Sahdeo Prasad and Bharat B. Aggarwal
INTRODUCTION
Extensive research over the last half a century indicates that inflammation plays an important role in cancer. Although acute inflammation can play a therapeutic role, low-level chronic inflammation can promote cancer. Different inflammatory cells, the various cell signaling pathways that lead to inflammation, and biomarkers of inflammation have now been well defined. These inflammatory pathways, which are primarily mediated through the transcription factors nuclear factor kappa B (NF-κB) and signal transducer and activator of transcription 3 (STAT3), have been linked to cellular transformation, tumor survival, proliferation, invasion, angiogenesis, and metastasis of cancer. These pathways have also now been linked with chemoresistance and radioresistance. This chapter considers the role of inflammation in cancer and its potential for cancer prevention and treatment.
Inflammation is the complex biologic responses of the body to irritation, injury, or infection. The recognition of inflammation dates back to antiquity. As documented by Aulus Cornelius Celsus, a Roman of the 1st century AD, inflammation is characterized by the tissue response to injury that results in rubor (redness, due to hyperemia), tumor (swelling, caused by increased permeability of the microvasculature and leakage of protein into the interstitial space), calor (heat, associated with increased blood flow and the metabolic activity of the cellular mediators of inflammation), and dolor (pain, in part due to changes in the perivasculature and associated nerve endings). Rudolf Virchow subsequently added functio laesa (dysfunction of the organs involved) in the 1850s. The process includes increased blood flow with an influx of white blood cells and other chemical substances that facilitate healing. Inflammation is also considered the body’s self-protective attempt to remove harmful stimuli, including damaged cells, irritants, or pathogens, and to begin the healing process.
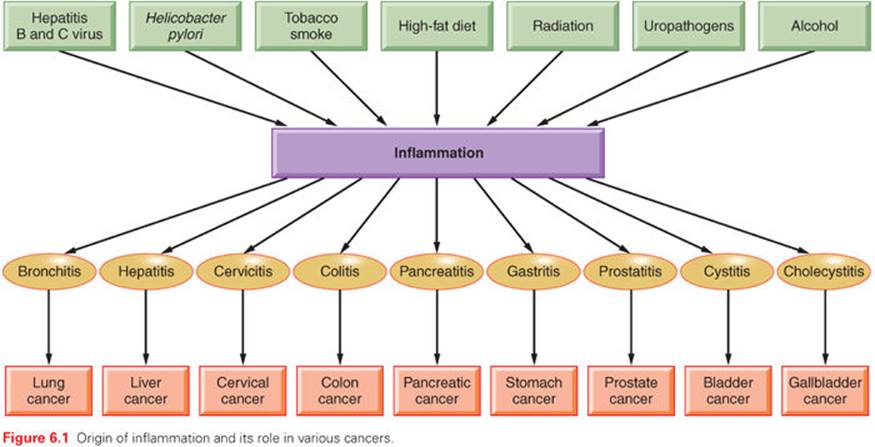
The word inflammation is derived from the Latin inflammo (meaning “I set alight, I ignite”). Because inflammation is a stereotyped response, it is considered a mechanism of innate immunity, as compared with adaptive immunity. On the basis of longevity, inflammation is classified as acute or chronic. When inflammation is short term, usually appearing within a few minutes or hours and ceasing upon the removal of the injurious stimulus, it is called acute. However, if it persists longer, it is called chronic inflammation, which leads to simultaneous destruction from the inflammatory process. Inflammation is beneficial when it is acute; however, chronic inflammation leads to several diseases, including cancer. Cancer is primarily a disease of lifestyle, with 30% of all cancers having been linked to smoking, 35% to diet, 14% to 20% to obesity, 18% to infection, and 7% to environmental pollution and radiation (Fig. 6.1).1 Smoking, obesity, infections, pollution, and radiation are all known to activate proinflammatory pathways.2 Therefore, understanding how inflammation contributes to cancer etiology is important for both cancer prevention and treatment.3
MOLECULAR BASIS OF INFLAMMATION
Although it is clear that inflammation and cancer are closely related, the mechanisms underlying persistent and chronic inflammation in chronic diseases remain unclear. Numerous cytokines have been linked with inflammation, including tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, IL-8, IL-17, and vascular endothelial growth factor (VEGF). Among various cytokines that have been linked with inflammation, TNF is a primary mediator of inflammation linked to cancer.4 However, it has been shown that proinflammatory transcriptional factors (activator protein [AP]-1, STAT3, NF-κB, hypoxia-inducible factor [HIF]-1, and β-catenin/Wnt) are ubiquitously expressed and control numerous physiologic processes, including development, differentiation, immunity, and metabolism in chronic diseases. Although these transcription factors are regulated by completely different signaling mechanisms, they are activated in response to various stimuli, including stresses and cytokines, and are involved in inflammation-induced tumor development and its metastasis.5 Interestingly, inflammation plays a role at all stages of tumor development: initiation, progression, and metastasis.2 In initiation, inflammation induces the release of a variety of cytokines and chemokines that promote the release of inflammatory cells and associated factors. This further causes oxidative damage, DNA mutations, and other changes in the tissue microenvironment, making it more conducive to cell transformation, increased survival, and proliferation. Inflammation also contributes to tissue injury, remodeling of the extracellular matrix, angiogenesis, and fibrosis in diverse target tissues. Among all the inflammatory cell signaling pathways, NF-κB has been shown to play a major role in cancer,6,7 and TNF is one of the most potent activators of NF-κB.8,9
ROLE OF INFLAMMATION IN TRANSFORMATION
Transformation is the process by which the cellular and molecular makeup of a cell is altered as it becomes malignant. Numerous factors are involved in the process of cell transformation, including inflammation. A clinical study has shown that chronic inflammation due to heavy metal deposition in lymph nodes leads to malignant transformation and, finally, to patient death.10 More recently, chronic exposure to cigarette smoke extract11 and arsenite12 has been shown to induce inflammation followed by epithelial–mesenchymal transition and transformation of human bronchial epithelial (HBE) cells. Furthermore, activation of NF-κB and HIF-2α increased the levels of the proinflammatory IL-6, IL-8, and IL-1β, which are essential for the malignant progression of transformed HBE cells. Sox2, another important molecular factor, cooperates with inflammation-mediated STAT3 activation, which precedes the malignant transformation of foregut basal progenitor cells.13 A clinical study reported that the p53 mutation is a critical event for the malignant transformation of sinonasal inverted papilloma. This p53 mutation resulted in cyclooxygenase (COX)-2–mediated inflammatory signals that contribute to the proliferation of advanced sinonasal inverted papilloma.14 In another study in patients, the YKL-40 protein was found to be involved in chronic inflammation and oncogenic transformation of human breast tissues.15 Inflammation-mediated transformation was also found to be regulated by MyD88 in a mouse model through Ras signaling.16 In addition, inflammation contributed to the activation of the epidermal growth factor receptor (EGFR) and its subsequent interaction with PKCδ, which leads to the transformation of normal esophageal epithelia to squamous cell carcinoma.17 Activation of Src oncoprotein triggers an inflammatory response mediated by NF-κB that directly activates Lin28 transcription and rapidly reduces let-7 microRNA levels. The inflammatory cytokine IL-6 mediates the activation of STAT3 transcription factor, which results in the transformation of cells.18
ROLE OF INFLAMMATION IN SURVIVAL
Numerous findings across different cancer populations have suggested that inflammation has an important role in carcinogenesis and disease progression.19,20 The important markers of systemic inflammatory response in both in vitro findings and clinical outcomes include plasma C-reactive protein (CRP) concentration,21,22 hypoalbuminemia,23 and the Glasgow Prognostic Score (GPS), which combines CRP and albumin.24,25 In addition to these, hematologic markers of systemic inflammatory response such as absolute white-cell count or its components (neutrophils, neutrophil-to-lymphocyte ratio [NLR]),26–28platelets, and a platelet-to-lymphocyte ratio29,30 are also prognostic indicators for cancer clinical outcomes. Whether these inflammatory biomarkers influence the survival of cancer patients is discussed in this section.
