Preparations that contain synthetic hormones identical to those secreted endogenously by endocrine glands may be administered as drugs. These synthetic hormones resemble the endogenous substances in structure and activity. Typically, the clinical application of these drugs is for hormone replacement to provide a physiologic effect. In certain patients, however, large doses of synthetic hormones are used to exert a pharmacologic effect. Recombinant DNA technology permits the incorporation of synthetic genes that code for the synthesis of specific human hormones by bacteria, thus permitting production of pure hormones devoid of allergic properties.
Corticosteroids
The actions of corticosteroids are classified according to the potencies of these compounds to (a) evoke distal renal tubular reabsorption of sodium in exchange for potassium ions (mineralocorticoid effect) or (b) produce an antiinflammatory response (glucocorticoid effect). Naturally occurring corticosteroids are cortisol (hydrocortisone), cortisone, corticosterone, desoxycorticosterone, and aldosterone (Fig. 40-1). Several synthetic corticosteroids are available, principally for use to produce antiinflammatory effects. Although it is possible to separate mineralocorticoid and glucocorticoid effects using synthetic drugs, it has not been possible to separate the various components of glucocorticoid effects. Consequently, all synthetic corticosteroids, when used in pharmacologic doses for their antiinflammatory effects, also produce less desirable effects, such as suppression of the hypothalamic-pituitary-adrenal (HPA) axis, weight gain, and skeletal muscle wasting.
Structure–Activity Relationships
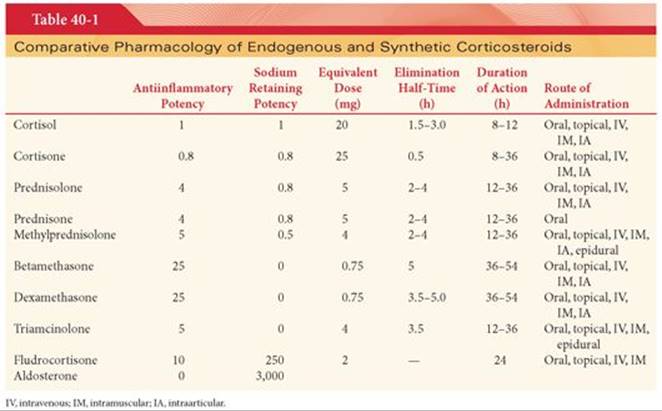
All corticosteroids are constructed on the same primary molecular framework, designated as the steroid nucleus (see Fig. 40-1). Changes in molecular structure may result in altered biologic responses due to changes in absorption, protein binding, rate of metabolism, and intrinsic effectiveness of the drug at receptors. Modifications of structure, such as introduction of a double bond in prednisolone and prednisone, have resulted in synthetic corticosteroids with more potent glucocorticoid effects than the two closely related natural hormones, cortisol and cortisone, respectively (Table 40-1). At the same time, mineralocorticoid effects and the rate of hepatic metabolism of these synthetic drugs are less than those of the natural hormones. Despite increased antiinflammatory effects, it has not been possible to separate this response from alterations in carbohydrate and protein metabolism. This suggests that the multiple manifestations of drug-induced glucocorticoid effects are mediated by the same receptor.
Mechanism of Action
Glucocorticoids attach to cytoplasmic receptors to enhance or suppress changes in the transcription of DNA and thus the synthesis of proteins. Glucocorticoids also inhibit the secretion of cytokines via posttranslational effects.1 Two distinct types of corticosteroid receptors have been identified (mineralocorticoid and glucocorticoid). Mineralocorticoid receptors are present in distal renal tubules, colon, salivary glands, and the hippocampus. In contrast, glucocorticoids receptors are more widely distributed and do not bind aldosterone, making these receptors glucocorticoid-selective. Local mechanisms that result in release of steroids from their carrier proteins serve to facilitate steroid entry into cells. Target cells also contain an enzyme, 11-β hydroxysteroid dehydrogenase that controls the interconversion of cortisol (active) and cortisone (inert). The concentration of glucocorticoids receptors may fluctuate and thus influence responsiveness to glucocorticoids.
Maintenance of Homeostasis
Permissive and protective effects of glucocorticoids are critical for the maintenance of homeostasis during severe stress. The permissive and protective actions of glucocorticoids are complementary and permit the individual to affect an appropriate stress response and to maintain homeostasis.
Permissive Actions
Permissive actions of glucocorticoids occur at low physiologic steroid concentrations and serve to prepare the individual for responding to stress. These permissive actions of glucocorticoids maintain basal activity of the HPA by providing negative feedback and by setting the threshold for a response to stress.
Protective Actions
The protective mode of glucocorticoids occurs when high plasma concentrations of steroids exert antiinflammatory and immunosuppressive effects. This protective response prevents the host-defense mechanisms that are activated during stress from overshooting and damaging the organism. Other important protective actions of glucocorticoids include redirection of metabolism to meet energy needs during stress.
Pharmacokinetics
Synthetic cortisol and its derivatives are effective orally (see Table 40-1). Antacids, but not food, interfere with the oral absorption of corticosteroids. Water-soluble cortisol succinate can be administered intravenously (IV) to achieve prompt increases in plasma concentrations. More prolonged effects are possible with intramuscular (IM) injection. Cortisone acetate may be given orally or intramuscularly but cannot be administered IV. The acetate preparation is a slow-release preparation lasting 8 to 12 hours. After release, cortisone is converted to cortisol in the liver. Corticosteroids are also promptly absorbed after topical application or aerosol administration.
Cortisol is highly bound (90% or more) in the plasma to corticosteroid-binding globulin. Cortisol also binds albumin and erythrocytes.2 Nevertheless, cortisol and related compounds readily cross the placenta. Small amounts of cortisol appear unchanged in the urine, but at least 70% is conjugated in the liver to inactive or poorly active metabolites. These water-soluble conjugated metabolites appear in the urine and bile. The elimination half-time of cortisol is 1.5 to 3.0 hours but its biologic effects persist for several hours. The half-lives of synthetic glucocorticoids range from 1 hour (prednisolone) to more than 4 hours (dexamethasone) and clearance may be prolonged in older individuals.3 Individuals who clear glucocorticoids slowly may be subject to an increased incidence of side effects.4
Cortisol is released from the adrenal glands in an episodic manner and the frequency of pulses follows a circadian rhythm that is linked to the sleep-wake cycle. Maximal plasma concentrations of cortisol occur just before awakening and the lowest levels occur 8 to 10 hours later. Stress-induced changes in the plasma concentrations of cortisol are superimposed on the background baseline release of cortisol. Synthesis of cortisol is governed by adrenocorticotrophic hormone (ACTH) that is controlled by the hypothalamic hormones, corticotropin-releasing hormone and arginine vasopressin.
Synthetic Corticosteroids
Synthetic corticosteroids administered for their glucocorticoid effects include prednisolone, prednisone, methylprednisolone, betamethasone, dexamethasone, and triamcinolone (see Table 40-1, Fig. 40-2). Fludrocortisone is a synthetic halogenated derivative of cortisol that is administered for its mineralocorticoid effect (see Table 40-1 and Fig. 40-2). Naturally occurring corticosteroids, such as cortisol and cortisone, are also available as synthetic drugs (see Table 40-1 and Fig. 40-1).
Prednisolone
Prednisolone is an analogue of cortisol that is available as an oral or parenteral preparation. The antiinflammatory effect of 5 mg of prednisolone is equivalent to that of 20 mg of cortisol. This drug and prednisone are suitable for sole replacement therapy in adrenocortical insufficiency because of the presence of glucocorticoid and mineralocorticoid effects.
Prednisone
Prednisone is an analogue of cortisone that is available as an oral or parenteral preparation. It is rapidly converted to prednisolone after its absorption from the gastrointestinal tract. Its antiinflammatory effect and clinical uses are similar to those of prednisolone.
Methylprednisolone
Methylprednisolone is the methyl derivative of prednisolone. The antiinflammatory effect of 4 mg of methylprednisolone is equivalent to that of 20 mg of cortisol. The acetate preparation administered intraarticularly has a prolonged effect. Methylprednisolone succinate is highly soluble in water and is used IV to produce an intense glucocorticoid effect.
Betamethasone
Betamethasone is a fluorinated derivative of prednisolone. The antiinflammatory effect of 0.75 mg is equivalent to that of 20 mg of cortisol. Betamethasone lacks the mineralocorticoid properties of cortisol and thus is not acceptable for sole replacement therapy in adrenocortical insufficiency. Oral or parenteral administration is acceptable.
Dexamethasone
Dexamethasone is a fluorinated derivative of prednisolone and an isomer of betamethasone. The antiinflammatory effect of 0.75 mg is equivalent to that of 20 mg of cortisol. Oral and parenteral preparations are available. The acetate preparation is used as a long-acting repository suspension. Dexamethasone sodium phosphate is water soluble, rendering it appropriate for parenteral use. This corticosteroid is commonly chosen to treat certain types of cerebral edema.
Triamcinolone
Triamcinolone is a fluorinated derivative of prednisolone. The antiinflammatory effect of 4 mg is equivalent to that of 20 mg of cortisol. Triamcinolone has less mineralocorticoid effect than does prednisolone. Oral and parenteral preparations are available. The hexacetonide preparation injected intraarticularly may provide therapeutic effects for 3 months or longer. This drug is often used for epidural injections in the treatment of lumbar disc disease.
During the first days of treatment with triamcinolone, mild diuresis with sodium loss may occur. Conversely, edema may occur in patients with decreased glomerular filtration rates. Triamcinolone does not increase urinary potassium loss except when administered in large doses.
