James C. Tsai,
Martin Wand
Over the past century, our knowledge about neovascular glaucoma (NVG) has followed an exponential growth curve, paralleling the accrual of knowledge in all of medicine. The first documentable allusion to this disorder was in 1871 when Pagenstecher[1] referred to an eye with intraocular bleeding and elevated intraocular pressure (IOP) as having hemorrhagic glaucoma. In the absence of any knowledge concerning the anatomy or pathophysiology of NVG, a confusing array of terms appeared in the literature to identify vastly different disorders with superficial similarities. Historically, the terms hemorrhagic glaucoma, thrombotic glaucoma, congestive glaucoma, rubeotic glaucoma, and diabetic hemorrhagic glaucoma all referred to NVG as we now understand it.
It was not until 1906 that Coats[2] put our knowledge of NVG on a sound anatomic basis, when he described the histologic findings of new vessels on the irises of eyes with central retinal vein occlusion (CRVO). In 1928, Salus[3] described similar new vessels on the irises of eyes of diabetic patients. In 1937, after the introduction of clinical gonioscopy, Kurz[4] correlated his clinical observation of fine new vessels in the angle (NVA) with the histologic finding of connective tissue along these vessels. He felt that the contraction of this connective tissue along these new vessels was the cause of synechial angle closure. In 1963, Weiss and colleagues[5] proposed the term neovascular glaucoma because the glaucoma is caused by the new vessels rather than the inconsistently present intraocular bleeding. This term has since found universal acceptance. Walton and Grant,[6] in 1968, proposed the more accurate and appropriate term neovascularization of the iris (NVI) rather than rubeosis iridis. Although this has not yet found wide acceptance, it is the correct terminology and NVI is used in this chapter instead of rubeosis iridis.
Since the 1960s, there has been a virtual explosion in our knowledge of the pathophysiology of NVG as well as great progress in our ability to treat this disorder. Starting with the introduction of modern panretinal photocoagulation (PRP) by Aiello and associates[7] in 1967 and continuing to the recent developments in glaucoma surgery, a significant number of cases of NVG have become treatable. As important, major advances in our understanding of the angiogenesis aspects of this disease have led to the introduction of novel treatments for this disease.[8]
|
Key Features |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
CLINICAL MANIFESTATION
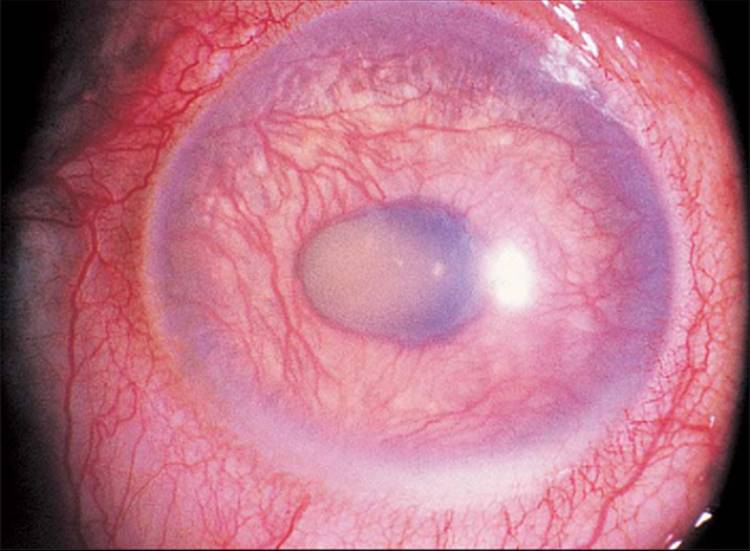
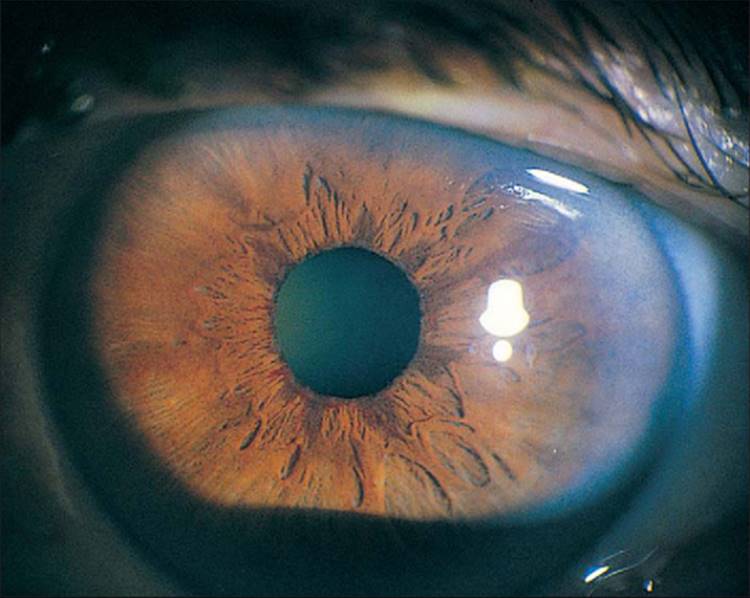
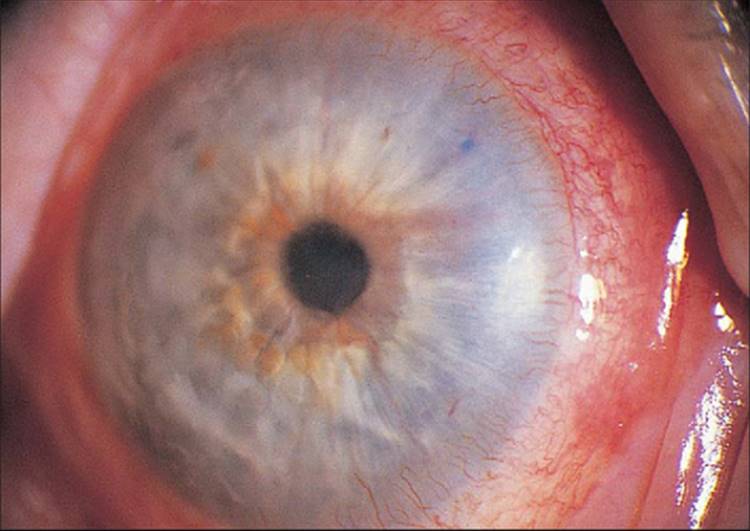
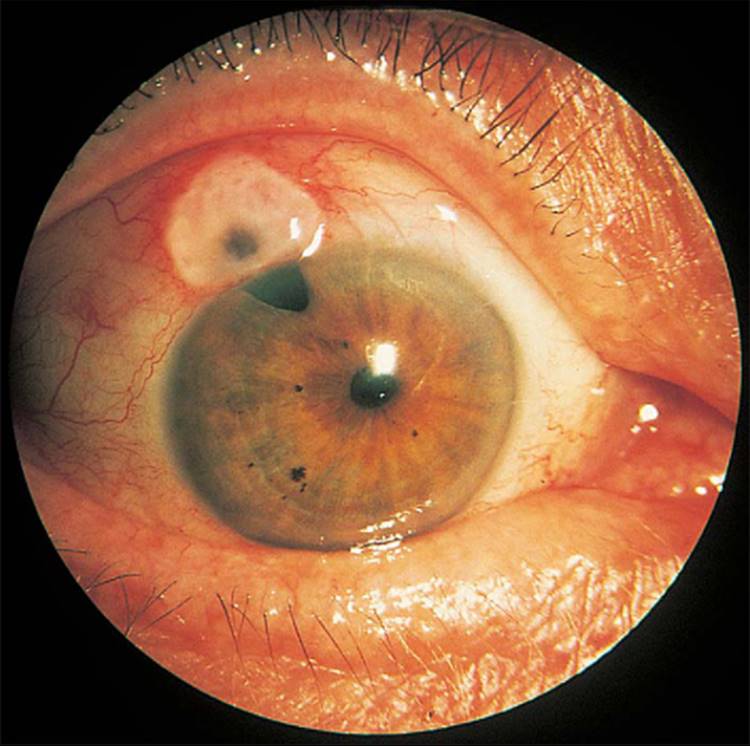
The classic picture of NVG is a patient with a painful and photophobic eye, moderate to marked conjunctival congestion, steamy cornea, florid NVI and ectropion uveae, and markedly elevated IOP (Fig. 213.1). However, the clinician must also be cognizant of diagnosing NVG in its earliest stages, thereby preventing progression of the disease to its prototypic stage. The first visible signs of incipient NVG may be subtle and consist of tiny tufts of new vessels at the pupillary margin. These vessels enlarge and become clinically visible knuckles of fine vessels, appearing similar to a glomerulus (Fig. 213.2). Unless one maintains a high index of suspicion and looks carefully under high magnification at the slit lamp, it is easy to overlook these vessels and forgo the opportunity to prevent the development of end-stage NVG. It is especially difficult to detect early pupillary NVI in darkly pigmented irises. If a contact gonioscopy lens is used at the initial examination, even light pressure on the lens is sufficient to collapse these neovascular tufts and render them clinically invisible.[9] One previous misconception was that NVA could not occur without NVI developing first in the pupillary margin.[10] However, dark irises may conceal early pupillary NVI on slit-lamp examination, and NVA may be visible only on gonioscopy. Thus, it is imperative to perform gonioscopy in all eyes that have the potential for the development of NVG.
|
|
|
|
FIGURE 213.1 Advanced fulminant neovascular glaucoma with marked conjunctival congestion, corneal haze from elevated IOP, neovascularization of the iris, and ectropion uvea. |
|
|
|
|
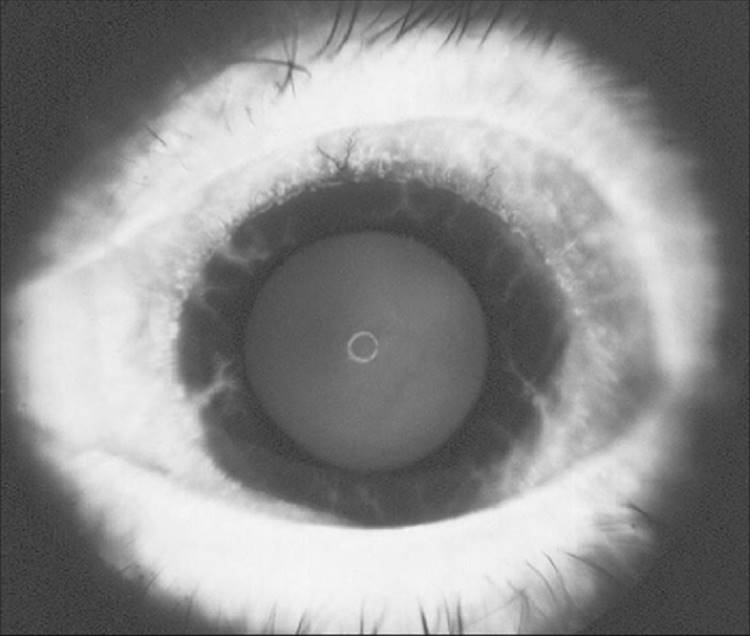
FIGURE 213.2 The earliest manifestations of NVI at the pupillary margin, although visible on high magnification with the slit lamp, are not visible in printed photographs. This eye has slightly more advanced NVI at the pupillary margin. |
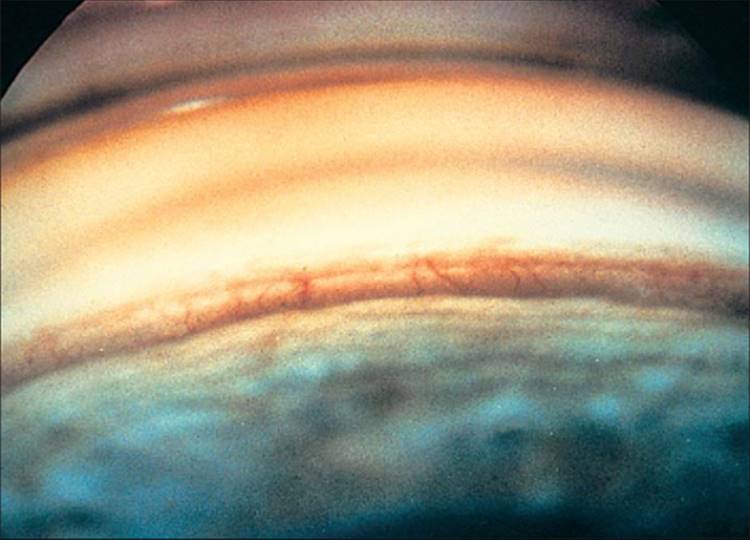
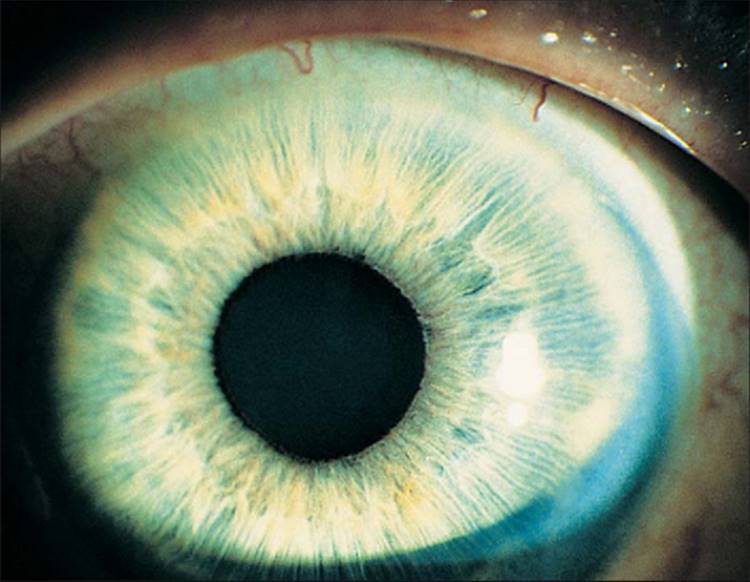
Besides the pupillary margin, NVI may present in other parts of the iris. In an elderly man with diabetes mellitus who had a previous surgical peripheral iridectomy, NVI was present at the edges of the surgical iridectomy before any NVI could be seen at the pupil.[9] As NVI progresses, new vessels extend from the pupillary tufts in an irregular, meandering manner. New vessels, at least clinically, appear on the surface of the iris. In elderly individuals with atrophic irises and in individuals with light irises, normal iris vessels are sometimes visible, but if one looks carefully, these vessels are within the stroma of the iris and have a more radial orientation. Occasionally, with inflammation and secondary engorgement, it may be difficult if not impossible to tell if iris vessels are abnormal. When new vessels reach the iris collarette, the collarette vessel, which is often present but normally not visible, may become engorged and become part of the rubeotic vasculature. At this stage, but usually at more advanced stages, it is possible to see effacement of the normal iris surface architecture, resulting in a relatively smooth iris. When these new vessels reach the angle, they cross the ciliary body band and scleral spur onto the trabecular meshwork (Fig. 213.3). Chandler and Grant[11] have long taught that if a blood vessel crosses over the scleral spur onto the trabecular meshwork, the vessel is abnormal; all normal vessels remain behind the scleral spur.[11] This clinical axiom has withstood the test of time. Occasionally, new vessels seem to arise from the major arterial circle of the iris and cross onto the trabecular meshwork.
|
|
|
|
FIGURE 213.3 Goniophotograph shows new angle vessels crossing over the ciliary body band and scleral spur and arborizing over the trabecular meshwork. |
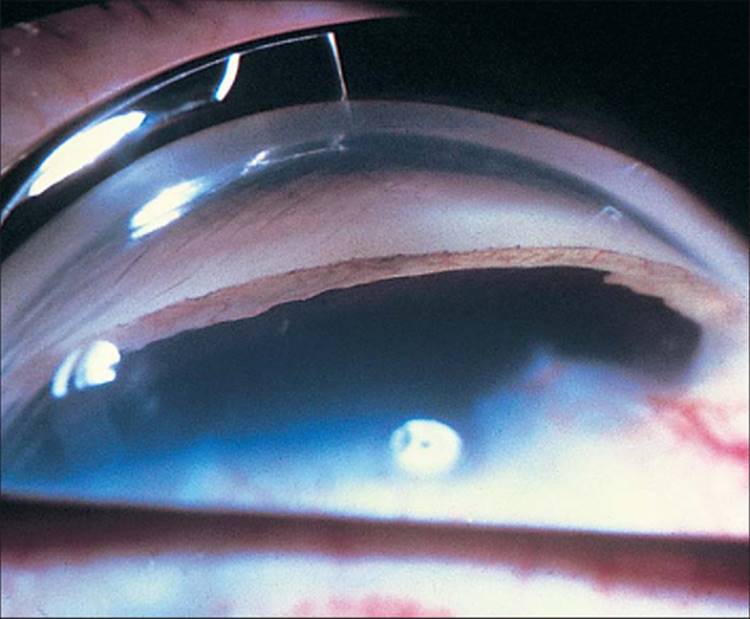
At the angle, larger abnormal vessels, after crossing the scleral spur, arborize with fine capillaries over several clock hours of the trabecular meshwork, similar to a tree trunk with branches overhead. Again, a high index of suspicion and careful examination are necessary to see these early fine NVA. The higher magnification and higher light intensity offered by the slit lamp with the Goldmann goniolens versus Koeppe's gonioscopy, facilitates this examination. Until NVA covers a significant portion of the trabecular meshwork, the IOP may be completely normal. A fibrovascular membrane, which is invisible on gonioscopy, accompanies NVA and may block enough of the trabecular meshwork to cause a secondary form of open-angle glaucoma (OAG) at this time. This fibrovascular membrane along these new angle vessels has a tendency to contract and pull the vessels taut, bridging the angle initially, and then tenting the iris toward the trabecular meshwork (Fig. 213.4). As these peripheral anterior synechiae coalesce, synechial angle closure occurs. The radial traction along the surface of the iris pulls the posterior pigment layer of the iris around the pupillary margin onto the anterior iris surface, commonly, although inaccurately, known as ectropion uveae.
|
|
|
|
FIGURE 213.4 Goniophotograph shows new vessels covering all the visible trabecular meshwork with early synechial angle closure at 1:30 o'clock. |
As the scattered areas of synechial closure coalesce, there is total angle closure. The picture of a smooth, zippered-up line of iridocorneal adhesion is pathognomonic (Fig. 213.5). After this end-stage of NVG is reached, there can be a remarkable decrease in the number of new vessels visible in the angle and on the iris. When a prominent pigmented Schwalbe line is present, a totally closed angle in NVG can be mistaken for a normal 'open' angle by even an experienced gonioscopist. Attempts have been made to classify and quantitate NVI.[12,13,14] Such a grading system could be valuable in clinical staging, in following progression, when comparing patients, and when evaluating the efficacy of therapy. Unfortunately, such a classification has not been widely accepted.
|
|
|
|
FIGURE 213.5 Late-stage neovascular glaucoma with total synechial angle closure and the typical picture of a smooth line of iridocorneal adhesion. The irregular dark line is the edge of the ectropion uvea. |
NVI may progress to total synechial angle closure within days, or it may remain stationary with no angle involvement for years. This quiescent stage may also suddenly become active after years of inactivity. Although the clinical picture of NVG from different primary causes is not distinguishable, and each case of NVG shows its individual pattern of progression, as a group, NVI resulting from CRVO seems to be more fulminant in appearance and rapid in course than that from other causes.
