Karen C. Bloch, MD, MPH
Infectious diseases remain one of the leading causes of death in both developed and developing countries. Infections cause significant morbidity and mortality, especially in individuals who are most vulnerable to illness: the very young, the elderly, the immunocompromised, and the disenfranchised.
The pathogenesis of infectious diseases reflects the relationship among the human host, the infectious agent, and the external environment. Figure 4-1 portrays a host-agent-environment paradigm for the study of infectious diseases. The infectious agent can be either exogenous (ie, not normally found on or in the body) or endogenous (ie, one that may be routinely cultured from a specific anatomic site but that does not normally cause disease in the host). Infection results when an exogenous agent is introduced into a host from the environment or when an endogenous agent overcomes innate host immunity to cause disease. Host susceptibility plays an important role in either of these settings.
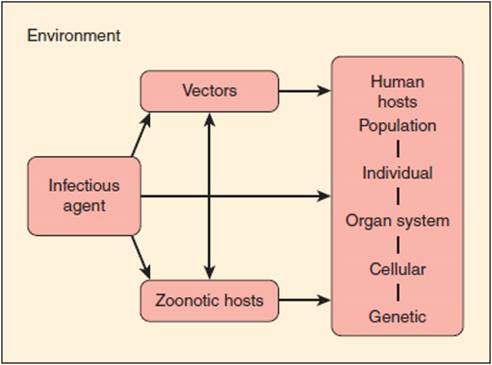
FIGURE 4-1 The fundamental relationships involved in the host-agent-environment interaction model. In the host, pathogenetic mechanisms extend from the level of populations (eg, person-to-person transmission) to the level of cellular and molecular processes (eg, genetic susceptibility).
The environment includes vectors (insects and other carriers that transmit infectious agents) and zoonotic hosts or reservoirs (animals that harbor infectious agents and often act to amplify the infectious agent). For example, the white-footed mouse (Peromyscus leucopus) serves as an animal reservoir for Borrelia burgdorferi, the bacterium that causes Lyme disease. The Ixodes tick serves as an insect vector. Infection in the mouse is asymptomatic, and the bacteria can multiply to high levels in this animal. When the tick larva feeds on an infected mouse, it becomes secondarily infected with B burgdorferi, and this infection persists when the tick molts into a nymph. Subsequently, when an infected nymph feeds on a human, the bacterium is transmitted through infected saliva into the host bloodstream, causing disease.
The study of infectious diseases requires understanding of pathogenesis at the level of the population, the individual, the cell, and the gene. For example, at the population level, the spread of tuberculosis in the community is related to the social interactions of an infectious human host. Outbreaks of tuberculosis have occurred in group settings such as homeless shelters, prisons, and nursing homes when an index case comes in close contact with susceptible persons. At the individual level, tuberculosis results from inhalation of respiratory droplets containing airborne tubercle bacilli. At the cellular level, these bacilli activate T cells, which play a critical role in containing the infection. Individuals with an impaired T-cell response (eg, those infected with human immunodeficiency virus [HIV]) are at particularly high risk for developing active tuberculosis at the time of the initial infection or for reactivation of latent tuberculosis as their immunity wanes. Finally, at the genetic level, individuals with specific polymorphisms in a macrophage protein gene may be at significantly higher risk for pulmonary tuberculosis.
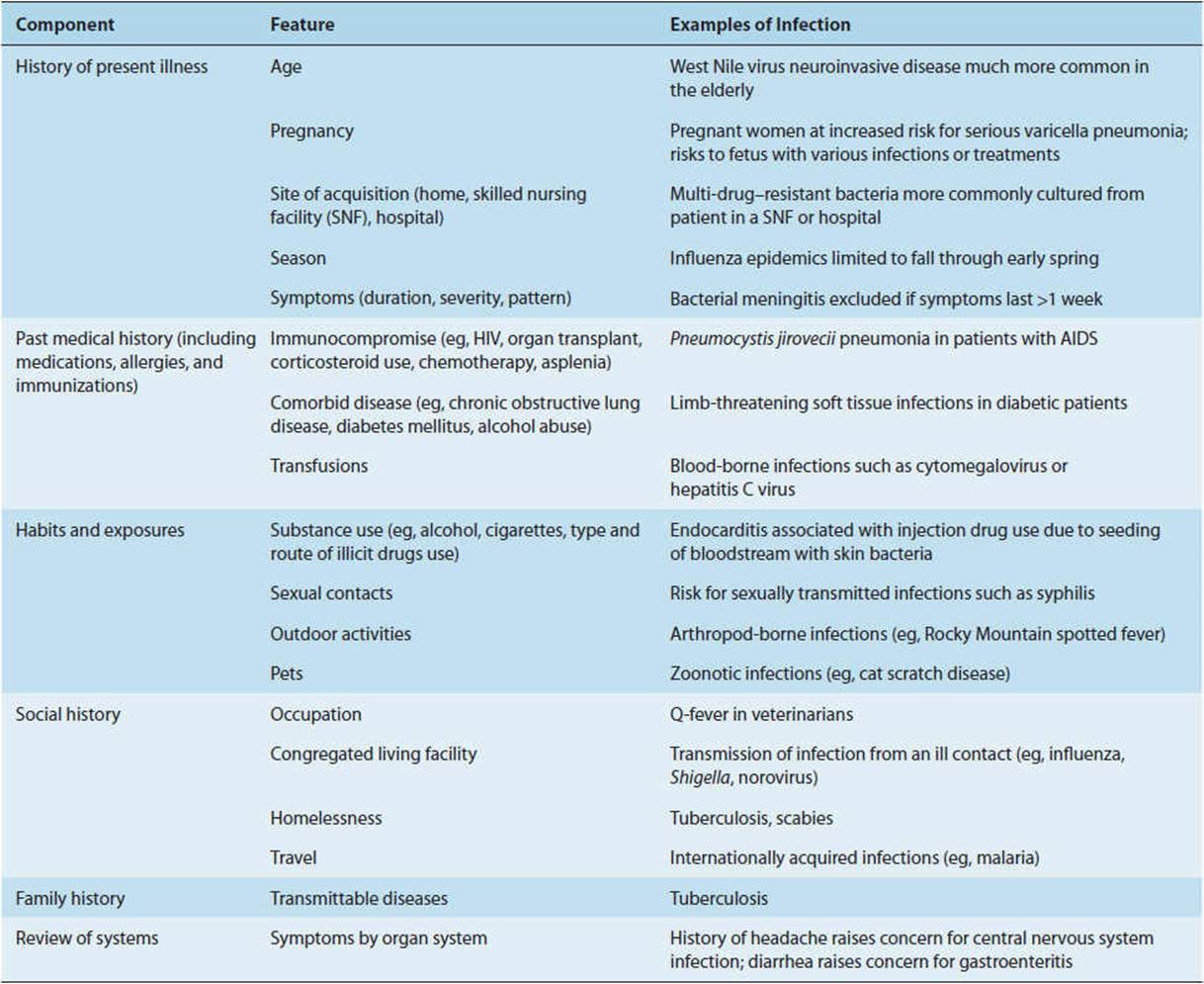
Specific microorganisms have a tendency to cause certain types of infections: Streptococcus pneumoniae commonly causes pneumonia, meningitis, and bacteremia but rarely causes endocarditis (infection of the heart valves); Escherichia coli is a common cause of gastrointestinal and urinary tract infections; Plasmodium species infect red blood cells and liver cells to cause malaria; Entamoeba histolytica causes amebic dysentery, liver abscesses, and so on. Table 4-1 presents a clinical approach to taking a patient history that considers features of the host and the environment in identifying the most likely microorganisms associated with specific clinical syndromes.
TABLE 4-1 Obtaining a history in the diagnosis of infectious diseases.
HOST DEFENSES AGAINST INFECTION
The human body has the ability to control infection through a number of different mechanisms. Physical barriers impede the entry of bacteria from the external environment and from normally colonized sites in the body into sterile anatomic areas. When these physical defenses are breached, the immune system is activated (Figure 4-2). Constitutive or innate immunity, provided by preformed proteins (eg, complement) and immune cells (eg, phagocytes) that are activated by nonspecific foreign proteins, allows an immediate response to foreign material. Induced or adaptive immunity includes both early and late adaptive responses activated by specific antigenic proteins (eg, production of antibodies active against the specific strains of S pneumoniae contained in the pneumococcal vaccine in a previously vaccinated individual). Induction of these specific immune receptor cells may take several days in the immunologically naive host. Protective immunity, which occurs after initial exposure (infection or vaccination) through generation of memory lymphocytes and pathogen-specific antibody, allows a much more rapid response to reinfection. These components of the immune response are discussed in detail later.

FIGURE 4-2 Phases of the host response to infection. During the earliest stage of initial infection, nonspecific mediators (complement, phagocytes) predominate. Adaptive immunity (production of antibody, stimulation of lymphocytes) requires clonal expansion after recognition of specific antigens. Once immunity toward a specific agent is induced, the immune response remains primed so that the response to reinfection is much more rapid.
NORMAL MICROBIAL FLORA
The human body normally harbors numerous species of bacteria, viruses, fungi, and protozoa, referred to as the human microbiota. The great majority of these are commensals, defined as organisms that live symbiotically on or within the human host but rarely cause disease (Figure 4-3). Anatomic sites where bacteria are normally found include the skin (staphylococci and diphtheroids), oropharynx (streptococci, anaerobes), large intestine (enterococci, enteric bacilli), and vagina (lactobacilli).
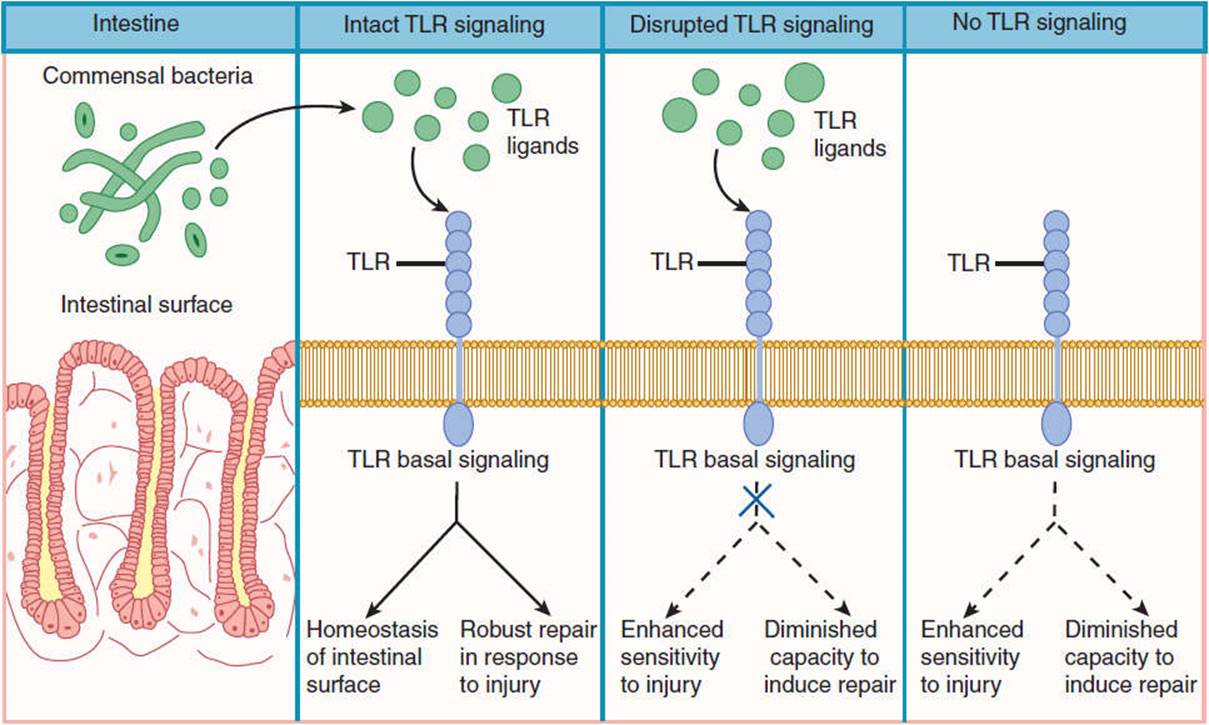
FIGURE 4-3 Commensal bacteria secrete toll-like receptor (TLR) ligands, which bind to TLR on the surface of normal intestinal tissue. This interaction stimulates basal signaling, which protects against cellular injury. Disruption of TLR signaling or antibiotic associated eradication of commensal bacteria result in compromised ability of the intestinal epithelium to withstand injury and repair cell damage. (Redrawn, with permission, from Madara J. Building an intestine—Architectural contributions of commensal bacteria. N Engl J Med. 2004;351:1686.)
Determining when an isolate is a component of the normal flora rather than an invasive pathogen may be difficult. For example, culture of staphylococci from a blood sample may represent skin contamination at the time of phlebotomy or may indicate a potentially life-threatening bloodstream infection. Helpful clues include burden of organism (eg, number of positive blood cultures), symptoms and signs of infection (eg, cough, fever), and the presence of inflammatory cells (eg, polymorphonuclear cells in the sputum and an increased proportion of immature neutrophils in the blood). Isolation of an obligate pathogen such as Mycobacterium tuberculosis from any anatomic site is diagnostic of infection. Fortunately, few microorganisms are absolute pathogens. For example, Neisseria meningitidis, a major bacterial cause of meningitis, can be cultured from the oropharynx of as many as 10% of asymptomatic individuals, in which case it represents transient normal flora. Even if asymptomatic, the host can serve as a carrier, transferring bacteria to susceptible individuals. Infections resulting from commensals that rarely cause disease (eg, Candida albicans) or organisms ubiquitous in the environment that are generally not considered human pathogens (eg, Aspergillus) are termed opportunistic infections. These infections occur almost exclusively in immunocompromised hosts such as HIV-infected patients or transplant recipients. The agents are opportunists in that they take advantage of impaired host immunity to cause infection but rarely cause disease in a healthy host.
The site from which an organism is cultured is important in differentiating colonization from infection. Growth of any microorganism from a normally sterile site such as blood, cerebrospinal fluid, synovial (joint) fluid, or deep tissues of the body is diagnostic of infection. For example, Bacteroides, the predominant genus of bacteria in the colon, may cause intra-abdominal abscesses and sepsis when the integrity of the colonic mucosa is breached. Staphylococcus epidermidis, a common skin commensal, can cause bacteremia after intravascular catheter placement. Knowledge of the common endogenous flora may be useful in determining the cause of an infection and may aid in the choice of empiric antibiotic therapy.
When the delicate symbiosis between the commensal and the host is disturbed, the normal flora may be overgrown by either endogenous or exogenous organisms. This phenomenon, which may be transient or persistent, is called colonization. For example, broad-spectrum antibiotics will destroy normal vaginal flora, such as lactobacilli, and allow overgrowth of Candida (yeast) species. When replacement of the normal flora occurs in the hospital environment, the organisms are said to be nosocomially acquired. The distinction between hospital-acquired and community-acquired infections has blurred in recent years, because of an increase in medical care in the home or skilled nursing facility among patients who previously would have required long-term hospitalization. For this reason, the broader term “healthcare-associated infections” is used to encompass both hospitalized patients and patients with frequent medical interactions (eg, residence in nursing home, outpatient hemodialysis, home intravenous antibiotics). Healthcare-associated infections are significant because the organisms are often resistant to multiple antibiotics. Not uncommonly, colonization will progress to symptomatic infection. For example, individuals hospitalized for extended periods often become colonized with gram-negative bacteria such as Pseudomonas aeruginosa. These individuals are then at increased risk for life-threatening infections such as pseudomonas pneumonia.
Host defense mechanisms that serve to inhibit colonization by pathogenic bacteria include: (1) mechanical clearance, (2) phagocytic killing, and (3) depriving organisms of necessary nutrients. Successful colonizers have adapted to evade or overcome these defenses. For example, gonococci, the bacteria that cause gonorrhea, avoid excretion in the urine by adhering to the mucosal epithelium of the urogenital tract with pili. Pneumococci resist phagocytosis by encapsulation within a slime layer that impairs uptake by neutrophils. Some staphylococci elaborate enzymes known as hemolysins that destroy host red blood cells, thus giving them access to a needed source of iron.
