Edward Valenstein
Matthew Musulin
Neuromuscular disorders are commonly encountered in the critical care setting. Patients with known neuromuscular disorders, such as Guillain-Barré syndrome or myasthenia gravis, are admitted when weakness compromises the patient's ability to protect his or her airway or threatens ventilatory failure. Patients admitted with ventilatory failure of unknown cause may be diagnosed with a neuromuscular disorder such as amyotrophic lateral sclerosis or myasthenia gravis. Rarely, treatment may unmask a pre-existing neuromuscular disorder, as when drugs with neuromuscular blocking properties are given to patients with myasthenia gravis. Finally, patients with nonneuromuscular critical illnesses frequently develop neuromuscular weakness as a result of their illness or its treatment (1).
Intensive care physicians need to be familiar with the principles underlying diagnosis and management of patients with neuromuscular disease. Because electrophysiologic testing is often necessary to diagnose neuromuscular disorders in the intensive care setting, they should also have a basic appreciation of the techniques and indications for these studies. They should be familiar with the common neuromuscular disorders that require intensive care; Guillain-Barré syndrome and myasthenia gravis are considered in some detail because they are prevalent and treatable, and because, with proper treatment, they often carry a favorable prognosis. Finally, the intensive care physician will be responsible for recognizing and managing neuromuscular disorders that result from critical illness, which are now the neuromuscular disorders most commonly encountered in the intensive care setting (2).
General Considerations in the Diagnosis of Neuromuscular Disorders
As with central nervous system (CNS) disorders, the first step in the diagnosis of neuromuscular disorders is localization. Neuromuscular disorders comprise disorders of the anterior horn cell, the peripheral nervous system (PNS), the neuromuscular junction, and muscle (Fig. 149.1). Disorders of the PNS are further subdivided into disorders of roots, plexus, and peripheral nerve; and disorders of peripheral nerve are further subdivided into focal neuropathies, multifocal neuropathies, and polyneuropathies. Polyneuropathies, in turn, can be either length dependent or non–length dependent. Each of these nine major localizations has a characteristic pattern of symptoms and physical findings (Table 149.1). Localization is not just an academic exercise; each localization allows the physician to narrow the range considerably of likely diagnoses (Fig. 149.1). For example, if deficits are restricted to the distribution of a single nerve, the diagnosis is probably nerve entrapment, compression, or trauma. Multiple individual peripheral nerve involvement, in the absence of multiple trauma, should suggest an autoimmune neuropathy, often a vasculitis. Length-dependent polyneuropathy affects the longest nerves first, and consequently presents with deficits in the feet and legs before affecting the hands. Proximal strength is preserved until late. The differential diagnosis is wide, but comprises principally metabolic, toxic, or inherited neuropathies. Non–length-dependent localization is suggested when proximal weakness is greater than would be expected with a length-dependent polyneuropathy. Non–length-dependent polyneuropathies are overwhelmingly caused by autoimmune processes, including Guillain-Barré syndrome, but critical illness polyneuropathy may also be considered non–length dependent.
Recognition that a patient's problem is related to a disorder of nerve, neuromuscular junction, or muscle is relatively straightforward in patients who can give a history of their illness, and who can be adequately examined. If the problem is exclusively neuromuscular, features suggestive of central nervous system localization should be absent. These include mental status changes, hemibody weakness, or numbness suggestive of hemispheric or lateralized brainstem lesions; truncal motor or sensory level suggestive of spinal cord localization; increased muscle tone; hyperactive tendon reflexes; and pathologic reflexes such as the Babinski response. Features consistent with localization to the peripheral nervous system include weakness, atrophy, decreased or absent tendon reflexes, and sensory loss. Most acquired myopathies present with symmetric proximal weakness, and sensory loss is not a feature of either myopathy or disorders of neuromuscular transmission. Fatigable ptosis, weakness of extraocular movement, dysphagia, dysarthria, and neck, respiratory, and proximal limb weakness should suggest myasthenia gravis, the most common nonpharmacologic disorder of neuromuscular transmission.
Rarely, the presentation of a neuromuscular disorder may be so restricted as to first suggest a nonneurologic problem. Acute pandysautonomia presents with autonomic failure that can mimic cardiovascular, gastrointestinal, or urinary disorders (3). Diabetic truncal neuropathy can present with abdominal pain that often leads to extensive evaluation for nonneurological abdominal disorders, including exploratory surgery (4). Respiratory failure can be the presenting feature of a number of neuromuscular diseases, including amyotrophic lateral sclerosis (ALS) (5,6), myasthenia gravis (7), and myopathies such as adult-onset acid maltase deficiency (8). These patients may be admitted for respiratory support before the cause is understood; it is left to the intensive care physician to suspect the correct diagnosis.
|
|
|
Figure 149.1. Localization of neuromuscular disorders, with the most important exemplars for each of the nine localizations. ALS, amyotrophic lateral sclerosis; CIDP, chronic inflammatory demyelinating polyneuropathy. |
Diagnosing neuromuscular disorders that develop in critically ill patients can be challenging, since the patients are often unable to provide a history or to cooperate with the examination. Neuromuscular problems are usually first suspected when patients recovering from critical illness cannot be weaned from the respirator or are not moving their limbs; both central and neuromuscular causes of weakness need to be considered. Central causes of weakness may be suggested by the history and physical examination. Hemiparesis should always suggest a central localization; however, bilateral weakness can also result from central causes. A history of severe hypotension suggests the possibility of bilateral watershed infarction (9); rapid correction of hyponatremia predisposes to central pontine myelinolysis (CPM), an osmotic demyelination syndrome that presents with bulbar weakness and quadriparesis (10); patients after cardiac surgery may have multiple cerebral emboli; and patients with aortic dissection may suffer spinal cord infarction. Brain imaging may help establish a diagnosis in stroke or CPM; in other conditions, such as hypoxia or even acute spinal cord ischemia, imaging may be normal or nondiagnostic. If the cause of apparent weakness remains in question, electrodiagnostic testing is of value in suggesting or helping to exclude a neuromuscular localization.
|
Table 149.1 Salient Features of Neuromuscular Disorders |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Electrodiagnostic Studies
Although it requires the expertise of a physician trained in both the clinical and electrophysiologic diagnosis of neuromuscular disorders to decide what studies are appropriate and to interpret the results, it is important for the treating physician to appreciate how these studies can assist in diagnosis and management. Electrodiagnostic studies can help to answer the following questions:
1. Is there a neuromuscular disorder causing weakness?
2. Is this a disorder of nerve, neuromuscular junction, or muscle?
3. If there is a disorder of nerve, is the pathology primarily demyelinating or axonal?
4. What is the distribution of the deficits?
Nerve conduction studies, late wave analysis, needle electromyography (EMG), and repetitive stimulation are the standard procedures available to assist in the diagnosis of neuromuscular disorders, and can be performed using portable equipment in the intensive care setting. Table 149.2 summarizes the typical electrodiagnostic findings in disorders of nerve, neuromuscular junction, and muscle.
Motor Nerve Conduction Studies
Figure 149.2A illustrates the typical motor nerve conduction procedure and normal response from stimulation of the tibial nerve. The nerve is stimulated at two or more sites, and the response is recorded with surface electrodes over a muscle innervated by that nerve. The muscle response is called a compound muscle action potential (CMAP), because it is the sum of all muscle fiber action potentials activated by nerve stimulation. The stimulus at each site is gradually increased until no further increase in CMAP amplitude is seen. This indicates that all of the motor neurons capable of generating an action potential have been activated. The latency to the onset of the CMAP reflects the speed of conduction in the fastest nerve fibers. Motor nerve conduction velocity is calculated by measuring the distance between the sites of stimulation, and dividing that distance by the proximal minus the distal latency, which is the time it takes for the stimulus to traverse that nerve segment. By itself, motor nerve conduction can be diagnostic of focal or generalized demyelinating neuropathy (Fig. 149.2C,D). Interpreting a low-amplitude response (Fig. 149.2B) requires additional studies, since low CMAP amplitude can reflect not only loss of nerve axons or conduction block, but also defective neuromuscular transmission or loss of muscle fibers.
Sensory Nerve Conduction Studies
In orthodromic sensory nerve conduction studies, the skin is stimulated and a response is recorded over the nerve. Antidromic studies are more commonly performed by stimulating the nerve and recording over the skin. The result in either case is a sensory nerve action potential (SNAP), with a latency that reflects conduction in the fastest nerve fibers, and an amplitude that is proportional to the number of functioning nerve fibers. The normal SNAP amplitude is small and measured in microvolts. Abnormal SNAPs are indicative of pathology distal to dorsal root ganglion—either plexopathy or neuropathy. SNAPs are normal in radiculopathy if the pathology spares the dorsal root ganglia in CNS disorders as well as in disorders of neuromuscular transmission and muscle. In a patient with weakness and abnormal motor nerve conduction studies, small or absent SNAPs suggest sensorimotor neuropathy or plexopathy.
F-wave Studies
Action potentials in motor neurons are normally generated at the axon hillock and travel distally to nerve terminals in muscle; but when the nerve is stimulated in the course of nerve conduction studies, action potentials travel both orthodromically (distally) and antidromically (proximally) from the point of stimulation. The antidromic action potential depolarizes the axon hillock and, in a minority of neurons, this depolarization generates another action potential that travels orthodromically down the nerve. This produces a small, late, muscle action potential, called the F wave, which can be recorded from most muscles. Increased F-wave latency, when combined with normal distal motor nerve conduction, indicates disease in the proximal portions of the nerve. Abnormal or absent F waves are the earliest electrophysiologic manifestation of Guillain-Barré syndrome (11,12).
|
Table 149.2 Electrodiagnostic Features in Nonfocal Neuromuscular Disorders |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Needle Electromyography
Needle EMG entails inserting a recording needle electrode into a muscle and observing (a) insertional activity consisting of action potentials generated by mechanical deformation of muscle fibers as the needle is inserted; (b) resting activity, which in normal muscles is only seen when the needle is near an end plate; and (c) voluntary activity, which consists of motor unit action potentials (MUPs). The motor unit consists of a motor neuron and all the muscle fibers innervated by it. The MUP is the sum of action potentials of all the muscle fibers in the vicinity of the recording needle electrode that are activated by a single motor neuron. Insertional and resting activity do not require patient cooperation, and can be observed in comatose or uncooperative patients. One to three weeks after denervation associated with axon damage, denervated muscle membranes become hypersensitive and discharge both spontaneously and in response to needle insertion, causing increased insertional activity and the appearance of positive sharp waves and fibrillations, which are spontaneous or needle movement–induced discharges of single muscle fibers. Similar abnormal spontaneous activity can be seen in disorders of muscle, particularly in the inflammatory myopathies, perhaps as a result of functional denervation caused by muscle fiber damage. If the patient is cooperative or happens to activate the muscle during the needle EMG study, additional information can be obtained by observing MUP activity. MUP morphology is of diagnostic value: high-amplitude, long-duration motor unit potentials indicate denervation with reinnervation, and low-amplitude, polyphasic motor unit potentials suggest primary disease of muscle or neuromuscular junction. Observation of motor unit recruitment with increasing effort can be diagnostic of denervation in the absence of any other abnormality. With denervation, MUP firing frequency increases with increasing effort, but insufficient numbers of different MUPs are recruited. Conversely, rapid recruitment of small polyphasic MUPs suggests myopathy or a disorder of neuromuscular transmission. Increased variability of the shape of a single MUP suggests a disorder of neuromuscular transmission.