DIFFERENTIAL DIAGNOSIS
As already stressed, the key aim in the treatment of NVG is early diagnosis so that an optimally effective treatment regimen can be instituted. In the differential diagnosis, there are two stages to consider: the early stage, in which only NVI is present, and the late stage, in which there is elevated IOP, a cloudy cornea, and vascular congestion. In both stages, a complete patient history and careful examination of both eyes usually provide the answer. For true NVG, the history is critical. Diabetes mellitus in the patient or family; history of previous loss of vision, suggesting old CRVO or retinal detachment; or history of hypertension or arteriosclerosis, suggesting possible carotid artery obstructive disease (CAOD), are all significant. Even a misleadingly benign-appearing posterior segment should not deter one from considering one of these major causes of NVI.
There are several entities to consider in the differential diagnosis of prominent iris vessels. NVI may be present in Fuchs' heterochromic cyclitis. The eye is usually white and quiet. The iris vessels tend to be extremely fine, thin-walled, and fragile. Spontaneous hyphemas may occur but do so more often with manipulation of the eye, such as during a gonioscopic examination or after paracentesis. These vessels may cross over the scleral spur onto the trabecular meshwork, but they only rarely cause synechial angle closure or NVG. Secondary glaucoma may occur, but probably on the basis of the trabeculitis.[15]Histologic studies show localized thickening of the iris vessel walls caused by hyalinization and proliferation of the endothelium, resulting in decreased lumen size and vascular perfusion.[16] Fluorescein studies have shown leakage from the iris vessels, narrowed radial iris vessels, and ischemic iris sectors, confirming localized areas of iris hypoxia as a cause of the NVI.[17]
NVI may also be present in pseudoexfoliation syndrome. Electron microscopic studies have shown endothelial thickening with decreased lumen size and fenestration of vessel walls,[18] accounting for the fluorescein leakage seen in the irises of some eyes.[19] True NVG has not been reported. In all likelihood, the NVI occasionally seen after retinal detachment or strabismus surgery is a result of trauma to the anterior ciliary vessels, which also results in localized anterior segment hypoxia.[17]
Finally, inflammatory conditions can cause prominent iris vessels, which are sometimes impossible to differentiate clinically from progressive NVI. This is especially true in diabetes mellitus after cataract extraction or other ocular surgery, when a profound iritis and secondary vascular engorgement can simulate fulminant NVG. In view of the important role that certain molecular components of inflammation plays in the angiogenesis process, it is not surprising that inflammation could cause engorgement and dilatation of iris vessels, especially in eyes that already have some compromised retinal circulation. In any case, with topical steroid therapy, pseudo-NVI from inflammation resolves, but true NVI persists.
There is another list of differential diagnoses in the late stages, when there is an elevated IOP and a cloudy cornea. Although the underlying cause of true NVG is usually of long-term duration, such as proliferative diabetic retinopathy (PDR) or CRVO, the presenting signs and symptoms are often precipitous. It is not unusual for a patient to present for the first time with an inflamed eye and an IOP of 60 mmHg or higher. Acute angle closure must be considered high on the differential diagnosis list. Gonioscopy may be impossible, even with the use of systemic hyperosmotic agents and topical glycerin. However, it is almost always possible to see the NVI through the hazy cornea. More important, gonioscopy of the fellow eye provides the clue because narrow angles and angle-closure glaucoma tend to be a bilateral disorder. Many surgical or laser iridotomies have been performed inadvertently on eyes with unrecognized NVG. Conversely, Figure 213.6 shows an elderly woman with all the findings of NVG in one eye and a normal fellow eye with a deep anterior chamber angle. She was found to have an intumescent cataract and phacomorphic secondary angle-closure glaucoma with inflammation. After a neodymium:yttrium-aluminum garnet (Nd:YAG) laser iridotomy and resolution of the angle closure, the 'NVI' completely disappeared.
|
|
|
|
FIGURE 213.6 An 86-year-old woman with hand motion vision, IOP of 76 mmHg, a congested eye with edematous cornea, and engorged vessels on the iris that appear to be neovascularization of the iris. The fellow eye had a normal-depth anterior chamber. She had an intumescent cataract with phacomorphic secondary angle-closure glaucoma mimicking neovascular glaucoma. |
An opaque media may prevent visualization of the posterior segment, and the diagnosis may be difficult. Hidden intraocular tumors,[6,20] chronic retinal detachment,[21] and other degenerative conditions[10]must be considered. Any cause of intraocular bleeding, especially with a hyphema, may be confused with NVG. Ghost-cell glaucoma, presenting after trauma or surgical procedures, should be considered. This diagnosis is usually made easier because there is a khaki-colored hypopyon in the anterior chamber that covers the trabecular meshwork on gonioscopy. The difficulty arises when a superimposed hyphema prevents this observation. The history and paracentesis with phase-contrast microscopy of the aspirate would confirm this diagnosis.
FLUORESCEIN STUDIES
There is good evidence that functional derangement of the vascular endothelium, as evidenced by leakage of fluorescein, occurs before any structural changes and well before clinically visible neovascularization. This is understandable because vascular endothelial growth factor (VEGF), a growth factor responsible for ocular neovascularization, is also a potent vasopermeability factor - 50 000 times more potent - than histamine.[22,23] Therefore, various fluorescein studies play an important clinical and research role in NVG.
Iris fluorescein angiography shows leakage from damaged iris vessels (Fig. 213.7) long before new vessels can be detected on slit-lamp examination (Fig. 213.8). It has been shown that normal eyes can have pupillary tufts with fluorescein leakage and that the incidence seems to increase with age.[24] In addition, patients with pseudoexfoliation,[19] myotonic dystrophy,[25] or abnormal insulin secretion[26] can also show abnormal pupillary tufts with fluorescein leakage. However, in these benign forms of vascular incompetence, the fluorescein leakage occurs only at the pupillary margins, the leakage is minimal, and the leakage fades rapidly after the injection.[27] In contrast, in pathologic states, fluorescein leakage occurs throughout the iris and persists and increases with time.[28]
|
|
|
|
FIGURE 213.7 Iris fluorescein angiogram showing leakage from new iris vessels that were missed initially on careful slit-lamp examination. Normal irises, especially in elderly individuals, may show leakage in the pupillary region, but these leaking vessels are more peripheral and are abnormal. |
|
|
|
|
FIGURE 213.8 Same eye as in Figure 213.7. Very early NVI that was initially missed on careful slit-lamp examination. |
Iris fluorescein angiography may be performed at the same time as the fundus angiography.[27] Newer fundus cameras have an anterior segment lens that can be flipped into position for anterior segment pictures. After taking the early transit fundus photographs, iris photographs can be taken. In one study, NVI could be detected on fluorescein iris angiography in 37% of eyes before the subsequent development of clinically visible new vessels.[27] However, in most eyes with NVI, a high index of suspicion with diligent observation makes iris fluorescein angiography unnecessary. Before vitrectomy for complications of diabetic retinopathy, 94% of eyes showed no clinical findings of NVI, but had fluorescein leakage from iris vessels.[29] If capillary dropout is noted on fundus angiography in eyes with diabetic retinopathy or CRVO, and NVI is not clinically detectable, iris angiography should be performed at the same time.
High-power fluorescein gonioscopy has demonstrated that significantly more diabetic eyes will have leakage of new angle vessels before they are readily visible on slit-lamp examination.[30,31] Leakage of fluorescein from the pupillary margin usually occurs before there is leakage in the angle, confirming clinical observations that new vessel formation at the pupillary margin generally occurs before angle involvement.[9] Clinically, however, the value of fluorescein iris angioscopy must still be proved.
Of greater potential clinical value is posterior vitreous fluorophotometry. There seems to be a correlation between alteration of the blood-retinal barrier (as detected by vitreous fluorophotometry) and the severity of the retinopathy, duration of diabetes mellitus, and metabolic control of the diabetes.[32,33,34] To date, however, the therapeutic implications of this test remain to be seen.
PATHOLOGY
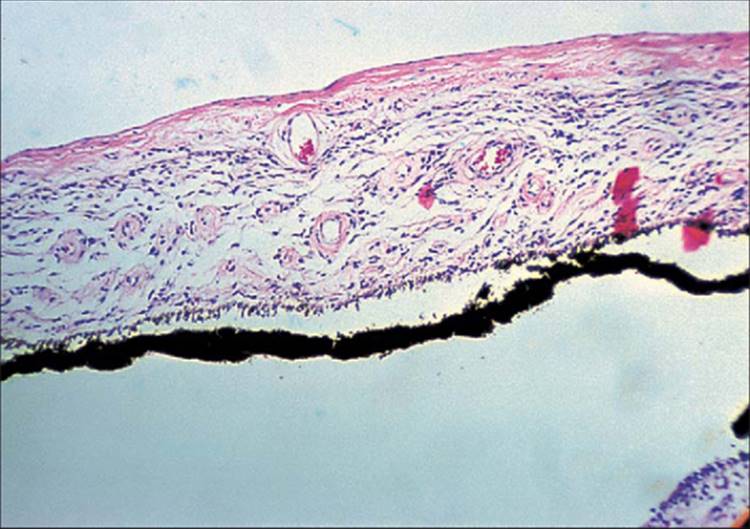
The anterior segment histopathologic features of NVG, regardless of the cause, are the same.[35,36] The only minor deviations are in CRVO, in which the new vessels may be more engorged, and in diabetes mellitus, in which there can be characteristic iris pigment epithelium cystoid changes from accumulation of glycogen.[33] The neovascularization process begins as endothelial budding from capillaries of the minor arterial circle at the pupil. Clinically, this neovascularization appears to progress sequentially from the pupil to the periphery. Histologically, however, once the process starts at the pupillary margin, new endothelial buds may appear from vessels anywhere in the iris, including the major arterial circle in the iris root.[33] These endothelial buds progress to become glomerulus-like vascular tufts. These capillary tufts are not unique to NVG. As mentioned earlier, elderly individuals without known diseases[24] and individuals with myotonic dystrophy may have pupillary tufts as well.[25] These tufts leak fluorescein, confirming that they are indeed new vessels.[26] These new vessels are essentially thin-walled endothelial cells without a muscular layer or much adventitia or supportive tissue. There are gaps between and fenestrations in these endothelial cells,[37,19] allowing leakage of fluorescein and presumably other substances as well. In vivo, these new vessels appear to be on the surface of the iris. Histologically, new vessels have a tendency to be thin-walled and located toward the surface of the iris, but they can be anywhere within the stroma of the iris (Fig. 213.9).[33]
|
|
|
|
FIGURE 213.9 H & E section of the iris with neovascularization of the iris. Note the new vessel on the iris surface but beneath the fibrovascular membrane. ×100. |
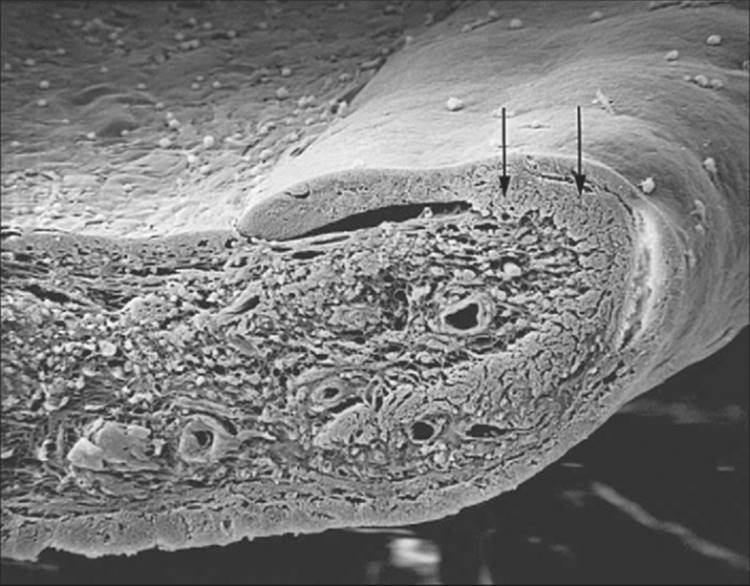
The existence of a fibrovascular membrane has been known for a long time. This membrane consists of proliferating myofibroblasts, which are fibroblasts with smooth muscle differentiation.[38] These cells are clinically transparent and are only hinted at by the aforementioned flattening of the usual iris surface architecture. Scanning electron microscopy shows the uniform presence of this membrane wherever there are new vessels. Anatomically, the new vessels are not on the surface of the iris, but are actually beneath this layer of myofibroblasts (see Fig. 213.9). The pervasiveness of this membrane explains why there can be increased IOP despite the gonioscopic appearance of a normal open angle or only slight NVA disproportionate to the degree of IOP elevation. The contractile smooth muscle components explain the effacement of the iris surface, the development of ectropion uveae, the formation of peripheral anterior synechiae and, ultimately, synechial angle closure (Fig. 213.10).[38] As this membrane continues to contract on the surface of the iris, the posterior pigment layer of the iris is pulled around the pupillary margin onto the anterior surface, causing ectropion uveae and pupillary distortion. The sphincter muscle can also be pulled anteriorly, resulting in ectropion of the sphincter (Fig. 213.11). Contraction may be so extensive that the iris is displaced forward or totally retracted from view. In such cases, the ciliary processes may be visible on gonioscopy. This contraction can also compress and embed the new vessels, hiding them from observation.[39] A fibrotic, nonresponsive iris and a fixed dilatated pupil are often seen in late NVG. As mentioned earlier, before total synechial angle closure, the membrane can obstruct the trabecular meshwork and produce a secondary OAG (Fig. 213.12). Further contraction results in synechial angle closure with iridocorneal touch. When the neovascularization process is stopped, such as with PRP, the new vessels regress, but synechial angle closure remains.
|
|
|
|
FIGURE 213.10 Scanning electron micrograph shows the fibrovascular membrane that has produced a smooth iris surface. The contraction of this membrane has completely closed the angle (arrow shows the compressed trabecular meshwork) and has started to pull the posterior pigment layer over the pupillary edge and onto the anterior iris surface (ectropion uvea). ×20. |
|
|
|
|
FIGURE 213.11 Scanning electron micrograph shows the marked ectropion uvea and ectropion of the sphincter muscle (arrows). ×160. |
|
|
|
|
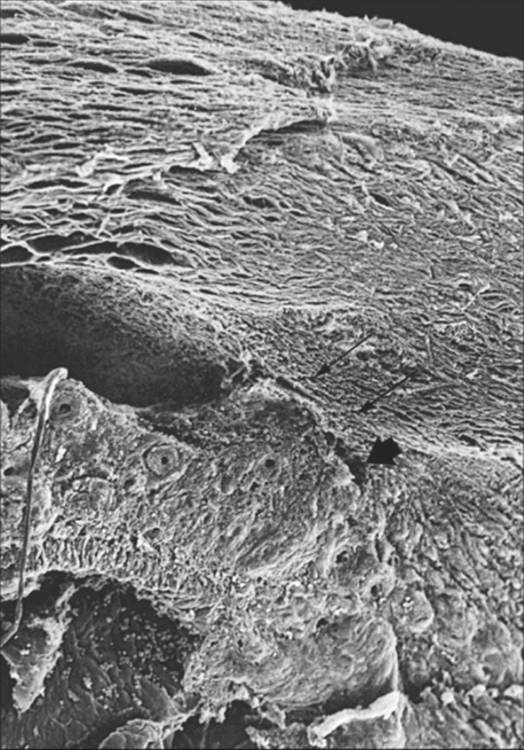
FIGURE 213.12 Scanning electron micrograph. Early synechial angle closure in neovascular glaucoma. The fibrovascular membrane is partially obstructing the anterior chamber angle that is still open (large single arrow). Part of the trabecular meshwork is already obstructed (double arrows); Schlemm's canal is superior to the trabecular meshwork. ×110. |
In some cases of synechial angle closure of long duration, the endothelium and Descemet's membrane may extend from the cornea across the synechiae onto the iris surface.[39] Scanning electron microscopic studies have confirmed that this endothelium is contiguous to and originates from the corneal endothelium.[40] Such endothelialization of the iris is also seen in the iridocorneal endothelial syndrome[41] and after trauma[39] and may represent a pathogenic mechanism that is common to violation of the barrier between the cornea and the iris, such as with synechial angle closure. This closed angle with few or no visible new vessels and covered with endothelium can easily be mistaken for a normal open angle, and the term pseudoangle is most appropriate.
PATHOGENESIS
Since the 1980s, our knowledge about angiogenesis has undergone exponential growth.[42-44,45] Fundamental to our understanding of angiogenesis is knowledge of how normal new blood vessels form. New vessel formation is one of the fundamental biologic processes necessary for embryogenesis and placenta formation, normal growth and development, and wound healing, as well as tumor growth and numerous pathologic disorders.[46] New vessel formation, whether in normal development, tumor growth, or NVI, appears to occur in the same sequence.[47] First, endothelial cells from venules or capillaries release enzymes that disrupt the adjacent basement membrane. Adjacent endothelial cells from existing vessels migrate toward the source of the angiogenic stimulus, whereas more distal cells undergo proliferation. The endothelial cells then elongate, and lumen formation occurs. Finally, new basement membrane forms, and pericytes surround the new capillaries to form mature new vessels.
Cutting across all medical specialties, there is now a group of angiogenic diseases including, among many, arthritis, atherosclerosis, psoriasis, scleroderma, Kaposi's sarcoma, and juvenile hemangiomas.[46]Ocular neovascularization per se is now recognized as the single greatest cause of blindness in the world.[48] All ischemic retinopathies, such as diabetic retinopathy, CRVO, retinopathy of prematurity (ROP), and their anterior segment sequelae, as well as subretinal and subchoroidal neovascularization, most ocular tumors, and many advanced corneal diseases would be considered angiogenic ocular diseases.