Colonization of sites that are normally sterile or have very few microbes is generally easier because there is no competition for nutrients from endogenous flora. However, host defenses at these sites are often vigorous. For instance, the stomach is normally sterile because few microbes can survive at the normal gastric pH of 4.0. However, if antacids are used to decrease gastric acidity, colonization of the stomach and trachea with gram-negative bacteria rapidly occurs.
The normal flora prevents colonization through numerous mechanisms. These organisms often have a selective advantage over colonizers in that they are already established in an anatomic niche. This means that they are bound to receptors on the host cell and are able to metabolize local nutrients. Many species of the normal flora are able to produce bacteriocins, proteins that are toxic to other bacterial strains or species. Finally, the normal flora promotes production of antibodies that may cross-react with colonizing organisms. For instance, an antibody produced against E coli, a gram-negative bacterium normally found in the large intestine, cross-reacts with the polysaccharide capsule of a meningitis-producing strain of N meningitidis. When the normal flora is altered (eg, by the administration of broad-spectrum antibiotics), one bacterial species may predominate or exogenous bacteria may gain a selective advantage, permitting colonization and predisposing the host to infection.
CONSTITUTIVE DEFENSES OF THE BODY
Constitutive defenses of the human body are nonspecific barriers against infectious diseases that do not require prior contact with the microorganism. These defenses consist of simple physical (eg, skin) and chemical (eg, acidic gastric secretions) barriers that prevent easy entry of microorganisms into the body. Some infectious agents use a vector (such as an insect) to bypass structural barriers and gain direct access to the blood or subcutaneous tissues of the body. Once an agent has entered the body, the major constitutive defenses are the acute inflammatory response and the complement system. These defenses can neutralize the agent, recruit phagocytic cells, and induce a more specific response through humoral and cell-mediated immunity. The constitutive defenses of the body are important from an evolutionary perspective in enabling humans to encounter and adapt to a variety of new and changing environments.
Physical & Chemical Barriers to Infection
The squamous epithelium of the skin is the first line of defense against microorganisms encountered in the outside world. As keratinized epithelial surface cells desquamate, the skin maintains its protective barrier by generating new epithelial cells beneath the surface. The skin is also bathed with oils and moisture from the sebaceous and sweat glands. These secretions contain fatty acids that inhibit bacterial growth. Poor vascular supply to the skin may result in skin breakdown and increased susceptibility to infection. For example, chronically debilitated or bedridden patients may suffer from decubitus ulcers as a result of constant pressure on dependent body parts, predisposing to severe infections by otherwise harmless skin flora.
The mucous membranes also provide a physical barrier to microbial invasion. The mucous membranes of the mouth, pharynx, esophagus, and lower urinary tract are composed of several layers of epithelial cells, whereas those of the lower respiratory tract, the GI tract, and the upper urinary tract are delicate single layers of epithelial cells. These membranes are covered by a protective layer of mucus, which traps foreign particles and prevents them from reaching the lining epithelial cells. Because the mucus is hydrophilic, many substances produced by the body easily diffuse to the surface, including enzymes with antimicrobial activity such as lysozyme and peroxidase.
Inflammatory Response
When a microorganism crosses the epidermis or the epithelial surface of the mucous membranes, it encounters other components of the host constitutive defenses. These responses are constitutive because they are nonspecific and do not require prior contact with the organism to be effective. Clinically, signs of inflammation (heat, erythema, pain, and swelling) are the characteristic features of localized infection, secondary tissue injury, and the body’s response to this injury. Blood supply to the affected area increases in response to vasodilation, and the capillaries become more permeable, allowing antibodies, complement, and white blood cells to cross the endothelium and reach the site of injury. An important consequence of inflammation is that the pH of the inflamed tissues is lowered, creating an inhospitable environment for the microbe. The increased blood flow to the area allows continued recruitment of inflammatory cells as well as the necessary components for tissue repair and recovery.
When a microorganism enters host tissue, it activates the complement system and components of the coagulation cascade and induces the release of chemical mediators of the inflammatory response. These mediators result in the increased vascular permeability and vasodilation characteristic of inflammation. For example, the anaphylatoxins C3a, C4a, and C5a, produced by the activation of complement, stimulate the release of histamine from mast cells. Histamine dilates the blood vessels and further increases their permeability. Bradykinin is also released, increasing vascular permeability.
Proinflammatory cytokines include interleukin-1 (IL-1), IL-6, tumor necrosis factor, and interferon-γ. These factors, singly or in combination, promote fever, produce local inflammatory signs, and trigger catabolic responses. During severe infection, a change in hepatic synthesis of proteins occurs, resulting in an increase in some proteins and a decrease in others. Most notable is the increase in “acute-phase reactants” that include rheumatoid factor, C-reactive protein, ferritin, and various proteinase inhibitors. The erythrocyte sedimentation rate, a nonspecific marker of inflammation, also rises, whereas the serum levels of various elements such as zinc and iron decrease. A catabolic state is further augmented by simultaneous increases in levels of circulating cortisol, glucagon, catecholamines, and other hormones.
Mild-to-moderate inflammatory responses serve important host defense functions. For example, elevated body temperature may inhibit viral replication. Inflammatory hyperemia and systemic neutrophilia optimize phagocyte delivery to sites of infection. The decreased availability of iron inhibits the growth of microbes such as Yersinia that require this element as a nutrient. However, when the inflammatory responses become extreme, extensive tissue damage can result, as in the case of sepsis.
Complement System
The complement system is composed of a series of plasma protein and cell membrane receptors that are important mediators of host defenses and inflammation (Figure 4-4). Most of the biologically significant effects of the complement system are mediated by the third component (C3) and the terminal components (C5–9). To carry out their host defense and inflammatory functions, C3 and C5–9 must first be activated. Two pathways of complement activation have been recognized and have been termed the classic and alternative pathways. The classic pathway is activated by antigen–antibody complexes or antibody-coated particles, and the alternative pathway is activated by mechanisms independent of antibodies, usually by interaction with bacterial surface components. Both pathways form C3 convertase, which cleaves the C3 component of complement, a key protein common to both pathways. The two pathways then proceed in identical fashion to bind late-acting components to form a membrane attack complex (C5–9), which results in target cell lysis.
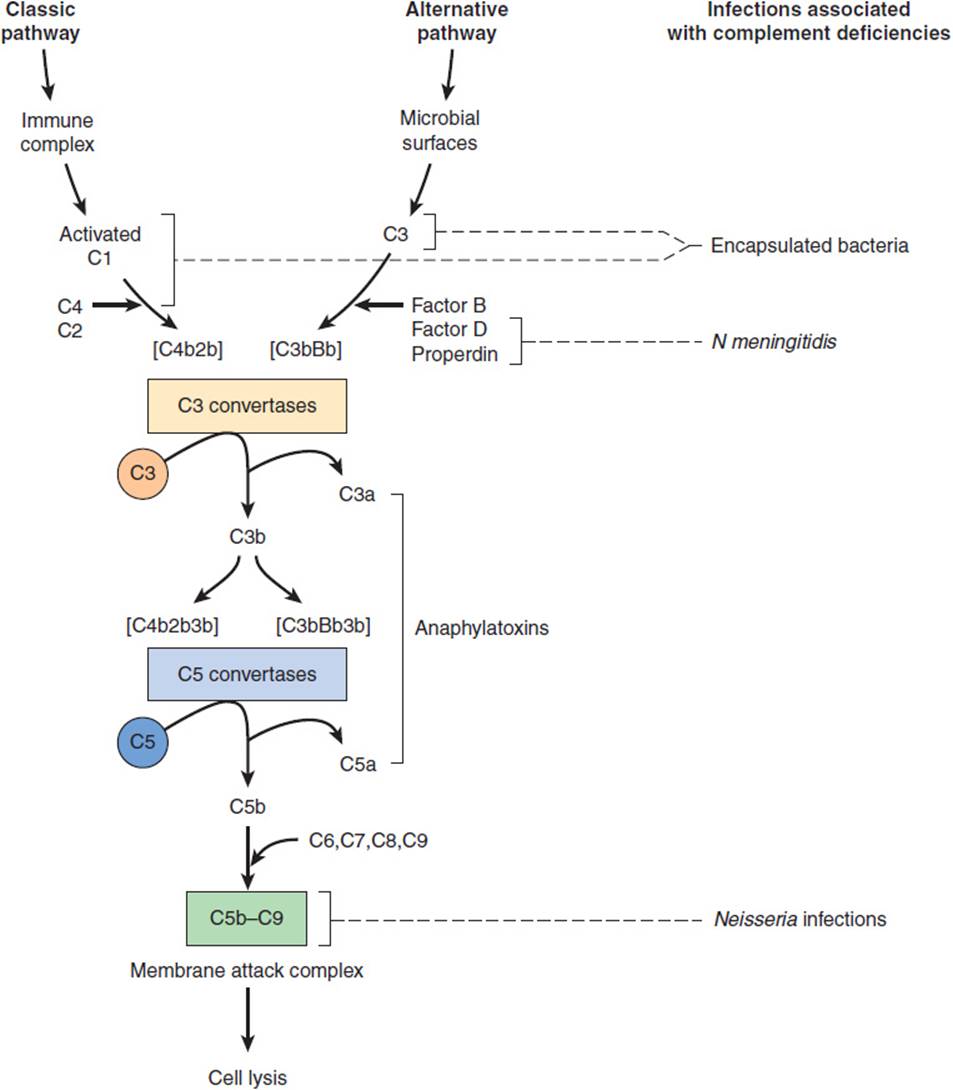
FIGURE 4-4 Complement reaction sequence and infections associated with deficiency states. (Redrawn, with permission, from Nairn R. Immunology. In: Brooks GF et al, eds. Jawetz, Melnick, and Adelberg’s Medical Microbiology, 23rd ed. McGraw-Hill, 2004.)
Once activated, complement functions to enhance the antimicrobial defenses in several ways. Complement facilitates phagocytosis through proteins called opsonins, which coat invading microorganisms, making them susceptible to engulfment and destruction by neutrophils and macrophages. The complement-derived membrane attack complex inserts itself into the membrane of a target organism, leading to increased permeability and subsequent lysis of the cell. Complement also acts indirectly through production of substances that are chemotactic for white blood cells and through promotion of the inflammatory response.
Inherited disorders of complement are associated with an increased risk of bacterial infection. The specific infections seen in complement-deficient patients relate to the biologic functions of the missing component (Figure 4-4). Patients with a deficiency of C3 or of a component in either of the two pathways necessary for the activation of C3 typically have increased susceptibility to infections with encapsulated bacteria such as S pneumoniae and Haemophilus influenzae. In contrast, patients with deficiencies of C5–9 have normal resistance to encapsulated bacteria because C3b-mediated opsonization is intact. These patients, however, are unusually susceptible to life-threatening infections with N meningitidis and N gonorrhoeae because they are unable to form a membrane attack complex and, therefore, cannot lyse the Neisseria cell membrane.
Phagocytosis
After the natural barriers of the skin or mucous membranes have been penetrated, the phagocytic cells—neutrophils, monocytes, and macrophages—constitute the next line of host defense. The process of internalizing organisms by these cells (phagocytosis) involves attachment of the organism to the cell surface. This triggers extension of a pseudopod to enclose the bacterium in an endocytic vesicle, or phagosome. The circulating polymorphonuclear neutrophil (PMN) is an important component of the host immune response that in the absence of infection circulates in a quiescent state. When chemotactic factors, arachidonic acid metabolites, or complement cleavage fragments interact with specific PMN membrane receptors, the neutrophil rapidly becomes activated and moves toward the chemoattractants. After phagocytosis, the mechanisms by which the phagolysosome destroys the microorganism can be divided into oxygen-independent and oxygen-dependent processes. Functional defects in circulating neutrophils or decreases in absolute number of neutrophils are important risk factors for infection.
Neutropenia, defined as an absolute neutrophil count of less than 1000 cells/μL, is a common predisposing factor for life-threatening bacterial and fungal infections. The risk of infection is inversely proportionate to the number of neutrophils, rising significantly with neutrophil counts less than 500 cells/μL. The longer the duration of profound neutropenia, the greater is the risk of infection. At the first sign of infection (eg, fever), these patients should be given broad-spectrum antibacterial agents to cover gram-negative bacterial pathogens. In addition to impaired immunity, neutropenic hosts often have additional risk factors for infection such as the need for long-term indwelling central venous catheters (predisposing infection with skin bacteria) and the frequent use of parenteral nutrition (predisposing to fungal infection).
Several inherited disorders of neutrophil function have been described. Chédiak-Higashi syndrome is a rare autosomal recessive hereditary disorder in which the neutrophils have a profound defect in the formation of intracellular granules. Opsonized bacteria such as Staphylococcus aureus are ingested normally, but viable bacteria persist intracellularly, presumably because of the inability of the neutrophil’s intracellular granules to fuse with phagosomes to form phagolysosomes. Patients with Chédiak-Higashi syndrome experience recurrent bacterial infections, most frequently involving the skin and soft tissues and the upper and lower respiratory tracts.
Myeloperoxidase deficiency is the most common neutrophil disorder, with a prevalence of one case per 2000 individuals. In this disorder, phagocytosis, chemotaxis, and degranulation are normal, but microbicidal activity for bacteria is delayed. In general, these patients do not suffer from recurrent infections. In contrast, chronic granulomatous disease is a genetically heterogeneous group of inherited disorders characterized by the failure of phagocytic cells to produce superoxides. The defect involves neutrophils, monocytes, eosinophils, and some macrophages. Oxygen-dependent intracellular killing is impaired, and these patients are susceptible to recurrent, often life-threatening infections. Patients with chronic granulomatous disease also tend to form granulomas in tissues, particularly in the lungs, liver, and spleen, and are particularly susceptible to infection with S aureus and Aspergillus species.
INDUCED DEFENSES OF THE BODY
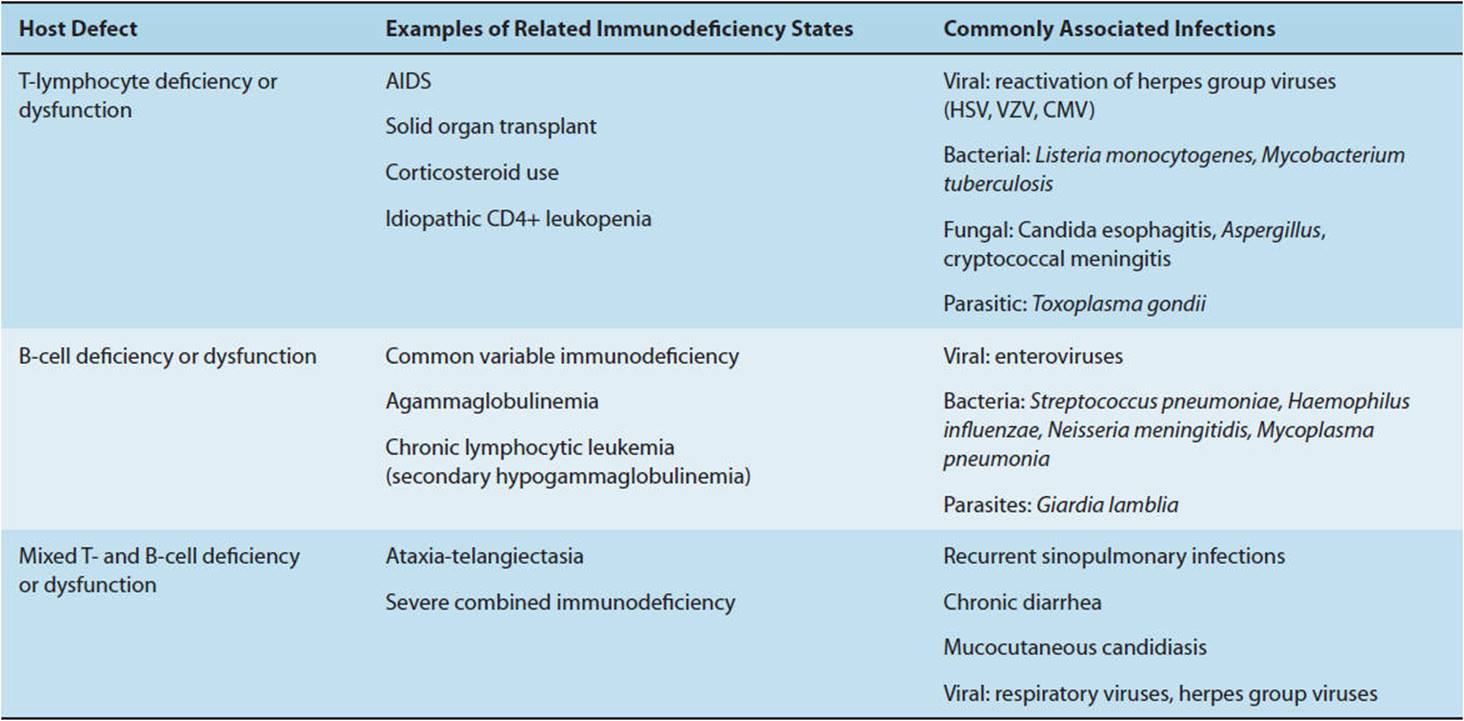
Although constitutive host defenses against infectious agents are generally nonspecific and do not require prior exposure to the invading agent, induced defenses are highly specific and are qualitatively and quantitatively altered by prior antigenic exposure. Details of the pathophysiology of the host immune system are covered in Chapter 3. Recurrent infections or infections with unusual organisms may be clues to an underlying defect in the induced immune response (Table 4-2).
