James E. Calvin, Jr.
Stephanie H. Dunlap
Heart failure is a complex syndrome physiologically characterized as low cardiac output leading to inadequate blood supply of tissues and vital organs. No matter the etiology, damage to the cardiocytes starts a complicated neurohormonal cascade that causes poor renal perfusion, leading to stimulation of the renin–angiotensin–aldosterone system and elevated levels of circulating catecholamines. These compensatory mechanisms lead to sodium and water retention and tachycardia that may cause pulmonary, hepatic, and splenic congestion, as well as peripheral and splanchnic bed constriction. The patient may experience symptoms such as dyspnea on exertion, lower extremity edema, early satiety, paroxysmal nocturnal dyspnea, fatigue, dizziness, and/or syncope.
Heart failure is a serious public health concern in the United States and other industrialized countries. It continues to increase in incidence and prevalence, with a prevalence of 5 million Americans having the syndrome in 2003 and with an incidence 550,000 new persons diagnosed annually (1). These derangements lead to frequent hospitalizations, totaling 1,093,000 hospital discharges (2) and 3.4 million visits to physicians, emergency departments, and hospital outpatient departments annually (3). The mortality rate remains high despite complex and expensive medical regimens, with self-assessment of the patient's quality of life rated as poor. The Framingham Heart Study now has 44 years of follow-up and, based upon these data, heart failure incidence approaches 10 per 1,000 population after age 65 (1). The lifetime risk for development of symptomatic heart failure is one in five for both men and women (4). The lifetime risk for heart failure is doubled for both sexes with blood pressure greater than or equal to 160/100 mm Hg compared to those without hypertension. Thus, given current demographics, cases of heart failure are predicted to continue to rise.
Risk factors for heart failure continue to include hypertension, with 75% of heart failure cases having antecedent hypertension (1). About 22% of male and 46% of female victims of myocardial infarction (MI) will become disabled with heart failure within 6 years of the MI (1). Diabetes mellitus is another significant risk factor for heart failure. In women with coronary heart disease, diabetes mellitus was found to be the strongest risk factor for the development of heart failure. Additionally, in this study, those with an elevated body mass index or depressed creatinine (Cr) clearance in diabetic women were noted to be at highest risk, with annual incidence rates of 7% and 13%, respectively (5). In another study of patients with heart failure, African American women with elevated body mass index and pre-existing hypertension were the cohort with the highest risk for the development of heart failure (6). With regard to mortality, deaths from heart failure based on the International Classification of Disease (Code 428) increased 20.5% from 1993 to 2003. In 2003, the overall death rate for heart failure was 19.1%. Examined by race and gender, death rates were 20.3% for Caucasian males, 22.9% for African American males, 18.3% for Caucasian females, and 19.0% for African American females. Heart failure discharges from hospitals were 1,093,000 in 2003.
Heart failure is a syndrome with diverse etiologies, anatomies, and physiologic presentations. The World Health Organization developed classification nomenclature for the cardiomyopathies. This nomenclature is based on anatomic and physiologic findings: restrictive, hypertrophic, and dilated. This chapter focuses on the dilated cardiomyopathies. The myocardial muscle disease causing dilated cardiomyopathy may be either a primary cardiocyte disorder or secondary to another disease process. Whether primary or secondary, the hallmark findings of dilated cardiomyopathy are a dilated left ventricle and a low left ventricular ejection fraction. The most common etiology of dilated cardiomyopathy (DCM) is secondary to ischemic heart disease. Ischemic cardiomyopathy accounts for nearly half of all cases of DCM in the United States. All the remaining dilated cardiomyopathies are considered primary muscle diseases. Although 75% of patients diagnosed with heart failure have antecedent hypertension, only about 22% of patients with DCM have hypertension as the only identifiable etiology (1). Nearly 25% of patients with dilated cardiomyopathy have no pre-existing risk factors for DCM and are given the diagnosis of idiopathic dilated cardiomyopathy. Nearly 25% of patients with idiopathic dilated cardiomyopathy have a familial component, likely related to abnormalities in cardiac β-adrenergic receptors (7). Once the common etiologies are excluded, a small but important number of other causes remains, including DCM secondary to toxins such as alcohol, anthracyclines, and cocaine. Endocrine diseases such as diabetes mellitus and thyroid disease may also cause heart failure. Other possible etiologies include human immunodeficiency virus (HIV) and Lyme disease infections, sarcoidosis, thiamine deficiency, peripartum cardiomyopathy, hemochromatosis, and underlying collagen vascular diseases.
Cellular Determinants of Myocardial Contraction
The Contractile Proteins
The interaction of the proteins actin, myosin, and the troponin complex is responsible for myocardial contraction.
Engagement of actin and myosin occurs when calcium (Ca2+) levels in the cytosol increase in the presence of adequate adenosine triphosphate (ATP). This process is regulated by a complex that consists of troponin C and other proteins. Troponin C contains a Ca2+-specific binding site with a variable affinity for Ca2+ (8). This binding of Ca2+ to troponin C is regulated by troponin I. When Ca2+ is bound to troponin C, tropomyosin—a protein, bound to the troponin complex by troponin T, which inhibits actin–myosin interaction in the absence of troponin C—undergoes a conformational change, allowing for actin–myosin interaction and contraction (Fig. 121.1).
|
|
|
Figure 121.1. A schematic representation of tropomyosin, troponin complex, actin, myosin, and calcium during relaxation (A), activation (B), and contraction (C). During relaxation, the myosin is prevented from interacting with actin by tropomyosin. Activation by the interaction of calcium with troponin C confers a configurational change in tropomyosin allowing the interaction of myosin and actin. |
Removal of Ca2+ from the cytosol results in dissociation from troponin C, with subsequent cessation of actin–myosin cross-linkage. This event signals the end of contractile activity and the start of relaxation—a process known as inactivation (9).
Calcium and Cyclic Adenosine Monophosphate
Alterations in the delivery, use, and myofibrillar sensitivity to Ca2+ and removal of Ca2+ from the myofibril and the myocyte cytosol constitutes the biologic basis for the vast majority, if not all, of the abnormalities in both contractility and relaxation. Entry of Ca2+ from extracellular locations occurs through either voltage-dependent, gated “slow channels” activated by membrane depolarization or via sodium–calcium (Na+–Ca2+) exchange across the sarcolemma. In addition, elevated levels of cyclic adenosine monophosphate (cAMP), the major intracellular second messenger, causes increased Ca2+ influx by recruitment of additional voltage-dependent channels on the sarcolemma, previously dormant. This is accomplished by cAMP-mediated transfer of phosphates to phospholamban, a protein linked to the voltage-gated channels. This Ca2+, rather than participating directly in activation of contraction, causes release of Ca2+ from the sarcoplasmic reticulum (SR) (10), termed Ca2 -dependent Ca2+ release. The Ca2+ released from the SR binds to troponin with subsequent contractile activity (Fig. 121.2).
Altered Ca2+ kinetics are responsible for the increases in contractility observed in other circumstances. The increased contractility of postextrasystolic beats (11,12), increased heart rate (HR) (13,14), and, during pharmacologic manipulation with cardiac glycosides, phosphodiesterase inhibitors (15), sympathomimetic amines (14), and caffeine (16) are dependent on changes in intracellular Ca2+ and/or cAMP levels.
Absolute blood Ca2+ levels—by raising cytosolic Ca2+ levels (17)—and hormonal changes such as hyperthyroidism increase contractility (18,19) secondary to increased troponin C affinity for Ca2+, increased ATPase activity with concomitant increased cAMP levels, and changes in intracellular Ca2+ handling.
Individual muscle units in the failing and hypertrophied ventricle have been found to have depressed function (20). The myocardial depression accompanying anoxia (21), acidosis (22), hypothyroidism (18), barbiturate use, administration of local and general anesthetics, Ca2+ antagonists (23), and ischemia (24) all result from abnormalities in the Ca2+-dependent mechanisms described here.
Cellular Determinants of Relaxation
Just as abnormalities of contraction have as their cellular basis derangements of Ca2+ handling, so too does relaxation. Cessation of the inward Ca2+ current—or inactivation—by closure of voltage-limited channels of the sarcolemma begins the period of relaxation, with the rate and extent of Ca2+ removal affecting the rate and extent of relaxation. As alluded to earlier, inactivation signals the end of actin–myosin interaction. However, this single term does not adequately describe the interplay of processes that are occurring at the cellular level to facilitate relaxation. The SR Ca2+ pump, in the presence of adequate ATP, pumps Ca2+ into the SR from the cytosol. Phospholamban, a protein within the SR, when phosphorylated by cAMP-dependent mechanisms, results in increased SR uptake of Ca2+ by the reticulum Ca2+ ATPase (SERCA) (16,25). This is noted to occur primarily after adrenergic stimuli. In addition, the Na+–Ca2+ exchange pump—also requiring ATP—allows the efflux of Ca2+ into the extracellular space from the cytosol (Fig. 121.2). The affinity of myofibrils for Ca2+ also affects relaxation. As the myofibril shortens, its affinity for Ca2+ decreases, limiting Ca2+effects (26). All of these mechanisms will be facilitated or inhibited by drugs or neurohumoral factors.
|
|
|
Figure 121.2. The role of calcium in excitation and contraction in the human myocyte. During depolarization, Ca2+ enters from the extracellular space through the voltage-dependent, gated slow channel, the so-called L-type channel. This entry is facilitated by β-receptor stimulation of a protein receptor, which in turn stimulates adenyl cyclase (AC) producing cyclic adenosine monophosphate (cAMP) and phosphorylation of the calcium channel. However, this intrusion of calcium into the cell does not moderate the interaction of actin and myosin. Rather, it acts as a trigger for the release of calcium from the sarcoplasmic reticulum through the Ca2+ release or ryanodine receptor channel. During repolarization, the sarcoplasmic reticulum (SR) Ca2+ ATPase (SERCA) mediates the uptake of cytosolic calcium into the SR. cAMP-mediated phosphorylation of phospholamban prevents the inhibition by phospholamban of SERCA activity. The Na–Ca2+ exchange pump is the dominant mechanism of Ca2+ extrusion out of the cytosol into the extracellular space. |
The process of relaxation has been extensively studied in DCM, left ventricular (LV) hypertrophy, ischemia, and hypertrophic cardiomyopathy (HCM). Impairment of relaxation in these patients occurs secondary to increased levels of Ca2+ within the myocyte in diastole (27,28), owing to diminished function of the SR uptake pump, decreased expression at the genetic level for this pump as evidenced by decreased mRNA levels (29), decreased phospholamban activity (30) and levels of phospholamban mRNA (31), down-regulation or uncoupling of β receptors (32,33), and inhibition of β-receptor function via G-protein inhibition (34).
Mediators of Contractility
Various mediators, acting independently by linking stimulated α, β, or acetylcholine receptors to intracytoplasmic enzyme systems, alter the contractile state of the myocardium. The best characterized of these systems, the guanine nucleotide regulatory proteins (35,36), are coupled to β, α, and acetylcholine receptors, and possibly to receptors for nitric oxide and endothelins.
β Receptors and Guanine Nucleotide Regulatory Proteins
As alluded to earlier, adrenergic stimulation of cardiac myocytes is a very important regulator of both Ca2+ influx and cAMP levels within the cell (Fig. 121.2) (37). The predominant (β) receptor of myocytes, when stimulated, increases the manufacture of cAMP. Via the mechanisms previously described, this action, in turn, results in increased Ca2+ influx. Ca2+ channels that are under the influence of β-receptor–mediated increases in cAMP are known as “receptor-operated channels” (38). Stimulation of β receptors on the cell surface results in activation of adenylate cyclase (AC) and subsequent increases in cAMP levels. This results in increased Ca2+ influx, with resultant increases in contractile force. The coupling between β receptors and AC occurs through guanine cyclic nucleotides, also known as G proteins (39). G proteins have both stimulatory and inhibitory influences on AC. The G-protein complex (GPC) in its active form contains guanosine triphosphate (GTP). When present, this protein “couples” the β receptor to AC, and when β-receptor adrenergic stimulation occurs, results in the formation of cAMP and subsequent increases in intracellular calcium. The GPC is capable of degrading its bound GTP to guanosine diphosphate (GDP) when β-receptor stimulation ceases, thereby no longer stimulating AC (40). G proteins also have a pivotal role in stimulation of cardiac contractility by α-receptor stimulation via a cAMP-independent mechanism (41), which results in increased contractility within a single heartbeat. cAMP-dependent mechanisms have demonstrated delays in response of 2 to 20 seconds, as opposed to G-protein pathway delays in response of 150 msec. This allows alterations in Ca2+ flux within a single heartbeat, explaining the observed phenomenon of increased contractility immediately after increased sympathetic stimulation.
Inhibitory G proteins, when activated by stimulating acetylcholine receptors, have been shown to inhibit Ca2+ influx (42). This has been demonstrated to occur via a cyclic guanosine monophosphate (cGMP)-mediated mechanism. A cGMP system, similar to the cAMP system outlined earlier, activates cGMP protein kinase (cGMP-PK), which inhibits calcium inward currents previously stimulated by cAMP (43). Additionally, cGMP-mediated inhibition of Ca2+ channels has been demonstrated not to be a result of cAMP hydrolysis or a result of inhibition of cAMP-PK, but a direct effect of cGMP-PK.
In addition to changes in receptor function, molecular alterations in β-receptor production in various disease states have been shown to occur. In dilated cardiomyopathies, both β1-receptor mRNA and absolute receptor levels were found to be depressed (32). At the same time, β-receptor kinase (βARK) levels were elevated. βARK, the molecule responsible for the phosphorylation of β-adrenergic receptors, is elevated when β receptors are dysfunctional (uncoupled). This may provide an explanation for the catecholamine insensitivity observed in failing hearts as well as after cardiopulmonary bypass (44).
Nitric Oxide
Nitric oxide (NO) has been shown to affect cardiac contractility. It has been found in many tissue types, including ventricular myocytes, where its physiologic effects are mediated via cGMP. Cholinergic myocardiac depressant effects, as evidenced by inhibition of the effect of the muscarinic agonist carbachol, were inhibited by antagonists of NO (methylene blue and oxyhemoglobin) as well as by L-arginine (the natural substrate of NO synthesis analogs), which inhibits NO. In addition, the positive inotropic action of the β agonist isoproterenol is enhanced by NO inhibition. These data indicate that the effect of NO is to activate the inhibitory receptor cyclic nucleotide interaction via cGMP mechanisms.
Nitric oxide has been implicated in the myocardial response to sepsis. It has been well documented that myocardial depression in septic shock occurs secondary to a yet undefined substance. One potential substance, tumor necrosis factor-α (TNF-α), has been shown in vitro to depress the activity of spontaneously beating rat cardiomyocytes in tissue culture. Inhibition of NO synthesis by N-methyl-L-arginine (NMA) blocked TNF-induced cardiomyocyte depression (45). Additionally, methylene blue, an inhibitor of guanylate cyclase (46), prevented TNF-induced cardiomyocyte depression.
Although the cellular mechanisms of the depression noted in septic myocardium have not been well characterized, it may be due to alterations in Ca2+ handling. Abnormalities of β-receptor function as measured by decreased levels of cAMP in peripheral lymphocytes in septic patients have been elicited, as well as possible abnormalities in septic patients (47). The exact cellular level of this defect, whether this abnormality of β-receptor function is receptor down-regulation or reduced transcription of β-receptor genes, has yet to be determined.