An unusual adverse side effect of triamcinolone is an increased incidence of skeletal muscle weakness. Likewise, anorexia rather than appetite stimulation, and sedation rather than euphoria may accompany administration of triamcinolone.
Clinical Uses
The only universally accepted clinical use of corticosteroids and their synthetic derivatives is as replacement therapy for deficiency states. With this exception, the use of corticosteroids in disease states is empirical and not curative, although antiinflammatory responses exert an intense palliative effect. The safety of corticosteroids is such that it is acceptable to administer a single large dose in a life-threatening situation on the presumption that unrecognized adrenal or pituitary insufficiency may be present.
Prednisolone or prednisone is recommended when an antiinflammatory effect is desired. The low mineralocorticoid potency of these drugs limits sodium and water retention when large doses are administered to produce the desired glucocorticoid effect. It must be recognized, however, that the antiinflammatory effect of corticosteroids is palliative because the underlying cause of the response remains. Nevertheless, suppression of the inflammatory response may be lifesaving in some situations. Conversely, masking of the symptoms of inflammation may delay diagnosis of life-threatening illness, such as peritonitis due to perforation of a peptic ulcer.
Deficiency States
Acute adrenal insufficiency requires electrolyte and fluid replacement as well as supplemental corticosteroids. Cortisol is administered at a rate of 100 mg IV every 8 hours after an initial injection of 100 mg. Management of chronic adrenal insufficiency in adults is with the daily oral administration of cortisone, 25.0 to 37.5 mg. A typical regimen is 25.0 mg in the morning and 12.5 mg in the late afternoon. This schedule mimics the normal diurnal cycle of adrenal secretion. An orally effective mineralocorticoid such as fludrocortisone, 0.1 to 0.3 mg daily, is required by most patients.
Allergic Therapy
Topical corticosteroids are capable of potent antiinflammatory effects and are the mainstay of allergic therapy. These medications interfere with the inflammatory response, induce cutaneous vasoconstriction, and have antimitotic activity.5 Corticosteroids work by inhibiting the production of inflammatory cytokines and chemokines, thus decreasing inflammation, cellular edema, and cellular recruitment to sites of disease. Oral administration of steroids is effective but the risk of unacceptable side effects with chronic treatment limits use by this route. Side effects, although possible with topical administration of corticosteroids, are usually not significant. Unlike antihistamines that provide pharmacologic effects within 1 to 2 hours, topical corticosteroids may require 3 to 5 days of treatment to produce a therapeutic effect.
Manifestations of allergic diseases that are of limited duration, such as hay fever, contact dermatitis, drug reactions, angioneurotic edema, and anaphylaxis, can be suppressed by adequate doses of corticosteroids. Life-threatening allergic reactions, however, must be treated with epinephrine, because the onset of the antiinflammatory effect produced by corticosteroids is delayed. Indeed, any beneficial effect of corticosteroids in the management of severe allergic reactions is probably related to suppression of the antiinflammatory response rather than to inhibition of production of immunoglobulins.
Asthma
Asthma is an inflammatory disease of the lungs and inhaled glucocorticoids (beclomethasone, budesonide, fluticasone, ciclesonide, and triamcinolone) are often recommended as first-line therapy for controlling the symptoms of asthma, improving quality of life and lung function, and in preventing exacerbations.6 Inhaled glucocorticoids are highly lipophilic and rapidly enter airway cells, where they have direct inhibitory effects on many of the cells involved in airway inflammation. One possible antiinflammatory mechanism is the modulation of the release of cytokines from inflammatory cells. It is estimated that 80% to 90% of the dose inhaled from the metered-dose inhaler is deposited in the oropharynx and swallowed. Inhaled glucocorticoids have oropharyngeal side effects that include dysphonia and candidiasis. Dysphonia occurs in approximately one-third of treated patients and may reflect myopathy of the laryngeal muscles that is reversible when treatment is stopped. Inhaled glucocorticoids, in doses of 1,500 µg per day or less in adults and 400 µg per day or less in children, have little, if any, effect on pituitary adrenal function.
Parenteral corticosteroids are important in the emergent preoperative preparation of patients with active reactive airway disease and in the treatment of intraoperative bronchospasm. Doses equivalent to 1 to 2 mg/kg of cortisol (or the equivalent dose of prednisolone) are commonly recommended. Preoperative corticosteroid administration 1 to 2 hours before induction of anesthesia is important because the beneficial effects of corticosteroids may not be fully manifest for several hours. Corticosteroids also enhance and prolong the responses to β-adrenergic agonists. Some enhancement of β-agonist effect may be present within 1 hour, but 4 to 6 hours are required for an antiinflammatory effect. In noncompliant or newly diagnosed patients with bronchial hyperactivity, preoperative treatment with combined corticosteroids (40 mg orally for 5 days) and salbutamol (0.2 mg puffs for 5 days) but not salbutamol alone minimizes intubation-evoked bronchoconstriction.7
Antiemetic Effect
Dexamethasone prevents postoperative nausea and vomiting only when administered near the beginning of surgery, probably by reducing surgery-induced inflammation due to inhibition of prostaglandin synthesis.8 In addition, dexamethasone may exert antiemetic effects by increasing the release of endorphins resulting in mood elevation and appetite stimulation. Prophylactic administration of dexamethasone 4 mg, ondansetron 4 mg, or droperidol 1.25 mg produced similar decreases (about 26%) in the incidence of postoperative nausea and vomiting.9 Because antiemetic interventions are similarly effective and act independently, it is recommended that the safest and least expensive antiemetic should be selected for prophylaxis. Prophylaxis is rarely warranted in low-risk patients, moderate-risk patients may benefit from a single intervention, and multiple interventions should be reserved for high-risk patients.9Rescue treatments are ineffective when the same drug has already been administered for prophylaxis. A suggested treatment strategy is to administer dexamethasone in conjunction with total intravenous anesthesia as first-line and second-line methods of prophylaxis against postoperative nausea and vomiting and to reserve serotonin antagonists as a rescue treatment.9 Administration of higher doses (8 to 10 mg) of dexamethasone has a similar clinical effect to lower doses (4 to 5 mg).10 Dexamethasone is also effective in suppressing chemotherapy-induced nausea and vomiting. The elimination half-time of dexamethasone is about 3 hours, but antiemetic effects, unlike other classes of antiemetics, often persist as long as 24 hours.
Postoperative Analgesia
Glucocorticoids peripherally inhibit phospholipase enzyme that is necessary for the inflammatory chain reaction along both the cyclooxygenase and lipoxygenase pathways.11 As a result, glucocorticoids may be effective in decreasing postoperative pain but with a different side effect profile than nonsteroidal antiinflammatory drugs. For example, administration of betamethasone 12 mg intramuscularly 30 minutes before induction of anesthesia for outpatient foot or hemorrhoid surgery, resulted in reductions in postoperative pain and the incidence of postoperative nausea and vomiting.12 A meta-analysis of perioperative intravenous dexamethasone suggests that dexamethasone at doses more than 0.1 mg/kg decreases acute postoperative pain and reduces opioid use, especially when administered preoperatively.13
Cerebral Edema
Corticosteroids in large doses are of value in the reduction or prevention of vasogenic cerebral edema and the resulting increases in intracranial pressure that may accompany intracranial tumors and metastatic lesions and bacterial meningitis.14 Dexamethasone, with minimal mineralocorticoid activity, is frequently selected to decrease cerebral edema and associated increases in intracranial pressure. Conversely, the administration of glucocorticoids to patients with severe head injury, cerebral infarction, and intracranial hemorrhage is not useful and can be associated with worse outcomes.15,16
Aspiration Pneumonitis
The use of corticosteroids in the treatment of aspiration pneumonitis is controversial. There is evidence in animals that corticosteroids administered immediately after the inhalation of acidic gastric fluid may be effective in decreasing pulmonary damage.17 Conversely, other data show no beneficial effect or suggest that the use of corticosteroids may enhance the likelihood of gram-negative pneumonia.18,19 Despite the absence of confirming evidence that corticosteroids are beneficial, it is not uncommon for the treatment of aspiration pneumonitis to include the empiric use of pharmacologic doses of these drugs.
Lumbar Disc Disease
An alternative to surgical treatment of lumbar disc disease is the epidural placement of corticosteroids.20 Corticosteroids may decrease inflammation and edema of the nerve root that has resulted from compression. A common regimen is epidural injection of 25 to 50 mg of triamcinolone, or 40 to 80 mg of methylprednisolone, in a solution containing lidocaine at or near the interspace corresponding to the distribution of pain. In animals, the epidural injection of triamcinolone, 2 mg/kg, interferes with the ability of the adrenal cortex to release cortisol in response to hypoglycemia for 4 weeks. Injection of triamcinolone, 80 mg, into the lumbar epidural space of patients with lumbar disc disease results in acute suppression of plasma concentrations of ACTH and cortisol between 15 minutes (midazolam sedation) and 45 minutes (midazolam not administered) of corticosteroid injection.21 Median suppression of the HPA axis was less than 1 month and all patients had recovered by 3 months. Exogenous corticosteroid coverage during this potentially vulnerable period should be considered in patients undergoing major stress, especially if the adrenocortical response to ACTH is subnormal. Although epidural injections of methylprednisolone may result in short-term improvement of symptoms (pain, sensory loss) due to sciatic nerve compression from a herniated nucleus pulposus, this treatment offers no significant functional benefit nor does it decrease the need for surgery.22
Immunosuppression
In organ transplantation, high doses of corticosteroids are often administered at the time of surgery to produce immunosuppression and decrease the risk of rejection of the newly transplanted organ. Smaller maintenance doses of corticosteroids are continued indefinitely, and the dosage is increased if rejection of the transplanted organ is threatened.
