Jeffrey C. Buchsbaum, James G. Douglas, Barnali Dasgupta, and Brian D. Lawenda
Primary tumors of the retroperitoneum present in unselected series with a frequency of 0.1% to 0.2%.1–3 They are notable for their widely disparate histologies and presentations. Morgagni published the first report of a retroperitoneal lesion in 1761 after an autopsy.4 However, the most comprehensive work on retroperitoneal lesions was done in 1954 by Pack and Tabah.3 They added 120 cases from the Memorial Cancer Center experience to a survey of the world’s literature, which contained 750 prior reports. Recent series are histology specific (e.g., sarcomas). Older series, based on clinical presentations, are composed of a variety of tumor types and because of this continue to provide the most valuable data on presenting signs and symptoms. However, because of the tremendous variations in histologies, widely disparate therapeutic recommendations came about. Radiographic interpretation of these lesions has been aided by more recent data and ever improving technology. The focus on this chapter is on lesions that are primary to the retroperitoneum rather than metastatic to it.
FIGURE 75.1. Sagittal view of trunk showing the retroperitoneal space (shaded area). The kidney is outlined by dots. (From Wasserman TH, Tepper JE. Retroperitoneum. In: Perez CA, Brady LW, eds. Principles and practice of radiation oncology, 3rd ed. Philadelphia, PA: Lippincott-Raven, 1997:1943–1956.)

![]() ANATOMY
ANATOMY
The retroperitoneal space is the potential space posterior to the peritoneum or abdominopelvic cavity (Fig. 75.1). The superior border of this space is the diaphragm, and its inferior border is the superior aspect of the pelvic diaphragm (the levator ani and the coccygeus muscles). In terms of vertebral levels, the retroperitoneum stretches from the 12th thoracic vertebral body to the distal coccyx. Bilaterally, the border was classically considered to be at the lateral edge of the quadratus lumborum. Pack and Tabah3 consider the lateral extent of the 12th rib to be more valuable because this is the origin of the transversus abdominis aponeurosis as well as the mid-iliac crest. The muscles of the posterior abdominal wall, the psoas and quadratus lumborum muscles, form the posterior border of the retroperitoneum. The renal fascia forms a cone that helps to protect the pelvis from perirenal disease.5,6
Most of the tissue in this space consists of lymphatics and loose connective tissue. Retroperitoneal organs, either partially or fully contained, include urinary organs (adrenals, kidneys, ureters, and bladder), vascular organs (aorta and inferior vena cava), alimentary canal organs (esophagus and the upper two-thirds of the rectum), and many nerves. Organs that moved from within the peritoneum to outside of it during organogenesis include parts of the pancreas (head, neck, and body), the nonproximal duodenum, and the ascending and descending portions of the colon. The tail of the pancreas is within the splenorenal ligament and is thus intraperitoneal.
Nethercliffe et al.7 described the surgical anatomy of the retroperitoneum in their review of retroperitoneal surgical techniques. The most common surgical approaches described include the following: (a) subcostal (below the 12th rib), (b) supracostal (above the 12th rib), (c) transcostal (removing the 11th or 12th rib), and (d) thoracoabdominal.7
Laparoscopic and other minimally invasive techniques that cause less postoperative morbidity often defer these approaches.
FIGURE 75.2. Age at diagnosis of malignant retroperitoneal tumors (pooled data from multiple series).
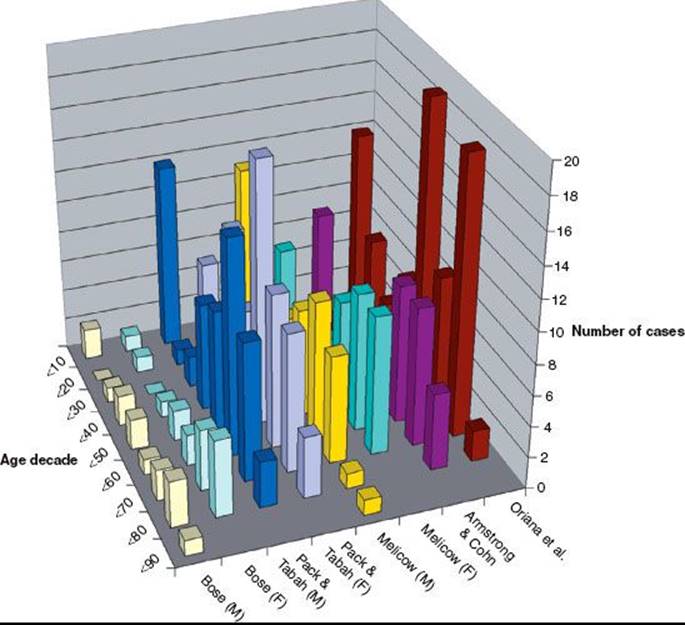
FIGURE 75.3. Percentage of retroperitoneal lesions by histology (pooled data from multiple published series).

![]() EPIDEMIOLOGY
EPIDEMIOLOGY
The population frequency for retroperitoneal tumors has been quoted as three per million persons in two population-based studies.1,2 The age range of patients varied between 3 and 83 years. Fifty percent of patients are diagnosed between 60 and 80 years of age. Males and females are equally likely to develop lesions of the retroperitoneum.2
Figure 75.2 graphs the age at diagnosis of several large series of patients with primary retroperitoneal tumors. There are two peak age periods. The first peak occurs during the first decade and is generally caused by neuroblastoma or germ cell tumors. The second peak occurs during the sixth decade of life and is generally caused by mesenchymal lesions. These histologies are outlined in Figure 75.3.
TABLE 75.1 PRESENTING COMPLAINTS (%) OF PATIENTS WITH RETROPERITONEAL LESIONS (POOLED FROM LITERATURE SOURCES)

TABLE 75.2 SURVEILLANCE, EPIDEMIOLOGY, AND END RESULTS DATA: RETROPERITONEAL MALIGNANCIES, 200216

![]() NATURAL HISTORY
NATURAL HISTORY
The natural history of these lesions varies by histology. Lesions of the peritoneum often present having achieved a substantial size owing to the anatomic properties of the region.
![]() CLINICAL PRESENTATION
CLINICAL PRESENTATION
Table 75.1 shows the summary of presenting symptoms and signs of tumors in the retroperitoneum at presentation.
![]() DIAGNOSTIC WORKUP
DIAGNOSTIC WORKUP
Melicow8 considered the ureter to be “the ‘weather vane’ of the retroperitoneum” because on intravenous urography it is often displaced by tumor. Hydroureter and hydronephrosis are relative late events caused by retroperitoneal pathology given that the ureter and kidney at baseline are highly mobile. Bose2 published that approximately 80% of these patients had such findings that were suggestive of a mass.
Two series demonstrated that the vascular bed is abnormal in patients with retroperitoneal masses. In the first of these series, which was made up of angiographic studies performed in 20 patients by Oriana et al.,9 aberrant circulation was noted in 75% and mass effect in 85%. All patients undergoing angiography reported by Bose2 had abnormal studies.
