Tina Scheufele,
Jay S. Duker,
David R. Guyer,
Evangelos S. Gragoudas
HISTORICAL PERSPECTIVE
The subretinal fibrosis and uveitis syndrome is a rare distinct posterior uveitis, which was first described by Palestine and associates in 1984.[1] These authors described three patients with the unusual findings of progressive subretinal fibrosis and uveitis. The condition most commonly occurs in healthy, young, myopic females, usually with only minimal signs of ocular inflammation. Early in the disorder, multiple small, whitish-yellow retinal pigment epithelial or choroidal lesions are observed in the posterior pole and mid-periphery. In the later stages of the disease, progressive subretinal fibrosis occurs. The condition usually becomes bilateral, but often initially presents unilaterally. The first histopathologic and immunohistopathologic features of this condition were subsequently described by the same group.[2,3]
When it was first being described, the subretinal fibrosis and uveitis syndrome was given many different names and was often grouped together with other chorioretinal inflammatory diseases. Most commonly, it was grouped with multifocal choroiditis, with some believing that this was a rarely observed late stage in the spectrum of this disease.[4-6] It was also classified as a 'disciform macular degeneration of young adults' by Doran and Hamilton.[7] However, in retrospect, many of these patients probably had choroidal neovascular membranes that lead to disciform scarring, rather than the subretinal fibrosis syndrome.[7] Now, it is considered a distinctly separate entity from multifocal choroiditis, punctate inner choroidopathy, and other chorioretinal inflammatory diseases.
PATIENT CHARACTERISTICS
The subretinal fibrosis and uveitis syndrome occurs predominantly in young, healthy, myopic females. The patients are usually less than 35 years of age.[1-11] In one series of 11 patients, the age range was 24-43 years, with a mean age of 30.2 years.[8] The disease has been described in patients as young as 6 years and as old as 76 years.[4,5,9] In several series, all of the patients were female,[1,2,6,8,10] although a meta-analysis of all large published series found that 86% of all reported cases were female.[11] The patients usually have a myopic refractive error.[7,9,11] In Morgan and Schatz's series,[8] 10 of 11 patients were myopic, with a range of ?2.75 to ?8.50 D. No racial predilection has been identified.[11] Systemic evaluation of these patients is almost always unremarkable.[1-9] Cases have been reported in patients with Reiter's syndrome; migraine; atopy; schizoid syndrome; gastric bypass surgery; positive skin, serologic, and biopsy findings of histoplasmosis; and a positive purified protein derivative test for tuberculosis.[1-9] In one series, six of 11 patients were taking oral contraceptives.[8] However, all of these associations are probably unrelated to the ocular condition.
SYMPTOMS
Patients usually complain of acute vision loss, often with metamorphopsia. Scotomas and photopsias also may be reported.
OBJECTIVE FINDINGS
The vision loss may be very mild or severe, depending on the stage of the disease at which the patient presents (see Table 166.1). Early in the disease course, the visual acuity can range from 20/20 to 20/400.[6,8] As the disease progresses and subretinal fibrosis develops, the visual acuity may be decreased severely to counting-figers, light perception, or even no light perception.[10-13] The condition is usually bilateral (45-100% of cases)[1-11]; however, the fellow eye is often asymptomatic initially and may not become involved until months later. For example, in one series of five patients, one case remained unilateral, one case presented with bilateral involvement, and three cases that were initially unilateral involved the second eye within 3-6 months.[6]
TABLE 166.1 -- Findings in the Subretinal Fibrosis and Uveitis Syndrome
|
Anterior uveitis |
|
Vitritis |
|
Multifocal choroiditis |
|
Subretinal fibrosis |
|
Optic disc edema |
|
Serous and hemorrhagic macular detachment |
|
Cystoid macular edema |
|
Choroidal neovascularization |
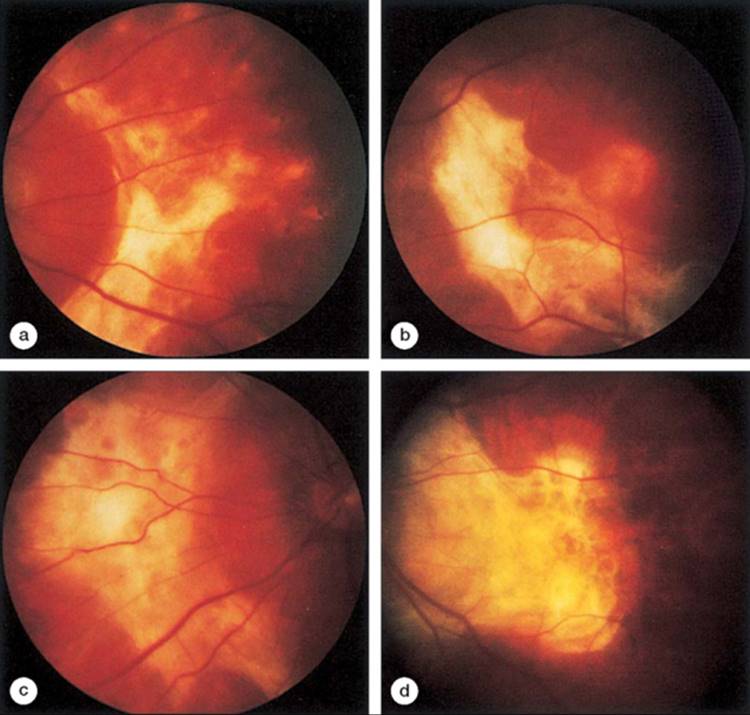
Slit-lamp examination may reveal an anterior or posterior uveitis (Fig. 166.1).[1-11] Usually, however, the inflammation is mild when present. A mild chronic vitritis is often associated with transient, multiple, small (100-500 ?m), round, discrete, yellowish white lesions of the retinal pigment epithelium or choriocapillaris in the posterior pole and mid-periphery (Fig. 166.1).[1-11] These lesions may fade or enlarge and coalesce to create multiple areas of whitish subretinal fibrosis (Fig. 166.2; see also Fig. 166.1).[1-11] The progression of subretinal fibrosis may occur over months to years, and relapses and recurrences are common.
