Asher Bashiri1 , Avishai Shemesh2, Angel Porgador3, Gershon Holcberg4 and Maor Kabessa5
(1)
Maternity C and Recurrent Pregnancy Loss Clinic, Department of Obstetrics and Gynecology, Faculty of Health Sciences, Soroka University Medical Center, Ben-Gurion University of the Negev, 151, Be’er Sheva, 84101, Israel
(2)
The Shraga Segal Department of Microbiology, Immunology and Genetics, Ben Gurion University of the Negev, Be’er Sehva, Israel
(3)
National Institute for Biotechnology in the Negev, Ben Gurion University of the Negev, Be’er Sheva, Israel
(4)
Placental Research Laboratory, Maternity-C Department and High Risk Pregnancy, Faculty of Health Sciences, Soroka University Medical Center, Ben Gurion University of the Negev, Be’er Sheva, Israel
(5)
Faculty of Health Sciences, Ben Gurion University of Negrev, Be’er Sheva, Israel
Asher BashiriDirector
Email: abashiri@bgu.ac.il
Keywords
Recurrent pregnancy lossHeparinAnnexin VLeptinMultifactorial etiologyNK cellsImmune system
Multifactorial Etiology of Recurrent Miscarriage
Despite the great advance in recurrent miscarriage (RM) research in the recent years, about 50 % of the cases are still without known etiology, demonstrating how much more information is needed.
Christiansen et al. [1] suggested changing the methods and goals of our research in RPL. Sporadic miscarriages happen in 10–20 % of the population and if recurrent miscarriage was solely due to three sporadic miscarriages we would expect a prevalence <0.5 %. However, the observed prevalence is 1.4–1.8 % (defined as three or more consecutive miscarriages). From the 50 % with known etiology, the following reasons were identified: uterine abnormalities , parental chromosome aberrations, various endocrine disturbances, and antiphospholipid antibodies. This multifactorial etiology is accepted at the population level, but at the individual level RM is considered to be monofactorial. Some of the pathologies indeed are found in RM couples in increased prevalence, but they are also found in couples with completely normal fecundity [2, 3]. This and the fact that none of the quoted etiologies exhibit a high sensitivity or specificity, led a group of researchers to propose that RM can be considered multifactorial in each couple.
Miscarriages Can Be Divided into Fetally Caused and Maternally Caused
Fetally caused miscarriages include all the chromosomal aberrations , and account for 43 % of the miscarriages in the normal population [4]. Abnormal trophoblast invasion and development are included in maternally caused miscarriages. This impairment of the trophoblast growth can be related to polycystic ovary syndrome (PCOS) [5, 6], excessive prothrombic events in the maternal vessels and the fetal–maternal interface [7, 8], and local or systemic immunological reaction to the fetus or trophoblast. Although anatomical abnormalities are considered to be maternally caused RMs they are not included in this model.
Biomarkers
Recognizing that autoimmune diseases, thromboembolic diseases, and PCOS are associated with RM led to the investigation of nongenetic and genetic biomarkers. Polymorphisms in approximately 100 genes have already been investigated. It is believed that those three diseases are caused by many genes and environmental factors—each of them contributes a little, and they add up to the overall risk of developing a disease. Once this total disease risk has exceeded the disease threshold, the disease will become a reality. An interesting finding is that many diseases are associated with several relatively common genetic polymorphisms associated with modest risk of disease and several rare polymorphisms associated with higher risk of disease [9], and that carrying two genetic biomarkers for RM results in a higher overall total risk for RM than the additive risk of each factor [10–12]. Despite some observations that recognized the connection between some polymorphisms and RM, no genetic polymorphism has so far proven unequivocally to be associated with RM. Apparently, RM inheritance happens through a multifactorial mode and is not simply Mendelian [1].
Rull et al. [13] explain that the difficulties in finding genetic biomarkers are due to differences in study designs, definitions of RPL and control group, focus on RM women instead of couples or placenta , low statistical power due to small sample size, ethnic difference in risk variants, population-specific low-impact gene variants increasing RM risk in consort, contribution of lifestyle and environmental factors on the pregnancy course, and secondary pathways affecting protein translation/metabolism leading to discrepancies between genotype and respective protein levels, e.g., Factor XII, Protein Z [14, 15].
Implications for Research
Apparently, discovering new genes and other risk factors that are strongly associated with RM will be impossible, and small studies that will succeed in doing so won’t be successfully replicated by subsequent studies. In order to successfully detect genetic polymorphism with a weak but statically significant association with RM, large sample size groups of patients and controls must be included. In order to screen for only one polymorphism, 1213 patients and 1213 controls are needed [8]. Researchers must also keep in mind the great genetic polymorphism diversity among different ethnic groups when doing meta-analyses , and include only patients from related ethnic backgrounds. The importance of combinations of genetic biomarkers for RM—immunological, thrombophilic, and endocrine—must be further investigated.
Implications for Clinical Practice
When a single risk factor for a patient with RM is detected, the explanation given to the patient is much easier, relieving for the patient and answering the patient’s wish. However, as we saw, still, a great percentage of patients are with unknown etiology.
According to Christiansen’s model, the optimal future scenario is the condition where for every couple, combining information about validated genetic biomarkers, their individual strengths of association with RM, and their degree of epistatic interaction, together with information about relevant clinical factors into a computer-based algorithm, will derive the etiological fractions that are immunological, thrombophilic, endocrine, and fetal.
This knowledge will provide the clinician with the optimal means of providing the treatment that has been proven to be most efficient in placebo-controlled trials in adequately selected patients as discussed.
Window of Implantation
Human reproduction is very inefficient compared to other mammalian species, with many pregnancies being complicated or lost due to different disorders . A new research field might shed light on the implantation molecular events and try to understand some of those pathologies and their connection to RPL.
Recent publications have shown that implantation is a complex process, with many perils waiting to happen, such as preterm birth, IUGR, preeclampsia, placenta accerta, placenta previa, and miscarriage, in a case implantation doesn’t occur in its usual precise way (Fig. 14.1) [16]. Implantation is composed of two key components: the embryo and the receptive endometrium . This complex process depends on cross-talk between the embryo and the endometrium, and the very well synchronized progesterone-dependent changes in the endometrium to render it responsive to the embryonic signals. The concept of the “passive” deciduas and the “active” or “invasive” embryo is being challenged by recent research. Due to ethics issues and inaccessibility of implantation sites in humans, most of our knowledge of early pregnancy events is based on animal models and in vitro models. The window of implantation is a short time span starting ~6 days after ovulation and can last up to 5 days, in which the blastocyst is competent and the endometrium is at its receptive stage [17, 18]. In humans, compared to other mammals, the trophoblast invasion is deep, thus ensuring the endometrium has decidualized and is now ready for the embryo, can prevent failure in implantation, and be “selective” for the embryo quality [19].
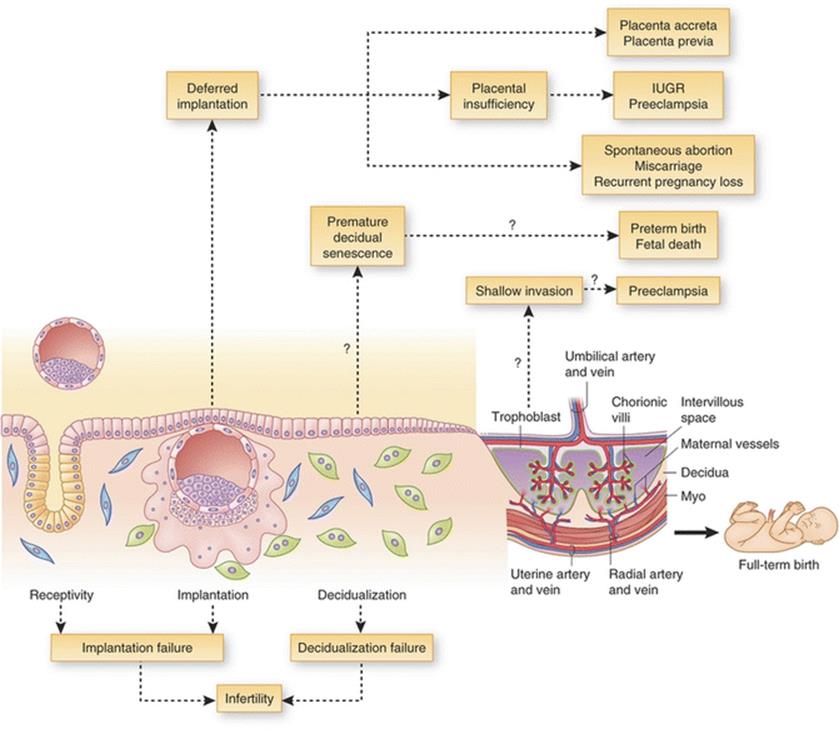
Fig. 14.1
Defective receptivity, implantation, and/or decidualization can lead to infertility. Deferred implantation past the window of receptivity can lead to misguided embryo placement and implantation, resulting in placenta previa, ectopic placentation (placenta accreta) or placental insufficiency resulting in intrauterine growth restriction (IUGR) and/or preeclampsia. Implantation beyond the normal window can also give rise to spontaneous abortion, miscarriage and recurrent pregnancy loss, leading to infertility. Premature decidual senescence can lead to preterm birth and fetal death , whereas shallow trophoblast invasion into maternal decidua and/or blood vessels can lead to preeclampsia. [Reprinted Cha, J., X. Sun, and S.K. Dey, Mechanisms of implantation: strategies for successful pregnancy. Nature medicine, 2012. 18(12): p. 1754–1767. With permission Nature Publishing Group]
Decidualization is a postovulatory process, driven mainly by the progesterone secretion from the corpus luteum , in which the endometrium is prepared for embryo implantation and pregnancy. Human endometrial stroma cells differentiate from fibroblast-like into secretory and receptive decidual endometrial stromal cells. It happens every cycle, during the mid-secretory phase, irrespective of pregnancy [20]. A novel hypothesis named “menstrual preconditioning” might have the answer for why the decidualization process happens each cycle, ending in most cases in menstrual bleeding and shedding of the decidua [21].
