Goran Augustin1, 2
(1)
Department of Surgery Division of Gastrointestinal Surgery, University Hospital Center Zagreb, Zagreb, Croatia
(2)
School of Medicine University of Zagreb, Zagreb, Croatia
Abstract
Acute pancreatitis (AP) during pregnancy is extremely rare. Schmitt in 1818 [1] reported the first case of pregnancy complicated by AP in a 30-year-old woman, who died in the 4th month of her eighth pregnancy, and Lawrence in 1838 [2] described the earliest series of 53 cases. Longmade and Edmondson [3] added nine cases of their own to an already existing 53 cases. In 1952 four additional cases were reported from France [4]. Acute fatty liver of pregnancy and AP were first recognized as a specific clinical entity by Sheehan in 1940. AP caused by primary hyperparathyroidism (PHPT) in general population was first described by Cope et al. in 1957 [5]. The earliest report of AP occurring in the postpartum period seems to be that of Haidlen in 1884 [6]. Another early cases were reported by Watts [7] and Deaver [8]. Joske collected personal series of six postpartum AP patients [9]. Since then there have been numerous single case reports and case series of various causes of AP during pregnancy.
3.1 History
Acute pancreatitis (AP) during pregnancy is extremely rare. Schmitt in 1818 [1] reported the first case of pregnancy complicated by AP in a 30-year-old woman, who died in the 4th month of her eighth pregnancy, and Lawrence in 1838 [2] described the earliest series of 53 cases. Longmade and Edmondson [3] added nine cases of their own to an already existing 53 cases. In 1952 four additional cases were reported from France [4]. Acute fatty liver of pregnancy and AP were first recognized as a specific clinical entity by Sheehan in 1940. AP caused by primary hyperparathyroidism (PHPT) in general population was first described by Cope et al. in 1957 [5]. The earliest report of AP occurring in the postpartum period seems to be that of Haidlen in 1884 [6]. Another early cases were reported by Watts [7] and Deaver [8]. Joske collected personal series of six postpartum AP patients [9]. Since then there have been numerous single case reports and case series of various causes of AP during pregnancy.
3.2 Incidence
3.2.1 Incidence in General Population
Patients in general population with alcohol-related AP are the youngest (mean age: 39–41.5 years and decreases with age), while those with gallstone AP were the eldest (mean age 64.1 years) [10, 11]. The incidence of AP in general population in England, Denmark, and the United States varies: 4.8–24.2/100,000 [12]. In developing countries, alcohol abuse has been reported to be the second most common factor. In these countries, alcohol abuse is associated with approximately 35 % of the cases [13].
3.2.2 Incidence in Pregnancy
Accurate assessment of disease incidence in pregnancy is difficult since mild disease may be missed.
3.2.2.1 Age
Also more and more women become pregnant in more advanced age, and it is known that the incidence of AP in general population increases with age [14]. Literature shows varying incidence in pregnancy ranging 1/1,000–1/12,000 cases usually late in the third trimester or in early postpartum period and rarely progresses to the necrotizing form [15–21]. The wide variation in the incidence is influenced by the prevalence of its most important etiological factor – gallstone disease. While biliary AP complicated 1/3,300 pregnancies at a large public hospital in Dallas, Texas [15], in Southern California 1/1,500 women were affected [22]. Incidence is race dependent, and Hispanic population has higher incidence (0.1 %) due to higher risk for gallstone disease [23]. Discrepancy in incidence is due to [24]:
· The rarity of disease
· Studies span different decades and countries
· Underestimation due to underreporting
· Small number of cases included in the studies
AP appears to be more prevalent with advanced gestational stage, occurring more commonly in the second or the third trimester [25–27]. Ramin et al. noted that 19 % of AP occurs in the first, 26 % in the second, 53 % in the third (consistent with a potential lithogenic effect of estrogen during pregnancy), and 2 % in the postpartum period, while some reported most of cases, 56 %, in the second trimester [15].
3.2.2.2 Hyperlipidemia/Dyslipidemia
AP secondary to hyperlipidemia/dyslipidemia has an estimated incidence of 1/25,000 births [28], or 10–50 % of all cases [29], which is much higher that the incidence of 4 % in nonpregnant population [30]. The association of hyperlipidemia with AP during pregnancy was first reported in 1818 [1]. By 1970 a total of only 101 reports had been published, the vast majority of which were case studies. Between 1.3 and 7 % of all cases of AP have been attributed to hypertriglyceridemia (HTG) [30, 31], with 1.7–6 % of cases of AP in pregnancy attributed to hyperlipidemia [32, 33]. Although there are a dozen cases of hypertriglyceridemia-induced AP in pregnancy in the literature, it is likely that the reported incidence has been underestimated secondary to lack of testing. Additionally, triglycerides universally increase during an episode of AP. Whether the elevated triglycerides are an epiphenomenon of the AP or the actual cause of the inflammation can be difficult to differentiate.
3.2.2.3 Alcohol
Idiopathic AP in pregnancy is considered in 16.5 and 12.3 % is due to alcohol consumption [24, 34]. Alcoholic AP was more prevalent in the study by Eddy et al. (17.8 % overall; 12.3 % of 89 AP cases vs. ≤7 % in other studies) [25, 35, 36], due to inclusion of chronic pancreatitis and perhaps also to selection of Midwestern states with a high prevalence of alcohol use [37].
In 1951, Lawrence and Edmondson found that there is a tendency for primipara to suffer from AP more often than the multipara. The primipara is even more subject to the disease if she has gallstones [3].
3.2.2.4 Primary Hyperparathyroidism
In general population, PHPT is considered a common disorder. Its greatest frequency is observed in postmenopausal women, in whom it reaches a prevalence of 2–3 % [38]. The incidence in women of childbearing age is approximately 8/100,000 per year [39]. The most frequent cause of PHPT in the general population is a single parathyroid adenoma (85 %), followed by parathyroid hyperplasia (15–20 %) and, albeit very rarely (less than 1 %), by carcinoma [40, 41].
The frequency of AP in pregnancy related to PHPT is higher (7–13 %) than in nonpregnant population (1–2 %) [42–44]. Others found wider incidence of PHPT-induced AP in general population of 1–12 % [45–48]. Jacob et al. have shown a 28-fold increased risk of AP in hyperparathyroid patients compared to the general population [49]. Interestingly, less than 200 of patients with PHPT have been described in gestation and during the postpartum period [42, 44, 50, 51]. The occurrence of PHPT during pregnancy was described by Hunter and Turnbul1 in 1931 [52]. It is estimated that for all women younger than the age of 40 years, eight new cases per 100,000 pregnant women occur annually [53]. This relative paucity of data may be explained by at least three different causes. Firstly, the average age of the initial manifestation of this disorder is higher than that of women of childbearing age [38, 40, 41]. Secondly, about 80 % of nonpregnant individuals with PHPT are characterized by an asymptomatic course of this disease [42]. Finally, some symptoms of PHPT may be misinterpreted as a simple consequence of pregnancy or other gestation-related disorders, while physiological changes during gestation may mask some abnormalities typical to PHPT [50, 54]. Whereas some reports – most of them based on case series – have suggested an association between PHPT and AP, a community-based study showed no increase in the incidence of AP among patients with PHPT as compared with matched controls [55].
The most frequent cause of PHPT in general population is a single parathyroid adenoma (85 %), followed by parathyroid hyperplasia (15–20 %) and, albeit very rarely (less than 1 %), by carcinoma [40, 41]. More than 100 cases reported in the English language literature between 1930 and 1990 were found [39] and less than 200 up to date during pregnancy and postpartum [42, 50, 51]. PHPT is a rare etiology of hypercalcemic-induced AP, causing anywhere from 0.4 to 1.5 % of cases in the general population and up to 13 % of cases during pregnancy [45, 56, 57]. This relative paucity of data may be explained by at least three different causes. Firstly, the average age of the initial manifestation of this disorder is higher than that of women of childbearing age [38, 40, 41]. Secondly, about 80 % of nonpregnant individuals with hyperparathyroidism are characterized by an asymptomatic course of this disease [42]. And finally, some symptoms of PHPT may be misinterpreted as a simple consequence of pregnancy or other gestation-related disorders, while physiological changes during gestation may mask some abnormalities typical to PHPT [50, 54]. The simultaneous occurrence of PHPT and AP in pregnancy has only been reported 13 times up to 1998 [43, 58–66]. There was no single aforementioned patient with presentation during the first trimester, and more than 90 % of patients had thyroid adenoma (89 %) and there was only one thyroid carcinoma.
3.2.2.5 Preeclampsia-Eclampsia
Hojo et al. reviewed a total of 15 cases in the literature of AP thought to be associated with preeclampsia dating from 1956 to 2007 [67].
3.3 Etiopathogenesis
3.3.1 Introduction
The role of pregnancy in the etiology of AP was stressed in the writings of the nineteenth century, for example, Mondière in 1836 [68]. In AP associated with pregnancy, Longmade and Edmondson suggested a time limit of 6 weeks postpartum [3]. In 1955, Ross suggested a rise in intra-abdominal pressure such as undue prolonged second stage of labor as etiology of AP in pregnancy [69]. Elevation of intra-abdominal pressure leading to the high pancreatic ductal pressure [70], and increased tonus of the Oddi’s sphincter [71] may be other possible mechanisms to induce AP.
The list of most of the causes of AP in general population which could also be a cause in pregnant population but with somewhat different incidence is shown in Table 3.1.
Table 3.1
Causes of acute pancreatitis in general and pregnant population
|
Alcohol or methanol abuse (>100 g/day for >3–5 years) |
|
Autoimmune diseases |
|
Choledochal cyst |
|
Cystic fibrosis |
|
Gallstones |
|
Hereditary (familial) pancreatitis (including an autosomal dominant mutation of the cationic trypsinogen gene which causes pancreatitis in 80 % of carriers) |
|
Hyperlipidemia or hypertriglyceridemia (1,000 mg/dl) |
|
Hypercalcemia (including hyperparathyroidism) |
|
Infection (Coxsackie B virus, cytomegalovirus, mumps) |
|
Ischemia from hypotension or atheroembolism |
|
Medications (ACE inhibitors, asparaginase, azathioprine, oral estrogens, antibiotics, 2′, 3′-dideoxyinosine, furosemide, 6-mercaptouride, pentamidine, sulfa drugs, valproate, thiazide diuretic, corticosteroids) |
|
Neoplasm |
|
Pancreatic or periampullary cancer |
|
Pancreas divisum |
|
Peptic ulcers |
|
Preeclampsia-eclampsia |
|
Post- ERCP |
|
Postoperative inflammation |
|
Post-renal transplant |
|
Sphincter of Oddi stenosis |
|
Blunt or penetrating trauma |
|
Surgery (ischemia/perfusion/mechanical) |
|
Tropical pancreatitis |
|
Vasculitis |
|
Viral infections |
|
Idiopathic |
Corlett and Mishell in 1972 assessed 52 patients with AP in pregnancy to classify their etiology into cholelithiasis 23 % (12/52), alcohol abuse 4 % (2/52), and idiopathic 65 % (34/52) [20]. There are too many idiopathic cases in this study. Probably not all investigations were performed in this and some other, especially older studies and this distribution is not precise. More than half or, in some studies, nearly 70 % of cases of AP during pregnancy are secondary to biliary stones or sludge, followed by hyperlipidemia and/or alcohol abuse in approximately 20 % of cases [13, 15, 25, 27, 72]. HTG is an uncommon but well-documented cause of AP, accounting for 1–4 % of cases [30]. Moreover, HTG was listed as causative in 56 % of gestational AP cases in one study [73]. Approximately 15 cases of AP, pregnancy, and hyperlipidemia have been described from 1956 to 1996 [74]. In developed countries other causes are hyperparathyroidism, iatrogenic (diuretics, antibiotics, and antihypertensive drugs), connective tissue diseases, abdominal surgery, infections (viral, bacterial, or parasitic), and blunt abdominal injuries [15, 25, 72]. Necrotizing AP is also reported in preeclampsia due to pancreatic microvascular alterations [75]. Post-ERCP is the cause in 3.45 % [27]. Today, it is still not clear, whether the pathogenesis of AP is one entity or whether it consists of a group of distinct pathogenetic mechanisms [76]. Idiopathic AP is the cause in 10 % of cases [27]. There is one case published with AP during pregnancy caused by mucinous cystic pancreatic neoplasm. Pancreatic cystic lesions in general are rare but are difficult to treat given problems in clarifying their malignancy. Mucinous cystic neoplasms are considered premalignant lesions and resection is recommended. Receptors for estrogen and progesterone receptors in these cysts may cause cystic growth during pregnancy [77].
During pregnancy, gallstones and sludge induce most of the cases of AP, causing duct obstruction with pancreatic hyperstimulation that increases pancreatic duct pressure, trypsin reflux, and activation of trypsin in the pancreatic acinar cells. This leads to enzyme activation within the pancreas and causes autodigestion of the gland, followed by local inflammation (Fig. 3.1).
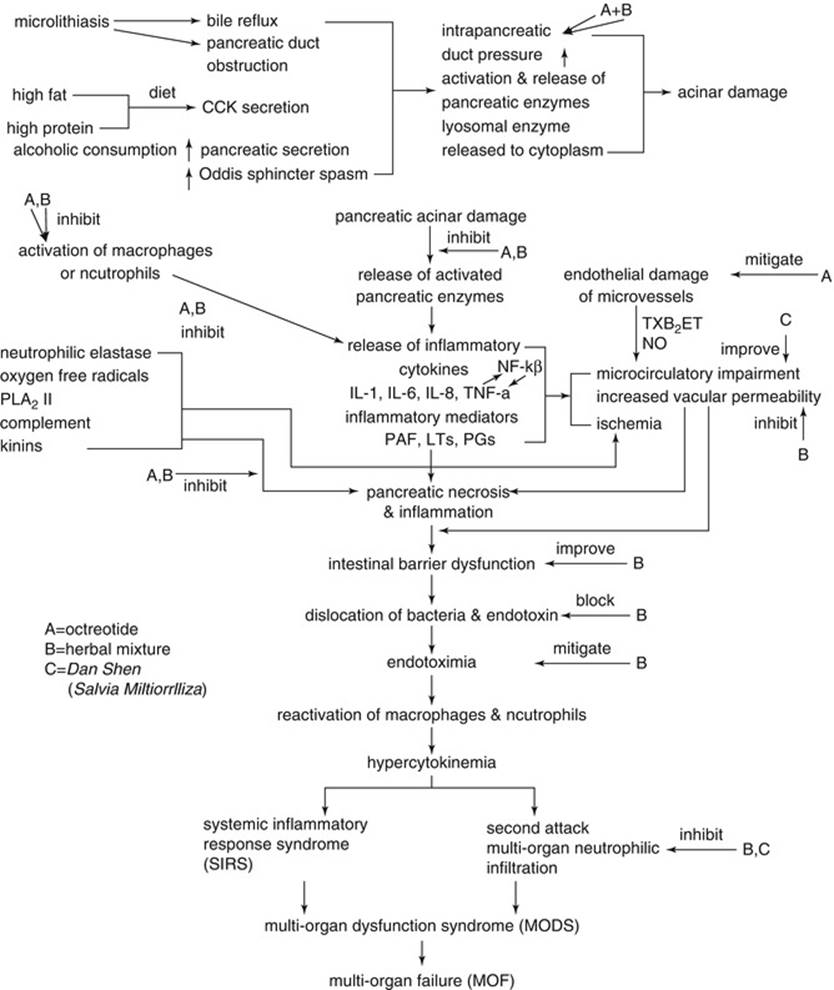
Fig. 3.1
Schematic representation of step-by-step pathogenesis of (biliary) pancreatitis with possible action of medications
Pregnancy does not primarily predispose the pregnant woman to AP, but it does increase the risk of cholelithiasis and biliary sludge formation (see Chap. 2) [15]. Gallstone AP occurs in relatively older age (28.2 years) as compared to non-gallstone AP (24.4 years). In both groups, pregnant women were usually multiparous and AP mostly presented in the third trimester [27].
The trigger events or precipitating factors for AP in 60 % of pregnant women were associated with excess high-fat/high-protein diet. The favored explanation for it may be that large amounts of bile/trypsin release can overwhelm the defense mechanism and activate other enzymes, resulting in local and systemic complications that are commonly seen in the course of the disease [78]. Severe diabetic ketoacidosis and hyperglycemia with associated dehydration are known risk factors of AP in general population [79]. Additional pathophysiologic phenomenon in pregnancy is gestational diabetes mellitus (DM). It could be a trigger for AP in pregnancy. It should be noted that the percentage of idiopathic AP is declining as knowledge of genetic etiologies and predispositions to AP accumulates.
3.3.2 Primary Hyperparathyroidism
3.3.2.1 Calcium-PTH Metabolism in Pregnancy
Pregnancy and lactation are characterized by important alterations in calcium homeostasis, being a consequence of pregnancy-induced changes in the synthesis, metabolism, and excretion of calcium and calcitropic hormones [50, 80]. There is an interesting alteration of the calcium-PTH dynamics during pregnancy. Early reports described a physiological hyperparathyroidism of pregnancy with an increase in serum immunoreactive PTH levels beginning in the second trimester [81]. However, the development of more accurate and specific immunoradiometric (IRMA) methods has discredited this notion by showing a reduction, rather than an elevation, of intact PTH levels [82]. In fact, one study found mean serum PTH levels in nonpregnant women to be 72 % higher than in pregnant women [83]. These changes in PTH during pregnancy may be a response to altered calcium metabolism. Intravascular fluid expansion and hypoalbuminemia (albumin falls by 20 %) make less protein available to bind calcium, thus lowering total (maternal serum calcium falls by about 10 %) but not ionized calcium [83, 84]. In addition, an increase in urinary excretion of calcium occurs due to increased glomerular filtration rate, and maternal calcium in the blood is actively transported across the placenta to fulfill the needs of the growing fetus [85]. Hypercalcemia is defined as a (corrected) total serum calcium above the standard laboratory reference range (2.2–2.6 mmol/l). In pregnancy, the reference range is marginally lower depending on the trimester (Table 3.2).
Table 3.2
Corrected serum calcium in pregnancy
|
Normal |
1st trimester |
2nd trimester |
3rd trimester |
|
2.20–2.60 mmol/l |
2.25–2.57 mmol/l |
2.30–2.50 mmol/l |
2.30–2.59 mmol/l |
The latter imposes an increased calcium requirement that is partly fulfilled by mobilization of calcium from the maternal skeleton. Together, these effects have a tendency to lower maternal calcium levels [86]. During pregnancy the placenta actively transports calcium ions to the fetus but does not allow transfer of parathormone [87]. Maternal hyperparathyroidism can therefore result in fetal hypercalcemia which substantially increases the risk of spontaneous abortion [88]. However, they are more than offset by a large increase in intestinal calcium absorption that occurs during pregnancy [89, 90]. Hormonal changes during pregnancy may also be responsible for an increase in the production or activity of the enzyme 1-a-hydroxylase in the kidney [91], which in turn may account for the observed differences between pregnant and nonpregnant women, including the elevation of 1,25-dihydroxyvitamin D [82], the slight increase in serum calcium levels [92], and the reduction in PTH levels [83]. Fetal 1,25-dihydroxyvitamin D, synthesized in fetal kidney and placenta, acts as the major stimulus and regulator of calcium transfer across the placenta. It increases maternal gastrointestinal absorption of calcium by 150–400 mg daily; additionally, maternal urinary excretion is also increased from 90 to 300 mg daily. Major fetal calcium demands of approximately 25–30 g are required in the third trimester for skeletal tissue mineralization. This requires an active transport of calcium across the placenta, and the fetal serum calcium remains higher than maternal blood. Conversely, after delivery, when the maternal transplacental supply of calcium ceases, neonatal hypocalcemia becomes the major problem. This may occur because the neonate is unable to mobilize calcium stores adequately as a result of prolonged parathyroid gland suppression. What is worth mentioning is that compared to the remaining subjects, pregnant ones with PHPT often experience a clinically overt course of this disease [42, 51].
3.3.2.2 Calcium-Induced Acute Pancreatitis
After eliminating all other causes, mean plasma calcium level seems to be the only predictive factor for AP development [46, 49, 93]. Its dosage must be included in the etiological work-up, although PHPT is found in <1 % of patients who present with AP [56]. Felderbauer et al. have stressed that genetic mutations constitute a greater risk factor for AP than serum calcium [48]. The pathophysiologic mechanism that leads to AP seems more related to hypercalcemia than to PHPT. It has been shown that hypercalcemia from any cause can lead to AP [94–96]. As confirmed by experimental studies, calcium ions cause calculus deposition within the pancreatic ductules, with consequent obstruction and inflammation [97]. Moreover, calcium can trigger the AP cascade by promoting conversion of trypsinogen to trypsin [98, 99].
Interrelation between AP and parathyroid function can be summarized as follows: (1) AP results in a tendency to hypocalcemia and secondary hyperparathyroidism [100, 101]. Compensation need is correlated to AP severity as shown by PTH level [102]; (2) severe and/or complicated AP can lead to overt hypocalcemia through relative deficiency in PTH secretion [101], because exogenous administration of PTH normalizes calcium level [103]; (3) in severe AP, resistance to PTH action in bones and kidneys may occur because of fluid sequestration and reduction in efficient arterial blood volume [100]; and (4) once the diagnosis of PHPT-induced AP is established, parathyroidectomy is mandatory because it prevents recurrence [46, 56].
The initiation and growth of kidney calculi may be attributed to overlapping of both increased calcium load, secondary to enhanced PTH synthesis and release, and a pregnancy-induced increase in urine calcium excretion (a typical symptom of physiological pregnancy). For these reasons clinical manifestations of parathyroid disorders in pregnancy are often different from those observed in nonpregnant women. As the symptoms experienced by patients with parathyroid disorders are not specific, their diagnosis during gestation and breastfeeding may be sometimes very difficult [50, 80]. In opposition to the general population, four of every five PHPT pregnant patients experience clinical manifestations of this disorder. The most frequent of them is the presence of nephrolithiasis [42, 104].
The fact that AP is present in three of four pregnancies complicated by PHPT, while never occurred before and between pregnancies, supports these statistical data that gestation makes hyperparathyroid patients particularly prone to the development of this complication. It is assumed that AP occurs more frequently in primiparous than in women who underwent multiple pregnancies and occurs mainly in the first and third trimester of gestation [51, 105]. PHPT can result in calcifications occurring in the pancreatic ducts thereby blocking secretions, which then damage the pancreatic tissues and result in AP [58]. Usually, these cases present with higher calcium levels compared with cases of PHPT without AP [58]. Hyperparathyroidism can result in calcifications occurring in the pancreatic ducts thereby blocking secretions, which then damage the pancreatic tissues and result in AP [58]. Although the absolute calcium level is an important predictor, it may attenuate during the pregnant state, and individual predisposing factors may be important in the manifestation of pancreatic inflammation [66].
3.3.3 Acute Fatty Liver of Pregnancy
Acute fatty liver of pregnancy (AFLP) occurs in 1/1,000–1/13,000 [106–109] pregnancies, and it usually complicates gestation as part of pregnancy-induced hypertension or HELLP syndrome [108]. In 39 % of the cases, it appears secondary to a urinary or respiratory infection [110, 111]. In all cases, multiple organs are involved.
The pathophysiology of the disease is obscure. The deficiency of long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) in the fetus may be implicated in the pathogenesis of AFLP [112, 113]. A fetus with this enzyme deficiency accumulates long-chain fatty acids that have not undergone oxidation. These fatty acids enter the mother’s serum and are hepatotoxic. Furthermore, the placenta itself may produce excess fatty acids and may further elevate the level of maternal free fatty acids. Mothers who are heterozygous for LCHAD deficiency also have a greater risk of developing AFLP [112]. Studies suggest that AFLP is caused by a mitochondrial defect. The long-chain 3-hydroxyacyl-CoA-dehydrogenase deficiency in the mitochondria determines long- and medium-chain fatty acid accumulation into the cell. This defective enzyme is determined by a gene mutation (E47Q) [114] with an incidence of 1:150–1:200 in population. Alternatively, pregnancy may itself affect mitochondrial function. Other hypotheses favor above-normal (for pregnancy) level of estrogens potentiating the effects of an otherwise tolerable hormonal insult to the mitochondria in the third trimester. Clinically, the onset is between the 30th and 38th weeks of gestation. Complications cited in the literature are hepatic encephalopathy (13 %), hypoglycemia (55 %), renal failure (50 %), coagulopathy (96 %), disseminated intravascular coagulopathy (55 %), and preeclampsia (50 %) [115].
The association of AFLP and AP in pregnancy is very rare, and in the last 15 years, only a few cases have been reported in the literature [116, 117] and currently only one published case of chronic pancreatitis after AFLP during pregnancy in a patient with gestational DM [118]. It was likely due to the serious insult to the pancreas during AFLP, despite normal abdominal CT with i.v. contrast on postpartum day 5 (serum amylase and lipase levels 3× elevated and serum calcium level below lower border) after Cesarean section due to placenta previa with fetal distress when yellow amniotic fluid was found.
3.3.4 Hyperlipidemia/Hypertriglyceridemia
3.3.4.1 Hereditary Causes
The total cholesterol and triglycerides often increase during the second and third trimester of pregnancy with triglyceride levels approximately doubled during the third trimester of pregnancy [119] due to the combination of reduced LPL activity and increased hepatic lipase activity affecting both triglyceride synthesis in the liver and catabolism of triglyceride-rich particles. The elevation in triglyceride levels is even greater than that of cholesterol, and triglyceride levels usually increase two- to three-fold during the third trimester of pregnancy (Fig. 3.2) [120, 121].
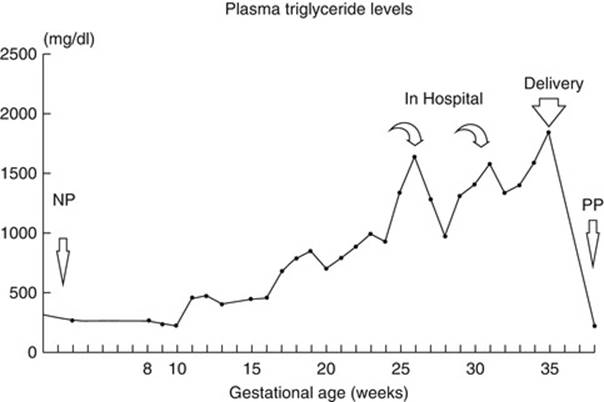
Fig. 3.2
Triglyceride levels at different gestational ages. Triglyceride levels were measured weekly beginning at 8 weeks of gestation of the patient’s second pregnancy. Two separate lipid measurements during the nonpregnant (NP) state and one measurement 6 months postpartum (PP) are also included [120]
Combination of a human placental lactogen-related increase in adipose tissue lipolysis and hepatic synthesis of very low-density lipoprotein results in increased production of triglyceride-rich lipoproteins [122]. These changes are thought to be adaptive for fetal-maternal requirements as triglycerides are thought to provide maternal fuel, sparing the glucose for the fetus. LPL is a key enzyme for the hydrolysis of TG from chylomicrons and VLDL particles of blood plasma [123]. They are likely to be mediated by the complex hormonal changes evolving during the second and third trimester of pregnancy. During pregnancy, there is a physiological increase in plasma levels of cholesterol and triglycerides; however, this increase is not sufficient to cause AP. These will start to rise, beginning in the third month and peak during the third trimester. Plasma triglyceride (TG) levels normally increase, often as much as threefold [124], but this is usually of little clinical consequence. The physiological increase in serum triglycerides in pregnancy will rarely exceed 300 mg/dl (3.3 mmol/l), a concentration that alone is not sufficient to cause AP. Preexisting genetic abnormalities in the lipid metabolism may be exacerbated during pregnancy and can cause gestational hyperlipidemic AP. Plasma lipid and lipoprotein concentrations are determined by complex interactions between genetic and environmental factors. This is well illustrated during pregnancy where fasting plasma triglyceride levels increase two- to fourfold by the third trimester, occurring predominantly through increased liver synthesis of triglyceride and very low-density lipoproteins (VLDL) in response to elevated estrogen levels [125]. This so-called physiological hyperlipidemia of pregnancy is thought to represent a generalized increase in substrate mobilization, both for the placenta and for the growing fetus [126, 127]. Fat depots increased during early pregnancy and later break down causing an accumulation of TGs [124]. The presence of lipoprotein receptors in the placenta together with LPL and intracellular lipase activities allows the release of fatty acids to the fetus [124].
