Chemotherapy is a term that was coined to refer to a broad range of chemicals (drugs) aimed at treating cancer by eradicating malignant cells anywhere in the body.1 Conventional wisdom is that the effectiveness of chemotherapy requires that there be complete destruction (total cell kill) of all cancer cells because a single surviving cell with the ability to divide can give rise to sufficient progeny to ultimately kill the host. The role of the immune system in identifying and eliminating foreign tumor cells has gained increasing recognition, and harnessing our intrinsic immune surveillance system has become more and more a part of contemporary investigations of cancer and its treatment.2 The use of several chemotherapeutic drugs (also called antineoplastic drugs) concurrently or in a planned sequence is commonly done in efforts to eradicate even small residual tumor cell populations that have survived treatment with a single or previous agent. In practice, combination chemotherapy regimens typically use the largest tolerated doses of each chemotherapeutic drug; drugs that work via different mechanisms and that do not share similar toxic effects are combined. Using a combination of agents that have different mechanisms also decreases the chances that drug-resistant tumor cell populations will emerge. Chemotherapeutic drugs used in combination are usually administered over short periods at specific treatment intervals rather than as continuous therapy. This approach is based on the empiric observation that normal cells usually recover more rapidly from a pulse of maximal chemotherapy than do malignant cells. Furthermore, immunosuppression is less profound with intermittent administration of chemotherapy. With rare exceptions, the optimal dose of chemotherapeutic drugs requires repetitive dosing because even if all cells in a tumor are sensitive to a drug, a single dose of the drug is not usually sufficient to kill the typically hundreds of millions of cells that are present in patients with cancer.
Malignant cells are often characterized by rapid division and synthesis of DNA. Most conventional chemotherapeutic drugs exert their antineoplastic effects on cells that are actively undergoing division (mitosis) or DNA synthesis. Many chemotherapeutic drugs act only at specific phases of the cell cycle (Fig. 42-1).1 The biology of the cancer under treatment and the cell cycle specificity of agents effect how drugs are scheduled and combined for maximal effect. Slow-growing malignant cells with a slow rate of division, like carcinoma of the lung and colon, are often unresponsive or at best partially responsive to conventional chemotherapy. Conversely, rapidly dividing normal cells, like the cells found in the bone marrow, gastrointestinal mucosa, skin, and hair follicles, are more vulnerable to the toxic effects of chemotherapeutic drugs. Thus, it is predictable that clinical manifestations of toxicity caused by chemotherapeutic drugs often include myelosuppression (leukopenia, thrombocytopenia, or anemia), nausea, vomiting, diarrhea, mucosal ulceration, dermatitis, and alopecia as these represent activity at normal rapidly dividing cells. Myelosuppression is the dose-limiting factor for many chemotherapeutic drugs and is the most common toxicity that leads to temporary or permanent withdrawal of therapy. Drug-induced myelosuppression is usually reversible with discontinuation of the chemotherapeutic agent.

Drug Resistance
Resistance to chemotherapeutic drugs often occurs and has many causes.3 Some chemotherapy agents lead to induction of drug-metabolizing enzymes in the liver, other tissues, or tumor cells, accelerating drug conversion to nontoxic metabolites. Many solid tumors grow so rapidly that portions of the tumor are poorly vascularized, preventing therapeutic concentrations from reaching many target cells. In poorly perfused areas of some tumors, cells remain resistant to chemotherapeutic drugs because of relative hypoxia. Indeed, hypoxia causes resistance to both radiation and most chemotherapeutic drugs (with the exception of malignancies susceptible to treatment with the mitomycins).
As in the treatment of infections, multiple drug resistance describes the clinical circumstance in which a tumor is no longer susceptible to several chemotherapeutic drugs. For a number of agents, P-glycoprotein spans the plasma membrane and acts to pump chemotherapeutic drugs (anthracyclines, vinca alkaloids, and taxanes but not alkylating drugs, platinating drugs, and antimetabolites) to the extracellular space such that an effective toxic intracellular concentration is not reached. In addition to P-glycoprotein, there is an additional family of multidrug resistance proteins (MRPs) that are located on plasma membranes and endoplasmic reticulum of some tumor cell types, which confer drug resistance via an adenosine triphosphate (ATP)–dependent decrease in cellular drug accumulation (with the exception of malignancies susceptible to treatment with the taxanes). Collectively, P-glycoprotein and the MRPs are members of the ATP-binding cassette (ABC) class of transporter proteins that protect normal tissues from a variety of toxicants and are overexpressed in some tumor cells. The breast cancer resistance protein (also known as the ATP-binding cassette subfamily G member 2 or ABCG2 protein or the mitoxantrone resistance–associated protein) is a specific example of an ABC protein that confers tumor resistance to certain chemotherapeutic agents via active transport of the offending agent out of cells expressing this transporter.