|
|
|
Figure 149.2. Typical motor nerve conduction findings. The sweep speed is 5 ms/div in all traces, but the gain is 5 mV/div in A and C, 1 mV/div in B, and 500 µV/div in D. A: Tibial motor nerve conduction in a normal subject. The drawing shows the two sites of stimulation over the posterior tibial nerve at the ankle and knee, and the sites of surface electrodes recording the response of the medial plantar muscle group. The two waveforms are the responses to stimulation at the ankle and knee. Conduction velocity is calculated as the distance between proximal and distal sites of stimulation divided by the time it takes for the stimulus to travel between the sites of stimulation (PL minus DL). B: Tibial motor nerve conduction in a patient with axonal sensorimotor neuropathy. CMAP amplitudes are less than 2 mV, with normal being greater than 4 mV. The amplitudes are similar with proximal (knee) and distal (ankle) stimulation. The conduction velocity is slightly slow, but is greater than 80% of the lower limits of normal. C: Peroneal motor nerve conduction study in focal nerve compression. CMAP amplitude drops sharply with nerve stimulation above the site of compression, at the fibular head. This conduction block is caused by focal nerve demyelination. Because there is no damage to axons, the nerve responds normally to stimulation distal to the area of demyelination. Conduction velocity is slow across the fibular head. D: Acquired diffuse demyelinating polyradiculoneuropathy. Multifocal demyelination affects some neurons more than others, resulting in prolonged and irregular CMAPs. Conduction velocities less than 70% of the lower limits of normal are also diagnostic of demyelination. DL, distal latency; PL, proximal latency; CMAP, compound muscle action potential. |
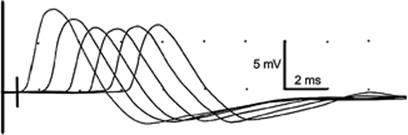
Repetitive Stimulation
Disorders of neuromuscular transmission may be postsynaptic, as is the case in myasthenia gravis or most pharmacologic neuromuscular blocking agents, or presynaptic, as in Lambert-Eaton myasthenic syndrome and with exposure to botulinum toxin. Repetitive supramaximal stimulation of a motor nerve at a constant rate for five to ten stimuli is a simple way to assess for deficits of neuromuscular transmission. The resultant CMAP amplitudes should be constant. Progressive decrements in amplitude across the first four to five responses with 3-Hz stimulation is seen in both postsynaptic and presynaptic disorders of neuromuscular transmission (Fig. 149.3). Progressive increments in response at rates of stimulation above 20 Hz suggest a presynaptic neuromuscular block, as in Lambert-Eaton myasthenic syndrome, or botulism. An alternative—and more tolerable—means to look for this incremental response is to stimulate the nerve supramaximally just before and just after 10 to 60 seconds of sustained voluntary contraction of the muscle being tested. An increase in amplitude after exercise—postexercise facilitation—is seen in pre- and postsynaptic disorders; however, increases greater than 100% are seen only in presynaptic disorders (13). Repetitive stimulation is important in documenting residual neuromuscular blockade as a cause of weakness in ICU patients.
|
|
|
Figure 149.3. Repetitive stimulation at 3 Hz of the median nerve, recording from the thenar muscles. Each response is displayed 1 ms to the right of the prior response to show a train of six responses. The progressive decrease in compound muscle action potential amplitude between the first and fourth responses indicates a partial defect in neuromuscular transmission. With normal neuromuscular transmission, each response would have the same amplitude. |
Electrodiagnostic studies are valuable extensions of the neurologic assessment, and need to be planned and interpreted in the context of the history and neurologic examination. For example, in some contexts, absent sensory nerve responses provide evidence that weakness is related to an acute disorder of nerve, but in other circumstances may reflect prior sensory neuropathy, focal entrapment, or technical difficulties when recording very small potentials in an electrically noisy environment. Well-trained technicians can perform routine nerve conduction and repetitive stimulation studies; however, needle EMG must be performed by a knowledgeable physician, and study planning and interpretation must be the responsibility of physicians expert in both clinical neurophysiology and the clinical assessment of neuromuscular disorders.
General Principles for the Intensive Care of Patients with Neuromuscular Disorders
Indications for intensive care of patients with neuromuscular disorders include airway protection, ventilatory failure, and autonomic instability. Principles of evaluation and care are covered elsewhere in this book; however, it is worth pointing out here that many of the signs typically associated with respiratory failure in patients with obstructive pulmonary disorders, such as increased airway noises and increased respiratory excursion, may not be seen in patients with neuromuscular causes of respiratory failure. Instead, air hunger, tachypnea, shallow breaths, and staccato speech are characteristic. An inability to lay supine and paradoxical respirations are manifestations of diaphragmatic weakness. Confusion and somnolence with rising PaCO2 may lessen respiratory distress and make recognition of respiratory insufficiency more difficult. Bedside tests of respiratory function that indicate a need for respiratory support include the inability to count to 20 on one breath, a forced vital capacity of less than 15 mL/kg, a negative inspiratory force of less than 25 cm H2O, and a positive expiratory force of less than 40 cm H2O (14,15,16,17). Patients with facial weakness may have difficulty performing bedside tests, necessitating greater reliance on blood gas monitoring. Intubation may be required earlier in patients with dysphagia in order to protect the airway.
Autonomic dysfunction and pain can be features of the Guillain-Barré syndrome and are discussed under that topic below. Patients who are immobile from weakness or other causes may suffer compressive neuropathies. The peroneal nerve at the head of the fibula and the ulnar nerve at the elbow are especially vulnerable. Constant attention to positioning is required to prevent pressure palsies. Pneumatic compression devices should not cover the proximal fibula.
Neuromuscular disorders do not affect mentation, and it is important to maintain communication to inquire about discomfort and pain, involve patients in care and decision making, and support them through what can be a terrifying experience. Bowes, a physician who recovered from ventilator-dependent Guillain-Barré syndrome, gives a personal account of her experience that is instructive for caregivers (18). Additional aspects of the intensive care of neuromuscular disorders are discussed below with reference to specific disorders.
Neuromuscular Disorders Requiring Intensive Care
Table 149.3 lists some of the neuromuscular conditions that can cause respiratory failure. In many of these conditions, weakness develops gradually, and management does not require intensive care, except when intercurrent illness, such as aspiration pneumonia, causes decompensation. ALS inevitably leads to respiratory failure, but alternative arrangements for care usually preclude intensive care admission. In a minority of patients with ALS, however, respiratory insufficiency is the presenting symptom, and leads to intensive care admission before the diagnosis is made. The diagnosis is suggested by finding fasciculations, atrophy, weakness, and hyperactive reflexes in the absence of sensory loss, extraocular muscle weakness, or autonomic dysfunction, and by excluding alternative causes, such as spinal cord compression or neuropathy. Electrophysiologic studies can be helpful. Nerve conduction studies document normal sensory nerve action potentials, and needle EMG reveals denervation in muscle of three or more extremities, or two extremities and bulbar muscles. Needle EMG can also document diaphragm denervation. Early in the course, clinical and electrophysiologic findings may not suffice for definitive diagnosis.
Respiratory weakness is seen in some muscle disorders, and is a terminal event in Duchenne muscular dystrophy. Respiratory weakness can be the presenting feature of adult-onset acid maltase deficiency, a glycogen storage disease (19), and it is a feature of some of the congenital myopathies and muscular dystrophies. Respiratory weakness can occur in inflammatory myopathies in the context of severe proximal limb weakness, and polymyositis associated with Jo-1 antibodies can be associated with interstitial pulmonary disease (20).
The two neuromuscular disorders that most commonly lead to admission for intensive care are Guillain-Barré syndrome and myasthenia gravis. They are both autoimmune disorders and can cause rapidly progressive weakness; they are both treatable and, with appropriate management, most affected patients recover.
|
Table 149.3 Neuromuscular Causes of Respiratory Failure |
|
|
Guillain-Barré Syndrome
History
In 1859, Landry (21) described ascending paralysis progressing to respiratory paralysis and death. Guillain, Barré, and Strohl described the classic features of progressive weakness, sensory symptoms without signs, and loss of tendon reflexes, but their patients recovered. They also described increased cerebrospinal fluid protein without cells (albuminocytologic dissociation), a finding that helped to distinguish this disorder from poliomyelitis (22). An association with prior infection was established (23,24), and the concept that the disease was of autoimmune pathogenesis was supported by the development of an animal model—experimental allergic neuritis, in which inflammation and demyelination of peripheral nerves followed exposure to components of peripheral nerves (25).
Definition and Subtypes
Guillain-Barré syndrome is an autoimmune disorder of peripheral nerves with progression over less than 4 weeks, which is manifested by weakness, reduced or absent tendon reflexes, and variable sensory and autonomic dysfunction (26). Subtypes have been distinguished based on the presence or absence of motor, sensory, and/or autonomic involvement, and on whether the predominant damage is to myelin sheaths or axons. Acute inflammatory demyelinating polyradiculoneuropathy (AIDP) is by far the most common subtype, accounting for more than 85% of cases in the United States (27). Patients have weakness and reflex loss, with varying degrees of sensory and autonomic involvement, and there is electrophysiologic and pathologic evidence of peripheral nerve demyelination. Patients who have no sensory loss and electrodiagnostic studies suggesting axonal rather than demyelinating pathology have acute motor axonal neuropathy (AMAN). This variety of Guillain-Barré syndrome was first described in Asia, where it occurs epidemically following gastroenteritis caused by Campylobacter jejuni (28). Acute motor sensory axonal neuropathy (AMSAN) is diagnosed when there is both motor and sensory involvement and nerve conduction studies are consistent with early axonal dysfunction (29). The axonal forms of Guillain-Barré comprise less than 5% of cases in Europe and North America, but up to 47% of cases in China, Japan, and Central and South America (30). Acute pandysautonomia (31,32) and some forms of acute sensory neuropathy (33) are sometimes considered to be subtypes of Guillain-Barré syndrome even though weakness is not a feature, since they run a similar time course, and are presumed to be autoimmune. The Miller-Fisher variant of Guillain-Barré syndrome features ophthalmoplegia, ataxia, and areflexia (34). The Miller-Fisher variant may overlap with other forms of Guillain-Barré syndrome (30,35).