Endothelin
Endothelin-1, a 21-amino acid vasoconstrictor peptide released from the vascular endothelium, was isolated by Yanagisawa et al. in 1988 (48). Since that time, four additional isoforms and a closely related substance, vasoactive intestinal contractor (VIC), have been isolated. These substances have been found ubiquitously in mammalian tissues. Their intracardiac site of genesis is unknown, but is believed to be the endothelium of the coronary arteries and microvasculature. Various vasodilator and vasoconstrictor substances released from the vascular endothelium alter blood flow and, in this way, indirectly affect cardiac contractility (49). Of these locally elaborated paracrine substances, the endothelins have been most extensively studied.
High-affinity receptors for endothelins in mammalian atria and ventricles have been isolated. In vitro, these substances have been shown to be potent vasoconstrictors and positive inotropes, acting via a yet incompletely defined mechanism. Kelly et al. (50) demonstrated that at constant cytosolic Ca2+ levels using the Ca2+-specific intracellular probe Fura-2, endothelins cause marked increases in the contractility of isolated rat ventricular myocytes. This observation suggests that endothelins may sensitize the myofibrils to calcium, although this finding has not been duplicated by others.
As previously discussed, an increase in intracellular pH results in increased myofibrillar sensitivity for Ca2+, thereby increasing contractility. Endothelin causes an increase in intracellular pH, with a subsequent increase in contraction when studied in rat ventricular myocytes. This effect is completely inhibited by pretreatment with amiloride, which inhibits Na+–H+ exchange across the sarcolemmal membrane and, hence, prevents increases in intracellular pH (51). Furthermore, the effect of endothelin appears to be mediated via G proteins participating in signal transduction after binding of endothelin to its sarcolemmal receptor.
The Cellular Basis of Heart Failure
Heart failure, either acute or chronic, results from loss of myocytes or loss of intrinsic contractility within individual myocytes. Several functional abnormalities involving excitation–contraction coupling of contractile proteins within myocytes and myocardial energetics have been identified.
The importance of Ca2+ in the regulation of myocardial contraction cannot be overstated. Evidence has mounted about the role of disturbances in calcium handling in heart failure (52,53,54,55,56,57,58). Prolonged elevation of Ca2+ concentration intracellularly is apparent in heart failure. Furthermore, there is a blunted rise of depolarization, causing slower activation and a slower rate of fall during repolarization. This is likely mediated via the impaired reuptake of calcium into the sarcoplasmic reticulum because Ca2+–adenosine triphosphatase activity (SERCA2) is reduced. The activity of SERCA2 activity is inhibited by phosphorylation of phospholamban through cAMP stimulated by β-adrenergic receptors.
The calcium release channel (CRC) of the sarcoplasmic reticulum mediates release of Ca2+ from the SR into the cytosol during contraction and is critical for activation of the contractile elements. It now appears that the CRC is hyperphosphorylated by protein kinase A in heart failure, resulting in a high rate of Ca2+ leakage from the SR (59) throughout the cardiac cycle. Other abnormalities identified in heart failure include increased activity in the Na+–Ca2+ exchanger, perhaps as a compensation for reduced SERCA2, and decreased mRNA and protein levels of the voltage-dependent Ca2+ channel.
Contractile Protein Alteration in the Failure Heart
Considerable data now exist (60,61) demonstrating alterations in contractile proteins in heart failure. Both myosin heavy and light chains have been demonstrated to revert to fetal phenotypes in chronic heart failure. In addition, troponin I and T have also been shown to revert to fetal phenotypes.
|
|
|
Figure 121.3. Typical Frank-Starling curves for normal and depressed myocardium. For a given end-diastolic volume, stroke volume is decreased with depressed contractility. |
Regulation of Interstitial Collagen
Collagen provides stents along which myocytes are aligned. The quantity and nature of the collagen in the extracellular matrix (ECM) are determined by the balance between synthesis and degradation. The latter is regulated by matrix metalloproteinases (MMPs) (62,63). In heart failure, there appears to be a maladaptation of collagen stents, perhaps mediated by increased activity of MMPs. Paradoxically, in hypertension and pressure overload, an increase in interstitial collagen has been noted.
Adaptations to Changes in Load or Myocardial Injury
In situations where an excessive load is placed on either ventricle, or where an injury has occurred that depresses myocardial contraction, the ventricle (14) can adapt to maintain cardiac output by doing the following:
1. Increase ventricular preload (so-called Frank Starling effect) (Fig. 121.3) (64)
2. Activation of neurohumeral systems such as the renin–angiotensin–aldosterone system (65)
3. The release of norepinephrine (66)
4. Myocardial remodeling via chamber dilatation and augmentation of myocardial mass (67)
The first adaptation increases cardiac output by increasing preload (end-diastolic pressure and volume). The second increases blood volume and blood pressure by increasing peripheral vascular resistance. The third attempts to increase contractility by shifting the Frank-Starling curves upward, and the fourth enhances contractility by restoring the number of contractile units. The next effect of these adaptations is maintenance of cardiac output and blood pressure in the face of increased afterload (peripheral vascular resistance [PVR]) at the expense of increasing ventricular dilatation.
Myocardial remodeling takes longer to develop and there are two distinct patterns (67,68): (a) concentric hypertrophy (increased mass without ventricular dilatation) and (b) eccentric hypertrophy (increased mass and ventricular dilatation) (69). Concentric hypertrophy usually occurs in response to pressure overload, and is characterized by parallel replication of myofibrils and thickening of individual myocytes. Eccentric hypertrophy occurs in response to volume overload, leading to increased diastolic stress with a series replication of sarcomeres, elongation of myocytes, and ventricular dilatation. In both of these cases, wall stress is returned toward normal, at least initially. There appears to be differences in patterns of gene activation for several peptide growth factors.
At the cellular level, there is an increase in the number of mitochondria, an increase in cell size, and an increase in the amount of collagen within the extracellular matrix. When the injury or stress exceeds the ability of these adaptive mechanisms to compensate, myocardial contractility decreases. The transition from a compensated state to failure involves the following processes:
1. Inadequate hypertrophy to maintain contractility
2. Re-expression of fetal genes and decreased expression of adult genes (70)
3. Alteration in proteins involved in excitation–contraction coupling
4. Myocardial death by necrosis or apoptosis (71)
5. Changes in myocardial energetics
Neurohormonal Abnormalities in Heart Failure
When low cardiac output occurs, renal perfusion pressure is decreased. This causes stimulation of renin in the juxtaglomerular apparatus. Renin is then converted in the kidney to angiotensinogen, and then to angiotensin I. Angiotensin I is released into the circulation and is converted in the lung by angiotensin-converting enzyme to angiotensin II, a potent vasoconstrictor. A major breakthrough in therapeutic options for heart failure was the discovery of this enzyme system that then led to pharmaceutical development of angiotensin-converting enzyme inhibitors to interrupt this pathway. Another major breakthrough was the realization that other enzymes, located in the myocardium, could cleave the decapeptide angiotensin I into the octapeptide angiotensin II. These enzymes are chymase, CAGE, and cathepsin G. This discovery cemented the idea that the heart was a neuroendocrine organ. The potent vasoconstrictor effects of angiotensin II were part of the cardiovascular system's compensatory mechanisms: angiotensin II increased renal perfusion pressure.
Elevated levels of aldosterone are also seen in patients with left ventricular systolic dysfunction. Stimulation of renin and angiotensin II leads to increased production of aldosterone by the adrenal gland. Additional mechanisms of increased aldosterone levels are decreased hepatic clearance of the hormone as well as stimulation by sodium-restricted diets. Since elevated aldosterone levels lead to sodium and water retention, plasma volume is increased. Low cardiac output also causes the adrenal gland to produce more norepinephrine. Elevated levels of circulating catecholamines in patients with heart failure have been shown to have prognostic significance, with higher norepinephrine levels predicting higher mortality (72). With increased levels of norepinephrine, heart rate is increased, thus aiding in maintenance of cardiac output since:
CO = SV × HR
where CO = cardiac output, SV = stroke volume, and HR = heart rate.
Elevated levels of norepinephrine, angiotensin II, and aldosterone result in maladaptive changes in LV structure. These neurohormones lead to dilatation of the left ventricle as well as changes in cardiocyte architecture, collagen type and content, and β-adrenergic receptor abnormalities. Collectively, these changes in LV architecture are known as remodeling. Complex signaling pathways have been shown to activate cellular responses during hypertrophy of cardiocytes in vivo. These include accumulation and assembly of contractile proteins, increase in size of cardiocytes, and expression of embryonic genes resulting in eccentric hypertrophy. Eccentric hypertrophy is found in DCM and is a result of contractile protein units being assembled in series, rather than in parallel, as seen in concentric hypertrophy. Scientific advances that have led to this knowledge were achieved by first identifying mutant genes, such as those that cause familial idiopathic cardiomyopathy, then being able to culture cardiocytes, followed by studying genetically engineered animals. Factors that led to the hypertrophic response include growth factors, peptides, and cytokines, with the most comprehensively studied substances being insulin growth factor I, angiotensin II, endothelin, and those that activate a form of GTP-binding protein (ras) signaling pathways, cytokines including interleukin-6, and heteromeric (Gq) (70).
The presence of these neurohormones in abnormal amounts has been intensively studied. These substances are produced as part of compensatory mechanisms used to alleviate the adverse effects of low cardiac output. The vasoconstrictors include endothelin and angiotensin II. These are the two strongest intrinsic vasoconstrictors discovered in humans, with endothelin being the most potent. Vasoconstrictors aid the failing ventricle by keeping perfusion pressure up. Another compensatory mechanism is stimulation of aldosterone. Increased production of aldosterone results in the kidney retaining sodium—and thus, water—in exchange for increased secretion of potassium, retention of sodium and water, and increased plasma volume. Other adverse effects of this compensatory release of aldosterone include vascular and myocardial fibrosis, baroreceptor dysfunction, and prevention of myocardial norepinephrine uptake. In response to decreased renal perfusion pressure, arterial baroreceptors are activated, leading to nonosmotic stimulation of arginine vasopressin from the supraoptic nucleus of the hypothalamus. This increase in arginine vasopressin stimulates both V1a vascular smooth muscle receptors and V2 receptors on the collecting duct. In the collecting duct, the V2 receptor stimulation activates the adenylate–cAMP pathway, increasing the aquaporin-2 water channel trafficking, which leads to increased water reabsorption and hyponatremia. The adrenal gland also produces more norepinephrine in response to low cardiac output, which results in an increase in heart rate, initially a compensatory mechanism to increase cardiac output. However, when β-adrenergic receptors have prolonged exposure to norepinephrine, they become down-regulated. The first response to prolonged exposure is for the β-adrenergic receptor to migrate into the cell membrane. As exposure continues, it is then transported intracellularly and becomes phagocytized (73), as was shown by Bristow et al. by performing endomyocardial biopsies of patients with systolic heart failure. This group noted that there were fewer β-adrenergic receptors in patients with heart failure and that the β receptors that were present were less responsive to isoproterenol, a β-adrenergic agonist (73). Mann et al. found that cultured cardiac myocytes exposed to norepinephrine first began to contract normally and then, after prolonged exposure, became irreversibly hypercontracted and cell viability decreased over time (74). Thus, with low cardiac output, a variety of neurohormones are produced in abnormal amounts, leading to left ventricular remodeling, apoptosis, resting tachycardia, β-receptor down-regulation, arrhythmias, increased systemic vascular resistance (elevated afterload), and edematous states (increased preload).
Another group of neurohormones produced in response to the failing heart has been determined to be beneficial. These include the natriuretic peptides made in response to left ventricular dilatation, especially brain natriuretic peptide (BNP). This hormone has natriuretic and vasodilatory properties that are beneficial in patients with heart failure, their function being an attempt to counteract the deleterious effects of aldosterone and angiotensin II. Levels of BNP are elevated in patients with heart failure, compared to those with normal LV function. Additionally, data suggest that BNP levels are more elevated as the patient becomes more symptomatic—that is, as the New York Heart Association Functional Class increases—and levels may be more elevated when patients present in decompensated heart failure compared to their compensated state. Despite elevated BNP production, the patient may continue to deteriorate, the current explanation being that BNP is rapidly overwhelmed by more potent vasoconstrictor substances present in the heart failure milieu.
Other substances with vasodilatory properties include bradykinin, nitric oxide, and prostaglandins. One mechanism for vasodilatation with angiotensin-converting enzyme (ACE) inhibitors is that degradation of bradykinin is inhibited by ACE inhibitors, since kininase, the enzyme that degrades bradykinin, is also known as angiotensin-converting enzyme. Cytokines are also produced in abnormal amounts, including interleukins and TNF-α. Once LV dysfunction is present, the overexpression of these compounds contributes to the progression of heart failure, promoting LV dilatation and remodeling. Moreover, elevated TNF-α levels are found in patients with advanced heart failure, and a trend toward higher mortality with higher TNF-α levels was found in an analysis of the Studies of Left Ventricular Dysfunction (SOLVD) trial (75,76).
Remodeling
Remodeling is a complex process in response to either acute or chronic injury to cardiocytes. The term encompasses LV dilatation, eccentric hypertrophy, apoptosis (programmed cell death not secondary to ischemia), changes in valvular structure, and arrhythmias. Increases in left ventricular end-diastolic and end-systolic dimensions occur, as well as increased size at the cellular level with cardiocytes developing increased cell volume and eccentric hypertrophy. White et al. (77) found that the most potent predictor of death following acute myocardial infarction was end-systolic volume and, in a multivariable model, was more potent than the extent of coronary artery disease. With the increase in chamber size, LV geometry is also altered, with the left ventricle losing its elliptical shape and developing a more spherical shape. The more spherical LV has been associated with higher end-systolic wall stress and abnormal distribution of fiber shortening (78). Additionally, one small study of patients with idiopathic dilated cardiomyopathy found poorer survival in those that had developed a more spherical ventricle and more uniform distribution of afterload (79).
Mitral regurgitation frequently accompanies LV remodeling. This occurs due to misalignment of papillary muscles and the subvalvular structures, as well as distortion at the mitral annulus secondary to LV enlargement. Left atrial enlargement and pulmonary venous hypertension are frequent sequelae of mitral regurgitation. Electrocardiographic abnormalities and arrhythmias are also common problems in patients with heart failure. These include left bundle branch block, P-R interval prolongation, atrial flutter and atrial fibrillation, and ventricular tachycardia. Development of atrial fibrillation is frequently seen in patients with decompensated heart failure and, in a chicken and egg type phenomenon, may either cause the decompensation or may occur as a result of the decompensation. Syncope, an ominous predictor for sudden cardiac death, may be secondary to ventricular tachycardia or atrial arrhythmias, with rapid ventricular response. Any patient, presenting with syncope should have a thorough investigation for its etiology.
