Eliot L. Berson
Retinitis pigmentosa (RP) is the name applied to a group of hereditary retinal degenerations that affect ~1 in 4000 people worldwide.[1-6] In the state of Maine in the United States, genetic types by families were found to be 19% autosomal dominant, 19% autosomal recessive, 8% X-chromosome-linked, 8% undetermined, and 46% isolates with only one affected member in a given family.[5] In England, the X-linked type has been reported to account for 16% of families and the autosomal recessive type for 7%.[4] The condition can rarely be inherited by a digenic or mitochondrial mode of transmission and uniparental isodisomy has also been reported.[7-10] Taking into account that most isolate cases are inherited by an autosomal recessive mode but some are X-linked and that most dominant and X-linked families have two or more affected members, one can estimate that 30-40% of cases are inherited as an autosomal dominant trait, 50-60% as an autosomal recessive trait, and ~10-15% as an X-linked trait in North America.
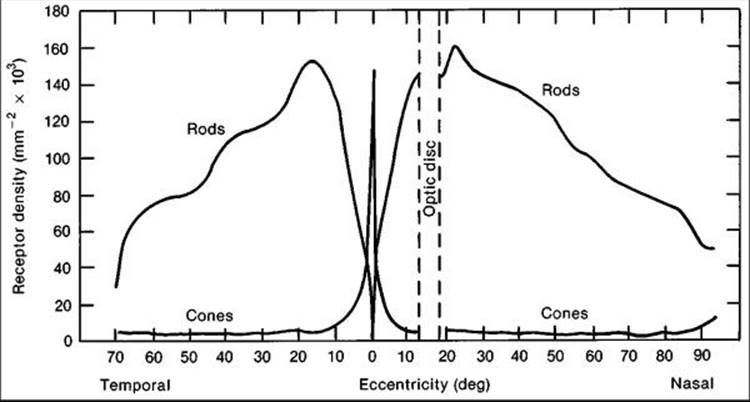
To understand the functional abnormalities that occur in these diseases, it is first important to know that the normal human retina has ~120 million photoreceptors; ~93% are rods and 7% are cones. Although cone density is highest in the central macula (Fig. 177.1), more than 90% of the cones are outside the central 10°. A patient with normal cone function and absent rod function has normal visual acuity and a full visual field even with a small (I-4e) white test light in the Goldmann perimeter. A patient with absent cone function and normal rod function has reduced visual acuity with a 1.2° diameter central scotoma but still retains a full peripheral visual field with a I-4e white test light. A patient with RP with a mid-peripheral scotoma has, by defiition, lost both cone and rod function in the area of the scotoma. When a patient reports 'night blindness' in the era of the electric light bulb, most often this symptom is related to impaired cone function. Because the cones are used most of the time, patients can lose all their rod function and not be aware of visual loss unless they are trying to see under starlight or moonlight conditions.[11]
The rapid expansion of knowledge about RP and allied night-blinding diseases precludes a comprehensive review in a single chapter. This chapter provides a framework for evaluating patients with these diseases (Table 177.1), information on some of the genes that, when mutated, cause these diseases, and a summary of treatments for some forms.
|
|
|
|
FIGURE 177.1 Distribution of rods and cones in the normal human retina. Corresponding perimetric angles from the fovea at 0° are given. |
TABLE 177.1 -- Retinitis Pigmentosa and Some Allied Diseases
|
Autosomal dominant forms of retinitis pigmentosa |
|
Autosomal recessive forms of retinitis pigmentosa |
|
Sex-linked (X-chromosome-linked) forms of retinitis pigmentosa |
|
Isolated (simplex) forms of retinitis pigmentosa |
|
Progressive cone-rod degeneration |
|
Atypical retinitis pigmentosa including sector, pericentral, paravenous, and unilateral forms |
|
Some syndromes or diseases of which retinitis pigmentosa is a part |
|
Bassen-Kornzweig syndrome (abetalipoproteinemia) |
|
Refsum disease |
|
Familial isolated vitamin E deficiency |
|
Usher syndrome types I, II, and III |
|
Laurence-Moon and Bardet-Biedl syndromes |
|
Kearns-Sayre syndrome |
|
Hereditary cerebroretinal degenerations including Batten disease |
|
Olivopontocerebellar atrophy |
|
Cockayne syndrome |
|
Alström disease |
|
Congenital amaurosis of Leber |
|
Choroideremia |
|
Gyrate atrophy of the choroid and retina |
|
Retinitis punctata albescens |
|
Clumped pigmentary retinal degenerations (enhanced S-cone syndrome in some cases) |
|
Stationary forms of night blindness |
SYMPTOMS AND SIGNS
|
Key Features: Retinitis Pigmentosa - Clinical Findings |
|||||||||||||||
|
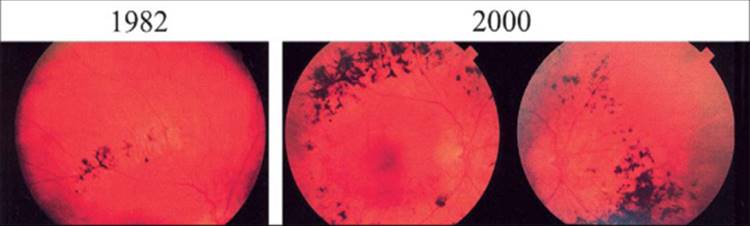
Patients with RP characteristically develop night blindness and difficulty with mid-peripheral visual field in adolescence; as their condition progresses, they develop a tendency to blue blindness, lose far-peripheral field, and eventually lose central vision often by age 60. Signs on ocular examination include narrowed retinal vessels, depigmentation of the retinal pigment epithelium, intraretinal bone spicule pigmentation, waxy pallor of the optic disks, and vitreous cells.[12-20] Posterior subcapsular cataracts develop in many cases,[21] and some patients show cystoid macular edema.[22,23] Refractive errors, including astigmatism and myopia, are common.[24,25] The characteristic bone spicule pigment is typically distributed around the mid-peripheral fundus (Fig. 177.2a) in the zone where rods are normally in maximal concentration. Histopathologic studies of autopsy eyes have shown loss of photoreceptors as well as photoreceptors with shortened or absent outer segments.[26-30]
|
|
|
|
FIGURE 177.2 Fundus photographs of moderately advanced retinitis pigmentosa (a), moderately advanced choroideremia (b), gyrate atrophy of the choroid and retina (c), Oguchi disease without dark adaptation (d), and fundus albipunctatus (e). |
ELECTRORETINOGRAMS
In 1945, Karpe discovered that patients with advanced RP had very small or nondetectable (<10 ?V) ERGs.[31] Subsequently, it was shown that patients with early RP could have subnormal but easily detectable ERGs.[32-35] Responses were not only reduced in amplitude but also delayed in b-wave implicit times,[36-39] and these ERG changes could be detected in some instances many years before diagnostic abnormalities were visible on fundus examination.[20,37] Figure 177.3 illustrates representative full-field ERGs from a normal subject and four children ages 9-14 years with early RP. Rod responses to dim blue light under dark-adapted conditions (left column) are reduced in all genetic types and when detectable are delayed in b-wave implicit times, as designated by horizontal arrows. Cone responses to 30-cps (i.e., 30Hz) white flickering light (right column) are normal or reduced in amplitude and normal or delayed in b-wave implicit times. In most cases, cone b-wave implicit times (displayed by arrows in the right column) are so delayed that a phase shift occurs between the stimulus artifacts (designated by the vertical lines) and the corresponding response peaks. Each stimulus flash elicits the next-plus-one response in contrast to the normal. In the mixed cone-rod responses to single flashes of white light under dark-adapted conditions (middle column), the cornea-negative a-wave generated by the photoreceptors is reduced in amplitude in all genetic types, pointing to the involvement of the photoreceptors in these early stages.[37]
|
|
|
|
FIGURE 177.3 Electroretinogram (ERG) responses for a normal subject and four patients with retinitis pigmentosa (RP) (ages 13, 14, 14, and 9 years). Responses were obtained after 45 min of dark adaptation to single flashes of blue light (left column) and white light (middle column). Responses (right column) were obtained to a 30-cps (or 30-Hz) white flickering light. Calibration symbol (lower right) signifies 50 msec horizontally and 100 ?V vertically. Rod b-wave implicit times in column 1 and cone implicit times in column 3 are designated with arrows. Auto. Rec., autosomal recessive. a, peak of cornea-negative a-wave; b, peak of cornea-positive b-wave. |
The subnormal responses with delayed b-wave implicit times seen in widespread progressive forms of RP contrast with the subnormal responses with normal b-wave implicit times seen in self-limited sector RP (Fig. 177.4). For example, a father and son with dominantly inherited sector RP, separated in age by almost 30 years, have comparably reduced amplitudes and normal b-wave implicit times. These patients usually have an area of intraretinal pigment confied to one or two quadrants in the periphery of each eye, with loss of peripheral rods and cones and consequent reductions in both rod and cone amplitudes. Rod b-wave implicit times are within the normal range (designated by the vertical bars), and cone b-wave implicit times are also within the normal range, as each stimulus elicits the succeeding response, as seen in the normal. The ERGs recorded from patients with sector RP are comparable to those recorded from patients with large peripheral chorioretinal scars.[36,38]
|
|
|
|
FIGURE 177.4 ERG responses of a normal subject and four patients with sector or stationary retinal disease. Horizontal arrows (column 1) designate the range of normal rod b-wave implicit times, and the vertical bar defining this range (mean ± SD) has been extended through responses of patients with sector retinitis pigmentosa (RP). Responses (middle column) from a patient with Oguchi's disease are interrupted by reflex blinking, so the latter part cannot be illustrated. Cone implicit times in column 3 are designated with arrows. SNB, stationary night blindness. |
Studies of patients with widespread progressive forms of RP with detectable rod ERGs and delayed cone b-wave implicit times have demonstrated that cone b-wave implicit time to white flicker is inversely proportional to the log amplitude of the dark-adapted rod b-wave to blue light. In these patients, cone b-wave implicit time did not vary with the amplitudes of the dark-adapted cone b-waves. A 10-fold reduction in rod amplitude was associated with ~5.5 msec slowing in cone b-wave implicit times.[39] These results would suggest that it is the loss of rod photoreceptors among remaining cones that apparently leads to abnormal rod-cone interaction,[40] which can account in large part for the delays in cone b-wave implicit times seen in progressive forms of RP.
The ERG can be used to identify not only which patients have widespread progressive forms of RP but also which patients are normal. Relatives of patients with RP, age 6 years or over, with normal rod and cone ERG amplitudes and implicit times have not been observed to develop this disease at a later time.[20,37,38,41]
CARRIERS OF X-LINKED RP
Female carriers of X-linked RP can show a patch of bone spicule pigmentation in the periphery or an abnormal tapetal reflex in the macula; among carriers of childbearing age, less than half show diagnostic abnormalities on fundus examination. ERG testing can be used as an aid in detecting female carriers of X-linked RP.[42,43] ERG testing of obligate carriers has shown that more than 90% have abnormal responses that either are reduced in amplitude to single flashes of blue or white light under dark-adapted conditions or are delayed in cone b-wave implicit times, or both, in one or both eyes. A few older obligate carriers with visual symptoms have had very small or even nondetectable full-field ERGs (Fig. 177.5).[42] Daughters of obligate carriers have had either normal ERGs or abnormal ERGs similar to those recorded from obligate carriers.
|
|
|
|
FIGURE 177.5 ERG responses from a normal subject and four obligate carriers of sex-linked retinitis pigmentosa (RP). Pt (patient) 15, age 39 years; Pt 8, age 41; Pt 4, age 51; Pt 22, age 70. Stimulus onset is designated by 'vertical hatched lines' for columns 1 and 2 and 'vertical shock artifacts' for column 3. Cornea-positivity is upward deflection. Arrows in column 3 designate cone b-wave implicit times. |
The abnormal ERGs of carriers of X-linked RP contrast with the normal full-field ERG amplitudes and normal fundi observed in obligate female carriers of autosomal recessive disease.[42] Once carrier females of X-linked RP have been detected by ophthalmoscopy or full-field ERG testing, or both, they would know that they have a 50% chance of having an affected son and a 50% chance of having a carrier daughter with each childbirth.
Female carriers of X-linked RP can have a slowly progressive retinal degeneration, although the natural course remains to be defied. Some female carriers have had considerable loss of visual field and substantial reductions in ERG amplitudes by age 70 years.
NATURAL COURSE OF RP
The ERG provides a quantitative measure of remaining retinal function and therefore can aid in defining the natural course of RP. For example, full-field ERGs have been recorded over a 20-year interval in young patients in a family with a dominant form. Amplitudes decreased over this period, and patients age 13 years or under with normal fundi and abnormal ERGs in 1967 showed fundus changes of RP and further reductions in their ERG amplitudes in 1977 (Fig. 177.6).[44] Examination of some members of this family showed further loss in 1987.[38]
|
|
|
|
FIGURE 177.6 (a) ERGs recorded in 1967 for a normal subject and four affected members from a family with dominant retinitis pigmentosa with reduced penetrance. One to three responses to the same stimulus are represented. Stimulus onset is designated by 'vertical hatched lines' in columns 1 and 2 and 'vertical shock artifacts' in column 3; cornea-positivity is upward deflection; arrows in column 3 designate cone implicit times. Calibration symbol (lower right) designates 50 msec horizontally for columns 1 and 2 and 25 msec for column 3 and 50 ?V vertically for column 1 and 100 ?V for columns 2 and 3. (b) ERGs recorded in 1977 for a normal subject and four affected members from the same family as in a for comparison with the ERGs recorded in 1967. Calibration symbol (lower right) designates 50 msec horizontally and 100 ?V vertically for all tracings. |
Signal averaging with a bipolar artifact reject buffer and bandpass filtering has extended the range of detectability of responses from affected patients. ERGs can now be recorded that are as low as 1 ?V to 0.5-Hz flashes of white light with signal averaging alone and, with bandpass filtering and signal averaging, as low as 0.05 ?V to 30-Hz white flicker. Full-field ERG function could be monitored with at least one test criterion in 90% of an outpatient population with a visual-field diameter greater than 8°, thereby making this technique useful in defining the natural course of the disease. ERGs in an adult male with X-linked RP are illustrated to show changes in ERG function over a 2-year period (Fig. 177.7).[20,45,46]
|
|
|
|
FIGURE 177.7 Computer-averaged full-field ERGs from a 26-year-old man with X-linked retinitis pigmentosa obtained in response to 10-?sec flashes of white light at 16000 ftL presented at 0.5 Hz (n = 64), 30 Hz (n = 256), and 42 Hz (n = 256) at baseline (year 0) and at 2-year follow-up (year 2). Three consecutive response averages are superimposed in each case. 'Vertical broken lines' denote flash onset for the 0.5-Hz condition and the onset of one of the trains of flashes for the 30-Hz and 42-Hz conditions. |
Among 94 patients, ages 6-49 years, with the common forms of RP, full-field ERGs declined significantly over a 3-year interval in 66 of 86 patients (77%) with detectable responses at baseline. Patients lost on average 16% of remaining full-field ERG amplitude per year to single flashes of white light (95% confidence limits 13.1-18.6%) and 18.5% of remaining amplitude per year to 30-Hz white flicker (95% confidence limits 15.1-21.5%). Patients lost on average 5.2% of remaining foveal cone ERG amplitude per year, indicating that loss of retinal function was primarily extrafoveal in these patients. They lost on average 4.6% of remaining visual-field area per year in the Goldmann perimeter with a V-4e white test light, whereas visual acuity and dark-adaptation thresholds remained relatively stable. Bone spicule pigment increased in 54% of patients for whom comparisons could be made over a 3-year interval, suggesting that observation of increased pigmentation as a means of following this condition was not as sensitive as full-field ERG testing.[45]
Caution must be exercised in applying these population ERG results to predict longitudinal patterns in individual patients, since standard deviations derived from standard errors indicated considerable variation around the mean for these patients.[45] However, these results, describing the natural course on a quantitative basis, provide a frame of reference for planning interventions in similar populations to stabilize or slow the course of RP, particularly if monitored with full-field ERG testing.
ABNORMAL ROD ERG DIURNAL RHYTHM IN RP
The changes that occur in RP can be considered not only in terms of the course over years but also in terms of abnormalities in rod function that can occur over the course of a day. Abnormal rod ERG diurnal rhythms have been observed in light-entrained patients with dominant RP; these patients had abnormal reductions in rod b-wave sensitivity 1½h and 8 h after light onset (Fig. 177.8). The abnormal reductions in rod b-wave sensitivity at 9:30a.m. and 4:00p.m. raised as one possibility that these patients with dominant RP shed abnormally large fractions of rod outer segments throughout the light period and that their rods were slow to renew their pre-light-onset outer segment length. Abnormal diurnal rhythms have also been reported in patients with autosomal recessive and isolate forms of RP. The pathogenetic mechanism that leads to these abnormal rod ERG diurnal rhythms remains to be defied.[47]
|
|
|
|
FIGURE 177.8 Mean log Ithreshold?1 of rod b-wave V = kln function versus time of day for 5 light-entrained normal subjects and 5 light-entrained patients with dominant retinitis pigmentosa (RP). Subjects were dark-adapted for 1 h before 9:30 a.m., 4 p.m., and 9 p.m. test sessions. log Ithresholds?1 were based on criterion amplitudes averaging 28.8 ?V for normal subjects and 10.6 ?V for patients with retinitis pigmentosa. 'Error bars' designate ±SEM derived from pooled estimates of population variances obtained from analyses of variance. |
MOLECULAR GENETIC STUDIES OF RP
|
Key Features: Retinitis Pigmentosa - Summary of Molecular Genetic Findings |
||||||||||||
|
More than 100 gene loci that cause RP have been mapped or identified; a current list of genes responsible for this group of diseases is maintained on the RetNet site at http://www.sph.uth.tmc.edu/Retnet/sum-dis.htm![]() . Estimated proportions of cases of RP, excluding syndromic RP other than Usher syndrome, caused by identified genes are listed in Table 177.2. The most common forms of RP are due to either mutations in the rhodopsin gene, the Usher IIA gene, or the RPGR gene. The genes listed in this table account for ~50-60% of the cases of RP in North America.
. Estimated proportions of cases of RP, excluding syndromic RP other than Usher syndrome, caused by identified genes are listed in Table 177.2. The most common forms of RP are due to either mutations in the rhodopsin gene, the Usher IIA gene, or the RPGR gene. The genes listed in this table account for ~50-60% of the cases of RP in North America.