Additionally, the clearance of VLDL and chylomicrons decreases due to a reduction of lipoprotein lipase (LPL) at the capillary endothelium [128, 129]. This appears to be due to a decrease in LPL synthesis in adipose tissue and possibly skeletal muscle, through the downregulation of LPL gene expression by estrogen [128, 130]. The direct correlation between estrogen levels and HTG has also been observed in premenopausal women using estrogen-containing oral contraceptives [131, 132]. A similar increase in triglyceride levels has been seen in estrogen-fed rats [133] and in estrogen-fed chicks [134].
The precise mechanisms for the pathogenesis of HTG-induced AP are not established. Cameron et al. found that 38 % of pregnant women with AP had hypertriglyceridemia compared with 9 % of pregnant women without AP [135]. Havel has suggested that hydrolysis of excessive triglyceride-rich lipoproteins by high levels of pancreatic lipase releases very high concentrations of free fatty acids (FFAs) which exceed the binding capacity of plasma albumin, thus resulting in self-aggregating FFA micellar structures with detergent properties [136]. The FFA micelles injure the vascular endothelium and acinar cells of the pancreas, producing a self-perpetuating ischemic/acidic environment with resultant toxicity. Another theory favors ischemia secondary to plasma hyperviscosity due to severe chylomicronemia [137, 138]. However, these two theories are not mutually exclusive.
The development of marked HTG, specifically in pregnancy, in the absence of factors such as DM, drug, or alcohol intake, raises the possibility of partial defects in triglyceride metabolism. While several genetic factors have been implicated, the best validated has been LPL where an impairment in function has been ascribed to the presence of mutations in the LPL gene. LPL is one of two intravascular lipases whose primary roles are the clearance of the triglyceride and phospholipid components of circulating lipoproteins. This enzyme can be assayed in plasma after release from the capillary endothelium by an intravenous bolus of heparin. Mutations in the LPL gene underlying severe hyperlipidemia in pregnancy were first reported in 1994 [139], and thereafter, two additional studies have examined LPL function in women presenting with severe HTG during pregnancy [122, 140]. Of the eight subjects studied in these combined reports, only three were known to be hyperlipidemic prior to their pregnancies. Postheparin plasma LPL activity was assayed in most of these patients in the nonpregnant state and was found to vary from 12 to 100 % of normal. DNA analysis revealed six different LPL gene mutations in this patient cohort. Two subjects, however, yielded normal coding sequence at the LPL gene. In vitro assessment of the catalytic activity of the six mutant lipases revealed three to be partially active (<50 % normal) while the remaining three manifested zero activity. While pregnancy is also recognized as a secondary factor, little data are available on the magnitude of the induced HTG in carriers of LPL gene mutations, where LPL levels most often approximate 50 % of normal. The paucity of reports of severe HTG or HTG-induced AP in founder communities with high carrier frequencies (1/169) such as the French Canadians [141] indicates, however, that pathologic levels rarely ensue and suggests that 50 % of normal activity may be sufficient to prevent hyperlipidemic crises.
Interestingly, Syed et al. stated that it is HTG itself that causes AP and not hypercholesterolemia [142]. The mechanism by which HTG causes AP is not completely understood. The mechanism probably involves the release of large amounts of toxic unbound free fatty acids by pancreatic lipase that damages the endothelium in the capillaries of the pancreas. This results in sludging of red blood cells, stasis, pancreatic ischemic injury, and eventually inflammation. In addition, physical damage by cholesterol crystals might cause microvascular endothelial cell disruption [136, 143]. There are several studies that show inflammatory effects of triglyceride-rich lipoproteins. Ting et al. increased expression of leukocyte adhesion molecules and monocyte adherence in response to the inflammatory cytokine tumor necrosis factor-α (TNF-α) by treating the endothelial cells with triglyceride-rich lipoproteins [144, 145]. Dandona et al. also observed that FFA in the plasma positively correlates with NF-κ and ROS generation [146]. Locally produced fatty acids might also alter endothelial reactivity by inhibiting the actions of eNOS [147].
It is possible that estrogen, aside from producing an alteration in plasma triglyceride concentrations, has toxic effects within the pancreas itself. Pancreatic acinar cells have significant amounts of an estradiol-binding protein [148]. Estrogen increases LDL receptors [149, 150] in some situations and conceivably could promote lipid uptake into acinar cells. Sufficient excess lipid uptake leads to lipotoxicity and cellular apoptosis, a process that is best characterized in muscle cells [151]. Direct effects of estrogens on pancreatic function are supported by the observation that pancreatic amylase release in the rat is stimulated by estrogen [152]. On the other hand, it could be seen that an elevation in estrogen may increase synthesis of triglyceride [153] and depresses plasma postheparin lipolytic activities (PHLA) lowering the removal efficiency of triglyceride during pregnancy. Actually the production rates of triglyceride and total cholesterol at the end of pregnancy are markedly increased to 140 and 50 %, respectively, as compared to that of nonpregnant period [154]. Additionally, triglycerides universally increase during an episode of AP. Whether the elevated triglycerides are an epiphenomenon of the AP or the actual cause of the inflammation can be difficult to differentiate. Mild-to-moderate elevations of triglyceride are seen in up to 50 % of all-cause AP and are generally regarded an epiphenomenon rather than a cause. However, the increase caused by AP alone is transient, peaking at 72 h and declining to near-normal values in 2 weeks [155]. Generally, reported cases of hypertriglyceridemia-induced AP have included levels in excess of 1,000 mg/dl. A more recent report on AP induced by hypertriglyceridemia actually indicates that the majority of cases of AP occurred only when levels exceeded 3,000 mg/dl [156].
Patients presenting with severe HTG should be evaluated for a genetic disorder in lipid metabolism. According to Fredrickson classification of dyslipidemias, types I, IV, and V are associated with severe HTG and predispose for AP [157]. Types I and V can present with AP without an exacerbating factor, whereas type IV usually requires secondary precipitating factors (e.g., poorly controlled DM, alcohol use, estrogen and pregnancy, or medications that can increase or raise TG levels) [158]. Type I hyperlipidemia (also known as familial chylomicronemia) often presents in infancy and is caused by an autosomal recessive trait resulting in lipoprotein lipase or apo C-II deficiency. Type IV, known as familial HTG or familial combined hyperlipidemia, is autosomal dominant and presents in adulthood.
A derangement in lipoprotein lipase (LPL) has been found in association with pregnancy-induced HTG and AP. More than 30 mutations have been identified in the lipoprotein lipase gene in women with gestational AP [139, 140, 159]. More than 60 mutations of the human LPL gene have been described since the complementary DNA sequence of normal human LPL was determined by Wion in 1987 [160]. Mutations of the LPL gene (Fig. 3.1) may result in partial or more rarely complete LPL deficiency. Complete deficiency usually but not invariably presents in childhood with hyperchylomicronemia, eruptive xanthomatosis, and recurrent AP [161]. Homozygosity for a missense Ser172 → cys mutation could be responsible for a 10-fold increase in plasma triglyceride levels [120]. The report of twin sisters shows a possible cause of the AP in pregnancy – a disorder of triglyceride clearance related to defective lipoprotein lipase. LPL is a key lipolytic enzyme that may be down- and upregulated by various influences, including insulin, estrogen, and medications [128, 162]. Both patients showed a defect at amino acid residue 188 of LPL and are heterozygous for the mutation. Patient 1 had a more classic variety during the last trimester and subsequently responded well to fat restriction and conservative management. Patient 2 had a more severe and protracted course, possibly because of the use of cholestyramine resin, a drug that may have elevated rather than decreased her triglyceride levels [163, 164]. Patient 2 had multiple medical and surgical complications related to the severity of her AP. Patient 1 had mild glucose intolerance that may have exacerbated the triglyceride level. Years before developing pregnancy-induced AP, patient 1 may have had hyperlipidemia and AP as a result of the use of birth control pills. Mutation at codon 188 appears throughout the world, perhaps most often in the French Canadian population. In Quebec, the carrier rate has been estimated to be as high as 1/169, with a total of 19,600 persons affected [141, 165]. The assumption is that the rarity of the clinical syndrome of hyperlipidemia and AP in pregnancy in patients with this specific genetic defect may be due to the presence of an undetected additional mutation in affected patients [140].
There is only one case report of the LPL W86R mutation causing serious exacerbation of the hyperlipidemia symptoms during pregnancy, and the first case in which the level of residual activity in a homozygous W86R mutation could be clearly documented. It is a case of a 25-year-old patient with several attacks of AP during the third trimester with the last episode of necrotizing AP. She was operated several times, and first, immediate surgery revealed diffuse peritonitis with 3 L of purulent fluid. The dead fetus was removed with the uterus, the pancreas then resected. Following this, a number of reoperations were performed due to omental abscesses and progressive intra-abdominal sepsis. Gallbladder was also removed and the abdominal wound treated in a semi-open way. She was discharged after 32 days [166].
Previous studies showed that approximately one-third of the women developing HTG-induced AP during pregnancy were nulliparous [15, 167].
3.3.4.2 Acquired Causes
Hypertriglyceridemia
HTG-induced AP occurs often in untreated or uncontrolled DM [30, 168]. In type I DM, the absence of insulin reduces the ability of LPL to reduce TG into FFA, resulting in elevated TG levels [169]. In type II DM, insulin resistance leads to enhanced production and reduced clearance of TG [170]. Diabetic ketoacidosis may pose a separate risk for HTG as evidenced by a prospective study of 100 patients presenting with diabetic ketoacidosis. Eleven percent of patients had AP, and in four cases, HTG was the only attributable cause [171]. The role of alcohol in HTG is unclear but may be attributed to alcohol competing with FFAs for oxidation, leaving more FFAs available for TG synthesis [158]. Some authors suspect that alcohol alone does not cause HTG, but more likely exacerbates an underlying genetic hyperlipidemia [168]. In addition, hypothyroidism has been documented as a cause of HTG [31, 172]. In a case report, a craniopharyngioma was implicated as it caused central hypothyroidism, leading to HTG (3,300 mg/dl) and eventual AP [31]. Medications such as estrogen [173] are known to raise the serum TG level. Accordingly, hormone therapy in women is not recommended when TG is >500 mg/dl due to heightened risk of HTG-induced AP [173].
Exogenous estrogens elevate triglycerides by increasing the production of triglyceride-carrying very low-density lipoproteins (VLDLs) by the liver and reducing the levels of LPL and hepatic lipase, thus reducing triglyceride clearance while also elevating triglycerides by augmentation of insulin resistance [174, 175]. As early as 1972, clinicians were alerted to cases of marked hyperlipidemia and AP associated with the use of birth control pills, although no genetic testing was available at that time [176]. Tamoxifen is known to cause a small, but significant decrease in high-density lipoprotein (HDL) cholesterol, unlike estrogen, which elevates HDL. In women with HTG, tamoxifen’s ability to increase the triglyceride level is especially pronounced, to an extent that may induce AP [174, 177, 178]. Clomiphene citrate is a synthetic estrogen analog with a biochemical structure similar to that of tamoxifen. Clomiphene has mixed agonistic but mainly antagonistic properties [179]. The effects of clomiphene on lipid metabolism have not been as well documented as the effects of tamoxifen because it is not used continuously and not as commonly as tamoxifen. Clomiphene elevates the triglyceride level mainly in women with a predisposed risk for HTG, due to mutations in enzymes such as LPL [143, 180].
Apoprotein E (apo E), particularly apo E allele 2, has been found in association with higher triglyceride levels in pregnant patients with chylomicronemia [139]. Authors did not find apo E alleles 2 or 4 in their twin patients; both had apo E genotype 3/3. Therefore, apo E was not a primary cause of the HTG in these patients [140]. Although it is known that pregnancy results in an increase in plasma triglyceride levels [119], how this increase occurs is not totally understood. In addition, certain persons express profound elevations in triglyceride values during pregnancy [74, 181].
There are many causes of secondary hypertriglyceridemia (Table 3.3):
Table 3.3
Acquired/secondary causes of hypertriglyceridemia [182]
|
Alcohol excess |
|
High-carbohydrate diet |
|
Obesity |
|
Type 2 diabetes mellitus |
|
Pregnancy |
|
Chronic renal failure, nephrotic syndrome |
|
Hypothyroidism, Cushing syndrome |
|
Acute pancreatitis |
|
Viral hepatitis |
|
Biliary cirrhosis |
|
Multiple myeloma, monoclonal gammopathy |
|
Glycogen storage disease |
|
Lipodystrophy |
|
Systemic lupus erythematosus |
|
Drugs – exogenous estrogens, tamoxifen, glucocorticoids, b-blockers, amiodarone, thiazide diuretics, ciclosporin, retinoids, bile acid binding resins, antiretrovirals (protease inhibitors), propofol, clozapine, parenteral lipid infusion |
3.3.5 Alcohol
The pathophysiologic role of alcohol in the etiology and occurrence of acute AP is complex, but increased oxidative stress [183, 184], disruption of cytosolic calcium homeostasis [185], and changes in gene expression [186] in the pancreas seem to be involved. Yet, only 1–3 % of heavy alcohol drinkers (4–5 standard drinks of alcohol per day) develop AP after 10–20 years of follow-up [187, 188]. For many years there has been an ongoing discussion on whether the type of alcoholic beverage might influence the occurrence of AP [189]. Indeed, a potential role for type of beverage was indicated by descriptive data from Stockholm County in Sweden showing a decline in the incidence of AP alongside a decline in the sales of spirits between 1971 and 1987, despite increased sales of beer and wine [190]. In Finland, there was also a decline in the incidence of alcoholic AP between 1989 and 2007 [191]. During the same period, the percentage of people drinking spirits each week dropped from 24 % in 1988 to 19 % in 2007 [191]. However, clinical studies have generally been too small to study the association between different alcoholic beverages and the risk of AP [192–194]. In a recent population-based study from Denmark, the risk of AP was found to be associated with the amount of beer consumed [187]. However, the information on alcohol use was limited to the consumption of alcoholic beverages assessed as total number of drinks per week, not including the amount of alcohol consumed on a single occasion or overall drinking frequency. The metabolism of alcohol (ethanol) is known to induce oxidative stress, which in turn depletes cellular glutathione storage and results in lipid peroxidation and damage to pancreatic tissue [183, 195]. However, in animal models it seems that ethanol alone is not enough to induce AP [196, 197]. Beer [198] and wine [199] include polyphenols with antioxidant capabilities. In experimental studies, other constituents in spirits such as long-chain alcohols have been shown to be more potent than ethanol in inducing oxidative stress [200]. Comparing the same amounts of alcohol, spirits deplete the antioxidant capacities more readily than beer or wine [201]. Thus, there might be other constituents in spirits that induce AP, in combination with ethanol or alone. Those drinking spirits might also have lower reserves of antioxidants at baseline, which could be depleted rapidly after intake of spirits [202].
Alcohol use is associated with increased risk of AP, in a dose-dependent manner [203], but the main point is that chemical analysis (using gas chromatography/mass spectrometry) of the consumed alcohols revealed the presence of other constituents (besides ethanol and water) are potential cause of injury of the pancreas, but, to date, remain largely unexplored [189, 204]. The results reported in the study must therefore be carefully interpreted as only tentative based on semiquantitative analysis. Also data for the patterns of drinking from the Global Information System on Alcohol and Health (http://apps.who.int/globalatlas/default.asp) indicate that the more harmful the pattern of drinking (i.e., the more heavy drinking), the higher the rates of alcohol-induced AP [205]. A recent systematic review and meta-analysis also detected the existence of a threshold at approximately four drinks daily [206].
3.3.6 Medications
AP in general population due to medications is an unusual event, although the incidence may be increasing. In a review of records from 45 German centers, only 1.4 % episodes of AP in 1993 were related to medication use [207]. Further confirming the rarity of this condition, only 0.3 % adverse drug reactions reported to the Swiss Drug Monitoring Center between 1981 and 1993 were drug-related AP [208]. The literature on drug-induced AP mostly consists of case reports and anecdotal accounts. Over 55 drugs have been implicated as etiological agents, and the list continues to grow. Proposed criteria for classifying drugs as having an association with AP include the following [209]:
· Pancreatitis develops during treatment with the drug.
· Other likely causes of pancreatitis are not present.
· Pancreatitis resolves upon discontinuing the drug.
· Pancreatitis usually recurs upon readministration of the drug.
Assignment to a particular category (definite, probable, or possible association) is often arbitrary due to inadequate and conflicting data and interpretation bias of the reviewers. Thus, the strength of the association has been interpreted differently, substantially so in some cases, by different reviewers of the subject. The pathogenesis of drug-induced AP may be due to an allergic response in some cases (6-mercaptopurine, aminosalicylates, sulfonamides) or to a direct toxic effect (diuretics, sulfonamides, steroids). AP associated with angiotensin-converting enzyme inhibitors is thought to reflect angioedema of the gland. Medications with the influence on estrogen activity are discussed in previous section of acquired causes of HTG.
Drug-induced AP in pregnancy is rare [15]. These facts would point out mifepristone or possibly gemeprost as the likely causative agents [210]. However, there have been a handful of published cases of AP complicating treatment with codeine [211–213]. A common feature of these reported cases are previous cholecystectomies [212, 213]. The patient reported here had not been cholecystectomized. Also, three cases of possible codeine-precipitated AP have been reported since 1965 to the Swedish Drug Information System (SWEDIS) handling reports on suspected adverse reactions to drugs used in Sweden [214]. Codeine is known to cause rapid but transient spasm of the sphincter of Oddi [212]. Laboratory studies have shown that codeine may cause a mild, transient hyperamylasemia [212]. AP following paracetamol overdose has been previously reported [212], but the doses taken in the present case are unlikely to have been causative. Gemeprost is a synthetic prostaglandin E1 (PGE1) analog. Studies indicate that PGE1 is a modulator of pancreatic blood flow and protein production. For instance, PGE1 stimulated the production and secretion of alpha-amylase from minces of porcine pancreas in vitro [215] and enzyme output in dogs in vivo [216]. PGE1 further increases mesenteric and pancreatic blood flow [216]. Thus, PGE1 is not devoid of actions on the pancreas. Progesterone has also been shown to exert modulating action on the pancreas. In rats, progesterone stimulated pancreatic cell proliferation in vivo [217]. Progesterone receptors have also been shown to be present in human pancreatic tissue [218]. However, the effect of a progesterone receptor antagonist such as mifepristone on the pancreas has not been studied. Evidence thus exists that both PGE1 and progesterone exert modulatory action on the pancreas, but currently no reports on AP following treatment with gemeprost or mifepristone have been published.
Diuretics can induce AP [219, 220]. Preeclampsia-associated AP can occur but is very rare [75, 221, 222]. Preeclampsia is associated with microvascular abnormalities that may involve cerebral, placental, hepatic, renal, and splanchnic circulation. It is likely that pancreatic vasculature was also altered and caused AP that resulted in organized pancreatic necrosis. In a review from 1995 [15], none of the 43 women had preeclampsia-associated AP, whereas an older review [16] reported nine of 98 cases of preeclampsia-associated AP but five of those received diuretics and another case also reported diuretic use [16]. There is a question: is it really preeclampsia-eclampsia the cause or is therapy with diuretics the real cause of AP during pregnancy in such situations?
Additional medications associated with elevated TG include protease inhibitors [223], propofol [224], olanzapine [225], mirtazapine [226], and isotretinoin [227].
3.3.7 In Vitro Fertilization
Currently, there is only one case report of AP during in vitro fertilization (IVF) pregnancy published. A 29-year-old female with Fredrickson type V dyslipidemia and BMI 26 at 32 weeks gestation with twins was treated medically. Emergency Cesarean section was performed after 48 h due to clinical deterioration with increasing metabolic acidosis. The Cesarean section was performed with successful outcome, and postoperatively her severe HTG settled. She was discharged on postoperative day 9 on a combination of fenofibrate and Omacor [228]. Another two case reports described patients developing AP during a routine IVF stimulation cycle [229, 230]. IVF probably represents high-risk group to the number of hormones, procedures, and medications used simultaneously which could increase the probability of developing AP.
3.3.8 Postpartum Acute Pancreatitis
Evidence is abundant of gallstones and biliary sludge during pregnancy causing AP. Also, due to hormonal changes, there is increase in incidence of other causes such as hyperlipidemic/hypertriglyceridemic AP. Most of the AP resolves during medical (or surgical) therapy, but some are resolved after Cesarean section when all the gestational (mostly hormonal) changes return to pregestational state. Findings of the study by Maringhini et al. are that AP associated with pregnancy occurred in the youngest women but only during the postpartum period and was associated with gallstones but not related to pregnancy per se [231]. The association of AP with gallstones in young postpartum women is most likely due to the known alterations of bile composition, gallbladder contractility, and gallstone or gallbladder sludge formation that occurs during and after pregnancy (see Chap. 2). Small gallstones may appear during pregnancy, but most of them disappear during the early postpartum period. The appearance of gallstones during pregnancy and their postpartum disappearance may be due to the impressive modifications of bile composition and gallbladder motility that occur during pregnancy. The changes in hepatic bile that occur in the last trimester of pregnancy are secondary to high estrogen levels, while gallbladder stasis during pregnancy is due to high progesterone levels. These phenomena produce the milieu for nucleation and crystal formation that finally generates sludge and stones. Bile composition and gallbladder motility return to normal after delivery; thus, sludge and small stones may be eliminated or dissolved. The data are consistent with the hypothesis that at least some of the gallstones disappear during the postpartum period because they are ejected from the gallbladder into the bile ducts and duodenum and may cause AP. The study did not confirm direct data on the role of biliary sludge in AP in pregnancy. No women with AP had documentation of biliary sludge, but the diagnosis of biliary sludge is often difficult, and a prospective study is needed. However, authors demonstrated that gallstones are the only etiology significantly associated with postpartum-related AP. The increased RR for gallstone AP in young postpartum women is in agreement with the increased incidence of gallstones in young pregnant women [232–236]. The age-dependent risk of developing gallstones associated with pregnancy (the risk being greatest for subjects younger than 29 years) has been shown in some Australians [234] and in Chippewa Indians [235]. Authors previously reported that the spontaneous disappearance of gallstones after delivery is significantly more frequent in older women [237]. Speculation is that a few young women eject small gallstones from the gallbladder during the postpartum period when gallbladder contraction is restored, and some of these women develop an attack of AP. In contrast, in older women with reduced gallbladder contractility, most gallstones likely remain in the gallbladder until dissolved by less lithogenic bile. Thus, AP associated with pregnancy usually occurs in young postpartum women and is usually due to gallstones [231]. Generally, AP can occur during any trimester but around 52 % of cases are found in the third trimester; it is rarely seen in the postpartum period [15].
3.3.9 Preeclampsia-Eclampsia
AP has also been reported in preeclampsia but with only several cases published [75, 221, 222, 238–242]. Preeclampsia is associated with microvascular abnormalities that may involve cerebral, placental, hepatic, renal, and splanchnic circulation. It is likely that the pancreatic vasculature was also altered and caused AP that resulted in organized pancreatic necrosis. In a review from 1995 [15], none of the 43 women had preeclampsia-associated AP, whereas an older review [16] reported nine of 98 cases of preeclampsia-associated AP but five of those received diuretics and another case also reported diuretic use [222]. Diuretics can induce AP [219]. The rise of amylase and lipase levels exceeded the expected increased values due to the slight reduction of the renal function in the preeclamptic patients and therefore indicates a concomitant injury of the pancreas [243].
3.3.10 Placental Abruption
Placental abruption is when the decidual spiral artery ruptures to cause a hematoma that separates the placenta from the uterus and is commonly associated with maternal hypertension [244]. In severe cases, blood prominently infiltrates the uterine myometrium up to the serosa, and this phenomenon is designated as uteroplacental apoplexy. Placental abruption induced by AP is very rare and was reported in 1962 by Pagliari et al. [245] and later Cheang et al. [246]. Placental abruption likely occurred in the first phase of AP, resulting from a systemic inflammatory response.
3.4 Clinical Features
AP presents essentially in the same way during pregnancy as in the nonpregnant state. However, it is difficult to diagnose AP by history and physical examination due to similarity to many acute abdominal illnesses and due to maternal changes during pregnancy.
3.4.1 History Taking
All relevant information about possible causes should be obtained. Family history about hyperlipidemias, DM, AP, etc. should be noted. It is important if these data could be noted before or during pregnancy planning to eliminate or minimize possibility of AP during pregnancy.
Since no safe level of alcohol has been established in pregnancy, it may not be socially acceptable for pregnant women to admit they consume alcohol. Assessing this risk accurately can be challenging. The T-ACE [Take (number of drinks), Annoyed, Cut down, Eye-opener] TWEAK (Tolerance, Worried, Eye-opener, Amnesia, Kut down), and AUDIT-C (AUDIT consumption) alcohol screening questionnaires show promise for use with pregnant women, but have not yet been validated as stand-alone tools in this population [247].
3.4.2 Clinical Presentation
The symptoms of AP in pregnancy could be nonspecific; the predominant symptom is upper abdominal pain which is usually midepigastric and could radiate to the back in about 40 % of the cases [15]. Pain is commonly accompanied by midepigastric tenderness, nausea, and vomiting [248, 249]. Fever may be present [249]. Some cases may have persistent vomiting, abdominal distension, and tenderness in the whole abdomen. The duration of symptoms may vary from 1 day up to 3 weeks. In severe cases sinus tachycardia, hyperventilation, and smell of acetone of the breath can also be present [250]. If accompanied by fever, unstable respiratory and circulatory function, shock, and gastrointestinal bleeding, these are strong indications for severe AP.
AP in pregnancy is mainly related to gallbladder disorders and correlates with cholelithiasis and biliary sludge (muddy sediment, precursor to gallstone formation) as the most likely predisposing causes [15]. The symptoms of gallbladder disease can be present or can precede the clinical presentation of AP. The symptoms include abdominal pain (colicky or stabbing) which may radiate to the right flank, scapula, and shoulder. Onset of pain is rapid, with maximal intensity in 10–20 min. Pain is steady and moderate to severe. Band-like radiation of the pain to the back occurs in half of patients. Other symptoms of gallbladder disease include anorexia, nausea, vomiting, dyspepsia, low-grade fever, tachycardia, and fatty food intolerance [15]. Vomiting is a common symptom. Marcus in 1930 emphasized that persistent vomiting in pregnancy could be related to AP and that blood and enzyme studies should be done [251].
Clinical signs are the same as in nonpregnant population. Cullen’s sign (Fig. 3.3) and Grey Turner’s sign are rare but among most common as in general population.
Fig. 3.3
Cullen’s sign [252]
3.4.2.1 Primary Hyperparathyroidism
Truly asymptomatic PHPT in general population is rare when thorough anamnesis looks for subtle symptoms. Most frequent digestive manifestations are constipation, heartburn, nausea, and appetite loss that occur in 33, 30, 24, and 15 % of cases, respectively [253]. The diagnosis should be suspected during pregnancy if the following conditions exist: appropriate clinical signs or symptoms (especially nephrolithiasis or AP), hyperemesis beyond the first trimester, history of recurrent spontaneous abortions/stillbirths or neonatal deaths, neonatal hypocalcemia or tetany, or a total serum calcium concentration greater than 10.1 mg/dl (2.52 mmol/l) or 8.8 mg/dl (2.2 mmol/l) during the second or third trimester, respectively. Symptomatic PHPT is rarely detected in pregnancy due to the physiological changes that mask the symptoms; this includes maternal blood volume expansion, hypoalbuminemia, increased fetal calcium requirements, and increased calcium clearance. The data obtained from analysis of so far described cases indicate that in opposition to the general population, four of every five hyperparathyroid pregnant patients experience clinical manifestations of this disorder. The most frequent of them is the presence of nephrolithiasis [42, 104]. The initiation and growth of kidney calculi may be attributed to overlapping of both increased calcium load, secondary to enhanced PTH synthesis and release, and a pregnancy-induced increase in urine calcium excretion (a typical symptom of physiological pregnancy).
3.4.2.2 Acute Fatty Liver of Pregnancy
The onset of AFLP occurs typically in the third trimester or the early postpartum period and is characterized by a nonspecific prodrome of symptoms: sudden onset of nausea, vomiting, and vague abdominal pain followed by jaundice, profound hepatic failure with encephalopathy or coma, coagulopathy, and frequent hypoglycemia. Due to the similarity of symptoms, AP during pregnancy cannot be always recognized clinically. In a series of 12 cases of AP in pregnant women with AFLP, Moldenhauser et al. [110] found the following complications: encephalopathy (50 %), respiratory failure (17 %), and acute renal failure (33 %).