In “menstrual preconditioning” the repetitive, short exposures to harmful stimuli to a degree below the threshold for tissue injury will provide some degree of protection from subsequent injury—as will happen when the trophoblast deeply invades into the endometrium during pregnancy. It appears that decidualization grants valuable characteristics to the endometrium, including embryo defense against environmental and oxidative stress, regulation of trophoblast invasion, and protection of the uterus from aggressive invasion by the embryo.
Only after the decidualization process do the endometrium stroma cells have the ability to act as biosensors of the embryo quality and react to low quality embryos by shutting down production of key implantation mediators and immunomodulators such as IL-1b, -6, -10, -17, -18, eotaxin, and heparin-binding EGF-like growth factor, thus preventing it from implanting [22]. This process is considered to be “embryo selection” in humans.
What Happens When There Is Impaired Embryo Selection?
Understanding decidualization’s important role led to the idea that aberrations during the decidualization and implantation might give rise to pregnancy with embryos who would otherwise be rejected by the decidua , resulting in early embryo loss. For example, a study of 221 women attempting to conceive demonstrated dramatic risk, increasing on a daily basis, of early miscarriage if pregnancy was established beyond the normal “implantation window” [23]. Correspondingly, recent observations succeeded in identifying several differences between women who suffer from RPL and no-RPL women: Women suffering from RPL express lower levels of mucin 1, an anti-adhesion molecule that contributes to the barrier function of luminal epithelium [24–26]. Another research showed different levels of two maker genes : higher levels of prok1, which encodes prok1, a cytokine that promotes endometrium receptivity, and lower levels of prolactin, a prototypic marker of decidualizing endometrial cells [27].
It was also shown that decidualized endometrial stromal cells from women who suffer from RPL fail to discriminate between low and high quality embryos when it comes to migrating towards the embryo at the site of implantation, unlike non-RPL women (as seen in Fig. 14.2) [28].
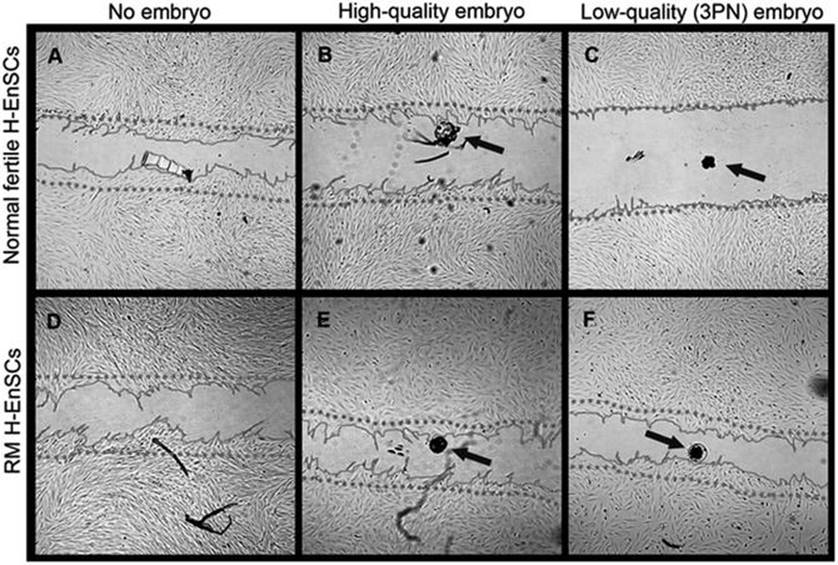
Fig. 14.2
The migration zone after adding a high-quality, low-quality or no embryo. The migratory response of decidualized H-EnSCs from normally fertile (a–c) and RM women (d–f) was analyzed in absence of a human embryo (a, d), in presence of a high-quality embryo (b, e) or a low-quality embryo (c, f). Phase contrast pictures were taken 18 h after creating the migration zone. The dotted line represents the front of the migration zone directly after its creation. As a reference for the position of the embryo, the bottom of the plate was marked. The arrows indicate the position of the embryo. All pictures were taken with 25× magnification. [Reprinted from Weimar, C.H., et al., Endometrial stromal cells of women with recurrent miscarriage fail to discriminate between high-and low-quality human embryos. PLoS One, 2012. 7(7): p. e41424. With permission from PLoS One]
The lack of embryo natural selection might provide an explanation for chromosomal and nonchromosomal pregnancy failures.
Superfertility and RPL
One could describe clinic al pregnancies as “the tip of the iceberg”—representing only 40 % of all conceptions, when 30 % of all conceptions are lost before implantation and an additional 30 % are lost before 6 weeks gestation [21].
The odds for a fertile couple to achieve pregnancy is ~20 % during one menstrual cycle, defined as the Monthly Fecundity Rate (MFR), odds that are considered to be very low compared to other mammalian species [29].
Using the MFR, a mathematical model predicts that 74, 93, and 100 % of normally fertile couples will achieve pregnancy in 6, 12, and 24 months.
Subfertility and superfertility have been defined by MFRs of 5 % or less, and 60 % or more, respectively [29]. According to the Tietze Model, it has been estimated that 79 % of the population is fertile, 18 % subfertile or infertile, and 3 % superfertile [30].
This relative inefficiency is sometimes considered to represent a strategy dealing with embryo chromosomal abnormalities, which is accepted as the most common cause for miscarriage, including recurrent miscarriage [31].
Recent observations have shown that as much as 40 % of the women with RPL can be defined as superfertile, with time-to-pregnancy (TTP) shorter than 3 months [27]. Rapid conceptions are associated with risk of early pregnancy loss even in low-risk populations, which can partly be explained by the lack of natural embryo selection.
In summary, we might learn from the low efficacy of human reproduction the role of cyclic decidualization of the endometrium and its importance.
Teklenburg et al. [22] coined the term “window of natural embryo selection,” as it reflects the functional role of decidualizing stromal cells in assessing the implanting embryo’s quality, and moreover the decidualizing stromal cells end the window of endometrial receptivity and enable the mother to dispose of embryos who are not “high quality.”
The most significant conclusion might be preventing early pregnancy complications and failures by targeting the endometrial decidu al response prior to pregnancy or immediately after implantation.
Heparin Use in Recurrent Pregnancy Loss
Heparin is a very widely used injectable anticoagulant and, according to the World Health Organization's List of Essential Medicines, it is one of the most important medications needed in a basic health system [32].
It was originally isolated from canine liver cells, hence its name (hepar is Greek for "liver") in 1916, but it wasn’t until the mid-30s that heparin was manufactured in a safe, nontoxic form, easily available, thus making it a popular anticoagulant. In the human body, heparin is stored exclusively in the granules of subsets of mast cells [33]. Although it is used principally in medicine for anticoagulation, its true physiological role in the body is still uncertain, since blood anticoagulation is achieved mostly by heparan sulfate proteoglycans derived from endothelial cells [34].
