Neal P. Barney,
Timothy J. Daley,
C. Stephen Foster
Toxoplasmosis is one of the leading causes of infectious retinitis.[1] It is produced by the coccidian parasite Toxoplasma gondii, and the cat is its definitive host. Humans and other animals act as intermediate hosts. Toxoplasmosis infection is particularly serious and potentially fatal in congenital disease as well as in immunocompromised patients.
T. gondii is ubiquitous in nature. The parasite has three forms: tachyzoite, bradyzoite, and sporozoite.[2] The tachyzoite is the infectious form of the parasite; it multiplies intracellularly and causes host cell death and the release of more tachyzoites. The host's immune response causes the tachyzoite to transform into the slowly dividing bradyzoite, the encysted intracellular form of the parasite found in tissue cysts. These cysts may remain in any tissue, such as the retina, for years without provoking an immune response in the host. When the cysts rupture, parasites are released into the surrounding tissues, resulting in a recurrence of clinical disease. The sporozoite forms from oocysts, which are produced exclusively in the intestinal enterocytes of cats. Millions of oocysts are released into the environment for 2-3 weeks after primary infection until the cat becomes immune.[3] The oocysts undergo sporulation within a few days and may remain infectious for up to 2 years.
Human infection may occur by either the congenital or the acquired route. The acquired disease can occur by the ingestion of either sporozoites or tissue cysts in inappropriately cooked infected meat and is usually asymptomatic in immunocompetent persons. Infected livestock are a prominent source of infection for humans. Recent evidence also suggests the disease can be acquired through inhalation of spores and ingestion of contaminated drinking water. Infection can also be acquired through contaminated blood transfusions and organ transplants.[4] Congenital infection occurs through transplacental transmission of tachyzoites from a mother infected just before or during pregnancy to the developing fetus.[5] The severity of congenital infection is highest when acquired during the first trimester of pregnancy, although the frequency of transmission to the fetus is greatest during the third trimester when contact of the maternal and fetal circulations is more likely to occur. Once maternal immunity has developed, it is believed that all future fetuses are protected from the development of congenital toxoplasmosis.
CLINICAL FEATURES
The majority of cases of ocular toxoplasmosis are congenital. In one study of 300 cases of congenital toxoplasmosis in newborns, ocular lesions were present in 76%, neurologic involvement was present in 51%, intracranial calcifications were present in 32%, and microcephaly or hydrocephalus was present in 26%.[6] Among the clinical manifestations of congenital ocular toxoplasmosis reported in infants are microphthalmia, enophthalmos, ptosis, nystagmus, choroidal colobomas, and strabismus.[7]
|
Key Features |
||||||||||||||||||||||||||||||||||||
|
Several reports of acquired ocular toxoplasmosis suggest that this route may actually be more important and more prevalent than previously thought.[8-11] A population study in southern Brazil reported a high prevalence of acquired ocular toxoplasmosis in subjects older than 12 years.[12] Similarly, a study after an outbreak of toxoplasmosis on Victoria Island, in British Columbia, Canada reported that 21% of confirmed cases of new infection had ocular disease.[13]
