investigate accordingly) |
MRI – may show spinal and cerebral white matter hyperintensity on T2 images Nerve conduction studies – may show axonal neuropathy
Treatment
Consider treatment for patients who have serum B12 level of less than 130 ng/l (neurological dysfunction normally occurs with levels < 100 ng/l).
Initiate treatment with vitamin B12, 1000 micrograms intramuscularly given daily for 3 to 7 days, then weekly for 4 weeks.
Continue maintenance therapy for life.
Course and progression
Untreated, the disorder is progressive, the patient eventually becoming bed-bound and comatose. If diagnosed and treated early (within 2 months of onset), complete recovery can be anticipated. In established cases, only progression may be halted.
Caution: When folic acid is prescribed alone, it will improve the haematological picture of B12 deficiency but cause rapid often irreversible neurological deterioration.
A clinically similar syndrome can be rarely caused by copper deficiency.
Tocopherol (vit. E).
In its active form – D α tocopherol – it acts as a membrane stabilizer and anti-oxidant. Deficiency occurs in chronic fat malabsorption (e.g. coeliac disease or cystic fibrosis) and results in widespread neurological disturbances – ataxia, ophthalmoplegia, seizures and corticospinal tract dysfunction. These are halted and often reversed by i.m. vit. E. It has been speculated that the antioxidant effect might make vitamin E a candidate for cytoprotection and repair within the nervous system. Studies in Parkinson’s disease, multiple sclerosis and stroke are disappointing.
Abetalipoproteinaemia (Bassen-Kornzweig syndrome) predisposes to vit. E deficiency (the vitamin is transported by low-density lipoproteins). These patients have acanthocytes in the peripheral blood and a pigmentary retinopathy.
NUTRITIONAL POLYNEUROPATHY
Deficiency of vitamin B complex – B1 (Thiamine), B2 (Riboflavin), B3 (Nicotinic acid), B5 (Pantothenic acid) or B6 (Pyridoxine) – results in peripheral nerve damage.
The combination of polyneuropathy and cardiac involvement is referred to as BERI-BERI. When oedema is also present it is termed wet beri-beri and, when absent, dry beri-beri. Beri-beri occurs in rice eating countries.
In Western countries, alcoholism is the major cause of nutritional polyneuropathy with or without cardiac involvement, otherwise world wide famine and starvation is responsible.
Pathology

The distal portions of nerves are initially affected.
Anterior horn cells and dorsal root ganglion cells undergo chromatolysis.
Vagus nerve and sympathetic trunk involvement occurs in severe cases.
Clinical features
Symptoms: Progressive distal weakness and sensory loss with painful tingling paraesthesia involving initially lower limbs.
Autonomic complaints – impotence, dizziness (orthostatic hypotension) and disordered sweating – are common.

Signs:
• Varying degrees of areflexia (only ankle reflexes are lost initially).
• Weakness which is more marked distally than proximally and initially involves the lower limbs.
• Sensory loss of a ‘stocking/glove’ type involving all modalities of sensation.
• Autonomic involvement results in sweating soles of feet and postural blood pressure drop.
• Vagus nerve involvement results in a hoarse voice and disturbance of swallowing.
Associated signs
Shiny skin on legs with poor distal hair growth. ‘Hyperpathic’ painful soles of feet. Evidence of liver failure.
Diagnosis
Suggested by nutritional/alcohol history.
Supported by investigation such as peripheral blood film (elevated MCV) and disturbed liver function tests.
Nerve conduction studies reveal mildly reduced motor and sensory conduction velocities.
Differential diagnosis
Consider other causes of subacute or chronic sensorimotor neuropathy (see page 436).
Treatment
A high calorie (3000) diet should be supplemented daily with Thiamine (25 mg), Niacin (100 mg). Riboflavin (10 mg), Pantothenic acid (10 mg) and Pyridoxine (5 mg).
Burning paraesthesia may respond to gabapentin, pregabalin or carbamazepine.
Recovery may be very slow and incomplete but with the withdrawal of alcohol and adequate vitamin supplementation some improvement should occur.
TOBACCO–ALCOHOL AMBLYOPIA
A large number of toxic substances can produce impaired vision. Methyl alcohol causes sudden and permanent blindness. Chronic painless visual loss from optic neuritis develops in malnourished patients with a high tobacco consumption (Tobacco-alcohol amblyopia). This is caused by exposure to cyanide from tobacco smoking associated with low vitamin levels due to poor nutrition and absorption associated with drinking alcohol. Other potential toxins include methyl alcohol (moonshine) and ethylene glycol (antifreeze).

