Chris L. Wells
INTRODUCTION
The goal of this chapter is to provide a review of pulmonary diseases and disorders that impact pulmonary function. The pulmonary system is responsible for the delivery of oxygen and the release of carbon dioxide, which is vital for normal cellular function. The lungs also assist the renal system in the regulation and maintenance of acid–base balance. When lung function becomes impaired, multiple systems may be affected. Consequently, it is important that physical therapists have an understanding of lung pathologies and their clinical presentation to perform a thorough evaluation, properly monitor the patient, and design an appropriate treatment plan.
This chapter is divided into sections that are based on common pathological impairments and clinical presentations. The first group of pathologies has been classified as chronic obstructive pulmonary diseases (COPD). The second section involves diseases that cause a pulmonary restrictive breathing pattern. Other smaller categories include infections, diseases that disrupt the pulmonary vascular system, and diseases that have pleural involvement. Separate chapters (Chapters 13 and 14) will address neuromuscular and musculoskeletal disorders that affect pulmonary functioning.
CHRONIC OBSTRUCTIVE PULMONARY DISEASES
Chronic obstructive pulmonary disease (COPD) is a generic term that refers to lung diseases that result in air trapping in the lungs, causing hyperinflation of the lungs, and a barrel-chest deformity. The American Thoracic Society and European Respiratory Society recently updated the definition of COPD, which commonly refers to emphysema and chronic bronchitis as “a preventable and treatable disease state characterized by airflow limitation that is not fully reversible. The airflow limitation is usually progressive and associated with an abnormal inflammatory response of the lungs to noxious particles/gases, primarily caused by cigarette smoking. Although COPD affects the lungs it also produces systemic consequences” (The text in bold has been added to this new definition in 2004).1,2 This classification of pulmonary disease can be further subdivided based on the presentation of chronic production of purulent sputum. Nonseptic obstructive diseases typically do not clinically present with chronic and consistent sputum production. Nonseptic obstructive disease includes diagnoses such as emphysema, α1-antitrypsin deficiency (α1-ATD), and asthma. Patients with a nonseptic disease may produce a small quantity of sputum, but it is not as significant as it is in diseases like cystic fibrosis (CF), chronic bronchitis, and bronchiectasis, which are classified as septic obstructive pulmonary diseases. These diseases are clinically associated with large volumes of sputum production, colonization of bacteria and fungus, and chronic infections. This division may assist the physical therapist in anticipating where bronchial hygiene techniques will be a primary focus of intervention.
Nonseptic Obstructive Airway Diseases
As a classification of pulmonary disease, COPD is the fourth leading cause of death in the United States, afflicting 16 million Americans with 20% of the population affected with some type of COPD. By 2010, it is estimated that COPD will be the third biggest cause of mortality within the world. Acute exacerbations of COPD account for $16 million spent annually in doctor visits. Forty thousand people die from COPD annually.3 This group of diseases is characterized by an increase in lung compliance with larger lung volumes and air trapping due to premature closure of the airways. The destruction of the lung architecture leads to hypoxia and hypercapnia (see Fig. 7-1).
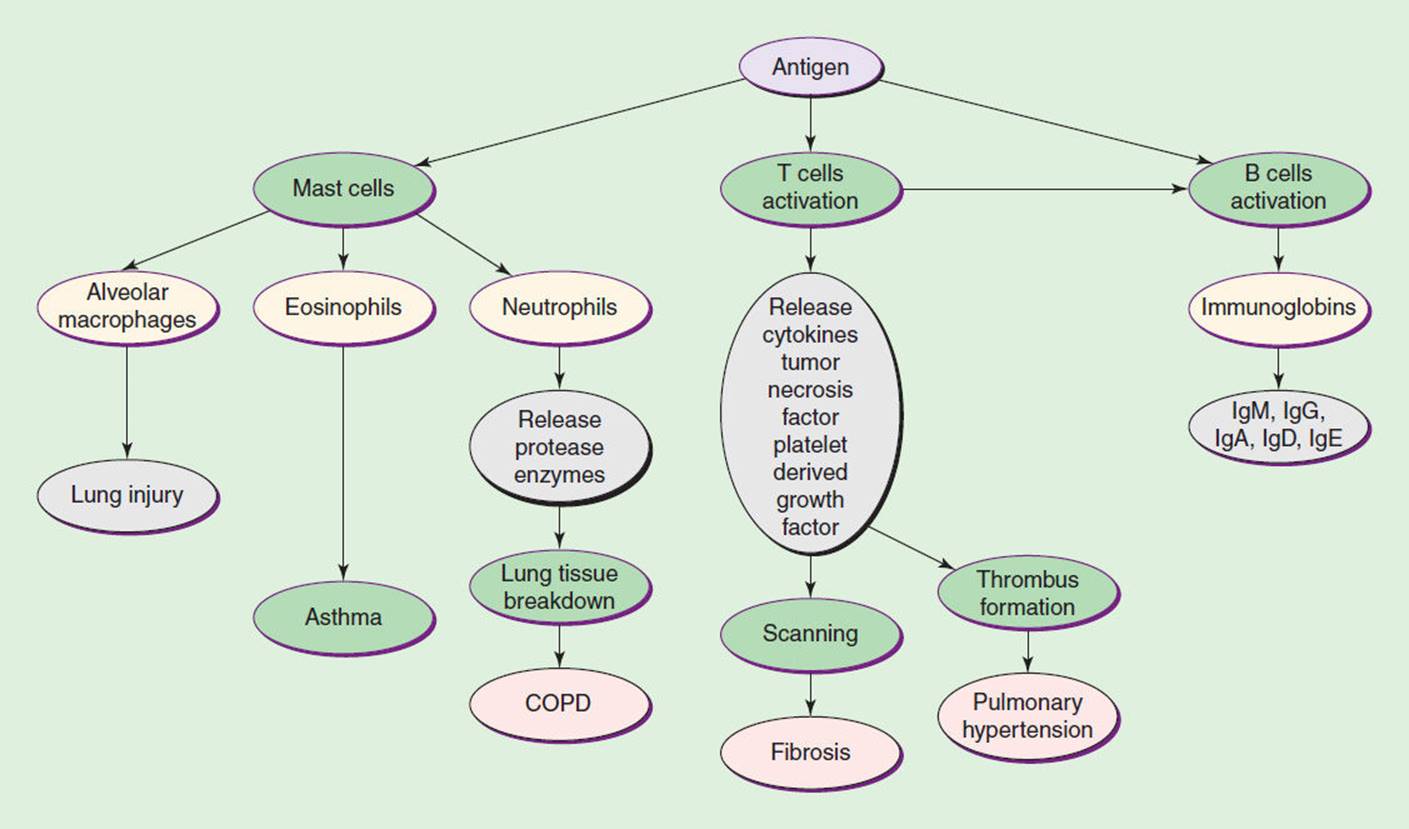
FIGURE 7-1 This diagram summarizes the general immune response that leads to various pulmonary diseases through destruction of lung tissue. This may lead to hypoxia and hypercapnia.
These diseases are classified under COPD because the patients’ presentations have similar characteristics (see Table 7-1). These patients present with hyperinflation, a barrel chest, and excessive use of accessory respiratory muscles. The pulmonary function test (PFT) demonstrates an increase in total lung capacity (TLC), inspiratory reserve capacity (IRC), and residual volume (RV) and a decrease in forced vital capacity (FVC), forced expiratory volume in 1 second (FEV1), diffusion capacity of carbon monoxide (DLCO), and an FEV1/FVC ratio (see Fig. 7-2 and Table 7-2).4 Blood gases typically show hypoxia with or without hypercapnia. In the case of septic obstructive disease, like CF or bronchiectasis, the patient will have a chronic productive cough with excessive sputum production. Finally, from the adverse effects of treatment and a decrease in activity, many of these patients will also have muscle weakness, both type 1 and type 2 muscle fiber atrophy, osteopenia, or osteoporosis and may develop right-sided heart failure.
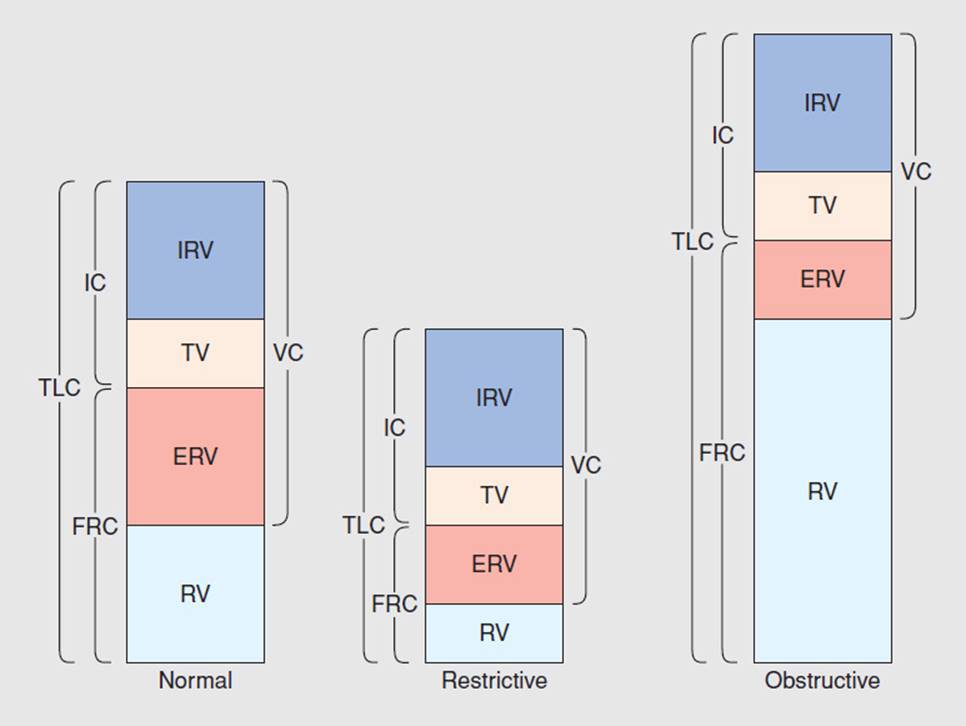
FIGURE 7-2 Pulmonary function tests. (Reprinted with permission from Ali J, Summer W, Levitzky M. Pulmonary Pathophysiology. New York: McGraw-Hill; 1999.)
TABLE 7-1 Clinical Summary
TABLE 7-2 Classification of Lung Disease by Pulmonary Function Test
Asthma (ICD-9-CM Code: 493)
Asthma is a chronic disease characterized by reversible obstruction to airflow within the lungs. Between asthmatic episodes lung function is relatively normal, and only exposure to a stimulant results in airway hyperreactivity and airway obstruction.5–7 It is estimated that there are 15 million people who suffer from asthma within the United States,8 and the World Health Organization reports an estimated 300 million with asthma worldwide.9 Asthma is the most common chronic disease in children with an incidence rate of 5.9% annually. There has been a 160% increase in asthma cases in children younger than 4 years since the 1980s.8 The increased incidence of asthma has been associated with increased exposure to nitric oxide, ozone, secondhand smoke, and indoor pollutants.10 It has been theorized that most cases of atopic or allergy-associated asthma is related to indoor exposure to irritants such as tobacco smoke, carbon monoxide, and nitric oxide from poorly vented heating systems, pesticides, dust mites, mold, rodents, cockroaches, and animal dander.8 There is a higher rate of asthma in females.6 Occupational exposure, which is related to the duration and intensity of exposure, is a factor for discussing adult-onset asthma. Asthma accounts for more than 10.4 million doctor appointments annually and more than $23 million is spent on medical management and on the loss of work time related to exacerbation of occupational asthma.8 Table 7-3 lists the most common risk factors associated with asthma.
TABLE 7-3 Risk Factors of Asthma
Thirty-four percent of children who develop asthma present with wheezing before the age of 3 years. The incidence rate in children younger than 3 years is 11.3%, whereas 15% present for medical evaluation between 3 and 6 years of age.11 Eighty percent of all asthmatic cases have an onset before the age of 5 years with 50% to 70% of these children reporting diminishing or absent symptoms in late adolescence or adulthood.7 Approximately 10% will continue to have symptoms, but these symptoms will be controllable, whereas 30% of children will remain symptomatic, with 17% of these children being classified as having severe asthma.12 Therefore, approximately 35% to 40% of children will be limited by lung disease in their activities and play. The most widely recognized phenotype of childhood asthma is atopic asthma. Atopic asthma is an allergy-associated form of asthma that accounts for 85% of all school-age asthma cases. It is associated with wheezing, cough, and shortness of breath, and the child may present with other atopic diseases such as eczema and hay fever.13 It has been suggested that children are most susceptible to atopic asthma due to the maturation process of lymphocytes as the immune system is developing along with the maturation and development of the lung and airway tissue.14
Asthma affects people across the lifespan— The majority of asthmatic children have a family history of asthma, particularly on their mother’s side. Maternal smoking compounds the risk of childhood asthma.11 There is a twofold increase in the risk of asthma development in the first year of life and a fourfold increase if the mother smokes and has allergies in the prenatal period.7 There is a 40% to 50% risk of a child developing asthma if one of the parents or the child’s primary caregiver smokes.10 There is an increase in obesity-related asthma with 6.6% of all childhood asthma cases related to obesity. Obesity-related asthma is characterized by low-grade inflammation with an increase in cytokines and tumor necrosis factor α (TNFα), which may upregulate airway inflammation.15 The persistence of childhood asthma is associated with a low FEV1 and a FEV1/vital capacity (VC) ratio less than 0.8 L, frequent asthmatic episodes, the need for anti-inflammatory medications, and atopy.11 Atopy is characterized by an immediate skin hypersensitivity reaction with wheal and flare.7 Approximately 3% of the elderly have a past medical history that includes asthma. It is estimated that 25% of the elderly are undiagnosed with asthma, even though the classic symptoms are present, and it takes an average of .5 years before a diagnosis is made. Only approximately one-third of the elderly are under appropriate medical care.16 Table 7-4 includes the guidelines for classifying asthma as mild, moderate, or severe.
TABLE 7-4 Classification of Asthma
The etiology is unknown, but several factors have been associated with the development of asthma. It has been theorized that children are most susceptible to the development of asthma because there is less epithelial cell differentiation leading to more fragile airways. The exposure to allergens and, particularly, respiratory viral infections lead to an increase in the proliferation and migration of endothelial cells, recruitment of perivascular supporting cells, an increase in fibroblastic cells, and less normal epithelial cells. The consequence is the development of a hyperactive respiratory immune response.9,14,17 Maternal smoking has been linked to the increased exacerbation of existing asthma in children. These children also have a higher incidence of infections that may contribute to the development of asthma. Early infections, particularly infections due to the respiratory syncytial virus (RSV) and ureaplasma urealyticum, are associated with asthma.7,14,18 There also appears to be a genetic link. Chromosomes 5, 11, and 14 appear to contribute to the inflammatory process of asthma.12,19
The stimulants that cause asthmatic episodes are similar whether the patient is a child or an adult. Stimulants may include air pollutants, pollen associated with allergies, respiratory infections, exertion, and medications. Children are more at risk because of allergies, whereas the elderly are more susceptible to pollutants and medications. Medications that have been linked to asthma include nonsteroidal anti-inflammatory drugs (NSAIDS), aspirin, nonselective β-blockers, and angiotensin-converting enzyme (ACE) inhibitors.16
Airways are characterized by the infiltration of inflammatory cells. There is evidence of remodeling in composition and organization of the walls of the airways. Smooth muscle hyperplasia and hypertrophy are present as well as increased proliferation of epithelial cells. There is hyperplasia of the goblet cells and hypertrophy of vascular tissue and finally, an increased accumulation of myofibroblast cells.9 The inflammation is associated with the increase in mast cells and eosinophils.20
Asthma may also be exacerbated by exercise— It is theorized that exercise-induced asthma (EIA) is due to loss of water and heat from the lower respiratory system. Breathing through the mouth during exercise bypasses the nasal passages that warm and humidify the inspired air. By bypassing the nasal passages, there is a resultant loss of heat and water from the mucosa and the lower airways, and the lower airways have to compensate. It is postulated that the loss of heat causes hyperemia, vascular engorgement, and bronchial edema, which reduce the lumen size of the bronchioles.21 The severity of EIA is determined by minute ventilation during exercise, temperature, humidity of air, and baseline airway reactivity.21
Whether antigens, allergies, or exercises stimulate the asthmatic episode, the result is the onset of an inflammatory process (see Fig. 7-1). There is a release of T cells that causes a cell-mediated immune response of particularly CD4 helper/inducer cells. The activation of T cells stimulates the release of antibodies from the humoral-mediated immune system. The elevation of immunoglobin E (IgE) antibodies is associated with asthma-activated mast cells and eosinophils, which in turn help to further promote this inflammatory process by releasing other proinflammatory mediators.7,12,19 This inflammatory process is associated with bronchoconstriction and airway obstruction.16
Asthma is associated with an increase in airway resistance. It is theorized that the drying of the airways, in the presence of EIA, stimulates the inflammatory process and leads to bronchoconstriction and an increase in airway resistance.22 The resistance is related to contraction of the smooth muscle of the bronchioles. There is edema and cellular infiltration of the airways that also contribute to airway obstruction.23
The classic symptoms of asthma are wheezing, dyspnea, chest pain, facial distress, and usually a nonproductive cough18 (see Fig. 7-3). Airways may become obstructed with viscous, tenacious mucus during acute exacerbation that leads to further hyperinflation. These symptoms are more severe in children than in adults. There is an increased risk of an asthmatic attack in children because of the natural lower lung compliance and less compliance with medication.18 Symptoms in adults may also include paroxysmal nocturnal dyspnea, morning chest pain, and increased symptoms with exposure to cold.16 In the case of a severe asthma attack that is refractory to bronchodilators, called status asthmaticus, the patient may present with decreased breath sounds, cyanosis, exhaustion, hypercapnia, and pending respiratory failure.24 It is important to note that EIA symptoms related to bronchoconstriction may present 6 to 8 hours after cessation of submaximal aerobic exercise or immediately after short intense bouts of exercise.22,21

FIGURE 7-3 Patient in an acute asthmatic attack. Note the shortness of breath, anxiety, and general increase in sympathetic discharge. (Image from www.netterimages.com. Reused with permission of Elsevier, Inc. All rights reserved.)
The guidelines for diagnosis vary and are based on the age of the patient. In infants, the diagnosis of asthma is made based on at least three episodes of wheezing observed by a physician.18 In children, asthma is diagnosed by a history of intermittent episodes of wheezing, coughing, shortness of breath, and chest tightness. These symptoms are worse at night or in the early morning hours. An allergen, pollutant, or exercise may be identified as stimulant.18In older children and adults, an improvement in FEV1 by 15% or more after use of a bronchodilator and sustained improvement in symptoms and lung function with corticosteroids are also consistent with the diagnosis of asthma.16,18Finally, the diagnosis of EIA requires the documentation of a 15% decrease in the peak expiratory flow recording following exercise.21
The first intervention for asthma is prevention. Smoking cessation, as well as minimizing the exposure to secondhand smoke, is important for any woman who is pregnant. It is also important to receive an annual flu shot and avoid stimulants that precipitate an asthmatic episode.21 The use of high-efficiency particulate air vacuums, mattress covers, and methods to improve heating and home ventilation systems may be effective in reducing the incidence of asthmatic episodes or atopic asthma.8
CLINICAL CORRELATE
EIA exacerbation can be minimized through medication and by performing a warm-up approximately 45 to 60 minutes before the exercise program. This warm-up exercise period should consist of 30-second exercise bouts with 2-minute rest periods. This reduces the severity of signs and symptoms of EIA.21
The goal of treatment is to minimize exacerbation25— The use of short- and long-acting bronchodilators is the main defense in controlling asthma.21 There are several types of bronchodilators that can be prescribed, and it is important that the patient and the therapist understand the proper use of the medication (refer to Table 7-5). β-Adrenergic agonists can be used to increase smooth muscle relaxation that results in bronchodilation and inhibits the release of mediators. Cromolyn, a corticosteroid, is also effective as a prophylactic but not as a rescue drug.21 More recently, there are other medications being trialled for management of asthma when more traditional methods are not sufficient. These include the use of leukotriene modifiers that block the proinflammatory mediators that promote smooth muscle contraction, vascular leakage, mucus secretion, and airway hyperactivity. IgE inhibitors such as malizurals and other medications like etanercept that block TNF-α are also under investigation. Finally, immunosuppressive medications, methotrexate, and cyclosporine are also being used for the chronic severe cases of asthma.20
TABLE 7-5 Common Medications Used for Asthma and Other COPD

Establishment of a routine exercise program is also important in the treatment of asthma. Fifty percent of children with asthma are severely deconditioned, but the level of deconditioning cannot be predicted by the history of asthma.5 People with asthma have a positive response to exercise with improved minute ventilation and oxygen consumption and decrease in blood lactate.21
Wheezing and breathlessness are poor predictors for asthma.16 Asthma that persists into adulthood can be associated with irreversible obstructive disease that may increase the incidence of deaths related to end-stage obstructive disease and increases the risk of pneumonia. Severe asthma is defined as asthmatic symptoms that persist despite maximized medical therapy with one or more exacerbation annually. There is a predominance of neutrophil cells associated with airway inflammation and there is a 30% to 50% loss in expiratory airflow, which appears to be associated with a fixed loss of the elastic recoil of the lung that is not associated with emphysema.26,27
Emphysema (ICD-9-CM Code: 492)
Emphysema is the second most prevalent disease within the category of COPD, with only asthma having a higher incidence. Emphysema is characterized by abnormal, irreversible enlargement of the airways distal to the terminal bronchioles, leading to decrease in driving pressure and intraluminal pressure, which leads to the impairment in expiratory airflow and maintenance of airway patency during inspiration28,29 (see Figs. 7-4 and 7-5). This may result in destruction of the acini, which are the functional units of the lungs for gas exchange. Each acinus is composed of one to three respiratory bronchioles and the alveolar ducts and sacs.
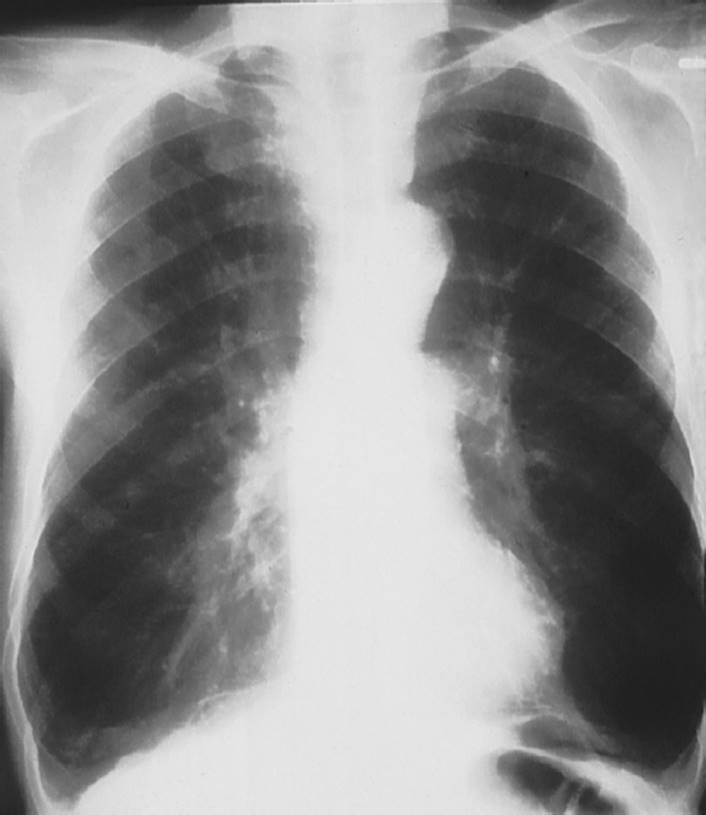
FIGURE 7-4 Emphysema. Notice the hyperinflation of the lungs on this P-A (posterior-anterior) X-ray with flattening of the diaphragm and elongation of the cardiac silhouette. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
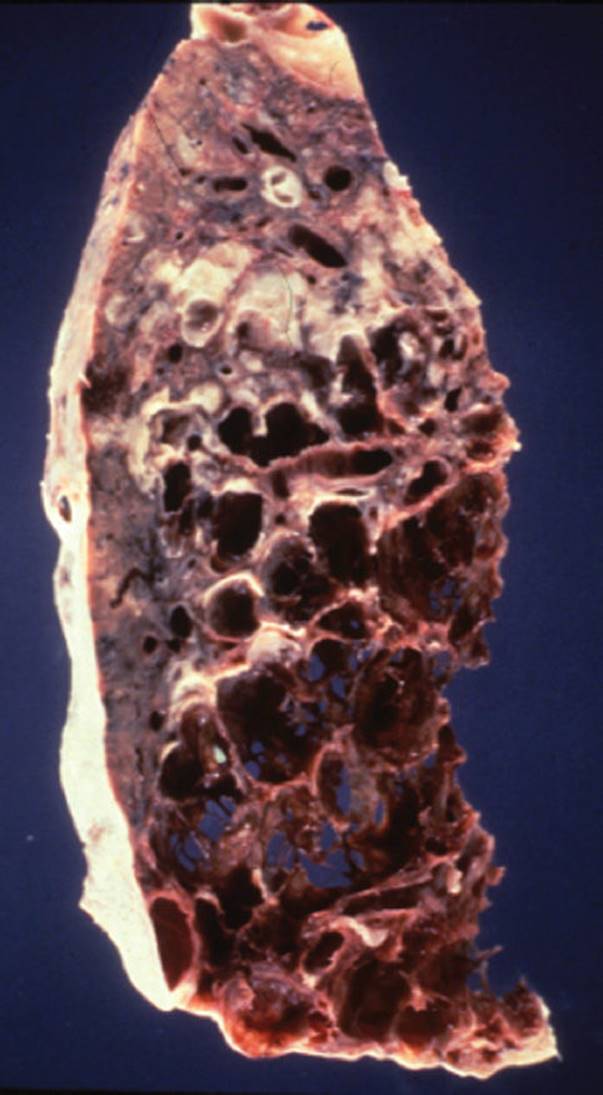
FIGURE 7-5 Emphysema. This disease results in the destruction of bronchioles and parenchymal tissue, which leads to the loss of elastic recoil properties of the lung. This results in the dilatation of airways, which leads to air trapping and hyperinflation. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
Emphysema can be classified as centriacinar, panacinar, or paraseptal, based on the location of the anatomical disruption. See Fig. 7-6 for the illustrations of different types of emphysema. The key structure in the classification of emphysema is the respiratory bronchiole. Centriacinar or centrilobular emphysema involves the enlargement and destruction of the first- and second-order respiratory bronchioles, and the alveoli remain intact. Centriacinar emphysema is most commonly associated with smoking. In contrast, the enlargement and destruction of the entire acinus are the defining characteristics of panacinar emphysema, where there is a more even distribution of destruction and dilatation of the entire acinus.30 Paraseptal emphysema involves the periphery of the secondary lobule along the septum. Paraseptal emphysema is not typically associated with the progression of end-stage COPD but can be associated with an increased risk of pneumothorax (PTX).28
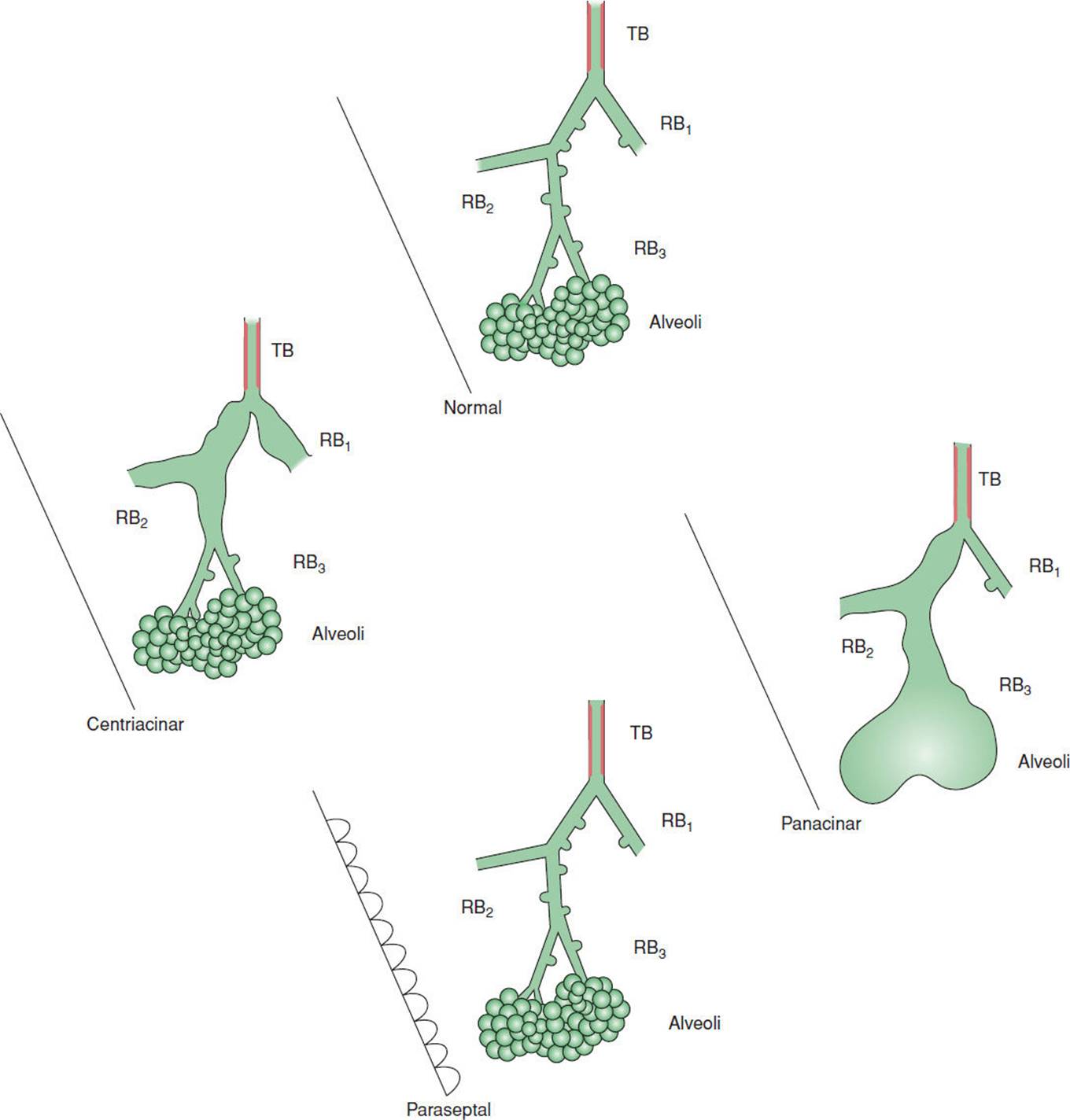
FIGURE 7-6 Types of emphysema. (Modified with permission from Gurney JW. Pathophysiology of obstructive airways disease. Radiol Clin North Am. 1998; 36(1):15-27.)