Almost 100 years after heparin’s discovery, nowadays best known for its first described anticoagulant ability, it is being researched for its other abilities as well. Heparin is useful in RPL in very specific indications but there is an option that some other less-known characteristics of the drug have impact on RPL outcome that come from other medical fields. Heparin is believed to possess many biological activities that include the ability to modulate embryonic development, neurite outgrowth, tissue homeostasis, wound healing, metastasis, cell differentiation, cell proliferation, and inflammation [33].
In this chapter we’ll see which of those attributes is pertinent in the RPL clinic through heparin’s effect on different molecules in in vitro studies and in vivo experiments.
Heparin’s Effect on Different Molecules
Although our knowledge about heparin’s anticoagulant properties has developed in the last century, discoveries about heparin’s anti-inflammatory features are comparatively new (see Chap. 2 for elaborated information). Heparin affects several molecules, including cytokines, growth factors, adhesion molecules, cytotoxic peptides, and tissue-destructive enzymes, many of which are crucially involved in the inflammatory process. Each of these proteins might be essential in better understanding the inflammatory component in RPL, and might help to decrease this phenomenon.
During the process of implantation heparin, and heparin-derived molecules, affect through expression of adhesion molecules, matrix-degrading enzymes, and trophoblast phenotype and apoptosis [35].
LMWH was shown to increase matrix metalloproteinase (MMPs) concentrations and activity and reduce their tissue inhibitor (TIMP) in a dose-dependent manner. Two of those MMPs, a group of matrix-degrading enzymes, were found to be necessary for the embryo’s ability to degrade the basement membrane of the uterine epithelium and to invade the uterine stroma [36]. Additionally, It was shown that LMWH induces an increased decidual expression and secretion of heparin-binding EGF-like growth factor (HB-EGF) and reduced TNF-alpha-induced apoptosis [37]. LMWH induces activation of a DNA-binding transcription factor that, once activated, enhances HB-EGF expression [38]; heparin has the ability to activate the EGF receptor in primary villous trophoblasts [39].
During the first trimester, fetal and placental development takes place in a low O2 tension environment, which is important to prevent complications related to exposure with normal concentrations of oxygen, such as preeclampsia, IUGR, and miscarriage [40]. HB-EGF has an important role in preventing hypoxic-induced apoptosis in the early stages of placentation. Heparin was also found to prevent apoptosis in human trophoblasts triggered by pathological and other stimuli ((IFN)-γ, (TNF)-α, thrombin, staurosporine) and activated survival signal transduction pathways [39].
More information on the effects of heparin on different molecules is elaborated in Table 14.1.
Table 14.1
Proteins involved in the inflammatory response that are bound by heparin and related molecules
|
Examples of heparin-binding inflammatory mediators |
(Patho)physiological significance |
Examples |
Comments |
|
Adhesion molecules |
Cell transport |
CD11b/CD18 [132] (MAC1) |
– |
|
– |
– |
P-selectin [133, 134] L-selectin [133] |
Specific residues that are found in P- and L- (but not E-)selectin are required for heparin/heparin-sulfate binding [135]. Inhibition of selectin-dependent leukocyte rolling by heparin and related molecules is directly related to sulphation [136] |
|
Chemokines [137] |
Inflammatory cell recruitment and activation; viral infection |
RANTES, IL-8, MIP1, MCP1, eotaxin |
– |
|
– |
– |
PF4 |
A minimum length of GAG chain is required for binding of PF4, a property that is exploited in improving the side-effect profile of heparin as an anticoagulant [138] |
|
Growth factors [137] |
Tissue repair and repai ring; angiogenesis |
PDGF, VEGF, TGF-β |
– |
|
– |
– |
FGF2 |
Binding affinity is related to both length and composition (L-iduronic acid content) of the GAG chain [139]. FGF2 signal transduction requires binding of heparin-sulfate (or heparin) to both FGF2 and its receptor. 2-O-sulphation is essential for the former and 6-O-sulphation for the latter. Therefore, selectively 6-O-desulphated heparin competitively inhibits FGF2-induced angiogenesis [140] |
|
Enzymes [137] |
Digestion of tissue/ECM structural components |
Heparanase69 MMPs |
– |
|
– |
– |
Elastase cathepsin |
As well as directly binding elastase and cathepsin G, heparin is thought to modulate the activity of these enzymes through potentiation of their natural inhibitor, SLPI. Heparin binds SLPI with greater affinity than less-sulphated GAGs, although within heparin, undersulphated chains bind with the highest affinity [141] |
|
Cytotoxic mediators [137] |
Destr uction of parasites; tissue damage |
ECP MBP |
– |
ECM extracellular matrix, ECP eosinophil cationic protein, FGF2 fibroblast growth factor 2, GAG glycosaminoglycan, IL-8 interleukin-8, MAC1 macrophage 1, MBP major basic protein, MCP1 monocyte chemotactic protein 1, MIP1 macrophage inflammatory protein 1, MMPs matrix metalloproteases, PDGF platelet-derived growth factor, PF4 platelet factor 4, RANTES regulated on activation, normal T-cell expressed and secreted, SLPI secretory leukocyte protease inhibitor, TGF-β transforming growth factor-β, VEGF vascular endothelial growth factor
Reprinted from Lever R, Page CP. Nonanticoagulant Effects of Heparin: An Overview. Handb Exp Pharmacol. 2012;(207):281–305. With permission from Springer Science
In Vitro Models Succeeded in Showing Some of Heparin’s Beneficial Effects
1.
2.
3.
In Vivo Experiments
Several small clinical trials have shown heparin can be helpful in several inflammatory diseases thanks to its anti-inflammatory qualities.
1.
2.
3.
Heparin and Recurrent Implantation Failure (RIF)
The term RIF has been used to describe IVF treatment failure due to embryos’ failure to implant. The ESHRE PGD consortium document mentioned that RIF can be considered after more than three high-quality embryo transfers or implantation failure with transfer of ≥10 embryos in multiple transfers with exact numbers to be determined by each center [48]. A meta-analysis with systemic review of the literature compared the use of LMWH with placebo or no adjuvant treatment in women with RIF undergoing IIVF/ICSI. The results have shown that in women with at least three RIFs, the use of LMWH during IVF treatment improved the live birth rate in 79 %. These results show that heparin might have a useful and important role during IVF treatment and more research should be done.
Heparin and Recurrent Pregnancy Loss
The role of heparin and LMWH has long been investigated and published, mainly in the context of antiphospholipid syndrome and thrombophilia. As was shown in the LIVE-ENOX study, prophylactic administration of enoxaparin to women with RPL and thrombophilia was found to be effective and safe [49]. Several studies that investigated heparin’s role when it comes to women who suffer with RPL with an unknown etiology didn’t show heparin to be beneficial, despite heparin attributes described hitherto.
One research done in Israel in 2006 compared live birth rates in women who suffered from RPL. One group was administered enoxaparin (LMWH) while the other received aspirin. No statistical difference was found between the two groups when comparing live birth rates, but a difference was shown between the live birth rates as expected from the literature (60 % after three pregnancy failures, 40 % after four pregnancy failures) and their results in both groups with more than 80 % live birth rate.
In a study where enoxaparin was compared to placebo when treating women with RPL with unknown cause no significant difference in live birth rate was found between the two groups (66.6 and 72.9 %, respectively) [50].
A study that compared different treatment methods for women with unexplained RPL found no difference in live birth rate (written in brackets after each group) when comparing treatment regimens with aspirin (50.8 %), aspirin + (LMWH) nadroparin (54.5 %), and placebo (57.0 %) [51].
In conclusion, heparin has a place of honor in RPL treatment thanks to its anticoagulant ability; heparin’s additional characteristics are the reason for the conclusion that it might be useful in RPL women who don’t have thrombophilia. Hitherto, no conclusive evidence has yet supported this idea. Further research and investigation are needed to be sure that we do not inject heparin for no reason.
Involvement of Immunity in Recurrent Pregnancy Loss
Pregnancy: A Balancing Act of the Maternal Immune System
Pregnancy success requires suppression of the mother’s immune system, enabling an immune-tolerant state [52]. The maternal immune system of endometrial and decidual tissues is primarily composed of immune cell populations that are myelomonocytic, T cells and decidual Natural Killer (dNK) cells . dNK cells are thought to play an important role, serving as the predominant cell type in this process [53]. These immune cell populations play an important role in placental tissue development, fetal growth, and establishment of immune-tolerance by secretion of various type-1 cytokines (IFN-γ, TNF-α, TNF-β, and IL-2) that contribute to cellular immunity, and type-2 cytokines (IL-4, IL-5, IL-6, IL-10, and IL-13) that encourage humoral immunity.