Given that LV remodeling is a harbinger for poor outcomes, attempts to delay or reverse the remodeling process have become therapeutic targets for altering the outcome and improving survival in patients afflicted with heart failure. The Cooperative Northern Scandinavian Enalapril Survival (CONSENSUS) (80) and SOLVD Treatment trials (75,76) determined that therapy with enalapril improved survival in patients with pre-existing mild to moderate and advanced heart failure, most likely by its impact on angiotensin II production. The Survival and Ventricular Enlargement (SAVE) trial (81) revealed that patients surviving myocardial infarction with resultant left ventricular enlargement—but not clinical heart failure—had better survival with the ACE inhibitor, captopril. Additionally, those patients receiving captopril had less morbidity and mortality secondary to cardiovascular events and less recurrent myocardial infarctions. The SOLVD Prevention trial showed that therapy with the ACE inhibitor, enalapril, delayed development of heart failure symptoms and decreased both all-cause hospitalizations and hospitalizations for heart failure, whether for patients with recent myocardial infarcts or those with pre-existing heart failure. Trials with β-adrenergic receptor therapy, in addition to ACE inhibitor therapy, revealed that these agents were capable of decreasing LV size and improving the ejection fraction. Cardiac resynchronization of therapy has also been shown to reverse remodeling the left ventricle.
Diastolic Dysfunction in Heart Failure
Left ventricular diastolic function is dependent on many factors, some intrinsic to the heart itself (e.g., the active energy-dependent process of relaxation and material properties of the myocardium) and some extrinsic to the LV (e.g., pericardial constraining forces and ventricular interaction). Furthermore, Gilbert and Glantz (82) have suggested that relaxation can be further divided into the extent of relaxation (i.e., the completeness of relaxation) and the rate of relaxation. Alterations in the extent, rate, or both characterize the abnormalities of relaxation and result in characteristic hemodynamic patterns (82).
Left Ventricular Compliance and the Diastolic Left Ventricular Pressure–Volume Relationship
A nonlinear relationship normally exists between pressure and volume during ventricular diastole. Shifts in this relationship are reflected either by a change in the slope of the relationship of filling pressure and volume—stiffness of the left ventricle—as filling volume increases (83), or by changes in either the slope or intercept of the relationship resulting from various disease states (82).
Extent of Relaxation
The extent of relaxation is the major determinant of end-diastolic volume (EDV) and end-diastolic pressure (EDP), because these are measurements made at the end of the relaxation process. Abnormalities in extent of relaxation affect the end-diastolic pressure–volume (P–V) relationship to the greatest extent. Abnormalities in the rate of relaxation, however, tend to have minimal effect on the end-diastolic P–V relationship, because of the fact that they occur early in diastole and therefore do not alter EDP and EDV to any appreciable extent.
The extent of relaxation, as stated earlier, may be viewed as the compliance properties of the LV at the point where relaxation is complete (i.e., end-diastole). Alterations in the determinants of this relationship, intrinsic to the myocardium, result in shifts of the diastolic P–V curve. LV geometry (i.e., thickness, size, and chamber dimension) in large part determines the LV end-diastolic P–V relationship, as determined by mathematic approximations based on Laplace's law (82). Alterations in the LV end-diastolic P–V relationship may occur secondary to the change in elastic properties as the ventricle stretches during filling. Changes in the diastolic P–V relationship that depend on the rate at which the LV deforms are known as viscoelasticity, a property that myocardium shares with most biomaterials (84). This property is manifest when filling rates are highest, occurring during the first half of diastole, or after atrial contraction. Stress relaxation—a decrease in the distending pressure of the ventricle over time—or creep, a rightward shift in the diastolic P–V relationship—are two experimental manifestations of viscosity. The clinical importance of viscoelasticity has been disputed, however.
Other dynamic changes in relaxation that occur during ventricular filling are due to alterations in the elastic properties and the rate of relaxation of the myocardium, mediated via changes in the load sensed by the LV during relaxation. These load-dependent relaxation phenomena cause instantaneous changes in the LV compliance as well as in the rate of relaxation (9), which are independent of heart rate when LV muscle is abruptly stretched.
An additional determinant of the diastolic P–V relationship previously alluded to is coronary vascular turgor. The effect of this condition on the extent of relaxation is primarily through its erectile effect on LV stiffness (85). This decreases LV diastolic compliance by increasing LV wall volume, resulting in a higher EDP for a given volume. This effect seems to be independent of pericardial influences and predominates in the late diastolic filling period, thereby influencing the extent of relaxation, albeit to a small degree. In addition, the constraining effect of the pericardium and the degree of ventricular interaction affect the extent of relaxation (discussed later in the text).
LV hypertrophy results in abnormalities of relaxation that are characteristic of the manner in which the hypertrophy developed and of the type of hypertrophy formed (86). Eccentric hypertrophy, as seen with mitral or aortic insufficiency, is characterized by increased ventricular volume but little or no change in elasticity. This results in little increase in pressure at increased volumes. In contrast, concentric hypertrophy, seen with aortic stenosis or chronic untreated hypertension (87), is characterized by increased elastic stiffness and an elevated EDP for a given volume. Geometrically, pressure overload or hypertrophy is characterized by additional myocytes in parallel with existing cells; volume overload (eccentric hypertrophy) results in increased length of existing myocytes. Alterations in Ca2+ metabolism, as discussed, result in elevated myocyte diastolic Ca2+ levels. These factors account for the elevated EDP seen in chronic pressure overload hypertrophy.
Ischemia affects the extent of relaxation as evidenced by upward shift in the end-diastolic P–V relationship when myocardial oxygen demand outstrips supply (88,89). Pacing-induced ischemia after the creation of a coronary stenosis in dogs results in such a shift in the P–V relationship at end-diastole. These effects are independent of pericardial, right ventricular (RV), or lung interactions, implying a change in the intrinsic myocardial elastic properties. As previously discussed, changes in diastolic properties secondary to changes in myocardial Ca2+ handling (90,91), as well as in hydrogen ion accumulation (92) and repeated systolic stretch of the ischemia segment (93), interact to produce the observed changes.
The changes in ventricular compliance seem to be restricted to the region of active ischemia (93). Furthermore, uninvolved areas show evidence of a proportional increase in regional size and pressure—with a resultant constant diastolic P–V relation—to maintain SV by the Frank-Starling mechanism. Hence, during acute ischemia, the remaining normal areas of myocardium appear to utilize the Frank-Starling mechanism to maintain SV in compensation for the effects of abnormal contractility or an upward shift of the regional diastolic P–V relationship within the ischemic areas.
Rate of Relaxation
The rate of relaxation, as stated earlier, results primarily in changes in the rate of diastolic early filling (82). The determinants of relaxation rate are many, and their interactions complicated. Increases in heart rate and inotropy (94) result in increased rates of relaxation. Alterations in end-diastolic loading conditions result in changes in the rate of relaxation during experimental conditions (9,95). Nonuniformity of relaxation (9), which describes a nonuniform distribution of load and electric inactivation during diastole in space and time, results in alterations in the rate of relaxation. Ventricular suction, or the ability of the ventricle to generate pressures below equilibrium diastolic pressures, may alter the rate and extent of LV filling (94). Finally, ischemia can alter the rate as well as the extent of relaxation. Resolution of ischemia results in reversal of these changes.
Extrinsic Influences on the Diastolic Pressure–Volume Relationship
External loads can also profoundly influence ventricular compliance properties. Specifically, the RV (96), the pericardium (97), and the lungs (98,99) all may acutely induce shifts of the LV diastolic P–V relationship. While not widely studied in acute decompensated heart failure, it is likely that each of these influences may be exerted when both ventricles are dilated and when the lungs are hyperinflated during ventilator therapy.
The influence of the pericardium in the diastolic P–V relationship is a function of both its stiffness and its ability to constrain the entire heart (100). An increase in size of one ventricle therefore causes an increase in the EDP for a given volume (i.e., a shift upward in the P–V relationship) (Fig. 121.4). The constraining effect of a normal pericardium is dependent on its intrinsic compliance and how it affects LV pressures. Just as dilatation of the RV affects the LV diastolic P–V relation (discussed later), dilatation of the LV (i.e., a high LV filling pressure) amplifies the pericardium's influence. This has been demonstrated by measurement of the diastolic P–V relation before and after removal of the pericardium (97,101). Little normal pericardial effect is observed at normal filling pressures.
In addition, the intact pericardium allows interaction between the atria and the LV as well as between the RV and LV. The effect of left atrial (LA) pressure was approximately one-fourth that of the RV pressure in determining the LV diastolic pressure (102). Studies of the influence of the RV on LV compliance (102,103,104) have demonstrated that an upward shift of the LV diastolic P–V curve (i.e., reduced compliance) accompanies RV volume increases at end-diastole (Fig. 121.4). Although this effect is present with the pericardium open, the coupling is much stronger when it is closed (97). Ventricular interaction is therefore an important mechanism underlying acute reductions in LV compliance, whether the RV is enlarged as a result of pressure or volume overload. Ventricular interaction may also be responsible for some of the improved LV compliance properties observed with the administration of vasoactive medications that reduce volume return to the RV, such as nitrates (105).
|
|
|
Figure 121.4. Diastolic portions of left ventricular pressure–area loops. Representative loops are depicted during both control and a microvascular injury (MVI) of the lungs at different right ventricular preloads or right ventricular end-diastolic pressures of 5, 10, and 15 mm Hg. The loops during both control and MVI are shifted upward by increasing right ventricular end-diastolic pressure (RVEDP), although left ventricular end-diastolic area is reduced at a given RVEDP during MVI. |
Alterations in LV geometry were also noted with increasing pulmonary hypertension. The LV septal/free wall axis appears disproportionately reduced when compared with either the base-to-apex or the anteroposterior axis (96,98). Acute pulmonary hypertension induced by glass bead embolization confirms that upward shifts in the LV diastolic P–V relationship occur with changes in RV afterload (i.e., a reduction in LV compliance) and that this effect is largely mediated via a reduction in the LV septum to the free wall dimension and an increase in intrapericardial pressure (98).
The Special Case of Acute Right Ventricular Failure
The established mechanisms for acute RV failure are shown in Table 121.1. As mentioned previously, an inverse relationship between the vascular load and stroke output has been previously demonstrated (106). Calvin et al. (98) demonstrated that RV stroke volume was inversely related to the pulmonary input resistance, which is a more precise measurement of vascular load. In another study, it was demonstrated that tripling of the pulmonary artery pressure (PAP) by glass bead embolism is well tolerated by the RV, with cardiac output being maintained by both the heart rate (chronotropic) response and the Frank-Starling mechanism (preload reserve) (107,108). However, further increases in PAP sufficient to decrease the cardiac output by 20% result in a disproportionate increase in end-systolic volume compared with end-diastolic volume (i.e., stroke volume and ejection fraction decrease as a result). At this particular point, the RV is performing largely pressure work and very little flow work. ATP and creatine phosphate levels are normal at this point (Table 121.2). These phenomena represent evidence of an afterload mismatch; further decreases in stroke volume because of RV afterload mismatches are associated with ATP and creatine phosphate depletion (Table 121.2) (109,110).
|
Table 121.1 Causes of right ventricular failure |
||||||||||||||||||||||||
|
The pericardium plays a significant role in mediating a direct interaction between the RV and the LV. As the RV dilates within an intact pericardium, RV end-diastolic pressure increases. The first implication of this observation is that the intrapericardial pressure increases and this external pressure is exerted on the LV and affects its distensibility (Fig. 121.4) (97,111). In an experimental model of acute pulmonary hypertension produced by ventricular glass bead embolism, it was determined that the LV diastolic pressure–segment length relationship was shifted upward, indicating decreased distensibility. This effect was found to be independent of any change in heart rate.
|
Table 121.2 Myocardial adenosine triphosphate (ATP) and creatine phosphate (CP) in open pericardia experiments (n = 8a) |
|||||||||||||||||||||
|
|||||||||||||||||||||
The second implication of these events is that the transeptal pressure gradient decreases or, in fact, reverses. Kingma et al., in a model of pulmonary hypertension produced by pulmonary artery banding, demonstrated that the septum shifts leftward, further impairing LV filling (112) as the RV dilates and septal curvature—normally rightward—is flattened. Kingma et al. also clearly demonstrated the inverse relationship between the transeptal pressure gradient and the LV septal–free wall dimension.
Evaluation and Treatment
With heart failure a growing public health and economic concern, rigorous and expert analysis of therapies and procedures with attention to their risks and benefits have led to publication of guidelines by the Heart Failure Society of America, joint guidelines by the American Heart Association (AHA) and the American College of Cardiology (ACC), with endorsement of the latter by the Heart Rhythm Society (1). The latest documents emphasize recognition of patients at risk to develop heart failure and target interventions to halt its development. Examples include patients with hypertension, diabetes mellitus, or coronary artery disease but without demonstrable abnormalities in cardiac structure or function; these individuals would be termed stage A. Stage B patients are defined as those with similar risk factors that are asymptomatic, but with cardiac abnormalities such as left ventricular hypertrophy (Fig. 121.5). The guidelines also give recommendations for initial evaluation of patients presenting with both systolic and diastolic heart failure, chronic outpatient management for the wide range of NYHA classes, and inpatient management of those presenting with acute decompensated heart failure (ADHF).
Individuals with risk factors for the development of heart failure (stage A) are those with hypertension, diabetes mellitus, metabolic syndrome, coronary artery disease, and obesity, as well as those with a family history of cardiomyopathy or who have exposure to cardiac toxins (alcohol, illicit drugs, anthracyclines, or tobacco abuse). These individuals should be encouraged to exercise regularly and lose weight, and counseled regarding smoking, alcohol, and illicit drug cessation.
Additionally, therapy for hypertension, dyslipidemia, and diabetes mellitus should be maximized according to the latest guidelines as published in the seventh report of the Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure (JNC VII) (113); the American Diabetes Association (114); and ACC/AHA (115) joint practice guidelines regarding hypercholesterolemia. Therapeutic options for stage A individuals include appropriate therapy for vascular disease and diabetes mellitus, and ACE inhibitors or angiotensin II receptor blockers (ARBs) as appropriate. Stage B patients remain asymptomatic, but exhibit abnormalities in cardiac structure and function such as LV remodeling, low LV ejection fraction (LVEF), or LV hypertrophy (LVH). These individuals should have all therapeutic measures as those in stage A and should receive ACE inhibitors, ARBs, and β-adrenergic antagonists as appropriate. Additionally, patients in stage B should receive implantable cardioverter defibrillators as appropriate for their LVEF and pre-existing disease. Individuals classified as stage C and D have overt heart failure; their care is subsequently discussed.
|
|
|
Figure 121.5. American College of Cardiology/American Heart Association 2005 guideline update for the diagnosis and management of chronic heart failure in the adult—summary article. A report of the American College of Cardiology Heart Association. Task Force in Practice Guidelines. (Writing Committee to Update the 2001 Guidelines for the Evaluation and Management of Heart Failure. J Am Coll Cardiol. 2005;46:1116–1143.) |
Initial Evaluation
Patients presenting with new-onset heart failure should have a thorough history and physical examination, with special attention to risk factors and noncardiac disorders that may aggravate their cardiac condition. Behaviors or therapies that may cause heart failure or exacerbate LV dysfunction should also be sought. Thorough laboratory data, such as a 12-lead electrocardiogram, complete blood count, blood urea nitrogen, creatinine, electrolytes, fasting lipid panel and glucose, hemoglobin A1C in diabetics, and thyroid-stimulating hormone, should be obtained. If suspicion is high that common etiologies of heart failure are not present, patients may be tested for HIV, hemochromatosis, sleep apnea, amyloidosis, pheochromocytoma, and rheumatologic disorders. BNP may be useful to obtain as a baseline. Endomyocardial biopsy should be considered only if the results would influence therapy.