TABLE 177.2 -- Estimated Proportions of Cases of Retinitis Pigmentosa (RP) Caused by Identified Genes (Excluding Syndromic RP Except for Usher Syndrome) (Genes currently identified account for 50-60% of cases of RP)
|
Inheritance Pattern/Gene |
Percent of all RP (including Usher Syndrome) |
|
|
Autosomal Dominant (?40% of all cases of RP) |
||
|
1. |
RHO (rhodopsin) (3q) |
10% |
|
2. |
RP1 (8q) |
2.2% |
|
3. |
PRPF31 (19q) |
2.0% |
|
4. |
PRPF3 (1q) |
1.6% |
|
5. |
peripherin/RDS (6p) |
?1% |
|
6. |
FSCN2 (17q) |
?1% |
|
7. |
PRPF8 (17p) |
0.8% |
|
8. |
IMPDH1 (7q) |
0.8% |
|
9. |
NRL (14q) |
0.4% |
|
10. |
CRX (19q) |
0.4% |
|
11. |
CA4 (carbonic anhydrase) (17q) |
? frequency in USA (?2% in UK) |
|
12. |
PAP1 (7p) |
<1% |
|
13. |
SEMA4A (semaphorin) (1q) |
<1% |
|
Autosomal Recessive (?50% of all cases of RP, including isolates) |
||
|
14. |
usherin (1q) |
?10% (based on analysis of all 72 exons in USH2A gene) |
|
15. |
PDE6B (4p) |
2% |
|
16. |
PDE6A (5q) |
1.5% |
|
17. |
CNGA1 (4p) |
1% |
|
18. |
RPE65 (1p) |
1% |
|
19. |
myosin VIIa (11q) |
?1% (Usher IB) |
|
20. |
LRAT (4q) |
<1% |
|
21. |
NR2E3 (15q) |
<1% |
|
22. |
MERTK (2q) |
<1% |
|
23. |
harmonin (11p) |
<1% (Usher IC) |
|
24. |
CDH23 (10q) |
<1% (Usher ID) |
|
25. |
PCDH15 (10q) |
<1% (Usher IF) |
|
26. |
VLGRI (5q) |
<1% (Usher IIC) |
|
27. |
clarin-1 (3q) |
<1% (Usher IIIA) |
|
28. |
SAG (arrestin) (2q) |
?0.5% |
|
29. |
CRALBP (15q) |
?0.5% |
|
30. |
RHO (rhodopsin) (3q) |
?0.5% |
|
31. |
TULP1 (6q) |
?0.5% |
|
32. |
ABCA4 (i.e., ABCR) (1p) |
<1% |
|
33. |
RGR (10q) |
<1% |
|
34. |
CRB1 (1q) |
<1% |
|
35. |
CERKL (2q) |
<1% |
|
36. |
CNGB1 (16q) |
<1% |
|
37. |
SANS (17q) |
<1% (Usher 1G) |
|
39. |
RP1 (8q) |
<1% |
|
X-linked (?10% of all cases of RP) |
||
|
39. |
RPGR (Xp21.1) |
8% (RPGR and RP2 combined account for ?90% of XLRP) |
|
40. |
RP2 (Xp11.3) |
|
|
Digenic (only a few families described to date) |
||
|
41. |
ROM1 (11q) and peripherin/RDS (6p) |
<1% |
|
Mitochondrial (only one family, with Usher type III, described to date) |
||
|
42. |
MTTS2 |
<1% |
|
Note: Percentages are approximate and are based on the breakdown of RP cases according to genetic type reported by Fishman (Arch Ophthalmol 96:822-826, 1978.), Macrae (Birth Defects: Original Article Series 18:175-185, 1982), and Bunker et al. (Am J Ophthalmol 97:357-365, 1984) and on the assumptions that all isolate cases are autosomal recessive and that about one third of cases of Usher syndrome type I (approximately 6% of all cases of RP) are caused by defects in myosin VIIa. The values are calculated based on frequency of cases in a published series multiplied by the proportion of RP with that inheritance pattern (e.g., dominant rhodopsin mutations were found in 90/363 cases of ADRP; ADRP accounts for about 40% of all cases of RP; accordingly, the percentage of cases caused by dominant rhodopsin mutations is 90/363 = 24.8% × 0.4 = 9.92 ? 10%). Table prepared 4/15/06. A current listing of genes causing retinitis pigmentosa and allied hereditary retinal diseases can be accessed on the world-wide web at:http://www.sph.uth.tmc.edu/RetNet |
The first gene abnormality found to cause RP was a single-base substitution in codon 23 of the rhodopsin gene found among 17 of 148 unrelated patients with autosomal dominant RP from across North America (Fig. 177.9). The nucleotide base change was a cytosine-to-adenine transversion (i.e., CCC to CAC) in codon 23, corresponding to a substitution of histidine for proline in the twenty-third amino acid of rhodopsin. Only clinically affected relatives showed this gene defect in the families studied. These results, coupled with the fact that proline-23 is highly conserved among normal opsins, suggested that this point mutation affected a critical amino acid in rhodopsin and that this mutation could be the cause of one form of autosomal dominant RP. This mutation was designated as rhodopsin, proline-23-histidine (i.e., Pro23His).[48]
|
|
|
|
FIGURE 177.9 Nucleotide sequence of codons 20-26 of the human rhodopsin gene derived from leukocyte DNA of a normal individual (N79) and of five representative patients with autosomal dominant retinitis pigmentosa included in this study and identified by their molecular genetic numbers AD12, AD160, AD133, AD87, and AD126. The normal subject and patient AD12 show the normal sequence, whereas the other four patients are heterozygous for the cytosine-to-adenine transversion within codon 23 (CCC to CAC). In these four patients with this mutation, the single base change can be seen as a band marked in brackets. |
Subsequent studies have revealed more than 100 additional mutations in the rhodopsin gene.[49-50] Only one mutation has been found in a given family, and each mutation has segregated perfectly with the disease in the families so far studied. All patients with rhodopsin gene mutations so far tested have had abnormal rod ERGs. None of these mutations has been found in unrelated normal subjects.
The site of the mutation in the rhodopsin gene has implications with respect to the clinical severity of the disease. A group of 17 patients from separate families with Pro23His (mean age 37 years) retained a mean visual acuity of 20/26, compared with 20/37 for the group of eight patients from separate families with Pro347Leu (mean age 32 years). Visual-field area was on average 3463 deg[2] in the Pro23His group to a V-4e white test light (normal >11399 deg[2]) and 1224 deg[2] in the Pro347Leu group. Mixed cone plus rod ERG responses to single 0.5-Hz flashes of white light were on average 14.4 ?V in the Pro23His group, almost 10-fold larger when compared with the mean amplitude of 1.7 ?V in the Pro347Leu group. Cone isolated responses to 30-Hz flicker were also 10-fold larger on average for the Pro23His group (i.e., 5.5 ?V) compared with the Pro347Leu group (i.e., 0.5 ?V). The ERG and other clinical findings taken together would support the idea that patients with Pro23His have on average a less severe disease at a given age than those with rhodopsin, Pro347Leu.[51] A study of the longitudinal course of the disease among patients with these mutations has shown that patients with Pro23His have, on average, a slower course than patients with Pro347Leu.[52]
Although patients with the Pro23His mutation retain more retinal function on average than patients with the Pro347Leu mutation, considerable variability in clinical expression exists at a given age among patients with these mutations, particularly among the group with Pro23His. This variability in clinical expression among patients with the same gene defect has raised the possibility that some factor or factors other than the gene defect itself are responsible for the expression of this condition. This could be the result of a modifier gene or environmental factors, or both. Risk factor analyses of patients with a given mutation and varying severity of disease may help to identify ameliorating or aggravating factors that may be affecting the course of this condition with possible implications for therapy.[20]
The rhodopsin (RHO) gene is ~7 kilobases (kb) in length with five exons; the gene normally encodes 348 amino acids in the rhodopsin protein. Normal rhodopsin molecules are thought to traverse the rod outer segment membrane seven times (Fig. 177.10) and are folded in three dimensions, with the first and seventh transmembrane segments in close proximity. Loops on the intradiscal side near the amino terminal tail are thought to be involved in the folding of the molecule, whereas some loops on the cytoplasmic side appear to interact with transducin as the first step in the phototransduction cascade. The normally folded rhodopsin molecule forms a pocket for vitamin A, which is bound to a lysine residue at position 296, designated by the letter K in the seventh transmembrane segment (Fig. 177.11). Mutations resulting in abnormal amino acids in the intradiscal or intramembranous domain (e.g., Pro23His) may interfere with the folding of rhodopsin and thereby modify the capacity of the pocket to hold vitamin A with consequent instability of opsin in the outer segment membranes. Mutations involving the intracytoplasmic domain (e.g., Pro347Leu) may result in abnormal transport of nascent rhodopsin to the outer segments. The reason mutations involving the rod system eventually also lead to cone photoreceptor cell death is not known. Studies of transgenic mice with these mutations as well as evaluation of cultured cell systems with these mutations should help to defie the mechanisms by which these mutations lead to photoreceptor cell death.[53]
|
|
|
|
FIGURE 177.10 Model of the normal rhodopsin protein in a rod outer segment disk membrane. Each letter signifies an amino acid residue, using the standard single-letter code. A, alanine; R, arginine; N, asparagine; D, aspartic acid; C, cysteine; E, glutamic acid; Q, glutamine; G, glycine; H, histidine; I, isoleucine; L, leucine; K, lysine; M, methionine; F, phenylalanine; P, proline; S, serine; T, threonine; W, tryptophan; Y, tryosine; V, valine. The protein, which contains 348 amino acids, traverses the outer segment disk membrane seven times, so that different portions of the molecule are in the cytoplasm, within the outer segment membrane, or in the intradiscal space. The lysine residue (K) in the seventh transmembrane domain is covalently bound to vitamin A aldehyde (i.e., 11-cis-retinal). Glycosylation sites are labeled near the N-terminal of the protein. A disulfide bond between two cysteines in separate intradiscal loops is indicated by a line. |
|
|
|
|
FIGURE 177.11 Schematic representation of a normal rhodopsin molecule folded in three dimensions to form a pocket to hold the vitamin A-derived chromophore (11-cis-retinal), which is covalently attached to a lysine residue, designated by the letter K in the seventh transmembrane segment. |
Mutations in the human retinal degeneration slow (RDS) gene on the short arm of chromosome 6 have been reported in other families with autosomal dominant RP.[54,55] All patients so far studied have had abnormal rod and cone ERGs. This gene encodes for the protein peripherin, thought to help maintain normal outer segment structure.[56] An abnormality in this gene is also known to be responsible for a retinal degeneration in a mouse model called rds. Abnormalities in the human RDS gene have been reported to be a cause not only of RP but also of retinal pattern dystrophy, retinitis punctata albescens, macular dystrophy, butterfly shaped macular dystrophy, and cone-rod dystrophy.[57-60] Even among relatives who share the same mutant RDS allele various phenotypes have been reported, including RP, pattern dystrophy, and fundus flavimaculatus.[60] Phenotypic variability may be due to an unexplained variation in the relative involvement of rods and cones and in the presence of yellow or yellow-white deposits at the level of the retinal pigment epithelium seen in only some patients.
Genes that cause RP can be subclassified into those that affect (1) the phototransduction cascade (RHO, PDE6A, PDE6B, CNGA1, CNGB1, and SAG), (2) the retinoid cycle (ABCA4, RLBP1, RPE65, LRAT, and RGR), (3) photoreceptor structure (RDS/peripherin, ROM-1, FSCN2, TULP1, CRB1, RP1, and USH2A), (4) cell-cell interactions (SEMA4A, CDH23, PCDH15, USH1C, MASS1, USH3A, and RP2), (5) RNA intron-splicing factors (PRPF31, PRPF8, PRPF3, and PAP1), (6) intracellular transport of proteins (MYO7A and SANS), (7) maintenance of cilia or ciliated cells with possible role in trafficking (BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, PTHB1, and RPGR), (8) regulation of carbon dioxide/bicarbonate balance (CA4), (9) phagocytosis (MERTK), and (10) other yet-to-be-defied functions of the photoreceptors and retinal pigment epithelium (CERKL, BBS10, and IMPDH1).[61]
TREATMENT OF THE COMMON FORMS OF RP
|
Key Features: Retinitis Pigmentosa - Treatment of Common Forms |
||||||||||||||||||
|
Many treatments have been attempted without proven benefit for the common forms of RP.[62-69] These include various vitamins and minerals, vasodilators, tissue therapy with placental extract, cortisone, cervical sympathectomy, injections of a hydrolysate of yeast RNA, ultrasound, transfer factor, dimethyl sulfoxide, ozone, muscle transplants, and subretinal injections of fetal retinal cells. A study of patients evaluated before and after receiving electrical stimulation, autotransfused ozonated blood, and ocular surgery in Cuba showed that this intervention provided no benefit; the results raised the possibility that this intervention was aggravating the course of the disease.[69]
Claims of success with one or another treatment by patients with RP that are based solely on subjective reporting of improved visual function should be interpreted with caution. Spontaneous fluctuations in acuity and field are well known in this condition. Given the slow course of the disease without treatment, it will usually require several years to assess whether or not any proposed treatment has an effect on stabilizing or slowing the course of the disease. The problem of assessing treatments may be further complicated by the genetic heterogeneity of this condition and the stage of disease at which treatment is initiated.
None of these attempts at treatment was conducted with a randomized, controlled, double-masked protocol, which is necessary to avoid possible patient or examiner biases. Most of these studies were performed without ERG data as an end-point for evaluating efficacy, so that the amount of remaining retinal function could not be quantitated in an objective manner.
Preliminary data obtained while conducting a natural history study of the course of RP revealed that those patients self-treating with a separate capsule of vitamin A or vitamin E or both were losing less retinal function than those not self-treating with these supplements.[70] This prompted a randomized, controlled, double-masked trial among 601 adult patients to determine whether supplementation with vitamin A or vitamin E, alone or in combination, would halt or slow the progression of the common forms of RP including Usher syndrome type II. Patients were randomly assigned to one of four treatment groups, as follows: vitamin A, 15000IU/day plus vitamin E, 3IU/day (Group A); vitamin A, 75IU/day plus vitamin E, 3IU/day (Group Trace); vitamin A, 15000IU/day plus vitamin E, 400IU/day (Group A + E); and vitamin A, 75IU/day plus vitamin E, 400IU/day (Group E). The main outcome variable was the 30-Hz cone flicker ERG.[70-72]
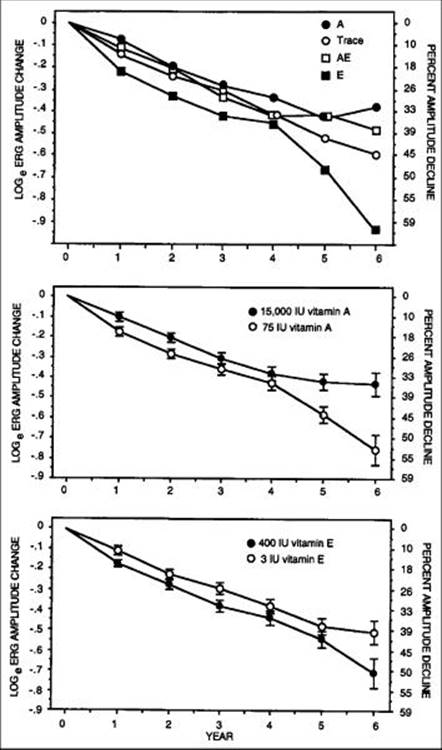
Mean annual rates of decline of remaining ERG amplitude were slowest for the group taking 15000IU/day of vitamin A and fastest for the group taking 400IU/day of vitamin E. These rates were observed among all randomized patients as well as among a subgroup of 354 patients with slightly higher initial ERG amplitudes (i.e., ? 0.68 ?V) who could be followed more reliably and who were designated 'the higher amplitude cohort'. Mean annual rates of decline of remaining 30-Hz ERG amplitude among this cohort were as follows: group A, 8.3%; group trace, 10%; group A + E, 8.8%; and group E, 11.8%. These results are summarized in Table 177.3. The treatment effect is plotted by year in Figure 177.12. Comparison of those taking vitamin A 15000IU/day versus those not on this dose revealed that the beneficial effect of vitamin A was significant at the P =.01 level for all randomized patients and at the P <.001 level for the higher-amplitude cohort. Comparison of those taking vitamin E 400IU/day versus those not on this dose revealed no significant treatment effect among all randomized patients but did show an adverse effect among those in the higher amplitude cohort (p=0.04).
TABLE 177.3 -- Mean Rates of Decline in Visual Function by Treatment Group Among Patients with the Common Forms of Retinitis Pigmentosa Supplemented with Vitamins A and E[*]
|
All Randomized Patients |
Higher Amplitude Cohort |
|||||||
|
Test |
A |
Trace |
A+E |
E |
A |
Trace |
A+E |
E |
|
30 Hz[*] |
6.1 |
7.1 |
6.3 |
7.9 |
8.3 |
10.0 |
8.8 |
11.8 |
|
0.5 Hz[*] |
8.7 |
9.6 |
9.1 |
10.6 |
8.1 |
9.4 |
8.4 |
10.9 |
|
Field Area[*] |
5.6 |
5.9 |
6.2 |
6.3 |
6.3 |
7.2 |
7.3 |
7.8 |
|
Visual Acuity[?] |
1.1 |
0.9 |
0.7 |
0.9 |
0.8 |
0.8 |
0.7 |
0.7 |
Modified from Berson EL, Rosner B, Sandberg MA, et al.: Arch Ophthalmol 1993; 111:761-772; copyright 1993, American Medical Association.
|
* |
Percent decline in remaining function per year |
|
? |
Letters lost per year |
|
|
|
|
FIGURE 177.12 Mean change from baseline over 6 years in 30-Hz ERG amplitude in the higher amplitude cohort by treatment group (top), by vitamin A main effect (middle); and by vitamin E main effect (bottom). Sample sizes for years 1 through 6, respectively, were n = 171, n = 167, n = 168, n = 164, n = 123, and n = 59 for patients receiving vitamin A, 15000 IU/day; and n = 178, n = 182, n = 172, n = 171, n = 125, and n = 64 for patients receiving vitamin A, 75 IU/day. Sample sizes for years 1-6, respectively, were n = 178, n = 177, n = 173, n = 168, n = 122, and n = 61 for the patients on vitamin E, 400 IU/day; and n = 171, n = 172, n = 167, n = 167, n = 126, and n = 62 for patients receiving vitamin E, 3 IU/day. |
Rates of decline for visual field area both for all randomized patients and for the higher-amplitude cohort showed similar trends (see Table 177.3), although the differences were not statistically significant. No significant differences were observed among groups with respect to rates of decline of visual acuity. For a subset of 125 patients with the lowest intervisit variability (i.e., ?5%), mean rates of decline of remaining visual field area per year were group A, 3.4%; group trace, 7.3%; group A + E, 4.0%; and group E, 5.3%. Comparisons of specific groups showed that the rate of decline of visual field area for group A differed significantly (p=0.03) from that of group trace, whereas the rates of decline of the A + E and E groups did not differ significantly from that of the group trace. Therefore, for this subset of 125 patients representing all genetic types, a significant beneficial effect of vitamin A supplementation on visual field area could be detected.[73]
The optimal total intake of vitamin A appeared to be ~18000 IU/day; that is, a supplement of 15000 units plus a regular diet of ~3500 units of vitamin A per day resulted in the smallest ERG decline (Fig. 177.13). Vitamin A intake greater than 18380 IU/day provided no greater benefit. Vitamin A intake of 25000 IU/day or greater is potentially toxic over the long term.[74-76]
|
|
|
|
FIGURE 177.13 Mean ± SE decline from baseline in 30-Hz ERG amplitude by total vitamin A intake (diet plus capsules) irrespective of randomization assignment for all patients in the higher-amplitude cohort. The mean decline was calculated as the mean of screening and baseline minus the mean of all follow-up visits by quintile of total vitamin A intake averaged over all visits. Sample sizes were 69, 72, 74, 65, and 74 for the lowest to highest quintiles of total vitamin A intake. Vertical bars indicate SEs. |
No case of toxicity in adults in good general health on this dose has been reported.[70-77] No significant differences in treatment effects were noted among different genetic types. The precise mechanism by which vitamin A supplementation provides its benefit is not known. It has been speculated that vitamin A rescues remaining cones, thereby explaining how one supplement may help a group of patients many of whom have different rod-specific gene defects. Vitamin E may lead to an adverse effect on the course of RP by inhibiting the absorption or transport of vitamin A.[70]
Based on the results of this trial, it is recommended that most adults with the common forms of RP should take 15000 units of vitamin A palmitate daily under the supervision of their ophthalmologists and avoid high-dose supplements of vitamin E such as the 400 units used in this trial. It is also recommended that patients continue on a regular diet without specifically selecting foods containing high levels of preformed vitamin A. The palmitate form of vitamin A is recommended, as this was the form used in this study. Other forms might be suitable, but some are probably not, for example beta-carotene, which is not predictably converted to vitamin A from one patient to another.