In 1948, Michaelson[49] postulated the existence of a vasoformative factor, 'X' factor, which controlled normal development of new vessels during embryogenesis. Ashton and associates[50] in 1954 suggested that the retinal ischemia in ROP might result in excess amounts of this factor, which, in turn, would lead to retinal neovascularization. In 1956, Wise[51] proposed that retinal capillary or venous obstruction resulted in hypoxia of the retinal cells; if the hypoxic cells did not die, they would produce a vasoformative factor. Ashton[52] agreed that the prerequisite for neovascularization was hypoxic metabolism, and he further speculated that the vasoformative factor from this hypoxic metabolism could diffuse anteriorly to stimulate NVI. Earlier, Ashton and associates[50] had demonstrated that the vasoobliteration seen in ROP was comparable to the capillary closure or nonperfusion seen in diabetic retinopathy. Clinically, it has been confirmed that widespread capillary occlusion and chronic tissue hypoxia are almost always present when there is neovascularization.[53,54]
In 1963, Folkman et al[55] hypothesized that solid tumors produce a substance that could stimulate new vessel formation. Subsequently, several investigators independently identified and isolated a soluble substance from solid neoplasms capable of stimulating neovascularization and popularized the term tumor angiogenesis factor.[56,55,57] A specific tumor angiogenesis factor has never been isolated and up to the early 1980s, not a single angiogenesis factor (AF) had been isolated. Then, in 1983, the fortuitous discovery that a growth factor from a rat chondrosarcoma had high affinity for heparin led to the isolation and purification of many heparin-binding growth factors by heparin-affinity chromatography.[58] At this time, at least nine different families of polypeptide growth factors have been identified, sequenced, and cloned. The growth factor currently of greatest interest in ophthalmology is VEGF, a secreted glycoprotein with a molecular weight of ~45 kDa. It is highly specific for endothelial cells, causing their mitosis and migration in vitro. In vivo, it can trigger the entire sequence of steps in neovascularization.[22,59,45] VEGF is a highly conserved protein found in four isoforms in humans[60] and three isoforms in mice.[61] In humans, its wide distribution in normal tissue and organs suggests that it is mainly a regulator of normal function and, in fact, may be necessary to maintain a tonic status of the microvasculature.[59]
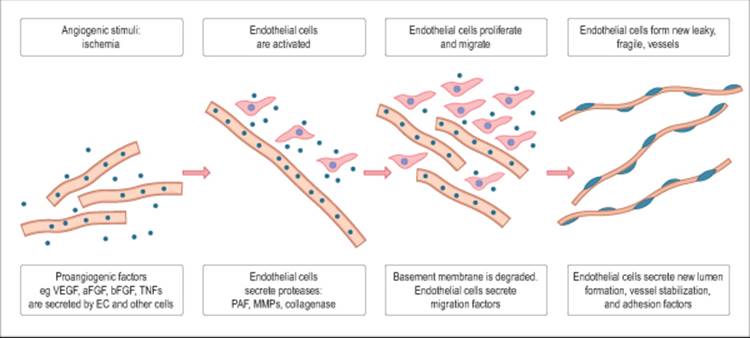
Ischemia of the retina has been postulated to be one of the most significant etiologic factors in the formation of NVI and NVA. The resulting hypoxia is known to cause the release of factors that both promote and inhibit new vessel growth.[62] Moreover, vascular endothelial cells play a crucial role in the entire process of angiogenesis. In response to tissue hypoxia, these endothelial cells secrete diffusible, proangiogenic factors, such as VEGF, basic fibroblast growth factor (bFGF), tumor necrosis factor-? (TNF-?), insulin-like growth factors, interleukin-6, and platelet-derived growth factor (Fig. 213.13).[63] This process stimulates a cascade, leading to the activation, proliferation, and migration of endothelial cells with formation of new, leaky, fragile blood vessels.
|
|
|
|
FIGURE 213.13 Key molecular events occurring during the process of angiogenesis. |
To prove that VEGF is indeed the long sought after 'X' or ocular AF, Frank,[64] in 1994, believed that five criteria had to be fulfilled. At present, all these criteria have been met. First, this AF must be present in the eye, especially in the retina. Expression of VEGF mRNA has now been found in many ocular cells, including retinal pigment epithelium, pericytes, astrocytes, Müller's cells, ganglion cells, smooth muscle cells, and endothelial cells.[65]
Second, receptors for this AF must be present on the vascular endothelial cells. The intracellular signal transduction pathway of VEGF has been identified as protein kinase C (PKC) activation.[45] More importantly, PKC inhibitors have been shown to abolish both VEGF-induced PKC activation and VEGF-induced endothelial cell proliferation.[45]
Third, hypoxia and hypoxic ocular conditions known to stimulate NV must enhance either the synthesis or the activation of this AF. In cell cultures, retinal pigment epithelium, pericytes, and microvascular endothelial cells under hypoxic conditions showed increased VEGF mRNA levels.[66] There was a 3- to 30-fold increase that was oxygen concentration-dependent. In another study, VEGF was shown to be the sole growth factor response in hypoxic retinal pigment epithelium cell cultures.[67] It was also shown that media conditioned by hypoxic retinal pericytes and retinal pigment epithelium stimulated endothelial cell growth.[66] In monkeys with experimental CRVO, VEGF mRNA is increased within 1 day after induction of the CRVO,[68] and there is a temporal and spatial relation between aqueous VEGF levels and the degree of NVI.[69] In humans, it has been shown that increased VEGF mRNA is found within the inner nuclear layer in eyes with CRVO and within the outer nuclear layer with retinal detachment, as might be expected with VEGF stimulation at the level of retinal hypoxia from these two respective conditions.[48] There is increased VEGF immunoreactivity in the retina and choroid of diabetic eyes.[70] Vitreous VEGF levels[71] and VEGF mRNA levels in neovascular membranes[72] have been found to be elevated in human eyes with PDR when compared with eyes without diabetes. In an impressively large study (210 samples from 164 patients) of vitreous obtained during vitrectomy, Aiello and colleagues[73] showed that vitreous VEGF levels were significantly elevated in eyes with PDR but not with quiescent, inactive, or no diabetic retinopathy. In 10 eyes in which simultaneous vitreous and aqueous samples were obtained, there was a VEGF gradient consistent with a posterior segment source and anterior segment diffusion. Furthermore, vitreous samples from eyes with PDR were able to stimulate endothelial cell culture growth.[73]
The fourth criterion is that this AF must cause ocular neovascularization after local infusion. Tolentino and colleagues[18] injected human recombinant VEGF intravitreally in six monkey eyes, with four eyes receiving inactivated recombinant VEGF. All six eyes receiving the bioactive VEGF had dilated, tortuous retinal vessels that leaked fluorescein. Eyes receiving multiple injections of VEGF had progressive retinopathy, including retinal edema, microaneurysms, intraretinal hemorrhages, and capillary closure with retinal ischemia. None of the four control eyes had any retinal vascular abnormalities. This important study also suggests that, rather than retinal ischemia being the only cause of increased VEGF expression, physiologically relevant levels of VEGF may be induced by other stimuli and may be sufficient to produce the retinal vascular changes seen in human ischemic retinopathies. This is consistent with a study of streptozotocin-induced diabetic rats, in which VEGF mRNA levels correlated with the extent of breakdown of the blood-retinal barrier.[74] Finally, in the most dramatic confirmation of VEGF as the cause of NVG, Tolentino and colleagues[75] gave intravitreal human recombinant VEGF to five monkey eyes, inactivated VEGF to two control eyes, and only the vehicle to one control eye. All five eyes receiving the bioactive VEGF had NVI that leaked fluorescein, whereas none of the control eyes had any NVI. A dose response was noted in one monkey that received a higher concentration of bioactive VEGF in one eye than in the contralateral eye. When VEGF injection was continued every 3 days for 30 days, full-blown NVG with ectropion uvea was seen.
The fifth and final criterion is that specific inhibition of this AF blocks ocular neovascularization. In a novel approach to VEGF inhibition, chimeric protein was made by joining the extracellular domain of murine and human high-affinity VEGF receptors with IgG protein.[76,67] When this chimeric protein was added to in vitro retinal endothelial cell cultures, VEGF stimulation of the endothelial cells was completely eliminated.[76] In murine ROP model eyes, these chimeric proteins reduced retinal neovascularization in 95-100% of the eyes.[74] In another study, Adamis and colleagues[77] gave intravitreal injections of neutralizing anti-VEGF monoclonal antibody to monkeys with experimental CRVO. None of the eyes receiving the VEGF antibody acquired NVI, but five of eight control eyes receiving inactive antibody acquired NVI. In endothelial cell cultures, the anti-VEGF antibody inhibited VEGF-driven endothelial cell proliferation.[77] In another novel approach, antisense phosphorothioate oligodeoxynucleotides were used to inhibit the synthesis of VEGF in murine ROP eyes.[78] This agent reduced VEGF levels by 40-60% and retinal NV by 25-31%. This treatment approach was not as effective as the other two methods of inhibiting VEGF action, probably because VEGF production was not completely suppressed.
CAUSES OF NVG
There have been a number of comprehensive reviews on NVG.[8,42] Each review has tried to list the disorders known to cause or at least be associated with NVG, with the highest count in the 40s.[33,8] There have since been more reports of entities that purportedly cause NVG. However, it has become abundantly clear that wherever NVI progresses to NVG, there is almost always widespread posterior segment hypoxia or localized anterior segment hypoxia.
Rather than compiling lists of entities that can cause NVI, it now seems more logical to review the major groups of disorders that can lead to NVG. In one review of patients with NVG admitted in the 1960s to a Danish hospital, 43% had glaucoma attributed to diabetic retinopathy, 37% had glaucoma attributed to CRVO, and the rest had glaucoma from miscellaneous causes.[79] Surprisingly, despite the widespread use of PRP, the picture has not changed greatly. In 1984, of 208 consecutive cases of NVG diagnosed over 4 years, 36% were caused by CRVO, 32% by diabetic retinopathy, and 13% by CAOD.[80] At present, probably one-third of the cases of NVG are attributable to CRVO, one-third to diabetic retinopathy, and one-third to diverse causes, with CAOD being prominent.
CENTRAL RETINAL VEIN OCCLUSION
It has been known since at least the time of von Graefe[81] that CRVO can be associated with NVG. Numerous studies have reported widely varying incidences of this complication, ranging from 15% to 65%, with an average of ~30%.[82] Investigators have found that CRVO can be divided into two distinct entities based on the presence or absence of retinal ischemia.[83] The group without retinal ischemia is termed nonischemic retinopathy and the group with retinal ischemia is termed ischemic retinopathy. This differentiation has been confirmed using fundus fluorescein angiography.[84,85]
The natural history of untreated CRVO is that essentially none of the nonischemic eyes progress on to NVG,[81,86,87] whereas 18[88] to 60%[86] of eyes in the ischemic group do so. The greater the degree of capillary nonperfusion (hence, retinal hypoxia), the greater the chances of neovascularization developing.[81,89] Studies with experimental branch retinal vein occlusion in miniature pigs have shown that the resultant vasoproliferative microangiopathy was associated with decreased preretinal oxygen levels, confirming that the ischemic areas were indeed hypoxic.[90] An overall incidence of NVG of 40% for the ischemic type of CRVO is supported by the largest, least-biased study.[81] NVG can appear at any time from 2 weeks to 2 years after the initial occlusion,[91] but in more than 80% of the cases, NVI and NVG appear within the first 6 months after CRVO.[81]
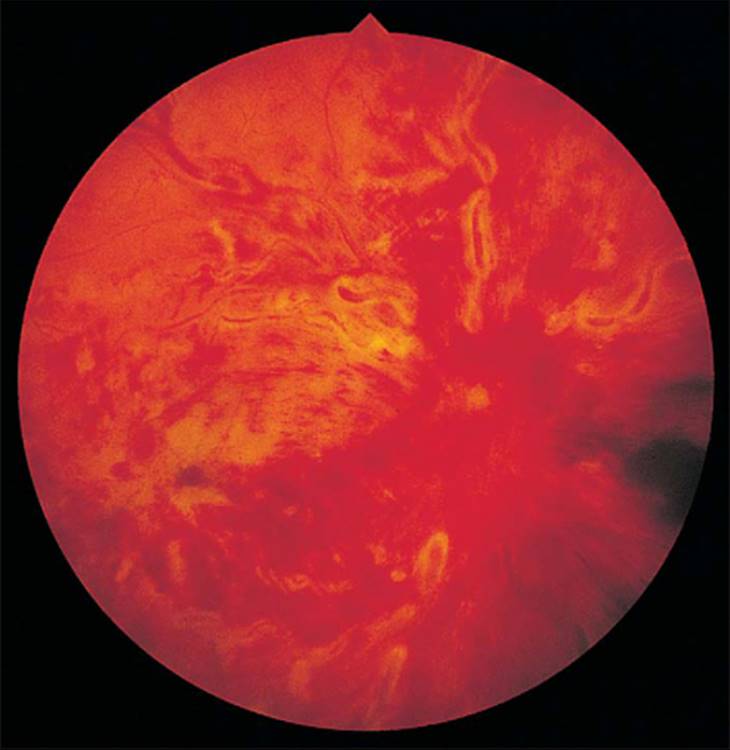
The clinical appearance of the retina can be deceiving in determining the perfusion status. A fluorescein angiogram is imperative in the diagnosis and, ultimately, the treatment of CRVO. Clinically, sometimes it is not possible to determine how much capillary nonperfusion occurs because retinal hemorrhages block out fluorescence in 30% of the initial fluorescein angiograms (Figs 213.14 and 213.15).[81] Almost half of CRVO eyes with indeterminate fluorescein angiograms due to retinal hemorrhages ultimately turn out to be the ischemic type and need to be followed-up carefully with repeat fluorescein angiograms as needed.[92] The nonischemic CRVO can convert to the ischemic type.[81] Approximately 1093 to 15%[94] convert within 8 months, and ultimately one-third[95] to one-half[16] convert to the ischemic type, and all nonischemic CRVO eyes need to be followed-up carefully as well.
|
|
|
|
FIGURE 213.14 Central retinal vein occlusion with extensive intraretinal hemorrhages. |
|
|
|
|
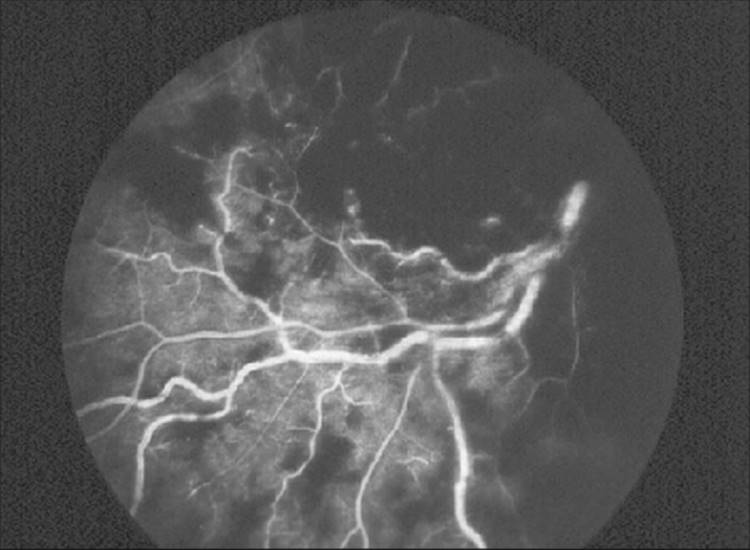
FIGURE 213.15 Fundus fluorescein angiogram of the eye shown in Figure 213.14. With the extensive intraretinal hemorrhages, it was not possible to determine the extent of retinal ischemia. Since up to half of these eyes ultimately turn out to be the ischemic type, these eyes need to be followed-up carefully with repeat fluorescein angiograms, as if they were ischemic. |
The current technique of choice in detecting retinal ischemia is fundus fluorescein angiography. Aside from the already mentioned problem of retinal hemorrhages obscuring the fluorescein angiogram, this technique is fraught with limitations. Foremost is the fact that the fluorescein angiogram is a measure of morphologic changes. It can show areas of nonperfusion but does not differentiate hypoxic from totally anoxic tissue. Furthermore, interpretation of the fluorescein angiogram may be difficult. In a classic study, it was shown that eight retinal specialists were accurate only 60% of the time in correctly identifying retinal ischemia on fluorescein angiograms.[96] Another study pointed out that up to 60% of fluorescein angiograms give false-positive results and that these eyes may be treated needlessly.[97] The electroretinogram measures the functional status of the retina and is not prone to the vagaries of human interpretation or to the handicap of retinal hemorrhages or opacities of the ocular media.[98]Unfortunately, despite recent attempts to standardize electroretinographic equipment and results,[99] it is not yet readily available to most clinicians. Objective and accurate measurement of the extent of retinal ischemia in CRVO may ultimately lead to a more rational approach to the prophylactic treatment of the neovascular complications of CRVO.
Twenty percent of human eyes have a two-trunked central retinal vein,[100] and either of the two trunks may become occluded (i.e., hemicentral retinal vein occlusion). When that happens, it appears to behave just like CRVO.[101,81] In general, there must be ischemia in more than at least half of the retina to cause NVI.[81] For that reason, NVG is extremely rare with branch retinal vein occlusion[102] and macular retinal vein occlusion.[81] However, cases of NVG have been reported after branch retinal vein occlusion.[103]
There appears to be an association between CRVO and preexisting OAG. This relationship has been confirmed in many studies, and the incidence ranges from 6% to 66%.[104] In the largest series published, involving 360 patients with CRVO, there was a 23% incidence of primary OAG or ocular hypertension.[81] The association between CRVO and primary OAG seems to be age-related, with up to 75% of CRVO patients older than 65 years of age having concomitant OAG.[105] There may be an explanation for this apparent difference between older and younger patients. Although in a study of 12 young (average age 29 years) patients with CRVO, none had daytime IOP greater than 21 mmHg, 11 of 12 had diurnal fluctuation greater than 21 mmHg, with some IOPs up to 30 mmHg.[106] In addition, eight patients had a maximum diurnal swing in the IOP greater than 8 mmHg. This study supports the belief that elevated IOPs may play an important role in the cause of CRVO in both young and elderly patients and points out that some cases of OAG, especially in young patients, may be missed if the applanation tension is not taken at different times of day. However, there is no consensus with regard to whether or not the presence of OAG influences the ultimate development of NVG.[81,86]
DIABETES MELLITUS
Diabetes mellitus is one of the leading causes of blindness in the United States today.[107] Despite the 1976 Diabetic Retinopathy Study, which showed the beneficial effects of PRP in preventing severe visual loss, there has not been a significant decrease in the incidence of NVG from diabetes mellitus;[80,79] in part, this may be due to the increasing incidence of diabetes.[107]
The association between NVI and diabetes mellitus was first made by Salus in 1928.[3] In 1939, Fehrmann[108] correlated NVI with the presence of retinal neovascularization. Subsequent studies on unselected diabetic populations found that the incidence of NVI ranged from 1%[109] to 17%.[79] In eyes with PDR, the incidence of NVI goes up to 65%.[110] In a histologic study of unselected eyes removed from patients with diabetes mellitus, 95% of the eyes were found to have NVI, and of these, 90% had retinal neovascularization.[111] It is now well accepted that NVI is associated with retinal hypoxia and PDR.[112] It was shown that the conjunctival oxygen tension in diabetics with PDR is significantly lower than that in diabetics with nonproliferative diabetic retinopathy, which is significantly lower than in diabetics with no retinopathy.[113]
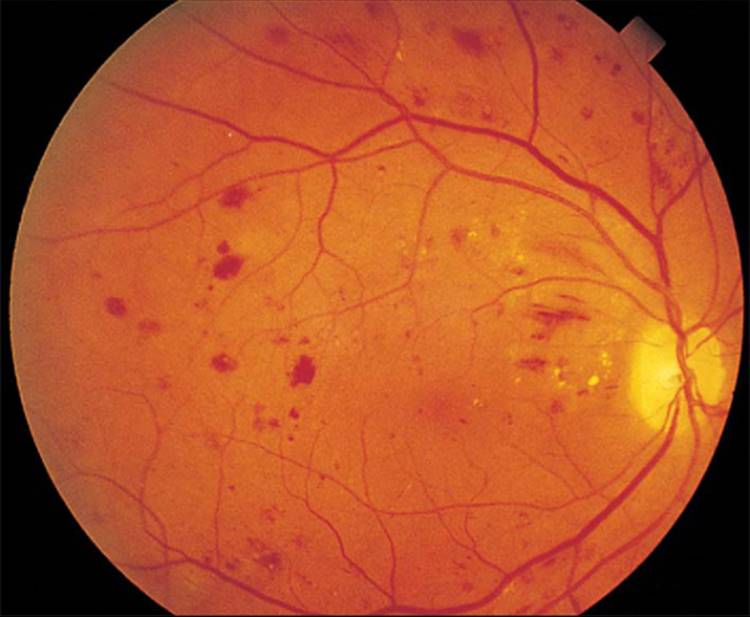
Clinically, there can be significant retinal hypoxia with few visible signs of PDR. Fundus fluorescein angiography is often necessary to show capillary nonperfusion. Even then, the posterior pole may appear remarkably benign, and it is imperative to examine the peripheral retina on angioscopy and to take peripheral photographs on the angiograms (Figs 213.16 and 213.17).
|
|
|
|
FIGURE 213.16 A 51-year-old woman with a 10-year history of type II diabetes mellitus. The fundus shows nonproliferative diabetic retinopathy. |
|
|
|
|
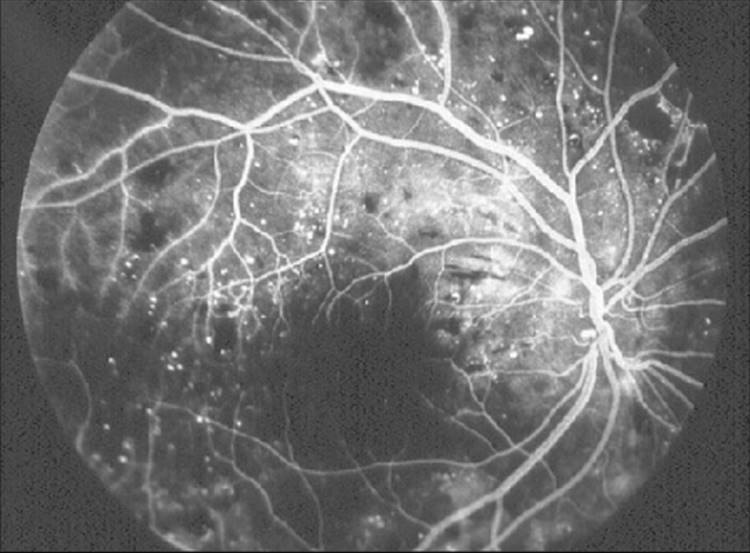
FIGURE 213.17 Fundus fluorescein angiogram of the eye shown in Figure 213.16, displaying extensive capillary dropout and retinal ischemia that was not apparent on funduscopy. |
There are two major groups of diabetes mellitus. Type I or insulin-dependent diabetes, previously known as juvenile-onset diabetes, makes up ~15% and type II or noninsulin-dependent diabetes, previously known as maturity or adult-onset diabetes, makes up ~80% of the total. The remaining 5% consists of other types of diabetes, such as secondary diabetes mellitus. Regardless of the type of diabetes, it seems that the major factor in the onset of diabetic retinopathy is the duration of diabetes. In type I diabetes, retinopathy occurs in 10% of patients after 10 years, in 50% of patients after 15 years, and in 90% of patients after 25 years.[114] In type II diabetes, retinopathy seems to occur sooner than in type I diabetes.[27] After 10 years, up to 50% of patients have retinopathy.[114]
Despite the difference in the onset of retinopathy between type I and type II diabetes, studies have shown that these long-term complications are due to the metabolic derangement (hyperglycemia) rather than to genetic differences between the two types of diabetes.[115,116] The importance of tight metabolic control of diabetes was answered by the Diabetes Control and Complication Trial (DCCT) Study.[117]With a mean follow-up of 6.5 years, 1441 patients were studied prospectively. Patients who had intensive insulin therapy showed a 76% reduction in the mean risk of retinopathy developing and in those with preexisting mild retinopathy, 54% showed a decreased progression of retinopathy.
