Local anesthetics are used to provide analgesia and anesthesia for various surgical and nonsurgical procedures. These drugs are also used for acute and chronic pain management, to reduce perioperative stress, to improve perioperative outcomes, and to treat dysrhythmias. Local anesthetics produce reversible conduction blockade of impulses along central and peripheral nerve pathways. With progressive increases in concentrations of local anesthetics, the transmission of autonomic, somatic sensory, and somatic motor impulses is interrupted, producing autonomic nervous system blockade, sensory anesthesia, and skeletal muscle paralysis in the area innervated by the affected nerve.
Karl Koller introduced cocaine as the first local anesthetic in 1884, for use in ophthalmology. Halsted recognized the ability of injected cocaine to interrupt nerve impulse conduction, leading to the introduction of peripheral nerve block anesthesia and spinal anesthesia. Cocaine (an ester of benzoic acid) is present in large amounts in the leaves of Erythroxylon coca, a plant growing in the Andes Mountains, where its cerebral-stimulating qualities are well known. Another unique feature of cocaine is its ability to produce localized vasoconstriction, making it useful to shrink the nasal mucosa in rhinolaryngologic procedures and nasotracheal intubation. The first synthetic local anesthetic was the ester derivative procaine, introduced by Einhorn in 1905. Lidocaine was synthesized as an amide local anesthetic by Lofgren in 1943. It produces more rapid, intense, and longer-lasting conduction blockade than procaine. Unlike procaine, lidocaine is effective topically and is a highly efficacious cardiac antidysrhythmic drug. For these reasons, lidocaine is the standard to which all other anesthetics are compared.
Molecular Structure
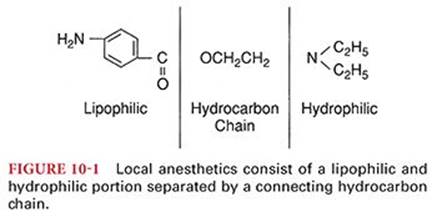
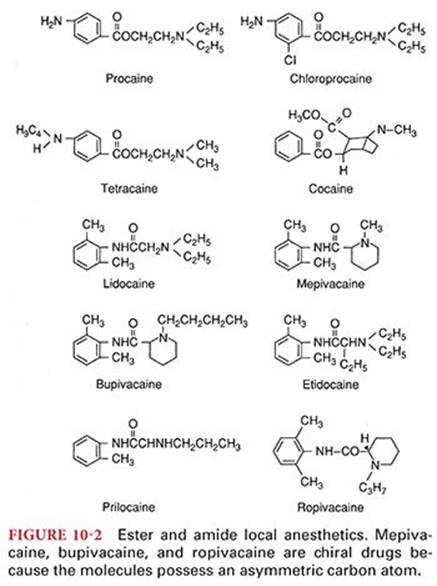
Local anesthetics consist of a lipophilic and a hydrophilic portion separated by a connecting hydrocarbon chain (Fig. 10-1). The hydrophilic group is usually a tertiary amine, such as diethylamine, whereas the lipophilic portion is usually an unsaturated aromatic ring, such as paraaminobenzoic acid. The lipophilic portion is essential for anesthetic activity, and therapeutically useful local anesthetics require a delicate balance between lipid solubility and water solubility. In almost all instances, an ester(–CO–) or an amide (–NHC–) bond links the hydrocarbon chain to the lipophilic aromatic ring. The nature of this bond is the basis for classifying drugs that produce conduction blockade of nerve impulses as ester local anesthetics or amide local anesthetics (Fig. 10-2). The important differences between ester and amide local anesthetics relate to the site of metabolism and the potential to produce allergic reactions.
Local anesthetics are poorly soluble in water and therefore are marketed most often as water-soluble hydrochloride salts. These hydrochloride salt solutions are acidic (pH 6), contributing to the stability of the local anesthetic. An acidic pH is also important if epinephrine is present in the local anesthetic solution, because this catecholamine is unstable at an alkaline pH. Sodium bisulfite, which is strongly acidic, may be added to commercially prepared local anesthetic–epinephrine solutions (pH 4) to prevent oxidative decomposition of epinephrine.
Structure–Activity Relationships
Modifying the chemical structure of a local anesthetic alters its pharmacologic effects. For example, lengthening the connecting hydrocarbon chain or increasing the number of carbon atoms on the tertiary amine or aromatic ring often results in a local anesthetic with a different lipid solubility, potency, rate of metabolism, and duration of action (Table 10-1). Substituting a butyl group for the amine group on the benzene ring of procaine results in tetracaine. Compared with procaine, tetracaine is more lipid soluble, is 10 times more potent, and has a longer duration of action corresponding to a 4- to 5-fold decrease in the rate of metabolism. Halogenation of procaine to chloroprocaine results in a 3- to 4-fold increase in the hydrolysis rate of chloroprocaine by plasma cholinesterase. This rapid hydrolysis rate of chloroprocaine limits the duration of action and systemic toxicity of this local anesthetic. Etidocaine resembles lidocaine, but substituting a propyl group for an ethyl group at the amine end and adding an ethyl group on the alpha carbon of the connecting hydrocarbon chain produces a 50-fold increase in lipid solubility and a 2- to 3-fold increase in the duration of action.
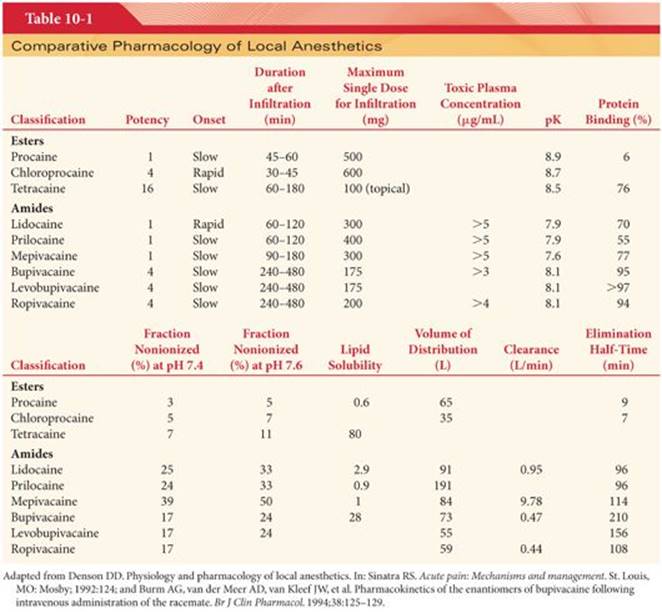
Mepivacaine, bupivacaine, and ropivacaine are characterized as pipecoloxylidides (see Fig. 10-2). Mepivacaine has a methyl group on the piperidine nitrogen atom (amine end) of the molecule. Addition of a butyl group to the piperidine nitrogen of mepivacaine results in bupivacaine, which is 35 times more lipid soluble and has a potency and duration of action 3 to 4 times that of mepivacaine. Ropivacaine structurally resembles bupivacaine and mepivacaine, with a propyl group on the piperidine nitrogen atom of the molecule.
Racemic Mixtures or Pure Isomers
The pipecoloxylidide local anesthetics (mepivacaine, bupivacaine, ropivacaine, levobupivacaine) are chiral drugs because their molecules possess an asymmetric carbon atom (see Fig. 10-2). As such, these drugs may have a left- (S) or right- (R) handed configuration. Mepivacaine and bupivacaine are available for clinical use as racemic mixtures (50:50 mixture) of the enantiomers. The enantiomers of a chiral drug may vary in their pharmacokinetics, pharmacodynamics, and toxicity.1 These differences in pharmacologic activity reflect the fact that individual enantiomers bind to receptors or enzymes that are chiral amino acids with stereoselective properties. The S enantiomers of bupivacaine and mepivacaine appear to be less toxic than the commercially available racemic mixtures of these local anesthetics.2 In contrast to mepivacaine and bupivacaine, ropivacaine and levobupivacaine have been developed as a pure S enantiomers.3 These S enantiomers are considered to produce less neurotoxicity and cardiotoxicity than racemic mixtures or the R enantiomers of local anesthetics, perhaps reflecting decreased potency at sodium ion channel.4
Liposomal Local Anesthetics
Various formulation and drug delivery system including liposomes, cyclodextrins, and biopolymers are studied to prolong the duration and to limit the toxicity of local anesthetics. The goal is to upload higher amount of local anesthetic into the molecule and to have a consistent release of local anesthetic in the tissues.5 Liposomes, hydrophobic-based polymer particles such as Poly(lactic-co-glycolic acid) microspheres and solid polymers such as Poly(sebacic-co-ricinoleic acid) P(SA:RA) and their combination with synthetic and natural local anesthetic are examples of delivery systems currently in development or in clinical use.6
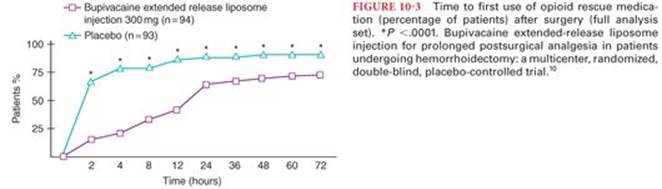
Drugs such as lidocaine, tetracaine, and bupivacaine have been incorporated into liposomes to prolong the duration of action and decrease toxicity.7 Bupivacaine extended release liposome injection was recently approved by U.S. Food and Drug Administration (FDA) for local infiltration anesthesia for hemorrhoidectomy and bunionectomy.8–10 Bupivacaine extended release liposome injection consists of microscopic, spherical, and multivesicular liposomes and each liposome particle is composed of a honeycomb-like structure of numerous internal aqueous chambers. Lipid membranes separate these aqueous chambers, and the chambers contain encapsulated bupivacaine. Bupivacaine is released from the liposome particles by a complex mechanism over an extended period (up to 96 hours). In a randomized, double-blind, placebo-controlled, parallel-group study, bupivacaine extended release liposome injection demonstrated a statistically significant reduction in pain through 72 hours, decreased opioid requirements, delayed time to first opioid use, and improved patient satisfaction compared with placebo after hemorrhoidectomy10 (Fig. 10-3). Bupivacaine extended release liposome injection has also been studied for use in nerve blocks,11 intraoperative administration in ileostomy reversal,12 but the results are inconclusive due to smaller sample size. At the time of this writing, bupivacaine extended release liposome injection has limited use, until more conclusive evidence is available for its clinical use.
Mechanism of Action
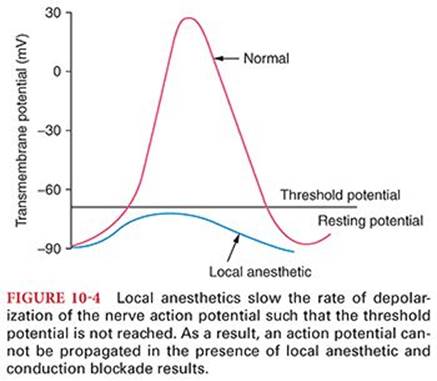
Local anesthetics bind to specific sites in voltage-gated Na+ channels. They block Na+ current, thereby reducing excitability of neuronal, cardiac or central nervous system tissue.13 Local anesthetics prevent transmission of nerve impulses (conduction blockade) by inhibiting passage of sodium ions through ion-selective sodium channels in nerve membranes.14 The sodium channel itself is a specific receptor for local anesthetic molecules. Failure of sodium ion channel permeability to increase slows the rate of depolarization such that threshold potential is not reached and thus an action potential is not propagated (Fig. 10-4). Local anesthetics do not alter the resting transmembrane potential or threshold potential.
Sodium Channels
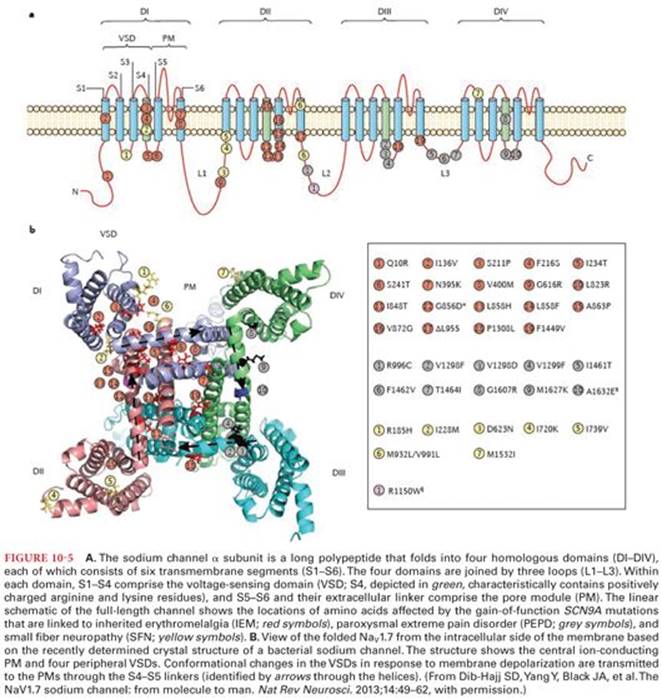
The sodium channel is a dynamic transmembrane protein consisting of the large sodium-conducting pore (α subunit) and varying numbers of adjacent smaller β subunits. Nine distinct functional subtypes of voltage-gated Na+channels are recognized, corresponding to nine genes for their pore-forming α subunits. These have different tissue distributions in the adult and are differentially regulated at the cellular level by receptor-coupled cell signaling systems.15 Different isoforms of voltage-gated Na+ channels, based on biophysical and pharmacologic studies, can provide distinct targets for interventions in various pain syndromes.16 The large polypeptide that forms the α subunit is further divided into four subunits (DI to DIV) (Fig. 10-5). H is the α subunit that allows ion conduction and binds to local anesthetics. However, β subunits may modulate local anesthetic binding to the α subunit. Binding affinities of local anesthetics to the sodium ion channels are stereospecific and depend on the conformational state of the sodium channel.17 Sodium channels exist in activated-open, inactivated-closed, and rested-closed states during various phases of the action potential.18 Voltage-gated Na+ channels undergo fast and slow inactivation processes and this is critical for membrane excitability. The structural changes associated with the inactivation process are poorly understood.19 In the resting nerve membrane, sodium channels are distributed in equilibrium between the rested-closed and inactivated-closed states. By selectively binding to sodium channels in inactivated-closed states, local anesthetic molecules stabilize these channels in this configuration and prevent their change to the rested-closed and activated-open states in response to nerve impulses. Sodium channels in the inactivated-closed state are not permeable to sodium, and thus conduction of nerve impulses in the form of propagated action potentials cannot occur. It is speculated that local anesthetics bind to specific sites located on the inner portion of sodium channels (internal gate or H gate) as well as obstructing sodium channels near their external openings to maintain these channels in inactivated-closed states.14 This binding appears to be weak and to reflect a relatively poor fit of the local anesthetic molecule with the receptor. This is consistent with the broad variety of chemical structures that exhibit local anesthetic activity on sodium channels.17
Frequency-Dependent Blockade
Sodium ion channels tend to recover from local anesthetic–induced conduction blockade between action potentials. Additional conduction blockade is developed each time sodium channels open during an action potential (frequency-dependent blockade). Local anesthetic molecules can gain access to receptors only when sodium channels are in activated-open states and local anesthetic binds more strongly to inactivated state. For this reason, selective conduction blockade of nerve fibers by local anesthetics may be related to the nerve’s characteristic frequencies of activity as well as to its anatomic properties such as diameter. Indeed, a resting nerve is less sensitive to local anesthetic-induced conduction blockade than is a nerve that has been repetitively stimulated. The pharmacologic effects of other drugs, including anticonvulsants and barbiturates in addition to local anesthetics, may reflect frequency-dependent blockade.
Other Site of Action Targets
In addition to sodium ion channels, local anesthetics block voltage-dependent potassium ion channels. Compared with sodium ion channels, local anesthetics exhibit a much lower affinity for potassium channels. However, blockade of potassium ion channels might explain broadening of the action potential in the presence of local anesthetics. Considering the structural similarity between voltage-dependent calcium ion channels and sodium ion channels, it is not surprising that calcium ion currents (L-type is most sensitive) may also be blocked by local anesthetics.20 Although local anesthetics are considered principally ion channel blockers, there is evidence that these drugs may also act on G protein–coupled receptors.21
Minimum Effective Concentration
The minimum concentration of local anesthetic necessary to produce conduction blockade of nerve impulses is termed the Cm. The Cm is analogous to the minimum alveolar concentration (MAC) for inhaled anesthetics. Nerve fiber diameter influences Cm, with larger nerve fibers requiring higher concentrations of local anesthetic for production of conduction blockade. An increased tissue pH or high frequency of nerve stimulation decreases Cm.
Each local anesthetic has a unique Cm, reflecting differing potencies of each drug. The Cm of motor fibers is approximately twice that of sensory fibers; thus, sensory anesthesia may not always be accompanied by skeletal muscle paralysis. Despite an unchanged Cm, less local anesthetic is needed for subarachnoid anesthesia than for epidural anesthesia, reflecting greater access of local anesthetics to unprotected nerves in the subarachnoid space.
Peripheral nerves are composed of myelinated A and B fibers and unmyelinated C fibers. A minimal length of myelinated nerve fiber must be exposed to an adequate concentration of local anesthetic for conduction blockade of nerve impulses to occur. For example, if only one node of Ranvier is blocked (site of change in sodium permeability), the nerve impulse can jump (skip) across this node and conduction blockade does not occur. For conduction blockade to occur in an A fiber, it is necessary to expose at least two and preferably three successive nodes of Ranvier (approximately 1 cm) to an adequate concentration of local anesthetic. Both types of pain-conducting fibers (myelinated A-δ and unmyelinated C fibers) are blocked by similar concentrations of local anesthetics, despite the differences in the diameters of these fibers. Preganglionic B fibers are more readily blocked by local anesthetics than any fiber, even though these fibers are myelinated.