Cigarette smoking has been linked to the development of centriacinar emphysema; however, a small proportion of smokers will develop panacinar emphysema. Approximately 10% to 15% of patients with a significant history of smoking will develop clinically significant obstructive disease. A large proportion of the small particles in cigarette smoke are distributed to the first- and second-order bronchioles. Many of the particles are removed through a well-developed lymphatic system of the lower lobes; however, the particles that are also deposited in the upper lobes are revoked at a slower removal rate due to the smaller size of its lymphatic system.28 The loss of FEV1function occurs more than twice as fast as the aging process and declines by 25% for each pack year of smoking.28,31 It is associated with a very insidious onset, which occurs over 30 to 40 years.
Cigarette smoking is associated with an increase in cellular apoptosis, early and excessive cell death. There is an increased accumulation of apoptotic cells and slow cell removal with macrophage dysfunction. There is also an increase in TNF-α and a decrease in surfactant protein. These changes lead to alteration of alveolar and small airway function, inflammatory and proteolytic activity, and changes in the endothelium and epithelium cells. The consequences of destruction of the alveolar wall, decrease in surface area, loss of functioning pulmonary capillary bed, and loss of the parenchyma lead to air trapping and ventilation–perfusion (![]() /Q.) mismatch.3,32
/Q.) mismatch.3,32
The etiology of emphysema is based on the protease–antiprotease hypothesis in which there is an imbalance between protease, which causes tissue breakdown, and antiprotease enzymes. This imbalance leads to the loss of lung parenchyma and elastic recoil, which the small airways depend on return to their resting states during exhalation. The elastic property of the parenchyma also provides a normal level of airway resistance during inspiration. This loss of parenchyma tissue results in the loss of radial traction on the airways. The end result is dilation of airways, premature airway closure and air trapping, and an increase in RV.28,29,33
The nicotine in cigarette smoke attracts neutrophils, activates alveolar macrophages, and inactivates the protective nature of antiprotease.34 The alveolar macrophages and neutrophils contain protease enzymes, which are capable of destroying the elastic property of the lung tissue, thus producing emphysema.28 The cigarette smoke also causes a proliferation of endothelial cells, smooth muscle cells, platelet aggregation, and destruction of pulmonary capillaries. The impairment to the small blood vessels within the pulmonary system may lead to the decrease in DLCO and the development of secondary hypertension.3,31,32
As a consequence of intrinsic pulmonary damage, hyper-inflation of the lungs occurs, which eventually leads to the compensatory changes of the chest wall. This disruption of normal chest wall mechanics leads to dysfunction of the inspiratory muscles, particularly of the diaphragm. The dysfunction of the diaphragm is an important cause of respiratory failure in patients with emphysema. Hyperinflation causes shortening of inspiratory muscles and flattening of the diaphragm with the loss of sarcomeres. The end result is a loss of diaphragmatic excursion and subsequent decline in the mechanical effectiveness of the diaphragm, and other respiratory muscles needed to support the increased demand of ventilation.35
The most common complaint of patients with emphysema is dyspnea on exertion (DOE). The result of a physical examination reveals the following findings: diminished breath sounds and wheezing, which are typically associated with exertion, and a prolonged expiratory phase.36 The patient will present with an enlarged anterior–posterior dimension of the chest wall, called a barrel chest, with an increase in rib angle. The accessory muscles are commonly hypertrophied from overuse. There is hyperresonance sound upon mediate percussion, which is consistent with the hyperinflation of the lungs. The presence of a chronic cough and sputum production will vary and depend on the infectious history of the patient. As the disease advances, many patients become cachectic, or emaciated, and begin to show signs of right-sided heart failure due to secondary pulmonary hypertension. The classic signs and symptoms of right-sided heart failure include peripheral pitted edema, weight gain, jugular vein distension, diminished appetite, right upper quadrant discomfort, and ventricular gallop, S3 heart sound (see Fig. 7-7). Emphysema is considered as a systemic disease with the increase of the inflammatory process. Patients with emphysema commonly suffer from osteoporosis, skeletal muscle disease, depression, and an increase in incidence of cardiovascular disease.29,37 Beyond the physical examination, PFT results are consistent with other obstructive airway diseases, which include a decline in FVC, FEV1, and FEV1/FVC ratio that indicates small airway disease. There is an increase in TLC and RV.38 The chest X-ray reveals hyperinflation with a flattened diaphragm, decreased vascular markings, and possible enlargement of the right side of the heart. Because of the destruction of the gas-exchange areas of the lungs, there is also a mismatch between ventilation and perfusion (![]() ) that is demonstrated on a
) that is demonstrated on a ![]() scan.36
scan.36

FIGURE 7-7 Patient with a diagnosis of emphysema. Note the generalized muscle wasting, shortness of breath with pursed-lip breathing, and use of accessory muscles with a forward-leaning posture. (Image from www.netterimages.com. Reused with permission of Elsevier, Inc. All rights reserved.)
Patients who present with the classic presentation of emphysema will have arterial blood gas analysis that typically reveals hypoxia and normal-to-slight hypocapnia. These patients present with tachypnea, labored breathing, and a normal-to-low body mass index (BMI). Some patients with emphysema will present with signs more associated with chronic bronchitis including hypoxemia, hypercapnia, signs of right-sided heart failure, copious secretions, and an above-normal BMI.3 Caution must be taken in this basic medical description because many patients with emphysema will present with a mixture of clinical features.
Smoking cessation is instrumental in the care of patients with emphysema because it leads to a slower decline in FEV1 when compared to the patients who continue to smoke.39 Smoking cessation has been the only treatment that has shown to slow the alterations in the natural progression of emphysema with patients with mild disease.29 In addition, there are several other treatment options. Pharmacology interventions include short-acting and long-acting β2-agonists that cause bronchodilation of the airways. Anticholinergic drugs can be used, but are not usually the first line of medications. These medications, such as Atrovent, block bronchoconstriction. Refer to Table 7-5 for a summary of medications used in treating emphysema. Xanthine derivatives (eg, theophylline) also produce bronchodilation, accelerate mucociliary transport, and limit the inflammatory response. Corticosteroids are common agents used for their anti-inflammatory effects. It is also important that the patients receive the preventive vaccinations against influenza and pneumococcus.20
Long-term oxygen therapy helps correct hypoxemia and minimizes secondary pulmonary hypertension. The threshold for oxygen prescription include a PaO2 less than 55 mm Hg, an oxygen saturation less than 88%, evidence of cor pulmonale, or a hemocrit greater than 56% (Table 7-6).39 Oxygen therapy has been shown to reduce the level of dyspnea, decrease maximal voluntary ventilation and polycythemia by correcting hypoxemia, decrease pulmonary hypertension, improve quality and quantity of sleep, and decrease nocturnal arrhythmias. Supplemental oxygen can also improve cognitive function and exercise tolerance. The use of BiPAP ventilation, which is a form of mechanical ventilation, provides airway pressure on both inspiration and expiration to decrease the work of breathing and prevent early airway closure. This minimizes air trapping and has also been found to reduce the retention of carbon dioxide.
TABLE 7-6 Indications for Supplemental Oxygen

CLINICAL CORRELATE
Pulmonary rehabilitation has become a widely accepted intervention in the care of patients with emphysema but is still not covered by many insurance plans, although research supports its positive effects of increasing maximal exercise tolerance, oxygen uptake, and exercise endurance. There is also an improvement of the perceived level of dyspnea and a decrease in muscle fatigue. There is an improvement in quality of life, including the improvement in self-worth, well-being, and an increased sense of self-control.39 Pulmonary rehabilitation should include general muscle strengthening with emphasis on the upper body; aerobic conditioning; and education about smoking cessation, nutrition, vaccinations, proper use of medications and supplemental oxygen, and the disease process.
Bullectomy is a common surgical intervention for a patient with emphysema with significant bullae disease. A bulla is a large air space greater than 1 cm in diameter, which is the result of destruction of the parenchyma. A bulla no longer participates in gas exchange or diffusion. A bulla may also cause compression of adjacent functional lung tissue, which further impairs diffusion. Bullae are associated with a 15% to 20% incidence of PTX. A bullectomy is indicated when there is a significant level of dyspnea, clear presence of bullae that compress viable tissue, and when there is a high incidence of PTX. A bullectomy results in the reduction of pulmonary vascular and airway resistance, a reduction of functional residual capacity (FRC), and less air trapping. If enough diseased tissue is removed, the diaphragm may return to a more normal position that will improve muscle contraction.39,40
Volume reduction is the surgical resection of approximately 20% of dysfunctional lung tissue, thus reducing the hyperinflated state (see Figs. 7-8 and 7-9). The most common complication is air leaking in which there is a disruption of subatmospheric pressure within the thorax.39 From the outcomes of the NIH National Emphysema Treatment Trial (NETT) study, it has been determined that the patients with the best outcome have primarily upper lobe disease and have a decrease in exercise capacity, less than 25 W for women and less than 40 W for men.40
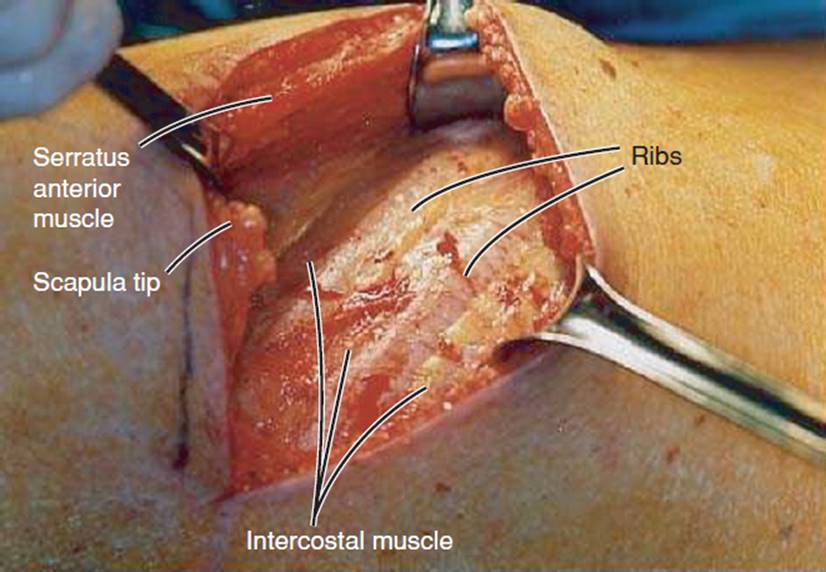
FIGURE 7-8 Surgical entrance into the chest wall via thoracotomy as a precursor to volume reduction surgery. The ribs and intercostal muscles are identified. The ribs are counted to ascertain the proper level for entering the chest cavity. (Courtesy of Peter Fergen, MD, University of Pittsburgh, PA.)

FIGURE 7-9 Volume reduction surgery. Rib-spreading retractors increase the exposure of the chest cavity. (Courtesy of Peter Fergen, MD, University of Pittsburgh, PA.)
Lung transplantation is a viable option for patients with end-stage disease who have maximized medical intervention. Patients with emphysema are potential candidates for either single- or double-lung procedures. Transplantation has the potential to significantly improve quality of life, but there are still questions as to whether transplantation extends the life of the recipient.39,40
In general, the prognosis of emphysema varies depending on the degree of obstruction, the presence of hypercapnia, the recurrence of infections, and the development of right-sided heart failure. It is generally accepted that an FEV1of less than 25% is associated with a 50% mortality rate in 2 years.39
α1-Antitrypsin Deficiency (Genetically Acquired Emphysema) (ICD-9-CM Code: 493.8)
α1-Antitrypsin is an enzyme that is predominantly synthesized by the liver parenchymal cells and counterbalances the degradation of tissue caused by protease, a proteolytic enzyme. α1-Antitrypsin primarily inhibits neutrophil elastase, which works to break down and remove bacteria from the airways.41 The normal level of α1-AT is 104 to 276 mg/dL. At an α1-AT level below 50 mg/dL, the genetic disorder α1-ATD should be suspected.42 Genetic emphysema or α1-ATD commonly presents as panacinar disease.42,43 The degree of deficiency is associated with the severity of the disease.
α1-ATD is the most common autosomal recessive genetic cause of liver disease in children43,44 and only second to CF in genetic pulmonary disease.44 Beyond the second decade of life, α1-ATD is primarily associated with lung disease.45 It is estimated that 1.29 million patients suffer from α1-ATD in the United States and another 1.1 million worldwide, which causes end-stage obstructive lung disease.44–46 There is an estimated 25 million carriers of α1-ATD worldwide. The prevalence is 1 in 1,500 people of European descent.41
α1-ATD can be divided into four categories based on variation in allele. An allele is one or more alternative forms of a gene on a chromosome. There have been more than 100 alleles identified with 34 associated with functional deficiency in the circulating α1-AT.41 Allele M involves a variation that carries little to no risk of disease, because the α1-AT level is sufficient enough not to cause cellular damage.42 The deficient variants that most commonly lead to liver or lung disease are allele S, allele Z, and allele null (0). Allele Z is the most common variant that is characterized by a normal level of α1-AT synthesis, but the secretion is only 15% of predicted levels. Allele Z accounts for 95% of severe α1-ATD. Allele S and allele 0 with the null variants are characterized by little α1-AT production, accounting for the most severe expression of the disease. The Z allele and null variants most predictably lead to premature lung disease.42,43
The onset of symptoms associated with pulmonary dys-function typically begins in the 30s with the diagnosis of 1-ATD occurring between 40 and 50 years of age.42,45 It is estimated that up to 13% of patients with emphysema actually have α1-ATD. Only approximately 10% of 1-ATD cases are actually diagnosed correctly. Most are initially diagnosed with asthma or nongenetic emphysema.43 A correct diagnosis takes 7.2 years on an average, and it is not uncommon that the patient is seen by 6 to 10 physicians before a correct diagnosis is made.45 Diagnosis prior to 20 years of age is typically related to liver dysfunction.44
Smoking has been associated with the acceleration of lung disease in the presence of α1-ATD. It has been estimated that cigarette smoking accelerates the progression of the lung disease by 19 years.45 Patients with α1-ATD or a family history of α1-ATD should not smoke! The tobacco smoke increases the level of oxidant exposure, alveolar macrophages, and neutrophils in the airways, along with other inflammatory cells. Cigarette smoke also inactivates 1-AT and leads to neutrophil protease being unopposed, thus causing the degradation of proteins within the lungs.43 An elastase–antielastase hypothesis has been proposed to explain the destructive changes that occur because of this deficiency. Neutrophil protease is capable of cleaving many of the proteins from connective tissue within the lung. Without the appropriate level of 1-AT, neutrophil protease is unopposed, which leads to an imbalance, with degradation occurring at a faster rate than repair and remodeling.43
The primary significance of α1-ATD is the premature development of emphysema, occurring in the third or fourth decade of life. Shortness of breath is typically the first symptom that causes the patient to present for medical intervention.43 Patients also report a chronic cough in 37% of all cases, sputum production (38%), wheezes (44%), and hyperre-active airways (20%).45 Many patients may present with weight loss, cor pulmonale, and polycythemia as the disease progresses to end stage.47 For up to 20% of patients, the clinical presentation of pulmonary impairment will also have liver disease, and up to 70% of patients will have abnormal liver enzymes.42
Upon examination of 1-ATD, the diagnostic testing shows similar findings to smoke-related emphysema. The results of PFTs demonstrate an obstructive pattern with a decline in FVC, FEV1, and FEV1/FVC ratio. DLCO is also diminished. There is an increase in TLC and RV. Radiological studies show the classic signs of hyperinflation and a decrease in vascular markings particularly of the lower lobes. Smoking-related emphysema shows more upper lobe or uniform disease throughout the lungs.42,44,47 High-resolution CT scan is the gold standard for diagnosis. When a patient younger than 50 years presents with signs and symptoms of emphysema or asthma with impairment more excessive than expected, blood testing should be conducted to test the serum 1-AT level.43
Most of the available treatment protocols are consistent with the treatments for emphysema such as bronchodilators, aerosolized or systemic corticosteroids, cessation of smoking, and preventive vaccinations. Pulmonary rehabilitation and supplemental oxygen therapy also are effective in the management of α1-ATD. It has been recommended that if the plasma level of α1-AT is less than 11 μmol/L, the patient should be given augmentation therapy (Prolastin, Aralast, and Zemaira), with the goal to increase α1-AT above 15 μmol/L (80 mg/dL), which appears to protect the lungs and slow down the decline of PFTs.41,42 Work is also being done to develop gene therapy for the treatment of α1-ATD. The most common surgical intervention for end-stage lung disease due to α1-ATD is lung transplantation. The clinical trials for lung volume reduction have shown that it is an ineffective procedure because of the primary presence of lower lobe disease. However, the effectiveness of this procedure is still in question.42
The prognosis of α1-ATD depends on the type of variant or allele, history of cigarette smoking, age of onset of symptoms, and the development of infectious bronchiectasis. Rapid decline in PFTs is associated with a worse prognosis.42 Sixty-two percent of patients with α1-ATD will die from respiratory failure, whereas 13% will die from end-stage liver disease.45,47
Chronic Bronchitis (ICD-9-CM Code: 491)
Chronic bronchitis is a clinical diagnosis that consists of a persistent cough that produces sputum for more than 3 months per year for at least two consecutive years in the absence of another definable medical cause. It is associated with obstruction of the airways and mucus plugging.1,23 It is estimated that 15 million people suffer from chronic bronchitis in the United States.48 Cigarette smoking is the most important risk factor in the development of chronic bronchitis.49
Smoking causes inflammation throughout the lung tissue with an increase in macrophages and T lymphocytes found within the airways. This inflammation is associated with airway remodeling, hypertrophy of submucosal glands, enlargement of smooth muscle cells, fibrosis of airway walls, and goblet cell hyperplasia.50 Polymorphonu-clear neutrophils are suspected to be the primary cause of the chronic airway inflammation.51 The increased activity of macrophages and neutrophils leads to the release of various enzymes such as interleukin 8, TNF-α, and elastase, which leads to further inflammation and airway destruction.52 The inflammation within the airways is correlated with alveolar wall destruction and rupture of the attachment between the outer airways and the alveoli. The end result is the loss of the elastic recoil within the lung tissue, which leads to airway obstruction and hyperinflation of the lungs.34
Approximately 15% of smokers will develop emphysema or chronic bronchitis. In the smokers who go on to develop chronic bronchitis, the exposure to nicotine causes an inflammatory response, as discussed previously, which stimulates mucus secretion, and disruption of the architecture of the airways and capillary system.34,53
There are other risk factors that have been identified in the development of chronic bronchitis. Age and the degree of airway obstruction along with the degree of hypoxia and hypercapnia are associated with chronic bronchitis. Aging is associated with a decline in B cells and T cells and a decrease in responsiveness to protect the airway. There is also a decrease in ciliary function and in the presence of chronic bronchitis, there is a further decline in CD4 and CD8 cells. This imbalance increases airway destruction and mucus production and retention.52 There is a greater risk of chronic bronchitis if the patient requires systemic steroids for medical management. At times of exacerbation, the patient may develop acute respiratory failure with secretion retention and severe abnormal blood gases requiring mechanical ventilation.54
Chronic bronchitis can be divided into subsets based on the degree of pulmonary dysfunction and sputum production. Acute tracheobronchitis is not associated with any pulmonary dysfunction but is typically associated with an acute viral infection. With simple chronic bronchitis, there is a mild-to-moderate decline in FEV1 and an increase in sputum production. Complicated chronic bronchitis pertains to a patient who is of advanced age, has an FEV1 of less than 50% of predicted, and has repeated exacerbations, poor nutrition, and comorbidities. Exacerbations that are associated with an increase in purulent secretions are likely from a bacteria infection such as streptococcus or Haemophilus influenzae. Finally, with chronic bronchitis, infection is distinct from the other subsets in that the patient has constant sputum production throughout the year.54
Upon inspection of the lung tissue in the presence of chronic bronchitis, there is hypertrophy and increased density of the secretory cells down the tracheobronchial tree. Along with airway obstruction from the loss of elastic recoil, there is inflammation of the respiratory bronchioles, hyper-trophy of the smooth muscle at the level of the small noncartilaginous airways, and mucus plugging that further narrows or occludes the small airways.24,49 The end result is a retention of mucus in the airways that lead to further airway obstruction and creates a vicious cycle of further pulmonary infections and destruction.54
These patients commonly present with an increase in shortness of breath and a productive cough during the acute exacerbation. There is an increase in sputum production and purulence along with a positive culture that confirms an infection. H. influenzae is the most common source of infection. A wheeze may be present upon auscultation. The physical examination and testing will reveal a barrel chest; decline in the FVC, FEV1, and FEV1/FVC ratio; decrease in DLCO; and commonly, hypercapnia and hypoxemia. The patients may also report anorexia associated with dyspnea while eating24,49 (see Fig. 7-10).

FIGURE 7-10 Patient with a diagnosis of chronic bronchitis. Note the cyanosis, use of accessory muscles, and sputum production. (Image from www.netterimages.com. Reused with permission of Elsevier, Inc. All rights reserved.)