There is also convincing evidence that concomitant hypertension in diabetic patients results in greater frequency and greater severity of diabetic retinopathy.[118] Therefore, controlling hypertension is potentially important in the prevention of ocular complications of diabetes mellitus.
After NVG develops in one eye of a diabetic patient, the natural history without treatment is that it almost inevitably develops in the fellow eye as well.[119] Bilateral NVI or NVG in an adult is almost always caused by diabetic retinopathy.[39] The time interval between the onset of diabetes mellitus and the development of retinopathy is known, but the time sequence between development of retinopathy and the appearance of NVI is not known. The time interval between the onset of NVI and NVG in untreated cases varies from 1 month to more than 3 years.[120] Furthermore, it is unpredictable as to which untreated eyes with NVI will ultimately progress to NVG. There are well-documented cases of spontaneous regression of diabetic retinopathy. In eyes with early changes of microaneurysms and hemorrhages, 45% were found to return to retinopathy-free status at least once during regular annual ophthalmic examinations.[121] In up to 26% of eyes, NVI may spontaneously disappear in 5 years.[120] This unpredictable course of NVI and NVG due to diabetic retinopathy makes the evaluation of any prophylactic treatment for NVI and NVG difficult at best.
POSTCATARACT EXTRACTION IN PATIENTS WITH DIABETES MELLITUS
Capillary dropout and retinal hypoxia make the eye with diabetic retinopathy especially vulnerable to further insult. Surgical procedures such as cataract extraction and vitrectomy add considerable risk for the development of NVG in such eyes. NVG has not been reported as a complication in large series of intracapsular cataract extraction in unselected populations.[122] However, we found that diabetic patients who had undergone intracapsular cataract extraction in one eye, with the fellow eye serving as the unoperated control, had a significant increase in postoperative NVI-NVG (7.8% vs 0%).[123] In patients with preoperative PDR, the risks of postoperative NVI-NVG developing was even greater (40% vs 0%). The preoperative presence of PDR also carries significantly greater risk of NVG developing after vitrectomy.[124,125] Extracapsular cataract extraction (ECCE) in diabetics does not seem to be associated with an increased incidence of postoperative NVG,[126] but primary capsulotomy at the time of ECCE does seem to predispose them toward the postoperative development of NVG.[126] Thus, it appears that both active PDR as a source of some AF and removal of the diffusion barrier, through intracapsular cataract extraction or ECCE with capsulotomy, are necessary predisposing factors toward the development of NVG.
There is some question whether the posterior capsule or the anterior hyaloid membrane is the critical diffusion barrier[127] because if only the lens capsule was the effective barrier, one would have to assume that the zonules are an effective functional, if not anatomic, barrier as well. If the hyaloid membrane were the effective barrier, the assumption about the zonules would not be necessary. Regardless, the importance of the posterior capsule-anterior hyaloid barrier was shown in a report of three cases of NVG developing after Nd:YAG laser capsulotomy in diabetic patients.[128] The fundus was not visualized before the capsulotomy in these patients, but when NVG was noted, two had PDR and one had nonproliferative diabetic retinopathy. Other concomitant causes of retinal ischemia in that case were not noted. In another report, in eight patients with nonproliferative diabetic retinopathy who had PDR after ECCE, only two patients developed NVG, and both had a broken posterior capsule that required a vitrectomy.[129] Clearly, the role of the posterior capsule versus the hyaloid has not yet been settled, but either or both are important barriers to the diffusion of AFs.
The posterior capsule-anterior hyaloid barrier is only a relative one[129] and can be overwhelmed if there is a great amount of AF; e.g., it is common to see NVG in phakic patients with extensive posterior segment ischemia. If this barrier is removed or disrupted, such as with intracapsular cataract extraction or ECCE with capsulotomy, enough AF may then diffuse forward to stimulate NVI. Inflammation from cataract surgery or laser capsulotomy may contribute as well. Whenever possible, adequate PRP should be performed in any patient with early PDR and/or ischemic CRVO before cataract extraction.[123]These patients should also be followed-up carefully after cataract surgery and laser capsulotomy so that NVI can be detected early and appropriate treatment can be started.
POSTVITRECTOMY FOR COMPLICATIONS OF DIABETIC RETINOPATHY
Modern pars plana vitrectomy heralded a new era in the treatment of complications of diabetic retinopathy.[130] Initially, concurrent lens surgery was often performed whether or not a cataract was present. It soon became clear that concurrent lensectomy significantly increased the risks of postoperative NVI and NVG developing. Large series of vitrectomies in diabetic patients showed a twofold[131] to threefold[132] increased risk of NVI developing if lensectomy was performed; however, not all studies found this association.[133,130]
Long before the advent of vitrectomy, it was known that chronic retinal detachment is a significant cause of NVG.[33] Studies have noted the importance of retinal detachment as a cause of postvitrectomy NVI and NVG.[134] However, the often-cited large studies did not consider the role of postoperative retinal detachment as a cause of postvitrectomy NVG.[132] The records of 255 consecutive vitrectomies were reviewed, 81 of which were performed for complications of diabetic retinopathy and 175 of which were performed for nondiabetes-related problems that served as controls in the study.[135] Sixty-five different preoperative, intraoperative, and postoperative parameters were statistically analyzed, and only postoperative retinal detachment had a significant correlation with the development of NVI and NVG after vitrectomy. In the diabetic group, 83% of the eyes with retinal detachment developed NVI-NVG versus 2% of the eyes with an attached retina. For comparison, it should be noted that 2% of eyes with severe PDR in the Diabetic Retinopathy Vitrectomy Study progressed on to NVG even without vitrectomy.[136] In the control group, 19% of the eyes with retinal detachment developed NVI and NVG, versus none of the eyes with attached retinas.[135] Aphakia alone did not correlate significantly with the development of NVI and NVG. However, aphakia combined with retinal detachment was associated with an even greater risk of NVI and NVG (92%) developing in the diabetic group.[135] In a series of 15 diabetic patients undergoing combined cataract extraction, posterior chamber intraocular lens implant, and pars plana vitrectomy, the only case of postoperative NVI was in an eye with chronic inoperable retinal detachment.[137]
It is clear that both diabetic retinopathy and retinal detachment are important causes of NVI and NVG, and when both conditions coexist, there is an even greater stimulus for neovascularization. As was pointed out earlier, the lens-posterior capsule-anterior hyaloid is a relative, but important, diffusion barrier for the anterior passage of the AF.[127,138] When the stimulus for neovascularization has not been eliminated, such as with reattachment of the retina and adequate PRP, the presence or absence of a barrier against the AF has clinical significance. However, if there is no source of this AF, the presence or absence of this lens-hyaloid barrier is academic. An attached retina does not mean that the stimulus for neovascularization is not present. Cases have been reported in which NVI was present after vitrectomy despite an attached retina.[137,135] With postvitrectomy PRP, there was regression of the NVI.[137,135] The conclusion is that after vitrectomy for complications of PDR, an attached retina with adequate PRP is of paramount importance in preventing NVG; lensectomy per se is of secondary importance.
CAROTID ARTERY OCCLUSIVE DISEASE
Despite numerous reports of NVG resulting from occlusion of the common carotid, internal carotid, and ophthalmic arteries,[139,140,141,142] it was believed that CAOD was not a common cause of NVG.[82] It is now known that CAOD is the third most common cause of NVG, accounting for at least 13% of cases.[80] This figure is probably low because CAOD may be an unrecognized component of CRVO and diabetic retinopathy. Demonstrable common or internal carotid artery atherosclerosis is present in up to 37% of patients with CRVO.[143] The incidence is 50% for the ischemic type of CRVO and 17% for the nonischemic type.[143] In one series, six of 12 patients who presented with CRVO had CAOD requiring endarterectomy.[144] Forty percent of patients diagnosed with CAOD presented with CRVO in the same study.
NVG actually represents only one manifestation of CAOD. The spectrum ranges from transient ischemic attack[145] to hypoperfusion retinopathy[35] (previously called venous stasis retinopathy[146]) to ocular ischemic syndrome. It has been proposed that the term chronic ocular ischemia should be used for this confusing array of terms.[147] It has been estimated that evidence of chronic ocular ischemia will be found in 4%[148] to 18%[149] of patients with CAOD. It has been shown that eyes with NVI have reversal of blood flow in the ophthalmic artery, suggesting that carotid obstruction alone is insufficient to cause the ocular sequelae of CAOD; there must be diversion of blood from the eye to the brain.[150] It is precisely these patients with ocular and visual symptoms who are usually seen by an ophthalmologist first and who are at risk of having a cerebrovascular accident. Thus, it is imperative to consider CAOD in a patient with NVG.
There are several unique aspects of NVG resulting from CAOD. Decreased perfusion of the ciliary body from CAOD significantly decreases aqueous production.[151,152,153] Thus, despite extensive synechial angle closure, the IOP may be normal or even low (see Fig. 213.17).[140,150,153] One study found that only one-third of the eyes with NVG from CAOD had elevated IOP.[154] In every case of NVG with low IOP that is disproportionate to the extent of angle closure, and there is no concurrent retinal detachment, CAOD must be considered.[155] After endarterectomy or carotid bypass surgery, aqueous production often returns to the normal pre-ischemic level. IOP can increase dramatically, despite the seemingly paradoxical regression of the NVI.[140,146,157]
CENTRAL RETINAL ARTERY OCCLUSION
The association between NVG and CRAO was first noted in 1874.[158] The incidence has been variously cited as between 1% and 17%.[159,160,161] If some of these studies are analyzed in greater detail, a more consistent figure emerges. In the largest (171 patients) reported series,[162] branch retinal artery occlusion and CRAO are included together in the analysis; if only CRAO is considered, the incidence of NVG in that study is 7%. In another series,[161] 10 of 64 eyes (16%) with CRAO developed NVG, but nine of them also had CAOD; one of 64 (2%) with only CRAO developed NVG. A number of factors suggest that the incidence might be lower than the reported high of 17%.[159] Some of the cases reported may not have been NVG, because the NVI was determined clinically, and gonioscopy was not performed. Also, some of the reported cases could have been ophthalmic artery obstruction and other cases may have been CAOD.[163] In a prospective study,[160] five of 33 (15%) consecutive patients with CRAO developed NVG. However, two of seven patients who had any ocular neovascularization (five patients with NVG, one patient with NVI only, and one patient with optic disk neovascularization) also had concurrent ipsilateral CAOD. If the two patients with CAOD had NVG, the incidence of NVG after CRAO alone would have been 9%. Thus, the true incidence of CRAO is probably higher than the previously accepted 2% and is lower than the extremely high 17%. Without a definitive study, it would be estimated that the incidence of NVG due to CRAO alone is between 5% and 10%.
With CRAO, there is destruction of the inner retinal layer and capillary endothelium.[164] This explains why there is generally no retinal neovascularization in CRAO. Cases in which there is neovascularization of the disk after CRAO are rare,[165,166,167] and the new vessels may come from the choroidal circulation.[167] A more important consideration is that with no viable retinal tissue, the eye should not be capable of inducing a neovascular response elsewhere.[168] If this is so, it is unclear why there is NVI-NVG at all with CRAO. It has been proposed that there is first CAOD, which causes anterior segment ischemia, leading to NVI and secondary NVG.[161] At the same time, the decreased perfusion of the central retinal artery makes it susceptible to occlusion, especially if there is increased IOP from the already existent NVG. In support of this theory, several studies of eyes with CRAO and NVG have found CAOD in 45%,[169] 90%, and 100%[154] of these eyes. Thus, rather than CRAO causing NVG, it is believed that it is more likely that NVG from CAOD contributes to the CRAO.
This is clearly not the only explanation of NVG resulting from CRAO. There are documented cases of NVG that did not have concomitant CAOD.[170,159,160] Although some believe that the NVG from CRAO is a different entity from CRVO,[171,45] the histologic appearance is exactly the same. Ultimately, it is believed that retinal ischemia or anterior segment ischemia from CAOD is the cause. The beneficial effect of PRP in NVG from CRAO also supports this concept.[172,128]
INTRAOCULAR TUMORS
RETINOBLASTOMA
The association between retinoblastoma and glaucoma was first noted in 1965.[173] The incidence of NVI in eyes enucleated for retinoblastoma has been reported to be from 30 to 72%.[39,6,20] There is a significant association between the presence of NVI and tumor involvement of the choroid[6] and the posterior pole.[20] In fact, the posterior pole involvement was so consistent that it was possible to predict the presence or absence of NVI by histologic examination of the posterior pole only.[20] It was also found that the duration of retinoblastoma was associated with the development of NVI.[6] In a recent study using retinoblastoma cells lines, VEGF mRNA was detected with in situ hybridization, there was immunostaining for VEGF, and hypoxic conditions resulted in increased VEGF production.[174] This confirms the clinical impression that retinoblastoma is a cause of NVI-NVG. When children present with opaque media and NVI or NVG, one must consider occult retinoblastoma high on the list. Ultrasonography, computed tomography, and other appropriate studies must be performed.
MALIGNANT MELANOMA
The association between NVG and untreated malignant melanoma was first noted in 1944.[175] The incidence in subsequent studies has ranged from 0.5% to 15%[176,177,178] not as high an association as with retinoblastoma. The occurrence of NVI correlates with increased tumor size, tumor necrosis, or extent of overlying retinal detachment.[179,176,180] With newer treatment modalities for uveal melanomas, specifically helium ion irradiation[180] and photocoagulation with hematoporphyrin,[181] the incidence of NVI-NVG does not seem to be any lower. Since various forms of irradiation are also known to cause NVI-NVG,[182,183,184] it is not possible to determine if the NVI-NVG in these reports was due to the tumor, the irradiation, or the secondary tumor necrosis. Interestingly, unlike retinoblastoma, VEGF and VEGF mRNA were not detectable by immunostaining and in situ hybridization studies in malignant melanomas.[174] Hypoxic conditions in melanoma cell lines did not increase VEGF production, in contrast to retinoblastoma cell lines.[174] However, one case report suggests that there must be some production of AF (e.g., VEGF) from an intraocular melanoma. An eye with a primary epithelioid melanoma of the iris showed complete resolution of NVI-NVG after the tumor was excised.[185]
Nevertheless, in an eye with NVG in which the posterior segment is not visible because of opaque media, malignant melanoma must still be considered, especially in the absence of diabetic retinopathy in the contralateral eye or a past history of CRVO in the ipsilateral eye.
OTHER TUMORS
NVI has also been described in eyes with metastatic tumors[186] and reticulum cell sarcoma,[187] and in these cases, the direct effect of tumor-secreted AF may be the cause of the new vessels.[185]
POSTRETINAL DETACHMENT SURGERY
Chronic retinal detachment is a well-known cause of NVG.[39] In one study, retinal detachment was found to be the second most common cause (23%) of NVI in a series of 105 enucleated eyes with NVG.[188]In children younger than 5 years of age, retinal detachment was also found to be the second most common cause (32%) of NVI.[6] As previously mentioned, persistent retinal detachment is the major cause of postvitrectomy NVI and NVG.[135] Several conditions thought to cause NVG may, in fact, act through the common pathway of secondary retinal detachment. For example, NVG associated with Coats' disease is often associated with retinal detachment.[140,188,6] Other reported causes, such as X-linked juvenile retinoschisis,[189] also have retinal detachment as a common denominator. Retinal detachment must be considered if an obvious cause of NVG cannot be found because the retina must be reattached, if possible, as the first step in the treatment of NVG.
MISCELLANEOUS CAUSES
Now that we have a better understanding of the pathophysiology of this condition, we know that there are several common pathways, such as chronic retinal detachment and/or extensive capillary nonperfusion with resultant retinal hypoxia. In addition to CRVO, sickle-cell retinopathy,[190,191] Stickler's syndrome,[192] autosomal dominant neovascular inflammatory vitreoretinopathy,[193] and other as yet unreported retinal diseases produce NVG by the retinal hypoxia pathway. Decreased ocular perfusion, such as with CAOD, is yet another common pathway. For instance, reported cases of optic disc glioma causing NVG probably are due to the enlarging tumor compressing ciliary and retinal vessels with resultant retinal hypoxia.[194] More localized anterior segment perfusion compromise has been shown to be the cause of NVI in Fuchs' heterochromic iridocyclitis[15,17] and the pseudoexfoliation syndrome.[195] There has not been any documented case of NVG from these two entities, perhaps because the vascular occlusion and secondary hypoxia are so limited. The association between scleritis and NVG is also probably due to secondary vascular occlusion.[196] Finally, direct stimulation of iris vessels by intraocular tumor-secreted AF is yet another pathway.[185]
|
Treatment Summary |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
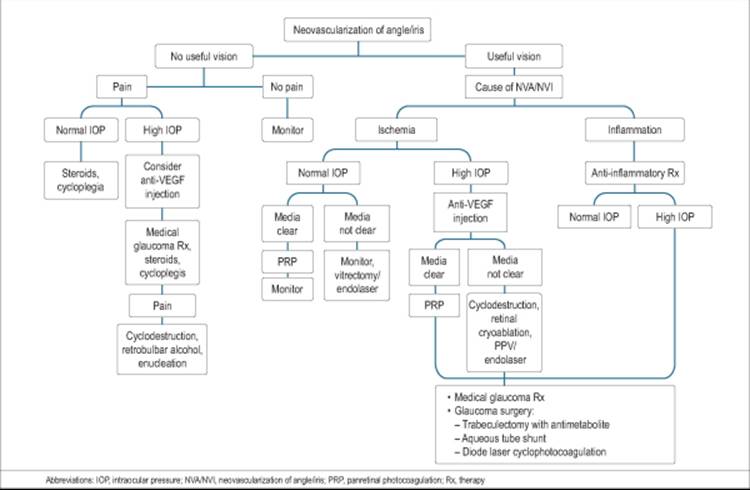
MANAGEMENT
As recently as 1974, no methods were known to prevent or treat NVG.[82,197] Since 1974, many review articles have appeared on the management of NVG,[198,8,199] but preventive treatment has received little attention.[198,200]
PROPHYLACTIC TREATMENT
Frequently, the causes of NVG may be multifactorial and include diabetes mellitus, hypertension, CAOD, or CRVO, or some combination thereof. Control of hypertension, blood glucose, and cholesterol levels, as well as a strong clinical suspicion for occult CAOD, is the best prophylaxis for the patient against NVG.