Differential Conduction Blockade
Differential conduction blockade is illustrated by selective blockade of preganglionic sympathetic nervous system B fibers using low concentrations of local anesthetics. Slightly higher concentrations of local anesthetics interrupt conduction in small C fibers and small- and medium-sized A fibers, with loss of sensation for pain and temperature. Nevertheless, touch, proprioception, and motor function are still present such that the patient will sense pressure but not pain with surgical stimulation. In an anxious patient, however, any sensation may be misinterpreted as failure of the local anesthetic.
Changes during Pregnancy
Increased sensitivity (more rapid onset of conduction blockade) may be present during pregnancy.22 Alterations in protein-binding characteristics of bupivacaine may result in increased concentrations of pharmacologically active unbound drug in the parturient’s plasma.23 Nevertheless, progesterone, which binds to the same α1-acid glycoprotein as bupivacaine, does not influence protein binding of this local anesthetic.23 This evidence suggests that bupivacaine and progesterone bind to discrete but separate sites on protein molecules.
Pharmacokinetics
Local anesthetics are weak bases that have pK values somewhat above physiologic pH (see Table 10-1). As a result, <50% of the local anesthetic exists in a lipid-soluble nonionized form at physiologic pH. For example, at pH 7.4, only 5% of tetracaine exists in a nonionized form. Acidosis in the environment into which the local anesthetic is injected (as is present with tissue infection) further increases the ionized fraction of drug. This is consistent with the poor quality of local anesthesia that often results when a local anesthetic is injected into an acidic infected area. Local anesthetics with pKs nearest to physiologic pH have the most rapid onset of action, reflecting the presence of an optimal ratio of ionized to nonionized drug fraction (see Table 10-1).
Intrinsic vasodilator activity will also influence apparent potency and duration of action. For example, the enhanced vasodilator action of lidocaine compared with mepivacaine results in the greater systemic absorption and shorter duration of action of lidocaine. Bupivacaine and etidocaine produce similar vasodilation, but plasma concentrations of bupivacaine after epidural placement exceed those of etidocaine. Presumably, the greater lipid solubility of etidocaine results in tissue sequestration and less available drug for systemic absorption. Occasional prolonged sensory blockade after injection of etidocaine has been attributed to tissue sequestration.
Absorption and Distribution
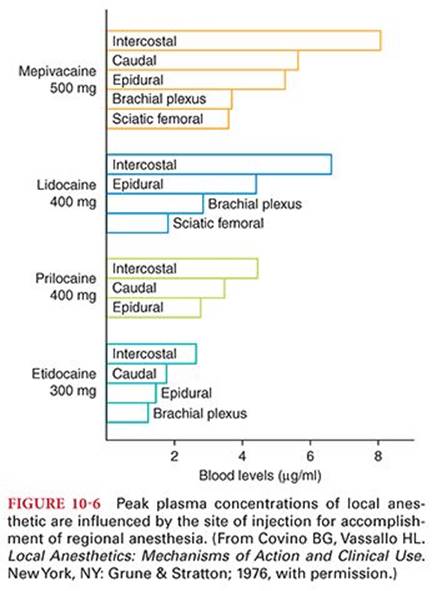
Absorption of a local anesthetic from its site of injection into the systemic circulation is influenced by the site of injection and dosage, use of epinephrine, and pharmacologic characteristics of the drug (Fig. 10-6). The ultimate plasma concentration of a local anesthetic is determined by the rate of tissue distribution and the rate of clearance of the drug. For example, the infusion of lidocaine for 1 minute is followed by a rapid decrease in the drug’s plasma concentration that is paralleled by an initial high uptake into the lungs and distribution of the local anesthetic to highly perfused tissues (brain, heart, and kidneys).24 Lipid solubility of the local anesthetic is important in this redistribution, as well as being a primary determinant of intrinsic local anesthetic potency. After distribution to highly perfused tissues, the local anesthetic is redistributed to less well perfused tissues, including skeletal muscles and fat. Consideration of cardiac output is important for describing the overall tissue distribution of local anesthetics and presumably their intercompartmental clearance.25 Ultimately, the local anesthetic is eliminated from the plasma by metabolism and excretion.
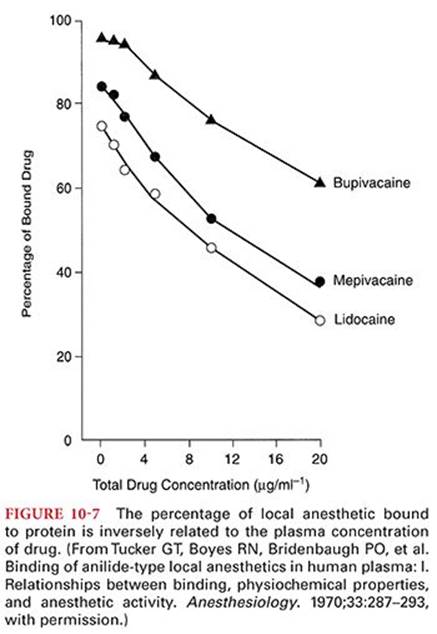
In addition to the tissue blood flow and lipid solubility of the local anesthetic, patient-related factors such as age, cardiovascular status, and hepatic function will also influence the absorption and resultant plasma concentrations of local anesthetics. Protein binding of local anesthetics will influence their distribution and excretion. In this regard, protein binding parallels lipid solubility of the local anesthetic and is inversely related to the plasma concentration of drug (see Table 10-1) (Fig. 10-7).26 Overall, after systemic absorption, amide local anesthetics are more widely distributed in tissues than ester local anesthetics.
Lung Extraction
The lungs are capable of extracting local anesthetics such as lidocaine, bupivacaine, and prilocaine from the circulation.27 After rapid entry of local anesthetics into the venous circulation, this pulmonary extraction will limit the concentration of drug that reaches the systemic circulation for distribution to the coronary and cerebral circulations. For bupivacaine, this first-pass pulmonary extraction is dose dependent, suggesting that the uptake process becomes saturated rapidly.28 Propranolol impairs bupivacaine extraction by the lungs, perhaps reflecting a common receptor site for the two drugs.29 Furthermore, propranolol decreases plasma clearance of lidocaine and bupivacaine, presumably reflecting propranolol-induced decreases in hepatic blood flow or inhibition of hepatic metabolism.30
Placental Transfer
There may be clinically significant transplacental transfer of local anesthetics between the mother and fetus. Plasma protein binding influences the rate and degree of diffusion of local anesthetics across the placenta (see Table 10-1). Bupivacaine, which is highly protein bound (approximately 95%), has an umbilical vein–maternal arterial concentration ratio of about 0.32 compared with a ratio of 0.73 for lidocaine (approximately 70% protein bound) and a ratio of 0.85 for prilocaine (approximately 55% protein bound).31 Ester local anesthetics, because of their rapid hydrolysis, are not available to cross the placenta in significant amounts. Acidosis in the fetus, which may occur during prolonged labor, can result in accumulation of local anesthetic molecules in the fetus (ion trapping) (Fig. 10-8).32
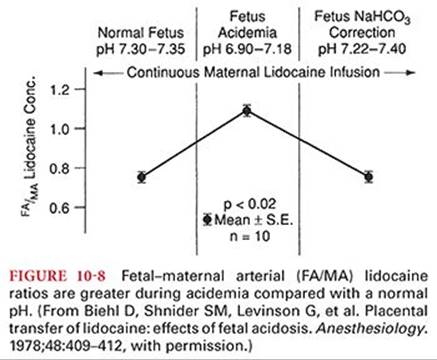
Renal Elimination and Clearance
The poor water solubility of local anesthetics usually limits renal excretion of unchanged drug to less than 5%.33 The exception is cocaine, of which 10% to 12% of unchanged drug can be recovered in urine. Water-soluble metabolites of local anesthetics, such as paraaminobenzoic acid resulting from metabolism of ester local anesthetics, are readily excreted in urine. Clearance values and elimination half-times for amide local anesthetics probably represent mainly hepatic metabolism, because renal excretion of unchanged drug is minimal (see Table 10-1). Pharmacokinetic studies of ester local anesthetics are limited because of a short elimination half-time due to their rapid hydrolysis in the plasma and liver.
Metabolism of Amide Local Anesthetics
Amide local anesthetics undergo varying rates of metabolism by microsomal enzymes located primarily in the liver. Prilocaine undergoes the most rapid metabolism; lidocaine and mepivacaine are intermediate; and etidocaine, bupivacaine, and ropivacaine undergo the slowest metabolism among the amide local anesthetics. The initial step is conversion of the amide base to aminocarboxylic acid and a cyclic aniline derivative. Complete metabolism usually involves additional steps, such as hydroxylation of the aniline moiety and N-dealkylation of the aminocarboxylic acid.
Compared with that of ester local anesthetics, the metabolism of amide local anesthetics is more complex and slower. This slower metabolism means that sustained increases of the plasma concentrations of amide local anesthetics, and thus systemic toxicity, are more likely than with ester local anesthetics. Furthermore, cumulative drug effects of amide local anesthetics are more likely than with ester local anesthetics.
Lidocaine
The principal metabolic pathway of lidocaine is oxidative dealkylation in the liver to monoethylglycinexylidide followed by hydrolysis of this metabolite to xylidide. Monoethylglycinexylidide has approximately 80% of the activity of lidocaine for protecting against cardiac dysrhythmias in an animal model. This metabolite has a prolonged elimination half-time, accounting for its efficacy in controlling cardiac dysrhythmias after the infusion of lidocaine is discontinued. Xylidide has only approximately 10% of the cardiac antidysrhythmic activity of lidocaine. In humans, approximately 75% of xylidide is excreted in the urine as 4-hydroxy-2,6-dimethylaniline.
Hepatic disease or decreases in hepatic blood flow, which may occur during anesthesia, can decrease the rate of metabolism of lidocaine. For example, the elimination half-time of lidocaine is increased more than fivefold in patients with liver dysfunction compared with normal patients. Decreased hepatic metabolism of lidocaine should be anticipated when patients are anesthetized with volatile anesthetics. Maternal clearance of lidocaine is prolonged in the presence of pregnancy-induced hypertension, and repeated administration of lidocaine can result in higher plasma concentrations than in normotensive parturients.34
Prilocaine
Prilocaine is an amide local anesthetic that is metabolized to orthotoluidine. Orthotoluidine is an oxidizing compound capable of converting hemoglobin to its oxidized form, methemoglobin, resulting in a potentially life-threatening complication, methemoglobinemia (see the section “Methemoglobinemia”). When the dose of prilocaine is >600 mg, there may be sufficient methemoglobin present (3 to 5 g/dL) to cause the patient to appear cyanotic, and oxygen-carrying capacity is decreased. Methemoglobinemia is readily reversed by the administration of methylene blue, 1 to 2 mg/kg intravenously, over 5 minutes (total dose should not exceed 7 to 8 mg/kg). The ability of prilocaine to cause dose-related methemoglobinemia limits its clinical usefulness, with the exception of intravenous (IV) regional anesthesia. Prilocaine causes less vasodilation than other local anesthetics and thus can be utilized without epinephrine added to the local anesthetic solution.
Mepivacaine
Mepivacaine has pharmacologic properties similar to those of lidocaine, although the duration of action of mepivacaine is somewhat longer. Clearance of mepivacaine is decreased in neonates, leading to a prolonged elimination half-time. In contrast to lidocaine, mepivacaine lacks vasodilator activity. As such, mepivacaine is an alternate selection when addition of epinephrine to the local anesthetic solution is not recommended.
Bupivacaine
Possible pathways for metabolism of bupivacaine include aromatic hydroxylation, N-dealkylation, amide hydrolysis, and conjugation.35 Only the N-dealkylated metabolite N-desbutylbupivacaine, has been measured in blood or urine after epidural or spinal anesthesia. The mean total urinary excretion of bupivacaine and its dealkylation and hydroxylation metabolites account for >40% of the total anesthetic dose.35 α1-Acid glycoprotein is the most important plasma protein-binding site of bupivacaine, and its concentration is increased in many clinical situations, including postoperative trauma.36
Ropivacaine
Ropivacaine is metabolized to 2,6-pipecoloxylidide and 3-hydroxyropivacaine by hepatic cytochrome P450 enzymes. Both metabolites have significantly less local anesthetic potency than ropivacaine. Because only a very small fraction of ropivacaine is excreted unchanged in the urine (about 1%) when the liver is functioning normally, dosage adjustments based on renal function do not seem necessary. However, in uremic patients, 2,6-pipecoloxylidide may accumulate and produce toxic effects.37 Overall, clearance of ropivacaine is higher than that determined for bupivacaine, and its elimination half-time is shorter.3 The higher clearance of ropivacaine may offer an advantage over bupivacaine in terms of systemic toxicity. The lipid solubility of ropivacaine is intermediate between lidocaine and bupivacaine. Ropivacaine is highly bound to α1-acid glycoprotein.
Dibucaine
Dibucaine is a quinoline derivative with an amide bond in the connecting hydrocarbon chain. This local anesthetic is metabolized in the liver and is the most slowly eliminated of all the amide derivatives. Dibucaine is better known for its ability to inhibit the activity of normal butyrylcholinesterase (plasma cholinesterase) by more than 70%, compared with only approximately 20% inhibition of the activity of atypical enzyme. Atypical plasma cholinesterases account for prolonged effects and toxicity of drugs such as succinylcholine and chloroprocaine that are metabolized by this enzyme. Laboratory evaluation of patients suspected of having atypical pseudocholinesterase is facilitated by measurement of the degree of enzyme suppression by dibucaine, a test termed the dibucaine number. See further discussion of dibucaine in Chapter 12, Neuromuscular Blocking Drugs and Reversal Agents.
Metabolism of Ester Local Anesthetics
Ester local anesthetics undergo hydrolysis by cholinesterase enzyme, principally in the plasma and to a lesser extent in the liver. The rate of hydrolysis varies, with chloroprocaine being most rapid, procaine being intermediate, and tetracaine being the slowest. The resulting metabolites are pharmacologically inactive, although paraaminobenzoic acid may be an antigen responsible for subsequent allergic reactions. The exception to hydrolysis of ester local anesthetics in the plasma is cocaine, which undergoes significant metabolism in the liver.
Systemic toxicity is inversely proportional to the rate of hydrolysis; thus, tetracaine is more likely than chloroprocaine to result in excessive plasma concentrations. Because cerebrospinal fluid contains little to no cholinesterase enzyme, anesthesia produced by subarachnoid placement of tetracaine will persist until the drug has been absorbed into the systemic circulation. Plasma cholinesterase activity and the hydrolysis rate of ester local anesthetics are slowed in the presence of liver disease or an increased blood urea nitrogen concentration. Plasma cholinesterase activity may be decreased in parturients and in patients being treated with certain chemotherapeutic drugs. Patients with atypical plasma cholinesterase may be at increased risk for developing excess systemic concentrations of an ester local anesthetic due to absent or limited plasma hydrolysis.
Procaine
Procaine is hydrolyzed to paraaminobenzoic acid, which is excreted unchanged in urine, and to diethylaminoethanol, which is further metabolized because only 30% is recovered in urine. Overall, <50% of procaine is excreted unchanged in urine. Increased plasma concentrations of paraaminobenzoic acid do not produce symptoms of systemic toxicity.
Chloroprocaine
Addition of a chlorine atom to the benzene ring of procaine to form chloroprocaine increases by 3.5 times the rate of hydrolysis of the local anesthetic by plasma cholinesterase, as compared with procaine. Resulting pharmacologically inactive metabolites of chloroprocaine are 2-chloro-aminobenzoic acid and 2-diethylaminoethanol. Maternal and neonatal plasma cholinesterase activity may be decreased up to 40% at term, but minimal placental passage of chloroprocaine confirms that even this decreased activity is adequate to hydrolyze most of the chloroprocaine that is absorbed from the maternal epidural space.38,39
Tetracaine
Tetracaine undergoes hydrolysis by plasma cholinesterase, but the rate is slower than for procaine.
Benzocaine
Benzocaine (ethyl aminobenzoate) is unique among clinically useful local anesthetics because it is a weak acid (pKa 3.5), so that it exists predominantly in the nonionized form at physiologic pH. As such, benzocaine is ideally suited for topical anesthesia of mucous membranes prior to tracheal intubation, endoscopy, transesophageal echocardiography, and bronchoscopy. Onset of topical anesthesia is rapid and lasts 30 to 60 minutes. A brief spray of 20% benzocaine delivers the recommended dose of 200 to 300 mg. Systemic absorption of topical benzocaine is enhanced by defects in the skin and mucosa as well as from the gastrointestinal tract should any of the local anesthetic be swallowed. The product Cetacaine is marketed as a combination of 14% benzocaine, 2% tetracaine, and 2% butamben in a topical applicator that acts as an atomizer. Methemoglobinemia is a rare but potentially life-threatening complication following topical application of benzocaine, especially when the dose exceeds 200 to 300 mg (see the section “Methemoglobinemia”).
Cocaine
Cocaine is metabolized by plasma and liver cholinesterases to water-soluble metabolites that are excreted in urine. Plasma cholinesterase activity is decreased in parturients, neonates, the elderly, and patients with severe underlying hepatic disease. Cocaine may be present in urine for 24 to 36 hours, depending on the route of administration and cholinesterase activity. Assays for the metabolites of cocaine in urine are useful markers of cocaine use or absorption (see the section “Cocaine Toxicity”).
Alkalinization of Local Anesthetic Solutions
Alkalinization of local anesthetic solutions shortens the onset of neural blockade, enhances the depth of sensory and motor blockade, and increases the spread of epidural blockade.40 The pH of commercial preparations of local anesthetic solutions ranges from 3.9 to 6.5 and is especially acidic if prepackaged with epinephrine (increased acidity prolongs the shelf life of epinephrine). The pKa of local anesthetics used clinically is near 8, so that only a small fraction (about 3%) of the local anesthetic exists in the lipid-soluble form. Alkalinization increases the percentage of local anesthetic existing in the lipid-soluble form that is available to diffuse across lipid cellular barriers. Adding sodium bicarbonate will speed the onset of peripheral nerve block and epidural block by 3 to 5 minutes.