In a study of 416 patients with renal cell carcinoma, with 362 patients included in the analysis, elevated neutrophil count, elevated platelet counts, and a high NLR were found. This inflammatory response was predictive for shorter overall patient survival.31 Another study in unresectable malignant biliary obstruction (UMBO) found that patients with low GPS (0 and 1) had better postoperative survivals than did patients with a higher GPS. The 6-month and 1-year survival rates were 58.1% to 27.3%, respectively, for patients with low GPS and 25% to 6.2%, respectively, for patients with a higher GPS.32 It has been also shown that prostate cancer patients with aggressive, clinically significant disease and an elevated GPS2 had a higher risk of death overall as well as high-grade disease.33 Other than GPS, age and gastrectomy have also been shown to independently influence the disease-specific and progression-free survival of gastric cancer patients.34 A biomarker of systemic inflammation, the blood NLR, predicted patient survival with hepatocellular carcinoma (HCC) after transarterial chemoembolization. Patients in whom the NLR remained stable or became normalized after transarterial chemoembolization showed improved overall survival compared with patients showing a persistently abnormal index of NLR.35
A further study found that inflammatory transcription factors and cytokines contribute to the overall survival of patients. One study found that 97% of patients with epithelial tumors of malignant pleural mesothelioma and 95% of patients with nonepithelial tumors expressed IL-4Rα protein, and this strong IL-4Rα expression was correlated with a worse survival. In response to IL-4, human malignant pleural mesothelioma cells showed increased STAT6 phosphorylation and increased production of IL-6, IL-8, and VEGF without any effect on proliferation or apoptosis. This finding indicates that high expression of STAT6 as well as STAT3 and cytokines is inversely correlated with survival in patients.36,37 NF-κB, along with IL-6, contributes to the survival of mammospheres in culture, because NF-κB and IL-6 were hyperactive in breast cancer–derived mammospheres.38 In addition, elevated CRP and serum amyloid A (SAA) were associated with reduced disease-free survival of breast cancer patients.39 In gastroesophageal cancer, proinflammatory cytokines IL-1β, IL-6, IL-8, and TNF-α and acute phase protein concentrations (CRP) were found to be elevated, and these levels were associated with reduced survival of patients.40 Additionally, the Bcl-2 family protein COX-2, which is regulated by inflammatory transcription factors, is also involved in the survival of cancer cells.41,42 Thus, we conclude that inflammation in general contributes to poor survival of patients.
In contrast to these findings, an in vivo study of dogs with osteosarcoma showed that survival improvement was apparent with inflammation or lymphocyte-infiltration scores >1, as well as in dogs that had apoptosis scores in the top 50th percentile.43 Also, in patients with epithelioid malignant pleural mesothelioma, a high degree of chronic inflammatory cell infiltration in the stromal component was associated with improved overall survival.44
ROLE OF INFLAMMATION IN PROLIFERATION
Several studies have shown that cell proliferation is affected by inflammation.45 More significantly, proliferation in the setting of chronic inflammation predisposes humans to carcinoma in the esophagus, stomach, colon, liver, and urinary bladder.46 In postgastrectomy patients, Helicobacter pylori induced inflammation and was associated with increased epithelial cell proliferation.47 Even in the mouse model, chronic infection with Helicobacter hepaticusinduced hepatic inflammation, which further led to hepatic cell proliferation.48 Other reports found an increased expression of the cell proliferative markers PCNA and Ki-67 in the linings of inflamed odontogenic keratocysts compared with noninflamed lesions.49,50 These findings suggest the existence of greater proliferative activity in the cells with inflammation. Wang et al.51 showed an increased expression of cell proliferative markers PCNA and Ki-67 in a sample of 45 patients with benign prostatic hyperplasia.