Neville and Herts10 have published a comprehensive review of computed tomography (CT) appearance of primary retroperitoneal lesions. CT can differentiate between well-differentiated and dedifferentiated retroperitoneal liposarcoma. A focal nodular/water density area has been found to be a very sensitive marker of dedifferentiated tumor (sensitivity, 97.8%). CT scan can identify most dedifferentiated tumors accurately.11 Liposarcomas frequently are large lesions with fatty components causing mass effect. Teratomas can have numerous types of tissue imaging characteristics, reflecting their capacity to be made up of differing stromal types. Nerve sheath tumors and other tumors of the nerves are commonly paraspinal. CT characteristics of retroperitoneal tumors have been used in pediatric patients as well.12
Magnetic resonance imaging (MRI) often demonstrates additional imaging details to further classify primary lesions of the retroperitoneum, as reported by two groups.13,14 CT-guided needle biopsy may be performed to obtain initial tissue for histologic diagnosis when an open or laparoscopic biopsy is not possible. Positron emission tomography (PET) scans can be helpful in evaluating the extent of distant metastases during the staging workup. Fluorodeoxyglucose (FDG)-PET can complement CT and MRI for detecting high- and intermediate-grade local recurrence of retroperitoneal soft tissue sarcoma (STS).15
![]() CONTEMPORARY POPULATION-BASED DEMOGRAPHICS
CONTEMPORARY POPULATION-BASED DEMOGRAPHICS
A comprehensive review of the Surveillance Epidemiology and End Results (SEER) public use registry focused on retroperitoneal tumors was performed by Lawenda and Johnstone.16 The SEER program is made up of 15 regions designed to collect information on cancer incidence, prevalence, and survival on a significant portion of the U.S. population. Table 75.2 shows the results of this review.
![]() GENERAL MANAGEMENT OF RETROPERITONEAL SARCOMA
GENERAL MANAGEMENT OF RETROPERITONEAL SARCOMA
Gross total resection is the primary treatment for retroperitoneal sarcoma (RPS) and is attempted when possible. Aggressive surgical management is the key for long-term survival for patients with retroperitoneal STS.17,18,19 Criteria for unresectable tumors may include spinal cord or major vessel involvement.20 Recent series report that the rate of resectability ranges from 65% to 85%.21–23 En bloc resection of the primary tumor has been suggested as necessary for tumor control in the retroperitoneum.23,24
Surgical resection is often classified by the final margin status or R status as follows:
R0—gross or macroscopically complete tumor resection with microscopically negative margins,
R1—gross or macroscopically complete tumor resection with microscopically positive margins, and
R2—incomplete or partial tumor resection with gross tumor residual.
Achieving an R1 or macroscopically negative margin is a significant prognostic factor in local control and survival; however, the significance of a microscopically positive margin is less clear.22,25 Even with an R0 resection, the long-term outcomes of tumors of the retroperitoneum are poor: 33% to 77% (local recurrence) and 35% to 63% (5-year overall survival).22,23,26 Local recurrence is the primary cause of death from RPS.23,24 Local disease in the absence of metastatic disease was associated with death in 75% of patients with primary RPS in one series.22
No prospective data exist suggesting that postoperative radiation after surgery is superior to surgery alone for local control; multiple retrospective series support adjuvant radiation therapy.23,26 The effect of adjuvant radiation on overall survival is less certain. Preoperative or postoperative radiation therapy can be employed. Other modalities such as hyperthermia have shown early promise and can be of use for increased local control.20
Postoperative radiation is felt to be more toxic than preoperative radiation therapy because the clinical target volume (CTV) may be larger and typical contains a larger amount of normal tissue. Preoperative radiation therapy may be less toxic because fields are smaller, there is less normal tissue in fields as the tumor displaces these tissues “out of the way,” the tumor may be more easily resectable owing to a “rind” forming, and oxygenation may be better with an intact vasculature.
Lower dose to the region and better delineation of tumor with preoperative treatment are perhaps the most critical reasons why the trend is moving toward preoperative chemotherapy in Europe and many academic centers in North America. The role of preoperative radiation therapy was studied in the American College of Surgeons Oncology Group (ACOSOG) protocol Z9031, a phase III prospective randomized study of preoperative radiation plus surgery versus surgery alone for patients with RPS. The study, which accrued 370 patients, closed in 2006. Results have not yet been published.
Children’s Oncology Group (COG) protocol ARST0332 allowed both pre- and postoperative radiation therapy as well as significant chemotherapy. It just recently closed at the time of this writing and contains significant cross sections of histologies. Most accrued cases on the study have been treated with preoperative radiation therapy and chemotherapy. It allowed enrollment for patients up to age 30 years with nonrhabdomyosarcoma STS histologies.
Completeness of resection, tumor volume, grade, and subtype are prognostic factors of RPS.27 Preoperative radiotherapy or chemotherapy may be employed in cases of unresectable tumor as a means to attempt to shrink the tumor and make it resectable.28 A 60% local control rate was reported in a series of unresectable RPS patients employing doses of radiation of at least 63 Gy by the Massachusetts General Hospital.29 Patients with locally extensive tumors that are not amenable to complete resection can be offered palliative debulking surgery to help control local symptoms.30
Following aggressive surgery, local recurrence is a key factor determining morbidity and mortality. Local recurrence growth rate >0.9 cm per month has been associated with a poorer outcome.31 The use of higher doses of external-beam radiation therapy (EBRT) or intraoperative radiation therapy (IORT) does not result in significant improvement in outcome in such cases.32 Locally recurrent RPS is potentially surgically salvageable, with longer disease-free interval to recurrence being associated with better prognosis.23 Patients who have not received prior radiation to the tumor bed should be considered for either pre- or postoperative radiation therapy. IORT has been reported to increase local control after resection in several series33–35; however, in a randomized prospective study at the National Institutes of Health (NIH), IORT was shown to not be significantly better than EBRT.36
There is no standard way to manage patients who present or recur with distant metastases. Guidelines presented by the National Comprehensive Cancer Network (NCCN) for sarcoma management (version 2.2011) suggest that the same approach used for localized sarcoma patients be employed to deal with the oligometastatic population.37 In the case of oligometastatic disease, chemotherapy may be considered. With disease that is extensively metastatic, palliative approaches are used so as to focus on quality of life.