Adjuvant Mixed with Local Anesthetics
Dexmedetomidine has been used as an adjuvant in local anesthetic admixtures and a central effect is postulated for prolongation of the local anesthetic affect. IV dexmedetomidine, in a recent systematic review and meta-analysis, showed increased duration of motor and sensory block and also increased duration for first analgesic request after spinal anesthesia.41 Magnesium has also shown promising initial results when introduced in to the intrathecal space as an addition to local anesthetic with or without opioids. Duration of spinal anesthesia was increased in magnesium group.42 In pediatric patients, addition of clonidine and ketamine to the regional anesthesia showed good pharmacokinetic and pharmacodynamic profiles of efficacy and safety, improving and prolonging the action of associated local anesthetics.43
Combinations of Local Anesthetics
Local anesthetics may be combined in an effort to produce a rapid onset (chloroprocaine) and prolonged duration (bupivacaine) of action. Nevertheless, placement of chloroprocaine in the epidural space may decrease the efficacy of subsequent epidural bupivacaine-induced analgesia during labor. It is speculated that the low pH of the chloroprocaine solution could decrease the nonionized pharmacologically active fraction of bupivacaine. Tachyphylaxis to the local anesthetic mixture could also reflect local acidosis due to the low pH of the bathing solution. For these reasons, adjustment of the pH of the chloroprocaine solution with the addition of 1 mL of 8.4% sodium bicarbonate added to 30 mL of chloroprocaine solution just before placement into the epidural space may improve the efficacy of the chloroprocaine-bupivacaine combination.44 Local anesthetic toxicity of combinations of drugs are additive rather than synergistic.45
Use of Vasoconstrictors
The duration of action of a local anesthetic is proportional to the time the drug is in contact with nerve fibers. For this reason, epinephrine (1:200,000 or 5 µg/mL) may be added to local anesthetic solutions to produce vasoconstriction, which limits systemic absorption and maintains the drug concentration in the vicinity of the nerve fibers to be anesthetized. Indeed, addition of epinephrine to a lidocaine or mepivacaine solution prolongs the duration of conduction blockade and decreases systemic absorption of local anesthetics by 20% to 30% (Fig. 10-9).46,47 For bupivacaine, addition of epinephrine also increases the duration of conduction blockade but to a lesser degree and the reduction in systemic absorption is by 10% to 20%. Most local anesthetics, with the exception of ropivacaine, possess intrinsic vasodilator properties, and it is possible that epinephrine-induced vasoconstriction will slow clearance from the injection site, thus prolonging the time the drug is in contact with nerve fibers.
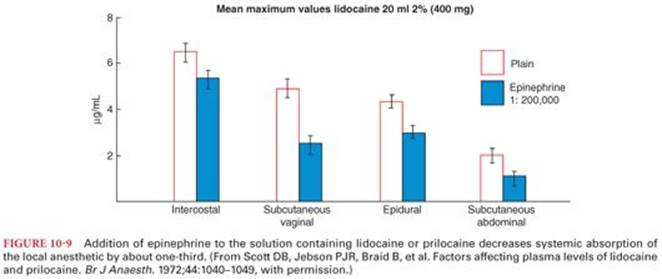
The impact of adding epinephrine to the local anesthetic solution is influenced by the specific local anesthetic selected and the level of sensory blockade required if a spinal or epidural anesthetic is chosen. For example, the impact of epinephrine in prolonging the duration of conduction blockade and decreasing systemic absorption of bupivacaine and etidocaine is less than that observed with lidocaine, presumably because the greater lipid solubility of bupivacaine and etidocaine causes them to bind avidly to tissues. The duration of sensory anesthesia in the lower extremities, but not the abdominal region, is extended when epinephrine (0.2 mg) or phenylephrine (2 mg) is added to local anesthetic solutions of bupivacaine or lidocaine placed into the subarachnoid space. Vasoconstrictors prolong the effect of tetracaine for spinal anesthesia. Epinephrine added to a low dose of tetracaine (6 mg) increases the success rate of spinal anesthesia, whereas the success rate is not altered by epinephrine when the subarachnoid dose of tetracaine is 10 mg.48 In addition to decreasing systemic absorption to prolong conduction blockade, epinephrine may also enhance conduction blockade by increasing neuronal uptake of the local anesthetic. The α-adrenergic effects of epinephrine may be associated with some degree of analgesia that could contribute to the effects of the conduction blockade. The addition of epinephrine to local anesthetic solutions has little, if any, effect on the onset rate of local anesthesia.
Decreased systemic absorption of local anesthetic due to vasoconstriction produced by epinephrine increases the likelihood that the rate of metabolism will match that of absorption, thus decreasing the possibility of systemic toxicity. Whenever local anesthetic solutions containing epinephrine are administered in the presence of inhaled anesthetics, the possibility of enhanced cardiac irritability should be considered. Systemic absorption of epinephrine may accentuate systemic hypertension in vulnerable patients.
Adverse Effects of Local Anesthetics
The principal side effects related to the use of local anesthetics are allergic reactions and systemic toxicity due to excessive plasma and tissue concentrations of the local anesthetic. Systemic toxicity in association with regional anesthesia is estimated to result in seizures in 1 to 4 per 1,000 patient exposures to local anesthetics, with bupivacaine being the drug most likely to be associated with this adverse response.49
Allergic Reactions
Allergic reactions to local anesthetics are rare despite the frequent use of these drugs. It is estimated that less than 1% of all adverse reactions to local anesthetics are due to an allergic mechanism.50 Instead, the overwhelming majority of adverse responses that are often attributed to an allergic reaction are instead manifestations of excess plasma concentrations of the local anesthetic.
Esters of local anesthetics that produce metabolites related to paraaminobenzoic acid are more likely than amide local anesthetics, which are not metabolized to paraaminobenzoic acid, to evoke an allergic reaction. An allergic reaction after the use of a local anesthetic may be due to methylparaben or similar substances used as preservatives in commercial preparations of ester and amide local anesthetics. These preservatives are structurally similar to paraaminobenzoic acid. As a result, an allergic reaction may reflect prior stimulation of antibody production by the preservative and not a reaction to the local anesthetic.
Cross-Sensitivity
Cross-sensitivity between local anesthetics reflects the common metabolite paraaminobenzoic acid. A similar cross-sensitivity, however, does not exist between classes of local anesthetics. Therefore, a patient with a known allergy to an ester local anesthetic can receive an amide local anesthetic without an increased risk of an allergic reaction. Likewise, an ester local anesthetic can be administered to a patient with a known allergy to an amide local anesthetic. It is important that the “safe” local anesthetic be preservative-free.
Documentation of Allergy
Documentation of allergy to a local anesthetic is based on the clinical history and perhaps the use of intradermal testing. The occurrence of rash, urticaria, and laryngeal edema, with or without hypotension and bronchospasm, is highly suggestive of a local anesthetic–induced allergic reaction. Conversely, hypotension associated with syncope or tachycardia when an epinephrine-containing local anesthetic solution is administered suggests an accidental intravascular injection of drug. Use of an intradermal test requires injection of preservative-free preparations of local anesthetic solutions to eliminate the possibility that the allergic reaction was caused by a substance other than the local anesthetic.
Local Anesthetic Systemic Toxicity
Local anesthetic systemic toxicity (LAST) is due to an excess plasma concentration of the drug. Plasma concentrations of local anesthetics are determined by the rate of drug entrance into the systemic circulation relative to their redistribution to inactive tissue sites and clearance by metabolism. Accidental direct intravascular injection of local anesthetic solutions during performance of peripheral nerve block anesthesia or epidural anesthesia is the most common mechanism for production of excess plasma concentrations of local anesthetics. A variety of factors influence the likelihood and severity of LAST, including individual patient risk factors, concurrent medications, location and technique of block, specific local anesthetic compound, total local anesthetic dose (the product of concentration and volume), timeliness of detection, and adequacy of treatment.51 The magnitude of this systemic absorption depends on the (a) dose administered into the tissues, (b) vascularity of the injection site, (c) presence of epinephrine in the solution, and (d) physicochemical properties of the drug (see Table 10-1). For example, systemic absorption of local anesthetics is greatest after injection for an intercostal nerve bock, intermediate for epidural anesthesia, and least for a brachial plexus block.52 Addition of 5 µg of epinephrine to every milliliter of local anesthetic solution (1:200,000 dilution) decreases systemic absorption of local anesthetics by approximately one-third47 (see the section “Use of Vasoconstrictors”). Systemic toxicity of local anesthetics involves the central nervous system (CNS) and cardiovascular system.
Local anesthetics differ with regard to their CNS toxicity and cardiac toxicity. The cardiovascular/central nervous system (CV/CNS) ratio describes the dose required to produce CV arrhythmias versus that required to produce seizures.53 If the ratio is lower, it implies a reduced safety margin for the local anesthetic compounds compared to the compound with higher ratio, to detect impending cardiotoxicity based on premonitory CNS signs. Bupivacaine is a more potent local anesthetic and generates arrhythmias at lower concentrations compared with lidocaine and mepivacaine. Also in animal studies (dogs), at comparable dosages, bupivacaine and etidocaine cause severe arrhythmias without decreased myocardial contractility, whereas lidocaine caused the opposite, that is, depressed myocardial contractility without arrhythmia.54
Central Nervous System Effects
Low plasma concentrations of local anesthetics are likely to produce numbness of the tongue and circumoral tissues, presumably reflecting delivery of drug to these highly vascular tissues (Table 10-2). As the plasma concentrations continue to increase, local anesthetics readily cross the blood–brain barrier and produce a predictable pattern of CNS changes. Restlessness, vertigo, tinnitus, and difficulty in focusing occur initially. Further increases in the CNS concentration of local anesthetic result in slurred speech and skeletal muscle twitching. Skeletal muscle twitching is often first evident in the face and extremities and signals the imminence of tonic-clonic seizures. Vivid fear of imminent death and a delusional belief of having died have been described in patients experiencing toxic reactions to local anesthetics administered for regional anesthesia and pain relief.55
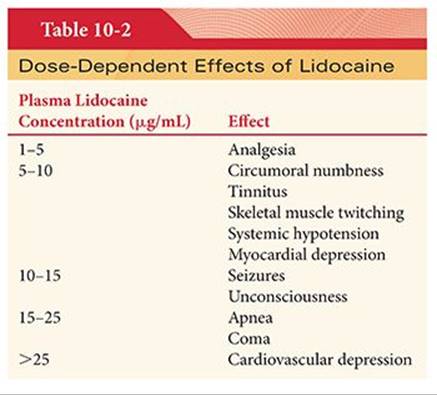
Lidocaine and other amide local anesthetics may cause drowsiness before the onset of seizures. Seizures are classically followed by CNS depression, which may be accompanied by hypotension and apnea. The onset of seizures may reflect selective depression of inhibitory cortical neurons by local anesthetics, leaving excitatory pathways unopposed. An alternative explanation for seizures is local anesthetic–induced inhibition of the release of neurotransmitters, particularly γ-aminobutyric acid. The precise site of local anesthetic–induced seizures is not known, although it appears to be in the temporal lobe or the amygdala.
Plasma concentrations of local anesthetics producing signs of central nervous system (CNS) toxicity depend on the specific drug involved. Lidocaine, mepivacaine, and prilocaine demonstrate effects on the CNS at plasma concentrations of 5 to 10 µg/mL. The typical plasma concentration of bupivacaine associated with seizures is 4.5 to 5.5 µg/mL.52 Ropivacaine and bupivacaine produce convulsions in awake animals at similar doses.3 The threshold plasma concentration at which CNS toxicity occurs may be related more to the rate of increase of the serum concentration than to the total amount of drug injected.33
The active metabolites of lidocaine, including monoethylglycinexylidide, may exert an additive effect in causing systemic toxicity after epidural administration of lidocaine. For this reason, it has been recommended that the plasma venous concentration of lidocaine be monitored when the cumulative epidural dose of lidocaine is >900 mg.56 The seizure threshold for lidocaine may be related to CNS levels of serotonin (5-hydroxytryptophan). For example, accumulation of serotonin decreases the seizure threshold of lidocaine and prolongs the duration of seizure activity.
There is an inverse relationship between the PaCO2 and seizure thresholds of local anesthetics, presumably reflecting variations in cerebral blood flow and resultant delivery of drugs to the brain. Increases in the serum potassium concentration can facilitate depolarization and thus markedly increase local anesthetic toxicity. Conversely, hypokalemia, by creating hyperpolarization, can greatly decrease local anesthetic toxicity. The threshold for neurotoxicity of lidocaine may be decreased when patients being treated with the antidysrhythmic drug mexiletine receive lidocaine during the perioperative period.57
Cardiovascular System Effects
The cardiovascular system is more resistant to the toxic effects of high plasma concentrations of local anesthetics than is the CNS. For example, lidocaine in plasma concentrations of <5 µg/mL is devoid of adverse cardiac effects, producing only a decrease in the rate of spontaneous phase 4 depolarization (automaticity). Nevertheless, plasma lidocaine concentrations of 5 to 10 µg/mL, and equivalent plasma concentrations of other local anesthetics, may produce profound hypotension due to relaxation of arteriolar vascular smooth muscle and direct myocardial depression (see Table 10-2). As a result, hypotension reflects both decreased systemic vascular resistance and decreased cardiac output.
Part of the cardiac toxicity that results from high plasma concentrations of local anesthetics occurs because these drugs also block cardiac sodium channels. At low concentrations of local anesthetics, this effect on sodium channels probably contributes to cardiac antidysrhythmic properties of these drugs. However, when the plasma concentrations of local anesthetics are excessive, sufficient cardiac sodium channels become blocked so that conduction and automaticity become adversely depressed. For example, excessive plasma concentrations of lidocaine may slow conduction of cardiac impulses through the heart, manifesting as prolongation of the P-R interval and QRS complex on the electrocardiogram. Effects of local anesthetics on calcium ion and potassium ion channels and local anesthetic-induced inhibition of cyclic adenosine monophosphate (cAMP) production may also contribute to cardiac toxicity.58
Selective Cardiac Toxicity
Accidental IV injection of bupivacaine may result in precipitous hypotension, cardiac dysrhythmias, and atrioventricular heart block.59 After accidental IV injection, the protein-binding sites (α1-acid glycoprotein and albumin) for bupivacaine are quickly saturated, leaving a significant mass of unbound drug available for diffusion into the conducting tissue of the heart. IV injection of bupivacaine or lidocaine to awake animals produces serious cardiac dysrhythmias only in animals receiving bupivacaine. Premature ventricular contractions, widening of the QRS complex, and ventricular tachycardia are the most common arrhythmias seen, though other arrhythmias including supraventricular tachycardia, atrioventricular heart block, and ST-T wave changes can also occur but are less common.60 Cardiotoxic plasma concentrations of bupivacaine are 8 to 10 µg/mL.61
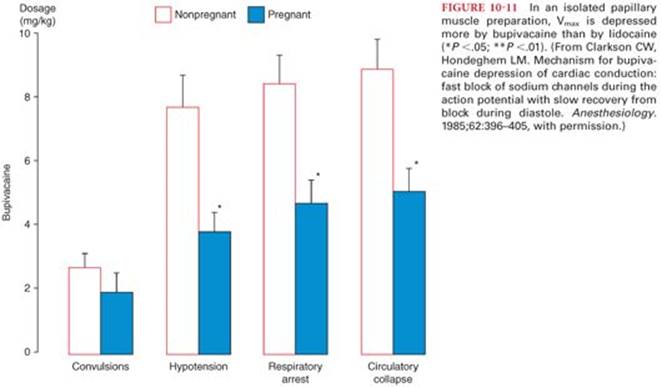
Physiologic changes and concomitant drug therapy may make patients more vulnerable to bupivacaine cardiac toxicity. For example, pregnancy may increase sensitivity to cardiotoxic effects of bupivacaine, but not ropivacaine, as emphasized by occurrence of cardiopulmonary collapse with a smaller dose of bupivacaine in pregnant compared with nonpregnant animals (Fig. 10-10).3,62 The threshold for cardiac toxicity produced by bupivacaine may be decreased in patients being treated with drugs that inhibit myocardial impulse propagation (β-adrenergic blockers, digitalis preparations, calcium channel blockers).63Indeed, in the presence of propranolol, atrioventricular heart block and cardiac dysrhythmias occurred at plasma bupivacaine concentrations of 2 to 3 µg/mL.61 This suggests that caution must be taken in the use of bupivacaine in patients who are on antidysrhythmic drugs or other cardiac medications known to depress impulse propagation. Epinephrine and phenylephrine may increase bupivacaine cardiotoxicity, reflecting bupivacaine-induced inhibition of catecholamine-stimulated production of cAMP.64 The cardiac toxicity of bupivacaine in animals is enhanced by arterial hypoxemia, acidosis, or hypercarbia.