Individuals should also receive posteroanterior (PA) and lateral chest radiographs initially, as well as echocardiography to assess for pulmonary congestion, LV dimensions, LVEF, LVH, and valvular and wall motion abnormalities. Radionuclide ventriculography may also be performed to assess LVEF and LV volumes, and may also be useful to assess right ventricular ejection fraction (RVEF). In patients with angina pectoris or ischemia, coronary angiography should be performed unless contraindicated, since maneuvers to reverse or halt progression of heart failure, such as revascularization, should always be entertained. Additionally, patients with chest pain either consistent or not consistent with cardiac ischemia who have not previously undergone coronary angiography, as well as patients without angina but with known coronary artery disease, should undergo coronary angiography if there are no contraindications. Noninvasive imaging to determine myocardial viability is reasonable to perform in patients with known coronary artery disease (CAD) in HF patients without angina.
Prognosis in Heart Failure
The diagnosis of heart failure has historically been associated with reduced long-term survival, although improvement with newer therapy has been gratifying. The overall 5-year survival is 50%, while the 1-year survival for end-stage heart failure is 75%. Many studies have identified prognostic factors for both long-term and short-term survival. Those associated negatively over the years include the presence of an S3, low pulse pressure, elevated jugular venous pulse, and high NYHA class (116,117,118,119,120). Other important comorbidities include diabetes mellitus, renal insufficiency, and depression.
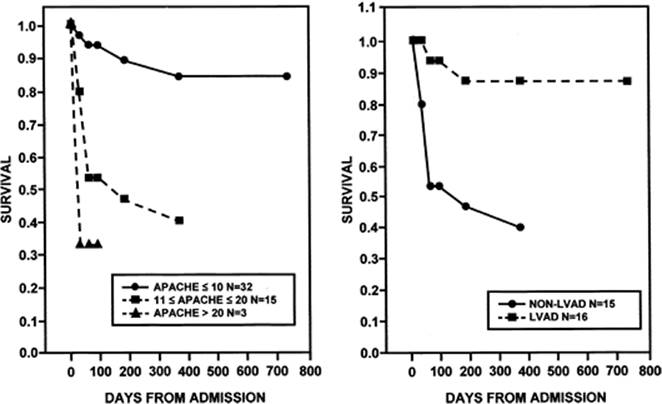
Cardiac testing plays an important role in prognostication (72). A simple cardiothoracic ratio measured by conventional chest radiograph correlates with survival. Ejection fraction continues as a very important marker for prognosis and as a target for new therapies such as implantable cardioverter defibrillator (ICD) and biventricular pacing. One of the more objective measurements is peak oxygen consumption. As noted in Figure 121.6, mortality rates vary from 20% per year if peak VO2 is greater than or equal to 14 but less than 18 mL/kg/minute to 60% per year if peak VO2 is less than or equal to 10 mL/kg/minute (121,122). An easier test to perform for chronic congestive heart failure is the 6-minute walk (123). In this test, a patient's distance walked after 6 minutes is measured and is predictive of morbidity and mortality (123).
Hemodynamic variables measured at heart catheterization, such as cardiac index, systemic and pulmonary vascular resistances, pulmonary artery pressures, and pulmonary capillary wedge pressures, are important indicators of prognosis as well as aiding in diagnosis. Stroke work index is an especially important predictor, as it incorporates both flow and pressure work.
Inverse relationships exist between survival and plasma norepinephrine, renin, vasopressin, aldosterone, atrial and B-type natriuretic peptides, and endothelin-1 (124,125,126,127) (Fig. 121.6). While many of these reflect abnormalities in pathophysiology, only B-type natriuretic peptide has become a routine laboratory test in suspected heart failure patients. Multivariate analyses of heart failure patients randomized in clinical trials have confirmed independent prognostic information from several factors including exercise tolerance parameters, plasma norepinephrine, pro-BNP, and BNP.
Studies of critically ill patients in the past have utilized multivariate models that can be used to assess the severity of acute decompensated heart failure. Teskey et al. (128) looked at the use of the Acute Physiology and Chronic Health Evaluation (APACHE) II score in patients admitted to the coronary care unit and found that it predicted mortality in acute heart failure. Survivors had lower APACHE II scores than nonsurvivors. This score weighs the degree of deviation from normal of selected clinical variables, as well as comorbid conditions. Later, Gracin et al. (129) demonstrated that APACHE II also predicted outcome after ventricular assist device implantation (Fig. 121.7). The Acute Decompensated Heart Failure Registry (ADHERE) has recently published an acuity model for heart failure based on 33,046 hospitalizations, which was subsequently validated prospectively on another 32,229 hospitalizations. Using recursive partitioning—a nonparametric multivariable technique—these investigators identified three predictors: blood urea nitrogen greater than or equal to 43 mg/dL, serum creatinine greater than 2.5 mg/dL, and systolic blood pressure less than 115 mm Hg. This allowed partitioning patients into one high-risk group (three factors present, crude mortality 21.9%), three intermediate-risk groups (varying combinations of risk factors present, crude mortality 5.5%–12.4%), and one low-risk group (no risk factors present, crude mortality 2.14%).
Therapeutic Trials and Findings
Unless otherwise stated, all studies of pharmacologic agents in humans, discussed below, were conducted in randomized, double-blind, placebo-controlled trials.
Angiotensin-converting Enzyme Inhibitors
Hypertension studies in animal models using ACE inhibitors revealed that animals receiving these drugs had less development of heart failure. This led to interest in vasodilator therapeutics for heart failure in animals and, eventually, in humans. The CONSENSUS trial (80) enrolled 253 patients with severe heart failure (NYHA class IV) already receiving digoxin and diuretics, then randomized them to receive enalapril versus placebo. Those randomized to enalapril had better survival, with a 40% risk reduction at 6 months (p = 0.002) and 31% at 1 year (p = 0.001). The risk reduction was due to reduction in deaths from progressive heart failure. Additionally, the NYHA functional class improved in a significant number of those receiving enalapril. Based on the CONSENSUS trial, and other studies of ACE inhibitors investigating symptoms and hemodynamic indexes, the National Institutes of Health sponsored the SOLVD trials (75,76). The SOLVD treatment trial enrolled 2,569 patients with left ventricular systolic dysfunction (LVEF less than or equal to 35%) and symptoms of heart failure, ranging from NYHA class I through IV, with the majority of enrollees being classes II and III. The primary end point was mortality. Subjects randomized to receive enalapril had better survival than those receiving placebo; risk reduction with enalapril was 16% (95% confidence interval [CI] 5%–26%; p = 0.0036). As seen in the CONSENSUS trial, the chief difference in mortality was in deaths due to progressive heart failure, with risk reduction 22% (95% CI 6%–35%; p <0.0045) (79).
The SOLVD prevention trial randomized 4,228 patients with LV systolic dysfunction, but without symptoms, to receive enalapril versus placebo. End points in the SOLVD prevention trial were total mortality and two composite end points: Death or development of heart failure requiring hospitalization, and death in the hospital from heart failure. In these asymptomatic patients, there was no significant decrease in total mortality in those receiving enalapril. However, for those receiving enalapril, the combined end point of death plus development of heart failure was reached less often than those receiving placebo (630 vs. 818; risk reduction 29%; 95% CI 21%–36%; p <0.001). Additionally, those receiving enalapril had fewer hospitalizations for heart failure or death (434 in the enalapril group vs. 518 in the placebo group; risk reduction 20%; 95% CI 9%–30%; p <0.001) (76).
|
|
|
Figure 121.6. Survival curves for heart failure patients by peak VO2 (upper panel), plasma norepinephrine (middle), and brain natriuretic peptide (BNP, lower panel). Survival is best with peak VO2greater than 18 mL/kg/minute, plasma norepinephrine less than 400 pg/mL, and BNP less than 100 pg/mL. (Reproduced from Mancin DM, Eisen H, Kausmaul W, et al. Circulation. 1991;83:778–786; Cohn JN, Levin B, Olivari MT, et al. N Engl J Med. 1984;311:819–823; and Anand IS, Fisher LD, Chiang Y, et al. Changes in brain natriuretic peptide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-HeFT) Circulation. 2003;107:1278–1283.) |
The SAVE trial enrolled 2,231 patients within 3 to 16 days of an acute myocardial infarction and with LV systolic dysfunction (LVEF less than 40%) to receive the ACE inhibitor captopril versus placebo (81). The primary end point was all-cause mortality. Other SAVE trial end points included cardiovascular deaths, treatment failure requiring open-label ACE inhibitor, and all-cause mortality plus progressive LV systolic dysfunction. All-cause mortality was significantly decreased in the captopril group compared to the placebo group (228 deaths vs. 275 deaths; risk reduction 19%; 95% CI 3%–32%; p = 0.019). The incidence of both fatal and nonfatal major cardiovascular events was less in those receiving captopril. For death from cardiovascular causes, the risk reduction was 21% (95% CI 5%–35%; p = 0.014) and for the development of severe heart failure, the risk reduction for those randomized to captopril was 37% (95% CI 20%–50%; p <0.001). Those receiving captopril also had fewer congestive heart failure end points requiring hospitalization and fewer recurrent myocardial infarctions (130).
The Assessment of Treatment with Lisinopril and Survival (ATLAS) (131) investigators randomized 2,006 patients with an acute myocardial infarction and clinical evidence of heart failure to receive ramipril versus placebo, with a primary end point of all-cause mortality. As in the SAVE trial, there were fewer deaths in those randomized to ramipril. Additionally, other events examined were grouped in a category called “any event” that included death, cerebrovascular accident, resistant heart failure, and recurrent myocardial infarction. There were fewer events in the ramipril as compared to the placebo group: 28% versus 35% (risk reduction 19%; p = 0.008).
|
|
|
Figure 121.7. Kaplan Meier survival curves for non–left ventricular assist device (LVAD)-treated heart failure patients demonstrates a strong relationship between survival and APACHE II scores (left panel) and survival curves for heart failure patients with APACHE II scores between 11 and 20 for both LVAD and non–LVAD-treated patients (right panel). (Right panel from Gracin N, Johnson MR, Spokes D, et al. The use of Apache II scores to select candidates for left ventricular assist device placement. J Heart Lung Transplant. 1998;17:1017–1023.) |
The Assessment of Treatment with Lisinopril and Survival (ATLAS) trial (131) differed from the other ACE inhibitor trials since it compared the safety and efficacy of low-dose and high-dose lisinopril on the outcomes of all-cause mortality, as well as cardiovascular mortality, cardiovascular hospitalizations, and combined end points (131). Those randomized to low-dose lisinopril received an average daily dose of 4.5 mg, and those randomized to the high-dose group received an average daily dose of 33.2 mg (129). Other ACE inhibitors showing survival benefit in heart failure patients include lisinopril, fosinopril, and quinapril. Trials of ACE inhibitors in survivors of myocardial infarction showing improved survival with the ACE inhibitors and less development of LV systolic dysfunction now tally approximately 100,000 patients.
Based on the above-cited overwhelming evidence of the impact of this class of drug in patients with symptomatic LV systolic dysfunction and in asymptomatic patients with LV dysfunction, ACE inhibitors are the foundation of therapy for heart failure. Thus, ACE inhibitors should be prescribed for all patients with asymptomatic LV systolic dysfunction and asymptomatic valvular heart disease (stage B), as well as in patients with symptomatic and/or refractory heart failure (stages C and D) unless the patient is intolerant to ACE inhibitors. In stage A patients, ACE inhibitors are indicated for those with diabetes mellitus and LVH.
In patients experiencing angioedema, intolerable cough, or other untoward effects of ACE inhibitors, those with stages B, C, and D LV dysfunction should be prescribed ARBs. Those patients with stage A disease meeting the requirements as above should also be prescribed ARBs if intolerant of ACE inhibitors.
Angiotensin II Receptor Blockers
Reviewing the data for the use of ARBs in heart failure begins with the Evaluation of Losartan in the Elderly (ELITE) trial. This trial (132) studied 722 ACE inhibitor–naïve patients older than age 65 years of age with LV systolic dysfunction and randomized them to receive either losartan 50 mg daily or captopril 50 mg three times a day, as tolerated. The primary end point of the ELITE trial was the tolerability measure of a persistent increase in serum creatinine of greater than or equal to 0.3 mg/dL (26.5 µmol/L) and the secondary end point was a combined one either of death or heart failure hospitalization. Analysis of the ELITE trial data determined that there was less cough in the losartan group, that the increases in serum creatinine were the same in both groups, but that the combined end point was less frequent in those randomized to losartan as compared to captopril, 9.4% vs. 13.2% (risk reduction 32%, 95% CI -4% to +55%, p = 0.075). The majority of decreased events in the losartan group were due to a decrease in all-cause mortality: 4.8% versus 8.7% (risk reduction 46%; 95% CI 5%–69%; p = 0.035). To confirm the findings found in the ELITE trial, the ELITE II trial (133) was designed as a mortality trial, this time enrolling 3,152 subjects and decreasing the enrollment age to 60 years. Patients were also stratified according to β-blocker use, and were once again randomized to the previous target dosages of losartan and captopril. With a median follow-up of 555 days, there was no difference in all-cause mortality—11.7% versus 10.4% (hazard ratio [HR] 1.13; 95% CI 0.95–1.35; p = 0.16), or in sudden death—9.0% versus 7.3% (HR 1.225; 95% CI 0.98–1.60; p = 0.08) between the two treatment groups. The investigators concluded that the differences in the first ELITE trial and ELITE II were likely different secondary to the small number of deaths seen in the original ELITE trial, and that ELITE II had four times as many study subjects and ten times more events. However, losartan was, again, well tolerated.
Valsartan, another ARB, was studied in mortality trials for both chronic heart failure and in heart failure following myocardial infarction. The Valsartan Heart Failure Trial (Val-HeFT) was designed to assess the efficacy of adding valsartan to standard therapy for heart failure (134). Val-HeFT examined 5,010 subjects with LVEFs less than 40% and NYHA class II through IV heart failure judged to be clinically stable by the investigator. Background therapy included ACE inhibitors (93%), diuretics, digoxin, and β-blockers (35%). Subjects were randomized to receive valsartan versus placebo, and randomization was stratified by β-blocker therapy. Thus, most study subjects in the valsartan group were also receiving ACE inhibitor therapy. Mortality was similar in the two treatment groups as was the adjudicated cause of death: 19.7% versus 19.4% (risk reduction 1.02; 95% CI 0.88–1.18; p = 0.80). For the combined end point of mortality and morbidity, subjects receiving valsartan had significantly fewer events than those receiving placebo: 28.8% versus 32.1% (0.87; 95% CI 0.77–0.97; p = 0.009). This end point was consistent across all prespecified subgroups: men and women, young and old, those with and without diabetes mellitus or coronary artery disease, LVEFs above and below the median, and in the different NYHA classes. The investigators did not find a difference in response to valsartan therapy based on background therapy with neurohormonal inhibitors. They examined four subgroups: those receiving ACE inhibitors or not, and those receiving β-blockers or not. In the three groups (group 1 = neither ACE inhibitor nor β-blockers, group 2 = ACE inhibitor but not β-blockers, group 3 = β-blockers but not ACE inhibitors), there was a favorable effect of valsartan on the combined end point of mortality and morbidity (p = 0.003, p = 0.002, and p = 0.037, respectively). However, in patients receiving both ACE inhibitors and β-blockers, valsartan had an adverse effect on mortality (p = 0.009) and was associated with a trend to more events in the combined end point of morbidity and mortality. The investigators were uncertain whether this finding was a chance or true interaction, as other ARB trials in heart failure have not found this same outcome. In a mortality trial of patients with myocardial infarction complicated by heart failure, 14,808 patients were randomized to receive valsartan, captopril, or valsartan plus captopril in addition to conventional therapy (135). In this trial (the VALIANT trial), valsartan was found to be noninferior to captopril with regard to mortality (p = 0.004), as well as with regard to the composite end point of fatal plus nonfatal cardiovascular events (p <0.001). The valsartan plus captopril group was found to have the most drug-related adverse events.