A histology-based system identifies RPS into three prognostic groups and can be used in both primary and recurrent RPS. Distinct risk stratification is necessary for specific assessment of prognosis and decisions regarding individualized adjuvant therapy.38 Some authors39 recommend the formulation of a liposarcoma-specific postoperative nomogram based on histologic subtypes, which provides more accurate survival predictions for patients with primary RPS. Well-differentiated and dedifferentiated liposarcoma differ significantly in their biologic behavior and are treated by different surgical approaches at the University of Texas MD Anderson Cancer Center.40 Researchers at the center recommend that the American Joint Committee on Cancer (AJCC) STS staging system needs the incorporation of primary site, histologic subtype, margin status, and recurrence to shed light on prognosis.41 Tumor size was found to have no correlation with survival.42 Patients with dedifferentiated retroperitoneal STS carry the worst prognosis according to a small series from Italy.43
Assessing the response to therapy is challenging and limited to either pathologic review of biopsy or resection specimens, or to serial imaging studies (i.e., CT or MRI with contrast enhancement). Based on the findings of a recently published meta-analysis, FDG-PET imaging may not accurately reflect the response to radiation therapy.44
![]() CHEMOTHERAPY
CHEMOTHERAPY
Adjuvant chemotherapy is controversial in the management of adult patients with macroscopically completely resected retroperitoneal STS. In a National Cancer Institute trial, patients with STS were randomized to chemotherapy or observation following resection; some patients had postoperative irradiation.45 Among patients who were assigned to chemotherapy, survival was favorably affected in those with extremity tumors. However, patients with head and neck and truncal lesions (including RPS) did not benefit; the 5-year survival rate was approximately 40% in both arms. Similar findings were reported in a large meta-analysis of patients with STS.46
The use of neoadjuvant or adjuvant chemotherapy is not standard in the management of nonmetastatic adult retroperitoneal STS. Neoadjuvant chemotherapy has been primarily studied in the setting of high-grade extremity STS. Pisters et al.47 demonstrated feasibility of using preoperative concurrent doxorubicin and EBRT (18 to 50.4 Gy) followed by resection and IORT (15 Gy) in patients with RPS. An R0 or R1 resection was possible in 90% of the patients who went to surgery (83%). Despite these promising results, concurrent neoadjuvant chemoradiotherapy is not recommended outside clinical protocols.48
![]() RADIATION THERAPY TECHNIQUES
RADIATION THERAPY TECHNIQUES
Three-dimensional conformal radiation therapy (3DCRT) planning is preferable to conventional planning techniques to more clearly define target and nontarget tissues and to optimize field arrangements. Intensity-modulated radiation therapy and helical tomotherapy, proton beam therapy, and IORT have reported benefits over standard 3DCRT in enhancing the delivery of higher radiation doses to the target volume while minimizing doses to normal tissues.33–35,49–51
We recommend preoperative radiation therapy for the reasons outlined previously. This usually requires that an initial procedure be done (CT-guided core needle biopsy, if adequate tissue can be obtained, or a small operative procedure) to obtain tissue for histologic study. Obtaining a contrast-enhanced CT scan or MRI prior to simulation will help to more clearly define tumor and normal tissues for planning purposes. The use of [F-18]FDG-PET in tumor localization has been reported and may differentiate tumor versus surrounding normal tissues.52 Hybrid FDG-PET/CT is becoming increasingly popular for diagnosis and monitoring of treatment for RPS.52 Most cases of RPS will require irradiating the ipsilateral kidney, except in the case of proton therapy, where some kidney may be spared on a case-by-case basis. Therefore, a renal perfusion study should be ordered as part of the initial radiation planning process to ascertain the degree of functionality of each kidney.
Target volumes are defined as follows (preoperative volumes as per ACOSOG Z9031 as an example):
1. The gross tumor volume (GTV) is the visualized GTV based on preradiotherapy imaging with CT and/or MRI. PET/CT can also be useful.
2. The CTV is defined as the tissues adjacent to the GTV that have a potential for microscopic disease and are not visible on radiographic imaging. The CTV should be contoured at least 1.5 cm outside the GTV but can be less in areas where there is minimal risk of direct invasion (i.e., peritoneum, bone, muscle).
3. The planning target volume (PTV) is an expansion volume outside the CTV that accounts for setup error and patient/organ movement.
This variable is institution dependent as well as patient dependent. A minimum of 0.5 cm expansion outside the CTV should be used. Infraction organ motion of retroperitoneal structures can be significant—that is, average movement between 11 and 19 mm (kidneys, normal breathing) and 18 and 22 mm (pancreas, normal breathing).53 Stereotactic ultrasound-based image-guided targeting, cone-beam CT imaging, active breathing control, and respiratory gating may be useful modalities in decreasing the required PTV expansion owing to organ motion. Four-dimensional (4D) CT simulation may be of value in helping to define a GTV during the breath cycle54 but his needs to be better evaluated on clinical trials as it has been used primarily in the lung.55
Critical normal structures should be contoured (i.e., liver, kidneys, spinal cord, bowel, stomach) and dose-volume histograms calculated. The spinal cord dose should be limited to 45 Gy in standard fractionation over 5 weeks. The current ACOSOG Z9031 trial limits liver doses as follows: no more than 20% of the liver volume should receive >50 Gy, and no more than 50% should receive >25 Gy. At least two-thirds of the volume of one functioning kidney should receive <20 Gy. Stomach and bowel should be limited to a maximum dose of 45 Gy. Volume expansions should be minimized in regions where tolerance doses to critical structures will be reached.
In photon-based therapy, conventional anteroposterior/posteroanterior or slightly oblique fields often provide the best target coverage with the least amount of normal tissue in the beam. The use of lateral fields can result in irradiating a substantial volume of normal tissue (i.e., liver and kidneys) and should be used sparingly.
Preoperative radiation doses of 45 to 50 Gy (in 1.8 to 2 Gy per fraction per day) are recommended. Surgical resection usually is usually delayed until 3 to 8 weeks after completion of the radiation therapy. Radiographic restaging is typically done prior to resection to assess for metastases and response to therapy.
IORT with either brachytherapy or EBRT may be used as a focal boost to deliver additional radiation to areas of concern (i.e., close or positive margins). The addition of an IORT boost has been shown to increase local control rates over resection and EBRT alone.34,56 Although not always possible, care should be taken to avoid including critical structures from the boost (i.e., bowel, ureters, nerves). A postoperative external-beam boost can also be delivered to an area of close or positive margin. We recommend asking the surgeon to place radiopaque markers (i.e., clips, metallic seeds) at the borders of the resection cavity and in the areas where the margin may be close or positive. This will be helpful in defining these areas later if a postoperative boost is given. The boost dose is typically 10 to 15 Gy and is delivered in either a single intraoperative dose (electron beam or brachytherapy) or in a once-daily, fractionated regimen (1.8 to 2 Gy per fraction) postoperatively. Both low and high dose rate brachytherapy have similar efficacy in terms of disease control when used alone or in combination with EBRT. Brachytherapy results in fewer complications compared with combination of brachytherapy and EBRT.57
High dose rate intraoperative brachytherapy has been described in the treatment for primary and locally recurrent RPS. A Harrison-Anderson-Mick applicator (an array of catheters spaced 1 cm apart in a silicone rubber pad), or similar device, can be used to deliver an intraoperative dose of 12 to 15 Gy to the tumor bed, using iridium-192 (192Ir) sources.33 Adjuvant interstitial postoperative brachytherapy provides local control of tumor in both low- and high-grade STS.58
Postoperative radiation therapy is recommended for patients who initially present after resection. These fields frequently are more extensive than preoperative fields; thus, a larger volume of normal tissue is usually included. Normal tissues (i.e., stomach and bowel) that were previously displaced by the tumor mass will subsequently fill the resection cavity after the tumor has been removed, increasing the toxicity of the treatment and limiting the dose that can be delivered. Although not commonly employed, silicone-filled implants have been used to displace bowel and other tissues out of the radiation field.33 Postoperative radiation fields should include the preoperative GTV (as defined on the preoperative CT or MRI) and the entire resection cavity. Defining the tumor bed can be challenging; therefore, the radiation oncologist generally needs to err on the side of treatment that affects more, rather than less, normal tissue in cases that are uncertain. Residual disease may be visible on postoperative scans; however, surgical clips placed at the time of the resection will remove much of the uncertainty of defining these areas. Boost fields can encompass areas of close margins or residual disease. Expansion volumes (CTV and PTV) should be limited in regions where the tumor did not violate fascial or peritoneal boundaries. A postoperative radiation dose of 45 Gy (1.8 Gy per fraction per day) is recommended. Limited boost fields (5.4 to 9 Gy per fraction) can be planned, although careful attention must be paid to the surrounding normal tissue tolerances. Particle therapy may be advantageous in terms of decreasing the integral dose to the abdomen and pelvis and is under study.51,59
![]() RESULTS OF THERAPY
RESULTS OF THERAPY
Despite poor local control rates with resection alone, complete surgical resection remains the only curative treatment modality in patients with RPS.25 In patients with nonmetastatic, completely excised RPS, 5-year overall survival is 49% to 70%24,25,60 (Table 75.3). Failure to achieve local control of disease is the major cause of death in patients with RPS.21 In a large, single-institution report of RPS, patients who successfully underwent a gross total resection (n = 185) had a median survival of 103 months versus 18 months (n = 46) for those who underwent an incomplete resection.21 Similarly, Stoeckle et al.22 published actuarial 5-year overall survival rates of 62% versus 26% for patients who had a complete (n = 94) versus incomplete (n = 50) excision, respectively.