ALCOHOL RELATED DISORDERS
ALCOHOL MYOPATHY
Muscle damage (elevated creatine phosphokinase) is not uncommon in alcoholics following acute ingestion. The cause of alcoholic myopathy is uncertain; mitochondrial disturbances, potassium depletion, rhabdomyolysis (due to seizures or local compression) have all been suggested.
There are two forms of alcoholic muscle disease
1. Acute necrotizing myopathy occurs after ‘binge’ drinking.
– Acute muscle necrosis ensues with pain/cramping and muscle tenderness/swelling.
– Myoglobin is excreted in the urine (myoglobinuria) after release from damaged muscles.
– Symptoms of alcohol withdrawal – delirium, etc. – coexist.
– Limb involvement may be markedly asymmetrical.
– Sometimes calf muscles are swollen and tender.
– Improvement occurs over weeks to months.
– Serum creatine phosphokinase (CPK) is elevated. Marked myoglobinuria when present may result in renal failure.
– Elevated serum K+ may provide cardiac arrhythmias.
2. Chronic myopathy Painless proximal weakness sometimes associated with cardiomyopathy. Muscle biopsy showing type 2 fibre atrophy.
Management is abstinence, vitamin supplementation and IV saline in acute necrotizing myopathy with myoglobinuria to prevent renal failure.
ALCOHOLIC DEMENTIA
Experimentally, chronic alcohol consumption results in neuronal loss. CT evidence of atrophy and neuropsychological impairment is common in alcoholics. However, whether or not these result from the direct toxic and dementing effect of alcohol remains uncertain.
ALCOHOLIC CEREBELLAR DEGENERATION
Probably the commonest cause of acquired ataxia, alcoholic patients may develop a chronic cerebellar syndrome either as a sequel of Wernicke’s syndrome or as a distinct clinical entity. A long history of alcohol abuse is obtained. Males are predominantly affected. Onset is gradual and symptoms often stabilise. Ataxia of gait with lower limb inco-ordination predominates. The upper limbs are spared. Nystagmus is rarely present. Cerebellar dysarthria is usually mild. Coexistent signs of peripheral neuropathy are often found.
|
Investigations: |
– Abnormal liver function tests e.g. elevation of enzymes – γ GT. |
|
– Macrocytosis in peripheral blood film. |
|
|
– CSF examination normal. |
|
|
– CT and MRI reveal cerebellar vermal atrophy. |
|
|
Progression |
may evolve rapidly and reverse with improved nutrition and alcohol withdrawal. |
|
may evolve subacutely. |
|
|
may evolve chronically and slowly progress over many years. |
|
|
Pathology: |
– All the cellular elements of the cerebellar cortex are affected, but particularly Purkinje cells of the anterior and superior vermis and the anterior portion of the anterior lobes. |
|
Pathogenesis: |
– The disorder may be due to nutritional deficiency, especially thiamine, or else result from the direct toxic effect of alcohol or electrolyte disturbance on the cerebellum. |
|
Differential diagnosis: |
– Distinguish from hereditary and other acquired ataxias, e.g. hypothyroidism. |
|
Treatment: |
– Alcohol withdrawal, a well balanced diet and adequate vitamin supplementation. |
CENTRAL PONTINE MYELINOLYSIS
Alcohol abuse, debilitating disease or rapid correction of hyponatraemia may precipitate presentation.
The lesion is one of demyelination with cavitation. Microscopically, myelin is lost, oligodendrocytes degenerate but neurons and axons are spared.
Clinically, an acute or subacute pontine lesion is suspected, evolving over a few days, with bulbar weakness and tetraparesis (locked-in syndrome).
The limbs are flaccid with extensor plantar responses.
With progression of the lesion, eye signs become evident and conscious level becomes depressed → coma → death.
Investigations:
Electrolytic disturbances (low sodium, low phosphate) are found.
Liver function is normal. CSF examination is normal.
MRI is more sensitive than CT showing an abnormality in the pons.
Recognition of this condition before death is important in view of its reversibility, though prior to CT/MRI availability it was diagnosed at autopsy. Vigorous supportive therapy with correction of metabolic abnormalities and vitamin supplementation is advised. In patients with severe hyponatraemia (< 110 mmol/l), especially alcoholics, slow correction is essential.

CORPUS CALLOSUM DEMYELINATION
(syn: Marchiafava–Bignami disease)
This is a rare disorder occurring in malnourished alcoholics. Occasionally diagnosed premortem by MRI, progressing to death over some weeks. The clinical picture is that of personality change with signs of frontal lobe disease. The condition occurs most commonly in persons of Italian origin.
NON-METASTATIC MANIFESTATIONS OF MALIGNANT DISEASE
Disturbance of neurological function can occur in association with malignancy without evidence of metastases (0.1% of all cancer patients). Brain, spinal cord, peripheral nerve and muscle may be affected, either separately or in combination.
Small cell carcinoma of the lung, gynaecological malignancy and lymphoma are the commonest associated disorders. Specific antibodies (anti-neuronal), are responsible for certain syndromes. These are directed towards antigens in the nervous system and the tumour and may explain the trend toward greater life expectancy in those with, rather than those without, such non-metastatic disorders.

These are not discreet, e.g. neuropathy and myopathy may coexist → carcinomatous neuromyopathy; encephalitis and myelopathy → carcinomatous encephalomyelitis.
LIMBIC ENCEPHALITIS
Associated commonly with small cell lung cancer (SCLC) usually before this becomes clinically manifest.