3.4.2.3 Hypertriglyceridemic Pancreatitis
Though the initial presentation of HTG-induced AP is similar to AP due to other etiologies, some features should lead to the consideration of HTG-induced AP. Poorly controlled DM, alcoholism, obesity, prior AP, and personal or family history of hyperlipidemia will suggest HTG-induced AP [30, 158]. Alcoholism or DM has been reported in 72 % of HTG-induced AP episodes [30, 158]. Lactescent serum on hospital admission was found in 45 % of patients [30].
3.4.2.4 Medication-Induced Acute Pancreatitis
Drug-induced AP in general population has no distinguishing clinical features. A high index of suspicion and careful drug history are therefore essential for making the diagnosis. The time course of developing the disorder depends upon the drug involved. As an example, AP may develop within a few weeks after beginning a drug associated with an immunologically mediated adverse reaction; in this setting, the patient may also have a rash and eosinophilia. In contrast, patients taking valproic acid, pentamidine, or didanosine may not develop AP until after many months of use, presumably due to the chronic accumulation of toxic metabolic products. Proving the association with a particular drug may not always be straightforward, even in suspected cases. Thus, patients restarted on their medications should be closely monitored and the drug promptly discontinued if symptoms recur. It is known that many medications are discontinued during pregnancy by mothers themselves or by physicians when not sure about teratogenicity.
3.4.3 Physical Examination
Physical findings vary with the severity of illness; in moderate to severe AP, the patient appears acutely ill and is found lying in the “fetal position” with flexed knees, hips, and trunk. Abdominal tenderness is often found; in diffuse peritonitis muscle rigidity can be present. Bowel sounds, secondary to paralytic ileus, are usually hypoactive or absent. In severe AP the general physical examination may reveal abnormal vital signs if there are third-space fluid losses and systemic toxicity. Due to hypovolemia, tachycardia up to 150/min and low blood pressure could be found. Also, because of severe retroperitoneal inflammatory process, temperature may increase. Dyspnea, tachypnea, and shallow respirations resulting in hypoxemia may be present. Altered maternal acid-base status can adversely affect fetal acid-base status. Acute fetal hypoxia activates some compensatory mechanisms for redistribution of blood that enable fetus to achieve a constancy of oxygen consumption in the fetal cerebral circulation and in the fetal myocardium. Redistribution of blood to vital organs enables fetus to survive for moderately long period of limited oxygen supply, but during more severe or sustained hypoxemia, these responses were no longer maintained and decompensation with fetal tissue damage and even fetal death may occur [167, 254]. Some physical findings point to a specific cause of AP: jaundice in biliary origin, spider angiomas in alcoholic, or xanthomata and lipemia retinalis in hyperlipidemic AP. HTG can lead to chylomicronemia syndrome, which can manifest with eruptive xanthomata over extensor surfaces of the arms, legs, buttocks, and back (Fig. 3.4) [255–258], lipemia retinalis (Fig. 3.5) [255, 260], and hepatosplenomegaly from fatty infiltration of the liver [255, 257, 258].
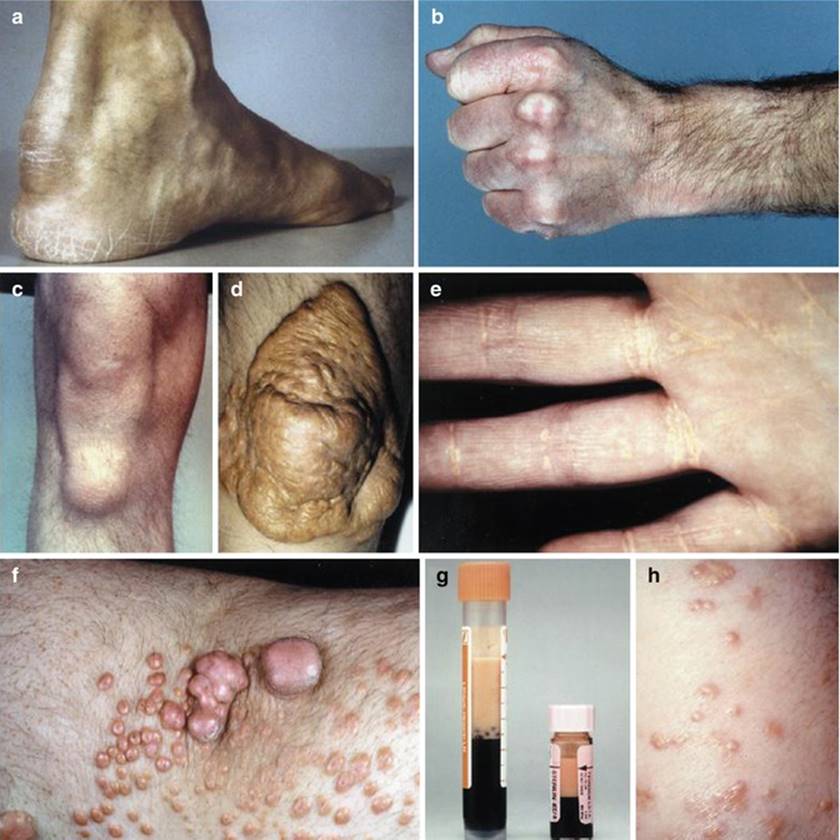
Fig. 3.4
Clinical manifestations of hyperlipidemia. (a) Achilles tendon xanthoma (heterozygous familial hypercholesterolemia – type IIa); (b) tendon xanthomata on dorsum of the hand (heterozygous familial hypercholesterolemia); (c) subperiosteal xanthomata (heterozygous familial hypercholesterolemia); (d) planar xanthoma in antecubital fossa (homozygous familial hypercholesterolemia); (e) striate palmar xanthomata (type III); (f) tuberoeruptive xanthomata on elbow and extensor surface of arm (type III); (g) milky plasma from patient with acute abdominal pain (severe hypertriglyceridemia); (h) eruptive xanthomata on extensor surface of the forearm (severe hypertriglyceridemia) [255]
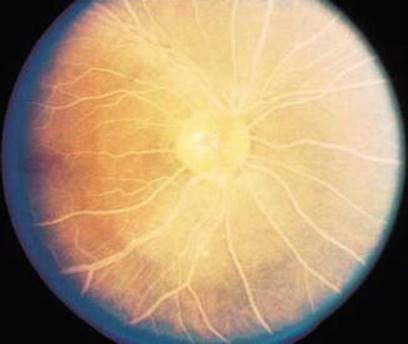
Fig. 3.5
Lipemia retinalis associated with hyperlipidemia. Rare and asymptomatic creamy white appearance of retinal vessels occurs when triglyceride value reaches more than 2,000 mg/dl (22.6 mmol/l) – the effect due to dispersion of light caused by high value of circulating chylomicrons in the blood, most commonly occurring in familial hyperchylomicronemia [259]
3.5 Diagnosis
AP in pregnancy is diagnosed by symptoms already described, by laboratory investigations, and by imaging methods. Some important normal values in pregnant and nonpregnant women are compared in Table 3.4.
Table 3.4
Normal laboratory values for blood and urine in pregnant and nonpregnant women [261]
|
Variable |
Nonpregnant women |
Women in third trimester |
|
Serum total calcium (mmol/l) |
2.2–2.63 |
2.15–2.3, mean 2.15–2.23 |
|
Serum albumin (g/dl) |
3.6–4.6 |
2.8–3.6 |
|
Serum ionized calcium (mmol/l) |
1.13–1.32 |
No change |
|
Parathyroid hormone (pg/ml) |
11–80 |
Middle of normal range |
|
25-hydroxyvitamin D (nmol/l) |
62–200 |
No change |
|
1,25-dihydroxyvitamin D (nmol/l) |
37–187 |
Doubled |
|
PTH-related protein (pg/ml) |
0–12 |
Increased |
|
Triglycerides (mmol/l) |
0.4–1.7 |
Doubled to quadrupled |
|
Urinary calcium (mmol/h) |
<6.24 |
Doubled to tripled |
3.5.1 Laboratory Findings
Marcus in 1930 is credited with the first clinical diagnosis of AP in pregnancy with the aid of diastase and amylase studies [251]. Pregnancy-related hematological and biochemical alterations interfere with the interpretation of diagnostic tests and assessment of severity of AP. Laboratory investigations are the same as in nonpregnant and rely on at least a threefold elevation of serum amylase and lipase levels in the blood. The total serum amylase level rises within 6–12 h of onset of the disease, usually remains elevated for 3–5 days. However, there are several conditions (i.e., pathologic processes in salivary glands, Fallopian tubes, bowel obstruction, cholecystitis, hepatic trauma, perforative duodenal ulcer, hyperamylasemia on familial basis, etc.) that may result in the elevation of serum amylase. Serum lipase is elevated on the first day of illness and remains elevated longer than the serum amylase. In terms of diagnostic accuracy, lipase has been proven to be superior to amylase in AP [262, 263]. However, lipase is also not specific to the pancreas, having been isolated in the tongue, esophagus, stomach, duodenum, small bowel, liver, lung, and adipose tissue [264, 265]. Consequently, hyperlipasemia has been reported to appear in the event of cholecystitis, esophagitis, peptic ulcer disease, enteritis, peritonitis, and bowel obstruction and infarction [263–265]. AP could not be ruled out if normal level of serum amylase is detected. One reason is that the serum amylase may not increase when pancreas have extensive necrosis. The other reason is that amylase and biochemical parameters cannot be checked truly with the significant increase of plasma triglycerides level. So, in nonpregnant patients, a normal amylase would usually exclude the diagnosis of AP, with the exception of AP secondary to hyperlipidemia, acute exacerbation of chronic pancreatitis, and when the estimation of amylase is delayed in the course of the disease [266]. Therefore, checking the urine amylase level may be more helpful. Caution in the interpretation of serum amylase and urinary diastase determination should be exercised if morphine has been given. Morphine has been shown to cause spasm of the sphincter of Oddi with obstruction of the pancreatic drainage. In nonpregnant population with AP, no enzyme assay has a predictive role in determining the severity or etiology of AP. Once the diagnosis of AP is established, daily measurements of enzymes have no value in assessing the clinical progress of the patient or ultimate prognosis and should be discouraged. A persistently raised serum amylase activity may suggest the presence of a pancreatic pseudocyst. For the early postpartum period, there are no data available about amylase and lipase dynamics due to the extreme rarity. An elevated serum amylase level has a diagnostic sensitivity of 81 %, and adding serum lipase increases the sensitivity to 94 %. However, amylase levels do not correlate with disease severity [267]. Typically, a serum amylase concentration greater than three times normal is seen at presentation, which peaks in the first 24 h and falls to baseline in 3–5 days. In contrast, serum lipase concentrations are elevated for up to 2 weeks, making it a more sensitive and specific diagnostic test. Karsenti et al. found that enzyme concentrations were similar in nonpregnant and pregnant women and concluded that an increase in either would be suggestive of AP in pregnancy [268].
Elevated amylase and/or lipase are the diagnostic hallmarks of AP; yet, in hypertriglyceridemia-induced AP, amylase levels may be reported as normal or even low in more than 50 % of patients. This phenomenon has been attributed to an interference of plasma lipids with the assay and/or to the presence of a circulating inhibitor of amylase in serum and urine [158, 269, 270]. In such cases, dilution or ultracentrifugation of the sample is recommended to ensure accurate analysis. Also, in general population, 16–25 % of patients with diabetic ketoacidosis may have elevated pancreatic enzymes and triglycerides, circumstance less reliable diagnosis of pancreatitis based only on biochemical parameters [171].
In addition, lipase is the pancreas-specific enzyme lasting in the blood for a long time. Despite the aforementioned, elevation of serum alanine aminotransferase levels to >3 times the upper limit of normal is a very sensitive biochemical marker of biliary AP in nonpregnant population [271, 272] and should be also suspected in pregnancy. Calculation of an amylase to creatinine clearance ratio may be helpful in pregnancy; ratio greater than 5 % suggests AP [273]. Gamma-glutamyl transpeptidase (GGTP) levels either are unchanged or fall slightly during gestation. An elevated GGTP level can help us to evaluate the history of alcohol use during pregnancy as patients might not be coming forth, due to stigmata associated with it [274].
HTG AP is conventionally thought to be triggered when triglyceride (TG) levels exceed 1,000 mg/dl (10 mmol/l) unless accompanied by lactescent serum [30, 168]. In severe HTG (serum TG level >2,000 mg/dl), there is an increased risk of aggravating preexisting AP [275, 276]. Chylomicrons are formed at these high TG levels and serum becomes lactescent (milky coloration). TG values of even 3,810 mg/dl were encountered [277].
Serum calcium level must be considered among the usual tests in patients with rare and/or nonspecific abdominal symptom, especially when AP is suspected [278]. Calcium metabolism in pregnancy is a dynamic process. Maternal serum calcium falls by about 10 % in pregnancy; however, as the serum albumin falls by 20 %, the ionized calcium remains unchanged. Making the correct diagnosis of PHPT in pregnancy is regarded crucial, because if this disorder remains unrecognized and left untreated, it may pose a significant risk to the mother and fetus, which is associated with increased perinatal and maternal morbidity and mortality [51, 279].
In a study by Tang et al. liver tests in pregnant women with biliary AP were frequently normal. The transaminase levels were less than 5× the upper normal limits in 89 % of patients and less than 3× the upper normal limits in 80 % of patients. Authors do not have a good explanation for this finding. One possibility is that increased metabolism of maternal transaminases by the placenta led to relatively normal maternal levels of liver enzymes [23].
Laboratory abnormalities consistent with AFLP include mild elevation of ALT and AST to 200–300 IU/l [108, 280], prolongation of prothrombin time and partial thromboplastin time, decreased fibrinogen, acute renal failure, severe hypoglycemia, a bilirubin level of 1–10 mg/dl, and leukocytosis. In a series of 12 cases of AP in pregnant women with AFLP, Moldenhauser et al. [110] found that elevated serum lipase was present in 91 %.
It should be noted that hyperparathyroidism may be falsely lowered due to hypoalbuminemia or suppressed by magnesium tocolysis [59].
Some authors recommend that a lipemic blood sample found at any stage during pregnancy should be considered as potentially indicative of partial LPL deficiency [159].
At present, serum CRP at 48 h is the best available laboratory marker of severity. Urinary trypsinogen activation peptides within 12–24 h of onset of AP are able to predict the severity but are not widely available. Serum IL-6 and IL-8 seem promising but remain experimental [266].
3.5.1.1 Confounding Laboratory Investigations in HTG of Nonpregnant Patients
Elevated TG levels can alter routine measurements of sodium, serum amylase, and LDL. Clinicians must be wary of pseudohyponatremia as the excess TG in a serum sample can displace water-containing sodium. During laboratory measurements, the sodium level appears lower than the actual value [281]. Ultracentrifugation is needed to separate the aqueous phase and measure the true sodium level [281]. HTG levels >500 mg/dl may cause a falsely normal amylase level, likely from HTG interference with calorimetric reading of the assay or presence of an interfering inhibitor. This problem can be partially overcome by assaying serial sample dilutions [282] or measuring serum lipase or amylase to creatinine ratio, neither of which is affected by HTG [158, 283]. There are no official recommendations on lipase utility in HTG-induced AP; however, serum lipase was found to have higher specificity and sensitivity for AP compared to amylase in a report by Treacy et al. [284]. Friedewald calculations used to determine LDL from triglyceride levels lose accuracy with high TG levels [285]. A commonly accepted upper limit is 400–500 mg/dl [286]. Lipid analysis requires direct measurement through centrifugation or immunoprecipitation.
3.5.1.2 Ranson Criteria
Ranson scoring system is calculated in several case reports but is not validated in pregnancy. With a Ranson prognostic index at 7, as calculated in one patient with AP due to preeclampsia-eclampsia, there was a high risk of death. However, recovery was prompt and uneventful [221]. Another patient with Ranson score 3 had two exploratory laparotomies and survived after complicated course [287].
3.5.2 Imaging Methods
Imaging in pregnancy remains a controversial issue with concern of the effect of radiation on the developing fetus. Abdominal ultrasound (US) with no radiation to the fetus is the initial imaging technique of choice to identify or exclude biliary etiology, the finding on which further therapy depends (Fig. 3.6). However, it is insensitive for the detection of common bile duct stones or sludge and the morphological changes of the pancreas. It is not accurate for detection of dilated pancreatic ducts but is good for pseudocysts and focal accumulations larger than 2–3 cm. US is limited by operator skill, patient obesity, and bowel dilation especially found in patients with peritonitis. Additional abdominal US role is for estimation of fetal vitality by measuring direct (femur) or indirect parameters (oligohydramnios) (Fig. 3.7).
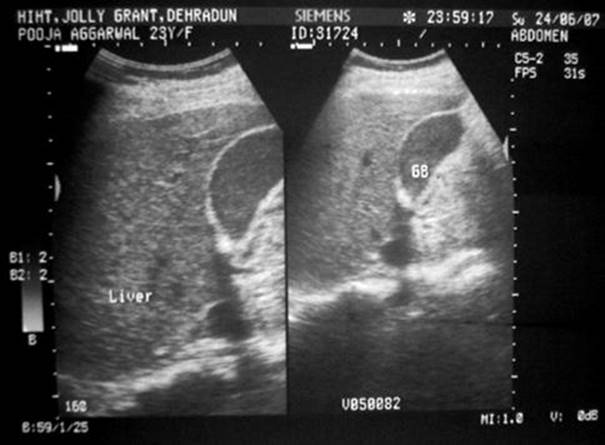
Fig. 3.6
Abdominal ultrasound of the abdomen showing gallbladder sludge in a 23-year-old nulliparous woman in 33 weeks of pregnancy [288]
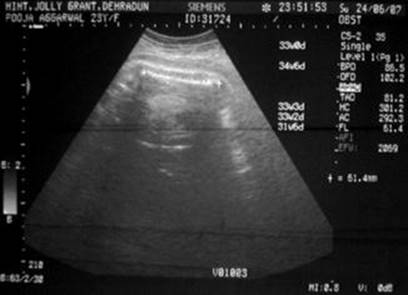
Fig. 3.7
Sonography of the same patient as in Fig. 3.6 showing femur length of 61.4 mm corresponding to 33 weeks of gestation with oligohydramnios [288]
Computed tomography (CT) should be avoided, especially during the first trimester, because of radiation exposure to the fetus, but has to be performed when benefits outweighed the risk. Imaging diagnostic modalities are used not only for the diagnosis but also to provide information about the severity in AP (Figs. 3.8, 3.9, 3.10, and 3.11) [290]. In a series of 12 cases of AP in pregnant women with AFLP, Moldenhauser et al. [110] found that imaging techniques (ultrasound and computed tomography) were accurate in only 58 %. In addition, if thyroid ultrasound is equivocal, a helical CT scan is helpful in mediastinal parathyroid adenoma localization, especially during pregnancy when radioisotope techniques are contraindicated.
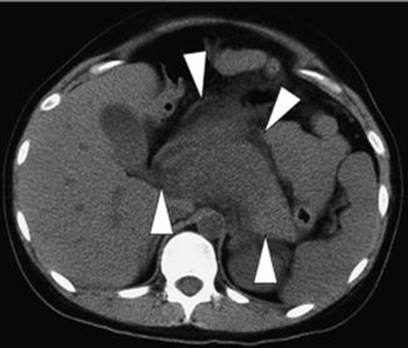
Fig. 3.8
Computed tomogram on admission of a 25-year-old primigravida at 35 weeks of gestation shows marked swelling of the pancreas (arrowheads) and effusion around the pancreas [289]
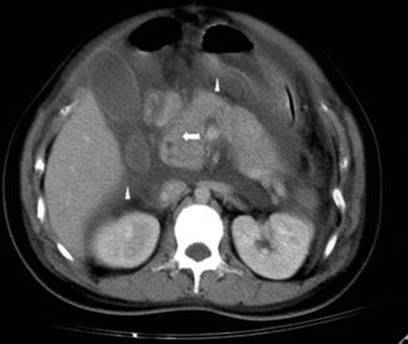
Fig. 3.9
Abdominal CT of a 28-year-old female in the 34th week of pregnancy with swelling of the pancreas and blurring of the mesenteric fat plane (arrow). Reactive paralytic ileus, fluid accumulation at bilateral anterior pararenal space, lesser sac, and extraperitoneal space are noted (arrowheads) [287]
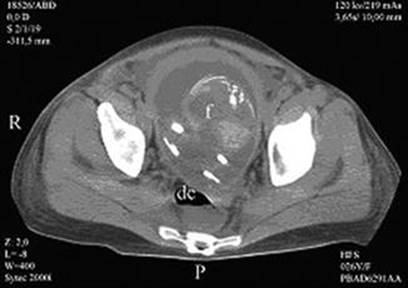
Fig. 3.10
Pregnant uterus (f fetus, dc Douglas collection) [111]
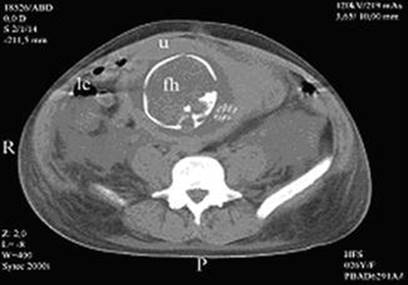
Fig. 3.11
Pregnant uterus (u uterus, fh fetal head, lc liquid collection) [111]
When a common bile duct stone is suspected, endoscopic ultrasound (EUS) has a high positive predictive value nearing 100 %, even for small stones ≤2 mm or sludge [291, 292]. EUS is considered the best imaging study to evaluate the common bile duct but requires expensive equipment, intravenous sedation, and technical expertise. It is superior to magnetic resonance cholangiopancreatography (MRCP), an imaging method providing multiplanar large field of view images of the biliopancreaticoductal system.
There are some concerns about the safety of MRCP in the first trimester of pregnancy because radiofrequency pulses result in energy deposition and could potentially result in tissue heating [293]. MR procedures are indicated in pregnancy if other nonionizing forms of diagnostic imaging studies are inadequate or if the examination provides information that would otherwise require exposure to ionizing radiation with excellent soft tissue contrast (Fig. 3.12) [295].

Fig. 3.12
Magnetic resonance cholangiopancreatography showing normal common bile duct and pregnancy in coronal section [294]
Endoscopic retrograde cholangiopancreatography (ERCP) as a diagnostic tool lost its value because of the risk of radiation and the availability of safer procedures (i.e., EUS or MRCP). ERCP should be used only as a therapeutic option in selected cases with confirmed bile duct stones. In cases of severe acute biliary pancreatitis (SABP) with or without cholangitis, early ERCP, preferably within 24 h, is recommended [296]. Decompression of the common bile duct and removal of gallstones with subsequent papillotomy could prevent complications and reduce mortality in SABP. Before proceeding to therapeutic ERCP, a less-invasive diagnostic method such as MRCP or EUS should be performed. In pregnancy it is necessary to minimize radiation exposure during ERCP, the procedure should be carried out only by a very experienced endoscopic and radiological team, and the fetus should be shielded all the time [297, 298]. With the advent of ERCP and MRCP, the need for intraoperative cholangiogram is minimal, although there have been no reports investigating the safety of IOC during pregnancy [254]. Laparoscopic US scan appears to be alternative to retained common bile duct stones [254].
3.6 Differential Diagnosis
Differential diagnosis contains the same diseases present in nonpregnant population with the addition of some specific diseases and states present in pregnancy. Interestingly, even a case of ruptured ectopic pregnancy mimicking AP is described with elevated serum amylase and lipase levels [299]. This is due to the fact that a variety of organs and secretions contain amylase activity, including the pancreas, salivary glands, Fallopian tubes and ovarian cyst fluids, testes, thyroid, tonsils, breast milk, sweat, tears, and some malignant neoplasms [300]. Therefore, hyperamylasemia has been reported to occur in mumps, parotitis, perforated peptic ulcer, perforated appendicitis, intestinal obstruction, mesenteric infarction, pulmonary embolism, pneumonia, myocardial infarction, lung cancer, breast cancer, lymphoma, and several tubo-ovarian disorders [264, 301–303]. The discovery that structures containing epithelium of Müllerian and mesonephric duct origin could produce amylase was made over 50 years ago [304]. In keeping with this, hyperamylasemia has been documented in association with several tubo-ovarian pathologies, including ruptured ectopic pregnancy, salpingitis, pelvic inflammatory disease, ovarian papillary serous cystadenocarcinoma, ovarian adenosquamous carcinoma, ovarian endometrioid carcinoma, mucinous tumors, and surface papillary carcinoma [305–309]. In all published cases of hyperamylasemia associated with tubo-ovarian disease in which isoenzyme analysis was performed, the predominant amylase has been shown to be either electrophoretically identical to s-type amylase or an acidic variant thereof [310–313]. However, predominantly p-type hyperamylasemia has up to this case been reported previously with tubo-ovarian disorders. In addition, according to Saruc et al. hemolysis of extravasated blood might have been the reason of the elevated pancreatic enzyme activity [314]. Ruptured ectopic pregnancy in rare situations can present with significantly elevated amylase and lipase levels mimicking the diagnosis. The problem is if hemorrhagic AP is suspected and conservative approach indicated, prolonged hemorrhage from ectopic pregnancy can lead to hemorrhagic shock [299].
In terms of diagnostic accuracy, lipase has been proven to be superior to amylase in AP [262, 315]. However, lipase is also not specific to the pancreas, having been isolated in the tongue, esophagus, stomach, duodenum, small bowel, liver, lung, and adipose tissue [264, 265]. Consequently, hyperlipasemia has been reported to appear in the event of cholecystitis, esophagitis, peptic ulcer disease, enteritis, peritonitis, and bowel obstruction and infarction [263–265]. Currently, there are only two published cases of hyperlipasemia occurring with tubo-ovarian disorders. One is aforementioned and another is Sinha et al. who presented a case of lipase activity elevation secondary to ruptured ovarian cyst [316].
3.7 Treatment
When a diagnosis of AP in pregnancy is made, assessment of severity based on clinical signs, blood tests, urinalysis, and imaging tests should be performed to determine the appropriate treatment for each patient. The treatment of AP is not standardized and is mainly supportive, and severe AP is still a significant clinical problem for all physicians. The treatment goals are to avoid organ failure and infectious complications which also influence on the fetal development. With the advances of diagnostic techniques and therapeutic methods, maternal and fetal outcomes have significantly improved over the last decades.
3.7.1 Conservative Treatment
3.7.1.1 Supportive Measures
It is important to stress that treatment includes the treatment of the AP itself and also specific treatment of the cause of AP. The initial management of AP during pregnancy is similar to management in nonpregnant patients. Treatment consists of fluid restoration, oxygen, analgesics, antiemetics, and monitoring of vital signs. An adequate volume of intravenous fluid should be administered promptly to correct the volume deficit and maintain basal fluid requirement [290, 317, 318]. Fluid resuscitation should be done carefully by closely monitoring the patient’s vital signs. Important additional measures during pregnancy include fetal monitoring, attention to the choice of medications, and positioning of the mother to avoid inferior vena cava obstruction.
Mild AP (MAP) treated conservatively usually resolves within 7 days. Ten percent of patients have severe course, and they are best managed in an intensive care unit (ICU). The third-space fluid sequestration is the most serious hemodynamic disorder leading to hypovolemia and organ hypoperfusion resulting in multiple organ failure. In volume-depleted patients, the essential treatment modality is initial infusion of 500–1,000 ml of fluid per hour [319]. Monitoring of hydration, cardiovascular, renal, and respiratory functions is important for early detection of volume overload and electrolyte disturbances [320]. In cases with severe AP (SAP), ICU should be considered, and intensive care is recommended to prevent both organ failures and infectious complications. Hemodynamic stabilization and respiratory support are the major parts of intensive care in the early period of SAP. Parenteral nutrition is considered to be safe and necessary in pregnancy [24]. Currently, there are still different opinions on the use of total parenteral nutrition (TPN) or enteral nutrition (EN). Pezzilli et al. showed that enteral feeding in SAP is better than TPN, because EN may help to maintain the immune function of the gastrointestinal mucosa, protect the mucosal barrier, and improve the blood supply to the small intestine [290].