The standard of care includes antibiotic treatment in the presence of an acute infection and short-acting β-agonists, long-acting bronchodilators, and inhaled corticosteroids. Frequently, these patients may be on chronic inhaled or intravenous antibiotics on a monthly basis to control infections. Smoking cessation is the most effective way to decrease mortality. The patients may also be taking expectorants and mucolytics to assist in management of sputum. Patients are encouraged to be well-hydrated. Bronchodilators may be used to manage bronchospasms. As this disease progresses, supplemental oxygen may be required to correct hypoxemia, and some form of pulmonary hygiene may need to be incorporated into the patient’s daily routine. Pulmonary rehabilitation is becoming recognized as a key component in the medical treatment plan. Finally, some pulmonologists are administrating an antiprotease, such as prolastin, to counteract the destruction of elastase caused by the chronic inflammatory process.49,55
The prognosis of chronic bronchitis is dependent on age, smoking, and the degree of airway obstruction. The 10-year mortality rate is 60% in smokers, whereas it is only 15% in nonsmokers. If the FEV1 is less than 1 L, the median survival rate is 4 years. Patients who spend at least 50% of the day in bed are four times more likely to die than patients who are mobilized early and frequently.49,54
Bronchiolitis Obliterans (ICD-9-CM Code: 496)
Bronchiolitis obliterans (OB) is an acute inflammatory injury usually characterized by a diffuse destruction of the bronchioles. There are multiple causes of OB in the adult population including an infectious process, toxic fume exposure, collagen vascular disease, chronic bronchitis, and lung transplantation. Most recently it has been recognized as the exposure to diacetyl, an additive used in artificial butter flavoring for popcorn, has been linked to OB.56 In pediatric patients, OB is primarily a complication of severe lower airway infection.57,58
The characteristics of OB may vary based on the underlying disease. In the presence of OB with organizing pneumonia (BOOP), an infection, there is fibroblastic proliferation in the small airways and mild chronic inflammatory infiltrates consistent with increased composition of alveolar macrophages in the alveoli.59 Polyp formation within the bronchioles consisting of granulation tissue can extend into the terminal bronchioles, which can partially or completely obstruct the airway. These polyps form at the site of epithelial injury. The proliferative form of OB is characterized by inflammation and infiltration of mesenchymal cells, which are composed of fibroblasts, myofibroblasts, and other extracellular substances, that leads to fibrosis and airway destruction.60 The proliferative form of OB has three principal processes that include an acute inflammatory phase with reversible fibrosis. In the first form, the basement membrane is intact, which allows for recovery. The second is described as an acute to subacute inflammatory presence with irreversible bronchiole fibrosis, and the most severe pattern is the chronic inflammatory picture with irreversible fibrosis. This third pattern is mostly associated with complications from transplantation and graft-versus-host disease.60 With obstructive pulmonary disease like emphysema and asthma, injury of the bronchioles is associated with mucus plugging, inflammatory infiltration, smooth muscle hypertrophy, goblet cell hyperplasia, and bronchial gland hypertrophy. This leads to narrowing of the airways that compounds the loss of elastic recoil and results in further airway obstruction.59
Constrictive OB is an uncommon idiopathic form of OB. The usual site of injury with constrictive OB is isolated to the bronchioles with preservation of distal airways. It is characterized by mural fibrosis, resulting in reduction in the lumen size of the bronchioles. Compared to nonconstrictive idiopathic OB, there is luminal narrowing due to scarring rather than smooth muscle hypertrophy.59,60
Recently, the most common cause of OB is associated with lung transplantation and is associated with the loss of graft function. It is speculated that OB is the result of chronic rejection. The hallmark sign is submucosal bronchiolar fibrosis preceded by bronchiolar inflammation resulting in epithelial necrosis through a process of lymphohistiocytic-mediated cytotoxicity that targets the respiratory bronchioles. The end process is the deposition of collagen and airway obliteration also referred to as vanishing airway disease. Transplanted OB is associated with frequency and severity of acute cellular rejection, ischemic and reperfusion injury, cytomegalovirus (CMV), and other bacterial or fungal respiratory infections. The incidence of OB associated with transplantation has been reported as high as 80% in recipients after 5 years with an onset of 16 to 20 months.58,61 More than 50% of recipients who survive beyond 3 months will develop OB.62 OB in transplant recipients can be classified as active or inactive, based on the presence or absence of lymphocytic infiltration, respectively.63 There are three typical patterns that OB can follow: a rapid and relentless decrease in FEV1 with death within 1 year of diagnosis, an insidious onset with a slow decline in FEV1, and finally, a rapid decrease in FEV1 onset followed by a stabilization over a prolonged period of time.61
In children and infants, OB is the primary cause of obstructive pulmonary disease. The primary site of injury involves the inflammation of the peripheral, small airways. The onset is typically the result of a viral infection, adenovirus, RSV, parainfluenza, influenza, and rhinovirus. Thirty-four percent of children who require mechanical ventilatory support as part of the medical therapy for severe respiratory infection will go on to develop OB as opposed to only 3% in cases where children did not require mechanical ventilation.57 Hyperactivity of the airways may develop as a result of this chronic inflammatory process.24,59
In general, there is a pronounced degree of fibroproliferative activity in the bronchioles that leads to or adds to further airway obstruction. There is the presence of derangement of epithelial function, local necrosis, fibropurulent exudate, and deposition of collagen.57,60 The damage is typically confined to the cartilaginous airways, with sparing of the respiratory bronchioles, alveolar ducts, pulmonary alveoli, and interstitium. In larger airways, there may be signs of bronchiectasis, mucus plugs, and chronic inflammatory infiltrates by lymphocytes, macrophages, and plasma cells.59,63
Frequently, there is an insidious onset with progressive dyspnea with exertion, often associated with a cough in the development of OB. Upon auscultation, wheezing and crackles are present when OB presents with an obstructive pattern. In the stage where there is an increase in sputum production, there may be the presence of low-pitched wheezing or rhonchi. Patients commonly complain of dyspnea, a low-grade fever, and a persistent cough.63 In children, the signs and symptoms will include hypoventilation and hypercapnia, intercostal retractions, tachypnea, grunting, expiratory wheezes, crackles, hyperinflation, and atelectasis.57,59 On examination of lung function, there is a decrease in FVC and FEV1 as well as an increase in RV57,61; in the presence of small airway involvement, there will also be a reduction of the FEV1/FVC ratio. A chest X-ray will illustrate hyperinflation and patchy atelectasis, and a high-resolution CT scan will reveal mosaic perfusion, vascular attenuation, and central bronchiectasis.57,58
Medical management begins with prevention, primarily to decrease the incidence of exposure to toxic gases, minerals, and organic particulates.60 Therapy also includes the use of supplemental oxygen, antiviral medications, and corticosteroids to suppress the inflammatory process. Bronchodilators may be used for the management of bronchospasm. For transplant recipients, prevention and early treatments of acute rejection and viral infection are the best medical approaches to preserve the function of the donor lung.63
Mortality is generally low for OB and is predominantly associated with the underlying pathology that is responsible for the inflammatory process. Death due to OB in children is approximately 1%.59 In the transplant population, the mortality rate has been reported as high as 56% with more than 60% of the deaths related to a respiratory infection.
Lymphangioleiomyomatosis (ICD-9-CM Code: 496)
Lymphangioleiomyomatosis (LAM) is a rare disease that affects women in their reproductive years. It is a multisystem disease that is characterized by nonneoplastic proliferation of atypical smooth muscle cells in the parenchyma and lymphatic system. It is also associated with the development of renal angiomyolipomas in approximately 50% of the cases.64,65
The etiology of LAM is unknown, but there may be a genetic link because LAM is present in the autosomal genetic disorder tuberous sclerosis complex on chromosome 16. Typically, the onset of LAM is in the early to mid-30s and has an incidence of 1 per 1 million.66 The pulmonary system is the primary site of dysfunction.64 There are two patterns of LAM: tumor sclerosis gene LAM, which is also associated with central nervous system involvement including seizures and cognitive impairments, and sporadic LAM, which does not have any neurological involvement.67
Upon examination of the lung tissue, there is diffuse formation of cysts that leads to degradation of supportive elastic fibers by an imbalance between 1-AT and elastase. There is a proliferation of immature smooth muscle cells (LAM cells) within the walls of the airways, which leads to the destruction of alveoli and obstruction of the small airways.64–66
This disease is frequently misdiagnosed as asthma, COPD, pulmonary fibrosis, tuberculosis (TB) infection, or sarcoidosis. On an average, there is a 4-year delay between the onset of symptoms and the diagnosis, unless the patient experiences a spontaneous PTX; then the average time to obtain a correct diagnosis is slightly more than 2 years.67 The chest X-ray demonstrates nonspecific changes with preserved or increased lung volumes, hyperinflation, diffuse reticular opacities, pleural effusions, and a PTX. High-resonance CT scan is the best tool for diagnosis because it is very sensitive in detecting cystic formation and honeycombing without fibrosis and dilatation of the thoracic duct. There may also be the dilatation of the thoracic duct. The results of PFTs may show an obstructive, restrictive, or mixed pattern.65 The majority of cases demonstrate an obstructive volume pattern. Approximately 35% of patients will have normal a PFT until late into the disease67; however, a reduction of DLCO is seen in most cases regardless of the pattern.65,66
The most common presenting signs and symptoms are DOE and PTX. The patient may also present with a nonproductive cough, hemoptysis, chylous pleural effusion, wheezing, chest pain, abdominal pain, and ascites, if associated with dilatation and cysts involving the lymphatic system of pelvis and abdomen. Symptoms may worsen with the use of oral contraceptives.64,66 Signs and symptoms of right-sided heart failure may be documented in association with pulmonary hypertension.65,67
Treatment includes counseling the patient to avoid pregnancy because the hormonal changes worsen the disease. The patient should also be encouraged to avoid labor-intensive jobs because of the risk of PTX. Corticosteroids and cytotoxin are typically not effective in alleviating signs and symptoms. It has been suggested that the use of progesterone and tamoxifen may stabilize the disease progression.66 More recently, antiestrogen and luteinizing hormone are being used to manipulate the endocrine system as well as oophorectomy, which appears to slow the progression of the disease.68
Death is usually due to respiratory failure. Mortality rates are variable and are greatly influenced by the degree of small airway obstruction and impairments in DLCO. On an average, there is a 50% to 80% survival rate at 8 to 10 years after the onset of symptoms.64,66 Patients whose primary complaint is dyspnea have a significantly higher mortality rate as opposed to those suffering from PTX; 10-year survival rates are 47% and 89%, respectively.67
Septic Obstructive Airway Diseases
This group of diseases is also under the umbrella term of COPD, but these diseases are classified as septic diseases because of the presence of purulent sputum production and a high incidence of pulmonary infections. The hallmark clinical feature is a productive cough with excessive secretion production. The PFT findings are similar with a decrease in expiratory effort despite an increase in TLC. Many of these patients develop hypercapnia, which leads to pulmonary hypertension and cor pulmonale.
Cystic Fibrosis (ICD-9-CM Code: 277)
CF is the most common autosomal recessively inherited disorder in Caucasians.69 Within the lungs, this genetic defect leads to excessive production of thick, dehydrated, hyperviscous mucus and impairment of the mucociliary blanket.70,71 The incidence is 1 in 3,000 births in the United States and Europe. Chronic bouts of inflammation and infection lead to the breakdown of protein in the lungs. Obstructions of small airways develop from mucus plugs and destruction of the cartilaginous support of the airways. The end result is bronchiectasis, which is a permanent dilatation of the bronchi that is characterized by inflamed airways, which are full of purulent sputum23 (see Figs. 7-11and 7-12).
FIGURE 7-11 Cystic fibrosis. Notice the hyperinflation of the lung, the fibrotic changes throughout the lung fields, particularly the upper lobes, and decreased aeration. (Courtesy of Joseph Pilewshi, MD, University of Pittsburgh, PA.)
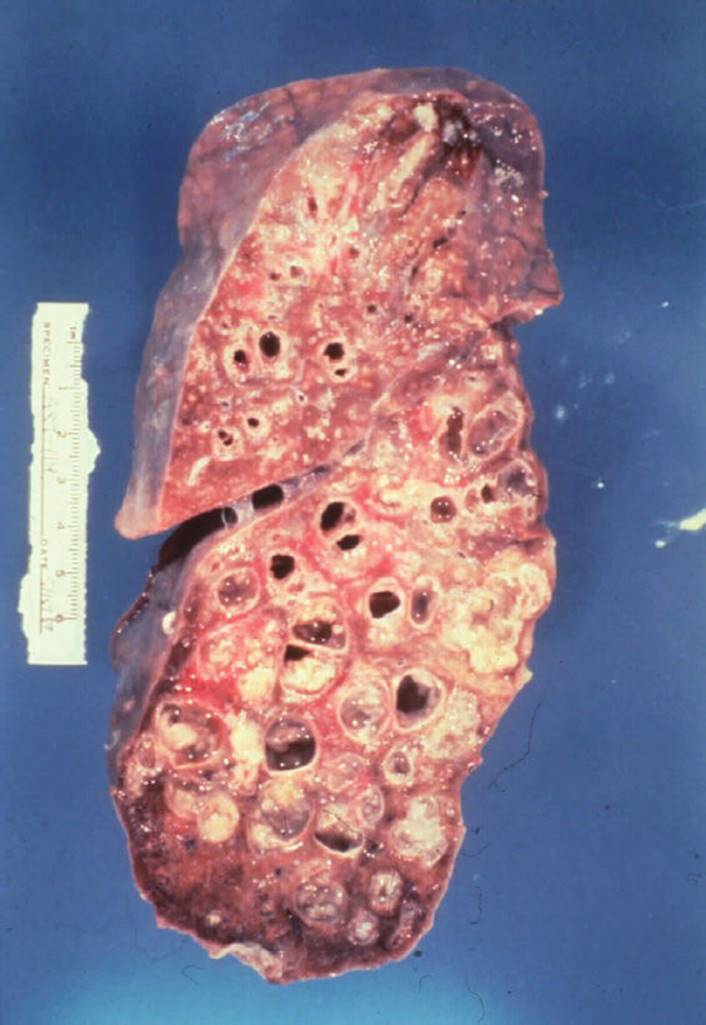
FIGURE 7-12 Cystic fibrosis. This gross pathology slide clearly illustrates destruction of the parenchymal tissue and the large cyst formation. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.).
CF is the result of mutation of the gene, CF transmembrane regulator (CFTR), which is associated with the failure of chloride secretion that results in dehydration of endo-bronchial secretions and cripples the mucociliary function as well as disrupts the function of the pancreas and reproductive system. This leads to an increased attraction to bacteria because of the decreased ability to contain and remove bacteria. CFTR is also associated with the transportation of bicarbonate and sodium and has been linked to the differentiation of osteoblastic cells.72,73
Ninety percent of people who are diagnosed with CF will also have pancreatic insufficiency.69 Recently, it has been recognized that early diagnosis and treatment is important to the aggressive nutritional support that aids in the management of musculoskeletal and pulmonary health.74 Through infancy and childhood, patients with CF will suffer from nasal polyps; failure to thrive syndrome; chronic or recurrent pneumonia; and a chronic cough, pancreatitis, and gastroesophageal reflux disease (GERD).69
The diagnostic findings are similar in patients with other obstructive lung diseases. A chest X-ray typically demonstrates hyperinflation and flattening of the diaphragm. High-resolution CT scans are more sensitive than conventional radiographic studies to detect airway changes and progression of bronchiectasis; there is a strong correlation between CT scan findings and PFTs.72 PFTs reveal an obstructive pattern and a decline in DLCO as the disease progresses. Abnormal arterial blood gases will be consistent with hypoxia and hypercapnia with advancement of the disease.
Most patients will present with a chronic productive cough, dyspnea with accessory muscle use, inspiratory crackles and wheezing, and clubbing of the nail beds.69,75 Patients also present with weight loss, decreased activity tolerance, pancreatic insufficiency, hemoptysis, and sputum production. This clinical picture may be complicated with osteoporosis, muscle wasting, diabetes mellitus, chronic back pain, and developmental delays.24,76 CF is associated with the following complications: massive hemoptysis and spontaneous PTX, which are associated with chronic infection and inflammation.77
Treatment primarily addresses pulmonary care and management of pancreatic insufficiency. Antibiotic and antifungal medications have become the mainstay in managing active infection and minimizing chronic colonization.78Research has shown that high dosages of ibuprofen have resulted in a reduction in inflammation and slowed the decline in FEV1. Clinically, the risk to renal and gastrointestinal system is too high, and most physicians will not prescribe the use of ibuprofen. Also, low-dose levels have also been linked to the increase of neutrophil migration into the lungs. Other pharmaceutical interventions are being directed at the primary CFTR defect or the direct consequences of its mutation.79 Sputum retention is managed by a variety of airway and pulmonary hygiene techniques.76,80 Additionally, the primary surgical intervention for CF is a double-lung transplantation. Therapy should also include management of osteoporosis and proper nutritional support. Finally, a well-rounded exercise program should be prescribed that addresses aerobic and endurance training, muscle strengthening, spine and osteoporosis care, and energy conservation as the disease progresses. See Chapter 17 for more information on the disease and treatment of CF.
The prognosis is dependent on the aggressiveness of the genetic expression of the disease as well as on the quality of the medical care. Certainly the lifespan of patients with CF has improved with advances in antibiotics, management of pancreatic insufficiency, hypoxia, and hypercapnia. The median life expectancy has increased over the years to 38 years of age.72 Eighty percent of people with CF will succumb to respiratory failure; others will die from complications of right-sided heart failure, severe hemoptysis, and spontaneous PTX.81,82
Bronchiectasis (ICD-9-CM Code: 494)
Bronchiectasis is the permanent dilatation of the bronchi from the destruction of the muscular and elastic properties of the lung. It is characterized by thickening of the bronchial walls, impairment of the mucociliary blanket, hypersecretion of purulent sputum, and bacterial colonization.23,83,84 Indeed, purulent overproduction of secretions is the hallmark of this pulmonary disease.85 There is a classification system for bronchiectasis that describes the distortion of the bronchi. Cylindrical bronchiectasis is associated with relatively uniform dilatation, whereas varicose bronchiectasis is characterized by local constrictions superimposed on cylindrical bronchiectasis. Finally, saccular or cystic bronchiectasis is associated with more severe disease and leads to the formation of bullae.84
Bronchiectasis is usually associated with other underlying pulmonary diseases, but there are rare cases of idiopathetic bronchiectasis that accounts for approximately 30% of the cases.86 Bronchiectasis is typically associated with CF, primary ciliary dyskinesia, and connective tissue disorders, such as rheumatoid arthritis (RA), lupus, α1-ATD, emphysema, and recurrent pulmonary infections.84
There is a higher prevalence of bronchiectasis in underdeveloped countries because of the lack of antibiotics. There is an increased number of patients being treated for bronchiectasis because people are living longer due to medical advances with various pulmonary diseases.87 It has been suggested that there may be a genetic predisposition, as well as environmental factors, that contributes to the development of bronchiectasis.84 The onset of bronchiectasis is commonly seen in the middle aged or in the elderly, but in the cases of congenital lung disease, the diagnosis may be made in childhood or early adulthood.85
There are two key factors that account for the development of bronchiectasis: the presence of intense and chronic inflammation and an inadequate defense mechanism to minimize the effects of infection resulting in tissue damage. These are the foundations for bronchial dilatation, inflammation, and weakening of the bronchial walls, which account for the impairment of the mucociliary escalator. Pooling of secretions creates an environment for bacterial colonization and infection. The increased levels of macrophages contribute to the influx of neutrophils. This increase in neutrophils stimulates phagocytosis; the production of reactive oxygen mediators; the release of proinflammatory mediators such as interleukin-1, interleukin-8, tumor necrosis factor; and the release of protease that causes irreversible loss of the elastin layer and causes the destruction of the smooth muscle and cartilaginous support of airways.85 There is a dysfunction of natural killer cells and the consequences of this cellular response leads to the increase in oxidative stress that causes further tissue damage. A vicious circle hypothesis has been proposed to describe the cycle of infection and chronic immune response, both of which cause lung tissue damage.88
The clinical presentation of bronchiectasis is associated with persistent production of large volumes of secretions, frequent hemoptysis, and recurrent infections. Secretions collect within the bronchioles in dependent positions.85Crackles, high- and low-pitched rhonchi, and pleural rubs may be heard on auscultation. The patient may also present with fever, fatigue, dyspnea, finger clubbing, and a chronic productive cough with foul-smelling sputum, which may have a bloody tinge to it.84,85 The remaining part of the physical examination is consistent with the underlying disease process. When the underlying disease is an obstructive process such as emphysema or CF, there will be a barrel chest and an obstructive pattern on the PFT. Bronchiectic changes can be seen on X-ray in the presence of a restrictive disease process but, typically, are not associated with overproduction of sputum.84
The diagnosis is primarily made upon clinical history and physical examination. The chest X-ray is relatively nonspecific, but usually illustrates hyperinflation with focal areas of atelectasis. A high-resolution CT scan is the gold standard for diagnosis, which documents dilatation of bronchi with or without bronchial wall thickening.84
The principal treatment for bronchiectasis involves the management of the underlying disease, which commonly includes the use of antibiotics, corticosteroids, and bronchodilators. Nutritional support, supplemental oxygen, airway clearance, and rehabilitation are also key components in the management of a patient with bronchiectasis. Surgical resection of the lung tissue that is the source of repeated infections or hemoptysis and lung transplantation may be an effective treatment plan to minimize recurrent exacerbations and further loss of lung function.85,87
The prognosis is dependent on the underlying disease and its severity, the quality and responsiveness to medical treatment, and the age of the patient. The majority of patients will succumb to respiratory failure or right-sided heart failure related to pulmonary hypertension.87,88
PULMONARY VASCULAR DISEASES
Pulmonary Embolism (ICD-9-CM Code: 415.1)
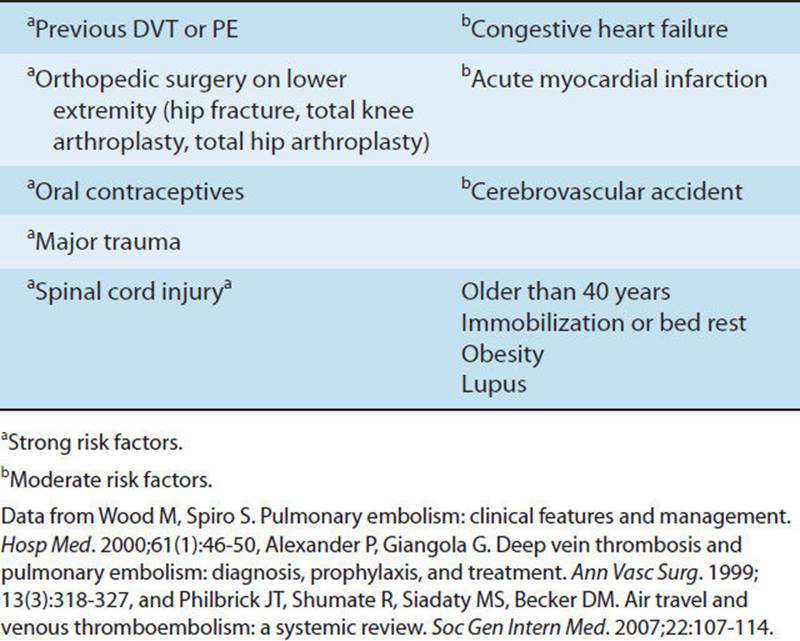
More than 600,000 patients suffer a pulmonary embolism (PE) annually in the United States.89 PE is closely linked to the presence of deep vein thrombus (DVT), blood clots, or a thrombus in the peripheral venous system, and it is the third leading cardiovascular cause of death, accounting for 200,000 deaths annually in the United States and Europe.90,91 Refer to Table 7-7 for risk factors associated with PE.92 The prevalence of suffering a PE is 28% and 74% with moderate and strong risk factors, respectively.90 A medical history that includes PE as a potential risk of another PE may be a fatal event.93 Typically, embolic events arise from the upper legs and pelvis. Air, fat, and amniotic fluid are also sources of embolisms.94 Small emboli may have little compromise to a healthy individual but may cause severe respiratory failure in an elderly individual with a reduced reserve of the cardiopulmonary system.94
TABLE 7-7 Risk Factors Associated with Pulmonary Embolism
PE accounts for 3% of deaths related to patients who had undergone surgical intervention, and a PE is found in 24% of surgical cases in an autopsy series. In one study of approximately 1,000 autopsies, a PE was reported as the primary cause of death in 26% of the cases; and in another 35%, PE was a primary contributor of death.95 Untreated PE accounts for 30% of hospital mortality, whereas, if treated, the mortality rate is reduced to 2%.85,96 Only one-third of patients diagnosed with a venous thromboembolism (VTE), which include a PE or DVT, are symptomatic and, if left untreated, can raise the mortality rate to as high as 25%.96,97 The formation of a thromboembolus is associated with three pathological features. Stasis, which typically occurs with immobility or bed rest, is due to a decrease in muscle contraction, lower cardiac output, and subsequent venodilation. There is usually the presence of endothelial injury that activates the inflammatory process, platelet aggregation, and the formation of the thrombus typically in the area of the venous valves. Finally, hypercoagulability may be related to the immobility.95
It is difficult to initially diagnose a PE in the elderly because the signs and symptoms are often vague and typically mimic the signs and symptoms of other comorbidities.90 The most pronounced clinical presentation of a PE includes an unexplained rapid onset of dyspnea (87% of all cases) and pleuritic chest pain (52%). Hemoptysis (44%) indicates pulmonary hemorrhage or infarction, cough (20%), leg pain and edema (37%), and syncope (14%). Tachycardia is present in 25% of all cases as well as tachypnea (65%), decreased breath sounds and abnormal lung sounds with rales (55%), and abnormal heart sounds (15%). Pleuritic chest pain, dyspnea, and tachypnea are present in 97% of all diagnosed cases of PE.89
During the process of evaluation, it is important to develop a differential diagnostic list as you proceed with testing to begin to formulate a clinical diagnosis. The differential diagnoses may include the following conditions: acute myocardial infarction, asthma, PTX, congestive heart failure, acute pulmonary edema, pleurisy, pericarditis, musculoskeletal trauma to the chest wall, sepsis, tamponade, and aortic dissection.94
Upon physical examination, the findings will vary based upon the size of the embolism. There may be a low-grade fever, cyanosis, tachycardia, jugular venous distension, tachypnea, and hypotension. Upon auscultation, there may be a pleural rub and a split of the S2 heart sound heard over the pulmonic valve. One-third of cases are associated with the presence of a pleural effusion. Ninety percent of all cases are associated with DVT, but what is alarming is that the DVT is only clinically present in 10% of all PE cases.89 The degree of respiratory compromise is dependent on the size of PE and on the preexisting cardiopulmonary reserves.
The clinical diagnosis is nonspecific, with false-negative physical examination findings in 50% of patients, and 50% confirmed false-positive findings in patients who present with symptoms related to conditions other than DVT.95The Homan Test is very nonspecific and lacks sensitivity in the diagnosis of a DVT. Examination of the arterial blood gases usually reveals hypoxia, hypocapnia, and a high alveolar–arterial gradient.45 An echocardiogram may be suggestive of right heart strain or ischemia, and the ECG may demonstrate a T-wave inversion in one or more precordial leads.93 Examination of the cardiac biomarkers may reveal an elevation in troponin, which indicated myocardial microinfarctions and release of brain-type natriuretic peptide from the myocytes because of increased workload and stress of the right ventricle.98
The usual first step when a PE is suspected is to obtain a ![]() /Q. scan,85 although the results may be inconclusive. Pulmonary angiography is the definitive study to evaluate the pulmonary artery system and assess the presence of a PE.93 The helical CT scan has largely replaced the use of
/Q. scan,85 although the results may be inconclusive. Pulmonary angiography is the definitive study to evaluate the pulmonary artery system and assess the presence of a PE.93 The helical CT scan has largely replaced the use of ![]() /Q. scan for rapid diagnosis.90 It is also important to determine the source of the PE. A contrast and compression venogram is the definitive study for DVT but is invasive and carries risks.93 Most recently, color flow duplex imaging has become an acceptable and sensitive tool for the detection of DVT.95
/Q. scan for rapid diagnosis.90 It is also important to determine the source of the PE. A contrast and compression venogram is the definitive study for DVT but is invasive and carries risks.93 Most recently, color flow duplex imaging has become an acceptable and sensitive tool for the detection of DVT.95
The key to quality medical care is the identification of patients who are at high risk and the implementation of effective prophylactic treatment. In one multicenter study, it was reported that only 32% of patients at high risk for DVT actually received some form of prophylactic intervention.95 Prophylaxis includes early mobilization and the use of graduated compression stockings, or compression stockings. Intermittent pneumatic compression stockings are used to provide a peripheral pump to enhance venous return and reduce venous stasis. Many patients are prescribed anticoagulants for the prevention and treatment of DVT formation. An inferior vena cava filter is the treatment of choice for patients who have a history of recurrent PEs, proximal DVT, or acute PE and who cannot be administered anticoagulation medications. The use of a filter is suggested for patients who have suffered multiple trauma and cancer.99In patients who cannot take anticoagulants, an inferior vena cava filter may be placed to decrease the risk of a PE occurring from a lower extremity or pelvic thrombus.