The balance between type-1 and type-2 cytokines is essential to the success of pregnancy [54–56]. IFN-γ supports remodeling of spiral arteries, promotes immune tolerance by inhibition of pro-inflammatory TH17 cells, and encourages indoleamine 2,3-dioxygenase (IDO) upregulation in myelomonocytic cells, which in turn induces Treg FOXP3+ and suppress CD8+ T cells [53, 57, 58]. IFN-γ also increases TRAIL-R expression on syncytiotrophoblasts, and human trophoblasts express FasL; both molecules can serve as a mechanism for protection against dNK and T cells. TNF-α mediates apoptosis of cytotrophoblasts and promotes the formation of syncytiotrophoblasts [59]. IL-10 inhibits the secretion of IFN-γ and TNF-α, modulating trophoblast invasion and suppressing TH17 cells [60]. IL-10 also regulates the number of myelomonocytic cells that are the main antigen presenting cells in the decidua tissue (20–30 % throughout pregnancy) and have a role in defending against microbes. Type 3 cytokines, such as TGF-β, can regulate the balance between type-1 and type-2 cytokines. Secretion of TGF-β by decidual myelomonocytic cells leads to inhibition of dNK and prevents the killing of the cytotrophoblast by dNK [61]. Other cytokines and chemokines additionally have a role in placenta development and immune tolerance. For example, MIP-1α (CCL3) and MIP-1β (CCL4) are important to attract and activate maternal immune cells, while IL-8 promotes trophoblast migration. GM-CSF regulates decidual leukocyte populations and enhances placental growth and differentiation [62, 63] thereby, both type-1 and type-2 cytokines have a major role in regulating the development of placental tissue and establishment of an immune-tolerance towards the fetus.
Recurrent Pregnancy Loss and NK Cells: Breaking the Balance
Recurrent pregnancy loss (RPL) was recently suggested to be associated with superfertility. In a retrospective analysis of 560 RPL patients, Salker et al. showed that 40 % of the women were considered “superfertile,” relative to the prevalence in the total population that is about 3 % [21, 64]. Superfertility refers to a longer “Implantation window ” [65]. This interval in the menstrual cycle is a transient endometrial state with a high receptivity for the adherence of a blastocyst that is dependent on paracrine signals from stromal cells to the immune cell populations and is part of the decidualization process[66]. This prolonged blastocyst receptivity correlates with early pregnancy loss and superfertility [21].
During the first trimester of pregnancy, dNK cells are the dominant cell population and are about 50–70 % of leukocytes (Fig. 14.3) [67–69]. dNK cells secrete IFN-γ, TNF-α, GM-CSF, IP-10, MIP-1α (CCL3), MIP-1β (CCL4), IL-8, VEGF, PLGF, Ang-1, Ang-2, IL-10, and IL-1RA [70, 71]. Decidual stromal cells can regulate myelomonocytic cells and dNK cells by the IL-33/ST2L/sST2 axis. Upon decidualization, a mechanism that was disordered in stromal cells of women with RPL [72], the IL-33/ST2 axis, also promotes the temporal expression of receptivity genes in stromal cells; failure to restrain this axis leads to a longer implantation window [72]. IL-33 promotes proliferation of stromal cells, macrophages, and trophoblasts [73]. ST2, a receptor for IL-33, is expressed on dNK but not on peripheral blood NK cells (see Chap. 6 for elaborated information). Culture medium from decidual stromal cells inhibits the cytolytic activity of dNKs. Furthermore, it shifts the cytokines’ balance of dNK from type-1 to type-2 by downregulating the secretion of IFN-γ and TNF-α, and upregulating IL-10 [74]. In mice, administration of IFN-γ and TNF-α promote abortions and both cytokines inhibit growth of human trophoblasts in vitro [55]. IL-33 also up regulates the expression of PCNA in stromal cells, a nuclear protein that was shown to inhibit NK cell function by interaction with NKp44 [75–77].
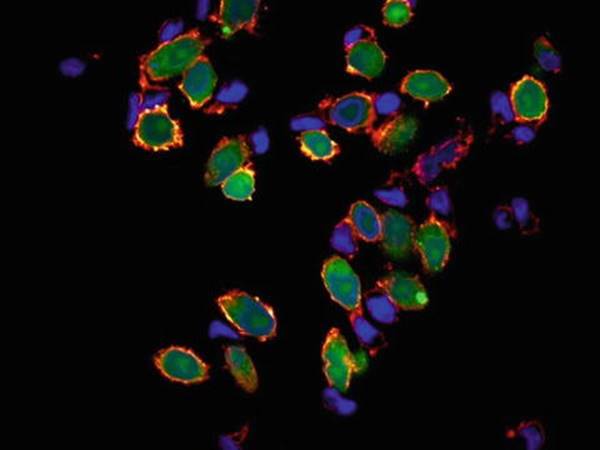
Fig. 14.3
Human natural killer cells interacting with CFSE-labeled cervical cancer (HeLa) cells (green) and nuclei dye (DAPI blue). Filamentus actin is labeled with Phalloidin (yellow to red) to show points of interaction. Acquires using FV1000 (Olympus) equipped with 100× oil objective. [Confocal Microscopy, Courtesy of Mr. Uzi Hadad, Ben-Gurion University of the Negev]
Moreover, it was shown that decidual stromal cells and trophoblasts express ligands to DNAM-1, NKp30, and NKp44, which are activation receptors expressed on NK cells [71, 78]. Recently, it was suggested that women with RPL have impaired regulation of pregnancy-related cytokines [79, 80]. Analysis of peripheral blood NK cells and their NKp30, NKp44, and NKp46 receptors (NCRs) expression compared to intracellular cytokine expression (TNF-a, IFN-γ, IL-4, IL-10) after activation revealed a negative correlation between NCRs’ protein expression and intracellular cytokine. Furthermore, the correlation between the mRNA expression of NKp30, NKp46, and pregnancy-related cytokines seems to be lost in placental tissue from RPL patients compared to elective abortions. Moreover, mRNA of IFN-γ, TNF-α, and IL-10 was higher in placenta tissue obtained from spontaneous miscarriage patients [81, 82].
Depletion of NK cells in a murine model did not have an effect on the outcome of pregnancy [83]. However, in an IL-10 KO murine model, NK cell activity was enhanced after LPS administration and led to pregnancy loss. Depletion of dNK or administration of anti-TNF-α or -IL-10 rescued pregnancies [84]. These observations point to the necessity of a balance between type-1 and type-2 cytokines, which in turn influence dNK cells activity and can lead to pregnancy loss [85].
Influencing the Balance: Target for Therapy
Immunologic etiologies can be attributed to 40 % of all RPL cases. There is a strong association between pregnancy loss and type-1 cytokines while a successful pregnancy is associated with type-2 cytokines [54–56]. In a murine model of antiphospholipid syndrome (APS) , antiphospholipid (aPL) Abs increased in decidual and systemic TNF-α levels, which promote trophoblast apoptosis, identifying TNF blockade as a potential therapy for the pregnancy complications [86]. TNF-α activity can be blocked with monoclonal antibodies against the TNF-α molecule (adalimumab) or against soluble TNF-α receptors (etanercept) [87, 88]. TNF-α blockers administered with or without anticoagulants, or anticoagulants + IVIG treatments (control groups), were given to women with RPL history during pregnancy. The live birth rate of patients who received TNF-α blockers was 71 %, whereas the control groups showed rates of 19 and 54 %, respectively [89]. The same trend was observed in women undergoing IVF; TNF-α blockers improve the implantation rate [90].
G-CSF was also found to have a positive effect on RPL patients. G-CSF reduces the cytotoxicity and IFN-γ secretion of dNK cells, increases the number of Treg cells, and reduces the synthesis of various cytokines, among them TNF-α [91]. A randomized controlled trial of women with RM treated with G-CSF or a placebo showed that G-CSF administrations increased the live birth rate from 48.5 % (placebo group) to 82.8 % (G-CSF group) [92]. In a second study, treatment of RPL patients undergoing assisted reproductive treatment (IVIG, LMWH, cortisone) with G-CSF increased the live birth rate from 13 to 32 % [93].
The results of these few clinical studies reveal the necessity of maintaining a balance between type 1 and type 2 cytokines in early pregnancy. TNF-α inhibition seems to have much potential in future clinical therapies.
RPL and Several Molecular Findings
Alijotas-Reig et al. list in their review three new risk factors : microparticles, glycoproteins, and leptin [94].