The inflammatory biomarker COX-2 was also associated with the proliferation of cells. The highest proliferation index was found in COX-2–positive epithelium.51 The association of COX-2 and proliferation was also reported in a rat model. The carcinogen dimethylhydrazine (DMH) induces an increase in epithelial cell proliferation and in the expression of COX-2 in the colon of rats.52 Erbb2, a kinase, regulates inflammation through the induction of NF-κB, Comp1, IL-1β, COX-2, and multiple chemokines in the skin by ultraviolet (UV) exposure. This inflammation has been shown to increase the proliferation of skin tissue after UV irradiation.53
ROLE OF INFLAMMATION IN INVASION
A characteristic of invasive cancer cells is survival and growth under nonadhesive conditions. This invasion of cancer cells causes the disease to spread, which results in poor patient survival.54 A strong relationship has been documented between inflammation and cancer cell invasion.55,56 In a study of 150 patients with HCC, a high GPS score was associated with a high vascular invasion of cancer cells.57Another study of colorectal cancer also supports the links between inflammation and the invasion of cancer cells, with a finding that a high GPS increased the invasion of colorectal cancer cells.58 In patients with esophageal squamous cell carcinoma, a high GPS score also showed a close relationship with lymphatic and venous invasion.59
At the molecular level, various proteins are known to be involved in tumor cell invasion. MMP-9, a gelatinase that degrades type IV collagen—the major structural protein component in the extracellular matrix and basement membrane—is thought to play an important role in facilitating tumor invasion, as it is highly expressed in various malignant tumors.60,61 Additionally, the high expression of HIF-1α has been proposed as being associated with a greater incidence of vascular invasion of HCC. This expression of HIF-1α was further correlated with high expression of the inflammatory molecule COX-2.62
Breast cancer invasion has been linked to proteolytic activity at the tumor cell surface. In inflammatory breast cancer (IBC) cells, high expression of cathepsin B, a cell surface proteolytic enzyme, has been shown to be associated with invasiveness of IBC. In addition, a high coexpression of cathepsin B and caveolin-1 was found in IBC patient biopsies. Thus, proteolytic activity of cathepsin B and its coexpression with caveolin-1 contributes to the invasiveness of IBC.63 In IBC, RhoC GTPase is also responsible for the invasive phenotype.64 In addition, the PI3K/Akt signaling pathway is crucial in IBC invasion. The molecules involved in cell motility are specifically upregulated in IBC patients compared with stage-matched and cell-type-of-origin–matched non-IBCs patients. Distinctively, RhoC GTPase is a substrate for Akt1, and its phosphorylation is absolutely essential for IBC cell invasion.65
ROLE OF INFLAMMATION IN ANGIOGENESIS
Angiogenesis—the formation of new blood vessels from existing vessels—is tightly linked to chronic inflammation and cancer. Angiogenesis is one of the molecular events that bridges the gap between inflammation and cancer. Angiogenesis results from multiple signals acting on endothelial cells. Mature vessels control exchanges of hematopoietic cells and solutes between blood and surrounding tissues by responding to microenvironmental cues, including inflammation. Although inflammation is essential to defend the body against pathogens, it has adverse effects on the surrounding tissue, and some of these effects induce angiogenesis. Inflammation and angiogenesis are thereby linked processes, but exactly how they are related has not been well understood. Both inflammation and angiogenesis are exacerbated by an increased production of chemokines/cytokines, growth factors, proteolytic enzymes, proteoglycans, lipid mediators, and prostaglandins.
A close relationship has been reported between inflammation and angiogenesis in breast cancer. Tissue section staining showed increased vascularity with the intensity of diffuse inflammation.66 Offersen et al.67 found that inflammation was significantly correlated in bladder carcinoma with microvessel density, which is a marker of angiogenesis. Leukocytes have been described as mediators of inflammation-associated angiogenesis. In addition, the stable expression of TNF-α in endothelial cells increased angiogenic sprout formation independently of angiogenic growth factors. Furthermore, in work using the Matrigel plug assay in vivo, increased angiogenesis was observed in endothelial TNF-α–expressing mice. Thus, chronic inflammatory changes mediated by TNF-α can induce angiogenesis in vitro and in vivo, suggesting a direct link between inflammation and angiogenesis.68 TNF-α–induced inhibitor of nuclear factor kappa kinase (IKK)-β activation also activates the angiogenic process. IKK-β activates the mammalian target of rapamycin (mTOR) pathway and enhances angiogenesis through VEGF production.66 In addition to TNF-α, proinflammatory cytokines IL-1 (mainly IL-1β) and IL-8 were also found to be major proangiogenic stimuli of both physiologic and pathologic angiogenesis.69,70 Recently, another cytokine macrophage migration inhibitory factor (MIF) was found to play a role in neoangiogenesis/vasculogenesis by endothelial cell activation along with inflammation.71
Benest et al.72 found that a well-known regulator of angiogenesis, angiopoietin-2 (Ang-2), can upregulate inflammatory responses, indicating a common signaling pathway for inflammation and angiogenesis. TGF-β induction was also reported in head and neck epithelia and human head and neck squamous cell carcinomas (HNSCC), with severe inflammation that leads to angiogenesis.73 The tumor-derived cytokine endothelial monocyte-activating polypeptide II (EMAP-II) has been shown to have profound effects on inflammation as well as on the processes involved in angiogenesis.74 NF-κB plays an important role in inflammation as well as in angiogenesis, because the suppression of NF-κB and IkB-2A blocks basic fibroblast growth factor–induced angiogenesis in vivo. NF-κB regulates the angiogenic protein VEGF promoted by α5β1 integrin, which coordinately regulates angiogenesis and inflammation.75 It has been also reported that a coculture of cancer cells with macrophages synergistically increased the production of various angiogenesis-related factors when stimulated by the inflammatory cytokine. This inflammatory angiogenesis was mediated by the activation of NF-κB and activator protein 1 (Jun/Fos), because the administration of either NF-κB–targeting drugs or COX-2 inhibitors or the depletion of macrophages blocked inflammatory angiogenesis.76
In a mouse model, cigarette smoke induced the inflammatory protein 5-lipoxygenase (5-LOX), and this induction activated matrix metalloproteinase 2 (MMP-2) and VEGF to induce the angiogenic process.77A cellular enzyme, Tank-binding kinase 1 (TBK-1), has been proposed as a putative mediator in tumor angiogenesis. TBK-1 mediates angiogenesis through the upregulation of VEGF and exerts proinflammatory effects via the induction of inflammatory cytokines. Thus, these pathways, including TBK-1, are an important cross-link between angiogenesis and inflammation.78
ROLE OF INFLAMMATION IN METASTASIS
Inflammation plays a regulatory role in cancer progression and metastasis. Chronic or tumor-derived inflammation and inflammation-related stimuli within the tumor microenvironment promote blood and lymphatic vessel formation and aid in invasion and metastasis.79,80 The association of inflammation and metastasis has been observed in several cancer types. In an immunohistochemical analysis of lung cancer tissues, a remarkably high level of metastasis was observed with severe inflammation.81 A mouse model of breast cancer found that mammary tumors increased the frequency of lung metastases, and this effect was associated with the recruitment of inflammatory cells to the lung as well as elevated levels of IL-6 in the lung airways.82 In another murine model, implanting human ovarian tumor cells into the ovaries of severe combined immunodeficient mice resulted in peritoneal inflammation and tumor cell dissemination from the ovaries. In addition, enhancement of the inflammatory response with thioglycolate accelerated the development of ascites and metastases, and its suppression with acetylsalicylic acid delayed metastasis.83Thus, it can be concluded that inflammation facilitates ovarian tumor metastasis by a mechanism largely mediated by cytokines.