Acute management for a PE includes the use of thrombolytic therapy, which is most effective if used within the first 48 hours, and surgical intervention. Alteplase and recombinant tissue plasminogen activator are most effective and used with 92% response rate. There is a 13% incident of major hemorrhage complications and 1.8% incidence of intracranial or fatal hemorrhage. Pulmonary embolectomy has become an effective intervention for a massive PE, with a 5% to 10% mortality rate with an increase in death rate as the pulmonary vascular resistance increases.91,98
The prognosis of patients suffering from PE is dependent on the size of the embolism, its impact on the cardiopulmonary system, primarily acute failure of the right ventricle because of rapid rise in pulmonary vascular resistance, and the promptness of medical care. It is estimated that PE has a 35% mortality rate.91
Pulmonary Hypertension (ICD-9-CM Code: 417)
The normal mean pressure within the pulmonary arterial system is less than 15 mm Hg. Pulmonary hypertension can be defined as a mean pulmonary arterial pressure (PAP) greater than 25 mm Hg at rest and greater than 30 mm Hg during exercise, and pulmonary capillary wedge pressure, which is the pressure to assess the delivery of blood to the left atria and left heart function, is of 15 mm Hg or less. More recently, it has become evident that the definition of pulmonary hypertension should also include an elevation in pulmonary vascular resistance.100,101 In the past, pulmonary hypertension was classified as primary, or idiopathic, and secondary, which was associated with a contributing disease or disorder. Recently, there has been a new classification of pulmonary hypertension put forth that clusters the clinical presentation by similarities in pathology, clinical presentation, and therapy options.102 See Table 7-8 to review the new classification of pulmonary hypertension.103
TABLE 7-8 Classification for Pulmonary Hypertensiona
Group I: Pulmonary arterial hypertension (PAH)
Idiopathic PAH (primary)
Familial PAH
PAH associated with
Collagen vascular disease
Congenital systemic to pulmonary shunts
Portal hypertension
HIV infections
Drugs and toxins
Other diseases: glycogen storage disease, Gaucher disease, etc.
PAH associated with significant venous or capillary involvement
Pulmonary venoocclusive disease
Pulmonary capillary hemangiomatosis
Group II: Pulmonary venous hypertension
Left-sided atrial or ventricular heart disease
Left-sided valvular disease
Group III: Pulmonary hypertension associated with lung diseases and/or hypoxemia
COPD
Interstitial lung disease
Sleep disordered breathing
Alveolar hypoventilation disorders
Chronic exposure to high altitudes
Group IV: Pulmonary hypertension due to chronic thrombotic and or embolic disease
Thromboembolic obstruction of proximal pulmonary arteries
Thromboembolic obstruction of distal pulmonary arteries
Nonthrombotic pulmonary embolism (tumor, parasites, foreign material)
Group V: Miscellaneous
Sarcoidosis
Histiocytosis X
Lymphangiomatosis
Compression of pulmonary vessels
aThird World Conference on Pulmonary Hypertension.
Data from Simonneau G, Galei N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S–12S.
Pulmonary arterial hypertension (PAH) is defined as elevated PAP with normal left atrial and/or ventricular pressure. The pathology stems from abnormal vascular proliferation and remodeling of the small pulmonary arteries and arterioles. These changes lead to progressive elevation in pulmonary vascular resistance and eventually, right-sided heart failure. PAH includes such pathologies as idiopathic and familial, which accounts for approximately 6% of all cases and is associated with a mutation of bone morphogenetic protein receptor II genes.101,102 This category also includes connective tissue disorders such as scleroderma, congenital systemic to pulmonary shunts, HIV, thyroid diseases, and portal pulmonary hypertension.
Idiopathic pulmonary hypertension (iPAH), formally referred to as primary pulmonary hypertension, is a rare disease that predominantly affects women in their mid-30s; however, children may also be affected. The annual incidence of iPAH is one to two cases per million. iPAH is associated with systemic increases in the inflammatory process leading to muscle dysfunction and osteoporosis.104 The diagnosis of iPAH involves the elimination of any known cause for the hypertension and has a survival rate of 2.8 years after diagnosis.100,105
Most patients present with nonspecific symptoms and diagnosis may be delayed for an average of 2 to 3 years for iPAH. Delayed diagnoses of pulmonary hypertension may also occur for patients with other causes, but the delay occurs at a low incidence. Shortness of breath is typically the first symptom and is usually attributed to physical deconditioning by the patient as well as by the physician (see Table 7-1). Other symptoms include chest pain from right ventricular ischemia, near syncope or syncope, fatigue, and peripheral edema. These symptoms intensify as the pressure rises and is related to right-sided heart failure. A decrease in quality and volume of voice as well as a weak, ineffective cough is caused by the enlarged pulmonary artery compressing the left recurrent laryngeal nerve.104,106,107
PAH also presents from the sequelae of a congenital heart defect, which is referred to as Eisenmenger syndrome.108 Hypertension related to a congenital heart defect is underestimated by at least 10% and is typically the result of the blood shunted from the left side of the heart to the right, therefore inducing high volume and pressure through the pulmonary system resulting in hypertension.109 There is a decrease in Eisenmenger syndrome associated with many types of congenital heart defects due to improved detection and medical care; however, there is a rise in Eisenmenger syndrome in children with more complex congenital heart defects because children are surviving longer despite these defects.105
Pulmonary hypertension can also be associated with intrinsic lung disease. With intrinsic lung disease there is a disruption of the capillary beds and a destruction of the gas-exchange area of the parenchymal tissue that leads to hypoxia and vasoconstriction, which can cause hypertension.110
Despite the underlying cause of pulmonary hypertension, there are three conditions that contribute to the development of hypertension. These include vasoconstriction, remodeling of the vascular wall, and the dysfunction of platelets. Vasoconstriction is associated with endothelial dys-function, overproduction of endothelin-1, and a reduction in vasodilatation enzymes such as nitric oxide and prostacyclin. Vascular remodeling is the key factor and involves the presence and proliferation of smooth muscle cells in the small pulmonary arteries of the respiratory acini. There is endothelial cell proliferation that contributes to the remodeling and formation of plexiform lesions that are characteristic of pulmonary hypertension. Platelet dysfunction leads to thrombosis formation and contributes to further increases in vascular resistance and hypertension.111
Physical examination typically reveals abnormal heart sounds including S4, or atrial gallop, and a split S2 due to the asynchronous closure of the semilunar valves. As the disease progresses, an S3, or ventricular gallop, can also be heard and is indicative of advanced right-sided heart failure. The point of maximal impulse will be shifted to the left, indicative of right ventricular hypertrophy. The electrocardiogram is consistent with right ventricular hypertrophy and includes changes in the T wave. As the disease progresses, there will be overt signs of right-sided heart failure, jugular vein distension, hepatic congestion, peripheral edema, ascites, and systemic hypotension in most cases.111
The gold standard for diagnosing and documenting the severity of pulmonary hypertension is heart catheterization. This diagnostic procedure can determine whether there is a congenital or acquired intracardiac shunt or any abnormalities of the valves. It can also rule out myocardial dysfunction through biopsy and can measure pressures within the heart and cardiac output. PFTs may show a mild restrictive pattern in cases of severe hypertension and a decrease in DLCO. Echocardiograms can be used to monitor the progression of the disease and the effectiveness of treatment. Pulmonary angiography is the gold standard for determining the pulmonary arterial anatomy and assists in the diagnosis of thromboembolic disease. ![]() /Q. scans and CT scans are also used to rule out thromboembolic disease. Finally, desaturation during the 6-minute walk test is associated with lower survival rates.102,111 There is no cure for iPAH or many of the other causes of pulmonary hypertension, but many medical advances have improved quality of life and prolonged survival. These medical interventions may also be applied to the treatment of PAH. The antihypertensive benefits of calcium channel blockers are effective in the early stages of iPAH. Today there are three major pharmacological options. The most common is oral or continuous intravenous prostacyclin, such as Flolan or treprostinil. Prostacyclin is a group of potent vasodilators that, when infused into the pulmonary arterial system, improve hemodynamics. In many cases, there is an immediate reduction in PAP, whereas in others, there may be no or minimal immediate response but with long-term use, there is an improvement in symptoms. The dosage of prostacyclin is limited by complaints of jaw pain and joint pain, particularly in the foot and ankles. Nitric oxide–derived medications, such as sildenafil, are effective vasodilators. Endothelin-antagonist medications, bosentan, sitaxsentan, and phosphodiesterase 5 inhibitors, and sildenafil, inhibit the release of endothelin and improve the effects of brain natriuretic peptide and nitric oxide to reduce pulmonary pressures. The most common positive effects of these drugs include the improvement in exercise tolerance and a decrease in symptoms experienced at rest and with exertion. Supplemental oxygen may be helpful in patients who have hypoxemia either at rest or with activity. The use of anticoagulants and diuretics is common to prevent and minimize further thrombus formation and for the management of heart failure, respectively.100,112
/Q. scans and CT scans are also used to rule out thromboembolic disease. Finally, desaturation during the 6-minute walk test is associated with lower survival rates.102,111 There is no cure for iPAH or many of the other causes of pulmonary hypertension, but many medical advances have improved quality of life and prolonged survival. These medical interventions may also be applied to the treatment of PAH. The antihypertensive benefits of calcium channel blockers are effective in the early stages of iPAH. Today there are three major pharmacological options. The most common is oral or continuous intravenous prostacyclin, such as Flolan or treprostinil. Prostacyclin is a group of potent vasodilators that, when infused into the pulmonary arterial system, improve hemodynamics. In many cases, there is an immediate reduction in PAP, whereas in others, there may be no or minimal immediate response but with long-term use, there is an improvement in symptoms. The dosage of prostacyclin is limited by complaints of jaw pain and joint pain, particularly in the foot and ankles. Nitric oxide–derived medications, such as sildenafil, are effective vasodilators. Endothelin-antagonist medications, bosentan, sitaxsentan, and phosphodiesterase 5 inhibitors, and sildenafil, inhibit the release of endothelin and improve the effects of brain natriuretic peptide and nitric oxide to reduce pulmonary pressures. The most common positive effects of these drugs include the improvement in exercise tolerance and a decrease in symptoms experienced at rest and with exertion. Supplemental oxygen may be helpful in patients who have hypoxemia either at rest or with activity. The use of anticoagulants and diuretics is common to prevent and minimize further thrombus formation and for the management of heart failure, respectively.100,112
Beyond the advances in medical treatment, the options of surgical intervention have also improved significantly. There have been improvements in the surgical correction for multiple congenital defects. Lung transplantation is the most common surgical intervention for iPAH and for many patients with PAH. More recently other surgical procedures including pulmonary thromboendarterectomy for thromboembolic disease and atrial septostomy are being conducted to decrease pulmonary hypertension. In children with congenital heart defects, the success of surgical intervention depends on the age at the time of the surgery, the degree of heart failure, and the reversibility of the endothelial damage.105,108
The prognosis for iPAH is very poor despite advances in care with a mean survival time of 2.8 years. The National Institute of Health reports a 1-year survival in 64% of the patients and a 3-year survival in 48% of the patients. The prognosis of the patients with PAH depends on the progression of the underlying disease including the progression of PAH and how responsive the patient is to intervention and management.104,105
Pulmonary Edema (ICD-9-CM Code: 518.8)
Pulmonary edema is defined as failure of the microvascular endothelium that leads to an abnormal accumulation of fluid in the extravascular components of the lungs.113,114 Pulmonary edema is the result of a breakdown of the capillary endothelium and the alveolar epithelium barriers that protect the respiratory system.53 Along with these barriers, the lymphatic system is impaired and is unable to eliminate excess fluid from the pulmonary system.113,114
Pulmonary edema can be classified based on the pathological changes that lead to the edema. Hydrostatic pulmonary edema is due to an imbalance between the intravascular and extravascular space that allows fluid to move into the interstitial space and eventually into the alveoli. Hydrostatic edema is mostly associated with postoperative fluid overload, near drowning, and PE.113,115 Pulmonary edema with diffuse alveolar disease (DAD) is associated with acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), and actually involves damage to the endothelial cells.113–115 Please refer to the section on ARDS for more specific information. Pulmonary edema without DAD is primarily associated with drug-induced reactions that may be related to hypoxia and acidosis. This edema typically occurs with reversible cellular damage with rapid recovery.113,115 Finally, mixed pulmonary edema is associated with severe head injury. Fifty percent of the patients will experience pulmonary edema that is most likely related to an increase in microvascular pressure that causes arterial hypertension and vasoconstriction.113,115 Mixed edema is also seen in high-altitude illness with hypoxic-induced vasoconstriction and reperfusion injury after following lung transplantation and pneumonectomy.116
There are several factors that may predispose the patient to developing pulmonary edema. The function of the cardiac, renal, and hepatic systems contributes to the homeostasis of the body fluid and blood volume. Body weight, age, vascular tone, and fluid overload related to surgery also contribute to pulmonary edema.117 Postoperative pulmonary edema has an incidence of 8% and accounts for 12% of the deaths in patients during the postoperative period.117
Damage to the endothelial lining is a key factor in the development of pulmonary edema because the endothelial cells regulate permeability, modulate vascular tone and ![]() /Q. matching, and interact with blood-borne cells. Injury to the endothelial cells leads to an excessive inflammatory response with the release in interleukin-8, leading to increased vascular leaking and further endothelial damage. Platelet aggregation and a subsequent clot increase oxidative stress. Finally, the angiotensin-converting enzymes impairment is associated with more severe lung injury and lower survival.114,118
/Q. matching, and interact with blood-borne cells. Injury to the endothelial cells leads to an excessive inflammatory response with the release in interleukin-8, leading to increased vascular leaking and further endothelial damage. Platelet aggregation and a subsequent clot increase oxidative stress. Finally, the angiotensin-converting enzymes impairment is associated with more severe lung injury and lower survival.114,118
The clinical presentation of pulmonary edema does not depend on the underlying cause. The patient could present with acute or subacute onset of dyspnea, tachypnea, restlessness, crackles, and eventual peripheral cyanosis with hypoxemia.115 When a significant amount of the gas-exchange area is impaired, the patient will succumb to respiratory distress and possibly to respiratory failure, which would require mechanical ventilatory support. The clinical presentation is the primary focus for the diagnosis of pulmonary edema.
It is important to diagnose the underlying cause of the edema because this determines prognosis and helps to identify a treatment. Diuretics will be used to minimize fluid but fluid status needs to be monitored carefully to prevent a decline in cardiac function. Medications are used to support cardiac function and blood pressure and to treat or prevent infection. Corticosteroids are used to minimize the inflammatory response in the care of patients with pulmonary edema.
PLEURAL DISEASES AND DISORDERS
Pneumothorax (ICD-9-CM Code: 512)
PTX is defined as the presence of air in the pleural cavity between the parietal and visceral pleura. PTX can be classified as primary, secondary, iatrogenic, traumatic, or tension. Primary PTX typically has a spontaneous occurrence particularly in young, tall, thin men with no underlying lung disease. Secondary PTX is associated with underlying disease, with COPD being the most common.119–121 In both primary and secondary spontaneous PTX, it appears that blebs and bullae play roles in the pathogenesis. There is an imbalance between protease and antiprotease enzymes and an increase in the number of neutrophils and macrophages that result in the development of bullae. These bullae and blebs can rupture under increased pressure, as with a cough or Valsalva maneuver, causing air to leak into the pleural space.119
Iatrogenic PTX is due to a complication from a diagnostic or treatment procedure. The introduction of a central line into the subclavian vein is the most common procedure associated with PTX. Traumatic PTX is caused by the entry of air through the chest wall or from a laceration of the lung because of the penetrating wound of a gunshot or from a nonpenetrating wound of a rib fracture. The pleural space can become filled with air or blood (hemothorax) or both.119,120
Finally, tension PTX may be a potentially life-threatening situation and is associated with many etiologies. In this case, air enters the pleural space, but cannot escape. The increasing pressure will cause a progressive collapse of the lung and will eventually displace the mediastinum to the contralateral side (see Fig. 7-13). This can compromise venous return and subsequently decrease cardiac output.

FIGURE 7-13 Left lung tension pneumothorax. When the right and left lung fields are compared, you can appreciate a loss of the lung markings of the left lung, the subtle outline of the compressed lung, and a shift of the mediastinum to the right. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
Signs and symptoms will vary based on the size of the PTX and any other underlying pulmonary dysfunction. If the PTX is small and there is no underlying pulmonary disease, the patient may be asymptomatic. The most common symptom is acute dyspnea and pleuritic chest pain. The chest pain may be initially described as sharp and later as a steady ache. Symptoms may resolve within 24 hours even though the PTX has not resolved. Patients with underlying pulmonary disease will be symptomatic with even a small loss of lung function. As more lung tissue is involved, the symptoms will escalate. Breath sounds will be absent or diminished, there will be a hyperresonant sound upon mediate percussion, and a decrease in tactile fremitus.
A chest X-ray at peak inspiration and peak expiration is recommended to establish the diagnosis and determine the size of the PTX.56 Large PTX is defined as a 2 cm or greater distance between the chest wall and rim of the lung. PFTs will demonstrate a decrease in VC and an increase in the alveolar–arterial gradient causing varying degrees of hypoxemia. In severe cases, the patient may become hemodynamically unstable with tachycardia, hypotension, cyanosis, and cardiovascular and/or pulmonary arrest.119,121
If the PTX has affected less than 15% of the lung, it can be treated conservatively with an estimated resolution of 1.8% daily. When a patient is symptomatic, it typically means that the PTX is larger than 15% of lung tissue. The lung can be reexpanded with placement of a needle or a chest tube to aspirate the air or blood from the pleural space.120 A more aggressive treatment approach is chemical pleurodesis, the placement of a chemical, talc, or tetracycline into the pleural space to adhere visceral pleura to parietal pleura. The chemical causes an inflammatory response with resultant fibrosis and scarring.119,120 Finally, through a thoracotomy, the bullae can be resected.119–121
The prognosis is related to the size of the PTX, to the presence of an underlying pulmonary disease, and to the extent of the trauma. There is approximately a 32% recurrence rate within the first 2 years after the initial PTX, and, in general, PTX has a 15% mortality rate.23
Pleural Effusion (ICD-9-CM Code: 518)
Pleural effusion is an excessive collection of fluid between the parietal and visceral pleurae. Pleural effusions can be classified into two types: transudate and exudate. Transudate effusion is generally noninfectious and typically results from mechanical factors influenced by the rate of formation or reabsorption of pleural fluid. Transudate effusions are the results of an increased pulmonary capillary pressure such as in congestive heart failure, a decrease in lymphatic drainage as in thoracic surgery, a decrease in osmotic pressure resultant from renal disease, or an increase in intrapleural pressure from atelectasis. Exudative pleural effusion is generally the result of an infectious process with the most common cause being pneumonia.122–124
In children, the most common causes of effusion are parapneumonia; however, 40% to 50% of all pneumonia cases having documented effusions also have a congenital heart disease.122,124,125 A small number of childhood pleural effusions are associated with malignancy and liver failure.126
The development of pleural effusion can be separated into phases. The first phase is the exudative phase in which the fluid is free flowing and contains a predominance of neutrophils, elevated proteins, and a normal level of glucose. The effusion may be the result of fluid overload, an alteration in the permeability of the pleura, or an inflammatory process. Parapneumonia, in which the infection involves the visceral pleura, may initiate an outpouring of fluid into the pleural space.127 The second phase is the fibrinopurulent stage with continued influx of inflammatory cells and proteins. The increased permeability of the pleura will permit bacteria to enter the pleural cavity when the effusion is associated with pneumonia and fibrin will begin to deposit over the visceral and parietal pleurae. The final stage is the organization stage with the development of an empyema. In this stage, the fluid becomes more viscous, the inflammatory process progresses, bacteria multiply, and there is a deposition of fibrin and subsequent formation of a pleural peel. This infected pleura, or peel, can cause restriction of ventilation.125,128
The most common symptom is dyspnea, cough, and pleuritic chest pain. The severity of the distress depends on the size of the effusion, the rate of fluid collection, and the condition of pulmonary function. Dyspnea is the consequence of the restrictive lung defect from the compression of the effusion, ![]() /Q. mismatch, and the decrease in cardiac output. The cough is dry and nonproductive and is the consequence of inflammation and compression of the bronchial walls. If the cough is productive, the clinician should suspect pneumonia, and if hemoptysis is present, it is more likely related to PE or cancer.123 If there is pleuritic pain, it is usually an initial symptom and worsens with deep inspiration or in the supine position, but it can suggest an inflammation or infiltration of the parietal pleura or the presence of a PE.123,124 If the pain is constant, the underlying pathology may be cancer. Pain on palpation of the chest wall may be an indication of an empyema or metastatic cancer.127 The most common symptoms in children are chest or abdominal pain and vomiting. If children have pneumonia along with effusion, they may also present with cough, fever, and dyspnea.126
/Q. mismatch, and the decrease in cardiac output. The cough is dry and nonproductive and is the consequence of inflammation and compression of the bronchial walls. If the cough is productive, the clinician should suspect pneumonia, and if hemoptysis is present, it is more likely related to PE or cancer.123 If there is pleuritic pain, it is usually an initial symptom and worsens with deep inspiration or in the supine position, but it can suggest an inflammation or infiltration of the parietal pleura or the presence of a PE.123,124 If the pain is constant, the underlying pathology may be cancer. Pain on palpation of the chest wall may be an indication of an empyema or metastatic cancer.127 The most common symptoms in children are chest or abdominal pain and vomiting. If children have pneumonia along with effusion, they may also present with cough, fever, and dyspnea.126
Results of physical examination are associated with the size of the effusion and may reveal a decrease in chest expansion, dullness on percussion, and diminished breath sounds over the effusion. A zone of bronchial breathing may be heard upon auscultation at the upper border of the effusion.124 A pleural rub may be appreciated in the early stages of the effusion.126 If the effusion is large, the fluid can compress the lung and displace the mediastinum to the contralateral side. There can be a decrease in chest wall expansion and mild hypoxemia, which can be corrected with supplemental oxygen.123,124
Chest X-ray will be positive for the presence of a fluid collection and distortion of the dome of the diaphragm if at least 300 mL of fluid is in the space. A CT scan or ultrasound is more sensitive for the diagnosis, particularly if there is a small effusion. The effusion can also cause a mismatch between ![]() /Q. because the fluid is compressing the lung tissue. Needle aspiration of the fluid can also be performed for diagnostic purposes.124
/Q. because the fluid is compressing the lung tissue. Needle aspiration of the fluid can also be performed for diagnostic purposes.124
If the effusion is small and the patient is asymptomatic, the patient is typically monitored closely. If the patient is symptomatic, the fluid can be drained either by thoracentesis or by placement of a pigtail catheter or chest tube. The primary focus for the treatment of pleural effusion, once the symptoms have been alleviated, is to treat the underlying process. The use of fibrinolytic therapy such as urokinase or streptokinase can also be used to decrease the viscosity of the effusion to allow for drainage with the goal to avoid the need for surgical intervention.125 The prognosis is dependent on the underlying disease.124
PULMONARY INFECTION
Empyema (ICD-9-CM Code: 510)
Empyema is the presence of pus in the pleural space. The pus is highly viscous with an opaque whitish yellow color and consists of fibrin, cellular debris, and dead and live bacteria.129 An empyema can develop if a pleural effusion is not treated or is not responsive to treatment, the effusion progresses into the organization phase, and the fluid is contaminated with bacteria.
Approximately 40% of bacterial pneumonias will develop pleural effusion and approximately 15% of these cases will go on to develop empyema. There is an increased risk of empyema for children diagnosed with bacterial pneumonia.122 Empyema can also be caused by contamination of the pleura during a surgical procedure, with trauma, or with aspiration pneumonia.124
If the patient does not quickly respond to the treatment for a pleural effusion, further diagnostic testing should be conducted to evaluate for the presence of an empyema, particularly if this slow recovery is associated with a persistent fever, weight loss, malaise, and an elevation of white blood cells.124
The empyema should be drained and antibiotics should be prescribed. If a simple aspiration is unsuccessful, use of fibrinolytic medications can improve the ability to drain infection. An open thoracotomy or open drainage with rib resection for debridement of the empyema and infusion of an antibiotic can also be completed to treat an empyema.124,125 The mortality rate associated with an empyema can range between 20% and 75%, particularly in the elderly and children and in patients who are severely debilitated.122,130
Pneumonia (ICD-9-CM Codes: 482.2, 483, 484, 486)
Pneumonia is defined as an acute inflammation of the lungs, which usually occurs when the normal defense mechanisms of the respiratory system fail to keep the lower respiratory tract sterile, causing the small bronchioles and alveoli to become plugged with fibrotic exudate. The cellular response gives rise to the appearance of consolidation on chest X-ray. There are many etiologies that underlie the infection including bacterial, viral, and fungal sources (see Figs. 7-14 and 7-15).23,131
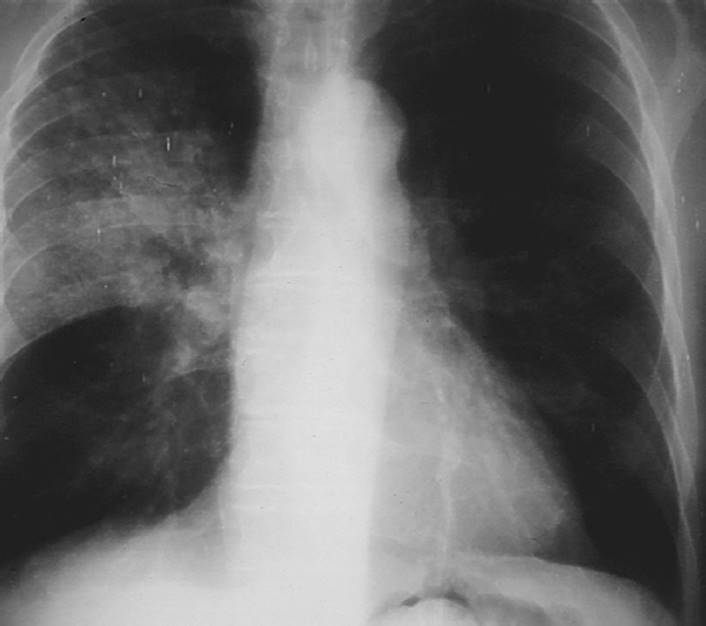
FIGURE 7-14 Right middle lobe (RML) pneumonia. This chest X-ray illustrates the consolidation of the RML with minimal involvement of right upper lobe (RUL), which is consistent with pneumonia. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)

FIGURE 7-15 Pneumonia. The examination of this lung reveals infiltration and consolidation of the right lower lobe (RLL) with relatively normal lung tissue in the superior lobes. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
Various reference points can be used to classify pneumonia for medical intervention and research. One common classification system uses the infectious agent, such as bacterial, viral, or fungal pneumonia. The infections can also be classified according to the incidence of the infectious agent for a particular population or region of the country, such as typical versus atypical pneumonia.