Arthritis
The criterion for initiating corticosteroid therapy in patients with rheumatoid arthritis is rapid control of symptomatic flares and progressive disability despite maximal medical therapy. Corticosteroids are administered in the smallest dose possible that provides significant but not complete symptomatic relief. The usual initial dose is prednisolone, 10 mg or its equivalent, in divided doses. Intraarticular injection of corticosteroids is recommended for treatment of episodic manifestations of acute joint inflammation associated with osteoarthritis. However, painless destruction of the joint is a risk of this treatment.
Collagen Diseases
Manifestations of collagen diseases, such as polymyositis, polyarteritis nodosa, and Wegener granulomatosis, but not scleroderma, are decreased and longevity is improved by corticosteroid therapy. Fulminating systemic lupus erythematosus is a life-threatening illness that is aggressively treated initially with large doses of prednisone, 1 mg/kg, or its equivalent. Large doses of corticosteroids are effective for inducing a remission of sarcoidosis. In temporal arteritis, corticosteroid therapy is necessary to prevent blindness, which occurs in about 20% of untreated patients. Some forms of nephrotic syndrome respond favorably to corticosteroids. Rheumatic carditis may be suppressed by large doses of corticosteroids.
Ocular Inflammation
Corticosteroids are used to suppress ocular inflammation (uveitis and iritis) and thus preserve sight. Instillation of corticosteroids into the conjunctival sac results in therapeutic concentrations in the aqueous humor. Topical and intraocular corticosteroid therapy often increases intraocular pressure and is associated with cataractogenesis. For this reason, it is recommended that intraocular pressure be monitored when topical corticosteroids are used for more than 2 weeks. Corticosteroids are not recommended in herpes simplex infections (dendritic keratitis) of the eye. Topical corticosteroids should not be used for treatment of ocular abrasions because delayed healing and infections may occur.
Cutaneous Disorders
Topical administration of corticosteroids is frequently effective in the treatment of skin diseases. Effectiveness is increased by application of the corticosteroid as an ointment under an occlusive dressing. Systemic absorption is also occasionally enhanced to the degree that suppression of the HPA axis occurs or manifestations of Cushing syndrome appear. Corticosteroids may also be administered systemically for treatment of severe episodes of acute skin disorders and exacerbations of chronic disorders.
Postintubation Laryngeal Edema
Treatment of postintubation laryngeal edema may include administration of corticosteroids, such as dexamethasone, 0.1 to 0.2 mg/kg IV. Nevertheless, the efficacy of corticosteroids for treatment of this condition has not been confirmed. Dexamethasone, 0.6 mg/kg orally is an effective treatment for children with mild croup.23
Ulcerative Colitis
Corticosteroid therapy is indicated in selected patients with chronic ulcerative colitis. A disadvantage of this therapy is that signs and symptoms of intestinal perforation and peritonitis may be masked.
Myasthenia Gravis
Corticosteroids are usually reserved for patients with myasthenia gravis who are unresponsive to medical or surgical therapy. These drugs seem to be most effective after thymectomy. The mechanism of beneficial effects produced by corticosteroids is not known but may reflect drug-induced suppression of the production of an immunoglobulin that normally binds to the neuromuscular junction.
Respiratory Distress Syndrome
Administration of corticosteroids at least 24 hours before delivery decreases the incidence and severity of respiratory distress syndrome in neonates born between 24 and 36 weeks’ gestation. Dexamethasone administered for prolonged periods (42 days) improves pulmonary and neurodevelopmental outcome of low-birth-weight infants at risk for bronchopulmonary dysplasia.24 Glucocorticoid administration in the setting of acute respiratory distress syndrome is controversial and the effect may vary according to timing. Early administration (≤72 hours) of methylprednisolone in one small study has been associated with improved outcomes.25 Later administration (>14 days) of glucocorticoids is associated with increased mortality, ventilator-free days, oxygenation, and compliance.26
Leukemia
The antilymphocytic effects of glucocorticoids are used to advantage in combination chemotherapy of acute lymphocytic leukemia and lymphomas, including Hodgkin disease and multiple myeloma. For example, prednisone and vincristine produce remissions in about 90% of children with lymphoblastic leukemia.
Cardiac Arrest
Cardiac arrest is associated with lower cortisol levels (relative adrenal insufficiency), vasoplegia, and myocardial dysfunction. Recent studies preliminarily suggest that the administration of glucocorticoids (along with vasopressin and epinephrine) during a cardiac arrest may improve survival and is associated with better neurologic outcomes.27 Potential explanations include attenuation of the systemic inflammatory response syndrome and enhancement of myocardial and vascular function.28
Side Effects
The side effects of chronic corticosteroid therapy include (a) suppression of the HPA axis, (b) electrolyte and metabolic changes, (c) osteoporosis, (d) peptic ulcer disease, (e) skeletal muscle myopathy, (f) central nervous system dysfunction, (g) peripheral blood changes, and (h) inhibition of normal growth. Increased susceptibility to bacterial or fungal infection accompanies treatment with corticosteroids. Corticosteroid administration is associated with greater clearance of salicylates and decreased effectiveness of anticoagulants. Systemic corticosteroids used for short periods of time (<7 days) even at high doses are unlikely to cause adverse side effects. Inhaled corticosteroids are unlikely to evoke adverse systemic effects.
Corticosteroid Supplementation in the Perioperative Period
Corticosteroid supplementation should be increased whenever the patient being treated for chronic hypoadrenocorticism undergoes a surgical procedure. This recommendation is based on the concern that these patients are susceptible to cardiovascular collapse because they cannot release additional endogenous cortisol in response to the stress of surgery. More controversial is the management of patients who may manifest suppression of the HPA axis because of current or previous administration of corticosteroids for treatment of a disease unrelated to pituitary or adrenal function. Recommendations that prescribe supraphysiologic doses have been advocated despite the absence of supporting scientific data.29 In adrenalectomized primates undergoing general anesthesia and surgery, the animals receiving physiologic replacement doses of cortisol were indistinguishable from those receiving supraphysiologic doses (10 times the normal production rate) of cortisol.30 Subphysiologically treated animals (one-tenth the normal production rate) were hemodynamically unstable during surgery and had a significantly higher mortality rate. Based on these animal data, it was concluded that there is no advantage in supraphysiologic glucocorticoid prophylaxis during surgical stress, and replacement doses of cortisol equivalent to the daily unstressed cortisol production rate are sufficient to allow homeostatic mechanisms to function during surgery.30
Patients taking greater than 20 mg per day of prednisone or its equivalent for more than 3 weeks have a suppressed HPA axis. Patients taking less than 5 mg per day of prednisone or its equivalent can be considered not to have suppression of their HPA axis. However, patients taking 5 to 20 mg per day of prednisone or its equivalent for more than 3 weeks may or may not have suppression of the HPA axis.
A rational regimen for corticosteroid supplementation in the perioperative period is to avoid steroid supplementation in patients who do not have a suppressed HPA axis (patients taking any dose of glucocorticoids for less than 3 weeks or a daily dose of prednisone <5 mg).
Patients taking 5 to 20 mg per day of prednisone or its equivalent for more than 3 weeks may or may not have suppression of the HPA axis. These patients may benefit from further assessment of their HPA axis. Glucocorticoid supplementation considers preoperative doses and the stress of surgery.
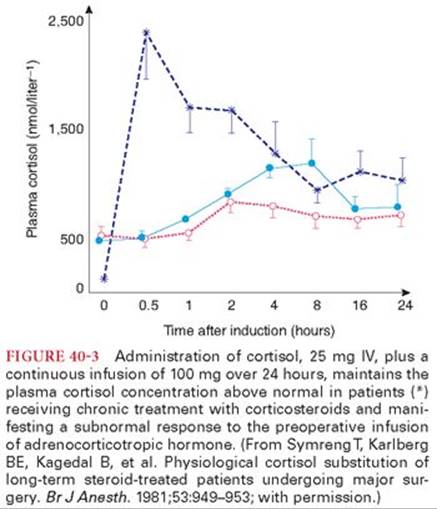
For patients with a suppressed HPA axis (patients taking >20 mg prednisone per day for more than 3 weeks), glucocorticoid supplementation should consider the stress of surgery. For minor surgical stress (inguinal hernia repair), the daily cortisol secretion rate and static plasma cortisol measurements suggest that a glucocorticoid replacement dose of 25 mg of hydrocortisone or 5 mg of methylprednisolone is sufficient. If the postoperative course is uncomplicated, the patient can be returned the next day to the prior glucocorticoid maintenance dose. For moderate surgical stress (nonlaparoscopic cholecystectomy, colon resection, total hip replacement), cortisol production rates suggest the glucocorticoid requirement is about 50 to 75 mg daily of hydrocortisone for 1 to 2 days. For major surgical stress (pancreatoduodenectomy, esophagectomy, cardiopulmonary bypass), the glucocorticoid dose should be 100 to 150 mg of hydrocortisone daily for 2 to 3 days. Even with this coverage, vascular collapse has been described in a patient experiencing massive hemorrhage during surgery.31 This approach maintains the plasma concentration of cortisol above normal during major surgery in patients receiving chronic treatment with corticosteroids and manifesting a subnormal response to the preoperative infusion of ACTH (Fig. 40-3).32 In those instances in which events such as burns or sepsis could exaggerate the need for exogenous corticosteroid supplementation, the continuous infusion of cortisol, 100 mg every 12 hours, should be sufficient. Indeed, endogenous cortisol production during stress introduced by major surgery or extensive burns is not greater than 150 mg daily.33,34 It is likely that patients undergoing minor operations will need minimal to no additional corticosteroid coverage during the perioperative period.