TABLE 4-2 Infections associated with common defects in humoral and cellular immune response.
ESTABLISHMENT OF INFECTION
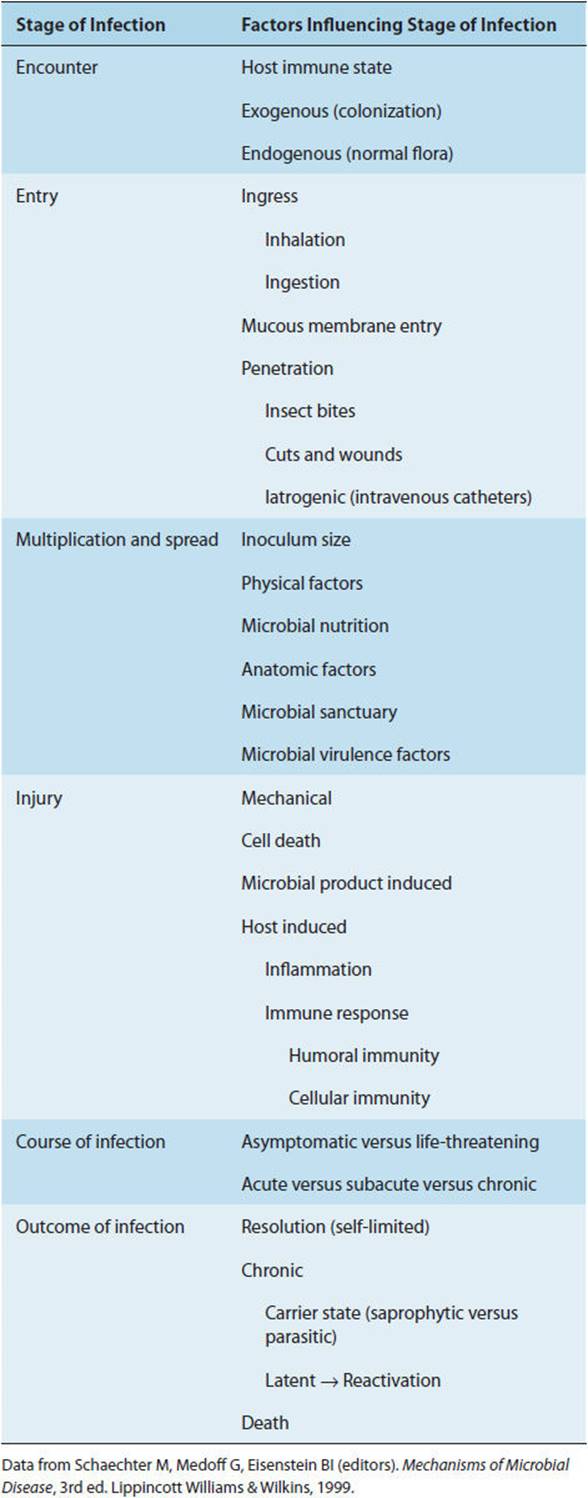
An infectious disease occurs when a pathogenic organism causes inflammation or organ dysfunction. This may be caused directly by the infection itself, as when the etiologic agent multiplies in the host, or indirectly as a result of the host’s inflammatory response. Many infections are subclinical, not producing any obvious manifestations of disease. To cause overt infection, all microorganisms must go through the following stages (Table 4-3): The microorganism must (1) encounter the host, (2) gain entry into the host, (3) multiply and spread from the site of entry, and (4) cause host tissue injury, either directly (eg, cytotoxins) or indirectly (host inflammatory response). The severity of infection ranges from asymptomatic to life-threatening, and the course may be characterized as acute, subacute, or chronic. Whether infection is subclinical or overt, the outcome is either (1) resolution (eg, eradication of the infecting pathogen), (2) chronic active infection (eg, HIV or hepatitis), (3) prolonged asymptomatic excretion of the agent (eg, carrier state with Salmonella typhi), (4) latency of the agent within host tissues (eg, latent tuberculosis or varicella zoster virus), or (5) host death from infection.
TABLE 4-3 The establishment and outcome of infectious diseases.
Except for congenital infections (acquired in utero) humans first encounter microorganisms at birth. During parturition, the newborn comes into contact with microorganisms present in the mother’s vaginal canal and on her skin. Most of the bacteria the newborn encounters do not cause harm, and for those that might cause infection, the newborn usually has passive immunity through antibodies acquired from the mother in utero. For example, neonates are protected against infection with H influenzae by maternal antibodies for the first 6 months of life until passive immunity wanes and the risk of infection with this bacterium increases. On the other hand, newborns whose mothers are vaginally colonized with group B streptococci are at increased risk in the perinatal period for serious infections such as sepsis or meningitis with this organism. For this reason, it is recommended that (1) vaginal cultures be done to screen for group B streptococci for all pregnant women and (2) intrapartum antibiotic prophylaxis be administered to those with positive detection of group B streptococci.
Direct entry of microorganisms into the host (ie, bypassing the usual chemical and physical barriers) may occur when (1) an insect vector directly inoculates the infectious agent into the host (mosquitoes transmitting malaria), (2) bacteria gain direct access to host tissues through loss of integrity of the skin or mucous membranes (trauma or surgical wounds), or (3) microbes gain access via instruments or catheters that allow communication between usually sterile sites and the outside world (indwelling venous catheters). Ingression occurs when an infectious agent enters the host via an orifice contiguous with the external environment. This primarily involves inhalation of infectious aerosolized droplets (M tuberculosis) or ingestion of contaminated foods (salmonella, hepatitis A virus).
Other infectious agents directly infect mucous membranes or cross the epithelial surface to cause infection. This commonly occurs in sexually transmitted diseases. For example, HIV can cross vaginal mucous membranes by penetration of virus-laden macrophages from semen.
After the initial encounter with the host, the infectious agent must successfully multiply at the site of entry. The process whereby the newly introduced microorganism successfully competes with normal flora and is able to multiply is termed colonization (eg, pneumococci colonizing the upper respiratory tract). When the microorganism multiplies at a normally sterile site, it is termed infection (eg, pneumococci multiplying in the alveoli, causing pneumonia). Factors that facilitate the multiplication and spread of infection include inoculum size (the quantity of infectious organisms introduced), host anatomic factors (eg, impaired ciliary function in children with cystic fibrosis), availability of nutrients for the microbe, physicochemical factors (eg, gastric pH), microbial virulence factors, and microbial sanctuary (eg, abscesses). An abscess is a special case in which the host has contained the infection but is unable to eradicate it, and these localized infections generally require surgical drainage. Once introduced, infections can spread along the epidermis (impetigo), along the dermis (erysipelas), along subcutaneous tissues (cellulitis), along fascial planes (necrotizing fasciitis), into muscle tissue (myositis), along veins (suppurative thrombophlebitis), into the blood (bacteremia, fungemia, viremia, etc), along lymphatics (lymphangitis), and into organs (eg, pneumonia, brain abscesses, hepatitis).
Infections cause direct injury to the host through a variety of mechanisms. If organisms are present in sufficient numbers and are of sufficient size, mechanical obstruction can occur (eg, children with roundworm gastrointestinal infections may present with bowel obstruction). More commonly, pathogens cause an intense secondary inflammatory response, which may result in life-threatening complications (eg, children with H influenzae epiglottitis may present with mechanical airway obstruction secondary to intense soft tissue swelling of the epiglottis). Some bacteria produce neurotoxins that affect host cell metabolism rather than directly causing cell damage (eg, tetanus toxin antagonizes inhibitory neurons, causing unopposed motor neuron stimulation, manifested clinically as sustained muscle rigidity). Host cell death can occur by a variety of mechanisms. Shigella produces a cytotoxin that causes death of large intestine enterocytes, resulting in the clinical syndrome of dysentery. Poliovirus-induced cell lysis of the anterior horn cells of the spinal cord causes flaccid paralysis. Gram-negative bacterial endotoxin can initiate a cascade of cytokine release, resulting in sepsis syndrome and septic shock.
The time course of an infection can be characterized as acute, subacute, or chronic, and its severity may vary from asymptomatic to life-threatening. Many infections that begin as mild and easily treatable conditions readily progress without prompt treatment. Small, seemingly insignificant skin abrasions superinfected with toxic shock syndrome toxin (TSST-1)–producing S aureus can result in fulminant infection and death. Even indolent infections, such as infective endocarditis resulting from Streptococcus viridans, can be fatal unless they are recognized and appropriately treated.
There are three potential outcomes of infection: recovery, chronic infection, and death. Most infections resolve, either spontaneously (eg, rhinovirus, the leading cause of the common cold) or with medical therapy (eg, after treatment of streptococcal pharyngitis with penicillin). Chronic infections may be either saprophytic, in which case the organism does not adversely affect the health of the host; or parasitic, causing tissue damage to the host. An example of the former is Salmonella typhi, which may be harbored asymptomatically in the gallbladder of about 2% of individuals after acute infection. Chronic infection with the hepatitis B virus may be either saprophytic, in which case the human host is infectious for the virus but has no clinical evidence of liver damage, or parasitic, with progressive liver damage and cirrhosis. A final form of chronic infection is tissue latency. Varicella-zoster virus, the agent causing chickenpox, survives in the dorsal root ganglia, with reactivation causing a dermatomal eruption with vesicles or shallow ulcerations, commonly known as shingles or zoster. When the ability of the immune system to control either the acute or the chronic infection is exceeded, the infection may result in host death.
All infectious agents, regardless of specific mechanisms, must successfully reproduce and evade host defense mechanisms. This knowledge helps the physician to prevent infections (eg, vaccinate against influenza virus); to treat and cure infection (eg, antibiotics for E coli urinary tract infection); and when infection cannot be cured, to prevent further transmission, recurrence, or reactivation (eg, barrier protection to reduce the sexual spread of genital herpes simplex infection).
CHECKPOINT
1. By what three general mechanisms do hosts resist colonization by pathogenic bacteria?
2. What are three ways in which the normal flora contributes to the balance between health and disease?
3. Which specific host defenses against infection do not require prior contact with the infecting organism?
4. What are the categories of outcomes from an infection?
PATHOPHYSIOLOGY OF SELECTED INFECTIOUS DISEASE SYNDROMES
INFECTIVE ENDOCARDITIS
Clinical Presentation
Infective endocarditis refers to a bacterial or, rarely, a fungal infection of the cardiac valves. Infection of extracardiac endothelium is termed “endarteritis” and can cause disease that is clinically similar to endocarditis. The most common predisposing factor for infective endocarditis is the presence of structurally abnormal cardiac valves. Consequently, patients with a history of rheumatic or congenital heart disease, a prosthetic heart valve, or a history of prior endocarditis are at increased risk for infective endocarditis. Infection involves the left side of the heart (mitral and aortic valves) almost exclusively, except in patients who are injection drug users or, less commonly, in patients with valve injury from a pulmonary artery (Swan-Ganz) catheter, in whom infection of the right side of the heart (tricuspid or pulmonary valve) may occur.
Etiology
The most common infectious agents causing native valve infective endocarditis are gram-positive bacteria, including viridians group streptococci, S aureus, and enterococci. The specific bacterial species causing endocarditis can often be anticipated on the basis of host factors. Injection drug users commonly introduce skin bacteria such as S aureus into the blood when nonsterile needles are used or the skin is not adequately cleaned before needle insertion. Patients with recent dental work are at risk for transient bacteremia with normal oral flora, particularly viridians group streptococci, with subsequent endocarditis. Genitourinary tract infections with enterococci may lead to bacteremia and subsequent seeding of damaged heart valves. Patients with prosthetic heart valves are also at increased risk for infective endocarditis resulting from skin flora such as S epidermidis or S aureus. Before the availability of antibiotics, infective endocarditis was a uniformly fatal disease. Even with antibiotics, the case fatality rate for endocarditis approaches 25%, and definitive cure often requires both prolonged intravenous antibiotic administration and urgent surgery to replace infected cardiac valves.
Pathogenesis
Several hemodynamic factors predispose patients to endocarditis: (1) a high-velocity jet stream causing turbulent blood flow, (2) flow from a high-pressure to a low-pressure chamber, and (3) a comparatively narrow orifice separating the two chambers that creates a pressure gradient. The lesions of infective endocarditis tend to form on the surface of the valve in the cardiac chamber with the lower pressure (eg, on the ventricular surface of an abnormal aortic valve and on the atrial surface of an abnormal mitral valve). Endothelium damaged by turbulent blood flow results in exposure of extracellular matrix proteins, promoting the deposition of fibrin and platelets, which form sterile vegetations (nonbacterial thrombotic endocarditis or marantic endocarditis). Infective endocarditis occurs when microorganisms are deposited onto these sterile vegetations during the course of bacteremia (Figure 4-5). Not all bacteria adhere equally well to these sites. For example, E coli, a frequent cause of urosepsis, is rarely implicated as a cause of endocarditis. Conversely, virulent organisms such as S aureus can invade intact endothelium, causing endocarditis in the absence of preexisting valvular abnormalities.
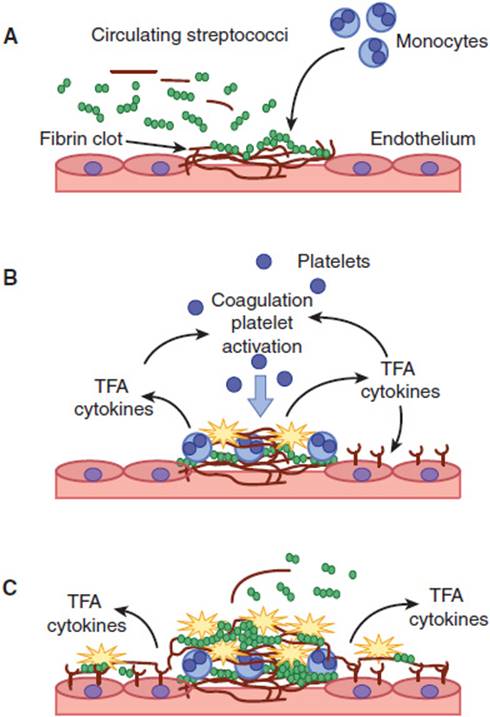
FIGURE 4-5 Pathogenesis of bacterial valve colonization. Viridans group streptococci adhere to fibrin-platelet clots that form at the site of damaged cardiac endothelium (A). The fibrin-adherent streptococci activate monocytes to produce tissue factor activity (TFA) and cytokines (B). These mediators activate the coagulation pathway, resulting in further recruitment of platelets and growth of the vegetation (C). (Redrawn, with permission, from Moreillon P et al. Pathogenesis of streptococcal and staphylococcal endocarditis. Infect Dis Clin North Am. 2002;16:297.)