It has been shown that metastatic tumor cells entering a distant organ such as the liver trigger a proinflammatory response involving the Kupffer cell–mediated release of TNF-α and the upregulation of vascular endothelial cell adhesion receptors, such as E-selectin.84 The physiologic expression of the selectins is tightly controlled to limit the inflammatory response, but dysregulated expression of selectins contributes to inflammatory and thrombotic disorders as well as tumor metastases.85 Using P-selectin knockout mice, the importance of P-selectin–mediated cell adhesive interactions in the pathogenesis of inflammation and metastasis of cancers has been clearly demonstrated.86
Tumor-associated inflammatory monocytes and macrophages are essential promoters of tumor cell migration, invasion, and metastasis.87 Macrophages and their mediators affect the multistep process of invasion and metastasis, from interaction with the extracellular matrix to the construction of a premetastatic niche. Monocytes are attracted by cytokines and chemokines (e.g., CSF-1, GM-CSF, and MCP-1), which are released by tumor cells or cells of the tumor microenvironment. These monocytes are then induced to express proangiogenic and metastatic factors, including VEGF, fibroblast growth factor (FGF)-2, platelet-derived growth factor (PDGF), intercellular adhesion molecule (ICAM)-1, vascular cell adhesion molecule (VCAM)-1, E-selectin, P- selectin, and MMP-9.88 Versican, a large extracellular matrix proteoglycan, has been shown to activate tumor-infiltrating myeloid cells through Toll-like receptor (TLR) 2 and its coreceptors TLR6 and CD14 and to elicit the production of proinflammatory cytokines (including TNF-α), which enhance tumor metastasis. TLR2 increases the secretion of IL-8, which potentiates metastatic growth. Ligation of TLR2 by versican induces inflammatory cytokine secretion, providing a link between inflammation and cancer metastasis.89
IKK-α has been shown to be important in the inflammation-associated metastasis of cancer cells. Luo et al.90 demonstrated that activation and nuclear localization of IKK-α by tumor-infiltrating immune cells in prostatic epithelial tumor cells leads to malignant prostatic epithelial cells with a metastatic fate. Src family kinases, when inappropriately activated, promote pathologic inflammatory processes and tumor metastasis, in part through their effects on the regulation of endothelial monolayer permeability.91 Platelet-activating factor (PAF), an inflammatory biolipid, has also been shown to increase metastasis. In particular, Melnikova et al.92 demonstrated that PAF receptor antagonists can effectively inhibit the metastatic potential of human melanoma cells in nude mice. Mesenchymal stem cells promote HCC metastasis under the influence of inflammation through TGF-β.93
EPIGENETIC CHANGES AND INFLAMMATION
Epigenetics considers the heritable changes in the activity of gene expression without the alteration of DNA sequences, and such changes have been linked to many human diseases, including cancer.94 DNA methylation and histone modification are well-known epigenetic changes that can lead to gene activation or inactivation.94–96 DNA methylation occurs primarily at cytosine-phosphate-guanine (CpG) dinucleotides as well as at transcriptional regulatory sites on the gene promoter.96–98 Epigenetic abnormalities result in dysregulated gene expression and function, which can further lead to cancer. Inflammation and epigenetic abnormalities in cancer are highly associated. Inflammation induces aberrant epigenetic alterations in a tissue early in the process of carcinogenesis, and accumulation of such alterations forms an epigenetic field for cancer. Yara et al.99 have shown that increased inflammation, as evidenced by the activation of NF-κB, production of IL-6 and COX-2, as well as the decrease of IκB, leads to the promoter’s methylation. However, preincubation of cells with a demethylating agent prevented inflammation.
Infectious agents also contribute to inflammation-induced epigenetic changes. Infectious agents such as H. pylori and hepatitis C virus as well as intrinsic mediators of inflammatory responses, including proinflammatory cytokines, induce genetic and epigenetic changes, including point mutations, deletions, duplications, recombinations, and methylation of various tumor-related genes. Interestingly, disturbances in cytokine and chemokine signals and the induction of cell proliferation are important ways that inflammation induces aberrant DNA methylation. A study has shown that infection of human gastric mucosae with H. pylori induces chronic inflammation and further gastric cancers.100 This inflammation is associated with high methylation levels or high incidences of methylation.101–103
Furthermore, numerous reports have documented the fact that inflammation is linked with epigenetic changes in carcinogenesis. Recently, Achyut104 reported that inflammation in stromal fibroblasts caused epigenetic silencing of p21 and further tumor progression. Chronic inflammation also led to epigenetic regulation of p16 and activation of DNA damage in a lung carcinogenesis model.105
A transient inflammatory signal has been shown to initiate an epigenetic switch from nontransformed cells to cancer cells via a positive feedback loop involving NF-κB, Lin28, let-7, and IL-6. This IL-6 induced STAT3, directly activated miR-21 and miR-181b-1, and further induced the epigenetic switch. Thus, STAT3 underlies the epigenetic switch of mir-21 and mir-181b-1 that links inflammation to cancer.106 Another report also showed that transient activation of Src oncoprotein mediates an epigenetic switch from immortalized breast cells to a stably transformed line that contained cancer stem cells. Thus, inflammation activates a positive feedback loop that maintains the epigenetic transformed state for many generations in the absence of the inducing signal.18
DNA hypermethylation at promoter CpG islands is an important mechanism by which carcinogenesis occurs through the inactivation of tumor-suppressor genes. Aberrant CpG island hypermethylation is also frequently observed in chronic inflammation and precancerous lesions, which again suggests links between inflammation and epigenetic change.107 In addition, inflammation induced the halogenation of cytosine nucleotide. Damage products of this inflammation-mediated halogenated cytosine interfere with normal epigenetic control by altering DNA-protein interactions that are critical for gene regulation and the heritable transmission of methylation patterns. These inflammation-mediated cytosine damage products also provide a mechanistic link between inflammation and cancer.108