Pathology
The encephalitic process selectively affects the limbic system – with neuronal loss, astrocytic proliferation and perivascular inflammatory changes.
Clinical features
Disturbance in behaviour precedes the development of complex partial (temporal lobe) seizures and memory impairment. Autonomic dysfunction and sensory neuropathy often co-exist. Progression is rapid.
Investigations.
Anti-voltage gated potassium channel antibodies (anti-VGKA) or anti-Hu antibodies are the most commonly found antibodies. Recently anti-NMDA receptor antibodies have been demonstrated in some patients with limbic encephalitis and prominent extrapyramidal movements. MRI may show temporal lobe abnormalities. EEG may show temporal lobe abnormalities. CSF reveals a mild lymphocytosis with protein elevation.
CEREBELLAR DEGENERATION (anti-Yo syndrome) associated with breast or ovarian carcinoma.

Pathology:
Characterised by Purkinje cell loss with some involvement of the dentate muscles. Brain stem changes also occur.
Clinical features:
The patient presents with a rapidly developing ataxia. Brain stem involvement results in nystagmus, opsoclonus and vertigo. The course is usually rapid.
Investigations
MRI shows cortical and vermal cerebellar atrophy.
CSF is mildly abnormal and anti-Yo antibodies are present in 50% of suspected cases.
NEUROPATHY (see page 430)
Sensory neuropathy: Destruction of the posterior root ganglion combined with axonal and demyelinative peripheral nerve damage causes progressive sensory symptoms. The neuropathy is subacute or chronic in evolution. Clinically dysaesthesia and numbness starts in extremities and spreads. Associated with SCLC and anti-Hu antibodies.
Sensorimotor neuropathy: A mixed neuropathy with weakness and sensory loss. The syndrome may predate the recognition of the underlying neoplasm. Rate of progression is slow and predominantly motor forms may be mistaken for ALS (page 555) associated with Hodgkin’s and other lymphomas.
Rarely an acute neuropathy indistinguishable from postinfectious polyneuropathy occurs.
NECROTISING MYELOPATHY:
Flaccid paraplegia develops subacutely. Spinal MRI may show a swollen cord. Mechanism is uncertain.
MYOPATHY
Muscle weakness in malignancy takes several forms.
Proximal myopathy: A slowly progressive syndrome with weakness of proximal limb muscles.
Inflammatory myopathy (polymyositis/dermatomyositis) (see page 474):
The overall incidence of associated neoplasm in inflammatory myopathy is 15%. The typical patient is in middle age with a proximal weakness, elevated ESR and muscle enzymes with or without the skin features of dermatomyositis.
Myopathy with endocrine disturbance: Ectopic hormone production (by malignant cells) may induce a myopathy characterised by chronic progressive proximal weakness, e.g. ectopic ACTH production from small cell carcinoma of lung.
Cachetic myopathy occurs in terminally ill, wasted patients.
Investigation and treatment of non-metastatic syndromes
Successful treatment of the underlying tumour offers the only hope of improvement. The search must be exhaustive and repeated where first negative. Tumour markers (AFP, CEA, PSA etc), chest and abdominal CT, pelvic ultrasound, mammography are advised with PET (FDG) if available. Treatment with steroids, immunosuppressants (AZT, cyclosporine, etc), IVIG or plasma exchange is of uncertain benefit.
THE MYASTHENIC SYNDROME (Lambert-Eaton syndrome)

In men the association with underlying malignancy warrants detailed investigation (see above) though a proportion of patients have no evidence of this.
Clinical features
The patient develops weakness of lower then upper limbs with a tendency to fatigue. Following brief exercise, power may paradoxically suddenly improve – second wind phenomenon. In contrast to myasthenia gravis ocular and bulbar muscles are rarely affected. Examination reveals a proximal pattern of wasting and weakness with diminished tendon reflexes. Up to 50% of patients experience symptoms of autonomic (cholinergic) dysfunction – impotence, dry mouth and visual disturbance.
Diagnosis
Confirmed electrophysiologically; the ‘second wind phenomenon’ is shown up as an incrementing response to repetitive nerve stimulation (as opposed to the decrementing response in myasthenia gravis,page 485). VGCCAs are detected in serum.
Treatment
3,4-diaminopyridine and pyridostigmine can improve symptoms. Immunosuppression with steroids, plasmapheresis or IVIG can suppress the underlying immunological abnormality.
This syndrome may respond to the removal of the underlying neoplasm if present.
DEGENERATIVE DISORDERS
Introduction
This heterogeneous group of neurological diseases characterised by selective neuronal loss, is grouped together by the lack of known aetiology. As causes of such disease are identified (e.g. metabolic, viral) they have been reclassified in their appropriate category. Of the remaining conditions many are age related or familial and in some there is an identifiable genetic basis.
Characteristically these disorders:
– are gradually progressive
– are symmetrical (bilateral symptoms and signs)
– may affect one or several specific systems of the nervous system
– may demonstrate a specific pathology or just show neuronal atrophy and eventual loss without other features.
Classification
Degenerative disoders are classified according to the specific part or parts of the central/peripheral nervous system affected and according to the ensuing clinical manifestations. These degenerative disorders may be alternatively termed the system degenerations because of their propensity to affect only part of the nervous system.