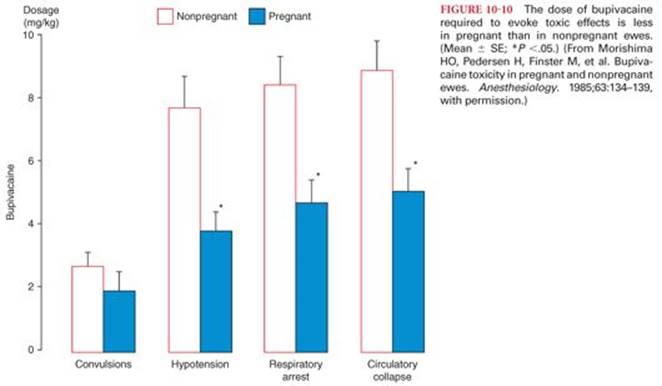
All local anesthetics depress the maximal depolarization rate of the cardiac action potential (Vmax) by virtue of their ability to inhibit sodium ion influx via sodium channels. In isolated papillary muscle preparations, bupivacaine depresses Vmax considerably more than lidocaine, whereas ropivacaine is intermediate in its depressant effect on Vmax (Fig. 10-11).3,65 The resulting slowed conduction of the cardiac action potential manifests on the electrocardiogram as prolongation of the P-R and QRS intervals and reentry ventricular cardiac dysrhythmias. Both bupivacaine and lidocaine block cardiac sodium ion channels during systole, whereas during diastole, highly lipid soluble bupivacaine dissociates off these channels at a slow rate compared with lidocaine, thus accounting for bupivacaine’s persistent depressant effect on Vmax and greater cardiac toxicity.66 At normal heart rates, diastolic time is sufficiently long for lidocaine dissociation but bupivacaine block intensifies and depresses electrical conduction, causing reentrant-type ventricular dysrhythmias. Less lipid-soluble lidocaine dissociates rapidly from cardiac sodium channels and cardiac toxicity is low. Furthermore, high plasma concentrations of bupivacaine may cause ventricular cardiac dysrhythmias through a direct brainstem effect. The R enantiomer of bupivacaine is more toxic than the S enantiomer. For example, seizure activity following an interscalene block with levobupivacaine was not associated with cardiac dysrhythmias or other signs of cardiovascular toxicity.67 In animals, levobupivacaine compared with bupivacaine was associated with a lower incidence of ventricular cardiac dysrhythmias, and successful resuscitation was more likely in the presence of levobupivacaine.68,69 Ropivacaine is a pure S enantiomer that is less lipid soluble and less cardiotoxic than bupivacaine but more cardiotoxic than lidocaine.70,71 Although, ropivacaine-induced cardiac arrest has been described following peripheral nerve block anesthesia, in contrast to bupivacaine, cardiac resuscitation is more likely to be successful.72–74
Tachycardia can enhance frequency-dependent blockade of cardiac sodium channels by bupivacaine, further contributing to the selective cardiac toxicity of this local anesthetic.75 Conversely, a low degree of frequency-dependent blockade may contribute to the antidysrhythmic properties of lidocaine. In anesthetized dogs, bretylium, 20 mg/kg intravenously, reverses bupivacaine-induced cardiac depression and increases the threshold for ventricular tachycardia.76 In an effort to decrease the potential for cardiotoxicity, in the event that accidental intravascular injection does occur, it may be prudent to limit the concentration of bupivacaine to be used for epidural anesthesia to 0.5%. In addition, slow or fractionated administration of all local anesthetics, but particularly bupivacaine, so as to detect systemic toxicity from accidental intravascular injection, should help decrease the risk of cardiotoxicity.77
Treatment of Local Anesthetic Systemic Toxicity
Treatment of LAST has undergone swift changes in last decade and includes prompt airway management, circulatory support, and mechanisms to remove local anesthetic from the receptor sites. Treatment should be instituted at the earliest suspicion of toxicity. Treatment of local anesthetic–induced seizures includes ventilation of the patient’s lungs with oxygen because arterial hypoxemia and metabolic acidosis occur within seconds.78 Equally important is the delivery of supplemental oxygen at the earliest sign of local anesthetic toxicity. IV administration of a benzodiazepine such as midazolam or diazepam is effective in suppressing local anesthetic–induced seizures. Propofol can be used for seizure treatment if hemodynamic stability is confirmed. For seizures that are not responsive to initial treatment, use of muscle relaxant such as succinylcholine or nondepolarizing blockers can help prevent acidosis and hypoxia associated with seizures. Early use of lipid emulsion for the treatment of local anesthetic toxicity is becoming standard of care. Multiple published cases have shown that intralipid can be successfully used for resuscitation, the mean total (bolus plus infusion) intralipid dose over the first 30 minutes was 3.8 mL/kg (range, 1.2 to 6.0 mL/kg). The American Society of Regional Anesthesia has published specific recommendations and checklist for treatment of LAST (Fig. 10-12).51,79,80 Use of lipid emulsion is recommended at the earliest sign of toxicity after airway management. Initial bolus of 1.5 mL/kg 20% lipid emulsion followed by 0.25 mL/kg per minute of infusion, continued for at least 10 minutes after circulatory stability is attained is recommended. Epinephrine should be used at a lower than typical dose 10 to 100 µg during resuscitation, and vasopressin use is not recommended. Calcium channel blockers and β-blockers should be avoided. Nonresponse to treatment should prompt institution of cardiopulmonary bypass (CPB), whenever this modality is available. Prompt institution of CPB, with support of circulation while drug clearance proceeds, has been associated with full recovery from LAST in a number of case reports.

Neural Tissue Toxicity (Neurotoxicity)
Neurotoxicity from placement of local anesthetic–containing solutions into the epidural or subarachnoid space can lead to various complications. The spectrum of this neurotoxicity may range from patchy groin numbness and persistent isolated myotomal weakness to cauda equina syndrome.81 Myofascial pain may be erroneously diagnosed as transient neurologic symptoms after intrathecal placement of local anesthetics.82 Overall, permanent neurologic injury after regional anesthesia remains a very rare event.83,84 Lidocaine-induced increases in intracellular calcium ion concentrations may be the mechanism for this toxicity.85
Transient Neurologic Symptoms
Transient neurologic symptoms (TNS) manifest as moderate to severe pain in the lower back, buttocks, and posterior thighs that appears within 6 to 36 hours after complete recovery from uneventful single-shot spinal anesthesia.86The etiology of transient neurologic symptoms is not known, but relief of pain with trigger point injections and nonsteroidal antiinflammatory drugs suggests a musculoskeletal component. In TNS, the sensory and motor neurologic examination is not abnormal and full recovery from symptoms usually occurs within 1 to 7 days.
The incidence of transient neurologic symptoms is greatest following the intrathecal injection of lidocaine (as high as 30%).87 Initial reports of transient neurologic symptoms involved spinal anesthesia produced by hyperbaric 5% lidocaine, suggesting that the observed neurotoxicity might be, at least in part, concentration dependent. Nevertheless, the incidence of transient neurologic symptoms is similar after intrathecal placement of 1 mg/kg of either 5% or 2% lidocaine in 7.5% glucose.88,89 For ambulatory patients undergoing arthroscopy, the incidence of transient neurologic symptoms is not altered by decreasing spinal lidocaine concentrations from 2% to 1% or 0.5% and are similar to the incidence of symptoms described with 5% lidocaine.90 The risk of transient neurologic symptoms associated with bupivacaine, tetracaine, mepivacaine, prilocaine, or procaine is significantly less than with lidocaine.91In a recent Cochrane review, spinal anesthesia with lidocaine was associated with significantly higher incidence of TNS when compared to bupivacaine, prilocaine, or procaine.92 Also, patients who experienced TNS were less satisfied and had more functional impairment compared to patients who did not experience TNS.
The lithotomy position,93 early ambulation,94 and the glucose concentration and osmolarity of the anesthetic solution do not influence the incidence of transient neurologic symptoms.93 Vasoconstrictors can decrease blood supply to the nerve and there is evidence that adding phenylephrine to the local anesthetic solution increases the incidence of transient neurologic symptoms after spinal anesthesia with tetracaine.93 There are some clinical data suggesting that addition of epinephrine to local anesthetic solutions does not alter the incidence of transient neurologic symptoms.95
Cauda Equina Syndrome
Cauda equina syndrome (CES) occurs when diffuse injury across the lumbosacral plexus produces varying degrees of sensory anesthesia, bowel and bladder sphincter dysfunction, and paraplegia. Cauda equina syndrome is most frequently associated with large central lumbar disc herniation, prolapse or sequestration with 50% to 60% patients having urinary retention on presentation.96 In the regional anesthesia literature, initial reports of cauda equina syndrome were associated with the use of hyperbaric 5% lidocaine for continuous spinal anesthesia.97,98 In these cases, it was postulated that microcatheters used during continuous spinal anesthesia (28 gauge or smaller) contributed to inhomogeneous distribution of the local anesthetic solution, with pooling of high concentrations of the local anesthetic solution on certain dependent or stretched (lithotomy position) nerves. Nevertheless, this same complication has also been reported after intrathecal injection of 100 mg of 5% lidocaine through a 25-gauge needle.99
Anterior Spinal Artery Syndrome
Anterior spinal artery syndrome consists of lower extremity paresis with a variable sensory deficit that is usually diagnosed as the neural blockade resolves. The etiology of this syndrome is uncertain, although thrombosis or spasm of the anterior spinal artery is possible, as well as effects of hypotension or vasoconstrictor drugs. Although the addition of epinephrine to local anesthetic solutions has been implicated as a theoretical cause, spinal cord perfusion studies do not show a deleterious effect of the catecholamine.100 Advanced age and the presence of peripheral vascular disease may predispose patients to development of anterior spinal artery syndrome. It may be difficult to distinguish symptoms due to anterior spinal artery syndrome from those caused by spinal cord compression produced by an epidural abscess or hematoma.
Methemoglobinemia
Methemoglobinemia is a rare but potentially life-threatening complication (decreased oxygen-carrying capacity) that may follow the administration of certain drugs or chemicals that cause oxidation of hemoglobin to methemoglobin more rapidly than methemoglobin is reduced to hemoglobin. Known oxidant substances include topical local anesthetics (prilocaine, benzocaine, and lidocaine), nitroglycerin, phenytoin, and sulfonamides.101 Neonates may be at greater risk because of more readily oxidized fetal hemoglobin.
Methemoglobin cannot bind oxygen or carbon dioxide, resulting in loss of the hemoglobin molecule’s transport function. Methemoglobin normally constitutes <1% of the total hemoglobin. Central cyanosis usually occurs when methemoglobin concentrations exceed 15%. The presence of methemoglobinemia is suggested by a difference between the calculated and measured arterial oxygen saturation. The diagnosis is confirmed by qualitative measurements of methemoglobin by cooximetry.
Methemoglobinemia is readily reversed by the administration of methylene blue, 1 to 2 mg/kg intravenously, over 5 minutes (total dose should not exceed 7 to 8 mg/kg). Methylene blue is reduced to leukomethylene blue, which then acts as an electron donor and nonenzymatically reduces methemoglobin to hemoglobin. Normal levels of methemoglobin should be achieved within 20 to 60 minutes after the administration of methylene blue. This therapeutic effect, however, is short-lived because methylene blue may be cleared before conversion of all the methemoglobin to hemoglobin. Furthermore, continued absorption of highly lipophilic local anesthetics such as benzocaine from adipose tissue stores may continue to occur after methylene blue plasma concentrations are no longer therapeutic.
Ventilatory Response to Hypoxia
Lidocaine at clinically useful plasma concentrations depresses the ventilatory responses to arterial hypoxemia.102 In this regard, patients with carbon dioxide retention whose resting ventilation depends on hypoxic drive may be at risk of ventilatory failure when lidocaine is administered for treatment of cardiac dysrhythmias. Conversely, systemic absorption of bupivacaine, such as follows a brachial plexus block, stimulates the ventilatory response to carbon dioxide.
Hepatotoxicity
Continuous or intermittent epidural administration of bupivacaine to treat postherpetic neuralgia has been associated with increased plasma concentrations of liver transaminase enzymes that normalized when bupivacaine infusion was discontinued or lidocaine was substituted for bupivacaine.103 The preservative present in both local anesthetics was the same. Drug-induced liver injury can be a direct toxic injury, an allergic reaction, or idiosyncratic metabolic abnormality. The hepatic dysfunction described seems most likely to represent an allergic reaction.104
Uses of Local Anesthetics
Local anesthetics are most often used to produce topical, infiltration, and regional anesthesia.52,105 Less common reasons to select local anesthetics are to prevent or treat cardiac dysrhythmias, prevent or treat increases in intracranial pressure, provide analgesia, and treat grand mal seizures. Antiinflammatory effects of local anesthetics may be responsible for beneficial effects in the perioperative period that are attributed to spinal or epidural anesthesia.106
Regional Anesthesia
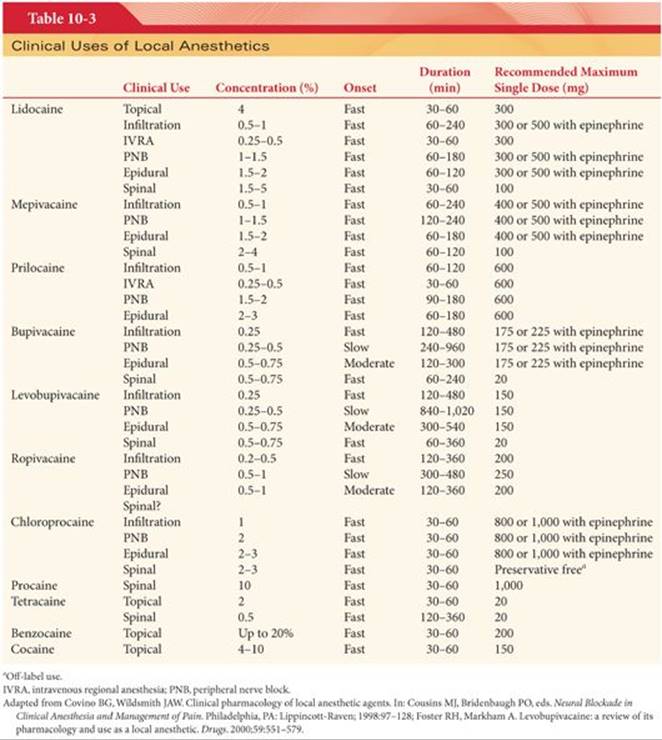
Regional anesthesia is classified according to the following six sites of placement of the local anesthetic solution: (a) topical or surface anesthesia, (b) local infiltration, (c) peripheral nerve block, (d) IV regional anesthesia (Bier block), (e) epidural anesthesia, and (f) spinal (subarachnoid) anesthesia (Table 10-3). Maximum doses of local anesthetics (based on body weight) as recommended for topical or peripheral nerve block anesthesia must be viewed as imprecise guidelines that often do not consider the pharmacokinetics of the drugs.71
Topical Anesthesia
Local anesthetics are used to produce topical anesthesia by placement on the mucous membranes of the nose, mouth, tracheobronchial tree, esophagus, or genitourinary tract. Cocaine (4% to 10%), tetracaine (1% to 2%), and lidocaine (2% to 4%) are most often used. It is estimated that topical cocaine anesthesia is used in >50% of rhinolaryngologic procedures performed annually in the United States.107 (see the section “Cocaine Toxicity”). Cocaine’s popularity for topical anesthesia reflects its unique ability to produce localized vasoconstriction, thus decreasing blood loss and improving surgical visualization. There is no difference between the intranasal anesthetic or vasoconstrictive effects of cocaine and those of a lidocaine-oxymetazoline or tetracaine-oxymetazoline mixture, emphasizing the usefulness of these combinations as substitutes for cocaine.108 Procaine and chloroprocaine penetrate mucous membranes poorly and are ineffective for topical anesthesia.
Nebulized lidocaine is used to produce surface anesthesia of the upper and lower respiratory tract before fiberoptic laryngoscopy and/or bronchoscopy and as a treatment for patients experiencing intractable coughing.109 The inhalation of local anesthetics by normal subjects does not alter airway resistance and may even produce mild bronchodilation.110 In contrast, inhalation of nebulized lidocaine can cause bronchoconstriction in some patients with asthma, which may become an important consideration when bronchoscopy is planned in these patients.109 Local anesthetics are absorbed into the systemic circulation after topical application to mucous membranes. Systemic absorption of tetracaine, and to a lesser extent lidocaine, after placement on the tracheobronchial mucosa produces plasma concentrations similar to those present after IV injection of the local anesthetic. For example, plasma lidocaine concentrations 15 minutes after laryngotracheal spray of the local anesthetic are similar to those concentrations present at the same time after an IV injection of a similar dose of lidocaine.111 This systemic absorption reflects the high vascularity of the tracheobronchial tree and the injection of the local anesthetic as a spray that spreads the solution over a wide surface area.
Eutectic Mixture of Local Anesthetics
The keratinized layer of the skin provides an effective barrier to diffusion of topical drugs, making it difficult to achieve anesthesia of intact skin by topical application. A popular use of prilocaine is for topical anesthesia when used in a eutectic mixture. A 5% lidocaine–prilocaine cream (2.5% lidocaine and 2.5% prilocaine) allows the use of high concentrations of the anesthetic bases without concern about local irritation, uneven absorption, or systemic toxicity.112,113 This combination of local anesthetics is considered a eutectic mixture of local anesthetics (EMLA), as the melting point of the combined drugs is lower than lidocaine or prilocaine alone. EMLA cream acts by diffusing through intact skin to block neuronal transmission from dermal receptors. Usually 1 to 2 g of EMLA cream are applied per a 10 cm2 area of skin and covered with an occlusive dressing. The duration of application varies according to the type of procedure being undertaken and the site of application. For example, skin-graft harvesting requires 2 hours, whereas cautery of genital warts can be undertaken after only a 10-minute application. EMLA cream is effective in relieving the pain of venipuncture, arterial cannulation, lumbar puncture, and myringotomy in children and adults. Pain during circumcision in neonates is attenuated by this topical anesthetic.113 Although 45 minutes has been suggested as the minimum effective onset time for decreasing the pain of IV cannulation, a significant decrease in pain scores may be noted after only 5 minutes. Low-frequency ultrasound pretreatment is effective in accelerating the onset of EMLA cream.114The addition of nitroglycerin ointment to EMLA cream increases the ease of venous cannulation by promoting venodilation.115 If EMLA cream is used to anesthetize the skin before blood sampling, the results of the analyses of the blood are not distorted. However, the use of EMLA cream to prevent the pain of intradermal skin tests decreases the flare response and may lead to false-negative interpretation of weakly positive tests.