Microparticles —Microparticles (MPs) are a heterogeneous group of submicronic phospholipid vesicles, 0.1–1 μm in size, derived from different cell types including platelets, endothelial cells, leukocytes, and red blood cells besides several other cell types and are found also in normal healthy conditions. They are released from the cytoplasmic membranes during activation or apoptosis [94, 95]. They represent subcellular elements for cell signaling and intracellular communication in inflammation and thrombosis [96] and were found in increased numbers in several prothrombotic conditions such as deep vein thrombosis, pulmonary embolism, and stroke [97]. This proinflammatory attribute is believed to be due to MPs’ expression of different anionic phospholipids such as phosphatidylserine [94]. MPs can also be found in increased numbers in normal pregnancy [98, 99] and in complicated pregnancy disorders, mainly severe preeclampsia [100, 101], which was recently discovered to be correlated to RPL in studies on trophoblastic circulating MPs [102, 103].
It is still unclear if this increase in MPs is a cause or consequence of RPL [94].
Glycoproteins —Glycoproteins expressed at the fetal–maternal interface have been shown to have immunomodulating effects. Human chorionic gonadotropin (hCG) and glycodelin (Gd) are glycoproteins secreted in large amounts by the trophoblast or the decidualized endometrium, mainly during the first trimester of pregnancy [104], but in RPL patients both proteins were found to be downregulated [105, 106].
It was found that glycodelin and hCG both inhibit the E-selectin-mediated cell adhesion, so this could indicate a possible role of these proteins in preventing leukocyte adhesion to the fetal trophoblast [107].
hCG—Our understanding of hCG has improved a lot during the last 15 years. Nowadays, we know that hCG consists of two different forms with different actions [108]. We will focus on the new immunological revelations: Lymphocytes from pregnant women express the hCG receptor gene [109]. High hCG levels at very early pregnancy stages ensure regulatory T-cell migration to the fetal–maternal interface, the contact site between paternal antigens and maternal immune cells, where it orchestrates the immunologic tolerance of the fetus [110]. Thus, reduced expression of hCG in patients with recurrent miscarriage affects the process of fetal tolerance.
Leptin —The hormone leptin, a 16 kDa polypeptide, is mainly synthesized and secreted by the white adipose tissue (WAT) . Leptin, acting on specific populations of neurons in the brain, including hypothalamic, midbrain, and brainstem neurons, plays a central role in weight control by suppressing food intake and increasing energy expenditure [111–113] and apparently it also plays an important role in reproduction. Mice that have leptin deficiency are infertile, but fertility can be restored by injections of recombinant leptin [114, 115]. Leptin and receptor transcripts were identified in the villous and extravillous trophoblast [116]. Interestingly, Lage et al. showed that a group of women who suffered spontaneous miscarriage showed leptin levels identical to women post-partum, but significantly reduced when compared with the control group and women in the first trimester of pregnancy, and significantly lower than nonpregnant control women. As these women were actually in the first trimester of pregnancy when the miscarriage occurred, higher levels of leptin should be expected [117]. The similar leptin values of post-partum and post-miscarriage groups suggests that leptin seems to be acting as an indicator that the pregnancy process has been stopped, either naturally at term or pathologically some time earlier. Larid et al. suggested that the significantly lower concentration of leptin in women who subsequently miscarried suggests that leptin may play a role in preventing miscarriage [118].
Annexin 5
As mentioned, among the leading candidates for the molecular basis of RPL are various inherited hypercoagulation disorders that promote thrombosis, collectively named “thrombophilias.” Among these disorders are carriers of either factor V Leiden (FVL) mutation or the factor 2 (Prothrombin) G20210A (PTm) mutation that have proved by meta-analyses to be association with RPL [7].
Annexin-V is a member of a family of calcium-dependent phospholipid binding proteins [119]. It shows the essential tetrad structure and calcium-dependent phospholipid binding and is one of the few annexins that can be found extracellularly [120]. The annexin-V gene (ANXA5) is located in chromosome 4q27, and has several transcription options [121]. Annexin-V has been isolated from human placenta [122], blood vessels [123], and other sites as well. Annexin-V has anticoagulant activity in vitro, which is based on its high affinity for anionic phospholipids and its capacity to displace coagulation factors from the phospholipid surface [124] and/or its ability to downregulate the cell surface presentation of tissue factor [124].
It seems that Annexin-V forms an antithrombotic shield on the apical surface of placental syncytiotrophoblasts, and that it might be interrupted by antiphospholipid antibodies [125].
Annexin-V and aPL
A well-known major risk factor f or RPL is the presence of circulating maternal antiphospholipid antibodies (aPL) [126]. APLA syndrome is marked by vascular thromboembolism or recurrent pregnancy losses, and by evidence for antibodies against anionic phospholipid–protein complexes in the plasma or serum of affected patients. The pathophysiologic pathways of this syndrome are not completely known [127].
Using atomic microscope, it was discovered that in the presence of aPL antibodies and cofactor, structures presumed to be aPL monoclonal antibody–antigen complexes, were associated with varying degrees of disruption to the Annexin-V structure, which is valuable for its anticoagulant activity [128].
Rand et al. [125] revealed that once trophoblasts and endothelial cells were exposed to antiphospholipid-antibody IgG, annexin-V levels were reduced. The antiphospholipid antibodies accelerated the coagulation of plasma on the trophoblasts and endothelial cells. The reduction of Annexin-V levels on vascular cells may be an impor tant pathway in the aPL syndrome.
Annexin-V and RPL
Bogdanova et al. [129] analyzed 70 German RPL patients, all known to carry neither factor V Leiden nor a prothrombin mutation , and found that carriers of genetic variant, haplotype M2, in the ANXA5 gene promoter have two to four times higher risk for RPL compared to two different groups of noncarriers. Apparently M2 haplotype reduces the in vitro activity of the ANXA5 promoter to 37–42 % of the normal range.
Additionally, carrying the M2 haplotype of the ANAX5 gene was also found to be associated with delivering small-for-gestational age newborns [130].
Miyamura et al. [131] genotyped 243 Japanese women who suffered from RPL and 119 fertile control women for 4 ANXA5 gene promoter polymorphisms. Very similar to the M2 haplotype for Western women, one haplotype was found at a significantly greater frequency in RPL women than in the control group. Homozygotes of the SNP5 minor allele were more frequent in the RPL group (p = 0.02), and this genotype conferred a sevenfold higher risk of RPL (OR = 7.76). These observations give rise to the thought that variations in the ANAX5 gene leading to Annexin-V structural change, or to its reduced expression, could be responsible for the immunological and hemostatic phenomena that, together, lead to fetal loss . Annexin-V is a potent anticoagulant that serves a thrombo-modulatory function in the placental circulation. One explanation might be that decreased expression of Annexin-V on the surface of the trophoblast might result in inefficient phospholipid shielding and hence in a potential enrichment of antigenic determinants, leading to aPL generation. Another explanation is that even in the absence of aPL, reduced expression of Annexin-V can cause a hyper-coagulable state in the intervillous placental space [129]. Annexin-V might act as a genetic marker for RPL, taking us one step further in understanding and preventing RPL.
References
1.
Christiansen OB, et al. Multifactorial etiology of recurrent miscarriage and its scientific and clinical implications. Gynecol Obstet Invest. 2008;66(4):257–67.PubMed
2.
Stephenson M. Frequency of factors associated with habitual abortion in 197 couples. Fertil Steril. 1996;66(1):24–9.PubMed
3.
Nielsen HS, Christiansen OB. Prognostic impact of anticardiolipin antibodies in women with recurrent miscarriage negative for the lupus anticoagulant. Hum Reprod. 2005;20(6):1720–8.PubMed
4.
Creasy R. The cytogenetics of spontaneous abortion in humans. Early pregnancy loss. London: Springer; 1988. p. 293–304.
5.
Bellver J, et al. Obesity and poor reproductive outcome: the potential role of the endometrium. Fertil Steril. 2007;88(2):446–51.PubMed
6.
Homburg R. Pregnancy complications in PCOS. Best Pract Res Clin Endocrinol Metab. 2006;20(2):281–92.PubMed
7.
Rey E, et al. Thrombophilic disorders and fetal loss: a meta-analysis. Lancet. 2003;361(9361):901–8.PubMed
8.
Robertson L, et al. Thrombophilia in pregnancy: a systematic review. Br J Haematol. 2006;132(2):171–96.PubMed
9.
Hattersley AT, McCarthy MI. What makes a good genetic association study? Lancet. 2005;366(9493):1315–23.PubMed
10.
Hviid T, et al. Association between human leukocyte antigen‐G genotype and success of in vitro fertilization and pregnancy outcome. Tissue Antigens. 2004;64(1):66–9.PubMed
11.