Most of these conditions are discussed in other chapters.
PROGRESSIVE BLINDNESS
LEBER’S HEREDITARY OPTIC NEUROPATHY (LHON)
Leber’s optic neuropathy is a familial disorder of maternal inheritance with a tendency to affect males significantly more than females. It is classified as a mitochondrial disorder due to DNA mutation (page 481). Most individuals have one of three point mutations of mitochondrial DNA (mtDNA).
Pathology


Clinical features
Onset of visual loss in late teens/early twenties.
– Both eyes are simultaneously affected (rarely one eye months before the other).
– Central vision is lost with large bilateral scotomata.
Characteristically, blue/yellow colour discrimination is affected before red/green. The optic disc initially appears pink and swollen with an increase in small vessels, eventually becoming pale and atrophic.
Visual impairment progresses with peripheral construction of the fields. Complete visual loss seldom occurs.
Associated symptoms and signs of a more generalised nervous system disorder occur in a proportion of cases – dementia, ataxia, progressive spastic paraplegia – and confusion with multiple sclerosis may arise. In contrast to bilateral optic neuritis, ‘leakage’ occurs with fluorescein angiography. Genetic counselling for LHON is complicated by the sex and age-dependent penetrance. The mother of an affected male has the mitochondrial mutation and may or may not have symptoms. No treatment exists. Quinone analogues (ubiquinone and idebenone) may help during periods of rapid visual worsening.
RETINITIS PIGMENTOSA
A heterogeneous hereditary disorder of the retina which may be inherited as an autosomal dominant, recessive or X-linked disorder. All layers of the retina are affected. Posterior pole cataracts and glaucoma are occasionally associated.

Clinical features
Onset of visual loss in childhood. Both eyes are simultaneously affected. Initially there is a failure of twilight vision. The patient has difficulty in making his/her way as darkness falls (nyctalopia). The retina around the macular area is first affected resulting in a characteristic ring scotoma. This gradually spreads outwards; eventually only a small ‘tunnel’ of central vision is left. Finally, complete blindness occurs. The majority of patients are completely blind by 50 years of age. The fundal appearance is diagnostic as a result of the superficial migration of pigment.

The electroretinogram – recording the electrical activity of the retina – is eventually lost.
Treatment
None. Vitamins and steroids have been tried unsuccessfully.
Associated conditions in retinitis pigmentosa
Several conditions are associated with retinitis pigmentosa:
• Hypogonadism/obesity/mental deficiency
• Spinocerebellar ataxis
• Laurence Moon syndrome
• Friedreich’s ataxia

The association with neuropathy and ataxia (NARP), or progressive external ophthalmoplegia and heart block (Kearns-Sayre syndrome) are due to mitochondrial disease (page 481).
PROGRESSIVE ATAXIA
The degenerative disorders manifested by progressive ataxia are termed spinocerebellar-ataxias.
These may be classified by age of onset, presence of associated features, but increasingly by mode of inheritance.
RECESSIVELY INHERITED ATAXIAS
ATAXIA TELANGIECTASIA (Louis-Barr Syndrome)
This multisystem disorder is characterised by progressive cerebellar ataxia, ocular and cutaneous telangiectasia and immunodeficiency.

The gene maps to chromosome 11q23 associated with mutations in the ATM gene. The ATM protein is a member of the family of proteins involved in DNA repair.
Pathologically, widespread cerebellar Purkinje and granular cell loss occurs.
A progressive ataxia develops in infancy. Telangiectasia develops later, becoming more obvious after exposure to the sun. Prevalence similar to Freidrich’s ataxia.
Patients are eventually confined to a wheelchair and, because of associated low serum immunoglobulin levels are susceptible to repetitive infections.
Malignant neoplasms (lymphoreticular tumours) occur in 10%.
Patients are unusually sensitive to X-rays. Treatment of malignancy with conventional dosages of radiation can prove fatal.
Death occurs in second or third decade from infection or malignancy (often lymphoma).
FRIEDREICH’S ATAXIA
Friedreich’s ataxia is the commonest inherited ataxia with an incidence of 1/50000 in European populations and carrier frequency of 1/20.
It is caused by mutations in the FRDA gene located on chromosome 9 which encodes the protein Frataxin. It is the first autosomal recessive disease identified in which a triplet repeat expansion (GAA) is responsible.
Pathology:

Spinal: The spinal cord is shrunken, especially in the thoracic region.
There is degeneration, demyelination and gliosis of:
1. – Posterior columns.
2. – Corticospinal tracts
3. – Dorsal spinocerebellar tracts
4. – Ventral spinocerebellar tracts.
Dorsal roots and peripheral nerves are shrunken in advanced cases.
Cerebellar: Changes in the cerebellum are less marked, there is Purkinje cell loss and atrophy of the dentate nucleus.
Peripheral nerves show loss of large myelinated axons and segmental demyelination. The corticobulbar tract and cerebrum are relatively spared.
RECESSIVELY INHERITED ATAXIA
FRIEDREICH’S ATAXIA
Clinical features
Friedreich’s ataxia is characterised by progressive gait ataxia and limb incordination, hypertrophic cardiomyopathy and increased incidence of diabetes mellitus/impaired glucose tolerance.
Sexes are equally affected. Onset of symptoms is normally around puberty, and always before 25 years of age; most patients become wheelchair bound by their late twenties. Cardio-pulmonary failure is the usual common cause of death.
Disturbance of balance is the initial symptom, often associated with the development of scoliosis. A spastic, ataxic gait develops with inco-ordination of the limbs.
Corticospinal tract involvement results in limb weakness with absent abdominal reflexes and extensor plantar responses.
Posterior column involvement results in loss of vibration and proprioception in the extremities.
Dorsal root and peripheral nerve involvement results in absent lower limb reflexes.
Involvement of myocardial muscle (cardiomyopathy) is common and results in cardiac failure or dysrhythmias. Musculoskeletal abnormalities occur in 80% of cases.