Skin blood flow, epidermal and dermal thickness, duration of application, and the presence of skin pathology are important factors affecting the onset, efficacy, and duration of EMLA analgesia. African Americans may be less responsive than Whites, presumably because of increased density of the stratum corneum.116 Blanching of the skin may be seen after 30 to 60 minutes, probably due to vasoconstriction. Plasma levels of lidocaine and prilocaine are below toxic levels, although methemoglobin concentrations reflecting the metabolism of prilocaine may be increased in children <3 months of age, reflecting immature reductase pathways. The enzyme capacity for red blood cell methemoglobin reductase in children <3 months of age can be overloaded when EMLA cream is administered concurrently with other methemoglobin-inducing drugs (sulfonamides, acetaminophen, phenytoin, nitroglycerin, nitroprusside).117 Likewise, EMLA cream should not be used in those rare patients with congenital or idiopathic methemoglobinemia. Local skin reactions, such as pallor, erythema, alterations in temperature sensation, edema, pruritus, and rash are common after EMLA cream application.
EMLA cream is not recommended for use on mucous membranes because of the faster absorption of lidocaine and prilocaine than through intact skin.112 Similarly, EMLA cream is not recommended for skin wounds, and the risk of wound infection may be increased.118 Patients being treated with certain antidysrhythmic drugs (mexiletine) may experience additive and potentially synergistic effects when exposed to EMLA cream. EMLA cream is contraindicated in patients with a known history of allergy to amide local anesthetics.
Other Topically Effective Local Anesthetics
Amethocaine, like EMLA cream, requires several minutes to become effective, and the cream must be covered with an occlusive dressing. A microemulsion of amethocaine increases skin penetration and shortens the time until cutaneous anesthesia is achieved.119 New commercial preparations for topical anesthesia are being developed. Tetracaine in a 4% gel is used to provide topical anaesthesia for venous cannulation and a patch containing 7% lidocaine and 7% tetracaine has been developed to provide topical anesthesia by heat-assisted delivery. They provide comparable pain relief for venous cannulation in 90% of the patients.120
Local Infiltration
Local infiltration anesthesia involves extravascular placement of local anesthetic in the area to be anesthetized. Subcutaneous injection of the local anesthetic in the area to be traversed for placement of an intravascular cannula is one example. Lidocaine is the local anesthetic most often selected for infiltration anesthesia. Infiltration of 0.25% ropivacaine or bupivacaine is equally effective in the management of pain at an inguinal operative site.121
The duration of infiltration anesthesia can be approximately doubled by adding 1:200,000 epinephrine to the local anesthetic solution. Epinephrine-containing solutions, however, should not be injected intracutaneously or into tissues supplied by end arteries (fingers, ears, and nose) because resulting vasoconstriction can produce ischemia and may result in tissue necrosis.
Peripheral Nerve Block Anesthesia
Peripheral nerve block anesthesia is achieved by injection of local anesthetic solutions into tissues surrounding individual peripheral nerves or nerve plexuses such as the brachial plexus. When local anesthetic solutions are deposited in the vicinity of a peripheral nerve, they diffuse from the outer surface (mantle) toward the center (core) of the nerve along a concentration gradient.122 Consequently, nerve fibers located in the mantle of the mixed nerve are anesthetized first. These mantle fibers usually are distributed to more proximal anatomic structures in contrast to distal structures innervated by nerve fibers near the core of the nerve. This explains the initial development of anesthesia proximally, with subsequent distal spread as local anesthetic solution diffuses to reach more central core nerve fibers. Conversely, recovery of sensation occurs in a reverse direction; nerve fibers in the mantle that are exposed to extraneural fluid are the first to lose local anesthetic, so that sensation returns initially to the proximal and last to the distal parts of the limb.
Skeletal muscle paralysis may precede the onset of sensory anesthesia if motor nerve fibers are distributed peripheral to sensory fibers in the mixed peripheral nerve.122,123 Indeed, the sequence of onset and recovery from blockade of sympathetic, sensory, and motor nerve fibers in a mixed peripheral nerve depends as much on anatomic location of the nerve fibers within the mixed nerve as on their sensitivity to local anesthetics. This differs from results of in vitro studies on single nerve fibers, in which diffusion distance does not play a role. In an in vitro model, nerve fiber size is most important, with the onset of conduction blockade being inversely proportional to fiber size. For example, the smallest sensory and autonomic nervous system fibers are anesthetized first, followed by larger motor and proprioceptive axons.
The rapidity of onset of sensory anesthesia after injection of a local anesthetic solution into tissues around a peripheral nerve depends on the pK of the drug. The pK determines the amount of local anesthetic that exists in the active nonionized form at the pH of the tissue (see Table 10-1). For example, the onset of action of lidocaine occurs in approximately 3 minutes, whereas onset after injection of bupivacaine, levobupivacaine, or ropivacaine requires approximately 15 minutes, reflecting the greater fraction of lidocaine that exists in the lipid-soluble nonionized form. The onset and duration of sensory anesthesia for brachial plexus block produced by 0.5% bupivacaine, levobupivacaine, or ropivacaine is similar. Ropivacaine, 33 mL of a 0.5% solution used for performance of a subclavian perivascular block, produces a rapid onset of sensory anesthesia (about 4 minutes) with prolonged sensory (>13 hours) and motor blockade.124 For ulnar nerve block, ropivacaine was found to be maximally effective at concentrations between 0.5% and 0.75%, and its onset and duration of action resembled those of bupivacaine.125 Tetracaine, with a slow onset of anesthesia and a high potential to cause systemic toxicity, is not recommended for local infiltration or peripheral nerve block anesthesia.
Duration of peripheral nerve block anesthesia depends on the dose of local anesthetic, its lipid solubility, its degree of protein binding, and concomitant use of a vasoconstrictor such as epinephrine. The duration of action is prolonged more safely by epinephrine than by increasing the dose of local anesthetic, which also increases the likelihood of systemic toxicity. Bupivacaine combined with epinephrine may produce peripheral nerve block anesthesia lasting up to 14 hours. Conversely, not all reports document a prolongation of the duration of action when epinephrine is added to bupivacaine or ropivacaine.126
Continuous Peripheral Nerve Blocks
The modern practice of regional anesthesia has moved toward ultrasound (US)-guided peripheral nerve blocks and using perineural catheters for continuous infusions. Use of ultrasound guidance increases the chances for successful block, takes less time to perform, hastens onset, and prolongs block duration when compared to blocks performed with the guidance of peripheral nerve stimulation without ultrasound.127 US guidance also decreased the risk of vascular puncture during block performance. Continuous nerve blocks have been shown to be associated with improved pain control, decreased need for opioid analgesics, less nausea, and greater patient satisfaction, when compared to single shot blocks.128 Commonly used medications and their dosages are shown in Table 10-4. Midazolam, magnesium, dexmedetomidine, and ketamine have been used as additives to local anesthetic solutions for peripheral nerve blocks, but they cannot be routinely recommended due to a dearth of supportive data, modest efficacy, and (in the case of ketamine) significant adverse effects.129

Intravenous Regional Anesthesia (Bier Block)
The IV injection of a local anesthetic solution into an extremity isolated from the rest of the systemic circulation by a tourniquet produces a rapid onset of anesthesia and skeletal muscle relaxation. This has been termed intravenous regional anesthesia, often referred to as a Bier block after August Bier, who first described the use of local anesthetic in this manner to produce anesthesia of the limb. The duration of anesthesia is independent of the specific local anesthetic and is determined by how long the tourniquet is kept inflated. The mechanism by which local anesthetics produce IV regional anesthesia is unknown but probably reflects action of the drug on nerve endings as well as nerve trunks. Normal sensation and skeletal muscle tone return promptly on release of the tourniquet, which allows blood flow to dilute the concentration of local anesthetic.
Ester and amide local anesthetics produce satisfactory effects when used for IV regional anesthesia. Lidocaine is the most frequently selected amide local anesthetic for producing this type of regional anesthesia. Alternatives to lidocaine include prilocaine, mepivacaine, and ropivacaine. The onset, duration, and quality of IV regional anesthesia produced by 50 mL of a 0.5% solution of lidocaine or prilocaine are similar, but plasma concentrations of prilocaine are lower than those of lidocaine after tourniquet deflation (Fig. 10-13).130 The associated degree of methemoglobinemia (3% of hemoglobin as methemoglobin) seen with prilocaine is far below the level needed to produce cyanosis (10% hemoglobin as methemoglobin). The significantly lower plasma prilocaine concentrations after tourniquet deflation may indicate a greater margin of safety for prilocaine compared to lidocaine in terms of potential systemic toxicity. Mepivacaine 5 mg/kg provided superior analgesia to lidocaine 3 mg/kg when utilized for IV regional anesthesia.131 Plasma concentrations of lidocaine decreased significantly in the first 60 minutes following tourniquet deflation, whereas blood concentrations of mepivacaine remained below toxic concentrations. Ropivacaine, 1.2 mg/kg and 1.8 mg/kg compared with lidocaine 3 mg/kg produced comparable IV regional anesthesia but residual analgesia was longer lasting with ropivacaine.132Chloroprocaine is not selected for IV regional anesthesia because of a high incidence of thrombophlebitis. Bupivacaine is not recommended for IV regional anesthesia considering its greater likelihood than other local anesthetics for producing cardiotoxicity when the tourniquet is deflated at the conclusion of the anesthetic. Ropivacaine, although less likely to produce cardiotoxicity than bupivacaine, is not recommended for IV regional anesthesia over similar fears that its increased potency may lead to cardiotoxicity at the time of tourniquet release.
Epidural Anesthesia
Local anesthetic solutions placed in the epidural or sacral caudal space produce epidural anesthesia by two presumed mechanisms. First, local anesthetic diffuses across the dura to act on nerve roots and the spinal cord as it does when injected directly into the lumbar subarachnoid space to produce spinal anesthesia. Second, local anesthetic also diffuses into the paravertebral area through the intervertebral foramina, producing multiple paravertebral nerve blocks. These slow diffusion processes account for the 15- to 30-minute delay in onset of sensory anesthesia after placement of local anesthetic solutions in the epidural space. Lidocaine is commonly used for epidural anesthesia because of its good diffusion through tissues. Despite a reasonable safety profile, bupivacaine is being replaced by levobupivacaine and ropivacaine, as these local anesthetics are associated with less risk for cardiac and central nervous system toxicity and are also less likely to result in unwanted postoperative motor blockade.
Bupivacaine and ropivacaine at similar concentrations (0.5% to 0.75%) produce similar prolonged sensory anesthesia (ropivacaine has a greater tendency to block A-δ and C fibers) when used for epidural anesthesia, but the motor anesthesia produced by ropivacaine is less intense and of shorter duration.133,134 These characteristics of ropivacaine may be advantageous for obstetric patients in labor and for those experiencing acute and chronic pain. The addition of epinephrine 1:200,000 to 0.5% or 0.75% bupivacaine or ropivacaine does not appear to offer an advantage in terms of duration of action.135 Use of 1% ropivacaine may provide longer sensory anesthesia than 0.75% bupivacaine, whereas the motor block is similar.126,136 The lower systemic toxicity of ropivacaine compared with bupivacaine enables ropivacaine to be used for surgical anesthesia in concentrations up to 1%.3 For postoperative analgesia, the epidural infusion of 0.2% ropivacaine at 6 to 10 mL per hour is effective.137 Intermittent epidural bolus technique produces comparable results in analgesia, local anesthetic consumption and maternal satisfaction, when compared to continuous epidural infusion for epidural use in labor analgesia.138
In children, there is no difference with regard to postoperative analgesia provided by bupivacaine, levobupivacaine, or ropivacaine but unwanted motor blockade was more frequent in patients receiving bupivacaine (Fig. 10-14).139 Epidural analgesia or anesthesia for labor or cesarean delivery is similar with 0.5% bupivacaine or ropivacaine but the duration of motor block is shorter in parturients receiving ropivacaine.140 Likewise, 0.25% ropivacaine and 0.25% bupivacaine administered as intermittent doses into the epidural space are equally efficacious in providing relief of labor pain.141 The conclusion is that ropivacaine and bupivacaine both provide excellent labor analgesia with no significant differences between the two drugs in the incidence of measured obstetric outcomes.142 Increased plasma concentrations of local anesthetics after epidural anesthesia are of special importance when this technique is used to provide anesthesia to the parturient. Local anesthetics cross the placenta and may produce detectable, although not necessarily adverse, effects on the fetus for 24 to 48 hours. The fetus and neonate are less able to metabolize mepivacaine, resulting in a prolonged elimination half-time compared with that of adults. Use of a more lipid-soluble and protein-bound local anesthetic such as bupivacaine may limit passage across the placenta to the fetus. Even low doses of lidocaine, such as those used for spinal anesthesia during labor, result in some systemic absorption, as reflected by the presence of lidocaine and its metabolites in neonatal urine for >36 hours.39 Conversely, bupivacaine is undetectable in neonatal plasma 24 hours after cesarean delivery using bupivacaine-induced spinal anesthesia.143 Indeed, maternal plasma concentrations of bupivacaine in mothers of those neonates is approximately 5% of that level present after epidural anesthesia, and plasma umbilical vein concentrations are approximately 7% of those present after epidural anesthesia.
In contrast to spinal anesthesia, during epidural anesthesia, there often is not a zone of differential sympathetic nervous system blockade, and the zone of differential motor blockade may average up to four rather than two segments below the sensory level. Another difference from spinal anesthesia is the larger dose required to produce epidural anesthesia, leading to substantial systemic absorption of the local anesthetic. For example, peak plasma concentrations of lidocaine are 3 to 4 µg/mL after placement of 400 mg into the epidural space. Bupivacaine, 70 to 100 mg of 0.5% with 1:200,000 epinephrine placed in the epidural space, results in peak average plasma concentrations of 0.335 µg/mL occurring about 30 minutes after instillation of the local anesthetic.144 Peak plasma concentrations of bupivacaine near 1 µg/mL occur when epinephrine is not added to the local anesthetic solution placed in the epidural space. Thus, addition of epinephrine to the local anesthetic solution may decrease systemic absorption of the local anesthetic by approximately one-third. The peak venous plasma concentration of ropivacaine is 1.3 µg/mL after epidural placement of 200 mg of the local anesthetic. Addition of 1:200,000 epinephrine solution decreases systemic absorption of ropivacaine by approximately one-third. Systemic absorption of epinephrine produces β-adrenergic stimulation characterized by peripheral vasodilation, with resultant decreases in systemic blood pressure, even though the inotropic and chronotropic effects of epinephrine increase cardiac output.
Addition of opioids to local anesthetic solutions placed in the epidural or intrathecal space results in improved analgesia.145 An exception to this analgesic synergy is 2-chloroprocaine, which appears to decrease the effectiveness of epidural opioids when administered with local anesthetic solutions placed into the epidural space.146 Combining local anesthetics and opioids for peripheral nerve blocks appears to be ineffective in altering the characteristics or results of the block.
Spinal Anesthesia
Spinal anesthesia is produced by injection of local anesthetic solutions into the lumbar subarachnoid space. Local anesthetic solutions placed into lumbar cerebrospinal fluid act on superficial layers of the spinal cord, but the principal site of action is the preganglionic fibers as they leave the spinal cord in the anterior rami. Because the concentration of local anesthetics in cerebrospinal fluid decreases as a function of distance from the site of injection, and because different types of nerve fibers differ in their sensitivity to the effects of local anesthetics, zones of differential anesthesia develop. Because preganglionic sympathetic nervous system fibers are blocked by concentrations of local anesthetics that are insufficient to affect sensory or motor fibers, the level of sympathetic nervous system denervation during spinal anesthesia extends approximately two spinal segments cephalad to the level of sensory anesthesia. For the same reasons, the level of motor anesthesia averages two segments below sensory anesthesia.
Dosages of local anesthetics used for spinal anesthesia vary according to the (a) height of the patient, which determines the volume of the subarachnoid space; (b) segmental level of anesthesia desired; and (c) duration of anesthesia desired. The total dose of local anesthetic administered for spinal anesthesia is more important than the concentration of drug or the volume of the solution injected. Tetracaine, lidocaine, bupivacaine, ropivacaine, and levobupivacaine are the local anesthetics most likely to be administered for spinal anesthesia.
Spinal anesthesia with lidocaine has been reported to produce a higher incidence of transient neurologic symptoms than spinal anesthesia produced by bupivacaine (see the section “Neural Tissue Toxicity [Neurotoxicity]”). For these reasons, bupivacaine has been proposed as an alternative local anesthetic to lidocaine for spinal anesthesia.147,148 If lidocaine is selected, it may be prudent to limit the dose to 60 mg.148 Bupivacaine used for spinal anesthesia is more effective than tetracaine in preventing lower extremity tourniquet pain during orthopedic surgery.149 This effectiveness may reflect the ability of bupivacaine to produce greater frequency-dependent conduction blockade of fibers than does tetracaine. In parturients, the intrathecal placement of bupivacaine, 2.5 mg, plus sufentanil, 10 µg, provided labor analgesia and allowed the patients to continue to ambulate.150 The addition of intrathecal fentanyl 5 µg provides a bupivacaine dose-sparing effect similar to that provided by 15 µg or 25 µg of IV fentanyl, resulting in less pruritus but a shortening of the duration of action.151
Ropivacaine, 3 mL of 0.5% or 0.75%, produces sensory anesthesia, although complete motor blockade was present in only about 50% of patients receiving the lower dose.152 Ropivacaine is an acceptable local anesthetic to produce spinal anesthesia for cesarean section, and decreased lower extremity blockade compared with bupivacaine may be a desirable feature.153,154 Levobupivacaine has equivalent clinical efficacy to bupivacaine for spinal anesthesia (Fig. 10-15).155 Dibucaine is 1.5 to 2.0 times as potent as tetracaine when used for spinal anesthesia. In the past, chloroprocaine was not recommended for placement in the subarachnoid space because of potential neurotoxicity.156–158 However, preservative-free 2-chloroprocaine solutions (2% and 3%) are available for intrathecal injection (“off-label use”) and have been shown to produce reliable sensory and motor blockade with a short duration and little or no risk of transient neurologic symptoms, making this local anesthetic an attractive selection for outpatient surgical procedures performed under spinal anesthesia (Fig. 10-16).159,160
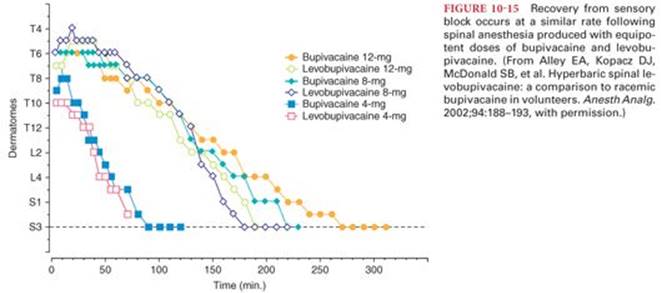
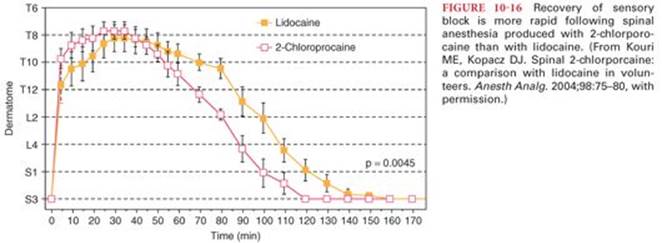
The specific gravity of local anesthetic solutions injected into the lumbar cerebrospinal fluid is important in determining spread of the drugs. Addition of glucose to local anesthetic solutions increases the specific gravity of local anesthetic solutions above that of cerebrospinal fluid (hyperbaric). Addition of distilled water lowers the specific gravity of local anesthetic solutions below that of cerebrospinal fluid (hypobaric). Cerebrospinal fluid does not contain significant amounts of cholinesterase enzyme; therefore, the duration of action of ester local anesthetics as well as amides placed in the subarachnoid space depends on systemic absorption of the drug.