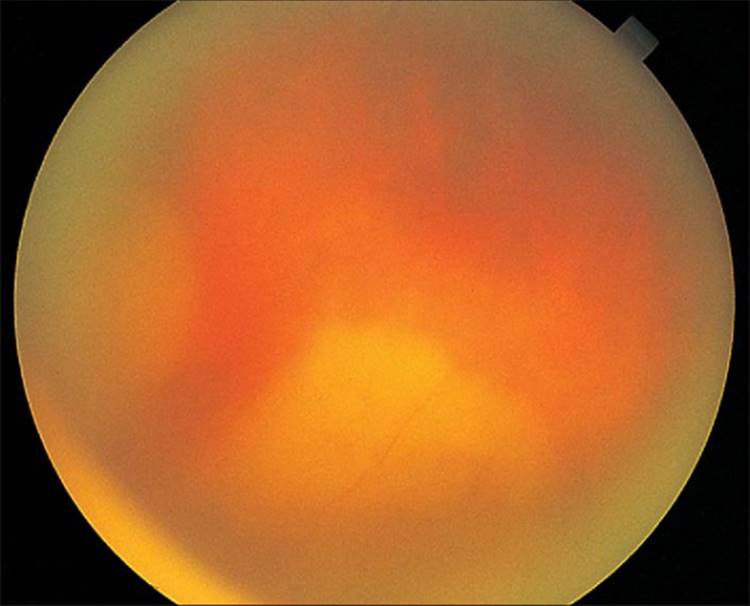
Ocular toxoplasmosis frequently presents as a focal necrotizing retinitis, usually adjacent to a large, atrophic chorioretinal scar (Fig. 162.1), which is often located in the macula in congenital cases. The scar is typically yellow-white and unevenly pigmented. Acquired cases of toxoplasmosis may present as a unilateral focal chorioretinitis with no preexisting retinal scars in either eye.[14] The areas of retinitis are the result of tissue cysts bursting and releasing bradyzoites that transform into tachyzoites, which in turn invade neighboring cells. These destructive lesions are usually larger than one disk diameter and appear as soft, white, fluffy infiltrates surrounded by retinal edema with subjacent choroiditis.[15] When the tachyzoites come under increasing attack by the host's immune response, they gradually transform back into bradyzoites. Inflammatory cells will be found in the vitreous overlying the active lesion (Fig. 162.2). Perivascular inflammatory exudates are frequently present around retinal vessels peripheral to an area of active inflammation. In cases where the retinal vessel transverses the active toxoplasmic lesion, regression of the vascular infiltrates may take longer to recede or may even fail to disappear.[16]Patients will often complain of pain, blurred vision, floaters, and photophobia. More than a third of patients with active ocular toxoplasmosis will have macular involvement resulting in severe visual loss.[17,18]Ocular toxoplasmosis may also present as gray-white punctate lesions in the outer retina and retinal pigment epithelium (Fig. 162.3).[19] Occasionally, patients will present initially with severe unilateral papillitis, macular hard exudates distributed in a star fashion, and vitreal inflammation (Fig. 162.4).[20,21] Abnormal intraocular pressure has also been associated with active disease.[22] Active toxoplasmosis simultaneously involving both the retina and the optic nerve is unusual but has been documented clinically in an immunocompetent patient using magnetic resonance imaging.[21] There has also been a report of active ocular toxoplasmosis presenting as hemorrhagic retinochoroiditis in an otherwise healthy adult.[23]
|
|
|
|
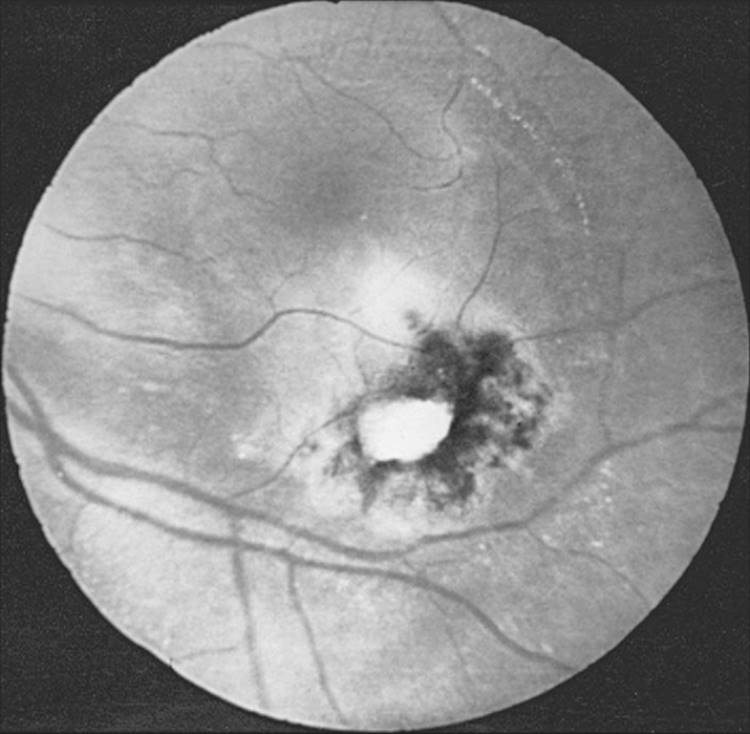
FIGURE 162.1 Atrophic chorioretinal scar with surrounding retinal pigment epithelial proliferation; a chorioretinal scar quite typical of Toxoplasma chorioretinitis. However, note also the fresh satellite lesion at the edge of the pigmented chorioretinal scar (at the 11 o'clock position) threatening the fovea. |
|
|
|
|
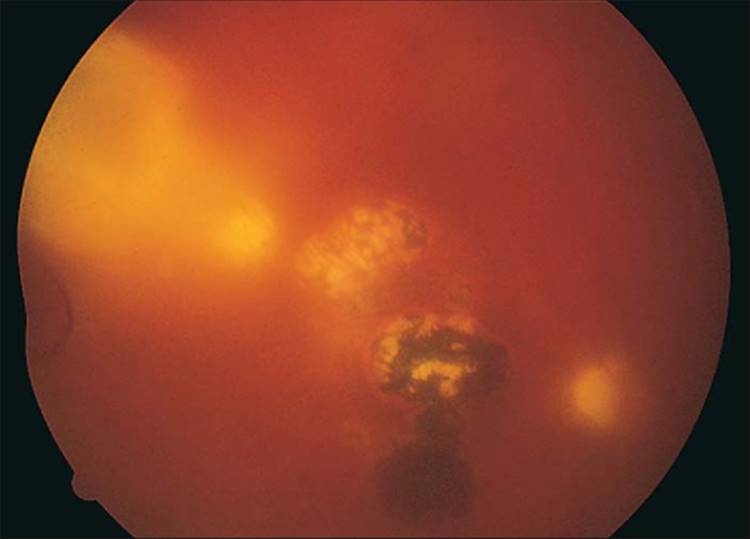
FIGURE 162.2 Multiple atrophic and hyperpigmented chorioretinal scars from previous toxoplasmosis. Note the fresh area of chorioretinitis with overlying vitreal inflammatory cells and vitreal 'haze'. |
|
|
|
|
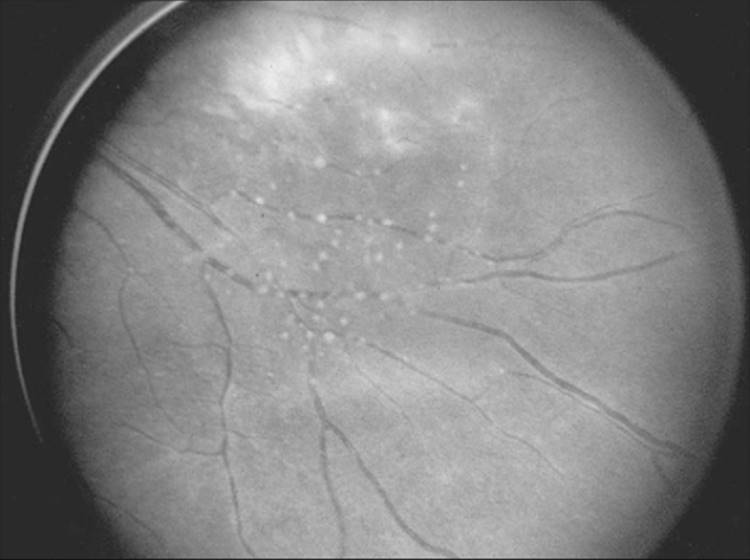
FIGURE 162.3 Toxoplasma retinitis without typical choroidal involvement. Note the multifocal nature of the lesions, with an area of active retinitis at the 11 o'clock position. Note also the associated vasculitis. |
|
|
|
|
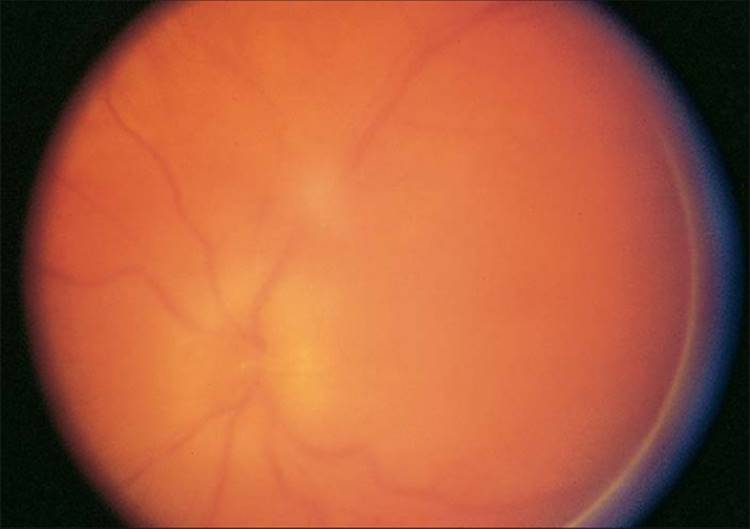
FIGURE 162.4 Toxoplasma papillitis with vitreal inflammatory cells anterior to the optic nerve, making visualization of the optic nerve slightly difficult. The ELISA for Toxoplasma antibodies gave the following results: The IgG titer was 1:496; the IgM titer at the time this photograph was taken was 1:64; 2 weeks later, the anti-Toxoplasma IgM antibody titer was 1:256. |