Epidemiology and Pathogenesis of Guillain-Barré Syndrome
The incidence of Guillain-Barré is less than 1 in 100,000 in children, averages 1 to 2 per 100,000 in adults, and increases to 4 in 100,000 in adults over 75 years of age (36,37). In about two thirds of patients, the disease follows an infection, most often a flulike syndrome or gastroenteritis, by 1 to 8 weeks (38). When serologic studies are performed, evidence of infection with C. jejuni, cytomegalovirus, Epstein-Barr virus, Haemophilus influenzae, and Mycoplasma pneumoniae are found to account for the majority of cases (39,40); 5% follow surgery (24). Although 3% of cases are attributed to vaccination (38), only rabies vaccine is associated with a definite increase in the incidence of Guillain-Barré (41); influenza vaccination causes little, if any, increase (41,42).
The pathology of AIDP consists of a multifocal inflammation of peripheral nerves, in which macrophages destroy the myelin sheath (43,44,45,46). This response is mediated by activated T cells (47). Direct binding of antibodies to Schwann cell membranes with subsequent complement-mediated destruction and secondary macrophage invasion may also play a role (48). Similar pathologic findings characterize experimental allergic neuritis (EAN), which is considered to be a good experimental model for Guillain-Barré syndrome (49). EAN can be initiated by injections of several myelin components, including myelin proteins P0 and P2, myelin basic protein, peripheral myelin protein-22 (PMP-22), and myelin-associated glycoprotein (MAG) (47). No specific myelin antibodies have been identified in the pathogenesis of AIDP.
The pathology of AMAN differs from that of AIDP in that macrophages target the axonal membrane in ventral (motor) roots and nerves at nodes of Ranvier, resulting in axonal rather than myelin damage (50,51). The pathology in AMSAN is similar to that of AMAN, except that the disorder affects both motor and sensory roots and nerves (29). Recent evidence suggests that AMAN may result from molecular mimicry. The capsule of the strains of C. jejuni implicated in patients with Guillain-Barré syndrome contains a lipopolysaccharide with a structure similar to the GM1 ganglioside present in peripheral nerves.
Antibodies to these strains of C. jejuni cross-react with peripheral nerve GM1 ganglioside, resulting in axonal dysfunction in experimental animals (52). Similarly, many patients with the Miller-Fisher variant have GQ1b and GT1a antibodies (53) that cross-react with Campylobacter lipopolysaccharides (54,55). Anti-GD1b antibodies have been associated with acute and chronic sensory neuropathies (30). Although there is evidence that molecular mimicry is a plausible explanation for some patients with Guillain-Barré polyneuropathy, the relationships are complex. Patients may have antibodies to several gangliosides, antibodies to a particular ganglioside can be associated with several different types of neuropathy, and infections with a specific organism may be followed by more than one kind of neuropathy (30).
Clinical Presentation and Diagnosis
AIDP, the most common form of Guillain-Barré, presents with weakness and decreased or absent tendon reflexes. It is a non–length-dependent polyneuropathy, with proximal as well as distal weakness that usually begins in the legs and ascends, but can affect other regions first. Bilateral facial weakness occurs in half of the patients. Since symptoms can progress to oropharyngeal and respiratory weakness in 33% of patients, all patients require careful monitoring, as discussed below. Eye movements are usually spared, the exception being the Miller-Fisher variant. Sensory symptoms occur in 80% of patients. Sensory signs are less frequent and often mild, but, when prominent, can contribute significantly to disability (56). Pain and temperature sensation mediated by small unmyelinated neurons may be affected less than position and vibratory sensation, mediated by large myelinated fibers. Fifty percent have pain, either back and muscle pain that can be aggravated by movement, or neuropathic pain, such as burning paresthesias, sometimes with allodynia (pathologic sensitivity to touch). Tendon reflexes are lost early in the course of the disease; indeed, the diagnosis should be questioned if reflexes are retained. Autonomic dysfunction is present in 66% of patients, and can manifest with tachyarrhythmias or bradyarrhythmias, spontaneous or orthostatic hypotension, paroxysmal hypertension, abnormalities of sweating, urinary retention, or ileus (57). Guillain-Barré syndrome is a subacute process: 50% of patients stop progressing in less than 2 weeks, 80% by 3 weeks, and more than 90% by 4 weeks (58).
The much less common axonal variants of Guillain-Barré are distinguished from AIDP by electrophysiologic studies. Nerve conduction studies should show low-amplitude motor CMAPs without marked conduction slowing, conduction block, or dispersion in both AMAN and AMSAN. AMAN is distinguished from AMSAN by preservation of sensory nerve action potential amplitudes (59).
The differential diagnosis of patients presenting with subacute weakness includes disorders of the CNS, PNS, neuromuscular junction, and muscle. Although Babinski responses, brisk reflexes, and sensory level will usually distinguish spinal cord from peripheral nerve diseases, there is overlap because reflexes may be depressed acutely in CNS disorders; bladder dysfunction, while suggestive of CNS lesions, can be seen in Guillain-Barré; and CNS demyelination may occasionally accompany Guillain-Barré syndrome (60). Conversely, Guillain-Barré syndrome should be in the differential of patients with the locked-in syndrome when tendon reflexes are depressed. Toxic, drug-induced, and metabolic neuropathies may occasionally present over a similar time course as Guillain-Barré syndrome. In patients without sensory disturbances, disorders of neuromuscular junction and muscle should be considered. Muscle disorders that can present with severe subacute or acute weakness include the periodic paralyses, hypokalemic myopathy, drug-induced myopathies, and inflammatory myopathies.
Evaluation should include appropriate imaging to exclude CNS disorders, routine laboratory studies, and lumbar puncture. The cerebrospinal fluid (CSF) is normal during the first days of illness, after which there is a transient elevation of protein with minimal or no pleocytosis. A significant pleocytosis should raise the question of infection such as Lyme disease or West Nile virus (61); when the clinical picture is otherwise classic for Guillain-Barré, CSF pleocytosis should suggest human immunodeficiency virus (HIV) infection in which immune dysregulation increases the incidence of autoimmune disorders (62). Blood should be sent for basic metabolic profile, B12, porphyrins, tests for Lyme disease and HIV, and heavy metal levels. Electrodiagnostic studies must be interpreted in the context of the clinical findings. Early in Guillain-Barré, nerve conduction and F-wave studies may be normal if recorded from a limb without severe weakness. Delayed or absent F waves, sometimes replaced by axon reflex waves, are the earliest abnormalities (63). When recorded from a paralyzed muscle, normal motor nerve conduction studies with normal F waves indicate that the weakness does not result from a peripheral cause, and should direct attention toward an alternative diagnosis with localization to the central nervous system.
Management of Guillain-Barré Syndrome
Patients with Guillain-Barré syndrome should be admitted and carefully monitored for respiratory function, swallowing ability, and autonomic function. Specific treatment can hasten recovery and reduce hospital costs, and is recommended for all patients whose weakness threatens loss of ambulation (64). Plasma exchange was the first treatment demonstrated to be effective in Guillain-Barré syndrome. Treated patients demonstrated greater improvement at 4 weeks than untreated controls, and treatment reduced the proportion of patients requiring mechanical ventilation at 4 weeks from 27% to 14% (28,65,66,67). Every-other-day treatment to a total of five plasma volumes is usually employed. Complications include pneumothorax and sepsis related to central venous access, hypocalcemia, coagulopathy, and hypotension. Intravenous immunoglobulin treatment (IVIG) was shown to be equally effective as plasma exchange in controlled trials (68), and has become the preferred treatment because of greater ease of administration and lesser risk of serious complications. The usual dose is 0.4 g/kg/day infused slowly for 5 days to a total dose of 2 g/kg. Complications include hypersensitivity in patients with immunoglobulin A (IgA) deficiency, acute renal failure, congestive failure, hypercoagulable state, and aseptic meningitis. Treatment is indicated in all but the mildest cases, and is best instituted early in the course of the disease. Treatment after 4 weeks is unlikely to be of benefit. High-dose corticosteroids are of no proven benefit, and in several trials treated patients had more disability than controls (69).
Patients with rapidly progressive weakness should be transferred to the ICU. Respiratory failure is often preceded by progression of weakness to involve the upper extremities, shoulders, neck, and face, but may occur before this weakness is evident. Tachypnea, paradoxical respirations, hypophonia, staccato speech, and a marked increase in respiratory dysfunction when supine are clinical indications of respiratory failure (14). Confusion from carbon dioxide retention is a late manifestation. Respiratory function should be monitored with bedside spirometry, and heart rate and blood pressure should be closely monitored. Indications for intubation are reviewed under General Principles, above.
Autonomic instability is common and usually not life threatening, unless it is manifested by asystole or ventricular fibrillation. Hypertension should be treated cautiously, as patients may be unusually sensitive to antihypertensive medications. Paroxysmal hypertension may sometimes be a manifestation of pain, in which case adequately treating the pain can be curative. Neuropathic pain can be treated with antiepileptic medications such as gabapentin (70), antidepressants such as amitriptyline, or analgesics.
The prognosis for recovery is good in all types of Guillain-Barré syndrome, but is poorest for patients with severe AMSAN. Poor recovery is correlated with age, severity of deficits at the nadir of the disease, more rapid progression of deficits (30), and, in some studies, evidence of axonal damage on electrophysiologic studies (71); however, even the most severely afflicted patients may recover fully. About half of patients with otherwise good recovery are left with mild sensory disturbances (72,73,74).
Myasthenia Gravis
History
The first description of myasthenia gravis is variously credited to Wilks in 1867 or to Erb in 1879, although Sir Thomas Willis provided a description of the symptoms more than 200 years earlier (75). Freidreich Jolly, who first described the decremental response to repetitive stimulation, called this disorder myasthenia gravis pseudoparalytica (76). Mary Walker, a physician working at St. Alfege's Hospital in Greenwich, was the first to use acetylcholinesterase inhibitors to treat myasthenic patients (77). The dramatic, albeit temporary, reversal of weakness by physostigmine was called the “Miracle of St. Alfege's Hospital.” The transient weakness manifested by infants born to myasthenic mothers, and the association of myasthenia with thymic tumors and with thyroiditis, led to the theory that myasthenia was an autoimmune disease (78). This was confirmed in the early 1970s with the demonstration that animals injected with concentrated acetylcholine receptor produced antibodies that caused weakness (79), as well as with the discovery of antibodies to acetylcholine receptors in the sera of myasthenic patients (80). Immunotherapy and improved critical care have lessened overall mortality from 20% to 30% in the 1950s (81) to nearly zero (82).
Epidemiology
Myasthenia gravis has a current prevalence of about 20 per 100,000 population. There are about 60,000 patients with myasthenia in the United States (83). Myasthenia gravis occurs throughout life with bimodal peaks in the second and third decade in women and in the fifth and six decades in men (84). As the population ages, the average age of onset has increased and men are now more often affected than women (83). Myasthenic crisis occurs in about 20% of patients with myasthenia; almost 33% of these patients can experience a second crisis (85,86).