Once infected, these vegetations continue to enlarge through further deposition of platelets and fibrin, providing the bacteria a sanctuary from host defense mechanisms such as polymorphonuclear leukocytes and complement. Consequently, once infection takes hold, the infected vegetation continues to grow in a largely unimpeded fashion. Prolonged administration (4–6 weeks) of bactericidal antibiotics is required to penetrate the vegetation and cure this disease. Bacteriostatic antimicrobial agents, which inhibit but do not kill the bacteria, are inadequate. Surgical removal of the infected valve is sometimes required for cure, particularly if there is mechanical dysfunction of the valve with resultant heart failure, abscess formation around the valve ring, or prosthetic valve infections.
A hallmark of infective endocarditis is sustained high-grade bacteremia, which stimulates both the humoral and cellular immune systems. A variety of immunoglobulins are expressed, resulting in immune complex formation, increased serum levels of rheumatoid factor, and nonspecific hypergammaglobulinemia. Immune complex deposition along the renal glomerular basement membrane may result in the development of acute glomerulonephritis and renal failure.
Clinical Manifestations
Infective endocarditis is a multisystem disease with protean manifestations. For these reasons, the symptoms can be nonspecific. Table 4-4 summarizes the important features of the history, physical examination, laboratory results, and complications of infective endocarditis. Cutaneous findings suggestive of endocarditis include Osler nodes, painful papules on the pads of the fingers and toes thought to be secondary to deposition of immune complexes; and Janeway lesions, painless hemorrhagic lesions on the palms and soles caused by septic microemboli (Figure 4-6). Symptoms and signs of endocarditis may be acute, subacute, or chronic. The clinical manifestations reflect primarily (1) hemodynamic changes from valvular damage; (2) end-organ symptoms and signs from septic emboli (right-sided emboli to the lungs, left-sided emboli to the brain, spleen, kidney, and extremities); (3) end-organ symptoms and signs from immune complex deposition; and (4) persistent bacteremia with metastatic seeding of infection (abscesses or septic joints). Death is usually caused by hemodynamic collapse or by septic emboli to the central nervous system (CNS), resulting in brain abscesses or mycotic aneurysms and intracerebral hemorrhage. Risk factors for a fatal outcome include patients with left-sided valvular infection, bacterial infection other than viridans group streptococci, medical comorbidities, complications from endocarditis (heart failure, valve ring abscess, or embolic disease), and delayed valvular surgery (for those with large vegetations and significant valvular destruction).
TABLE 4-4 Diagnosis of infective endocarditis.
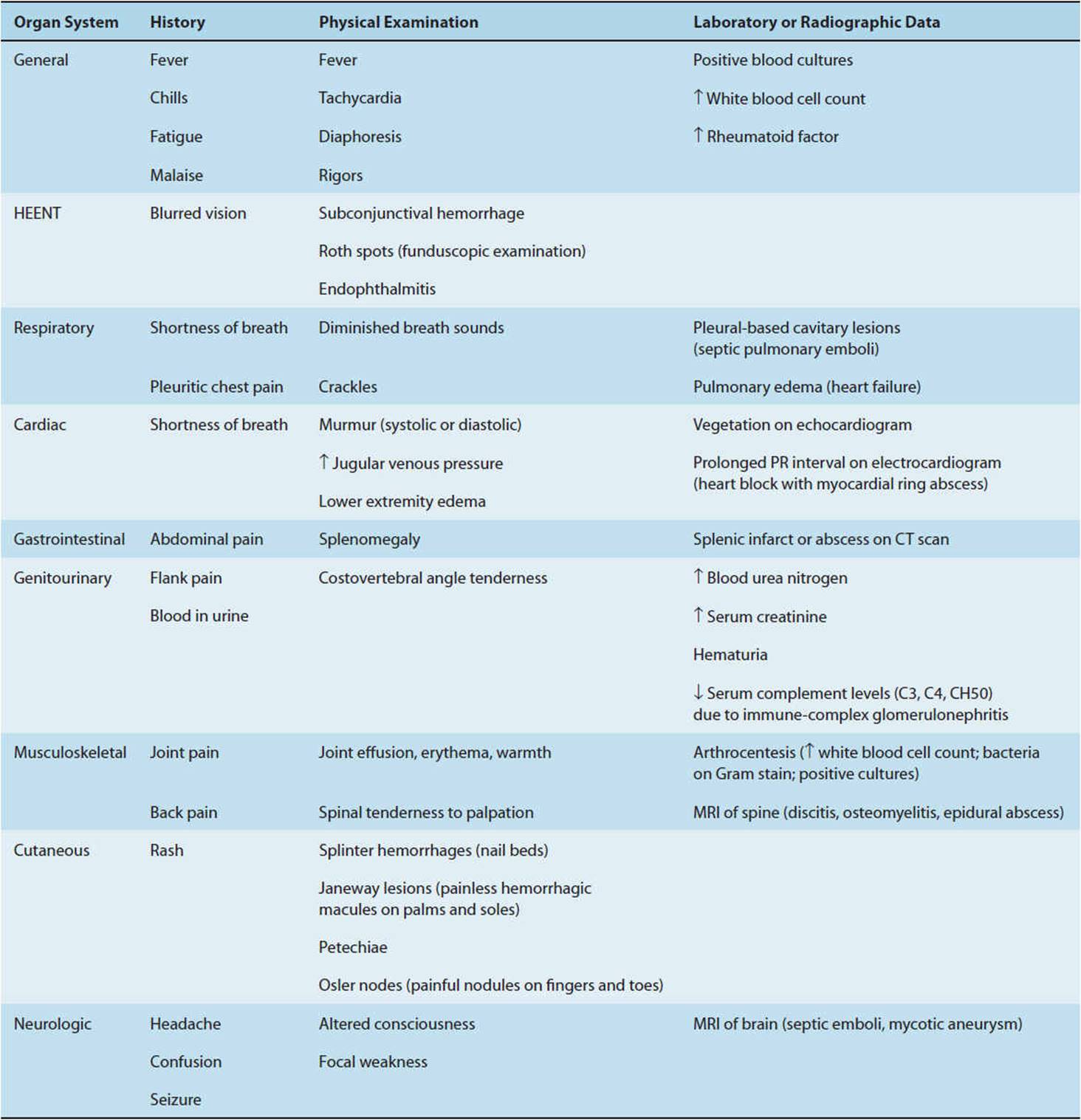
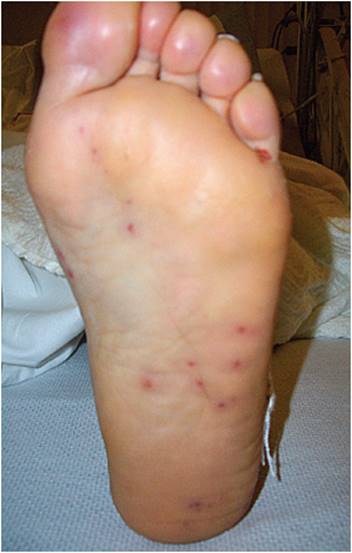
FIGURE 4-6 Osler node causing pain within pulp of the big toe in a woman hospitalized with acute bacterial endocarditis. (Osler nodes are painful: remember “O” for Ouch and Osler.) Note the multiple painless flat Janeway lesions over the sole of the foot. (Used, with permission, from David A. Kasper, DO, MBA. Originally published in: Chumley H. Bacterial endocarditis. In: Usatine RP et al, eds. The Color Atlas of Family Medicine. McGraw-Hill, 2009:205–9.)
CHECKPOINT
5. Which patients are at highest risk for infective endocarditis?
6. What are the leading bacterial agents of infective endocarditis?
7. What features characterize infective endocarditis in intravenous drug users? In patients with prosthetic heart valves?
8. What hemodynamic features predispose to infective endocarditis?
9. What is the outcome of untreated bacterial endocarditis?
10. What are the risk factors for a fatal outcome? What are the most common causes of death in untreated infective endocarditis?
MENINGITIS
Clinical Presentation
Symptoms commonly associated with both bacterial and viral meningitis include acute onset of fever, headache, neck stiffness (meningismus), photophobia, and confusion. Bacterial meningitis causes significant morbidity (neurologic sequelae, particularly sensorineural hearing loss) and mortality and thus requires immediate antibiotic therapy. With rare exceptions, only supportive care with analgesics is necessary for viral meningitis.
Because the clinical presentations of bacterial and viral meningitis may be indistinguishable, laboratory studies of the cerebrospinal fluid are critical in differentiating these entities. Cerebrospinal fluid leukocyte pleocytosis (white blood cells in the cerebrospinal fluid) is the hallmark of meningitis. Bacterial meningitis is generally characterized by neutrophilic pleocytosis (predominance of polymorphonuclear neutrophils in the cerebrospinal fluid). Common causes of lymphocytic pleocytosis include viral infections (eg, enterovirus, West Nile virus), fungal infections (eg, cryptococcus in HIV-infected persons), and spirochetal infections (eg, neurosyphilis or Lyme neuroborreliosis). Noninfectious causes such as cancer, connective tissue diseases, and hypersensitivity reactions to drugs can also cause lymphocytic pleocytosis. The cerebrospinal fluid in bacterial meningitis is generally characterized by marked elevations in protein concentration, an extremely low glucose level, and, in the absence of previous antibiotic treatment, a positive Gram stain for bacteria. However, there is often significant overlap between the cerebrospinal fluid findings in bacterial and nonbacterial meningitis, and differentiating these entities at presentation is a significant clinical challenge.
Etiology
The microbiology of bacterial meningitis in the United States has changed dramatically following the introduction of the Haemophilus influenzae conjugate vaccine. The routine use of this vaccine in the pediatric population has resulted in a more than 95% decrease in the incidence of H influenzae meningitis in the United States.
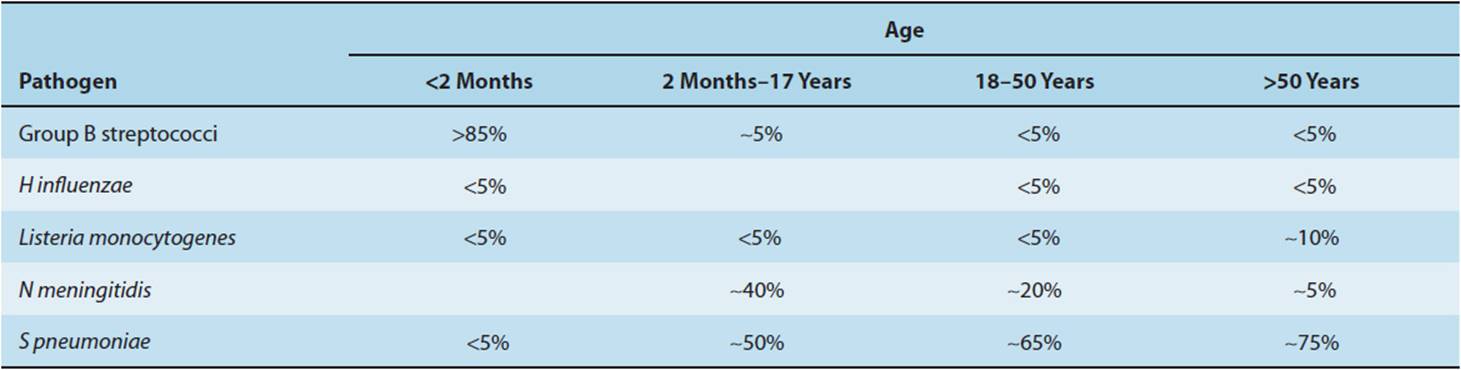
Bacterial agents causing meningitis vary according to host age (Table 4-5). Additional bacteria must be considered for postneurosurgery patients (S aureus, gram-negative bacilli, P aeruginosa), patients with ventricular shunts (S epidermidis, S aureus, gram-negative bacilli), pregnant patients (Listeria), or neutropenic patients (gram-negative bacilli, including P aeruginosa). Subacute or chronic meningitides may be caused by M tuberculosis, fungi (eg, Coccidioides immitis, Cryptococcus neoformans), and spirochetes such as Treponema pallidum (the bacterium causing syphilis) or Borrelia burgdorferi (the bacterium causing Lyme disease). The diagnosis of meningitis caused by these organisms may be delayed because many of these pathogens are difficult to culture and require special serologic or molecular diagnostic techniques.
TABLE 4-5 Proportion of cases of bacterial meningitis in the United States by host age, 2003–2007.
Pathogenesis
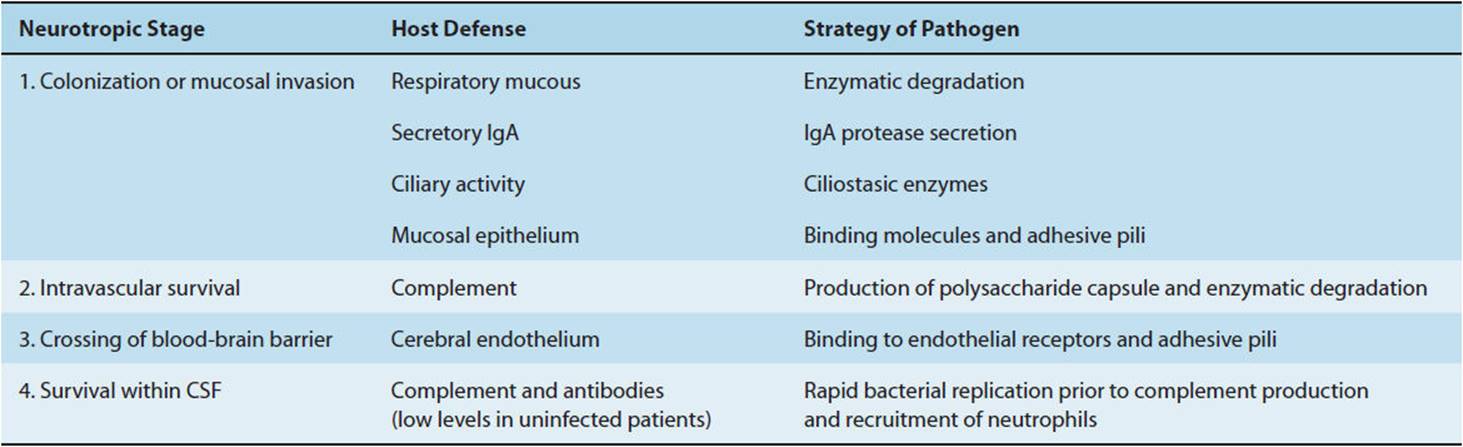
The pathogenesis of bacterial meningitis involves a sequence of events in which virulent microorganisms overcome the host defense mechanisms (Table 4-6).
TABLE 4-6 Pathogenetic sequence of bacterial neurotropism.
Most cases of bacterial meningitis begin with bacterial colonization of the nasopharynx (Figure 4-7, panel A). An exception is Listeria, which enters the bloodstream through ingestion of contaminated food. Pathogenic bacteria such as S pneumoniae and N meningitidis secrete an IgA protease that inactivates host antibody and facilitates mucosal attachment. Many of the causal pathogens also possess surface characteristics that enhance mucosal colonization. N meningitidis binds to nonciliated epithelial cells by finger-like projections known as pili.