As a precaution, patients should be advised to have a pretreatment assessment of fasting serum vitamin A and liver function and annual evaluations thereafter. Because of the potential for birth defects, women who are pregnant or planning to become pregnant are advised not to take this dose of vitamin A. Because patients under 18 years were not evaluated, no formal recommendation for patients under this age can be made.[70]
In older adults, long-term vitamin A supplementation has been associated with a decrease in bone density and up to a 1% increased risk of hip fractures.[78,79] Therefore, postmenopausal women and men aged 49 and over who are taking vitamin A should be advised to consult with their physician about their bone health and be followed regularly in this regard. Vitamin A should not be given to patients with RP who have had renal transplants as they have excessive renal re-absorption of vitamin A and, therefore are more susceptible to toxicity. Patients on chronic doxycycline should not take vitamin A because the combination has been associated with increased intracranial pressure.
While conducting the clinical trial on vitamins A and E, data were obtained raising the possibility that docosahexaenoic acid (DHA) in addition to vitamin A could further slow the course of RP. This led to a second randomized, controlled, double-masked trial for the typical forms of RP. Patients were randomly assigned to DHA capsules (1200 mg/day) or control fatty acid capsules; all participants were given vitamin A palmitate 15000 IU/day. In this trial the main outcome variable was the total point score for the 30-2 program of the Humphrey field analyzer. Among all participants taken together, DHA supplementation by capsules did not, on average, slow the course of RP over a 4-year interval.[80] However, a subgroup analysis showed that those already taking vitamin A who also ate an omega-3 rich diet (?0.2 g/day) of which DHA is a major constituent, had a 40-50% slowing of annual decline in visual field sensitivity.[81] This omega-3 rich diet consisted of two or more 3-ounce servings of oily fish per week (i.e., salmon, tuna, herring, mackerel, or sardines). DHA is thought to facilitate the release of vitamin A from its carrier protein (interphotoreceptor retinoid binding protein, IRBP) in the subretinal space. It has been estimated that an average patient age 37 with typical RP already on vitamin A palmitate 15000IU/day who maintains this oily fish DHA-rich diet would be expected to lose virtually all central visual field sensitivity by age 78, whereas an average patient who eats less than two servings per week would be expected to lose all central visual field sensitivity by age 59.[81] In summary, whereas vitamin A alone is thought to provide seven additional years of vision, vitamin A with an oily fish diet provides almost 20 years of visual preservation for the average patient with RP who starts this regimen in their mid-30s.
About 3 months after starting an oily fish diet (thereby allowing sufficient time for red blood cell (RBC) turnover), patients with RP should have a measurement of their fasting RBC DHA to confirm that the RBC DHA level is at least 4% of total RBC fatty acids as it has been reported that such patients have, on average, a slower rate of decline of visual field sensitivity than those with lower levels.[81] If the RBC DHA level is not at least 4%, patients should be advised to consult with their physician on how to best reach this level through food. If the level is 4% or greater, this test should be repeated annually thereafter.
Although high DHA supplementation (1200 mg/day) has no benefit over a 4-year interval among patients taking vitamin A,[80] it has been reported that this dose can shorten the interval for vitamin A to achieve its benefit among those taking vitamin A for the first time.[81] Therefore, adults with RP starting vitamin A for the first time should be advised to take vitamin A palmitate 15000IU/day and DHA capsules 600 mg twice per day for 2 years (i.e., three 200 mg capsules both morning and night with meals). After 2 years, patients should discontinue DHA supplementation by capsules because no evidence was found for continued benefit and because a slight tendency toward adversity on ocular function was observed over the long-term among patients on high DHA with vitamin A.[81] After stopping DHA capsules after 2 years, patients should continue the vitamin A and eat two 3-ounce servings per week of omega- 3 rich fish. About 3 months thereafter, RBC DHA levels should be checked as described above. It should be noted that combining an omega-3 rich diet with DHA capsules did not provide any additional benefit.
Sources of vitamin A palmitate and DHA capsules as well as laboratories that perform measurements of RBC DHA are maintained on the website of the Foundation Fighting Blindness (www.blindness.org![]() ).
).
RP ASSOCIATED WITH HEREDITARY ABETALIPOPROTEINEMIA
|
Key Features: Retinitis Pigmentosa - Treatment of Rare Forms |
|||||||||
|
In 1950, Bassen and Kornzweig described an 18-year-old girl, born of first cousins, who had a malabsorption syndrome, a generalized retinal degeneration, a diffuse neuromuscular disease similar to Friedreich ataxia, and a peculiar crenation of the RBCs, now called acanthocytosis. In 1956, low serum cholesterol (<50 mg/dL) was observed.[82-86] Soon thereafter, an absence of low-density plasma lipoproteins or so-called beta-lipoproteins was found, and the term abetalipoproteinemia was assigned to this recessively inherited disorder.[87-89] Other classes of lipoproteins have also been found to be abnormal.[90]
Patients with hereditary abetalipoproteinemia can assimilate fat into the intestinal mucosa, but a defect exists in its removal from this site because of the lack of chylomicra. Intestinal biopsies have revealed normal-sized villi filled with lipid droplets that are essentially triglycerides. Mutations in the gene encoding a microsomal triglyceride transfer protein have been found in patients with this condition.[91] It appears that the liver and then the retina become depleted of vitamin A. Abnormal ERGs have been reported in children in whom the fundi were still normal.[92] The original case described by Bassen and Kornzweig showed multiple white dots in the early stages, but by age 31 years, the patient developed multiple areas of pigment epithelial cell atrophy. In other cases, the typical intraretinal pigment associated with RP has been noted in the retinal periphery.
Patients with this condition are treated with a low-fat diet and supplements of the fat-soluble vitamins A, E, and K. Vitamin A supplementation has been shown to restore elevated dark-adaptation thresholds and reduced ERG responses to normal in two patients in the early stages (Fig. 177.14).[93,94] More advanced cases have not responded, but in one such case, in which the retina was examined after the death of the patient, widespread loss of photoreceptor cells was observed.[95] Vitamin A therapy may not maintain retinal function over the long term, as patients have been reported in whom vitamin A levels have been restored to normal and yet the retinal degeneration has appeared to progress.[96] Since these patients have low serum vitamin E levels, supplementation with vitamin E in addition to vitamin A has been advocated with reported stabilization of retinal function.[97-101]
|
|
|
|
FIGURE 177.14 Full-field ERGs to red (top) and blue (middle) light, equal for rod vision, and brighter white stimulus (bottom) from a patient with hereditary abetalipoproteinemia (dark-adapted). Responses in the left column were obtained before vitamin A therapy, those in middle column at 6 h, and those on right at 24 h after vitamin A therapy. Two to three responses to the same stimulus are superimposed. Light stimulus begins with each trace. Calibration (lower right) signifies 0.06 mV vertically and 60 msec horizontally. |
RP ASSOCIATED WITH REFSUM DISEASE
Refsum disease is a recessively inherited condition in which the patient accumulates exogenous phytanic acid.[102,103] Findings include a peripheral neuropathy, ataxia, an increase in cerebrospinal fluid protein with a normal cell count, and RP. All patients have elevated serum phytanic acid. Some cases have anosmia, neurogenic impairment of hearing, electrocardiogram abnormalities, and skin changes resembling ichthyosis. The fundus can appear granular with areas of depigmentation around the periphery with a subnormal ERG in the early stages or can show more typical RP with a nondetectable ERG in more advanced stages.[104]
A defect exists in the conversion of phytanic acid to ?-hydroxy phytanic acid, specifically in the introduction of a hydroxyl group on the ?-carbon of phytanic acid (Fig. 177.15).[105] The pathogenesis appears to involve accumulation of phytanic acid in a variety of tissues, including the retinal pigment epithelium.[106]
|
|
|
|
FIGURE 177.15 Phytanic acid, its immediate precursors and metabolites, and site of enzyme defect in Refsum disease (Rd). |
Treatment consists of restricting not only animal fats and milk products (i.e., foods that contain phytanic acid) but also green leafy vegetables containing phytol.[107] Success of treatment depends on the patient's maintaining his or her body weight; if body weight becomes reduced, phytanic acid is released from tissue stores, resulting in an increase of phytanic acid in serum and exacerbation of symptoms. Refsum reported two patients whose serum phytanic acid levels were lowered to normal and who showed improvement in motor nerve conduction velocity, some relief of ataxia, and return of the cerebrospinal fluid protein to normal. Moreover, the RP and hearing impairment did not progress; one of these patients was followed for 10 years and the other for many years.[108] One young adult with a mild form of this disorder has been followed on a low- phytol, low-phytanic acid diet for 4 years; full-field ERGs, reduced ~75% below normal before commencement of this diet, have remained about the same over this period (Fig. 177.16). Long-term effects of this diet on retinal function continue to be studied.[38]
|
|
|
|
FIGURE 177.16 Full-field ERGs from a normal subject and a patient with a mild form of Refsum disease before and 4 years after treatment with a low-phytol, low-phytanic acid diet. Pretreatment responses were recorded at age 31 years. |
FAMILIAL ISOLATED VITAMIN EDEFICIENCY
Another rare recessively inherited form of ataxia associated with RP has recently been termed familial isolated vitamin E deficiency. These patients present in adulthood with Friedreich-like ataxia, dysarthria, hyporeflexia, and decreased proprioceptive and vibratory sensation as well as markedly decreased serum vitamin E levels. In later stages, patients can develop the fundus changes of RP and abnormal ERGs. Molecular genetic analysis has revealed a mutation in the alpha-tocopherol-transfer protein (alpha-TTP) gene. Oral administration of vitamin E restored serum vitamin E levels to normal and appeared to halt or slow the progression of the neurologic abnormalities and RP in three patients followed for 1, 4, and 10 years, respectively.[109,110]
OTHER FORMS OF RP
Some 15-20% of individuals with RP have associated hearing loss, sometimes referred to as Usher syndrome in recognition of the British ophthalmologist, C. H. Usher, who observed that this condition was recessively inherited.[111,112] Patients with Usher syndrome type I typically have night blindness in the first or second decade of life, profound congenital deafness (i.e., >70 dB loss at all frequencies) with unintelligible speech, and vestibular ataxia, whereas those with Usher syndrome type II usually report night blindness in the second to fourth decades, have a partial high-tone hearing loss with intelligible speech, and do not show ataxia.[113] Patients with Usher syndrome type II have abnormalities in the cilia of sperm and in the connecting cilia in many remaining photoreceptors in autopsy eyes.[114,115]Patients with Usher syndrome type III have RP with onset of hearing loss in mid-adulthood that can progress to profound deafness; some have vestibular ataxia.[116] All forms of Usher syndrome appear to be slowly progressive over the long term. Patients with Usher syndrome type II were included in the clinical trial of vitamin A[70] and DHA[80,81] and, therefore, adult patients are being treated with vitamin A palmitate 15000IU/day and an omega-3 rich diet as described earlier in this chapter.
Molecular genetic analyses have revealed gene loci for Usher syndrome type I (further subdivided into forms B through F); for Usher syndrome type II (forms A through D); and for Usher syndrome type III (form A only so far detected) (see Table 177.2). The authors who originally reported Usher syndrome type IA have subsequently found that this form does not exist as initially described.[117]
Some 2-6% of congenitally deaf children will develop Usher syndrome type I. If a child presents with profound deafness and a balance disorder manifested by late onset of walking, usually after 15 months of age, the possibility of Usher syndrome type I should be considered.[118] The diagnosis of RP as part of Usher syndrome can be made in early life with ERG testing. No tests of retinal function are yet available to identify carriers of Usher syndrome.
The Laurence-Moon-Bardet-Biedl syndrome includes RP, mental retardation, polydactylism, truncal obesity, and hypogonadism as the most frequent features.[119-122] Some authors have subdivided this syndrome into two disorders: polydactylism is found primarily in patients with the Bardet-Biedl form, whereas neurologic findings are typically observed in patients with the Laurence-Moon form.[123-124]However, some patients have been described with both polydactylism and neurologic abnormalities and therefore would seem to qualify for inclusion in both syndromes.[125,126] Variability of clinical expression is well known, and some patients may have this condition without mental retardation or polydactylism.[127] Genetic heterogeneity also exists, as the condition is caused by at least 12 different genes (BBS1, BBS2, ARL6, BBS4, BBS5, MKKS, BBS7, TTC8, PHTB1, BBS10, BBS11, and BBS12). These genes in the aggregate account for only about half of the cases of this syndrome. Renal disease can be a part of this syndrome,[127] and therefore, patients should have their blood pressure and urine checked periodically, with appropriate treatment instituted for their renal disease if detected.
Retinal degeneration is the most common feature of the Laurence-Moon-Bardet-Biedl syndrome, occurring in ~90% of cases.[127] The macula is often involved early, leading to the suggestion that these patients have an inverse form of RP. The fundus in early life may be granular without pigment formation, so that the diagnosis is established only after ERG testing.[128] A child born with polydactylism to normal parents should be a suspect for this syndrome. Once this syndrome is identified in one individual, parents would know that they have as much as a 25% chance with each succeeding childbirth of having another child with this condition. The inheritance of the Bardet-Biedl syndrome can be unusual as mutations in two unlinked Bardet-Biedl genes at one locus have been detected in some families and expression of this condition may be modified by a mutation at a second locus that may not alone cause disease.[129-131] No treatment is currently known for the retinal degeneration associated with this syndrome.
Congenital amaurosis of Leber is an autosomal recessive disorder associated with severe reduction in vision near birth and very reduced ERGs.[132-133] Patients characteristically have hyperopia and nystagmus, and fundus examination reveals granularity, white flecks, or intraretinal bone spicule pigment, or some combination of these. Carrier parents have a 25% chance with each childbirth of having a child affected with this condition. Mental retardation has been reported in some patients, possibly secondary to visual impairment. Leber congenital amaurosis involving the retina and pigment epithelium should not be confused with Leber optic atrophy, a maternally inherited condition involving the optic nerve associated in some families with an abnormality in mitochondrial DNA.[134,135]
Abnormalities in 12 genes have been so far identified as causes of Leber congenital amaurosis. Genes so far implicated are GUCY2D, CRB1, IMPDH1, RPE65, AIPL1, RDH12, RPGRIP1, CRX, TULP1, LRAT, CEP290, and LCA5. These genes account for more than half of the known cases. In one form of this condition mutations in GUCY2D (retinal guanylate cyclase 1) suggest that cGMP production in photoreceptors is abolished. Consequently, photoreceptor excitation would be expected to be impaired due to closure of cGMP-gated cation channels with hyperpolarization of the photoreceptors. It has been proposed that the cGMP concentration in photoreceptor cells cannot be restored to the dark level, leading to a situation equivalent to constant light exposure during photoreceptor development.[136] A model for this disease exists in the rd/rd chicken, which is functionally blind at hatching, with an extinguished ERG, absent guanylate cyclase activity, and a mutation in the guanylate cyclase gene.[137] Mutations in another gene, designated as RPE65, affect vitamin A metabolism in the retinal pigment epithelium.[138,139] A canine model of this condition with an RPE65 gene deficiency has been rescued with subretinal injection of the RPE65 gene carried by an attenuated adenoviral (AAV) vector.[140] Another model of Leber congenital amaurosis has been produced in mice with knockout of the RPGRIP1 gene, and rod structure and function have been restored with subretinal injection of the RPGRIP1 gene also carried in an AAV vector.[141] It remains to be established whether gene therapy can be safely used to reverse retinal malfunction in humans with Leber congenital amaurosis with remaining photoreceptor cells.
Retinitis punctata albescens can be associated with some signs and symptoms characteristic of RP.[142] Patients usually present with profound adaptational problems and gradual loss of peripheral vision. Examination with a direct ophthalmoscope reveals multiple punctate white deposits in the macula and around the mid-periphery at the level of the pigment epithelium, for which this condition was named. Patients have retinal arteriolar attenuation and some develop round areas of atrophy of the retinal pigment epithelium as well as intraretinal bone spicule pigment in the midperiphery. This raises the possibility that this condition is a variant of RP.[143] This form of retinal degeneration may affect the eyes of a patient asymmetrically; therefore, these patients often receive a neurologic evaluation in search of an intracranial abnormality before it is realized that the visual loss is due to a widespread retinal degeneration. Full-field ERGs of such patients are invariably abnormal with reductions in amplitude and delays in implicit times. The condition is usually slowly progressive, although the course appears to vary from one individual to another. This condition has been thought to be inherited by an autosomal recessive mode, but a dominant mode of transmission can occur. Therefore, ERG testing of relatives of affected patients is recommended to help establish the mode of transmission. A null mutation in the peripherin/RDS gene has been reported in one family with this condition with a dominant mode of transmission[144]; other families with an autosomal recessive mode of transmission have shown mutations in the cellular retinaldehyde-binding protein (CRALBP) gene.[145-147]
Another atypical form of RP has been designated as "progressive cone-rod degeneration."[148] Patients with this form present with reduced visual acuity, photophobia, color deficiency, and night deficiency and show signs of macular degeneration, retinal arteriolar attenuation, and in some cases, intraretinal bone spicule pigment in the peripheral fundus. Dark-adaptation testing shows elevated fial dark-adaptation thresholds across the fundus, and full-field ERGs show a profound loss of cone function and some reduction in rod function. This condition is usually inherited by an autosomal recessive mode but can also be inherited by an autosomal dominant mode. Mutations have been found in the retina-specific ATP-binding cassette transporter (ABCA4) gene,[149] the same gene that has been found to be abnormal in patients with juvenile macular degeneration (Stargardt disease).[150-152] No treatment is yet known for progressive cone-rod degeneration.
