The mammalian neuromuscular junction is one of the most studied and best understood synapses.1 The neuromuscular junction is a synapse that develops between a motor neuron and a muscle fiber and is made up of several components: the presynaptic nerve terminal, the postsynaptic muscle membrane, and the intervening cleft (or gap). In the 19th century and early years of the 20th century, there was broad support for the concept that impulses in nerves acted directly on muscle, resulting in muscular contraction—the “electrical theory.” This theory was eventually refuted with the discovery of the role of acetylcholine in neuromuscular transmission,2,3 and in 1936, the Nobel Prize in Physiology or Medicine was awarded to Sir Henry Hallett Dale and Otto Loewi “for their discoveries relating to chemical transmission of nerve impulses.” This was followed by the discovery that a muscle membrane–bound allosteric protein, the nicotinic acetylcholine receptor.4
Muscle Types
Muscle is generally classified as skeletal, smooth, or cardiac. Both skeletal and cardiac muscles are striated muscles sharing a common basic organization of the contractile filaments. There are, however, distinct histologic and functional differences between these two muscles. Skeletal muscle cells are multinucleated and tubular in shape, whereas cardiac muscle cells may be mono- or binucleated and are branched and contain intercalated discs. Skeletal muscle is responsible for voluntary actions, whereas smooth muscle and cardiac muscle subserve functions related to the cardiovascular, respiratory, gastrointestinal, and genitourinary systems. Muscle composes 45% to 50% of total body mass, with skeletal muscles accounting for approximately 40% of body mass. An estimated 250 million cells are present in the more than 400 skeletal muscles of humans. Muscle cells are highly specialized cells for the conversion of chemical energy into mechanical energy. Inappropriate activity of smooth muscle is involved in many illnesses including hypertension, atherosclerosis, asthma, and disorders of the gastrointestinal tract.
Motor Units
Vertebrate skeletal muscles are innervated by large myelinated α motor neurons that originate from cell bodies located in the brainstem or ventral (anterior) horns of the spinal cord (Fig. 11-1).5 The myelinated nerve axon reaches the muscle through mixed peripheral nerves. Motor nerves branch in the skeletal muscle with each nerve terminal innervating a single muscle cell.
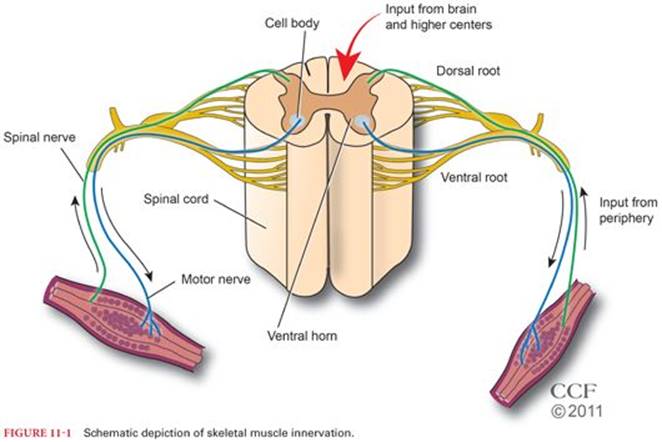
The motor unit is the functional contractile unit and is composed of a single myelinated α motor neuron and all muscle fibers that receive innervation from this single neuron. Motor units vary in size. A large motor nerve innervates more muscle fibers than a smaller motor nerve does. In general, small motor units innervate the “red slow” muscle fibers, whereas large motor units innervate the “white or pale fast” muscle fibers. The slow muscle fibers appear red as a result of high contents of myoglobin, mitochondria, and capillaries compared with white muscle fibers. Unlike the white muscle fibers, the red fibers are resistant to fatigue.6 Muscles contain a varying mixture of motor units depending on their function.