In addition to intravenous supplementation with cortisol, patients receiving daily maintenance doses of a corticosteroid should also receive this dose with the preoperative medication on the day of surgery. There is no objective evidence to support increasing the maintenance dose of corticosteroid preoperatively.
Electrolyte and Metabolic Changes and Weight Gain
Hypokalemic metabolic alkalosis reflects mineralocorticoid effects of corticosteroids on distal renal tubules, leading to enhanced absorption of sodium and loss of potassium. Edema and weight gain accompany this corticosteroid effect. Corticosteroids inhibit the use of glucose in peripheral tissues and promote hepatic gluconeogenesis. The resulting corticosteroid-induced hyperglycemia can usually be managed with diet, insulin, or both. The dose requirement for oral hypoglycemics may be increased by corticosteroids. There is a redistribution of body fat characterized by deposition of fat in the back of the neck (buffalo hump), supraclavicular area, and face (moon facies) and loss of fat from the extremities. The mechanism by which corticosteroids elicit this redistribution of fat is not known. Peripherally, corticosteroids mobilize amino acids from tissues. This catabolic effect manifests as decreased skeletal muscle mass, osteoporosis, thinning of the skin, and a negative nitrogen balance.
Osteoporosis
Osteoporosis, vertebral compression fractures, and rib fractures are common and serious complications of corticosteroid therapy that can be found in patients of all ages. Corticosteroids appear to inhibit the activities of osteoblasts and stimulate osteoclasts by inhibition of calcium absorption from the gastrointestinal tract, which causes an increased secretion of parathyroid hormone. Osteoporosis is an indication for withdrawal of corticosteroid therapy. Evidence of osteoporosis should be sought on radiographs of the spines of patients being treated chronically with corticosteroids. The presence of osteoporosis could predispose patients to fractures during positioning in the operating room. Bisphosphonates are effective in decreasing vertebral fractures in patients taking corticosteroids.35
Peptic Ulcer Disease
Although a cause-and-effect relationship has not been proved, the incidence of peptic ulcer disease seems to be increased by chronic corticosteroid therapy. Indeed, corticosteroids may decrease the normal protective barrier provided by gastric mucus.
Skeletal Muscle Myopathy
Skeletal muscle myopathy characterized by weakness of the proximal musculature is occasionally observed in patients taking large doses of corticosteroids. In some patients, this skeletal muscle weakness is so severe that ambulation is not possible and corticosteroid therapy must be discontinued.
Central Nervous System Dysfunction
Corticosteroid therapy is associated with an increased incidence of neuroses and psychoses. Behavioral changes include manic depression and suicidal tendencies. Cataracts develop in almost all patients who receive prednisone, 20 mg daily, or its equivalent for 4 years.
Peripheral Blood Changes
Corticosteroids tend to increase the hematocrit and number of circulating leukocytes. Conversely, a single dose of cortisol decreases by almost 70%—the number of circulating lymphocytes, and by more than 90%—the number of circulating monocytes in 4 to 6 hours. This acute lymphocytopenia most likely reflects sequestration from the blood rather than destruction of cells.
Inhibition of Normal Growth
Inhibition or arrest of growth can result from the administration of relatively small doses of glucocorticoids to children. The mechanism of this effect is presumed to be the generalized inhibitory effect of glucocorticoids on DNA synthesis and cell division.
Inhibitors of Corticosteroid Synthesis
Metyrapone
Metyrapone decreases cortisol synthesis by inhibition of the 11-β-hydroxylation reaction, resulting in accumulation of 11-deoxycortisol. Metyrapone may induce acute adrenal insufficiency in patients with decreased adrenocortical function. A deficiency of mineralocorticoids does not occur, because metyrapone-induced inhibition of 11-β-hydroxylation results in increased production of the mineralocorticoid 11-desoxycorticosterone.
Metyrapone has been used in the diagnosis of adrenal insufficiency and treatment of excessive adrenocortical function that results from adrenal neoplasms that function autonomously or as a result of ectopic production of ACTH by tumors.
Aminoglutethimide
Aminoglutethimide inhibits the conversion of cholesterol to 20-α-hydroxycholesterol, which interrupts production of both cortisol and aldosterone. Thus, this drug is effective in decreasing the excessive secretion of cortisol in autonomously functioning adrenal tumors and in hypersecretion resulting from ectopic production of ACTH.
Drugs that Regulate Calcium
Calcium is ingested and absorbed in the gastrointestinal tract; resorbed by bone; and filtered and reabsorbed by the kidney. The effects of parathyroid hormone, calcitonin, and vitamin D metabolites regulate calcium homeostasis (see Chapter 37). Parathyroid hormone (PTH) regulates extracellular calcium concentration through action on the bone, kidney, and intestine. PTH secretion is activated by hypocalcemia and elevated phosphorous levels. The net effect of PTH is to increase extracellular calcium. An excess or deficiency of calcium can disrupt coagulation, neurotransmitter and hormone secretion, neuromuscular excitability, muscle contraction, hormone action, and enzyme function.
Hypercalcemia
Hypercalcemia can be categorized as either parathyroid dependent or non–parathyroid dependent. Disorders of the parathyroid gland that result in hypercalcemia include primary and tertiary hyperparathyroidism, familial hypocalciuric hypercalcemia, and lithium-induced hypercalcemia. Hypercalcemia of malignancy is usually associated with destructive bone lesions or secretion of a PTH-like tumor peptide (PTH-RP). Hypercalcemia from parathyroid disease is associated with bone loss and osteoporosis. Management of hypercalcemia includes intravenous fluids, bisphosphonates, calcitonin (see Chapter 37), glucocorticoids and other less commonly used medications such as cinacalcet and denosumab.
Bisphosphonates
Bisphosphonates (pamidronate, zoledronate, alendronate, etc) are pyrophosphate analogues that lower calcium levels by inhibiting osteoclastic-mediated bone reabsorption. Hypercalcemia from malignancy, primary hyperparathyroidism, vitamin A intoxication, granulomatous disease, and Paget’s disease has been successfully managed with bisphosphonate therapy. Renal injury and jaw osteonecrosis has been reported in patients who take bisphosphonates. These medications should be prescribed early in the course of hypercalcemia because clinically significant reductions in calcium levels may not be observed for 2 days.
Glucocorticoids
In the setting of hypercalcemia from solid tumors and primary hyperparathyroidism, glucocorticoids are minimally effective agents. Glucocorticoids decrease synthesis of 1,25-dihydroxyvitamin D to decrease intestinal absorption of calcium and increase renal excretion of calcium.
Hypocalcemia
Preoperative patients with rhabdomyolysis, pancreatitis, sepsis, burns, fat embolism syndrome, recent massive transfusion, hypoalbuminemia, hypomagnesemia, or renal insufficiency are at risk for hypocalcemia. Chronic hypocalcemia may have few clinical signs or symptoms, whereas rapidly developing hypocalcemia may have impressive clinical effects. The most common setting for symptomatic hypocalcemia is within 12 to 24 hours after surgery, particularly after total or subtotal thyroidectomy or four-gland parathyroid exploration or removal.
Long-standing hypocalcemia with hyperphosphatemia and PTH deficiency is associated with calcification of the basal ganglia with extrapyramidal signs. Hypocalcemia can cause neuromuscular irritability, arrhythmias, congestive heart failure (decreased myocardial contractility), and hypotension. Acute, severe hypocalcemia (total serum calcium levels <7.5 mg/dL, normal albumin) is a medical emergency associated with death from laryngeal spasm or grand mal seizures. Intravenous calcium is indicated for acute symptomatic hypocalcemia. Ten percent calcium gluconate contains less elemental calcium than calcium chloride but is less likely to cause tissue necrosis during an extravasation.
Drugs for Pituitary Function
Anterior Pituitary Hormones
Anterior pituitary hormones include (a) growth hormone; (b) prolactin; (c) gonadotropins, including luteinizing hormone and follicle-stimulating hormone; (d) ACTH; and (e) thyroid-stimulating hormone (TSH). Growth hormone, gonadotropins, and ACTH can be administered in the form of synthetic drugs.
Perioperative replacement of anterior pituitary hormones may be necessary for patients receiving exogenous hormones because of a prior hypophysectomy. For example, cortisol must be provided continuously. Conversely, thyroid hormones have such a long elimination half-time that they can be omitted for several days without adverse effects. Likewise, the loss of other anterior pituitary hormones has no immediate physiologic implications.
Growth Hormone
Recombinant growth hormone is administered subcutaneously and daily to treat growth hormone deficiency. Growth hormone is also used to manage growth failure from chronic kidney disease and short stature from Turner syndrome, Prader-Willi syndrome, Noonan’s syndrome, and mutations in the Short Stature Homeobox gene. Radioimmunoassays for growth hormone are used to measure plasma concentrations of the hormone. Treatment is maintained and titrated for months to years in response to growth velocity and insulin-like growth factor 1 (IGF-1) levels (which are associated with growth velocity) and is often discontinued when linear growth decreases to less than 1 in per year.36,37
Octreotide
Octreotide is a somatostatin analogue that inhibits the release of growth hormone, making it an effective treatment for patients with acromegaly.38 Long-term treatment with octreotide (>1 month) is associated with an increased incidence of cholesterol gallstones (occurring in 20% to 30% of treated patients). Because somatostatin analogues inhibit the secretion of insulin, decreased glucose tolerance and even overt hyperglycemia might be expected during treatment with octreotide. Octreotide may be a lifesaving treatment in patients experiencing an acute carcinoid crisis although bolus injection of this somatostatin analog may be accompanied by bradycardia and second- and third-degree heart block.39
Gonadotropins
Gonadotropins are used most often for the treatment of infertility and cryptorchism. Induction of ovulation can be stimulated in females who are infertile because of pituitary insufficiency. Excessive ovarian enlargement and maturation of many follicles, leading to multiple births, is a possibility. Gonadotropins are effective only by parenteral injection. Radioimmunoassays are useful in measuring plasma and urine concentrations of gonadotropins.