COMPLICATIONS
Secondary complications arising from ocular toxoplasmosis include choroidal neovascularization,[24] retinochoroidal vascular anastomosis,[25] and branch retinal artery[26] and vein occlusion,[27,28] cystoid macular edema, retinal detachment, cataracts, secondary glaucoma, and optic atrophy.[17] The most frequent complication is secondary glaucoma. There is a well-known association between Fuchs' heterochromic iridocyclitis and ocular toxoplasmosis, but the reasons for this are unclear.[29]
HISTOPATHOLOGIC AND IMMUNOLOGIC FEATURES
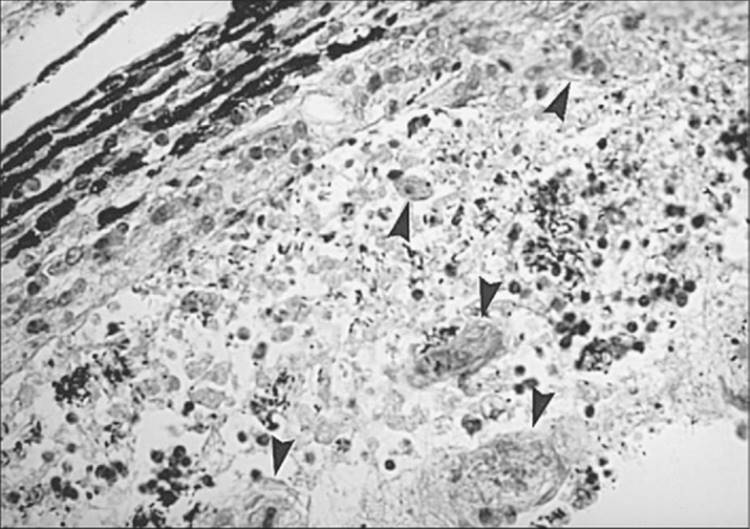
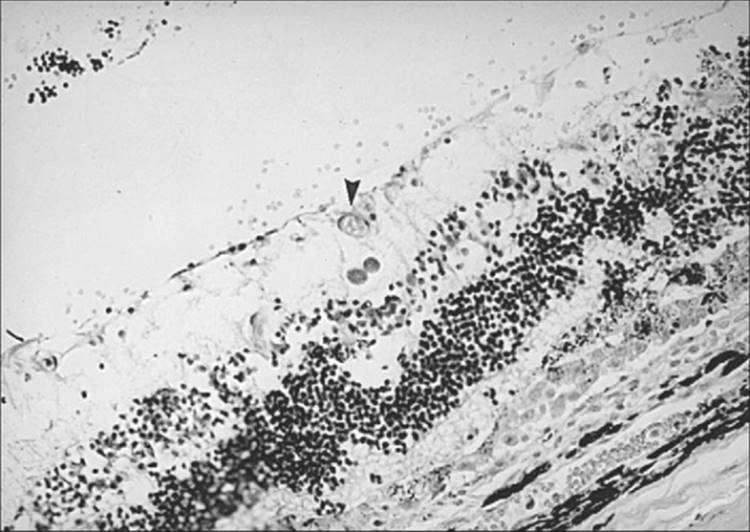
Histopathologically, there is necrosis of the involved retina with destruction of the retinal architecture and the underlying choroid.[30] Since the parasite has a propensity for attacking neural tissue, tissue cysts and trophozoites are usually found in the superficial layers of the retina within the area of necrosis (Figs 162.5 and 162.6). The infiltrate consists predominantly of lymphocytes, macrophages, and epitheloid cells, with plasma cells found in the periphery of the lesion.
|
|
|
|
FIGURE 162.5 Toxoplasma feline retinochoroiditis. Histopathologic appearance of the retina and choroid. Note the Toxoplasma cysts (arrowheads). |
|
|
|
|
FIGURE 162.6 Same specimen as in Figure 162.5. Note the Toxoplasma cyst (arrowhead). |
Cell-mediated immunity is felt to be the major defense mechanism against Toxoplasma infection.[31] Several purified antigens from tachyzoites have been identified, including the major surface protein antigen p30, which can participate in antibody-dependent complement-mediated lysis of the tachyzoite.[32,33] In patients with ocular toxoplasmosis, the cellular immune response appears to be directed predominantly against the cell surface protein p22.[34] Additionally, gamma interferon and tumor necrosis factor both appear to play an important role in inducing and maintaining the interconversion of the organism's tachyzoite form to the bradyzoite form during active infection.[35] When neutralizing doses of antibodies to either gamma interferon or tumor necrosis factor are given to animal models with chronic infection, free tachyzoites and an increased number of cysts are observed.[36] However, some evidence also indicates that part of the disease may be mediated by an autoimmune mechanism directed against certain retinal antigens.
The role of the humoral response to toxoplasmosis is unclear. Whereas an intact cellular component of the immune system is necessary for the resolution of active disease, antibody may be important in establishing a state of immunity in the host.[37] Antibodies directed against the cytoplasmic protein F3G3[38] can protect passively immunized mice from a lethal challenge of T. gondii.[39]
OCULAR TOXOPLASMOSIS IN THE IMMUNOCOMPROMISED HOST
Although T. gondii is a common opportunistic pathogen with a penchant for attacking the central nervous system in immunocompromised patients,[40-42] including those with acquired immunodeficiency syndrome (AIDS),[43-45] ocular toxoplasmosis appears to be uncommon. Ocular toxoplasmosis occurs in ~1% of patients with AIDS.[46] Systemically, these patients are prone to the development of Toxoplasma encephalitis, myocarditis, or pneumonitis.