Injected intrathecally, tetracaine produces a significant increase in spinal cord blood flow, an effect that can be prevented or reversed by epinephrine.161 Vasodilation is less prominent with lidocaine, and bupivacaine produces vasoconstriction. Predictably, vasoconstrictors appear to be most effective in prolonging tetracaine-induced spinal anesthesia (up to 100%) and less effective at prolonging lidocaine spinal anesthesia, whereas the effect on bupivacaine spinal anesthesia remains controversial and is, at best, minimal.
Physiologic Effects
The goal of spinal anesthesia is to provide sensory anesthesia and skeletal muscle relaxation. It is the accompanying level of sympathetic nervous system blockade that produces physiologic alterations. Plasma concentrations of local anesthetics after subarachnoid injection are too low to produce physiologic changes.
Cardiac Arrest
Cardiac arrest may accompany hypotension and bradycardia associated with spinal anesthesia.84,162,163 The incidence of hypotension is about 33%, and the incidence of bradycardia approximates 13% in nonobstetric populations. Risk factors for hypotension include sensory anesthesia above T5 and baseline systolic blood pressure of less than 120 mm Hg. Risk factors for bradycardia include sensory anesthesia above T5, baseline heart rate less than 60 beats per minute, prolonged P-R interval on the electrocardiogram, and concomitant treatment with β-blocking drugs. Common features of cardiac arrest in patients receiving spinal anesthesia have included administration of sedation to produce a sleeplike state without spontaneous verbalization and lack of early administration of epinephrine. Even when therapy is promptly administered, patients may be refractory to treatment, because local anesthetic-induced sympathetic nervous system blockade, which decreases circulating blood volume, may also cause a defective neuroendocrine response to stress. Even early administration of epinephrine in patients being previously monitored with pulse oximetry and capnography may not guarantee a good outcome following cardiac arrest during spinal anesthesia.84
Sympathetic nervous system blockade results in arteriolar dilatation, but systemic blood pressure does not decrease proportionally because of compensatory vasoconstriction in areas with intact sympathetic nervous system innervation. Compensatory vasoconstriction occurs principally in the upper extremities and does not involve the cerebral vasculature. Even with total sympathetic nervous system blockade produced by spinal anesthesia, the decrease in systemic vascular resistance is <15%. This change is minimal because smooth muscles of arterioles retain intrinsic tone and do not dilate maximally.
The most important cardiovascular responses produced by spinal anesthesia are those that result from changes in the venous circulation. Unlike arterioles denervated by sympathetic nervous system blockade, venules do not maintain intrinsic tone and thus dilate maximally during spinal anesthesia. The resulting increased vascular capacitance decreases venous return to the heart, leading to decreases in cardiac output and systemic blood pressure. The physiologic effect of spinal anesthesia on venous return emphasizes the risk of extreme systemic hypotension if this technique is instituted in hypovolemic patients. Blockade of preganglionic cardiac accelerator fibers (T1 to T4) results in heart rate slowing, particularly if decreased venous return and central venous pressure decrease the stimulation of intrinsic stretch receptors in the right atrium (Bezold Jarisch reflex). For example, heart rate will increase with a head-down position that increases venous return and central venous pressure so as to stimulate these receptors. During spinal anesthesia, myocardial oxygen requirements are decreased as a result of decreased heart rate, venous return, and systemic blood pressure.
Apnea
Apnea that occurs with an excessive level of spinal anesthesia probably reflects ischemic paralysis of the medullary ventilatory centers due to profound hypotension and associated decreases in cerebral blood flow. Concentrations of local anesthetics in ventricular cerebrospinal fluid are usually too low to produce pharmacologic effects on the ventilatory centers. Rarely is the cause of apnea due to phrenic nerve paralysis.
Analgesia
Lidocaine and procaine have been demonstrated to produce intense analgesia when injected intravenously. Use of local anesthetics for this purpose, however, is limited by the small margin of safety between IV analgesic doses and those that produce systemic toxicity. Nevertheless, continuous low-dose infusion of lidocaine to maintain a plasma concentration of 1 to 2 µg/mL decreases the severity of postoperative pain and decreases requirements for opioids without producing systemic toxicity.164 Lidocaine administered intravenously also decreases anesthetic requirements for volatile drugs.165 Lidocaine may also be administered intravenously in the perioperative period as a cough suppressant. In this regard, the cough reflex during intubation of the trachea is suppressed by plasma concentrations of lidocaine >2 µg/mL.166Stump pain (neuromas) but not phantom pain (cortical reorganization) following amputation is temporarily diminished by IV administration of lidocaine.167
Suppression of Ventricular Cardiac Dysrhythmias
In addition to suppressing ventricular cardiac dysrhythmias, the IV administration of lidocaine may increase the defibrillation threshold. Failure to recognize this effect could lead to unnecessary revision of the lead system of an implantable cardioverter defibrillator.168
Suppression of Grand Mal Seizures
Grand mal seizures have been suppressed by IV administration of low doses of lidocaine or mepivacaine. Presumably, these and perhaps other local anesthetics, when present at low plasma concentrations, are effective in suppressing seizures through initial depression of hyperexcitable cortical neurons. Nevertheless, inhibitory neurons are usually more sensitive to depressant actions of local anesthetics than are excitatory neurons, and excitatory phenomena predominate.
Antiinflammatory Effects
Local anesthetics modulate inflammatory responses and may be useful in mitigating perioperative inflammatory injury.106 For example, overreactive inflammatory responses that destroy rather than protect are critical in the development of several perioperative phenomena including postoperative pain, adult respiratory distress syndrome, systemic inflammatory response syndrome, and multiple organ failure. Beneficial effects attributed to epidural anesthesia (e.g., pain relief, decreased thrombosis from hypercoagulability) may reflect antiinflammatory effects of local anesthetics. Adverse effects of local anesthetic–induced antiinflammatory effects include retardation of wound healing and increased risk of infection. Nevertheless, there is evidence that local anesthetics also possess significant antibacterial effects (tetracaine > bupivacaine > lidocaine).169 IV lidocaine has been postulated to decrease perioperative stress and improve anesthetic depth. Effect on depth of anesthesia, of IV lidocaine, as measured by bispectral index (BIS) was studied in the presence or absence of midazolam. IV lidocaine decreased BIS by modulating the effect of midazolam, suggesting an increase in the depth of anesthesia.170
Mechanism
Antiinflammatory effects of local anesthetics are not dependent on the sodium ion channel blockade that is responsible for the anesthetic effects of these drugs. Local anesthetics may modulate inflammatory responses by inhibiting inflammatory mediator signaling. For example, local anesthetics inhibit platelet-activating factor (an inflammatory mediator), which is an established signaling mechanism in early acute respiratory distress syndrome, a typical postoperative inflammatory disorder.171 Many of the mediators (thrombin, thromboxane, platelet-activating factor, and interleukins) of the inflammatory and hemostatic systems act through G protein–coupled receptors. Local anesthetics may inhibit G proteins, resulting in antiinflammatory effects.172 In addition, local anesthetics inhibit neutrophil accumulation at sites of inflammation and impair free radical and mediator release.173 Local anesthetics in clinically relevant concentrations inhibit superoxide anion production of platelet-activating factor–primed neutrophils.171 Priming is the process whereby the response of neutrophils to a subsequent activating stimulus is potentiated. Levobupivacaine is more effective than bupivacaine and other local anesthetics in suppressing neutrophil priming.174 Decreased generation of reactive oxygen radicals is associated with decreases in ischemic damage after myocardial infarction.
Bronchodilation
Inhaled lidocaine and ropivacaine attenuate histamine-induced bronchospasm and induce airway anesthesia. This response most likely reflects topical airway anesthesia, as bronchial reactivity is inhibited at plasma concentrations that are lower than those needed to attenuate bronchial reactivity. Nevertheless, dyclonine, a longer lasting and more potent local anesthetic, does not reliably attenuate bronchial hyperreactivity, suggesting that other properties of local anesthetics may be important.175
Tumescent Liposuction
The “tumescent” technique for liposuction is carried out via the subcutaneous infiltration of large volumes (5 or more liters) of solution containing highly diluted lidocaine (0.05% to 0.10%) with epinephrine (1:100,000). The taut stretching of overlying blanched skin by the large volume of solution and epinephrine-induced vasoconstriction is the origin of the term tumescent technique.
The result is sufficient local anesthesia for the liposuction, virtually bloodless aspirates, and prolonged postoperative analgesia. Slow and sustained release of lidocaine into the circulation is associated with plasma concentrations <1.5 µg/mL that peak 12 to 14 hours after injection and then decline gradually over the next 6 to 14 hours.176 Plasma concentrations of epinephrine peak at three to five times the upper normal limits approximately 3 hours following injection of the solution and return to normal after about 12 hours.177 The recommended adult dose of lidocaine with epinephrine for regional anesthesia is about 7 mg/kg. When highly diluted lidocaine solutions are administered for tumescent liposuction, the dose of lidocaine may range from 35 to 55 mg/kg (“mega-dose lidocaine”).178 It is estimated that 1 g of subcutaneous tissue can absorb up to 1 mg of lidocaine (“tissue buffering system”). The injection of additional undiluted lidocaine for concomitant cosmetic procedures must be considered in estimating the likely plasma concentrations of lidocaine that will occur.
Tumescent liposuction is commonly used in aesthetic contouring of thigh, abdomen, hip and buttocks and its use is increasing in reconstructive plastic surgery procedures. Despite the popularity and presumed safety of tumescent liposuction, there are reports of increased mortality associated with this technique (greater than mortality associated with automobile accidents).179 Causes of death may include lidocaine toxicity or local anesthetic–induced depression of cardiac conduction and contractility. Overall complication rate in a nationwide quality improvement study was 0.7% in which 0.57% was minor complications and 0.14% was major complications.180,181
Cocaine Toxicity
Cocaine abuse and intoxication is widespread across the globe. In the United States, 1.5 million (2010) adults use cocaine; in Europe, 4 million (2009) adults use cocaine.182 Cocaine produces sympathetic nervous system stimulation by blocking the presynaptic uptake of norepinephrine and dopamine, thus increasing their postsynaptic concentrations. Because of this blocking effect, dopamine remains at high concentrations in the synapse and continues to affect adjacent neurons, producing the characteristic cocaine “high.”183,184 Chronic exposure to cocaine is postulated to affect adversely dopaminergic function in the brain due to dopamine depletion.
Pharmacokinetics
Once cocaine is absorbed, the pharmacokinetics, regardless of the route of administration, are similar.185 Conversely, the route of administration is important in the rate of onset as well as the intensity and duration of cocaine’s effects. For example, peak venous plasma concentrations of cocaine are achieved at approximately 30 to 40 minutes after intranasal administration and approximately 5 minutes after IV and smoked cocaine administration. The maximum physiologic effects of intranasal cocaine occur within 15 to 40 minutes, and the maximum subjective effects occur within 10 to 20 minutes. The duration of effects is approximately 60 minutes or longer after peak effects. The subjective effects occur within minutes of IV or smoked cocaine use, and the duration of effect is approximately 30 to 45 minutes. The elimination half-time of cocaine is 60 to 90 minutes, and metabolism is principally by plasma esterases (see the section “Metabolism of Ester Local Anesthetics”). Urinary excretion of unchanged cocaine (typically <1% of the total dose but may reach 10% to 12%) and metabolites (benzoylecgonine and ecgonine methyl ester representing about 65% of the dose) is similar regardless of the route of administration.
Adverse Physiologic Effects
Acute cocaine administration has local anesthetic, vasoconstrictive and sympathomimetic effects. Cardiac function is adversely affected and cocaine is known to cause coronary vasospasm, myocardial ischemia, myocardial infarction, and ventricular cardiac dysrhythmias, including ventricular fibrillation.186 Associated hypertension and tachycardia further increase myocardial oxygen requirements at a time when coronary oxygen delivery is decreased by the effects of cocaine on coronary blood flow. Even remote cocaine use can result in myocardial ischemia and hypotension for as long as 6 weeks after discontinuing cocaine use.187,188 Presumably, delayed episodes of myocardial ischemia are due to cocaine-induced coronary artery vasospasm. In animals, chronic cocaine exposure sensitizes the left anterior descending coronary artery to catecholamines, even in the absence of circulating cocaine. Excessive sensitivity of the coronary vasculature to catecholamines after chronic exposure to cocaine may be due in part to cocaine-induced depletion of dopamine activity. Cocaine-abusing parturients are at higher risk for interim peripartum events such as hypertension, hypotension, and wheezing episodes.189 Cocaine produces dose-dependent decreases in uterine blood flow that result in fetal hypoxemia.190
Cocaine may produce hyperpyrexia, which could contribute to seizures. Unexpected patient agitation in the perioperative period may reflect the effects of cocaine ingestion.191 There is a temporal relationship between the recreational use of cocaine and cerebrovascular accidents.192 Administration of topical cocaine plus epinephrine, or the presence of volatile anesthetics that sensitize the myocardium, may exaggerate the cardiac-stimulating effects of cocaine. Cocaine should be used with caution, if at all, in patients with hypertension or coronary artery disease and in patients receiving drugs that potentiate the effects of catecholamines, such as monoamine oxidase inhibitors.
Treatment
Nitroglycerin has been used to treat cocaine-induced myocardial ischemia.193 Although esmolol has been recommended to treat tachycardia due to cocaine overdose, there is also evidence that β-adrenergic blockade accentuates coronary artery vasospasm in the setting of acute cocaine overdose.107,194 Whether β-adrenergic blockade is harmful for coronary vasospasm in the setting of chronic cocaine use is not known. Furthermore, administration of β-blocking drugs in the presence of catecholamine-induced hypertension and tachycardia has been associated with profound cardiovascular collapse and cardiac arrest that is unresponsive to aggressive cardiopulmonary resuscitation.195 In this situation, administration of a vasodilating drug such as nitroprusside may be the safest intervention. α-Adrenergic blockade may be effective in treatment of coronary vasoconstriction due to cocaine, but in the presence of hypotension, this intervention is questionable.
In 2008, the American Heart Association published a statement for management of cocaine-associated chest pain and myocardial infarction196 (Fig. 10-17). In addition to their beneficial effect in seizure control, benzodiazepines also help in acute coronary syndromes by relieving chest pain and improving hemodynamic profile. Nitroglycerin and phentolamine is useful in reversing coronary vasoconstriction.
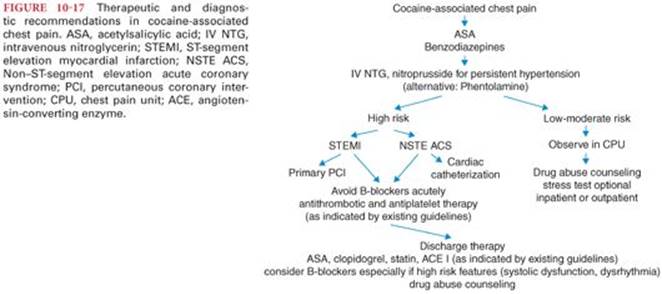
References
1. Ehrlich GE. Racemic mixtures: harmless or potentially toxic? Am J Hosp Pharm. 1992;49:S15–S18.
2. Burm AG, Cohen IM, van Kleef JW, et al. Pharmacokinetics of the enantiomers of mepivacaine after intravenous administration of the racemate in volunteers. Anesth Analg. 1997;84:85–89.
3. McClure JH. Ropivacaine. Br J Anaesth. 1996;76:300–307.
4. Vladimirov M, Nau C, Mok WM, et al. Potency of bupivacaine stereoisomers tested in vitro and in vivo: biochemical, electrophysiological, and neurobehavioral studies. Anesthesiology. 2000;93:744–755.
5. De Paula E, Cereda CM, Fraceto LF, et al. Micro and nanosystems for delivering local anesthetics. Exp Opin Drug Deliv. 2012;9:1505–1524.
6. Weiniger CF, Golovanevski L, Domb AJ, et al. Extended release formulations for local anaesthetic agents. Anaesthesia. 2012;67:906–916.