TABLE 75.3 COMPLETE RESECTION AND SURVIVAL IN PATIENTS WITH NONMETASTATIC (M0) RETROPERITONEAL SARCOMA AND SURVIVAL

TABLE 75.4 LOCAL RECURRENCE IN PATIENTS WITH OR WITHOUT POSTOPERATIVE RADIATION THERAPY FOLLOWING A COMPLETE RESECTION

Retrospective studies demonstrate an improvement in local control with postoperative radiation therapy (Table 75.4). In the report by Stoeckle et al.,22 patients with complete excision had a significant reduction in local recurrence risk when they received postoperative radiation therapy (median dose 50 Gy) than when they did not (relative risk [RR] 3.36; p = .0002); there was no improvement in overall survival in multivariate analysis. Catton et al.25 found that adjuvant radiation therapy increased the time to locoregional relapse from 30 months (no radiation) to 103 months (p = .06). Similar to the trial by Stoeckle et al.,22 radiation did not exert an effect on survival.
The optimal dose for treating RPS after resection is not known. Fein et al.61 reported improved local control rates with adjuvant radiation doses >55 Gy (using shrinking photon fields and/or an IORT boost): 25% local failure with doses >55 Gy versus 38% local failure with doses <55 Gy. The National Cancer Institute demonstrated higher locoregional control in patients who underwent a gross total resection followed by IORT (20 Gy) and EBRT (35 to 40 Gy) compared to postoperative EBRT alone (50 to 55 Gy) of 60% versus 20%, respectively56 (Table 75.5). Alektiar et al.33 reported higher local control rates in patients who received IORT (12 to 15 Gy) and postoperative EBRT (45 to 50.4 Gy) compared to those who received IORT alone of 66% versus 50%, respectively.
Prospective trials have shown that intermediate- or high-grade RPS patients treated with preoperative radiotherapy and complete resection had a median survival >60 months.62 In trials of preoperative radiation therapy, researchers from the Princess Margaret Hospital published results using preoperative EBRT (median dose 45 Gy) followed by resection and postoperative brachytherapy implant (low dose rate, median dose 25 Gy).63 Local recurrence was 19.6% and overall survival at 2 years was 88% in the 46 patients resected with curative intent. In a Massachusetts General Hospital series, 35 patients received preoperative EBRT (45 to 50 Gy) followed by resection (79% complete resections, 11% partial resections, and 5% unresectable) and IORT (n = 20/37) using 9- to 15-MeV electrons: 10 Gy (complete resection), 12.5 to 15 Gy (microscopically involved margins), and 15 to 20 Gy for macroscopic residual disease.34 In those patients who underwent a complete resection, there was a nonsignificant improvement for local control if they received IORT (83% vs. 61%; p = .2). Petersen et al.35 reported the Mayo Clinic experience of primary and recurrent RPS or intrapelvic STS treated with preoperative EBRT (n = 53), postoperative EBRT (n = 12), or both (n = 12); the median EBRT dose was 47.6 Gy. IORT (median dose 15 Gy, electron beam) was also employed. Local control at 5 years for patients with gross residual disease (n = 15) was 41%; for those with microscopic residual disease (n = 56), it was 60%; and for those with complete resections (n = 16), it was 100%. The 5-year overall survival rates were 37% (in patients with gross residual disease) and 52% (in patients with microscopic and no residual disease).
TABLE 75.5 OUTCOMES WITH EXTERNAL-BEAM RADIOTHERAPY AND INTRAOPERATIVE RADIOTHERAPY BOOST FOLLOWING RESECTION OF PRIMARY AND RECURRENT RETROPERITONEAL SARCOMA

![]() TREATMENT SEQUELAE
TREATMENT SEQUELAE
The most common acute symptoms from EBRT of tumors in the retroperitoneum are nausea, vomiting, diarrhea, skin redness, and fatigue. Anemia, neutropenia, and thrombocytopenia may occur, especially with large radiation fields that often involve the adjacent spine. Weekly monitoring of the patient’s complete blood count and daily vital signs should be performed. Reported postoperative complications include bleeding, impaired wound healing or dehiscence, infection, myocardial infarction, and death.35,63
The major long-term sequelae of surgery and radiation are small bowel enteritis, stricture, perforation, fistula, and obstruction. Development of nephritis is possible after radiation doses >30 Gy, with resultant hypertension. Late complications are associated with the number of laparotomies to which the patient has been subjected and to the radiation dose and volume.29 A lower incidence of enteritis has been reported with the use of EBRT and an IORT boost compared with EBRT alone, as the bowel is subjected to lower radiation doses when it is able to retracted out of the field56 (Table 75.5). One must pay careful attention to potential areas of overlap when using abutting IORT boost fields to decrease the risk of neuropathy and ureteral injury.34,35,63 Studies of preoperative radiation therapy have demonstrated that this approach is well tolerated and appears to be less toxic than postoperative radiation therapy.34,35,63 As mentioned previously, the role of preoperative radiation therapy is the focus of a current phase III study (ACOSOG Z9031). Proton therapy may allow decreased sequelae in that less bowel anterior to the retroperitoneum is in the treatment field.51
![]() LYMPHOMAS
LYMPHOMAS
Lymphomas are the most frequent malignant tumors of the retroperitoneum, with non-Hodgkin lymphoma the predominant histologic variant accounting for approximately 94%. CT, MRI, and PET imaging are used in the staging workup of these tumors. CT, ultrasound, or MRI guided-needle biopsy is preferable to fine-needle aspiration for diagnostic purposes of the retroperitoneal mass if no other superficial lymph nodes are involved.64,65 Flow cytometry and immunohistochemical stains are used to confirm the diagnosis of lymphoma. The diagnosis, staging, and management of retroperitoneal lymphoma are similar to that of other lymphoma sites and are discussed elsewhere. As with all retroperitoneal tumors, radiation doses and fields are often limited by surrounding normal tissues.