The Neuromuscular Junction
The neuromuscular junction (NMJ) or the endplate is a highly specialized synapse at which presynaptic motor nerve endings meet the postsynaptic membranes of skeletal muscles (motor endplates) (Fig. 11-2). The formation, differentiation, and function of the NMJ requires a proper interaction (cross-talk) between the nerve terminal and muscle cell (for a review, see Naguib et al.1). Failure of this cross-talk will result in a wide spectrum of neuromuscular disorders. It should be noted that in early postnatal period, muscle fibers receive multiple innervation. It has been shown in mice that on the second postnatal day, approximately 75% of muscle fibers are multiply innervated (>95% by two axons).7 However, the transition from multiple to single innervation at the NMJ occurs within few days.7 The motor nerve ending branches to form a complex of nerve terminals that invaginate into the skeletal muscle fiber but lie outside the sarcolemma. As each motor neuron approaches its target muscle fiber, it loses its myelin sheath and makes a contact with a single muscle fiber to form an NMJ. The naked motor nerve terminal that is not in contact with the muscle fiber is capped by Schwann cells. The importance of Schwann cells for development, survival, and repair of several aspects of NMJ is reviewed elsewhere.1
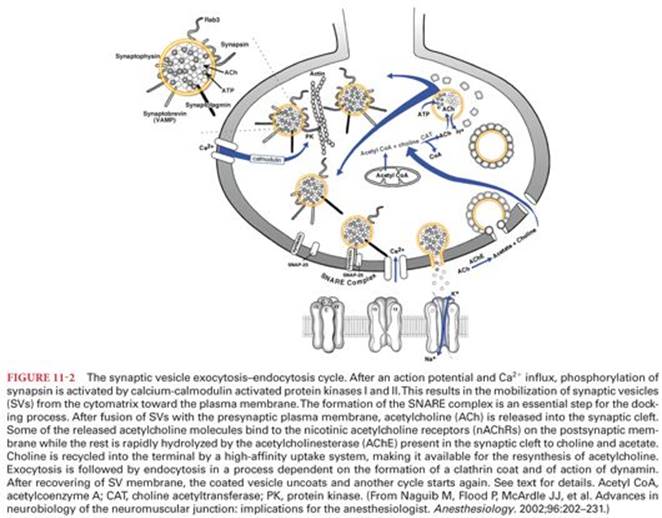
The NMJ is designed to transmit electrical impulses from the nerve terminal to the skeletal muscle via the chemical transmitter, acetylcholine (ACh). Structurally, the NMJ is consisted of a three components: (a) the presynaptic(or prejunctional) nerve terminal containing synaptic vesicles (filled with ACh) and mitochondria; (b) the synaptic cleft that contains basal lamina to which acetylcholinesterase enzyme responsible for hydrolysis of free ACh is attached; and (c) the postsynaptic (or postjunctional) muscle membrane that opposes the nerve terminal, which is highly infolded and these folds are called secondary folds (or secondary postsynaptic cleft). Membrane infoldings increase the surface area of the muscle plasma membrane in the postsynaptic region. Nicotinic acetylcholine receptors (nAChRs) are concentrated at the crests of these folds (directly opposing the active zones of the presynaptic membrane in which synaptic vesicles are clustered) and voltage-gated sodium channels (VGSC) are present in the troughs of the folds.
The plasticity of neuromuscular transmission is dependent on a highly orchestrated mechanism involving (a) synthesis, storage, and release of acetylcholine from the presynaptic region at the NMJ; (b) binding of acetylcholine to nicotinic receptors on the muscle membrane (postsynaptic region) and generation of action potentials; (c) rapid hydrolysis of acetylcholine by the enzyme acetylcholinesterase, which is present in the synaptic cleft; and (d) adaptation of the muscle contractile proteins to functional demands.1 Synaptic plasticity is the “ability of individual synaptic junctions to respond [i.e., to change in strength in response] to either use or disuse.”8
Presynaptic Region
Synaptic Vesicles
Synaptic vesicles (SVs) are specialized secretory organelles (see Fig. 11-2). SVs are synthesized in the neuronal cell body in the endoplasmic reticulum and transported to the nerve terminal via the microtubule system. SVs are then loaded with ACh in the motor nerve endings. Acetylcholine is first synthesized in the cytoplasm of the nerve terminal from acetyl coenzyme A and choline in a reaction catalyzed by the soluble enzyme choline acetyltransferase. An energy-dependent “transporter” then accumulates acetylcholine within vesicles. Each vesicle appears to contain 5,000 to 10,000 molecules of acetylcholine. The acetylcholine contained in a single vesicle is often referred to as a “quantum” of transmitter. The synaptic vesicles possess a diverse set of specialized proteins that can be divided into two functional classes: proteins involved in the uptake of neurotransmitters (transport proteins) and proteins that mediate SV membrane traffic such as docking, fusion, and budding (for review, see Naguib et al.1). Ca2+ signal plays a pivotal role in the process of acetylcholine vesicles exocytosis (see Fig. 11-2). There are two pools of vesicles that differ in the probability of mobilization to the active site: a readily releasable store (active pool) and a reserve store. The active pool is aligned near the active zones.