7. Mowat JJ, Mok MJ, MacLeod BA, et al. Liposomal bupivacaine. Extended duration nerve blockade using large unilamellar vesicles that exhibit a proton gradient. Anesthesiology. 1996;85:635–643.
8. Viscusi ER, Sinatra R, Onel E, et al. The safety of liposome bupivacaine, a novel local analgesic formulation. Clin J Pain. 2014;30:102–110.
9. Candiotti K. Liposomal bupivacaine: an innovative nonopioid local analgesic for the management of postsurgical pain. Pharmacotherapy. 2012;32:19s–26s.
10. Barreveld A, Witte J, Chahal H, et al. Preventive analgesia by local anesthetics: the reduction of postoperative pain by peripheral nerve blocks and intravenous drugs. Anesth Analg. 2013;116:1141–1161.
11. Ilfeld BM, Malhotra N, Furnish TJ, et al. Liposomal bupivacaine as a single-injection peripheral nerve block: a dose-response study. Anesth Analg. 2013;117:1248–1256.
12. Marcet JE, Nfonsam VN, Larach S. An extended pain relief trial utilizing the infiltration of a long-acting multivesicular liposome formulation of bupivacaine, EXPAREL (IMPROVE): a phase IV health economic trial in adult patients undergoing ileostomy reversal. J Pain Res. 2013;6:549–555.
13. Nau C, Wang GK. Interactions of local anesthetics with voltage-gated Na+ channels. J Membr Biol. 2004;201:1–8.
14. Butterworth JF IV, Strichartz GR. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990;72:711–734.
15. Docherty RJ, Farmer CE. The pharmacology of voltage-gated sodium channels in sensory neurones. Handb Exp Pharmacol. 2009;519–561.
16. Dib-Hajj SD, Yang Y, Black JA, et al. The NaV1.7 sodium channel: from molecule to man. Nat Rev Neurosci. 2013;14:49–62.
17. Lee-Son S, Wang GK, Concus A, et al. Stereoselective inhibition of neuronal sodium channels by local anesthetics. Evidence for two sites of action? Anesthesiology. 1992;77:324–335.
18. Bahring R, Covarrubias M. Mechanisms of closed-state inactivation in voltage-gated ion channels. J Physiol. 2011;589:461–479.
19. Payandeh J, Gamal El-Din TM, Scheuer T, et al. Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature. 2012;486:135–139.
20. Sugiyama K, Muteki T. Local anesthetics depress the calcium current of rat sensory neurons in culture. Anesthesiology. 1994;80:1369–1378.
21. Hollmann MW, Wieczorek KS, Berger A, et al. Local anesthetic inhibition of G protein-coupled receptor signaling by interference with Galpha(q) protein function. Mol Pharmacol. 2001;59:294–301.
22. Datta S, Lambert DH, Gregus J, et al. Differential sensitivities of mammalian nerve fibers during pregnancy. Anesth Analg. 1983;62:1070–1072.
23. Denson DD, Coyle DE, Thompson GA, et al. Bupivacaine protein binding in the term parturient: effects of lactic acidosis. Clin Pharmacol Therapeut. 1984;35:702–709.
24. Benowitz N, Forsyth FP, Melmon KL, et al. Lidocaine disposition kinetics in monkey and man. I. Prediction by a perfusion model. Clin Pharmacol Ther. 1974;16:87–98.
25. Kuipers JA, Boer F, de Roode A, et al. Modeling population pharmacokinetics of lidocaine: should cardiac output be included as a patient factor? Anesthesiology. 2001;94:566–573.
26. Tucker GT, Boyes RN, Bridenbaugh PO, et al. Binding of anilide-type local anesthetics in human plasma. I. Relationships between binding, physicochemical properties, and anesthetic activity. Anesthesiology. 1970;33:287–303.
27. Jorfeldt L, Lewis DH, Lofstrom JB, et al. Lung uptake of lidocaine in man as influenced by anaesthesia, mepivacaine infusion or lung insufficiency. Acta Anaesthesiol Scand. 1983;27:5–9.
28. Rothstein P, Cole J, Pitt B. Pulmonary extraction of bupivacaine is dose dependent. Anesthesiology. 1984;61:A236.
29. Rothstein P, Pitt B. Pulmonary extraction of bupivacaine and its modification by propranolol. Anesthesiology. 1983;59:A189.
30. Bowdle TA, Freund PR, Slattery JT. Propranolol reduces bupivacaine clearance. Anesthesiology. 1987;66:36–38.
31. Thomas J, Long G, Moore G, et al. Plasma protein binding and placental transfer of bupivacaine. Clin Pharmacol Ther. 1976;19:426–434.
32. Biehl D, Shnider SM, Levinson G, et al. Placental transfer of lidocaine: effects of fetal acidosis. Anesthesiology. 1978;48:409–412.
33. Tucker GT, Mather LE. Clinical pharmacokinetics of local anaesthetics. Clin Pharmacokinet. 1979;4:241–278.
34. Ramanathan J, Bottorff M, Jeter JN, et al. The pharmacokinetics and maternal and neonatal effects of epidural lidocaine in preeclampsia. Anesth Analg. 1986;65:120–126.
35. Pihlajamaki K, Kanto J, Lindberg R, et al. Extradural administration of bupivacaine: pharmacokinetics and metabolism in pregnant and non-pregnant women. Br J Anaesth. 1990;64:556–562.
36. Dauphin A, Gupta RN, Young JE, et al. Serum bupivacaine concentrations during continuous extrapleural infusion. Can J Anaesth. 1997;44:367–370.
37. Pere P, Salonen M, Jokinen M, et al. Pharmacokinetics of ropivacaine in uremic and nonuremic patients after axillary brachial plexus block. Anesth Analg. 2003;96:563–569, table of contents.
38. Kuhnert BR, Kuhnert PM, Philipson EH, et al. The half-life of 2-chloroprocaine. Anesth Analg. 1986;65:273–278.
39. Kuhnert BR, Philipson EH, Pimental R, et al. Lidocaine disposition in mother, fetus, and neonate after spinal anesthesia. Anesth Analg. 1986;65:139–144.
40. Curatolo M, Petersen-Felix S, Arendt-Nielsen L, et al. Adding sodium bicarbonate to lidocaine enhances the depth of epidural blockade. Anesth Analg. 1998;86:341–347.
41. Abdallah FW, Abrishami A, Brull R. The facilitatory effects of intravenous dexmedetomidine on the duration of spinal anesthesia: a systematic review and meta-analysis. Anesth Analg. 2013;117:271–278.
42. Morrison AP, Hunter JM, Halpern SH, et al. Effect of intrathecal magnesium in the presence or absence of local anaesthetic with and without lipophilic opioids: a systematic review and meta-analysis. Br J Anaesth. 2013;110:702–712.
43. Mossetti V, Vicchio N, Ivani G. Local anesthetics and adjuvants in pediatric regional anesthesia. Curr Drug Targets. 2012;13:952–960.
44. Chestnut DH, Geiger M, Bates JN, et al. The influence of pH-adjusted 2-chloroprocaine on the quality and duration of subsequent epidural bupivacaine analgesia during labor: a randomized, double-blind study. Anesthesiology. 1989;70:437–441.
45. Munson ES, Paul WL, Embro WJ. Central-nervous-system toxicity of local anesthetic mixtures in monkeys. Anesthesiology. 1977;46:179–183.
46. Liu S. Local anesthetics and analgesia. In: Ashburn M, Rice L, eds. The Management of Pain. New York, NY: Churchill Livingstone; 1997:141–170.
47. Scott DB, Jebson PJ, Braid DP, et al. Factors affecting plasma levels of lignocaine and prilocaine. Br J Anaesth. 1972;44:1040–1049.
48. Carpenter RL, Smith HS, Bridenbaugh LD. Epinephrine increases the effectiveness of tetracaine spinal anesthesia. Anesthesiology. 1989;71:33–36.
49. Brown DL, Ransom DM, Hall JA, et al. Regional anesthesia and local anesthetic-induced systemic toxicity: seizure frequency and accompanying cardiovascular changes. Anesth Analg. 1995;81:321–328.
50. Brown DT, Beamish D, Wildsmith JA. Allergic reaction to an amide local anaesthetic. Br J Anaesth. 1981;53:435–437.
51. Neal JM, Bernards CM, Butterworth JF IV, et al. ASRA practice advisory on local anesthetic systemic toxicity. Reg Anesth Pain Med. 2010;35:152–161.
52. Covino B, Wildsmith J. Clinical pharmacology of local anesthetic agents. In: Cousins M, Bridenbaugh P, eds. Neural Blockade in Clinical Anesthesia and Management of Pain. Philadelphia, PA: Lippincott-Raven; 1998:97–128.
53. Butterworth JF IV. Models and mechanisms of local anesthetic cardiac toxicity: a review. Reg Anesth Pain Med. 2010;35:167–176.
54. Feldman HS, Arthur GR, Covino BG. Comparative systemic toxicity of convulsant and supraconvulsant doses of intravenous ropivacaine, bupivacaine, and lidocaine in the conscious dog. Anesth Analg. 1989;69:794–801.
55. Marsch SC, Schaefer HG, Castelli I. Unusual psychological manifestation of systemic local anesthetic toxicity. Anesthesiology. 1998;88:531–533.
56. Inoue R, Suganuma T, Echizen H, et al. Plasma concentrations of lidocaine and its principal metabolites during intermittent epidural anesthesia. Anesthesiology. 1985;63:304–310.
57. Christie JM, Valdes C, Markowsky SJ. Neurotoxicity of lidocaine combined with mexiletine. Anesth Analg. 1993;77:1291–1294.
58. Butterworth J, James RL, Grimes J. Structure-affinity relationships and stereospecificity of several homologous series of local anesthetics for the beta2-adrenergic receptor. Anesth Analg. 1997;85:336–342.
59. Albright GA. Cardiac arrest following regional anesthesia with etidocaine or bupivacaine. Anesthesiology. 1979;51:285–287.
60. Kotelko DM, Shnider SM, Dailey PA, et al. Bupivacaine-induced cardiac arrhythmias in sheep. Anesthesiology. 1984;60:10–18.
61. Timour Q, Freysz M, Couzon P, et al. Possible role of drug interactions in bupivacaine-induced problems related to intraventricular conduction disorders. Reg Anesth. 1990;15:180–185.
62. Morishima HO, Pedersen H, Finster M, et al. Bupivacaine toxicity in pregnant and nonpregnant ewes. Anesthesiology. 1985;63:134–139.
63. Roitman K, Sprung J, Wallace M, et al. Enhancement of bupivacaine cardiotoxicity with cardiac glycosides and beta-adrenergic blockers: a case report. Anesth Analg. 1993;76:658–661.
64. Butterworth JF IV, Brownlow RC, Leith JP, et al. Bupivacaine inhibits cyclic-3′,5′-adenosine monophosphate production. A possible contributing factor to cardiovascular toxicity. Anesthesiology. 1993;79:88–95.
65. Clarkson CW, Hondeghem LM. Mechanism for bupivacaine depression of cardiac conduction: fast block of sodium channels during the action potential with slow recovery from block during diastole. Anesthesiology. 1985;62:396–405.
66. Atlee JL III, Bosnjak ZJ. Mechanisms for cardiac dysrhythmias during anesthesia. Anesthesiology. 1990;72:347–374.
67. Crews JC, Rothman TE. Seizure after levobupivacaine for interscalene brachial plexus block. Anesth Analg. 2003;96:1188–1190.
68. Groban L, Deal DD, Vernon JC, et al. Cardiac resuscitation after incremental overdosage with lidocaine, bupivacaine, levobupivacaine, and ropivacaine in anesthetized dogs. Anesth Analg. 2001;92:37–43.
69. Huang YF, Pryor ME, Mather LE, et al. Cardiovascular and central nervous system effects of intravenous levobupivacaine and bupivacaine in sheep. Anesth Analg. 1998;86:797–804.
70. Moller R, Covino BG. Cardiac electrophysiologic properties of bupivacaine and lidocaine compared with those of ropivacaine, a new amide local anesthetic. Anesthesiology. 1990;72:322–329.
71. Scott DB, Lee A, Fagan D, et al. Acute toxicity of ropivacaine compared with that of bupivacaine. Anesth Analg. 1989;69:563–569.
72. Chazalon P, Tourtier JP, Villevielle T, et al. Ropivacaine-induced cardiac arrest after peripheral nerve block: successful resuscitation. Anesthesiology. 2003;99:1449–1451.
73. Klein SM, Pierce T, Rubin Y, et al. Successful resuscitation after ropivacaine-induced ventricular fibrillation. Anesth Analg. 2003;97:901–903.
74. Polley LS, Santos AC. Cardiac arrest following regional anesthesia with ropivacaine: here we go again! Anesthesiology. 2003;99:1253–1254.
75. Kendig JJ. Clinical implications of the modulated receptor hypothesis: local anesthetics and the heart. Anesthesiology. 1985;62:382–384.
76. Kasten GW, Martin ST. Bupivacaine cardiovascular toxicity: comparison of treatment with bretylium and lidocaine. Anesth Analg. 1985;64:911–916.
77. Xuecheng J, Xiaobin W, Bo G, et al. The plasma concentrations of lidocaine after slow versus rapid administration of an initial dose of epidural anesthesia. Anesth Analg. 1997;84:570–573.
78. Moore DC, Crawford RD, Scurlock JE. Severe hypoxia and acidosis following local anesthetic-induced convulsions. Anesthesiology. 1980;53:259–260.
79. Weinberg GL. Treatment of local anesthetic systemic toxicity (LAST). Reg Anesth Pain Med. 2010;35:188–193.
80. Neal JM, Mulroy MF, Weinberg GL; American Society of Regional Anesthesia and Pain Medicine. American Society of Regional Anesthesia and Pain Medicine checklist for managing local anesthetic systemic toxicity: 2012 version. Reg Anesth Pain Med. 2012;37:16–18.
81. Horlocker TT, McGregor DG, Matsushige DK, et al. A retrospective review of 4767 consecutive spinal anesthetics: central nervous system complications. Perioperative Outcomes Group. Anesth Analg. 1997;84:578–584.
82. Naveira FA, Copeland S, Anderson M, et al. Transient neurologic toxicity after spinal anesthesia, or is it myofascial pain? Two case reports. Anesthesiology. 1998;88:268–270.
83. Eisenach JC. Regional anesthesia: vintage Bordeaux (and Napa Valley). Anesthesiology. 1997;87:467–469.
84. Lee LA, Posner KL, Domino KB, et al. Injuries associated with regional anesthesia in the 1980s and 1990s: a closed claims analysis. Anesthesiology. 2004;101:143–152.
85. Kuboyoma N, Nakoa S, Moriya Y, et al. Bupivacaine-included Ca2+ release on intracellular Ca2+ stores in rat DRG neurons. Jpn J Pharmacol. 1997;73:98–103.
86. Schneider M, Ettlin T, Kaufmann M, et al. Transient neurologic toxicity after hyperbaric subarachnoid anesthesia with 5% lidocaine. Anesth Analg. 1993;76:1154–1157.
87. Hodgson PS, Neal JM, Pollock JE, et al. The neurotoxicity of drugs given intrathecally (spinal). Anesth Analg. 1999;88:797–809.
88. Hampl KF, Schneider MC, Pargger H, et al. A similar incidence of transient neurologic symptoms after spinal anesthesia with 2% and 5% lidocaine. Anesth Analg. 1996;83:1051–1054.
89. Liu SS, Ware PD, Allen HW, et al. Dose-response characteristics of spinal bupivacaine in volunteers. Clinical implications for ambulatory anesthesia. Anesthesiology. 1996;85:729–736.
90. Pollock JE, Liu SS, Neal JM, et al. Dilution of spinal lidocaine does not alter the incidence of transient neurologic symptoms. Anesthesiology. 1999;90:445–450.
91. Schneider MC, Birnbach DJ. Lidocaine neurotoxicity in the obstetric patient: is the water safe? Anesth Analg. 2001;92:287–290.
92. Zaric D, Pace NL. Transient neurologic symptoms (TNS) following spinal anaesthesia with lidocaine versus other local anaesthetics. Cochrane Database Syst Rev. 2009;(2):CD003006.
93. Sakura S, Sumi M, Sakaguchi Y, et al. The addition of phenylephrine contributes to the development of transient neurologic symptoms after spinal anesthesia with 0.5% tetracaine. Anesthesiology. 1997;87:771–778.
94. Silvanto M, Tarkkila P, Makela ML, et al. The influence of ambulation time on the incidence of transient neurologic symptoms after lidocaine spinal anesthesia. Anesth Analg. 2004;98:642–646, table of contents.
95. Pollock JE, Neal JM, Stephenson CA, et al. Prospective study of the incidence of transient radicular irritation in patients undergoing spinal anesthesia. Anesthesiology. 1996;84:1361–1367.
96. Gardner A, Gardner E, Morley T. Cauda equina syndrome: a review of the current clinical and medico-legal position. Eur Spine J. 2011; 20:690–697.
97. Lambert DH, Hurley RJ. Cauda equina syndrome and continuous spinal anesthesia. Anesth Analg. 1991;72:817–819.
98. Rigler ML, Drasner K, Krejcie TC, et al. Cauda equina syndrome after continuous spinal anesthesia. Anesth Analg. 1991;72:275–281.
99. Gerancher JC. Cauda equina syndrome following a single spinal administration of 5% hyperbaric lidocaine through a 25-gauge Whitacre needle. Anesthesiology. 1997;87:687–689.
100. Kozody R, Palahniuk RJ, Wade JG, et al. The effect of subarachnoid epinephrine and phenylephrine on spinal cord blood flow. Can Anaesth Soc J. 1984;31:503–508.
101. Nguyen ST, Cabrales RE, Bashour CA, et al. Benzocaine-induced methemoglobinemia. Anesth Analg. 2000;90:369–371.
102. Gross JB, Caldwell CB, Shaw LM, et al. The effect of lidocaine infusion on the ventilatory response to hypoxia. Anesthesiology. 1984;61:662–665.