Adrenocorticotrophic Hormone
The physiologic and pharmacologic effects of ACTH result from this hormone’s stimulation of secretion of corticosteroids from the adrenal cortex, principally cortisol. An important clinical use of ACTH is as a diagnostic aid in patients with suspected adrenal insufficiency. For example, a normal increase in the plasma concentration of cortisol in response to the administration of ACTH rules out primary adrenocortical insufficiency. Furthermore, ACTH may be administered therapeutically to evoke the release of cortisol. Treatment of disease states with ACTH is not physiologically equivalent to administration of a specific hormone because ACTH exposes the tissues to a mixture of glucocorticoids, mineralocorticoids, and androgens. Indeed, there may be associated retention of sodium, development of hypokalemic metabolic alkalosis, and appearance of acne, which are unlikely to accompany selective-acting corticosteroids.
Absorption of ACTH after IM injection is prompt. After intravenous injection, ACTH disappears rapidly from the plasma, with an elimination half-time of about 15 minutes. Allergic reactions ranging from mild fever to life-threatening anaphylaxis may be associated with administration of ACTH.
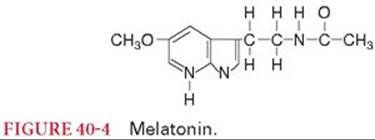
Melatonin
Melatonin (N-acetyl-5-methoxytryptamine) is the principal substance secreted by the pineal gland (Fig. 40-4).40 The mammalian pineal gland is a neuroendocrine transducer. Photic information from the retina is transmitted to the pineal gland through the suprachiasmatic nucleus of the hypothalamus and the sympathetic nervous system. The neural input to the gland is norepinephrine and the output is melatonin. The synthesis and release of melatonin are stimulated by darkness and inhibited by light. As the synthesis of melatonin increases, the hormone enters the bloodstream through passive diffusion. Melatonin is rapidly metabolized, chiefly in the liver, by hydroxylation to 6-hydroxymelatonin, and, after conjugation with sulfuric or glucuronic acid, is excreted in the urine. Intravenous melatonin is rapidly distributed, and the elimination half-time is 0.5 to 5.6 minutes. The bioavailability of orally administered melatonin varies widely.
Dose-dependent physiologic effects of melatonin include biologic regulation of circadian rhythms, sleep, mood, and perhaps reproduction, tumor growth, and aging.40 In humans, the circadian rhythm for the release of melatonin from the pineal gland is closely synchronized with the habitual hours of sleep. Alterations in synchronization due to phase shifts (acute change in time zones or working hours) are correlated with sleep disturbances. Ingestion of melatonin affects the speed of falling asleep as well as the duration and quality of sleep and has hypnotic effects. The circadian cycle of body temperature is linked to the 24-hour cycle of subjective sleepiness and inversely related to serum melatonin concentrations. Nevertheless, sleep-promoting doses of melatonin do not have any effect on body temperature. It is unclear whether the beneficial effect of exogenous melatonin on symptoms of jet lag is due to a hypnotic effect or resynchronization of the circadian rhythm.
Posterior Pituitary Hormones
Arginine vasopressin (AVP) (also known as antidiuretic hormone [ADH]) and oxytocin are the two principal hormones secreted by the posterior pituitary. AVP targets the renal collecting ducts to increase permeability of cell membranes to water and promote passive water reabsorption from renal collecting ducts into extracellular fluid. AVP also elicits intense arterial vasoconstriction via activation of vascular V1a receptors.41 Oxytocin elicits contractions of the uterus, which are indistinguishable from those that occur in spontaneous labor.
Arginine Vasopressin
Vasopressin is the exogenous preparation of AVP used for (a) treatment of AVP-sensitive diabetes insipidus, (b) management of refractory hypotension during anesthesia, (c) management of uncontrolled hemorrhage from esophageal varices, (d) hemodynamic stabilization in the presence of hemorrhagic and septic shock, and (e) management of refractory cardiac arrest. This drug is not effective in the management of patients with nephrogenic diabetes insipidus.
Diabetes Insipidus
Inadequate secretion of vasopressin by the posterior pituitary causes diabetes insipidus. Excessive water loss and hypernatremia via polyuria follow. Neurotrauma and surgery of the pituitary and hypothalamus, cerebral ischemia, or cerebral malignancy can cause diabetes insipidus.41 Nephrogenic diabetes insipidus resulting from an inability of the renal tubules to respond to adequate amounts of centrally produced AVP does not respond to exogenous administration of the hormone or its congeners.
Vasopressin administered IV is used for the initial evaluation of patients with suspected diabetes insipidus, which may follow head trauma or hypophysectomy. Under these circumstances, polyuria may be transient, and a longer antidiuretic effect (1 to 3 days) as produced by IM vasopressin tannate in oil could produce water intoxication. Oral administration of vasopressin is followed by rapid inactivation by trypsin, which cleaves a peptide linkage. Likewise, intravenous administration of vasopressin results in a brief effect because of rapid enzymatic breakdown of peptides in the tissues, especially the kidneys.
Administration of the synthetic selective V2 receptor agonist, desmopressin (DDAVP), treats central diabetes insipidus. DDAVP has an intense antidiuretic (V2) effect and decreased pressor (V1) effect. Through its V2 effects, DDAVP also causes endothelial cells to release von Willebrand factor, tissue-type plasminogen activator, and prostaglandins. The elimination half-time of DDAVP is 2.5 to 4.4 hours.42There are fewer side effects produced by DDAVP than are associated with vasopressin, although nausea and increases in systemic blood pressure can occur. DDAVP, which is not inactivated by trypsin can be administered orally (0.3 to 0.6 mg per day), IV (1 to 4 µg per day), or nasally (5 to 40 µg per day).41
Administered intranasally twice daily, using a calibrated catheter (Rhinyle), DDAVP is the drug of choice in the treatment of diabetes insipidus due to inadequate production of AVP by the posterior pituitary. DDAVP, like all the AVP analogues, is not effective in the treatment of nephrogenic diabetes insipidus. Increased release of von Willebrand factor accounts for the hemostatic activity of DDAVP in patients with uremia, chronic liver disease, and certain types of hemophilia by promoting platelet adhesiveness to the vascular endothelium. DDAVP has also been reported to minimize intraoperative blood loss in patients undergoing cardiac surgery with cardiopulmonary bypass, whereas other reports find no effect on blood loss in patients undergoing cardiac surgery or spinal fusion surgery.43–45 DDAVP does not decrease bleeding following cardiopulmonary bypass in patients who were maintained on aspirin therapy until the day before surgery.46 DDAVP administered IV may decrease systemic vascular resistance leading to hypotension.43
Lypressin is a synthetic analogue of AVP that produces antidiuresis for about 4 hours after intranasal administration. Its short duration of action limits its usefulness in the treatment of diabetes insipidus.
Hypotension During Anesthesia
Perioperative administration of angiotensin-converting enzyme inhibitors (ACE-I) or angiotensin II receptor blockers (ARB) inhibits the renin-angiotensin system and can cause refractory hypotension after administration of anesthesia.47 In these cases, catecholamine administration may be unsuccessful.48 The synthetic vasopressin analog, terlipressin (1 mg), has been used to manage refractory hypotension in patients who have taken ACE- I or ARB.49,50 Vasopressin may be effective to treat hypotension from anaphylaxis and from severe catecholamine deficiency after resection of a pheochromocytoma.51,52
Septic Shock
Excess generation of nitric oxide, activation of the renin-angiotensin system, and low plasma concentrations of vasopressin contribute to progressive loss of vascular tone during sepsis.53 Vasopressin levels are initially elevated with the onset of sepsis but decrease to normal levels after 24 hours producing a relative vasopressin deficiency.54 Vasopressin infusion (0.01 to 0.04 unit per minute) can reverse systemic hypotension and decrease norepinephrine dosages in catecholamine-resistant septic shock.55
Refractory Cardiac Arrest
Vasopressin is an alternative to epinephrine for vasopressor therapy during cardiopulmonary resuscitation. Vasopressin, 40 units IV, was similar to epinephrine 1 mg IV for management of ventricular fibrillation and pulseless electrical activity. Vasopressin was more effective than epinephrine for management of asystole and the treatment of refractory cardiac arrest.56 The American Heart Association recommends that vasopressin, at a one-time only dose of 40 units IV be considered instead of epinephrine 1 mg IV every 3 to 5 minutes for patients who are being treated for cardiac arrest. Vasopressin functions as a vasoconstrictor when it is administered in supraphysiologic doses which serve to displace peripheral blood volume to the central circulation without some of the adverse effects produced by epinephrine. This drug needs to only be administered once during cardiopulmonary resuscitation because of its 10- to 20-minute elimination half-time. Vasopressin administered during cardiac arrest and hemorrhagic shock may improve vital organ blood flow during cardiopulmonary resuscitation and stabilize cardiocirculatory function after successful resuscitation.57
Esophageal Varices
Vasopressin may serve as an adjunct in the control of bleeding esophageal varices and during abdominal surgery in patients with cirrhosis and portal hypertension. Infusion of 20 units over 5 minutes results in marked decreases in hepatic blood flow lasting about 30 minutes. Only a moderate increase in systemic blood pressure occurs. This effect on the portal circulation is attributable to marked splanchnic vasoconstriction. An alternative to systemic administration is the infusion of vasopressin directly into the superior mesenteric artery. It has not been established whether selective arterial administration is safer than systemic administration with respect to cardiac and vascular side effects.