Another classification system is based on the environment in which the patient becomes infected with the agent that produces the pneumonia. This system allows health care professionals to identify specific interventions to minimize, prevent, and treat the common characteristics of the environmental setting. Community-acquired pneumonia (CAP) is an infection that occurs while the patient is living out in the community or the infection manifests itself within the first 72 hours of a hospitalization. CAP accounts for 1.7 million hospital admissions at the cost of $9 million annually and is the sixth leading cause of death in the United States.132 The incidence of CAP is 34 out of 1,000 cases for patients older than 75 years with up to 40% requiring hospitalization and 10% requiring admission into an intensive care unit with a 5% to 10% mortality rate.133 Hospital-acquired pneumonia (HAP) is the second leading cause of nosocomial infections, only exceeded by urinary tract infections, and is defined as an acute infection that is neither present nor incubated at the time of hospital admission up to 48 hours postadmission. The source of the infection is introduced to the patient within the hospital and accounts for 5 to 10 cases per 1,000 hospital admissions annually. Approximately 25% to 50% of hospital-acquired pneumonia is associated with mechanical ventilation (ventilator-associated pneumonia [VAP]).134,135 More recently a third large category, nursing home–acquired pneumonia, has been included in this environmental classification system because of the increasing number of people who are living in nursing care facilities and the higher incidence of pneumonia within this group. Nursing home–acquired pneumonia has an annual incidence rate of 32 cases per 1,000.136
Many risk factors have been identified that are associated with the development of pneumonia. These risk factors can be divided into medical and environmental conditions and respiratory function. A medical condition includes such factors as age; the integrity of the immune system; the presence of acute or residual effects of head, neck, or chest trauma; and surgery. The environmental situation takes into account the increased risk of pulmonary infection due to admission into a hospital and particularly into an intensive care unit, placement or use of tracheal or gastric devices, and exposure of other individuals. Finally, respiratory function risk factors include the increased infection rate associated with the use of mechanical ventilation and the need for aerosolized breathing treatments.132,137
Risk factors that have been associated with nosocomial infections include surgery and the depressive effects of the anesthesia, smoking within 8 weeks of admission, age, number and severity of comorbidities, recent hospital admissions, wound care or infusion care including hemodialysis, immunosuppressive medications, and the presence of atelectasis from immobility and aspiration.136,138 Age is a key factor because the elderly demonstrate a decrease in the function of the mucociliary cells, a decrease in lung compliance with a lower FEV1 and VC, as well as a decrease in muscle mass and an increase in comorbidities.138,139
The most common pathogens found in sputum identified in pneumonia are streptococcus, pneumococcus, H. influenzae, Pseudomonas aeruginosa, Acinetobacter, Staphylococcus aureus, and anaerobes obtained from aspiration. Less common sources of infections include legionella, aspergillus and candida fungal infections, CMV, RSV, and protozoan-like Pneumocystis carinii, most commonly seen in immunocom-promised patients from HIV infections, transplant recipients, and the very ill.130,140 There has been a rise in the incidence of multiple-resistant S. aureus that accounts for up to 50% of VAP with an associated mortality rate reported between 25% and 76%.141
The infection actually occurs because an agent, bacterial, viral or fungal, has reached the lower respiratory track where it has multiplied to a point the mucociliary blanket or macrophages cannot cleanse the system. An inflammatory process will be activated along with the immune response, causing localized edema and collateral cellular damage from toxins released by the pathogen, reactive oxidative enzymes, and lysosomal enzymes released by neutrophils and macrophages. The moist, warm environment of the lung is a breeding ground for growth of the pathogen and a vicious cycle between the immune response and the infection. The result is the consolidation of the lung tissue impairing the lungs’ ability to perform proper gas exchange. This infection can also spread to other segments of the lungs as well as to the pleural space and pericardium.142
When pneumonia in patients with HIV is examined, the infection appears to be related to the CD4 count. If the CD4 count is above 200/μL, the pneumonia is more likely to be caused by more typical agents such as streptococcus pneumococcal, H. influenzae, and S. aureus. If the CD4 count is less than 200/μL, patients with HIV are more likely to become infected with disseminated mycobacterium TB, fungal, CMV, and pneumocystis pneumonia (PCP).143,144
Aspiration has been clearly identified as a common contributing factor to the development of pneumonia. There is an increased risk of aspiration for the patient who has nasal or oral gastric tubes, an endotracheal tube, head and neck trauma, and a depressed mental status.141 Aspiration is also associated with malnutrition, tube feeding, contracture of cervical extensors, and use of CNS depressant medications.140 Other events have been linked to aspiration including dysphagia due to loss of dentition and poor hygiene, decreased saliva production, and weakening of muscles of mastication. Aging is associated with a delay in the neural processing to perform the proper swallowing sequence and a decrease in sensation of the oral cavity. Finally, there is an increased incidence of aspiration in the presence of some diseases such as Parkinson disease, cerebral vascular accident, GERD, connective tissue disorders, and Alzheimer disease.145
There are three clinical presentations for aspiration. The first syndrome occurs if the aspiration causes airway obstruction. If the obstruction is in the large bronchus, it will produce stridor and dysphonia. If the obstruction is more distal, a high-pitched wheeze is likely to be present. Clinically, the patient may present with fever, hemoptysis, and pleuritic chest pain. There will also be a decrease in breath sounds to the obstructed area. The second aspiration syndrome is a chemical pneumonitis, which is typically associated with gastric content aspiration but can also be caused by aspiration of bile, medications, and alcohol. These patients will present with dyspnea, high-pitched wheezing, cyanosis, and hypoxemia. If extensive lung tissue is involved, the presentation will include cough, pulmonary congestion, and shock. There may also be crackles upon auscultation along with dyspnea and respiratory distress. The last aspiration syndrome is pleuropulmonary infection, which includes acute pneumonitis, necrotizing pneumonia, lung abscess, empyema, and infectious pneumonia. These patients present with fever, productive cough, crackles and/or low-pitched wheezes, and an increase in fremitus and dullness on percussion over the area of consolidation.133,145
The typical clinical presentation for pneumonia includes fever and a productive cough with sputum production that is usually yellowish green or rust colored. There is also an elevation in the WBC count and a positive sputum culture identifying the infectious agent in most cases.135 The patient may report an increased level of fatigue and weight loss. If a substantial amount of lung tissue is involved, the patient may also present with dyspnea, tachycardia and tachypnea, and hypoxemia with desaturation upon exertion. Unfortunately, the elderly present with atypical signs. The health care professional should be monitoring for unexplained changes in mental status and functional impairments including an increase in falls, anorexia, incontinence, low-to-normal WBC count, tachypnea, and tachycardia.133,140 The health care professional should also be concerned with aspiration pneumonia if the patient presents with drooling, poor oral motor control, poor phonation quality, and choking. The patient may also complain of dry mouth, difficulty in chewing, hoarseness, and throat discomfort.145
The diagnosis for pneumonia is made based on a positive X-ray showing infiltration or consolidation of the infected segment for at least 48 hours and at least two clinical signs and symptoms of dyspnea, fever, productive cough, leukocytosis, or leukopenia. The accuracy of diagnosis is dependent upon the analysis of the sputum.146 The severity of the infection can be graded by the use of the CURB 65 (confusion, urea nitrogen, respiratory rate, blood pressure, 65 years of age and older) scoring system. This is a scale graded from 0 to 5 with a point given for each of the positive response to the categories: confusion, increased respiratory rate, low blood pressure (<90/<60), urea levels greater than 7 mmol/L, and 65 years of age or older. A score of 3 or higher is associated with an increase in mortality.133
The primary focus of intervention should first begin with prevention. Annual flu vaccination is an important preventative measure. Facilities should properly clean all respiratory equipment and engage in good hand-washing techniques. If aspiration is a concern, the patient should be positioned in a semirecumbent position and should avoid overfeeding. Good dental hygiene will decrease the colonization of bacteria in the upper airway.
CLINICAL CORRELATE
Early mobilization that encourages an increase in tidal volume and secretion mobilization is critical.135,140 In patients who cannot be mobilized, the facility should implement chest physical therapy, a bed-positioning schedule, and use of incentive spirometers. Many of these preventive techniques can also be implemented in the home.
Once the diagnosis of pneumonia has been made, treatment should include administration of the proper pharmacological therapy. Typically for bacterial infections, the patient is placed on a wide-spectrum antibiotic. If the signs and symptoms do not resolve or become recurrent, a sputum culture should be done. These results will allow the physician to prescribe a more precise and effective antibiotic. Treatment should also include early mobilization and chest physical therapy as mentioned earlier.140
There are several factors that have been identified as prognostic indicators. They include age greater than 65 years, inability to protect the airway, a respiratory rate greater than 30 breaths/min, fever, X-ray showing more than one lobe involvement, and the need for mechanical ventilation. The clinical outcome is also negatively affected by the presence of comorbidities such as COPD, chronic renal failure, diabetes mellitus, chronic heart failure, malnutrition, recent splenectomy, and diminished mental status.132,147
Pneumonia is the sixth leading cause of death.132 CAP mortality is 1% to 2% for mild pneumonia, 5% to 10% for patients who require hospitalization, 30% in the elderly, and 20% to 50% in cases of severe pneumonia. In community dwellers who are ambulatory, the mortality rate is 5% whereas it is 37% for patients in an ICU.130 In the cases of nursing home–acquired pneumonia, mortality rates are as high as 48% for patients who are dependent in activities of daily living (ADL) and are nonambulatory.140 Thirty-two percent of all nursing home residents who survive the pneumonia will die within the next 24 months because of associated functional impairments and severity of the comorbidities.136
Mycobacterium Tuberculosis (ICD-9-CM Code: 486)
According to the World Health Organization, it is estimated that one-third of the world’s population is infected with TB and 1.8 million deaths annually are TB related. Globally, there are 8.3 million new cases of TB diagnosed every year. These data support that more individuals are being diagnosed with TB at the same death rate in the past decade.148 Ninety-five percent of individuals with a new diagnosis and 98% of all deaths are related to TB and HIV coinfection.149 In the United States, 15 million people are infected with TB. There has been a rise in TB since the early 1980s because of the HIV/AIDS epidemic. TB is a social disease that disproportionately affects the under-privileged who are malnourished, homeless, substance abusers, and people who live in underdeveloped countries or who are institutionalized in extended care facilities and prisons.148,150,151
Mycobacterium TB is primarily an infection in which the lung is the primary site of incubation (see Figs. 7-16 and 7-17). This mycobacterium is an immobile organism that thrives in warm, well-oxygenated tissues. The mode of transmission is the inhalation of small, dry droplet nuclei that becomes airborne from the cough or sneeze of an infected person. To become infected, the person needs to be exposed to an actively infected person for an extended period of time, to a person who is infected with laryngeal TB, or to a person who has extensive pulmonary disease. The particles need to reach the alveoli to replicate. The lung is usually the site of the primary infection. The risk of infection is dependent on the concentration of organism in the air particles, the length of exposure time to the infected person, and the host’s immune system.151

FIGURE 7-16 TB. This patient presented with a primary pulmonary infection of TB. The chest X-ray illustrates unilateral, patchy parenchymal infiltrates with pleural effusion. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
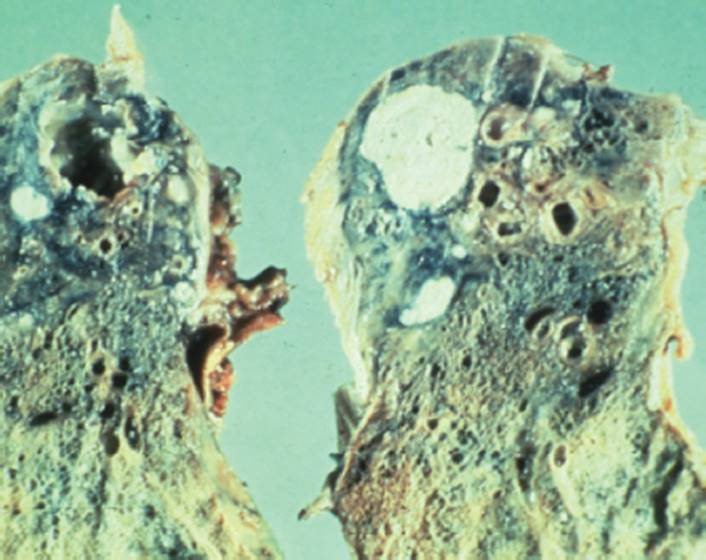
FIGURE 7-17 TB. In the RUL there is a large granuloma. A necrotic cavitation is present in the LUL. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
The incubation period is 2 to 12 weeks. At approximately 6 to 8 weeks, the TB skin test, purified protein derivative (PPD), will be positive because the count of the organism is high enough to cause an immune response to the invasion. Diagnosis by a sputum smear is difficult owing to the difficulty in obtaining sufficient sample. Diagnosis is made by an increase in interferon gamma, early-secreted antigen, and culture filtrate protein 10 (CFP-10). Diagnosis in children is very difficult with nonspecific signs and symptoms of failure to thrive, persistent fever, and malaise.148
The infection can spread via the blood and lymphatic system to other organs such as the kidneys, bones, and brain. The immune system will surmount an attack against the organism and will surround the TB to form a granuloma.151Healing will cause fibrosis and calcification of the granulomas. If the patient is immunocompromised, the TB will not be contained within the lungs and can cause a disseminated disease.149,151
Reactivation or secondary infection can occur from an encapsulated lesion containing virulent TB. The secondary infection usually occurs at a time when the immune system is in a compromised state due to an illness or to aging. The site of reactivation is usually the upper lobes of the lungs or an extrapulmonary site.151
During the primary infection, most patients are asymptomatic. If the patient presents with signs and symptoms, they are typically similar to the clinical presentation of pneumonia, such as an unproductive cough and a fever. If the pleura is involved, the patient may also experience dyspnea and pleuritic pain. Crackles may be present in the area of infection along with bronchial breath sounds if there is consolidation. X-ray will be abnormal with fluffy shadows of the upper lobes, atelectasis, enlarged lymph nodes, and cavitations, mainly in the upper lobes. There is also scarring of the lungs with a loss of tissue function.148
The secondary infection is associated with a cough, which becomes increasingly productive as the disease progresses, accompanied by night sweats, weight loss, low-grade fever, and sometimes pleuritic pain. There will be subtle inspiratory crackles, a decrease in tactile fremitus, and adventitious breath sounds over areas of pleural thickening and cavitation.151 The signs and symptoms of extrapulmonary disease are dependent on the tissue infected. Mycobacterium osteomyelitis of the upper thoracic spine is associated with chronic hypercapnic respiratory insufficiency or failure.152
The first line of defense is prevention of the transmission of this disease. Universal precautions should be used around anyone who has a cough. One should avoid overcrowded housing as in homeless shelters and prisons. Finally, people should be vaccinated and screened for TB exposure. If the purified protein derivative test is positive, the person should undergo at least 6 months of treatment to minimize the risk of a secondary infection. During the primary infection, respiratory isolation is important to minimize the spread of the disease. Patients are usually given rifampin and isoniazid (INH) for 6 months to suppress the infection followed by another 2 months of pyrazinamide and ethambutol administration. Further medical or surgical intervention will depend on the site and severity of the extrapulmonary infections.148,151
The mortality rate in patients who go untreated is as high as 80%, and a median time period to death is 2.5 years. In the HIV population, mortality exceeds 80%.151 Respiratory failure associated with acute hypoxia may require mechanical ventilation and is associated with a mortality of 70%.152
Meconium Aspiration (ICD-9-CM Code: 770.1; Practice Pattern 5B, 6G)
Meconium aspiration syndrome (MAS) is one of the primary causes of neonatal respiratory distress that frequently leads to respiratory failure and death.153 Meconium is the green viscous fluid that consists of fetal gastrointestinal secretions, cellular debris, mucus, blood, and other waste products. This material appears in the 10th to 16th week of gestation. In approximately 12% of deliveries, the meconium is passed in the later weeks of the pregnancy and contaminates the amniotic fluid.153,154 Over the past decade there has been a significant decrease in the incidence of MAS due to early detection, amniotransfusion, and caesarean deliveries.155 Approximately one-third of these infants will require mechanical ventilation. It is estimated that 1.5% of all infants will suffer from MAS, and mortality rates vary based on gestational time of MAS with the mortality rate as high as 30% in the second trimester and 22% in the third trimester.155 The reader may refer to Chapter 21 for a more detailed description of this disease.
RESTRICTIVE PULMONARY DISEASES (PULMONARY FIBROSIS)
This next group of pathologies has more than 200 different diseases. Pulmonary fibrosis has been linked to immune disorders, occupational exposures, genetic and hormonal abnormalities, and a complication of lung injury. These diseases are classified together because they have similar clinical features such as a shallow, rapid breathing pattern due to a loss in the compliance of the lungs and the chest wall. One of the most pronounced features is significant hypoxemia with rapid desaturation on exertion. Many patients develop pulmonary hypertension and cor pulmonale (Table 7-1). The PFTs consistently show decreased levels in FVC and FEV1 with the FEV1/FVC within normal limits. There is a significant reduction in VC and TLC and the DLCO is usually quite diminished (Fig. 7-2 and Table 7-2).
Idiopathic Pulmonary Fibrosis (ICD-9-CM Code: 518)
Idiopathic pulmonary fibrosis (IPF) is a type of interstitial pulmonary fibrosis, which encompasses a large heterogeneous group of diseases that involves an inflammatory to fibrotic process of the parenchyma. IPF is also referred to as cryptogenic fibrosis alveolitis and includes such pneumonias as non-specific, desquamative, cryptogenic, and lymphocytic interstitial.156 The onset of IPF occurs in mid- to late life, and the prevalence is approximately 14 to 15 per 1,000 in the general population. There is a slightly higher incidence in men and there appears to be no racial differences. The cause is unknown, but much work has been done to develop a classification system of IPF that is based on pathological findings and clinical presentation. In general, usual interstitial pneumonia (UIP) is characterized by patchy, nonuniform, and variable destruction of interstitial tissue (see Figs. 7-18 and 7-19) and accounts for 60% of the cases of IPF. There is also a minimal inflammatory component to this disease with collagen deposition that thickens the alveolar septum.157 Desquamative interstitial pneumonia (DIP) is another form of IPF that presents with little fibrosis but a significant inflammatory response with an accumulation of alveolar macrophages within the alveolar spaces and interstitium.

FIGURE 7-18 Interstitial pulmonary fibrosis. Note the relatively small lung volumes with the diffuse infiltrates, mostly apparent in the lower lobes. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
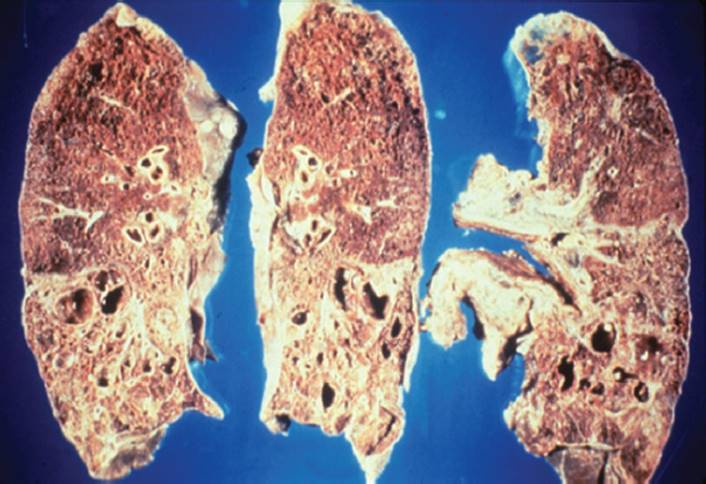
FIGURE 7-19 Interstitial pulmonary fibrosis. The progression of this inflammatory and fibrotic process leads to large cyst formation, which is referred to as honeycombing. This destructive process typically predominates in the lower lobes. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
The initial injury appears to damage the alveolar and epithelial cells. It has been suggested that the immune response to some unknown stimulant is what starts a cascade of events. There also appears to be an autosomal dominant genetic link for some patients with the prevalence of familial IPF being 5.9 per million.158 The damage causes inflammatory cells to release cytokines, tumor necrosis factor, and platelet-derived growth factor. These inflammatory chemicals result in smooth muscle and fibroblast proliferation, degradation of the alveoli, and collagen deposition.159
Open lung biopsy is the most definitive method for the diagnosis of IPF because there are many diseases that have a similar clinical presentation. It is important to rule out occupational or environmental exposure as the cause for the pulmonary fibrosis. Chest X-ray and high-resonance CT scan will document bilateral interstitial infiltrates typically starting in the upper lobes. PFTs will be consistent with a restrictive pattern that includes a reduction in lung volumes and a low diffusing capacity. However, if the patient has a significant history of cigarette smoking, the PFTs will show both restrictive and obstructive patterns.160,161 The majority of these patients will also be hypoxic, and many will require high levels of supplemental oxygen.162
The patients with usual interstitial pneumonia or desquamative interstitial pneumonia primarily present with progressive but insidious onset of breathlessness and a nonproductive cough. Patients may also complain of systemic symptoms such as low-grade fever, malaise, arthralgias, weight loss, and clubbing of the fingers and toenails.160,161,163 These patients usually desaturate quickly, even when attempting to complete activities of daily living. Severe desaturation may also be associated with light-headedness, dizziness, and arrhythmias. Exertion is commonly associated with pleuritic chest pain as the patient struggles to increase tidal volume and the respiratory pattern is shallow and rapid. Along with the interstitial fibrosis, up to 84% of these patients will also develop pulmonary hypertension. The hypertension is multifactorial as it relates to chronic hypoxia leading to vascular remodeling, vascular obstruction and destruction associated with the inflammatory and fibrotic processes, and also heart dysfunction. These patients may present with chest pressure, arrhythmias, dizziness upon exertion, and signs and symptoms of right-sided heart failure.164
Treatment is very limited in its effectiveness in stopping the progression of this disease, although there are new drugs currently under clinical trials, such as growth factor β and endothielin-1, which have been shown to interfere with the adverse effects of the immune response.156 High levels of systemic corticosteroids are commonly used as the first line of intervention. Cyclophosphamide impairs the function of neutrophils that eventually decreases fibroblastic and collagen proliferation. Azathioprine and cyclosporine suppress the production and maturation of T and B cells involved in the immune response.165 More recently, patients with moderate-to-severe pulmonary hypertension may be administered Flolan or UT15. These medications are very potent vasodilators that may be effective in lowering the level of hypertension and making the patient less symptomatic.
The prognosis is dependent on the rate of progression of the disease. Death is usually the result of respiratory failure or heart failure in the presence of pulmonary hypertension. The mean survival of IPF is 5 to 6 years but will vary based on the aggressiveness and type of IPF, duration of symptoms, presence of pulmonary hypertension, and responsiveness to therapy.156
Acute Respiratory Distress Syndrome (ICD-9-CM Code: 769)
ARDS has more recently been referred to as ALI to reflect the cellular status. The ALI results in pulmonary infiltrates, severe refractory oxygenation, and fibrin deposition leading to lung stiffness and a subsequent decrease in compliance. It has been suggested that ALI/ARDS is the most severe form of pulmonary edema in which diffuse alveolar involvement contributes to further injury.118 A diagnosis of ALI/ARDS is made when the ratio of the partial pressure of oxygen (PaO2) and the fraction of oxygen inhaled (FiO2) (PaO2/FiO2) is less than 200 mm Hg and when all cardiac sources for pulmonary edema have been ruled out. A low value of 200 mmHg represents severe lung injury (normal PaO2/FiO2 is 380–486 mmHg).
The development of ARDS can be the result of direct injury to the lung tissue as with a blunt chest trauma or as a result of an indirect injury that is associated with systemic inflammation and elevations in inflammatory mediators such as in sepsis and mechanical VAP, which can precipitate further lung injury.118,166 The causes of ALI/ARDS are multiple, but the development of ARDS is associated with damage to the alveolar epithelial cells and the disruption of the pulmonary vascular endothelium.114,118 Barotrauma, volutrauma, and oxygen toxicity caused by the use of mechanical ventilator to compensate for respiratory dysfunction or failure can also lead to further inflammation and cellular damage.167
There are several factors that predispose an individual to ARDS, which can include, but are not limited to, pneumonia, aspiration, lung contusion, fat emboli, near drowning, inhalation injury, sepsis, severe trauma, and blood product transfusion.168 It has been estimated that the incidence of ARDS is 75 per 100,000. Despite advancements in medicine, the incidence has not really declined in the last 30 years.168 Forty-two percent of patients diagnosed with ARDS will be associated with a complication of sepsis, whereas orthopedic trauma accounts for 11% of the cases. Twenty-two percent of trauma cases that are complicated with ARDS will involve a lung contusion. Finally, 50% of aspiration cases will progress to ARDS, with 85% of the cases showing signs of ARDS within 72 hours.169
ARDS is characterized as a heterogeneous disorder that changes over time. There are three stages in the process of ARDS. The first stage is an exudate phase, which is characterized by pulmonary edema, hemorrhage, and hyaline membrane formation. Clinically, there is a rapid onset of respiratory failure that is refractory to supplemental oxygen.167 Damage to the type I alveolar epithelial cells leads to an increased risk of infection, alveolar remodeling, alveolar edema, and a decrease in antioxidative function. Damage to the type II cells impairs the ability of the epithelium to transport fluid that accumulates and causes additional and persistent edema. The dysfunction of type II cells also causes the reduction of surfactant, which increases airway resistance and pressure to ventilate, and is associated with the increased risk of bacterial infection and colonization.118 The inflammatory mediators, such as tumor necrosis factor α and interleukin-1 and interleukin-6, lead to an increased vascular permeability and thrombus formation.114 The second phase involves cellular proliferation with the elevation of neutrophils and other inflammatory cells. The influx of neutrophils, whose duration and severity of the level of neutrophils is a predictor of mortality, leads to intrafibrin deposition and pulmonary vascular thrombi.118 This phase is characterized by DAD, which is associated with cellular necrosis, epithelial hyperplasia, and further inflammation that leads to destruction of the delicate structures of the lung. The third phase is fibroproliferation, which is the result of chronic inflammation whereby injured lung tissue is replaced with fibrotic tissue.168 The inflammatory response leads to a reduction in protein C and antithrombinase, and an increase in plasminogen activator inhibitor and angiotensin II, which leads to vascular remodeling and thrombi.114,166 If the remodeling and destruction of the pulmonary vascular bed are significant, the patient will also develop pulmonary hypertension and will present clinically with right-sided heart failure.167,168
The initial clinical presentation is characterized by acute respiratory failure that typically requires mechanical ventilatory support. Upon examination, diffuse crackles can be observed, along with pink frothy secretions, a sign of alveolar edema. This respiratory failure is associated with difficulty in ventilating the patient and is refractory to supplemental oxygen.168
Once the patient has been weaned from the mechanical ventilator, a restrictive breathing pattern is common for the first 12 months. Some patients will have a complete recovery, whereas others will continue to demonstrate a mild-to-moderate restrictive breathing process with a decrease in TLC, VC, and low diffusion capacity. The recovery is associated with the degree of lung injury, inflammatory response, particularly level and duration of increased neutrophil levels, and vascular remodeling.114 Patients with significant residual deficits will present with DOE, hypoxemia, functional impairments, hypertrophy of accessory respiratory muscles, and a shallow, rapid breathing pattern. The duration on the ventilator and severity of illness are associated with a higher level of pulmonary impairment as well as mild-to-moderate decline in quality of life.115,168
The diagnosis of ALI/ARDS is based on the clinical presentation of respiratory failure with severe hypoxia and a decrease in lung compliance. The chest X-ray will illustrate a patchy, diffuse airspace disease. The CT scan is consistent with diffuse damage due to injury and edema, which compresses uninvolved tissue that further impairs the respiratory system.115,170 In patients with residual pulmonary dysfunction, the PFTs will show a restrictive process with a decline in lung volumes.
There is a delicate art involved in the treatment of patients with pulmonary edema and ALI/ARDS. High ventilation pressures and high oxygen concentration need to be avoided because these factors can actually contribute to further cellular injury. The common approach to mechanical ventilator support is to use a low tidal volume to minimize barotrauma and a low percentage of oxygen to avoid tissue damage from oxygen toxicity. The ventilator is usually set with a high positive end-expiratory pressure (PEEP) to keep airways open and allow for easier ventilation.167 The newer mechanical ventilators have two modes of ventilation, which are beginning to be recognized as valuable components in the treatment of ARDS. BiVent ventilation or airway pressure release ventilation (APRV) is combining high and low positive end-expiratory pressure levels over two separate time intervals with the goal to recruit alveoli and improve gas exchange with a decreased exposure to high airway pressure.171 The other mode of ventilation is pressure-regulated volume control (PRVC) mode of ventilation in which the ventilator adjusts pressure to deliver a prescribed tidal volume under the lowest pressure.