103. Yokoyama M, Ohashi I, Nakatsuka H, et al. Drug-induced liver disease during continuous epidural block with bupivacaine. Anesthesiology. 2001;95:259–261.
104. Craft DV, Good RP. Delayed hypersensitivity reaction of the knee after injection of arthroscopy portals with bupivacaine (marcaine). Arthroscopy. 1994;10:305–308.
105. Foster RH, Markham A. Levobupivacaine: a review of its pharmacology and use as a local anaesthetic. Drugs. 2000;59:551–579.
106. Hollmann MW, Durieux ME. Local anesthetics and the inflammatory response: a new therapeutic indication? Anesthesiology. 2000;93:858–875.
107. Lange RA, Cigarroa RG, Flores ED, et al. Potentiation of cocaine-induced coronary vasoconstriction by beta-adrenergic blockade. Ann Intern Med. 1990;112:897–903.
108. Noorily AD, Noorily SH, Otto RA. Cocaine, lidocaine, tetracaine: which is best for topical nasal anesthesia? Anesth Analg. 1995;81:724–727.
109. McAlpine LG, Thomson NC. Lidocaine-induced bronchoconstriction in asthmatic patients. Relation to histamine airway responsiveness and effect of preservative. Chest. 1989;96:1012–1015.
110. Kirkpatrick MB, Sanders RV, Bass JB Jr. Physiologic effects and serum lidocaine concentrations after inhalation of lidocaine from a compressed gas-powered jet nebulizer. Am Rev Respir Dis. 1987;136:447–449.
111. Viegas O, Stoelting RK. Lidocaine in arterial blood after laryngotracheal administration. Anesthesiology. 1975;43:491–493.
112. Gajraj NM, Pennant JH, Watcha MF. Eutectic mixture of local anesthetics (EMLA) cream. Anesth Analg. 1994;78:574–583.
113. Taddio A, Stevens B, Craig K, et al. Efficacy and safety of lidocaine-prilocaine cream for pain during circumcision. N Engl J Med. 1997;336:1197–1201.
114. Katz NP, Shapiro DE, Herrmann TE, et al. Rapid onset of cutaneous anesthesia with EMLA cream after pretreatment with a new ultrasound-emitting device. Anesth Analg. 2004;98:371–376, table of contents.
115. Teillol-Foo WL, Kassab JY. Topical glyceryl trinitrate and eutectic mixture of local anaesthetics in children. A randomised controlled trial on choice of site and ease of venous cannulation. Anaesthesia. 1991;46:881–884.
116. Hymes J, Spraker M. Racial differences in the effectiveness of a topically applied mixture of local anesthetics. Reg Anesth. 1986;11:11–13.
117. Jakobson B, Nilsson A. Methemoglobinemia associated with a prilocaine-lidocaine cream and trimetoprim-sulphamethoxazole. A case report. Acta Anaesthesiol Scand. 1985;29:453–455.
118. Powell DM, Rodeheaver GT, Foresman PA, et al. Damage to tissue defenses by EMLA cream. J Emerg Med. 1991;9:205–209.
119. Arevalo MI, Escribano E, Calpena A, et al. Rapid skin anesthesia using a new topical amethocaine formulation: a preclinical study. Anesth Analg. 2004;98:1407–1412, table of contents.
120. Ravishankar N, Elliot SC, Beardow Z, et al. A comparison of Rapydan® patch and Ametop® gel for venous cannulation. Anaesthesia. 2012;67:367–370.
121. Erichsen CJ, Vibits H, Dahl JB, et al. Wound infiltration with ropivacaine and bupivacaine for pain after inguinal herniotomy. Acta Anaesthesiol Scand. 1995;39:67–70.
122. Winnie AP, Tay CH, Patel KP, et al. Pharmacokinetics of local anesthetics during plexus blocks. Anesth Analg. 1977;56:852–861.
123. Winnie AP, La Vallee DA, Pe Sosa B, et al. Clinical pharmacokinetics of local anaesthetics. Can Anaesth Soc J. 1977;24:252–262.
124. Hickey R, Candido KD, Ramamurthy S, et al. Brachial plexus block with a new local anaesthetic: 0.5 per cent ropivacaine. Can J Anaesth. 1990;37:732–738.
125. Nolte H, Fruhstorfer H, Edstrom HH. Local anesthetic efficacy of ropivacaine (LEA 103) in ulnar nerve block. Reg Anesth. 1990;15:118–124.
126. Niesel HC, Eilingsfeld T, Hornung M, et al. Ropivacaine 1% versus bupivacaine 0.75% without a vasoconstrictor. A comparative study of epidural anesthesia in orthopedic surgery [in German]. Anaesthesist. 1993;42:605–611.
127. Abrahams MS, Aziz MF, Fu RF, et al. Ultrasound guidance compared with electrical neurostimulation for peripheral nerve block: a systematic review and meta-analysis of randomized controlled trials. Br J Anaesth. 2009;102:408–417.
128. Bingham AE, Fu R, Horn JL, et al. Continuous peripheral nerve block compared with single-injection peripheral nerve block: a systematic review and meta-analysis of randomized controlled trials. Reg Anesth Pain Med. 2012;37:583–594.
129. Bailard NS, Ortiz J, Flores RA. Additives to local anesthetics for peripheral nerve blocks: Evidence, limitations, and recommendations. Am J Health Syst Pharm. 2014;71:373–385.
130. Bader AM, Concepcion M, Hurley RJ, et al. Comparison of lidocaine and prilocaine for intravenous regional anesthesia. Anesthesiology. 1988;69:409–412.
131. Prieto-Alvarez P, Calas-Guerra A, Fuentes-Bellido J, et al. Comparison of mepivacaine and lidocaine for intravenous regional anaesthesia: pharmacokinetic study and clinical correlation. Br J Anaesth. 2002;88:516–519.
132. Chan VW, Weisbrod MJ, Kaszas Z, et al. Comparison of ropivacaine and lidocaine for intravenous regional anesthesia in volunteers: a preliminary study on anesthetic efficacy and blood level. Anesthesiology. 1999;90:1602–1608.
133. Brown DL, Carpenter RL, Thompson GE. Comparison of 0.5% ropivacaine and 0.5% bupivacaine for epidural anesthesia in patients undergoing lower-extremity surgery. Anesthesiology. 1990;72:633–636.
134. Feldman HS, Covino BG. Comparative motor-blocking effects of bupivacaine and ropivacaine, a new amino amide local anesthetic, in the rat and dog. Anesth Analg. 1988;67:1047–1052.
135. Cederholm I, Anskar S, Bengtsson M. Sensory, motor, and sympathetic block during epidural analgesia with 0.5% and 0.75% ropivacaine with and without epinephrine. Reg Anesth. 1994;19:18–33.
136. Wood MB, Rubin AP. A comparison of epidural 1% ropivacaine and 0.75% bupivacaine for lower abdominal gynecologic surgery. Anesth Analg. 1993;76:1274–1278.
137. Erichsen CJ, Sjovall J, Kehlet H, et al. Pharmacokinetics and analgesic effect of ropivacaine during continuous epidural infusion for postoperative pain relief. Anesthesiology. 1996;84:834–842.
138. George RB, Allen TK, Habib AS. Intermittent epidural bolus compared with continuous epidural infusions for labor analgesia: a systematic review and meta-analysis. Anesth Analg. 2013;116:133–144.
139. De Negri P, Ivani G, Tirri T, et al. A comparison of epidural bupivacaine, levobupivacaine, and ropivacaine on postoperative analgesia and motor blockade. Anesth Analg. 2004;99:45–48.
140. Alahuhta S, Rasanen J, Jouppila P, et al. The effects of epidural ropivacaine and bupivacaine for cesarean section on uteroplacental and fetal circulation. Anesthesiology. 1995;83:23–32.
141. Muir HA, Writer D, Douglas J, et al. Double-blind comparison of epidural ropivacaine 0.25% and bupivacaine 0.25%, for the relief of childbirth pain. Can J Anaesth. 1997;44:599–604.
142. Polley LS, Columb MO. Ropivacaine and bupivacaine: concentrating on dosing! Anesth Analg. 2003;96:1251–1253.
143. Kuhnert BR, Zuspan KJ, Kuhnert PM, et al. Bupivacaine disposition in mother, fetus, and neonate after spinal anesthesia for cesarean section. Anesth Analg. 1987;66:407–412.
144. Reynolds F. A comparison of the potential toxicity of bupivacaine, lignocaine and mepivacaine during epidural blockade for surgery. Br J Anaesth. 1971;43:567–572.
145. Solomon RE, Gebhart GF. Synergistic antinociceptive interactions among drugs administered to the spinal cord. Anesth Analg. 1994;78:1164–1172.
146. Coda B, Bausch S, Haas M, et al. The hypothesis that antagonism of fentanyl analgesia by 2-chloroprocaine is mediated by direct action on opioid receptors. Reg Anesth. 1997;22:43–52.
147. Carpenter RL. Hyperbaric lidocaine spinal anesthesia: do we need an alternative? Anesth Analg. 1995;81:1125–1128.
148. Drasner K. Lidocaine spinal anesthesia: a vanishing therapeutic index? Anesthesiology. 1997;87:469–472.
149. Stewart A, Lambert DH, Concepcion MA, et al. Decreased incidence of tourniquet pain during spinal anesthesia with bupivacaine. A possible explanation. Anesth Analg. 1988;67:833–837.
150. Campbell DC, Banner R, Crone LA, et al. Addition of epinephrine to intrathecal bupivacaine and sufentanil for ambulatory labor analgesia. Anesthesiology. 1997;86:525–531.
151. Stocks GM, Hallworth SP, Fernando R, et al. Minimum local analgesic dose of intrathecal bupivacaine in labor and the effect of intrathecal fentanyl. Anesthesiology. 2001;94:593–598; discussion 5A.
152. van Kleef JW, Veering BT, Burm AG. Spinal anesthesia with ropivacaine: a double-blind study on the efficacy and safety of 0.5% and 0.75% solutions in patients undergoing minor lower limb surgery. Anesth Analg. 1994; 78:1125–1130.
153. Halpern SH, Breen TW, Campbell DC, et al. A multicenter, randomized, controlled trial comparing bupivacaine with ropivacaine for labor analgesia. Anesthesiology. 2003;98:1431–1435.
154. Khaw KS, Ngan Kee WD, Wong EL, et al. Spinal ropivacaine for cesarean section: a dose-finding study. Anesthesiology. 2001;95:1346–1350.
155. Alley EA, Kopacz DJ, McDonald SB, et al. Hyperbaric spinal levobupivacaine: a comparison to racemic bupivacaine in volunteers. Anesth Analg. 2002;94:188–193, table of contents.
156. Covino BG, Marx GF, Finster M, et al. Prolonged sensory/motor deficits following inadvertent spinal anesthesia. Anesth Analg. 1980;59:399–400.
157. Ravindran RS, Bond VK, Tasch MD, et al. Prolonged neural blockade following regional analgesia with 2-chloroprocaine. Anesth Analg. 1980;59:447–451.
158. Reisner LS, Hochman BN, Plumer MH. Persistent neurologic deficit and adhesive arachnoiditis following intrathecal 2-chloroprocaine injection. Anesth Analg. 1980;59:452–454.
159. Drasner K. Chloroprocaine spinal anesthesia: back to the future? Anesth Analg. 2005;100:549–552.
160. Kouri ME, Kopacz DJ. Spinal 2-chloroprocaine: a comparison with lidocaine in volunteers. Anesth Analg. 2004;98:75–80, table of contents.
161. Kozody R, Palahniuk RJ, Cumming MO. Spinal cord blood flow following subarachnoid tetracaine. Can Anaesth Soc J. 1985;32:23–29.
162. Liu SS, McDonald SB. Current issues in spinal anesthesia. Anesthesiology. 2001;94:888–906.
163. Pollard JB. Cardiac arrest during spinal anesthesia: common mechanisms and strategies for prevention. Anesth Analg. 2001;92:252–256.
164. Cassuto J, Wallin G, Hogstrom S, et al. Inhibition of postoperative pain by continuous low-dose intravenous infusion of lidocaine. Anesth Analg. 1985;64:971–974.
165. DiFazio CA, Neiderlehner JR, Burney RG. The anesthetic potency of lidocaine in the rat. Anesth Analg. 1976;55:818–821.
166. Yukioka H, Yoshimoto N, Nishimura K, et al. Intravenous lidocaine as a suppressant of coughing during tracheal intubation. Anesth Analg. 1985;64:1189–1192.
167. Wu CL, Tella P, Staats PS, et al. Analgesic effects of intravenous lidocaine and morphine on postamputation pain: a randomized double-blind, active placebo-controlled, crossover trial. Anesthesiology. 2002;96:841–848.
168. Peters RW, Gilbert TB, Johns-Walton S, et al. Lidocaine-related increase in defibrillation threshold. Anesth Analg. 1997;85:299–300.
169. Pere P, Lindgren L, Vaara M. Poor antibacterial effect of ropivacaine: comparison with bupivacaine. Anesthesiology. 1999;91:884–886.
170. Gottschalk A, McKay AM, Malik ZM, et al. Systemic lidocaine decreases the Bispectral Index in the presence of midazolam, but not its absence. J Clin Anesth. 2012;24:121–125.
171. Hollmann MW, Gross A, Jelacin N, Durieux ME. Local anesthetic effects on priming and activation of human neutrophils. Anesthesiology. 2001;95:113–122.
172. Hollmann MW, McIntire WE, Garrison JC, et al. Inhibition of mammalian Gq protein function by local anesthetics. Anesthesiology. 2002;97:1451–1457.
173. Kiefer RT, Ploppa A, Krueger WA, et al. Local anesthetics impair human granulocyte phagocytosis activity, oxidative burst, and CD11b expression in response to Staphylococcus aureus. Anesthesiology. 2003;98:842–848.
174. Hollmann MW, Kurz K, Herroeder S, et al. The effects of S(−)-, R(+)-, and racemic bupivacaine on lysophosphatidate-induced priming of human neutrophils. Anesth Analg. 2003;97:1053–1058, table of contents.
175. Groeben H, Grosswendt T, Silvanus MT, et al. Airway anesthesia alone does not explain attenuation of histamine-induced bronchospasm by local anesthetics: a comparison of lidocaine, ropivacaine, and dyclonine. Anesthesiology. 2001;94:423–428; discussion 5A-6A.
176. Klein JA. Tumescent technique for regional anesthesia permits lidocaine doses of 35 mg/kg for liposuction. J Dermatol Surg Oncol. 1990;16:248–263.
177. Burk RW III, Guzman-Stein G, Vasconez LO. Lidocaine and epinephrine levels in tumescent technique liposuction. Plast Reconstr Surg. 1996;97:1379–1384.
178. Ostad A, Kageyama N, Moy RL. Tumescent anesthesia with a lidocaine dose of 55 mg/kg is safe for liposuction. Dermatol Surg. 1996;22:921–927.
179. Rao RB, Ely SF, Hoffman RS. Deaths related to liposuction. N Engl J Med. 1999;340:1471–1475.
180. Hanke W, Cox SE, Kuznets N, et al. Tumescent liposuction report performance measurement initiative: national survey results. Dermatol Surg. 2004;30:967–977; discussion 978.
181. Svedman KJ, Coldiron B, Coleman WP III, et al. ASDS guidelines of care for tumescent liposuction. Dermatol Surg. 2006;32:709–716.
182. Zimmerman JL. Cocaine intoxication. Crit Care Clin. 2012;28:517–526.
183. Mendelson JH, Mello NK. Management of cocaine abuse and dependence. N Engl J Med. 1996;334:965–972.
184. Leshner AI. Molecular mechanisms of cocaine addiction. N Engl J Med. 1996;335:128–129.
185. Hatsukami DK, Fischman MW. Crack cocaine and cocaine hydrochloride. Are the differences myth or reality? JAMA. 1996;276:1580–1588.
186. Hollander JE, Hoffman RS, Burstein JL, et al. Cocaine-associated myocardial infarction. Mortality and complications. Cocaine-Associated Myocardial Infarction Study Group. Arch Intern Med. 1995;155:1081–1086.
187. Weicht GT, Bernards CM. Remote cocaine use as a likely cause of cardiogenic shock after penetrating trauma. Anesthesiology. 1996;85:933–935.
188. Nademanee K, Gorelick DA, Josephson MA, et al. Myocardial ischemia during cocaine withdrawal. Ann Intern Med. 1989;111:876–880.
189. Kain ZN, Mayes LC, Ferris CA, et al. Cocaine-abusing parturients undergoing cesarean section. A cohort study. Anesthesiology. 1996;85:1028–1035.
190. Woods JR Jr, Plessinger MA, Clark KE. Effect of cocaine on uterine blood flow and fetal oxygenation. JAMA. 1987;257:957–961.
191. Bernards CM, Teijeiro A. Illicit cocaine ingestion during anesthesia. Anesthesiology. 1996;84:218–220.
192. Levine SR, Brust JC, Futrell N, et al. Cerebrovascular complications of the use of the “crack” form of alkaloidal cocaine. N Engl J Med. 1990;323:699–704.
193. Hollander JE, Hoffman RS, Gennis P, et al. Nitroglycerin in the treatment of cocaine associated chest pain—clinical safety and efficacy. J Toxicol Clin Toxicol. 1994;32:243–256.
194. Pollan S, Tadjziechy M. Esmolol in the management of epinephrine- and cocaine-induced cardiovascular toxicity. Anesth Analg. 1989;69:663–664.
195. Groudine SB, Hollinger I, Jones J, et al. New York State guidelines on the topical use of phenylephrine in the operating room. The Phenylephrine Advisory Committee. Anesthesiology. 2000;92:859–864.
196. McCord J, Jneid H, Hollander JE, et al; American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Management of cocaine-associated chest pain and myocardial infarction: a scientific statement from the American Heart Association Acute Cardiac Care Committee of the Council on Clinical Cardiology. Circulation. 2008;117:1897–1907.