|
|
|
|
FIGURE 166.1 This 26-year-old woman presented with decreased vision, redness, and pain in both eyes. (a) Slit-lamp examination revealed conjunctival injection, anterior chamber reaction, posterior synechiae, and a cataract. (b and c) Examination of the fundus showed subretinal fibrosis. |
|
|
|
|
FIGURE 166.2 (a-d) The extensive subretinal fibrosis that occurs in this syndrome is illustrated in this patient. |
Other findings may include optic disk edema, serous and hemorrhagic macular detachment, macular hole, cystoid macular edema, and choroidal neovascularization (CNV).[1-11] CNV may occur in up to 44% of patients at some time during the course of the disease.[5,6,8,11] In one study, CNV occurred 2-6 months after the onset of the disease, during the fibrotic stage of the disease.[5-8]
FLUOROANGIOGRAPHIC AND ELECTROPHYSIOLOGIC FINDINGS
Retinal pigment epithelial window defects or mottled hyperfluorescence of the acute lesions are observed by fluorescein angiography in the early stages of this disorder.[1-11] The subretinal fibrosis shows late staining.[1,2,6] Optic disk leakage and macular edema may be present. CNV is commonly observed later in the disease course.[5,6,8,11]
Electroretinographic signals may be depressed.[1,2] The electrooculogram may be markedly decreased[1,2] or normal.[6,8] Three patients in one series[8] had normal color vision testing results.
NATURAL HISTORY
In all but one series,[8] the visual prognosis of these patients was poor. Many of these patients had final visual acuities of 20/200 to counting-figers in the more severely affected eye.[5,6,8,10,11] Two case reports documented visual decline to no light perception.[12,13] This poor visual prognosis was confirmed in all of the other reports except for the series of Morgan and Schatz.[8] These authors noted a final vision of 20/20 to 20/70 in patients treated with oral or periocular corticosteroids and vision of 20/400 in patients who were not treated. The beneficial effect of steroids or other immunosuppressants is still controversial. Although some have noted an improvement in vision and halting of the fibrotic response when steroids were started early in the disease course, most authors agree that once subretinal fibrosis occurs, the response to corticosteroids is minimal at best and the visual prognosis is generally poor.[5,6,10,11] The eye affected first usually has the worst final visual acuity, except in cases where a subfoveal CNV develops in the second eye.[5,6,10]
Recurrences are common in this disorder and have been documented to range from two to seven recurrences per patient.