The Candesartan in Heart failure Assessment of Reduction in Mortality and morbidity (CHARM) program evaluated the effects of candesartan on mortality and morbidity in a variety of patients with chronic heart failure (136). Unlike the previous heart failure trials, the CHARM-Preserved arm of the program investigated the use of ARBs in heart failure patients with preserved LV systolic dysfunction (e.g., diastolic dysfunction) as well as in patients intolerant to ACE inhibitors. Similar to other trials, the CHARM-Added arm examined the results of adding candesartan to patients still symptomatic despite the presence of ACE inhibitors and other conventional therapy. In the CHARM-Preserved trial, subjects were NYHA class II through IV and baseline LVEF was greater than 40%. The primary outcome of cardiovascular death or admission to hospital for heart failure was not different for those in the candesartan versus the placebo group, although there was a trend toward fewer events in the candesartan group. As could be expected in patients intolerant to ACE inhibitors (CHARM-Alternative), patients receiving candesartan had fewer events than those receiving placebo; 33% of patients receiving candesartan met the combined end point of cardiovascular death or hospitalization for heart failure versus 40% in the placebo group (HR 0.77; 95% CI 0.60–0.81; p <0.0001). In the CHARM-Added trial, patients with LVEF <40% already on ACE inhibitors but still experiencing heart failure symptoms were randomized to candesartan versus placebo. For the primary outcome of cardiovascular death or hospitalization for heart failure, fewer events occurred in the candesartan group versus the placebo group, 38% versus 42%, respectively (HR 0.85; 95% CI 0.75–0.96; p = 0.010). The CHARM investigators did not uncover the same untoward effects as did the Val-HeFT investigators with regard to patients receiving ARBs (candesartan) plus ACE inhibitors plus β-blockers. The frequency of new diabetes was lower in the candesartan group than in the placebo group.
β-Adrenergic Receptor Antagonists
Early reports of treating heart failure patients with β-adrenergic receptor antagonists were met with skepticism (137). Then, the initial randomized, double-blind, placebo-controlled trials did not meet the primary end point of decreased mortality but did have some success with decreased need for transplantation in the Metoprolol in Dilated Cardiomyopathy (MDC) trial (138) and improvement in ejection fraction in both the MDC and Cardiac Insufficiency Bisoprolol Study (CIBIS) trials (139). The above trials used different β-adrenergic blockers with different mechanisms of action: metoprolol, a β1-selective β-blocker in the MDC trial, and bisoprolol, a nonselective β-blocker, in the CIBIS trial. Carvedilol was studied in four efficacy, dosing, and safety trials that were combined into a single report appearing in the literature in 1996. Although mortality was not a primary end point, the trial was not a single trial designed with the power to detect changes in mortality, seven deaths in the open-label run-in period were not included in the analysis, and there were a small number of deaths reported in a short followup time, the results were definitive enough for decreasing hospitalization and improving quality of life that the drug was approved for use in patients with NYHA class II and III heart failure for the end points that the program did have the power to detect. Thus, carvedilol, a nonselective β-blocker as well as an α-blocker, was the first drug approved by the Food and Drug Administration (FDA) for the treatment of heart failure, 21 years after the first report with practolol appeared in the literature.
Carvedilol was further studied in two other trials. The first of these, the Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial studied the outcome of carvedilol on survival in 2,289 patients with severe heart failure already receiving standard heart failure therapy (ACE inhibitors or ARBs plus diuretic) (140). Study subjects had an LVEF of less than 25% and had symptoms of dyspnea or fatigue at rest or on minimal exertion (NYHA class IIIB to IV) for 2 months prior to randomization. Unlike the prior studies with carvedilol, COPERNICUS found that patients randomized to carvedilol had fewer deaths (130) than those randomized to placebo (190). This was a 35% decrease in the risk of death with carvedilol (95% CI 19%–48%; p = 0.00013 and p = 0.00014 after adjustment for interim analyses). Subjects in the carvedilol group also had significantly fewer events for the combined end point of death or hospitalization, and the results with carvedilol for both the primary end point and the combined end point were the same across all prespecified groups of age, gender, LVEF, heart failure etiology, study center location, and history of prior hospitalization for heart failure within 1 year prior to randomization. Additionally, fewer patients in the carvedilol group required discontinuation of study drug for adverse events than did the placebo group. A second trial with carvedilol following acute myocardial infarction with left ventricular dysfunction Carvediol Post Infarct Survival Control in Left Ventricular Dysfuntion (CAPRICORN) study had reduced all-cause mortality, cardiovascular mortality, and recurrent, nonfatal myocardial infarctions (141). The CIBIS II trial enrolled 2,647 symptomatic heart failure patients to receive bisoprolol versus placebo, in addition to standard therapy with ACE inhibitors, to determine if bisoprolol, a selective β1 antagonist, could decrease all-cause mortality. After a follow-up of 1.3 years, bisoprolol significantly decreased all-cause mortality as well as sudden death (HR 0.56; 95% CI 0.39–0.80; p = 0.0011) (142).
Another selective β1 antagonist, extended-release metoprolol (Toprol-XL) was examined in the Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF) trial (144) for possible benefits to survival in heart failure patients. When randomized to extended-release metoprolol, in addition to optimum standard therapy for heart failure, all-cause mortality and sudden death were once again significantly decreased. However, these data should not be extrapolated to mean that any β-blocker can be used in heart failure patients. The warning bell was sounded with the Beta-blocker Evaluation of Survival Trial (BEST) trial (143), which examined the effects of bucindolol on mortality in a group of patients very similar to the study subjects in the COPERNICUS trial. Bucindolol, a nonselective β-blocker and a weak α-blocker, did not improve survival, unlike carvedilol, bisoprolol, and extended-release metoprolol. Unlike the other β-blockers studied in heart failure patients in the late 1990s, bucindolol has sympatholytic activity, meaning that it is able to decrease norepinephrine levels. Another explanation for the failure of bucindolol to improve survival is that the makeup of its heart failure cohort was designed to enroll more African Americans and women. Not only did bucindolol not improve survival, but it also worsened mortality in African Americans and those with NYHA class IV versus class III heart failure, and, although the hazard ratio for women favored bucindolol, the 95% confidence interval was wide and crossed the line of unity. Another β-blocker trial, the Carvedilol or Metoprolol European Trial (COMET), randomized subjects to receive either carvedilol or short-acting metoprolol. In this trial, carvedilol was found to improve survival and short-acting metoprolol did not. Therefore, short-acting metoprolol has been found ineffective in two trials: the COMET (145) and the MDC trial. Use of β-adrenergic receptor antagonists should be limited to those shown to improve survival in large, randomized clinical trials: carvedilol and extended-release metoprolol in the United States and bisoprolol in Europe.
Aldosterone Receptor Antagonists
Since aldosterone promotes retention of sodium, sympathetic activation, inhibition of the parasympathetic nervous system, myocardial and vascular fibrosis, and loss of magnesium and potassium, it was reasonable to propose that blockade of aldosterone receptors could alter the course of progressive LV dysfunction and, thereby, mortality. Although some may think that ACE inhibitors suppress aldosterone production, there is evidence suggesting that they do it transiently. Thus, investigators postulated that there might be a role for spironolactone, an aldosterone receptor antagonist, in the therapy for heart failure, and the Randomized Aldactone Evaluation Study (RALES) was formed (146). A total of 1,663 patients were randomized to receive spironolactone in addition to standard therapy for heart failure, including ACE inhibitors and diuretics, and sometimes digoxin and other vasodilators. Patients with hyperkalemia (K+ greater than 5.5 mmol/L) and serum creatinine greater than 2.5 mg/dL were excluded. Spironolactone successfully decreased mortality versus placebo: 35% versus 46%, respectively (relative risk of death 0.70; 95% CI 0.60–0.82; p <0.001).
Other Vasodilators
The combination of hydralazine and oral nitrates for heart failure therapy has been studied in three large randomized clinical trials. The first of these was the Veterans Administration Cooperative Vasodilator Heart Failure Trial (VeHeFT-I) (147), which enrolled 642 patients taking standard heart failure therapy—at the time, digoxin and a diuretic—to three other groups: placebo, prazosin 20 mg/day, or combination of hydralazine (300 mg/day) and isosorbide dinitrate (160 mg/day). This study determined that prazosin was equal to placebo, and that combination vasodilator therapy with hydralazine and isosorbide dinitrate reduced mortality at 2 years with a risk reduction of 34% (p <0.028). Following the publication of the CONSENSUS trial, questions arose about superiority for heart failure therapy: enalapril versus combination hydralazine and isosorbide dinitrate. Study investigators enrolled 804 men—already receiving digoxin and diuretic—in the Veterans Administration Cooperative Vasodilator Heart Failure comparison of enalapril with hydralazine-isosorbide dinitrate Trial (VeHeFT-II) to receive either enalapril 20 mg/day or combination of hydralazine 300 mg/day plus isosorbide dinitrate 160 mg/day. Mortality after 2 years was 18% in the enalapril group and 25% in the hydralazine-isosorbide dinitrate arm (RR 28%; p = 0.016) (148). Following retrospective analyses of the combined data sets of VeHeFT-I and -II, interest arose in different treatments based on racial findings in the original trials, with the understanding that race is likely a phenotypic marker of a particular genotype. These analyses showed an absence of treatment effect in Caucasians in VeHeFT-I no matter the assignment arm, but a mortality benefit for African Americans receiving combination therapy. In contrast, Caucasians had survival benefit from enalapril but not combination therapy in VeHeFT-II, whereas mortality was similar in the same trial in the two treatment groups for African Americans. Some clinical investigations found that persons who identify themselves as African American may have a less active renin–angiotensin system (149,150) and lower nitric oxide bioavailability than other racial groups (151,152). This led to the hypothesis that a survival benefit with combination therapy—hydralazine plus isosorbide dinitrate—was possible for NYHA class III and IV African Americans who remained symptomatic with heart failure despite standard therapy, including ACE inhibitors or ARBs in ACE-intolerant patients, β-blockers, digoxin, spironolactone, and diuretics. The African-American Heart Failure Trial (A-HeFT) enrolled 1,050 subjects, at which point randomization stopped, secondary to the independent data and safety monitoring board determining a significantly higher mortality rate in those receiving placebo: 54 patients in the placebo group died versus 32 patients in the combination therapy group. With combination therapy there was a 43% survival improvement (HR 0.57; p = 0.01) (153).
Digoxin
Digitalis glycosides have been used for more than 200 years since first described by Sir William Withering in his account of foxglove.1 Following acceptance of ACE inhibitors for the treatment of heart failure, the efficacy of digoxin became controversial. Two small withdrawal trials in the early 1990s studied the efficacy of digoxin in heart failure patients; in both, patients were already receiving digoxin as part of their heart failure regimen. The Randomized Study assessing the Effect of Digoxin Withdrawal in Patients with Mild to Moderate Chronic Congestive Heart Failure (PROVED) trial withdrew digoxin from 42 subjects receiving diuretic and digoxin and 46 subjects continued with both drugs. In patients withdrawn from digoxin, maximal exercise capacity worsened and patients had more treatment failure with a decreased time-to-treatment failure (154). In the Randomized Assessment of the Effect of Digoxin on Inhibitors of the Angiotensin Converting Enzyme (RADIANCE) trial, digoxin was once again withdrawn, in a randomized fashion, from patients receiving ACE inhibitors and diuretics. Exercise capacity was assessed using treadmill testing and exercise endurance was assessed using the 6-minute walk test. Functional capacity deteriorated in patients withdrawn from digoxin compared with those continuing to receive digoxin (p = 0.019 for NYHA class, p = 0.033 for maximal exercise tolerance, and p = 0.01 for submaximal exercise endurance). Additionally, left ventricular ejection fractions decreased in the placebo group and those in the placebo group also had increases in both weight and heart rate (155). Despite the evidence from the PROVED and RADIANCE trials demonstrating the efficacy of digoxin in the treatment of heart failure, concerns remained regarding the effect of digoxin on survival. The Digitalis Investigation Group trial studied the long-term effect of digoxin on all-cause mortality and heart failure hospitalizations. Study subjects were required to have left ventricular ejection fractions less than 45% and the majority were receiving diuretics and ACE inhibitors at baseline. Patients enrolled into the digoxin arm numbered 3,397, and those randomized to placebo numbered 3,403. There was no difference in mortality between those randomized to digoxin or placebo (34.8% vs. 35.1%; p = 0.80), but there were fewer hospitalizations for worsening heart failure in those randomized to digoxin than in those in the placebo group: 910 versus 1,180 (0.72; 95% CI 0.66–0.79; p <0.001). Total hospitalizations were also less frequent in those randomized to the digoxin group, but there were no differences in hospitalizations for ventricular arrhythmias or cardiac arrest between the two groups. Thus, in the era prior to β-blockers, digoxin was shown to be efficacious for treatment of heart failure and to have a neutral effect on mortality.
Acute Decompensated Heart Failure
Although acute decompensated heart failure has a variety of presentations, common symptoms include fatigue, shortness of breath, and congestion of the lungs and abdominal organs. Many disease states may present with all or some of the above symptoms, such as pneumonia, pulmonary embolus, sepsis, and other volume overload states. Patients with preserved systolic function and systemic hypertension comprise the majority of hospitalized patients. The minority are those with poor end-organ perfusion, low ejection fractions, and hypotension. Despite advances in therapeutics, both pharmacologic and devices, risk of death and rehospitalization continues to be high for patients admitted with acute decompensated heart failure. Cost of hospitalization for ADHF remains the bulk of the cost in caring for those afflicted with heart failure. The ability to measure BNP, a neurohormone secreted by the LV in response to volume overload, has become a useful diagnostic test in those presenting with symptoms of ADHF. Normal values range from 0 to 100 pg/mL, and those over 400 have a high probability for heart failure, whether systolic or diastolic in origin (156). The decision to hospitalize patients for ADHF continues to be based on clinical judgment. The Heart Failure Society of America guidelines recommend hospitalization for patients with altered mentation, worsening renal function, hypotension, resting dyspnea, hemodynamically significant arrhythmias, and acute coronary syndromes (157). They further state that hospitalization should be considered when congestion has worsened with evidence of weight gain of greater than or equal to 5 kg, and when there are signs and symptoms of abdominal or pulmonary congestion, even in the absence of weight gain. Other considerations include major electrolyte abnormalities, repeated ICD discharges, and associated comorbid conditions. Recently, the ADHERE collected data for hospitalized patients in community, tertiary, and academic centers in the United States (158,159): 33,046 hospitalization episodes from the ADHERE were analyzed for predictors of in-hospital mortality and were then subjected to a classification and regression tree (CART) analysis to develop the best predictors of in-hospital mortality as well as a risk stratification model (160). Thirty-nine variables were identified and, following the CART method, blood urea nitrogen level greater than 43 mg/dL at hospital admission was identified as the best single discriminator between survivors and nonsurvivors of hospitalization for ADHF. The second best predictor was systolic blood pressure less than 115 mm Hg.