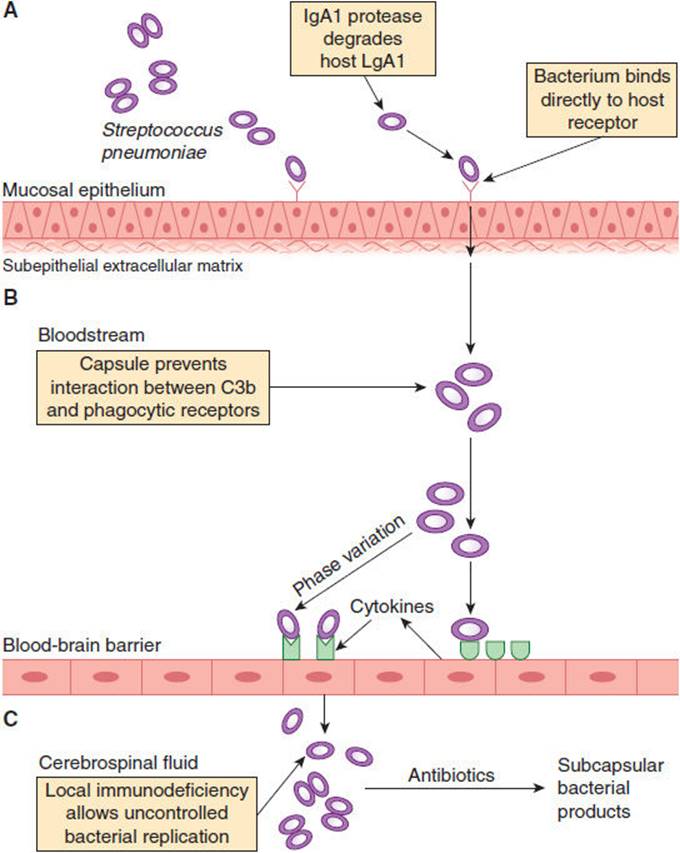
FIGURE 4-7 Pathogenic steps leading to pneumococcal meningitis. The pneumococcus adheres to and colonizes the nasopharynx. IgA1 protease protects the pneumococcus from host antibody (A). Once in the bloodstream, the bacterial capsule helps the pneumococcus to evade opsonization (B). The pneumococcus accesses the cerebrospinal fluid through receptors on the endothelial surface of the blood-brain barrier (C). (Redrawn, with permission, from Koedel U et al. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. 2002;2:731.)
Once the mucosal barrier is breached, bacteria gain access to the bloodstream, where they must overcome host defense mechanisms to survive and invade the CNS (Figure 4-7, panel B). The bacterial capsule, a feature common to N meningitidis, H influenzae, and S pneumoniae, is the most important virulence factor in this regard. Host defenses counteract the protective effects of the pneumococcal polysaccharide capsule by activating the alternative complement pathway, resulting in C3b activation, opsonization, phagocytosis, and intravascular clearance of the organism. This defense mechanism is impaired in patients who have undergone splenectomy, and such patients are predisposed to the development of overwhelming bacteremia and meningitis with encapsulated bacteria. Activation of the complement system membrane attack complex is an essential host defense mechanism against invasive disease by N meningitidis, and patients with deficiencies of the late complement components (C5–9) are at increased risk for meningococcal meningitis.
The mechanisms by which bacterial pathogens gain access to the CNS are largely unknown. Experimental studies suggest that receptors for bacterial pathogens are present on cells in the choroid plexus, which may facilitate movement of these pathogens into the subarachnoid space (Figure 4-7, panel C). Invasion of the spinal fluid by a meningeal pathogen results in increased permeability of the blood–brain barrier, where local host defense mechanisms are inadequate to control the infection. Normally, complement components are minimal or absent in the cerebrospinal fluid. Meningeal inflammation leads to increased, but still low, concentrations of complement, inadequate for opsonization, phagocytosis, and removal of encapsulated meningeal pathogens. Immunoglobulin concentrations are also low in the cerebrospinal fluid, with an average blood to cerebrospinal fluid IgG ratio of 800:1.
The ability of meningeal pathogens to induce a marked subarachnoid space inflammatory response contributes to many of the pathophysiologic consequences of bacterial meningitis. Although the bacterial capsule is largely responsible for intravascular and cerebrospinal fluid survival, the subcapsular surface components (ie, the cell wall and lipopolysaccharide) of bacteria are more important determinants of meningeal inflammation. The major mediators of the inflammatory process are thought to be IL-1, IL-6, matrix metalloproteinases, and tumor necrosis factor (TNF). Within 1–3 hours after intracisternal inoculation of purified lipopolysaccharide in an animal model, there is a brisk release of TNF and IL-1 into the cerebrospinal fluid, preceding the development of inflammation. Indeed, direct inoculation of TNF and IL-1 into the cerebrospinal fluid produces an inflammatory cascade identical to that seen with experimental bacterial infection. In contrast, experimental injection of purified pneumococcal capsular polysaccharide proteins directly into the cerebrospinal fluid does not result in significant inflammation in animals.
Cytokine and proteolytic enzyme release leads to loss of membrane integrity, with resultant cellular swelling. The development of cerebral edema contributes to an increase in intracranial pressure, potentially resulting in life-threatening cerebral herniation (Figure 4-8). Vasogenic cerebral edema is principally caused by the increase in blood-brain barrier permeability. Cytotoxic cerebral edema results from swelling of the cellular elements of the brain because of toxic factors from bacteria or neutrophils. Interstitial cerebral edema reflects obstruction of flow of cerebrospinal fluid, as in hydrocephalus. Neuronal cell death or apoptosis is caused both by the immune inflammatory response and by direct toxicity of bacterial components, and clinically may be associated with cognitive impairment as a long-term sequela of meningitis. Cerebrovascular complications including infarction or hemorrhage are common and may be due to localized intravascular coagulation.
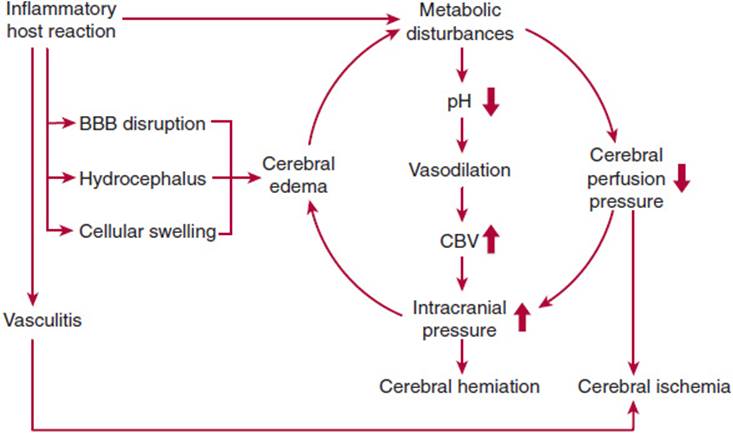
FIGURE 4-8 Pathophysiological alterations leading to neuronal injury during bacterial meningitis. BBB, blood-brain barrier; CBV, cerebral blood volume. (Redrawn, with permission, from Koedel U et al. Pathogenesis and pathophysiology of pneumococcal meningitis. Lancet Infect Dis. 2002;2:731.)
Understanding the pathophysiology of bacterial meningitis has therapeutic implications. Although bactericidal antibiotic therapy is critical for adequate treatment, rapid bacterial killing releases inflammatory bacterial fragments, potentially exacerbating inflammation and abnormalities of the cerebral microvasculature. In animal models, antibiotic therapy has been shown to cause rapid bacteriolysis and release of bacterial endotoxin, resulting in increased cerebrospinal fluid inflammation and cerebral edema.
The importance of the immune response in triggering cerebral edema has led researchers to study the role of adjuvant anti-inflammatory medications for bacterial meningitis. The use of corticosteroids has been shown to decrease the risk of sensorineural hearing loss among children with H influenzae meningitis and mortality among adults with pneumococcal meningitis. The benefit of adjuvant corticosteroids for other types of meningitis is unproven.
Clinical Manifestations
Among patients who develop community-acquired bacterial meningitis, an antecedent upper respiratory tract infection is common. Patients with a history of head injury or neurosurgery, especially those with a persistent cerebrospinal fluid leak, are at particularly high risk for meningitis. Manifestations of meningitis in infants may be difficult to recognize and interpret; therefore, the physician must be alert to the possibility of meningitis in the evaluation of any febrile neonate.
Most patients with meningitis have a rapid onset of fever, headache, lethargy, and confusion. Fewer than half complain of neck stiffness, but nuchal rigidity is frequently noted on physical examination. Other clues seen in a variable proportion of cases include nausea or vomiting, photophobia, Kernig sign (resistance to passive extension of the flexed leg with the patient lying supine), and Brudzinski sign(involuntary flexion of the hip and knee when the examiner passively flexes the patient’s neck). More than half of patients with meningococcemia develop a characteristic petechial or purpuric rash, predominantly on the extremities.
Although a change in mental status (lethargy, confusion) is common in bacterial meningitis, up to one-third of patients present with normal mentation. From 10% to 30% of patients have cranial nerve dysfunction, focal neurologic signs, or seizures. Coma, papilledema, and the Cushing triad (bradycardia, respiratory depression, and hypertension) are ominous signs of impending herniation (brain displacement through the foramen magnum with brain stem compression), heralding imminent death.
Despite advances in treatment, the case fatality rate of meningitis remains approximately 15%, and neurologic impairment is common among survivors. Morbidity and mortality may be decreased by rapid initiation of appropriate antibiotics. Any patient suspected of having meningitis requires prompt medical assessment and emergent lumbar puncture for Gram stain and culture of the cerebrospinal fluid, followed immediately by the administration of antibiotics (and corticosteroids if pneumococcal meningitis is suspected).
CHECKPOINT
11. What is the typical presentation of bacterial meningitis?
12. What are the major etiologic agents of meningitis, and how do they vary with age or other characteristics of the host?
13. What is the sequence of events in development of meningitis, and what features of particular organisms predispose to meningitis?
14. What are the diverse causes of cerebral edema in patients with meningitis?
15. Why is rapid bacteriolysis theoretically dangerous in meningitis?
16. What are the associated clinical manifestations of untreated bacterial meningitis?
PNEUMONIA
Clinical Presentation
The respiratory tract is the most common site of infection by pathogenic microorganisms. Pneumonia accounts for >1 million hospitalizations each year in the United States, and >50,000 deaths. Pneumonia, together with influenza, is the leading cause of death from an infectious disease in the United States.
Diagnosis and management of pneumonia require knowledge of host risk factors, potential infectious agents, and environmental exposures. Pneumonia is an infection of the lung tissue caused by a number of different bacteria, viruses, parasites, and fungi, resulting in inflammation of the lung parenchyma and accumulation of an inflammatory exudate in the airways. Infection typically begins in the alveoli, with secondary spread to the interstitium, resulting in consolidation and impaired gas exchange. Infection can also extend to the pleural space, causing pleuritis (inflammation of the pleura, characterized by pain on inspiration). The exudative inflammatory response of the pleura to pneumonia is termed a parapneumonic effusion; when bacterial infection is present in the pleura, this is termed empyema.
Etiology
Despite technologic advances in diagnosis, no causative agent is identified in approximately 50% of cases of community-acquired pneumonia. Even in cases in which a microbiologic diagnosis is made, there is usually a delay of several days before the pathogen can be identified and antibiotic susceptibility determined. Symptoms are nonspecific and do not reliably differentiate the various causes of pneumonia. Therefore, knowledge of the most common etiologic organisms is crucial in determining rational empiric antibiotic regimens. Bacterial causes of community pneumonia vary by comorbid disease and severity of pulmonary infection (Table 4-7).
TABLE 4-7 Common etiologic agents of community-acquired pneumonia as determined by severity of illness.
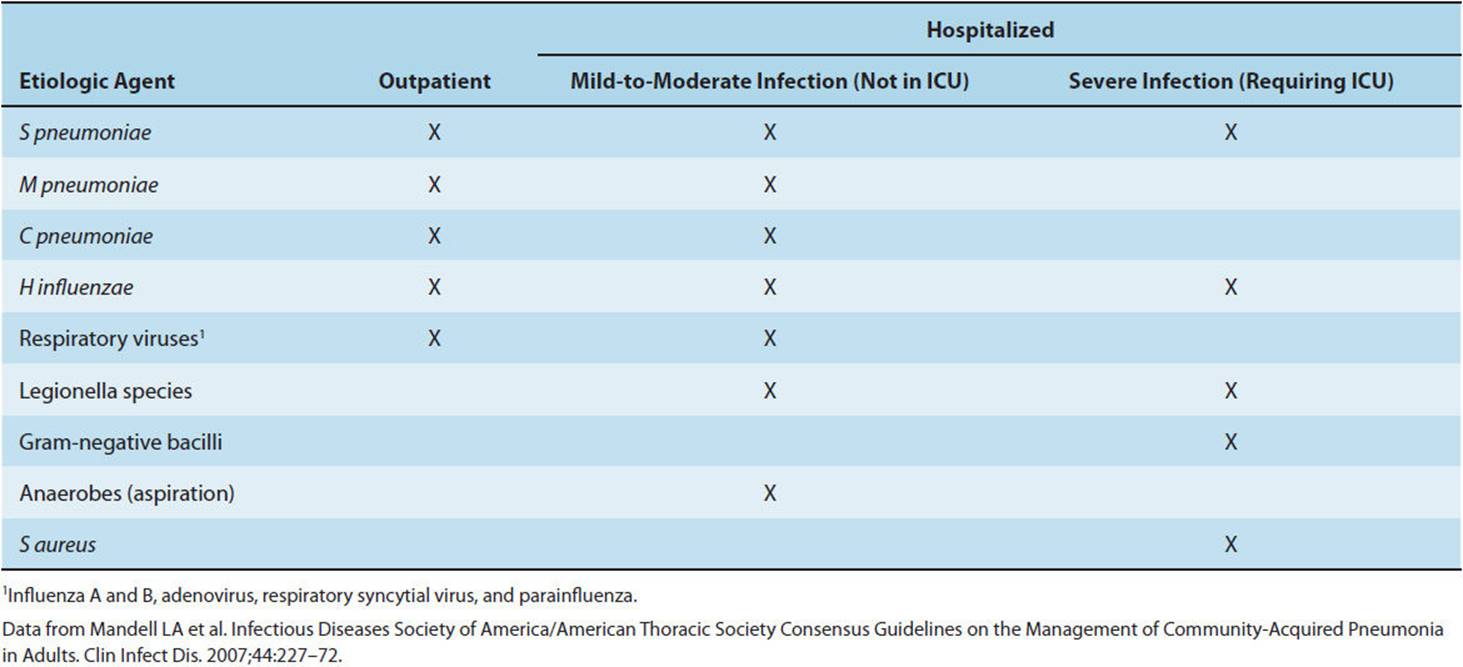
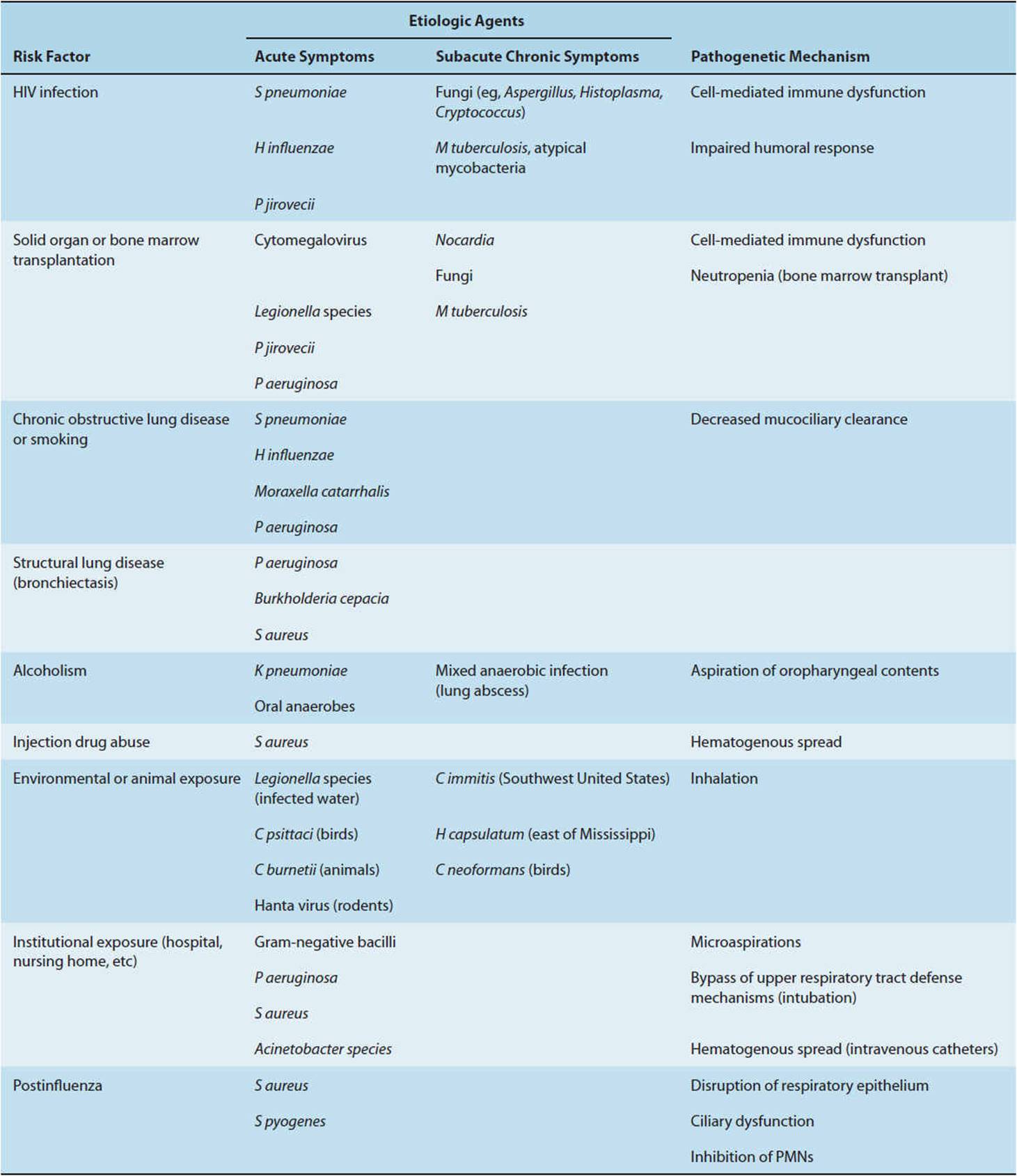
S pneumoniae is the most common organism isolated in community-acquired pneumonia in both immunocompetent and immunocompromised individuals. Several additional organisms require special consideration in specific hosts or because of public health importance (Table 4-8). Understanding and identifying patient risk factors (eg, smoking, HIV infection) and host defense mechanisms (cough reflex, cell-mediated immunity) focuses attention on the most likely etiologic agents, guides empiric therapy, and suggests possible interventions to decrease further risk. For example, patients who have suffered strokes and have impaired ability to protect their airways are at risk for aspirating oropharyngeal secretions. Precautions such as avoiding thin liquids in these patients may decrease the risk of future lung infections. Likewise, an HIV-infected patient with a low CD4 lymphocyte count is at risk for pneumocystis pneumonia and should be given prophylactic antibiotics.