![]() OTHER LESIONS
OTHER LESIONS
Neuroblastoma is the most common non–central nervous system solid tumor in children and the most common malignancy in infancy. In the United States, approximately 650 new cases are diagnosed per annum with an incidence of 0.9 per 100,000 population. Ninety-eight percent are diagnosed in children <10 years of age, with almost half (40.1%) in infants <1 year of age.66 This tumor is extraordinarily uncommon in adults, with an estimated incidence of 0.2 per million population in those aged 30 to 39 years.67 The paradigm for neuroblastoma treatment includes combination therapy with chemotherapy, surgery, and radiotherapy in advanced-staged disease, which is most commonly found in adults.68,69
Wilms tumor (WT), or nephroblastoma, is the most common intra-abdominal malignancy of childhood, with an incidence of eight per million population <16 years of age. It is the second most common extracranial solid neoplasm of childhood and the most common renal malignancy in children.70 In contrast, it is an uncommon malignancy in patients ≥16 years of age, with an estimated incidence of 0.2 per million population.71,72 Adults frequently present with flank pain, whereas children are more likely to present with a painless hematuria and/or abdominal distention.66 The treatment and outcomes of patients with adult Wilms Tumor (AWT) remain controversial. Kalapurakal et al.73reported the outcomes of 23 adults (>16 years of age) treated on COG protocols (National Wilms Tumor Study Group; NWTS 4 and 5) and found no difference between the adults and their pediatric counterparts.73 Similarly, the International Society of Paediatric Oncology (SIOP) published results from an SIOP retrospective review of 30 AWT patients >16 years of age, with comparable results between adults and children.74,75 Conversely Izawa et al.76reported on 128 AWT patients from SIOP, as well as individual institutional reports, and concluded that the outcomes in adults were inferior to that of children. Recently, Ali et al.,77,78 using the U.S. SEER database, concluded that the outcome in 152 AWT patients was worse than in children. Overall survival was 69% in AWT versus 88% in children (p <.001). By multivariate analysis, adult status (≥16 years), SEER stage, treatment era (before 1990), and lack of surgical staging of lymph nodes were significant prognostic factors in this cohort. A consensus statement by an international group of childhood renal experts has been recently published that recommends adoption of the pediatric paradigm for treatment for AWT patients, including surgical resection, chemotherapy agents depending on stage and histologic subtype, and local therapy with irradiation depending on the stage of disease.79 Additionally, the current COG WT studies have revised the inclusion age criteria upward to age 30 years. The toxicities of treatment (severe acute neuropathy, grade 4 hematologic toxicity) in AWT patients may be somewhat higher than in the pediatric population74 but are thought to be reasonable in view of the high response rates.
Retroperitoneal schwannomas are a rare tumor, accounting for approximately 4% of retroperitoneal tumors, and the most common benign tumor found is this location.80,81 They belong in the family of peripheral nerve sheath tumors, which in addition to schwannomas includes neurofibromas, solitary circumcised neuromas, and perineuriomas.82 Schwannomas may occur in any nerve trunk in the body, with exception of cranial nerves 1 and 2 (which are not covered by Schwann cells), and are most commonly found at peripheral nerve sites of the upper extremities and cranial nerves; only 0.3% to 3.2% of all schwannomas occur in the retroperitoneum. Benign schwannomas are associated with Von Recklinghausen’s disease or neurofibromatosis 1 (NF-1) in approximately 5% to 18% of NF-1 patients, presenting most commonly between 20 and 50 years of age.83 They are slow-growing nonaggressive tumors that displace rather than invade normal tissues80,82 and often form large, well-circumscribed masses. They may display cystic degeneration, calcification, hemorrhage, and hyalinization on imaging studies. In several moderate to large series, there appears to be a slight female patient propensity, and the tumors often are diagnosed incidentally during radiologic examinations for unrelated symptoms.84,85 Boney changes may occur in perispinal tumors, and invasion into nerve roots or the spinal canal may lead to neurologic symptoms including paresthesias, weakness, and pain. MRI examinations are the preferred method for imaging these soft tissue neoplasms, although CT scanning may be necessary to better visualize potential boney abnormalities, particularly in the spine. Because these tumors are frequently quite vascular, many authors do not recommend the use of CT-guided biopsies because of the risk of hemorrhage. Whenever feasible, complete surgical resection with negative margins is the treatment of choice, although adjuvant radiation therapy may be necessary for incompletely excised sacral schwannomas and malignant peripheral nerve sheath tumors (MPNST).86 NF-1 patients have a particularly high risk of developing STS, particularly MPNST, often with an aggressive clinical presentation and poor outcome.87
Aggressive fibromatosis, also referred to as desmoid tumors, is a rare fibroblastic neoplasm that arises from deep musculoaponeurotic connective tissue and has an incidence of two to four cases per million population, occurring either sporadically or associated with familial adenomatous polyposis (FAP).88–92 Ten to 30% of patients with FAP eventually develop desmoid tumors, most occurring either in the extremities or abdomen/retroperitoneum (De Carmago et al.,93 Meazza et al.94,95). They occur more frequently in fertile women than in men (1.5 to 2.5:1). Although benign in nature, they are locally aggressive and may infiltrate critical structures. Complete surgical resection is the mainstay of treatment, although local recurrence is common even in the setting of a wide local resection in up to 50% of patients. Postoperative radiotherapy and/or chemotherapy may be indicated when a complete surgical resection is not achieved or not feasible or in multiply recurrent disease. The data is variable regarding the dose and treatment volumes; however, most reported series suggest that a dose of 50 to 60 Gy with margins of 5 to 7 cm is appropriate therapy. Doses >56 Gy may not be necessary to control gross disease.96 A recent meta-analysis that included data from 22 studies suggested that local control is improved with adjuvant radiation therapy compared to surgery alone; local control after surgery alone was 72% (R0) and 41% (R1 and R2) compared to 94% (R0) and 75% (R1 and R2) after surgery and adjuvant radiation.97 In patients with unresectable disease, radiation therapy alone is effective in providing long-term local control in up to 80% of patients.97,98 Chemotherapy or antiestrogen therapy may also play a role in nonresectable, incompletely resected, or recurrent disease. Regimens containing anthracyclines and antiestrogens appear to be the most effective chemotherapeutic options,93 although more recently, the tyrosine kinase inhibitor imatinib has shown activity in desmoid tumors.99,100
TABLE 75.6 THE INTERNATIONAL GERM CELL CANCER COLLABORATIVE GROUP CLASSIFICATION SYSTEM FOR ASSESSING PROGNOSIS IN NSGCT AND SGCT103

FIGURE 75.4. Proton treatment plan for retroperitoneal tumor in a young adult patient. Because of concerns specific to the case, the bowel and retroperitoneal region as shown was treated to 36 Gy. The retroperitoneal space plus a small margin was then treated to 54 Gy. Concurrent high-dose chemotherapy was employed.