There are many other treatments used in the management of ARDS. Treatment also includes management of fluid balance to minimize pulmonary edema and still maintain cardiac output. The administration of surfactant replacement therapy is undergoing clinical trials in adults. Nitric oxide gas mixture is used to promote bronchodilation to reduce the positive pressure required to ventilate the lungs, thus reducing risk of barotrauma.166 Antibiotics and systemic corticosteroids are used to minimize infection and inflammation, respectively.168 In extreme cases an extracorporeal membranous oxygenator (ECMO) may be used to completely support the respiratory system and allow the lungs to rest (see Figs. 7-20 and 7-21). Finally, it is suggested to try positioning the patient in prone. Prone positioning seems to improve oxygenation in approximately 50% of cases. The reason for this improvement is unclear, but it may be related to a reduction in pleural pressure, more uniform ventilation, decreased atelectasis, or reduced abdominal pressure on the thoracic cavity. In addition, lying prone appears to promote postural drainage and promote the redistribution of perfusion.170
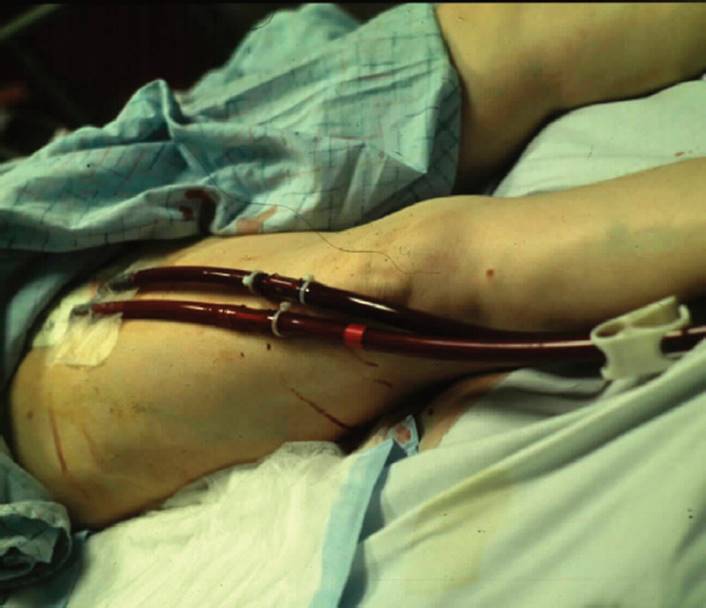
FIGURE 7-20 Extracorporeal membranous oxygenator. This patient is being supported on ECMO because of cardiopulmonary failure. The cannulas are connected to the femoral artery and vein. (Courtesy of Chris L. Wells, University of Pittsburgh, PA.)

FIGURE 7-21 ECMO. This device is used to oxygenate the blood outside the body (white canisters) and then pump the blood back into the patient’s body (pump is in the right upper corner). The patient is in critical condition and cannot be supported by mechanical ventilation alone. (Courtesy of Chris L. Wells, University of Pittsburgh, PA.)
There is a 40% to 60% mortality rate associated with ARDS.168 Mortality is highly associated with sepsis, especially when there is a high concentration of neutrophilic activity.118 These deaths are primarily associated with respiratory failure among the elderly. This fact should support early detection and aggressive medical management of respiratory infections in elderly patients. This is different in the younger patient, whose death is primarily due to multisystem organ failure, including right-sided heart failure due to pulmonary hypertension.114,169
Bronchopulmonary Dysplasia (ICD-9-CM Code: 769)
Bronchopulmonary dysplasia (BPD) is a chronic, restrictive pulmonary disorder that is a consequence of unresolved or abnormally repaired lung disease. BPD is most prevalent in premature infants who are exposed to high concentrations of supplemental oxygen and positive-pressure mechanical ventilation in order to compensate for immature lungs.172 The reader may refer to Chapter 21 for a more detailed description of this disease.
Hypersensitivity Pneumonitis (ICD-9-CM Codes: 503, 505)
Hypersensitivity pneumonitis (HP), or external allergic alveolitis, is an immunologically mediated disease that is typically associated with sensitivity from repeated exposure or large dosage exposure to an antigen. Further exposure results in an inflammatory response that involves the distal airways and alveoli.173–175 There are numerous agents that can be the impetus for HP including organic material such as agents from moldy hay or grains; fungi from water reservoirs; and bird serum, feathers, and excreta. HP can also be caused from exposure of mining dust and pharmacological products such as gold, amiodarone, and minocycline.173,175–177 Acute HP is a nonprogressive and intermittent inflammatory response to the exposure of an antigen. There is spontaneous improvement in the individual’s condition once the person is removed from the antigen. The onset of symptoms usually occurs 4 to 8 hours after exposure and there is a resolution of signs and symptoms within a month of onset.173,175,176 Subacute HP is more common than acute HP and it is caused by intermittent or continuous exposure to the antigen with symptoms presenting within weeks to months after exposure. Chronic HP is from persistent and recurrent exposure to low levels of the antigen, which can be divided into two cases. The first case is where the individual is suffering recurrent acute bouts of acute HP triggered by repeated exposures and the second case involves the individual who has low dosage exposures that progress in an insidious manner without any history of acute symptoms. The presentation and progression of this disease are dependent on the exposure dosage and duration as well as the individual’s genetic susceptibility.173
Host factors are important for the pathogenesis of this disease process, because many individuals are exposed to the same materials and do not develop HP. It is unclear exactly what are these host factors that allow for the induction of the inflammatory process, but hypersensitivity testing is generally negative for the antigen. In the acute phase, there is infiltration of macrophages, lymphocytes, and neutrophils into the alveolar spaces and small vessel vasculitis. In the subacute stage, the histological changes are consistent with three findings: interstitial inflammatory cell infiltrates, poorly formed non-necrotizing granuloma formation, and cellular bronchiolitis with destruction and intra-alveolar fibrosis.173 If the disease progresses into the chronic phase, the granulomas will disappear and be replaced by peribronchial fibrosis, smooth muscle hyperplasia, and fibrotic formation predominantly in the respiratory bronchioles.175
Four to eight hours after exposure, the individual will present with fever, chills, cough, dyspnea, and inspiratory crackles. In the acute phase there will be an increase in leukocytes and neutrophils in the bronchoalveolar fluid. In severe cases, hypoxemia may also be present and there will be infiltrates seen on the chest X-ray, which can be mistaken for a viral pneumonia.175,178 A low-level, long-term exposure is characteristic of the exposure history of individuals that progress to the chronic phase. There is extensive fibrosis and a honeycombing pattern on radiologic studies as well as severe hypoxia.178 Along with the classic symptoms presented in the acute and subacute phases, these individuals also present with malaise, weight loss, and significant fatigue and weakness. Hypoxemia is commonly present from diffuse infiltrates, hilar lymphadenopathy, and pleural effusion as can be seen on a chest X-ray.89,162,178 There will be a decrease in lung volumes on pulmonary function studies and a low DLCO, which is consistent with a restrictive lung pattern.
Part of the diagnosis of HP is the elimination of other granulomatous diseases such as sarcoidosis, viral pneumonia, aspergillus infection, and other collagen vascular diseases. The ratio of the CD4 (T-helper cells, which assist the B-cell response and interact with macrophages to strengthen the immune system) to the CD8 count (T-suppressor cells which assist in the body’s defense) may also assist in the diagnosis, in that elevation of CD8 cells and a decrease in the CD4/CD8 ratio are associated with an increase in cytokines that modulate inflammation and granuloma formation.175 An increase in the CD4/CD8 ratio is associated with lymphocytic infiltration.173,174
The first line of treatment is the prevention of HP with removal of the antigen when possible or the use of individual protective equipment to minimize exposure to harmful materials. With the presentation of HP, exposure to the antigen should be eliminated. The patient is typically treated with a high dose of corticosteroids followed by a slow taper.
Prognosis is dependent on the responsiveness to corticosteroids in the acute and subacute phases. If the inflammatory process persists, even despite aggressive treatment, the prognosis is poor. The progression of the fibroblastic activity destroys the small airways and alveoli and leads to refractory hypoxemia and progressive respiratory failure.174 Prognosis is also associated with the presence of pulmonary hypertension with higher mortality rates with up to 80% mortality at 5 years of onset of symptoms.162
Occupational Diseases (ICD-9-CM Codes: 502, 503)
There is a subset of pulmonary interstitial disorders that result from the inhalation of inorganic dusts (pneumoconioses), organic particles (hypersensitivity pneumoconioses), and industrial gases, fumes, and smoke. These occupational lung diseases are associated with a chronic inflammatory process that leads to scarring and pulmonary fibrosis.179
Pneumoconioses involve the permanent deposition of inorganic material (coal, asbestos, silica, beryllium, etc) within the pulmonary system. Pneumoconiosis is also referred to as allergic alveolitis and is considered an immunologically mediated disease.173 These diseases are typically associated with occupations such as miners, pipe fitters, welders, cutters, stonecutters, and fabric mill and quarry workers.180,181 This section focuses on asbestos and silica exposure, but the different exposures tend to be clinically similar.
Recently, attention has been focused on reports of pulmonary disease related to work in microwave popcorn production. A cluster of people who work in the butter-flavoring room or the packing room have been diagnosed with fixed OB (fixed meaning unresponsive to bronchodilators). They have a history of inhalation exposure to diacetyl, which is a volatile butter-flavoring chemical. The reader is referred to the section on OB for a more detailed clinical presentation.56
Asbestos is a generic term for a group of naturally occurring complex crystalline mineral fibers that are ideal for a variety of construction purposes because of their tensile strength.181 Small particles from these fibers can deposit in the distal airways depending upon the size of these particles and pressure gradient within the lung.182 This deposition is associated with a high incidence of pulmonary carcinomas. Mesothelioma is a highly aggressive tumor associated with asbestos exposure. It has a high mortality rate due to the late diagnosis and resistance to treatment.183 Other fibers have a high concentration of iron and are associated with scarring and pulmonary fibrosis. Finally, the most common fiber type, which is used commercially for building material, is frequently associated with pleural disease.182,183
The clinical presentation of asbestosis depends upon the type, length, and concentration of the fibers inhaled and the immunological response. For example, crocidolite asbestos fibers are associated with more fibrogenic changes and mesotheliomas due to greater cytotoxicity, cell proliferation and inflammation, and the production of reactive oxygen molecules.184–186 In many cases, the pulmonary fibrosis and malignancies are associated with a history of 20-plus years of exposure.187 The incidence of pleural disease and fibrosis varies, but it is reported that the parenchyma is involved in up to 82% of cases. Isolated fibrosis is diagnosed in approximately 15% cases. Pleural disease is found in 48% and presents as either effusion, fibrotic plaque, or malignant mesothelioma.187,188
Silica is a group of naturally occurring minerals with quartz being the most common form used in construction. Silicosis is associated with progressive fibrotic pneumoconiosis and formation of nodules (see Fig. 7-22).180Although the environmental control of exposure has improved over the last two decades, the incidence of lung disease associated with silica is still on the rise. The increase in the documented onset of pneumoconiosis is due to the long latency period between the time of exposure and the actual onset of disease. The latency between exposure and disease can extend for 20 to 40 years, and it has been estimated that the incidence will not peak until 2030. The risk factors involved in the development of pulmonary disease include the duration and intensity of the exposure to the inorganic material and to the size and water solubility of the particles.189,190

FIGURE 7-22 Silicosis. This disease is associated with deposits of foreign material that causes inflammation, progressive scarring, and destruction of the lung. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
Regardless of the fine particles that are inhaled, these inorganic particles trigger the accumulation of alveolar macrophages and inflammation that extends into the terminal respiratory bronchioles and adjacent alveolar interstitium. The release and activation of macrophages cause the release of inflammatory mediators such as cytokines, tumor necrosis, tumor growth factor, platelet-derived growth factor, and fibroblasts. This histological response amplifies cellular injury, fibroblastic proliferations, and collagen deposition.191 Toxic oxygen and nitrogen species are released and further lead to cell mutation and death. Neutrophils and lymphocytes are also released and this immune response can be characterized by the loss of alveolar type I and type II cells, an increase in WBCs, fibroblast proliferation, and collagen accumulation. Damage to type I cells is regarded as an early sign of fibrosis followed by hyperplasia and hypertrophy of type II epithelial cells.180 Type II alveolar cells, which produce surfactant, play a role in the repair of the injured alveolar epithelium and determine the extent of the lung disease. This cascade of events leads to the alteration of biological function of lipids, damage and mutation of cellular DNA and RNA resulting in cellular dysfunction, fibrosis, and malignant transformation.182,189
Pneumoconiosis is associated with radical oxygen and nitrogen species, which are linked to cellular mutation, RNA, and DNA alterations and apoptosis. The damage to the DNA leads to neoplastic transformation and apoptosis. Apoptosis, the disposal of cellular debris, is the major pathway responsible for alveolar type II cell hyperplasia in acute lung. Normally, there exists a balance between apoptosis and cell proliferation. When proliferation exceeds apoptosis, it leads to the triggering of the inflammatory process or malignant cell formation. If the apoptosis rate exceeds proliferation, there is epithelial dysfunction and lung injury. Apoptosis and tumor growth are stimulated by the presence of the reactive oxygen molecules.182,188
Pneumoconiosis is classified in two forms: acute and classic. Acute pneumoconiosis is associated with a high concentration exposure leading to severe alveolitis. Neutrophils and eosinophils infiltrate the alveolar spaces and cause small vessel vasculitis.173 Acute alveolitis clinically presents with progressive respiratory failure, bilateral pulmonary consolidation, and a high incidence of deaths related to respiratory failure or right-sided heart failure.190The classic form has a more insidious clinical presentation with pulmonary nodules, collagen deposition, and macrophage infiltrates. The classic form can be divided into simple and complicated subtypes. In the simple form, the radiological findings show multiple nodules with well-defined borders. The nodules are less than 10 mm in diameter. The complicated form is associated with massive fibrosis, nodules are greater than 1 cm in diameter, and have irregular margins and calcifications.189,190
Regardless of the various causes of pneumoconioses, the patient presents with a similar clinical picture of a slowly progressive respiratory and functional decline, with exertional dyspnea, a dry nonproductive cough, and cyanosis. The disease can progress regardless of any further exposure. There is usually the presence of expiratory wheezing, bilateral inspiratory crackles in the lower lung fields, and clubbing of the nail beds. In the advanced stages of the disease, the patient may develop cor pulmonale typically due to pulmonary hypertension.180
The radiological findings by chest X-ray reveal a ground-glass appearance, and as the disease progresses, honeycombing will develop throughout the lungs. With asbestosis, the abnormality is first documented in the lower lung fields, but with silicosis most of the pathology is first seen in the upper lobes and posterior lung fields.189,190 Plaque formation may occur in the parietal or visceral pleura. Pleural disease is the hallmark of asbestosis and is used to help differentiate asbestosis from other fibrotic pathologies.187 There may also be signs of pleura-based masses that may resemble cancerous tumors.180,181 Occasionally, there will be calcification of the hilar nodes with silicosis.191
In the simple form of pneumoconiosis, typically, lung function is preserved, but in the complicated form, there is a restrictive pattern with a reduction in lung volumes, especially VC and TLC; a decrease in DLCO; and a decrease in pulmonary compliance with a normal FEV1/FVC ratio. The earliest physiological abnormalities include a reduction in VC, DLCO, and desaturation with exertion. If FEV1 is <75% of the age- and gender-matched predictive value, it is usually associated with a history of smoking. Occasionally, in the presence of silicosis, there will initially be a normal PFT result that can progress into an obstructive airflow pattern or a milder restrictive pattern.181,191
The best defense against pneumoconioses is prevention. The exposure to inorganic dust should be avoided by using a proper respiratory filter device if the exposure risk is high and the ventilation in the work area is poor. Prevention should also include proper protective clothing and employee education. Employees should be encouraged to undergo annual physical check ups that include chest X-rays and spirometry. Once the diagnosis has been made, medical management should include the use of corticosteroids to minimize the inflammatory response. Monitoring the progression of the disease should be done through radiological studies, PFTs, and exercise testing. With the progression of the disease, medical care may include the use of supplemental oxygen and prostacyclin drugs for right-sided heart failure due to pulmonary hypertension; chemotherapy, radiation and surgical resection for cancer; and ventilatory support for respiratory failure. In the presence of isolated pulmonary fibrosis, lung transplantation should be considered on a case-by-case basis.181,191
With the insidious onset of the clinical signs and symptoms and the long latency between exposure and pathology, many clients are diagnosed with advanced disease and therefore, prognosis is poor. Twenty percent of the deaths from asbestos exposure are due to pulmonary fibrosis. Deaths related to cancer account for 39% and malignant mesothelioma accounts for 9% of the deaths associated with asbestos exposure. Compared to age-matched smokers, clients with a history of smoking and asbestos exposure have a 4.5-fold increased risk of developing bronchogenic carcinoma.181
IMMUNOLOGICAL DISEASES
This is a group of diseases that are suspected to have been mediated by an immune response, which activates an inflammatory process. RA is a systemic disease that is characterized by persistent inflammation of the synovial joints with hyper-plasia of the synovium resulting in destruction of the joint.192 RA can be associated with pleural disease, OB, chronic pulmonary infections, and interstitial pulmonary fibrosis.23,192,193 The etiology of systemic lupus erythematosus (SLE) is unknown, but it has been suggested that there is a genetic, environmental, and hormonal influence that leads to a variety of pulmonary diseases and dysfunctions. Besides the pulmonary diseases mentioned earlier, lupus is also associated with pulmonary hypertension, alveolar hemorrhage, and respiratory muscle weakness.194,195 Scleroderma; calcinosis, Raynaud phenomenon, esophageal dysfunction, sclerodactyly telangiectasia (CREST); and mixed connective tissue disease (MCTD) are three diseases that are associated with dysfunction of collagen tissue throughout the body.196–198 Pulmonary involvement typically presents with pulmonary fibrosis and/or hypertension.23,196 Finally, Sjögren syndrome is a chronic autoimmune inflammatory disease that is usually associated with another immune disease like RA.199 This disease is characterized by dryness of the airways with mucus retention and pulmonary fibrosis.200
This group of diseases has similar pulmonary manifestations based on the presenting pulmonary disease. Many patients will present with a chronic cough that worsens with exertion, dyspnea, Raynaud syndrome, pulmonary crackles, fever, and signs of right-sided heart failure.192,193 PFTs typically document a restrictive breathing pattern with low DLCO, hypoxia, and cyanosis.192,194,201,202
Treatment and prognosis are also dependent on the clinical signs and symptoms, aggressiveness of the disease, and the specific underlying immunological disease. Functional impairment and dysfunction are primarily linked to hypoxia due to fibrosis, right-sided heart failure from pulmonary hypertension, and pulmonary infections with mortality rates exceeding 50%, 2 years after presentation of pulmonary involvement.145,194,201,203 Please refer to Chapter 13 for more specific details about these diseases, their clinical presentation, and management.
Drug Toxicity (ICD-9-CM Code: 518.82)
Many of the medications that are prescribed to treat systemic autoimmune diseases and oncological disorders can lead to pulmonary toxicity. The most common pulmonary complications include bronchospasm, OB, interstitial pneumonitis, pulmonary edema, and lupus.111
Pulmonary interstitial pneumonitis and fibrosis are associated with methotrexate, gold, and NSAIDS.204 The patient usually presents with DOE and a nonproductive cough. The symptoms may have an acute onset with a clinical presentation that is similar to HP. However, symptoms may also have a gradual presentation like pulmonary fibrosis.23 Acute reaction is associated with fever and cough. The clinician may also observe tachypnea and cyanosis. The chronic reaction is associated with inspiratory crackles, hypoxemia, and infiltrates can be seen on X-ray. PFTs illustrate a restrictive pattern in the chronic process with a decline in FVC, IRC, and TLC.204
Pulmonary edema is associated with the overuse of salicylates, narcotics, and chemotherapeutic agents. This edema is noncardiac in origin and patients usually present with chest tightness, coughing, wheezing, dyspnea, and respiratory depression and distress. The symptoms may present 24 to 48 hours after drug administration.204
A patient can present with pulmonary hemorrhage if the patient is taking anticoagulants or has been given thrombolytic therapy. Hemorrhage may also be a complication of cocaine abuse. The patient may present with crackles, dyspnea, a bloody productive cough, and respiratory distress.23
The presence of bronchospasm is common in the presence of NSAIDS, salicylates, and β-blocker administration. It is theorized that the drug causes an imbalance between bronchodilation and bronchoconstriction by inhibiting the production of prostaglandin, which is a potent bronchodilator. The patient may present clinically with wheezing, dyspnea, and a cough.23,204
With the use of gold and penicillamine for RA, the patient can potentially develop OB, which is characterized by the inflammation of the small airways. If the drug is not discontinued, OB will progress to destruction of the bronchioles and an obstructive breathing pattern with dyspnea at rest and on exertion, and hyperinflation of the lungs will be seen.23,204
SLE may be induced from the side effects of penicillamine, gold, procainamide, and phenylbutazone. The most common clinical feature is pleural effusion.23,204 The degree of symptoms and signs will depend on size of the effusion. With a small effusion, the patient may be asymptomatic, whereas a large effusion may cause dyspnea, tachypnea, oxygen desaturation, and pain on inspiration.
The primary treatment in any of these pulmonary complications, first and foremost, is to monitor the patient for signs and symptoms of the above diseases, once the drug has been administered, and to educate the patient on self-examination for signs and symptoms. It is important to take a thorough history including examining the patient’s medications when the patient presents with pulmonary symptoms with no history of disease. Once the signs and symptoms have been evaluated and a diagnosis of drug-induced pulmonary dysfunction has been made, the medications should be discontinued. Further medication intervention should be provided to support the respiratory system and to treat the clinical presentation. This intervention may include bronchodilators, corticosteroids, and supplemental oxygen.
PULMONARY ONCOLOGY (ICD-9-CM Code: 239.9)
Lung cancer is the leading cause of cancer-related deaths in the United States, only surpassed by cardiac disease.205 The rate of diagnosis and mortality has not declined over the past decade despite advances in diagnostic procedures and treatment. In 2007, 213,380 new cases of lung cancer were diagnosed and accounted for 160,390 deaths.206
Lung cancer has also been linked to occupational exposures of asbestos, arsenic, beryllium, cadmium, radon, and silica, just to name a few; but 80% to 90% of all lung cancer cases can be attributed to smoking.207,208 A genetic propensity has been identified in 71% of the cases of patients diagnosed with lung cancer before the age of 50 years. This is compounded by a history of smoking. In patients who are older than 70 years, smoking alone has been linked to 72% of these cancer cases.209 There has been a decline in lung cancer in men, which is associated with a decline in smoking, but smoking is on the rise in women, particularly young women, with lung cancer diagnosis also on the rise.210 Smoking accounts for more cases of cancer in men, with a higher incidence in black men, but the rise in cancer in women is associated not only with the increase in smoking behavior, but also with a family history, a genetic link, of lung cancer that is a dominant factor in incidence and deaths.209
Smoking behavior is an important information to obtain during the interview process. It is vital to determine the number of cigarettes smoked per day, years of smoking, frequency of breaths per cigarette, and the depth of inhalation. All these factors contribute to risk of developing lung cancer.211
There have been several risk factors linked to lung caner. Passive or secondhand smoke exposure needs to be investigated during the interview because patients who do not smoke themselves, but have a spouse who smokes, have a 20% higher death rate from lung cancer than those who live with nonsmokers.207 Approximately 12,200 people die annually from lung cancer that is not linked directly to a personal history of smoking. Of those cases, as many as 8,400 deaths are attributed to passive exposure to tobacco smoke.212 Besides tobacco smoke, radiation exposure to the chest, asbestos, radon, chromium, arsenic, air, and air pollutants have also been identified as risk factors.213,214
There are several components in tobacco that are carcinogenic, such as polyaromatic hydrocarbons, and N-nitroso compounds from the nicotine and nicotine-like tobacco compounds. There are reactive metabolites from the smoke that bind to the DNA and damage the cell’s genetic composition. As the cell repairs itself, the mutation is incorporated into the DNA sequence. If this process occurs in cells that naturally undergo mitosis frequently, like the epithelium of the bronchi, the mutation is replicated rapidly.209 The carcinogenetic process is a complex series of events driven by the accumulation of DNA changes. These DNA changes or mutations occur in three stages: initiation, promotion, and progression. The initiation is the phase where there is an actual genetic change due to exposure causing injury. This is followed by the promotion phase in which there may be further mutations and reproduction of the changes. Finally, the progression of the mutations determines the aggressiveness of the tumor growth and determines the characteristics of malignancy.209
Lung carcinomas are divided into small-cell and non–small-cell cancers (see Figs. 7-23 and 7-24). Small-cell carcinoma is diagnosed if the tumor cells are smaller than the diameter of the mature lymphocytes.215 Small-cell lung cancer accounts for 15% of all new diagnoses and 25% of lung cancer deaths annually.210 Ninety-five percent of small-cell lung cancer cases are linked to smoking, and 70% of patients with small-cell cancers are metastatic at the time of diagnosis and are generally considered inoperable.216 There are two distinct types of small-cell lung cancer: small-cell carcinoma or oat-cells cancer and combined small cell. Staging is classified as limited, which means tumors are involving one lung, tissue between lung and nearby lymph nodes; extensive disease where the cancer has spread outside the lungs; recurrent disease of the thorax or metastatic lesions to the bone and 15% will have lesions within the brain. Frequently, small-cell carcinomas are classified as limited or extensive.214

FIGURE 7-23 Lung cancer. On this chest X-ray, notice the irregular, mass located in the left upper lobe (LUL) that has been diagnosed as an adenocarcinoma. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)

FIGURE 7-24 Metastatic cancer. This gross specimen clearly illustrated multiple tumor formation throughout this lung. (Courtesy of Dana Gryzbicki, MD, University of Pittsburgh, PA.)