TABLE 4-8 Common risk factors and causes of pneumonia in specific adult hosts.
Pathogenesis
Pneumonia is disproportionally a disease of the elderly and impaired host; it occurs infrequently in immunocompetent individuals. This can be attributed to the effectiveness of host defenses, including anatomic barriers and cleansing mechanisms in the nasopharynx and upper airways and local humoral and cellular factors in the alveoli. Normal lungs are sterile below the first major bronchial divisions.
Pulmonary pathogens reach the lungs by one of four routes: (1) direct inhalation of infectious respiratory droplets into the lower airways, (2) aspiration of oropharyngeal contents, (3) direct spread along the mucosal membrane surface from the upper to the lower respiratory system, and (4) hematogenous spread. The pulmonary antimicrobial defense mechanisms are shown in Figure 4-9. Incoming air with suspended particulate matter is subjected to turbulence in the nasal passages and then to abrupt changes in direction as the airstream is diverted through the pharynx and along the branches of the tracheobronchial tree. Particles larger than 10 mm are trapped in the nose or pharynx; those with diameters of 2–9 mm are deposited on the mucociliary blanket; only smaller particles reach the alveoli. M tuberculosis and Legionella pneumophila are examples of bacteria that are deposited directly in the lower airways through inhalation of small airborne particles. Bacteria trapped in the upper airways can colonize the oropharynx and subsequently be transported into the lungs either by “microaspiration” or by overt aspiration through an open epiglottis (eg, in patients who lose consciousness after excessive alcohol intake).
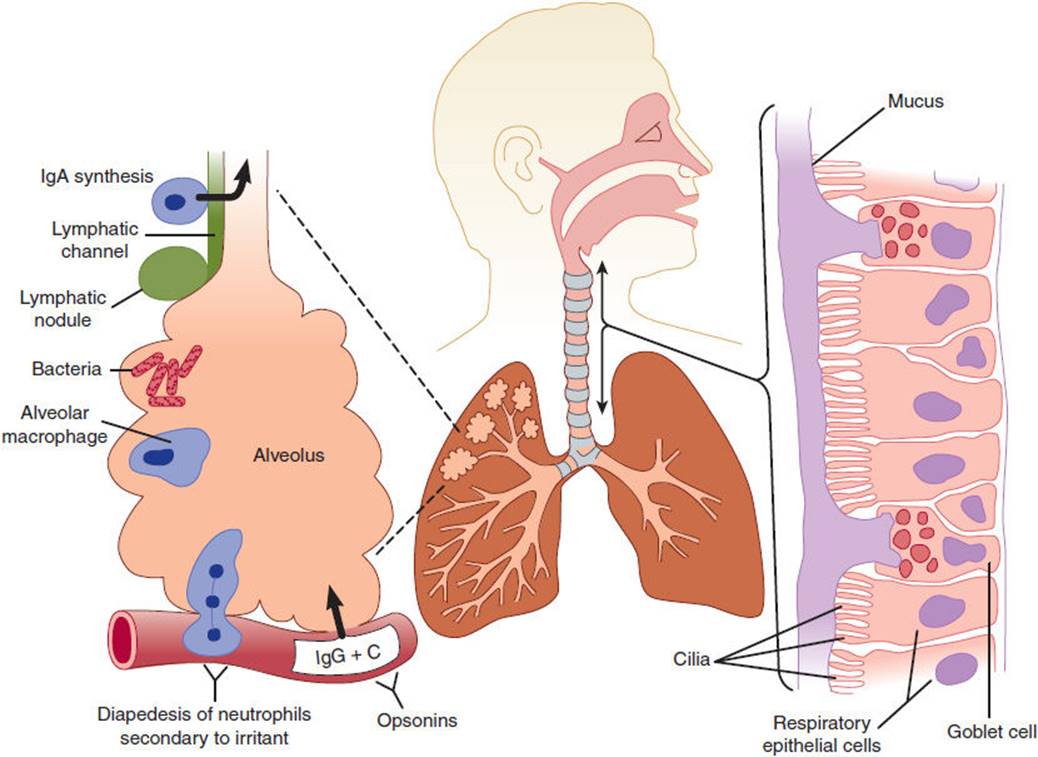
FIGURE 4-9 Pulmonary defense mechanisms. Abrupt changes in direction of airflow in the nasal passages can trap potential pathogens. The epiglottis and cough reflex prevent introduction of particulate matter in the lower airway. The ciliated respiratory epithelium propels the overlying mucous layer (right) upward toward the mouth. In the alveoli, cell-mediated immunity, humoral factors, and the inflammatory response defend against lower respiratory tract infections. (C, complement.) (Redrawn, with permission, from Storch GA. Respiratory system. In: Schaechter M et al, eds. Mechanisms of Microbial Disease, 4th ed. Lippincott Williams & Wilkins, 2007.)
The respiratory epithelium has special properties for fighting off infection. Epithelial cells are covered with beating cilia blanketed by a layer of mucus. Each cell has about 200 cilia that beat up to 500 times/min, moving the mucus layer upward toward the larynx. The mucus itself contains antimicrobial compounds such as lysozyme and secretory IgA antibodies. Chronic cigarette smokers have decreased mucociliary clearance secondary to damage of cilia and therefore compensate through the cough reflex to clear aspirated material, excess secretions, and foreign bodies.
Bacteria that reach the terminal bronchioles, alveolar ducts, and alveoli are inactivated primarily by alveolar macrophages and neutrophils. Opsonization of the microorganism by complement and antibodies enhances phagocytosis by these cells.
Impairment at any level of host defenses increases the risk of developing pneumonia. Children with cystic fibrosis have defective ciliary activity and are prone to develop recurrent sinopulmonary infections, particularly with S aureusand P aeruginosa. Patients with neutropenia, whether acquired or congenital, are also susceptible to lung infections with gram-negative bacteria and fungi. Antigenic stimulation of T cells leads to the production of lymphokines that activate macrophages with enhanced bactericidal activity. HIV-infected patients have depleted CD4 T lymphocyte counts and are predisposed to a variety of bacterial (including mycobacterial) and fungal infections.
Clinical Manifestations
Most patients with pneumonia have fever, cough, tachypnea, tachycardia, and an infiltrate on chest x-ray film. Extrapulmonary manifestations that may provide clues to the etiologic agents include pharyngitis (Chlamydia pneumoniae), erythema nodosum rash (fungal and mycobacterial infections), and diarrhea (Legionella).
The following considerations aid in guiding empiric therapy for a patient who presents with symptoms consistent with pneumonia: (1) Is this pneumonia community acquired or healthcare acquired (eg, hospital, nursing home)? (2) Is this patient immunocompromised (HIV infected, a transplant recipient)? (3) Is this patient an injection drug user? (4) Has this patient had a recent alteration in consciousness (suggestive of aspiration)? (5) Are the symptoms acute (days) or chronic (weeks to months)? (6) Has this patient lived in or traveled through geographic areas associated with specific endemic infections (histoplasmosis, coccidioidomycosis)? (7) Has this patient had recent zoonotic exposures associated with pulmonary infections (psittacosis, Q fever)? (8) Could this patient have a contagious infection of public health importance (tuberculosis)? (9) Could this patient’s pulmonary infection be associated with a common source exposure (Legionella or influenza outbreak)? (10) Does the illness necessitate hospitalization or intensive care admission (eg, pneumonia due to Legionella, S pneumonia, S aureus)?
CHECKPOINT
17. What are the important pathogens for patients with community-acquired pneumonia based on severity of illness and site of care?
18. What host features influence the likelihood of particular causes of pneumonia?
19. What are the four mechanisms by which pathogens reach the lungs?
20. What are the defenses of the respiratory epithelium against infection?
INFECTIOUS DIARRHEA
Clinical Presentation
Each year throughout the world more than 5 million people—most of them children younger than 1 year—die of acute infectious diarrhea (see also Chapter 13). Although death is a rare outcome of infectious diarrhea in the United States, morbidity is substantial. It is estimated that there are more than 200 million episodes each year, resulting in 1.8 million hospitalizations at a cost of $6 billion per year. The morbidity and mortality attributable to diarrhea are largely due to loss of intravascular volume and electrolytes, with resultant cardiovascular failure. For example, adults with cholera can excrete more than 1 L of fluid per hour. Contrast this with the normal volume of fluid lost daily in the stools (150 mL), and it is clear why massive fluid losses associated with infectious diarrhea can lead to dehydration, cardiovascular collapse, and death.
Gastrointestinal (GI) tract infections can present with primarily upper tract symptoms (nausea, vomiting, crampy epigastric pain), small intestine symptoms (profuse watery diarrhea), or large intestine symptoms (tenesmus, fecal urgency, bloody diarrhea). Sources of infection include person-to-person transmission (fecal-oral spread of Shigella), water-borne transmission (Cryptosporidium), food-borne transmission (Salmonella or S aureus food poisoning), and overgrowth after antibiotic administration (Clostridium difficile infection).
Etiology
A wide range of viruses, bacteria, fungi, and protozoa can infect the GI tract. However, in the majority of cases, symptoms are self-limited, and diagnostic evaluation is not performed. Patients presenting to medical attention are biased toward the subset with more severe symptoms (eg, high fevers or hypotension), immunocompromise (eg, HIV or neutropenia), or prolonged duration (eg, chronic diarrhea defined as lasting 14 days). An exception is large outbreaks of food-borne illness, in which epidemiologic investigations may detect patients with milder variants of disease.
Pathogenesis
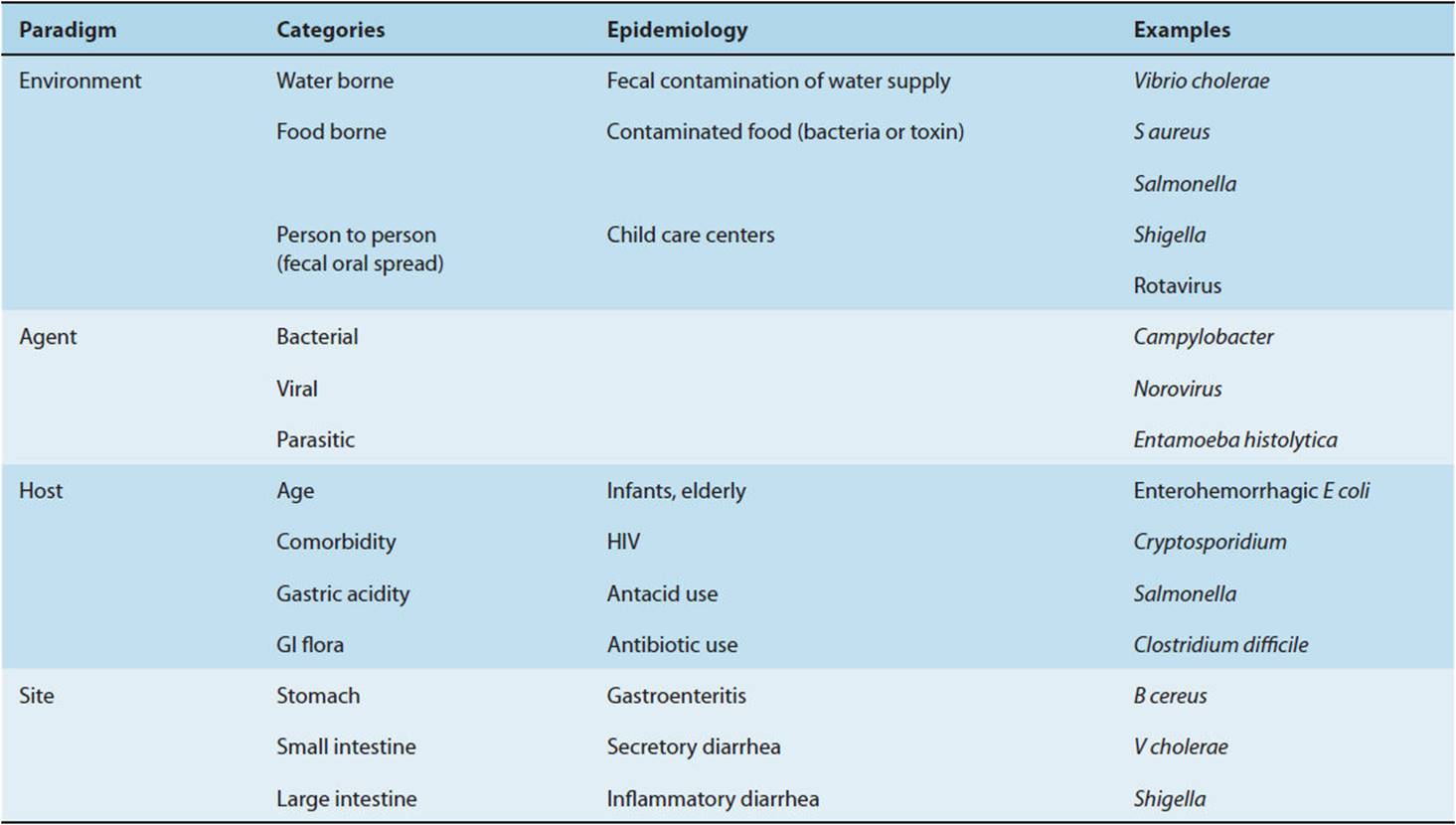
A comprehensive approach to GI tract infections starts with the classic host-agent-environment interaction model. A number of host factors influence GI tract infections. Patients at extremes of age and with comorbid conditions (eg, HIV infection) are at higher risk for symptomatic infection. Medications that alter the GI microenvironment or destroy normal bacterial flora (eg, antacids or antibiotics) also predispose patients to infection. Microbial agents responsible for GI illness can be categorized according to type of organism (bacterial, viral, protozoal), propensity to attach to different anatomic sites (stomach, small bowel, colon), and pathogenesis (enterotoxigenic, cytotoxigenic, enteroinvasive). Environmental factors can be divided into three broad categories based on mode of transmission: (1) water borne, (2) food borne, and (3) person to person. Table 4-9 summarizes these relationships and provides a framework for assessing the pathogenesis of GI tract infections.