Other atypical forms of RP include pericentral, paravenous, or unilateral RP. The pericentral form is characterized by a near mid-peripheral scotoma extending from the 5-30° isopter (Fig. 177.17) (in contrast to typical RP where the field loss initially extends from the 20-40° isopter), single flash (0.5Hz) ERGs greater than 100 ?V and 30-Hz cone ERGs greater than 15 ?V initially (Fig. 177.18), and a normal or nearly normal fial dark adaptation threshold in the periphery. This form progresses at a slower rate than typical RP; in later life they can lose central vision but they retain considerable peripheral vision.[153]The paravenous form is usually characterized by slightly reduced full-field ERGs and intraretinal pigment and atrophy of the pigment epithelium confied to the distribution of the retinal veins in each eye. In some cases the macula degenerates as well (Fig. 177.19). Patients with paravenous disease may lose central vision but, like pericentral RP, they retain peripheral vision into later life.[154] Unilateral RP is characterized by fundus changes of RP in one eye with no evidence of RP in the fellow eye. Full-field ERGs are substantially reduced in the affected eye and normal in the fellow eye. Patients with unilateral RP have progressive disease in the affected eye but have not been observed to develop a generalized form of RP in the fellow eye at a later time. Pericentral RP can be inherited by a recessive or dominant mode while paravenous and unilateral RP have been found characteristically in patients with no family history of this condition. No evidence exists that patients with pericentral, paravenous, or unilateral RP will benefit from vitamin A treatment.
|
|
|
|
FIGURE 177.17 Fundus photographs for a patient with pericentral retinitis pigmentosa to show an increase in pericentral bone-spicule pigmentation over an 18-year interval. |
|
|
|
|
FIGURE 177.18 Visual fields to the V-4e white test light and full-field electroretinograms to 0.5 Hz and 30 Hz flashes spanning 22 years from a patient with pericentral retinitis pigmentosa to illustrate disease progression. |
|
|
|
|
FIGURE 177.19 Fundus photographs of the right eye of a patient at age 22 years (left) and at age 39 years (right) with pigmented paravenous retinochoroidal atrophy. At age 22 years, the fundus showed paravenous intraretinal bone spicule deposits overlying areas of pigment epithelial and choroidal atrophy with some disturbance of the retinal pigment epithelium in the macula. At age 39 years, progression can be seen with more paravenous pigment and some clumped pigment in the macula. |
An estimated 0.5% of patients with RP have a clumped pattern of pigmentation in the mid-peripheral fundus, sometimes referred to as 'clumped pigmentary degeneration'. These patients characteristically have hyperopia, night blindness, and reduced and delayed full-field ERGs. Humphrey static perimetry with blue stimuli on a yellow background to evaluate short-wave (S) blue-cone function and white stimuli on a white background to evaluate function mediated by all three cone types has revealed that some of these patients with clumped pigment have relatively enhanced blue-cone function and such patients are therefore considered to have the enhanced S-cone syndrome.[155] Molecular genetic analyses have revealed mutations in the NR2E3 gene which is thought to encode a ligand-dependent transcription factor[156] or in the NRL gene which encodes a basic motif-leucine zipper protein.[157] Patients with enhanced S-cone syndrome have been shown to have shared mutations in the NR2E3 gene with patients with Goldmann-Favre syndrome. Patients with NR2E3 mutations appear to inherit this condition by an autosomal recessive mode while those with NRL mutations can inherit this condition by a dominant or recessive mode. Patients with the enhanced S-cone syndrome appear to have a slowly progressive disease with gradual loss of peripheral field. No treatment is yet known.
CHOROIDEREMIA
Young males with choroideremia characteristically have normal visual acuities, minimally increased dark-adaptation thresholds, and full visual fields to large test lights at a time when granularity and depigmentation of the retinal pigment epithelium are seen around the fundus periphery. ERGs are reduced in amplitude with delays in b-wave implicit time (Fig. 177.20). In more advanced stages in adulthood, dark-adaptation thresholds are further increased and the visual fields become constricted at a time when ERGs are profoundly reduced or nondetectable and extensive choroidal atrophy and clumped pigment are visible around the peripheral fundus (see Fig. 177.2b). Males usually retain little, if any, central vision beyond the age of 60 years.[158,159]
|
|
|
|
FIGURE 177.20 ERG responses of four males with choroideremia. Cone flicker responses to white 30-cps flicker remained detectable in the oldest male when rod responses to blue light were not detectable. Normal responses are shown for comparison. Time of stimulus onset is designated by 'vertical hatched lines' in columns 1 and 2 and vertical shock artifacts in column 3. Arrows and vertical bar on rod responses to blue light show the range of normal b-wave implicit times. Responses to single flashes of white light show reduced a- and b-wave amplitudes in all males. Arrows on white flicker responses show delayed implicit times in the affected males. |
Obligate female carriers of this X-linked disease may demonstrate fundus changes that include patchy depigmentation of the retinal pigment epithelium and coarse pigment granularity or even pigment clumps in the periphery. However, carriers typically retain normal visual acuities and normal fial dark-adapted rod thresholds.[159-161] Generally, carriers have been thought to have a nonprogressive course, but carriers with severe disease have been described.[159] In contrast to the carrier state of X-linked RP, ERGs of carriers of choroideremia usually are normal,[159] although reduced amplitudes have been reported in some cases.[159,161]
Study of a patient with choroideremia with a large deletion helped to assign the choroideremia gene to a small segment of the long arm of the X-chromosome, specifically in the Xq21 band.[162,163] The gene (CHM) responsible for this condition, encodes Rab escort protein 1 (REP-1).[164] Many mutations in this gene have been described subsequently.[165,166] Loss of the REP-1 protein is thought to compromise Rab protein prenylation; the lack of prenylation, in turn, would affect protein trafficking inside the retinal pigment epithelium or choroidal cells triggering the degenerative process in this condition.[167-169] An animal model of this condition has been created and is under study.[170]
Interfamilial and intrafamilial variability of clinical expression is known to exist in this disease. The natural course of choroideremia on a year-to-year basis is being investigated. Correlations, if any, between specific mutations and clinical expression of the disease remain to be completed. No treatment is yet known.
GYRATE ATROPHY OF THE CHOROID AND RETINA
|
Key Features: Gyrate Atrophy of the Choroid and Retina - Diagnosis and Management |
||||||||||||
|
Gyrate atrophy of the choroid and retina is a rare chorioretinal degeneration inherited by an autosomal recessive mode of transmission.[171,172] Patients usually report night deficiency and loss of peripheral vision in adolescence. Ocular findings include myopia, constricted visual fields, elevated dark-adaptation thresholds, very small or nondetectable ERG responses, and chorioretinal atrophy distributed circumferentially around the peripheral fundus (see Fig. 177.2c) and sometimes near the optic disk. In addition to the ocular findings, abnormalities in electroencephalograms, muscle and hair morphology, and mitochondrial structure in the liver have been reported.[173] Enlargement, coalescence, and posterior extension of areas of atrophy have been observed in young patients within 2 years.[174] Patients develop cataracts in young adulthood and often require surgery. If untreated, patients usually become virtually blind between the ages of 40 and 55 years as a consequence of extensive chorioretinal atrophy.[175]
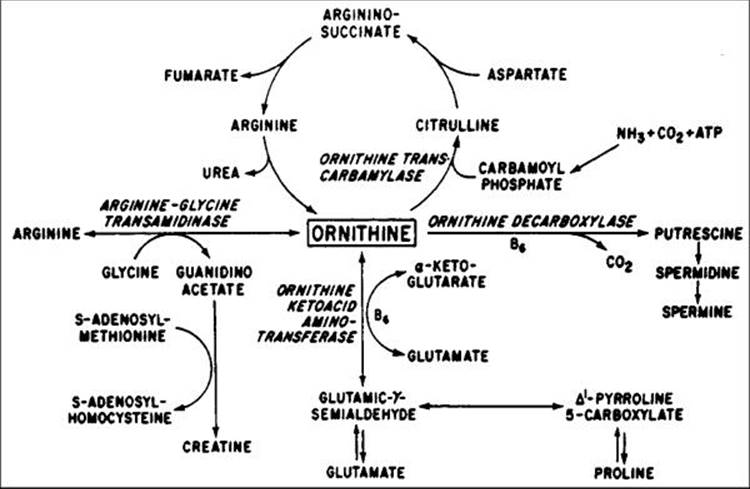
Patients with this condition have plasma ornithine concentrations elevated 10- to 20-fold above normal[176,177] due to a deficiency of ornithine aminotransferase (OAT) activity (Fig. 177.21).[178-180] Affected individuals cannot convert ornithine to pyrroline-5-carboxylic acid (PCA); this deficiency can be detected in extracts of cultured skin fibroblasts. Patients have virtually no OAT activity in contrast to normal persons, whereas carrier parents have a partial deficiency. Cultured cells from some patients have shown increased OAT activity when the cofactor for OAT, namely pyridoxal phosphate or vitamin B6, is added to the media in increased concentration. Plasma lysine[181] and glutamate and glutamine,[182] as well as serum and urine creatine,[183] have also been found to be reduced. More than 60 mutations have been discovered in the OAT gene on chromosome 10 in affected patients.[184]
|
|
|
|
FIGURE 177.21 Pathways of ornithine metabolism. |
Since arginine is a precursor of ornithine, and since arginine, but not ornithine, is a constituent of food protein, it has been suggested that dietary restriction of protein and arginine will reduce plasma ornithine levels in these patients.[185] OAT-deficient mice produced by gene targeting develop a retinal degeneration over several months that is amenable to treatment postweaning with an arginine-restricted diet.[186]
The hyperornithinemia associated with human disease has been lowered toward normal with a low-protein, low-arginine diet in all cases so far studied[185,187,188] and with vitamin B6 (300 to 500 mg/day) in some cases.[189-191] However, extreme protein restriction (10-15 g/day) with substantial lowering of plasma ornithine, accomplished under supervision in the hospital, has been difficult to achieve at home, and therefore, many of the patients have followed modified (20-35 g/day) protein restriction with slight rises in their plasma ornithine levels. Some investigators have reported improvements in visual acuity, visual fields, dark-adaptation thresholds, or ERGs in patients with gyrate atrophy after onset of either the diet or vitamin B6,[187] whereas others have not documented any significant improvement in visual function despite substantial reductions in plasma ornithine concentrations.[191] Improvement in muscle morphology after creatine supplementation has been reported in several patients.[192]
The effect of severe arginine restriction has been evaluated in two pairs of siblings younger than 10 years who were followed for 5-7 years.[193] The plasma ornithine levels were reduced to approximately the normal range (106 and 121 ?mol/L) in one pair of siblings and reached twice the upper limit of normal (251 and 313 ?mol/L) in the other pair. These younger siblings had significantly less atrophy than their elder siblings when they reached or approached the same age at which their elders had begun the diet. Thus, there is evidence that a low-protein, low-arginine diet can slow progression of the chorioretinal degeneration, but only a few patients have so far been studied.
The goal of treatment is to maintain plasma ornithine levels as close to normal as possible. Since some patients may respond to supplementation with pyridoxine (vitamin B6), all patients are initially given a trial of this vitamin to determine to what extent, if any, it will lower plasma ornithine levels. Both pyridoxine-responsive and nonresponsive patients are then placed on a low-protein, low-arginine diet. Based on plasma ornithine levels, biochemical control has been classified as good to excellent (<200 ?mol/L), fair (200 to 400 ?mol/L), or poor (>400 ?mol/L). In the management of children, expertise is required to be certain that growth and development remain normal while lowering ornithine levels with a low-protein diet. An arginine-free essential amino acid mixture (e.g., Cyclinex-1 or -2 [Ross Laboratories], depending on the patient's age) is used to provide sufficient nitrogen and meet essential amino acid requirements. In adults, a low-protein diet is also likely to result in amino acid deficiency. Thus, adults should also be placed on an arginine-free essential amino acid mixture. Lysine supplementation may be necessary, depending on plasma levels. As a precaution, all patients are placed on a multivitamin preparation with minerals. In addition to a regular ocular examination, all patients on this treatment regimen should have their amino acid and protein levels monitored periodically.[194]
OTHER RETINAL DEGENERATIONS
Retinal degenerations involving the photoreceptors across the retina can exist in association with other systemic findings. If a patient presents with ophthalmoplegia and retinal degeneration, the Kearns-Sayre syndrome should be considered. Abnormalities include ptosis, chronic progressive external ophthalmoplegia, a disturbance of the retinal pigment epithelium, ERGs usually reduced in amplitude, and respiratory distress. Some patients develop heart block and may require a pacemaker.[195,196] The diagnosis is confirmed by the observation of ragged red fibers on muscle biopsy. This syndrome has been associated with mitochondrial DNA mutations; treatment of one patient with coenzyme Q10 and succinate resulted in clinical improvement of respiratory distress.[197] The value, if any, of this treatment for retinal malfunction remains to be established.
Patients who present with a widespread loss of photoreceptor function with abnormal ERGs and symptoms of cerebral deterioration may have a cerebroretinal degeneration, grouped under the overall heading of neuronal-ceroid lipofuscinosis, or Batten disease. This group of diseases, which are recessively inherited, is characterized by accumulation of lipopigments (lipofuscin and ceroid) in neurons and other cell types. Batten disease can be subdivided into an infantile form (psychomotor deterioration by age 2 years, ataxia, microcephaly, granular inclusions on conjunctival biopsy), a late-infantile form (seizures, rapid mental deterioration in early childhood, curvilinear inclusions on biopsy), a juvenile form (mental deterioration beginning around age 6 years, bull's-eye maculopathy, figerprint inclusions on conjunctival biopsy), and an adult-onset form (seizures, slow dementia, abnormal inclusions best seen on muscle biopsy).
Molecular genetic studies have revealed that the juvenile onset form of Batten disease results from defects in the CLN3 gene on chromosome 16p; the majority of patients have a 1.2-kb deletion in this gene on at least one chromosome.[198] The gene encodes a novel protein of unknown function. Studies have suggested that the mechanism of apoptosis is involved in the demise of both neurons and photoreceptors.[199] No treatment exists for the retinal degeneration in this condition.
Patients with olivopontocerebellar atrophy can present with a history of tremors, ataxia, and dysarthria and show findings of oculomotor impairment and retinal degeneration. This condition is inherited by a dominant mode with variable penetrance. Some patients have retinal arteriolar attenuation, a diffuse disturbance of the retinal pigment epithelium, and very reduced ERGs. Some patients may show only neurologic findings and have normal fundi and normal retinal function, whereas others have had abnormal ERGs with no diagnostic findings on fundus examination and no history of neurologic disease. Of all the known forms of spinocerebellar atrophy, if there is associated photoreceptor degeneration, almost all are due to mutations in the SCA-7 gene. In general, each succeeding generation is more severely affected and has an earlier onset and increase in severity is more common if the condition is inherited from the father. This condition is presently untreatable.[200-202]
Other rare syndromes associated with retinal degeneration include Alström disease and Cockayne syndrome. Patients with Alström disease have RP with profound loss of vision in the first decade of life and very reduced ERGs. Systemic findings include diabetes mellitus, obesity, deafness, renal failure, baldness, and hypogenitalism.[203,204] Patients with Cockayne syndrome show extensive loss of vision by the second decade of life and have RP, dwarfism, deafness, mental deterioration, and premature aging. No treatments are known for these conditions.[205,206]
STATIONARY FORMS OF NIGHT BLINDNESS
|
Key Features: Stationary Forms of Night Blindness |
||||||||||||
|
Stationary night blindness can be inherited by an autosomal dominant, autosomal recessive, or X-linked mode. Some patients can have a normal fundus appearance. Two forms, Oguchi disease and fundus albipunctatus, have characteristic fundus abnormalities (see Fig. 177.2d and e). It is the normality or near-normality of the cone system in the full-field ERGs that allows separation of most stationary forms of night blindness from practically all forms of night blindness associated with the early stages of RP (see Fig. 177.4).
The original classification of these diseases was based on the family of origin (e.g., Nougaret type) or by the initial description of the ERG waveforms associated with the condition (e.g., Riggs or Schubert-Bornschein type), or by the ophthalmologist who first described the disease (e.g., Oguchi disease). These diseases are now being reclassified by molecular genetic analysis.[207]
Missense mutations in three different genes encoding members of the rod phototransduction cascade have been reported to cause dominantly inherited stationary night blindness. Specifically, the mutations are the Ala292Glu and Gly90Asp mutations in the rhodopsin gene;[208,209] the His258Asp mutation in the beta-subunit of rod phosphodiesterase gene;[210] and the Gly38Asp mutation in the alpha-subunit of rod transducin gene.[211] The latter mutation has been seen only in the descendants of Jean Nougaret. Whereas the Ala292Gly rhodopsin mutation apparently causes a complete loss of rod function (i.e., Riggs type),[208] the other mutations cause an incomplete loss of rod function.[211,212]
Some evidence exists from psychophysical and ERG testing that the rods are constitutively active in the dark in some forms of stationary night blindness. In the rhodopsin, Gly90Asp mutation, psychophysical measures with Stiles two-color increment threshold technique show the equivalent of light adaptation in the dark owing to rhodopsin activation in darkness.[208] The ERG abnormalities in patients with the Gly38Asp mutation in the alpha-subunit of rod transducin can be simulated by light-adapting the normal retina, compatible with the idea that these patients have mutant rod transducin that is constitutively active in the dark.[212] Evidence that patients with stationary night blindness have rod photoreceptors despite their night blindness is supported by fundus reflectometry studies that have shown normal levels of rhodopsin and normal rhodopsin kinetics.[213]
Oguchi disease is a recessively inherited form of stationary night blindness. Patients usually require 2-12 h to attain normal dark-adapted rod thresholds and show a characteristic change from a golden brown color of the fundus in the light-adapted state (see Fig. 177.2d) to a fundus of normal color in the dark-adapted state (the 'Mizuo phenomenon'). After 1 h of dark adaptation, patients with Oguchi disease have no rod b-wave in response to dim blue light flashes, a cornea-negative response to white light, and a normal cone response to 30-Hz white flicker. Following complete dark-adaptation after 12 h, some patients have a normal rod b-wave amplitude and normal rod b-wave implicit time, but only in response to one or two flashes of light.[214] Apparently, the test flash used to elicit the ERG, although relatively dim, can be intense enough to light-adapt the rod system. Molecular genetic analyses have shown a null mutation, Asn309(1-bp del), in the gene encoding arrestin in some patients of Japanese descent.[215] Several null mutations have also been described in the gene encoding rhodopsin kinase in some patients of Eastern European descent.[216] Both arrestin and rhodopsin kinase participate in the deactivation of photoactivated rhodopsin. The lack of either arrestin or rhodopsin kinase would be expected to have the same physiologic effect, namely the prolongation of photoactivated rhodopsin with a consequent abnormality in rod sensitivity.
Fundus albipunctatus is another form of recessively inherited stationary night blindness.[217,218] Patients have yellow-white deposits in the deep retina outside the macula (see Fig. 177.2e). Some develop macular degeneration in later life. Patients with fundus albipunctatus have a delay in rod visual pigment and foveal cone pigment regeneration as monitored with fundus reflectometry.[217,219] Changes in visual pigment levels parallel the prolonged cone and rod limbs of the dark-adaptation curve. Full-field ERGs have been found to be normal with respect to both cone and rod amplitudes and b-wave implicit times after full dark-adaptation.[218] The findings in fundus albipunctatus suggest an abnormality in visual pigment regeneration secondary to some abnormality in the relationship between the photoreceptors and the pigment epithelium. The composition of the yellow-white lesions and their relationship to the abnormality in visual pigment regeneration are not known. Mutations in the RDH5 gene compromising the function of 11 cis-retinol-dehydrogenase have been found to be the cause of this condition.[220,221]
Another form of stationary night blindness is congenital nyctalopia with myopia. This condition (i.e., Schubert-Bornschein type) is inherited by either an X-linked or an autosomal recessive mode.[222-224] The extent of the myopia is characteristically ?3.5 to ?14.5 D. Patients cannot attain normal dark-adapted rod thresholds and have fundus findings of myopia. Their rod ERG b-wave responses to dim blue light are nondetectable, and they show a characteristic cornea-negative response to white light in the dark-adapted state and normal or nearly normal cone responses to 30-Hz white flicker (see Fig. 177.4). The preservation of the a-wave from the rod photoreceptors with loss of the b-wave from neurons proximal to the photoreceptors suggests some abnormality in intraretinal transmission of the response from the rod photoreceptors to proximal retinal cells. ERG amplitudes are reduced in congenital nyctalopia with myopia compared with those from normal emmetropes; this probably occurs because of the known reductions of ERG amplitude seen in patients with moderate axial myopia as the only fiding.[225] Patients with X-linked congenital nyctalopia have been further subdivided into the complete or incomplete form. In the complete form, the rod a-wave is normal and the rod b-wave is absent and responses are mediated by the cone system. This form appears to be caused by abnormalities in the NYX gene, whose gene product appears to be involved in ON-bipolar cell signaling.[226,227] In the incomplete form rod ERG responses, although reduced in amplitude, are still measurable to dim blue light, and the cone ERG is also subnormal. The incomplete form appears due to mutations in the CACNAIF gene; patients with this condition have ERG abnormalities due to malfunction of both the ON- and OFF-bipolar pathways. Whereas patients with the complete form have moderate or high myopia, those with the incomplete form are thought to have mild myopia or slight hyperopia.[228] Whereas the complete form appears to be stationary, the long-term course of patients with the incomplete form remains to be established.