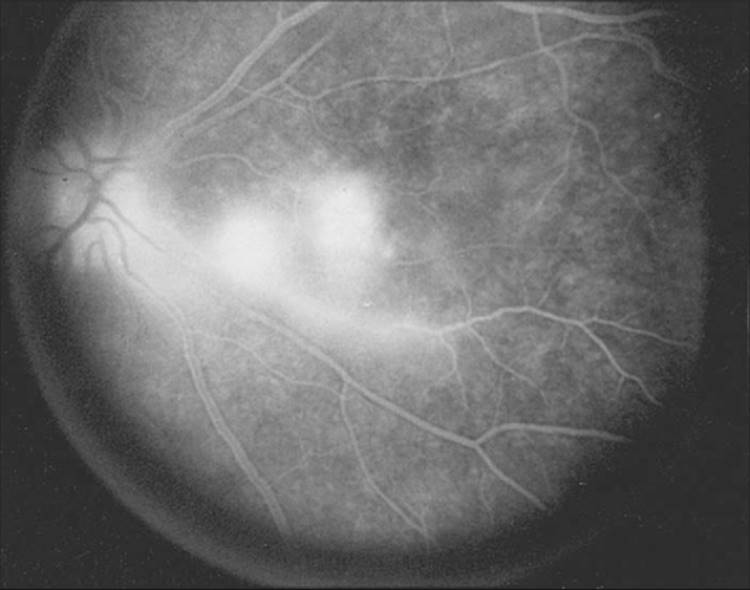
AIDS-related Toxoplasma retinochoroiditis has several atypical clinical manifestations, including bilateral involvement and single or multifocal discrete lesions[47-49] or diffuse areas of retinal necrosis (Fig. 162.7).[50] Furthermore, unlike immunocompetent individuals, who rarely have more than one focus of active retinal disease, even if there are multiple retinochoroidal scars, immunocompromised individuals can have multiple active lesions in one or both eyes.[51] Vascular sheathing may be present, and prominent inflammatory reactions in the vitreous and anterior chambers are common (Fig. 162.8). Lesions may occur adjacent to retinal blood vessels and are usually not associated with preexisting retinochoroidal scars. These findings suggest that the ocular lesions in AIDS patients may be the result of newly acquired disease or from organisms that have disseminated to the eye from extraocular sites. The retina can be infected simultaneously by both T. gondii and cytomegalovirus in patients with AIDS.[52,53]Anti-Toxoplasma immunoglobulin G (IgG) antibodies are present in these patients, although IgM antibodies against Toxoplasma are uncommon.
|
|
|
|
FIGURE 162.7 Diffuse retinitis with probable underlying choroiditis in an immunocompromised patient with toxoplasmosis. Note the overlying vitreal inflammatory cellular response with vitreal 'haze'. |
|
|
|
|
FIGURE 162.8 Fluorescein angiogram in a patient with active toxoplasmosis. Note not only the dye accumulation in the two foci representing the areas of active Toxoplasma chorioretinitis but also the papillitis with dye staining of the nerve head and the associated retinal vasculitis with late vascular staining. |
Infection usually produces a full-thickness retinal necrosis, but early necrosis may be confined to either the inner or the outer layers. Large numbers of trophozoites and cysts with scanty inflammatory reaction are seen within the necrotic retina and optic nerve head. Although T. gondii organisms may involve anterior segment structures and uveal tissue in patients with AIDS, parasites are rarely found in these structures in otherwise healthy patients.[54] Toxoplasma retinochoroiditis may rarely progress to panophthalmitis and orbital cellulitis in patients with AIDS.[55] Finally, not only do patients with AIDS develop more extensive disease, but lesions are more likely to reactivate if treatment is discontinued.[56]
DIAGNOSIS
The definitive diagnosis of ocular toxoplasmosis requires the demonstration of tachyzoites in ocular tissues.[57] Cytologic evaluation of vitrectomy specimens to detect the presence of Toxoplasma organisms is especially useful in patients with necrotizing retinitis where the cause for the retinitis is uncertain.[58] The presence of cysts in tissue samples, however, only suggests, but does not prove acute infection. Toxoplasma retinochoroiditis is, therefore, usually a clinical diagnosis based on a compatible lesion in the fundus and positive serologic results for anti-Toxoplasma antibodies. Additionally, other causes of focal exudative retinitis, including syphilis, tuberculosis, sarcoidosis, cytomegalovirus, and fungal infections, must be excluded.
Because of their ease of performance and their relatively high sensitivity and specificity, serologic methods are often preferred for the diagnosis of toxoplasmosis. Serologic diagnosis is complicated by the high prevalence of T. gondii-specific antibodies in the human population persisting for years and therefore frequently reflecting past infection. Classic serodiagnosis of an acute systemic infection requires the demonstration of a seroconversion, a significant rise in specific antibody titers in paired sera taken 4-6 weeks apart, or the presence of anti-Toxoplasma IgM antibody in a single serum sample. If retinochoroiditis develops subsequently, it is presumed to be acquired. However, because systemic toxoplasmosis infection is often subclinical in its manifestations, the diagnosis of acquired ocular toxoplasmosis may be missed or confused with reactivation of the congenital type if ocular involvement occurs months or years after a systemic infection. Because sera are often submitted late after infection, it is difficult to detect an antibody titer rise. Establishing the presence of Toxoplasma antibodies in a patient's serum is considered essential in the diagnosis of Toxoplasma retinochoroiditis. In this case, any titer of antibody is significant because no correlation exists between the serum level of anti-Toxoplasma titers and the activity of the ocular disease in recurrent ocular toxoplasmosis.[59]
The most widely used serologic methods for detecting anti-Toxoplasma antibodies are the Sabin-Feldman test, the complement fixation test, the agglutination tests, the indirect immunofluorescence assay (IFA), and the enzyme-linked immunosorbent assay (ELISA). Although the Sabin-Feldman dye test represents the standard against which all other tests are measured, it is no longer performed routinely because it requires the constant maintenance of virulent organisms in the laboratory with the associated risks to laboratory personnel.