TABLE 4-9 Approach to GI tract infections.
GI tract infections can involve the stomach, causing nausea and vomiting, or affect the small and large bowel, with diarrhea as the predominant symptom. The term “gastroenteritis” classically denotes infection of the stomach and proximal small bowel. Causative organisms include Bacillus cereus, S aureus, and a number of viruses (rotavirus, norovirus). B cereus and S aureus produce a preformed neurotoxin that, even in the absence of viable bacteria, is capable of causing disease. Although the exact mechanisms are poorly understood, it is thought that neurotoxins act locally, through stimulation of the sympathetic nervous system with a resultant increase in peristaltic activity, and centrally, through activation of emetic centers in the brain.
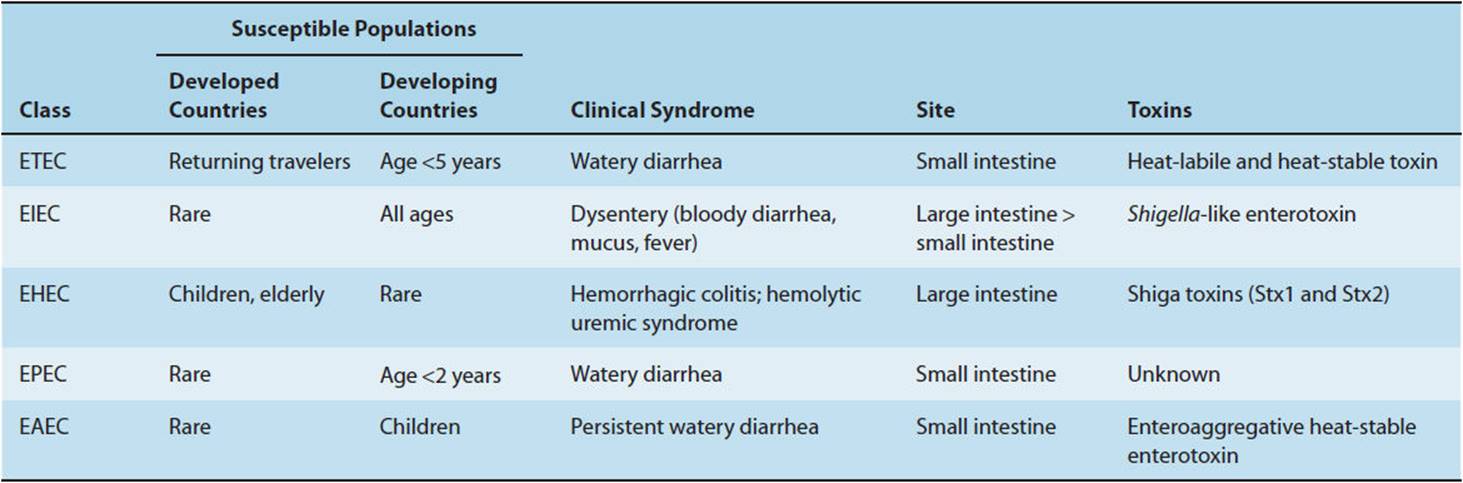
The spectrum of diarrheal infections is typified by the diverse clinical manifestations and mechanisms through which E coli can cause diarrhea. Colonization of the human GI tract by E coli is universal, typically occurring within hours after birth. However, when the host organism is exposed to pathogenic strains of E coli not normally present in the bowel flora, localized GI disease or even systemic illness may occur. There are five major classes of diarrheogenic E coli (and several newer proposed subgroups): enterotoxigenic (ETEC), enteropathogenic (EPEC), enterohemorrhagic (EHEC), enteroaggregative (EAEC), and enteroinvasive (EIEC) (Table 4-10). Features common to all pathogenic E coli are evasion of host defenses, colonization of intestinal mucosa, and multiplication with host cell injury. This organism, like all GI pathogens, must survive transit through the acidic gastric environment and be able to persist in the GI tract despite the mechanical force of peristalsis and competition for scarce nutrients from existing bacterial flora. Adherence can be nonspecific (at any part of the intestinal tract) or, more commonly, specific, with attachment occurring at well-defined anatomic areas.
TABLE 4-10 Escherichia coli in diarrheal disease.
Once colonization and multiplication occur, the stage is set for host injury. Infectious diarrhea is clinically differentiated into secretory, inflammatory, and hemorrhagic types, with different pathophysiologic mechanisms accounting for these diverse presentations. Secretory (watery) diarrhea is caused by a number of bacteria (eg, Vibrio cholerae, ETEC, EAEC), viruses (rotavirus, norovirus), and protozoa (Giardia, Cryptosporidium). These organisms attach superficially to enterocytes in the lumen of the small bowel. Stool examination is notable for the absence of fecal leukocytes, although in rare cases there is occult blood in the stools. Some of these pathogens elaborate enterotoxins, proteins that increase intestinal cyclic adenosine monophosphate (cAMP) production, leading to net fluid secretion. The classic example is cholera. The bacterium V cholerae produces cholera toxin, which causes prolonged activation of epithelial adenylyl cyclase in the small bowel, leading to secretion of massive amounts of fluid and electrolytes into the intestinal lumen (Figure 4-10). Clinically, the patient presents with copious diarrhea (“rice-water stools”), progressing to dehydration and vascular collapse without vigorous volume resuscitation. ETEC, a common cause of acute diarrheal illness in young children and the most common cause of diarrhea in travelers returning to the United States from developing countries, produces two enterotoxins. The heat-labile toxin (LT) activates adenylyl cyclase in a manner analogous to cholera toxin, whereas the heat-stable toxin (ST) activates guanylyl cyclase activity.
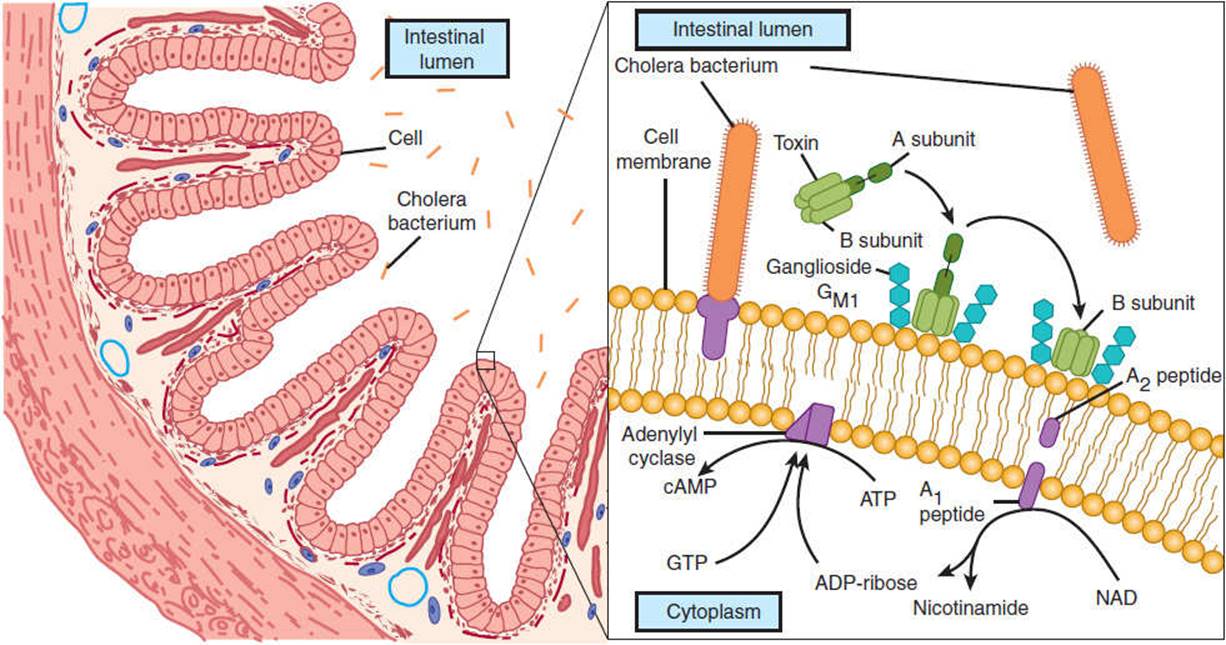
FIGURE 4-10 Pathogenesis of Vibrio cholerae and enterotoxigenic E coli (ETEC) in diarrheal disease. V cholerae and ETEC share similar pathogenetic mechanisms in causing diarrheal illness. The bacteria gain entry to the small intestinal lumen through ingestion of contaminated food (left). They elaborate an enterotoxin that is composed of one A subunit and five B subunits. The B subunits bind to the intestinal cell membrane and facilitate entry of part of the A subunit (right). Subsequently, this results in a prolonged activation of adenylyl cyclase and the formation of cyclic adenosine monophosphate (cAMP), which stimulates water and electrolyte secretion by intestinal endothelial cells. (Redrawn, with permission, from Vaughan M. Cholera and cell regulation. Hosp Pract. 1982;17(6):145–52.)
Inflammatory diarrhea is a result of bacterial invasion of the mucosal lumen, with resultant cell death. Patients with this syndrome are usually febrile, with complaints of crampy lower abdominal pain as well as diarrhea, which may contain visible mucous. The term dysentery is used when there are significant numbers of fecal leukocytes and gross blood. Pathogens associated with inflammatory diarrhea include EIEC, Shigella, Salmonella, Campylobacter, and Entamoeba histolytica. Shigella, the prototypical cause of bacillary dysentery, invades the enterocyte through formation of an endoplasmic vacuole, which is lysed intracellularly. Bacteria then proliferate in the cytoplasm and invade adjacent epithelial cells. Production of a cytotoxin, the Shiga toxin, leads to local cell destruction and death. EIEC resembles Shigella both clinically and with respect to the mechanism of invasion of the enterocyte wall through a similar toxin, termed Shigella-like enterotoxin.
Hemorrhagic diarrhea, a variant of inflammatory diarrhea, is primarily caused by EHEC. Infection with E coli O157:H7 has been associated with a number of deaths from the hemolytic-uremic syndrome, with several well-publicized outbreaks related to contaminated foods. EHEC causes a broad spectrum of clinical disease, with manifestations including (1) asymptomatic infection, (2) watery (nonbloody) diarrhea, (3) hemorrhagic colitis (bloody, noninflammatory diarrhea), and (4) hemolytic-uremic syndrome (an acute illness, primarily of children, characterized by anemia and renal failure). EHEC does not invade enterocytes; however, it does produce two Shiga-like toxins (Stx1 and Stx2) that closely resemble the Shiga toxin in structure and function. After binding of EHEC to the cell surface receptor, the A subunit of the Shiga toxin catalyzes the destructive cleavage of ribosomal RNA and halts protein synthesis, leading to cell death.
Clinical Manifestations
Clinical manifestations of GI tract infections vary depending on the site of involvement (Table 4-9). For instance, in staphylococcal food poisoning, symptoms develop several hours after ingestion of food contaminated with neurotoxin-producing S aureus. The symptoms of staphylococcal food poisoning are profuse vomiting, nausea, and abdominal cramps. Diarrhea is variably present with agents causing gastroenteritis. Profuse watery (noninflammatory, nonbloody) diarrhea is associated with bacteria that have infected the small intestine and elaborated an enterotoxin (eg, Clostridium perfringens, V cholerae). In contrast, colitis-like symptoms (lower abdominal pain, tenesmus, fecal urgency) and an inflammatory or bloody diarrhea occur with bacteria that more commonly infect the large intestine. The incubation period is generally longer (>3 days) for bacteria that localize to the large intestine, and colonic mucosal invasion can occur, causing fever, bacteremia, and systemic symptoms.
CHECKPOINT
21. How many individuals in the world die yearly of infectious diarrhea?
22. What are different modes of spread of infectious diarrhea? Give an example of each.
23. What are the different mechanisms by which infectious organisms cause diarrhea?
SEPSIS & SEPTIC SHOCK
Clinical Presentation
Sepsis is a leading cause of death in the United States, with more than 34,000 deaths occurring annually and an overall case fatality rate approaching 20%. The medical costs of sepsis in the United States exceed $17 billion annually. Rates of sepsis continue to rise secondary to medical advances such as the widespread use of indwelling intravascular catheters, increased implantation of prosthetic material (eg, cardiac valves and artificial joints), and administration of immunosuppressive drugs and chemotherapeutic agents. These interventions serve to increase the risk of infection and subsequent sepsis.
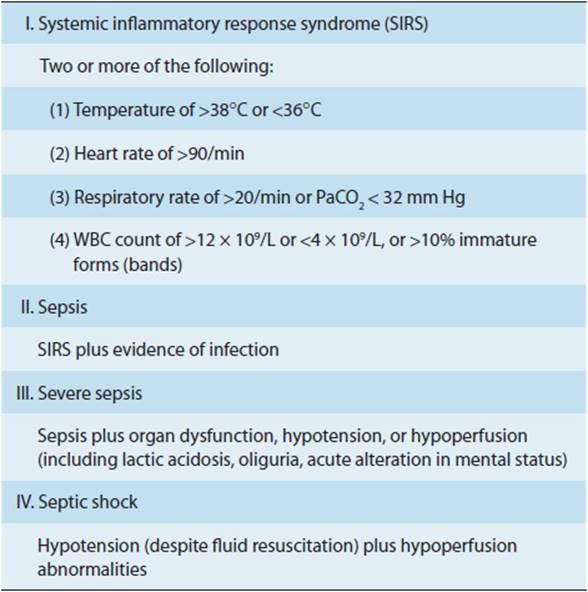
The study of sepsis has been facilitated by establishment of standardized case definitions (Table 4-11). The systemic inflammatory response syndrome (SIRS) is a nonspecific inflammatory state that may be seen with infection as well as with noninfectious states such as pancreatitis, pulmonary embolism, and myocardial infarction. Leukopenia and hypothermia, included in the SIRS case definition, are predictors of a poor prognosis when associated with sepsis. Sepsis is defined as the presence of SIRS associated with an infectious precipitant. Severe sepsis occurs when there is objective evidence of organ dysfunction (eg, renal failure, hepatic failure, altered mentation), usually associated with tissue hypoperfusion. The final stage of sepsis is septic shock, defined as hypotension (systolic blood pressure <90 mm Hg or a 40 mm Hg decrease below the baseline systolic blood pressure) unresponsive to fluid resuscitation.
TABLE 4-11 Clinical definition of sepsis.
Etiology
Although evidence of infection is a diagnostic criterion for sepsis, only 28% of patients with sepsis have bacteremia, and slightly more than 10% will have primary bacteremia, defined as positive blood cultures without an obvious source of bacterial seeding. Common sites of infection among patients with sepsis syndrome (in decreasing order of frequency) include the respiratory tract, the genitourinary tract, abdominal sources (gall bladder, colon), device-related infections, and wound or soft tissue infections.