Extragonadal germ cell tumors (EGCTs) account for approximately 1% to 5% of all germ cell tumors and occur in the retroperitoneum as the second most common extragonadal site in adults after the mediastinum.101 Primary retroperitoneal germ cell tumors account for approximately 10% of all primary malignant retroperitoneal tumors in adults and about 30% to 40% of all EGCTs. It is believed that these tumors arise from primordial germ cells, which are displaced during their migration along the urogenital ridge to the gonads.101 When they occur in the retroperitoneum, they are considered to be metastases from an occult or “burned out” gonadal primary until proven otherwise. EGCTs are most typically found in children or young adults and mostly arise in midline locations. Most of these tumors occur in young men. In young men (in whom most of these tumors occur), histologically, seminomatous germ cell tumors (SGCTs) comprise approximately 30% to 40%, whereas the remaining tumors are nonseminomatous germ cell tumors (NSGCTs). In young women, the histologic varieties are dysgerminomas and nondysgerminomas. NSGCTs include the following histologies: teratoma, embryonal carcinoma, endodermal sinus tumor (yolk sac tumor), and choriocarcinoma, as well as mixed histologies. Any proportion of nonseminomatous components is enough to classify a tumor as an NSGCT. NSGCTs generally have a much more aggressive course than do SGCTs. Serum markers, although nonspecific, may help to categorize the histology and can be useful for following both response to therapy and the presence of recurrence. Beta human chorionic gonadotropin (β-hCG) is elevated in choriocarcinoma and embryonal carcinoma, as well as in approximately 10% to 15% of SGCTs. Serum α-fetoprotein is elevated in endodermal sinus tumors and embryonal carcinomas. These tumors often present as large masses in the retroperitoneum and frequently displace, compress, or encase abdominal vessels. There are no distinguishing features by imaging to differentiate germ cell tumors from other retroperitoneal masses; thus, biopsy by ultrasound, CT, or MRI guidance is warranted. Gonadal primaries must be ruled out using high-resolution ultrasound. A biopsy of the gonads appears to be unnecessary with a negative ultrasound. Common sites of metastases are liver, bone, brain, and lungs. The prognosis of retroperitoneal SGCTs and their mediastinal counterparts are similar; however, retroperitoneal NSGCTs actually have a better prognosis than those occurring in the mediastinum.102
The treatment for EGCTs has evolved over the past 10 to 15 years from the use of primary radiotherapy for SGCTs to the more commonly recommended treatment using platinum-based chemotherapy regimens.101 Treatment paradigms for early-stage SGCT and NSGCT are covered elsewhere in this book. The International Germ Cell Cancer Collaborative Group established a classification system for assessing prognosis in NSGCT and SGCT103 (Table 75.6), which is used for treatment decisions. Some controversy remains as to the management of residual masses after three to four cycles of platinum-based chemotherapy in more advanced-stage disease or bulky retroperitoneal disease after chemotherapy or radiotherapy alone. Some authors favor surgical resection for residual masses >3 cm, particularly for NSGCT, whereas others favor observation alone. Subsequent chemotherapy is recommended for patients with viable SGCT on resection of any residual masses if radiotherapy was the sole treatment modality.104
![]() CONCLUSIONS
CONCLUSIONS
The retroperitoneum presents the clinician with a huge variety of histologies in a complex anatomic space. Treatment is driven by the histology in general. Newer technology will play a critical role in delivering dose with fewer side effects or perhaps allowing dose escalation with the same late effects. Image-guided therapy and intensity-modulated radiotherapy are mainstays in the treatment to the retroperitoneum when normal tissue toxicity is a concern. Particle therapy can now deliver dose to large tumor volumes while sparing normal tissue via gantry systems in place in many centers, as shown in Figure 75.4. New agents will continue to come forward that will allow more individualized treatment based on histology and genetic profiling. Despite our newest radiation and chemotherapeutic technology, complete surgical resection remains the backbone of successful therapy for these lesions.105 The rarity and poor results associated with these tumors points to a need to conduct prospective, multi-institutional trials.
![]() REFERENCES
REFERENCES
1. Armstrong JR, Cohn I Jr. Primary malignant tumors of the retroperitoneum. Nebr State Med J 1965;50(10):520–524.
2. Bose B. Primary malignant retroperitoneal tumours: analysis of 30 cases. Can J Surg 1979;22(3):215–220.
3. Pack GT, Tabah EJ. Primary retroperitoneal tumors: a study of 120 cases. Int Abstr Surg 1954;99(4):313–341.
4. Pemberton JD, Whitlock M. Large retroperitoneal lipoma. Surg Clin North Am 1934;14:601.
5. Raptopoulos V, et al. Why perirenal disease does not extend into the pelvis: the importance of closure of the cone of the renal fasciae. AJR Am J Roentgenol 1995;164(5):1179–1184.
6. Raptopoulos V, et al. Medial border of the perirenal space: CT and anatomic correlation. Radiology 1997;205(3):777–784.
7. Nethercliffe J, et al. Retroperitoneal and transthoracic anatomy and surgical approaches. BJU Int 2004;94(5):705–718.
8. Melicow MM. Primary tumors of the retroperitoneum; a clinicopathologic analysis of 162 cases; review of the literature and tables of classification. J Int Coll Surg 1953;19(4):401–449.
9. Oriana S, Bonardi P, Preda F. Primary retroperitoneal tumors. Tumori 1977;63(4):397–405.
10. Neville A, Herts BR. CT characteristics of primary retroperitoneal neoplasms. Crit Rev Comput Tomogr 2004;45(4):247–270.
11. Lahat G, et al. Computed tomography scan-driven selection of treatment for retroperitoneal liposarcoma histologic subtypes. Cancer 2009;115(5):1081–1090.
12. Xu Y, et al. CT characteristics of primary retroperitoneal neoplasms in children. Eur J Radiol 2010;75(3):321–328.
13. Song T, et al. Retroperitoneal liposarcoma: MR characteristics and pathological correlative analysis. Abdom Imaging 2007;32(5):668–674.
14. Crema MD, et al. [MR imaging of large and rare pelvic masses from non-gynecological etiology]. J Radiol 2008;89(7–8 Pt 1):853–861.
15. Schwarzbach MH, et al. Clinical value of [18-F] fluorodeoxyglucose positron emission tomography imaging in soft tissue sarcomas. Ann Surg 2000;231(3):380–386.
16. Ries LAG, Eisner MP, Kosary CL, et al., eds. SEER cancer statistics review, 1975–2002. Bethesda, MD: National Cancer Institute. Available at: http://seer.cancer.gov/csr/1975_2002/, based on November 2004 SEER data submission, posted to the SEER web site 2005.
17. Anaya DA, et al. Postoperative nomogram for survival of patients with retroperitoneal sarcoma treated with curative intent. Ann Oncol 2010;21(2):397–402.
18. Pacelli F, et al. Retroperitoneal soft tissue sarcoma: prognostic factors and therapeutic approaches. Tumori 2008;94(4):497–504.
19. Mendenhall WM, et al. The management of adult soft tissue sarcomas. Am J Clin Oncol 2009;32(4):436–442.
20. Schwarzbach MH, Hohenberger P. Current concepts in the management of retroperitoneal soft tissue sarcoma. Recent Results Cancer Res 2009;179:301–319.
21. Hassan I, et al. Operative management of primary retroperitoneal sarcomas: a reappraisal of an institutional experience. Ann Surg 2004;239(2):244–250.
22. Lewis JJ, et al. Retroperitoneal soft-tissue sarcoma: analysis of 500 patients treated and followed at a single institution. Ann Surg 1998;228(3):355–365.
23. Stoeckle E, et al. Prognostic factors in retroperitoneal sarcoma: a multivariate analysis of a series of 165 patients of the French Cancer Center Federation Sarcoma Group. Cancer 2001;92(2):359–368.