The IFA test, which has largely replaced the Sabin-Feldman dye test, has become the most widely used test for the detection of anti-Toxoplasma IgG or IgM antibodies.[60] Although it is easy to perform, the interpretations of its results can be misleading. False-negative IgM titers may occur because of competitive inhibition by anti-Toxoplasma IgG if present simultaneously.[61] False-positive IgM titers can occur because of the presence of rheumatoid factor.[62] Rheumatoid factor, an anti-IgG IgM antibody, can bind to specific anti-Toxoplasma IgG. Finally, the presence of antinuclear antibody may produce false-positive IgM and IgG titers.[63] This occurs because Toxoplasma organisms possess antigens that are indistinguishable from those found in human leukocyte nuclei using immunologic laboratory testing techniques. Because IgM titers in ocular toxoplasmosis become significant only in cases of acquired retinochoroiditis,[9,64] the specificity limitations of the IFA tests are restricted, for practical purposes, to false-positive IgG titers produced by the presence of antinuclear antibodies.
Commercially available IFA tests for Toxoplasma begin with a 1:16 serum dilution because specificity decreases when more concentrated dilutions are used. Since any titer of antibody is considered significant in establishing the diagnosis of Toxoplasma retinochoroiditis, it is logical to expect a significant percentage of false-negative titers using this serologic method. One report cited three documented cases of Toxoplasma retinochoroiditis with positive Sabin-Feldman dye titers and negative immunofluorescent titers (titer <1:16).[65]
The double-sandwich ELISA for anti-Toxoplasma antibodies eliminates IgM specificity problems associated with the IFA test.[66,67] False-positive reactions resulting from rheumatoid factor and false-negative reactions because of competing IgG are avoided because only IgM of the test serum adheres to plates precoated with anti-IgM antiserum. The ELISA for anti-Toxoplasma antibodies is as specific and sensitive as the Sabin-Feldman dye test. Therefore, in cases where ocular toxoplasmosis is strongly suspected despite negative immunofluorescent titers, a Sabin-Feldman titer or an ELISA titer should be obtained before excluding the diagnosis of toxoplasmosis.
Some experts advocate measuring anti-Toxoplasma antibody titers in intraocular fluids,[68] or even tears,[69] but this has not been widely adopted at the present time. Polymerase chain reaction (PCR) techniques have been used to detect the presence of toxoplasma genes in ocular tissues and fluids.[70,71] However, its precise role in the diagnosis of ocular toxoplasmosis remains to be defined by controlled clinical trials. It is quite expensive, not widely available, and requires special equipment and highly specialized technical skills to perform properly. Although, with the advent of real-time PCR and its advantages of higher sensitivity, improved quantitation, and fewer manual manipulations required, it could prove to be useful in determining disease burden, therapy efficacy, and epidemiology of the disease.[72]
THERAPY
Since available agents against T. gondii are ineffective against tissue cysts, the major aim of therapy has been to stop the multiplication of tachyzoites during episodes of active retinochoroiditis. It is important for the clinician to realize this and to inform the patient that drug therapy does not totally eradicate the parasite.
Complicating matters is that little is known about the natural history of active disease. There have been surprisingly few prospective, randomized, placebo-controlled clinical trials for the treatment of ocular toxoplasmosis in immunocompetent individuals. Interestingly, none of them demonstrated that short-term drug therapy was effective in treating active toxoplasmic retinochoroiditis, reducing recurrent disease, or shortening duration of disease.[73]
This is in contrast to numerous animal studies, which have found antimicrobial agents to be highly efficacious for active toxoplasmosis treatment. In fact, some newer drugs such as atovaquone and azithromycin, have even been shown to reduce the number of tissue cysts in animal models.[74] Furthermore, while treatment efficacy has been difficult to determine in immunocompetent patients, antimicrobial therapy can be highly effective in patients with AIDS. Long-standing active infections rapidly become inactive with treatment in these individuals. Finally, observational experience suggests that therapy is beneficial in immunocompetent patients, despite difficulties to quantify their efficacy. Most clinicians agree that antimicrobial therapy is warranted in active disease.
Toxoplasma organisms lack the transmembrane transport systems for physiologic folates and thus synthesize this substance instead. Folic acid antagonists inhibit this biochemical pathway and prevent the organism from replicating by impairing DNA synthesis.[75] A combination of sulfadiazine (1 g PO qid for first day, then 500 mg PO qid for 2-8 weeks) with pyrimethamine (50 mg PO bid for first day, then 25 mg PO qid for 2-8 weeks) and folinic acid (5 mg PO three times a week) has classically been considered to be the most effective treatment for toxoplasmosis.[76] If necessary, repeated courses of drugs may be administered. However, many toxic side effects occur with this regimen, most notably bone marrow depression with subsequent hematologic complications. Weekly white blood cell counts and platelet counts are therefore mandatory in these patients.
A less toxic means of treatment consists of clindamycin (300 mg PO tid or qid) in combination with sulfadiazine (500 mg PO qid). Patients are generally treated for 4-6 weeks on this drug regimen. However, diarrhea, colitis, and pseudomembranous colitis remain potential problems with clindamycin use.
Co-trimoxazole (trimethoprim 160 mg with sulfamethoxazole 800 mg) given twice daily for 2-6 weeks either alone or in combination with clindamycin (300 mg PO qid) has also been suggested for the treatment of toxoplasmosis in humans.[77,78] The side effects are similar to those of the sulfonamides.
Anterior uveitis associated with ocular toxoplasmosis may be treated with topical corticosteroids and cycloplegic agents along with concurrent systemic antitoxoplasmosis therapy. Increased intraocular pressure can be managed with standard glaucoma medical therapy.
The addition of systemic corticosteroids to the therapeutic regimen may diminish the degree of collateral damage from the inflammatory response. Some experts advocate adding prednisone 12-24 h after initiating the antiparasitic therapy if the Toxoplasma lesion is in the posterior pole, threatening the optic nerve head, or if the vitreal response is extreme, starting with 40-80 mg of prednisone with breakfast daily for 1 week, then rapidly tapering the prednisone before discontinuing the antitoxoplasmosis therapy. Under no circumstances, however, should systemic corticosteroids be used without concurrent antitoxoplasmosis therapy, nor should they be used in patients with necrotizing retinitis where the diagnosis of ocular toxoplasmosis has not been ruled out. Several cases of fulminant ocular toxoplasmosis have been described after the use of corticosteroids alone.[79,80] Other recommendations for ocular toxoplasmosis therapy in various situations are summarized in Table 162.1.