The miniature endplate potential (MEPP) amplitude represents the depolarization of the postsynaptic membrane produced by the contents of a single vesicle. Endplate potential (EPP) results from summation of MEPP produced by ACh contents of ~50 to 300 SVs. Nearly 50% of the released acetylcholine is rapidly hydrolyzed by the acetylcholinesterase during the time of diffusion across the synaptic cleft before reaching nAChRs. The products of this hydrolysis are choline and acetate. Choline is recycled into the terminal by a high-affinity uptake system, making it available for the resynthesis of acetylcholine. The drug hemicholinium-3 inhibits the later mechanism. After exocytosis, the membrane components of the SVs are recovered by endocytosis and recycled for future use.
Synaptic Cleft
The synaptic cleft is ~20 to 50 nm wide. It separates nerve and muscle fiber plasma membranes and encompasses the synaptic basal lamina and is filled with extracellular fluid. Acetylcholinesterase enzyme is bound to the basal lamina at the cleft (Fig. 11-3).
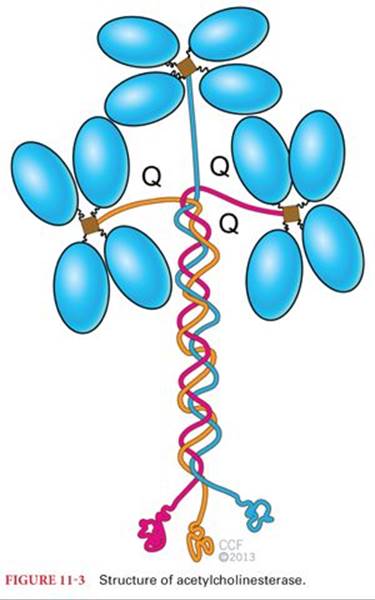
Acetylcholinesterase ranks as one of the highest catalytic efficiencies known. The efficiency of acetylcholinesterase depends on its fast catalytic activity. It can catalyze acetylcholine hydrolysis (4,000 molecules of acetylcholine hydrolyzed per active site per second) at near diffusion-limited rates.9 Acetylcholinesterase is highly concentrated at the NMJ but present in a lower concentration throughout the length of muscle fibers.10 Acetylcholinesterase is regulated, in part, by muscle activity and by the spontaneous or nerve-evoked depolarization of the plasma membrane.11 After denervation, there is a large decrease in the density of acetylcholinesterase molecules at the NMJ. In addition to hydrolysis of acetylcholine, acetylcholinesterase has other functions such as nerve growth–promoting activities12 and modulation of nAChRs.