24. Gronchi A, et al. Retroperitoneal soft tissue sarcomas: patterns of recurrence in 167 patients treated at a single institution. Cancer 2004;100(11):2448–2455.
25. Singer S, et al. Histologic subtype and margin of resection predict pattern of recurrence and survival for retroperitoneal liposarcoma. Ann Surg 2003;238(3):358–370; discussion 370–371.
26. Catton CN, et al. Outcome and prognosis in retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys 1994;29(5):1005–1010.
27. Chen CQ, et al. Prognostic factors of retroperitoneal soft tissue sarcomas: analysis of 132 cases. Chin Med J (Engl) 2007;120(12):1047–1050.
28. Meric F, et al. Impact of neoadjuvant chemotherapy on postoperative morbidity in soft tissue sarcomas. J Clin Oncol 2000;18(19):3378–3383.
29. Kepka L, et al. Results of radiation therapy for unresected soft-tissue sarcomas. Int J Radiat Oncol Biol Phys 2005;63(3):852–859.
30. Shibata D, et al. Is there a role for incomplete resection in the management of retroperitoneal liposarcomas? J Am Coll Surg 2001;193(4):373–379.
31. Park JO, et al. Predicting outcome by growth rate of locally recurrent retroperitoneal liposarcoma: the one centimeter per month rule. Ann Surg 2009;250(6):977–982.
32. Ballo MT, et al. Retroperitoneal soft tissue sarcoma: an analysis of radiation and surgical treatment. Int J Radiat Oncol Biol Phys 2007;67(1):158–163.
33. Alektiar KM, et al. High-dose-rate intraoperative radiation therapy (HDR-IORT) for retroperitoneal sarcomas. Int J Radiat Oncol Biol Phys 2000;47(1):157–163.
34. Gieschen HL, et al. Long-term results of intraoperative electron beam radiotherapy for primary and recurrent retroperitoneal soft tissue sarcoma. Int J Radiat Oncol Biol Phys 2001;50(1):127–131.
35. Petersen IA, et al. Use of intraoperative electron beam radiotherapy in the management of retroperitoneal soft tissue sarcomas. Int J Radiat Oncol Biol Phys 2002;52(2):469–475.
36. Kinsella TJ, et al. Preliminary results of a randomized study of adjuvant radiation therapy in resectable adult retroperitoneal soft tissue sarcomas. J Clin Oncol 1988;6(1):18–25.
37. National Comprehensive Cancer Network. NCCN practice guidelines in oncology, Version 2.2011. Available at: http://www.nccn.org/professionals/physician_gls/pdf/sarcoma.pdf.
38. Anaya DA, et al. Establishing prognosis in retroperitoneal sarcoma: a new histology-based paradigm. Ann Surg Oncol 2009;16(3):667–675.
39. Dalal KM, et al. Subtype specific prognostic nomogram for patients with primary liposarcoma of the retroperitoneum, extremity, or trunk. Ann Surg 2006;244(3):381–391.
40. Lahat G, et al. Resectable well-differentiated versus dedifferentiated liposarcomas: two different diseases possibly requiring different treatment approaches. Ann Surg Oncol 2008;15(6):1585–1593.
41. Lahat G, et al. New perspectives for staging and prognosis in soft tissue sarcoma. Ann Surg Oncol 2008;15(10):2739–2748.
42. Nathan H, et al. Predictors of survival after resection of retroperitoneal sarcoma: a population-based analysis and critical appraisal of the AJCC staging system. Ann Surg 2009;250(6):970–976.
43. Mussi C, et al. The prognostic impact of dedifferentiation in retroperitoneal liposarcoma: a series of surgically treated patients at a single institution. Cancer 2008;113(7):1657–1665.
44. Bastiaannet E, et al. The value of FDG-PET in the detection, grading and response to therapy of soft tissue and bone sarcomas; a systematic review and meta-analysis. Cancer Treat Rev 2004;30(1):83–101.
45. Rosenberg SA. Prospective randomized trials demonstrating the efficacy of adjuvant chemotherapy in adult patients with soft tissue sarcomas. Cancer Treat Rep 1984;68(9):1067–1078.
46. Adjuvant chemotherapy for localised resectable soft-tissue sarcoma of adults: meta-analysis of individual data. Sarcoma Meta-analysis Collaboration. Lancet 1997;350(9092):1647–1654.
47. Pisters PW, et al. Phase I trial of preoperative concurrent doxorubicin and radiation therapy, surgical resection, and intraoperative electron-beam radiation therapy for patients with localized retroperitoneal sarcoma. J Clin Oncol2003;21(16):3092–3097.
48. Adjuvant chemotherapy for localised resectable soft tissue sarcoma in adults. Cochrane Database Syst Rev 2000(4):CD001419.
49. DeLaney TF, et al. Advanced-technology radiation therapy in the management of bone and soft tissue sarcomas. Cancer Control 2005;12(1):27–35.
50. Musat E, et al. [Comparison of intensity-modulated postoperative radiotherapy with conventional postoperative conformal radiotherapy for retroperitoneal sarcoma]. Cancer Radiother 2004;8(4):255–261.
51. Hug EB, et al. Conformal proton radiation treatment for retroperitoneal neuroblastoma: introduction of a novel technique. Med Pediatr Oncol 2001;37(1):36–41.
52. Schramm N, et al. [Combined functional and morphological imaging of sarcomas: significance for diagnostics and therapy monitoring]. Radiologe 2010;50(4):339–348.
53. Langen KM, Jones DT. Organ motion and its management. Int J Radiat Oncol Biol Phys 2001;50(1):265–278.
54. Eom J, et al. Modeling respiratory motion for cancer radiation therapy based on patient-specific 4DCT data. Med Image Comput Comput Assist Interv 2009;12(Pt 2):348–355.
55. Liao ZX, et al. Influence of technologic advances on outcomes in patients with unresectable, locally advanced non-small-cell lung cancer receiving concomitant chemoradiotherapy. Int J Radiat Oncol Biol Phys 2010;76(3):775–781.
56. Sindelar WF, et al. Intraoperative radiotherapy in retroperitoneal sarcomas. Final results of a prospective, randomized, clinical trial. Arch Surg 1993;128(4):402–410.
57. Laskar S, et al. Perioperative interstitial brachytherapy for soft tissue sarcomas: prognostic factors and long-term results of 155 patients. Ann Surg Oncol 2007;14(2):560–567.
58. Mierzwa ML, et al. Interstitial brachytherapy for soft tissue sarcoma: a single institution experience. Brachytherapy 2007;6(4):298–303.
59. Blattmann C, et al. Non-randomized therapy trial to determine the safety and efficacy of heavy ion radiotherapy in patients with non-resectable osteosarcoma. BMC Cancer 2010;10:96.
60. Ferrario T, Karakousis CP. Retroperitoneal sarcomas: grade and survival. Arch Surg 2003;138(3):248–251.
61. Fein DA, et al. Management of retroperitoneal sarcomas: does dose escalation impact on locoregional control? Int J Radiat Oncol Biol Phys 1995;31(1):129–134.
62. Pawlik TM, et al. Long-term results of two prospective trials of preoperative external beam radiotherapy for localized intermediate- or high-grade retroperitoneal soft tissue sarcoma. Ann Surg Oncol 2006;13(4):508–517.