TABLE 162.1 -- Recommended Therapy for Ocular Toxoplasmosis
|
Clinical Features |
Recommended Treatment |
|
Peripheral lesion: mild to moderate vitreal cells |
Clindamycin, 300 mg PO tid or qid and sulfadiazine, 1 g initially then 500 mg PO qid |
|
Peripheral lesion: moderate to severe vitreal cells |
As above plus prednisone 1 mg/kg PO/day with taper based on clinical response |
|
Juxtamacular lesion |
Clindamycin and sulfadiazine as above plus pyrimethamine, 50 mg bid during the first day then 25 mg PO/day plus folinic acid, 5 mg PO three times a week prednisone use based on vitreal cells, as in the case of peripheral lesions |
Standard antitoxoplasmosis drug regimens can usually control AIDS-related ocular lesions. Treatment is given until 4-6 weeks after resolution of all signs and symptoms. However, as many as 20% of immunocompromised patients with toxoplasmosis may fail to respond to medical therapy.[43] A combination of pyrimethamine and clindamycin was reported to be more effective than either pyrimethamine alone or in combination with sulfadiazine for patients with AIDS.[81] Toxoplasmosis in patients with AIDS frequently recurs when medical treatment is discontinued.[49,82,83] It may be necessary to continue treatment indefinitely to maintain control of the disease, but adequate maintenance treatment regimens have not been established. Zidovudine (azidothymidine (AZT) antagonizes the anti-Toxoplasma activity of pyrimethamine both in vivo and in vitro and may have to be discontinued.[84] Co-trimoxazole has been employed in AIDS patients sensitive to pyrimethamine, but this has been reported to be associated with treatment failure.[85] Comanagement with an infectious disease specialist is recommended when treating these patients. Atovaquone (Mepron), 750 mg tid, plus clarithromycin (Biaxin), 500 mg bid, has been used for long-term suppressive therapy of recurrent toxoplasmosis in selected individuals with AIDS, especially in the context of sensitivity to the more classic antitoxoplasma agents. Azithromycin (Zithromax) and Spiramycin (Rovamycine) are additional agents with antitoxoplasma activity that have had limited use thus far with promising results.[86,87]
The role of laser photocoagulation in the treatment of active ocular toxoplasmosis and for prophylaxis against the spread of lesions is limited.[88] Although photocoagulation can destroy Toxoplasma cysts and tachyzoites, Toxoplasma cysts can reside in ophthalmoscopically normal-appearing retina. Furthermore, photocoagulation of active lesions can be complicated by retinal or vitreous hemorrhage or even by retinal detachment. Vitrectomy and lensectomy can be performed to remove vitreous and lens opacities in these patients.[89] Patients undergoing surgery should probably be treated preoperatively with anti-Toxoplasma agents, and the medications should be continued postoperatively.
REFERENCES
1. Perkins ES: Ocular toxoplasmosis. Br J Ophthalmol 1973; 57:1-17.
2. Krick JA, Remington JS: Toxoplasmosis in the adult - an overview. N Engl J Med 1978; 298:550-553.
3. van Knapen F: Toxoplasmosis, old stories and new facts. Int Ophthalmol 1989; 13:371-375.
4. Holland GN: Reconsidering the pathogenesis of ocular toxoplasmosis. Am J Ophthalmol 1999; 128:502-505.
5. Swartzberg JE, Remington JS: Transmission of toxoplasma. Am J Dis Child 1975; 129:777-779.
6. Couvreur J, Desmonts G: Congenital and maternal toxoplasmosis. A review of 300 congenital cases. Dev Med Child Neurol 1962; 4:519-530.
7. de Jong PT: Ocular toxoplasmosis; common and rare symptoms and signs. Int Ophthalmol 1989; 13:391-397.
8. Teutsch SM, Juranek DD, Sulzer A, et al: Epidemic toxoplasmosis associated with infected cats. N Engl J Med 1979; 300:695-699.
9. Akstein RB, Wilson LA, Teutsch SM: Acquired toxoplasmosis. Ophthalmology 1982; 89:1299-1302.
10. Silveira C, Belfort Jr R, Burnier Jr. M, et al: Acquired toxoplasmic infection as the cause of toxoplasmic retinochoroiditis in families. Am J Ophthalmol 1988; 106:362-364.
11. Couvreur J, Thulliez P: Acquired toxoplasmosis of ocular or neurologic site: 49 cases. Presse Med 1996; 25:438-442.
12. Glasner PD, Silveria C, Kruszon-Moran D, et al: An unusually high prevalence of ocular toxoplasmosis in southern Brazil. Am J Ophthalmol 1992; 114:136-144.
13. Burnett AJ, Shortt SG, Isaac-Renton J, et al: Multiple cases of acquired toxoplasmosis retinitis presenting in an outbreak. Ophthalmology 1998; 105:1032-1037.
14. Ronday MJ, Luyendijk L, Baarsma GS, et al: Presumed acquired ocular toxoplasmosis. Arch Ophthalmol 1995; 113:1524-1529.
15. Tabbara KF: Ocular toxoplasmosis: toxoplasmic retinochoroiditis. Int Ophthalmol Clin 1995; 35:15-29.
16. Theodossiadis P, Kokolakis S, Ladas I, et al: Retinal vascular involvement in acute toxoplasmic retinochoroiditis. Int Ophthalmol 1995; 19:19-24.
17. Friedmann CT, Knox DL: Variations in recurrent active toxoplasmic retinochoroiditis. Arch Ophthalmol 1969; 81:481-493.
18. Stanford MR, Tomlin EA, Comyn O, et al: The visual field in toxoplasmic retinochoroiditis. Br J Ophthalmol 2005; 89:812-814.
19. Doft BH, Gass JD: Outer retinal layer toxoplasmosis. Graefes Arch Clin Exp Ophthalmol 1986; 224:78-82.
20. Folk JC, Lobes LA: Presumed toxoplasmic papillitis. Ophthalmology 1984; 91:64-67.
21. Borruat FX, Kapoor R, Sanders MD: Simultaneous retinal and optic nerve lesions in toxoplasmosis: the advantages of magnetic resonance imaging. Br J Ophthalmol 1993; 77:450-452.
22. Westfall AC, Lauer AK, Suhler EB, et al: Toxoplasmosis retinochoroiditis and elevated intraocular pressure: a retrospective study. J Glaucoma 2005; 14:3-10.