Pathophysiology
The neuromuscular junction is composed of a presynaptic motor neuron membrane, the synaptic cleft, and the postsynaptic muscle membrane. In the normal neuromuscular junction, the arrival of an action potential at the nerve terminal leads to the influx of calcium through voltage-gated calcium channels, which triggers release of acetylcholine into the synaptic cleft (87). Acetylcholine diffuses across the synaptic cleft to bind with acetylcholine receptors on the postsynaptic muscle membrane. This binding generates end-plate potentials that depolarize the muscle membrane. When the depolarization exceeds threshold, a muscle action potential is generated, which leads to a cascade of events that results in muscle contraction (88). Neuromuscular transmission fails if the end-plate potential fails to reach the threshold for muscle action potential generation. In Lambert-Eaton myasthenic syndrome, the end-plate potential is small because antibodies to voltage-gated calcium channels on the presynaptic membrane interfere with acetylcholine release. In myasthenia gravis, the end-plate potential is small because antibodies to the acetylcholine receptor or related sites on the postsynaptic membrane decrease the depolarization caused by normal amounts of acetylcholine.
The acetylcholine receptor is composed of four subunits (89). Antibodies to the acetylcholine receptor can be found in 80% to 90% of patients with generalized myasthenia (90). These antibodies can cause neuromuscular transmission failure by binding to the acetylcholine receptor and altering function, by increasing the rate of degradation of the receptors, and by complement-mediated destruction of the postsynaptic membrane (91). About 30% of patients with generalized myasthenia who are seronegative for acetylcholine receptor antibodies have antibodies to muscle-specific kinase (MuSK) (92). Patients with MuSK antibodies demonstrate more severe disease than patients with antibodies against the acetylcholine receptor (93).
The thymus gland has been implicated in the pathogenesis of myasthenia gravis. Thymic abnormalities are seen in 75% of patients with myasthenia gravis. Of these, 75% have lymphoid follicular hyperplasia and 15% have thymomas (94). Acetylcholine receptors are found on myoid cells of the thymus (95). The majority of myasthenic thymuses contain B cells that can produce antibodies to the acetylcholine receptor (96). Antistriated muscle antibodies in patients with myasthenia gravis correlate highly with thymoma (97).
Patients with myasthenia gravis often have other autoimmune disorders, including thyroiditis, scleroderma, Sjögren syndrome, rheumatoid arthritis, pernicious anemia, polymyositis, and lupus erythematosus (98).
Clinical Presentation
Patients with myasthenia gravis present with fluctuating weakness that improves with rest. The most common complaints are ocular—namely, ptosis and diplopia related to weakness of the levator palpebrae and the extraocular muscles, respectively. Ptosis is the presenting symptom in 50% to 90% of patients, and 90% to 95% of patients complain of diplopia during the disease. Weakness can remain purely ocular in up to 15% of cases, but the majority of patients with myasthenia gravis develop generalized symptoms (76). Dysarthria and dysphagia are common complaints. Neck extensors and flexors are often more involved than other muscles in generalized disease (76). Patients may have to support their chin with their hand to prevent head drop because of neck extensor weakness or to keep their jaw shut because of weakness of the temporalis and masseter muscles. Proximal limb weakness is usually present when the disease is not purely ocular. It is sometimes asymmetric, is occasionally very focal, and can affect distal muscles. In less than 10% of cases, myasthenia presents with proximal weakness without bulbar or ocular weakness.
|
Table 149.4 Myasthenic Weakness |
||
|
Myasthenic crisis is diagnosed when weakness progresses to cause dysphagia with loss of airway protection or hypoventilation. Myasthenic crisis can be precipitated by infection, surgical procedures, pregnancy, treatment with corticosteroids, sepsis, cholinergic crisis, and the rapid withdrawal of immune-suppressing medications (86). Pulmonary system infection, including aspiration pneumonia, is the most common identifiable precipitating factor (86). Many medications can also worsen myasthenic symptoms (Table 149.4) (99), often because they impair neuromuscular transmission. Aminoglycoside antibiotics, quinidine, and procainamide are particularly likely to cause weakness, and should be avoided in patients with myasthenia. Limited experience with telithromycin suggests that it should also be avoided (100). Penicillamine and α-interferon may cause autoimmune myasthenia. Evidence that other medications exacerbate myasthenia gravis is less convincing, but in patients with increasing weakness, all medications must be scrutinized against the long list of potentially offending agents (99).
Myasthenic crisis can involve muscles of inspiration, including the diaphragm and the external intercostals, along with accessory muscles of inspiration, including the scalene and sternocleidomastoid muscles. Respiratory strength can be measured by vital capacity and negative inspiratory force (101). As respiratory weakness leads to ventilatory failure, tidal volumes decline. Patients in myasthenic crisis have an intact respiratory drive, and therefore respond to the decrease in tidal volume with an increase in respiratory rate (102). Oropharyngeal muscle weakness may result in obstruction with upper airway collapse. Secretions can be difficult to control and can also obstruct the airway and lead to aspiration. In the 1950s and 1960s, mortality from myasthenic crisis was as high as 70% to 80% (103,104). In more recent reports, mortality is closer to 4% (85,86), related to improvements in intensive care as well as effective immunomodulatory treatment.
Diagnosis
The history and examination often strongly suggest the diagnosis. A history of fluctuating ptosis, diplopia, fatigable dysarthria, dysphagia, and neck weakness are highly suggestive. Sometimes the presentation may be misleading, as when ocular myasthenia presents with isolated eye muscle weakness or with a pattern of eye movements that mimics internuclear ophthalmoplegia from brainstem lesions, or when patients have proximal limb weakness without eye or bulbar weakness, suggesting a primary disorder of muscle. Conversely, patients with purely ocular findings should be worked up for other causes of ophthalmoplegia, including thyroid eye disease and retrobulbar or cavernous sinus lesions. ALS may present with bulbar symptoms, but patients with ALS do not have ptosis or eye movement weakness, and often have pseudobulbar dysarthria, characterized by slow labored speech and poorly modulated emotional expression, with laughing or crying that is disproportionate to the situation.
Many tests are available to confirm the diagnosis of myasthenia. Definitive diagnosis is important, as most patients will require years of immunomodulatory therapy. Pharmacologic testing with edrophonium (Tensilon), a rapid-acting, short-duration acetylcholinesterase, can result in transient increase in strength. Caution should be used in using edrophonium because anticholinergic side effects, such as bradycardia and bronchospasm, can develop. A test dose of 2 mg of edrophonium, given intravenously, should be administered, followed by increments of 2 mg, for a total dose of 10 mg. Atropine should be available to counteract muscarinic side effects (105). Definite improvement in strength, as demonstrated by improvement in ptosis, dysarthria, or resistance against the examiner's testing, is supportive of the diagnosis. The test is, therefore, most helpful in patients with clear weakness that can be evaluated in an objective manner (105). In patients with cardiac disease, especially those on β-blockers or drugs that cause atrioventricular block, edrophonium should be avoided, as it can cause asystole (106). Fortunately, there are many alternative tests to confirm the diagnosis of myasthenia.
Serum acetylcholine receptor antibodies are found in 80% to 90% of patients with generalized myasthenia gravis, and have very high diagnostic specificity. Increased titers are also found in patients with thymoma without myasthenia, and rarely in other diseases, including systemic lupus, ALS, liver disease, and inflammatory neuropathies, and in patients with Lambert-Eaton myasthenic syndrome (107). Antibodies to striated muscle are present in about 30% of patients with myasthenic gravis, and are highly associated with thymoma (108). About 30% of patients with myasthenia gravis who lack antibodies to acetylcholine receptor have antibodies to MuSK. Other antibodies directed against muscle antigens can be found in patients with myasthenia gravis (109).
Repetitive nerve stimulation can be performed to rule out a decrement in the amplitude or area of the fourth or fifth potential compared to the first potential (Fig. 149.3). A train of five to ten stimuli is delivered at a rate of 2 to 3 Hz. If the decrement is more than 10%, it is considered abnormal. The yield can be increased by testing clinically weak muscles and with repeat testing after exercise of the specific muscle. Because typically myasthenia affects proximal more than distal muscles, the test is more likely to be abnormal when testing proximal or facial muscles than when testing hand muscles (110). Single-fiber EMG is abnormal in nearly all patients with generalized myasthenia (111), but it is not widely available because it is time consuming, is technically demanding, and requires expensive needle electrodes.
Patients diagnosed with myasthenia should have additional testing for associated conditions. Computed tomography (CT) or magnetic resonance imaging (MRI) of the chest is required to evaluate for thymoma (82). Tests of thyroid function are important, because about 10% of myasthenics have associated autoimmune thyroid disease, and because proximal weakness and restriction of extraocular movements can be caused by thyroid disease. Antinuclear antibody (ANA) and rheumatoid factor may detect much less commonly associated autoimmune disorders. Tuberculin test and fasting glucose should be obtained prior to treatment with steroids.
Management
The majority of myasthenic patients respond well to treatment and never require intensive care. Although anticholinesterase medications can produce dramatic improvement in strength, they do not address the underlying autoimmune pathogenesis of the disease, and tend to become ineffective as the disease progresses. They may be sufficient, however, for patients with mild, nonprogressive disease, and they can be helpful as adjunctive treatment in patients on immunomodulatory treatment. Pyridostigmine, which does not cross the blood–brain barrier, is the most commonly used oral agent. Sustained-release pyridostigmine is appropriate for use at bedtime to mitigate weakness during the night and in the morning; however, multiple daytime doses of sustained-release preparations should be avoided, as accumulation may lead to cholinergic weakness.
Oral prednisone is the most commonly used agent to induce remission in myasthenic patients, and is effective in 80% to 90% of patients. High-dose prednisone—60 to 100 mg/day—may cause transient worsening of myasthenia, and should not be initiated unless patients can be closely observed. Lower doses appear safer, but are not entirely free from this complication. Most patients experience gradual improvement in strength beginning 2 or 3 weeks after initiation of therapy. Once a good response has been achieved, the dose can be tapered over several months to low-dose alternate-day treatment. Doses less than 40 mg every other day are generally well tolerated. Doses of 40 mg or above every other day are associated with many long-term side effects, including diabetes, hypertension, cataracts, avascular necrosis, and increased susceptibility to infection. Azathioprine may reduce steroid dependence. Treatment with IVIG can produce rapid improvement in strength, and may be useful to provide short-term improvement while waiting for steroids to become effective.