Optic atrophy and deafness coexist in many cases.
There is a clinical resemblance to mitochondrial encephalopathies as well as reduced respiratory enzyme activities in some patients (Friedreich’s has been suspected to involve some degree of disturbance of mitochondrial respiration).
Investigation
Identification of the gene and availability of diagnostic testing has limited the value of other ancillary investigations such as imaging and neurophysiology. Regular cardiac assessment and monitoring of blood glucose is important.
Treatment
Although there is no specific treatment for Friedreich’s ataxia, many of its symptoms can be managed. Orthopaedic intervention can alleviate scoliosis, and orthopaedic appliances and physical therapy help maintain ambulation. Cardiac problems can be successfully treated pharmacologically and insulin therapy may be necessary to control diabetes mellitus.
Other causes of areflexic ataxia
Abetalipoproteinaemia (Bassen Kornzweig disease)
• Malabsorption syndrome
• Acanthocytes (thorn-shaped red blood cells)
• Low serum cholesterol, triglycerides and fatty acids
• Low vitamin E.
Hexosaminidase deficiency
• Accumulation of GM2 gangliosides in brain and skin.
Xeroderma pigmentatosum
• Sensitive to ultraviolet light
• Keratosis and skin cancer.
DOMINANTLY INHERITED AND OTHER ATAXIAS
Classification of the dominantly inherited, late-onset, cerebellar ataxias is complex and controversial. The term ‘late-onset’ is misleading given that these disorders may present in childhood and adolescence. Commonly other neurological features co-exist: ophthalmoplegia, optic atrophy, retinal pigmentation, deafness, dysarthria, dysphagia, dementia, extra pyramidal and pyramidal signs and peripheral neuropathy. This bewildering condition is classified into 3 different clinical phenotypes.

Many different gene loci have been reported to be responsible – the spinocerebellar ataxia or SCA mutations. SCA1, SCA2, SCA3 (also known as Machado-Joseph disease), cause ADCA type 1, SCA7 causes ADCA type 2, SCA4, SCA5, SCA6 and SCA11 cause ADCA type 3, though there remains considerable phenotypic variation even within families. Causative genes have been identified as expansions of trinucleotide CAG repeat for SCA1, SCA2, SCA3, SCA6, SCA7, and SCA12, and the CTG repeat for SCA8. DNA testing is diagnostic though new loci remain to be discovered.
IDIOPATHIC LATE ONSET ATAXIA
Some may be new mutations of ADCA. For diagnosis all other causes of acquired ataxia – inflammatory, infective, nutritional, metabolic, endocrine and non-metastatic – must be excluded by appropriate investigations.
|
Type 1 – Age of onset 35–55 years – ataxia ± dementia, spasticity |
|
Type 2 – Age of onset > 55 years – mid-line ataxia sparing speech/limbs |
|
Type 3 – Age of onset 50–60 years – ataxia, titubation and tremor |
THE HEREDITARY INTERMITTENT ATAXIAS
These disorders are characterized by brief paroxysmal episodes with no neurological impairment between attacks. Two types can be distinguished on the basis of the length of the attacks, the presence of myokymia (facial twitching), precipitating factors, response to acetazolamide and the nature of the genetic defect.
Type 1, attacks are precipitated by sudden movements, emotional stress, fatigue, exercise, or hunger. Stiffness, generalized myokymia, vertigo, nausea, diplopia and tremor also occur. The attacks last 10 minutes or less. This disorder is associated with a variety of point mutations in the voltage-gated potassium channel gene, KCNA1, located on chromosome 12p13.
Type 2, myokymia is absent, the prominent symptoms being ataxia of gait and limbs, dysarthria, and gaze-evoked nystagmus. The attacks begin abruptly and last from 15 minutes to a few hours though sometimes days. Emotional stress, physical exertion, but not movement trigger attacks. The carbonic anhydrase inhibitor acetazolamide is very effective in preventing attacks. This disorder is associated with mutations in CACNL1A4 (subunit of a voltage-gated calcium channel gene) located on chromosome 19p. SCA-6 is also associated with a small expansion of CAG repeats in this gene as is familial hemiplegic migraine, a condition sharing similar features.
MOTOR NEURON DISEASE/AMYOTROPHIC LATERAL SCLEROSIS (ALS)
Motor neuron disease (MND) is a progressive condition characterised by degeneration of upper and lower motor neurons.