23. Baglivo E, Safran AB: Haemorrhagic toxoplasmic retinochoroiditis: description of an unusual clinical presentation. Br J Ophthalmol 2003; 87:1051-1052.
24. Fine SL, Owens SL, Haller JA, et al: Choroidal neovascularization as a late complication of ocular toxoplasmosis. Am J Ophthalmol 1981; 91:318-322.
25. Kennedy JE, Wise GN: Retinochoroidal vascular anastomoses in uveitis. Am J Ophthalmol 1971; 71:1221-1225.
26. Braunstein RA, Gass JD: Branch artery obstruction caused by acute toxoplasmosis. Arch Ophthalmol 1980; 98:512-513.
27. Rose GE: Papillitis, retinal neovascularisation and recurrent retinal vein occlusion in toxoplasma retinochoroiditis: a case report with uncommon clinical signs. Aust N Z J Ophthalmol 1991; 19:155-157.
28. Gentile RC, Berinstein DM, Oppenheim R, et al: Retinal vascular occlusions complicating acute toxoplasmic retinochoroiditis. Can J Ophthalmol 1997; 32:354-358.
29. Toledo de Abreu Jr M, Belfort R, Hirata PS: Fuchs' heterochromic cyclitis and ocular toxoplasmosis. Am J Ophthalmol 1982; 93:739-744.
30. Jabs DA: Ocular toxoplasmosis. Int Ophthalmol Clin 1990; 30:264-270.
31. Williams DM, Grumet FC, Remington JS: Genetic control of murine resistance to toxoplasma gondii. Infect Immun 1978; 19:416-420.
32. Kasper LH, Crabb JH, Pfefferkorn ER: Purification of a major membrane protein of toxoplasma gondii by immunoabsorption with a monoclonal antibody. J Immunol 1983; 130:2407-2412.
33. Kasper LH, Crabb JH, Pfefferkorn ER: Isolation and characterization of a monoclonal antibody-resistant antigenic mutant of toxoplasma gondii. J Immunol 1982; 129:1694-1699.
34. Nussenblatt RB, Mittal KK, Fuhrman S, et al: Lymphocyte proliferative responses of patients with ocular toxoplasmosis to parasite and retinal antigens. Am J Ophthalmol 1989; 107:632-641.
35. Bohne W, Heesemann J, Gross U: Induction of bradyzoite-specific toxoplasma gondii antigens in gamma interferon-treated mouse macrophages. Infect Immun 1993; 61:1141-1145.
36. Denkers EY, Gazzinelli RT: Regulation and function of T-cell-mediated immunity during toxoplasma gondii infection. Clin. Microbiol. Rev 1998; 11:569-588.
37. Hafizi A, Modabber FZ: Effect of cyclophosphamide on toxoplasma gondii infection: reversal of the effect by passive immunization. Clin Exp Immunol 1978; 33:389-394.
38. Naot Y, Remington JS: Use of enzyme-linked immunosorbent assays (ELISA) for detection of monoclonal antibodies: experience with antigens of toxoplasma gondii. J Immunol Methods 1981; 43:333-341.
39. Sharma SD, Araujo FG, Remington JS: Toxoplasma antigen isolated by affinity chromatography with monoclonal antibody protects mice against lethal infection with toxoplasma gondii. J Immunol 1984; 133:2818-2820.
40. Cohen SN: Toxoplasmosis in patients receiving immunosuppressive therapy. JAMA 1970; 211:657-660.
41. Ruskin J, Remington JS: Toxoplasmosis in the compromised host. Ann Intern Med 1976; 84:193-199.
42. Ryning FW, Mills J: Pneumocystis carinii, toxoplasma gondii, cytomegalovirus and the compromised host. West J Med 1979; 130:18-34.
43. Wong B, Gold JW, Brown AE, et al: Central-nervous-system toxoplasmosis in homosexual men and parenteral drug abusers. Ann Intern Med 1984; 100:36-42.
44. Luft BJ, Brooks RG, Conley FK, et al: Toxoplasmic encephalitis in patients with acquired immune deficiency syndrome. JAMA 1984; 252:913-917.
45. Araujo FG, Remington JS: Toxoplasmosis in immunocompromised patients. Eur J Clin Microbiol 1987; 6:1-2.
46. Jabs DA, Green WR, Fox R, et al: Ocular manifestations of acquired immune deficiency syndrome. Ophthalmology 1989; 96:1092-1099.
47. Friedman AH: The retinal lesions of the acquired immune deficiency syndrome. Trans Am Ophthalmol Soc 1984; 82:447-491.
48. Schuman JS, Friedman AH: Retinal manifestations of the acquired immune deficiency syndrome (AIDS): cytomegalovirus, candida albicans, cryptococcus, toxoplasmosis and Pneumocystis carinii. Trans Ophthalmol Soc UK 1983; 103(Pt 2):177-190.
49. Heinemann MH, Gold JM, Maisel J: Bilateral toxoplasma retinochoroiditis in a patient with acquired immune deficiency syndrome. Retina 1986; 6:224-227.
50. Parke 2nd DW, Font RL: Diffuse toxoplasmic retinochoroiditis in a patient with AIDS. Arch Ophthalmol 1986; 104:571-575.
51. Holland GN: Ocular toxoplasmosis: a global reassessment. Part II. Disease manifestations and management. Am J Ophthalmol 2004; 137:1-17.
52. Cochereau-Massin I, LeHoang P, Lautier-Frau M, et al: Ocular toxoplasmosis in human immunodeficiency virus-infected patients. Am J Ophthalmol 1992; 114:130-135.
53. Pivetti-Pezzi P, Accorinti M, Tamburi S, et al: Clinical features of toxoplasmic retinochoroiditis in patients with acquired immunodeficiency syndrome. Ann Ophthalmol 1994; 26:73-84.
54. Zimmerman LE: Ocular pathology of toxoplasmosis. Surv Ophthalmol 1961; 6:832-856.
55. Moorthy RS, Smith RE, Rao NA: Progressive ocular toxoplasmosis in patients with acquired immunodeficiency syndrome. Am J Ophthalmol 1993; 115:742-747.
56. Holland GN, Engstrom Jr RE, Glasgow BJ, et al: Ocular toxoplasmosis in patients with the acquired immunodeficiency syndrome. Am J Ophthalmol 1988; 106:653-667.