Side Effects
Vasoconstriction and increased systemic blood pressure occur only with doses of vasopressin that are much larger than those administered for the treatment of diabetes insipidus. This response is because of a direct and generalized effect on vascular smooth muscles that is not antagonized by denervation or adrenergic-blocking drugs. Facial pallor due to cutaneous vasoconstriction may also accompany large doses of vasopressin. The magnitude of increase in systemic blood pressure caused by vasopressin depends, to some extent, on the reactivity of the baroreceptor reflexes. For example, when baroreceptor reflexes are depressed by anesthesia, smaller amounts of vasopressin are capable of evoking a pressor response. Pulmonary artery pressures are also increased by vasopressin. Higher doses of vasopressin may cause splanchnic, digit, and coronary ischemia.58
Vasopressin, even in small doses, may produce selective vasoconstriction of the coronary arteries, with decreases in coronary blood flow manifesting as angina pectoris, electrocardiographic evidence of myocardial ischemia, and, in some instances, myocardial infarction. Ventricular cardiac dysrhythmias may accompany these cardiac effects.
Large doses of vasopressin stimulate gastrointestinal smooth muscle, and the resulting increased peristalsis may manifest as abdominal pain, nausea, and vomiting. Smooth muscle of the uterus is also stimulated by large doses of vasopressin.
A decrease in platelet count has been attributed to AVP-mediated platelet aggregation via V1 receptors.55 AVP infusion in advanced vasodilatory shock does not increase plasma concentrations of factor VIII, von Willebrand factor antigen, and ristocetin cofactor. Allergic reactions ranging from urticaria to anaphylaxis may occasionally follow the administration of vasopressin. Prolonged use of vasopressin may result in antibody formation and a shortened duration of action of the drug.
Oxytocin
Oxytocin stimulates uterine muscle and is administered to induce labor at term, reduce and prevent uterine atony, and decrease hemorrhage in the postpartum or postabortion period.59 By stimulating smooth muscle uterine contraction, blood loss at the site of placental attachment is reduced. All preparations of oxytocin used clinically are synthetic, and their potency is described in units. These synthetic preparations are identical to the hormone normally released from the posterior pituitary but devoid of contamination by other polypeptide hormones and proteins found in natural proteins.
For induction of labor, a continuous infusion is preferred, because the low dose of oxytocin needed can be precisely controlled. Indeed, the sensitivity of the uterus to oxytocin increases as pregnancy progresses. To induce labor, a dilute solution (10 mU/mL) is administered by a constant infusion pump beginning at 1 to 2 mU per minute. This infusion rate is increased 1 to 2 mU per minute every 15 to 30 minutes until an optimal response (uterine contraction every 2 to 3 minutes) is obtained. The average dose of oxytocin to induce labor is 8 to 10 mU per minute. Infusion rates up to 40 mU per minute of oxytocin may be necessary to treat uterine atony initially after delivery. IM injections of oxytocin are commonly used to provide sustained uterine contractions in the postpartum period.
To prevent uterine atony, slow administration (to reduce adverse side effects) of 1 to 3 International Unit of oxytocin over 30 seconds is recommended. Coadministration with phenylephrine may be required if higher doses are used. To manage uterine atony and postpartum hemorrhage, 3 to 5 International Unit of intravenous oxytocin over 30 seconds is recommended.60 Prior oxytocin exposure promotes oxytocin receptor downregulation and desensitization and may be a risk factor for postpartum hemorrhage from uterine atony.61,62
Side Effects
High and bolus doses of oxytocin are more likely to decrease systolic and diastolic blood pressure via a direct relaxant effect on vascular smooth muscles.63 Reflex tachycardia and increased cardiac output accompany the transient decrease in systemic blood pressure.64 The amounts of oxytocin administered for most obstetric purposes are inadequate to produce marked alterations in systemic blood pressure. A marked decrease in blood pressure, however, may occur if oxytocin is administered to patients with blunted compensatory reflex responses, as may be produced by anesthesia. Likewise, hypovolemic patients may be particularly susceptible to oxytocin-induced hypotension. The hemodynamic effects of a second dose of oxytocin are diminished compared to the initial dose.65
In the past, oxytocin preparations were often contaminated with ergot alkaloids, resulting in exaggerated systemic blood pressure increases when administered to patients previously treated with a sympathomimetic. Modern synthetic commercial preparations are pure oxytocin and do not introduce the risk of exaggerated vasoconstriction when administered in the presence of a sympathomimetic drug.
Oxytocin exhibits a slight AVP-like activity when administered in high doses, introducing the possibility of water intoxication, hyponatremia, and neurologic dysfunction if an excessive volume of fluid is administered.66 The risk of this complication can be minimized by infusion of oxytocin in an electrolyte-containing solution rather than glucose in water.
Drugs for Reproductive Regulation
Ovarian Hormones
An understanding of the synthesis and action of ovarian hormones, including estrogens and progesterone, permits therapeutic interventions in certain disease states. Equally important is the therapeutic use of drugs that can mimic effects of these hormones and act as contraceptives.
Estrogens
Estrogens are effective in treating unpleasant side effects of menopause (Fig. 40-5). Hormone replacement therapy may reduce the depressive symptoms during menopause.67 Senile or atrophic vaginitis responds to topical estrogen. There is no evidence that administration of estrogens delays the progression of atherosclerosis in postmenopausal women. There is abundant evidence that administration of estrogen to postmenopausal women prevents bone loss (protects against osteoporosis) and also prevents vertebral and femoral bone fractures.68 Estrogens are administered to decrease milk production in the postpartum period. The presence of receptors for estrogen increases the likelihood of a palliative response to estrogen therapy in women with metastatic breast cancer. An important use of estrogens is in combination with progestins as oral contraceptives.
Route of Administration
The absorption of most estrogens and their derivatives from the gastrointestinal tract is prompt and nearly complete. Metabolism in the liver, however, limits the effectiveness of orally administered estrogens. Topical and IM administration of estrogens is also effective. Radioimmunoassay methods are highly specific and sensitive for measuring the plasma concentrations of estrogens.
Side Effects
The most frequent unpleasant symptom associated with the use of estrogens is nausea. Large doses of estrogens may cause retention of sodium and water, which is particularly undesirable in patients with cardiac or renal disease. There is an increased incidence of vaginal and cervical adenocarcinoma in daughters of mothers treated with diethylstilbestrol or other synthetic estrogens during the first trimester of pregnancy. Most of the affected women have been 20 to 25 years old when diagnosed. Use of estrogen by postmenopausal women increases the risk of developing endometrial cancer.
Antiestrogens
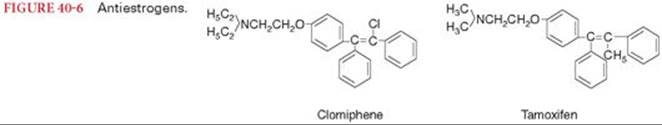
Clomiphene and tamoxifen act as antiestrogens by binding to estrogen receptors (Fig. 40-6). Tamoxifen is administered for a period of 5 years to postmenopausal women with breast cancer that was characterized by estrogen-responsive receptors. It is of interest that tamoxifen has estrogenic activity in some tissues, including bone. The loss of normal feedback inhibition of estrogen synthesis causes an increased secretion of gonadotropins. The most prominent effect on increased plasma concentrations of gonadotropins is the enlargement of the ovaries and enhancement of fertility in otherwise infertile women. Endometrial stimulation and an increased incidence of temperature disturbances (“hot flashes”) may accompany treatment with tamoxifen.
Tissue-Specific Estrogens
Raloxifene is a nonsteroidal benzothiophene that acts as a selective estrogen-receptor modulator.69 In this regard, raloxifene preserves the beneficial effects of estrogens (prevention of bone loss and lowering of plasma cholesterol concentrations) without any associated effects on reproductive organs. For example, endometrial stimulation does not accompany treatment with raloxifene. Tissue-specific estrogen agonist or antagonist actions of raloxifene may be related to estrogen receptor-mediated gene activation.
Progesterone
Orally active derivatives of progesterone are designated progestins (Fig. 40-7). Progestins are often combined with estrogens as oral contraceptives. Dysfunctional uterine bleeding can be treated with small doses of a progestin for a few days, with the goal being induction of progesterone-withdrawal bleeding. Progestins, like estrogens, are effective in suppressing lactation in the immediate postpartum period. Palliative treatment of metastatic endometrial carcinoma is achieved with progestins. Absorption of progestins from the gastrointestinal tract is rapid, but hepatic first-pass metabolism is extensive.
Antiprogestins
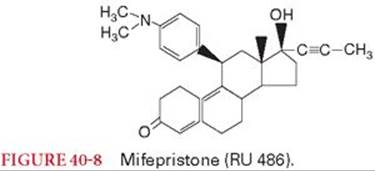
Antiprogestins inhibit the hormonal effects of progesterone and are the most effective and safest means of medical abortion.70 In this regard, mifepristone (RU 486) can be administered in a single oral dose to produce termination of pregnancy (Fig. 40-8). The combination of mifepristone with a prostaglandin administered 48 hours later by IM injection (sulprostone), by vaginal suppository (gemeprost), or orally (misoprostol) has resulted in a rate of complete abortion approaching 100%. Mifepristone has been used as a postcoital contraceptive within 72 hours of unprotected intercourse. In addition to its antiprogesterone properties, mifepristone has antiglucocorticoid activity and is useful in the treatment of patients with hypercortisolism. Side effects of mifepristone include vaginal bleeding, nausea, vomiting, abdominal pain, and fatigue.
Oral Contraceptives
Oral contraceptives are most often a combination of an estrogen and a progestin. This combination inhibits ovulation, presumably by preventing release of follicle-stimulating hormone by estrogen and luteinizing hormone by progesterone.