Topoisomerases, enzymes that regulate the overwinding or underwinding of DNA during replication, are the targets for many chemotherapeutic drugs. Resistance to chemotherapeutic drugs can also occur through mutations in the drug-binding domain of the target enzyme. Resistance to the drug methotrexate may reflect mutations in the drug target, the enzyme dihydrofolate reductase; dihydrofolate reductase converts dihydrofolate into tetrahydrofolate and is required for the de novo synthesis of purines and thymidylic acid, which are important for cell growth and proliferation. Resistance to alkylating drugs occurs through overexpression of drug-neutralizing substances and metabolizing proteins.
Classification
Chemotherapeutic drugs are classified according to their mechanism of action (Table 42-1)1; adverse effects associated with these drugs are generally similar among drugs with similar mechanisms of action (Table 42-2).4–6Knowledge of drug-induced adverse effects and evaluation of appropriate laboratory tests (hemoglobin, platelet count, white blood cell count, coagulation profile, arterial blood gases, blood glucose, plasma electrolytes, liver and renal function tests, electrocardiogram [ECG], and radiograph of the chest) are useful in the preoperative evaluation of patients being treated with specific chemotherapeutic drugs. Immunosuppression makes these patients susceptible to iatrogenic infections, thus asepsis and the use of appropriate prophylactic antibiotics is critical. A history of severe vomiting or diarrhea may be associated with electrolyte disturbances and decreased intravascular fluid volume. The existence of mucositis makes placement of pharyngeal airways, laryngeal mask airways, and esophageal catheters questionable. The response to inhaled and injected anesthetic drugs may be altered by drug-induced cardiac, hepatic, or renal dysfunction and induction of hepatic enzymes. The response to older nondepolarizing neuromuscular blocking drugs may be altered by impaired renal function. Theoretically, the effects of succinylcholine may be prolonged if plasma cholinesterase activity is decreased by chemotherapeutic drugs.






Toxicities
Chemotherapeutic drugs typically target proteins or nucleic acids, which are common to malignant and nonmalignant cells and thus possess a narrow therapeutic index. Indeed, using the standard definition of therapeutic index (the dose that causes toxicity divided by the minimum effective dose) is not useful, as these agents all produce significant, even life-threatening toxicities at doses which may not reach levels that are high enough to eradicate cancer (therapeutic index <1). Furthermore, chemotherapeutic drugs are usually administered at maximum tolerated doses. Although toxicities may be unique for specific drugs, many toxicities are shared (nausea and vomiting, myelosuppression, mucositis, alopecia) (see Table 42-2).4 Nausea and vomiting result from local gastrointestinal effects as well as activation of the chemoreceptor trigger zone in the central nervous system. Patients who have a history of chemotherapy-induced nausea and vomiting are not necessarily prone to postoperative nausea and vomiting, as there is only weak positive association between the two; however, patients who have a history of tolerating emetogenic chemotherapy regimens are unlikely to develop postoperative nausea and vomiting.7Development of serotonin antagonists as effective antiemetics in addition to combination antiemetic regimens has facilitated the tolerance of emetogenic chemotherapeutic drugs. Mucositis and diarrhea are common gastrointestinal toxicities that reflect the high proliferative rate of gastrointestinal tissues, which makes these tissues more susceptible to the cytotoxic effects of certain chemotherapeutic drugs. Myelosuppression and alopecia reflect similar chemotherapeutic drug effects on highly proliferative tissues. Chemotherapeutic drugs that damage DNA (alkylating drugs, topoisomerases) are associated with secondary malignancies.