The bacteriology of sepsis has evolved in the last decade. Gram-negative bacteria (Enterobacteriaceae and Pseudomonas), previously the most common cause of sepsis, have been surplanted by gram-positive organisms, which now cause more than 50% of cases. Staphylococci are the most common bacteria cultured from the bloodstream, presumably because of an increase in the prevalence of chronic indwelling venous access devices and implanted prosthetic material. For similar reasons, the incidence of fungal sepsis due to Candida species has risen dramatically in the last decade. Sepsis associated with P aeruginosa, Candida, or mixed (polymicrobial) organisms is an independent predictor of mortality.
Pathogenesis
The different stages of sepsis (SIRS to septic shock) represent a continuum, with patients often progressing from one stage to the next within days or even hours after admission. Sepsis generally starts with a localized infection. Bacteria may then invade the bloodstream directly (leading to bacteremia and positive blood cultures) or may proliferate locally and release toxins into the bloodstream. These toxins can arise from a structural component of the bacteria (eg, endotoxin) or may be exotoxins, which are proteins synthesized and released by the bacteria. Endotoxin is defined as the lipopolysaccharide (LPS) moiety contained in the outer membrane of gram-negative bacteria. Endotoxin is composed of an outer polysaccharide chain (the O side chain), which varies between species and is not toxic, and a highly conserved lipid portion (lipid A), which is embedded in the outer bacterial membrane. Injection of either purified endotoxin or lipid A is highly toxic in animal models, causing a syndrome analogous to septic shock in the absence of viable bacteria.
Sepsis was initially considered to be a result of overstimulation of the host inflammatory response and uncontrolled release of inflammatory mediators. The failure of a number of pharmacologic interventions aimed at blocking endotoxin or the resultant inflammatory cascade suggests that other factors, such as host immunosuppression, play a critical role. Specific stimuli such as organism, inoculum, and site of infection stimulate CD4 T cells to secrete cytokines with either inflammatory (type 1 helper T-cell) or anti-inflammatory (type 2 helper T-cell) properties (Figure 4-11). Among patients who die of sepsis, there is significant loss of cells essential for the adaptive immune response (B lymphocytes, CD4 T cells, dendritic cells). Genetically programmed cell death, termed apoptosis, is thought to play a key role in the decrease in these cell lines and downregulates the surviving immune cells. The clinical consequences of sepsis include hemodynamic changes (tachycardia, tachypnea), inappropriate vasodilation, and poor tissue perfusion, with resultant organ dysfunction (Figure 4-11).

FIGURE 4-11 Pathogenic sequence of the events in septic shock. Activation of macrophages by endotoxin and other proteins leads to release of inflammatory mediators and immune modulation resulting in host tissue damage and, in some cases, death. (Redrawn, with permission, from Horn DL et al. What are the microbial components implicated in the pathogenesis of sepsis? Clin Infect Dis. 2000;31:852.)
A. Hemodynamic Alterations
All forms of shock result in inadequate tissue perfusion and subsequent cell dysfunction and death (see Chapter 11). In noninfectious forms (such as cardiogenic shock and hypovolemic shock), systemic vascular resistance is elevated as a compensatory mechanism to maintain blood pressure. In the hypoperfused tissues, there is enhanced extraction of oxygen from circulating red blood cells, leading to decreased pulmonary artery oxygenation. In contrast, early in septic shock there is hypovolemia from inappropriate arterial and venous dilation (low systemic vascular resistance) and leakage of plasma into the extravascular space. Even with correction of the hypovolemia, systemic vascular resistance remains low despite a compensatory increase in cardiac output. Inefficient oxygen extraction and tissue hypoperfusion result in an increased pulmonary artery oxygen content.
A hyperdynamic circulatory state, described as distributive shock to emphasize the maldistribution of blood flow to various tissues, is the common hemodynamic finding in sepsis. The release of vasoactive substances (including nitric oxide) results in loss of normal mechanisms of vascular autoregulation, producing imbalances in blood flow with regional shunting and relative hypoperfusion of some organs. Animal studies have documented predictable changes in organ blood flow, with a marked reduction in blood flow to the stomach, duodenum, small bowel, and pancreas; a moderate reduction in blood flow to the myocardium and the skeletal muscles; and relative preservation of perfusion to the kidneys and CNS.
Myocardial depression is a common finding in early septic shock. Initially, patients have low cardiac filling pressures and low cardiac output secondary to volume depletion and vasodilation. After fluid replacement, cardiac output is normal or increased, but ventricular function is abnormal. From 24 to 48 hours after the onset of sepsis, left and right ventricular ejection fractions are reduced, and end-diastolic and end-systolic volumes are increased. This myocardial depression has been attributed to direct toxic effects of nitric oxide, TNF, and IL-1. Reduced ejection fraction and consequent myocardial depression are reversible in patients who survive the initial period of septic shock.
B. Vascular and Multiorgan Dysfunction
Most patients who die of septic shock have either refractory hypotension or multiple-organ failure. Refractory hypotension can occur from two mechanisms. First, some patients cannot sustain high cardiac output in response to the septic state and develop progressive high-output cardiac failure. Second, circulatory failure may be associated with severe vasodilation and hypotension refractory to intravenous fluid resuscitation and vasopressor therapy.
The development of multiple-organ failure represents the terminal phase of a hypermetabolic process that begins during the initial stages of shock. Organ failure results from microvascular injury induced by local and systemic inflammatory responses to infection. Maldistribution of blood flow is accentuated by impaired erythrocyte deformability, with microvascular obstruction. Aggregation of neutrophils and platelets may also reduce blood flow. Demargination of neutrophils from vascular endothelium results in further release of inflammatory mediators and subsequent migration of neutrophils into tissues. Components of the complement system are activated, attracting more neutrophils and releasing locally active substances such as prostaglandins and leukotrienes. The net result of all of these changes is microvascular collapse and, ultimately, organ failure.
The outcome of sepsis depends on the number of organs that fail: The mortality among patients with multiorgan failure (three or more organ systems) averages 70%. Respiratory failure develops in 18% of patients with sepsis. At the most severe end of the spectrum is acute respiratory distress syndrome, characterized by refractory hypoxia, decreased lung compliance, noncardiogenic pulmonary edema, and pulmonary hypertension. Renal failure, seen in 15% of cases, is usually a multifactorial process, with additive injury from intra-renal shunting, renal hypoperfusion, and administration of nephrotoxic agents (antibiotics and radiologic imaging dye). Other organs affected by sepsis include the CNS (altered mentation, coma) and the blood (disseminated intravascular coagulation).
Clinical Manifestations
The clinical manifestations of sepsis include those related to the systemic response to infections (tachycardia, tachypnea, alterations in temperature and leukocyte count) and those related to specific organ system dysfunction (cardiovascular, respiratory, renal, hepatic, and hematologic abnormalities). Sepsis sometimes begins with very subtle clues that can be easily confused with more common and less serious illnesses. Awareness of these early signs of sepsis can lead to early recognition and intervention. Clinical guidelines emphasize the use of a systematic approach to the recognition and early treatment of sepsis. Initial responses should include obtaining cultures of blood and other body fluids, empiric administration of broad-spectrum antibiotics, determination of serum lactate as a marker of hypoperfusion, and use of intravenous fluid and vasopressor therapy for patients with sustained hypotension.
CHECKPOINT
24. What is the mortality rate of sepsis and septic shock in the United States?
25. What factors contribute to hospital-related sepsis?
26. Which organisms are most commonly associated with sepsis?
27. What is the role of the host immune system in the pathogenesis of sepsis?
28. What activates the immune response?
29. What are some distinctive hemodynamic features of septic shock versus noninfectious shock syndromes?
CASE STUDIES
Yeong Kwok, MD
(See Chapter 25, p. 700 for Answers)
CASE 11
A 55-year-old man who recently emigrated from China presents to the emergency department with fever. He states that he has had recurring fevers over the past 3 weeks, associated with chills, night sweats, and malaise. Today he developed new painful lesions on the pads of his fingers, prompting him to come to the emergency department. His medical history is remarkable for “being very sick as a child after a sore throat.” He has recently had several teeth extracted for severe dental caries. He is taking no medications. On physical examination, he has a fever of 38.5°C, blood pressure 120/80 mm Hg, heart rate 108 bpm, respiratory rate 16/min, with an oxygen saturation of 97% on room air. Skin examination is remarkable for painful nodules on the pads of several fingers and toes. He has multiple splinter hemorrhages in the nail beds and painless hemorrhagic macules on the palms of the hands. Ophthalmoscopic examination is remarkable for retinal hemorrhages. Chest examination is clear to auscultation and percussion. Cardiac examination is notable for a grade 3/6 holosystolic murmur heard loudest at the left lower sternal border, with radiation to the axilla. Abdominal and back examinations are unremarkable.
Questions
A. What is the likely diagnosis? What are some common predisposing factors to this disease? Which is most likely in this patient?
B. Which infectious agents are most likely to be involved?
C. What hemodynamic factors predispose to this disease? How do these factors contribute to the establishment of this disease and resistance to normal host immune responses?
D. What is the name given to the various lesions found on this man’s hands and feet? What is the pathogenetic mechanism responsible for their formation?
E. What are some other common clinical manifestations of this disease? What are the most common causes of death in this disease? What factors are predictive of a fatal outcome?
CASE 12
A 25-year-old man presents to the emergency department with fever and in a confused, irrational state. He is accompanied by his wife, who provides the history. She states that he had been well until approximately 1 week ago, when he developed symptoms of upper respiratory tract infection that were slow to improve. On the morning of admission, he complained of progressive severe headache and nausea. He vomited once. He became progressively lethargic as the day progressed, and she brought him to the hospital. He has no other medical problems and takes no medications.
On examination, he is febrile to 39°C, with a blood pressure of 95/60 mm Hg, heart rate of 100 bpm, and respiratory rate of 18/min. He is lethargic and confused, lying with his hand over his eyes. Funduscopic examination shows no papilledema. The neck is stiff, with a positive Brudzinski sign. Heart, lung, and abdominal examinations are unremarkable. Neurologic examination is limited by the patient’s inability to cooperate but appears to be nonfocal. The Kernig sign (resistance to passive extension of the flexed leg with the patient lying supine) is negative.
Questions
A. What infectious diagnosis is suggested? What are the most likely etiologic agents in this patient? What would they be if he were a newborn? If he were a child?
B. What is the pathophysiologic sequence of events in the development of this disease? What features of the pathogens involved facilitate their ability to produce this disease?
C. What are the possible causes of cerebral edema in this patient?
D. What tests should be performed to confirm the diagnosis? What kinds of treatments should be started or considered? Why?
CASE 13
A 68-year-old man presents to the hospital emergency department with acute fever and persistent cough. He has had cough productive of green sputum for 3 days, associated with shortness of breath, left-sided pleuritic chest pain, fever, chills, and night sweats. His medical history is notable for chronic obstructive pulmonary disease (COPD), requiring intermittent steroid use. His medications include albuterol, ipratropium bromide, and corticosteroid inhalers. The patient lives at home and is active. On examination, he is febrile to 38°C, with a blood pressure of 110/50 mm Hg, heart rate of 98 bpm, and respiratory rate of 20/min. Oxygen saturation is 92% on room air. He is a thin man in moderate respiratory distress, speaking in sentences of three or four words. Lung examination is notable for rales in the left lung base and left axilla, and diffuse expiratory wheezes. The remainder of the examination is unremarkable. Chest x-ray film reveals a left lower lobe and lingular infiltrate. A diagnosis of pneumonia is made, and the patient is admitted to the hospital for administration of intravenous antibiotics.
Questions
A. On the basis of this patient’s underlying condition and severity of illness, what are the likely pathogens involved in this case? How would your differential change if he required ICU admission?
B. What are the mechanisms by which pathogens reach the lungs?
C. What are the normal host defenses against pneumonia?
D. What are some common host risk factors for pneumonia? What are the pathogenetic mechanisms by which they increase the risk of pneumonia? Which of these risk factors are present in this patient?
CASE 14
A 21-year-old woman presents with the complaint of diarrhea. She returned from Mexico the day before her visit. The day before that, she had an acute onset of profuse watery diarrhea. She denies blood or mucus in the stools. She has had no associated fever, chills, nausea, or vomiting. She has no other medical problems and is taking no medications. Examination is remarkable for diffuse, mild abdominal tenderness to palpation without guarding or rebound tenderness. Stool is guaiac negative. Infectious diarrhea is suspected.
Questions
A. What are the different modes of spread of infectious diarrhea? Give an example of each.
B. What is the likely anatomic site of infection in this case? Why?
C. What is the most likely pathogen in this case? What is the pathogenetic mechanism by which it causes diarrhea?
CASE 15
A 65-year-old woman is admitted to the hospital with community-acquired pneumonia. She is treated with intravenous antibiotics and is given oxygen by nasal cannula. A Foley catheter is placed in her bladder. On the third hospital day she is switched to oral antibiotics in anticipation of discharge. On the evening of hospital day 3, she develops fever and tachycardia. Blood and urine cultures are ordered. The following morning, she is lethargic and difficult to arouse. Her temperature is 35°C, blood pressure 85/40 mm Hg, heart rate 110 bpm, and respiratory rate 20/min. Oxygen saturation is 94% on room air. Head and neck examinations are unremarkable. Lung examination is unchanged from admission, with rales in the left base. Cardiac examination is notable for a rapid but regular rhythm, without murmurs, gallops, or rubs. Abdominal examination is normal. Extremities are warm. Neurologic examination is nonfocal. The patient is transferred to the ICU for management of presumed sepsis and given intravenous fluids and antibiotics. Blood and urine cultures are positive for gram-negative rods.
Questions
A. What factors contribute to hospital-related sepsis?
B. By what mechanism do gram-negative rods result in sepsis? What role does the immune response play in the pathogenesis of sepsis?
C. Describe the hemodynamic changes that result in septic shock?
D. By what mechanisms does sepsis result in multiorgan failure?
E. What factors predict a poor outcome in patients with sepsis?
REFERENCES
General
Brodsky IE et al. Targeting of immune signalling networks by bacterial pathogens. Nat Cell Biol. 2009 May;11(5):521–6. [PMID: 19404331]
Diacovich L et al. Bacterial manipulation of innate immunity to promote infection. Nat Rev Microbiol. 2010 Feb;8(2):117–28. [PMID: 20075926]
Infective Endocarditis
Chorianopoulos E et al. The role of endothelial cell biology in endocarditis. Cell Tissue Res. 2009 Jan;335(1):153–63. [PMID: 19015889]
Fernández Guerrero ML et al. Endocarditis caused by Staphylococcus aureus: a reappraisal of the epidemiologic, clinical, and pathologic manifestations with analysis of factors determining outcome. Medicine (Baltimore). 2009 Jan;88(1):1–22. [PMID: 19352296]
Que YA et al. Infective endocarditis. Nat Rev Cardiol. 2011 Jun;8(6):322–36. [PMID: 21487430]
Meningitis
Brouwer MC et al. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2010 Sep 8;(9):CD004405. [PMID: 20824838]
Mook-Kanamori BB et al. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. 2011 Jul;24(3):557–91. [PMID: 21734248]
Thigpen MC et al; Emerging Infections Programs Network. Bacterial meningitis in the United States, 1998–2007. N Engl J Med. 2011 May 26;364(21):2016–25. [PMID: 21612470]
Pneumonia
Dockrell DH et al. Pneumococcal pneumonia: mechanisms of infection and resolution. Chest. 2012 Aug;14(2):482–91. [PMID: 22871758]
Mandell LA et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007 Mar 1;44(Suppl 2):S27–72. [PMID: 17278083]
Infectious Diarrhea
Clements A et al. Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes. 2012 Mar–Apr;3(2):71–87. [PMID: 22555463]
Sepsis, Sepsis Syndrome, and Septic Shock
Dellinger RP et al; Surviving Sepsis Campaign Guidelines Committee including The Pediatric Subgroup. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 2013 Feb;39(2):165–228. [PMID: 23361625]
Hotchkiss RS et al. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013 Mar;13(3):260–8. [PMID: 23427891]
Zanotti-Cavazzoni SL et al. Cardiac dysfunction in severe sepsis and septic shock. Curr Opin Crit Care. 2009 Oct;15(5):392–7. [PMID: 19633546]