The Nicotinic Acetylcholine Receptor at the Neuromuscular Junction
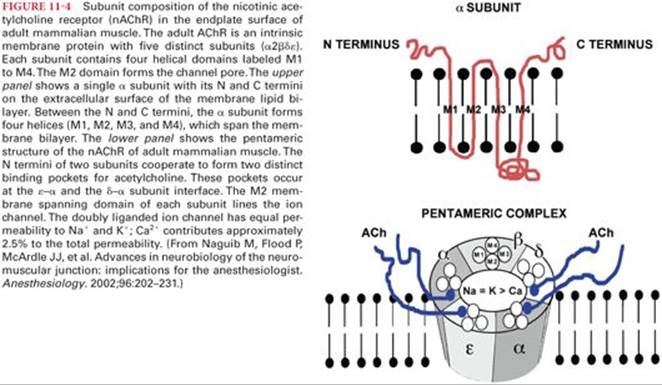
In the adult mammalian skeletal muscle the nicotinic acetylcholine receptor (nAChR) is a pentameric complex of two α subunits in association with a single β, δ, and ε subunit (Fig. 11-4). These subunits are organized to form a transmembrane pore (a channel) as well as the extracellular binding pockets for acetylcholine and other agonists or antagonists.1 Each of the two α subunits has an acetylcholine-binding site. These sites are proteins located in pockets approximately 3.0 nm above the surface membrane at the interfaces of the α-ε and α-δ subunits.13 The fetal nAChR contains a γ subunit instead of an ε adult subunit. The mature nAChR has shorter burst duration and a higher conductance to Na+, K+, and Ca2+ than the fetal nAChR.1,14
The fetal nAChR is a low-conductance channel in contrast to the high-conductance channel of the adult nAChR (Fig. 11-5). Thus acetylcholine release causes brief activation and reduced probability of channel opening.1Upregulation of nicotinic acetylcholine receptors (nAChRs), found in states of functional or surgical denervation, is characterized by the spreading of predominantly fetal type nAChRs. These receptors are resistant to nondepolarizing neuromuscular blockers and more sensitive to succinylcholine (SCh).15 When depolarized, the immature isoform has a prolonged open channel time that exaggerates the K+ efflux.16
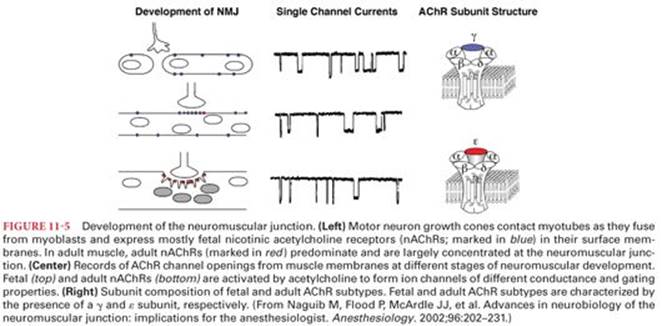
Simultaneous binding of two acetylcholine molecules to the two α subunits of nAChRs initiates conformational changes that open a channel through the center of the receptor, allowing sodium and calcium ions to move into the skeletal muscle and potassium ions to leave. Each NMJ contains several million postjunctional receptors, and a burst of acetylcholine from the nerve ending opens at least 400,000 receptors. As a result, sufficient current flows through these open receptors to depolarize the endplate and create the action potential that triggers contraction of the skeletal muscle. It is the flow of ions that is the basis of normal neuromuscular transmission.
The two α subunits, in addition to being the binding sites for acetylcholine, are the sites occupied by neuromuscular blocking drugs. Nondepolarizing neuromuscular blocking drugs bind to one or both α subunits, but unlike acetylcholine, lack agonist activity (competitive blockade). As a result, conformational changes do not occur, and the receptor channel remains closed. Therefore, ions do not flow through these channels, and depolarization cannot occur at these sites. If enough channels remain closed, there is blockade of neuromuscular transmission. A nondepolarizing neuromuscular-blocking drug may show preference for one of the two α subunits. This may result in synergism if two nondepolarizing neuromuscular-blocking drugs with different selective preferences for each α subunit are administered simultaneously. The probability of binding is dependent on the concentration of acetylcholine and nondepolarizing neuromuscular-blocking drug at the receptor and the affinity of the receptor for the neurotransmitter or drug. When the neuromuscular-blocking drug diffuses from the nAChRs, the probability of receptor binding of acetylcholine increases, and the effect of the nondepolarizing neuromuscular-blocking drug decreases.