Different levels of the nervous system are involved:
1. Frontal atrophy in the precentral gyrus
2. The corticobulbar pathway
3. The cranial nerve nuclei
4. The corticospinal tract
5. The anterior horn cell
The term AMYOTROPHIC LATERAL SCLEROSIS (ALS) is used synonymously with motor neuron disease.
Epidemiology
Incidence: 2 per 100 000 per year, with a prevalence of 6 per 100000. Clusters and conjugal cases have been reported.
Familial ALS accounts for 5% of cases and is usually inherited as a dominant trait.
Sex ratio: male/female – 1.5:1
Mean age of onset – 55 years.
Mean survival – 3 years (50%).
Pathology

Microscopic: Loss of neurons in motor cortex.
Loss of neurons in cranial nerve nuclei and anterior horns.
Section of brain stem: reduction of corticobulbar and corticospinal fibres.
No evidence of inflammatory response is seen in involved structures.
MOTOR NEURON DISEASE/ALS
AETIOLOGY
The cause of motor neuron disease is unknown. Several possibilities have been suggested:
– Genetic: Mutations in the SOD1 gene (responsible for producing the enzyme superoxide dismutase) are found in 20% of familial cases of ALS. Superoxide dismutase is important in removing toxic superoxide radicals and converting them into non-harmful substances. Defects in the enzyme lead to accumulation and anterior horn cell death. These account for about 2% of patients with ALS.
– Viruses: Chronic virus infection has been proposed, partly because neurotropic viruses such as polio have a devastating effect on anterior horn cells. However, no serological or virological evidence for any infection has been found.
– Toxins: No evidence of toxic cause has been demonstrated.
– Minerals: Clinical similarities between MND and neurological involvement in hyperparathyroidism and phosphate deficiency suggest a relationship with chronic calcium deficiency.
The final common pathway of anterior horn cell death, irrespective of what actually triggers the process, is a complex interaction of genetic factors, oxidative stress and glutamate excess (excitatory injury). Abnormal clumps of proteins (neurofilaments) can be found in motor neurons that may themselves be toxic or by-products of overwhelming cell injury.
CLINICAL FEATURES At onset:
Asymmetric weakness and wasting of extremities – 75%
Bulbar or pseudobulbar features – 25% – dysphagia or dysarthria
In both limb-onset and bulbar-onset disease the key feature is the mixture of upper and lower motor neuron involvement with normal sensation.
Frontal lobe involvement
Frontal dementia occurs in 3–5% of all patients, but is more prevalent in familial cases. Emotional lability – unprovoked outbursts of laughing or crying occur.
Limb-onset disease
Limb-onset ALS results from involvement of corticospinal tracts and anterior horn cells.
Signs of corticospinal tract degeneration lead to:
– increased tone
– brisk reflexes
– extensor plantar responses
– distinctive distribution of weakness (extensors in upper limbs; flexors in lower limbs). Spasticity is rarely severe (intact extrapyramidal inhibition). Primary lateral sclerosis is a slowly progressive form of MND restricted to the cortical spinal tract.
Anterior horn cell involvement leads to muscle atrophy, weakness and fasciculations.
The patient may be aware of fasciculation.
Muscle cramps are common. Weakness is not as severe as the degree of wasting suggests.
In the hand: wasting is evident.

MOTOR NEURON DISEASE/ALS
Bulbar-onset disease = Progressive bulbar palsy
Progressive bulbar palsy presents with a combination of corticobulbar degeneration and lower cranial nerve motor nuclei involvement.
Degeneration of corticobulbar pathways to V, VII, X, XI and XII cranial nerve motor nuclei (with sparing of III, IV and VI) leads to an apparent weakness of the muscles of mastication and expression, the patient has difficulty in chewing and the face is expressionless. The jaw jerk (page 15) is exaggerated.
Food and fluid enter nasopharynx when swallowing – palatal weakness (X).


As the disease progresses, all levels of the motor system become involved. Patients with limb-onset develop bulbar symptoms and vice versa. Respiratory muscle weakness ultimately occurs and is the usual cause of death.
Less common clinical presentations
Occasionally patients can present with:
– breathlessness from respiratory muscle failure
– repeated chest infections from occult aspiration or
– weight loss.
Uncommon clinical variants
Primary lateral sclerosis is a very slowly progressive purely upper motor neuron syndrome that presents with asymmetrical spasticity.
‘Flail arm’ variant is when there is marked weakness and wasting of the arms with only modest weakness in the legs. This generally progresses more slowly.