Few trials for those with ADHF admitted to the hospital have been published. To date, only two trials of therapeutics for ADHF have been published: these are the Outcome of a Prospective Trial of Intravenous Milronone for Exacerbations (OPTIME) trial and the Vasodilation in the Management of Acute Congestive Heart Failure (VMAC) trials. Another two trials, the Evaluation Study of Congestive heart Failure And Pulmonary artery catheterization Effectiveness (ESCAPE) and the Pulmonary Artery Catheters in patient MANagement (PAC-MAN) trials, assess the effectiveness of pulmonary artery catheters in the management of ADHF. A variety of patients were enrolled in the PAC-Man trial (161). Diagnoses at enrollment included multiorgan failure, decompensated heart failure, and respiratory failure; surgical as well as medical patients were enrolled. The investigators noted no differences in hospital mortality in those managed with or without pulmonary artery catheters. Additionally, there were no differences in length of stay in either intensive care unit settings or in hospital. A 10% complication rate was noted in those receiving pulmonary artery catheters, with the most frequent complication being hematoma at the insertion site and arrhythmias requiring treatment within 1 hour of insertion. The ESCAPE trial randomized 433 subjects with decompensated heart failure to receive therapy guided by clinical assessment and a pulmonary artery catheter or by clinical assessment alone. The primary end point of the ESCAPE trial was days alive out of hospital during the first 6 months. Secondary end points included quality of life, exercise, and biochemical and echo changes. Those randomized to pulmonary artery catheters did not impact the primary end point of number of days alive and out of the hospital during 6 months of follow-up, mortality, or the number of days hospitalized. There were no deaths due to use of pulmonary artery catheters, but there were more complications, with the most common being infection (162).
The Vasodilator in the Management of Acute Heart Failure trial randomized 489 patients hospitalized with ADHF to a complicated schema testing nitroglycerin, nesiritide, and standard therapy for the treatment; based on clinical judgment, half of patients received a pulmonary artery catheter. The primary end point of the trial was a change in pulmonary capillary wedge pressure (PCWP) from baseline and change in dyspnea score from baseline at 3 hours (163). Nesiritide lowered pulmonary capillary wedge pressure from baseline by 6.5 mm Hg, and statistically significantly better than placebo (p <0.001) and nitroglycerin (p = 0.03).
Based on the paucity of data for treating patients with ADHF admitted to the hospital, current guidelines include identification of precipitating factors and etiology, optimizing volume status, treating with recommended vasodilator therapy, and minimizing side effects. Educating patients about lifestyle changes, including sodium and fluid restriction, as well as self-management techniques are recommended general measures. Precipitating factors include development of arrhythmias, acute infections, and nonadherence with medications and dietary restrictions. Optimization of medical therapy should also be attempted during hospitalization. During hospitalization, the patient should be weighed daily and accurate recordings of intake and output should be kept. Loop diuretics administered intravenously are recommended and, in those not achieving adequate diuresis, continuous intravenous infusion of loop diuretics or addition of other diuretics, such as metolazone, should be considered. In those not responding to the above measures, and without symptomatic hypotension, intravenous nitroglycerin, nitroprusside, or nesiritide may be used in addition to intravenous diuretics. Intravenous inotropes should be reserved for those with advanced heart failure and evidence of end-organ dysfunction and/or decreased peripheral perfusion, especially if systolic blood pressure is <90 mm Hg. Small studies have investigated ultrafiltration (UF) as an alternative to diuretics for volume removal since the procedure does not require central venous access but instead is able to utilize peripheral veins, such as in the antecubital fossa, with a 16-gauge, 35-cm catheter. In one study, after 24 hours 4,650 mL was removed via UF, whereas 2,838 mL was removed in the usual care group, which included diuretics. Those in the ultrafiltration group were also able to receive diuretics after the 8 hours of UF was completed. The authors concluded that UF was feasible and well tolerated (164).
Nonpharmacologic and Nonsurgical Therapy for Chronic Heart Failure
Electrophysiologic therapy is a new addition to the heart failure management arsenal and is having enormous impact. Defibrillator therapy with an Automatic Implantable Cardioverter Defibrillator (AICD) was first shown to be effective at improving long-term survival in survivors of cardiac arrest (165) and later it was proven beneficial in post-MI patients with low ejection fraction and ventricular dysrhythmia (166). Recent studies have shown its benefits in both ischemic and nonischemic dilated cardiomyopathy (167). Biventricular pacing is a newer electrophysiologic therapy that attempts to improve cardiac efficiency by reducing dyssynchrony in cardiac contraction. It also has been shown to improve NYHA class (168), exercise tolerance (169), and ejection fraction (170); mortality benefits have also been demonstrated. In this section, we will review both defibrillator and biventricular pacing and their role in managing heart failure.
Implantable Cardioverter Defibrillators
Data from the Antiarrhythmic versus Implantable Defibrillator (AVID) trial (165), the Multicenter Automatic Defibrillator Implantation Trial (MADIT) (166), the Multicenter Unstrained Tachycardia Trial (MUSTT) (171), and the Multicenter Automatic Defibrillator Implantation Trial II (172) support the use of ICDs in patients who have survived symptomatic ventricular tachycardia or cardiac arrest, patients with ischemic cardiomyopathy with ejection fraction less than 30%, and patients with asymptomatic ventricular tachycardia that is inducible.
Recently, data from the Sudden Cardiac Death in Heart Failure Trial (SCD-HEFT) (167) and Comparison of Medical Therapy, Pacing and Defibrillation in Chronic Heart Failure (COMPANION) trial (168,173) have broadened the indications to other heart failure patients, including nonischemic cardiomyopathy patients who had QRS duration greater than 120 msec, whose LVEF was less than 35%, and who had an admission for heart failure in the previous year. Patients were randomized to (a) medical therapy, (b) medical therapy and biventricular pacing, and (c) medical therapy, biventricular pacing, and defibrillator implantation. In this study, the addition of the combination therapies reduced mortality by 50% in the nonischemic group and 27% in the ischemic group.
At present, the indications for ICD for primary prevention of sudden cardiac death in heart failure (174) are:
· As primary prevention in patients with prior MI greater than 40 days old, with an ejection fraction between 30% and 40%, and who are NYHA class II or III on optimal therapy
· Patients with prior MI greater than or equal to 40 days previously, or ejection fraction between 30% and 35%
· Patients with nonischemic cardiomyopathy, ejection fraction between 30% and 35%, and NYHA class II or III on optimal medical therapy
Biventricular Pacing
Biventricular pacing is achieved by implantation of a left ventricular lead, generally via the coronary sinus to the great cardiac vein, in addition to a right ventricular lead. This strategy is based on the fact that most patients with intraventricular conduction delay have dyssynchronous left ventricular contraction, which reduces cardiac efficiency (decreased contractile force and impaired myocardial energetics). Studies have shown that biventricular pacing increases dp/dt, ejection fraction, and cardiac index (61,175,176,177,178). The COMPANION trial (noted above) demonstrated a significant decrease in mortality (50%) with the combination of biventricular pacing plus ICD compared to biventricular pacing alone (9% mortality reduction).
Cardiac Transplantation and Left Ventricular Assist Devices
When patients continue to experience advanced heart failure symptoms despite maximization of medical therapy, cardiac transplantation and left ventricular assist devices may be considered. There are currently three left ventricular assist devices available for use in the United States. The Abiomed system is available for temporary use as either right, left, or biventricular assist systems. It is most commonly used after high-risk coronary artery bypass surgery in patients who fail to wean from the cardiopulmonary bypass machine. Cannulae are placed in the atria and the pumps are external to the body. Since the cannulae are implanted in the atria, removal is feasible should the patient recover. Additionally, one ventricular system may be removed at a time; this system requires full anticoagulation. The Thoratec Heart Mate device is approved for use as a bridge to transplantation and as destination therapy in those not considered transplant candidates. The outflow cannula is placed in the proximal ascending aorta, and the inflow cannula is placed in the left ventricular apex. Cannulae are named with respect to the pump that is commonly implanted in the left upper abdominal quadrant preperitoneally. This device does not require anticoagulation, although antiplatelet therapy with aspirin is recommended. The World Heart left ventricular assist device is implanted in a similar fashion to the Heart Mate device and is currently FDA approved as a bridge to transplantation. Currently, a clinical trial is under way for its potential use as destination therapy. The World Heart left ventricular assist device requires full anticoagulation with warfarin, aspirin, and clopidogrel.
There have been 40,192 cardiac transplant procedures between 1988 and August 31, 2006; 2,125 of these procedures were performed in 2005. Kaplan-Meier survival rates for heart transplant procedures performed between 1997 and 2004 ranged from 85.7% to 90.6% at 1 year, 75.2% to 81.8% at 3 years, and 68.8% to 72.6% at 5 years. Ranges are given since the data were based on the United Network of Organ Sharing (UNOS) status at the time of transplantation. Criteria to be considered for transplantation include advanced symptoms despite maximization of medical therapy—including tailoring therapy via hemodynamic monitoring with pulmonary artery catheterization—and a peak oxygen consumption less than 15.0 mL/kg/minute during exercise testing. Exclusion criteria include fixed pulmonary hypertension with greater than 4 Wood units not responsive to vasodilator therapy; tobacco, alcohol, or illicit drug use, or other life-threatening illnesses such as cancer or advanced peripheral arterial disease; noncompliance with physician visits, diet, and medications or psychiatric or personality disorders likely to become exacerbated by, or that would interfere with, posttransplant care; and lack of social support.
Therapeutic Guidelines for Symptomatic Left Ventricular Systolic Dysfunction
Polypharmacy has become standard therapy for patients with symptomatic systolic dysfunction. ACE inhibitors are recommended for all patients fitting the above parameters. In those patients with contraindications to ACE inhibitor therapy—or if intolerable side effects develop—treatment should be with ARBs. After the introduction of ACE inhibitor or ARB therapy, a patient should be started on β-adrenergic receptor blockade consisting of either carvedilol or long-acting metoprolol (Toprol XL). β-Blockers should be instituted in patients with euvolemia, starting with low doses and titrating up the dose every 2 weeks as tolerated, to doses achieved in clinical trials; achievement of these doses may take 8 to 12 weeks. For patients being discharged from the hospital with ADHF, low-dose β-blockers may be instituted in-hospital once euvolemia has been achieved. In patients on stable doses of β-blockers who then experience an acute decompensation requiring hospitalization, continuation of β-blocker therapy is recommended. ARBs are recommended for patients intolerant to ACE inhibitors for reasons other than renal insufficiency or hyperkalemia. Hydralazine and oral nitrate combination may be considered in those not tolerating ACE inhibitors or ARBs. Aldosterone antagonists are recommended for patients with NYHA class III or IV symptoms in addition to standard therapy, including diuretics. Aldosterone antagonists are not recommended in patients with creatinine clearance less than 30 mL/minute, serum potassium greater than 5.0 mmol/L, or serum creatinine greater than 2.5 mg/dL. Guidelines recommend frequent monitoring of serum potassium levels following initiation of or change in dose of aldosterone antagonists. The combination of hydralazine and oral nitrates is recommended for African American patients—in addition to ACE inhibitors and β-blockers—who remain NYHA class III or IV despite these drugs. Loop and distal tubule diuretics should be viewed as necessary adjuncts to relieve sodium and water retention. Guidelines recommend that digoxin should be considered for symptomatic patients receiving standard therapy and that the dose should be 0.125 mg in the majority of patients. Serum digoxin level should be less than 1.0 ng/mL. Amiodarone and other antiarrhythmic medications should not be used for primary prevention of sudden death, but amiodarone may be considered in those with ICDs to decrease the frequency of repetitive ICD discharges should this become an issue with the devices. Patients with mild heart failure symptoms (NYHA class II) should restrict their sodium intake to 2 to 3 g daily. Those with more advanced symptoms (NYHA class III to IV) should restrict their sodium intake to 2 g daily. Patients with severe hyponatremia—serum sodium less than 130 mEq/L—should restrict their fluid intake to less than 2 liters per day, as should individuals in whom fluid balance is difficult to maintain despite sodium restriction and high-dose diuretics.
Summary
Heart failure is a complex syndrome characterized by both insufficient cardiac output to meet the body's energy requirements and elevated filling pressures leading to systemic and pulmonary edema. In this chapter, the pathophysiology at a biochemical, cellular, organ, and total body level was reviewed. Evidence-based therapies such as ACE inhibitors, β-blockers, ARBs, and aldosterone antagonists, and the clinical studies supporting their use have been reviewed. Novel therapies such as implantable defibrillators, biventricular pacing, and ventricular assist devices have been presented along with present recommendations for their use.
Acknowledgment
The authors would like to thank Geri Byrd for her patience, expertise, and tireless effort in the preparation of this manuscript. Thanks to Kristen Wienandt Marzejon for preparation of the figures included in the text.
References
1. Thom T, Haase N, Rosamond W, et al. Heart disease and stroke statistics–2006 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2006;113(6):e85–151.
2. National Hospital Discharge Survey CDC/National Center for Health Statistics (NCHS). Vital Health Stat. 2005;225 (July 10).
3. National Center for Health Statistics (NCHS). Vital Health Stat. 2004;13:157.
4. Lloyd-Jones DM, Larson MG, Lup EP, et al. Lifetime risk for developing congestive heart failure. The Framingham Heart Study. Circulation. 2002;106:3068–3072.
5. Bibbins-Domingo K, Lin F, Vittinghoff E, et al. Predictors of heart failure among women with coronary disease. Circulation. 2004;110:1424–1430.
6. Dunlap SH, Sueta CA, Tomasko L, et al. Association of body mass, gender and race with heart failure primarily due to hypertension. J Am Coll Cardiol. 1999;34:1602–1608.
7. Liggett SB, Mialet-Perez J, Thaneemit-Chen S, et al. A polymorphism within a conserved beta (1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proc Nat Acad Sci USA. 2006;103(30):11288–11293.
8. Housmans PR, Lee NKM, Blinks JR. Active shortening retards the decline of the intracellular calcium transient in mammalian heart muscle. Science. 1983;221:159.
9. Brutsaert DL, Rademakers FE, Sys SU. Triple control of relaxation: implications in cardiac disease. Circulation. 1984;69:190.
10. Kohmoto O, Spitzer KW, Movsesian MA, et al. Effects of intracellular acidosis on [Ca2+]i transients, transsarcolemmal C2+ fluxes, and contraction in ventricular myocytes. Circ Res. 1990;66:622.