Many pharmacological agents (somatostatin, octreotide, N-acetylcysteine, gabexate mesylate, lexipafant, and probiotics) have been investigated in AP, but because most of them have failed to show a positive effect, they should be avoided in pregnancy. The use of Stilamin (Somatostatin) may be effective to improve pathophysiology of the pancreas, especially in the early stage of AP. Applying Stilamin (Somatostatin) to inhibit both exocrine and endocrine portions of the pancreas is a very important part of nonsurgical treatment. However, it has not yet been proved whether somatostatin should be applied in pregnancy because of its potential effect on the fetus. In study by Li et al. Stilamin (Somatostatin) was administered in ten cases with good outcomes without malformations and abnormal newborn [78]. Treatment strategy cases with SAP is as follows: when the benefits of the drug may outweigh its risks with the permission of the patient and the agreement and signing of her family member, Stilamin (Somatostatin) is used in the early stage of SAP. Stilamin (Somatostatin) should be infused continuously with intravenous syringe infusion pumps with a low dose (150–250 μg/h) for 24–72 h and then reduced or withdrawn timely when the condition improves with hemodynamic stabilization. Despite the encouraging results, the data are limited, and somatostatin is not recommended for routine use.
Cessation of oral feeding has been thought to suppress the exocrine function of the pancreas and to prevent further pancreatic autodigestion. Bowel rest is associated with increased infectious complications, and TPN and EN have an important role in the management of AP. Keeping the patients nil by mouth with the use of TPN has been for years a traditional treatment of AP but carries a significant risk of infections and metabolic distress. EN is physiological, helps the gut flora maintain the gut mucosal immunity, and reduced translocation of bacteria, while simultaneously avoiding all the risks of TPN. Parenteral nutrition in pregnancy is considered safe and necessary when adequate oral nutrition is not possible, although the frequency of complications from centrally inserted catheters is higher than in nonpregnant patients [321]. Mild cases of AP do not need nutritional support, as the clinical course is usually uncomplicated and a low-fat diet can be started within 3–5 days. Treatment of SAP should include enteral feeding by nasojejunal tube and, if needed, should be supplemented by parenteral nutrition [322].
3.7.1.2 Antibiotics
Prophylactic use of antibiotics is very controversial and the choice of antibiotic in pregnancy is difficult. There are concerns with regard to the antibiotic being transplacentally transferred to the fetus with a risk of teratogenicity. Antibiotics have no role in the treatment of mild AP, normal common bile duct size, and no evidence for cholangitis, while the control of infection in the treatment of SAP plays an important role [290, 317, 318]. The use of prophylactic broad-spectrum antibiotics could reduce infection rates in necrotizing form of SAP [290]. The use of prophylactic antibiotics in severe AP remains controversial. The available evidence demonstrates that antibiotic prophylaxis might have a protective effect against non-pancreatic infections but failed to show a benefit on reduction of mortality, infected necrosis, and need for surgical intervention [296, 323, 324]. But the choice of antibiotic in pregnancy is difficult, and literatures show that penicillins and cephalosporins are preferred. There is no benefit in the prophylactic use of antibiotics in AP complicated by CT-proven pancreatic necrosis in the general population [323, 325]. In a meta-analysis, only imipenem significantly reduced the risk of pancreatic infection [325]. The use of imipenem/cilastatin is indicated in necrotizing AP, but dose adjustment in pregnancy should be considered even though there are currently no studies proposing the optimal dose [326]. Imipenem (N-formimidoyl thienamycin) is classified as category C although limited animal studies have shown no teratogenic risk or adverse fetal effects; data in humans are not available [327]. The pharmacokinetics of imipenem will change during pregnancy with a larger volume of distribution and faster total clearance from plasma [328]. The dose adjustment during pregnancy should be considered. Metronidazole passes freely across the placenta. However, studies from the 1990s do not show any association with an increased risk of teratogenic effects with metronidazole [329, 330]. Quinolones have been classified as category C because adverse effects have been noted in some animal studies. Due to the lack of evidence on beneficial effect of antibiotics, an even more conservative approach is recommended in pregnancy. Regardless of initial drug regimen, therapy should be modified to reflect the organisms recovered in blood cultures and the clinical status of the patient.
3.7.1.3 Continuous Renal Replacement Therapy
Continuous renal replacement therapy (CRRT), including a variety of blood purification techniques, which can remove water, nitrogenous wastes, and even inflammatory mediators, slowly and steadily, has been widely used in patients with critical conditions such as severe AP. CRRT is associated with significant improvement in pulmonary gas exchange, hemodynamic instability, azotemia control, fluid overload, and nutritional support in patients with MODS and acute renal failure [331]. Yekebas et al. [332] investigated the impact of different modalities on sepsis-induced alterations in the course of experimental AP, finding that CVVH (continuous veno-venous hemofiltration) can prevent sepsis and improve survival. Wang et al. [333] applied continuous high-volume hemofiltration in the treatment of patients with severe AP complicated with MODS and achieved satisfactory results. However, CVVH does not allow large molecules to pass through the hemofilter. Ronco et al. [334] proposed a peak concentration hypothesis of MODS and found that CVVH can be combined with plasma filtration absorption techniques to remove the excess circulating inflammatory mediators. HP (hemoperfusion) is another blood purification modality which can absorb pathogenic molecules in the blood flow circuit by sorbent materials installed in the hemoperfusion cartridge. Unlike CVVH, HP is more effective for removing middle and large molecules and toxins bound to proteins. For this reason, HP is wildly applied in drug overdose or intoxication cases [335]. Saotome et al. [336] reported a case of severe AP induced by alcohol abuse using CTR-001 direct HP cartridge to perform cytokine apheresis and demonstrated that this treatment can effectively reduce the serum levels of pro-inflammatory cytokines during severe AP. A pilot study performed by Kobe et al. [337] using direct HP (CYT-860) in patients with hypercytokinemia reported significant decrease in blood level of cytokines and improvement of PaO2/FiO2. Besides, a prospective, pilot, before-and-after self-crossover clinical trial carried out by Mao et al. [338] investigated the effect of coupled plasma filtration adsorption (CPFA) on the immune function of patients with MODS, finding that CPFA (using the resin cartridge HA-330I) was better than high-volume hemofiltration (HVHF) in increasing the ratios of anti-inflammatory to pro-inflammatory mediators, improving antigen presentation ability, and restoring leukocyte responsiveness. In only pregnant patient with AP, authors combined CVVH and broad-spectrum HP, assuming that HP can effectively remove excess endogenous and exogenous pathogenic molecules. The treatment was successful. After the first 3 days of treatment, the patient’s general condition significantly improved and her laboratory parameters virtually normalized [339]. Several studies suggest that toxic free fatty acids derived from plasma triglycerides induce local inflammation, leading to AP [144, 145]. HP may be more effective in clearing these fat-soluble factors because of its specific design. After receiving HP and CVVH treatments, the TG, CHOL, amylase, and lipase levels decreased dramatically (Fig. 3.13), explaining rapid recovery [339]. Moreover, the patient developed no side effects such as coagulopathy, hypotension, thrombocytopenia, or hypocalcemia.
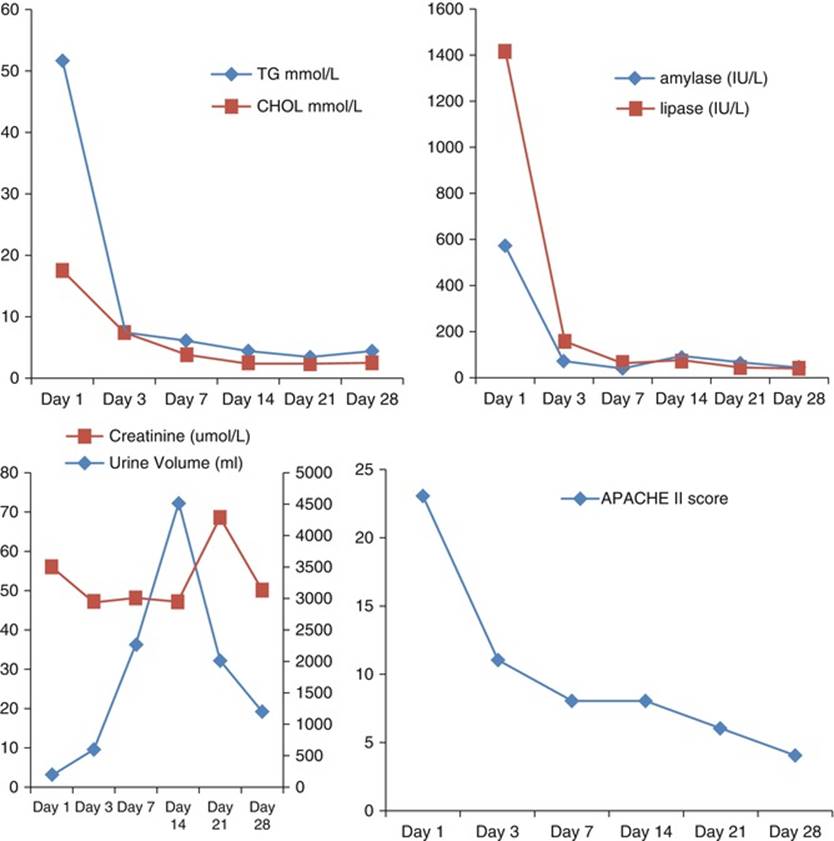
Fig. 3.13
The changing tendency of triglyceride (TG), cholesterol (CHOL), amylase, lipase, renal
Function, and APACHE II score during the treatment. CVVH and HP were initiated on day 1 and discontinued HP on day 3. CVVH was stopped on day 7. Normal range: TG 0.29–1.83 mmol/l, CHOL 2.8–5.7 mmol/l, amylase 25–125 IU/l, lipase 13–60 IU/l [339]
3.7.1.4 Intensive Care Unit Referral
In the general population, indicators for admission to ICU are [340]:
· Need for fluid resuscitation
· BMI >30 kg/m2
· Pleural effusions
· CRP >150 mg/dl at 48 h
· Necrosis of over 30 % of the pancreas
· Ranson criteria ≥3
Organ failure may occur in 50 % of patients. Early admission and management of critically ill obstetric patients in the ICU decreases maternal mortality and morbidity [341].
3.7.1.5 Hyperlipidemic/Hypertriglyceridemic Pancreatitis
While a moderate increase in plasma lipids in pregnancy is seen normally, severe HTG, predominantly chylomicrons and VLDL, is rarely encountered. This can however be a serious health problem if undetected as the predisposition to AP poses a significant risk of mortality for both mother and fetus [258]. Multiple treatment modalities have been suggested in the management of HTG-induced AP, but no clear consensus or accepted guidelines have been established [283].
The goals of management of pregnant patients with HTG-induced AP should include decreasing the serum triglyceride concentration and pancreatic activity while supplying maternal and fetal nutritional needs. Preconceptional control of TG levels may prevent or shorten the course of AP. Achieving these goals may be quite challenging. Therapies like plasma exchange, use of gemfibrozil, and extracorporeal lipid elimination may be effective in controlling triglyceridemia but do not meet the nutritional requirements of the mother and child [342, 343].
Avoidance of oral diet and intravenous administration of 5 % dextrose along with insulin often lead to a dramatic reduction of serum triglycerides [344]. A favorable effect of intravenous hyperalimentation (IVH) with nothing by mouth on AP with hyperlipidemia and pregnancy was referred by Weinberg et al. in 1982 [345]. The patient’s symptoms and triglyceride levels were only controlled after initiation of lipid-free total parenteral nutrition; however, the fetus developed intrauterine fetal growth retardation, while in a case by Ihimoyan et al. the patient delivered a healthy neonate with no compromise on fetal growth [346]. However, intravenous 5 % dextrose does not supply enough calories and could not be used for the extended duration required for enteric rest.
Dietary modification with a minimal fat oral diet should theoretically reduce chylomicron levels and decrease serum triglyceride concentration. However, it paradoxically increases the synthesis of very low-density lipoproteins which leads to enhanced production of triglycerides by the liver elevating the serum triglyceride levels [345]. The growing fetus requires essential fatty acids and amino acids for development and maturity of vital organs like the brain and lungs. TPN is an effective means of providing the necessary calories and essential amino acids for the growing fetus while controlling the maternal triglyceride concentrations and preventing the induction of AP. TPN with up to 10 % of calories of fat does not significantly increase maternal serum triglycerides. This is because of the systemic delivery of lipids which bypasses the liver where production of triglyceride-rich lipoproteins occurs. It also enables the supply of other nutritional supplements that are required by the fetus. The first reported case of TPN use in gestational HTG-induced AP was described by Weinberg et al. [345]. The patient’s symptoms and triglyceride levels were only controlled after initiation of lipid-free total parenteral nutrition; however, the fetus developed intrauterine fetal growth retardation. Parenteral nutrition in pregnancy should be managed preferably by an experienced clinical nutrition support staff. Other case of TPN resulted in the delivery of healthy neonate [346]. The major complications are related to central venous catheter placement and include pneumothorax, hemorrhage, and rarely death [347]. There has been a trend toward the use of peripherally inserted central catheters (PICC) because of lower rate of major complications and relative ease of insertion compared to central venous catheters [347]. PICCs should always be considered particularly in high-risk populations like pregnant women. Several studies however have shown a higher rate of minor complications like thrombophlebitis in patients with PICCs [321, 348]. PICC insertion is highly operator dependent, and lowest complication rates have been seen in the most experienced centers [349].
Additionally, lipid-lowering medications or plasma exchange has also been described in the literature as alternative therapies [277, 350, 351].
Here is one example of clinical course during lipid-lowering diet of hyperlipidemic AP during first and second pregnancy of the same patient (Figs. 3.14 and 3.15).
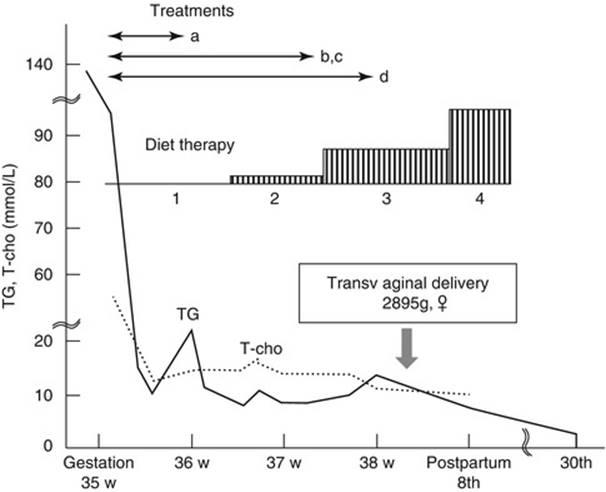
Fig. 3.14
Clinical course of the first pregnancy with type V hyperlipidemia (complicated with AP). Treatments were dietary therapy ((1), fasting for 11 d; (2), fat-restricted diet including 2.0 g of fat for 6 d; (3), fat-restricted diet including 10.0 g of fat; and (4), fat-restricted diet including 20.0 g of fat) and (a) a protease inhibitor (ulinastatin), (b) antibiotics (piperacillin, sulbactam/cefoperazone), (c) H2-blocker (famotidine), and (d) anticoagulation therapy (heparin) from admission. T-cho total cholesterol, TG triacylglycerol [289]
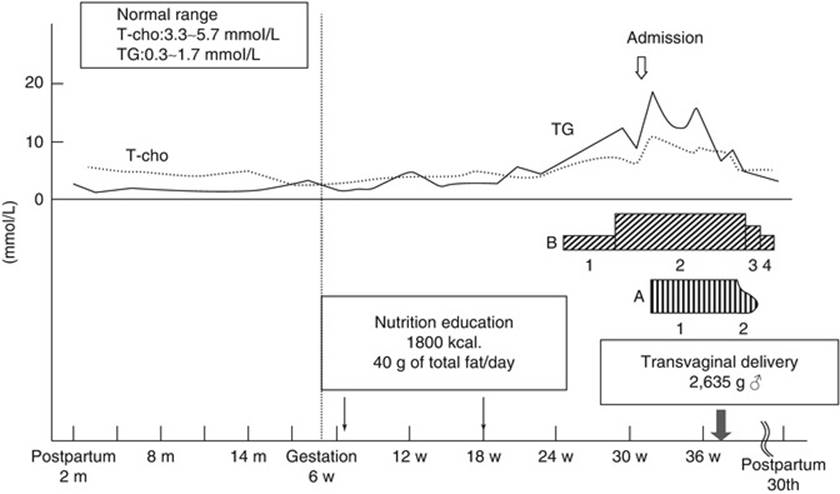
Fig. 3.15
Clinical course of the second pregnancy with type V hyperlipidemia of the same patient (without AP). Treatments were (A) dietary therapy ((1), fat-restricted diet including 20 g of fat daily; (2), fat-restricted diet including 15 g of fat daily) and (B) u-3 fatty acids ((1), 0.9 g; (2), 2.7 g; (3), 1.8 g; (4), 0.9 g). T-cho total cholesterol, TG triacylglycerol [289]
Insulin
Insulin activates LPL, an enzyme that accelerates chylomicron degradation into glycerol and FFAs. Insulin has also been shown to increase messenger RNA levels of LPL in animal adipose cells in vitro [352]. A few case series have demonstrated successful management with insulin monotherapy in the setting of HTG-induced AP [353–355]. Insulin (with administration of glucose) is safe and effective in the treatment of HTG-induced AP, even in patients without DM [353, 354]. Intravenous insulin may be more effective than subcutaneous insulin in severe cases given the potential limitations of absorption with the subcutaneous route. Intravenous insulin may be given as a continuous infusion starting with 0.1–0.3 U/kg per hour with titration as required.
Heparin
LPL is a ubiquitous, endothelially bound lipolytic enzyme. Intravenous heparin uncouples the enzyme from its endothelial anchor, thus stimulating the release of endothelial LPL into the circulation [356]. It has been used without insulin to successfully manage HTG [351, 357, 358]. A heparin dose of 10,000 U/day seems to be safe, as evidenced by a normal APTT [351]. Despite the success of intravenous heparin in combination with insulin in HTG management, heparin has come under greater scrutiny. Intravenous heparin does cause an initial rise in circulating LPL levels but is quickly followed by increased hepatic degradation [359]. This degradation contributes to further depletion in plasma stores of LPL and may ultimately potentiate the subsequent accumulation of circulating chylomicrons [360]. A further potential hazard is the risk of transformation into hemorrhagic AP. Heparin has been used to lower triglyceride levels. There are reports in the literature in which heparin therapy for AP has been seen to be both therapeutic and safe [158, 351]. Although heparin is an option to control triglyceride levels, women with AP are at risk for life-threatening hemorrhage within the pancreas, and therefore heparin could conceivably worsen the ultimate outcome.
Extracorporeal Elimination of Triglyceride-Rich Lipoproteins
Plasma apheresis for severe HTG was first reported in 1978 [361]. There are several controversial aspects to plasma apheresis including technical controversies as to whether plasma exchange is more efficacious than double membrane filtration. In clinical practice, appropriate and timely access to plasma apheresis is rare, and the evidence supporting its utility in HTG-induced AP has been confined to isolated case reports [283]. Plasmapheresis is successful in lowering TG levels. However, in the absence of a comparison with standard treatment (heparin or insulin infusion and lipid-lowering agents), the effect of plasmapheresis on lowering the morbidity and length of stay of patients with HTG-induced severe AP is uncertain and warrants further investigation into its efficacy. Another problem is the small number of patients for making strong prospective studies [142].
In the follow-on phase, as the triglyceride levels usually rapidly decrease within 48 h of the onset of AP, an accurate diagnosis outlining the etiology of the HTG and prevention of further attacks takes priority. Treatment modalities include dietary intervention initially, followed by drug therapy that includes a spectrum of pharmacological agents such as the fibric acid derivatives, omega-3 fatty acids, and nicotinic acid derivatives, insulin, and/or heparin treatment. When the etiology is LPL deficiency, pharmacological therapy is relatively ineffective. In this setting, the role of dietary restriction is central.
First reports of long-term extracorporeal elimination of triglyceride-rich lipoproteins by three modes of treatment (plasma exchange, immunospecific apheresis, and a combination of both treatments) were by Swoboda et al. [342, 362]. The loss of immunoglobulins remained acceptable.
Drug Therapy
Fibrates
Fibrates effectively lower triglyceride levels by 40–60 % and raise HDL-C levels [363, 364]. The triglyceride-lowering effects of fibrates have been attributed to enhanced catabolism of triglyceride-rich particles and reduced secretion of VLDL [363]. Nicotinic acid lowers triglyceride levels by 30–50 % [364] by reducing VLDL secretion, but flushing and gastric upset are prominent side effects [137]. Omega-3 fatty acids, studied in a prospective, double-blind, placebo-controlled trial, have proved capable of lowering high triglycerides (5.5–22.5 mmol/l, 500–2,000 mg/dl) by 45 % [365]. Fenofibrate 200 mg and niacin 500 mg daily and omega-3 fatty acids are relatively ineffective in patients with primary (genetic) HTG.
All fibric acid derivatives are renally excreted and can consequently display a prolonged plasma half-life of several days in severe renal impairment cases. Since all fibric acid derivatives have a high degree of protein binding (>95 %), none are removed by hemodialysis [366]. Gemfibrozil is the most frequently prescribed lipid-lowering agent for patients with renal insufficiency, as it is least dependent on renal excretion [367]. Gemfibrozil has however been reported to cause rhabdomyolysis in patients with compromised renal function. Although most incidences of rhabdomyolysis occur in patients taking both gemfibrozil and HMG-CoA reductase inhibitor [368, 369], rhabdomyolysis induced by gemfibrozil alone has also been reported [370]. The pathophysiologic mechanism of gemfibrozil toxicity remains unknown [367, 370], despite its relatively frequent occurrence.
Statins
Although statins have been identified as potential teratogens on the basis of theoretical considerations and small case series, the available evidence is far from conclusive. In fact, epidemiological data collected to date suggest that statins are not major teratogens. The actual risk for an exposed pregnancy seems to be small, if present at all, and does not by itself warrant termination of pregnancy. Nevertheless, given the scarcity of available data, it is still advisable to avoid use of these drugs in patients who are planning pregnancy in order to reduce the risks as much as possible [371, 372]. Statins are used for lowering cholesterol in general population but there are also cases of statin-induced AP (see Sect. 3.3).
Prophylactic Plasma Apheresis
Despite legitimate safety concerns regarding the use of fibrates, nicotinic acid, and omega-3 fatty acids in pregnancy, case reports of patients being prescribed these drugs suggest that they may be safe from both the maternal and fetal perspective [373]. As always, a careful risk-benefit analysis needs to be made when considering these therapeutic agents. Other treatment options for unresponsive patients include insulin/heparin therapy and plasma apheresis [374]. “Prophylactic” plasma apheresis has been proposed as a preventive treatment for severe uncontrolled HTG resistant to diet and drug therapy, and Piolot et al. have reported a reduced incidence of AP in such patients treated with prophylactic plasma apheresis at 4-week intervals [375].
Because pregnancy might lead to the exacerbation of hypertriglyceridemia in patients with familial hyperlipidemia, delivery is advocated because it is estimated to lower lipid levels by 15–20 % within 24 h and return them to prepregnant levels by 6 weeks postpartum [129, 342, 376, 377]. Others believe that there is no improvement in maternal outcome and that fetal and maternal health may be jeopardized by complicated delivery. In preterm pregnancies, delivery is recommended only if maternal or fetal condition deteriorates taking into account history of episodes of AP, gestational age, and the presence of persistent disease. The mode of delivery should be determined by obstetric factors [378].
Dietary Approach
The patient will be required to work closely with the dietician who must focus on a low-fat diet; this is primarily to cut off the production of chylomicrons [379]. Severe HTG can be exacerbated during pregnancy, imposing an even greater need for strict adherence to a low-fat diet, usually ≤20 g/day and sometimes as low as ≤10 g/day. Total caloric intake should be adequate, and what little fat is ingested should contain omega-3 and omega-6 fatty acids [289]. Medium-chain triglyceride-rich foods, such as coconut oil, can be used for cooking, as they are absorbed directly into the portal vein without becoming incorporated into the chylomicron triglyceride. The success of this therapy depends on the individual’s acceptance of the fat restriction, including both unsaturated and saturated fat.
Antioxidant therapy has been used by some experts in the management of HTG in patients with recurrent AP due to familial LPL deficiency [380]. This antioxidant cocktail significantly reduced the recurrence of AP in these individuals. It is thought that they neutralize free radicals resulting from chylomicronemia-related microvascular ischemia and thereby prevent the potential damage to pancreatic acinar cells and the resulting AP. A suggested pharmacological dose utilizes Antox® version 1.2 that contains antioxidants such as α-tocopherol, β-carotene, vitamin C, organic selenium, and methionine. The dose is two tablets three times a day.
3.7.1.6 Pancreatitis Secondary to Hyperparathyroidism
Hypercalcemia is the underlying cause of many of the maternal and fetal risks associated with hyperparathyroidism, and management normalizes the calcium level, decreasing these complications. Rajala et al. theorized that high-dose magnesium might be a therapeutic alternative for hyperparathyroidism in pregnancy [59].
In the reported cases, the hypercalcemia was managed with medical intervention, usually with magnesium infusion [44, 64]. When magnesium proves ineffective, other agents such as phosphate-of-soda enemas, oral phosphates, calcitonin, and loop diuretics have been used with varied success [43, 59]. In the past mithramycin was used but is currently contraindicated secondary to teratogenic effects [57]. Corticosteroids have also been used to decrease the absorption by the gastrointestinal tract but have shown minimal effect when the hypercalcemia is secondary to hyperparathyroidism [57]. Patient in one case report had undetectable levels of parathyroid hormone while receiving magnesium therapy, serum calcium never normalized, and serum vitamin D-1,25 was elevated. It is possible that a compensatory increase in serum vitamin D-1,25 in response to the decrease in the parathyroid hormone level caused by the magnesium increased calcium absorption from the gastrointestinal tract. Therefore, for some patients, because of persistent hypercalcemia, magnesium sulfate might not be a viable treatment option for hyperparathyroidism during pregnancy [44].
3.7.2 Surgical Treatment
3.7.2.1 Anesthetic Considerations
Before surgical treatment, anesthetic considerations are important in pregnant population. The choice of anesthesia for Cesarean delivery is not clearly defined. Hemodynamic stability may be better with general anesthesia and may be of benefit in the presence of sepsis and use of anticoagulants such as heparin. General anesthesia was chosen to allow greater hemodynamic control and facilitate management of the acid-base status especially in patients receiving epoprostenol for CVVH. Postoperative ventilatory support on ICU permits resolution of the acidosis.
3.7.2.2 Surgical Principles
Surgical treatment has two aspects, which include operative intervention for the AP itself and surgical management of associated local (biliary tract disease) or distant (PHPT, HTG, etc.) cause of the AP during attack or once acute inflammation subsides.
Since the first study published in 1963 [381] (see Chap. 2), the dilemma, whether or not to treat pregnant patients with gallstones conservatively, still exists. Risk of conservative treatment includes risk to the fetus due to recurrent episodes, complications of gallstones, and risk of malnutrition caused by lack of oral intake. Conversely, surgical treatment carries risk to the fetus from surgery and anesthesia and risk specific to laparoscopic surgery. Laparoscopic cholecystectomy (once considered contraindicated during pregnancy) [382] is currently the best treatment for the patients who failed to respond to conservative management or because of recurrent episodes [315, 383]. Benefits of laparoscopy during pregnancy appear similar to those nonpregnant patients including less postoperative pain, less postoperative ileus, significantly reduced hospitalization, and decreased narcotic use and quick return to a regular diet and faster recovery. Other advantages of laparoscopy include less manipulation of the uterus and detection of other pathology that may be present and because of early mobility reduced risk of postoperative deep vein thrombosis [254]. Cholecystectomy is considered safe at all stages of pregnancy and may be performed in any trimester of pregnancy without any increased risk to the mother or fetus [315, 383]. Historical recommendations to delay surgery until the second trimester or gestational age limit of 26–28 weeks of pregnancy have been refuted. Laparoscopy in pregnancy was connected with the fear of damage to the gravid uterus upon Veress or trocar insertion; technical difficulty in performing the surgery with the presence of an enlarged, gravid uterus; and the concern of fetal acidemia due to decreased uterine blood flow because of increased intra-abdominal pressure from insufflation and possible fetal carbon dioxide absorption [13]. Also, maternal venous return secondary to increased intraperitoneal pressure from CO2 insufflation could be present. The use of a uterine manipulator is contraindicated in pregnancy. At the beginning of 2011, The Society of American Gastrointestinal and Endoscopic Surgeons (SAGES) updated its guidelines for laparoscopy during pregnancy [383]. A report from 1999 suggests that the risk of fetal wasting and teratogenicity from gastrointestinal operation during pregnancy is minimal [384]. However, some precautions should be followed: the use of an open technique for the insertion of the umbilical port, avoiding high intraperitoneal pressures, using of left lateral position to minimize aortocaval compression, avoiding rapid changes in the position of the patient, and using electrocautery cautiously and away from the uterus [254].