Non–small-cell lung cancer is the leading cause of cancer deaths worldwide. These carcinomas include squamous cell, adenocarcinoma, and large cell cancer. Non–small-cell cancer comprises approximately 80% to 85% of the newly diagnosed cases with primary lung cancer annually.217 The high death rate is due to advanced stage of non–small-cell cancer when the diagnosis is made. Approximately 40% of patients with newly diagnosed non–small-cell cancer have a local, advanced, and inoperable cancer.218,219 Non–small-cell lung cancer encompasses squamous cell, large cell, or adrenocarcinoma, which is the most common of non–small-cell lung cancer.214
Non–small-cell cancers are staged based on the tumor node malignancy (TNM) system (see Tables 7-9 and 7-10).220,221 T indicates site and size of tumor, N is related to lymph node involvement, and M indicates the presence or absence of malignancy.222 Staging is the evaluation of the tumor for classification, which is used for prognosis, and medical or surgical intervention. This method to stage the cancer is still evolving particularly with the use of computer tomography and other pathological procedures.223
TABLE 7-9 TNM Classification for Cancer
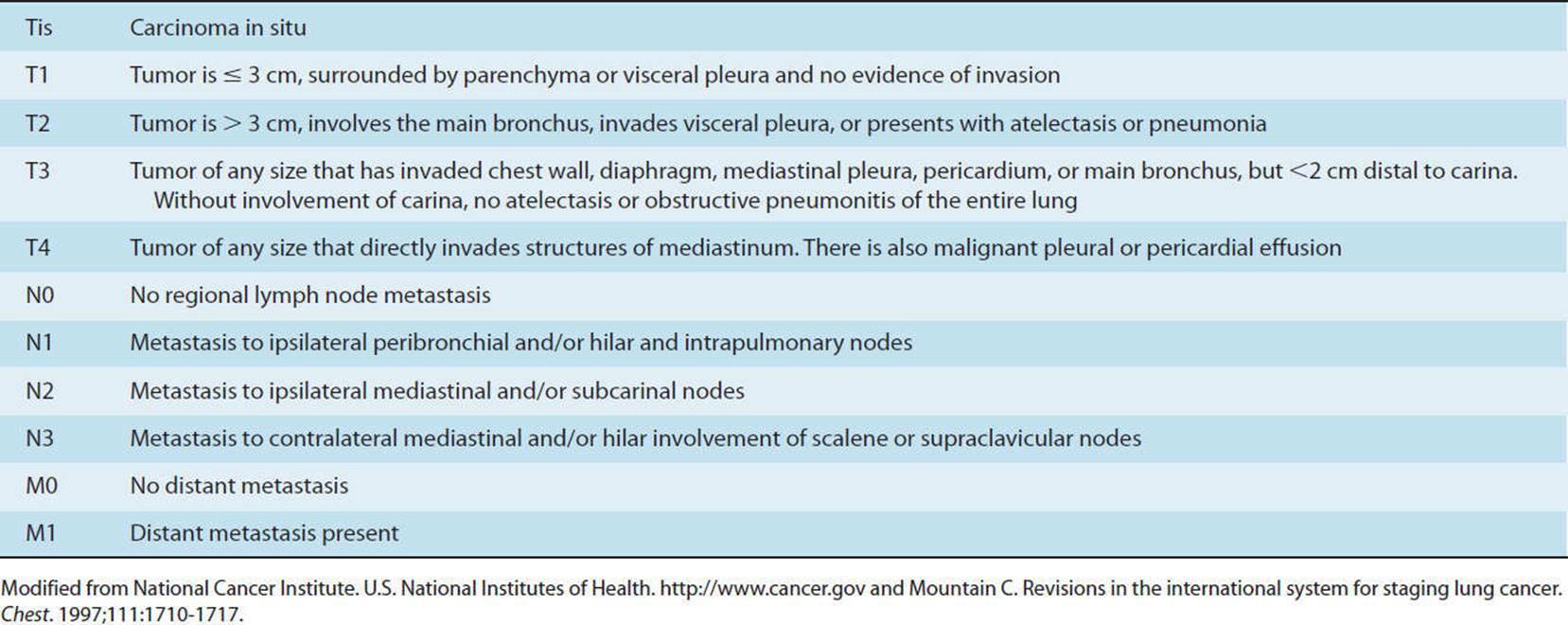
TABLE 7-10 Revised Stage Grouping
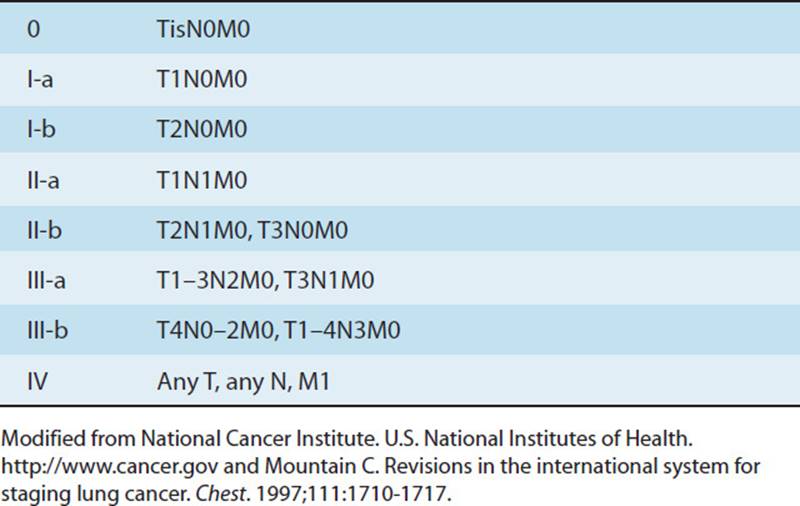
Approximately 5% of patients with lung cancer are asymptomatic at the time of diagnosis with the finding occurring during some medical screening for another medical procedure. The majority of patients will present with symptoms related to the primary impairment of lung function or symptoms related to dysfunction of distant sites due to metastases. The most common symptoms related to pulmonary involvement include dyspnea, persistent nonproductive cough, and hemoptysis. Some patients will complain of a dull aching chest pain.210,214 If the lesion obstructs a main airway, then the patient may present with dyspnea, postobstructive atelectasis, pneumonia, wheezing, or stridor. The patient may complain of pleuritic chest pain if there is involvement of the chest wall or pleura. The patient may present with signs or symptoms that are related to tissue dysfunction from metastasis such as headaches or seizures with CNS metastasis or jaundice with liver metastatic cancer. Finally, the performance of a thorough interview may reveal unexplained weight loss, night sweats, a decrease in activity, fatigue, anorexia, and pain that cannot be attributed to musculoskeletal or neuromuscular dysfunction.210,214
An important part of the diagnostic process is determination of the tumor type. There are certain characteristics that are associated with benign and malignant tumor cells. The presence of fat or a calcification pattern on CT scan and stability of tumor growth over a 2-year period are characteristics of a benign tumor. The border of a benign tumor is typically smooth and well-defined. Benign tumors are also more likely to occur in patients who are younger than 35 years with no smoking history and no occupational exposure to carcinogenic substances.220 Common indicators of malignancy include spiculated or non–smooth edges of the tumor. There is an absence of a calcification pattern and the tumor typically doubles in size within 400 days. The rate of tumor growth is a poorer prognostic indicator than the size of the tumor at the time of diagnosis.224 Tumors that are greater than 3 cm have a greater tendency of being malignant. Finally, the patients diagnosed with a malignant tumor usually have a history of smoking or occupational exposure and are around 65 years of age.220
There are various tests to assist in the diagnosis and staging of lung cancer. A chest X-ray is not sensitive enough to detect small nodules until they are greater than 1 cm in diameter.220 CT or MRI can be used to detect calcification and fat composition of the tumor that may be used for the diagnosis of malignant versus benign tumor.208 Bronchoscopy is the procedure of choice to obtain a biopsy when the lesion is centrally located.220 Transthoracic needle biopsy through the chest wall, or needle biopsy, can be obtained via bronchoscopy, or video-assisted thoracotomy, which is the most effective procedure for evaluation of peripheral parenchymal lung tissue.208,220 The thoracic surgeon may also choose to perform endoscopic ultrasonography with fine-needle aspiration to obtain enough tissue for both diagnosis and staging.220 If a pleural effusion is present, a thoracentesis can be performed to examine the cellular makeup of a pleural effusion, which is also helpful for diagnostic purposes.220
Lung cancer can be primary, with the lung as the initial site of the carcinoma, or secondary, meaning the cancer has spread or metastasized from another site other than the lungs. Metastasis requires a complex series of events to occur including cell growth, vascularization of the tumor, and transportation of the cancerous cells to other systems. The malignant cells must adhere to a distant site and there must be a presence of various cytokines and other tumor and host factors to promote the survival and growth of these new cells in distant sites.225
The most common sites of metastasis from the lungs are the brain (10%), bone (7%), liver (5%), and adrenal gland (3%).216,222 Metastasis from malignant lung cancer to the brain can present with signs and symptoms of headaches, hemiplegia, seizures, and behavioral changes. Metastasis to the skeletal system can present with complaints of dull, deep pain, muscle weakness, and pathological fractures. If the cancer invades the spinal cord, the patient may become paraplegic and have bowel and bladder dysfunction.220,225
Metastatic cancer to the lungs is typically from the primary sites: colorectum, breast, kidney, thyroid, or skin. The patients are usually asymptomatic until the advanced stages. If the cancer is adrenocarcinoma, non–small-cell cancer, with lymphatic involvement leading to breast, stomach, colon, prostate, or pancreas cancers, the patient usually presents with progressive dyspnea.225
The options for treatment have expanded significantly in the past decade and aggressive research continues to develop new interventions. Treatment decisions are dependent on the patient’s goals, staging of the tumor and extent of the disease, adverse effects of the treatment, respiratory function, operative risk, and the size of tumor growth.217,225,226
Surgical intervention is dependent on many factors including preoperative and predicted postoperative pulmonary function. Postoperative pulmonary function (PPO) can be quantified by assuming that each segment of the lung contributes to 5.2% of total lung function. Prior to surgical intervention, the surgeon must determine the effects of the resection on pulmonary function. Each segment can result in approximately a 5% decline in preoperative FEV1. Postoperative pulmonary complications can be expected if there is a pre-operative decrease in FVC and FEV1, presence of hypercapnia, and a low ![]() O2 capacity. If the FEV1 is less than 80% of the predicted value, a postoperative predicted value for FEV1 multiplied by the DLCO should be calculated. If this calculation is less than 30%, the patient would be considered a nonoperative candidate.227 Also, the patient would be nonoperative if an exercise test revealed a
O2 capacity. If the FEV1 is less than 80% of the predicted value, a postoperative predicted value for FEV1 multiplied by the DLCO should be calculated. If this calculation is less than 30%, the patient would be considered a nonoperative candidate.227 Also, the patient would be nonoperative if an exercise test revealed a ![]() O2 of less than 10 mL/kg/min. Surgery is the treatment of choice for non–small-cell cancer stages 0 through IIIA. Surgical intervention is commonly offered in conjunction with chemotherapy and/or radiation.214 The preoperative assessment is more complex when the patient has an underlying airway obstructive disease like emphysema. In cases where the patient has emphysema, the surgical resection may actually improve pulmonary function because the resection may decompress viable tissue to assist in ventilation and diffusion.218
O2 of less than 10 mL/kg/min. Surgery is the treatment of choice for non–small-cell cancer stages 0 through IIIA. Surgical intervention is commonly offered in conjunction with chemotherapy and/or radiation.214 The preoperative assessment is more complex when the patient has an underlying airway obstructive disease like emphysema. In cases where the patient has emphysema, the surgical resection may actually improve pulmonary function because the resection may decompress viable tissue to assist in ventilation and diffusion.218
Besides surgical intervention, the other leading treatment intervention includes the use of chemotherapy or radiation therapy (XRT) for the surgical and the nonsurgical candidates. Small-cell lung cancer has traditionally been treated with a combination of at least three chemotherapy drugs. The most common agents include cyclophosphamide, vincristine, cisplatin, etoposide, doxorubicin, methotrexate, and lomustine. Etoposide plus cisplatin in conjunction with XRT has shown to be an effective intervention for the limited stage of small-cell cancer.210 Seventy percent to ninety percent of the patients with small-cell cancer will initially respond to chemotherapy but more suffer recurrence and death within 2 years.210 The median survival for limited and extensive small-cell cancer is 14 to 16 months and 6 to 8 months, respectively, after intervention.23,216
Intervention for non–small-cell lung cancer depends upon the stage of the disease. Besides the surgical intervention, XRT is recommended in most cases. There are multiple clinical chemotherapy trials that are being studied in non–small-cell cancers of various stages. With the advances in treatment, there has been an increase in survival rates with a mean survival of 36 months and the 5-year survival of 29% for stage III cancer.226 Platinum-based chemotherapy followed by docetaxel appears to be the chemotherapy of choice but docetaxel is associated with high incidence of central nervous system or pulmonary toxicity.226 Carboplatin and paclitaxel along with traditional radiation have been effective in the management of locally advanced tumors.228 The combination of cisplatin and radiation improves survival by improving the local control of tumor growth.219
Survival depends on the staging of the tumor at the time of diagnosis. It is generally viewed that stages I and II tumors are surgically resectable and carry a higher survival rate than stages III and IV tumors, which are not resectable. If the tumor involves the mediastinal nodes, the 5-year survival rate is only 10%, whereas it is 50% with nonmediastinal involvement in stage I or II.218 Appropriately there is a 25% 5-year survival rate for stage III lung cancers.210,214,226 The mortality rate is higher in smokers and increases as the number of cigarettes smoked per day increases. There is also a higher mortality rate in male over female smokers.207
MECHANICAL PULMONARY DISORDERS (ICD-9-CM CODE: 518.83)
This group of extrapulmonary disorders can impair the pulmonary system since these disorders can lead to hypoxemia and pulmonary hypertension. They can also lead to a restrictive breathing pattern that may eventually lead to functional impairments and ultimately, high levels of morbidity and mortality.
Obstructive Sleep Apnea Syndrome
Obstructive sleep apnea syndrome (OSAS) is defined as recurrent episodes of apnea or the temporary cessation of ventilation during sleep. The obstruction is commonly due to occlusion of the upper airway. Obstructive sleep apnea is commonly associated with obesity, nasal obstruction, facial bony abnormalities such as retrognathia or micrognathia, hypertrophy of the uvula, and enlargement of the adenoids/tonsils. This syndrome affects approximately 4% of adults and 9% of children but is on the rise due to the elevating incidence in obesity in both groups in the United States.228,229 There is also an increased risk of developing OSAS in patients with a history of smoking.23,230
The hallmark signs of OSAS are snoring and daytime somnolence. Sleep apnea is associated with fragmented sleep, repeated arousal, intermittent hypoxemia, hypercapnia, and nocturnal hypertension. Apneic periods occurring more than 30 times/min is classified as severe sleep apnea and is associated with desaturation and a decrease in FEV1/VC ratio and a forced expiratory flow at 25% and 75% of vital capacity FEF of 25% to 75%.230A long-term consequence of sleep apnea includes daytime or chronic pulmonary hypertension in 42% of OSAS cases, which contributes to the increase in cardiovascular and cerebral morbidity.228,231
In children, sleep apnea is associated with sinus disease, which may be related to elevated airway resistance due to an inflamed upper airway. This may be caused by the higher incidence of bronchial hyperreactivity in children. Sleep apnea may also trigger nocturnal asthmatic exacerbations. Obesity in children increases the risk of sleep apnea threefold.124
Pregnancy
Pregnancy is associated with several temporary cardiopulmonary changes that support the fetal development. There is a 50% increase in cardiac output and oxygen consumption to support the increased work of breathing by the end of the gestational period. There is a drop in pulmonary vascular resistance, which is likely due to dilation and expansion of the pulmonary arterioles and capillaries.232
As the fetus grows, there are chest wall changes that occur early and progress throughout the pregnancy. There is an increase in the transverse diameter and in the circumference of the mother’s chest. Although the thorax expands, there is a decrease in chest wall compliance, which accounts for the increased work of breathing. The level of the diaphragm raises approximately 4 cm and diaphragm excursion increases. The elevated level of progesterone, which acts as a respiratory stimulant, leads to hyperventilation and chronic respiratory alkalosis.232
Clinically, dyspnea is the most common respiratory complaint during pregnancy. Approximately 50% of pregnant women complain about dyspnea at 20 weeks of gestation. By 31 weeks, this complaint rate increases to 75%. Women also commonly report fatigue and lower extremity edema. Upon auscultation, there is a decrease in breath sounds in the lower lung fields that is consistent with bibasilar atelectasis. Lung volumes are also altered during the pregnancy. Tidal volume increases as much as 40%, which accounts for the increased minute ventilation. VC remains unchanged, but inspiratory capacity increases slightly, which is consistent with the increase in diaphragmatic excursion. Expiratory reserve volume decreases by a mean of 15%, RV decreases by 20%, and functional reserve capacity decreases by 20% at the end of gestation. Finally, FEV1 remains stable. When the relationship between ![]() /Q. is examined there is a mismatch that can be three times higher than the 2% to 5% found in nonpregnant women. This mismatch is associated with an alteration in the distribution of ventilation.232
/Q. is examined there is a mismatch that can be three times higher than the 2% to 5% found in nonpregnant women. This mismatch is associated with an alteration in the distribution of ventilation.232
Obesity (ICD-9-CM Code: 278.0;Practice Pattern 6A)
Obesity has reached epidemic proportions in adults and children in the United States. Obesity is associated with several comorbidities including cardiovascular disease and diabetes. Severe obesity can also lead to pulmonary impairments, as it is associated with obstructive sleep apnea, obesity hypoventilation, atelectasis, and respiratory failure.233
In cases of mild-to-moderate obesity, it is uncommon to see deficits in VC and TLC, but severe or morbid obesity is likely to have decreased lung volumes. There is an inverse relationship between VC and torso circumference. It has been suggested that the distribution of fat is an important factor is determining pulmonary impairment. A decrease in FEV1 and TLC is associated with the weight distribution, primarily in the upper body. There is a decrease in chest wall compliance and diaphragmatic excursion. With substantial weight loss there is a normalization of lung volumes.234
Obesity hypoventilation syndrome (OHS) may develop in the most markedly obese individuals. This syndrome is associated with chronic hypercapnia. There are several factors that may contribute to the development of hypoventilation, including a decrease in chest wall compliance and an increase in work of breathing. It also appears that these individuals have a blunted respiratory drive with a diminished sensitivity to carbon dioxide. It has been suggested that OSAS may contribute to the development of obesity hypoventilation syndrome. The clinician should be alerted if the patient reports fragmented sleep and morning headaches that resolve when awakening.235
Diaphragmatic Hernia (ICD-9-CM Code: 553.3)
Congenital diaphragmatic hernia (CDH) may occur through a defect in various areas of the diaphragm. The congenital defect allows herniation of abdominal contents into the thoracic cavity. The most common sites for hernia ion are posterolateral, anterior, and adjacent to the esophagus. The posterolateral defect or herniation through the foramen of Bochdalek is the most common form of CDH. CDH is associated with a high mortality because approximately 40% of the infants with CDH also have other congenital or acquired defects that may be fatal. It also has a high morbidity, associated with the development of BPD, neurological deficits, and GERD. CDH occurs in approximately one infant per 2,000 births with 80% of the hernias involving the left diaphragm. The lungs of these infants are typically underdeveloped, which may be caused by the disruption of lung development during a critical growth period.236
Clinical presentation ranges from being asymptomatic to signs of pulmonary dysfunction due to entrapment of bowel in the thoracic cavity. In the newborn, the most common signs and symptoms are associated with respiratory insufficiency or failure occurring within minutes to hours after birth. Within the first 24 hours the infant’s respiratory function may deteriorate with the development of pulmonary hypertension, which can lead to a shunting of blood through the foramen ovale causing cyanosis. Upon examination, there will be absent breath sounds on the affected side. Chest X-ray may reveal the displacement of bowel or other visceral organs into the thoracic cavity.236
Medical management initially focuses on achieving hemo-dynamic stability. This is typically accomplished with high-frequency oscillation or intratracheal mechanical ventilation. The use of surfactant and liquid ventilation shows some promise in managing these patients during the acute critical phase. In critical cases, an extracorporeal membrane oxygenator may be utilized to rest the lungs. These invasive interventions are associated with some serious complications including intracranial bleeding and sepsis. Surgical repair of the diaphragm is warranted ideally when the infant is medically stable, which is usually after the first 24 hours.236
SUMMARY
This chapter has described most of the pulmonary disease processes that the physical therapist may encounter. After the physical therapist completes the examination, he or she should classify the patient into the most appropriate practice pattern. The selection of the practice pattern will be based on the documented findings of impairments, functional limitations, and disability. It is important that the physical therapist remembers that not every impairment leads to a limitation, but there are impairments underlying every functional limitation. This is also true for disabilities; there may be impairments or limitations, but these deficits do not necessarily lead to the perception of a disability by the patient. A patient may be classified into more than one practice pattern based on the severity of the pathology, or the patient may require reclassification into another practice pattern based on the progression of the disease or disorder or the effects of intervention. The process of selecting a practice pattern must be based on a priority system, which is the integration of the evaluation findings and the patient’s goals for rehabilitation. For most of the patients with a primary lung disease, the focus of treatment will center on the pulmonary impairment. Under these conditions, the physical therapist will use the cardiopulmonary Practice Patterns A, C, G, and H, which address impairments in ventilation, respiration, and a decline in aerobic capacity with or without airway clearance dysfunction. In the presence of respiratory failure, Pattern I or J may be the most appropriate selection. In some patients, the focus of physical therapy may be to address musculoskeletal problems, such as muscle weakness, osteoporosis, and arthralgias. If the primary physical therapy focus is nonpulmonary, the therapist may best select a musculoskeletal practice pattern that is included in A, B, C, or H.237 The goal of utilizing the Guide to Physical Therapist Practice is to complete a thorough evaluation, identify the disabilities and functional limitations, and select the most appropriate practice pattern based on the findings.
REFERENCES
1.Grossman R. Guidelines for the treatment of acute exacerbations of chronic bronchitis. Chest. 1997;112(suppl 6):310S-313S.
2.Celli BR, MacNee W. Standards for the diagnosis and treatment of patients with COPD: a summary of the ATS/ERS position paper. Eur Respir J. 2004;23:932-946.
3.Banning M. Chronic obstructive pulmonary disease: clinical signs and infections. Br J Nurs. 2006;15(16):874-880.
4.Vedantam R, Crawford A. The role of preoperative pulmonary function tests in patients with adolescent idiopathic scoliosis undergoing posterior spinal fusion. Spine. 1997;22(23):2731-2734.
5.Clark C, Cochrane L. Physical activity and asthma. Curr Opin Pulm Med. 1999;5:68-75.
6.Hartert T, Peebles RS. Epidemiology of asthma: the year in review. Curr Opin Pulm Med. 2000;6:4-9.
7.Weiss S. Environmental risk factors in childhood asthma. Clin Exp Allergy. 1998;28(suppl 5):29-34.
8.Wu F, Takaro TK. Childhood asthma and environmental interventions. Environ Health Prospect. 2007;115:971-975.
9.Warner SM, Knight DA. Airway modeling and remodeling in the pathogenesis or asthma. Curr Opin Allergy Clin Immunol. 2008;8(1):44-48.
10.Eggleston PA. The environment and asthma in US inner cities. Chest. 2007;132(suppl 5):782S-788S.
11.Sears M. Evolution of asthma through childhood. Clin Exp Allergy. 1998;28(suppl 5):82-89.
12.Cullinan P, Newman Taylor A. Aetiology of occupational asthma. Clin Exp Allergy. 1997;27(suppl 1):41-46.
13.Townshend J, Hails S, McKean M. Diagnosis of asthma in children. Br Med J. 2007;335(7612):198-202.
14.Holt PG, Sly PD. Prevention of allergic respiratory disease in infants: current aspects and future perspectives. Curr Opin Allergy Clin Immunol. 2007;7(6):547-555.
15.Story RE. Asthma and obesity in children. Curr Opin Pediatr. 2007;19(6):680-684.
16.Dow L. Asthma in older people. Clin Exp Allergy. 1998;28(suppl 5):195-202.
17.Walters EH, Soltani A, Reid DW, Ward C. Vascular remodeling in asthma. Curr Opin Allergy Clin Immunol. 2008;8(1):39-43.
18.Grimfeld A, Just J. Clinical characteristics of childhood asthma. Clin Exp Allergy. 1998;28(suppl 5):67-70.
19.Kim H, Tsai P, Oh C. The genetics of asthma. Curr Opin Pulm Med. 1998;4:46-48.
20.Hanania NA. Targeting airway inflammation in asthma: current and future therapies. Chest. 2008;133(4):989-998.
21.Cypcar D, Lemanske R. Asthma and exercise. Chest. 1994;15(2):351-365.
22.Beck K. Control of airway function during and after exercise in asthmatics. Med Sci Sports Exerc. 1999;31(suppl 1):S4-S11.
23.Fishman A. Pulmonary Diseases and Disorders: Companion Handbook. 2nd ed. New York: McGraw-Hill; 1994.
24.Ali J, Summer W, Levitzky M. Pulmonary Pathophysiology. New York: McGraw-Hill; 1999.
25.Kemp J. Comprehensive asthma management: guidelines for clinicians. J Asthma. 1998;35(8):601-620.
26.Reddy RC. Severe asthma: approach and management. Postgrad Med J. 2008;84(989):115-120.
27.Gelb AF, Zamel N, Kristnan A. Physiological similarities and differences between asthma and chronic obstructive pulmonary disease. Curr Opin Pulm Med. 2008;14(1):24-30.
28.Gurney J. Pathophysiology of obstructive airways disease. Radiol Clin North Am. 1998;36(1):15-27.
29.Rennard SI. Chronic obstructive pulmonary disease: linking outcomes and pathology of disease modification. Proc Am Thorac Soc. 2006;3:276-280.
30.MacNee W. Pathogenesis of chronic obstructive of pulmonary disease. Proc Am Thorac Soc. 2005;2:258-266.
31.O’Byrne P, Postma D. The many faces of airway inflammation. Am J Resp Crit Care Med. 1999;159:S41-S66.
32.Hensen PM, Vandivier W, Douglas IS. Cell death, remodeling and repair in chronic obstructive pulmonary disease? Proc Am Thorac Soc. 2006;3:713-717.
33.Churg A, Coslo M, Wright JL. Mechanism of cigarette smoke induced COPD: an insight from animal model. Am J Physiol Lung Cell Mol Physiol. 2008;294:L612-L631.
34.Cosio M, Guerassimov A. Chronic obstructive pulmonary disease: inflammation of small airways and lung parenchyma. Am J Resp Crit Care Med. 1999;160(5, pt 2):S21-S25.
35.Poole D, Sexton W, Farkas G, et al. Diaphragm structure and function in health and disease. Med Sci Sports Exerc. 1997;29(6):738-754.
36.Martinez F. Diagnosing chronic obstructive pulmonary disease. Postgrad Med. 1998;103(4):112-125.
37.Agusti A. Effects of chronic obstructive pulmonary disease: what we know and what we don’t know (but should). Proc Am Thorac Soc. 2007;4:522-525.
38.Martinez F. Diagnosing chronic obstructive pulmonary disease: the importance of differentiating asthma, emphysema, and chronic bronchitis. Postgrad Med. 1998;103(4):112-117, 121-122, 125.
39.Dasgupta A, Maurer J. Late stage emphysema: when medical therapy fails. Cleve Clin J Med. 1999;65(7):415-424.
40.Bennditt JO. Surgical options for patients with COPD: sorting out the choices. Respir Care. 2006;51(2):173-182.
41.Sandhaus RA. a-1 Antitrypsin deficiency. 6: new and emerging treatments for a-1 antitrypsin deficiency. Thorax. 2004;59:904-909.
42.Richmond RJ, Zellner KM. a-1 Antitrypsin deficiency. Dimens Crit Care Nurs. 2005;24(6):255-260.
43.Schwaiblmair M, Vogelmeier C. Alpha 1 antitrypsin: hope on the horizon for emphysema suffers? Drugs Aging. 1998;12(6):429-437.
44.Hogarth DK, Rachelefsky G. Screening and familial testing of patients for a-1 antitrypsin deficiency. Chest. 2008;133:981-988.
45.Stoller J. Clinical features and natural history of severe alpha 1 antitrypsin deficiency. Chest. 1997;111:123S-128S.
46.Luisetti M. Seersholm N. a-1 Antitrypsin deficiency. 1: epidemiology of α-1 antitrypsin deficiency. Thorax. 2004;59:164-169.