SCh, which is structurally two molecules of acetylcholine bound together, is a partial agonist at nAChRs and depolarizes (opens) the ion channels. This opening requires the binding of only one molecule of SCh to the α subunit. The other α subunit can be occupied by either acetylcholine or SCh. Because SCh is not hydrolyzed by acetylcholinesterase, the channel remains open for a longer period of time than would be produced by acetylcholine, resulting in a depolarizing block (sustained depolarization prevents propagation of an action potential). Furthermore, SCh can diffuse from nAChRs and repeatedly bind to other nAChRs until it is cleared from the area of the NMJ and is exposed to hydrolysis by plasma cholinesterase.
Large doses of nondepolarizing neuromuscular-blocking drugs may also prevent normal flow of ions by entering the channels formed by nAChRs to produce blockade within the channel. Similar blockade of sodium ion channels is produced by local anesthetics.
Neuromuscular Transmission and Excitation-Contraction Coupling
Motor Nerve
Depolarization of the motor nerve will open the voltage-gated Ca2+ channels that trigger both mobilization of synaptic vesicles and the fusion machinery in the nerve terminal to release acetylcholine.
Several forms of K+ channel present in the nerve terminal serve to limit the extent of Ca2+ entry and transmitter release (i.e., initiate repolarization of nerve terminal).17
Muscle
The released acetylcholine binds to α subunits of the nAChRs. These ligand-gated cation channels open almost instantaneously when two acetylcholine molecules bind cooperatively to sites on the extracellular surface of the protein, causing a conformational shift in the subunits. When the channel opens, sodium ions flow down their electrochemical gradient and into the muscle cell and depolarize the muscle cell membrane at the NMJ, whereas potassium simultaneously exits the cytosol of the fiber.17,18 This depolarization activates voltage-gated sodium channels that present in the muscle membrane, which mediate the initiation and propagation of action potentials across the surface of the muscle membrane and into the transverse tubules (T-tubules) resulting in the upstroke of the action potential.17–19
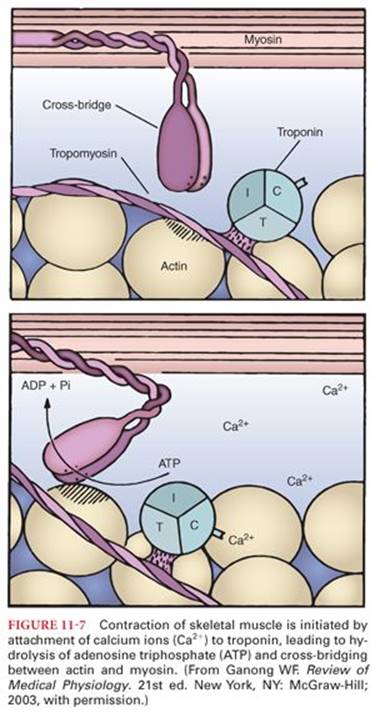
There are two types of calcium channels, dihydropyridine receptor (DHPR) in the T-tubules and the ryanodine receptor (RyR1) in the sarcoplasmic reticulum (Fig. 11-6). The DHPRs act as “voltage sensors”20,21 and are activated by membrane depolarization, which in turn activate RyR1 receptors. DHPR-RyR1 interaction22 releases large amounts of Ca2+ from the sarcoplasmic reticulum that result in a transient increase in myoplasmic free Ca2+, which binds to troponin C. This initiates movement of tropomyosin on the thin filament (actin) and allows cross bridges between the myosin with the binding sites on actin, which eventually result in force development. This process is known as excitation-contraction coupling (Fig. 11-7).23 Shortly after releasing calcium, the sarcoplasmic reticulum begins to reaccumulate this ion by an active transport process (calcium pump). This transport mechanism can concentrate calcium up to 2,000-fold inside the sarcoplasmic reticulum. Adenosine triphosphate (ATP) provides the energy for calcium ion transport. Once the calcium concentration in the sarcoplasm has been lowered sufficiently, cross-bridging between myosin and actin ceases and the skeletal muscle relaxes. Failure of the calcium ion pump results in sustained skeletal muscle contraction and marked increases in heat production, leading to malignant hyperthermia. The gene for this calcium ion channel (ryanodine receptor) is on chromosome 19. A mutation in this gene is associated with malignant hyperthermia susceptibility in some patients.