Kruse C, et al. A study of HLA‐DR and‐DQ alleles in 588 patients and 562 controls confirms that HLA‐DRB1* 03 is associated with recurrent miscarriage. Hum Reprod. 2004;19(5):1215–21.PubMed
12.
Christiansen OB, et al. Association between HLA-DR1 and-DR3 antigens and unexplained repeated miscarriage. Hum Reprod Update. 1999;5(3):249–55.PubMed
13.
Rull K, Nagirnaja L, Laan M. Genetics of recurrent miscarriage: challenges, current knowledge, future directions. Front Genet. 2012;3:34.PubMedPubMedCentral
14.
Topalidou M, et al. Low protein Z levels, but not the intron F G79A polymorphism, are associated with unexplained pregnancy loss. Thromb Res. 2009;124(1):24–7.PubMed
15.
Iinuma Y, et al. Coagulation factor XII activity, but not an associated common genetic polymorphism (46C/T), is linked to recurrent miscarriage. Fertil Steril. 2002;77(2):353–6.PubMed
16.
Cha J, Sun X, Dey SK. Mechanisms of implantation: strategies for successful pregnancy. Nat Med. 2012;18(12):1754–67.PubMed
17.
Horcajadas J, Pellicer A, Simon C. Wide genomic analysis of human endometrial receptivity: new times, new opportunities. Hum Reprod Update. 2007;13(1):77–86.PubMed
18.
Macklon NS, Geraedts JP, Fauser BC. Conception to ongoing pregnancy: the “black box” of early pregnancy loss. Hum Reprod Update. 2002;8(4):333–43.PubMed
19.
Ledbetter DH. Chaos in the embryo. Nat Med. 2009;15(5):490–1.PubMed
20.
Gellersen B, Brosens IA, Brosens JJ. Decidualization of the human endometrium: mechanisms, functions, and clinical perspectives. Semin Reprod Med. 2007;25(6):445–53.
21.
Teklenburg G, et al. The molecular basis of recurrent pregnancy loss: impaired natural embryo selection. Mol Hum Reprod. 2010;16(12):886–95.PubMed
22.
Teklenburg G, et al. Natural selection of human embryos: decidualizing endometrial stromal cells serve as sensors of embryo quality upon implantation. PLoS One. 2010;5(4):e10258.PubMedPubMedCentral
23.
Wilcox AJ, Baird DD, Weinberg CR. Time of implantation of the conceptus and loss of pregnancy. N Engl J Med. 1999;340(23):1796–9.PubMed
24.
Serle E, et al. Endometrial differentiation in the peri-implantation phase of women with recurrent miscarriage: a morphological and immunohistochemical study. Fertil Steril. 1994;62(5):989–96.PubMed
25.
Aplin J, Hey N, Li T. MUC1 as a cell surface and secretory component of endometrial epithelium: reduced levels in recurrent miscarriage. Am J Reprod Immunol. 1996;35(3):261–6.PubMed
26.
Hey N, et al. MUC1 in secretory phase endometrium: expression in precisely dated biopsies and flushings from normal and recurrent miscarriage patients. Hum Reprod. 1995;10(10):2655–62.PubMed
27.
Salker M, et al. Natural selection of human embryos: impaired decidualization of endometrium disables embryo-maternal interactions and causes recurrent pregnancy loss. PLoS One. 2010;5(4):e10287.PubMedPubMedCentral
28.
Weimar CH, et al. Endometrial stromal cells of women with recurrent miscarriage fail to discriminate between high-and low-quality human embryos. PLoS One. 2012;7(7):e41424.PubMedPubMedCentral
29.
Evers JL. Female subfertility. Lancet. 2002;360(9327):151–9.PubMed
30.
Tietze C, Guttmacher AF, Rubin S. Time required for conception in 1727 planned pregnancies. Fertil Steril. 1950;1(4):338.PubMed
31.
Brosens JJ, Gellersen B. Something new about early pregnancy: decidual biosensoring and natural embryo selection. Ultrasound Obstet Gynecol. 2010;36(1):1–5.PubMed
32.
WHO. Model list of essential medicines. In: World Health Organization. World Health Organization; 2014.
33.
Tyrrell DJ, Kilfeather S, Page CP. Therapeutic uses of heparin beyond its traditional role as an anticoagulant. Trends Pharmacol Sci. 1995;16(6):198–204.
34.
Marcum J, et al. Anticoagulantly active heparin-like molecules from mast cell-deficient mice. Am J Physiol. 1986;250(5):H879–88.PubMed
35.
Quaranta M, et al. The physiologic and therapeutic role of heparin in implantation and placentation. PeerJ. 2015;2:e691.
36.
Staun-Ram E, et al. Expression and importance of matrix metalloproteinase 2 and 9 (MMP-2 and -9) in human trophoblast invasion. Reprod Biol Endocrinol. 2004;2:59.PubMedPubMedCentral
37.
Di Simone N, et al. Low-molecular-weight heparins induce decidual heparin-binding epidermal growth factor-like growth factor expression and promote survival of decidual cells undergoing apoptosis. Fertil Steril. 2012;97(1):169–77 e1.PubMed
38.
D’Ippolito S, et al. Emerging nonanticoagulant role of low molecular weight heparins on extravillous trophoblast functions and on heparin binding-epidermal growth factor and cystein-rich angiogenic inducer 61 expression. Fertil Steril. 2012;98(4):1028–36 e1-2.PubMed
39.
Hills FA, et al. Heparin prevents programmed cell death in human trophoblast. Mol Hum Reprod. 2006;12(4):237–43.PubMed
40.
Jauniaux E, et al. Trophoblastic oxidative stress in relation to temporal and regional differences in maternal placental blood flow in normal and abnormal early pregnancies. Am J Pathol. 2003;162(1):115–25.PubMedPubMedCentral
41.
Wang X-F, et al. Low molecular weight heparin relieves experimental colitis in mice by downregulating IL-1β and inhibiting syndecan-1 shedding in the intestinal mucosa. PloS One. 2013;8(7):e66397.PubMedPubMedCentral
42.
Black SC, et al. Cardioprotective effects of heparin or N-acetylheparin in an in vivo model of myocardial ischaemic and reperfusion injury. Cardiovasc Res. 1995;29(5):629–36.PubMed
43.
Preuss JM, Page CP. Effect of heparin on antigen‐induced airway responses and pulmonary leukocyte accumulation in neonatally immunized rabbits. Br J Pharmacol. 2000;129(8):1585–96.PubMedPubMedCentral
44.
Ahmed T, Garrigo J, Danta I. Preventing bronchoconstriction in exercise-induced asthma with inhaled heparin. N Engl J Med. 1993;329(2):90–5.PubMed
45.
Evans R, et al. Treatment of corticosteroid‐resistant ulcerative colitis with heparin – a report of 16 cases. Aliment Pharmacol Ther. 1997;11(6):1037–40.PubMed
46.
Chande N, McDonald JW, MacDonald, JK. Unfractionated or low-molecular weight heparin for induction of remission in ulcerative colitis. Cochrane Database Syst Rev. 2008;2.
47.
Brown RA, et al. Additional clinical benefit of enoxaparin in COPD patients receiving salmeterol and fluticasone propionate in combination. Pulm Pharmacol Ther. 2006;19(6):419–24.PubMed
48.
Thornhill AR, ESHRE PGD Consortium, et al. Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS). Hum Reprod. 2005;20(1):35–48.PubMed
49.
Brenner B, et al. Efficacy and safety of two doses of enoxaparin in women with thrombophilia and recurrent pregnancy loss: the LIVE‐ENOX study. J Thromb Haemost. 2005;3(2):227–9.PubMed
50.
Pasquier E, et al. Enoxaparin for prevention of unexplained recurrent miscarriage: a multicenter randomized double-blind placebo-controlled trial. Blood. 2015;125(14):2200–5.PubMedPubMedCentral
51.
Kaandorp SP, et al. Aspirin plus heparin or aspirin alone in women with recurrent miscarriage. N Engl J Med. 2010;362(17):1586–96.PubMed
52.
Warning JC, McCracken SA, Morris JM. A balancing act: mechanisms by which the fetus avoids rejection by the maternal immune system. Reproduction. 2011;141(6):715–24.PubMed
53.
Vacca P, et al. Origin, phenotype and function of human natural killer cells in pregnancy. Trends Immunol. 2011;32(11):517–23.PubMed
54.
Raghupathy, Raj. Pregnancy: success and failure within the Th1/Th2/Th3 paradigm. Seminars in immunology. Vol. 13. No. 4. Academic Press, 2001.
55.
Chaouat G. The Th1/Th2 paradigm: still important in pregnancy? Seminars in Immunopathol. Vol. 29. No. 2. New York: Springer-Verlag; 2007.