Hexosaminidase deficiency (autosomal recessive disorder) may mimic ALS.
An ALS like syndrome can occur with elevated serum paraproteins, lymphoproliferative disease, lead poisoning and HIV infection.
Hyperthyroidism and hyperparathyroidism produce muscle wasting and hyperreflexia.
Pseudobulbar palsy, a pure upper motor neuron deficit reflecting corticobulbar involvement, may result also from cerebrovascular disease or multiple sclerosis.
Progressive muscular atrophy may be confused with a spinal muscular atrophy, multifocal motor neuropathy with conduction block, limb girdle dystrophy, diabetic amyotrophy or lead neuropathy.
N.B. IN MOTOR NEURON DISEASE:
– Sensory signs do not occur
– Bladder is never involved
– Ocular muscles are never affected.
Investigations
EMG reveals denervation with fibrillation.
Nerve conduction studies shows normal velocities and exclude in all limbs multifocal neuropathy with conduction block.
MRI (or myelography) where appropriate excludes foramen magnum or spinal cord compression.
Thyroid and calcium studies exclude endocrine or metabolic disease.
In selected cases screen for paraproteinaemia, lymphoreticular disease and hexosaminidase deficiency.
Diagnostic criteria (El Escorial criteria for MND/ALS – World Federation of Neurology)
Presence of –
LMN signs in at least 2 limbs.
UMN signs in at least 1 region (bulbar/cervical/lumbosacral)
Progression of disease.
Absence of –
Sensory signs.
Neurogenic sphincter disturbance.
Other clinically evident CNS/PNS disease.
Exclusion of ALS-like syndromes
TREATMENT
Treatment is primarily that of managing symptoms and supporting both patient and family as these progress and their needs change.
Counselling is essential to a full understanding of the illness and its natural history. Support from a Nurse Specialist is invaluable to meeting the challenges of each phase of illness and issues of feeding and methods of ventilatory support are best discussed well in advance so that informed decisions can be made. The comprehensive care of patients is challenging with medical, legal and ethical considerations.
Symptomatic treatment:
Anarthria and dysarthria: – Speech assessment and communication aids when indicated.
Dysphagia and aspiration: – Percutaneous endoscopic gastrostomy (PEG).
Nutrition: – Estimate calorific content and supplement diet with vitamins.
Muscle weakness: – Physiotherapy, walking aids. Splints, etc.
Respiratory failure: – As vital capacity drops respiratory failure becomes inevitable. Non-invasive ventilatory assistance should be considered when this falls below 75% or orthopnoea develops in patients without severe bulbar involvement. Recent trials indicate this can provide improvements to quality of life. The role for invasive mechanical ventilation is more uncertain. Rarely ALS can present with early respiratory failure before treatment issues have been discussed. This creates a major management dilemma.
Disease-modifying treatment
Riluzole is a drug with energy buffering and anti-glutamate properties. It is the only approved treatment and in a dose of 100 mg daily is safe with a marginal effect in prolonging survival by 2 months.
INHERITED MOTOR NEURON DISORDERS
SPINAL MUSCULAR ATROPHIES (SMAs)
Spinal muscular atrophy is the second most common fatal, autosomal recessive disease in Caucasians (after cystic fibrosis). The disorder is characterised by degeneration of the anterior horn cells and symmetrical muscle weakness and wasting.
Depending on the age of onset, degree of muscular involvement and length of survival, 3 types of recessive SMA are recognised: All map to the gene locus 5q12.2-q13.3.
With an incidence of 1/10000, the offspring of patients have a disease risk of approximately 1%.
Type I – Werdnig Hoffman disease (Acute Infantile SMA)
This is an autosomal recessive disorder.
Incidence 1:25 000 births

Clinical features:
Reduced fetal movements in late pregnancy with weakness and hypotonia at birth.

All motor milestones are delayed; 95% of all patients are dead by 18 months.
Type II – Kugelberg Welander disease (Late infantile or juvenile SMA)
Pathological features similar to Werdnig Hoffman disease.
Clinical features:
Limb girdle muscles affected.
It is slowly progressive with great variability even within the same family. Median age at death 12 years. Survival to adulthood occurs in the dominant form.
Type III (Adult onset SMA)
Onset between 2nd and 5th decade with progressive limb girdle weakness. Distinction from progressive muscular atrophy form of ALS is difficult. A benign course supports the former.
Distal and scapuloperoneal forms
Differentiation from CMT types I and II (page 444) and scapuloperoneal dystrophy (page 470) is clinically difficult and separation may only be possible on histological and neurophysiological grounds.
Spinal and bulbar muscular atrophy (Kennedy’s syndrome)
X-linked adult-onset neurogenic muscular atrophy with late distal and bulbar involvement (Gene Locus: Xq11-q12). Onset of fasciculations followed by muscle weakness and wasting occur at approximately 40 years of age. Bulbar signs and facial fasciculations are characteristic. Babinski sign is negative. The disorder is compatible with long life.
Management of spinal muscular atrophies
There is no specific treatment. Care is supportive. Genetic counselling is essential.
NEUROCUTANEOUS SYNDROMES
Previously called Phakomatoses – Phakos Greek: birthmark
These disorders are hereditary, characterised by multiorgan malformations and tumours. The literature includes many varieties of such conditions; most are extremely rare. Only the more major disorders are described below.
NEUROFIBROMATOSIS
Two distinct types occur:
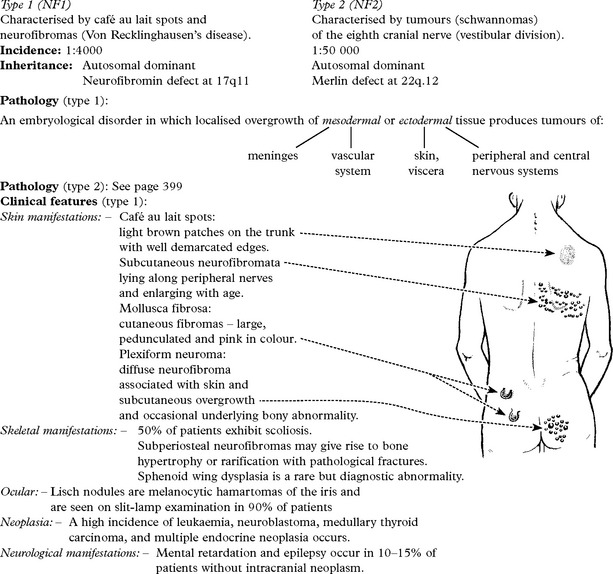
Cerebrovascular accidents as a consequence of intimal hyperplasia are not uncommon. Three patterns of neurological neoplasia are recognised:
|
1. Intracranial neoplasms: Optic nerve glioma Multiple meningioma. |
2. Intraspinal neoplasms: Meningioma Neurofibroma Glioma. |
3. Peripheral nerve neoplasms: Neurofibroma – a proportion of which become sarcomatous. |
Clinical features (type 2)
Skeletal manifestations are absent. Café au lait spots rare. Posterior subcapsular cataracts occur in 50% of cases. The condition is defined by bilateral vestibular schwannomas but may present as early unilateral acoustic neuroma plus a family history of NF2. Other intracranial and intraspinal neoplasms occur.
Diagnosis
A family history is obtained in over 50% of patients. In type 1, the cutaneous manifestations are characteristic, though they may be extremely mild with only café au lait spots (more than 6 in an individual is diagnostic). As a rule, the more florid the cutaneous manifestations the less likely is there nervous system involvement. CT scanning, MRI and myelography may be necessary when nervous system involvement is suspected. Type 2 is diagnosed when imaging (MRI) confirms bilateral vestibular schwannomas. The recent cloning of the type 2 gene to chromosome 22 may lead to direct gene testing in persons at risk.
Treatment
Plexiform neuromas may be removed for cosmetic reasons. The management of intracranial and intraspinal tumours has already been discussed.
TUBEROUS SCLEROSIS
Incidence: 1:30 000.
Autosomal dominant inheritance with high sporadic mutation rate. TSC1 is caused by a mutation on chromosome 9 in the hamartin, and TSC2 on chromosome 16 in tuberin.
Characterised by cutaneous, neurologic, renal, skeletal, cardiac and pulmonary abnormalities.
Pathology