ROLE OF INFLAMMATION IN CANCER DIAGNOSIS
Chronic inflammation plays an important role in the etiology and progression of chronic diseases, including cancer. Hence, chronic inflammation may have an important diagnostic role in cancer. Inflammation induced by inflammatory cells such as infiltrating cells and mesothelial cells is mediated via the release of various mediators and proteins, including PDGF, IL-8, monocyte chemotactic peptide (MCP-1), nitric oxide (NO), collagen, antioxidant enzymes, and the plasminogen activation inhibitor (PAI). Furthermore, several inflammatory mediators have been shown to be detected at increased concentrations, thereby aiding in the disease diagnosis.109
In one study, numerous inflammatory disorders were detected based on inflammation measured in gastric biopsies of patients by Fourier transform infrared spectroscopy (FT-IR). Using endoscopic samples, gastritis and gastric cancer were diagnosed.110 Furthermore, the degree of prostate inflammation has been used to determine the level of incidental prostatitis.111 An assessment of the expression of cytokines and other immune stimulatory molecules that drive B-cell activation provides insight into the etiology of cancers. It has been shown that the dysregulation of cytokine production precedes the diagnosis of non-Hodgkin lymphoma.112
Inflammation parameters have been used to diagnose cancer in patients. Inflammation parameters, including CRP, were found to differ in patients with cancer and in those without. In clinical practice, however, such parameters are considered to have modest diagnostic value for cancer.113 In a study with 1,275 patients, granulomatous inflammation was identified in 154 patients (12.1%), of whom 12 out of 154 (7.8%) had a concurrent diagnosis of cancer.114 In another study with 173 patients, 52% had lung adenocarcinoma. Patients with high systemic inflammation were more likely to have more than two sites of metastatic disease and to have poor performance status and less likely to receive any chemotherapy. Systemic inflammation at diagnosis is considered to be an independent marker of poor outcome in patients with advanced non-small cell lung cancer (NSCLC).115
INFLAMMATION AND GENOMICS
Recently, the genomic landscape of the most common forms of human cancer have been examined.116 Almost 140 genes and 12 cell signaling pathways have been linked with most cancers. Several of these genes and pathways are directly or indirectly linked with inflammation. A cytokine pattern in patients with cancer has been identified.117
INFLAMMATION AND TARGETED THERAPIES
That inflammation can be used as a target for cancer prevention and treatment is indicated by the fact that several drugs approved by the U.S. Food and Drug Administration (FDA) actually modulate proinflammatory pathways. For instance, EGFR, HER2, VEGF, CXCR4, and proteasome have been shown to activate NF-κB–mediated proinflammatory pathways, and their inhibitors have been approved by the FDA for the treatment of various cancers. Similarly, steroids such as dexamethasone, nonsteroidal anti-inflammatory drugs (NSAIDs), and statins that are currently used for prevention or treatment have also been found to suppress the NF-κB pathway. Thus, these observations indicate that inflammatory pathways are excellent targets for cancer.
CONCLUSIONS
According to Colditz et al.,118 almost 50% of all cancers can be prevented based on what we know today. All the studies summarized previously suggest that inflammation is closely linked to cancer, and the incidence of most cancers can be reduced by controlling inflammation. Proinflammatory conditions such as colitis, bronchitis, hepatitis, and gastritis can all eventually lead to cancer. Thus, one must find ways to treat these conditions before the appearance of cancer. All these studies indicate that an anti-inflammatory lifestyle could play an important role in both the prevention and treatment of cancer.
REFERENCES
1. Anand P, Kunnumakkara AB, Sundaram C, et al. Cancer is a preventable disease that requires major lifestyle changes. Pharm Res 2008;25:2097–2116.
2. Aggarwal BB, Gehlot P. Inflammation and cancer: how friendly is the relationship for cancer patients? Curr Opin Pharmacol 2009;9:351–369.
3. Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science 2013;339:286–291.
4. Sethi G, Sung B, Aggarwal BB. TNF: a master switch for inflammation to cancer. Front Biosci 2008;13:5094–5107.
5. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature 2006;441:431–436.
6. Aggarwal BB. Nuclear factor-kappaB: the enemy within. Cancer Cell 2004;6:203–208.
7. Chaturvedi MM, Sung B, Yadav VR, et al. NF-kappaB addiction and its role in cancer: ‘one size does not fit all’. Oncogene 2011;30:1615–1630.
8. Aggarwal BB. Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 2003;3:745–756.
9. Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 2012;119:651–665.
10. Iannitti T, Capone S, Gatti A, et al. Intracellular heavy metal nanoparticle storage: progressive accumulation within lymph nodes with transformation from chronic inflammation to malignancy. Int J Nanomed 2010;5:955–960.
11. Zhao Y, Xu Y, Li Y, et al. NF-kappaB-mediated inflammation leading to EMT via miR-200c is involved in cell transformation induced by cigarette smoke extract. Toxicol Sci 2013;135:265–276.
12. Xu Y, Zhao Y, Xu W, et al. Involvement of HIF-2alpha-mediated inflammation in arsenite-induced transformation of human bronchial epithelial cells. Toxicol Appl Pharmacol 2013;272:542–550.
13. Liu K, Jiang M, Lu Y, et al. Sox2 cooperates with inflammation-mediated Stat3 activation in the malignant transformation of foregut basal progenitor cells. Cell Stem Cell 2013;12:304–315.
14. Yoon BN, Chon KM, Hong SL, et al. Inflammation and apoptosis in malignant transformation of sinonasal inverted papilloma: the role of the bridge molecules, cyclooxygenase-2, and nuclear factor kappaB. Am J Otolaryngol 2013;34:22–30.
15. Roslind A, Johansen JS. YKL-40: a novel marker shared by chronic inflammation and oncogenic transformation. Methods Mol Biol 2009;511:159–184.
16. Coste I, Le Corf K, Kfoury A, et al. Dual function of MyD88 in RAS signaling and inflammation, leading to mouse and human cell transformation. J Clin Invest 2010;120:3663–3667.
17. Parthasarathy S, Dhayaparan D, Jayanthi V, et al. Aberrant expression of epidermal growth factor receptor and its interaction with protein kinase C delta in inflammation associated neoplastic transformation of human esophageal epithelium in high risk populations. J Gastroenterol Hepatol 2011;26:382–390.
18. Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009;139:693–706.
19. Colotta F, Allavena P, Sica A, et al. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis 2009;30:1073–1081.
20. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–674.
21. Canna K, McMillan DC, McKee RF, et al. Evaluation of a cumulative prognostic score based on the systemic inflammatory response in patients undergoing potentially curative surgery for colorectal cancer. Br J Cancer 2004;90:1707–1709.
22. Hilmy M, Bartlett JM, Underwood MA, et al. The relationship between the systemic inflammatory response and survival in patients with transitional cell carcinoma of the urinary bladder. Br J Cancer 2005;92:625–627.
23. Forrest LM, McMillan DC, McArdle CS, et al. Evaluation of cumulative prognostic scores based on the systemic inflammatory response in patients with inoperable non-small-cell lung cancer. Br J Cancer 2003;89:1028–1030.
24. Ramsey S, Lamb GW, Aitchison M, et al. Evaluation of an inflammation-based prognostic score in patients with metastatic renal cancer. Cancer 2007;109:205–212.
25. Crumley AB, Stuart RC, McKernan M, et al. Comparison of an inflammation-based prognostic score (GPS) with performance status (ECOG-ps) in patients receiving palliative chemotherapy for gastroesophageal cancer. J Gastroenterol Hepatol 2008;23:e325–329.
26. Yamanaka T, Matsumoto S, Teramukai S, et al. The baseline ratio of neutrophils to lymphocytes is associated with patient prognosis in advanced gastric cancer. Oncology 2007;73:215–220.
27. Halazun KJ, Aldoori A, Malik HZ, et al. Elevated preoperative neutrophil to lymphocyte ratio predicts survival following hepatic resection for colorectal liver metastases. Eur J Surg Oncol 2008;34:55–60.
28. Huang ZL, Luo J, Chen MS, et al. Blood neutrophil-to-lymphocyte ratio predicts survival in patients with unresectable hepatocellular carcinoma undergoing transarterial chemoembolization. J Vasc Interv Radiol 2011;22:702–709.
29. Heng DY, Xie W, Regan MM, et al. Prognostic factors for overall survival in patients with metastatic renal cell carcinoma treated with vascular endothelial growth factor-targeted agents: results from a large, multicenter study. J Clin Oncol 2009;27:5794–5799.
30. Smith RA, Bosonnet L, Raraty M, et al. Preoperative platelet-lymphocyte ratio is an independent significant prognostic marker in resected pancreatic ductal adenocarcinoma. Am J Surg 2009;197:466–472.
31. Fox P, Hudson M, Brown C, et al. Markers of systemic inflammation predict survival in patients with advanced renal cell cancer. Br J Cancer 2013;109:147–153.
32. Iwasaki Y, Ishizuka M, Kato M, et al. Usefulness of an inflammation-based prognostic score (mGPS) for predicting survival in patients with unresectable malignant biliary obstruction. World J Surg 2013;37:2222–2228.
33. Shafique K, Proctor MJ, McMillan DC, et al. Systemic inflammation and survival of patients with prostate cancer: evidence from the Glasgow Inflammation Outcome Study. Prostate Cancer Prostatic Dis 2012;15:195–201.
34. Kunisaki C, Takahashi M, Ono HA, et al. Inflammation-based prognostic score predicts survival in patients with advanced gastric cancer receiving biweekly docetaxel and s-1 combination chemotherapy. Oncology 2012;83:183–191.
35. Pinato DJ, Sharma R. An inflammation-based prognostic index predicts survival advantage after transarterial chemoembolization in hepatocellular carcinoma. Transl Res 2012;160:146–152.
36. Burt BM, Bader A, Winter D, et al. Expression of interleukin-4 receptor alpha in human pleural mesothelioma is associated with poor survival and promotion of tumor inflammation. Clin Cancer Res 2012;18:1568–1577.
37. Sethi G, Shanmugam MK, Ramachandran L, et al. Multifaceted link between cancer and inflammation. Biosci Rep 2012;32:1–15.
38. Papi A, Guarnieri T, Storci G, et al. Nuclear receptors agonists exert opposing effects on the inflammation dependent survival of breast cancer stem cells. Cell Death Differ 2012;19:1208–1219.
39. Pierce BL, Ballard-Barbash R, Bernstein L, et al. Elevated biomarkers of inflammation are associated with reduced survival among breast cancer patients. J Clin Oncol 2009;27:3437–3444.
40. Deans DA, Wigmore SJ, Gilmour H, et al. Elevated tumour interleukin-1beta is associated with systemic inflammation: a marker of reduced survival in gastro-oesophageal cancer. Br J Cancer 2006;95:1568–1575.
41. Chen LS, Balakrishnan K, Gandhi V. Inflammation and survival pathways: chronic lymphocytic leukemia as a model system. Biochem Pharmacol 2010;80:1936–1945.
42. Sharma-Walia N, Paul AG, Bottero V, et al. Kaposi’s sarcoma associated herpes virus (KSHV) induced COX-2: a key factor in latency, inflammation, angiogenesis, cell survival and invasion. PLoS Pathog 2010;6:e1000777.
43. Modiano JF, Bellgrau D, Cutter GR, et al. Inflammation, apoptosis, and necrosis induced by neoadjuvant fas ligand gene therapy improves survival of dogs with spontaneous bone cancer. Mol Ther 2012;20:2234–2243.
44. Suzuki K, Kadota K, Sima CS, et al. Chronic inflammation in tumor stroma is an independent predictor of prolonged survival in epithelioid malignant pleural mesothelioma patients. Cancer Immunol Immunother 2011;60:1721–1728.
45. Hu B, Elinav E, Flavell RA. Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle 2011;10:1936–1939.
46. Sugar LM. Inflammation and prostate cancer. Can J Urol 2006;13(Suppl 1):46–47.
47. Safatle-Ribeiro AV, Ribeiro U, Jr., Clarke MR, et al. Relationship between persistence of Helicobacter pylori and dysplasia, intestinal metaplasia, atrophy, inflammation, and cell proliferation following partial gastrectomy. Dig Dis Sci 1999;44:243–252.
48. Ihrig M, Schrenzel MD, Fox JG. Differential susceptibility to hepatic inflammation and proliferation in AXB recombinant inbred mice chronically infected with Helicobacter hepaticus. Am J Pathol 1999;155:571–582.