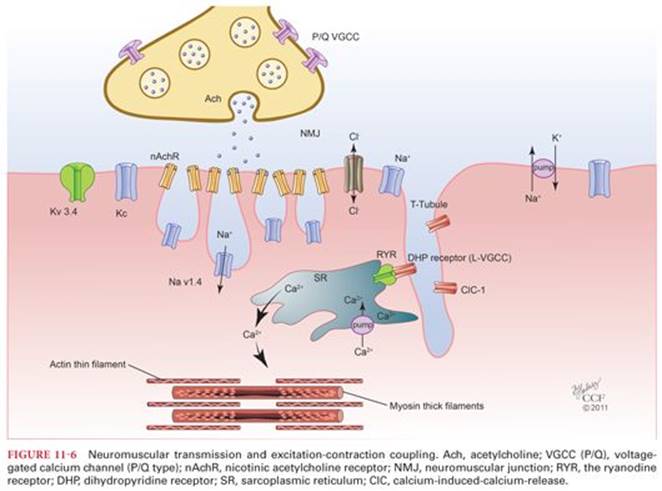
Repolarization of the muscle membrane is initiated by the closing of the sodium channels and by the opening of potassium ion channel that conduct an outward K+ current.24 The return the muscle membrane potential to its resting level (approximately −70 to −90 mV) is achieved by allowing Cl− to enter the cell through voltage-sensitive chloride channels.17
Blood Flow
Skeletal muscle blood flow can increase more than 20 times (a greater increase than in any other tissue of the body) during strenuous exercise. At rest, only 20% to 25% of the capillaries are open, and skeletal muscle blood flow is 3 to 4 mL/100 g per minute. During strenuous exercise, almost all skeletal muscle capillaries become patent. Opening of previously collapsed capillaries diminishes the distance that oxygen and other nutrients must diffuse from capillaries to skeletal muscle fibers and contributes an increased surface area through which nutrients can diffuse from blood. Presumably, exercise lowers the local concentration of oxygen, which in turn causes vasodilation because the vessel walls cannot maintain contraction in the absence of adequate amounts of oxygen. Alternatively, oxygen deficiency may cause release of vasodilator substances such as potassium ions and adenosine. The increase in cardiac output that occurs during exercise results principally from local vasodilation in active skeletal muscles and subsequent increased venous return to the heart. Among inhaled anesthetics, isoflurane is a potent vasodilator, producing marked increases in skeletal muscle blood flow.
Exercise is associated with a centrally mediated stimulation of the sympathetic nervous system manifesting as vasoconstriction in nonmuscular tissues and increases in systemic blood pressure. Excessive increases in systemic blood pressure, however, are prevented by vascular vasodilation that occurs in the large tissue mass represented by skeletal muscles. Exceptions to nonmuscular tissue vasoconstriction induced by exercise are the coronary and cerebral circulations. This is teleologically understandable because the heart and brain are essential to the response to exercise, as are the skeletal muscles.
Smooth Muscle
Smooth muscle is distinguished anatomically from skeletal and cardiac muscle because it lacks visible cross-striations (actin and myosin are not arranged in regular arrays). Smooth muscle is categorized as multiunit or visceralsmooth muscle. Multiunit smooth muscle contraction is controlled almost exclusively by nerve signals, and spontaneous contractions rarely occur. Examples of multiunit smooth muscles are the ciliary muscles of the eye, iris of the eye, and smooth muscles of many large blood vessels. Smooth muscle must develop force or shorten to provide motility or to alter the dimensions of an organ.
Smooth muscle cells lack T-tubules that provide electrical links to sarcoplasmic reticulum. However, the sarcolemma of smooth muscle contains saclike inpocketings (caveoli) that may be sites where calcium ions enter the cells through voltage-gated calcium ion channels. Calcium ions are released from the sarcoplasmic reticulum into the myoplasm when stimulatory neurotransmitters, hormones, or drugs bind to receptors on the sarcolemma. Calcium ion channels on the sarcoplasmic reticulum of smooth muscles includes ryanodine receptors (similar to that present in skeletal muscles) and inositol 1,4,5-triphosphate (IP3)-gated calcium ion channels. Neurotransmitters or hormones that act via receptors in the sarcolemma can activate phospholipase C followed by the generation of the second messenger IP3. IP3 channels are activated when hormones bind to calcium-mobilizing receptors in the sarcoplasmic reticulum in smooth muscle cells.