Congenital night blindness associated with a negative ERG waveform has also been described in some patients with mutations in the GRM6 gene encoding the glutamate receptor mGluR6. This receptor is present only in the synapses of the ON-cell bipolar dendrites and it mediates synaptic transmission from rod and cone photoreceptors to this second order neuron. Patients are night blind at an early age and have acuities that are normal or slightly reduced. In addition to reduced b-waves, ERG responses to saw-tooth flickering light indicate a reduced ON-response and a nearly normal OFF-response. The condition appears inherited by an autosomal recessive mode and is thought to be stationary or very slowly progressive.[229]
The ERG abnormality in congenital nyctalopia with myopia has been reported as an acquired defect in patients with cutaneous malignant melanoma, who report an acute onset of night blindness with selective reduction in the rod b-wave response (Fig. 177.22).[230] Development of an antibody to the melanoma that also reacts to retinal cells with an interruption in rod signal transmission has been considered as a probable mechanism to account for the sudden onset of night blindness in these patients.
|
|
|
|
FIGURE 177.22 Full-field ERG responses from a normal subject, from the right (OD) and left (OS) eyes of a patient with malignant melanoma, and from a patient with congenital stationary night blindness with myopia (CSNB). Stimulus onset is designated by the 'vertical hatched lines' in columns 1, 2, and 3 and the 'vertical lines' superimposed on the responses in column 4. Two or three consecutive responses are illustrated, and cornea-positivity is an upward deflection. The peak of the cornea-negative a-wave, generated by photoreceptors, and the peak of the cornea-positive b-wave, generated by activity of cells proximal to the photoreceptors, are designated in the response of the normal subject to single flashes of white light. Calibration symbol (lower right) designates 50 msec horizontally, 200 ?V vertically for the top recording in column 3, and 100 ?V vertically for all other tracings. Both patients lack the cornea-positive b-wave from the rod system in columns 1, 2, and 3 in contrast to the normal, while retaining normal cone amplitudes (?50 ?V) in their responses to 30-Hz white flickering light in column 4. |
Another form of congenital night blindness is that seen in patients with impaired cone adaptation and elevated fial rod thresholds which has been termed 'bradyopsia'.[231] Patients with this condition have mutations in the genes encoding RGS9 or its anchor protein R9AP with consequent slow photoreceptor deactivation. RGS9 specifically deactivates the G-proteins (transducins) in the rod and cone phototransduction cascades. It is anchored to photoreceptor membranes by the transmembrane protein R9AP which enhances RGS9 activity up to 70-fold. Patients present in childhood with photophobia and have difficulty adapting to sudden changes in luminance levels under photopic conditions. Visual acuity is either normal or moderately subnormal and can fluctuate. Patients have difficulty seeing moving objects especially with low contrast despite full visual fields. Fundus findings are unremarkable. Diminution in cone 30-Hz flicker ERG responses to successive flashes of light should suggest the possibility of this condition. Findings in patients followed for as long as 25 years suggest that this condition is stationary.
It is of interest that stationary forms of night blindness, with impaired rod function in early life but no evidence of rod cell death, are not associated with intraretinal pigment migration or attenuation of the retinal vessels, whereas these latter findings are characteristically seen in patients with rod cell degeneration and RP. Although the stationary or near stationary forms of night blindness are rare, it is important to identify these conditions when patients present with night blindness on a retinal basis, as the long-term prognosis in these patients is relatively good, in contrast to the progressive retinal degeneration of rods and cones seen in patients with night blindness associated with RP.
APPROACH TO THE PATIENT
Patients with known or suspected RP or an allied disease should be asked to provide a history of the age of onset of night blindness, visual-field loss, and loss of visual acuity. Often symptoms of night blindness reflect problems in adaptation under dim photopic conditions, and therefore suggest cone malfunction. Patients with a sudden onset of night blindness, particularly after the age of 50 years, should be considered suspects for a paraneoplastic process (i.e., nonocular malignancy with associated retinal malfunction), or so-called cancer-associated retinopathy.[232-234] Difficulties in reading may reflect a generalized loss of cone function or loss of cone function confied to the macula.
The medical history should include information on operations and illnesses as well as current medications and nutritional supplements. Intestinal surgery or liver disease may have affected the absorption or metabolism of vitamin A. Chronic intake of some medications (e.g., hydroxychloroquine, phenothiazines) may have compromised retinal function. The medical history should include information on the status of hearing, as some 10-15% of patients with RP have a high-tone partial hearing loss, and some 5% have profound congenital deafness. A history of polydactyly, mental retardation, or renal disease, if elicited, should raise the possibility of an association of these abnormalities with a retinal degeneration. A detailed family history is always warranted to determine whether other members of the family are affected with the retinal degeneration and to determine whether or not consanguinity exists in the parents, as this raises the likelihood of a hereditary disease.
The ocular examination should include a refraction; some 50% of patients with RP have 1 or more diopters of astigmatism in the less astigmatic eye.[25] Uncorrected myopia can be a cause of night blindness, and therefore it is important to establish that the patient has the correct prescription. Reading vision should be tested and lenses prescribed as needed to ensure that the patient can read at least 8-point print. After an assessment of acuity and refractive error, a visual field should be performed with either the Goldmann perimeter or the Humphrey field analyzer. If the Goldmann perimeter is used, it is recommended to test with both a small (e.g., I-4e or II-4e) and large (e.g., IV-4e or V-4e) white test light, as many patients do not have sufficient remaining function to detect the small light. If the Humphrey field analyzer is used, use of the size III or size V white target is advised using both the 30-2 and 60-2 programs to assess the central and the mid-peripheral visual fields. Color vision testing should then be performed, preferably without dilation and under standard lighting conditions. The Farnsworth panel D15 and the Ishihara plates are adequate for initial screening for red and green color deficiencies; a tritan axis of confusion (i.e., blue blindness) can occur in patients with RP. After these tests, applanation pressures are measured; elevated pressures (>35 mm Hg) can result in diminished ERG amplitudes.
Pupils are then maximally dilated with phenylephrine hydrochloride and cyclopentolate hydrochloride and the patient patched. After 30-45 min of dark adaptation, fial dark-adaptation thresholds are measured to establish whether the patient has impaired rod function. After dark-adaptation testing, full-field ERG testing is performed. If the responses are easily detected without computer averaging, the test can be accomplished within 10-15 min per eye; if computer averaging is required, the testing may take 20-30 min per eye. This sequence of evaluations helps to ensure that the patients have their dark-adaptation and ERG testing before more extensive light exposure associated with ophthalmoscopy that might light-adapt the patient and thereby affect the dark-adaptation and ERG measurements.
After the results of dark-adaptation and ERG testing are obtained, patients are examined with pupils fully dilated, at which time the refractive error can be confirmed by retinoscopy and glasses modified, if necessary. A slit-lamp examination and direct and indirect ophthalmoscopy are performed; fie retinal deposits (as, e.g., those seen in retinitis punctata albescens) and slight retinal arteriolar attenuation may be appreciated with direct but not indirect ophthalmoscopy. In the case of patients with cataracts, a retinal acuity can be obtained with a retinal potential acuity meter or equivalent instrument to compare retinal acuity with distance acuity.
In the case of isolate males with RP when X-linked disease is suspected, the patient's mother should be evaluated, if possible, with both a fundus examination and ERG testing, to determine whether she shows signs of a carrier state of X-linked disease and, if affected, how much retinal function remains. Carriers of X-linked RP have abnormal full-field ERGs in one or both eyes in more than 90% of cases, whereas carriers of autosomal recessive disease have normal fundi and normal ERG amplitudes. Establishment of a diagnosis of X-linked disease can be helpful in determining the visual prognosis of a male patient with RP, since untreated males with X-linked disease often become virtually blind by age 30-45 years, whereas males with autosomal recessive disease often become virtually blind after age 50-60 years.
If the diagnosis of a common form of RP is established, adult patients should be advised that their condition is treatable with vitamin A palmitate.[70-73] Before treatment is initiated, eligible patients should have a fasting serum vitamin A level and liver function profile performed to be certain these values are normal before starting vitamin A. Information should be provided as to available sources of the correct dose of vitamin A palmitate (i.e., 15000 IU/day for adults). (For current sources of vitamin A palmitate see www.blindness.org![]() ). The patient should be advised that higher doses provide no additional benefit and that doses of 25000 IU/day or greater are potentially toxic over the long term. All patients should be advised of the possible adverse effect of high-dose (i.e., ? 400 IU/day) vitamin E supplementation.[70] Adult patients already taking vitamin A should be advised to eat two 3-ounce servings of oily fish per week of which DHA is a major constituent. Adult patients starting vitamin A for the first time should be encouraged to take DHA gelcaps 600 mg twice a day for 2 years at which time they should stop the DHA supplement, continue to take vitamin A, and begin to eat two 3-ounce servings of oily fish (e.g., salmon, tuna, mackerel, or sardines of which DHA is a major constituent) per week. Once oily fish is begun, they should have a fasting red blood cell DHA (RBC DHA) level 3 months after starting consumption of oily fish (i.e., to give time for the red blood cells to renew themselves) to confirm that this level is at least 4% of total RBC fatty acids. Patients should have a fasting serum vitamin A, liver function profile, and RBC DHA annually thereafter. Whereas vitamin A alone is thought to slow the annual rate of decline of remaining retinal function by 20%, the combination of vitamin A plus an oily fish diet is thought to slow the amount of remaining function by an additional 40-50%.[81] Precautions with regard to the safe use of vitamin A are reviewed earlier in this chapter in the section on The Treatment of Retinitis Pigmentosa.
). The patient should be advised that higher doses provide no additional benefit and that doses of 25000 IU/day or greater are potentially toxic over the long term. All patients should be advised of the possible adverse effect of high-dose (i.e., ? 400 IU/day) vitamin E supplementation.[70] Adult patients already taking vitamin A should be advised to eat two 3-ounce servings of oily fish per week of which DHA is a major constituent. Adult patients starting vitamin A for the first time should be encouraged to take DHA gelcaps 600 mg twice a day for 2 years at which time they should stop the DHA supplement, continue to take vitamin A, and begin to eat two 3-ounce servings of oily fish (e.g., salmon, tuna, mackerel, or sardines of which DHA is a major constituent) per week. Once oily fish is begun, they should have a fasting red blood cell DHA (RBC DHA) level 3 months after starting consumption of oily fish (i.e., to give time for the red blood cells to renew themselves) to confirm that this level is at least 4% of total RBC fatty acids. Patients should have a fasting serum vitamin A, liver function profile, and RBC DHA annually thereafter. Whereas vitamin A alone is thought to slow the annual rate of decline of remaining retinal function by 20%, the combination of vitamin A plus an oily fish diet is thought to slow the amount of remaining function by an additional 40-50%.[81] Precautions with regard to the safe use of vitamin A are reviewed earlier in this chapter in the section on The Treatment of Retinitis Pigmentosa.
At the initial examination, every effort should be made to establish the diagnosis; consider the best possible optical devices to maximize remaining vision; explain the genetic implications, where indicated; and offer referral to appropriate psychologic, social, and community resources. In most cases, follow-up examinations are conducted every 2 years to help determine the course of the disease as monitored by visual acuity, visual fields, and ERGs (computer averaged in most cases); to adjust glasses and provide low-vision aids, if required; and to consider whether cataract surgery is indicated. Cataract surgery is usually deferred in the case of RP until a time when the patient can-not read with either eye. A trial with dilating drops (e.g., homatropine 2% taken at dusk) may be attempted to see whether the patient can meet her or his visual needs without surgery. Cataract extraction has the greatest potential for improving vision among those patients who have large enough cataracts to impair visualization of the fundus with the ophthalmoscope, detectable ERGs with computer averaging as a measure of some remaining retinal function, and evidence of retinal acuity that is better than distance acuity.
|
Key Features: Retinitis Pigmentosa - Other Treatment Considerations |
|||||||||||||||
|
Some 10-25% of patients with RP develop cystoid macular edema. In some cases the edema can be subclinical and only detected with optical coherence tomography (OCT). Acetazolamide and Acular drops have been advocated for patients with RP with cystoid macular edema; some given these medications have shown an improvement in acuity and others have claimed subjective improvement without measurable change in acuity within 1 month. Topical dorzolamide 2% can provide benefit. Intravitreal steroid injections have also been used for severe edema. The value of these treatments over the longer term for such patients remains to be established.[235,236]
For patients under age 40 years with autosomal recessive RP, serum lipoprotein electrophoresis and serum phytanic acid can be assessed after fasting for 12 h if Bassen-Kornzweig syndrome or Refsum disease is suspected. A fasting serum vitamin A level can be obtained if nutritional deficiency or intestinal malabsorption is suspected. A serologic test for syphilis should be performed if this diagnosis is suspected, as RP has been associated with this disease. A fasting serum vitamin E level should be considered in patients with the signs of RP who also report the onset of ataxia in adulthood, as some success has been reported with vitamin E supplementation for this rare disorder.
Discussion of results of the ocular examination with the patient, and with the patient's family if indicated, must be individualized for each case; but in any event, every effort should be made to answer the patient's questions faithfully and provide a clear description of not only what abnormalities exist but also how much function remains. In this regard, the measurement of any ERG function is an advantage, as the patient can be shown in positive terms how much function remains. The use of computer averaging and narrow bandpass filtering is particularly important, as most patients with remaining vision have detectable cone ERGs with use of this specialized technology (which can detect responses as low as 0.05 ?V) but will show nondetectable responses with conventional full-field ERG systems (which can detect responses only as low as 10 ?V). The demonstration of remaining retinal function has prognostic implications. For example, assuming an exponential rate of decline of remaining cone ERG function, the average patient with 1.3 ?V at age 32 would be expected to retain some useful vision (i.e., greater than or equal to 0.05 ?V) until age 63 without treatment or until age 83 with vitamin A supplementation plus an oily fish diet. A patient with 2.6 ?V at age 32 would be expected to retain useful vision until age 69 without treatment or until age 89 with vitamin A and oily fish. Knowledge of the amount of remaining cone function quantified by full-field ERG testing is therefore helpful in estimating the long-term visual prognosis.[237]
It is important to impress on the patient that these conditions in general progress at a very slow rate. Remarks such as "Your vision may last for 2 years or 30 years" should be avoided, as the patient hears only the more severe prognosis and does not understand that in most instances he or she may well retain vision for 20 to 40 years or more after the initial evaluation, particularly in the case of RP or choroideremia. It is often useful to hear what the patient believes he or she was told previously and to try to address any misconceptions. For example, some patients believe that retinal dystrophy and degeneration are 'different diseases' and erroneously conclude that none of the physicians agree on the diagnosis.
Literature provided to the patient at the time of the visit is extremely important, as most patients cannot assimilate all the information given to them verbally. Printed information describing their condition, patterns of inheritance, reasons why they should be followed up over time by their ophthalmologist, the significance of molecular genetic studies, and the like should be available for distribution in the office. Patients may wish to receive copies of the results of their ocular examinations; visual displays of their ERG responses as part of this report can be helpful in demonstrating to patients the amount of retinal function remaining.
When a child is detected with RP or an allied retinal degeneration, parents should be provided with a full written report. If the parents agree, then the child should be provided with sufficient verbal information at the time of the examination to allow the child to understand her or his own symptoms. For example, if the child is night deficient, the child should be advised to be careful when engaging in activities under conditions of dim illumination. If the child has a deficiency in side vision, the child should be advised that it may be difficult if not unsafe to play certain sports (e.g., baseball or hockey). A child may well be legally blind due to loss of acuity and yet still be able to read with magnifying lenses; if this child has sufficient visual field, he or she may well be able to continue functioning in a regular school. It is psychologically helpful to both the child and the parents to emphasize how much visual function remains rather than how much has been lost and to defer conclusions about long-term visual prognosis until the child has been examined several times over 2-year intervals. In the case of children aged 6-18 years with the common forms of RP and normal serum vitamin A and liver function, reduced doses of vitamin A palmitate adjusted for age and weight may be considered on a trial basis with the agreement of the parents and close follow-up by the pediatrician and ophthalmologist. As a general guideline, for children above the fifth percentile in weight for a given age, sex, and height, 5000 IU/day of vitamin A palmitate may be tried for those between ages 6 and 10 years, 10000 IU/day for those between 10 and 15 years of age, and 15000 IU/day thereafter. Sources of reduced doses of vitamin A palmitate can be found at www.blindness.org![]() . As noted earlier in this chapter in the section on treatment of RP, beta-carotene is not a suitable substitute for vitamin A palmitate in the context of this treatment as it is not predictably converted into vitamin A. Children should also be encouraged to eat 1-2 servings of oily fish per week. However, it must be re-emphasized that patients younger than age 18 were not included in our randomized trials and therefore no formal recommendations can be made.
. As noted earlier in this chapter in the section on treatment of RP, beta-carotene is not a suitable substitute for vitamin A palmitate in the context of this treatment as it is not predictably converted into vitamin A. Children should also be encouraged to eat 1-2 servings of oily fish per week. However, it must be re-emphasized that patients younger than age 18 were not included in our randomized trials and therefore no formal recommendations can be made.
NIGHT VISION POCKETSCOPE
Advances in electro-optical technology have resulted in night vision devices that allow patients with RP, choroideremia, stationary night blindness, and allied diseases to use their cones to function under scotopic (starlight or moonlight conditions) or, if necessary, under dim photopic conditions that exist at night near streetlights or in a slightly darkened room.[238,239] These light-amplifying devices have been incorporated into a monocular pocketscope or binoculars that can be used as an aid to alleviate the symptom of night blindness. The instruments contain bright-point-source protection so that a patient can view a dimly illuminated scene in the presence of automobile headlights or street lights. Figure 177.23 illustrates a hand-held monocular pocketscope. The instrument provides a patient with a 40-degree field of view and gives the patient her or his best daylight vision at night in one eye.
|
|
|
|
FIGURE 177.23 Top, Model LV6015 night vision pocketscope adapted for patients with night blindness (top and left). The instrument can also be used when held in the hand (right). Middle, Schematic diagram of a generation III night vision pocketscope. The pocketscope contains a light-emitting diode to provide supplementary illumination within 3 m and is powered by two AA batteries. It weighs 13.8 oz and is 4.5 inches long × 2 inches wide × 2.25 inches high. Bottom, Single-stage image intensifier tube, 3 cm in length, contained in the night vision pocketscope. |
Candidates for these devices should have a best-corrected vision of better than 20/200 and a central visual-field diameter of greater than 20° in at least one eye. Patients considering a monocular device should also have good mobility in daylight with one eye patched. Patients who depend heavily on a far-temporal crescent for mobility are not good candidates; nor are patients who, because of a central scotoma, cannot view the output screen in these devices. Patients should be advised to assess the value of the device for their routine activities before making a decision as to whether or not to obtain it.