Early cholecystectomy should be performed in patients with mild biliary AP, while patients with SABP should undergo this procedure within 4–6 weeks after hospital discharge [320].
Early surgery for necrotizing AP is not recommended, and it should be delayed as long as possible [25, 385]. Antibiotics are not indicated. The data suggest that remission could be achieved in most patients (78.9 %) with the conservative treatment. Therefore, the indications for surgery [25, 290, 317, 318, 340, 385] and antibiotics are as follows:
· Pancreatic necrosis and infection (3–4 weeks after the onset of symptoms)
· Large intra-abdominal exudates
· Clinical deterioration (Fig. 3.16)
Fig. 3.16
Prominent pus coating over the omentum, intestine, and colon, peripancreatic fatty necrosis, and excessive solid mass of swollen inflamed pancreas are noted. An area of impending perforation (arrow) over the transverse colon is also visible [287]
Minimal invasive surgical techniques are new in the management of AP with only a few relatively small series reported to date [386].
Percutaneous drainage, endoscopic drainage, or surgical procedures can be selected in accordance with the conditions of individual cases. Some reports show that the decompression and percutaneous drainage help avoid or delay surgery in most patients with SAP [25, 318, 385]. For patients with pancreatic abscess, drainage is recommended [317, 387]. Necrosectomy should be performed as late as possible and it can be also performed in the Cesarean section.
Pancreatic Pseudocysts
In nonpregnant patients, pancreatic pseudocysts are most frequently associated with alcoholic AP. The recommended management is to observe asymptomatic pseudocysts and those present for less than 6 weeks, as these have a 30–40 % rate of spontaneous resolution [388]. Internal drainage is preferred for symptomatic cysts >5 cm in diameter or those present for over 6 weeks, as these have a 3 % chance of resolution and a 57 % risk of rupture, infection, hemorrhage, or obstruction [389]. Internal drainage can be accomplished by anastomosis to the duodenum, a Roux-en-Y limb of the jejunum, or the posterior wall of the stomach. When the pseudocyst is infected, located greater than 1 cm from the bowel, or has a cyst wall insufficiently thick to permit anastomosis, percutaneous drainage can be considered despite a recurrence rate of 20–70 % [389, 390] and the potential risks of infection [391] and fistulization [390].
Endoscopy is used to avert surgery in high-risk cases [392]. An indwelling transgastric or transduodenal pigtail catheter may be placed or a more permanent internal fistula created by cautery between the cyst and the adjacent viscus. This last procedure, while reportedly effective, carries a significant risk of bleeding and depends on the skill of the endoscopist. Endoscopic transpapillary stenting is another alternative for patients with a partial pancreatic duct disruption and a communicating pseudocyst [388]. Limited information is available to guide the management of pancreatic pseudocysts in pregnancy. Many case reports of pancreatic pseudocysts occurring during pregnancy are limited by the lack of ultrasound measurements. Recommendation is that uncomplicated pseudocysts should be surgically managed in the postpartum period [393, 394].
Table 3.5 compiles the available information about presentation, etiology, management, and outcomes in these cases since 1980, when ultrasound, amylase, and lipase became widely available [395].
Table 3.5
Characteristics of pancreatic pseudocysts in pregnancy [395]
|
Author |
Age/gravida, para/EGA |
EGA when cyst was found |
Size and location |
Etiology |
History of cyst |
Management |
Delivery |
Morbidity and mortality |
|
Nies |
26 years old/gravida 2, para 0/34 weeks |
PP |
6 cm, tail |
Lipid |
Remained the same size |
Conservative |
Vaginal PT |
Pleural effusion versus pneumonia; ICU |
|
Glueck |
gravida 1, para 0/39 weeks |
PP |
N/A |
Lipid |
N/A |
Conservative |
Vaginal T |
Ruptured cyst, shock, ICU |
|
Chen |
28 years old/gravida 2, para 1/31 weeks |
31 weeks |
4 cm, tail |
Lipid |
Shrunk to 2 cm spontaneously |
Conservative |
N/A |
None |
|
Bar-David |
28 years old/gravida 1, para 0/17 weeks |
29 weeks |
N/A |
Lipid |
Infected pancreatic pseudocyst noted at 29 weeks disappeared by 37 weeks |
Conservative |
Vaginal T |
Septic shock, pneumonia with mechanical ventilation, TPN, ICU |
|
Stowell |
37 years old/gravida 3, para 2/31 weeks |
31 weeks |
5 cm, head; 8 cm, tail |
Alcohol |
Cyst in head resolved; tail cyst remained the same size |
Conservative; PP cystgastrostomy |
Vaginal T |
TPN for 5 weeks; FGR |
|
Ryan |
35 years old/6 weeks |
Present before pregnancy |
6 cm, tail |
Idiopathic (alcohol?) |
Grew to 7 cm; shrunk to 2 cm after stent; grew to 4 cm when stent fell out; stent replaced and cyst stable until 2 months PP |
Cystogastric stent placed by ERCP at 17 weeks; transpapillary stent placed at 35 weeks; PP pancreatectomy |
Vaginal T |
None |
|
Beattie |
20 years old/gravida 1, para 0/24 weeks |
27 weeks |
13 cm, posterior to stomach |
Anatomic |
1 l of fluid aspirated, then daily aspirations |
Percutaneous drainage at 24 weeks; PP hepatico-jejunostomy |
C/S PT |
Cyst collapsed around drain |
|
Eddy |
24 years old/gravida 3, para 0/26 weeks |
26 weeks |
6 cm, tail |
GS |
Grew to 15 cm |
Conservative; PP pancreatectomy |
C/S T |
TPN for 3 weeks; splenectomy |
|
Swisher |
N/A |
N/A |
6 cm |
Non-GS (alcohol or idiopathic) |
N/A |
Percutaneous drainage |
N/A |
N/A |
|
Hess |
25 years old/gravida 1, para 0/31 weeks |
PP |
N/A |
Hyperparathyroidism |
N/A |
Conservative |
Induced Vaginal PT |
Initial mental status changes, hypocalcemia after neck surgery renal failure |
G gravity, P parity, EGA estimated gestational age, N/A information not available, GS gallstone pancreatitis, PP postpartum, C/S cesarean section, T term, PT preterm, TPN total parenteral nutrition, ICUtransfer to intensive care unit, FGR fetal growth restriction
Pancreatic pseudocysts complicate 6.9 % of cases of AP and are almost never found in association with gallstone AP in pregnancy [396]. Six of the eight women of known gestation were primiparous. Alcohol was the etiology in less than a quarter of the cases. Gallstones, by far the most common cause of AP during pregnancy [15], accounted for only one case. Hyperlipidemia accounted for almost half of the cases. One patient was found to have Type I familial hyperlipoproteinemia [74] and another type V [257]. Two other cases were not fully diagnosed [397, 398]. Although their values are suggestive of type V hyperlipoproteinemia, it would be unwise to presume the diagnosis because type I may masquerade as type V in times of stress [257]. It can be concluded that non-gallstone AP in pregnancy has been shown to be significantly more prone to pseudocyst formation [24].
The natural history of pancreatic pseudocysts appears similar to that in nongravid patients because pseudocysts less than 5 cm shrank or resolved while those greater than 5 cm remained the same size or enlarged. In three cases, intervention was performed antepartum: two patients underwent percutaneous drainage and one was stented endoscopically. Percutaneous drainage of one 6 cm cyst had an unspecified result [35]; another, of a 13 cm cyst at 24 weeks gestation, showed initial relief of symptoms and reduction in cyst size with daily aspirations. However, in the latter case, the cyst subsequently collapsed around the drain, the cyst reaccumulated, and the patient had a preterm delivery [399]. One patient had a cystogastric stent placed with ERCP at 17 weeks gestation. In this case, the cyst shrank from 7 to 2 cm after the stent placement but grew rapidly to 4 cm once the stent fell out. A transpapillary stent was then placed at 35 weeks gestation, which stabilized the cyst until delivery at term [392]. Two cases of successful vaginal delivery were reported in women with 8 and 4 cm pseudocysts [392, 400], but concern over the risks of Valsalva during labor seems warranted in light of one report of a pancreatic pseudocyst rupturing during vaginal delivery, causing hypotension and shock and necessitating intensive care [257]. With the exception of this case and Tang et al.’s [395] patient’s splenectomy, all cases of morbidity reported in Table 3.5 resulted from AP or its underlying cause, rather than from the pseudocyst. Cystic neoplasms are even less common than pancreatic pseudocysts but can occur during pregnancy [401–403] and may mimic inflammatory fluid collections. Especially notable is papillary cystic neoplasm, which occurs predominantly in young women and whose growth may be enhanced by progesterone [402]. Factors to help differentiate inflammatory from neoplastic fluid collections include serum amylase level, which is elevated in 50–75 % of pseudocysts [404] and 5 % of pancreatic neoplasms[390]; cyst amylase level, which is normal in neoplasm and elevated in inflammatory fluid collections; ultrasonography demonstrating multiple cysts or internal septa that suggest neoplasm rather than inflammation [404]; and a history of AP or antecedent factors suggesting an inflammatory cause such as gallstones, hyperlipidemia, or alcoholism. There is only one case published of mucinous cystadenoma of the pancreas in pregnancy causing AP [77]. The presence of estrogen and progesterone receptors in the stroma of these cysts may cause accelerated cystic growth during pregnancy [405].
Primary Hyperparathyroidism
PHPT in pregnancy represents a significant risk for maternal and fetal complications that cannot be predicted by duration of symptoms or serum calcium levels. Persistency of symptoms and mainly calcium levels above 11 mg/dl are considered indications for surgical treatment, regardless of the trimester of pregnancy. Truong et al. favor this approach, underlying the need to weigh the benefits and risks of the surgical procedure [406]. Successful surgical management of PHPT eliminates the risk of PHPT deterioration postpartum and the risk of neonatal tetany. Surgery during the third trimester has been reported to increase the risk for preterm labor [406] along with other severe complications, although the occurrence of these complications could be due to the long-standing hypercalcemic status of both the mother and the fetus [406, 407]. Surgical treatment should be considered early, and a minimally invasive approach with ultrasound is best suited to mitigating the risk to the mother and fetus. Equally important, Tc-99 m sestamibi imaging may be used safely for localization of the parathyroids after negative cervical explorations. Although the treatment of choice in PHPT is the surgical intervention performed preferably during the second trimester of pregnancy, the patient declined this option of treatment because of the fear of potential complications to the fetus [105, 408]. Some authors claim that parathyroidectomy in pregnancy is associated with a slightly increased risk of a spontaneous abortion [105, 408]. Others did not have any maternal or fetal complications after surgery [407].
Medical therapy in pregnancy for symptomatic PHPT has been discouraged, due to safety issues of drug therapy and the suboptimal control of serum calcium which leads to a high fetal loss rate [42]. However, in symptom-free patients or those with no radiologically identifiable parathyroid adenoma or those with mild hypercalcemia diagnosed in third trimester may be managed medically, postponing the operation until after delivery. If conservative management is considered intensive, maternal and fetal surveillance should be initiated. Medical therapy primarily involves stabilizing the patient with hydration; limiting calcium intake; correcting electrolyte imbalance; and administration of magnesium sulfate, oral phosphate, and calciuretic diuretics [39, 80]. If the patient with the indication for operative treatment chooses medical therapy for PHPT, calcitonin is administered. It is considered the safest conservative treatment option in patients with hypercalcemia in pregnancy [42]. The safety of calcitonin during gestation probably results from its negligible passage through the placenta [409, 410]. The choice of this agent was also supported by suggested but not fully supported beneficial effects of calcitonin in the management of AP observed in nonpregnant patients [411]. Although presently no parameters are known that may predict the outcome of PHPT, in order to limit the risk of serious complications, most recommend maintaining total plasma calcium <3.0 mmol/l [412]. Exceeding this level is considered an indication for calcitonin administration. Interestingly, although calcitonin treatment may be associated with the development of tachyphylaxis [413], only tendency to tachyphylaxis was observed in the third trimester, and it disappeared after introduction of oral phosphates. This fact may be explained by administration of this agent only if plasma calcium levels exceeded the established threshold. Moreover, there was no nausea, vomiting, diarrhea, flushing, injection site reactions, and any other side effects associated with calcitonin treatment [413]. Although some animal studies suggested low birth weight in offspring of dams treated during pregnancy with high doses of calcitonin [411], this was not a published case, and infant’s birth mass was within normal limits. Interestingly, Horjus et al. have shown the benefits of combined administration of calcitonin and cinacalcet in pregnancy and puerperium [412]. In one case the only symptom experienced by the neonate after delivery was transient hypocalcemia, which disappeared shortly after the beginning of intravenous calcium administration. The presence of transient mild hypocalcemia probably resulted from increased calcium levels in fetal plasma, inhibiting parathyroid PTH synthesis and release during the pregnancy [409, 410, 414]. Interestingly, neonatal hypocalcemia was absent in the patient’s subsequent pregnancy, taking place after parathyroidectomy, which supports the recommendations that surgery should be considered the treatment of choice in young hyperparathyroid pregnant women or desiring pregnancy [105, 408]. The effectiveness of surgical intervention in the prevention of fetal and neonatal complications clearly indicates that PHPT, if successfully treated, cannot be regarded as a contraindication for consecutive pregnancies.
Rajala et al. theorized that high-dose magnesium might be a therapeutic alternative for hyperparathyroidism in pregnancy [59]. It is possible that a compensatory increase in serum vitamin D-1,25 in response to the decrease in the PTH level caused by the magnesium increased calcium absorption from the gastrointestinal tract. Therefore, for some patients, because of persistent hypercalcemia, magnesium sulfate might not be a viable treatment option for hyperparathyroidism during pregnancy [44].
Identification and removal of a hyperfunctioning parathyroid tumor, including most mediastinal tumors, can usually be accomplished by cervical exploration [415]. Occasionally, because of their deep location in the chest, median sternotomy is necessary. In a series of 38 patients from the Mayo Clinic who underwent median sternotomy for removal of a parathyroid adenoma, 21 % had postoperative chest complications [416, 417].
Six of nine (67 %) reported cases up to 1996 were diagnosed during the third trimester of pregnancy, of which four underwent neck exploration after delivery. The three cases diagnosed before the third trimester underwent surgery during the second trimester. Parathyroid adenoma was the underlying disease in eight cases; one case was caused by a parathyroid carcinoma [43].
3.7.3 Therapeutic Delivery
Because pregnancy might lead to the exacerbation of hypertriglyceridemia in patients with familial hyperlipidemia, delivery is advocated because it is estimated to lower lipid levels by 15–20 % within 24 h and return them to prepregnant levels by 6 weeks postpartum [129, 342, 376, 377]. Both cholesterol and triglyceride concentrations decreased significantly within 24 h of delivery, and this was reflected in all lipoproteins. However, while triglyceride levels continued to decrease rapidly returning to nonpregnant levels during the puerperium, cholesterol in low-density lipoprotein remained elevated for at least 6–7 weeks postpartum [121]. Others believe that there is no improvement in maternal outcome and that fetal and maternal health may be jeopardized by complicated delivery. Early delivery may allow more aggressive treatment of the mother, including the use of lipid-lowering agents or plasmapheresis. Delivery also makes it easier to resuscitate the mother and may prevent fetal distress if AP deteriorates rapidly. Hypotension during Cesarean section, however, can exacerbate AP due to hypoxia and should be avoided. Also, some lipid-lowering medications, such as statins, which are contraindicated in pregnancy (FDA Class X), can be introduced after delivery.
In preterm pregnancies, delivery is recommended only if maternal or fetal condition deteriorates taking into account history of episodes of AP, gestational age, and the presence of persistent disease. The mode of delivery should be determined by obstetric factors.
In AP due to gallstones or of unknown cause, rapid resolution has been described after intrauterine death of the fetus [21] and after vaginal delivery with a live birth [418]. In AP associated with hyperlipidemia, the effect of delivery on the decline of plasma triglyceride levels can be immediate and dramatic [419]. However, the morbidity from AP does not always fall proportionally. Rapid improvement [419] and worsening of hyperlipidemic AP after delivery [397] have both been described, and AP can progress to maternal death [258].
Stimulation of fetal lung maturation in the critical period for delivery is important. Standard therapy includes corticosteroids. In experimental animals, administration of 17 beta-estradiol accelerates fetal lung maturation and stimulates surfactant production: the hormone increases the amount of surfactant in fetal lung lavage, increases the rate of phosphatidylcholine synthesis, depletes fetal lung glycogen, and accelerates morphological maturation of the fetal lung. Both estrogens and glucocorticoids stimulate fetal lung choline-phosphate cytidylyltransferase in a number of in vivo and in vitro systems, and there is increasing evidence that this enzyme may be of particular importance in the regulation of phosphatidylcholine synthesis. Estrogen appears to increase the catalytic activity rather than the amount of choline-phosphate cytidylyltransferase. This action of estrogen is mediated by phospholipids [420].
3.7.4 Obstetric Treatment
Recommendation is to inhibit uterine contractions to reduce premature labor [78]. If the disease was improved, we would terminate the pregnancy to ensure the safety of the mother and fetus. Indications for termination of pregnancy included (1) obvious signs of miscarriage or premature birth, (2) fetal distress or intrauterine deaths, and (3) if the fetus can survive after birth, usually chosen Cesarean section timely; if the fetus is dead, make an induction of labor [78].
Mode of delivery in patients with associated pancreatic pseudocysts should be determined on a case-by-case basis [395]. While a case of pseudocyst rupture during Valsalva efforts during delivery is reported, there is a paucity of data to guide delivery recommendations.
3.8 Prognosis
3.8.1 General Considerations
The outcome and prognosis depend, as in general population, on the severity of the AP, cause of AP, and fetal outcome partly depending on the trimester of occurrence of AP. In one series of 53 patients with AP during pregnancy published in the medical literature before 1951, the vast majority of diagnoses were made during surgery and/or on autopsy. In only three patients was the diagnosis made based on clinical grounds [421]. In 1973 Wilkinson reviewed 98 cases of AP during pregnancy, 30 patients died (37 %), significantly higher than that in nonpregnant patients then: 12–33 % [16]. In the same study, the perinatal mortality was high -37 % [16]. Joupilla et al. in 1974 have quoted a maternal mortality rate of 5–15 % [422]. Maternal and perinatal mortality due to AP during pregnancy is variously reported to vary from 20 to 50 %, and most occur during the third trimester [15, 16, 20]. A more recent reviews found a maternal mortality rate of less than 1 % for AP during pregnancy of all etiologies [23, 24, 73, 78]. Swisher et al. (30 pregnant women) and Ramin et al. (43 pregnant patients) reported no maternal deaths [15].
There was a 72 % relapse rate during the same pregnancy [35]. Non-gallstone AP as a whole had worse outcomes than simple gallstone AP. One of the explanations for better maternal and fetal outcomes is higher trend of cholecystectomy in pregnant women who developed AP or symptomatic cholelithiasis/cholecystitis especially in early trimesters [23, 423]. This supports the high relapse of biliary colic and its complications during pregnancy [424]. Also, in earlier studies, the recurrent AP risks were reported to be 50–70 % during the same pregnancy [25, 35]. Traumatic, hyperlipidemic, and alcohol-induced AP had particularly poor outcomes [24].
Aging is associated with increased severity of AP characterized by augmented and prolonged pancreatic inflammation and the presence of multiple extra-pancreatic sequels including thrombosis [425]. Incidence rates for AP in general population in England with admission to hospital rose in both men and women from 1963 to 1998, particularly among younger age groups. This probably reflects, at least in part, an increase in alcoholic AP. Mortality after admission has not declined since the 1970s. This presumably reflects the fact that no major innovations in the treatment of AP have been introduced. AP remains a disease with a poor prognosis during the acute phase [426].
Fetal mortality rates quoted in the literature have improved in the last 20 years, as earlier studies reflected fetal deaths after preterm delivery which has reduced as a result of improved neonatal care [15]. One study found that 74 % of patients suffering AP delivered full-term healthy infants with 10.5 % of fetal mortality rate noted overall. A more recent study found a perinatal mortality rate of 3.6 % [24]. It is, however, important to note that these publications do not distinguish between the various causes of AP during pregnancy. In an 11-year retrospective study from 1995 describing 43 pregnant women with AP, there was no maternal mortality, but there were six preterm deliveries, and only two of these six infants survived [15]. ERCP had not been incorporated into the standard management at that time. Recently, the percentage of fatal outcomes of AP in general population has been less than 5 % [427], and, currently, data show no maternal mortality and 19 % of preterm labor but with fetal mortality of 5 % (single-center experience over 10 years with 21 patients with 34 attacks of AP) [25].
Patients who developed AP in the first trimester had the lowest percentage probability to reach term pregnancy (60 %), highest risk of fetal loss (20 %), and preterm delivery (16 %) [23, 27]. Maternal and perinatal mortality due to AP during pregnancy is variously reported to vary from 20 to 50 % and most occur during the third trimester [15, 16, 20]. It should be kept in mind that preterm labor may occur in as many as 60 % of patients who have AP in late pregnancy; therefore, gestational age is a primary determinant of perinatal outcome. In the past decades, high perinatal mortality rate, up to 50 % [16], secondary to AP resulted from neonatal deaths after preterm delivery but with early recognition and better supportive treatment of AP and improvements in neonatal intensive and supportive care play important role in premature babies’ survival. More recent publications report no maternal mortality and 0.57–4.7 % fetal mortality [25, 254]. The mechanisms of demise include, also, placental abruption and profound metabolic disturbance, including acidosis. Serious maternal pulmonary complications are often associated with AP. The destruction of pulmonary surfactant by degradation of lecithin accompanied with an increase of serum PLA2 results in increased capillary permeability of the lung and the elevation of surface tension. This leads to pulmonary edema which is considered an important etiopathogenic factor of acute respiratory insufficiency [428]. It is not known whether this mechanism is responsible for perinatal morbidity and mortality. All this highlights the importance of regular fetal monitoring and consideration of delivery if the maternal disease is deteriorating. AP complicated by DIC usually occurs in the third trimester and is particularly associated with poor fetal and maternal outcomes [23]. Post-ERCP AP does not adversely affect pregnancy-related outcomes [23, 423]. Hepatobiliary diseases can result in maternal and fetal physiological dysfunction, leading to adverse pregnancy outcomes, such as prematurity and low birth weight [423]. Thus, it is particularly important to identify hepatobiliary disease early during pregnancy and to intervene appropriately as early as possible.
Another point for discussion is AP in the postpartum period. It is known that estrogen is one of the initiators of different causes of acute pancreatitis and that some of them are cured with Cesarean section as additional measure not only to save the newborn. Opinion is that postpartum period should be analyzed separately. One of the examples is that biliary sludge and gallstones form in up to 31 and 3 % of pregnant women, respectively, with the sludge frequently resolving postpartum [237, 429, 430]. Langmade and Edmondson in 1951 observed a 73 % recurrence rate in the puerperium [421].
3.8.2 Primary Hyperparathyroidism
3.8.2.1 Maternal Outcome
Increased risk for mother complications apart from AP is also present including nephrolithiasis, weakness, lethargy, muscle cramps, and bone disease [57]. If the mother is treated medically to term (or if spontaneous or elective abortion occurs), the mother should be monitored for hyperparathyroid crisis postpartum. It consists of nausea, vomiting, weakness, and central nervous system retardation which could progress to uremia, coma, and death [57]. Sudden worsening of hypercalcemia can result from the loss of the placenta (active placental calcium transport may be somewhat protective) and dehydration [57]. Up to 1998 and from the 13 patients, maternal mortality was 15 % [66]. Mortality seems to be related to delayed resection of parathyroid tumor [66].
3.8.2.2 Fetal Outcome
Studies suggest that the fetal mortality rate when PHPT without AP is present can be reduced by a factor of four if operative cure is achieved [42, 431]. Hyperparathyroidism during pregnancy poses fetal risks of intrauterine demise, second-trimester loss, neonatal demise, and generalized tetany after delivery [57]. Up to 1998 and from the 13 patients, fetal mortality was 23 % [66]. Mortality seems to be related to delayed resection of parathyroid tumor [66]. PHPT results in high concentrations of fetal serum calcium that acts to suppress the parathyroid glands. Fetal calcitonin concentrations are high to encourage bone mineralization. At birth, however, the neonate is suddenly deprived of this source of calcium. It is incapable of mobilizing calcium from the bone owing to the low concentrations of parathyroid hormone and high concentrations of calcitonin. Acute neonatal hypocalcaemia results in neonatal tetany and convulsions, usually at 5–14 days of age. If the infant is breast-fed, tetany can be delayed by 1 month or more [415, 432]. Up to 1970, 17 children born of nine mothers have been reported to exhibit this phenomenon since the first description by Friderichsen in 1939 [433–437].
3.8.3 Acute Fatty Liver of Pregnancy
Report from 1980 demonstrated a very high maternal and perinatal mortality of 75 and 85 %, respectively [107]. More recent reports, however, indicate that with prompt diagnosis and treatment, the maternal and perinatal mortality rates have greatly decreased to approximately 18 and 23 %, respectively [438, 439]. In a series of 12 cases of AP in pregnant women with AFLP, Moldenhauser et al. found the following complications: encephalopathy (50 %), respiratory failure (17 %), and acute renal failure (33 %) [110].
3.8.4 Hypertriglyceridemia
3.8.4.1 Maternal Outcome
Interestingly, beyond the apparent significance of a TG threshold level to initiate AP (approximately 1,000 mg/dl), the severity of HTG-induced AP does not seem to correlate directly with TG level. In a series of 43 patients with HTG-induced AP, there was no relationship between TG level and severity of disease course or complications [440]. HTG-induced AP in pregnancy is a serious complication, and earlier studies noted that it is associated with a significant risk of death for both mothers (21 %) and fetuses (20 %) [376]. However, in 15 cases of gestational hyperlipidemic AP, no maternal death was reported [74, 346]. Though severity and complication rates with HTG-induced AP have been reported as higher in comparison to AP from other etiologies, mortality rates have not been found to differ. In a study of the clinical course of HTG-induced AP, the disease course of HTG-induced AP (19 patients) was compared to biliary AP (19 patients). Patients with HTG-induced AP had significantly more prior episodes of AP (possibly due to HTG) and more complications such as pancreatic necrosis, abscess formation, sepsis, or renal insufficiency, though there were no deaths in either group [441].
3.8.4.2 Fetal Outcome
Previously reported high perinatal mortality rates secondary to AP are due to neonatal deaths after preterm delivery. In recent series, perinatal mortality rates were improved with 74 % of infants delivered at term [15], or even preterm babies (five cases) delivered by Cesarean section could be saved due to better perinatal care [167], but studies still confirm of high premature delivery with even 100 % [78]. Pancreatic hemorrhage following pancreatic necrosis was also reported during pregnancy [442].
Outcomes for the mother and fetus in 1970 were similar with 21 % mortality rate for the mother and 20 % mortality rate for the fetus [376].
3.8.5 Medications
The prognosis of drug-induced AP in general population is generally excellent. In one report of 22 cases, for example, 19 were associated with interstitial AP; none of the patients with pancreatic necrosis on abdominal CT or ultrasound had over 33 % of the pancreas involved, and none died [207]. Mortality has also been rare in other reviews, although there are reports of a few deaths directly related to drug-induced AP.
3.8.6 Alcohol
3.8.6.1 Maternal Outcome
Patients with alcoholic AP were more likely to have recurrence of AP during pregnancy (75 % vs. 29 %) compared to cases where alcohol was not a factor [24]. Pseudocysts were almost exclusively associated with non-gallstone AP [24].
3.8.6.2 Fetal Outcome
Alcoholic AP was associated with significantly higher rates of preterm delivery and recurrence than all-cause AP. Patients with alcoholic AP were more likely to have preterm delivery (67 % vs. 26 %) compared to cases where alcohol was not a factor [24].