Side Effects
Estrogens in combined preparations are believed to be responsible for most, if not all of the side effects of oral contraceptives. For example, estrogens seem to be responsible for the increased incidence of thromboembolism. Indeed, patients taking estrogens manifest increased blood concentrations of some clotting factors as well as increased platelet aggregation. Nausea, vomiting, weight gain, and breast discomfort resembling early pregnancy are attributed to the estrogen component of oral contraceptives. The incidence of myocardial infarction and stroke is increased in patients who chronically take oral contraceptives.71 Hypertension occurs in about 5% of women taking oral contraceptives chronically.72 This response probably reflects estrogen-induced increases in circulating plasma concentrations of renin and angiotensin, with associated retention of sodium and water.
Oral contraceptives containing high doses of estrogen may produce alterations in the glucose tolerance curves of patients with preclinical diabetes mellitus. These drugs increase the concentration of cholesterol in bile, which is consistent with an increased incidence of cholelithiasis. Benign hepatomas have been associated with the use of oral contraceptives. An increased incidence of breast cancer in patients taking oral contraceptives has not been documented. Depression of mood and fatigue have been attributed to the progestin component of oral contraceptives.
Androgens
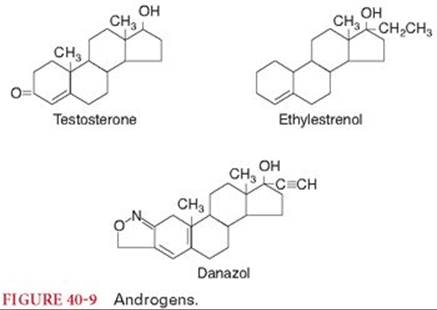
Androgens are administered to males to stimulate the development and maintenance of secondary sexual characteristics (Fig. 40-9). Testosterone is also prescribed to hypogonadal men who have evidence of androgen deficiency and a low serum testosterone concentration. The most common indication of androgen therapy in females is palliative management of metastatic breast cancer. Androgens enhance erythropoiesis by stimulation of renal production of erythropoietin as well as by direct dose-related stimulation of erythropoietin-sensitive elements in bone marrow. In addition, there is a drug-induced increase in 2,3-diphosphoglycerate levels, which decreases hemoglobin affinity for oxygen, thus enhancing the availability of oxygen to tissues. For these reasons, androgen therapy is often instituted in patients with aplastic anemia or hemolytic anemia. Androgen-anabolic steroids have been used in the treatment of chronic debilitating diseases. These drugs promote a feeling of well-being and may improve appetite when administered to patients with terminal illnesses. The efficacy of anabolic steroids to improve athletic performance is not documented and is condemned on ethical grounds. Certain androgens may be useful in the treatment of hereditary angioedema.
Route of Administration
About 99% of testosterone circulating in the plasma is bound to sex hormone–binding globulin. As a result, this globulin determines the concentration of free testosterone in the plasma and thus its elimination half-time, which is 10 to 20 minutes. Testosterone administered orally is readily absorbed but is metabolized so extensively by the liver that therapeutic effects do not occur. Alkylation of androgens at the 17 position retards their hepatic metabolism and permits such derivatives to be effective (see Fig. 40-9). Alkylated testosterones are rarely prescribed because of their association with hepatic dysfunction. Intramuscular injection of esters of testosterone (i.e., testosterone enanthate and testosterone cypionate), which are more lipophilic than testosterone alone, prolongs the duration of time that testosterone is present in the blood. Testosterone can also be delivered via patch and gels.
Side Effects
Dose-related cholestatic hepatitis and jaundice are particularly likely to accompany androgen therapy for palliation in neoplastic disease. Increases in the plasma alkaline phosphatase, hematocrit, and transaminase enzymes are also likely. Prolonged therapy (>1 year) with androgens, as for management of anemia, is associated with an increased incidence of hepatic cancer. Retention of sodium and water is also likely to accompany palliative treatment of cancer with high doses of androgens. Androgens increase the potency of coumarin anticoagulants and the likelihood of spontaneous hemorrhage. Androgens can decrease the concentration of thyroid-binding globulin in plasma and thus influence thyroid function tests.
Danazol
The low androgenic activity of danazol makes it the preferred androgen for treatment of hereditary angioedema (see Fig. 40-9). In treated patients, there is a remission of symptoms as well as increased production of previously deficient plasma protein factors. As with other androgens, danazol therapy has been associated with abnormal liver function tests and jaundice. Danazol also decreases breast pain and nodularity in many women with fibrocystic breast disease. Symptoms of endometriosis are decreased, and fertility may be restored in danazol-treated women. In patients with hemophilia A, danazol increases factor VIII activity and decreases the incidence of hemorrhage.73
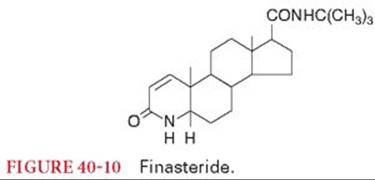
Finasteride
Finasteride is a competitive 5-α-reductase inhibitor that does not bind to the androgen receptor (Fig. 40-10).74 As a result of this drug-induced enzyme inhibition, dihydrotestosterone production from testosterone does not occur. In the absence of dihydrotestosterone, the androgen effects on the prostate and skin do not occur. Finasteride is administered orally (5 mg once daily) for the treatment of benign prostatic hyperplasia. Treatment of male pattern baldness, hirsutism, and acne may represent other potentially useful applications for finasteride. There is no evidence that finasteride is beneficial in men with established prostate cancer. The elimination half-time after oral administration is 6 to 8 hours. The only important side effects of finasteride are related to decreased sexual function. Finasteride has no effect on serum lipids or bone density. Prostate-specific antigen concentrations are decreased by treatment with finasteride, introducing the concern that detection of prostate cancer could be masked in patients treated with this drug.
References
1. Tobler A, Meier R, Seitz M, et al. Glucocorticoids downregulate gene expression of GM-CSF, NAP-1/IL-8, and IL-6, but not of M-CSF in human fibroblasts. Blood. 1992;79:45–51.
2. Migeon CJ, Lawrence B, Bertrand J, et al. In vivo distribution of some 17-hydroxycorticoids between the plasma and red blood cells of man. J Clin Endocrinol Metab. 1959;19:1411–1419.
3. Tornatore KM, Logue G, Venuto RC, e t al. Cortisol pharmacodynamics after methylprednisolone administration in young and elderly males. J Clin Pharmacol. 1997;37:304–311.
4. Kozower M, Veatch L, Kaplan MM. Decreased clearance of prednisolone, a factor in the development of corticosteroid side effects. J Clin Endocrinol Metab. 1974;38:407–412.
5. Cornell RC, Stoughton RB. Correlation of the vasoconstriction assay and clinical activity in psoriasis. Arch Dermatol. 1985;121:63–67.
6. Bel EH. Mild asthma. New Engl J Med. 2013;369:549–557.
7. Silvanus M-T, Groeben H, Peters J. Corticosteroids and inhaled salbutamol in patients with reversible airway obstruction markedly decrease the incidence of bronchoconstriction after tracheal intubation. Anesthesiology. 2004;100:1052–1057.
8. Wang JJ, Ho ST, Tzeng JI, et al. The effect of timing of dexamethasone administration on its efficacy as a prophylactic antiemetic for postoperative nausea and vomiting. Anesth Analg. 2000;91:136–139.
9. Apfel CC, Korttila K, Abdalla M, et al. A factorial trial of six interventions for the prevention of postoperative nausea and vomiting. N Engl J Med. 2004;350:2441–2451.
10. De Oliveira GS, Santana Castro-Alves LJ, Ahmad S, et al. Dexamethasone to prevent postoperative nausea and vomiting: an updated meta-analysis of randomized controlled trials. Anesth Analg. 2013;116:58–74.
11. Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative pain. Anesth Analg. 2005;21:55–89.
12. Aasboe V, Raeder JC. Groegaard B. Betamethasone reduces postoperative pain and nausea after ambulatory surgery. Anesth Analg. 1998;87:319–323.
13. De Oliveira GS, Almeida M, Benzon H, et al. Perioperative single dose systemic dexamethasone for postoperative pain: a meta-analysis of randomized controlled trials. Anesthesiology. 2011;115:575–588.
14. De Gans J, van de Beek D; European Dexamethasone in Adulthood Bacterial Meningitis Study Investigators. Dexamethasone in adults with bacterial meningitis. N Engl J Med. 2002;347:1549–1556.
15. Roberts I, Yates D, Sandercock P, et al. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): a randomised placebo-controlled trial. Lancet. 2004;364:1321–1328.
16. Edwards P, Arango M, Balica L, et al. Final results of MRC CRASH, a randomized placebo-controlled trial of intravenous corticosteroid in adults with head-injury-outcomes at 6 months. Lancet. 2005;365:1957–1959.
17. Dudley WR, Marshall BE. Steroid treatment for acid-aspiration pneumonia. Anesthesiology. 1974;40:136–141.
18. Downs JB, Chapman RL, Modell JH, et al. An evaluation of steroid therapy in aspiration pneumonitis. Anesthesiology. 1974;40:129–135.
19. Wynne JW, DeMarco FJ, Hood CI. Physiological effects of corticosteroids in foodstuff aspiration. Arch Surg. 1981;116:46–49.
20. Haddox JD. Lumbar and cervical epidural steroid therapy. Anesth Clin North Am. 1992;10:179–203.
21. Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg. 1994;79: 501–505.
22. Carette S, Leclaire R, Marcoux S, et al. Epidural corticosteroid injections for sciatica due to herniated nucleus pulposus. N Engl J Med. 1997;336:1634–1640.