As well as skin lesions, primitive renal tumours and cystic lung hamartomas occur.
Clinical features
Skin manifestations

Neurological manifestations: – Mental retardation is present in 60% of patients, though the onset and its recognition may be delayed.
Seizures occur in almost all patients, often as early as the 1st week of life. Attacks are initially focal motor and eventually become generalised. The response to anticonvulsants is variable.
Intracranial neoplasms – astrocytomas – arise from tubers usually close to the ventricles and may result in an obstructive hydrocephalus.
Neoplasia: – Renal carcinoma occurs in 50% of patients. Retinal tumours (hamartomas) and muscle tumours (rhabdomyomas) are common, the latter often involving the heart.
Diagnosis:
The presence of epilepsy and adenoma sebaceum is diagnostic.
CT scan may show subependymal areas of calcium deposition. MRI shows uncalcified subependymal tubers. Other developmental abnormalities may be evident, e.g. microgyria.
Treatment:
Anticonvulsant therapy for epilepsy. Surgical removal of symptomatic lesions. High mutation rate indicates that antenatal diagnosis will not significantly reduce incidence.
STURGE-WEBER SYNDROME
This disorder is characterised by a facial angioma associated with a leptomeningeal venous angioma. There is no clear pattern of inheritance. Practically all cases are sporadic.

Skull X-rays show parallel linear calcification (tram-line sign) and CT scan, in addition, shows the associated atrophic change. Angiography demonstrates dilated deep cerebral veins with decreased cortical drainage. Arteriovenous and dural venous sinus malformations are present in 30%.
Treatment
Intractable epilepsy may require lobectomy, or even hemispherectomy. Some recommend early excision of the surface lesion, but the rarity of the condition prevents thorough treatment evaluation.
VON HIPPEL-LINDAU (VHL) DISEASE
An autosomal dominant disorder due to mutations in VHL gene on chromosome 3 where haemangioblastomas are found in the cerebellum, spinal canal and retina, and are associated with various visceral pathologies:
– Renal angioma
– Renal cell carcinoma
– Phaeochromocytoma
– Pancreatic adenoma/cyst
– Cysts and haemangiomas in liver and epididymis.
Mutation in a tumour suppressor gene is found in 60% of affected families. Any of the above may produce signs and symptoms.

Cerebellar haemangioblastoma presents with progressive ataxia. Compression of the fourth ventricle may cause hydrocephalus with a subsequent rise in intracranial pressure.
Spinal canal haemangioblastoma – intradural or intramedullary lesion presenting with signs and symptoms of cord or root compression.
Diagnosis is established from family history and cranial imaging (MRI or CT). Renal ultrasound, abdominal CT and urinary amine estimations are required to complete the evaluation. In patients at risk, regular screening for renal, adrenal, pancreatic and intracranial tumours is recommended.
 Retinal pigment epithelium becomes hyperplastic – densely pigmented areas result.
Retinal pigment epithelium becomes hyperplastic – densely pigmented areas result.