With CRVO, most if not all patients should undergo fluorescein angiography. If it is of the ischemic type, PRP should be considered, depending on the degree of retinal ischemia and the reliability of the patient regarding timely follow-up examinations. If there are retinal hemorrhages preventing photocoagulation, the patient should be followed-up carefully and treated as for ischemic CRVO. Nonischemic CRVO should also be followed-up carefully, because it can become ischemic. Without PRP, ~40% of the ischemic type of CRVO proceeds on to NVG.[81] The largest, and most convincing, study to date consists of a series of 100 eyes with ischemic-type CRVO.[201] All eyes received early argon PRP, and none developed NVG.
However, a more recent study suggested that rather than performing prophylactic PRP, careful follow-up of ischemic eyes would yield the same results if PRP were performed when NVI is first detected.[94]This seems to be in conflict with the practice of most clinicians. Part of this dilemma may be explained by the aforementioned lack of an objective and accurate measurement of the extent of retinal ischemia. Until such a test is available, each clinician must determine for each CRVO patient how severe the ischemia is and, based on that patient's compliance and reliability for continued follow-up, whether to perform prophylactic PRP or to follow-up that patient carefully.
In general, the visual outcome is unchanged whether or not PRP is performed, since the visual acuity has been determined by the primary vascular accident.[202] However, the majority of treated patients show some improvement in visual acuity.[203] OAG should also be well controlled, especially in higher risk elderly patients. Since there is frequently low IOP in eyes after CRVO, all eyes with CRVO should be considered glaucoma suspects and followed-up accordingly.
For NVG to develop in diabetes mellitus, there must be retinal hypoxia associated with PDR.[112] It has been well documented that the major factor in the onset of PDR is the duration of the diabetes mellitus.[114,204] With the publication of the DCCT results, there is now convincing evidence that near-normal glycemia is associated with later development and lesser severity of diabetic retinopathy.[117] Thus, the achievement of euglycemia is an important preventative measure. Once diabetic retinopathy is present, many studies have shown the beneficial effects of PRP on causing regression of NVI.[205] It has been shown that PRP in diabetic patients prevents the later development of NVI and NVG.[9]
Prophylactic therapy in CAOD is not aimed at preventing NVI since this is generally not possible, but rather at considering this diagnosis as a cause of NVG. CAOD should always be considered if there is asymmetric severity of diabetic retinopathy between two eyes, in any case of NVG in which the IOP is normal or subnormal, in any case in which adequate PRP has not caused regression of the NVI, or when there is no other apparent cause. If unsuspected CAOD is detected, and appropriate therapy is instituted, potential stroke or death could be averted for that patient. Since NVG has been reported to occur anywhere from 1 week to 5 months after CRAO,[160,161] patients with CRAO should be followed-up carefully for at least 6 months, and PRP should be instituted when NVI is first noted.
ANTIANGIOGENESIS
Probably the most exciting prospect in the treatment of NVG is the development of specific inhibitors of VEGF, but there are a number of obstacles along the way. VEGF, comprised of 4 homodimeric polypeptides, is the most extensively studied of the proangiogenic factors in the pathogenesis of NVG.[76,206] Müller cells are thought to represent a significant source of VEGF under conditions of retinal hypoxia. Elevated levels of VEGF have been identified in the aqueous humor of patients with NVG.[207] Experimental studies in nonhuman primates have also shown that intravitreal injections of human recombinant VEGF are sufficient to produce noninflammatory NVI, ectropion uveae, and NVG.[75]
Therapy targeted for VEGF is complex, owing to endothelial cell diversity, which includes variation in tissue expression of the gene encoding VEGF and the complexity of the VEGF family isoforms and receptors. Furthermore, VEGF therapy alone would likely not be sufficient to fully counteract the angiogenesis process due to the contribution of dozens of other factors in the angiogenesis cascade.[208]These other proangiogenic factors include insulin-like growth factors I and II, insulin-like growth factor binding proteins 2 and 3, bFGF, TNF-?, interleukin-6, and platelet-derived growth factor.
New vessel formation in the eye is affected to a large extent by the homeostatic balance between VEGF and other proangiogenic factors and the antiangiogenic factor, pigment-epithelium-derived factor (PEDF).[209,208] PEDF is a natural occurring and extremely potent angiogenesis inhibitor that not only targets new vessel growth but also has potent neuroprotective activities. In support of a VEGF-PEDF homeostatic equilibrium theory, studies have shown increased levels of VEGF and decreased levels of PEDF in the vitreous humor of patients with PDR. Observations of reduced PEDF levels in the vitreous of patients with active diabetic retinopathy, compared with inactive retinopathy, further support this theory.[210,211]
EARLY-STAGE THERAPY
In the therapy of NVG, the critical aspect is early detection of NVI. Thus, every patient with diabetes, regardless of duration, as well as every patient who has had CRVO or a CRAO, should have a careful slit-lamp examination of the iris, especially the pupillary margin before the pupil is dilated. Once NVI is discovered, and there is little or no angle involvement, the mainstay in early therapy is PRP.
How exactly PRP produces its beneficial effects remains unclear. Aiello and colleagues[73] obtained vitreous samples from six eyes at the time of vitrectomy for complications of diabetic retinopathy both before and after PRP had been performed and found a 60% decrease in vitreous VEGF levels after the laser treatment. PRP therefore definitely decreases ocular VEGF levels, but how it accomplishes this remains unclear. The effectiveness of various lasers is more dependent on the amount of retina treated than on the type of laser employed.[212] This is in agreement with studies that have shown that other forms of retinal destruction, such as xenon arc photocoagulation,[155] retinal diathermy,[109] and retinal cryotherapy,[213,214] also have beneficial effects on preventing and causing regression of NVI.
In practice, the most common cause of 'failed' PRP is inadequate photocoagulation. It is estimated that during standard PRP, only 13% of the total retinal area is treated.[215] According to the guidelines of the Diabetic Retinopathy Study, a total of 1200-1600 500-?m-sized burns should be applied randomly over the peripheral retina.[155] However, more than this number of photocoagulation burns may be necessary to effect regression of neovascularization. The Diabetic Retinopathy Study report showed that eyes that had more PRP had less risk of visual loss than did eyes with less PRP.[216] Twelve hundred spots represents less than one-quarter of the total confluent 500-?m spots that can be placed on the average extramacular retinal area of a human eye.[217] Many retinal specialists believe that a minimum of 1500-2000 spots using the Rodenstock wide-angle fundus contact lens is necessary to constitute adequate PRP. The Rodenstock lens produces a 670-?m-sized retinal lesion,[218] so a correspondingly greater number of lesions need to be produced if the Goldmann lens (500-?m retinal spot) is employed (Fig. 213.18). If there is some opacity in the media, or if there are retinal hemorrhages present, the krypton laser is frequently more effective than argon laser treatment.
|
|
|
|
FIGURE 213.18 Example of adequate panretinal photocoagulation. Note the almost confluent areas of photocoagulation spots. A total of 1500-2000 spots should be applied if using the Rodenstock lens (800-?m spot size); if the Goldmann lens (500-?m spot size) is used, proportionately more spots should be applied. |
When adequate PRP is performed early in the course of NVI, there is ample documentation that there is regression of the NVI in CRVO[219] and PDR.[129,220] PRP has also been shown to cause regression of NVI and appears to reduce the incidence of NVG after CRAO.[172] However, the results of PRP in CRAO are not nearly as impressive as in CRVO or PDR.[160] In eyes with radiation-induced ocular ischemia, PRP decreased the risk of NVG developing 6.4 times when compared with similar eyes not receiving PRP.[221] Even with CAOD, in which the hypoxia may not be limited to the retina alone, PRP can still produce a diminution of NVI until more definitive therapy can be instituted.[222,223] Thus, PRP is the most important early treatment for NVI and NVG. Obviously, if the cause is a detached retina or an ocular tumor, the therapy must be directed at these primary causes.
Endophotocoagulation
There are situations in which it is not possible to perform preoperative PRP in an eye with NVI, and intraocular surgery (e.g., cataract extraction or vitrectomy) must be performed. Increased postoperative risks of NVG developing in such a situation have already been noted, and postoperative vitreous hemorrhages could prevent any postoperative photocoagulation. Argon endophotocoagulation is an effective method of performing intraoperative PRP.[224] A method for performing endophotocoagulation with indirect ophthalmoscopy has been described; this technique allows better visualization of the fundus.[225]Endophotocoagulation can be just as effective as standard photocoagulation, and this procedure is now used extensively, especially during vitrectomy.[226] In high-risk eyes, endophotocoagulation is valuable in providing a 'head start' on retinal photocoagulation. In many cases, further photocoagulation needs to be performed via the slit lamp at a later time.[200]
Panretinal Cryotherapy
In cases in which the cornea, lens, or vitreous is too hazy to allow adequate PRP, there is a role for panretinal cryotherapy (PRC).[213,214] In the largest series to date, 27 eyes with NVG (nine of which had previous PRP) were treated, and 55% had stabilization or improvement of vision, 55% had reduction of IOP, and 70% had stabilization or regression of NVI after 12 months.[213] As might be expected, cryopexy over an area of retinal detachment has no effect because the retina is not affected by freezing. This is a major operation that produces significantly more inflammation and blood-retinal barrier breakdown than does PRP, as shown by vitreous fluorophotometry.[227] Potential complications include traction and exudative retinal detachment and vitreous hemorrhage.[213,228] However, PRC seems to have an added, although as yet unexplained, benefit of facilitating the clearance of existing vitreous blood.[228] Increased influx of macrophages after PRC may be an explanation.[229]
Goniophotocoagulation
Direct laser treatment of NVI before development of NVG has been investigated by several researchers with mixed results.[230,231] A 73% overall success rate as defined by elimination of angle vessels in the treated area, prevention of further angle closure in the treated area, and maintenance of IOP at less than 25 mmHg was reported by Simmons and associates.[231] However, 38% of these eyes also had concurrent PRP or PRC, and there was no breakdown of the success rate with and without this adjuvant therapy. The natural history of untreated diabetic retinopathy is rather unpredictable, and spontaneous arrest and even regression of NVI of up to one-third of the eyes is well known.[120] One study evaluated preoperative goniophotocoagulation on the success of filtration surgery but found no beneficial effect.[232]
When goniophotocoagulation was first introduced in 1977, the role and efficacy of PRP in the treatment of NVG were still being evaluated. It was believed that there might be cases in which goniophotocoagulation might provide "a period of grace to delay imminent synechial angle-closure until the definitive effect of the PRP can manifest itself."[200] However, since then, in a limited number of patients in whom it was not possible to perform PRP, goniophotocoagulation alone has not proved to be beneficial in preventing synechial angle closure and, at times, has caused increased inflammation and seemingly more rapid progression of angle NVA. At this stage, until a prospective randomized study is performed, the role of goniophotocoagulation in the treatment of NVG remains unclear.
Medical Therapy
Before synechial angle closure has occurred, it is possible to have a secondary OAG from the fibrovascular membrane accompanying the angle vessels blocking the trabecular meshwork. Concomitant OAG may be present as well, especially in a patient with CRVO or diabetic retinopathy. Under these circumstances, all the standard antiglaucoma medications (including miotics or adrenergic agents) will be effective to some degree in lowering the IOP, but one should not be lulled into a false sense of security with medical control. Unless PRP is performed, the angle can relentlessly close. Although there is no scientific evidence, the two medications that are of the greatest benefit clinically during this period are topical atropine 1% twice per day to decrease ocular congestion and topical steroids four times per day to decrease ocular inflammation.[10] Intravitreal injection of crystalline triamcinolone and/or bevacizumab (Avastin®) have also been studied as potential treatments to cause regression of iris neovasculature.[233]
LATE-STAGE THERAPY
Panretinal Photocoagulation
When synechial angle closure has already occurred, it is considered the late stage. If at all possible, PRP or PRC should still be performed to eliminate the stimulus for new vessel formation. Synechial angle closure cannot be reversed with PRP, but further closure can be prevented. Regression of NVI can occur within days of completed PRP. Without elimination of the stimulus for new vessel formation, filtration surgery is more likely to fail. Once PRP has been completed, it is important to allow adequate time to elapse to permit regression of the NVI. At least 1 week, and preferably 3-4 weeks, should elapse between completed PRP and filtration surgery. Generally, the optic nerve heads are not as compromised in eyes with NVG in the absence of concomitant OAG as in advanced chronic OAG and pressures less than 50 mmHg can be tolerated for this time span.[234] If possible, adequate disk perfusion should be confirmed by observing for the absence of central retinal artery pulsation.[234]
Medical Therapy
With extensive synechial angle closure, any of the medications acting on aqueous outflow (e.g., pilocarpine, echothiophate iodide (phospholine iodide), or adrenergic agents) are useless, and in fact, contraindicated because they increase hyperemia and inflammation. Medications that decrease aqueous production, such as topical ?-blockers and carbonic anhydrase inhibitors, are beneficial but do not lower the IOP to a normal range in the face of a closed angle.[235] Topical apraclonidine, an ?-adrenergic agonist, may be used on a short-term basis (days to weeks), but the effectiveness of long-term use has not yet been reported. At this time, the effectiveness of brimonidine or prostaglandin analogues (e.g., latanoprost) in late-stage NVG has not yet been evaluated. Osmotic agents can be used intermittently to clear the cornea enough for treatment or diagnosis. The most important medications remain topical atropine and topical steroids to decrease congestion and inflammation and prepare the eye for definitive surgery.[235]
Conventional Surgery
If there are engorged iris vessels at the time of surgery, intraoperative and postoperative hemorrhages are likely to occur. In addition, the presence of active neovascularization leads to late bleb failure through conjunctival scarring at the filtration site. Previously, the main thrust of surgery was to control intraoperative bleeding through different types of cautery or lasers[42] or even to bypass the whole anterior segment completely with a pars plana filtration technique.[236] To prevent postoperative scarring of the filtration bleb, ?-irradiation,[237] subconjunctival injection of 5-fluorouracil,[238,239,240] and intraoperative mitomycin C[241,242] have been tried. There have been many other interesting and even novel surgical techniques, but the number of cases involved is small and the follow-up has been too short to provide any meaningful information. These reports have been reviewed[82,10] and are of historical interest only. It is well accepted now that whatever the surgical procedure, preoperative PRP or PRC should be performed whenever possible.[232,12,243,8]
Filtration Technique
After adequate PRP, topical atropine, steroids, and enough time for the eye to quiet down, the patient is ready for surgery. If the IOP is high, osmotic agents may be used preoperatively (if the general medical status permits) to avoid sudden surgical decompression of a hard eye. Peribulbar or retrobulbar anesthesia with epinephrine plus orbital massage is preferred to soften the eye further and to reduce vascular congestion.
A full-thickness procedure (e.g., a trephine or posterior lip sclerectomy) or a guarded procedure (e.g., a trabeculectomy) may be performed, depending on the surgeon's choice. The results appear to be the same regardless of which procedure is used.[232] The authors' preference is to perform trabeculectomy with intraoperative application of mitomycin C. If some new vessels remain on the iris when the peripheral iridectomy is performed, preiridectomy cautery to the iris is possible when the iris is lifted by the forceps. Not infrequently, even with this precaution, some bleeding occurs with the iris surgery. Irrigation of the anterior chamber with balanced saline solution via the previously made paracentesis incision usually keeps the anterior chamber clear until the bleeding stops spontaneously. Occasionally, topical epinephrine may be necessary, but postiridectomy cautery to the iris or ciliary body should be avoided. Topical steroids and antibiotics are applied, and a single eye patch and protective shield are placed over the eye. Postoperative medications include topical cycloplegic agents two to three times per day, topical steroids four to six times per day, a topical nonsteroidal antiinflammatory drug four times per day, and topical antibiotics four times per day. Using standard filtration techniques with preoperative PRP, surgical success (IOP less than 25 mmHg on one medication or less[232]) has been reported as 67%,[232] 77%,[244] and 100%.[245]
Postoperatively, the filtration bleb tends to be more limited in size, appears less succulent, and often has a characteristic ring of conjunctival and episcleral vessels that delineate the base of the bleb (Fig. 213.19).[234] Since PRP would not be expected to ablate all the ischemic retina, it is logical that some AFs would still be present. The base of the filtration bleb is probably the new main interface between the aqueous-borne AFs and the potential vascular bed. The IOP in successful filtration with NVG tends to be somewhat higher than with OAG, but since the optic nerve usually is not as compromised in NVG, a slightly higher postoperative IOP is often tolerable.
|
|
|
|
FIGURE 213.19 Even with adequate panretinal photocoagulation, some angiogenesis factors may still be present in the eye. With successful filtration surgery, there is often a ring of injected episcleral vessels around the base of the filtration bleb. |
Antifibrotic Agents
With the introduction of antifibrotic agents intra- or postoperatively, the chances of successful filtration surgery in high-risk eyes such as those with NVG has been significantly increased. In most studies, however, the number of eyes with NVG are small and the follow-up short.[63] One study reported a surgical success rate of 68% at 3 years with filtration surgery augmented by postoperative subconjunctival 5-fluorouracil injections.[238] However, longer term follow-up of 34 eyes from this same group revealed 4- and 5-year success rates of only 41% and 28%, respectively.[240] In both studies, surgical success was defined as IOP less than or equal to 21 mmHg with maintenance of vision; the extent of preoperative PRP was also not evaluated. Although intraoperative mitomycin C has been shown to be more effective than postoperative 5-fluorouracil injections in IOP control in straight trabeculectomies,[241,242] there have been no long-term follow-up studies involving mitomycin C in NVG. Also, unfortunately, the major primary disorders that result in NVG also result in progressive loss of vision. In evaluating success of filtration surgery, comparing only IOP control in NVG may be more meaningful. Until prospective studies involving large numbers of eyes with NVG are available, the current protocol is to perform PRP, delay surgery until the eye is quiet and the NVG stimulus diminished or eliminated, and perform trabeculectomy with intraoperative application of mitomycin C.
Glaucoma Tube Implant Surgery
Filtration surgery in NVG should be reserved for eyes that have useful vision, bearing in mind that almost any vision is worth preserving in eyes with PDR. Because one of the major problems with filtration surgery in NVG is failure of the filtration bleb through episcleral scarring, some physical means to maintain an opening between the anterior chamber and the subconjunctival space has formed the basis for various seton surgical techniques. Countless different materials have been tried, including many kinds of sutures, metals, and plastics in various sheet, tube, or wire forms. None of them worked very well, and their history has been reviewed.[10] With the development of newer nonallergenic plastics, and the knowledge about how previous stents and valves have failed, a new group of implants has been developed for use with NVG and other refractory glaucomas. When conventional surgery fails or is not possible because of excessive conjunctival scarring, insertion of a glaucoma drainage device may be indicated.