HISTOPATHOLOGIC AND IMMUNOHISTOPATHOLOGIC FINDINGS
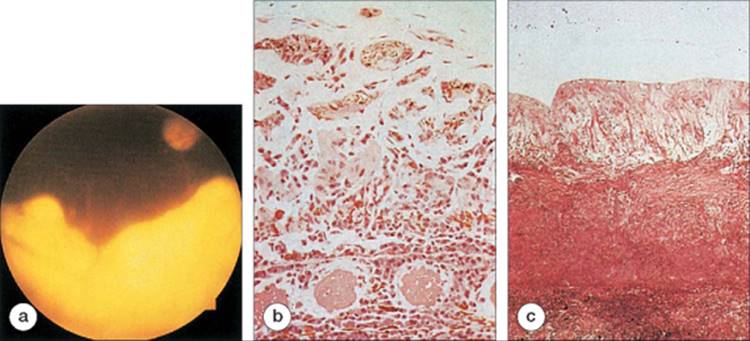
Histopathologic studies of chorioretinal biopsies from patients with this condition demonstrated a uveal inflammatory infiltrate, retinal gliosis, and subretinal fibrosis (Fig. 166.3).[2,3,12,13] The inflammatory infiltrate was composed of plasma cells and lymphocytes.[2,3,12] The subretinal space and choroid contained complement C3, immunoglobulin G, epithelioid histiocytes, multinucleated giant cells, noncaseating granulomas, and fibrin.[2,3,12] Stains for bacteria and fungi were negative, and polymerase chain reaction (PCR) did not detect any herpes viruses.[3,12]
|
|
|
|
FIGURE 166.3 (a) This 18-year-old woman presented with the subretinal fibrosis and uveitis syndrome and underwent a chorioretinal biopsy. (b) Histopathologic examination revealed a thickened choroid that contained numerous lymphocytes and plasma cells. Connective tissue replaced the normal retina. (c) Histopathologic study in another patient with the subretinal fibrosis and uveitis syndrome revealed thick, fibrous tissue interposed between the markedly gliotic retina and the choroid. |
Whereas T-cells normally make up 90% of the peripheral blood lymphocytes, T- and B-cells were found in equal numbers in the iris, ciliary body, and choroid of patients with this condition, indicating a large increase in the number of B-cells.[3] Within the T-cell population, there was a relative increase in the number of T-helper cells, whose functions include B-cell recruitment and granuloma formation.[3] Some authors have proposed an association of the subretinal fibrosis and uveitis syndrome with sympathetic ophthalmia, on the basis of histologic similarities.[12,13] However, the predominance of T-cells in sympathetic ophthalmia and B-cells in the subretinal fibrosis syndrome argues against a close association.[12,13]
By light microscopy, the retina was gliotic and markedly attenuated due to loss of photoreceptors and thinning of the bipolar cell layer.[3] The Müller cells were thickened and expressed class II antigen, suggestive of active proliferation.[3] The retinal pigment epithelium was absent and replaced by amorphous connective tissue.[3] Electron microscopy showed that this subretinal tissue contained retinal pigment epithelial cells.[3] It has been proposed that both the retinal pigment epithelial cells and the Muller cells are responsible for the subretinal fibrosis.[3]
PATHOPHYSIOLOGY
This condition is probably due to a localized autoimmune antibody-mediated inflammatory process that destroys the retinal pigment epithelium and may cause massive subretinal fibrosis.[1-3,6,12] These patients have no evidence of systemic disease. Antibodies against the photoreceptors and the retinal pigment epithelium have been detected in the serum of affected patients.[12] These antibodies, likely produced by the infiltrating plasma cells, lead to destruction of the retinal pigment epithelium and formation of fibrotic tissue.[3,12] Activated Müller cells likely stimulate retinal gliosis and further subretinal fibrosis.[3]
TREATMENT
Once subretinal fibrosis occurs, there appears to be no beneficial treatment.[6,10,11] Although controversial, steroids may be useful early in the disease. Some have documented visual improvement and halting of the fibrotic response when steroids were started during the acute phase.[5,6,8] However, many more cases have been reported where patients showed no improvement and their disease continued to progress.[10-13] In one series, there was a dramatic response to systemic and periocular corticosteroids.[8] All nine treated patients showed visual improvement after treatment (final visual acuity 20/20 to 20/70), whereas the two untreated patients had a poor visual prognosis (visual acuity of 20/400).[8] In this same series, one patient developed a CNV that appeared to respond to steroid treatment. In another case series, six of 18 patients responded with an improvement in visual acuity after treatment with steroids, but steroids failed to prevent the subfoveal progression of CNV in the one patient who declined laser treatment for an extrafoveal CNV.[5] Thus, it seems reasonable to consider steroid treatment for acute cases but not for cases in which subretinal fibrosis has already occurred.[10] Other immunosuppressants have been tried as well, but experience using these for the subretinal fibrosis and uveitis syndrome is limited. Cyclophosphamide and azathioprine have each been used in one reported case, but failed to halt the progression of disease in these two patients, who both already had extensive subretinal fibrosis at the time of treatment.[3,13]
DIFFERENTIAL DIAGNOSIS
The acute phase of this condition can mimic many entities, although the disease is easy to identify once progressive subretinal fibrosis occurs (Table 166.2). Acutely, the differential diagnosis includes sarcoidosis, presumed ocular histoplasmosis syndrome, tuberculosis, syphilis, birdshot chorioretinopathy, toxoplasmosis, fungal infections, acute posterior multifocal placoid pigment epitheliopathy, serpiginous choroidopathy, multiple evanescent white dot syndrome, punctate inner choroidopathy, diffuse unilateral subacute neuroretinitis, punctate outer retinal toxoplasmosis, sympathetic ophthalmia, acute retinal pigment epitheliitis, acute macular neuroretinopathy, and inflammatory pseudohistoplasmosis. Recently, a few authors proposed that this condition may, in fact, be a variant of sympathetic ophthalmia based on histopathologic similarities; however, the immunohistopathology is very different in these two conditions and both of the patients described in these case reports had prior intraocular surgery.[12,13] The clinical history, inflammatory reaction, and progression to subretinal fibrosis can distinguish the subretinal fibrosis and uveitis syndrome from each of these conditions. In the later stages of this disease, choroidopathies with CNV and disciform scarring, such serpiginous choroidopathy, may be confused with this entity.