Myasthenic weakness can progress very rapidly; therefore, patients with worsening myasthenic symptoms should be hospitalized for definitive management. Patients with progressive bulbar weakness who have difficulty controlling secretions, an ineffective cough, or vital capacities less than 20 to 25 mL/kg should be admitted to the ICU (112,113). When the vital capacity reaches 15 mL/kg, endotracheal intubation should be strongly considered. Elective intubation can help patients avoid abrupt respiratory failure and mucous plugging (114). Intermittent mandatory ventilation with pressure support is used to reduce alveolar collapse or atelectasis. Positive end-expiratory pressure can reduce complications related to atelectasis (115). If mechanical ventilation is required for more than 2 weeks, tracheostomy should be considered. The most common complications of myasthenic crisis are fever, pneumonia, and atelectasis (86). Aggressive respiratory treatment with suctioning, bronchodilator treatments, sighs, chest physiotherapy, and intermittent positive pressure can reduce the complications of mechanical ventilation and shorten the period that patients are in the intensive care unit (116). Weaning trials should be attempted with patients who show clear improvement of respiratory muscle strength, with the goal to eliminate mandatory ventilation and then reduce the amount of pressure support. Cholinesterase inhibitors are generally discontinued after intubation because of increased secretions and the potential for depolarization blockade. After a definite response to treatment, cholinesterase inhibitors can be restarted at a low dose.
Plasma exchange has been shown to be effective for the treatment of myasthenic crisis and to prepare patients for surgery (117). Many different protocols are used, but a series of five to six exchanges of 2 to 4 liters every other day is most commonly employed. Improvement can usually be seen in 3 to 4 days and can last for several weeks, but is not sustained without the addition of an immune suppressant agent. Side effects of plasma exchange, which can be life threatening, include hypotension, bradycardia, congestive heart failure, venous thrombosis, infection, and dramatic shifts in electrolytes, including calcium. Controlled randomized trials have demonstrated that IVIG is as effective as plasma exchange in patients with myasthenic crises (118). This trial used 1.2 and 2.0 g/kg of IVIG over 2 to 5 days. Patients who fail to respond to IVIG may subsequently respond to plasma exchange (119). Intravenous steroids have been used in high doses in myasthenic crisis in patients who do not respond to IVIG or plasma exchange (120), but this may be complicated by transient worsening of myasthenic weakness that may precipitate the need for, or prolong, mechanical ventilation. Other immune-suppressant medications can be useful as steroid-sparing drugs and as sole therapy for myasthenia, but they take weeks or months to produce improvement, and therefore are not appropriate choices to treat myasthenic crisis.
Thymectomy is indicated in patients with thymoma, and is also accepted therapy for myasthenia gravis without thymoma, particularly in younger patients without thymic involution. However, the absence of class I evidence and the shortcomings of existing case-control or cohort trials have led to a call for prospective randomized controlled clinical trials (121).
Perioperative Care
Myasthenic crisis is common in patients undergoing surgical procedures. The main risk factors for postoperative complications include severe bulbar weakness, chronic myasthenia, respiratory illness, and decreased vital capacity preoperatively (122). Whenever possible, preoperative treatment to improve strength should be undertaken. Plasma exchange or IVIG can be performed preoperatively in patients with bulbar weakness or generalized myasthenia. A response is often seen within 3 to 6 days, although preoperative IVIG has shown a variable time to maximal response (123). Preoperative pulmonary function testing can be performed to assess respiratory function. Drugs that are known to exacerbate myasthenia (Table 149.4) should be avoided whenever possible.
Critical Illness Polyneuropathy and Myopathy
History
Although earlier references are cited (see Lindsay et al. [124,125]), recognition that critically ill patients frequently develop new neuromuscular dysfunction was not widespread until the last decade of the 20th century. In 1984, Bolton et al. (126) described five patients who failed to wean from the ventilator after treatment of sepsis, and were found to have neuromuscular weakness related to an axonal sensorimotor polyneuropathy. They called this critical illness polyneuropathy (CIP). In 1992, Sagredo et al. (127) attributed prolonged paralysis, seen in patients in whom vecuronium had been weaned, to persistent plasma levels of vecuronium metabolites in those patients with impaired renal function. Hirano et al. (128) described severe weakness in two critically ill asthmatic patients treated with steroids and nondepolarizing blocking agents. Muscle biopsy demonstrated depletion of thick myosin filaments. They called this acute quadriplegic myopathy. Other terms for the same or similar illness have included hydrocortisone myopathy, necrotizing myopathy, thick filament myopathy, postparalysis syndrome, and acute myopathy of intensive care; we will refer to it by the most common designation: critical illness myopathy (CIM). The term critical illness neuromyopathy (CINM) is used to refer to patients in whom both neuropathy and myopathy coexist, or when it is uncertain which condition is causing the patient's weakness. The term CINM could also be applied to patients with persistent neuromuscular blockade; however, electrophysiologic studies should exclude this as a contributing factor in most patients.
Epidemiology
The incidence of critical illness neuromyopathy is estimated to be as high as 70% of mechanically ventilated ICU patients with sepsis and multiorgan failure. Witt et al. (129) found, in a prospective study, that 70% of 43 ICU patients with sepsis and failure of more than two organs developed electrophysiologic evidence of peripheral neuropathy, and half of these patients had clinically evident neuropathy with difficulty being weaned from the ventilator. De Jonghe et al. (130) found that 25.3% of ICU patients intubated for 7 or more days developed neuromuscular weakness. All had sensorimotor axonopathy, and all who were biopsied also had myopathy. Bednarik et al. (131) prospectively studied 61 ICU patients with evidence of multiorgan failure; 27% had clinical and 54% electrophysiologic evidence for critical illness neuromyopathy. Bercker et al. (132) found that 60% of 50 consecutive patients with acute respiratory distress syndrome studied retrospectively developed critical illness neuromyopathy.
Pathophysiology
Critical Illness Polyneuropathy
Sepsis and multiorgan failure are the major risk factors for CIP (131,133,134); other risk factors include duration of mechanical ventilation, hyperglycemia, hyperosmolality, and decreased serum albumin (129). The peripheral nervous system is one of the organ systems damaged in systemic inflammatory response syndrome (SIRS). Decreased capillary flow and increased capillary permeability related to sepsis-related cytokine release, and other factors such as a generally high catabolic rate, result in a reduced supply of nutrients, decreased clearance of toxic factors (including medications), and tissue edema. Pathologically, there is axonal degeneration without inflammation, and there is an increase in adhesion molecule expression in nerve (133).
Critical Illness Myopathy
Risk factors for CIM overlap with those for CIP; however, there is more emphasis on primary pulmonary disease (asthma and acute respiratory distress syndrome [ARDS]) and on the use of neuromuscular blocking agents and corticosteroids (130,135). Apoptotic activity may be increased. High corticosteroid levels may increase expression of ubiquitin, leading to increased proteolytic activity (136). Denervation from coexisting CIP may also make the muscle more susceptible. Muscle biopsy shows loss of thick (myosin) filaments; there may also be fiber-type atrophy and/or necrosis (128,137). Whether the different pathologies represent a spectrum of severity or different entities remains unsettled.
Most patients on ventilators are given neuromuscular blocking agents. Normally, the effects of these agents wear off within hours of discontinuation; however, it is not unusual for residual blockade to last 1 to 2 days (127). Early reports of more prolonged weakness after neuromuscular blockade did not consider the effects of CIP and CIM. In a recent review, Murray et al. (135) concluded that nondepolarizing neuromuscular blocking agents may potentiate muscle weakness by a number of mechanisms, including up-regulation of acetylcholine receptors with fetal-type receptors that are less responsive to acetylcholine, presynaptic inhibition of exocytosis of acetylcholine, and potentiation of muscle damage by corticosteroids and by sepsis-induced ischemia.
Clinical Presentation
CIP is preceded by sepsis and failure of more than one organ. Sepsis leads to SIRS, which is manifested by two or more of the following (138):
· Elevated (greater than 38°C) or reduced (less than 36°C) temperature
· Tachycardia (greater than 90 beats/minute)
· Tachypnea (greater than 20 breaths/minute)
· PaCO2 less than 32 mm Hg
· High (greater than 12,000) or low (less than 4,000 cells/µL) white blood cell count
Most patients have a septic encephalopathy manifested by confusion and reduced responsiveness, and most require ventilatory assistance. With antibiotic treatment, sepsis and septic encephalopathy improve, and patients awaken, tracking the examiner with the eyes and demonstrating comprehension with eye movements or weak limb movements; in patients who cannot cooperate for the examination, weakness is manifested by decreased or absent spontaneous limb movement and difficulty weaning from the ventilator. The diagnosis of CIP is further supported if reflexes are absent and if a sensory loss can be demonstrated; however, not all patients with CIP have absent reflexes or sensory loss on examination, and, in many patients, an accurate sensory examination cannot be done. Electrodiagnostic studies are, therefore, key to diagnosis.
CIM may have similar antecedents, but is more likely in the setting of ventilatory failure from asthma (128,137) or ARDS (132). The use of neuromuscular blocking agents (139) and corticosteroids (130) appears to predispose to CIM, although CIM has been reported without either one (140). Clinically, the presentation is similar to that of CIP. Sensation is normal if neuropathy does not coexist, but often confusion and poor communication make it difficult to assess sensation. Here again, electrophysiologic testing is key to diagnosis.
Diagnosis
Critical illness neuromyopathy should be suspected in any critically ill patient whose medical condition is improving but who appears to have trouble moving or cannot be weaned from the ventilator. A diagnostic algorithm is presented in Figure 149.4. The diagnosis is likely in patients with risk factors, particularly sepsis, SIRS, multiorgan failure, prolonged artificial ventilation, hyperglycemia, and exposure to neuromuscular blocking agents or corticosteroids (141). Alternative causes for weakness in the ICU must be considered and, if possible, excluded. These include medications that cause myopathy, neuromuscular blockade, or neuropathy; electrolyte disorders such as hypokalemia or hypophosphatemia; undiagnosed neuromuscular disorders such as myasthenia gravis, Guillain-Barré syndrome, or ALS; and a variety of central nervous system disorders, including ischemic myelopathy, anterior horn cell loss from hypoxia (142), central pontine myelinolysis that most often follows rapid correction of hyponatremia (143), and hemispheric stroke from hypotension, emboli, or hemorrhage (144). West Nile encephalitis (WNE) should be considered in patients who present with fever and encephalopathy, and in those who develop neuromuscular weakness, since WNE can affect anterior horn cells and, less often, peripheral nerves (145). Depending on clinical suspicions, imaging of the brain and spinal cord may be appropriate. If clinical findings suggest neuromuscular weakness rather than a CNS disorder, it may be more efficient to confirm with electrodiagnostic studies than to pursue studies for CNS disorders.
Laboratory studies should include a basic metabolic profile to assess blood sugar and electrolytes. Elevation of creatine kinase (CK) may suggest myopathy, and very high CK suggests rhabdomyolysis. Normal CK does not exclude critical illness myopathy, and mild elevations in CK can be seen with acute denervation. Tests for myasthenia (see above) are indicated when there is a clinical suspicion. Spinal fluid findings are not often reported in CIP. Lumbar puncture is indicated principally if there is clinical suspicion of infection.