The number of patients involved in some of the studies involving eyes with NVG is too small to evaluate adequately. In 59 eyes using a simple silicone tube, there was a reported 63% success in controlling IOP (<25 mmHg).[246] With the Molteno implant, there was an 83% success in controlling the IOP (<20 mmHg) in 24 eyes with NVG.[243] In another series using the single-plate Molteno technique, there was a 47% success rate in 15 eyes of patients with NVG (11 eyes had preoperative PRP).[247] Of significance is that nine of the 15 eyes (60%) had some type of severe complication, including choroidal hemorrhage (two eyes), phthisis or hypotony (two eyes), and traction retinal detachment (one eye).[247]
Another large series of tube implant surgery used the Krupin-Denver valve.[248] In this study, 79 eyes with NVG were operated on with a 67% success rate in controlling IOP (<24 mmHg), with a mean follow-up of 23 months. The Ahmed and Baerveldt glaucoma valve implant may also be useful in the surgical management of refractory NVG.[249] Improved success rates have also been reported in patients with refractory NVG when the drainage tube is implanted through the pars plana, combined with pars plana vitrectomy.[63]
However, long-term results of implant surgery are discouraging. In one study of single-plate Molteno implants in 60 eyes with NVG, success, as defined by IOP less than or equal to 21 mmHg and maintenance of vision, was only 10.3% at 5 years.[250] A more recent long-term prospective study of 145 eyes (130 patients) reported 1, 2, and 5 year success probability of 72%, 60%, and 40%, respectively.[251] Vision was maintained or improved in 39% of eyes, whereas 12% were eventually enucleated.
The problems with all stent and shunt procedures in the treatment of NVG remain the same: early postoperative hypotony, blockage of the internal fistula, blockage of the external filtration site, and a vigorous postoperative fibrous encapsulation response.[252,249] Postoperative hypotony and flat anterior chamber, with all its attendant complications, are problems common to all filtration procedures. Modifications of the Molteno technique using two stages[243] or suture intubation of the silicone tube[253] and the introduction of the valved implants (e.g., Krupin, Ahmed) are valid attempts to avoid early postoperative hypotony, but unfortunately they are not uniformly successful. Obstruction of the internal fistula is unique to the use of all drainage devices and this is especially frequent in NVG, in which intraocular blood and fibrovascular membranes can block the opening of the tube.[254,247] When the blockage is at the tube opening, a combination of the argon and Nd:YAG lasers can be used to pull and cut the iris or membrane away from the opening.
CILIODESTRUCTIVE PROCEDURES
Cyclocryotherapy
When medical therapy does not provide symptomatic relief in end-stage NVG, one of the ciliodestructive procedures should be considered. Cyclocryotherapy (CCT) produces its hypotensive effect by destruction of the secretory ciliary epithelium.[255] In the first large series of CCT on eyes with NVG, there was a 63% success in 38 eyes in controlling the IOP, but almost 40% had post-treatment hypotony.[256] This high complication rate was confirmed in another large series in which 34% of eyes achieved an IOP of 25 mmHg or less, but 34% acquired phthisis and 57% lost all light perception.[257] These studies employed either 360 degrees of CCT or nonstandardized techniques. When a standardized treatment protocol was introduced in 1973,[258] sensible comparison of data among studies became possible. With this standardized protocol, only 180 degrees of the ciliary body is treated at one time, employing six spots of freezing, 2.5 mm posterior to the limbus. Employing this technique, 45% of 43 eyes with NVG had good IOP control, but 33% lost all light perception and another 25% had a significant decrease in vision.[259] Thus, the complication is lower with this technique but is still unacceptably high. In addition, there have been two reports of sympathetic ophthalmia after CCT.[260,261] At this time, we believe that CCT should be reserved for the final effort in the treatment of NVG. If saving functional vision is not a factor, and relief of pain is the main consideration, CCT is a useful technique.
Cyclodiathermy
Cyclodiathermy has been tried in several small series and in one large series of 100 eyes.[262] The success rate (<25 mmHg) matched the phthisis rate: 5%, an unacceptable benefit: complication ratio. Diathermy under direct visualization of the ciliary body through scleral flap dissection has also been tried.[263] Unfortunately, the benefit:complication ratio is not any better. At this stage, cyclodiathermy appears to be more destructive than CCT without offering any real advantages.
Laser Cyclophotocoagulation
In the treatment of refractory NVG, the diode and Nd:YAG lasers, using slit-lamp or fiberoptic delivery systems, have proven useful.[264,265] In the majority of the cases, the diode laser is utilized.[264] However, standardization of a protocol for cyclophotocoagulation in NVG has not been established, and while the IOP can often be controlled, visual results are poor, with long-term visual loss in patients with NVG reaching 46.6%.[264] In severe cases of elevated IOP with concurrent florid rubeosis, combined transscleral diode cyclophotocoagulation and diode laser retinopexy may be considered.[266]
The Nd:YAG lasers have two modes for delivery for transscleral cyclophotocoagulation: noncontact[267] and contact.[268,68] A case of sympathetic ophthalmia has been reported after noncontact Nd:YAG transscleral cyclophotocoagulation. This particular patient had had two failed trabeculectomies, but the temporal sequence was consistent with the laser treatment as the inciting event.[269] Malignant glaucoma has been reported after both noncontact and contact Nd:YAG transscleral cyclophotocoagulation.[270]
Direct visualization and treatment of the ciliary processes with an endoscopic diode laser has also been studied.[271] Advantages include better control and titration of the ciliary processes destroyed, retreatment directed at the functional ciliary processes, and little if any damage of the trabecular meshwork. This procedure can be performed transpupil, though in the majority of cases it is done via endoscopy.[271] With the former approach, the cornea must be clear, the pupil must be markedly dilated from ectropion uveae, or a large-sector iridectomy must already be present so that the ciliary processes are visible with a goniolens. Even then, the treatment does not usually control the IOP. While the endoscopic approach allows for more complete treatment of the ciliary processes, it has the disadvantages of requiring an aphakic or pseudophakic eye and an intraocular procedure with vitrectomy has to be performed with all the potential complications of an invasive procedure.[272,273] At the present time, the role of direct cyclophotocoagulation is secondary and could be tried if other procedures fail.
Other Cyclodestructive Procedures
High-intensity ultrasonography[274] and intraocular carbon dioxide photocoagulation[275] have been used to destroy the ciliary processes as well. The success and complications for ultrasonography have been similar to CCT.[274] There has not been a prospective randomized controlled study comparing the different ciliodestructive procedures. It is our belief that all these procedures should be reserved as the last attempt in saving an eye. At the present time, our ciliodestructive treatment of choice for NVG is transscleral cyclophotocoagulation with the diode laser.[264]
RETROBULBAR INJECTION
We have not found any case in which pain could not be controlled with a combination of topical atropine and steroid or, infrequently, a cyclodestructive procedure. However, others have used retrobulbar alcohol (or chlorpromazine) injection with long-lasting relief of pain.[276] The major complications are temporary blepharoptosis or external ophthalmoplegia.[277] Rarely, enucleation may have to be performed for intractable pain.
CLINICAL ALGORITHM
Based on an extensive review of the literature, Figure 213.20 presents a proposed treatment algorithm for the management of patients with NVI and/or NVA. For eyes with useful vision, this algorithm focuses on treatment of the underlying retinal ischemia and/or inflammation. Once the appropriate retinal ablation procedure has been performed, the residual glaucoma should be managed with aggressive medical and/or surgical treatment. Surgical options include trabeculectomy with mitomycin C, aqueous tube shunt implant, or diode laser cyclophotocoagulation. For eyes with no useful vision, the goal of treatment becomes patient comfort (initially with medical management). In recalcitrant cases, surgical options include cyclodestruction, retrobulbar alcohol, and enucleation.
|
|
|
|
FIGURE 213.20 Proposed treatment algorithm for neovascular glaucoma. |
FUTURE THERAPEUTIC OPTIONS
Future therapeutic approaches will be based increasingly on successful modulation of the angiogenesis cascade. Inhibition of VEGF with neutralizing antibodies has been shown to prevent iris neovascularization in a nonhuman primate model of retinal vein occlusion.[77] Novel anti-VEGF compounds, which include bevacizumab (i.e., Avastin), small interfering RNA (siRNA) directed against VEGF or VEGF receptor 1, and VEGF trap, are being considered.[221,134,278] Recently, rapid resolution of iris and retinal neovascularization has been demonstrated following intravitreal injection of bevacizumab.[221]This may replace the role of PRP in eliminating NVI and/or NVA and result in greater success in filtration surgery in eyes with NVG.
Endogenous angiogenesis inhibitors, particularly those that act broadly at the earliest stages of the angiogenesis cascade, could prove useful in combating NVI/NVA. To date, an extensive number of antiangiogenic factors have been described (Table 213.1).[208] In a primate model of NVI, systemic treat-ment with ?-interferon, a polypeptide that inhibits proliferation and migration of endothelial cells and new vessel growth, resulted in regression of the neovascularization.[279] Moreover, the protein kinase C beta inhibitor has been shown to suppress tumor-induced angiogenesis and is currently in clinical development based on these properties.[280]
TABLE 213.1 -- Antiangiogenic Factors (22)
|
Constitutive |
Cryptic Fragments |
|
Angiogenin |
Angiostatin (38 kDa plasminogen fragment; Kringe 1-4) |
|
Antiangiogenic antithrombin III |
Angiotensinogen fragments |
|
Brain angiogenesis inhibitor 1 |
Arresten (fragment of a1 chain of type IV collagen) |
|
Interferon-? |
Canstatin (fragment ?2 chain of collagen type IV) |
|
Interferon-inducible protein |
Endostatin (20 kDa fragment of XVIII collagen) |
|
Interleukin-12 |
Fibronectin (20 kDa N-terminal fragment) |
|
PEDF |
Fibronectin type 111 peptide |
|
Placental ribonuclease inhibitor |
Fibronectin (40 kDa C-terminal fragment) |
|
Plasminogen-activator inhibitor |
Heparin hexasaccharide fragment |
|
Proliferin-related protein |
Kringle 1-5 (fragment of plasminogen) |
|
Protamine |
NC1 domain of type VIII collagen ?1 |
|
Somatostatin analog octreotide |
PEX (metalloproteinase fragment) |
|
SPARC (43 kDa secreted protein acidic and rich in cysteine) |
Platelet factor 4 fragment |
|
Thrombospondin-1 |
Prolactin (16 kDa N-terminal fragment) |
|
Thrombospondin-2 |
Prolactin fragments |
|
Tissue inhibitors of metalloproteinases 1, 2, and 3 |
Restin (22 kDa fragment of human collagen XV) |
|
Vasculostatin |
Trp-tRMA synthetase splice variant |
From Tombran-Tink J, Barnstable CJ: Therapeutic prospects for PEDF: more than a promising angiogenesis inhibitor. Trends Mol Med 2003; 9:244-250.
PEDF, a potent endogenous angiogenesis inhibitor with neuroprotective properties, also demonstrates promise in the future treatment of NVI. The molecule has remarkable specificity for causing regression of new vessels, with no known deleterious effect on mature vessels.[281] Experimental studies have shown that PEDF can be administered therapeutically as a soluble protein or by viral-mediated gene transfer.[208] In a mouse model of ischemia-induced retinal neovascularization, elevated concentrations of PEDF inhibited VEGF-induced retinal vascular endothelial cell growth and migration and retinal neovascularization.[282]
SUMMARY
NVG is a devastating ocular disease with a guarded visual prognosis, and no current medical or surgical treatment appears to have a high success rate. The most effective treatment involves sufficient retinal ablation to reduce the level of retinal hypoxia and retard the subsequent angiogenic cascade. However, this method is only effective when performed at an early stage of the disease process and has technical limitations. The best hope for effective treatment of NVG is increased understanding of the angiogenesis pathway, which will hopefully lead to the development of novel pharmacologic agents (e.g., bevacizumab, PEDF) to prevent and/or reverse the neovascularization process.
REFERENCES
1. Pagenstecher M: Beitrage zur Lehre vom hamorrhagischen Glaucom. Graefes Arch Clin Exp Ophthalmol 1871; 17:98.
2. Coats G: Further cases of thrombosis of the central vein. Roy Lond Ophthalmic Hosp Rep 1906; 16:516.
3. Salus R: Rubeosis iridis diabetica, eine bisher unbekannte diabetische Irisveranderung. Med Klin 1928; 24:256.
4. Kurz O: Zur Rubeosis iridis diabetica. Arch Augenheilkd 1937; 110:284.
5. Weiss DI, Shaffer RN, Nehrenberg TR: Neovascular glaucoma complicating carotid-cavernous fistula. Arch Ophthalmol 1963; 69:304.
6. Walton DS, Grant WM: Retinoblastoma and iris neovascularization. Am J Ophthalmol 1968; 65:598.
7. Aiello LM, Beetham WP, Balodimos MC, et al: Ruby laser photocoagulation and treatment of diabetic proliferating retinopathy. In: Goldberg MF, Fine SL, ed. Symposium on the treatment of diabetic retinopathy, Pub. No. 1890, Washington, DC: US Public Health Service; 1969:437-463.
8. Sivak-Callcott JA, O'Day DM, Gass JDM, Tsai JC: Evidenced-based recommendations for the diagnosis and treatment of neovascular glaucoma. Ophthalmology 2001; 108:1767.
9. Wand M, Dueker DK, Aiello LM, et al: Effects of panretinal photocoagulation on rubeosis iridis, angle neovascularization, and neovascular glaucoma. Am J Ophthalmol 1978; 86:332.
10. Wand M: Neovascular glaucoma. In: Ritch R, Shields MB, Krupin T, ed. The secondary glaucomas, St. Louis: CV Mosby; 1982:162-193.
11. Chandler PA, Grant WM: Lectures on glaucoma, Philadelphia: Lea & Febiger; 1965:268.
12. Laatikainen L: Development and classification of rubeosis iridis in diabetic eye disease. Br J Ophthalmol 1979; 63:156.
13. Teich SA, Walsh JB: A grading system for neovascularization: Prognostic implications for treatment. Ophthalmology 1981; 88:1102.
14. Weiss DI, Gold D: Neofibrovascularization of iris and anterior chamber angle: A clinical classification. Ann Ophthalmol 1978; 10:488.
15. Perry HD, Yanoff M, Scheie HG: Rubeosis in Fuchs heterochromic iridocyclitis. Arch Ophthalmol 1975; 93:337.
16. Glacet-Bernard A, Coscas C, Chabanol A, et al: Prognostic factors for retinal vein occlusion. Ophthalmology 1996; 103:551.
17. Saari M, Vuorre I, Nieminen H: Fuchs's heterochromic cyclitis: A simultaneous bilateral fluorescein angiographic study of the iris. Br J Ophthalmol 1978; 62:715.
18. Tolentino M, Miller JW, Gragoudas ES, et al: Intravitreal injections of vascular endothelial growth factor produce retinal ischemia and microangiopathy in an adult primate. Ophthalmology 1996; 103:1820.
19. Vannas A: Fluorescein angiography of the vessels of the iris in pseudo-exfoliation of the lens capsule, capsular glaucoma, and some other forms of glaucoma. Acta Ophthalmol 1969; 105(Suppl):9.
20. Yoshizumi MO, Thomas JV, Smith TR: Glaucoma-inducing mechanisms in eyes with retinoblastoma. Arch Ophthalmol 1978; 96:105.
21. Georgiades G: Les lesions de l'iris éérochromique en ééral. Bull Mem Soc Fr d'Ophthalmologie 1964; 77:465.
22. Caldwell RB, Bartoli M, Behzadian MA, et al: Vascular endothelial growth factor and diabetic retinopathy: role of oxidative stress. Curr Drug Targets 2005; 6:511-524.
23. Senger D, Connolly D, Van de Water L, et al: Purification and NH2-terminal amino acid sequence of guinea pig tumor-secreted vascular permeability factor. Cancer Res 1990; 50:1774.
24. Bagessen LH: Fluorescein angiography of the iris in diabetics and nondiabetics. Acta Ophthalmol 1969; 47:449.
25. Cobb B, Shilling JS, Chisholm IH: Vascular tufts at the pupillary margin in myotonic dystrophy. Am J Ophthalmol 1970; 69:573.
26. Mason GI: Iris neovascular tufts: Relationship to rubeosis, insulin, and hypotony. Arch Ophthalmol 1976; 97:2346.
27. Sanborn GE, Symes DJ, Magargal LE: Fundus-iris fluorescein angiography: Evaluation of its use in the diagnosis of rubeosis iridis. Ann Ophthalmol 1986; 18:52.
28. Jensen VA, Lundbock K: Fluorescein angiography of the iris in recent and long-term diabetes: Preliminary communication. Acta Ophthalmol 1968; 46:584.
29. Ehrenberg M, McCuen BW, Schindler RH, et al: Rubeosis iridis: Preoperative iris fluorescein angiography and periocular steroids. Ophthalmology 1984; 91:321.
30. Kimura R: Fluorescein goniophotography. Glaucoma 1980; 2:359.
31. Ohnishi Y, Ishibashi T, Sagawa T: Fluorescein gonioangiography in diabetic neovascularization. Graefes Arch Clin Exp Ophthalmol 1994; 232:199.
32. Cunha-Vaz JG: Blood-retinal barriers in health and disease. Trans Ophthalmol Soc UK 1980; 100:337.
33. Gartner S, Henkind P: Neovascularization of the iris (rubeosis iridis). Surv Ophthalmol 1978; 22:291.
34. Jackson WE, Chase P, Garg SK, et al: Vitreous fluorophotometry in insulin-dependent diabetes mellitus: Correlation with microalbuminuria and diastolic blood pressure. Arch Ophthalmol 1990; 108:1733.
35. Hedges TR: Ophthalmoscopic findings in internal carotid artery occlusion. Johns Hopkins Med J 1962; 111:89.
36. Weber PA: Neovascular glaucoma, current management. Surv Ophthalmol 1981; 25:149.
37. Tamara T: Electron microscopic study on the small blood vessels in rubeosis diabetica. Jpn J Ophthalmol 1969; 13:65.
38. John T, Sassani JW, Eagle Jr RC: The myofibroblastic component of rubeosis iridis. Ophthalmology 1983; 90:721.
39. Gartner S, Taffet S, Friedman AH: The association of rubeosis iridis with endothelialisation of the anterior chamber: Report of a clinical case with histopathological review of 16 additional cases. Br J Ophthalmol 1977; 61:267.
40. Nomura T: Pathology of anterior chamber angle in diabetic neovascular glaucoma: Extension of corneal endothelium onto iris surface. Jpn J Ophthalmol 1983; 27:193.
41. Campbell DG, Shields MB, Smith TR: The corneal endothelium and the spectrum of essential iris atrophy. Am J Ophthalmol 1978; 86:317.
42. Wand M: Neovascular glaucoma. In: Ritch R, Shields MB, Krupin T, ed. The glaucomas, St. Louis: CV Mosby; 1989:1063.
43. Wand M: Neovascular glaucoma: New approaches to inhibition of angiogenesis. J Glaucoma 1994; 3:178.
44. Wand M: Neovascular glaucoma: You've come a long way baby!. In: Van Buskirk EM, Shields MB, ed. 100 Years of progress in glaucoma, Philadelphia: Lippincott-Raven; 1997:204-222.
45. Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO: Vascular endothelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res 2003; 22:1-29.