OTHER OPTICAL AIDS
Some patients with RP have reduced acuity. A closed-circuit television for magnification is helpful in many instances, particularly since patients can view the reading material as white on a black background, thereby reducing glare. For those who have difficulty tracking a line, a ruler or cutout window may be of value. Patients with night deficiency should be encouraged to carry a small penlight. Magnifying lenses put into a glasses frame are also helpful. Bifocals are to be avoided in patients with very constricted visual fields, as the lower segment may cut the visual field such that patients fid them less than useful. In the author's experience, field wideners have had limited if any value for patients with RP and allied conditions.
GENETIC COUNSELING
If RP is dominantly inherited (i.e., three consecutive generations with father-to-son transmission), each affected patient has a one in two chance with each childbirth of having a child of either sex with this condition. If RP is inherited by an autosomal recessive mode (i.e., at least two comparably affected female siblings or male and female siblings comparably affected, with normal parents, or an isolate case with a family history of consanguinity), an affected patient has about a one in 80 chance of marrying a carrier with this condition and the carrier has a one in two chance of passing on the abnormal gene. Therefore, the chance of a patient with an autosomal recessive form of RP having an affected child is ~1 in 160 with each childbirth (1/80 × 1/2). If a male has X-linked RP or choroideremia, all of his sons will be normal and all of his daughters will be carriers. If a woman is a carrier of X-linked RP or choroideremia, she has a 50% chance of having an affected son and a 50% chance of having a carrier daughter with each childbirth. Patients with isolate (simplex) RP (i.e., no known affected family members) can be considered to be recessive, although exceptions exist. All offspring of males or females with autosomal recessive RP are carriers of this condition; carriers enjoy normal vision but have about a one in 320 chance of having an affected child with each childbirth (i.e., one in 80 chance of marrying a carrier and, if married to a carrier, one in four chance of having an affected child; 1/80 × 1/4 = 1/320). In the case of Usher syndrome type I, Usher syndrome type II, Leber congenital amaurosis, or other autosomal recessive diseases, once a couple has one affected child they have a one in four chance with each succeeding childbirth of having another affected child.
In families with only one affected male, RP can be inherited by an autosomal recessive or X-linked mode; ERG testing and fundus examination of the mother can help to determine whether she is a carrier of X-linked disease. A careful family history and examination of relatives of an affected patient with ERG testing can aid in establishing this mode of transmission. It is important to recognize that RP may skip generations and yet be transmitted by a dominant mode; the dominant mode with variable penetrance should be suspected in patients who have large cone ERGs with substantial delays in implicit time under age 20 years.
If a pedigree with multiple affected members is inconclusive as to whether the disease is transmitted by a recessive or a dominant mode, a digenic mode of inheritance should be considered. In digenic transmission, two unlinked mutations (neither of which results in RP) cause this disease in patients with both mutations. Affected individuals can have asymptomatic parents but a 25% chance of having an affected child with each birth. The first example of digenic transmission of RP involved a mutation in the peripherin/RDS gene on chromosome 6 (Leu185Pro) and one or another mutation in the rod outer segment membrane 1 (ROM1) gene on chromosome 11; both genes encode proteins responsible for maintaining photoreceptor outer segment structure. The discovery of this mode of transmission provides an explanation on a molecular genetic basis for the pattern of inheritance described clinically as dominant RP with reduced or incomplete penetrance.[7]
Patients often state that their children have had normal routine eye examinations and therefore must not have RP or an allied disease. Some young people may have these diseases and yet appear normal on routine ocular examination. By age 30 years, more than 90% of patients can be diagnosed with the ophthalmoscope, but under age 30 years, ERG testing is indicated if the patient wants to be certain whether any relatives are affected. The ERG identifies those who are affected and those who are normal; in families with RP, patients aged 6 years and over with normal ERGs would not be expected to develop the condition at a later time.
Recent advances with molecular genetic techniques undoubtedly will facilitate genetic counseling. For example, some 20-25% of patients with autosomal dominant RP in the United States have rhodopsin gene mutations detectable through analysis of leukocyte DNA. Patients identified through analysis of DNA would have an ocular examination including ERG testing to confirm the diagnosis and to determine the amount of remaining retinal function, as variable clinical expression at a given age is known to exist among patients with the same rhodopsin gene mutation. Patients with dominant RP due to rhodopsin Pro23His can differ by up to 1000-fold in their ERG amplitudes at a given age; therefore defiition of severity of disease cannot be determined simply by a molecular genetic analysis. Leukocyte DNA analysis should be considered not only for families with a known dominant mode of transmission over three consecutive generations but also for families with transmission over two consecutive generations or for isolate cases with presumably normal parents. Some of these patients may have a rhodopsin gene mutation described in dominant pedigrees, thereby helping to establish that the mode of inheritance is dominant. It is important to establish the correct genetic type in families with RP, not only for the sake of providing accurate genetic counseling but also as a guideline in establishing long-term visual prognoses of affected patients, as patients with dominantly inherited disease generally retain vision longer than those with autosomal recessive or X-linked disease.
PSYCHOLOGIC AND VOCATIONAL COUNSELING
Patients identified with progressive retinal degenerations often need psychologic counseling to help adjust to the knowledge that they can expect to lose their vision over time. Initial expressions of anger, frustration, despair, or depression are normal reactions in such patients, and they may need counseling in order to come to terms with their disease. Similarly, parents of affected patients may feel profound guilt when they learn about the genetic implications of the disease and may need psychologic counseling. Patients and their family members frequently benefit from a support group where they can talk to others who have similar problems. Patients may also need guidance in selecting an appropriate vocation that they can pursue with their present vision and can continue to pursue in the event their vision fails. To this end, guidance provided by a trained specialist can be extremely helpful. It is very important that the patient be advised to return every 1-2 years to see an ophthalmologist. Patients with these diseases need periodic contact with their ophthalmologists to receive information on the course of their disease, particularly if they are being treated with vitamin A plus an omega-3 rich fish diet; to obtain, wherever possible, optical aids to maximize use of remaining vision; and in some cases, to receive psychologic support.
SUNGLASSES
General agreement exists that patients with hereditary degenerations of the retina should avoid excessive exposure to bright sunlight outdoors until more is known about whether light stress will modify the course of these conditions.[240,241] Two patients with RP who wore an opaque scleral contact lens over one eye for 8-10 h per day for 5 years showed comparable degeneration in both eyes, suggesting that this type of protection from light did not alter the course of their disease.[241] Some patients have expressed a preference for orange photochromic sunglasses (e.g., Corning CPF 550) with tinted side shields or dark amber sunglasses (e.g., NoIR brand worn over an existing prescription) with tinted side shields. However, no particular type of sunglasses has been shown to alter the course of retinal degeneration. Until more is known, patients should be advised to select sunglasses for outdoor use that provide maximal comfort without compromising vision.
SOME FUTURE DIRECTIONS WITH REGARD TO THERAPY
|
Key Features: Retinitis Pigmentosa - Some Future Directions that May Lead to Treatments |
||||||||||||||||||
|
Some preliminary evidence exists that neuroprotection with a ciliary neurotrophic growth factor (CNTF) will substantially slow the course of retinal degeneration in animal models of RP. A phase I study in humans with RP that used encapsulated cell technology with intravitreal sustained release of CNTF showed that CNTF could be well-tolerated over 6 months with a few patients reporting improved vision.[242]This has set the stage for a phase II-III study. Preliminary work has also been done with implantation of light sensitive microchips on or under the retina in patients with advanced RP; it is not yet established whether patients can achieve useful vision with these devices.[243,244] Retinal transplantation of photoreceptor cells under the retina has also been advocated, but no convincing data exists that the transplanted cells form viable synaptic connections with the inner retina.[245]
Increased knowledge of the gene abnormalities and biochemical defects involved in these conditions provide new opportunities to discover additional therapies for hereditary retinal diseases. In the case of recessively inherited mutations which usually result in 'loss of function', patients typically develop disease due to loss of a critical protein, and gene replacement therapy could result in restoration of the missing protein. For example a deficiency in the RPE65 gene results in loss of the isomerase needed for production of 11-cis-retinal in the retinal pigment epithelium; this deficiency leads to a form of Leber congential amaurosis or early onset recessive RP. Gene therapy has been achieved in animal models of this condition[140,246,247] and trials of human therapy have begun. In dominantly inherited mutations or so-called 'gain of function mutations', the encoded amino acid sequence is altered and can result in accumulation of abnormal proteins leading to toxicity in retinal cells. In this instance treatment is aimed at eliminating the mutant gene with the expectation that the normal copy of the gene will encode sufficient normal protein. Research is being done involving ribosome-based or RNAi-based therapy to reduce expression or inactivate specific abnormal alleles.[248] With appropriate preliminary data continued study of nutritional interventions is also warranted.
Other strategies to promote photoreceptor survival include attempts to interfere with apoptosis and thereby prevent photoreceptor degeneration or use of a calcium channel blocker to counter the effects of cyclic GMP elevation that occurs in some degeneration with consequent opening of cGMP-gated channels. In the former case some success has been achieved with animal models[249,250] but none so far in humans. In the latter case an initial claim of success with a calcium channel blocker in an rd mouse model of retinal degeneration[251] was not confirmed by further studies of this channel blocker, namely D-cis-diltiazem, in this and other animal models.[252-254] Some research has been done with embryonic stem cells in mice and rats that were found to integrate into the host retina with reported protection of host retinal neurons.[255,256] It is not known whether stem cells will have clinical applications in the treatment of these diseases.
ERG testing, using computer averaging and narrow bandpass filtering, provides a basis for objectively monitoring the course of retinal degeneration in most patients with RP and allied diseases. Variability of clinical severity at a given age among patients with the same gene defects suggests that a modifier gene or some factor(s) other than the gene defects alone may be involved in the clinical expression of some forms of these diseases.[257] Evaluation of subgroups of patients with known gene defects prospectively over a period of years may reveal ameliorating or aggravating factors with possible implications for therapy. Collaborations between scientists and clinicians should enhance the likelihood of developing new treatments for these diseases.[258]
ACKNOWLEDGMENTS
This work is supported in part by the National Eye Institute, EY00169, Bethesda, MD and in part by the Foundation Fighting Blindness, Owings Mills, MD.
REFERENCES
1. Ammann F, Klein D, Franceschetti A: Genetic and epidemiological investigation of pigmentary degeneration of the retina and allied disorders in Switzerland. J Neurol Sci 1965; 2:183-196.
2. Boughman JA, Conneally PM, Nance WE: Population genetic studies of retinitis pigmentosa. Am J Hum Genet 1980; 32:223-235.
3. Hu DN: Genetic aspects of retinitis pigmentosa in China. Am J Med Genet 1982; 12:51-58.
4. Jay M: On the hereditary of retinitis pigmentosa. Br J Ophthalmol 1982; 66:405-416.
5. Bundy S, Crews SJ: Wishes of patients with retinitis pigmentosa concerning genetic counseling. J Med Genet 1982; 19:317-318.
6. Bunker CH, Berson EL, Bromley WC, et al: Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol 1984; 97:357-365.
7. Kajiwara K, Berson EL, Dryja TP: Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994; 264:1604-1608.
8. Mansergh FC, Millington-Ward S, Kennan A, et al: Retinitis pigmentosa and progressive sensorineural hearing loss caused by a C12258A mutation in the mitochondrial MTTS2 gene. Am J Hum Genet 1999; 64:971-985.
9. Rivolta C, Berson EL, Dryja TP: Paternal uniparental heterodisomy with partial isodisomy of chromosome 1 in a patient with retinitis pigmentosa without hearing loss and a missense mutation in the Usher syndrome type II gene USH2A. Arch Ophthalmol 2002; 120:1566-1571.
10. Thompson DA, McHenry CL, Li Y, et al: Retinal dystrophy due to paternal isodisomy for chromosome 1 or chromosome 2, with homoallelism for mutations in RPE65 or MERTK, respectively. Am J Hum Genet 2002; 70:224-229.
11. Berson EL: Visual function testing: clinical correlations. In: Tasman W, Jaeger E, ed. Foundations of clinical ophthalmology, Philadelphia: J.B. Lippincott; 1991:1-10.reprinted in J Clin Neurophys 11:472-481, 1994; updated Vol. 2, Chapter 14. Hagerstown, MD: Lippincott Williams & Wilkins; 2001: 1-12.
12. Donders FC: Beiträge zur pathologischen Anatomie des Auges. Graefes Arch Clin Exp Ophthalmol 1857; 2:106-118.1855; 1:139;
13. Leber T: Die Pigmentdegeneration der Netzhaut und mit ihr verwandte Erkrankungen. In: Graefe A, Saemisch T, Hess C, ed. Handbuch der Gesamten Augenheilkunde, 2nd edn, Leipzig: W Engelmann; 1916:1076-1225.
14. Bell J: Retinitis pigmentosa and allied diseases of the eye. In: Pearson K, ed. Treasury of human inheritance, 2. London: Cambridge University; 1922:1-28.
15. Franceschetti A, François J, Babel J: Les Héredodégenerescences Chorio-rétiniennes, Paris, Masson, 1963.
16. Duke-Elder S, Dobrie JH: Diseases of the retina. In: Duke-Elder S, ed. System of ophthalmology, Vol. 10. St. Louis: CV Mosby; 1967.
17. Marmor MF, Aguirre G, Arden G, et al: Retinitis pigmentosa, a symposium on terminology and methods of examination. Ophthalmology 1983; 90:126-131.
18. Pruett RC: Retinitis pigmentosa: Clinical observations and correlations. Trans Am Ophthalmol Soc 1983; 81:693-735.
19. Heckenlively JR: Retinitis pigmentosa, Philadelphia, JB Lippincott, 1988.
20. Berson EL: Retinitis pigmentosa. The Friedenwald lecture. Invest Ophthalmol Vis Sci 1993; 34:1659-1676.
21. Heckenlively JR: The frequency of posterior subcapsular cataract in the hereditary retinal degenerations. Am J Ophthalmol 1982; 93:733-738.
22. Fetkenhour CL, Choromokos E, Weinstein J, et al: Cystoid macular edema in retinitis pigmentosa. Trans Am Acad Ophthalmol Otolaryngol 1977; 83:515-521.
23. Fishman GA, Maggiero JM, Fishman M: Foveal lesions seen in retinitis pigmentosa. Arch Ophthalmol 1977; 95:1993-1996.
24. Sieving PA, Fishman GA: Refractive errors of retinitis pigmentosa patients. Br J Ophthalmol 1978; 52:625-633.
25. Berson EL, Rosner B, Simonoff EA: Risk factors for genetic typing and detection in retinitis pigmentosa. Am J Ophthalmol 1980; 89:763-775.
26. Kolb H, Gouras P: Electron microscopic observations of human retinitis pigmentosa dominantly inherited. Invest Ophthalmol Vis Sci 1974; 13:487-498.
27. Szamier RB, Berson EL: Retinal ultrastructure in advanced retinitis pigmentosa. Invest Ophthalmol Vis Sci 1977; 16:947-962.
28. Szamier RB, Berson EL, Klein R, et al: Sex-linked retinitis pigmentosa: ultrastructure of photoreceptors and pigment epithelium. Invest Ophthalmol Vis Sci 1979; 18:145-160.
29. Bunt-Milam AH, Kalina RE, Pagon RA: Clinical-ultrastructural study of a retinal dystrophy. Invest Ophthalmol Vis Sci 1983; 24:458-469.
30. Ben-Arie-Weintrob Y, Berson EL, Dryja TP: Histopathologic-genotypic correlations in retinitis pigmentosa and allied diseases. Ophthalm Genet 2005; 26:91-100.
31. Karpe G: The basis of clinical electroretinography. Acta Ophthalmol 1945; 23(Suppl):1-118.
32. Goodman G, Gunkel RD: Familial electroretinographic and adaptometric studies in retinitis pigmentosa. Am J Ophthalmol 1958; 46:142-172.
33. Gouras P, Carr RE: Electrophysiological studies in early retinitis pigmentosa. Arch Ophthalmol 1964; 72:104-110.
34. Berson EL, Gouras P, Gunkel RD: Rod responses in retinitis pigmentosa, dominantly inherited. Arch Ophthalmol 1968; 80:58-67.
35. Berson EL, Gouras P, Gunkel RD, et al: Dominant retinitis pigmentosa with reduced penetrance. Arch Ophthalmol 1969; 81:226-234.
36. Berson EL, Gouras P, Hoff M: Temporal aspects of the electroretinogram. Arch Ophthalmol 1969; 81:207-214.
37. Berson EL: Retinitis pigmentosa and allied diseases: Electrophysiologic findings. Trans Am Acad Ophthalmol Otolaryngol 1976; 81:659-666.
38. Berson EL: Electroretinographic findings in retinitis pigmentosa. Jpn J Ophthalmol 1987; 31:327-348.
39. Birch DG, Sandberg MA: Dependence of cone b-wave implicit time on rod amplitude in retinitis pigmentosa. Vision Res 1987; 27:1105-1112.
40. Sandberg MA, Berson EL, Effron MH: Rod-cone interaction in the distal human retina. Science 1981; 212:829-831.
41. Berson EL: Retinitis pigmentosa, the electroretinogram, and Mendel's laws. Trans Pa Acad Ophthalmol Otolaryngol 1973; 26:109-113.
42. Berson EL, Rosen JB, Simonoff EA: Electroretinographic testing as an aid in detection of carriers of X-chromosome-linked retinitis pigmentosa. Am J Ophthalmol 1979; 87:460-468.
43. Fishman GA, Weinberg AB, McMahon TT: X-linked recessive retinitis pigmentosa: Clinical characteristics of carriers. Arch Ophthalmol 1986; 104:1329-1335.
44. Berson EL, Simonoff EA: Dominant retinitis pigmentosa with reduced penetrance: Further studies of the electroretinogram. Arch Ophthalmol 1979; 97:1286-1291.
45. Berson EL, Sandberg MA, Rosner B, et al: Natural course of retinitis pigmentosa over a three-year interval. Am J Ophthalmol 1985; 99:240-251.
46. Andréasson SOL, Sandberg MA, Berson EL: Narrow-band filtering for monitoring low-amplitude cone electroretinograms in retinitis pigmentosa. Am J Ophthalmol 1988; 105:500-503.
47. Sandberg MA, Baruzzi CM, Hanson AH III, et al: Rod ERG diurnal rhythm in some patients with dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci 1988; 29:494-498.
48. Dryja TP, McGee TL, Reichel E, et al: A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature 1990; 343:364-366.
49. Dryja TP, McGee TL, Hahn LB, et al: Mutations within the rhodopsin gene in patients with autosomal dominant retinitis pigmentosa. N Engl J Med 1990; 323:1302-1307.
50. Berson EL, Rosner B, Sandberg MA, et al: Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (Pro23His). Arch Ophthalmol 1991; 109:92-101.
51. Berson EL, Rosner B, Sandberg MA, et al: Ocular findings in patients with autosomal dominant retinitis pigmentosa and rhodopsin, proline-347-leucine. Am J Ophthalmol 1991; 111:614-623.
52. Berson EL, Rosner B, Weigel-DiFranco C, et al: Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest Ophthalmol Vis Sci 2002; 43:3027-3036.
53. Hargrave PA, O'Brien PJ: Speculations on the molecular basis of retinal degeneration in retinitis pigmentosa. In: Anderson RE, Hollyfield JG, LaVail MM, ed. Retinal degenerations, Boca Raton, FL: CRC; 1991:517-518.