Visceral smooth muscle is characterized by cell membranes that contact adjacent cell membranes, forming a functional syncytium that often undergoes spontaneous contractions as a single unit in the absence of nerve stimulation. These spontaneous action potentials are particularly prominent in tubular structures, accounting for peristaltic motion in sites such as the bile ducts, ureters, and gastrointestinal tract, especially when they are distended. Plateaus in the action potentials of visceral smooth muscle lasting up to 30 seconds may occur in the ureters and uterus. The normal resting transmembrane potential is approximately −60 mV, which is approximately 30 mV less negative than in skeletal muscles.
In addition to stimulation in the absence of extrinsic innervation, smooth muscles are unique in their sensitivity to hormones or local tissue factors. For example, smooth muscle spasm may persist for hours in response to norepinephrine or antidiuretic hormone, whereas local factors such as lack of oxygen or accumulation of hydrogen ions cause vasodilation. It is believed that local factors and hormones cause smooth muscle contraction by activating the calcium ion transport mechanism. Drugs relax smooth muscle by increasing the intracellular concentration of cyclic adenosine monophosphate or cyclic guanosine monophosphate.
Mechanism of Contraction
Smooth muscles contain both actin and myosin but, unlike skeletal muscles, lack troponin. In contrast to skeletal muscles, in which calcium binds to troponin to initiate cross-bridging, in smooth muscle the calcium-calmodulin complex activates the enzyme necessary for phosphorylation of myosin. This myosin has ATPase activity, and actin then slides on myosin to produce contraction.
The source of calcium in smooth muscle differs from that in skeletal muscle because the sarcoplasmic reticulum of smooth muscle is poorly developed. Most of the calcium that causes contraction of smooth muscles enters from extracellular fluid at the time of the action potential. The time required for this diffusion is 200 to 300 ms, which is approximately 50 times longer than for skeletal muscles. Subsequent relaxation of smooth muscles is achieved by a calcium ion transport system that pumps these ions back into extracellular fluid or into the sarcoplasmic reticulum. This calcium ion pump is slow compared with the sarcoplasmic reticulum pump in skeletal muscles. As a result, the duration of smooth muscle contraction is often seconds rather than milliseconds as is characteristic of skeletal muscles.
Smooth muscles, unlike skeletal muscles, do not atrophy when denervated, but they do become hyperresponsive to the normal neurotransmitter. This denervation hypersensitivity is a general phenomenon that is largely due to synthesis or activation of more receptors.
An NMJ similar to that present on skeletal muscles does not occur in smooth muscles. Instead, nerve fibers branch diffusely on top of a sheet of smooth muscle fibers without making actual contact. These nerve fibers secrete their neurotransmitter into an interstitial fluid space a few microns from the smooth muscle cells. Two different neurotransmitters, acetylcholine and norepinephrine, are secreted by the autonomic nervous system nerves that innervate smooth muscles. Acetylcholine is an excitatory neurotransmitter for smooth muscles at some sites and functions as an inhibitory neurotransmitter at other sites. Norepinephrine exerts the reverse effect of acetylcholine. It is believed that the presence of specific excitatory or inhibitory receptors in the membranes of smooth muscle fibers determines the response to acetylcholine or norepinephrine. When the neurotransmitter interacts with an inhibitory receptor instead of an excitatory receptor, the membrane potential of the smooth muscle fiber becomes more negative (hyperpolarized).
Uterine Smooth Muscle
Uterine smooth muscle is characterized by a high degree of spontaneous electrical and contractile activity. Unlike the heart, there is no pacemaker, and the contraction process spreads from one cell to another at a rate of 1 to 3 cm/s. Contractions of labor result in peak intrauterine pressures of 60 to 80 mm Hg in the second stage. Resting uterine pressure during labor is approximately 10 mm Hg. Movement of sodium ions appears to be the primary determinant in depolarization, whereas calcium ions are necessary for excitation-contraction coupling. Availability of calcium ions greatly influences the response of uterine smooth muscle to physiologic and pharmacologic stimulation or inhibition. Alpha excitatory and beta inhibitory receptors are also present in the myometrium.