49. de Paula AM, Carvalhais JN, Domingues MG, et al. Cell proliferation markers in the odontogenic keratocyst: effect of inflammation. J Oral Pathol Med 2000;29:477–482.
50. Kaplan I, Hirshberg A. The correlation between epithelial cell proliferation and inflammation in odontogenic keratocyst. Oral Oncol 2004;40:985–991.
51. Wang W, Bergh A, Damber JE. Chronic inflammation in benign prostate hyperplasia is associated with focal upregulation of cyclooxygenase-2, Bcl-2, and cell proliferation in the glandular epithelium. Prostate 2004;61:60–72.
52. Demarzo MM, Martins LV, Fernandes CR, et al. Exercise reduces inflammation and cell proliferation in rat colon carcinogenesis. Med Sci Sports Exerc 2008;40:618–621.
53. Madson JG, Lynch DT, Tinkum KL, et al. Erbb2 regulates inflammation and proliferation in the skin after ultraviolet irradiation. Am J Pathol 2006;169:1402–1414.
54. Bondong S, Kiefel H, Hielscher T, et al. Prognostic significance of L1CAM in ovarian cancer and its role in constitutive NF-kappaB activation. Ann Oncol 2012;23:1795–1802.
55. Wu Y, Zhou BP. Inflammation: a driving force speeds cancer metastasis. Cell Cycle 2009;8:3267–3273.
56. Aggarwal BB, Vijayalekshmi RV, Sung B. Targeting inflammatory pathways for prevention and therapy of cancer: short-term friend, long-term foe. Clin Cancer Res 2009;15:425–430.
57. Kinoshita A, Onoda H, Imai N, et al. The Glasgow Prognostic Score, an inflammation based prognostic score, predicts survival in patients with hepatocellular carcinoma. BMC Cancer 2013;13:52.
58. Toiyama Y, Miki C, Inoue Y, et al. Evaluation of an inflammation-based prognostic score for the identification of patients requiring postoperative adjuvant chemotherapy for stage II colorectal cancer. Exp Ther Med 2011;2:95–101.
59. Kobayashi T, Teruya M, Kishiki T, et al. Inflammation-based prognostic score, prior to neoadjuvant chemoradiotherapy, predicts postoperative outcome in patients with esophageal squamous cell carcinoma. Surgery 2008;144:729–735.
60. Nelson AR, Fingleton B, Rothenberg ML, et al. Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol 2000;18:1135–1149.
61. Clark ES, Weaver AM. A new role for cortactin in invadopodia: regulation of protease secretion. Eur J Cell Biol 2008;87:581–590.
62. Dai CX, Gao Q, Qiu SJ, et al. Hypoxia-inducible factor-1 alpha, in association with inflammation, angiogenesis and MYC, is a critical prognostic factor in patients with HCC after surgery. BMC Cancer 2009;9:418.
63. Victor BC, Anbalagan A, Mohamed MM, et al. Inhibition of cathepsin B activity attenuates extracellular matrix degradation and inflammatory breast cancer invasion. Breast Cancer Res 2011;13:R115.
64. van Golen KL, Bao LW, Pan Q, et al. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin Exp Metastasis 2002;19:301–311.
65. Lehman HL, Van Laere SJ, van Golen CM, et al. Regulation of inflammatory breast cancer cell invasion through Akt1/PKBalpha phosphorylation of RhoC GTPase. Mol Cancer Res 2012;10:1306–1318.
66. Lee DF, Kuo HP, Chen CT, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 2007;130:440–455.
67. Offersen BV, Knap MM, Marcussen N, et al. Intense inflammation in bladder carcinoma is associated with angiogenesis and indicates good prognosis. Br J Cancer 2002;87:1422–1430.
68. Rajashekhar G, Willuweit A, Patterson CE, et al. Continuous endothelial cell activation increases angiogenesis: evidence for the direct role of endothelium linking angiogenesis and inflammation. J Vasc Res 2006;43:193–204.
69. Voronov E, Carmi Y, Apte RN. Role of IL-1-mediated inflammation in tumor angiogenesis. Adv Exp Med Biol 2007;601:265–270.
70. Qazi BS, Tang K, Qazi A. Recent advances in underlying pathologies provide insight into interleukin-8 expression-mediated inflammation and angiogenesis. Int J Inflam 2011;2011:908468.
71. Asare Y, Schmitt M, Bernhagen J. The vascular biology of macrophage migration inhibitory factor (MIF). Expression and effects in inflammation, atherogenesis and angiogenesis. Thromb Haemost 2013;109:391–398.
72. Benest AV, Kruse K, Savant S, et al. Angiopoietin-2 is critical for cytokine-induced vascular leakage. PLoS One 2013;8: e70459.
73. Lu SL, Reh D, Li AG, et al. Overexpression of transforming growth factor beta1 in head and neck epithelia results in inflammation, angiogenesis, and epithelial hyperproliferation. Cancer Res 2004;64:4405–4410.
74. Berger AC, Tang G, Alexander HR, et al. Endothelial monocyte-activating polypeptide II, a tumor-derived cytokine that plays an important role in inflammation, apoptosis, and angiogenesis. J Immunother 2000;23:519–527.
75. Klein S, de Fougerolles AR, Blaikie P, et al. Alpha 5 beta 1 integrin activates an NF-kappa B-dependent program of gene expression important for angiogenesis and inflammation. Mol Cell Biol 2002;22:5912–5922.
76. Ono M. Molecular links between tumor angiogenesis and inflammation: inflammatory stimuli of macrophages and cancer cells as targets for therapeutic strategy. Cancer Sci 2008;99:1501–1506.
77. Ye YN, Liu ES, Shin VY, et al. Contributory role of 5-lipoxygenase and its association with angiogenesis in the promotion of inflammation-associated colonic tumorigenesis by cigarette smoking. Toxicology 2004;203:179–188.
78. Czabanka M, Korherr C, Brinkmann U, et al. Influence of TBK-1 on tumor angiogenesis and microvascular inflammation. Front Biosci 2008;13:7243–7249.
79. Solinas G, Marchesi F, Garlanda C, et al. Inflammation-mediated promotion of invasion and metastasis. Cancer Metastasis Rev 2010;29:243–248.
80. Affara NI, Coussens LM. IKKalpha at the crossroads of inflammation and metastasis. Cell 2007;129:25–26.
81. Kayser K, Bulzebruck H, Ebert W, et al. Local tumor inflammation, lymph node metastasis, and survival of operated bronchus carcinoma patients. J Natl Cancer Inst 1986;77:77–81.