11. Hoffman BF, Bindler E, Suckling EE., et al. Postextrasystolic potentiation of contraction in cardiac muscle. Am J Physiol. 1956;185:95.
12. Ross J, Sonnenblick EH, Kaiser GA, et al. Electroaugmentation of ventricular performance and oxygen consumption by repetitive application of paired electrical stimuli. Circ Res. 1965;16:332.
13. Higgins CB, Vatner SF, Franklin D, et al. Extent of regulation of the heart's contractile state in the conscious dog by alteration in the frequency of contraction. J Clin Invest. 1973;52:1187.
14. Katz AM. Regulation of myocardial contractility 1958–1983: an odyssey. J Am Coll Cardiol. 1983;1:42.
15. Colluci WS, Wright RF, Braunwald E. New positive inotropic agents in the treatment of congestive heart failure (second of two parts). N Engl J Med. 1986;314:349.
16. Lee HC, Smith N, Mohabir R, et al. Cytosolic calcium transients from the beating mammalian heart. Physiol Sci. 1987;84:7793.
17. Lang RM, Fellner SK, Neumann A, et al. LV contractility varies directly with blood ionized calcium. Ann Intern Med. 1988;108:524.
18. MacKinnon R, Gwathmey JK, Allen PD, et al. Modulation by the thyroid state of intracellular calcium and contractility in ferret ventricular muscle. Circ Res. 1988;63:1080.
19. Morkin E, Flink IL, Goldman S. Biochemical and physiologic effects of thyroid hormone on cardiac performance. Prog Cardiovasc Dis. 1983;25:435.
20. Braunwald E, Sommenblick EH, Ross J Jr. Mechanisms of cardiac contraction and relaxation in heart disease. In: Braunwald E, ed. A Textbook of Cardiovascular Medicine. 4th ed. Philadelphia: W.B. Saunders Co.; 1992:351.
21. Beierholm EA, Grantham RN, O'Keefe DD, et al. Effects of acid-base changes, hypoxia, and catecholamines on ventricular performance. Am J Physiol. 1975;228:1555.
22. Williamson JR, Schaffer SW, Ford C, et al. Contribution of tissue acidosis to ischemic injury in the perfused rat heart. Circulation. 1976;53(Suppl 1):3.
23. Braunwald E. Mechanism of action of calcium-channel-blocking agents. N Engl J Med. 1982;307:1618.
24. Levine MJ, Harada K, Meuse AJ, et al. Excitation contraction uncoupling during ischemia in the blood perfused dog heart. Biochem Biophys Res Comm. 1991;179:502.
25. Hicks MJ, Shigekawa M, Katz AM. Mechanism by which cyclic adenosine 3′:5′-monophosphate-dependent protein kinase stimulates calcium transport in cardiac sarcoplasmic reticulum. Circ Res. 1979;44:384.
26. Gwathmey JK, Slawsky MT, Hajjar RJ, et al. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85:1599.
27. Gwathmey JK, Copelas L, MacKinnon R, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70.
28. Limas CJ, Olivari M, Goldenberg IF, et al. Calcium uptake by cardiac sarcoplasmic reticulum in human dilated cardiomyopathy. Cardiovasc Res. 1987;21:601.
29. Mercadier J, Lompré A, Duc P, et al. Altered sarcoplasmic reticulum CA2+/ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85:305.
30. Feldman MD, Copelas L, Gwathmey JK, et al. Deficient production of cyclic AMP: pharmacologic evidence of an important cause of contractile dysfunction in patients with end-stage heart failure. Circulation. 1987;75:331.
31. Feldman AM, Ray PE, Silan CM, et al. Selective gene expression in failing human heart. Circulation. 1991;83:1866.
32. Ungerer M, Bohm M, Elce JS, et al. Altered expression of β-adrenergic receptor kinase and β1-adrenergic receptors in the failing human heart. Circulation. 1993;87:454.
33. Bristow MR, Hershberger RE, Port JD, et al. β-adrenergic pathways in nonfailing and failing human ventricular myocardium. Circulation. 1990;82(Suppl 1):12.
34. Feldman AM, Cates AE, Veazey WB, et al. Increase of the 40,000-mol wt pertussis toxin substrate (G protein) in the failing human heart. J Clin Invest. 1988;82:189.
35. Gilman AG. G proteins: transducers of receptor-generated signals. Ann Rev Biochem. 1987;56:615.
36. Spiegel AM, Gierschik P, Levine MA, et al. Clinical implications of guanine nucleotide-binding proteins as receptor-effector couplers. N Engl J Med. 1985;312:26.
37. Katz AM. Cyclic adenosine monophosphate effects on the myocardium: a man who blows hot and cold with one breath. J Am Coll Cardiol. 1983;2:143.
38. Homcy CJ, Graham RM. Molecular characterization of adrenergic receptors. Circ Res. 1985;56:635.
39. Neer EJ, Clapham DE. Roles of G protein subunits in transmembrane signaling. Nature. 1988;333:129.
40. Colluci WS, Wright RF, Braunwald E. New positive inotropic agents in the treatment of congestive heart failure (first of two parts). N Engl J Med. 1986;314:290.
41. Yatani A, Brown AM. Rapid β-adrenergic modulation of cardiac calcium channel currents by a fast G protein pathway. Science. 1989;245:71.
42. Mery PF, Lohmann SM, Walter U, et al. Ca2+ current is regulated by cyclic GMP-dependent protein kinase in mammalian cardiac myocytes. Proc Natl Acad Sci. 1991;88:1197.
43. Nawrath H. Cyclic AMP and cyclic GMP may play opposing roles in influencing force of contraction in mammalian myocardium. Nature. 1976;262:509.
44. Schranz D, Droege A, Broede A, et al. Uncoupling of human cardiac β-adrenoceptors during cardiopulmonary bypass with cardioplegic cardiac arrest. Circulation. 1993;87:422.
45. Kumar A, Kosuri R, Kandula P, et al. Tumor necrosis factor-induced myocardial cell depression is mediated by nitric oxide and cyclic GMP generation. Crit Care Med. 1994;22:A191.
46. Schneider F, Lutun P, Hasselmann M, et al. Methylene blue increases systemic vascular resistance in human septic shock. Int Care Med. 1992;18:309.
47. Silverman HJ, Penaranda R, Orens JB. Impaired β-adrenergic receptor stimulation of cyclic adenosine monophosphate in human septic shock: association with myocardial hyporesponsiveness to catecholamines. Crit Care Med. 1993;21:31.
48. Yanagisawa M, Kurihara H, Kimura S, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411.
49. Kramer BK, Nishida M, Kelly RA, et al. Myocardial actions of a new class of cytokines. Circulation. 1992;85:350.
50. Kelly RA, Eid H, Kramer BK, et al. Endothelin enhances the contractile responsiveness of adult rat ventricular myocytes to calcium by a pertussis toxin-sensitive pathway. J Clin Invest. 1990;86:1164.
51. Kramer BK, Smith TW, Kelly RA, et al. Endothelin and increased contractility in adult rat ventricular myocytes: role of intracellular alkalosis induced by activation of the protein kinase C-dependent Na+-H+. Circ Res. 1991;68:269.
52. del Monte F, Hajjar RJ. Targeting calcium cycling proteins in heart failure through gene transfer. J Physiol. 2003;546(Pt 1):49–61.
53. Placentino V III, Weber CR, Chen X, et al. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651.
54. Hasenfuss G, Mulieri LA, Leavitt BJ, et al. Alteration of contractile function and excitation-contraction coupling in dilated cardiomyopathy. Circ Res. 1992;70(6):1225–1232.
55. Flesch M, Schwinger RH, Schnabel P, et al. Sarcoplasmic reticulum Ca2+ATPase and phospholamban mRNA and protein levels in end-stage heart failure due to ischemic or dilated cardiomyopathy. J Mol Med. 1996;74(6):321–332.
56. Hobai IA, O'Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103(11):1577–1584.
57. Meyer M, Bluhm WF, He H, et al. Phospholamban-to-SERCA2 ratio controls the force-frequency relationship. Am J Physiol. 1999;276(3 Pt 2):H779–785.
58. Marks AR, Reiken S, Marx SO. Progression of heart failure: is protein kinase a hyperphosphorylation of the ryanodine receptor a contributing factor? Circulation. 2002;105(3):272–275.
59. Shannon TR, Ginsburg KS, Bers DM. Quantitative assessment of the SR Ca++ leak load relationship. Circ Res. 2002;91:594.
60. Abraham WT, Gilbert EM, Lowes BD, et al. Coordinate changes in Myosin heavy chain isoform gene expression are selectively associated with alterations in dilated cardiomyopathy phenotype. Mol Med. 2002;8(11):750–760.
61. Lowes BD, Minobe W, Abraham WT, et al. Changes in gene expression in the intact human heart. Down regulation of alpha myosin heavy chain in hypertrophied failing ventricular myocardium. J Clin Invest. 1997;100:2362.
62. Thomas CV, Coker ML, Zellner JL, et al. Increased matrix metalloproteinase activity and selective up regulation in LV myocardium from patients with end-stage dilated cardiomyopathy. Circulation. 1998;97(17):1708–1715.
63. Coker ML, Thomas CV, Clair MJ, et al. Myocardial matrix metalloproteinase activity and abundance with congestive heart failure. Am J Physiol. 1998;274:H1516–1523.
64. Meerson, FZ. The myocardium in hyperfunction, hypertrophy, and heart failure. Circ Res. 1998;25(Suppl 2):1.
65. Chaudhry PA, Anagnostopouls PV, Mishima T, et al. Acute ventricular reduction with the acorn cardiac support device: effect on progressive left ventricular dysfunction and dilation in dogs with chronic heart failure. J Card Surg. 2001;16(2):118–126.
66. Francis GS, Cohn JN, Johnson G, et al. Plasma norepinephrine plasma renin activity and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. Circulation. 1993;87:VI-400.
67. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. 2000;35(3):569–582.
68. Grossman W, Jones D, McLaurin LP. Wall stress and patterns of hypertrophy in the human left ventricle. J Clin Invest. 1975;56(1):56–64.
69. Calderone A, Takahashi N, Izzo NJ, et al. Pressure- and volume-induced left ventricular hypertrophies are associated with distinct myocyte phenotypes and differential induction of peptide growth factor mRNAs. Circulation. 1995;92(9):2385–2390.
70. Hunter JJ, Chien K. Signaling pathways for cardiac hypertrophy and failure. N Engl J Med. 1999;341(17):1276–1283.
71. Kang PM, Yue P, Izumo S. New insights into the role of apoptosis in cardiovascular disease. Circ J. 2002;66(1):1–9.
72. Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–823.
73. Bristow MR, Ginsburg R, Minobe W, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307(4):205–222.
74. Mann DL, Kent RL, Parsons B, et al. Adrenergic effects on the biology of the adult mammalian cardiocyte. Circulation. 1992;85:790–804.
75. The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1992;325:293–302.
76. The SOLVD Investigators. Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. N Engl J Med. 1992;327:685–691.
77. White HD, Norris RM, Brown MA, et al. Left ventricular and end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76:44–51.
78. Borow KM, Lang RM, Neumann A, et al. Physiologic mechanisms governing hemodynamic responses to positive inotropic therapy in dilated cardiomyopathy. Circulation. 1988;77:625–637.
79. Douglas PS, Morrow R, Ioli A, et al. Left ventricular shape, afterload and survival in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 1989;13:311–315.
80. The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316:1429–1435.
81. Pfeffer MA, Braunwald E, Moye LA, et al.; SAVE Investigators. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction: results of the Survival and Ventricular Enlargement Trial. N Engl J Med. 1992;327:669–677.
82. Gilbert JC, Glantz SA. Determinants of LV filling and of the diastolic P-V relation. Circ Res. 1989;64:827.
83. Gaasch WH, Levine HJ, Quinones MA, et al. LV compliance: mechanisms and clinical implications. Am J Cardiol. 1976;38:645.
84. Nikolic SD, Tamura K, Tamura T, et al. Diastolic viscous properties of the intact canine left ventricle. Circ Res. 1990;67:352.
85. Momomura S, Ingwall JS, Parker JA, et al. The relationships of high energy phosphates, tissue pH, and regional blood flow to diastolic distensibility in the ischemic dog myocardium. Circ Res. 1985;57:822.
86. Lorell BH, Grossman W. Cardiac hypertrophy: the consequences for diastole. J Am Coll Cardiol. 1987;9:1189.
87. Cuocolo A, Sax FL, Brush JE, et al. LV hypertrophy and impaired diastolic filling in essential hypertrophy and impaired diastolic filling in essential hypertension. Circulation. 1990;81:978.
88. Apstein CS, Grossman W. Opposite initial effects of supply and demand ischemia on LV diastolic compliance: the ischemia-diastolic paradox. J Mol Cell Cardiol. 1987;19:119.
89. Paulus WJ, Grossman W, Serizawa T, et al. Different effects of two types of ischemia on myocardial systolic and diastolic function. Am J Physiol. 1985;248:H719.
90. Gordon AM, Huxley AF, Julian FJ. The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol. 1966;184:170.
91. Milnor WR. Cardiac dynamics. In: Milnor WR, ed. Hemodynamics. Baltimore: Williams & Wilkins Co.; 1982:244.
92. Ikenouchi H, Kohmoto O, McMillan M, et al. Contributions of [Ca2+]i, [Pi], and pHi to altered diastolic myocyte tone during partial metabolic inhibition. J Clin Invest. 1991;88:55.
93. Sasayama S, Nonogi H, Miyazaki S, et al. Changes in diastolic properties of the regional myocardium during pacing-induced ischemia in human subjects. J Am Coll Cardiol. 1985;5:599.
94. Miyazaki S, Guth BD, Miura T, et al. Changes of LV diastolic function in exercising dogs without and with ischemia. Circulation. 1990;81:1058.
95. Nikolic S, Yellin EL, Tamura K, et al. Effect of early diastolic loading on myocardial relaxation in the intact canine left ventricle. Circ Res. 1990;66:1217.
96. Stool EW, Mullins CB, Leshin SJ, et al. Dimensional changes of the left ventricle during acute pulmonary arterial hypertension in dogs. Am J Cardiol. 1974;33:868.
97. Glantz SA, Misbach GA, Moores WY, et al. The pericardium substantially affects the LV diastolic P-V relationship in the dog. Circ Res. 1978;42:433.
98. Calvin JE, Baer RW, Glantz SA. Pulmonary injury depresses cardiac systolic function through Starling mechanism. Am J Physiol. 1986;251:H722.
99. Cassidy SS, Ramanathan M. Dimensional analysis of the left ventricle during PEEP: relative septal and lateral wall displacements. Am J Physiol. 1984;246:H792.
100. Tyson GS, Maier GW, Olsen CO, et al. Pericardial influences on ventricular filling in the conscious dog. Circ Res. 1984;54:173.
101. Slinker BK, Glantz SA. End-systolic and end-diastolic ventricular interaction. Am J Physiol. 1986;251:H1062.
102. Calvin JE. Optimal RV filling pressures and the role of pericardial constraint in RV infarction in dogs. Circulation. 1991;84:852.
103. Calvin JE, Langlois S, Garneys G. Ventricular interaction in a canine model of acute pulmonary hypertension and its modulation by vasoactive drugs. J Crit Care. 1988;3:43.