References
1. Naguib M, Flood P, McArdle JJ, et al. Advances in neurobiology of the neuromuscular junction: implications for the anesthesiologist. Anesthesiology. 2002;96:202–231.
2. Loewi O. Über humorale Übertragbarkeit der Herznervenwirkung. Pflugers Arch Gesamte Physiol Menschen Tiere. 1921;189:239–242.
3. Dale HH, Feldberg W, Vogt M. Release of acetylcholine at voluntary motor nerve endings. J Physiol. 1936;86:353–380.
4. Changeux JP. The acetylcholine receptor: an “allosteric” membrane protein. Harvey Lect. 1981;75:85–254.
5. Berne RM, Levy MN, Koeppen BM, et al. Physiology. 5th ed. St Louis: Mosby; 2004.
6. Burke RE, Levine DN, Zajac FE III. Mammalian motor units: physiological-histochemical correlation in three types in cat gastrocnemius. Science. 1971;174:709–712.
7. Colman H, Nabekura J, Lichtman JW. Alterations in synaptic strength preceding axon withdrawal. Science. 1997;275:356–361.
8. Hughes JR. Post-tetanic potentiation. Physiol Rev. 1958;38:91–113.
9. Rosenberry TL. Acetylcholinesterase. Adv Enzymol Relat Areas Mol Biol. 1975;43:103–218.
10. Cresnar B, Crne-Finderle N, Breskvar K, et al. Neural regulation of muscle acetylcholinesterase is exerted on the level of its mRNA. J Neurosci Res. 1994;38:294–299.
11. Rossi SG, Vazquez AE, Rotundo RL. Local control of acetylcholinesterase gene expression in multinucleated skeletal muscle fibers: individual nuclei respond to signals from the overlying plasma membrane. J Neurosci. 2000;20:919–928.
12. Sung JJ, Kim SJ, Lee HB, et al. Anticholinesterase induces nicotinic receptor modulation. Muscle Nerve. 1998;21:1135–1144.
13. Machold J, Weise C, Utkin Y, et al. The handedness of the subunit arrangement of the nicotinic acetylcholine receptor from Torpedo californica. Eur J Biochem. 1995;234:427–430.
14. Villarroel A, Sakmann B. Calcium permeability increase of endplate channels in rat muscle during postnatal development. J Physiol (Lond). 1996;496:331–338.
15. Martyn JA. Basic and clinical pharmacology of the acetylcholine receptor: implications for the use of neuromuscular relaxants. Keio J Med. 1995;44:1–8.
16. Kallen RG, Sheng ZH, Yang J, et al. Primary structure and expression of a sodium channel characteristic of denervated and immature rat skeletal muscle. Neuron. 1990;4:233–242.
17. Cooper EC, Jan LY. Ion channel genes and human neurological disease: recent progress, prospects, and challenges. Proc Natl Acad Sci U S A. 1999;96:4759–4766.
18. Jurkat-Rott K, Lerche H, Lehmann-Horn F. Skeletal muscle channelopathies. J Neurol. 2002;249:1493–1502.
19. Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1372.
20. Rios E, Pizarro G. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiol Rev. 1991;71:849–908.
21. Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol Rev. 2002;82:893–922.
22. MacKrill JJ. Protein-protein interactions in intracellular Ca2+-release channel function. Biochem J. 1999;337(pt 3):345–361.
23. Hoffman EP. Voltage-gated ion channelopathies: inherited disorders caused by abnormal sodium, chloride, and calcium regulation in skeletal muscle. Annu Rev Med. 1995;46:431–441.
24. Lehmann-Horn F, Jurkat-Rott K, Rudel R. Periodic paralysis: understanding channelopathies. Curr Neurol Neurosci Rep. 2002;2: 61–69.