56.
Costeas PA, et al. Th2/Th3 cytokine genotypes are associated with pregnancy loss. Hum Immunol. 2004;65(2):135–41.PubMed
57.
Sones JL, et al. Role of decidual natural killer cells, interleukin-15, and interferon-γ in placental development and preeclampsia. Am J Physiol Regul Integr Comp Physiol. 2014;307(5):R490–2.PubMedPubMedCentral
58.
Fu B, et al. Natural killer cells promote immune tolerance by regulating inflammatory TH17 cells at the human maternal–fetal interface. Proc Natl Acad Sci. 2013;110(3):E231–40.PubMedPubMedCentral
59.
Jerzak M, Bischof P. Apoptosis in the first trimester human placenta: the role in maintaining immune privilege at the maternal-foetal interface and in the trophoblast remodelling. Eur J Obstet Gynecol Reprod Biol. 2002;100(2):138–42.PubMed
60.
Brogin Moreli J, et al. Interleukin 10 and tumor necrosis factor-alpha in pregnancy: aspects of interest in clinical obstetrics. ISRN Obstet Gynecol. 2012;2012:230742.PubMedPubMedCentral
61.
Gormley M, et al. Maternal decidual macrophages inhibit NK cell killing of invasive cytotrophoblasts during human pregnancy. Biol Reprod. 2013;88(6):155.PubMedPubMedCentral
62.
Robertson SA. GM-CSF regulation of embryo development and pregnancy. Cytokine Growth Factor Rev. 2007;18(3):287–98.PubMed
63.
Rahmati M, et al. Colony stimulating factors 1, 2, 3 and early pregnancy steps: from bench to bedside. J Reprod Immunol. 2015;109:1–6.PubMed
64.
Orlando J, Coulam C. Is superfertility associated with recurrent pregnancy loss? Am J Reprod Immunol. 2014;72(6):549–54.PubMed
65.
Aplin JD. The cell biological basis of human implantation. Best Pract Res Clin Obstet Gynaecol. 2000;14(5):757–64.
66.
Weimar CH, et al. The motile and invasive capacity of human endometrial stromal cells: implications for normal and impaired reproductive function. Hum Reprod Update. 2013;19(5):542–57.PubMed
67.
Bansal AS. Natural killer cells and their activation status in normal pregnancy. Int J Reprod Med. 2013;2013.
68.
Hanna J, Mandelboim O. When killers become helpers. Trends Immunol. 2007;28(5):201–6.PubMed
69.
Moffett-King A. Natural killer cells and pregnancy. Nat Rev Immunol. 2002;2(9):656–63.PubMed
70.
El Costa H, et al. Critical and differential roles of NKp46-and NKp30-activating receptors expressed by uterine NK cells in early pregnancy. J Immunol. 2008;181(5):3009–17.PubMed
71.
Hanna J, et al. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med. 2006;12(9):1065–74.PubMed
72.
Salker MS, et al. Disordered IL-33/ST2 activation in decidualizing stromal cells prolongs uterine receptivity in women with recurrent pregnancy loss. PLoS One. 2012;7(12):e52252.PubMedPubMedCentral
73.
Fock V, et al. Macrophage-derived IL-33 is a critical factor for placental growth. J Immunol. 2013;191(7):3734–43.PubMed
74.
Hu W-T, et al. Decidual stromal cell-derived IL-33 contributes to Th2 bias and inhibits decidual NK cell cytotoxicity through NF-κB signaling in human early pregnancy. J Reprod Immunol. 2015;109:52–65.PubMed
75.
Hu W-T, et al. IL-33 enhances proliferation and invasiveness of decidual stromal cells by up-regulation of CCL2/CCR2 via NF-κB and ERK1/2 signaling. Mol Hum Reprod. 2014;20(4):358–72.PubMed
76.
Rosental B, et al. Proliferating cell nuclear antigen is a novel inhibitory ligand for the natural cytotoxicity receptor NKp44. J Immunol. 2011;187(11):5693–702.PubMedPubMedCentral
77.
Rosental B, et al. A novel mechanism for cancer cells to evade immune attack by NK cells: the interaction between NKp44 and proliferating cell nuclear antigen. Oncoimmunology. 2012;1(4):572–4.PubMedPubMedCentral
78.
Vacca P, et al. Regulatory role of NKp44, NKp46, DNAM-1 and NKG2D receptors in the interaction between NK cells and trophoblast cells. Evidence for divergent functional profiles of decidual versus peripheral NK cells. Int Immunol. 2008;20(11):1395–405.PubMed
79.
Lash GE, Ernerudh J. Decidual cytokines and pregnancy complications: focus on spontaneous miscarriage. J Reprod Immunol. 2015;108:83–9.PubMed
80.
Carp H. Cytokines in recurrent miscarriage. Lupus. 2004;13(9):630–4.PubMed
81.
Fukui A, et al. Correlation between natural cytotoxicity receptors and intracellular cytokine expression of peripheral blood NK cells in women with recurrent pregnancy losses and implantation failures. Am J Reprod Immunol. 2009;62(6):371–80.PubMed
82.
Shemesh A, et al. First trimester pregnancy loss and the expression of alternatively spliced NKp30 isoforms in maternal blood and placental tissue. Front Immunol. 2015;6:189.PubMedPubMedCentral
83.
Barber EM, Pollard JW. The uterine NK cell population requires IL-15 but these cells are not required for pregnancy nor the resolution of a Listeria monocytogenes infection. J Immunol. 2003;171(1):37–46.PubMed
84.
Murphy SP, et al. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol. 2005;175(6):4084–90.PubMed
85.
Sharma S. Natural killer cells and regulatory T cells in early pregnancy loss. Int J Dev Biol. 2014;58:219.PubMedPubMedCentral
86.
Berman J, Girardi G, Salmon JE. TNF-α is a critical effector and a target for therapy in antiphospholipid antibody-induced pregnancy loss. J Immunol. 2005;174(1):485–90.PubMed
87.
Weinblatt ME, et al. Adalimumab, a fully human anti–tumor necrosis factor α monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: the ARMADA trial. Arthritis Rheum. 2003;48(1):35–45.PubMed
88.
Moreland LW, et al. Etanercept therapy in rheumatoid arthritis: a randomized, controlled trial. Ann Intern Med. 1999;130(6):478–86.PubMed
89.
Winger EE, Reed JL. Treatment with tumor necrosis factor inhibitors and intravenous immunoglobulin improves live birth rates in women with recurrent spontaneous abortion. Am J Reprod Immunol. 2008;60(1):8–16.PubMed
90.
Winger EE, et al. Treatment with adalimumab (Humira®) and intravenous immunoglobulin improves pregnancy rates in women undergoing IVF*. Am J Reprod Immunol. 2009;61(2):113–20.PubMed
91.
Würfel W. Treatment with granulocyte colony-stimulating factor in patients with repetitive implantation failures and/or recurrent spontaneous abortions. J Reprod Immunol. 2015;108:123–35.PubMed
92.
Scarpellini F, Sbracia M. Use of granulocyte colony-stimulating factor for the treatment of unexplained recurrent miscarriage: a randomised controlled trial. Hum Reprod. 2009;24(11):2703–8.PubMed
93.
Santjohanser C, et al. Granulocyte-colony stimulating factor as treatment option in patients with recurrent miscarriage. Arch Immunol Ther Exp (Warsz). 2013;61(2):159–64.
94.
Alijotas-Reig J, Garrido-Gimenez C. Current concepts and new trends in the diagnosis and management of recurrent miscarriage. Obstet Gynecol Surv. 2013;68(6):445–66.PubMed
95.
Patil R, et al. Elevated procoagulant endothelial and tissue factor expressing microparticles in women with recurrent pregnancy loss. PLoS One. 2013;8(11):e81407.PubMedPubMedCentral
96.
Distler JH, et al. Microparticles as regulators of inflammation: novel players of cellular crosstalk in the rheumatic diseases. Arthritis Rheum. 2005;52(11):3337–48.PubMed
97.
Zahra S, et al. Microparticles, malignancy and thrombosis. Br J Haematol. 2011;152(6):688–700.PubMed
98.
Bretelle F, et al. Circulating microparticles: a marker of procoagulant state in normal pregnancy and pregnancy complicated by preeclampsia or intrauterine growth restriction. Thromb Haemost. 2003;89(3):486–92.PubMed
99.
Alijotas-Reig J, Palacio-Garcia C, Vilardell-Tarres M. Circulating microparticles, lupus anticoagulant and recurrent miscarriages. Eur J Obstet Gynecol Reprod Biol. 2009;145(1):22–6.PubMed
100.