46. Folkman J, Klagsbrun M: Angiogenic factors. Science 1987; 23:442.
47. Zetter BR: Angiogenesis. State of the art. Chest 1988; 93:159S.
48. Pe'er J, Shweiki D, Itin A, et al: Hypoxia-induced expression of vascular endothelial growth factor by retinal cells is a common factor in neovascularizing ocular diseases. Lab Invest 1995; 72:638.
49. Michaelson IC: The mode of development of the vascular system of the retina with some observations of its significance in certain retinal diseases. Trans Ophthalmol Soc UK 1948; 68:137.
50. Ashton N, Ward B, Serpell G: Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br J Ophthalmol 1954; 38:397.
51. Wise GN: Retinal neovascularization. Trans Am Ophthalmol Soc 1956; 54:729.
52. Ashton N: Retinal vascularization in health and disease. Am J Ophthalmol 1957; 44:7.
53. Branch Vein Occlusion Study Group: Argon laser scatter photocoagulation for prevention of neovascularization and vitreous hemorrhage in branch vein occlusion. Arch Ophthalmol 1986; 104:34.
54. Patz A, Yassur Y, Fine SL, et al: Branch retinal venous occlusion. Trans Am Acad Ophthalmol Otolaryngol 1977; 83:373.
55. Folkman J, Merler E, Abernathy C, et al: Isolation of a tumor factor responsible for angiogenesis. J Exp Med 1971; 133:275.
56. Folkman J, Haudenschild C: Angiogenesis by capillary endothelial cell in culture. Trans Ophthalmol Soc UK 1980; 100:346.
57. Greenblatt M, Shubik P: Tumor angiogenesis: Transfilter diffusion studies in the hamster by the transparent chamber technique. J Natl Cancer Inst 1968; 41:111.
58. Shing Y, Folkman J, Sullivan R, et al: Heparin affinity: Purification of a tumor-derived capillary endothelial cell growth factor. Science 1984; 223:1296.
59. Ferrara N, Houck K, Jakeman L, et al: Molecular and biological properties of the vascular endothelial growth factor family of proteins. Endocrine Rev 1992; 13:18.
60. Houck K, Ferrara N, Winer J, et al: The vascular endothelial growth factor family: Identification of a fourth molecular species and characterization of alternative splicing. Mol Endocrinol 1991; 5:806.
61. Shima D, Kuroki M, Deutsch Y, et al: The mouse gene for vascular endothelial growth factor. Genomic structure, definition of the transcriptional unit, characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem 1996; 271:3877.
62. Casey R, Li WW: Factors controlling ocular angiogenesis (review). Am J Ophthalmol 1997; 124:521-529.
63. Tsai JC, Shields MB: Neovascular glaucoma. In: Tombran-Tink J, Barnstable CJ, ed. Ophthalmology: ocular angiogenesis: diseases, mechanisms, and therapeutics, Totowa, NJ: Humana Press; 2006.(in press).
64. Frank RN: Vascular endothelial growth factor-Its role in retinal vascular proliferation. N Engl J Med 1994; 331:1519.
65. Miller JW, Adamis AP, Aiello LP: Vascular endothelial growth factor, ocular neovascularization, and proliferative diabetic retinopathy. Diabetes Metab Rev 1997; 13:37.
66. Aiello LP, Northrup J, Keyt B, et al: Hypoxic regulation of vascular endothelial growth factor in retinal cells. Arch Ophthalmol 1995; 113:1538.
67. Shima D, Adamis AP, Ferrara N, et al: Hypoxic induction of endothelial cell growth factor in retinal cells: Identification and characterization of vascular endothelial growth factor (VEGF) as the mitogen. Mol Med 1995; 1:182.
68. Shima DT, Gougos A, Miller JW, et al: Cloning and mRNA expression of VEGF in ischemic retinas of Meccaca fascicularis. Invest Ophthalmol Vis Sci 1996; 37:1334.
69. Miller JW, Adamis AP, Shima D, et al: Vascular endothelial growth factor/vascular permeability factor is temporally and spatially correlated with ocular angiogenesis in a private model. Am J Pathol 1994; 145:574.
70. Lutty G, McLeod S, Merges C, et al: Localization of vascular endothelial growth factor in human retina and choroid. Arch Ophthalmol 1996; 114:971.
71. Adamis AP, Miller JW, Bernal M-T, et al: Increased vascular endothelial growth factor levels in the vitreous of eyes with proliferative diabetic retinopathy. Am J Ophthalmol 1994; 118:445.
72. Malecaze F, Clamens S, Simorre-Pinatel V, et al: Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferative diabetic retinopathy. Arch Ophthalmol 1994; 12:1476.
73. Aiello LP, Avery RL, Arrigg PG, et al: Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N Engl J Med 1994; 331:1480.
74. Murata T, Nakagawa K, Khalil A, et al: The relation between expression of vascular endothelial growth factor and breakdown of the blood-retinal barrier in diabetic rat retinas. Lab Invest 1996; 74:819.
75. Tolentino M, Miller JW, Gragoudas ES, et al: Vascular endothelial growth factor is sufficient to produce iris neovascularization and neovascular glaucoma in a nonhuman primate. Arch Ophthalmol 1996; 114:964.
76. Aiello LP, Pierce E, Foley E, et al: Suppression of retinal neovascularization endothelial growth factor (VEGF) using soluble VEGF-receptor chimeric proteins. Proc Natl Acad Sci USA 1995; 92:10457.
77. Adamis AP, Shima DT, Tolentino MJ, et al: Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a non-human primate. Arch Ophthalmol 1996; 114:66.
78. Robinson G, Pearce EA, Rook SL, et al: Oligodeoxynucleotides inhibit retinal neovascularization in a murine model of proliferative retinopathy. Proc Natl Acad Sci USA 1996; 93:4851.
79. Madsen PH: Haemorrhagic glaucoma: Comparative study in diabetic and non-diabetic patients. Br J Ophthalmol 1971; 55:444.
80. Brown GC, Magargal LE, Schachat A, et al: Neovascular glaucoma: Etiologic considerations. Ophthalmology 1984; 91:315.
81. Hayreh SS, Rojas P, Podhajsky P, et al: Ocular neovascularization with retinal vascular occlusion. III: Incidence of ocular neovascularization with retinal vein occlusion. Ophthalmology 1983; 90:488.
82. Grant WM: Management of neovascular glaucoma. In: Leopold IH, ed. Symposium on ocular therapy, St. Louis: CV Mosby; 1974:36-61.
83. Hayreh SS: Classification of central retinal vein occlusion. Ophthalmology 1983; 90:458.
84. Laatikainen L, Blach RK: Behavior of the iris vasculature in central retinal vein occlusion: A fluorescein angiographic study of the vascular response of the retina and the iris. Br J Ophthalmol 1977; 61:272.
85. Laatikainen L, Kohner EM: Fluorescein angiography and its prognostic significance in central retinal vein occlusion. Br J Ophthalmol 1976; 64:411.
86. Magargal LE, Brown GC, Augsburger JJ, et al: Neovascular glaucoma following central retinal vein obstruction. Ophthalmology 1981; 88:1095.
87. Tasman W, Magargal LE, Augsberger JJ: Effects of argon laser photocoagulation on rubeosis iridis and angle neovascularization. Ophthalmology 1980; 87:400.
88. Laatikainen L, Kohner EM, Khoury D, et al: Panretinal photocoagulation in central retinal vein occlusion: A randomized controlled clinical study. Br J Ophthalmol 1977; 61:741.
89. Kohner EM, Shilling JS, Hamilton AM: The role of avascular retina in new vessel formation. Metab Ophthalmol 1976; 1:15.
90. Pournaras CJ, Tsacopoulos M, Strommer K, et al: Experimental retinal branch vein occlusion in miniature pigs induces local tissue hypoxia and vasoproliferative microangiopathy. Ophthalmology 1990; 97:1321.
91. Magargal LE, Donoso LA, Sanborn GE: Retinal ischemia and risk of neovascularization following central retinal vein obstruction. Ophthalmology 1982; 89:1241.
92. Central Vein Occlusion Study Group: Baseline and early natural history report. Arch Ophthalmol 1993; 111:1087.
93. Quinlan PM, Elman MJ, Bhatt AK, et al: The national course of central retinal vein occlusion. Am J Ophthalmol 1990; 110:118.
94. The Central Vein Occlusion Study Group: Natural history and clinical management of central retinal vein occlusion. Arch Ophthalmol 1997; 115:486.
95. The Central Vein Occlusion Study Group Report: A randomized clinical trial of early panretinal photocoagulation for ischemic central vein occlusion. Ophthalmology 1995; 102:1434.
96. Welch JC, Augsburger JJ: Assessment of angiographic retinal capillary nonperfusion in central retinal vein occlusion. Am J Ophthalmol 1987; 103:761.
97. Breton ME, Quinn GE, Keene SS, et al: Electroretinogram parameters at presentation as predictors of rubeosis in central retinal vein occlusion patients. Ophthalmology 1989; 96:1343.
98. Bresnick GH: Following up patients with central retinal vein occlusion. Arch Ophthalmol 1988; 106:324.
99. Marmor MF: An updated standard for clinical electroretinography. Arch Ophthalmol 1995; 113:1375.
100. Hayreh SS, Hayreh MS: Hemi-central retinal vein occlusion. Arch Ophthalmol 1980; 98:1600.
101. Appiah AP, Trempe CL: Differences in contributory factors among hemicentral, central, and branch retinal vein occlusions. Ophthalmology 1989; 96:365.
102. Magargal LE, Sanborn GE, Kimmel AS, et al: Temporal branch retinal vein obstruction: A review. Ophthalmic Surg 1986; 17:240.
103. Magargal LE, Brown GC, Augsburger JJ, et al: Neovascular glaucoma following branch retinal vein obstruction. Ophthalmology 88:1095.
104. Luntz M, Schenker HI: Retinal vascular accidents in glaucoma and ocular hypertension. Surv Ophthalmol 1980; 25:163.
105. Vannas S: Discussion of Bertelsen TI: The relationship between thrombosis in the retinal vein and primary glaucoma. Acta Ophthalmol 1961; 39:603.
106. Chew EY, Trope GE, Mitchell BJ: Diurnal intraocular pressure in young patients with central retinal vein occlusion. Ophthalmology 1987; 94:1545.
107. Centers for Disease Control and Prevention (CDC) : Prevalence of visual impairment and selected eye diseases among persons ? 50 years with and without diabetes - United States, 2002. MMWR Morb Mortal Wkly Rep 2004; 53:1069-1071.
108. Fehrmann H: Uber rubeosis iridis diabetica and ihre allegemein-medizinische Bedeutung; mit anatomischem Befund. Graefes Arch Clin Exp Ophthalmol 1939; 140:354.
109. Armaly MF, Baloglou PJ: Diabetes and the eye. I: Changes in the anterior segment. Arch Ophthalmol 1967; 77:485.
110. Ohrt V: The frequency of rubeosis iridis in diabetic patients. Acta Ophthalmol 1971; 49:301.
111. Yanoff M, Fine BS: Ocular pathology: a text and atlas, New York: Harper & Row; 1975:339.
112. Hohl RD, Barnett DM: Diabetic hemorrhagic glaucoma. Diabetes 1970; 19:994.
113. Isenberg SJ, McRee WE, Jedrzynski MS: Conjunctival hypoxia in diabetes mellitus. Invest Ophthalmol Vis Sci 1986; 27:1512.
114. Caird RI, Pirie A, Ramsell TG: Diabetes and the eye, Oxford: Blackwell Scientific; 1968:76.
115. Bloodworth Jr JMB, Engerman RL: Diabetic microangiopathy in the experimentally-diabetic dog and its prevention by careful control with insulin. Diabetes 1973; 22:290.
116. Steffes MW, Sutherland DER, Goetz FC, et al: Studies of kidney and muscle biopsy specimens from identical twins discordant for type I diabetes mellitus. N Engl J Med 1985; 312:1282.
117. Diabetes Control and Complications Research Group: The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993; 329:977.
118. Kornerup T: Blood pressure and diabetic retinopathy. Acta Ophthalmol 1957; 35:163.
119. Ohrt V: Glaucoma due to rubeosis iridis diabetica. Ophthalmologica 1961; 142:356.
120. Madsen PH: Rubeosis of the iris and haemorrhagic glaucoma in patients with proliferative diabetic retinopathy. Br J Ophthalmol 1971; 55:369.
121. Murphy RP, Maguire M, Vitale S, et al: Retrogression of early diabetic retinopathy. Invest Ophthalmol Vis Sci 1988; 29(Suppl):259.
122. Jaffe NS: Cataract surgery and its complications, St. Louis, CV Mosby, 1972.
123. Aiello LM, Wand M, Liang G: Neovascular glaucoma and vitreous hemorrhage following cataract surgery in patients with diabetes mellitus. Ophthalmology 1983; 90:814.
124. Michels RG: Vitrectomy for complications of diabetic retinopathy. Arch Ophthalmol 1978; 96:237.
125. Oldendoerp J, Spitznas M: Factors influencing the results of vitreous surgery in diabetic retinopathy. I: Iris rubeosis and/or active neovascularization at the fundus. Graefes Arch Clin Exp Ophthalmol 1989; 227:1.
126. Poliner LS, Christianson DJ, Escoffery RF, et al: Neovascular glaucoma after intracapsular and extracapsular cataract extraction in diabetic patients. Am J Ophthalmol 1985; 100:637.
127. Wand M: Hyaloid membrane vs. posterior capsule as a protective barrier. Arch Ophthalmol 1985; 103:1112.
128. Vander JF, Brown GC, Benson WE: Iris neovascularization after central retinal artery obstruction despite previous panretinal photocoagulation for diabetic retinopathy. Am J Ophthalmol 1990; 109:464.
129. Jaffe GJ, Burton TC: Progression of nonproliferative diabetic retinopathy following cataract extraction. Arch Ophthalmol 1988; 106:745.
130. Chu KM, Chen TT, Lee PY: Clinical results of pars plana vitrectomy in posterior-segment disorders. Ann Ophthalmol 1985; 17:686.
131. Blankenship G, Cortez R, Machemer R: The lens and pars plana vitrectomy for diabetic retinopathy complications. Arch Ophthalmol 1979; 97:1263.
132. Rice TA, Michels RG, McQuire MG, et al: The effect of lensectomy on the incidence of iris neovascularization and neovascular glaucoma after vitrectomy for diabetic retinopathy. Am J Ophthalmol 1983; 85:1.
133. Aaberg TM: Pars plana vitrectomy for diabetic traction retinal detachment. Ophthalmology 1981; 88:639.
134. Campochiaro PA: Potential applications for RNAi to probe pathogenesis and develop new treatments for ocular disorders. Gene Ther 2006; 6:559.
135. Wand M, Madigan JC, Gaudio AR, et al: Neovascular glaucoma following pars plana vitrectomy for complications of diabetic retinopathy. Ophthalmic Surg 1990; 21:113.
136. Diabetic Retinopathy Vitrectomy Study Group: Two-year course of visual acuity in severe proliferative diabetic retinopathy with conventional management. Ophthalmology 1985; 92:492.
137. Kokame GT, Flynn HW, Blankenship GW: Posterior chamber intraocular lens implantation during diabetic pars plana vitrectomy. Ophthalmology 1989; 96:603.
138. Weinreb RN, Wasserstrom JP, Parker W: Neovascular glaucoma following neodymium:YAG laser posterior capsulotomy. Arch Ophthalmol 1986; 104:730.
139. Yoshinami M, Hamanaka T, Kawano H, et al: A case report of neovascular glaucoma due to carotid occlusive disease. Diagnosis of neovascular glaucoma and histopathological characteristics. Jpn J Ophthalmol 2000; 44:576-577.
140. Coppeto J, Wand M, Bear L, et al: Neovascular glaucoma and carotid vascular occlusion. Am J Ophthalmol 1985; 99:567.
141. Smith JL: Unilateral glaucoma in carotid occlusive disease. JAMA 1962; 182:683.
142. Yablonski ME, Jacobson J, Goldfarb M: Glaucoma and bilateral carotid artery occlusion. Glaucoma 1985; 7:19.
143. Brown GC, Shah HG, Magargal LE, et al: Central retinal vein obstruction and carotid artery disease. Ophthalmology 1984; 91:1627.
144. Lazzaro EC: Retinal-vein occlusions: Carotid artery evaluation indicated. Ann Ophthalmol 1986; 18:116.
145. Hollenhorst RW: Ocular manifestations of insufficiency or thrombosis of the internal carotid artery. Trans Am Ophthalmol Soc 1958; 56:472.
146. Kearns TP, Hollenhorst RW: Venous-stasis retinopathy of occlusive disease of the carotid artery. Mayo Clin Proc 1963; 38:304.
147. Sturrock GD, Mueller HR: Chronic ocular ischaemia. Br J Ophthalmol 1984; 68:716.
148. Kiser WD, Gonder J, Magargal LE, et al: Recovery of vision following treatment of the ocular ischemic syndrome. Ann Ophthalmol 1983; 15:305.
149. Kearns TP, Siekert RG, Sundt TM: The ocular aspects of bypass surgery of the carotid artery. Mayo Clin Proc 1979; 54:3.
150. Huckman MS, Haas J: Reversed flow through the ophthalmic artery as a cause of rubeosis iridis. Am J Ophthalmol 1972; 74:1094.
151. Barany E: Influence of local arterial blood pressure on aqueous humor and intraocular pressure. Acta Ophthalmol 1946; 24:337.
152. Kaplan BH, Kalina PH, Larson L-I, et al: Aqueous humor flow in unilateral carotid stenosis. J Glaucoma 1996; 5:237.
153. Swan KC, Raff J: Changes in the eye and orbit following carotid ligation. Trans Am Ophthalmol Soc 1951; 49:435.
154. Brown GC, Magargal LE, Simeone FA, et al: Arterial obstruction and ocular neovascularization. Ophthalmology 1982; 89:139.
155. Diabetic Retinopathy Study Research Group: Preliminary report on effects ofphotocoagulation therapy. Am J Ophthalmol 1976; 81:383.
156. Rosenberg PR, Walsh JB, Zimmerman RD: Neovascular glaucoma and carotid bruits. J Clin Neuroophthalmol 1984; 4:459.
157. Young LHY, Appen RE: Ischemic oculopathy: A manifestation of carotid artery disease. Arch Neurol 1981; 18:358.
158. Loring EG: Remarks on embolism. Am J Med Sci 1874; 67:313.
159. Duker JS, Brown GC: Iris neovascularization associated with obstruction of the central retinal artery. Ophthalmology 1988; 95:1244.
160. Duker JS, Sivalingam A, Brown GC: A prospective study of acute central retinal artery obstruction: The incidence of secondary ocular neovascularization. Arch Ophthalmol 1991; 109:339.
161. Hayreh SS, Podhajsky P: Ocular neovascularization with retinal vascular occlusion. II: Occurrence in central and branch retinal artery occlusion. Arch Ophthalmol 1982; 100:1585.
162. Katjalainen K: Occlusion of the central retinal artery and retinal branch arterioles: A clinical, tomographic, and fluorescein angiographic study of 175 patients. Acta Ophthalmol Suppl 1971; 109:9.
163. Schachat AP: Discussion of iris neovascularization associated with obstruction of the central retinal artery. Ophthalmology 1988; 95:1249.
164. Reinecke RD, Kuwabara T, Cogan DG, et al: Retinal vascular patterns. V: Experimental ischemia of the cat eye. Arch Ophthalmol 1962; 67:470.
165. Bovino JA, Marcus DF, Nelsen PT: Optic disk neovascularization and rubeosis iridis after surgical resection of the optic nerve. Am J Ophthalmol 1988; 106:231.
166. Duker JS, Brown GC: Neovascularization of the optic disc associated with obstruction of the central retinal artery. Ophthalmology 1989; 96:87.