54. Farrar GJ, Kenna P, Jordan S, et al: A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature 1991; 354:478-480.
55. Kajiwara K, Hahn L, Mukai S, et al: Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 1991; 354:480-483.
56. Molday RS: Peripherin/rds and rom-1: Molecular properties and role in photoreceptor cell degeneration. In: Osborne N, Chader G, ed. Progress in retinal and eye research, Oxford: Pergamon; 1994:271-299.
57. Weleber RG, Carr RE, Murphey WH, et al: Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch Ophthalmol 1993; 111:1531-1542.
58. Fossarello M, Bertini C, Galantuomo MS, et al: Deletion in the peripherin/RDS gene in two unrelated Sardinian families with autosomal dominant butterfly-shaped macular dystrophy. Arch Ophthalmol 1996; 114:448-456.
59. Nakazawa M, Kikawa E, Chida Y, et al: Autosomal dominant cone-rod dystrophy associated with mutations in codon 244 (Asn244His) and codon 184 (Tyr184Ser) of the peripherin RDS gene. Arch Ophthalmol 1996; 114:72-78.
60. Apfelstedt-Sylla E, Theischen M, Rüther K, et al: Extensive intrafamilial and interfamilial phenotypic variation among patients with autosomal dominant retinal dystrophy and mutations in the human RDS/peripherin gene. Br J Ophthalmol 1995; 79:28-34.
61. Hartong D, Berson EL, Dryja TP: Retinitis pigmentosa. Lancet 2006; 368:1795-1809.
62. Biro I: Therapeutic experiments in cases of retinitis pigmentosa. Br J Ophthalmol 1939; 23:332-342.
63. Gordon DM: The treatment of retinitis pigmentosa with special reference to the Filatov method. Am J Ophthalmol 1947; 30:565-580.
64. Campbell DA, Harrison R, Tonks EL: Retinitis pigmentosa: Vitamin A serum levels in relation to clinical findings. Exp Eye Res 1964; 3:412-426.
65. Chatzinoff A, Nelson E, Stahl N, et al: Eleven-CIS vitamin A in the treatment of retinitis pigmentosa: A negative study. Arch Ophthalmol 1968; 80:417-419.
66. Bergsma DR, Wolf ML: A therapeutic trial of vitamin A in patients with pigmentary retinal degeneration: A negative study. In: Landers MA, Wolbarsht ML, Laties AM, et al ed. Retinitis pigmentosa, New York: Plenum; 1976:197-209.
67. Katznelson LA, Khoroshilova-Maslova IP, Eliseyeva RF: A new method of treatment of retinitis pigmentosa/pigmentary abiotrophy. Ann Ophthalmol 1990; 22:167-172.
68. Duke-Elder S, Dobrie JH: System of ophthalmology. In: Duke-Elder S, ed. Diseases of the retina, 10. St. Louis: CV Mosby; 1967:574-622.
69. Berson EL, Remulla JFC, Rosner B, et al: Evaluation of patients with retinitis pigmentosa receiving electric stimulation, ozonated blood, and ocular surgery in Cuba. Arch Ophthalmol 1996; 114:560-563.
70. Berson EL, Rosner B, Sandberg MA, et al: A randomized trial of vitamin A and vitamin E supplementation for retinitis pigmentosa. Arch Ophthalmol 1993; 111:761-772.
71. Massof RW, Finkelstein D: Supplemental vitamin A retards loss of ERG amplitude in retinitis pigmentosa. Arch Ophthalmol 1993; 111:751-754.
72. Berson EL, Rosner B, Sandberg MA, et al: Vitamin A supplementation for retinitis pigmentosa. Correspondence Arch Ophthalmol 1993; 111:1456-1459.
73. Berson EL: Treatment of retinitis pigmentosa with vitamin A. Proceedings of the Fernström Symposium on Tapetoretinal Degenerations, Lund, Sweden. Digital J Ophthalmol 1998.4.
74. Bauernfeind JC: The safe use of vitamin a: a report of the International Vitamin A Consultative Group (IVACG), Washington, DC, The Nutrition Foundation, 1980.
75. Hathcock JN, Hattan DG, Jenkins MY, et al: Evaluation of vitamin A toxicity. Am J Clin Nutr 1990; 52:183-202.
76. Geubel AP, De Galocsy C, Alves N, et al: Liver damage caused by therapeutic vitamin A administration: Estimate of dose-related toxicity in 41 cases. Gastroenterology 1991; 100:1701-1709.
77. Sibulesky L, Hayes KC, Pronczuk A, et al: Safety of <7500 RE (<25000 IU) vitamin A daily in adults with retinitis pigmentosa. Am J Clin Nutr 1999; 69:656-663.
78. Feskanich D, Singh V, Willett WC, Colditz GA: Vitamin A intake and hip fractures among postmenopausal women. JAMA 2002; 287:47-54.
79. Michaelsson K, Lithell H, Vessby B, Melhus H: Serum retinol levels and the risk of fracture. N Engl J Med 2003; 348:287-294.
80. Berson EL, Rosner B, Sandberg MA, et al: Clinical trial of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment. Arch Ophthalmol 2004; 122:1297-1305.
81. Berson EL, Rosner B, Sandberg MA, et al: Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: Subgroup analyses. Arch Ophthalmol 2004; 122:1306-1314.
82. Bassen FA, Kornzweig AL: Malformation of the erythrocytes in a case of atypical retinitis pigmentosa. Blood 1950; 5:381-387.
83. Singer K, Fisher B, Perlstein MA: Acanthrocytosis: A genetic erythrocytic malformation. Blood 1952; 7:577-591.
84. Druez G: Un nouveau cas d'acanthocytose; Dysmorphie érythrocytaire congénitale avec rétinite, troubles nerveux et stigmates dégenératifs. Rev Hématol 1959; 14:3-11.
85. Jampel RS, Falls HF: Atypical retinitis pigmentosa, acanthocytosis and heredodegenerative neuromuscular disease. Arch Ophthalmol 1956; 59:818-820.
86. Salt HB, Wolff OH, Lloyd JK, et al: On having no betalipoprotein. A syndrome comprising A-beta-lipoproteinemia, acanthocytosis and steatorrhea. Lancet 1960; 2:325-329.
87. Druez G, Lamy M, Frézal J, et al: L'acanthocytose: Ses rapports avec l'absence congénitale de betalipoproteines. Presse Med 1961; 69:1546-1548.
88. Di-George AM, Mabry CC, Auerbach VH: A specific disorder of lipid transport (acanthocytosis). Treatment with intravenous lipids. Am J Dis Child 1961; 102:580.
89. Bach C, Polonovski J, Polonovski C, et al: L'absence congénitale de alpha-lipoproteines: Une nouvelle observation. Arch Fr Pediatr 1967; 25:1093-1111.
90. Scanu AM, Aggerbeck LP, Kruski AW, et al: A study of the abnormal lipoproteins in abetalipoproteinemia. J Clin Invest 1974; 53:440-446.
91. Narcisi TME, Shoulders CC, Chester SA, et al: Mutations of the microsomal triglyceride-transfer-protein gene in abetalipoproteinemia. Am J Hum Genet 1995; 57:1298-1310.
92. Lamy M, Frézal J, Polonovski J, et al: Congenital absence of beta-lipoproteins. Pediatrics 1962; 31:277-289.
93. Carr RE: Abetalipoproteinemia and the eye. Birth Defects: Original Article Series 1976; 12:385-408.
94. Gouras P, Carr RE, Gunkel RD: Retinitis pigmentosa in abetalipoproteinemia: Effects of vitamin A. Invest Ophthalmol Vis Sci 1971; 10:784-793.
95. von Sallmann L, Gelderman AH, Laster L: Ocular histopathologic changes in a case of a-beta-lipoproteinemia (Bassen-Kornzweig syndrome). Doc Ophthalmol 1969; 26:451-460.
96. Wolff OH, Lloyd JK, Tonks EL: A-beta-lipoproteinämia with special reference to the visual defect. Exp Eye Res 1964; 3:439-442.
97. Muller DR, Harries JT, Lloyd JK: Vitamin E therapy in abetalipoproteinaemia. Arch Dis Child 1970; 45:715.
98. Bishara S, Merin S, Cooper M, et al: Combined vitamin A and E therapy prevents retinal electrophysiological deterioration in abetalipoproteinemia. Br J Ophthalmol 1982; 66:767-770.
99. Runge P, Muller DP, McAllister J, et al: Oral vitamin E supplements can prevent the retinopathy of abetalipoproteinemia. Br J Ophthalmol 1986; 70:166-173.
100. Schaefer EJ: Diagnosis and management of ocular abnormalities in abetalipoproteinemia. In: Zrenner E, Krastel H, Goebel HH, ed. Research in Retinitis Pigmentosa, Advances in Biosciences, Oxford: Pergamon; 1987:579-581.
101. Rader DJ, Brewer Jr HB: Abetalipoproteinemia. New insights into lipoprotein assembly and vitamin E metabolism from a rare genetic disease. JAMA 1993; 270:865-869.
102. Refsum S: Heredopathia atactica polyneuritiformis: A familial syndrome not hitherto described. Acta Psychiatr Neurol Scand 1946; 38(Suppl):1.
103. Klenk E, Kahlke W: Über das Vorkommen der 3,7,11,15-tetramethylhexadecansäure (Phytansäure) in den Cholesterinestern und anderen Lipoidfraktionen der Organe bei einem Krankheitsfall unbekannter Genese: Verdacht auf Heredopathia atactica polyneuritiformis (Refsum syndrome). Hoppe Seylers Z Physiol Chem 1963; 333:133-139.
104. Berson EL: Nutrition and retinal degenerations: Vitamin A, taurine, ornithine and phytanic acid. Retina 1982; 2:236-255.
105. Eldjarn L, Stokke O, Try K: ?-Oxidation of branched chain fatty acids in man and its failure in patients with Refsum's disease showing phytanic acid accumulation. Scand J Clin Lab Invest 1966; 18:694-695.
106. Toussaint D, Danis P: An ocular pathologic study of Refsum's syndrome. Am J Ophthalmol 1971; 72:342-347.
107. Eldjarn L, Stokke O, Try K: Biochemical aspects of Refsum's disease and principles for the dietary treatment. In: Vinken PJ, Bruyn GW, ed. Handbook of Clinical Neurology, Amsterdam: North Holland; 1976:519-541.
108. Refsum S: Heredopathia atactica polyneuritiformis, phytanic acid storage disease. Refsum's disease: A biochemically well-defined disease with a specific dietary treatment. Arch Neurol 1981; 38:605-606.
109. Yokota T, Shiojiri T, Gotoda T, et al: Retinitis pigmentosa and ataxia caused by a mutation in the gene for the alpha-tocopherol-transfer protein. N Engl J Med 1996; 335:1770-1771.
110. Yokota T, Shiohiri T, Gotoda T, et al: Friedreich-like ataxia with retinitis pigmentosa caused by the His101Gln mutation of the alpha-tocopherol transfer protein gene. Ann Neurol 1997; 41:826-832.
111. Usher CH: On the inheritance of retinitis pigmentosa, with notes of cases. R Lond Ophthalmol Hosp Rep 1914; 19:130-236.
112. Usher CH: The Bowman lecture: On a few hereditary eye affections. Trans Ophthal Soc UK 1935; 55:164-245.
113. Fishman GA, Kumar A, Joseph ME, et al: Usher's syndrome: Ophthalmic and neuro-otologic findings suggesting genetic heterogeneity. Arch Ophthalmol 1983; 101:1367-1374.
114. Hunter DG, Fishman GA, Mehta RS, et al: Abnormal sperm and photoreceptor axonemes in Usher's syndrome. Am J Ophthalmol 1986; 104:385-389.
115. Berson EL, Adamian M: Ultrastructural findings in an autopsy eye from a patient with Usher's syndrome, type II. Am J Ophthalmol 1992; 114:748-757.
116. Sankila EM, Pakarinen L, Kaariainen H, et al: Assignment of an Usher syndrome type III (USH3) gene to chromosome 3q. Hum Mol Genet 1995; 4:93-98.
117. Gerer S, Bonneau D, Gilbert B, et al: USH1A: Chronicle of a slow death. Correspondence. Am J Hum Genet 2006; 78:357-359.
118. Vernon M: Usher's syndrome-Deafness and progressive blindness. Clinical cases, prevention, theory and literature survey. J Chron Dis 1969; 22:133-151.
119. Laurence JZ, Moon RC: Four cases of 'retinitis pigmentosa', occurring in the same family, and accompanied by general imperfections of development. Ophthalmic Rev (old series) 1866; 2:32-41.
120. Hutchinson J: Slowly progressive paraplegia and disease of the choroids with defective intellect and arrested sexual development in several brothers and a sister. Arch Surg (Lond) 1900; 11:118-122.
121. Bardet G: Sur un syndrome d'obésité congénitale avec polydactylie et rétinite pigmentaire (contribution à l'étude des formes cliniques de l'obésité hypophysaire). Université de Paris, Thesis no. 470, Legrand, 1920.
122. Biedl A: Ein Geschwisterpaar mit adiposo-genitaler Dystrophie. Dtsch Med Wochenschr 1922; 48:1630.
123. Schachat AP, Maumenee IH: Bardet-Biedl syndrome and related disorders. Arch Ophthalmol 1982; 100:285-288.
124. Campo RV, Aaberg TM: Ocular and systemic manifestations of the Bardet-Biedl syndrome. Am J Ophthalmol 1982; 94:750-756.
125. Björk A, Lindblom U, Wadensten L: Retinal degeneration in hereditary ataxia. J Neurol Neurosurg Psychiatry 1956; 19:186-193.
126. Rizzo J, Berson EL, Lessell S: Retinal and neurological findings in the Laurence-Moon-Bardet-Biedl phenotype. Ophthalmology 1986; 93:1452-1456.
127. Bell J: The Laurence-Moon syndrome. In: Penrose LS, ed. The Treasury of Human Inheritance, Cambridge: Cambridge University Press; 1958:51-96.
128. Krill AE, Folk E, Rosenthal IM: Electroretinography in the Laurence-Moon-Biedl syndrome: An aid in diagnosis of the atypical case. Am J Dis Child 1961; 102:205-209.
129. Katsanis N, Ansley SJ, Badano JL, et al: Triallelic inheritance in Bardet-Biedl syndrome, a Mendelian recessive disorder. Science 2001; 293:2256-2259.
130. Beales PL, Badano JL, Ross AJ, et al: Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome. Am J Hum Genet 2003; 72:1187-1199.
131. Badano JL, Leitch CC, Ansley SJ, et al: Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature 2006; 439:326-330.
132. Leber T: Über Retinitis pigmentosa und angeborene Amaurose. Graefes Arch Clin Exp Ophthalmol 1869; 15:1-25.
133. Heher KL, Traboulsi EI, Maumenee IH: The natural history of Leber's congenital amaurosis: Age-related findings in 35 patients. Ophthalmology 1992; 99:241-245.
134. Wallace DC, Singh G, Lott MT, et al: Mitochondrial DNA mutation associated with Leber's hereditary optic neuropathy. Science 1988; 242:1427-1430.
135. Neuman NJ, Lott MT, Wallace DC: The clinical characteristics of pedigrees of Leber's hereditary optic neuropathy with the 11778 mutation. Am J Ophthalmol 1991; 111:750-762.
136. Perrault I, Rozet JM, Calvas P, et al: Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet 1996; 14:461-464.
137. Semple-Rowland SL, Buczylko J, Palczewski K, et al: Abnormal expression of GCAP1 and photoreceptor guanylate cyclase in the rd chicken retina. Invest Ophthalmol Vis Sci 1996; 37(Suppl):3729.
138. Morimura H, Fishman GA, Grover SA, et al: Mutations in the RPE65 gene in patients with autosomal recessive retinitis pigmentosa or Leber congenital amaurosis. Proc Natl Acad Sci USA 1998; 95:3088-3093.
139. Gu S, Thompson DA, Srikumari CRS, et al: Mutations in RPE65 cause autosomal recessive childhood-onset severe retinal dystrophy. Nat Genet 1997; 17:194-197.
140. Acland GM, Aguirre GD, Ray J, et al: Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 2001; 28:92-95.
141. Pawlyk BS, Smith AJ, Buch PK, et al: Gene replacement therapy rescues photoreceptor degeneration in a murine model of Leber congenital amaurosis lacking RPGRIP. Invest Ophthalmol Vis Sci 2005; 46:3039-3045.
142. Mooren A: Fünf Lustren ophthalmologischer Wirksamkeit, Wiesbaden, Bergmann, 1882.
143. Kurozumi T, Ohara M, Yasui T, et al: Case of retinitis punctata albescens in a boy and degeneratio pigmentosa retinae sine pigmento in his sister. J Clin Ophthalmol 1963; 17:177.
144. Kajiwara K, Sandberg MA, Berson EL, et al: A null mutation in the human peripherin/RDS gene in a family with autosomal dominant retinitis punctata albescens. Nat Genet 1993; 3:208-212.
145. Maw MA, Kennedy B, Knight A, et al: Mutation of the gene encoding cellular retinaldehyde-binding protein in autosomal recessive retinitis pigmentosa. Nat Genet 1997; 17:198-200.
146. Burstedt MSI, Sandgren O, Holmgren G, Forsman-Semb K: Bothnia dystrophy caused by mutations in the cellular retinaldehyde-binding protein gene (RLBP1) on chromosome 15q26. Invest Ophthalmol Vis Sci 1999; 40:995-1000.
147. Morimura H, Berson EL, Dryja TP: Recessive mutations in the RLBP1 gene encoding cellular retinaldehyde-binding protein in a form of retinitis punctata albescens. Invest Ophthalmol Vis Sci 1999; 40:1000-1004.
148. Berson EL, Gouras P, Gunkel RD: Progressive cone-rod degeneration. Arch Ophthalmol 1968; 80:68-76.
149. Martinez-Mir A, Paloma E, Allikmets R, et al: Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat Genet 1998; 18:11-12.
150. Anderson KI, Baird I, Lewis RA, et al: A YAC contig encompassing the recessive Stargardt disease gene (STGD) on chromosome 1p. Am J Hum Genet 1995; 57:1351-1363.
151. Allikmets R, Singh N, Sun H, et al: A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997; 15:236-246.
152. Briggs CE, Rucinski D, McEvoy J, et al: Mutations in ABCR (ABCA4) in patients with Stargardt macular degeneration or cone-rod degeneration. Invest Ophthalmol Vis Sci 2001; 42:2229-2236.
153. Sandberg MA, Gaudio AR, Berson EL: Disease course of patients with pericentral retinitis pigmentosa. Amer J Ophthalmol 2005; 140:100-106.
154. Choi J, Sandberg MA, Berson EL: Natural course of ocular function in pigmented paravenous retinochoroidal atrophy. Am. J. Ophthalmol 2006; 141:763-765.
155. Jacobson SG, Marmor MF, Kemp CM, Knighton RW: SWS (blue) cone hypersensitivity in newly identified retinal degeneration. Invest Ophthalmol Vis Sci 1990; 31:827-838.
156. Sharon D, Sandberg MA, Caruso RC, et al: Shared mutations in NR2E3 in enhanced S-cone syndrome, Goldmann-Favre syndrome, and many cases of clumped pigmentary retinal degeneration. Arch Ophthalmol 2003; 121:1316-1323.
157. Nishiguchi KM, Friedman JS, Sandberg MA, et al: Recessive NRL mutations in patients with clumped pigmentary retinal degeneration and relative preservation of blue cone function. Proc Natl Acad Sci USA 2004; 101:17819-17824.