Electrodiagnostic testing is helpful to confirm a neuromuscular disorder and to distinguish among disorders of nerve, neuromuscular junction, and muscle (for a more detailed synopsis, see Dhand [146]). Repetitive nerve stimulation at 2 to 3 Hz will show a decremental response (Fig. 149.3) in patients with disorders of neuromuscular transmission. If the patient has been given neuromuscular blocking agents, repeat testing in 1 to 2 days should demonstrate resolution of neuromuscular blockade. Other causes of neuromuscular blockade should be considered if decremental responses persist. Motor nerve conduction studies will show a decreased amplitude CMAP in patients with CIP within several days of the onset of weakness (to allow for Wallerian degeneration). In patients with CIM, the CMAP is also of low amplitude, but may also be prolonged (147). Sensory nerve action potentials are often reduced or absent in CIP, and should be normal in CIM, unless there is coexistent CIP. It should be kept in mind that pre-existing neuropathy may account for some of the deficits found on electrophysiologic testing, as, for example, in patients with diabetic neuropathy. Guillain-Barré polyneuropathy can follow surgery or trauma, and could therefore occur in settings similar to the critical illness neuromyopathies. Electrophysiologic findings diagnostic of demyelinating neuropathy—not found in CIP or CIM—would suggest this possibility, leading to a trial of IVIG or plasma exchange. In theory, an axonal form of Guillain-Barré would be electrophysiologically indistinguishable from CIP. This highly unlikely occurrence should be considered in patients who lack the usual risk factors for CIP. Needle EMG studies may show abnormal spontaneous activity in patients with neuropathy, but only after 2 to 3 weeks of denervation. Voluntary activity may be difficult to study in poorly cooperative patients; however, when the patient can make a full effort, there will be reduced recruitment in neuropathy and small polyphasic motor unit potentials in myopathy, helping to differentiate CIP from CIM.
Additional electrodiagnostic studies that may be useful in diagnosis include phrenic nerve stimulation, diaphragmatic EMG, and direct muscle stimulation. While these studies do not require specialized equipment, many electromyographers do not have sufficient experience with them to be confident in their performance or interpretation. Phrenic nerve stimulation in the neck with surface recording over the diaphragm shows a normal response in patients with pulmonary causes of respiratory failure, but a low amplitude or absent response in patients with neuromuscular respiratory weakness. Needle EMG of the diaphragm can be done in patients without severe chronic obstructive pulmonary disease (COPD), ileus, or coagulopathy, and may diagnose denervation or myopathy affecting the diaphragm (148). CIP and CIM may be distinguished by comparing the muscle response to direct muscle stimulation with the response to nerve stimulation. If the response to muscle stimulation is larger than to nerve stimulation, CIP is likely; when both are small, CIM is likely (149,150).
In patients suspected of having CINM, nerve and muscle biopsy may document axonal neuropathy or myopathy with loss of myosin filaments; however, they will only change management if they demonstrate an alternative diagnosis that may be amenable to treatment, such as a demyelinating or vasculitic neuropathy, or an inflammatory myopathy. In patients whose neuromuscular deficits are clearly related to the usual risk factors for CINM, treatable conditions are not sufficiently likely to warrant biopsy (151).
Management
Of the factors that may contribute to the development of CINM, many—like the response to sepsis and the incidence of multiorgan failure—are related to the severity of the presenting critical illness, and are not easily modifiable. It remains to be demonstrated whether the reduced use of corticosteroids or neuromuscular blocking agents will result in a decreased incidence of CIM. It makes sense, however, to eliminate continued exposure to either type of agent whenever possible. It also makes sense to avoid medications that can be neurotoxic, or those that can interfere with neuromuscular transmission. Intensive insulin therapy in critically ill patients has been demonstrated to reduce the risk of CIP by 49% over standard therapy (152). The safety and efficacy of intensive insulin therapy remains controversial, however (153,154), and is currently the subject of several prospective controlled multicenter studies.
Patients with neuropathy will be more prone to pressure palsies, and thus, great care must be taken in positioning and support to avoid this complication. Physical therapy should be directed toward avoiding contractures until the patient is able to participate in strengthening exercises.
|
|
|
Figure 149.4. Algorithm for diagnostic evaluation and management of weakness developing in the intensive care setting. CIDP, chronic inflammatory demyelinating polyneuropathy; CIM, critical illness myopathy; CINM, critical illness neuromyopathy; CIP, critical illness polyneuropathy; CK, creatine kinase; CNS, central nervous system; GBS, Guillain-Barré syndrome; LEMS, Lambert-Eaton myasthenic syndrome; LP, lipid peroxidation; MG, myasthenia gravis; NM, neuromuscular. |
Prognosis
There is no comprehensive study on prognosis in CINM. Mortality is high early in the course, and is related to the underlying critical illness (155). Patients with CINM who survive the acute illness and who begin to recover strength are likely to continue to recover over the weeks to months that follow; it is sufficient to monitor improvement with clinical assessment of strength. Patients with CINM who fail to show clinical improvement in strength can be evaluated with repeat electrophysiologic testing. Patients with electrophysiologic evidence of severe axonal loss, with small or absent responses to nerve stimulation and abundant positive waves and fibrillations on needle EMG, will recover slowly if at all, and are likely to have residual deficits—sometimes severe (129,155,156,157). Although the task of regenerating severely damaged axons may be more daunting than regenerating muscle, it is not clear whether the prognosis for recovery from CIM is better than from CIP (158,159).
Summary
Neuromuscular diseases may necessitate admission for intensive care to provide airway protection and respiratory support. Disorders such as Guillain-Barré syndrome and myasthenia gravis can lead to precipitous weakness, and require careful monitoring so that assistance can be given before hypoxemia occurs. Management must also address autonomic dysfunction, sensory loss, pain, and psychological distress. Neuromuscular disorders of autoimmune pathogenesis often respond to specific treatment, and a favorable outcome can be expected for the majority of such patients. The critical illness neuromyopathies have become the most common cause of weakness that develop in the intensive care setting, requiring critical care physicians to be familiar with their presentation, diagnosis, and management. Although no specific treatment is available for CINM, most patients improve with supportive care. A minority, however, are left with irreversible weakness. Attempts to address the factors that predispose to CINM may eventually reduce the burden of this recently discovered group of neuromuscular disorders.
References
1. Bolton CF, Gilbert JJ, Hahn AF, et al. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiatry. 1984;47:1223.
2. Lacomis D, Petrella JT, Giuliani MJ. Causes of neuromuscular weakness in the intensive care unit: a study of ninety-two patients. Muscle Nerve. 1998;21:610.
3. Young RR, Asbury AK, Corbett JF, et al. Pure pandysautonomia with recovery: description and discussion of diagnostic criteria. Brain. 1975;98:613.
4. Waxman SG, Sabin TD. Diabetic truncal polyneuropathy. Arch Neurol. 1981;38:46.
5. Fromm GB, Wisdom PJ, Block AJ. Amyotrophic lateral sclerosis presenting with respiratory failure: diaphragmatic paralysis and dependence on mechanical ventilation in two patients. Chest. 1977;71:612.
6. Chen R, Grand'Maison F, Strong MJ, et al. Motor neuron disease presenting as acute respiratory failure. A clinical and pathological study. J Neurol Neurosurg Psychiatry. 1996;60:455.
7. Dushay KM, Zibrak JD, Jensen WA. Myasthenia gravis presenting as isolated respiratory failure. Chest. 1990;97:232.
8. Rosenow EC 3rd, Engel AG. Acid maltase deficiency in adults presenting as respiratory failure. Am J Med. 1978;64:485.
9. Mohr JP. Distal field infarction. Neurology. 1969;12:279.
10. Riggs JE. Neurologic manifestations of electrolyte disturbances. Neurol Clin. 2002;20:227.
11. Kimura J, Butzer JF. F-wave conduction velocity in Guillain-Barré syndrome. Assessment of nerve segment between axilla and spinal cord. Arch Neurol. 1975;32:524.
12. Kuwabara S, Ogawara K, Mizobuchi K, et al. Isolated absence of F waves and proximal axonal dysfunction in Guillain-Barré syndrome with antiganglioside antibodies J Neurol Neurosurg Psychiatry. 2000;68:191.
13. Maddison P, Newsom-Davis J, Mills KR. Distribution of electrophysiological abnormality in Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 1998;65:213.
14. Chalela JA. Pearls and pitfalls in the intensive care management of Guillain-Barré syndrome. Semin Neurol. 2004;21:399.
15. Borel CO, Guy J. Ventilatory management in critical neurologic illness. In: Jordan KG ed. Neurologic Clinics. Philadelphia: WB Saunders; 1995:627.
16. Ropper AJ, Wijdicks EFM, Truax BT. General care. In: Ropper AH, Wijdicks EFM, Truax BT, eds. Guillain-Barré Syndrome. Philadelphia: FA Davis; 1991:237.
17. Fulgham JR, Wijdicks EFM. Guillain-Barré syndrome. In: Diringer M, ed. Critical Care Clinics: Update on Neurologic Critical Care. Philadelphia: WB Saunders; 1997:1.
18. Bowes D. The doctor as a patient: an encounter with Guillain-Barré syndrome. Can Med Assoc J. 1984;131:1343.
19. Winkel LP, Hagemans ML, van Doom PA, et al. The natural course of non-classic Pompe's disease; a review of 225 published cases. J Neurol. 2005;252:875.
20. Dalakas MC, Hohlfeld R. Polylmyositis and dermatomyositis. Lancet. 2003;362:1762.
21. Landry O. Note sur la paralysie ascendante aiguë. Gaz Hebd Med Paris. 1859;6:472.
22. Guillain G, Barré JA, Strohl A. Sur un syndrome de radiculonévrite avec hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire. Remarques sur les caractéres cliniques et graphiques des réflexes tendineux. Bull Mem Soc Med Hop Paris. 1916;40:1462.
23. Melnick SC, Flewett TH. Role of infection in the Guillain-Barre syndrome. J Neurol Neurosurg Psychiatry. 1964;27:385.
24. Hurwitz ES, Holman RC, Nelson DB, et al. National surveillance for Guillain-Barre syndrome: January 1978–March 1979. Neurology. 1983; 33:150.
25. Waksman BH, Adams RD. A comparative study of experimental allergic neuritis in the rabbit, guinea pig, and mouse. J Neuropathol Exp Neurol. 1956;15:293.
26. National Institute of Neurological and Communicative Disorders and Stroke ad hoc Committee (NINCDS). Criteria for diagnosis of Guillain-Barré syndrome. Ann Neurol. 1978;3:565.
27. Griffin JW, Sheikh K. The Guillain-Barré syndromes. In: Dyck PJ, Thomas PK, eds. Peripheral Neuropathy. 4th ed. Philadelphia: Elsevier Saunders; 2005:2197.