3.8.7 Preeclampsia-Eclampsia
Hojo et al. reported five maternal deaths and four cases of intrauterine fetal demise, all before 1973 [67, 75, 221, 222]. Only one case was with maternal death when deterioration was rapid, but amylase and lipase were not taken during the course of the disease and also authors did not perform the abdominal CT scan [238].
References
1.
Schmitt WJ. Sammlung zweifelhafter Schangerschaft faille nebst eines kritischen Einleitung über die Methode des Untersuchens, zum Gebrauche für angehende Geburtshelfer. Wien: Wimmer; 1818. p. 172–80.
2.
Lawrence W. History of a case in which on examination after death the pancreas was found in a state of active inflammation. Tr Med Chirurg Soc Lond. 1831;16:367–76.
3.
Longmade CF, Edmondson HA. Acute pancreatitis during pregnancy and post-partum; a report of nine cases. Surg Gynecol Obstet. 1951;92:43–52.
4.
France P. Sudden death during delivery caused by acute necrotic hemorrhagic pancreatitis. Rev Fr Gynec Obst. 1952;47:379.
5.
Cope O, Culver PJ, Mixter CG, Nardi GL. Pancreatitis, a diagnostic clue to hyperparathyroidism. Ann Surg. 1957;145:857–63.PubMedCentralPubMed
6.
Haidlen R. Zbl Gynäk. 1884;8:609. Cited by: Roberts NJ, Baggenstoss AH, Comfort MW. Am J Clin Pathol. 1950;20:742.
7.
Watts SH. Acute and subacute pancreatitis. Report of seven cases. Ann Surg. 1918;67:278–92.PubMedCentralPubMed
8.
Deaver JB. Acute pancreatitis. Ann Surg. 1918;68:281–8.PubMedCentralPubMed
9.
Joske RA. Pancreatitis following pregnancy. Br Med J. 1955;1:124–8.PubMedCentralPubMed
10.
Chang MC, Su CH, Sun MS, et al. Etiology of acute pancreatitis – a multi-center study in Taiwan. Hepatogastroenterology. 2003;50:1655–7.PubMed
11.
Lindkvist B, Appelros S, Manjer J, Borgström A. Trends in incidence of acute pancreatitis in a Swedish population: is there really an increase? Clin Gastroenterol Hepatol. 2004;2:831–7.PubMed
12.
Go VL, Everhart JE. Pancreatitis. In: Everhart JE, editor. Digestive diseases in the United States: epidemiology and impact. Washington, DC: US Department of Health and Human Services, Public Health Service, National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases. US Government Printing Office NIH Publication no. 94-1447; 1994. p. 693.
13.
Wang GJ, Gao CF, Wei D, et al. Acute pancreatitis: etiology and common pathogenesis. World J Gastroenterol. 2009;15:1427–30.PubMedCentralPubMed
14.
Yadav D, Lowenfels AB. Trends in the epidemiology of the first attack of acute pancreatitis: a systemic review. Pancreas. 2006;33:323–30.PubMed
15.
Ramin KD, Ramin SM, Richey SD, Cunningham FG. Acute pancreatitis in pregnancy. Am J Obstet Gynecol. 1995;173:187–91.PubMed
16.
Wilkinson EJ. Acute pancreatitis in pregnancy: a review of 98 cases and a report of 8 new cases. Obstet Gynecol Surg. 1973;28:281–303.
17.
Walker BE, Diddle AW. Acute pancreatitis in gynaecologic and obstetric practice. Am J Obstet Gynaecol. 1969;105:206–11.
18.
Glasgow RE, Visser BC, Harris HW, et al. Changing management of gall stone disease during pregnancy. Surg Endosc. 1998;12:241–6.PubMed
19.
Robertson KW, Stewart IS, Imrie CW. Severe acute pancreatitis and pregnancy. Pancreatology. 2006;6:309–15.PubMed
20.
Corlett Jr RC, Mishell Jr DR. Pancreatitis in pregnancy. Am J Obstet Gynecol. 1972;113:281–90.PubMed
21.
Jouppila P, Mokka R, Larmi TKI. Acute pancreatitis in pregnancy. Surg Gynec Obstet. 1974;139:879–82.PubMed
22.
Swisher SG, Schmit PJ, Hunt KK, et al. Biliary disease during pregnancy. Am J Surg. 1994;168:576–81.PubMed
23.
Tang SJ, Rodriguez-Frias E, Singh S, et al. Acute pancreatitis during pregnancy. Clin Gastroenterol Hepatol. 2010;8:85–90.PubMed
24.
Eddy JJ, Gideonsen MD, Song JY, et al. Pancreatitis in pregnancy. Obstet Gynecol. 2008;112:1075–81.PubMedCentralPubMed
25.
Hernandez A, Petrov MS, Brooks DC, et al. Acute pancreatitis in pregnancy: a 10-year single center experience. J Gastrointest Surg. 2007;11:1623–7.PubMed
26.
Ramin KD, Ramsey PS. Disease of the gallbladder and pancreas in pregnancy. Obstet Gynecol Clin North Am. 2001;28:571–80.PubMed
27.
Rakshit A, Dey R, De M, Biswas RR, Biswas SC. Pancreatitis in pregnancy – a scenario in a tertiary care centre. Al Ameen J Med Sci. 2010;3:332–6.
28.
Herfort K, Fialova V, Srp B. Acute pancreatitis in pregnancy. Mater Med Pol. 1981;13:15–7.PubMed
29.
Ewald N, Hardt PD, Kloer HU. Severe hypertriglyceridemia and pancreatitis: presentation and management. Curr Opin Lipidol. 2009;20:497–504.PubMed
30.
Fortson MR, Freedman SN, Webster 3rd PD. Clinical assessment of hyperlipidemic pancreatitis. Am J Gastroenterol. 1995;90:2134–9.PubMed
31.
Gan SI, Edwards AL, Symonds CJ, et al. Hypertriglyceridemia-induced pancreatitis: a case-based review. World J Gastroenterol. 2006;12:7197–202.PubMed
32.
Sekimoto M, Takada T, Kawarada Y, et al. JPN guidelines for the management of acute pancreatitis: epidemiology, etiology, natural history, and outcome predictors in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2006;13:10–24.PubMedCentralPubMed
33.
Kayatao SE, Eser M, Cam C, et al. Acute pancreatitis associated with hypertriglyceridemia: a life-threatening complication. Arch Gynecol Obstet. 2010;281:427–9.
34.
Kim EJ, Baik JC, Chung JY, et al. A case of acute pancreatitis of the pregnancy. Kor J Obstet Gynecol. 2005;48:1967–70.
35.
Swisher SO, Hunt KK, Schmit PL, et al. Management of pancreatitis complicating pregnancy. Am Surg. 1994;60:759–62.PubMed
36.
Block P, Kelly TR. Management of gallstone pancreatitis during pregnancy and the postpartum period. Surg Gynecol Obstet. 1989;168:426–8.PubMed
37.
Centers for Disease Control and Prevention (CDC). Alcohol consumption among women who are pregnant or who might become pregnant – United States, 2002. Morb Mortal Wkly Rep. 2004;53:1178–81.
38.
Fraser WD. Hyperparathyroidism. Lancet. 2009;374:145–58.PubMed
39.
Kelly TR. Primary hyperparathyroidism during pregnancy. Surgery. 1991;110:1028–34.PubMed
40.
Rodgers SE, Lew JI, Solórzano CC. Primary hyperparathyroidism. Curr Opin Oncol. 2008;20:52–8.PubMed
41.
Silverberg SJ, Bilezikian JP. The diagnosis and management of asymptomatic primary hyperparathyroidism. Nat Clin Pract Endocrinol Metab. 2006;2:494–503.PubMed
42.
Schnatz PF, Curry SL. Primary hyperparathyroidism in pregnancy: evidence-based management. Obstet Gynecol Surv. 2002;57:365–76.PubMed
43.
Inabnet WB, Baldwin D, Daniel RO, Staren ED. Hyperparathyroidism and pancreatitis during pregnancy. Surgery. 1996;119:710–3.PubMed
44.
Dahan M, Chang RJ. Pancreatitis secondary to hyperparathyroidism during pregnancy. Obstet Gynecol. 2001;98(5 Pt 2):923–5.PubMed
45.
Bess MA, Edis AJ, van Heerden JA. Hyperparathyroidism and pancreatitis. Chance or a causal association? J Am Med Assoc. 1980;243:246–7.
46.
Carnaille B, Oudar C, Pattou F, et al. Pancreatitis and primary hyperparathyroidism: forty cases. Aust N Z J Surg. 1998;68:117–9.PubMed
47.
Shepherd JJ. Hyperparathyroidism presenting as pancreatitis or complicated by postoperative pancreatitis. Aust N Z J Surg. 1996;66:85–7.PubMed
48.
Felderbauer P, Karakas E, Fendrich V, et al. Pancreatitis risk in primary hyperparathyroidism: relation to mutations in the SPINK1 trypsin inhibitor (N34S) and the cystic fibrosis gene. Am J Gastroenterol. 2008;103:368–74.PubMed
49.
Jacob JJ, John M, Thomas N, et al. Does hyperparathyroidism cause pancreatitis? A South Indian experience and a review of published work. ANZ J Surg. 2006;76:740–4.PubMed
50.
Kovacs CS, El-Hajj Fuleihan G. Calcium and bone disorders during pregnancy and lactation. Endocrinol Metab Clin North Am. 2006;35:21–51.PubMed
51.
Mestman JH. Parathyroid disorders of pregnancy. Semin Perinatol. 1998;6:485–96.
52.
Hunter D, Turnbull H. Hyperparathyroidism: generalized osteitis fibrosa with observations upon bones, parathyroid tumors, and the normal parathyroid glands. Br J Surg. 1931;19:203–6.
53.
Heath H, Hodgson SF, Kennedy MA. Primary hyperparathyroidism: incidence, morbidity, and potential economic impact in a community. N Engl J Med. 1980;302:189–93.PubMed
54.
Murray JA, Newman 3rd WA, Dacus JV. Hyperparathyroidism in pregnancy: diagnostic dilemma? Obstet Gynecol Surv. 1997;52:202–5.PubMed
55.
Khoo TK, Vege SS, Abu-Lebdeh HS, et al. Acute pancreatitis in primary hyperparathyroidism: a population-based study. J Clin Endocrinol Metab. 2009;94:2115–8.PubMedCentralPubMed
56.
Prinz RA, Aranha GV. The association of primary hyperparathyroidism and pancreatitis. Am Surg. 1985;51:325–9.PubMed
57.
Carella M, Gossain V. Hyperparathyroidism and pregnancy: case report and review. J Gen Intern Med. 1992;7:448–53.PubMed
58.
Fabrin B, Eldon K. Pregnancy complicated by concurrent hyperparathyroidism and pancreatitis. Acta Obstet Gynecol Scand. 1986;65:651–2.PubMed
59.
Rajala B, Abbasi RA, Hutchinson HT, Taylor T. Acute pancreatitis and primary hyperparathyroidism in pregnancy: treatment of hypercalcemia with magnesium sulfate. Obstet Gynecol. 1987;70:460–2.PubMed
60.
Soyannwo SM, McGowan M. A case of acute hyperthyroidism, with thyrotoxicosis and pancreatitis, presenting as hyperemesis gravidarum. Postgrad Med. 1968;44:861–6.
61.
Bronsky D, Weisbeyl MG, Gross MC, Barton JJ. Hyperparathyroidism and acute postpartum pancreatitis with neonatal tetany in the child. Am J Med Sci. 1970;269:1604.
62.
Levine G, Tsin D, Risk A. Acute pancreatitis and hyperparathyroidism in pregnancy. Obstet Gynecol. 1979;54:246–8.PubMed
63.
Hess HM, Dickson J, Fox HE. Hyperfunctioning parathyroid carcinoma presenting as acute pancreatitis in pregnancy. J Reprod Med. 1980;25:83–7.PubMed
64.
Thomason JL, Sampson MB, Farb HF, Spellacy WN. Pregnancy complicated by concurrent primary hyperparathyroidism and pancreatitis. Obstet Gynecol. 1981;57:34S–6.PubMed
65.
Warneke G, Henning HV, Isemer FE, et al. Primary hyperparathyroidism with acute pancreatitis during pregnancy. Dtsch Med Wochenschr. 1988;113:641–3.PubMed
66.
Kondo Y, Nagai H, Kasahara K, Kanazawa K. Primary hyperparathyroidism and acute pancreatitis during pregnancy. Report of a case and a review of the English and Japanese literature. Int J Pancreatol. 1998;24:43–7.PubMed
67.
Hojo S, Tsukimori K, Hanaoka M, et al. Acute pancreatitis and cholecystitis associated with postpartum HELLP syndrome: a case and review. Hypertens Pregnancy. 2007;26:23–9.PubMed
68.
Mondière JT. Recherches pour servir a I’histoire pathologique du pancreas. Memoires et observations. Arch Gen de Med. 1836;2:36–58, 2:265–94, 3:133–64.
69.
Ross G. Pancreatitis following pregnancy. Br Med J. 1955;1:349–50.PubMedCentral
70.
Berk JE, Smith BH, Akrawi MM. Pregnancy pancreatitis. Am J Gastroenterol. 1971;56:216–26.PubMed
71.
Kehrer E. Anatomie and Physiologie der Schwangerschaft. In: Seitz-Amreich H, editor. Biologie and Patologie des Weibes, vol. 7. Munich: Urban Schwarzenberg; 1952.
72.
Gabryelewicz A. Etiology and pathogenesis of acute pancreatitis: current view. Rocz Akad Med Bialymst. 1995;40:218–26.PubMed
73.
Chang CC, Hsieh YY, Tsai HD, et al. Acute pancreatitis in pregnancy. Zhonghua Yi Xue Za Zhi. 1998;61:85–92.PubMed
74.
Nies BM, Dreiss RJ. Hyperlipidemic pancreatitis in pregnancy: a case report and review of the literature. Am J Perinatol. 1990;7:166–9.PubMed
75.
Parmar MS. Pancreatic necrosis associated with pre-eclampsia – eclampsia. JOP – J Pancreas (Online). 2004;5:101–4.
76.
Stimac D, Stimac T. Acute pancreatitis during pregnancy. Eur J Gastroenterol Hepatol. 2011;23:839–44.PubMed
77.
Asciutti S, Kanninen TT, Clerici G, et al. Acute pancreatitis with a mucinous cystoadenoma of the pancreas in pregnancy. Anticancer Res. 2010;30:1025–8.PubMed
78.
Li HP, Huang YJ, Chen X. Acute pancreatitis in pregnancy: a 6-year single center clinical experience. Chin Med J (Engl). 2011;124:2771–5.
79.
Shigetazu N, Masutoshi M. Acute pancreatitis with diabetic ketoacidosis associated with hypermyoglobinaemia. J Gastroenterol. 1996;31:623–6
80.
Kohlmeier L, Marcus R. Calcium disorders of pregnancy. Endocrinol Metab Clin North Am. 1995;24:15–39.PubMed
81.
Drake TS, Kaplan RA, Lewis TA. The physiologic hyperparathyroidism of pregnancy. Is it primary or secondary? Obstet Gynecol. 1979;53:746–9.PubMed
82.
Seely EW, Brown EM, DeMaggio DM, et al. A prospective study of calciotropic hormones in pregnancy and post partum: reciprocal changes in serum intact parathyroid hormone and 1,25-dihydroxyvitamin D. Am J Obstet Gynecol. 1997;176:214–7.PubMed
83.
Davis OK, Hawkins DS, Rubin LP, et al. Serum parathyroid hormone (PTH) in pregnant women determined by an immunoradiometric assay for intact PTH. J Clin Endocrinol Metab. 1988;67:850–2.PubMed
84.
Mimouni F, Tsang RC, Hertzberg V, et al. Parathyroid hormone and calcitriol changes in normal and insulin dependent diabetic pregnancies. Obstet Gynecol. 1989;74:49–54.PubMed
85.
Ammann P, Irion O, Gast J, et al. Alterations of calcium and phosphate metabolism in primary hyperparathyroidism during pregnancy. Acta Obstet Gynecol Scand. 1993;72:488–92.PubMed
86.
Hosking DJ. Calcium homeostasis in pregnancy. Clin Endocrinol. 1996;45:1–6.
87.
Pitkin RM, Reynolds WA, Williams GA, Hargis GK. Calcium metabolism in normal pregnancy: longitudinal study. Am J Obstet Gynecol. 1979;133:781–90.PubMed
88.
Ludwig GD. Hyperparathyroidism in relation to pregnancy. New Engl J Med. 1962;267:637–42.PubMed
89.
Kent GN, Price RI, Gutteridge DH, et al. The efficiency of intestinal calcium absorption is increased in late pregnancy but not in established lactation. Calcif Tissue Int. 1991;48:293–5.PubMed
90.
Gertner JM, Coustan DR, Kliger AS, et al. Pregnancy as a state of physiologic absorptive hypercalciuria. Am J Med. 1986;81:451–6.PubMed
91.
Zerwekh J, Breslau N. Human placental production of 1-alpha, 25-dihydroxyvitamin D3: biochemical characterization and production in normal subjects and patients with pseudohypoparathyroidism. J Clin Endocrinol Metab. 1986;62:192–6.PubMed
92.
Payne RB, Little AJ, Evans RT. Albumin-adjusted calcium concentrations in serum increases during normal pregnancy. Clin Chem. 1990;36:142–4.PubMed
93.
Curto C, Caillard C, Desurmont T, et al. Acute pancreatitis and primary hyperparathyroidism: a multicentric study by the Francophone Association of Endocrine Surgeons. J Chir (Paris). 2009;146:270–4.
94.
Brandwein SL, Sigman KM. Case report: milk-alkali syndrome and pancreatitis. Am J Med Sci. 1994;308:173–6.PubMed
95.
Gafter U, Mandel EM, Har-Zahav L, Weiss S. Acute pancreatitis secondary to hypercalcemia. Occurrence in a patient with breast carcinoma. J Am Med Assoc. 1976;235:2004–5.
96.
Hochgelerent EL, David DS. Acute pancreatitis secondary to calcium infusion in a dialysis patient. Arch Surg. 1974;108:218–9.PubMed
97.
Ward JB, Petersen OH, Jenkins SA, Sutton R. Is an elevated concentration of acinar cytosolic free ionised calcium the trigger for acute pancreatitis? Lancet. 1995;346:1016–9.PubMed
98.
Mithöfer K, Fernández-del Castillo C, Frick TW, et al. Acute hypercalcemia causes acute pancreatitis and ectopic trypsinogen activation in the rat. Gastroenterology. 1995;109:239–46.PubMed
99.
Frick TW, Fernández-del Castillo C, Bimmler D, Warshaw AL. Elevated calcium and activation of trypsinogen in rat pancreatic acini. Gut. 1997;41:339–43.PubMedCentralPubMed
100.
Hauser CJ, Kamrath RO, Sparks J, Shoemaker WC. Calcium homeostasis in patients with acute pancreatitis. Surgery. 1983;94:830–5.PubMed
101.
Condon JR, Ives D, Knight MJ, Day J. The aetiology of hypocalcaemia in acute pancreatitis. Br J Surg. 1975;62:115–8.PubMed
102.
McKay C, Beastall GH, Imrie CW, Baxter JN. Circulating intact parathyroid hormone levels in acute pancreatitis. Br J Surg. 1994;81:357–60.PubMed
103.
Robertson GM, Moore EW, Switz DM, et al. Inadequate parathyroid response in acute pancreatitis. N Engl J Med. 1976;294:512–6.PubMed
104.
Krysiak R, Wilk M, Okopien B. Recurrent pancreatitis induced by hyperparathyroidism in pregnancy. Arch Gynecol Obstet. 2011;284:531–4.PubMedCentralPubMed
105.
Schnatz PF, Thaxton S. Parathyroidectomy in the third trimester of pregnancy. Obstet Gynecol Surv. 2005;60:672–82.PubMed
106.
Ch’ng CL, Morgan M, Hainsworth I, Kingham JG. Prospective study of liver dysfunction in pregnancy in Southwest Wales. Gut. 2002;51:876–80.PubMedCentralPubMed
107.
Varner M, Rinderknecht NK. Acute fatty metamorphosis of pregnancy. A maternal mortality and literature review. J Reprod Med. 1980;24:177–80.PubMed
108.
Castro MA, Fassett MJ, Reynolds TB, et al. Reversible peripartum liver failure: a new perspective on the diagnosis, treatment, and cause of acute fatty liver of pregnancy, based on 28 consecutive cases. Am J Obstet Gynecol. 1999;181:389–95.PubMed
109.
Pockros PJ, Peters RL, Reynolds TB. Idiopathic fatty liver of pregnancy: findings in ten cases. Medicine (Baltimore). 1984;63:1–11.
110.
Moldenhauer JS, O’Brien JM, Barton JR, Sibai B. Acute fatty liver of pregnancy associated with pancreatitis: a life threatening complication. Am J Obstet Gynecol. 2004;190:502–5.PubMed
111.
Cruciat G, Stamatian F, Puscas M, et al. Acute pancreatitis in a pregnant woman with acute fatty liver dystrophy. A case report. J Gastrointestin Liver Dis. 2007;16:193–6.PubMed
112.
Treem WR, Shoup ME, Hale DE, et al. Acute fatty liver of pregnancy, hemolysis, elevated liver enzymes, and low platelets syndrome, and long chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Am J Gastroenterol. 1996;91:2293–300.PubMed
113.
Treem WR, Rinaldo P, Hale DE, et al. Acute fatty liver of pregnancy and long-chain 3-hydroxyacyl-coenzyme A dehydrogenase deficiency. Hepatology. 1994;19:339–45.PubMed
114.
Ijlst L, Oostheim W, Ruiter JP, Wanders RJ. Molecular basis of long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: identification of two new mutations. J Inherit Metab Dis. 1997;20:420–2.PubMed
115.
Pereira SP, O’Donohue J, Wendon J, Williams R. Maternal and perinatal outcome in severe pregnancy-related liver disease. Hepatology. 1997;26:1258–62.PubMed
116.
Minakami H, Kimura K, Tamada T, et al. Acute fatty liver of pregnancy: report of a case complicating DIC and acute pancreatitis (author’s transl). Nippon Sanka Fujinka Gakkai Zasshi. 1982;34:637–40.PubMed
117.
Minakami H, Kimura K, Kanazawa T, et al. Acute fatty liver of pregnancy with hyperlipidemia, acute hemorrhagic pancreatitis and disseminated intravascular coagulation. Asia Oceania J Obstet Gynaecol. 1985;11:371–6.PubMed
118.
Apiratpracha W, Yoshida EM, Charles SH, et al. Chronic pancreatitis: a sequela of acute fatty liver of pregnancy. Hepatobiliary Pancreat Dis Int. 2008;7:101–4.PubMed
119.
Gillett MP, Silva TB, Begerman EM. Plasma lipids and HDL cholesterol during pregnancy. Atherosclerosis. 1979;33:495–7.PubMed
120.
Ma Y, Liu MS, Ginzinger D, et al. Gene-environment interaction in the conversion of a mild to severe phenotype in a patient homozygous for a ser172 → cys mutation in the lipoprotein lipase gene. J Clin Invest. 1993;91:1953–8.PubMedCentralPubMed
121.
Potter JM, Nestel PJ. The hyperlipidemia of pregnancy in normal and complicated pregnancies. Am J Obstet Gynecol. 1979;133:165–70.PubMed
122.
Hsia SH, Connelly PW, Hegele RA. Successful outcome in severe pregnancy-associated hyperlipemia: a case report and literature review. Am J Med Sci. 1995;309:213–8.PubMed
123.
Gagne C, Brun LD, Julien P, et al. Primary lipoprotein-lipase-activity deficiency: clinical investigation of a French Canadian population. CMAJ. 1989;140:405–11.PubMedCentralPubMed
124.
Herrera E. Lipid metabolism in pregnancy and its consequences in the fetus and newborn. Endocrine. 2002;19:43–55.PubMed
125.
Salameh WA, Mastrogiannis DS. Maternal hyperlipidemia in pregnancy. Clin Obstet Gynecol. 1994;37:66–77.PubMed
126.
Perrone G, Critelli C. Severe hypertriglyceridemia in pregnancy. A clinical case report. Minerva Ginecol. 1996;48:573–6.PubMed
127.
Knopp R, Bergelin R, Wahl P, et al. Population-based lipoprotein lipid reference values for pregnant women compared to nonpregnant women classified by sex hormone usage. Am J Obstet Gynecol. 1982;143:626–37.PubMed
128.
Iverius P-H, Brunzell JD. Relationship between lipoprotein lipase activity and plasma sex steroid levels in obese women. J Clin Invest. 1988;82:1106–12.PubMedCentralPubMed
129.
Knopp RH, Warth MR, Charles D, et al. Lipid metabolism in pregnancy, fat transport to the foetus, and the effects of diabetes. Biol Neonate. 1986;50:297–317.PubMed
130.
Sorva R, Kuusi T, Dunkel L, Taskinen MR. Effects of endogenous sex steroids on serum lipoproteins and post-heparin plasma lipolytic enzymes. J Clin Endocrinol Metab. 1988;66:408–13.PubMed
131.
Kekki M, Nikkila EA. Plasma triglyceride turnover during use of oral contraceptives. Metab Clin Exp. 1971;20:878–89.PubMed
132.
The lipid research clinics population studies data book, vol 1. The prevalence study. US Department of Health and Human Services, NIH Publication No. 80-1527, Bethesda, Maryland; 1980. p. 4246.
133.
Kim HJ, Kalkhoff RK. Sex steroid influence on triglyceride metabolism. J Clin Invest. 1975;56:888–96.PubMedCentralPubMed
134.
Park JR, Cho BHS. Effects of estrogen on very-low-density lipoprotein triacylglycerol metabolism in chicks. Biochim Biophys Acta. 1990;1045:180–6.PubMed
135.
Cameron JL, Capuzzi DM, Zuidema GD. Acute pancreatitis with hyperlipidaemia: incidence of lipid abnormalities in acute pancreatitis. Ann Surg. 1973;177:479–83.
136.
Havel RJ. Pathogenesis, differentiation and management of hypertriglyceridemia. Adv Intern Med. 1969;15:117–45.PubMed
137.
Saharia P, Margolis S, Zuidema MD, Cameron JL. Acute pancreatitis with hyperlipidemia: studies with an isolated perfused canine pancreas. Surgery. 1977;82:60–7.PubMed
138.
Kimura W, Mössner J. Role of hypertriglyceridemia in the pathogenesis of experimental acute pancreatitis in rats. Int J Pancreatol. 1996;20:177–84.PubMed
139.
Ma Y, Ooi TC, Liu MS, et al. High frequency of mutations in the lipoprotein lipase gene in pregnancy-induced chylomicronemia: possible association with the apolipoprotein E2 isoform. J Lipid Res. 1994;35:1066–75.PubMed
140.
Keilson LM, Calvin PH, Sprecher DL, Renfrew R. Hyperlipidemia and pancreatitis during pregnancy in two sisters with a mutation in the lipoprotein lipase gene. Ann Intern Med. 1996;124:425–8.PubMed
141.
Bergeron J, Normand T, Barucha A, et al. Prevalence, geographical distribution and genealogical investigations of mutation 188 of lipoprotein lipase gene in the French Canadian population. Clin Genet. 1992;41:206–10.PubMed
142.
Syed H, Bilusic M, Rhondla C, Tavaria A. Plasmapheresis in the treatment of hypertriglyceridemia-induced pancreatitis: a community hospital’s experience. J Clin Apher. 2010;25:229–34.PubMed
143.
Castro MR, Nguyen TT, O’Brien T. Clomiphene-induced severe hypertriglyceridemia and pancreatitis. Mayo Clin Proc. 1999;74:1125–8.PubMed
144.
Ting HJ, Stice JP, Schaff UY, et al. Triglyceride-rich lipoproteins prime aortic endothelium for an enhanced inflammatory response to tumor necrosis factor-alpha. Circ Res. 2007;100:381–90.PubMed
145.
Libby P. Fat fuels the flame: triglyceride-rich lipoproteins and arterial inflammation. Circ Res. 2007;100:299–301.PubMed
146.
Tripathy D, Mohanty P, Dhindsa S, et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52:2882–3.PubMed
147.