167. Hutchins RK, Gittinger JW, Weiter JJ: Optic disk and iris neovascularization after surgical interruption of the retinal circulation. [Letter] Am J Ophthalmol 1986; 101:616.
168. Henkind P, Wise GN: Retinal neovascularization, collaterals, and vascular shunts. Br J Ophthalmol 1974; 58:413.
169. Shah HG, Brown GC, Goldberg RE: Digital subtraction carotid angiography and retinal arterial obstruction. Ophthalmology 1985; 92:68.
170. Brown GC: Isolated central retinal artery obstruction in association with ocular neovascularization. Am J Ophthalmol 1983; 96:110.
171. Holm A, Sachs J, Wilson A: Glaucoma secondary to occlusion of central retinal artery. Am J Ophthalmol 1959; 48:530.
172. Duker JS, Brown GC: The efficacy of panretinal photocoagulation for neovascularization of the iris after central retinal artery obstruction. Ophthalmology 1989; 96:92.
173. Howard GM, Ellsworth RM: Differential diagnosis of retinoblastoma. Am J Ophthalmol 1965; 60:618.
174. Kavanta A, Steen B, Seregard S: Expression of vascular endothelial growth factor (VEGF) in retinoblastoma but not in posterior uveal melanoma. Exp Eye Res 1996; 63:511.
175. Ellett EC: Metastatic carcinoma of choroid. III: Rubeosis iridis with melanoma of the choroid and secondary glaucoma. Am J Ophthalmol 1944; 27:726.
176. Cappin JM: Malignant melanoma and rubeosis iridis. Br J Ophthalmol 1973; 57:815.
177. Makley TA, Teed RW: Unsuspected intraocular malignant melanoma. Arch Ophthalmol 1950; 60:475.
178. Yanoff M: Mechanisms of glaucoma in eyes with uveal melanoma. Int Ophthalmol Clin 1972; 12:51.
179. Bujara K: Necrotic malignant melanomas of the choroid and ciliary body: A clinicopathological and statistical study. Graefes Arch Clin Exp Ophthalmol 1982; 291:40.
180. Kim MK, Char DH, Castro JL, et al: Neovascular glaucoma after helium ion irradiation for uveal melanoma. Ophthalmology 1986; 93:189.
181. Lewis RA, Tse DT, Phelps CD, et al: Neovascular glaucoma after photoradiation therapy for uveal melanoma. Arch Ophthalmol 1984; 102:839.
182. Gragoudas ES, Seddon J, Goitein M, et al: Current results of proton beam irradiation of uveal melanomas. Ophthalmology 1985; 92:284.
183. Jones RF: Glaucoma following radiotherapy. Br J Ophthalmol 1958; 42:636.
184. Packer S, Rotman M, Salanitro P: Iodine-125 irradiation of choroidal melanoma: Clinical experience. Ophthalmology 1984; 91:1700.
185. Shields MB, Proia AD: Neovascular glaucoma associated with an iris melanoma: A clinicopathologic report. Arch Ophthalmol 1987; 105:672.
186. Ferry AP, Font RL: Carcinoma metastatic to the eye and orbit. Arch Ophthalmol 1975; 93:472.
187. Sullivan ST, Dallow RL: Intraocular reticulum cell sarcoma. Ann Ophthalmol 1977; 9:401.
188. Schulze RR: Rubeosis iridis. Am J Ophthalmol 1967; 63:487.
189. Hung Y, Hilton GF: Neovascular glaucoma in a patient with X-linked juvenile retinoschisis. Ann Ophthalmol 1980; 12:1054.
190. Boniuk M: Cryotherapy in neovascular glaucoma. Trans Am Acad Ophthalmol Otolaryngol 1974; 78:337.
191. Galinos S, Rabb MF, Goldberg MF, et al: Hemoglobin SC disease and iris atrophy. Am J Ophthalmol 1973; 75:421.
192. Young NJA, Hitchings RA, Sehmi K, et al: Stickler's syndrome and neovascular glaucoma. Br J Ophthalmol 1979; 63:826.
193. Bennett SR, Folk JC, Kimura AE, et al: Autosomal dominant neovascular inflammatory vitreoretinopathy. Ophthalmology 1990; 97:1125.
194. Buchanan TAS, Hoyt WF: Optic nerve glioma and neovascular glaucoma: Report of a case. Br J Ophthalmol 1982; 66:96.
195. Ringvold A, Davanger M: Iris neovascularisation in eyes with pseudoexfoliation syndrome. Br J Ophthalmol 1981; 65:138.
196. Wilhelmus KR, Grierson I, Watson PG: Histopathologic and clinical associations of scleritis and glaucoma. Am J Ophthalmol 1981; 91:697.
197. Hoskins HD: Neovascular glaucoma: Current concepts. Trans Am Acad Ophthalmol Otolaryngol 1974; 78:330.
198. Cairns JE: Rationale for therapy in neovascular glaucoma. Trans Ophthalmol Soc UK 1981; 101:184.
199. Weber PA: Neovascular glaucoma, current management. Surv Ophthalmol 1981; 25:149.
200. Wand M: Treatment of neovascular glaucoma. In: Jakobiec FA, Sigelman J, ed. Advanced techniques in ocular surgery, Philadelphia: WB Saunders; 1984:169-182.
201. Magargal LE, Brown GC, Augsburger JJ, et al: Efficacy of panretinal photocoagulation in preventing neovascular glaucoma following ischemic central retinal vein obstruction. Ophthalmology 1982; 89:780.
202. Kohner EM, Laatikainen L, Oughton J: The management of central retinal vein occlusion. Ophthalmology 1983; 90:484.
203. Romem M, Isakow I: Photocoagulation in retinal vein occlusion. Ann Ophthalmol 1981; 13:1057.
204. Nathan DM, Singer DE, Godine JE, et al: Retinopathy in older type II diabetics: Association with glucose control. Diabetes 1986; 35:797.
205. Jacobson DR, Murphy RP, Rosenthal AR: The treatment of angle neovascularization with panretinal photocoagulation. Ophthalmology 1979; 86:1270.
206. Robinson CJ, Stringer SE: The splice variants of vascular endothelial growth factors (VEGF) and their receptors. J Cell Sci 2001; 114:853-865.
207. Tripathi RC, Li J, Tripathi BJ, et al: Increased levels of vascular endothelial growth factor in aqueous humor of patients with neovascular glaucoma. Ophthalmology 1998; 105:232-237.
208. Tombran-Tink J, Barnstable CJ: Therapeutic prospects for PEDF: more than a promising angiogenesis inhibitor. Trends Mol Med 2003; 9:244-250.
209. Tombran-Tink J, Barnstable CJ: PEDF: a multifaceted neurotrophic factor. Nat Rev 2003; 4:1-10.
210. Ogata N, Nishikawa M, Nishimura T, et al: Unbalanced vitreous levels of pigment-epithelium-derived factor and vascular endothelial growth factor in diabetic retinopathy. Am J Ophthalmol 2002; 134:348-353.
211. Ogata N, Tombran-Tink J, Nishikawa M, et al: Pigment epithelium-derived factor in the vitreous is low in diabetic retinopathy and high in rhegmatogenous retinal detachment. Am J Ophthalmol 2001; 132:378-382.
212. Hercules B, Bayed I, Lucas S, et al: Peripheral retinal ablation in the treatment of proliferative diabetic retinopathy: A three-year interim report of a randomized, controlled study using the argon laser. Br J Ophthalmol 1977; 61:555.
213. Brodell LP, Olk RJ, Arribas NP, et al: Neovascular glaucoma: A retrospective analysis of treatment with peripheral panretinal cryotherapy. Ophthalmic Surg 1987; 18:200.
214. May DR, Bergstrom TJ, Parmet AJ, et al: Treatment of neovascular glaucoma with transcleral panretinal cryotherapy. Ophthalmology 1980; 87:1106.
215. Ogden TE, Rickhof FT, Benkwith SM: Correlation of histologic andelectroretinographic changes in peripheral retinal ablation in the rhesus monkey. Am J Ophthalmol 1976; 81:1272.
216. Kaufman SC, Ferris FL III, Seigel DG, et al: Factors associated with visual outcome after photocoagulation for diabetic retinopathy. Diabetic retinopathy study report no. 13. Invest Ophthalmol Vis Sci 1989; 30:23.
217. Barr CC: Estimation of the maximum number of argon laser burns possible in panretinal photocoagulation. Am J Ophthalmol 1984; 97:697.
218. Reddy VM, Zamora VM, Olk RJ: A comparison of the size of the burn produced by Rodenstock and Goldmann contact lenses. Am J Ophthalmol 1991; 112:212.
219. Callahan MA, Hilton GF Letter: Photocoagulation and rubeosis iridis. Am J Ophthalmol 1974; 78:873.
220. Kaufman SC, Ferris FL III, Swartz M: Diabetic Retinopathy Study Research Group: Intraocular pressure following panretinal photocoagulation for diabetic retinopathy. Arch Ophthalmol 1987; 105:807.
221. Avery RL: Regression of retinal and iris neovascularization after intravitreal bevacizumab (avastin) treatment. Retina 2006; 26:352-354.
222. Carter JE: Panretinal photocoagulation for progressive ocular neovascularization secondary to occlusion of the common carotid artery. Ann Ophthalmol 1984; 16:572.
223. Eggleston TF, Bohling CA, Eggleston HC, et al: Photocoagulation for ocular ischemia associated with carotid artery occlusion. Ann Ophthalmol 1980; 12:84.
224. Fleischman JA, Swartz M, Dixon JA: Argon laser endophotocoagulation: An intraoperative trans-pars plana technique. Arch Ophthalmol 1981; 99:1610.
225. Hampton GR: Argon endophotocoagulation with indirect ophthalmolscopy. Arch Ophthalmol 1987; 105:132.
226. Michels RG: Vitrectomy techniques in retinal re-attachment surgery. Ophthalmology 1979; 86:556.
227. Jaccoma EH, Conway BP, Campochiaro PA: Cryotherapy causes extensive breakdown of the blood-retinal barrier. Arch Ophthalmol 1985; 103:1728.
228. Vernon SA, Cheng H: Panretinal cryotherapy in neovascular disease. Br J Ophthalmol 1988; 72:401.
229. Williams DF, Burke JW, Williams EA: Clearance of experimental vitreous hemorrhage after panretinal cryotherapy related to macrophage influx. Arch Ophthalmol 1986; 108:585.
230. Davidorf FH, Mouser JG, Derick RJ: Rapid improvement of rubeosis iridis from a single bevacizumab (avastin) injection. Retina 2006; 26:354-356.
231. Simmons RJ, Depperman SR, Dueker DK: The role of goniophotocoagulation in neovascularization of the anterior chamber angle. Ophthalmology 1980; 87:79.
232. Allen RC, Bellows AR, Hutchinson BT, et al: Filtration surgery in the treatment of neovascular glaucoma. Ophthalmology 1982; 89:1181.
233. Jonas JB, Hayler JK, Sofker A, Panda-Jonas S: Regression of neovascular iris vessels by intravitreal injection of crystalline cortisone. J Glaucoma 2001; 10:284-287.
234. Wand M, Hutchinson BT: The surgical management of neovascular glaucoma. Perspect Ophthalmol 1980; 4:147.
235. Tsai JC and Forbes M: Medical Management of Glaucoma. 2nd edn.; 2004:224-225.
236. Sinclair SH, Aaberg TM, Meredith TA: A pars plana filtering procedure combined with lensectomy and vitrectomy for neovascular glaucoma. Am J Ophthalmol 1982; 93:185.
237. Cameron ME: Thrombotic glaucoma successfully treated. Trans Ophthalmol Soc UK 1973; 93:537.
238. Rockwood EJ, Parrish RK, Heuer DK, et al: Glaucoma filtering surgery with 5-fluorouracil. Ophthalmology 1987; 94:1071.
239. The Fluorouracil Filtering Surgery Study Group: Fluorouracil filtering surgery study one-year follow-up. Am J Ophthalmol 1989; 108:625.
240. Tsai JC, Feuer WJ, Parrish RK II, et al: 5-Fluorouracil filtering surgery in neovascular glaucoma. Long-term follow-up of the original pilot study. Ophthalmology 1995; 102:887.
241. Lamping KA, Belkin JK: 5-Fluorouracil and mitomycin-C in pseudophakic patients. Ophthalmology 1995; 102:70.
242. Skuta GL, Beeson CC, Higginbotham EJ, et al: Intraoperative mitomycin versus postoperative 5-fluorouracil in high risk glaucoma filtering surgery. Ophthalmology 1992; 99:438.
243. Molteno ACB, Haddad PJ: The visual outcome in cases of neovascular glaucoma. Aust N Z J Ophthalmol 1985; 13:329.
244. Fernandez-Vigo J, Castro J, Cordido M: Treatment of diabetic neovascular glaucoma by panretinal ablation and trabeculectomy. Acta Ophthalmol 1988; 66:612.
245. Clearkin LG: Recent experience in the management of neovascular glaucoma by pan-retinal photocoagulation and trabeculectomy. Eye 1987; 1:397.
246. Honrubia FM, Gomez ML, Hernandez A, Grijalbo MP: Long term results of silicone tube filtering surgery for eyes with neovascular glaucoma. Am J Ophthalmol 1984; 97:501.
247. Minckler DS, Heuer DK, Hasty B, et al: Clinical experience with the single-plate Molteno implant in complicated glaucomas. Ophthalmology 1988; 95:1181.
248. Krupin T, Kaufman P, Mandell AI, et al: Long-term results of valve implants in filtering surgery for eyes with neovascular glaucoma. Am J Ophthalmol 1983; 95:775.
249. Tsai JC, Johnson CC, Dietrich MS: The Ahmed shunt versus the Baerveldt shunt for refractory glaucoma: a single-surgeon comparison of outcome. Ophthalmology 2003; 110:1814-1821.
250. Mermoud A, Salmon JF, Alexander P, et al: Molteno tube implantation for neovascular glaucoma. Long term results and factors influencing the outcome. Ophthalmology 1993; 100:897.
251. Every SG, Molteno AC, Bevin TH, Herbison P: Long-term results of Molteno implant insertion in cases of neovascular glaucoma. Arch Ophthalmol 2006; 124:355-360.
252. Tsai JC, Grajewski AL, Parrish RK II: Surgical Revision of Glaucoma Shunt Implants. Ophthalmic Surg Lasers 1999; 30:41-46.
253. Egbert PR, Lieberman MF: Internal suture occlusion of the Molteno glaucoma implant for the prevention of post-operative hypotony. Ophthalmol Surg 1989; 20:53.
254. Krupin T, Ritch R, Camras CB, et al: A long Krupin-Denver valve implant attached to a 180 degrees scleral explant for glaucoma surgery. Ophthalmology 1988; 95:1174.
255. Quigley HA: Histologic and physiologic studies of cyclocryotherapy in primate and human eyes. Am J Ophthalmol 1976; 82:722.
256. Feibel RM, Bigger JF: Rubeosis iridis and neovascular glaucoma. Am J Ophthalmol 1972; 74:862.
257. Krupin T, Mitchell KB, Becker B: Cyclocryotherapy in neovascular glaucoma. Am J Ophthalmol 1978; 86:24.
258. Bellows AR, Grant WM: Cyclocryotherapy in advanced inadequately controlled glaucoma. Am J Ophthalmol 1973; 75:679.
259. Bellows AR: Cyclocryotherapy for glaucoma. Int Ophthalmol Clin 1981; 21:99.
260. Harrison TJ: Sympathetic ophthalmia after cyclocryotherapy of neovascular glaucoma with ocular penetration. Ophthalmic Surg 1993; 24:44.
261. Sabates R: Choroiditis compatible with the histopathologic diagnosis of sympathetic ophthalmia following cyclocryotherapy of neovascular glaucoma. Ophthalmic Surg 1988; 19:176.
262. Walton DS, Grant WM: Penetrating cyclodiathermy for filtration. Arch Ophthalmol 1970; 83:47.
263. Riaskoff S, Pameyer JH: Results of the treatment of neovascular glaucoma by diathermy of the dissected ciliary body. Doc Ophthalmol 1980; 48:277.
264. Bloom PA, Tsai JC, Sharma K, et al: 'Cyclodiode'. Transscleral diode laser cyclophotocoagulation in the treatment of advanced refractory glaucoma. Ophthalmology 1997; 104:1508-1519.discussion 1519-1520.
265. Shields MB, Shields SE: Noncontact transscleral Nd:YAG cyclophotocoagulation: a long-term follow-up of 500 patients. Trans Am Ophthalmol Soc 1994; 92:271-283.discussion 283-287.
266. Tsai JC, Bloom PA, Franks WA, Khaw PT: Combined transscleral diode laser cyclophotocoagulation and transscleral retinal photocoagulation for refractory neovascular glaucoma. Retina 1996; 16:164-166.
267. Hampton C, Shields MB, Miller KN, et al: Evaluation of a protocol for transscleral neodymium:YAG cyclophotocoagulation in one hundred patients. Ophthalmology 1990; 97:910.
268. Allingham RR, de Kater AW, Bellows AR, et al: Probe placement and power levels in contact transscleral neodymium YAG cyclophotocoagulation. Arch Ophthalmol 1990; 108:738.
269. Edward DP, Brown SVL, Higginbotham E, et al: Sympathetic ophthalmia following neodymium:YAG cyclotherapy. Ophthalmic Surg 1989; 20:544.
270. Wand M, Schuman JS, Puliafito CA: Malignant glaucoma after contact transscleral Nd:YAG laser cyclophotocoagulation. J Glaucoma 1993; 2:110.
271. Uram M: Ophthalmic laser microendoscope ciliary process ablation in the management of neovascular glaucoma. Ophthalmology 1992; 99:1823-1828.
272. Blacharski PA, Charles S: Endocyclophotocoagulation. Ophthalmic Laser Surg 1987; 2:13.
273. Patel A, Thompson JT, Michels RG, et al: Endolaser treatment of the ciliary body for uncontrolled glaucoma. Ophthalmology 1986; 93:825.
274. Silverman RH, Vogelsang B, Rondeau MJ, et al: Therapeutic ultrasound for the treatment of glaucoma. Am J Ophthalmol 1991; 111:327.
275. Miller JB, Smith MR, Boyer DS: Intraocular carbon dioxide laser photocautery. Ophthalmology 1980; 87:1112.
276. Michaels RG, Maumenee AE: Retrobulbar alcohol injection in seeing eyes. Trans Am Acad Ophthalmol Otol 1973; 77:164.
277. Olurin O, Osuntokun O: Complications of retrobulbar alcohol injections. Ann Ophthalmol 1978; 10:474.
278. Lau SC, Rosa DD, Jayson G: Technology evaluation: VEGF Trap (cancer), Regeneron/sanofi-aventis. Curr Opin Mol Ther 2005; 7:493-501.
279. Miller JW, Stinson WG, Folkman J: Regression of experimental iris neovascularization with systemic alpha-interferon. Ophthalmology 1993; 100:9.
280. Graff JR, McNulty AM, Hanna KR, et al: The protein kinase Cbeta-selective inhibitor, Enzastaurin (LY317615.HC1), suppresses signaling through the AKT pathway, induces apoptosis, and suppresses tumor growth of human colon cancer and glioblastoma xenografts. Cancer Res 2005; 65:7462-7469.
281. Dawson DW, Volpert OV, Gillis P, et al: Pigment epithelium-derived factor: a potent inhibitor of angiogenesis. Science 1999; 285:245-248.
282. Duh EJ, Yang HS, Suzuma I, et al: Pigment epithelium-derived factor suppresses ischemia-induced retinal neovascularization and VEGF-induced migration and growth. Invest Ophthalmol Vis Sci 2002; 43:821-829.