28. McKhann GM, Cornblath DR, Griffin JW, et al. Acute motor axonal neuropathy: a frequent cause of acute flaccid paralysis in China. Ann Neurol. 1993;33:333.
29. Griffin JW, Li CY, Ho TW, et al. Pathology of the motor-sensory axonal Guillain-Barré syndrome. Ann Neurol. 1996;39:17.
30. Hughes RAC, Cornblath DR. Guillain-Barré syndrome. Lancet. 2005; 366:1653.
31. Young RR, Asbury AK, Adams RD, et al. Pure pandysautonomia with recovery. Trans Am Neurol Assoc. 1969;94:355.
32. Young RR, Asbury AK, Corbett JL, et al. Pure pandysautonomia with recovery: description and discussion of diagnostic criteria. Brain. 1975; 98:613.
33. Pan CL, Yuki N, Koga M, et al. Acute sensory ataxic neuropathy associated with monospecific anti-GD1b IgG antibody. Neurology. 2001;57:1316.
34. Fisher CM. An unusual variant of acute idiopathic polyneuritis: syndrome of ophthalmoplegia, ataxia and areflexia. N Engl J Med. 1956;255:57.
35. Shimamura H, Miura H, Iwaki Y, et al. Clinical, electrophysiological, and serological overlap between Miller Fisher syndrome and acute sensory ataxic neuropathy. Acta Neurol Scand. 2002;105:411.
36. Chio A, Cocito D, Leone M, et al. Guillain-Barré syndrome: a prospective, population-based incidence and outcome survey. Neurology. 2003; 60:1146.
37. Alter M. The epidemiology of Guillain-Barré syndrome. Ann Neurol. 1990; 27(Suppl):S7.
38. Hadden RDM, Karch H, Hartung H-P, et al. Preceding infections, immune factors, and outcome in Guillain-Barre syndrome. Neurology. 2001;56:758.
39. Van Koningsveld R, Schmitz PIM, Ang CW, et al. Infections and course of disease in mild forms of Guillain-Barre syndrome. Neurology. 2002;58:610.
40. Hemachudha T, Griffin DE, Chen WW, et al. Immunologic studies of rabies vaccination-induced GBS. Neurology. 1988;38:375.
41. Kaplan JE, Katona P, Hurwitz ES, et al. Guillain-Barré syndrome in the United States, 1989–1980 and 1980–1981. Lack of an association with influenza vaccination. JAMA. 1982;248:698.
42. Lasky T, Terracciano GJ, Magder L, et al. The Guillain-Barré syndrome and the 1992–1993 and 1993–1994 influenza vaccines. N Engl J Med. 1998;339:1797.
43. Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine. 1969;48:173.
44. Prineas JW. Acute idiopathic polyneuritis. An electronmicroscope study. Lab Invest. 1972;26:133.
45. Prineas JW. Pathology of the Guillain-Barré syndrome. Ann Neurol. 1981; 9(suppl):6.
46. Lampert PW. Electron microscopic studies on ordinary and hyperacute experimental allergic encephalomyelitis. Acta Neuropathol. 1967;9:99.
47. Kieseier BC, Kiefer R, Gold R, et al. Advances in understanding and treatment of immune-mediated disorders of the peripheral nervous system. Muscle Nerve. 2004;30:131.
48. Waksman NH, Adams RD. Allergic neuritis: an experimental disease in rabbits induced by the injection of peripheral nervous tissue and adjuvant. J Exp Med. 1955;102:213.
49. Hafer-Macko CE, Sheikh KA, Li CY, et al. Immune attack on the Schwann cell surface in acute inflammatory demyelinating polyneuropathy. Ann Neurol. 1996;39:627.
50. Griffin JW, Li CY, Macko C, et al. Early nodal changes in the acute motor axonal neuropathy pattern of the Guillain-Barré syndrome. J Neurocytol. 1996;25:33.
51. Hafer-Macko C, Hsieh ST, Li CY, et al. Acute motor axonal neuropathy: an antibody-mediated attack on axolemma. Ann Neurol. 1996;40:635.
52. Yuki N, Susuki K, Koga M, et al. Carbohydrate mimicry between human ganglioside GM1 and Campylobacter jejuni lipooligosaccharide causes Guillain-Barré syndrome. Proc Natl Acad Sci U S A. 2004;101:11404.
53. Chiba A, Kusunoki S, Shimizu T, et al. Ann Neurol 1992;31:6779.
54. Willison HJ. The immunobiology of Guillain-Barre syndromes. J Peripher Nerv Syst. 2005;10:94.
55. Koga M, Gilbert M, Li J, et al. Antecedent infections in Fisher syndrome: a common pathogenesis of molecular mimicry. Neurology. 2005;64:1605.
56. Bernsen RAJAM, de Jager AEJ, Schmitz PIM, et al. Long-term sensory deficit after Guillain-Barré syndrome. J Neurol. 2001;248:483.
57. Ropper AH, Wijdicks EFM, Truax, BT. Guillain-Barré Syndrome. Philadelphia: FA Davis; 1991.
58. Masucci EF, Kurtzke JG. Diagnostic criteria for the Guillain-Barré syndrome. An analysis of 50 cases. J Neurol Sci. 1971;13:483.
59. Kuwabara S, Ogawara K, Misawa S, et al. Sensory nerve conduction in demyelinating and axonal Guillain-Barre syndromes. Eur Neurol. 2004; 51:196.
60. Maier H, Schmidbauer M, Pfausler B, et al. Central nervous system pathology in patients with the Guillain-Barré syndrome. Brain. 1997;120:451.
61. Jeha LE, Sila CA, Lederman RJ, et al. West Nile virus infection: a new acute paralytic illness. Neurology. 2003;61:55.
62. Brannagan TH, Zhou Y. HIV-associated Guillain-Barré syndrome. J Neurol Sci. 2003;208:39.
63. Olney RK, Aminof MJ. Electrodiagnostic features of the Guillain-Barré syndrome: the relative sensitivity of different techniques. Neurology. 1990; 40:471.
64. Hughes RAC, Wijdicks E, Barohn RJ, et al. Practice parameter: immunotherapy for Guillain-Barré syndrome: report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2003;61:736.
65. Osterman PO, Fagius J, Lundemo G, et al. Beneficial effects of plasma exchange in acute inflammatory polyradiculoneuropathy. Lancet. 1984; 2:1296.
66. The Guillain-Barré Syndrome Study Group. Plasmapheresis and acute Guillain-Barré syndrome. Neurology. 1985;35:1096.
67. Raphaël JC, Chevret S, Hughes RA, et al. Plasma exchange for Guillain-Barré syndrome. Cochrane Database Syst Rev. 2002;2:CD001798.
68. van der Meché FGA, Schmitz PIM, Dutch Guillain-Barré Study Group. A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med. 1992;326:1123.
69. Hughes RAC, van der Meché FG. Corticosteroids for treating Guillain-Barré syndrome. Cochrane Database Syst Rev. 2000;2:CD001446.
70. Pandey CK, Bose N, Garg G, et al. Gabapentin for the treatment of pain in Guillain-Barré syndrome: a double-blinded, placebo-controlled, crossover study. Anesth Analg. 2002;95:1719.
71. Cornblath DR, Mellits ED, Griffin JW, et al. Motor conduction studies in Guillain-Barré syndrome: description and prognostic value. Ann Neurol. 1988;23:354.
72. De la Cour CD, Jakobsen J. Residual neuropathy in long-term population-based follow-up of Guillain-Barré syndrome. Neurology. 2005;64:246.
73. Bernsen RAJAM, de Jager AEJ, Schmitz PIM, et al. Long-term sensory deficits after Guillain-Barré syndrome. J Neurol. 2001;248:483.
74. Forsberg A, Pressc R, Einarssonb U, et al. Impairment in Guillain-Barré syndrome during the first 2 years after onset: a prospective study. J Neurol Sci. 2004;227:131.
75. Pearce JMS. Mary Broadfoot Walker (1888–1974): a historic discovery in myasthenia gravis. Eur Neurol. 2005;53:51.
76. Drachman DB. Myasthenia gravis: an illustrated history (book review). N Engl J Med. 2003;348:181.
77. Walker MB. Treatment of myasthenia gravis with physostigmine. Lancet. 1934;i:1200.
78. Simpson JA. Immunological disturbances in myasthenia gravis with a report of Hashimoto's disease developing after thymectomy. J Neurol Neurosurg Psychiatry. 1964;27:485.
79. Patrick J, Lindstrom J. Autoimmune response to acetylcholine receptor. Science. 1973;180:871.
80. Almon RR, Andrew CG, Appel SH. Serum globulin in myasthenia gravis: inhibition of a-bungarotoxin binding to acetylcholine receptors. Science. 1974;186:55.
81. Oosterhuis HJ. The natural course of myasthenia gravis: a long-term follow-up study. J Neurol Neurosurg Psychiatry. 1989;52:1121.
82. Drachman DB. Myasthenia gravis. N Engl J Med. 1994;330:1797.
83. Phillips LH II. The epidemiology of myasthenia gravis. Ann N Y Acad Sci. 2003;998:407.
84. Richman DP, Agius MA. Myasthenia gravis: pathogenesis and treatment. Semin Neurol. 1994;14:106.
85. Gracy DR, Divertie MB, Howard FM. Mechanical ventilation for respiratory failure in myasthenia gravis: two-year experience with 22 patients. Mayo Clin Proc. 1983;58:597.
86. Thomas CE, Mayer SA, Gungor Y, et al. Myasthenic crisis: clinical features, mortality, complications, and risk factors for prolonged intubation. Neurology. 1997;48:1253.
87. Engel AG. Anatomy and molecular architecture of the neuromuscular junction. In: Engel Ag, ed. Myasthenia Gravis and Myasthenic Disorders. Oxford: Oxford University Press; 1999:2.
88. Ruff RL. Neurophysiology of the neuromuscular junction: overview. Ann N Y Acad Sci. 2003;998:1.
89. Lindrstrom J. Acetylcholine receptor structure. In: Kaminiski HL, ed. Myasthenia Gravis and Related Disorders. Totowa, NJ: Humana Press; 2003:15.
90. Lindstrom JM, Seybold MD, Lennon VA, et al. Antibody to acetylcholine receptor in myasthenia gravis: prevalence, clinical correlates, and diagnostic value. Neurology. 1976;26:1054.
91. Drachman D, Angus CW, Adams RN, et al. Effect of myasthenic patients' immunoglobulin on acetylcholine receptor turnover: selectivity of degradation process. Proc Natl Acad Sci U S A. 1978;75:3422.
92. Hoch W, McConville J, Helms S, et al. Auto-antibodies to the receptor tyrosine kinase MuSK in patients with myasthenia gravis without acetylcholine receptor antibodies. Nat Med. 2001;7:365.