Du X, Edelstein D, Obici S, et al. Insulin resistance reduces arterial prostacyclin synthase and eNOS activities by increasing endothelial fatty acid oxidation. J Clin Invest. 2006;116:1071–80.PubMedCentralPubMed
148.
Grossman A, Oppenheim J, Grondin G, et al. Immunocytochemical localization of the estradiol-binding protein in rat pancreatic acinar cells. Endocrinology. 1989;124:2857–66.PubMed
149.
Heiss G, Tamir I, Davis CE, et al. Lipoprotein-cholesterol distributions in selected North American populations: the lipid research clinics program prevalence study. Circulation. 1980;61:302–15.PubMed
150.
Staels B, Jansen H, van Tol A, et al. Development, food intake, and ethinylestradiol influence hepatic triglyceride lipase and LDL-receptor mRNA levels in rats. J Lipid Res. 1990;31:1211–81.PubMed
151.
Hoefler G, Noehammer C, Levak-Frank S, et al. Muscle-specific overexpression of human lipoprotein lipase in mice causes increased intracellular free fatty acids and induction of peroxisomal enzymes. Biochimie. 1997;79:163–8.PubMed
152.
Blevins Jr GT, Huang HS, Tangoku A, et al. Estrogens influence cholecystokinin stimulated pancreatic amylase release and acinar cell membrane cholecystokinin receptors in rat. Life Sci. 1991;48:1565–74.PubMed
153.
Glueck CJ, Fallat RW, Scheel D. Effects of estrogenic compounds on triglyceride kinetics. Metabolism. 1975;24:537–45.PubMed
154.
Yoshioka T, Koike S, Sugiue A, Eguchi K. Hyperlipidemia in pregnancy, clinical observation of the newborn and mother. Nippon Sanfujinka Gakkai Zasshi. 1976;28:369–77.
155.
Dominguez-Munoz JE, Malfertheiner P, Dischuneit HH, et al. Hyperlipidemia in acute pancreatitis. Relationship with etiology, onset, and severity of disease. Int J Pancreatol. 1991;10:261–7.PubMed
156.
Lloret Linares C, Pelletier AL, Czernichow S, et al. Acute pancreatitis in a cohort of 129 patients referred for severe hypertriglyceridemia. Pancreas. 2008;37:13–7.PubMed
157.
Fredrickson DS. An international classification of hyperlipidemias and hyperlipoproteinemias. Ann Intern Med. 1971;75:471–2.PubMed
158.
Yadav D, Pitchumoni CS. Issues in hyperlipidemic pancreatitis. J Clin Gastroenterol. 2003;36:54–62.PubMed
159.
Henderson H, Leisegang F, Hassan F, et al. A novel Glu42 1Lys substitution in the lipoprotein lipase gene in pregnancy-induced hypertriglyceridemic pancreatitis. Clin Chim Acta. 1998;269:1–12.PubMed
160.
Wion KL, Kirchessner TG, Lusis AJ, et al. Human lipoprotein lipase complementary DNA sequence. Science. 1987;235:1638–41.PubMed
161.
Nikkila EA. Familial lipoprotein lipase deficiency and related disorders of chylomicron metabolism. In: Stanbury JB, Wyngaarden JB, Frederickson DS, Goldstein JL, Brown MS, editors. The metabolic basis of inherited disease. 5th ed. New York: McGraw-Hill; 1983. p. 622–42.
162.
Eckel RH. Lipoprotein lipase. A multifunctional enzyme relevant to common metabolic diseases. N Engl J Med. 1989;320:1060–8.PubMed
163.
Hunninghake DB. Bile acid sequestrants. In: Rifkind BM, editor. Drug treatment of hyperlipidemia. New York: Marcel Dekker; 1991. p. 92.
164.
Spence JD, Huff MW, Heidenheim P, et al. Combination therapy with colestipol and psyllium mucilloid in patients with hyperlipidemia. Ann Intern Med. 1995;123:493–9.PubMed
165.
Monsalve MV, Henderson H, Roederer G, et al. A missense mutation at codon 188 of the human lipoprotein lipase gene is a frequent cause of lipoprotein lipase deficiency in persons of different ancestries. J Clin Invest. 1990;86:728–34.PubMedCentralPubMed
166.
Bartha I, Dinya T, Seres I, et al. Acute hypertriglyceridemic pancreatitis during pregnancy due to homozygous lipoprotein lipase gene mutation. Clin Chim Acta. 2009;400:137–8.PubMed
167.
Crisan LS, Steidl ET, Rivera-Alsina ME. Acute hyperlipidemic pancreatitis in pregnancy. Am J Obstet Gynecol. 2008;198:e57–9.PubMed
168.
Toskes PP. Hyperlipidemic pancreatitis. Gastroenterol Clin North Am. 1990;19:783–91.PubMed
169.
Havel RJ. Approach to the patient with hyperlipidemia. Med Clin North Am. 1982;66:319–33.PubMed
170.
Rivellese AA, De Natale C, Di Marino L, et al. Exogenous and endogenous postprandial lipid abnormalities in type 2 diabetic patients with optimal blood glucose control and optimal fasting triglyceride levels. J Clin Endocrinol Metab. 2004;89:2153–9.PubMed
171.
Nair S, Yadav D, Pitchumoni CS. Association of diabetic ketoacidosis and acute pancreatitis: observations in 100 consecutive episodes of DKA. Am J Gastroenterol. 2000;95:2795–800.PubMed
172.
O’Brien T, Dinneen SF, O’ Brien PC, et al. Hyperlipidemia in patients with primary and secondary hypothyroidism. Mayo Clin Proc. 1993;68:860–6.PubMed
173.
Goldenberg NM, Wang P, Glueck CJ. An observational study of severe hypertriglyceridemia, hypertriglyceridemic acute pancreatitis, and failure of triglyceride-lowering therapy when estrogens are given to women with and without familial hypertriglyceridemia. Clin Chim Acta. 2003;332:11–9.PubMed
174.
Glueck CJ, Lang J, Hamer T, Tracy T. Severe hypertriglyceridemia and pancreatitis, when estrogen replacement therapy is given to hypertriglyceridemic women. J Lab Clin Med. 1994;123:59–64.PubMed
175.
Morr MM, Norman J. Acute pancreatitis. In: Greenfield LJ, editor. Surgery, scientific principles and practice. 3rd ed. Philadelphia: Lippincott Williams & Wilkins; 2001. p. 867.
176.
Davidoff F, Tishler S, Rosoff C. Marked hyperlipidemia and pancreatitis associated with oral contraceptive therapy. N Engl J Med. 1973;289:552–5.PubMed
177.
Hozumi Y, Kawano M, Saito T, Miyata M. Effect of tamoxifen on serum lipid metabolism. J Clin Endocrinol Metab. 1998;8:1633–5.
178.
Alagozlu H, Cindoruk M, Unal S. Tamoxifen-induced severe hypertriglyceridemia and acute pancreatitis. Clin Drug Investig. 2006;26:297–302.PubMed
179.
Brunzell JD, Schrott HG. The interaction of familial and secondary causes of hypertriglyceridemia: role in pancreatitis. Trans Assoc Am Physicians. 1973;86:245–54.PubMed
180.
Arbel Y, Weinstein D, Yogev R, Halevy A. Acute pancreatitis following clomiphene citrate treatment: case report and review of the literature. Int J Surg. 2008;6:483–4.PubMed
181.
Watts GF, Morton K, Jackson P, Lewis B. Management of patients with severe hypertriglyceridemia during pregnancy: report of two cases with familial lipoprotein lipase deficiency. Br J Obstet Gynaecol. 1992;99:163–6.PubMed
182.
Parulekar PS, Gidden F, Yavari A. A sticky situation. Q J Med. 2012;105:887–9.
183.
Palmieri VO, Grattagliano I, Palasciano G. Ethanol induces secretion of oxidized proteins by pancreatic acinar cells. Cell Biol Toxicol. 2007;23:459–64.PubMed
184.
Siech M, Zhou Z, Zhou S, et al. Stimulation of stellate cells by injured acinar cells: a model of acute pancreatitis induced by alcohol and fat (VLDL). Am J Physiol Gastrointest Liver Physiol. 2009;297:G1163–71.PubMed
185.
Sutton R, Criddle D, Raraty MG, et al. Signal transduction, calcium and acute pancreatitis. Pancreatology. 2003;3:497–505.PubMed
186.
Kubisch CH, Gukovsky I, Lugea A, et al. Long-term ethanol consumption alters pancreatic gene expression in rats: a possible connection to pancreatic injury. Pancreas. 2006;33:68–76.PubMed
187.
Kristiansen L, Gronbaek M, Becker U, Tolstrup JS. Risk of pancreatitis according to alcohol drinking habits: a population-based cohort study. Am J Epidemiol. 2008;168:932–7.PubMed
188.
Lankisch PG, Lowenfels AB, Maisonneuve P. What is the risk of alcoholic pancreatitis in heavy drinkers? Pancreas. 2002;25:411–2.PubMed
189.
Barreto SG, Saccone GT. Alcohol-induced acute pancreatitis: the ‘critical mass’ concept. Med Hypotheses. 2010;75:73–6.PubMed
190.
Schmidt DN. Apparent risk factors for chronic and acute pancreatitis in Stockholm County. Spirits but not wine and beer. Int J Pancreatol. 1991;8:45–50.PubMed
191.
Sand J, Välikoski A, Nordback I. Alcohol consumption in the country and hospitalizations for acute alcohol pancreatitis and liver cirrhosis during a 20-year period. Alcohol Alcohol. 2009;44:321–5.PubMed
192.
Wilson JS, Bernstein L, McDonald C, et al. Diet and drinking habits in relation to the development of alcoholic pancreatitis. Gut. 1985;26:882–7.PubMedCentralPubMed
193.
Whitcomb DC, Yadav D, Adam S, et al. North American Pancreatic Study Group. Multicenter approach to recurrent acute and chronic pancreatitis in the United States: the North American Pancreatitis Study 2 (NAPS2). Pancreatology. 2008;8:520–31.PubMedCentralPubMed
194.
Jaakkola M, Sillanaukee P, Löf K, et al. Amount of alcohol is an important determinant of the severity of acute alcoholic pancreatitis. Surgery. 1994;115:31–8.PubMed
195.
Altomare E, Grattagliano I, Vendemiale G, et al. Acute ethanol administration induces oxidative changes in rat pancreatic tissue. Gut. 1996;38:742–6.PubMedCentralPubMed
196.
Siech M, Weber H, Letko G, et al. Similar morphological and intracellular biochemical changes in alcoholic acute pancreatitis and ischemic acute pancreatitis in rats. Pancreas. 1997;14:32–8.PubMed
197.
Wittel UA, Bachem M, Siech M. Oxygen radical production precedes alcohol-induced acute pancreatitis in rats. Pancreas. 2003;26:e74–80.PubMed
198.
Rivero D, Pérez-Magarinõ S, González-Sanjosé ML, et al. Inhibition of induced DNA oxidative damage by beers: correlation with the content of polyphenols and melanoidins. J Agric Food Chem. 2005;53:3637–42.PubMed
199.
Howard A, Chopra M, Thurnham D, et al. Red wine consumption and inhibition of LDL oxidation: what are the important components? Med Hypotheses. 2002;59:101–4.PubMed
200.
Gorelick FS. Alcohol and zymogen activation in the pancreatic acinar cell. Pancreas. 2003;27:305–10.PubMed
201.
Addolorato G, Leggio L, Ojetti V, et al. Effects of short-term moderate alcohol administration on oxidative stress and nutritional status in healthy males. Appetite. 2008;50:50–6.PubMed
202.
Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health. 2003;27:220–31.PubMed
203.
Vonlaufen A, Wilson JS, Apte MV. Molecular mechanisms of pancreatitis: current opinion. J Gastroenterol Hepatol. 2008;23:1339–48.PubMed
204.
Barreto SG, Rodrigues J. Acute pancreatitis in Goa – a hospital-based study. J Indian Med Assoc. 2008;106:575–6, 578.PubMed
205.
Lachenmeier DW, Taylor B, Rehm J. Lack of evidence for a causal association between the type of alcoholic beverage and the incidence of acute pancreatitis. Pancreas. 2011;40:1143–5.PubMed
206.
Irving HM, Samokhvalov AV, Rehm J. Alcohol as a risk factor for pancreatitis. A systematic review and meta-analysis. JOP. 2009;10:387–92.PubMedCentralPubMed
207.
Lankisch PG, Droege M, Gottesleben F. Drug-induced acute pancreatitis: incidence and severity. Gut. 1995;37:565.PubMedCentralPubMed
208.
Werth B, Kuhn MK, Reinhart WH. Medikamentoes induzierte Pankreatiden: Erfahrungen der Scweizerischen Arzneimittel-Nebenswirkungenszentrale (SANZ) 1981–1993. Schweiz Med Wochenschr. 1995;125:731.PubMed
209.
Mallory A, Kern Jr F. Drug-induced pancreatitis: a critical review. Gastroenterology. 1980;78:813.PubMed
210.
Hallberg P, Hallberg E, Amini H. Acute pancreatitis following medical abortion: case report. BMC Womens Health. 2004;4:1–4.PubMedCentralPubMed
211.
Andersen V, Sonne J, Andersen M. Spontaneous reports on drug-induced pancreatitis in Denmark from 1968 to 1999. Eur J Clin Pharmacol. 2001;57:517–21.PubMed
212.
Hastier P, Buckley MJ, Peten EP, et al. A new source of drug-induced acute pancreatitis: codeine. Am J Gastroenterol. 2000;95:3295–8.PubMed
213.
Hastier P, Longo F, Buckley M, et al. Pancreatitis induced by codeine: a case report with positive rechallenge. Gut. 1997;41:705–6.PubMedCentralPubMed
214.
SWEDIS: http://sweweb.mpa.se.
215.
Rosenfeld MG, Abrass I, Chang B. Hormonal stimulation of alpha-amylase synthesis in porcine pancreatic minces. Endocrinology. 1976;99:611–8.PubMed
216.
Rudick J, Gonda M, Dreiling DA, Janowitz HD. Effects of prostaglandin E1 on pancreatic exocrine function. Gastroenterology. 1971;60:272–8.PubMed
217.
Nieuwenhuizen AG, Schuiling GA, Liem SM, et al. Progesterone stimulates pancreatic cell proliferation in vivo. Eur J Endocrinol. 1999;140:256–63.PubMed
218.
Singh S, Baker PR, Poulsom R, et al. Expression of oestrogen receptor and oestrogen-inducible genes in pancreatic cancer. Br J Surg. 1997;84:1085–9.PubMed
219.
Wilmink T, Frick TW. Drug-induced pancreatitis. Drug Saf. 1996;14:406–23.PubMed
220.
Greenberg A. Diuretic complications. Am J Med Sci. 2000;319:10–24.PubMed
221.
Badja N, Troche G, Zazzo JF, Benhamou D. Acute pancreatitis and preeclampsia-eclampsia. Am J Obstet Gynecol. 1997;176:707–9.PubMed
222.
Marcovici I, Marzano D. Pregnancy-induced hypertension complicated by postpartum renal failure and pancreatitis: a case report. Am J Perinatol. 2002;19:177–9.PubMed
223.
Chapman SJ, Woolley IJ, Visvanathan K, et al. Acute pancreatitis caused by tipranavir/ritonavir-induced hypertriglyceridemia. AIDS. 2007;21:532–3.PubMed
224.
Devlin JW, Lau AK, Tanios MA. Propofol-associated hypertriglyceridemia and pancreatitis in the intensive care unit: an analysis of frequency and risk factors. Pharmacotherapy. 2005;25:1348–52.PubMed
225.
Waage C, Carlsson H, Nielsen EW. Olanzapine-induced pancreatitis: a case report. JOP. 2004;5:388–91.PubMed
226.
Chen JL, Spinowitz N, Karwa M. Hypertriglyceridemia, acute pancreatitis, and diabetic ketoacidosis possibly associated with mirtazapine therapy: a case report. Pharmacotherapy. 2003;23:940–4.PubMed
227.
Flynn WJ, Freeman PG, Wickboldt LG. Pancreatitis associated with isotretinoin-induced hypertriglyceridemia. Ann Intern Med. 1987;107:63.PubMed
228.
Bondugulapati LR, Munigoti SP, Rees A. Hypertriglyceridemia-induced acute pancreatitis in the third trimester of pregnancy. Br J Diabetes Vasc Dis. 2011;11:155–6.
229.
Steinmetz OK, Hashim E, Falcone T, et al. Recurrent pancreatitis associated with in vitro fertilization. Obstet Gynecol. 1993;81:890–2.PubMed
230.
Lee J, Goldberg IJ. Hypertriglyceridemia-induced pancreatitis created by oral estrogen and in vitro fertilization ovulation induction. J Clin Lipidol. 2008;2:63–6.PubMedCentralPubMed
231.
Maringhini A, Lankisch MR, Zinsmeister AR, et al. Acute pancreatitis in the postpartum period: a population-based case-control study. Mayo Clin Proc. 2000;75:361–4.PubMed
232.
Barbara L, Sama C, Morselli Labate AM, et al. A population study on the prevalence of gallstone disease: the Sirmione study. Hepatology. 1987;7:913–7.PubMed
233.
Rome Group for the Epidemiology and Prevention of Cholelithiasis (GREPCO). Prevalence of gallstone disease in an Italian adult female population. Am J Epidemiol. 1984;119:796–805.
234.
Scragg RK, McMichael AJ, Seamark RF. Oral contraceptives, pregnancy, and endogenous oestrogen in gall stone disease – a case-control study. Br Med J. 1984;288:1795–9.
235.
Thistle JL, Eckhart KL, Nensel RE, et al. Prevalence of gallbladder disease among Chippewa Indians. Mayo Clin Proc. 1971;46:603–8.PubMed
236.
Bernstein RA, Werner LH, Rimm AA. Relationship of gallbladder disease to parity, obesity, and age. Health Serv Rep. 1973;88:925–36.PubMedCentralPubMed
237.
Maringhini A, Ciambra M, Baccelliere P, et al. Biliary sludge and gallstones in pregnancy: incidence, risk factors, and natural history. Ann Intern Med. 1993;119:116–20.PubMed
238.
Swank M, Nageotte M, Hatfield T. Necrotizing pancreatitis associated with severe preeclampsia. Obstet Gynecol. 2012;120(2 Pt 2):453–5.PubMed
239.
Opatrny L, Michon N, Ray E. Preeclampsia as a cause of pancreatitis: a case report. J Obstet Gynaecol Can. 2004;26:594–5.PubMed
240.
Yamada K, Hamatani K, Nishimoto H. HELLP (hemolysis, elevated liver enzymes and low platelets) syndrome and acute pancreatitis complicated with severe preeclampsia. Masui. 1997;46:373–5.PubMed
241.
Bahloul M, Ayedi M, Dammak H, et al. Pregnancy-induced hypertension complicated by acute pancreatitis. Ann Fr Anesth Reanim. 2004;23:157–9.PubMed
242.
Sipahi HM, Gullo L, Scolari MP, et al. Acute pancreatitis associated with eclampsia: a case report. Ital J Gastroenterol. 1994;26:148–50.PubMed
243.
Haukland HH, Florholmen J, Oian P, et al. The effect of severe pre-eclampsia on the pancreas: changes in the serum cationic trypsinogen and pancreatic amylase. Br J Obstet Gynaecol. 1987;94:765–7.PubMed
244.
Pritchard JA, Cunningham FG, Pritchard SA, Mason RA. On reducing the frequency of severe abruptio placentae. Am J Obstet Gynecol. 1991;165:1345–51.PubMed
245.
Pagliari M. On an unusual case of uterine apoplexy preceded by an acute necrosis of the pancreas. Quad Clin Ostet Ginecol. 1962;17:589–94.PubMed
246.
Cheang CU, Ho SW, Tee YT, et al. Acute necrotizing pancreatitis complicating uteroplacental apoplexy. Taiwan J Obstet Gynecol. 2007;46:64–7.PubMed
247.
Burns E, Gray R, Smith LA. Brief screening questionnaires to identify problem drinking during pregnancy: a systematic review. Addiction. 2010;105:601–14.PubMed
248.
Angelini DJ. Gallbladder and pancreatic disease during pregnancy. J Perinat Neonatal Nurs. 2002;15:1–12.PubMed
249.
Kennedy A. Assessment of acute abdominal pain in the pregnant patient. Semin Ultrasound CT MR. 2000;21:64–77.PubMed
250.
Kayatas SE, Eser M, Cam C, et al. Acute pancreatitis associated with hypertriglyceridemia: a life-threatening complication. Arch Gynecol Obstet. 2010;281:427–9.PubMed
251.
Marcus M. Akute Pankreaserkrankungen und Gestationvorgaenge. Beit Klin Chir. 1930;149:129–41.
252.
http://www.odermatol.com/issue-in-html/2011-2-11-eponyms-c/.
253.
Chan AK, Duh QY, Katz MH, et al. Clinical manifestations of primary hyperparathyroidism before and after parathyroidectomy. A case-control study. Ann Surg. 1995;222:402–14.PubMedCentralPubMed
254.
Date RS, Kaushal M, Ramesh A. A review of the management of gallstone disease and its complications in pregnancy. Am J Surg. 2008;196:599–608.PubMed
255.
Durrington P. Dyslipidaemia. Lancet. 2003;362:717–31.PubMed
256.
Nayak KR, Daly RG. Eruptive xanthomas associated with hypertriglyceridemia and new-onset diabetes mellitus. N Engl J Med. 2004;350:1235.PubMed
257.
Glueck CJ, Christopher C, Mishkel MA, et al. Pancreatitis, familial hypertriglyceridemia, and pregnancy. Am J Obstet Gynecol. 1980;136:755–61.PubMed
258.
De Chalain TM, Michell WL, Berger GM. Hyperlipidemia, pregnancy and pancreatitis. Surg Gynecol Obstet. 1988;167:469–73.PubMed
259.
Fred HL, Accad M. Lipemia retinalis. Images in clinical medicine. N Engl J Med. 1999;340:1969.PubMed
260.
Kumar J, Wierzbicki AS. Lipemia retinalis. N Engl J Med. 2005;353:823.PubMed
261.
Chamarthi B, Greene MF, Dluhy RG. A problem in gestation. N Engl J Med. 2011;365:843–8.PubMed
262.
Matull W, Pereira S, O’Donohue J. Biochemical markers of acute pancreatitis. J Clin Pathol. 2006;59:340–4.PubMedCentralPubMed
263.
Vissers R, Abu-Laban R, McHugh D. Amylase and lipase in the emergency department evaluation of acute pancreatitis. J Emerg Med. 1999;17:1027–37.PubMed
264.
Pacheco R, Nishioka Sde A, de Oliveira L. Validity of serum amylase and lipase in the differential diagnosis between acute/acutized chronic pancreatitis and other causes of acute abdominal pain. Arq Gastroenterol. 2003;40:233–8.PubMed
265.
Serrano N. Increased lipase plasma levels in ICU patients: when are they critical? Chest. 2005;127:7–10.PubMed
266.
Yadav D, Agarwal N, Pitchumoni CS. A critical evaluation of laboratory tests in acute pancreatitis. Am J Gastroenterol. 2002;97:1309–18.PubMed
267.
Sreelatha S, Nataraj VN. Acute pancreatitis in pregnancy. Indian J Clin Pract. 2012;23:231–2.
268.
Karsenti D, Bacq Y, Bréchot JF, et al. Serum amylase and lipase activities in normal pregnancy: a prospective case-control study. Am J Gastroenterol. 2001;96:697–9.PubMed
269.
Wickus GG, Dukerschein RO, Pierce JR, Davis KD. Interference in a chromogenic alpha-amylase assay caused by dyelabeled oligosaccharide-induced precipitation of lipoprotein. Clin Chem. 1982;28:2131–4.PubMed
270.
Warshaw AL, Bellini CA, Lesser PB. Inhibition of serum and urine amylase activity in pancreatitis with hyperlipemia. Ann Surg. 1975;182:72–5.PubMedCentralPubMed
271.
Wang SS, Lin XZ, Tsai YT, et al. Clinical significance of ultrasonography, computed tomography, and biochemical tests in the rapid diagnosis of gallstone-related pancreatitis: a prospective study. Pancreas. 1988;3:153–8.PubMed
272.
Neoptolemos JP, Hall AW, Finlay DF, et al. The urgent diagnosis of gallstones in acute pancreatitis: a prospective study of three methods. Br J Surg. 1984;71:230–3.PubMed
273.
Devore GR, Braken M, Berkowitz RL. The amylase/creatinine clearance ratio in normal pregnancies and pregnancies complicated by pancreatitis, hyperemesis, and toxemia. Am J Obstet Gynecol. 1980;136:747–54.PubMed
274.
Boakye MK, Macfoy D, Rice C. Alcoholic pancreatitis in pregnancy. J Obstet Gynaecol. 2006;26:814.PubMed
275.
Hofbauer B, Friess H, Weber A, et al. Hyperlipaemia intensifies the course of acute oedematous and acute necrotizing pancreatitis in the rat. Gut. 1996;38:753–8.PubMedCentralPubMed
276.
Harris WS, Rothrock DW, Fanning A, et al. Fish oils in hypertriglyceridemia: a dose – response study. Am J Clin Nutr. 1990;51:399–406.PubMed
277.
Abu Musa AA, Usta IM, Rechdan JB, Nassar AH. Recurrent hypertriglyceridemia-induced pancreatitis in pregnancy: a management dilemma. Pancreas. 2006;32:227–8.PubMed
278.
Abboud B, Daher R, Boujaoude J. Digestive manifestations of parathyroid disorders. World J Gastroenterol. 2011;17:4063–6.PubMedCentralPubMed
279.
Kort KC, Schiller HJ, Numann PJ. Hyperparathyroidism and pregnancy. Am J Surg. 1999;177:66–8.PubMed
280.
Castro MA, Goodwin TM, Shaw KJ, et al. Disseminated intravascular coagulation and antithrombin III depression in acute fatty liver of pregnancy. Am J Obstet Gynecol. 1996;174:211–6.PubMed
281.
Howard JM, Reed J. Pseudohyponatremia in acute hyperlipemic pancreatitis. A potential pitfall in therapy. Arch Surg. 1985;120:1053–5.PubMed
282.
Fallat RW, Vester JW, Glueck CJ. Suppression of amylase activity by hypertriglyceridemia. J Am Med Assoc. 1973;225:1331–4.
283.
Tsuang W, Navaneethan U, Ruiz L, et al. Hypertriglyceridemic pancreatitis: presentation and management. Am J Gastroenterol. 2009;104:984–91.PubMed
284.
Treacy J, Williams A, Bais R, et al. Evaluation of amylase and lipase in the diagnosis of acute pancreatitis. ANZ J Surg. 2001;71:577–82.PubMed
285.
Sniderman AD, Blank D, Zakarian R, et al. Triglycerides and small dense LDL: the twin Achilles heels of the Friedewald formula. Clin Biochem. 2003;36:499–504.PubMed
286.
Warnick GR, Knopp RH, Fitzpatrick V, et al. Estimating low-density lipoprotein cholesterol by the Friedewald equation is adequate for classifying patients on the basis of nationally recommended cutpoints. Clin Chem. 1990;36:15–9.PubMed
287.
Chuang SC, Lee KT, Wang SN, et al. Hypertriglyceridemia-associated acute pancreatitis with chylous ascites in pregnancy. J Formos Med Assoc. 2006;105:583–7.PubMed
288.
Sahu SK, Raghuvanshi S, Bahl DV, Sachan P. Acute pancreatitis in pregnancy. Internet J Surg. 2007;11(2).
289.
Takaishi K, Miyoshi J, Matsumura T, et al. Hypertriglyceridemic acute pancreatitis during pregnancy: prevention with diet therapy and omega-3 fatty acids in the following pregnancy. Nutrition. 2009;25:1094–7.PubMed
290.
Pezzilli R, Zerbi A, Di Carlo V, et al. Practical guidelines for acute pancreatitis. Pancreatology. 2010;10:523–35.PubMed
291.
Pitchumoni CS, Yegneswaran B. Acute pancreatitis in pregnancy. World J Gastroenterol. 2009;45:5641–6.
292.
Lee YT, Chan FK, Leung WK, et al. Comparison of EUS and ERCP in the investigation with suspected biliary obstruction in patients with choledocholithiasis: a randomized study. Gastrointest Endosc. 2008;67:660–8.PubMed
293.