82. Hobson J, Gummadidala P, Silverstrim B, et al. Acute inflammation induced by the biopsy of mouse mammary tumors promotes the development of metastasis. Breast Cancer Res Treat 2013;139:391–401.
83. Robinson-Smith TM, Isaacsohn I, Mercer CA, et al. Macrophages mediate inflammation-enhanced metastasis of ovarian tumors in mice. Cancer Res 2007;67:5708–5716.
84. Khatib AM, Auguste P, Fallavollita L, et al. Characterization of the host proinflammatory response to tumor cells during the initial stages of liver metastasis. Am J Pathol 2005;167:749–759.
85. McEver RP. Selectin-carbohydrate interactions during inflammation and metastasis. Glycoconj J 1997;14:585–591.
86. Geng JG, Chen M, Chou KC. P-selectin cell adhesion molecule in inflammation, thrombosis, cancer growth and metastasis. Curr Med Chem 2004;11:2153–2160.
87. Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006;124:263–266.
88. Siegel G, Malmsten M. The role of the endothelium in inflammation and tumor metastasis. Int J Microcirc Clin Exp 1997;17:257–272.
89. Wang W, Xu GL, Jia WD, et al. Ligation of TLR2 by versican: a link between inflammation and metastasis. Arch Med Res 2009;40:321–323.
90. Luo JL, Tan W, Ricono JM, et al. Nuclear cytokine-activated IKKalpha controls prostate cancer metastasis by repressing Maspin. Nature 2007;446:690–694.
91. Kim MP, Park SI, Kopetz S, et al. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res 2009;335:249–259.
92. Melnikova V, Bar-Eli M. Inflammation and melanoma growth and metastasis: the role of platelet-activating factor (PAF) and its receptor. Cancer Metastasis Rev 2007;26:359–371.
93. Jing Y, Han Z, Liu Y, et al. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS One 2012;7:e43272.
94. Jones PA, Baylin SB. The epigenomics of cancer. Cell 2007;128:683–692.
95. Esteller M. Aberrant DNA methylation as a cancer-inducing mechanism. Annu Rev Pharmacol Toxicol 2005;45:629–656.
96. Thiagalingam S, Cheng KH, Lee HJ, et al. Histone deacetylases: unique players in shaping the epigenetic histone code. Ann N Y Acad Sci 2003;983:84–100.
97. Li E, Beard C, Jaenisch R. Role for DNA methylation in genomic imprinting. Nature 1993;366:362–365.
98. Antequera F, Bird A. Number of CpG islands and genes in human and mouse. Proc Natl Acad Sci U S A 1993;90:11995–11999.
99. Yara S, Lavoie JC, Beaulieu JF, et al. Iron-ascorbate-mediated lipid peroxidation causes epigenetic changes in the antioxidant defense in intestinal epithelial cells: impact on inflammation. PLoS One 2013;8:e63456.
100. Uemura N, Okamoto S, Yamamoto S, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001;345:784–789.
101. Maekita T, Nakazawa K, Mihara M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res 2006;12:989–995.
102. Nakajima T, Maekita T, Oda I, et al. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev 2006;15:2317–2321.
103. Perri F, Cotugno R, Piepoli A, et al. Aberrant DNA methylation in non-neoplastic gastric mucosa of H. Pylori infected patients and effect of eradication. Am J Gastroenterol 2007;102:1361–1371.
104. Achyut BR, Bader DA, Robles AI, et al. Inflammation-mediated genetic and epigenetic alterations drive cancer development in the neighboring epithelium upon stromal abrogation of TGF-beta signaling. PLoS Genet 2013;9:e1003251.
105. Blanco D, Vicent S, Fraga MF, et al. Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway. Neoplasia 2007;9:840–852.
106. Iliopoulos D, Jaeger SA, Hirsch HA, et al. STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 2010;39:493–506.
107. Suzuki H, Toyota M, Kondo Y, et al. Inflammation-related aberrant patterns of DNA methylation: detection and role in epigenetic deregulation of cancer cell transcriptome. Methods Mol Biol 2009;512:55–69.
108. Valinluck V, Sowers LC. Inflammation-mediated cytosine damage: a mechanistic link between inflammation and the epigenetic alterations in human cancers. Cancer Res 2007;67:5583–5586.
109. Kroegel C, Antony VB. Immunobiology of pleural inflammation: potential implications for pathogenesis, diagnosis and therapy. Eur Respir J 1997;10:2411–2418.
110. Li QB, Sun XJ, Xu YZ, et al. Use of Fourier-transform infrared spectroscopy to rapidly diagnose gastric endoscopic biopsies. World J Gastroenterol 2005;11:3842–3845.
111. Difuccia B, Keith I, Teunissen B, et al. Diagnosis of prostatic inflammation: efficacy of needle biopsies versus tissue blocks. Urology 2005;65:445–448.
112. Vendrame E, Martinez-Maza O. Assessment of pre-diagnosis biomarkers of immune activation and inflammation: insights on the etiology of lymphoma. J Proteome Res 2011;10:113–119.
113. Baicus C, Caraiola S, Rimbas M, et al. Utility of routine hematological and inflammation parameters for the diagnosis of cancer in involuntary weight loss. J Investig Med 2011;59:951–955.
114. DePew ZS, Gonsalves WI, Roden AC, et al. Granulomatous inflammation detected by endobronchial ultrasound-guided transbronchial needle aspiration in patients with a concurrent diagnosis of cancer: a clinical conundrum. J Bronchology Interv Pulmonol 2012;19:176–181.
115. Jafri SH, Shi R, Mills G. Advance lung cancer inflammation index (ALI) at diagnosis is a prognostic marker in patients with metastatic non-small cell lung cancer (NSCLC): a retrospective review. BMC Cancer 2013;13:158.
116. Vogelstein B, Papadopoulos N, Velculescu VE, et al. Cancer genome landscapes. Science 2013;339:1546–1558.
117. Lippitz BE. Cytokine patterns in patients with cancer: a systematic review. Lancet Oncol 2013;14:e218–228.
118. Colditz GA, Wolin KY, Gehlert S. Applying what we know to accelerate cancer prevention. Sci Transl Med 2012;4(127):127rv4.