93. Evoli A, Tonali PA, Padua L, et al. Clinical correlates with anti-MuSk antibodies in generalized myasthenia gravis. Brain. 2003;126:2304.
94. Hohlfeld R, Wekerle H. The thymus in myasthenia gravis. Neurol Clin. 1994;12:331.
95. Kao I, Drachman DB. Thymic muscle cells bear acetylcholine receptors: possible relation to myasthenia gravis. Science. 1977;195:74.
96. Kaminski HJ, Fenstermaker RA, Abdul-Karim FW, et al. Acetylcholine receptor subunit gene expression in thymic tissue. Muscle Nerve. 1993; 16:1332.
97. Iwasa K. Striational autoantibodies in myasthenia gravis mainly react with ryanodine receptor. Muscle Nerve. 1997;20:753.
98. Behan PO. Immune disease and HLA associations with myasthenia gravis. J Neurol Neurosurg Psychiatry. 1980;43:611.
99. Pascuzzi RM. Medications and myasthenia gravis (A reference for health care professionals). Myasthenia Gravis Foundation of America. 2007. http://www.myasthenia.org/hp_edmaterials.cfm. Accessed June 12, 2007.
100. Perrot X, Bernard N, Vial C, et al. Myasthenia gravis exacerbation or unmasking associated with telithromycin treatment. Neurology. 2006;67: 2256.
101. Garrity ER. Respiratory failure due to disorders of the chest wall and respiratory muscles. In: MacDonnell KF, Fahey PJ, Segal MS, eds. Respiratory Intensive Care. Boston: Little Brown; 1987:312.
102. Borel CO, Teitelbaum JS, Hanley DF. Ventilatory failure and carbon monoxide response in ventilatory failure due to myasthenia gravis and Guillain-Barre syndrome. Crit Care Med. 1993;21:1717.
103. Tether JE. Management of myasthenic and cholinergic crisis. Am J Med. 1955;19:740.
104. Ashworth B, Hunter AR. Respiratory failure in myasthenia gravis. Proc R Soc Med. 1971;64:489.
105. Daroff RB. The office Tensilon test for ocular myasthenia gravis. Arch Neurol. 1986;43:843.
106. Okun MS, Charriez CM, Bhatti MR, et al. Asystole induced by edrophonium following beta blockade. Neurology. 2001;57:739.
107. Lennon VA. Serological diagnosis of myasthenic gravis and Lambert-Eaton myasthenic syndrome. In: Lisak RP, ed. Handbook of Myasthenic Gravis and Myasthenic Syndromes. New York: Marcel Dekker; 1994:149.
108. Cikes N, Momi MY, Williams CL, et al. Striational autoantibodies: quantitative detection by enzyme immunoassay in myasthenia gravis, thymoma, and recipients of D-penicillamine or allogenic bone marrow. Mayo Clinic Proc. 1998;63:474.
109. Skeie Go, Romi F, Aarli JA, et al. Pathogenesis of myositis and myasthenia associated with titin ryanodine receptor antibodies. Ann N Y Acad Sci. 2003;998:343.
110. Howard JF, Sanders DB, Massey JM. The electrodiagnosis of myasthenia gravis and the Lambert-Eaton myasthenic syndrome. Neurol Clin. 1994; 12:305.
111. Oh SJ, Kim DE, Kuruoglu R, et al. Diagnostic sensitivity of the laboratory tests in myasthenia gravis. Muscle Nerve. 1992;15:720.
112. Bedlack RS, Sanders DB. On the concept of myasthenic crisis. J Clin Neuromusc Dis. 2002;4:40.
113. Rabinstein AA, Wijdicks EFM. Warning signs of imminent respiratory failure in neurological patients. Semin Neurol. 2003;23:97.
114. Bennet DA, Bleck TP. Recognizing impending respiratory failure from neuromuscular causes. J Crit Illness. 1998;3:46.
115. Juel VC. Myasthenia gravis: management of myasthenic crisis and perioperative care. Semin Neurol. 2004;24:75.
116. Varelas PN, Chua HC, Natterman J, et al. Ventilatory care in myasthenia gravis crisis: assessing the baseline adverse event rate. Crit Care Med. 2002;30:2663.
117. Antozzi C, Gemma M, Regi B, et al. A short plasma exchange protocol is effective in severe myasthenia gravis. J Neurol. 1991;238:103.
118. Gajdos P, Chevert S, Clair B, et al. Myasthenia Gravis Clinical Study Group. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Ann Neurol. 1997;41:789.
119. Stricker RB, Kwiatkowska BJ, Habis JA, et al. Myasthenic crisis: response to plasmapheresis following failure of intravenous gamma-globulin. Arch Neurol. 1993;50:837.
120. Arsura El, Brunner NG, Namba T, et al. High-dose intravenous methylprednisolone in myasthenia gravis. Arch Neurol. 1985;42:1149.
121. Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:7.
122. Leventhal SR, Orkin FK, Hirsh RA. Prediction for postoperative mechanical ventilation in myasthenia gravis. Anesthesiology. 1980;53:26.
123. Huang C-s, Hsu H-s, Kao K-P, et al. Intravenous immunoglobulin in the preparation of thymectomy for myasthenia gravis. Acta Neurol Scand. 2003;108:136.
124. Lindsay AE, Dimachkie M, Pulley MT, et al. Critical illness myopathy and polyneuropathy. Medlink Neurol. 2006.
125. Gorson KC. Approach to neuromuscular disorders in the intensive care unit. Neurocrit Care. 2006;3:195.
126. Bolton CF, Gilbert JJ, Hahn AF, et al. Polyneuropathy in critically ill patients. J Neurol Neurosurg Psychiatry. 1984;47:1223.
127. Sagredo V, Caldwell JE, Matthay MA, et al. Persistent paralysis in critically ill patients after long-term administration of vecuronium. N Engl J Med. 1992;327:524.
128. Hirano M, Ott BR, Raps EC. Acute quadriplegic myopathy: complication of treatment with steroids, nondepolarizing blocking agents, or both. Neurology. 1992;42:2082.
129. Witt NJ, Zochodne DW, Bolton CF, et al. Peripheral nerve function in sepsis and multiple organ failure. Chest. 1991;99:176.
130. De Jonghe B, Sharshar T, Lefaucheur J-P, et al. Paresis acquired in the intensive care unit. A prospective multicenter study. JAMA. 2002;288:2859.
131. Bednarik J, Vondracek P, Dusek L, et al. Risk factors for critical illness polyneuropathy. J Neurol. 2005;252:343.
132. Bercker S, Weber-Carstens S, Deja M, et al. Critical illness polyneuropathy and myopathy in patients with acute respiratory distress syndrome. Crit Care Med. 2005;33:711.
133. Zochodne DW, Bolton CF, Wells GA, et al. Critical illness polyneuropathy: a complication of sepsis and multiple organ failure. Brain. 1987;110: 819.
134. de Letter MA, Schmitz PI, Visser LH, et al. Risk factors for the development of polyneuropathy and myopathy in critically ill patients. Crit Care Med. 2001;29:2281–2286.
135. Murray MJ, Brull SJ, Bolton CF. Brief review: nondepolarizing neuromuscular blocking drugs and critical illness myopathy. Can J Anesth. 2006;53:1148.
136. Minetti C, Hirano M, Morreale G, et al. Ubiquitin expression in acute steroid myopathy with loss of thick myosin filaments. Muscle Nerve. 1996; 19:94.
137. Danon MJ, Carpenter S. Myopathy and thick filament (myosin) loss following prolonged paralysis with vecuronium during steroid treatment. Muscle Nerve. 1991;14:1131.
138. Bone RC, Sprung CL, Sibbald WJ. Definitions for sepsis and organ failure. Crit Care Med. 1992;20:724.
139. Adnet F, Dhissi G, Borron SW, et al. Complication profiles of adult asthmatics requiring paralysis during mechanical ventilation. Intensive Care Med. 2001;27(11):1729.
140. Hoke A, Rewcastle NB, Zochodne DW. Acute quadriplegic myopathy unrelated to steroids or paralyzing agents: quantitative EMG studies. Can J Neurol Sci. 1999;26:325.
141. Deem S. Intensive-care-unit-acquired muscle weakness. Respir Care. 2006; 51:1042.
142. Azzarelli B, Roessmann U. Diffuse “anoxic” myelopathy. Neurology. 1977; 27:1049.
143. Riggs JE. Neurological manifestations of electrolyte disturbances. Neurol Clin. 2002;30:227.
144. Maramattom BV, Wijdicks EFM. Acute neuromuscular weakness in the intensive care unit. Crit Care Med. 2006;34:2835.
145. Glass JD, Samuels O, Rich MM. Poliomyelitis due to West Nile virus. N Engl J Med. 2002;347:1280.
146. Dhand UK. Clinical approach to the weak patient in the intensive care unit. Respir Care. 2006;51:1024.
147. Parak EJ, Nishida T, Sufit RL, et al. Prolonged compound muscle action potential duration in critical illness myopathy. Report of nine cases. J Clin Neuromusc Dis. 2004;5:176.
148. Bolton CF. AAEM minimonograph #40: clinical neurophysiology of the respiratory system. Muscle Nerve. 1993;16:809.
149. Rich MM, Teener JW, Raps EC, et al. Muscle is electrically inexcitable in acute quadriplegic myopathy. Neurology. 1996;46:731.
150. Lefaucheur JP, Nordine T, Rodriguez P, et al. Origin of ICU acquired paresis determined by direct muscle stimulation. J Neurol Neurosurg Psychiatry. 2006;77:500.
151. Lacomis D, Zochodne D, Bird SJ. Critical illness myopathy. Muscle Nerve. 2000;23:1785.
152. Van den Berghe G, Schoonheydt K, Becx P, et al. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64:1348.
153. Malhotra A. Intensive insulin in intensive care. N Engl J Med. 2006; 354:516.
154. Brunkhorst FM, Kuhnt E, Engel C, et al. Intensive insulin therapy in patient with severe sepsis and septic shock is associated with an increased rate of hypoglycemia: results from a randomized multicenter study (VISEP). Infection. 2005;33(Suppl):19.
155. Lacomis D, Petrella JT, Giuliani MJ. Causes of neuromuscular weakness in the intensive care unit: a study of ninety-two patients. Muscle Nerve. 1998;21:610.
156. Zifko UA. Long-term outcome of critical illness polyneuropathy. Muscle Nerve. 2000;9(Suppl):S49.
157. Latronico N, Shehu I, Seghelini E. Neuromuscular sequelae of critical illness. Curr Opin Crit Care. 2005;11:381.
158. Latronico N, Fenzi F, Boniotti C, et al. Acute reversible paralysis in critically ill patients. Acta Anaesthesiol Ital. 1993;44:157.
159. Latronico N, Fenzi F, Recupero D, et al. critical illness myopathy and neuropathy. Lancet. 1996;347:1579.