González-Quintero VH, et al. Elevated plasma endothelial microparticles: preeclampsia versus gestational hypertension. Am J Obstet Gynecol. 2004;191(4):1418–24.PubMed
101.
Alijotas-Reig J, et al. Circulating cell‐derived microparticles in women with pregnancy loss. Am J Reprod Immunol. 2011;66(3):199–208.PubMed
102.
Kaptan K, et al. Platelet-derived microparticle levels in women with recurrent spontaneous abortion. Int J Gynecol Obstet. 2008;102(3):271–4.
103.
Van der Post JA, et al. The functions of microparticles in pre-eclampsia. Seminars in Thrombosis and Hemostasis. Vol. 37. No. 2. 2011.
104.
Jeschke U, et al. Stimulation trials of trophoblast cells in vitro using PP14. Z Geburtshilfe Neonatol. 1995;200(5):199–201.
105.
Toth B, et al. Glycodelin protein and mRNA is downregulated in human first trimester abortion and partially upregulated in mole pregnancy. J Histochem Cytochem. 2008;56(5):477–85.PubMedPubMedCentral
106.
Salim R, et al. A comparative study of glycodelin concentrations in uterine flushings in women with subseptate uteri, history of unexplained recurrent miscarriage and healthy controls. Eur J Obstet Gynecol Reprod Biol. 2007;133(1):76–80.PubMed
107.
Jeschke U, et al. Glycodelin and amniotic fluid transferrin as inhibitors of E-selectin-mediated cell adhesion. Histochem Cell Biol. 2003;119(5):345–54.PubMed
108.
Carp H. Recurrent miscarriage and hCG supplementation: a review and metaanalysis. Gynecol Endocrinol. 2010;26(10):712–6.PubMed
109.
Lin J, et al. Lymphocytes from pregnant women express human chorionic gonadotropin/luteinizing hormone receptor gene. Mol Cell Endocrinol. 1995;111(1):R13–7.PubMed
110.
Schumacher A, et al. Human chorionic gonadotropin attracts regulatory T cells into the fetal-maternal interface during early human pregnancy. J Immunol. 2009;182(9):5488–97.PubMed
111.
Maffei M, et al. Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med. 1995;1:1155–61.PubMed
112.
Myers MG, et al. Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab. 2010;21(11):643–51.PubMedPubMedCentral
113.
Panariello F, et al. The role of leptin in antipsychotic-induced weight gain: genetic and non-genetic factors. J Obes. 2012;2012:572848.PubMedPubMedCentral
114.
Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12(3):318–20.PubMed
115.
Cunningham MJ, Clifton DK, Steiner RA. Leptin’s actions on the reproductive axis: perspectives and mechanisms. Biol Reprod. 1999;60(2):216–22.PubMed
116.
Henson MC, Swan KF, O’Neil JS. Expression of placental leptin and leptin receptor transcripts in early pregnancy and at term. Obstet Gynecol. 1998;92(6):1020–8.PubMed
117.
Lage M, et al. Serum leptin levels in women throughout pregnancy and the postpartum period and in women suffering spontaneous abortion. Clin Endocrinol (Oxf). 1999;50(2):211–6.
118.
Laird S, et al. Leptin and leptin-binding activity in women with recurrent miscarriage: correlation with pregnancy outcome. Hum Reprod. 2001;16(9):2008–13.PubMed
119.
Cookson BT, et al. Organization of the human annexin V (ANX5) gene. Genomics. 1994;20(3):463–7.PubMed
120.
Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6(6):449–61.PubMed
121.
CARCEDO M, et al. Functional analysis of the human annexin A5 gene promoter: a downstream DNA element and an upstream long terminal repeat regulate transcription. Biochem J. 2001;356:571–9.PubMedPubMedCentral
122.
Reutelingsperger CP, et al. Purification and characterization of a novel protein from bovine aorta that inhibits coagulation. Eur J Biochem. 1988;173(1):171–8.PubMed
123.
Creutz CE. The annexins and exocytosis. Science. 1992;258(5084):924–31.PubMed
124.
Ravassa S, et al. Annexin A5 down-regulates surface expression of tissue factor a novel mechanism of regulating the membrane receptor repertoir. J Biol Chem. 2005;280(7):6028–35.PubMed
125.
Rand JH, et al. Pregnancy loss in the antiphospholipid-antibody syndrome – a possible thrombogenic mechanism. N Engl J Med. 1997;337(3):154–60.PubMed
126.
Empson M, et al. Recurrent pregnancy loss with antiphospholipid antibody: a systematic review of therapeutic trials. Obstet Gynecol. 2002;99(1):135–44.PubMed
127.
Rand JH. Antiphospholipid antibody-mediated disruption of the annexin-V antithrombotic shield: a thrombogenic mechanism for the antiphospholipid syndrome. J Autoimmun. 2000;15(2):107–11.PubMed
128.
Rand JH, et al. Human monoclonal antiphospholipid antibodies disrupt the annexin A5 anticoagulant crystal shield on phospholipid bilayers: evidence from atomic force microscopy and functional assay. Am J Pathol. 2003;163(3):1193–200.PubMedPubMedCentral
129.
Bogdanova N, et al. A common haplotype of the annexin A5 (ANXA5) gene promoter is associated with recurrent pregnancy loss. Hum Mol Genet. 2007;16(5):573–8.PubMed
130.
Tiscia G, et al. Haplotype M2 in the annexin A5 (ANXA5) gene and the occurrence of obstetric complications. Thromb Haemost. 2009;102(2):309–13.PubMed
131.
Miyamura H, et al. Polymorphisms in the annexin A5 gene promoter in Japanese women with recurrent pregnancy loss. Mol Hum Reprod. 2011;17(7):447–52.PubMed
132.
Diamond MS, et al. Heparin is an adhesive ligand for the leukocyte integrin Mac-1 (CD11b/CD1). J Cell Biol. 1995;130(6):1473–82.PubMed
133.
Koenig A, et al. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins. Implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J Clin Invest. 1998;101(4):877.PubMedPubMedCentral
134.
Skinner MP, et al. GMP-140 binding to neutrophils is inhibited by sulfated glycans. J Biol Chem. 1991;266(9):5371–4.PubMed
135.
Revelle BM, Scott D, Beck PJ. Single amino acid residues in the E-and P-selectin epidermal growth factor domains can determine carbohydrate binding specificity. J Biol Chem. 1996;271(27):16160–70.PubMed
136.
Ley K, Cerrito M, Arfors K-E. Sulfated polysaccharides inhibit leukocyte rolling in rabbit mesentery venules. Am J Physiol. 1991;260(5):H1667–73.PubMed
137.
Tyrrell DJ, et al. Heparin in inflammation: potential therapeutic applications beyond anticoagulation. Adv Pharmacol. 1999;46:151–208.PubMed
138.
Petitou M, et al. Synthesis of thrombin-inhibiting heparin mimetics without side effects. Nature. 1999;398(6726):417–22.PubMed
139.
Tabeur C, et al. Oligosaccharides corresponding to the regular sequence of heparin: chemical synthesis and interaction with FGF-2. Bioorg Med Chem. 1999;7(9):2003–12.PubMed
140.
Lundin L, et al. Selectively desulfated heparin inhibits fibroblast growth factor-induced mitogenicity and angiogenesis. J Biol Chem. 2000;275(32):24653–60.PubMed
141.
Fath MA, et al. Interaction of secretory leukocyte protease inhibitor with heparin inhibits proteases involved in asthma. J Biol Chem. 1998;273(22):13563–9.PubMed
142.
Darien BJ, et al. Low molecular weight heparin prevents the pulmonary hemodynamic and pathomorphologic effects of endotoxin in a porcine acute lung injury model. Shock. 1998;9(4):274–81.PubMed
143.
Li L-F, et al. Unfractionated heparin and enoxaparin reduce high-stretch ventilation augmented lung injury: a prospective, controlled animal experiment. Crit Care. 2009;13(4):R108.PubMedPubMedCentral
144.
Kennedy T. Use of heparin to inhibit interleukin-8. International patent application, WO94/18989, 1994.
145.
Gaffney A, Gaffney P. Rheumatoid arthritis and heparin. Rheumatology. 1996;35(8):808–9.
146.
Lilly JD, Parsons CL. Bladder surface glycosaminoglycans is a human epithelial permeability barrier. Surg Gynecol Obstet. 1990;171(6):493–6.PubMed
147.
Parsons CL. Epithelial coating techniques in the treatment of interstitial cystitis. Urology. 1997;49(5):100–4.PubMed