104. Visner MS, Arentzen CE, O'Connor MJ, et al. Alterations in LV three-dimensional dynamic geometry and systolic function during acute RV hypertension in the conscious dog. Circulation. 1983;67:353.
105. Ludbrook PA, Byrne JD, McKnight RC. Influence of RV hemodynamics on LV diastolic P-V relations in man. Circulation. 1979;59:21.
106. Calvin JE, Baer RW, Glantz SA. Pulmonary artery constriction produces a greater RV dynamic afterload than lung microvascular injury in the open chest dog. Circ Res. 1985;56:40.
107. Calvin JE, Quinn B. RV pressure overload during acute lung injury: cardiac mechanics and the pathophysiology of RV systolic dysfunction. J Crit Care. 1989;4:251.
108. Calvin JE. RV afterload mismatch during acute pulmonary hypertension and its treatment with dobutamine: a pressure segment length analysis in a canine model. J Crit Care. 1989;4:239.
109. Vlahakes GJ, Turley K, Hoffman JIE. The pathophysiology of failure in acute RV hypertension: hemodynamic and biochemical correlations. Circulation. 1981;63:87.
110. Gold FL, Bache RJ. Transmural right ventricular blood flow during acute pulmonary artery hypertension in the sedated dog. Circ Res. 1982;51:196–204.
111. Taylor R, Covell J, Sonnenblick E, et al. Dependence of ventricular distensibility on filling of the opposite ventricle. Am J Physiol. 1967;213:711–718.
112. Kingma I, Tyberg J, Smith E, et al. Effects of diastolic transeptal pressure gradient on ventricular septal position and motion. Circulation. 1983;68(6):1304–1314.
113. Chobanian AV, Bakris GL, Black HR, et al. Seventh Report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension. 2003;42:1206–1252.
114. American Diabetes Association. Position Statement: Standards of Medical Care in Diabetes—2006. Diabetes Care. 2006;29(Supp 1):S4–S42.
115. Smith SC, Allen J, Blair SN, et al. AHA/ACC Guidelines for Secondary Prevention for Patients with Coronary and Other Atherosclerotic Vascular Disease: 2006 update: endorsed by the National Heart, Lung, and Blood Institute. Circulation. 2006;113:2363–2372.
116. Levy D, Kenchaiah S, Larson MG, et al. Long-term trends in the incidence of and survival with heart failure. N Engl J Med. 2002;347(18):1397–1402.
117. Ho KK, Anderson KM, Kannel WB, et al. Survival after the onset of congestive heart failure in Framingham Heart Study subjects. Circulation. 1993;88(1):107–115.
118. Rector TS, Cohn JN. Prognosis in congestive heart failure. Ann Rev Med. 1994;45:341–350.
119. Deedwania PC. The key to unraveling the mystery of mortality in heart failure: an integrated approach. Circulation. 2003;107(13):1719–1721.
120. Bart BA, Shaw LK, McCants CB, et al. Clinical determinants of mortality in patients with angiographically diagnosed ischemic or nonischemic cardiomyopathy. J Am Coll Cardiol. 1997;30(4):1002–1008.
121. Metra M, Faggiano P, D'Aloia A, et al. Use of cardiopulmonary exercise testing with hemodynamic monitoring in the prognostic assessment of ambulatory patients with chronic heart failure. J Am Coll Cardiol. 1999;33(4):943–950.
122. Myers J, Gullestad L, Vagelos R, et al. Clinical, hemodynamic, and cardiopulmonary exercise test determinants of survival in patients referred for evaluation of heart failure. Ann Intern Med. 1998;129(4):286–293.
123. Bittner V, Weiner DH, Yusuf S, et al. Prediction of mortality and morbidity with a 6 minute walk test in patients with left ventricular dysfunction. JAMA. 1993;270:1702.
124. Francis GS, Cohn JN, Johnson G, et al. Plasma norepinephrine, plasma renin activity, and congestive heart failure. Relations to survival and the effects of therapy in V-HeFT II. The V-HeFT VA Cooperative Studies Group. Circulation. 1993;87(6 Suppl):VI40–48.
125. Anand IS, Fisher LD, Chiang YT, et al. Changes in brain natriuretic peptide and norepinephrine over time and mortality and morbidity in the Valsartan Heart Failure Trial (Val-HeFT). Circulation. 2003;107(9):1278–1283.
126. Berger R, Stanek B, Frey B, et al. B-type natriuretic peptides (BNP and PRO-BNP) predict long-term survival in patients with advanced heart failure treated with atenolol. J Heart Lung Transplant. 2001;20(2):251.
127. Hulsmann M, Stanek B, Frey B, et al. Value of cardiopulmonary exercise testing and big endothelin plasma levels to predict short-term prognosis of patients with chronic heart failure. J Am Coll Cardiol. 1998;32(6):1695–1700.
128. Teskey RJ, Calvin JE, McPhail I. Disease severity in the coronary care unit. Chest. 1991;100(6):1637–1642.
129. Gracin N, Johnson MR, Spokas D, et al. The use of APACHE II scores to select candidates for left ventricular assist device placement. J Heart Lung Transplant. 1998;17:1017–1023.
130. The Acute Infarction Ramipril Efficacy (AIRE) Study Investigators. Effect of ramipril on mortality and morbidity of survivors of acute myocardial infarction with clinical evidence of heart failure. Lancet. 1993;342(8875):821–828.
131. Packer M, Poole-Wilson PA, Armstrong PW, et al. Comparative effects of low and high doses of the angiotensin-converting enzyme inhibitor, lisinopril, on morbidity and mortality in chronic heart failure. Circulation. 1999;100:2312–2318.
132. Pitt B, Segal R, Martinez FA, et al.; ELITE Study Investigators. Randomized trial of losartan versus captopril in patients over 65 with heart failure (Evaluation of Losartan in the Elderly study, ELITE). Lancet. 1997;349:747–752.
133. Pitt B, Poole-Wilson PA, Segal R, et al.; ELITE II Investigators. Effect of losartan compared with captopril on mortality in patients with symptomatic heart failure; randomized trial—the Losartan Heart Failure Survival study ELITE II. Lancet. 2000;355:1582–1587.
134. Cohn JN, Tognoni G; Valsartan Heart Failure Trial Investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–1675.
135. Pfeffer MA, McMurray JJV, Velazquez EJ, et al. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction or both. N Engl J Med. 2003;349:1893–1906.
136. Pfeffer MA, Swedberg K, Granger CB, et al.; CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–766.
137. Waagstein F, Hjalmarson A, Varnauskas E, et al. Effect of chronic beta-adrenergic receptor blockade in congestive cardiomyopathy. Br Heart J. 1975;37:1022–1036.
138. Waagstein F, Bristow MR, Swedberg K, et al. Beneficial effects of metoprolol in idiopathic dilated cardiomyopathy. Lancet. 1993;342:1441–1446.
139. CIBIS Investigators and Committees. A randomized trial of beta-blockade in heart failure: the Cardiac Insufficiency Bisoprolol Study (CIBIS). Circulation. 1994;90:1765–1773.
140. Packer M, Coats AJS, Fowler MB, et al.; Carvedilol Prospective Randomized Cumulative Survival Study Group. Effect of carvedilol on survival in severe chronic heart failure. N Engl J Med. 2001;344:1651–1658.
141. The CAPRICORN Investigators. Effect of carvedilol on outcome after myocardial infarction in patients with left ventricular dysfunction: the CAPRICORN randomised trial. Lancet. 2001;357:1385–1390.
142. CIBIS-II Investigators and Committees. The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II) a randomised trial. Lancet. 1999;353:9–13.
143. Domanski MJ, Krause-Steinrauff H, Massie BM, et al. A comparative analysis of the results from 4 trials of beta-blocker therapy for heart failure: BEST, CIBIS-II, MERIT-HF, and Copernicus. J Card Fail. 2003;9(5):354–363.
144. Goldstein S, Fagerberg B, Hjalmarson A, et al. Metoprolol controlled release/extended release in patients with severe heart failure: analysis of the experience in the MERIT-HF study. J Am Coll Cardiol. 2001;38(4):932–938.
145. Poole-Wilson PA, Swedberg K, Cleland JGF, et al., for the COMET Investigators. Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13.
146. Pitt B, Zannad F, Remme WJ, et al.; Randomized Aldactone Evaluation Study Investigators. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N Engl J Med. 1999;341:709–717.
147. Cohn JN, Archibald DG, Ziesche S, et al. Effect of vasodilator therapy on mortality in chronic congestive heart failure. results of a veterans administration cooperative study. N Engl J Med. 1986;314:1547–1552.
148. Cohn JN, Johnson G, Ziesche S, et al. A comparison of enalapril with hydralazine-isosorbide dinitrate in the treatment of chronic congestive heart failure. N Engl J Med. 1991;325:303–310.
149. Yancy CW. Heart failure in African Americans: a cardiovascular enigma. J Card Fail. 2000;6:183–186.
150. Gillum RF. Pathophysiology of hypertension in blacks and whites: a review of the basis of racial blood pressure differences. Hypertension. 1979;1:468–475.
151. Hinderliter AL, Sager AR, Sherwood A, et al. Ethnic differences in forearm vasodilator capacity. Am J Cardiol. 1996;78:208–211.
152. Stein CM, Lang CC, Nelson R, et al. Vasodilation in black Americans: attenuated nitric oxide-mediated responses. Clin Pharmacol Ther. 1997;62:436–443.
153. Taylor AL, Ziesche S, Yancy C, et al.; African-American Heart Failure Trial Investigators. Combination of isosorbide dinitrate and hydralazine in blacks with heart failure. N Engl J Med. 2004;351:2049–2057.
154. Uretsky BF, Young JB, Shahidi FE, et al. Randomized study assessing the effect of digoxin withdrawal in patients with mild to moderate chronic congestive heart failure: results of the PROVED trial. J Am Coll Cardiol. 1993;22:955–962.
155. Packer M, Gheorghiade M, Young JB, et al.; The RADIANCE Study. Withdrawal of digoxin from patients with chronic heart failure treated with angiotensin-converting–enzyme inhibitors. N Engl J Med. 1993;329:1–7.
156. Maisel AS, McCord J, Nowak RM, et al. Bedside B-type natriuretic peptide in the emergency diagnosis of heart failure with reduced or preserved ejection fraction. Results from the breathing not properly multinational study. J Am Coll Cardiol. 2003;41:2010–2017.
157. Heart Failure Society of America 2006 comprehensive heart failure practice guideline. J Card Fail. 2006;12.
158. Fonarow GC; ADHERE Scientific Advisory Committee. The Acute Decompensated Heart Failure National Registry (ADHERE): opportunities to improve care of patients hospitalized with acute decompensated heart failure. Rev Cardiovasc Med. 2003;4(suppl 7):S21–S30.
159. Adams KF, Fonarow GC, Eneman CL, et al. Characteristics and outcomes of patients hospitalized for heart failure in the United States (2001–2003): rationale, design and preliminary observations from the Acute Decompensated Heart Failure National Registry (ADHERE). Am Heart J. 2005;149(2):209–216.
160. Fonarow GC, Adams KF, Abraham WT, et al.; ADHERE Scientific Advisory Committee, Study Group, and Investigators. Risk stratification for in-hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA. 2005;293:572–580.
161. Harvey S, Harrison DA, Singer M, et al.; PAC-Man study collaboration. Assessment of the clinical effectiveness of Pulmonary Artery Catheters in Management of patients in intensive care (PAC-Man); a randomized controlled trial. Lancet. 2005;366:472–477.
162. The ESCAPE Investigators and ESCAPE Study Coordinators. Evaluation study of congestive heart failure and pulmonary artery catheterization effectiveness: the ESCAPE trial. JAMA. 2005;294:1625–1633.
163. Publication Committee for the JMAC Investigators. Intravenous nesiritide vs. nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA. 2002;287:1531–1540.
164. Bart BA, Boyle A, Bank AJ, et al. Ultrafiltration versus usual care for hospitalized patients with heart failure: the Relief for Acutely Fluid-overloaded Patients with Decompensated Congestive Heart Failure (RAPID-CHF) trial. J Am Coll Cardiol. 2005;46:2043–2046.
165. AVID Investigator. A comparison of antiarrhythmic-drug therapy with implantable defibrillators in patients resuscitated from near-fatal ventricular arrhythmias. The Antiarrhythmics versus Implantable Defibrillators. N Engl J Med. 1997;337(22):1576–1583.
166. MADIT Investigator. Improved survival with an implanted defibrillator in patients with coronary artery disease at high risk for ventricular arrhythmia. N Engl J Med. 1996;335:1933–1940.
167. Mark DB, Nelson CL, Anstrom KJ, et al. SCD-HeFT Investigators. Cost-effectiveness of defibrillator therapy or a amiodarone in chronic stable heart failure: results from the sudden cardiac death in heart failure trial (SCD-Heft). Circulation. 2006;114(2):135–142.
168. Bristow MR, Feldman AM, Saxon LA. Heart failure management using implantable devices for ventricular resynchronization: Comparison of Medical Therapy, Pacing, and Defibrillation in Chronic Heart Failure (COMPANION) trial. COMPANION Steering Committee and COMPANION clinical investigators. J Card Fail. 2000;6(3):276–285.
169. Leclercq C, Cazeau S, Le Breton H, et al. Acute hemodynamic effects of biventricular DDD pacing in patients with end-stage heart failure. J Am Coll Cardiol. 1998;32(7):1825–1831.
170. Kass DA, Chen CH, Curry C, et al. Improved left ventricular mechanics from acute VDD pacing in patients with dilated cardiomyopathy and ventricular conduction delay. Circulation. 1999;99:1567–1573.
171. Buxton AE, Lee KL, Fisher JD, et al. A randomized study of the prevention of sudden death in patients with coronary artery disease. N Engl J Med. 1999;341:1882–1890.
172. Moss AJ, Zareba W, Hall WJ, et al. Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med. 2002;346(12):877–883.
173. Bristow MR, Saxon LA, Boehmer, et al. Cardiac resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure: the COMPANION trial. N Engl J Med. 2004;350:2140.
174. Zipes, DP, Camm AJ, Borggrefe M, et al.; Writing Committee Members. ACC/AHA/ESC 2006 guidelines for management of patients with ventricular arrhythmias and the prevention of sudden cardiac death—executive summary. Circulation. 2006;114:1008–1132.
175. Kerwin WF, Botvinick EH, O'Connell JW. Ventricular contraction abnormalities in dilated cardiomyopathy: effect of biventricular pacing to correct interventricular dyssynchrony. J Am Coll Cardiol. 2000;35:1221–1227.
176. Cazeau S, Leclercq C, Lavergne T, et al. Effects of multisite biventricular pacing in patients with heart failure and intraventricular conduction delay. N Engl J Med. 2001;344(12):873–880.
177. Saxon LA, De Marco T, Schafer J, et al. Effects of long-term biventricular stimulation for resynchronization on echocardiographic measures of remodeling. Circulation. 2002;105(11):1304–1310.
178. St John Sutton MG, Plappert T, Abraham WT, et al. Effect of cardiac resynchronization therapy on left ventricular size and function in chronic heart failure. Circulation. 2003;107(15):1985–1990.