63. Jones JJ, et al. Initial results of a trial of preoperative external-beam radiation therapy and postoperative brachytherapy for retroperitoneal sarcoma. Ann Surg Oncol 2002;9(4):346–354.
64. Zangos S, et al. MR-guided biopsies of lesions in the retroperitoneal space: technique and results. Eur Radiol 2006;16(2):307–312.
65. Chen TC, et al. Solitary extramedullary plasmacytoma in the retroperitoneum. Am J Hematol 1998;58(3):235–238.
66. Pizzo P, Poplack D. Principles and practice of pediatric oncology, 6th ed. Philadelphia, PA: Lippincott Williams & Wilkins, 2010:1600.
67. Davis S, Rogers MA, Pendergrass TW. The incidence and epidemiologic characteristics of neuroblastoma in the United States. Am J Epidemiol 1987;126(6):1063–1074.
68. Ben Moualli S, et al. [Retroperitoneal neuroblastoma in the adult: case report and review of the literature]. Ann Urol (Paris) 2001;35(1):51–55.
69. Loeser A, Gerharz EW, Riedmiller H. Recurrent pelvic neuroblastoma in an adult patient. Gynecol Oncol 2007;106(1):257–258.
70. Breslow N, et al. Epidemiology of Wilms tumor. Med Pediatr Oncol 1993;21(3):172–181.
71. Merten DF, Yang SS, Bernstein J. Wilms’ tumor in adolescence. Cancer 1976;37(3):1532–1538.
72. Mitry E, et al. Incidence of and survival from Wilms’ tumour in adults in Europe: data from the EUROCARE study. Eur J Cancer 2006;42(14):2363–2368.
73. Kalapurakal JA, et al. Treatment outcomes in adults with favorable histologic type Wilms tumor—an update from the National Wilms Tumor Study Group. Int J Radiat Oncol Biol Phys 2004;60(5):1379–1384.
74. Reinhard H, et al. Wilms’ tumor in adults: results of the Society of Pediatric Oncology (SIOP) 93–01/Society for Pediatric Oncology and Hematology (GPOH) Study. J Clin Oncol 2004;22(22):4500–4506.
75. Reinhard H, et al. [Wilms’ tumor in adults]. Urologe A 2007;46(7):748–753.
76. Izawa JI, et al. Prognostic variables in adult Wilms tumour. Can J Surg 2008;51(4):252–256.
77. Ali AN, et al. A Surveillance, Epidemiology and End Results (SEER) program comparison of adult and pediatric Wilms’ tumor. Cancer 2012;118(9):2541–2451.
78. Ali EM, Elnashar AT. Adult Wilms’ tumor: review of literature. J Oncol Pharm Pract 2012;18(1):148–151.
79. Segers H, et al. Management of adults with Wilms’ tumor: recommendations based on international consensus. Expert Rev Anticancer Ther 2011;11(7):1105–1113.
80. Theodosopoulos T, et al. Special problems encountering surgical management of large retroperitoneal schwannomas. World J Surg Oncol 2008;6:107.
81. Nah YW, et al. Benign retroperitoneal schwannoma: surgical consideration. Hepatogastroenterology 2005;52(66):1681–1684.
82. Strauss DC, et al. Management of benign retroperitoneal schwannomas: a single-center experience. Am J Surg 2011;202(2):194–198.
83. Antiheimo J, et al. Population based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 2000;54:71–76.
84. Hughes MJ, et al. Imaging features of retroperitoneal and pelvic schwannomas. Clin Radiol 2005;60(8):886–893.
85. Li Q, et al. Analysis of 82 cases of retroperitoneal schwannoma. ANZ J Surg 2007;77(4):237–240.
86. Carli M, et al. Pediatric malignant peripheral nerve sheath tumor: the Italian and German soft tissue sarcoma cooperative group. J Clin Oncol 2005;23(33):8422–8430.
87. Ferrari A, et al. Soft-tissue sarcomas in children and adolescents with neurofibromatosis type 1. Cancer 2007;109(7):1406–1412.
88. Reitamo JJ, Scheinin TM, Hayry P. The desmoid syndrome. New aspects in the cause, pathogenesis and treatment of the desmoid tumor. Am J Surg 1986;151(2):230–237.
89. Nieuwenhuis MH, et al. A nation-wide study comparing sporadic and familial adenomatous polyposis-related desmoid-type fibromatoses. Int J Cancer 2011;129(1):256–261.
90. Latchford AR, et al. A 10-year review of surgery for desmoid disease associated with familial adenomatous polyposis. Br J Surg 2006;93(10):1258–1264.
91. Ferenc T, et al. Aggressive fibromatosis (desmoid tumors): definition, occurrence, pathology, diagnostic problems, clinical behavior, genetic background. Pol J Pathol 2006;57(1):5–15.
92. Lahat G, et al. Surgery for sporadic abdominal desmoid tumor: is low/no recurrence an achievable goal? Isr Med Assoc J 2009;11(7):398–402.
93. De Camargo VP, et al. Clinical outcomes of systemic therapy for patients with deep fibromatosis (desmoid tumor). Cancer 2010;116(9):2258–2265.
94. Meazza C, et al. Aggressive fibromatosis in children and adolescents: the Italian experience. Cancer 2010;116(1):233–240.
95. Meazza C, Alaggio R, Ferrari A. Aggressive fibromatosis in children: a changing approach. Minerva Pediatr 2011;63(4):305–318.
96. Ballo MT, Zagars GK, Pollack A. Radiation therapy in the management of desmoid tumors. Int J Radiat Oncol Biol Phys 1998;42(5):1007–1014.
97. Nuyttens JJ, et al. Surgery versus radiation therapy for patients with aggressive fibromatosis or desmoid tumors: a comparative review of 22 articles. Cancer 2000;88(7):1517–1523.
98. Micke O, Seegenschmiedt MH. Radiation therapy for aggressive fibromatosis (desmoid tumors): results of a national Patterns of Care Study. Int J Radiat Oncol Biol Phys 2005;61(3):882–891.
99. Penel N, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol 2011;22(2):452–457.
100. Bhama PK, et al. Gardner’s syndrome in a 40-year-old woman: successful treatment of locally aggressive desmoid tumors with cytotoxic chemotherapy. World J Surg Oncol 2006;4:96.
101. Bokemeyer C, et al. Extragonadal germ cell tumors of the mediastinum and retroperitoneum: results from an international analysis. J Clin Oncol 2002;20(7):1864–1873.
102. Jadhav AS, Pathare DB, Shingare MS. A validated stability indicating high performance reverse phase liquid chromatographic method for the determination of cilostazol in bulk drug substance. Drug Dev Ind Pharm2007;33(2):173–179.
103. International Germ Cell Consensus Classification: a prognostic factor-based staging system for metastatic germ cell cancers. International Germ Cell Cancer Collaborative Group. J Clin Oncol 1997;15(2):594–603.
104. Lavery HJ, Bahnson RR, Sharp DS, Pohar KS. Management of the residual post-chemotherapy retroperitoneal mass in germ cell tumors. Ther Adv Urol 2009;1(4):199–207.
105. Mullinax JE, Zager JS, Gonzalez RJ. Current diagnosis and management of retroperitoneal sarcoma. Cancer Control 2011;18(3):177–187.