Leyendecker JR, Gorengaut V, Brown JJ. MR imaging of maternal diseases of the abdomen and pelvis during pregnancy and the immediate postpartum period. Radiographics. 2004;24:1301–16.PubMed
294.
Chaundhry S, Hussain R. Acute pancreatitis in pregnancy: a rare presentation. Prof Med J. 2012;19:747–50.
295.
Shellock FG, Kana LE. Policies, guidelines, and recommendations for MR imaging safety and patient management. SMRI Safety Committee. J Magn Reson Imaging. 1991;1:97–101.PubMed
296.
Banks PA, Freeman ML. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101:2379–400.PubMed
297.
Chong VH, Jalihal A. Endoscopic management of biliary disorders during pregnancy. Hepatobiliary Pancreat Dis Int. 2010;9:180–5.PubMed
298.
O’Mahony S. Endoscopy in pregnancy. Best Pract Res Clin Gastroenterol. 2007;21:893–9.PubMed
299.
Mitura K, Romanczuk M. Ruptured ectopic pregnancy mimicking acute pancreatitis. Ginekol Pol. 2009;80:383–5.PubMed
300.
Pieper-Bigelow C, Strocchi A, Levitt M. Where does serum amylase come from and where does it go? Gastroenerol Clin North Am. 1990;19:793–810.
301.
Christensen H, Larsen M, Schebye O. Serum amylase levels in gynecologic patients with acute abdominal pain. Surg Gynecol Obstet. 1992;175:355–6.PubMed
302.
Um J, Kim K, Kang M, et al. Macroamylasemia in a patient with acute appendicitis: a case report. J Korean Med Sci. 1999;14:679–81.PubMedCentralPubMed
303.
Shapiro R, Dropkin R, Finkelstein J, et al. Ovarian carcinomatosis presenting with hyperamylasemia and plural effusion. Am J Gastroenterol. 1981;76:365–8.PubMed
304.
Green CL. Identification of alpha-amylase as a secretion of the human fallopian tube and “tubelike” epithelium of mullerian and mesonephric duct origin. Am J Obstet Gynecol. 1974;73:402–8.
305.
Clavien PA, Moossa AR. Serum enzymes and other laboratory tests in acute pancreatitis. Br J Surg. 1989;76:1234–43.PubMed
306.
Chow AW, Soll BA, Targan SR, Guze LB. Hyperamylasemia associated with gonococcal salpingitis and perihepatitis. Obstet Gynecol. 1976;48 Suppl 1:29s–30.PubMed
307.
Norwood SH, Torma MJ, Fontenelle LJ. Hyperamylasemia due to poorly differentiated adenosquamous carcinoma of the ovary. Arch Surg. 1981;116:225–6.PubMed
308.
Srivastava R, Fraser C, Gentleman D, et al. Hyperamylasaemia: not the usual suspects. Br Med J. 2005;331:890–1.
309.
Jacobs E, Jennette JC, Reavis RA. Chronic hyperamylasemia and chronic pelvic inflammatory disease. Clin Chem. 1983;29:887–8.PubMed
310.
Berk JE, Fridhandler L. Hyperamylasemia: interpretation and newer approaches to evaluation. Adv Intern Med. 1980;26:235–64.PubMed
311.
Van Kley H, Cramer S, Bruns DE. Serous ovarian neoplastic amylase (SONA): a potentially useful marker for serous ovarian tumors. Cancer. 1981;48:1444–9.PubMed
312.
Zakowski JJ, Gregory MR, Bruns DE. Amylase from human serous ovarian tumors: purification and characterization. Clin Chem. 1984;30:62–8.PubMed
313.
Brophy CM, Morris J, Sussman J, Modlin IM. “Pseudoascites” secondary to an amylase-producing serous ovarian cystadenoma: a case study. J Clin Gastroenterol. 1989;11:703–6.PubMed
314.
Saruc M, Yuceyar H, Turkel N, et al. The role of heme in hemolysis-induced acute pancreatitis. Med Sci Monit. 2007;13:67–72.
315.
Cosenza CA, Saffari B, Jabbour N, et al. Surgical management of biliary gallstone disease during pregnancy. Am J Surg. 1999;178:545–8.PubMed
316.
Sinha S, Khan H, Timms PM, Olagbaiye OA. Pancreatic-type hyperamylasemia and hyperlipasemia secondary to ruptured ovarian cyst: a case report and review of the literature. J Emerg Med. 2010;38:463–6.PubMed
317.
Wada K, Takada T, Hirata K, et al. Treatment strategy for acute pancreatitis. J Hepatobiliary Pancreat Sci. 2010;17:79–86.PubMed
318.
Hasibeder WR, Torgersen C, Rieger M. Critical care of the patient with acute pancreatitis. Anaesth Intensive Care. 2009;37:190–206.PubMed
319.
Gardner TB, Vege SS, Pearson RK, Chari ST. Fluid resuscitation in acute pancreatitis. Clin Gastroel Hepatol. 2008;6:1070–6.
320.
Forsmark CE, Baillie J. AGA Institute technical review on acute pancreatitis. Gastroenterology. 2007;132:2022–44.PubMed
321.
Russo-Stieglitz KE, Levine AB, Wagner BA, Armenti VT. Pregnancy outcoming patient requiring parenteral nutrition. J Matern Fetal Med. 1999;8:164–7.PubMed
322.
Meier R, Ockenga J, Pertkiewicz M, et al. ESPEN guidelines on enteral nutrition: pancreas. Clin Nutr. 2006;25:275–84.PubMed
323.
Bai Y, Gao J, Zou DW, Li ZS. Antibiotics prophylaxis in acute necrotizing pancreatitis: an update. Am J Gastroenterol. 2010;103:705–7.
324.
Jafri NS, Mahid SS, Idstein SR, et al. Antibiotic prophylaxis is not protective in severe acute pancreatitis: a systematic review and meta-analysis. Am J Surg. 2009;197:806–13.PubMed
325.
Villatoro E, Mulla M, Larvin M. Antibiotic therapy for prophylaxis against infection of pancreatic necrosis in acute pancreatitis. Cochrane Database Syst Rev. 2010;(5):CD002941.
326.
Jain P. Acute pancreatitis in pregnancy: an unresolved issue. World J Gastroenterol. 2010;16:2065–6.PubMedCentralPubMed
327.
Dinsmoor MJ. Imipenem-cilastatin. Obstet Gynecol Clin North Am. 1992;19:475–82.PubMed
328.
Heikkilä A, Renkonen OV, Erkkola R. Pharmacokinetics and transplacental passage of imipenem during pregnancy. Antimicrob Agents Chemother. 1992;36:2652–5.PubMedCentralPubMed
329.
Burtin P, Taddio A, Ariburnu O, et al. Safety of metronidazole in pregnancy: a meta-analysis. Am J Obstet Gynecol. 1995;172:525–9.PubMed
330.
Caro-Patón T, Carvajal A, Martin de Diego I, et al. Is metronidazole teratogenic? A meta-analysis. Br J Clin Pharmacol. 1997;44:179–82.PubMedCentralPubMed
331.
Dunham MC. Clinical impact of continuous renal replacement therapy on multiple organ failure. World J Surg. 2001;25:669–76.PubMed
332.
Yekebas EF, Strate T, Zolmajd S, et al. Impact of different modalities of continuous venovenous hemofiltration on sepsis-induced alterations in experimental pancreatitis. Kidney Int. 2002;62:1806–18.PubMed
333.
Wang H, Li WQ, Zhou W, et al. Clinical effects of continuous high volume hemofiltration on severe acute pancreatitis complicated with multiple organ dysfunction syndrome. World J Gastroenterol. 2003;9:2096–9.PubMed
334.
Ronco C, Tetta C, Mariano F, et al. Interpreting the mechanisms of continuous renal replacement therapy in sepsis: the peak concentration hypothesis. Artif Organs. 2003;27:792–801.PubMed
335.
Jung J, Eo E. A case of hemoperfusion and L-carnitine management in valproic acid overdose. Am J Emerg Med. 2008;26:388.e3–4.
336.
Saotome T, Endo Y, Sasaki T, et al. A case of severe acute pancreatitis treated with CTR-001 direct hemoperfusion for cytokine apheresis. Ther Apher Dial. 2005;9:367–71.PubMed
337.
Kobe Y, Oda S, Matsuda K, et al. Direct hemoperfusion with a cytokine-adsorbing device for the treatment of persistent or severe hypercytokinemia: a pilot study. Blood Purif. 2007;25:446–53.PubMedCentralPubMed
338.
Mao HJ, Yu S, Yu XB, et al. Effects of coupled plasma filtration adsorption on immune function of patients with multiple organ dysfunction syndrome. Int J Artif Organs. 2009;32:31–8.PubMed
339.
Tang Y, Zhang L, Fu P, et al. Hemoperfusion plus continuous veno-venous hemofiltration in a pregnant woman with severe acute pancreatitis: a case report. Int Urol Nephrol. 2012;44:987–90.PubMed
340.
Nathens AB, Curtis JR, Beale RJ, et al. Management of the critically ill patient with severe acute pancreatitis. Crit Care Med. 2004;32:2524–36.PubMed
341.
Demirkiran O, Dikmen Y, Utku T, Urkmez S. Critically ill obstetric patients in the intensive care unit. Int J Obstet Anesth. 2003;4:266–70.
342.
Swoboda K, Derfler K, Koppensteiner R, et al. Extracorporeal lipid elimination for treatment of gestational hyperlipidemic pancreatitis. Gastroenterology. 1993;104:1527–31.PubMed
343.
Saadi HF, Kurlander DJ, Erkins JM, Hoogwerf BJ. Severe hypertriglyceridemia and acute pancreatitis during pregnancy: treatment with gemfibrozil. Endocr Pract. 1999;5:33–6.PubMed
344.
Sanderson SL, Iverius PH, Wilson DE. Successful hyperlipemic pregnancy. J Am Med Assoc. 1991;265:1858–60.
345.
Weinberg RB, Sitrin MD, Adkins GM, Lin CC. Treatment of hyperlipidemic pancreatitis in pregnancy with total parenteral nutrition. Gastroenterology. 1982;83:1300–5.PubMed
346.
Ihimoyan A, Chelimilla H, Kalakada N, et al. Hypertriglyceridemia induced pancreatitis in a non-diabetic pregnant patient requiring the use of total parenteral nutrition. Gastroenterol Res. 2011;4:88–91.
347.
Smith JR, Friedell ML, Cheatham ML, et al. Peripherally inserted central catheters revisited. Am J Surg. 1998;176:208–11.PubMed
348.
Ogura JM, Francois KE, Perlow JH, Elliott JP. Complications associated with peripherally inserted central catheter use during pregnancy. Am J Obstet Gynecol. 2003;188:1223–5.PubMed
349.
Ng PK, Ault MJ, Ellrodt AG, Maldonado L. Peripherally inserted central catheters in general medicine. Mayo Clin Proc. 1997;72:225–33.PubMed
350.
Gosnell JE, O’Neill BB, Harris HW. Necrotizing pancreatitis during pregnancy: a rare cause and review of the literature. J Gastrointest Surg. 2001;5:371–6.PubMed
351.
Loo CC, Tan JYL. Decreasing the plasma triglyceride level in hypertriglyceridemia-induced pancreatitis in pregnancy: a case report. Am J Obstet Gynecol. 2002;187:241–2.PubMed
352.
Ong JM, Kirchgessner TG, Schotz MC, et al. Insulin increases the synthetic rate and messenger RNA level of lipoprotein lipase in isolated rat adipocytes. J Biol Chem. 1988;253:12,933–8.
353.
Jabbar MA, Zuhri-Yafi MI, Larrea J. Insulin therapy for a non-diabetic patient with severe hypertriglyceridemia. J Am Coll Nutr. 1998;17:458–61.PubMed
354.
Mikhail N, Trivedi K, Page C, et al. Treatment of severe hypertriglyceridemia in nondiabetic patients with insulin. Am J Emerg Med. 2005;23:415–7.PubMed
355.
Tamez-Perez HE, Saenz-Gallegos R, Hernandez–Rodriguez K, et al. Insulin therapy in patients with severe hypertriglyceridemia. Rev Med Inst Mex Seguro Soc. 2006;44:235–7.PubMed
356.
Korn ED. Clearing factor, a heparin-activated lipoprotein lipase. I. Isolation and characterization of the enzyme from normal rat heart. J Biol Chem. 1955;215:1–14.PubMed
357.
Sharma P, Lim S, James D, et al. Pancreatitis may occur with a normal amylase concentration in hypertriglyceridemia. Br Med J. 1996;313:1265.
358.
Sleth JC, Lafforgue E, Servais R, et al. A case of hypertriglycideremia-induced pancreatitis in pregnancy: value of heparin. Ann Fr Anesth Reanim. 2004;23:835–7.PubMed
359.
Nasstrom B, Olivecrona G, Olivecrona T, et al. Lipoprotein lipase during continuous heparin infusion: tissue stores become partially depleted. J Lab Clin Med. 2001;138:206–13.PubMed
360.
Weintraub M, Rassin T, Eisenberg S, et al. Continuous intravenous heparin administration in humans causes a decrease in serum lipolytic activity and accumulation of chylomicrons in circulation. J Lipid Res. 1994;35:229–38.PubMed
361.
Betteridge DJ, Bakowski M, Taylor KG, et al. Treatment of severe diabetic hypertriglyceridemia by plasma exchange. Lancet. 1978;1:1368.PubMed
362.
Ahner R, Speiser P, Swoboda K, et al. Hyperlipidemia-induced pancreatitis in pregnancy. Successful long-term treatment by extracorporeal elimination of triglyceride-rich lipoproteins. Gynakol Geburtshilfliche Rundsch. 1993;33 Suppl 1:337.PubMed
363.
Staels B, Dallongeville J, Auwerx J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–93.PubMed
364.
Third Report of the National Cholesterol Education Program (NCEP). Expert panel on detection, evaluation, and treatment of high blood cholesterol in adults (Adult Treatment Panel III): final report. NIH publication no. 02-5215. Bethesda: National Heart, Lung, and Blood Institute; 2002.
365.
Harris WS, Ginsberg HN, Arunakul N, et al. Safety and efficacy of Omacor in severe hypertriglyceridemia. J Cardiovasc Risk. 1997;4:385–91.PubMed
366.
Miller DB, Spence JD. Clinical pharmacokinetics of fibric acid derivatives (fibrate). Clin Pharmacokinet. 1998;34:155–62.PubMed
367.
Evans JR, Forland SC, Cutler RE. The effect of renal function on the pharmacokinetics of gemfibrozil. J Clin Pharmacol. 1987;27:994–1000.PubMed
368.
Oldemeyer JB, Lund RJ, Koch M, Meares AJ. Rhabdomyolysis and acute renal failure after changing statin-fibrate combinations. Cardiology. 2000;94:127–8.PubMed
369.
Ozdemir O, Boran M, Gokce V, et al. A case with severe rhabdomyolysis and renal failure associated with cerivastatin gemfibrozil combination therapy. Angiology. 2000;51:695–7.PubMed
370.
Magarian GJ, Lucas LM, Colley C. Gemfibrozil-induced myopathy. Arch Intern Med. 1991;151:1873–4.PubMed
371.
Kazmin A, Garcia-Bournissen F, Koren G. Risks of statin use during pregnancy: a systematic review. J Obstet Gynaecol Can. 2007;29:906–8.PubMed
372.
Taguchi N, Rubin ET, Hosokawa A, et al. Prenatal exposure to HMG-CoA reductase inhibitors: effects on fetal and neonatal outcomes. Reprod Toxicol. 2008;26:175–7.PubMed
373.
Tsai EC, Brown JA, Veldee MY, et al. Potential of essential fatty acid deficiency with extremely low fat diet in lipoprotein lipase deficiency during pregnancy: a case report. BMC Pregnancy Childbirth. 2004;4:27.PubMedCentralPubMed
374.
Basaran A. Pregnancy-induced hyperlipoproteinemia: review of the literature. Reprod Sci. 2009;16:431–7.PubMed
375.
Piolot A, Nadler F, Cavallero E, et al. Prevention of recurrent acute pancreatitis in patients with severe hypertriglyceridemia: value of regular plasmapheresis. Pancreas. 1996;13:96–9.PubMed
376.
Montgomery WH, Miller FC. Pancreatitis and pregnancy. Obstet Gynecol. 1970;35:658–64.PubMed
377.
Ahrens Jr EH, Hirsch J, Oette K, et al. Carbohydrate-induced and fat-induced lipemia. Trans Med Soc Lond. 1961;74:134–46.
378.
Sunamura M, Yamauchi H, Takeda K, et al. A case of acute pancreatitis associated with hyperlipidemia and pregnancy with reference to plasma exchange as a therapeutic intervention. Nihon Shokakibyo Gakkai Zasshi. 1985;82:2139–43.PubMed
379.
Connor WE. The dietary treatment of hypertriglyceridemia. Med Clin North Am. 1982;66:485.PubMed
380.
Healy AP, Sharer N, Rameh B, et al. Prevention of recurrent pancreatitis in familial lipoprotein lipase deficiency with high-dose antioxidant therapy. J Clin Endocrinol Metab. 1999;84:1203–5.
381.
Greene J, Rogers A, Rubin L. Fetal loss after cholecystectomy during pregnancy. Can Med Assoc J. 1963;88:576–7.PubMedCentralPubMed
382.
Gadacz TR, Talamini MA. Traditional versus laparoscopic cholecystectomy. Am J Surg. 1991;161:336–8.PubMed
383.
Soper NJ. SAGES’ guidelines for diagnosis, treatment, and use of laparoscopy for surgical problems during pregnancy. Surg Endosc. 2011;25:3477–8.PubMed
384.
Barone JE, Bears S, Chen S, et al. Outcome study of cholecystectomy during pregnancy. Am J Surg. 1999;177:232–6.PubMed
385.
Amano H, Takada T, Isaji S, et al. Therapeutic intervention and surgery of acute pancreatitis. J Hepatobiliary Pancreat Sci. 2010;17:53–9.PubMed
386.
Van Santvoort HC, Besselink MG, Horvath KD, et al. Videoscopic assisted retroperitoneal debridement in infected necrotizing pancreatitis. HPB (Oxford). 2007;9:156–9.
387.
Wig JD, Gupta V, Kochhar R, et al. The role of non-operative strategies in the management of severe acute pancreatitis. JOP. 2010;11:553–9.PubMed
388.
Goodley CD, Rattner DW. Pancreatic pseudocyst. In: Cameron JL, editor. Current surgical therapy. 6th ed. St. Louis: Mosby; 1988. p. 507–9.
389.
Bradley EL. Pancreatic pseudocysts. In: Bradley EL, editor. Complications of pancreatitis: medical and surgical management. Philadelphia: Saunders; 1982. p. 124–53.
390.
Reber HA. The pancreas. In: Schwartz SI, Shiver GT, Spencer FC, Daly JM, Fischer JE, Galloway AC, editors. Principles of surgery. New York: McGraw -Hill; 1999. p. 1485–7.
391.
Geokas MC, Baltaxe HA, Banks PA, et al. Acute pancreatitis. Ann Intern Med. 1985;103:86–100.PubMed
392.
Ryan ME. Endoscopic management of a pancreatic pseudocyst during pregnancy. Gastroint Endosc. 1992;38:605–8.
393.
Ko C. Biliary sludge and acute pancreatitis during pregnancy. Nat Clin Pract Gastroenterol Hepatol. 2006;3:53–7.PubMed
394.
Pai PR, Shah HK, Samsi AB. Post-partum pancreatitis. J Postgrad Med. 1993;39:93–4.PubMed
395.
Eddy JJ, Lynch GE, Treacy DE. Pancreatic pseudocysts in pregnancy: a case report and review of the literature. J Perinatol. 2003;23:69–72.PubMed
396.
Eddy JJ, Lynch GE, Treacy DE. Pancreatic pseudocysts in pregnancy: a case report and review of the literature. J Perinatol. 2004;29:242–3.
397.
Chen CP, Wang KG, Su TH, Yang YC. Acute pancreatitis in pregnancy. Acta Obstet Gynecol Scand. 1995;74:607–10.PubMed
398.
Bar-David J, Mazor M, Leiberman JR, et al. Gestational diabetes complicated by severe hypertriglyceridemia and acute pancreatitis. Arch Obstet Gynecol. 1996;258:101–4.
399.
Beattie GJ, Muir BB. Acute pancreatitis with pseudocyst formation complicating pregnancy in a patient with a coexistent choledochal cyst. Br J Obstet Gynecol. 1993;100:957–9.
400.
Stowell JC, Bottsford JE, Rubel HR. Pancreatitis with pseudocyst and cholelithiasis in third trimester of pregnancy. South Med J. 1984;77:502–4.PubMed
401.
Orlando CA, Bowman RL, Loose JH. Multicentric papillary–cystic neoplasm of the pancreas. Arch Pathol Lab Med. 1991;115:958–60.PubMed
402.
Morales A, Ruiz Molina JM, Esteves HO, et al. Papillary–cystic neoplasm of the pancreas, a sex – steroid dependent tumor. Int J Pancreatol. 1998;24:219–25.PubMed
403.
Olsen ME, Greer MS, Feintuch TA. Pancreatic mucinous cystadenoma during pregnancy. Am J Gynecol Health. 1993;7:27–30.
404.
Warshaw AL, Rutledge PL. Cystic tumors mistaken for pancreatic pseudocysts. Ann Surg. 1987;205:393–8.PubMedCentralPubMed
405.
Tanaka M, Chari S, Adsay V, et al. International Consensus Guidelines for management of intraductal papillary mucinous neoplasms and mucinous cystic neoplasms of the pancreas. Pancreatology. 2006;6:17–32.PubMed
406.
Truong MT, Lalakea ML, Robbins P, Friduss M. Primary hyperparathyroidism in pregnancy: a case series and review. Laryngoscope. 2008;118:1966–9.PubMed
407.
McMullen TP, Learoyd DL, Williams DC. Hyperparathyroidism in pregnancy: options for localization and surgical therapy. World J Surg. 2010;34:1811–6.PubMed
408.
Pothiwala P, Levine SN. Parathyroid surgery in pregnancy: review of the literature and localization by aspiration for parathyroid hormone levels. J Perinatol. 2009;29:779–84.PubMed
409.
Mitchell DM, Jüppner H. Regulation of calcium homeostasis and bone metabolism in the fetus and neonate. Curr Opin Endocrinol Diabetes Obes. 2010;17:25–30.PubMed
410.
Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium and lactation. Endocr Rev. 1997;18:832–72.PubMed
411.
Wolfe HJ. Calcitonin: perspectives in current concepts. J Endocrinol Invest. 1982;5:423–32.PubMed
412.
Horjus C, Groot I, Telting D, et al. Cinacalcet for hyperparathyroidism in pregnancy and puerperium. J Pediatr Endocrinol Metab. 2009;22:741–9.PubMed
413.
Nussbaum SR. Pathophysiology and management of severe hypercalcemia. Endocrinol Metab Clin North Am. 1993;22:343–62.PubMed
414.
Steffensrud S. Parathyroids: the forgotten glands. Neonatal Netw. 2000;19:9–16.PubMed
415.
Nathaniels EK, Nathaniels AM, Wang C. Mediastinal parathyroid tumors: a clinical and pathological study of 84 cases. Ann Surg. 1970;171:165–70.PubMedCentralPubMed
416.
Russell CFJ, Edis AJ, Scholz DA, et al. Mediastinal parathyroid tumors. Experience with 38 tumors requiring mediastinotomy for removal. Ann Surg. 1981;193:805–9.PubMedCentralPubMed
417.
Rooney DP, Traub AI, Russell CFJ, Hadden DR. Cure of hyperparathyroidism in pregnancy by sternotomy and removal of a mediastinal parathyroid adenoma. Postgrad Med J. 1998;74:233–6.PubMedCentralPubMed
418.
Young KR. Acute pancreatitis in pregnancy: two case reports. Obstet Gynecol. 1982;60:653–7.PubMed
419.
Lykkesfeldt G, Bock JE, Pdersen FD, et al. Excessive hypertriglyceridaemia and pancreatitis in pregnancy: association with lipoprotein lipase deficiency. Acta Obstet Gynecol Scand. 1981;60:79–82.PubMed
420.
Chu AJ, Rooney SA. Estrogen stimulation of surfactant synthesis. Pediatr Pulmonol. 1985;1(3 Suppl):S110–4.PubMed
421.
Langmade CF, Edmondson HA. Acute pancreatitis during pregnancy and post-partum period: report of 9 cases. Surg Gynecol Obstet. 1951;92:43–52.PubMed
422.
Joupilla P, Mokka R, Larmi TKI. Acute pancreatitis in pregnancy. Surg Gynaecol Obstet. 1974;139:879–82.
423.
Tang SJ, Mayo ML, Rodriguez-Frias E, et al. Safety and utility of ERCP during pregnancy. Gastrointest Endosc. 2009;69:453–61.PubMed
424.
Dixon NP, Faddis DM, Silberman H. Aggressive management of cholecystitis during pregnancy. Am J Surg. 1987;154:292–4.PubMed
425.
Okamura D, Starr ME, Lee EY, et al. Age-dependent vulnerability to experimental acute pancreatitis is associated with increased systemic inflammation and thrombosis. Aging Cell. 2012;11:760–9.PubMedCentralPubMed
426.
Goldacre MJ, Roberts SE. Hospital admission for acute pancreatitis in an English population, 1963–98: database study of incidence and mortality. Br Med J. 2004;328:1466–9.
427.
Talukdar R, Vege SS. Recent developments in acute pancreatitis. Clin Gastroenterol Hepatol. 2009;7(11 Suppl):S3–9.PubMed
428.
Sunamura M, Matsuno S, Takeda K, et al. Experimental study on the pathogenesis of pulmonary insufficiency in acute pancreatitis and changes in the pulmonary surfactant. Nihon Geka Gakkai Zasshi. 1988;89:1049–57.PubMed
429.
Ko CW. Risk factors for gallstone-related hospitalization during pregnancy and the postpartum. Am J Gastroenterol. 2006;101:2263–8.PubMed
430.
Valdivieso V, Covarrubias C, Siegel F, Cruz F. Pregnancy and cholelithiasis: pathogenesis and natural course of gallstones diagnosed in early puerperium. Hepatology. 1993;17:1–4.PubMed
431.
Delmonico FL, Neer RM, Cosimi AB, et al. Hyperparathyroidism during pregnancy. Am J Surg. 1976;131:328–37.PubMed
432.
Sauer M, Steere A, Parsons MT. Hyperparathyroidism in pregnancy with sonographic documentation of a parathyroid adenoma: a case report. J Reprod Med. 1985;30:615–7.PubMed
433.
Friderichsen C. Tetany in a suckling: with latent osteitis fibrosa in the mother. Lancet. 1939;233:85–6.
434.
Hutchnin P, Kessner DM. Neonatal tetany. Diagnostic lead to hyperparathyroidism in the mother. Ann Intern Med. 1964;61:1109–15.
435.
McGeown MG, Field CM. Asymptomatic hyperparathyroidism. Lancet. 1960;2:1268–9.PubMed
436.
Vanarsdel Jr PP. Maternal hyperparathyroidism as a cause of neonatal tetany: case report and review of 29 cases of parathyroid adenoma. J Clin Endocrinol Metab. 1955;15:680–4.PubMed
437.
Walton RL. Neonatal tetany in two siblings: effects of maternal hyperparathyroidism. Pediatrics. 1954;13:227–34.PubMed
438.
Kaplan MM. Acute fatty liver of pregnancy. N Engl J Med. 1985;313:367–70.PubMed
439.
Knox TA, Olans LB. Liver disease in pregnancy. N Engl J Med. 1996;335:569–76.PubMed
440.
Balachandra S, Virlos IT, King NK, et al. Hyperlipidemia and outcome in acute pancreatitis. Int J Clin Pract. 2006;60:156–9.PubMed
441.
Navarro S, Cubiella J, Feu F, et al. Hypertriglyceridemic acute pancreatitis. Is its clinical course different from lithiasic acute pancreatitis? Med Clin (Barc). 2004;123:567–70.
442.
Koscica KL, Nwaubani U, Nazir M, Gimovsky M. Severe hyperlipidemia induced hemorrhagic pancreatitis during pregnancy. Obstet Gynecol Int. 2009;2009:383942.PubMedCentralPubMed