47.Mahadeva R, Lomas D. Genetics and respiratory disease. 2: alpha 1 antitrypsin deficiency, cirrhosis and emphysema. Thorax. 1998;53(6):501-505.
48.Boucher RC. Relationship of airway epithelial ion transport to chronic bronchitis. Proc Am Thorac Soc. 2004;1:66-70.
49.Heath J, Mongia R. Chronic bronchitis: primary care management. Am Fam Physician. 1998;57(10):2365-2372, 2376-2378.
50.Guddo F, Vignola AM, Saetta M, et al. Upregulation of basic fibroblast growth factor in smokers with chronic bronchitis. Eur Respir J. 2006;27:L957-L963.
51.Kim JS, Okamoto K, Rubin BK. Pulmonary function is negatively correlated with sputum inflammatory markers and cough clearability in subjects with cystic fibrosis but not those with chronic bronchitis. Chest. 2006;129:1148-1154.
52.Hayes D, Meyer KC. Acute exacerbations of chronic bronchitis in elderly patients: pathogenesis, diagnosis and management. Drugs Aging. 2007;24(7):555-572.
53.Saetta M. Airway inflammation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1999;160(5, pt 2):S17-S20.
54.Wilson R, Wilson C. Defining subsets of patients with chronic bronchitis. Chest. 1997;112(suppl 6):303S-309S.
55.Jones K, Robbins R. Alternative therapies for chronic bronchitis. Am J Med Sci. 1999;318(2):96-98.
56.Kanwal R. Bronchiolitis obliterans in workers exposed to flavoring chemicals. Curr Opin Pulm Med. 2008;14:141-146.
57.Moonnumakal SP, Fan LL. Bronchiolitis obliterans in children. Curr Opin Pediatr. 2008;20:272-278.
58.Kreiss K. Flavoring-related bronchiolitis obliterans. Curr Opin Allergy Clin Immunol. 2007;7:162-167.
59.Colby T. Bronchiolitis: pathologic considerations. Am J Clin Pathol. 1997;109:101-109.
60.Cordier JF. Challenges in pulmonary fibrosis: bronchiolocentric fibrosis. Thorax. 2007;62:638-649.
61.Chan A, Allen R. Bronchiolitis obliterans: an update. Curr Opin Pulm Med. 2004;10:133-141.
62.Boehler A, Kesten S, Weder W, et al. Bronchiolitis obliterans after lung transplantation. Chest. 1998;114(5):1412-1421.
63.Kelly K, Hertz M. Obliterative bronchitis. Clin Chest Med. 1997;18(2):319-333.
64.Johnson S. Lymphangioleiomyomatosis: clinical features, management and basic mechanisms. Thorax. 1999;54(3): 254-264.
65.Niku S, Stark P, Levin DL, Friedman PJ. Lymphangioleiomyomatosis: clinical, pathologic, and radiologic manifestations. J Thorac Imaging. 2005;20:98-102.
66.Sullivan E. Lymphangioleiomyomatosis: a review. Chest. 1998;114(6):1689-1703.
67.McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133:507-516.
68.Alalawi R, Whelan T, Bajwa RS, Hodges TN. Lung transplantation and interstitial lung disease. Curr Opin Pulm Med. 2005;11:461-466.
69.Rosenstein B. What is a cystic fibrosis diagnosis? Clin Chest Med. 1998;19(3):423-441.
70.Dinwiddie R. Pathogenesis of lung disease in cystic fibrosis. Respiration. 2000;67:3-8.
71.Rowe SM, Clancy JP. Advances in cystic fibrosis therapies. Curr Opin Pulm Med. 2006;18:604-613.
72.Elizur A, Cannon CL, Ferkol TW. Airway inflammation in cystic fibrosis. Chest. 2008;133:489-495.
73.Boyle MP. Update on maintaining bone health in cystic fibrosis. Curr Opin Pediatr Med. 2006;12:453-458.
74.Kalnins D, Durie PR, Pencharz P. Nutritional management of cystic fibrosis patients. Curr Opin Clin Nutr Metab Care. 2007;10: 348-354.
75.Parasa R, Maffulli N. Musculoskeletal involvement in cystic fibrosis. Bull Hosp Joint Dis. 1999;58(1):37-43.
76.Prasad S, Main E. Finding evidence to support airway clearance techniques in cystic fibrosis. Disabil Rehabil. 1998;20(6/7):235-246.
77.Ferkol T, Rosenfeld M, Milla CE. Cystic fibrosis pulmonary exacerbations. J Pediatr. 2006;148:259-264.
78.Rubin B. Emerging therapies for cystic fibrosis lung disease. Chest. 1999;115:1120-1126.
79.Ratjen F. New pulmonary therapies for cystic fibrosis. Curr Opin Pediatr. 2007;13:541-546.
80.Langenderfer B. Alternatives to percussion and postural drainage. J Cardiopulm Rehabil. 1998;18:283-289.
81.Rosenstein B, Cutting G. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr. 1998;132(4):589-595.
82.Stenbit A, Flume PA. Pulmonary complications in adult patients with cystic fibrosis. Am J Med Sci. 2008;355(1):55-59.
83.Ilowite J, Spiegler P, Chawla S. Bronchiectasis: new findings in the pathogenesis and treatment of this disease. Curr Opin Infect Dis. 2008;21:163-167.
84.Hansell D. Bronchiectasis. Radiol Clin North Am. 1998;36(1):107-125.
85.Mysliwiee V, Pina J. Bronchiectasis: the “other” obstructive lung disease. Postgrad Med. 1999;106(1):123-131.
86.Spencer DA. From hemp seed and porcupine quill to HRCT: advances in the diagnosis and epidemiology of bronchiectasis. Arch Dis Child. 2005;90:712-714.
87.Chang AB, Bilton D. Exacerbation in cystic fibrosis: 4. Non-cystic fibrosis bronchiectasis. Thorax. 2008;63:269-276.
88.Fushillo S, De Felice A, Balzano G. Mucosal inflammation in idiopathic brnochiectasis: cellular and molecular mechanisms. Eur Respir J. 2008;31:396-406.
89.Porcel JM, Light RW. Pleural effusions due to pulmonary embolism. Curr Opin Pulm Med. 2008;14:337-342.
90.Bounameaux H, Perrier A. Diagnosis of pulmonary embolism: in transition. Curr Opin Hematol. 2006;13:344-350.
91.Konstantinides SV. Acute pulmonary embolism revisited. Heart. 2008;94:795-802.
92.Philbrick JT, Shumate R, Siadaty MS, Becker DM. Air travel and venous thromboembolism: a systemic review. Soc Gen Intern Med. 2007;22:107-114.
93.Tia N, Atwal A, Hamilton G. Modern management of pulmonary embolism. Br J Surg. 1999;86:853-868.
94.Wood M, Spiro S. Pulmonary embolism: clinical features and management. Hosp Med. 2000;61(1):46-50.
95.Alexander P, Giangola G. Deep vein thrombosis and pulmonary embolism: diagnosis, prophylaxis, and treatment. Ann Vasc Surg. 1999;13(3):318-327.
96.Segal JB, Streiff MB, Hoffman LV, Thornton K, Bass EB. Management of venous thromboembolism: a systemic review for a practice guideline. Ann Intern Med. 2007;146:211-222.
97.McRae SJ, Ginsberg JS. Update in the diagnosis of deep vein thrombosis and pulmonary embolism. Curr Opin Anaesthesiol. 2006;19:44-51.
98.Perrot M, Granton J. Pulmonary hypertension after pulmonary embolism: an underrecognized condition. Can Med Assoc J. 2006;174:1706-1707.
99.Young T, Tang H, Aukes J, Hughes R. Vena cavel filters for the prevention of pulmonary embolism. Cochrane Database Syst Rev. 2007;4:1-18.
100.Archer SL, Michelakis ED. An evidence based approach to the management of pulmonary arterial hypertension. Curr Opin Cardiol. 2006;21:385-392.
101.Fox DJ, Khattar RS. Pulmonary arterial hypertension: classification, diagnosis, and contemporary management. Postgrad Med J. 2006;82:717-722.
102.Highland KB. Pulmonary arterial hypertension. Am J Med Sci. 2008;335(1):40-45.
103.Simonneau G, Galei N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol. 2004;43:5S-12S.
104.Desai SA, Channick RN. Exercise in patients with pulmonary arterial hypertension. J Cardiopulm Rehabil Prev. 2008;28:12-16.
105.Diller GP, Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation. 2007;115:1039-1050.
106.Rubin L. Current concepts: primary pulmonary hypertension. New Engl J Med. 1997;336(2):111-117.
107.Gaine S, Rubin L. Primary pulmonary hypertension. Lancet. 1998;353(9129):719-725.
108.Barst R. Recent advances in the treatment of pediatric pulmonary artery hypertension. Pediatr Clin North Am. 1999;46(2):331-345.
109.Auger W, Channick R, Kerr K, et al. Evaluation of patients with suspected chronic thromboembolic pulmonary hypertension. Semin Thorac Cardiovasc Surg. 1999;11(2):179-190.
110.Rabinovitch M. Pulmonary hypertension: pathophysiology as a basis for clinical decision making. J Heart Lung Transplant. 1999;18(11):1041-1053.
111.Minai OA, Budev MM. Diagnostic strategies for suspected pulmonary arterial hypertension: a primer for the internist. Cleve Clin J Med. 2007;74(10):736-747.
112.Traiger GL. Pulmonary arterial hypertension. Crit Care Nurs Q. 2007;30(1):20-41.
113.Gluecker T, Capasso P, Schnyder P, et al. Clinical and radiologic features of pulmonary edema. Radiographics. 1999;19:1507-1531.
114.Maniatis NA, Orfanos SE. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2008;14:22-30.
115.Ketai L, Godwin D. A new view of pulmonary edema and acute respiratory distress syndrome. J Thorac Imaging. 1998;13:147-171.
116.Clarke C. Acute mountain sickness: medical problems associated with acute and subacute exposure to hypobaric hypoxia. Postgrad Med J. 2006;82:748-753.
117.Zwischenberger J, Alpand S, Bidani A. Early complication: respiratory failure. Chest Surg Clin North Am. 1999;9(3):543-559.
118.Gropper MA, Wiener-Kronish J. The epithelium in acute lung injury/acute respiratory distress syndrome. Curr Opin Crit Care. 2008;14:11-15.
119.Sahn S, Heffner J. Spontaneous pneumothorax. New Engl J Med. 2000;342(12):868-875.
120.Schramel F, Postmus P, Vanderschueren R. Current aspects of spontaneous pneumothorax. Eur Resp J. 1997;10:1372-1379.
121.Currie GP, Alluri R, Christie GL, Legge JS. Pneumothorax: an update. Postgrad Med J. 2007; 83:461-465.
122.Beers SL, Abramo TJ. Pleural effusions. Pediatr Emerg Care. 2007;23(5):330-339.
123.Rolston D, Diaz-Guzman E, Budev MM. Accuracy of the physical examination in evaluating pleural effusion. Cleve Clin J Med. 2008;75(4):297-304.
124.Parfrey H, Chilvers E. Pleural disease: diagnosis and management. Practitioner. 1999;243:412-423.
125.Cameron RJ, Davies HRHR. Intra-pleural fibrinolytic therapy versus conservation management in the treatment of adult parapneumonic effusions and empyema. Cochrane Database Syst Rev. 2008;2:1-48.
126.Givan D, Eigen H. Common pleural effusions in children. Clin Chest Med. 1998;19(2):363-371.
127.Colice G, Rubins J. Practical management of pleural effusions: when and how should fluid accumulations be drained? Postgrad Med. 1999;105(7):67-78.
128.Anthony V, Mohammed K. Pathophysiology of pleural space infections. Semin Respir Infect. 1999;14(1):9-17.
129.Strange C, Sahn S. The definitions and epidemiology of pleural space infection. Semin Respir Infect. 1999;14(1):3-8.
130.Heffner J. Infection of the pleural space. Clin Chest Med. 1999;20(3):607-618.
131.Scott JAG, Brooks A, Peiris JSM, Holtzman D, Mullholland EK. Pneumonia research to reduce childhood mortality in the developing world. J Clin Invest. 2008;118(4):1291-1301.
132.Talwar A, Lee H, Fein A. Community acquired pneumonia: what is relevant and what is not? Curr Opin Pulm Med. 2007;13:177-185.
133.Hoarse Z, Lim S. Pneumonia: update on diagnosis and management. Br Med J. 2006;323:1077-1080.
134.McNabb B, Isakow W. Probiotics for the prevention of nosocomial pneumonia: current evidence and opinions. Curr Opin Pulm Med. 2008;14:168-175.
135.Kharana P, Litaker D. The dilemma of nosocomial pneumonia: what primary care physicians should know. Cleve Clin J Med. 2000;67(1):25-41.
136.Ewing S. Community-acquired pneumonia: definition, epidemiology, and outcome. Semin Respir Infect. 1999;14(2):94-102.
137.Croce M. Postoperative pneumonia. Am Surg. 2000;66:133-138.
138.Jackson WL, Shorr AF. Update in ventilator associated pneumonia. Curr Opin Anaesthesiol. 2006;19:117-121.
139.Feldman C. Pneumonia in the elderly. Clin Chest Med. 1999;3:563-574.
140.Medina-Walpole A, Katz P. Nursing home-acquired pneumonia. J Am Geriatr Soc. 1999;47:1005-1015.
141.Bratzler DW, Nsa W, Houck PM. Performance measures for pneumonia: are they valuable, and are process measures adequate? Curr Opin Infect Dis. 2007;20:182-189.
142.Tsai KS, Grayson MH. Pulmonary defense mechanisms against pneumonia and sepsis. Curr Opin Pulm Med. 2008;14:260-265.
143.Barry S, Lipman M, Johnson M, et al. Respiratory infections in immunocompromised patients. Curr Opin Pulm Med. 1999;5: 168-173.
144.Wallace J. HIV and the lung. Curr Opin Pulm Med. 1998;4:135-141.
145.Lee-Chiong T. Pulmonary aspiration. Compr Ther. 1997;23(6):371-377.
146.Soto GJ. Diagnostic strategies for nosocomial pneumonia. Curr Opin Pulm Med. 2007;13:186-191.
147.Boersma W. Assessment of severity of community-acquired pneumonia. Semin Respir Infect. 1999;14(2):103-114.
148.Campbell IA, Bah-Sow O. Pulmonary tuberculosis: diagnosis and treatment. Br Med J. 2006;332:1194-1197.
149.Furin JJ, Johnson JL. Recent advances in the diagnosis and management of tuberculosis. Curr Opin Pulm Med. 2005;11:189-194.
150.Hirsch C, Johnson J, Ellner J. Pulmonary tuberculosis. Curr Opin Pulm Med. 1999;5:143-150.
151.ATS, CDC, IDS. Diagnostic standards and classification of tuberculosis in adults and children. Am J Respir Crit Care Med. 2000;161:1376-1395.
152.Shneerson JM. Respiratory failure in tuberculosis; a modern perspective. Clin Med J Royal Coll Physicians. 2004;4(1):72-76.
153.Srinivasan H, Vidyasagar D. Meconium aspiration syndrome: current concepts and management. Compr Ther. 1999;25(2):82-89.
154.Klingner M, Kruse J. Meconium aspiration syndrome: pathophysiology and prevention. J Am Board Fam Pract. 1999;12:450-466.
155.Ahanya SN, Lakshmanan J, Morgan BLG, Ross MG. Meconium passage in utero: mechanisms, consequences, and management. Obstet Gynecol Surv. 2004;60(1):45-57.
156.Afshar K, Sharma OP. Interstitial lung disease: trials and tribulations. Curr Opin Pulm Med. 2008;14:427-433.
157.Dempsey OJ, Kerr KM, Gomersall L, et al. Interstitial pulmonary fibrosis: an update. Q J Med. 2006;99:643-654.
158.Allam JS, Limper AH. Idiopathic pulmonary fibrosis: is it a familiar disease? Curr Opin Pulm Med. 2006;12:312-317.
159.Patel NM, Lederer DJ, Borczuk AC, et al. Pulmonary hypertension in idiopathic pulmonary fibrosis. Chest. 2007;132:998-1006.
160.Nicod L. Recognition and treatment of idiopathic pulmonary fibrosis. Drugs. 1998;55(4):55-62.
161.Ryu J, Colby T, Hartman T. Idiopathic pulmonary fibrosis: current concepts. Mayo Clin Proc. 1998;73(11):1085-1101.
162.Myers JL, Tazelaar HD. Challenges in pulmonary fibrosis: problematic granulomatous lung disease. Thorax. 2008;63;78-84.
163.Katzenstein A, Myers J. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157(4, pt 1):1301-1315.
164.Ryu JH, Krowka MJ, Pellikka PA, et al. Pulmonary hypertension in patients with interstitial lung diseases. Mayo Clin Proc. 2007; 82(3):342-350.
165.Egan J. Pharmacologic therapy of idiopathic pulmonary fibrosis. J Heart Lung Transplant. 1998;17(11):1039-1044.
166.Schultz MJ, Haitsma JJ, Zhang H, Slutsky AS. Pulmonary coagulopathy as a new target in therapeutic students of acute lung injusry: a review. Crit Care Med. 2006;34:871-877.
167.ATS, ERS, ESICM, et al. International consensus conferences in intensive care medicine. Am J Respir Crit Care Med. 1999;160:2118-2124.
168.Ware L, Matthay M. The acute respiratory distress syndrome. N Engl J Med. 2000;342(18):1334-1346.
169.Hudson L, Steinberg K. Epidemiology of acute lung injury and ARDS. Chest. 1999;116:74S-82S.
170.Wyncoll D, Evans T. Acute respiratory distress syndrome. Lancet. 1999;354:497-501.
171.Frawley PM, Habashi NM. Airway pressure regulated ventilation and pediatrics: theory and practice. Crit Care Nurs Clin North Am. 2004;16(3):337-348.
172.Farrell P, Fiascone J. Bronchopulmonary dysplasia in the 1990s: a review for the pediatrician. Curr Probl Pediatr. 1997;27(4): 129-163.
173.Takemura T, Askahi T, Ohtani Y, et al. Pathology of hypersensitivity pneumonitis. Curr Opin Pulm Med. 2008;14:440-454.
174.Daroowala F, Raghu G. Hypersensitivity pneumonitis. Compr Ther. 1997;23(4):244-248.
175.Ando M, Suga M, Kohrogi H. A new look at hypersensitivity pneumonitis. Curr Opin Pulm Med. 1999;5:299-304.
176.Selman M, Vargas M. Airway involvement in hypersensitivity pneumonitis. Curr Opin Pulm Med. 1998;4:9-15.
177.Craig T. Update on hypersensitivity pneumonitis. Compr Ther. 1996;22(9):559-564.
178.Salvaggio J. Extrinsic allergic alveolitis (hypersensitivity pneumonitis): past, present and future. Clin Exp Allergy. 1997;27(suppl 1):18-25.
179.Erdogdu G, Hasirci V. An overview of the role of mineral solubility in silicosis and asbestosis. Environ Res. 1998;78(1):38-42.
180.Mossman B, Churg A. Mechanisms in the pathogenesis of asbestos and silicosis. Am J Respir Crit Care Med. 1998;157(5, pt 1):1666-1680.
181.Kamp D, Weitzman S. Asbestos: clinical spectrum and pathogenic mechanisms. Proc Soc Exp Biol Med. 1997;214(1):12-26.
182.Miserocchi G, Sancini G, Mantegazza F, et al. Translocation pathways for inhaled asbestos fiber. Environ Health. 2008;7(4): 1-8.
183.Schneider J, Hoffman H, Dienemann H, et al. Diagnostic and prognostic value of soluble mesothelin-related proteins in patients with malignant pleural mesothelioma in comparison with benign asbestosis and lung cancer. J Thorac Oncol. 2008;3:1317-1324.
184.Robledo R, Mossman B. Cellular and molecular mechanisms of asbestos induced fibrosis. J Cell Physiol. 1999;180(2):158-166.
185.Rudd R. New developments in asbestos related pleural disease. Thorax. 1996;51(2):210-216.
186.Nishimura S, Broaddus V. Asbestos induced pleural disease. Clin Chest Med. 1998;19(2):311-329.
187.Murlidhar V, Kanhere V. Asbestosis composite mill at Mumbai: a prevalence study. Environ Health. 2005;4(24):1-7.
188.Kamp D, Weitzman S. Molecular basis of asbestos induced lung injury. Thorax. 1999;54(7):638-652.
189.Huaux F. New developments in the understanding of immunology in silicosis. Curr Opin Allergy Clin Immunol. 2007;7:163-173.
190.Chong S, Lee KS, Chung MJ, et al. Pneumoconiosis: comparison of imaging and pathological findings. Radiographics. 2006;26:59-77.
191.Steenland K, Goldsmith D. Silica exposure and autoimmune disease. Am J Ind Med. 1995;28(5):603-608.
192.Anaya J, Diethelm L, Ortiz L, et al. Pulmonary involvement in rheumatoid arthritis. Semin Arthritis Rheum. 1995;24(4):242-254.
193.Tanoue L. Pulmonary manifestations of rheumatoid arthritis. Clin Chest Med. 1998;19(4):667-683.
194.Murin S, Weidemann H, Matthay R. Pulmonary manifestations of systemic lupus erythematosus. Clin Chest Med. 1998;19(4): 641-665.
195.Godfrey T, Khamashta M, Hughes G. Therapeutic advances in systemic lupus erythematosus. Curr Opin Rheumatol. 1998;10(5):435-441.
196.Minai O, Dweik R, Arroliga A. Manifestations of scleroderma pulmonary disease. Clin Chest Med. 1998;19(4):713-727.
197.Silman A. Epidemiology of scleroderma. Ann Rheum Dis. 1991;50:846-853.
198.Steen V. Clinical manifestations of systemic sclerosis. Semin Cutan Med Surg. 1998;17(1):48-54.
199.Cain H, Noble P, Matthay R. Pulmonary manifestations of Sjogren’s Syndrome. Clin Chest Med. 1998;19(4):687-697.
200.Tavoni A, Cirigliano C, Frigelli S, et al. Shrinking lung in primary Sjogren’s syndrome. Arthritis Rheum. 1999;42(10):2249-2250.
201.Wiedemann H, Matthay R. Pulmonary manifestations of the collagen vascular disease. Clin Chest Med. 1989;10(4):677-715.
202.Corley D, Winterbauer R. Collagen vascular disease. Semin Respir Infect. 1995;10(2):78-85.
203.Bulpitt K, Clements P, Lachenbruch P, et al. Early undifferentiated connective tissue disease: III. Outcome and prognostic indicators in early scleroderma (systemic sclerosis). Ann Intern Med. 1993;118:602-609.
204.Libby D, White D. Pulmonary toxicity of drugs used to treat systemic autoimmune disease. Clin Chest Med. 1998;19(4):809-821.
205.Bradbury PA, Shepherd FA. Immunotherapy for lung cancer. J Thorac Oncol. 2008;3(suppl 2):S164-S170.
206.Gomez M, Silvestri GA. Lung cancer screening. Am J Med Sci. 2008;335(1):46-50.
207.Osann K. Epidemiology of lung cancer. Curr Opin Pulm Med. 1998;4:198-204.
208.McLoud T, Swenson S. Lung carcinoma. Clin Chest Med. 1999;20(4):697-714.
209.Christini D. Smoking and the molecular epidemiology of lung cancer. Clin Chest Med. 2000;21(1):87-93.
210.Sher T, Dy GK, Adjei AA. Small cell lung cancer. Mayo Clin Proc. 2008;83(3):355-367.
211.Lillington G. Neoplasms of the lung. Curr Opin Pulm Med. 1999;5:185-188.
212.Leonard C, Sachs D. Environmental tobacco smoke and lung cancer incidence. Curr Opin Pulm Med. 1999;5:189-193.
213.Smith RA, Cokkinides V, Brawley OW. Cancer screening in the United States, 2009. CA Cancer J Clin. 2009;59:27-41.
214.National Cancer Institute. U.S. National Institutes of Health. http://www.cancer.gov.
215.Franklin W. Diagnosis of lung cancer: pathology of invasive and preinvasive neoplasia. Chest. 2000;117:80S-89S.
216.Adjei A. Management of small cell cancer of the lung. Curr Opin Pulm Med. 2000;6:384-390.
217.Bunn PA, Thatcher N. Introduction. Oncologist. 2008;13(suppl 1):1-4.
218.Leonard C, Whyte R, Lillington G. Primary non-small-cell lung cancer: determining the suitability of the patient and tumor for resection. Curr Opin Pulm Med. 2000;6:391-395.
219.Johnson D. Locally advanced, unresectable non-small cell lung cancer. Chest. 2000;117:123S-126S.
220.Hyer J, Silvestri G. Diagnosis and staging of lung cancer. Clin Chest Med. 2000;21(1):95-108.
221.Mountain C. Revisions in the international system for staging lung cancer. Chest. 1997;111:1710-1717.
222.Deslauriers J, Gregoire J. Clinical and surgical staging of non-small-cell lung cancer. Chest. 2000;117:96S-103S.
223.Jacobs PCA, Mali W, Grobbee DE, van der Graaf Y. Prevalence of incidental findings in computed tomographic screening of the chest. J Comput Assist Tomogr. 2008;32:214-221.
224.Hillerdal G. Indolent lung cancers: time for a paradigm shift. J Thorac Oncol. 2008;3:208-211.
225.Yoneda K, Louie S, Shelton D. Approach to pulmonary metastases. Curr Opin Pulm Med. 2000;6:356-363.
226.Govindan R, Bogart J, Vokes EE. Locally advanced non-small cell lung cancer: the past, present and future. J Thorac Oncol. 2008;3:917-928.
227.Brunelli A, Salati M. Preoperative evaluation of lung cancer: predicting the impact of surgery on physiology and quality of life. Curr Opin Pulm Med. 2008;14:275-281.
228.Belani C. Combined modality therapy for unresectable stage III non-small-cell lung cancer. Chest. 2000;117:127S-132S.
229.Redline S, Tishler P, Schluchter M, et al. Risk factors for sleep-disordered breathing in children: associations with obesity, race, and respiratory problems. Am J Respir Crit Care Med. 1999;159:1527-1532.
230.Zerha-Lancner F, Lofaso F, Coste A, et al. Pulmonary function in obese snorers with or without sleep apnea syndrome. Am J Respir Crit Care Med. 1997;156:522-527.
231.Kay J. Hypoxia, obstructive sleep apnea syndrome, and pulmonary hypertension. Hum Pathol. 1997;28(3):261-263.
232.O’Day M. Cardiopulmonary physiological adaptation of pregnancy. Semin Perinatol. 1997;21(4):268-275.
233.Sue D. Obesity and pulmonary function: more or less? Chest. 1997;111(4):891-898.
234.Chen Y, Rennie D, Cormier Y, Dosman J. Waist circumference is associated with pulmonary function in normal-weight, overweight, and obese subjects. Am J Clin Nutr. 2007;85:35-39.
235.Klein S, Burke L, Bray G, Blair S, Allison D. Clinical implications of obesity with specific focus on cardiovascular disease. Circulation. 2004;110:2952-2967.
236.Langer J. Congenital diaphragmatic hernia. Chest Surg Clin North Am. 1998;8(2):295-311.
237.American Physical Therapy Association. Guide to Physical Therapist Practice. Phys Ther. 1997;77(11):1231-1619.