158. McCulloch C, McCulloch RJP: A hereditary and clinical study of choroideremia. Trans Am Acad Ophthalmol; 1948; 52:160-190.
159. Sieving PA, Niffenegger AB, Berson EL: Electroretinographic findings in selected pedigrees with choroideremia. Am J Ophthalmol 1986; 101:361-367.
160. Sorsby A: Ophthalmic Genetics, 2nd edn.. New York, Appleton-Century-Crofts, 1970.
161. Harris GS, Miller JR: Choroideremia. Arch Ophthalmol 1968; 80:423-429.
162. Cremers FPM, Sankila EM, Brunsmann F, et al: Deletions in patients with classical choroideremia vary in size from 45 kb to several megabases. Am J Hum Genet 1990; 47:622-628.
163. Cremers FPM, van de Pol DJR, Wieringa B, et al: Chromosomal jumping from the DXS165 locus allows molecular characterization of four microdeletions and a de novo chromosome X/13 translocation associated with choroideremia. Proc Natl Acad Sci USA 1989; 86:7510-7514.
164. Cremers FPM, van de Pol DJR, van Kerkhoff LPM, et al: Cloning of a gene that is rearranged in patients with choroideraemia. Nature 1990; 347:674-677.
165. Van den Hurk JA, Schwartz M, van Bokhoven H, et al: Molecular basis of choroideremia (CHM): Mutations involving the Rab escort protein-1 (REP-1) gene. Hum Mutat 1997; 9:110-117.
166. Nesslinger N, Mitchell G, Strasberg P, et al: Mutation analysis in Canadian families with choroideremia. Ophthalmic Genet 1996; 17:47-52.
167. Seabra MC, Brown MS, Goldstein JL: Retinal degeneration in choroideremia: Deficiency of Rab geranylgeranyl transferase. Science 1993; 259:377-381.
168. Seabra MC: New insights into the pathogenesis of choroideremia: A tale of two REPs. Ophthalmic Genet 1996; 17:43-46.
169. Seabra MC, Brown MS, Slaughter CA, et al: Purification of component A of Rab geranylgeranyl transferase: Possible identity with the choroideremia gene product. Cell 1992; 70:1049-1057.
170. Tolmachova T, Anders R, Abrink M, et al: Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest 2006; 116:386-394.
171. Lauber H: Die sogenannte Retinitis punctata albescens. Klia Monatsbl Augenheilkd 1910; 48:133-148.
172. Takki K, Simell O: Genetic aspects in gyrate atrophy of the choroid and retina withhyperornithinaemia. Br J Ophthalmol 1974; 58:907-916.
173. Kaiser-Kupfer MI, Kuwabara T, Askanas V, et al: Systemic manifestations of gyrate atrophy of the choroid and retina. Ophthalmology 1981; 88:302.
174. Berson EL, Schmidt SY, Shih VE: Ocular and biochemical abnormalities in gyrate atrophy of the choroid and retina. Ophthalmology 1978; 85:1018-1027.
175. Takki KK, Milton RC: The natural history of gyrate atrophy of the choroid and retina. Ophthalmology 1981; 88:292-301.
176. Simmel O, Takki K: Raised plasma-ornithine and gyrate atrophy of the choroid and retina. Lancet 1973; 2:1030-1033.
177. Takki K: Gyrate atrophy of the choroid and retina associated with hyperornithinaemia. Br J Ophthalmol 1974; 58:3-23.
178. Valle D, Kaiser-Kupfer M, Del Valle LA: Gyrate atrophy of the choroid and retina: Deficiency of ornithine aminotransferase transformed lymphocytes. Proc Natl Acad Sci USA 1977; 74:5159-5161.
179. Kennaway NG, Weleber RG, Buist NRM: Gyrate atrophy of the choroid and retina: Deficient activity of ornithine ketoacid aminotransferase in cultured skin fibroblasts. N Engl J Med 1977; 297:1180.
180. Trijbels JMF, Sengers RCA, Bakkeren JAJM, et al: l-Ornithine-ketoacid-transaminase deficiency in cultured fibroblasts of a patient with hyperornithemia and gyrate atrophy. Clin Chem Acta 1977; 79:371-377.
181. Berson EL, Schmidt SY, Rabin AR: Plasma amino-acids in hereditary retinal disease: Ornithine, lysine, and taurine. Br J Ophthalmol 1976; 60:142-147.
182. Arshinoff SA, McCulloch JC, Matuk Y, et al: Amino-acid metabolism and liver ultrastructure in hyperornithinemia with gyrate atrophy of the choroid and retina. Metabolism 1979; 28:979-988.
183. Sipilä I: Inhibition of arginine-glycine amidinotransferase by ornithine: A possible mechanism for the muscular and chorioretinal atrophies in gyrate atrophy of the choroid and retina with hyperornithinemia. Biochim Biophys Acta 1980; 613:79-84.
184. Valle D, Simell O: The hyperornithinaemias. In: Scriver C, Beaudet A, Sly W, et al ed. The metabolic and molecular bases of inherited disease, New York: McGraw-Hill; 2001:1859-1895.
185. Valle D, Walser M, Brusilow SW, et al: Gyrate atrophy of the choroid and retina: Amino acid metabolism and correction of hyperornithinemia with an arginine-restricted diet. J Clin Invest 1980; 65:371-378.
186. Wang T, Milam AH, Valle D: Correction of ornithine accumulation prevents retinal degeneration in a mouse model of gyrate atrophy of the choroid and retina. Am J Hum Genet 1996; 59(Suppl):A15.(70)
187. Kaiser-Kupfer MI, deMonasterio FM, Valle D, et al: Gyrate atrophy of the choroid and retina: Improved visual function following reduction of plasma ornithine by diet. Science 1980; 210:1128-1131.
188. Berson EL, Shih VE, Sullivan PL: Ocular findings in patients with gyrate atrophy on pyridoxine and low-protein, low-arginine diets. Ophthalmology 1981; 88:311-315.
189. Weleber RG, Kennaway NG, Buist NRM: Vitamin B6 in management of gyrate atrophy of the choroid and retina. Lancet 1978; 2:1213.
190. Kaiser-Kupfer MI, Valle D, Bron AJ: Clinical and biochemical heterogeneity in gyrate atrophy. Am J Ophthalmol 1980; 89:219-222.
191. Berson EL, Hanson AH, Rosner B, et al: A two-year trial of low-protein, low-arginine diets of vitamin B6 for patients with gyrate atrophy. In: Cotlier E, Maumenee I, Berman E, ed. Birth defects: original article series, New York: ARLiss; 1982:209-218.
192. Sipila I, Rapola J, Simell O, et al: Supplemental creatine as a treatment for gyrate atrophy of the choroid and retina. N Engl J Med 1981; 304:867-870.
193. Kaiser-Kupfer MI, Caruso RC, Valle D, Reed GF: Use of an arginine-restricted diet to slow progression of visual loss in patients with gyrate atrophy. Arch Ophthalmol 2004; 122:982-1044.
194. Acosta PB, Yannicelli S: Protocol 16-Gyrate atrophy of the choroid and retina. Nutrition support of infants, children, and adults with CyclinexT-1 and CyclinexT-2 amino acid-modified medical foods. The Ross metabolic formula system. nutrition support protocols, Columbus, OH: Ross Laboratories; 1993:359-392.
195. Kearns TP, Sayre GP: Retinitis pigmentosa, external ophthalmoplegia, and complete heart block: Unusual syndrome with histologic study in one of two cases. Arch Ophthalmol 1958; 60:280-289.
196. Kearns TP: External ophthalmoplegia, pigmentary degeneration of the retina, and cardiomyopathy: A newly recognized syndrome. Trans Am Ophthalmol Soc 1965; 63:559-625.
197. Shoffner JM, Lott MT, Voljavec AS, et al: Spontaneous Kearns-Sayre/chronic external ophthalmoplegia plus syndrome associated with a mitochondrial DNA deletion: A slip-replication model and metabolic therapy. Proc Natl Acad Sci USA 1989; 86:7952-7956.
198. The International Batten Disease Consortium: Isolation of a novel gene underlying Batten disease, CLN3. Cell 1995; 82:949-957.
199. Monroe PB, Mitchison HM, O'Rawe AM, et al: Spectrum of mutations in the Batten disease gene, CLN3. Am J Hum Genet 1997; 61:310-316.
200. Traboulsi EI, Maumenee IH, Green WR, et al: Olivopontocerebellar atrophy with retinal degeneration: A clinical and ocular histopathologic study. Arch Ophthalmol 1988; 106:801-806.
201. To KW, Adamian M, Jakobiec FA, et al: Olivopontocerebellar atrophy with retinal degeneration: An electroretinographic and histopathologic investigation. Ophthalmology 1993; 100:15-23.
202. McLaughlin ME, Dryja TP: Ocular findings in spinocerebellar ataxia 7. Arch Ophthalmol 2002; 120:655-659.
203. Alström CH, Hallgren B, Nilsson LB: Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: A specific syndrome (not hitherto described) distinct from the Laurence-Moon-Bardet-Biedl syndrome-Clinical, endrocrinological, and genetic examination based on a large pedigree. Acta Psychiatr Neurol Scand 1959; 34(Suppl 129):1-35.
204. Millay RH, Weleber RG, Heckenlively JR: Ophthalmologic and systemic manifestations of Alström's disease. Am J Ophthalmol 1986; 102:482-490.
205. Cockayne EA: Dwarfism with retinal atrophy and deafness. Arch Dis Child 1936; 11:1-8.
206. Pearce WG: Ocular and genetic features of Cockayne's syndrome. Can J Ophthalmol 1972; 7:435-444.
207. Dryja TP: Molecular genetics of Oguchi disease, fundus albipunctatus, and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol 2000; 130:547-563.
208. Dryja TP, Berson EL, Rao VR, Oprian DD: Heterozygous missense mutation in the rhodopsin gene as a cause of congenital stationary night blindness. Nat Genet 1993; 4:280-283.
209. Sieving PA, Richards JE, Naarendorp F, et al: Dark-light: Model for nightblindness from the human rhodopsin Gly-90?Asp mutation. Proc Natl Acad Sci USA 1995; 92:880-884.
210. Gal A, Orth U, Baehr W, et al: Heterozygous missense mutation in the rod cGMP phosphodiesterase beta-subunit gene in autosomal dominant stationary night blindness. Nat Genet 1994; 7:64-67.
211. Dryja TP, Hahn LB, Reboul T, et al: Missense mutation in the gene encoding the alpha-subunit of rod transducin in the Nougaret form of congenital stationary night blindness. Nat Genet 1996; 13:358-360.
212. Sandberg MA, Pawlyk BS, Arnaud B, et al: Rod and cone function in the Nougaret form of stationary night blindness. Arch Ophthalmol 1998; 116:867-872.
213. Carr RE, Ripps H, Siegel IM, et al: Rhodopsin and the electrical activity of the retina in congenital night blindness. Invest Ophthalmol 1966; 5:497-507.
214. Gouras P: Electroretinography: Some basic principles. Invest Ophthalmol 1970; 9:557-569.
215. Fuchs S, Nakazawa M, Maw M, et al: A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet 1995; 10:360-362.
216. Yamamoto S, Sippel KC, Berson EL, et al: Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat Genet 1997; 15:175-178.
217. Carr RE, Ripps H, Siegel IM: Visual pigment kinetics and adaptation in fundus albipunctatus. Documenta Ophthalmologica Proceedings Series 9. Eleventh International Society for Clinical Electroretinography Symposium, Bad Nauheim, West Germany, The Hague: The Netherlands: Dr. W. Junk BV; 1974:193-204.
218. Marmor MF: Defining fundus albipunctatus. In: Lawwill T, ed. Documenta Ophthalmologica Proceedings Series 13. Fourteenth International Society for Clinical Electroretinography Symposium, Louisville, KY, May 10-14, 1976. The Hague, The Netherlands: Dr. W. Junk BV; 1976:227-234.
219. Ripps H: Night blindness revisited: From man to molecules. Invest Ophthalmol Vis Sci 1982; 23:588-609.
220. Yamamoto H, Simon A, Eriksson U, et al: Mutations in the gene encoding 11-cis retinol dehydrogenase cause delayed dark adaptation and fundus albipunctatus. Nat Genet 1999; 22:188-191.
221. Niwa Y, Kondo M, Ueno S, et al: Cone and rod dysfunction in fundus albipunctatus with RDH5 mutation: an electrophysiological study. Invest Ophthalmol Vis Sci. 2005; 46:1480-1485.
222. Schubert G, Bornschein H: Beitrag zur Analyse des Menschlichen Elektroretinograms. Ophthalmologica 1952; 123:396-413.
223. Carr RE: Congenital stationary night blindness. Trans Am Ophthalmol Soc 1974; 72:448-487.
224. Goodman G, Bornschein H: Comparative electroretinographic studies in congenital night blindness and total color blindness. Arch Ophthalmol 1957; 58:174-182.
225. Hill DA, Arbel KF, Berson EL: Cone electroretinograms in congenital nyctalopia with myopia. Am J Ophthalmol 1974; 78:127-136.
226. Miyake Y, Yagasaki K, Horiguchi M, Kawase Y: On- and off-responses in photopic electroretinogram in complete and incomplete types of congenital stationary night blindness. Jpn J Ophthalmol 1987; 31:81-87.
227. Khan NW, Kondo M, Hiriyanna KT, et al: Primate retinal signaling pathways: Suppressing ON-pathway activity in monkey with glutamate analogues mimics human CSNB1-NYX genetic night nlindness. J Neurophys 2005; 93:481-492.
228. Miyake Y: Establishment of the concept of new clinical entities - complete and incomplete form of congenital stationary night blindness. Nippon Ganka Gakkai Zasshi 2002; 106:737-755.
229. Dryja TP, McGee TL, Berson EL, et al: Night blindness and abnormal cone electroretinogram ON responses in patients with mutations in the GRM6 gene encoding mGluR6. Proc Natl Acad Sci USA 2005; 102:4884-4889.
230. Berson EL, Lessell S: Paraneoplastic night blindness with malignant melanoma. Am J Ophthalmol 1988; 106:307-311.
231. Nishiguchi KM, Sandberg MA, Kooijman AC, et al: Defects in RGS9 or its anchor protein R(AP in pateints with slow photoreceptor deactivation. Nature 2004; 427:75-78.
232. Thirkill CE, FitzGerald P, Sergott RC, et al: Cancer-associated retinopathy (CAR syndrome) with antibodies reacting with retinal, optic-nerve and cancer cells. N Engl J Med 1989; 321:1589-1594.
233. Keltner JL, Thirkill CE, Tyler NK, et al: Management and monitoring of cancer-associated retinopathy. Arch Ophthalmol 1992; 110:48-53.
234. To K, Thirkill CE, Jakobiec FA, et al: Lymphoma-associated retinopathy. Ophthalmology 2002; 109:2149-2153.
235. Cox SN, Hay E, Bird AC: Treatment of chronic macular edema with acetazolamide. Arch Ophthalmol 1988; 106:1190-1195.
236. Grover S, Apushkin MA, Fishman GA: Topical Dorzolamide for the treatment of cystoid macular edema in patients with retinitis pigmentosa. Am J Ophthalmol 2006; 141:850-858.
237. Berson EL: Long-term visual prognoses in patients with retinitis pigmentosa: The Ludwig von Sallmann lecture. Exp Eye Res 2007; 85:7-14.
238. Berson EL, Rabin AR, Mehaffey L: Advances in night vision technology: A pocketscope for patients with retinitis pigmentosa. Arch Ophthalmol 1973; 90:427-431.
239. Hartong DT, Jorritsma FF, Neve JJ, et al: Improved mobility and independence of night-blind people using night-vision goggles. Invest Ophthalmol Vis Sci 2005; 45:1725-1731.
240. Newsome DA, Berson EL, Bonner R, et al: Possible role of optical radiation in retinal degenerations. In: Waxler M, Hitchins VM, ed. Optical Radiation and Visual Health, Boca Raton, FL: CRC; 1986:89-101.
241. Berson EL: Light deprivation and retinitis pigmentosa. Vision Res 1980; 20:1179-1184.
242. Sieving P, Caruso RC, Tao W, et al: Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci USA 2006; 103:3896-3901.
243. Weiland JD, Humayun MS: A biomimetic retinal stimulating array. IEEE Eng Med Biol Mag 2005; 24:14-21.
244. Jensen RJ, Rizzo JF: Thresholds for activation of rabbit retinal ganglion cells with a subretinal electrode. Exp Eye Res 2006; 83:367-373.
245. Berson EL, Jakobiec FA: Guest Editorial: Neural retinal cell transplantation: Ideal versus reality. Ophthalmology 1999; 106:445-446.
246. Acland GM, Aguirre GD, Bennett J, et al: Long-Term Restoration of Rod and Cone Vision by Single Dose rAAV-Mediated Gene Transfer to the Retina in a Canine Model of Childhood Blindness. Mol Ther 2005; 12:1072-1082.
247. Bennett J, Tanabe T, Sun D, et al: Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med 1996; 2:649-654.
248. LaVail MM, Yasumura D, Matthes MT, et al: Ribozyme rescue of photoreceptor cells in P23H transgenic rats: long-term survival and late-stage therapy. Proc Natl Acad Sci U S A 2000; 97:11488-11493.
249. Chen J, Flannery JG, LaVail MM, et al: bcl-2 overexpression reduces apoptotic photoreceptor cell death in three different retinal degenerations. Proc Natl Acad Sci U S A 1996; 93:7042-7047.
250. Nir I, Kedzierski W, Chen J, Travis GH: Expression of Bcl-2 protects against photoreceptor degeneration in retinal degeneration slow (rds) mice. J Neurosci 2001; 20:2150-2154.
251. Frasson M, Sahel JA, Fabre M, et al: Retinitis pigmentosa: rod photoreceptor rescue by a calcium-channel blocker in the rd mouse. Nat Med 1999; 5:1183-1187.
252. Pearce-Kelling SE, Aleman TS, Nickle A, et al: Calcium channel blocker D-cis-diltiazem does not slow retinal degeneration in the PDE6B mutant rcd1 canine model of retinitis pigmentosa. Mol Vis 2001; 7:42-47.
253. Bush RA, Kononen L, Machida S, Sieving PA: The effect of calcium channel blocker diltiazem on photoreceptor degeneration in the rhodopsin Pro213His rat. Invest Ophthalmol Vis Sci 2000; 41:2697-2701.
254. Pawlyk BS, Li T, Scimeca MS, et al: Absence of photoreceptor rescue with D-cis-diltiazem in the rd mouse. Invest Ophthalmol Vis Sci 2002; 43:1912-1915.
255. Meyer JS, Katz ML, Maruniak JA, Kirk MD: Embryonic stem cell-derived neural progenitors incorporate into degenerating retina and enhance survival of host photoreceptors. Stem Cells 2006; 24:274-283.
256. Banin E, Obolensky A, Idelson M, et al: Retinal incorporation and differentiation of neural precursors derived from human embryonic stem cells. Stem Cells 2006; 24:246-257.
257. Berson EL, Sandberg MA, Dryja TP: Search for correlations between severity of retinitis pigmentosa and primary mutations. Proceedings of the Fernström Symposium on Tapetoretinal Degenerations, Lund, Sweden. Digital J Ophthalmol 1998.
258. Berson EL: Retinal degenerations: Planning for the future. In: Anderson RE, LaVail MM, Hollyfield JG, ed. Recent Advances into Retinal Degeneration Research, New York: Springer; 2007.in press.