57. Remington JS, Miller MJ, Brownlee I: IgM antibodies in acute toxoplasmosis. II. Prevalence and significance in acquired cases. J Lab Clin Med 1968; 71:855-866.
58. Greven CM, Teot LA: Cytologic identification of toxoplasma gondii from vitreous fluid. Arch Ophthalmol 1994; 112:1086-1088.
59. Schlaegel Jr TF: Ocular toxoplasmosis and par planitis, New York: Grune & Stratton; 1978:138-172.
60. Kelan AE, Ayllon-Leindl L, Labzoffsky NA: Indirect fluorescent antibody method in serodiagnosis of toxoplasmosis. Can J Microbiol 1962; 8:545.
61. Pyndiah N, Krech U, Price P, et al: Simplified chromatographic separation of immunoglobulin M from G and its application to toxoplasma indirect immunofluorescence. J Clin Microbiol 1979; 9:170-174.
62. Fuccillo DA, Madden DL, Tzan N, et al: Difficulties associated with serological diagnosis of toxoplasma gondii infections. Diagn Clin Immunol 1987; 5:8-13.
63. Araujo FG, Barnett EV, Gentry LO, et al: False-positive anti-toxoplasma fluorescent-antibody tests in patients with antinuclear antibodies. Appl Microbiol 1971; 22:270-275.
64. Asbell PA, Vermund SH, Hofeldt AJ: Presumed toxoplasmic retinochoroiditis in four siblings. Am J Ophthalmol 1982; 94:656-663.
65. Weiss MJ, Velazquez N, Hofeldt AJ: Serologic tests in the diagnosis of presumed toxoplasmic retinochoroiditis. Am J Ophthalmol 1990; 109:407-411.
66. Naot Y, Desmonts G, Remington JS: IgM enzyme-linked immunosorbent assay test for the diagnosis of congenital toxoplasma infection. J Pediatr 1981; 98:32-36.
67. Tomasi JP, Schlit AF, Stadtsbaeder S: Rapid double-sandwich enzyme-linked immunosorbent assay for detection of human immunoglobulin M anti-toxoplasma gondii antibodies. J Clin Microbiol 1986; 24:849-850.
68. Davis JL, Feuer W, Culbertson WW, et al: Interpretation of intraocular and serum antibody levels in necrotizing retinitis. Retina 1995; 15:233-240.
69. Lynch MI, Cordeiro F, Ferreira S, et al: Lacrimal secretory IgA in active posterior uveitis induced by toxoplasma gondii. Mem Inst Oswaldo Cruz 2004; 99:861-864.
70. Aouizerate F, Cazenave J, Poirier L, et al: Detection of toxoplasma gondii in aqueous humour by the polymerase chain reaction. Br J Ophthalmol 1993; 77:107-109.
71. de Boer JH, Verhagen C, Bruinenberg M, et al: Serologic and polymerase chain reaction analysis of intraocular fluids in the diagnosis of infectious uveitis. Am J Ophthalmol 1996; 121:650-658.
72. Contini C, Seraceni S, Cultrera R, et al: Evaluation of a real-time PCR-based assay using the lightcycler system for detection of toxoplasma gondii bradyzoite genes in blood specimens from patients with toxoplasmic retinochoroiditis. Int J Parasitol 2005; 35:275-283.
73. Stanford MR, See SE, Jones LV, et al: Antibiotics for toxoplasmic retinochoroiditis: an evidence-based systematic review. Ophthalmology 2003; 110:926-931.quiz 931-932.
74. Huskinson-Mark J, Araujo FG, Remington JS: Evaluation of the effect of drugs on the cyst form of toxoplasma gondii. J Infect Dis 1991; 164:170-171.
75. Giles CL: Pyrimethamine (Daraprim) and the treatment of toxoplasmic uveitis. Surv Ophthalmol 1971; 16:88-91.
76. Smith RL, Nozik RA: Toxoplasmic retinochoroiditis. In: Smith RE, Nozik RA, ed. Uveitis: a clinical approach to diagnosis and management, Baltimore: Williams & Wilkins; 1989:128-134.
77. Norrby R, Eilard T, Svedhem A, et al: Treatment of toxoplasmosis with trimethoprim-sulphamethoxazole. Scand J Infect Dis 1975; 7:72-75.
78. Opremcak EM, Scales DK, Sharpe MR: Trimethoprim-sulfamethoxazole therapy for ocular toxoplasmosis. Ophthalmology 1992; 99:920-925.
79. Nicholson DH, Wolchok EB: Ocular toxoplasmosis in an adult receiving long-term corticosteroid therapy. Arch Ophthalmol 1976; 94:248-254.
80. Sabates R, Pruett RC, Brockhurst RJ: Fulminant ocular toxoplasmosis. Am J Ophthalmol 1981; 92:497-503.
81. Rolston KV, Hoy J: Role of clindamycin in the treatment of central nervous system toxoplasmosis. Am J Med 1987; 83:551-554.
82. Luft BJ, Conley F, Remington JS, et al: Outbreak of central-nervous-system toxoplasmosis in western Europe and North America. Lancet 1983; 1:781-784.
83. Levy RM, Bredesen DE, Rosenblum ML: Neurological manifestations of the acquired immunodeficiency syndrome (AIDS): experience at UCSF and review of the literature. J Neurosurg 1985; 62:475-495.
84. Israelski DM, Tom C, Remington JS: Zidovudine antagonizes the action of pyrimethamine in experimental infection with Toxoplasma gondii. Antimicrob Agents Chemother 1989; 33:30-34.
85. Holliman RE: Toxoplasmosis and the acquired immune deficiency syndrome. J Infect 1988; 16:121-128.
86. Chang HR: The potential role of azithromycin in the treatment of prophylaxis of toxoplasmosis. Int J STD AIDS 1996; 7(Suppl 1):18-22.
87. Hacker M, Richter R, Gumbel H, et al: Toxoplasmosis retinochorioiditis, a therapy comparison between spiramycin and pyrimethamine/sulfadiazine. Klin Monatsbl Augenheilkd 1998; 212:84-87.
88. Ghartey KN, Brockhurst RJ: Photocoagulation of active toxoplasmic retinochoroiditis. Am J Ophthalmol 1980; 89:858-864.
89. Fitzgerald CR: Pars plana vitrectomy for vitreous opacity secondary to presumed toxoplasmosis. Arch Ophthalmol 1980; 98:321-323.