TABLE 166.2 -- Differential Diagnosis in the Subretinal Fibrosis and Uveitis Syndrome
|
Sarcoidosis |
|
Presumed ocular histoplasmosis syndrome |
|
Tuberculosis |
|
Syphilis |
|
Birdshot chorioretinopathy |
|
Toxoplasmosis |
|
Fungal infections |
|
Acute posterior placoid pigment epitheliopathy |
|
Serpiginous choroidopathy |
|
Multiple evanescent white dot syndrome |
|
Diffuse unilateral subacute neuroretinitis |
|
Punctate outer retinal toxoplasmosis |
|
Sympathetic ophthalmia |
|
Acute retinal pigment epitheliitis |
|
Acute macular neuroretinopathy |
|
Inflammatory pseudohistoplasmosis |
CONCLUSIONS
The subretinal fibrosis and uveitis syndrome is a condition that generally affects young, otherwise healthy, myopic females in which progressive subretinal fibrosis occurs some time after the onset of a posterior uveitis and choroiditis. CNV and macular edema may also occur. The visual prognosis is generally poor. Corticosteroid treatment may be beneficial during the acute stages of the disorder, and chemotherapy may be considered in severe cases. However, once subretinal fibrosis occurs, the visual prognosis and response to treatment is generally poor.
The disease is probably caused by a localized autoimmune reaction in which antibodies destroy the retinal pigment epithelium and produce subretinal fibrosis. The condition is now considered a distinctly separate entity from multifocal choroiditis and panuveitis, punctate inner chorioretinopathy, and other chorioretinal inflammatory diseases. Although the acute stages of this condition may be confused with other types of choroiditis, the progressive subretinal fibrosis observed during the late stages distinguishes this disorder from most other diseases.
REFERENCES
1. Palestine AG, Nussenblatt RB, Parver LM, Knox DL: Progressive subretinal fibrosis and uveitis. Br J Ophthalmol 1984; 68:667-673.
2. Palestine AG, Nussenblatt RB, Chan CC, et al: Histopathology of the subretinal fibrosis and uveitis syndrome. Ophthalmology 1985; 92:838-844.
3. Kim MK, Chan CC, Belfort R, et al: Histopathologic and immunohistopathologic features of subretinal fibrosis and uveitis syndrome. Am J Ophthalmol 1987; 104:15-23.
4. Gass JDM, Margo CE, Levy MH: Progressive subretinal fibrosis and blindness in patients with multifocal granulomatous chorioretinitis. Am J Ophthalmol 1996; 122:76-85.
5. Dreyer RF, Gass JDM: Multifocal choroiditis and panuveitis: a syndrome that mimics ocular histoplasmosis. Arch Ophthalmol 1984; 102:1776-1784.
6. Cantrill HL, Folk JC: Multifocal choroiditis associated with progressive subretinal fibrosis. Am J Ophthalmol 1986; 101:170-180.
7. Doran RML, Hamilton AM: Disciform macular degeneration in young adults. Trans Ophthalmol Soc UK 1982; 102:471-480.
8. Morgan C, Schatz H: Recurrent multifocal choroiditis. Ophthalmology 1986; 93:1138-1143.
9. Nozik RA, Dorsch W: A new chorioretinopathy associated with anterior uveitis. Am J Ophthalmol 1973; 76:758-762.
10. Brown J, Folk JC, Reddy CV, et al: Visual prognosis of multifocal choroiditis, punctuate inner choroidopathy, and the diffuse subretinal fibrosis syndrome. Ophthalmology 1996; 103:1100-1105.
11. Kaiser PK, Gragoudas ES: The subretinal fibrosis and uveitis syndrome. Int Ophthalmol Clinics 1996; 36:145-152.
12. Wang RC, Zamir E, Dugel PU, et al: Progressive subretinal fibrosis and blindness associated with multifocal granulomatous chorioretinitis. Ophthalmology 2002; 109:1527-1531.
13. Lim W, Chee S, Sng I, et al: Immunopathology of progressive subretinal fibrosis: a variant of sympathetic ophthalmia. Am J Ophthalmol 2004; 138:475-477.