23. Bjornson CL, Klassen TP, Williamson J, et al. A randomized trial of a single dose of oral dexamethasone for mild croup. N Engl J Med. 2004;351:1306–1313.
24. Cummings JJ, D’Eugenio DB, Gross SJ. A controlled trial of dexamethasone in preterm infants at high risk for bronchopulmonary dysplasia. N Engl J Med. 1989;320:1505–1510.
25. Meduri GU, Golden E, Freire AX et al. Methyprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest. 2007;131:954–963.
26. Steinberg JP, Hudson LD, Goodman RB, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. N Engl J Med. 2006;354:1671–1684.
27. Mentzelopoulos SD, Malachias S, Chamos C, et al. Vasopressin, steroids, and epinephrine and neurologically favorable survival after in-hospital cardiac arrest. JAMA. 2013;310:270–279.
28. Mentzelopoulos SD, Zakynthinos SG, Tzoufi M, et al. Vasopressin, epinephrine, and corticosteroids for in-hospital cardiac arrest. Arch Intern Med. 2009;169:15–24.
29. Salem M, Tainsh RE Jr, Bromberg J, et al. Perioperative glucocorticoid coverage. A reassessment 42 years after emergence of a problem. Ann Surg. 1994;219:416–425.
30. Udelsman R, Ramp J, Gallucci WT, et al. Adaptation during surgical stress: a reevaluation of the role of glucocorticoids. J Clin Invest. 1986;77:1377–1381.
31. Ratner EF, Allen R, Mihm F, et al. Failure of steroid supplementation to prevent operative hypotension in a patient receiving chronic steroid therapy. Anesth Analg. 1996;82:1294–1296.
32. Symreng T, Karlberg BE, Kagedal B, et al. Physiological cortisol substitution of long-term steroid-treated patients undergoing major surgery. Br J Anaesth. 1981;53:949–953.
33. Hardy JD, Turner MD. Hydrocortisone secretion in man: studies of adrenal vein blood. Surgery. 1957;42:194–201.
34. Hume DM, Bell C, Bartter FC. Direct measurement of adrenal secretion during operative trauma and convalescence. Surgery. 1962;52: 174–187.
35. O’Dell JR. Therapeutic strategies for rheumatoid arthritis. N Engl J Med. 2004;350:2591–2602.
36. Silvers JB, Marinova D, Mercer MB, et al. A national study of physician recommendations to initiate and discontinue growth hormone for short stature. Pediatrics. 2010;126:468–476.
37. Cohen P, Germak J, Rogol AD, et al. Variable degree of growth hormone (GH) and insulin-like growth factor (IGF) sensitivity in children with idiopathic short stature compared with GH-deficient patients: evidence from an IGF-based dosing study of short children. J Clin Endocrinol Metab. 2010;95:2089–2098.
38. Lamberts SWJ, van der Lely AJ, de Herder WW, et al. Octreotide. N Engl J Med. 1996;334:246–254.
39. Dilger JA, Rho EH, Que FG, et al. Octreotide-induced bradycardia and heart block during surgical resection of a carcinoid tumor. Anesth Analg. 2004;98:318–320.
40. Brzezinski A. Melatonin in humans. N Engl J Med. 1997;336:186–195.
41. Treschan T, Jurgen P. The vasopressin system: physiology and clinical strategies. Anesthesiology. 2006;105:599–612.
42. Horrow JC. Desmopressin and antifibrinolytics. Int Anesthesiol Clin. 1990;28:230–236.
43. Frankville DD, Harper GB, Lake CL, et al. Hemodynamic consequences of desmopressin administration after cardiopulmonary bypass. Anesthesiology. 1991;74:988–996.
44. Guay J, Reinberg C, Poitras B, et al. A trial of desmopressin to reduce blood loss in patients undergoing spinal fusion for idiopathic scoliosis. Anesth Analg. 1992;75:405–410.
45. Mongan PD, Hosking MP. The role of desmopressin acetate in patients undergoing coronary artery bypass surgery: a controlled clinical trial with thromboelastographic risk stratification. Anesthesiology. 1992;77:38–46.
46. Pleym H, Stenseth R, Waqhba A, et al. Prophylactic treatment with desmopressin does not reduce postoperative bleeding after coronary surgery in patients treated with aspirin before surgery. Anesth Analg 2004;98:578–584.
47. Bertrand M, Godet G, Meersschaert K, et al. Should the angiotensin II antagonists be discontinued before surgery. Anesth Analg. 2001;92:26–30.
48. Brabant SM, Eyraud D, Bertrand M, et al. Refractory hypotension after induction of anesthesia in a patient chronically treated with angiotensin receptor antagonists. Anesth Analg. 1999;89:887–888.
49. Boccara G, Outtara A, Godet G, et al. Terlipressin versus norepinephrine to correct refractory arterial hypotension after general anesthesia in patients chronically treated with renin-angiotensin system inhibitors. Anesthesiology. 2003;98:1338–1344.
50. Eyraud D, Brabant S, Nathalie D, et al. Treatment of intraoperative refractory hypotension with terlipressin in patients chronically treated with an antagonist of the renin-angiotensin system. Anesth Analg. 1999;88:980–984.
51. Augoustides JG, Abrams M, Berkowitz D, et al. Vasopressin for hemodynamic rescue in catecholamine-resistant vasoplegic shock after resection of massive pheochromocytoma. Anesthesiology. 2004;101:1022–1024.
52. Schummer C, Wirsing M, Schummer W. The pivotal role of vasopressin in refractory anaphylactic shock. Anesth Analg. 2008;107:620–624.
53. Landry DW, Levin HR, Gallant EM, et al. Vasopressin deficiency contributes to the vasodilation of septic shock. Circulation. 1997; 95:1122–1125.
54. Sharshar T, Blanchard A, Paillard M, et al. Circulating vasopressin levels in septic shock. Crit Care Med. 2003;31:1752–1758
55. Dünser MW, Mayr AJ, Ulmer H, et al. Arginine vasopressin in advanced vasodilatory shock: a prospective, randomized, controlled study. Circulation. 2003;107:2313–2319.
56. Wenzel V, Krismer AC, Arntz HR, et al. A comparison of vasopressin and epinephrine for out-of-hospital cardiopulmonary resuscitation. N Engl J Med. 2004;350:105–113.
57. Voelckel WG, Lurie KG, Lindner KH, et al. Vasopressin improves survival after cardiac arrest in hypovolemic shock. Anesth Analg. 2000;91:627–634.
58. Dünser MW, Mayr AJ, Tür A, et al. Ischemic skin lesions as a complication of continuous vasopressin infusion in catecholamine-resistant vasodilatory shock: incidence and risk factors. Crit Care Med. 2003;31:1394–1398.
59. Winkioff B, Dabash R, Durocher J, et al. Treatment of postpartum haemorrhage with sublingual misoprostol versus oxytocin in women not exposed to oxytocin during labour: a double-blind, randomized, non-inferiority trial. Lancet. 2010;375:210–216.
60. Dyer RA, Butwick AJ, Carvalho B. Oxytocin for labour and caesarean delivery: implications for the anaesthesiologist. Curr Opin Anesthesiol. 2011;24:255–261.
61. Phaeneuf S, Rodriguez Linares B, TambyRaja RL, et al. Loss of myometrial oxytocin receptors during oxytocin-induced and oxytocin-augmented labour. J Reprod Fertil. 2000;120:91–97.
62. Grotegut CA, Paglia MJ, Johnson LN, et al. Oxytocin exposure during labor among women with postpartum hemorrhage secondary to uterine atony. Am J Obstet Gynecol. 2011;204:56.e1–56.e6.
63. Thomas JS, Koh SH, Cooper GM. Haemodynamic effects of oxytocin given as i.v. bolus or infusion on women undergoing Caesarean section. Br J Anaesth. 2007;98:116–119.
64. Dyer R, Reed A, van Dyk D, et al. Hemodynamic effects of ephedrine, phenylephrine, and the coadministration of phenylephrine with oxytocin during spinal anesthesia for elective cesarean delivery. Anesthesiology. 2009;111:753–765.
65. Langesaeter E, Rosseland LA, Stubhaug A. Haemodynamic effects of repeated doses of oxytocin during Caesarean delivery in healthy parturients. Br J Anaesth. 2009;103:260–262.
66. Bergum D, Lonne H, Hakli TF. Oxytocin infusion: acute hyponatremia, seizures, and coma. Acta Anaesthesiol Scand. 2009;53:826–827.
67. Zweifel JE, O’Brien WH. A meta-analysis of the effect of hormone replacement therapy upon depressed mood. Psychoneuroendrocrinology. 1997;22:189–212.
68. Belchetz PE. Hormonal treatment of postmenopausal women. N Engl J Med. 1994;330:1062–1072.
69. Delmas PD, Bjarnason NH, Mitlak BH, et al. Effects of raloxifene on bone mineral density, serum cholesterol concentrations, and uterine endometrium in postmenopausal women. N Engl J Med. 1997;337:1641–1647.
70. Spitz IM, Bardin CW. Mifepristone (RU 486)—a modulator of progestin and glucocorticoid action. N Engl J Med. 1993;329:404–412.
71. Kaplan NM. Cardiovascular complications of oral contraceptives. Ann Rev Med. 1978;29:31–40.
72. Laragh JH. Oral contraceptive-induced hypertension: nine years later. Am J Obstet Gynecol. 1976;126:141–147.
73. Gralnick HR, Maisonneuve P, Sultan Y, et al. Benefits of danazol treatment in patients with hemophilia A (classic hemophilia). JAMA. 1985;253:1151–1153.
74. Rittmaster RS. Finasteride. N Engl J Med. 1994;330:120–126.