Alkylating Agents
Alkylating drugs include nitrogen mustards, alkyl sulfonates, nitrosoureas, and triazenes. These chemotherapeutic drugs form covalent alkyl bonds with nucleic acid bases, resulting in intrastrand or interstrand DNA cross-links which are toxic to cells undergoing division. By altering the structure of DNA, these drugs inhibit DNA replication and transcription. DNA damage produced by alkylating chemotherapeutic drugs is more likely to kill malignant cells than nonmalignant cells because rates of proliferation are greater for the cancer cells. Acquired resistance to alkylating drugs is a common occurrence and may reflect decreased cell membrane permeability to the drugs and increased production of nucleophilic substances that can compete with target DNA for alkylation.
Side Effects
Bone marrow suppression is the most important dose-limiting factor in the clinical use of alkylating drugs, especially busulfan. Cessation of mitosis is evident within 6 to 8 hours. Lymphocytopenia is usually present within 24 hours. Variable degrees of depression of platelet and erythrocyte counts may occur. Hemolytic anemia is predictably present.
Treatment with alkylating drugs is often associated with gonadal dysfunction, including oligospermia and amenorrhea. Hemorrhagic cystitis can result from irritation by the acrolein metabolite of cyclophosphamide or ifosfamide. Gastrointestinal mucosa is sensitive to the effects of alkylating drugs, manifesting as mitotic arrest, cellular hypertrophy, and desquamation of the epithelium. Damage to hair follicles, often leading to alopecia, is a common side effect. Increased skin pigmentation is frequent. All alkylating drugs are powerful central nervous system (CNS) stimulants, manifesting most often as nausea and vomiting. Skeletal muscle weakness and seizures may be present. Pneumonitis and pulmonary fibrosis are potential adverse effects of alkylating drugs. Symptomatic patients may demonstrate a decreased pulmonary diffusing capacity. Inhibition of plasma cholinesterase activity may be present for as long as 2 to 3 weeks after administration of chemotherapy regimens that include an alkylating agent and can lead to prolonged skeletal muscle paralysis after administration of succinylcholine.8,9
Rapid drug-induced destruction of malignant cells can produce increased purine and pyrimidine breakdown, leading to uric acid–induced nephropathy. To minimize the likelihood of this complication, it is recommended that adequate fluid intake, alkalinization of the urine, and administration of allopurinol be established before drug treatment.
Nitrogen Mustards
The most commonly used nitrogen mustards are mechlorethamine, cyclophosphamide, melphalan, and chlorambucil.
Mechlorethamine
Mechlorethamine is a rapidly acting nitrogen mustard administered intravenously (IV) to minimize local tissue irritation. This drug must be freshly prepared before each administration. Mechlorethamine and other nitrogen mustards are intensely powerful vesicants, requiring that gloves be worn by personnel handling the drug. A course of therapy with mechlorethamine consists of the injection of a total dose of 0.4 mg/kg. The drug undergoes rapid chemical transformation in tissues such that active drug is no longer present after a few minutes. For this reason, it is possible to prevent tissue toxicity from the drug by isolating the blood supply to that tissue. Alternatively, it is theoretically possible to localize the action of mechlorethamine in a specific tissue by injecting the drug into the arterial blood supply to the tissue.
Clinical Uses
Mechlorethamine produces beneficial effects in the treatment of Hodgkin disease and, less predictably, in other lymphomas. The drug is most often used in combination with vincristine, procarbazine, and prednisone (MOPP regimen) for the treatment of Hodgkin disease.
Side Effects
The major side effects of mechlorethamine include nausea, vomiting, and myelosuppression. Leukopenia and thrombocytopenia constitute the principal limitation on the amount of drug that can be given. Herpes zoster is a type of skin lesion frequently associated with nitrogen mustard therapy. Latent viral infections may be unmasked by treatment with mechlorethamine. Thrombophlebitis is a potential complication, and extravasation of the drug results in severe local tissue reactions, with brawny and tender induration that may persist for prolonged periods.
Cyclophosphamide
Cyclophosphamide is well absorbed after oral administration and is subsequently activated in the liver to aldophosphamide for transport to target tissues. Parenteral administration is also effective. Target cells are able to convert aldophosphamide to highly cytotoxic metabolites, phosphoramide, and acrolein that then alkylate DNA. Maximal plasma concentrations of cyclophosphamide are achieved about 1 hour after oral administration, and the elimination half-time is 6 to 7 hours. Urinary elimination accounts for approximately 14% of this drug in an unchanged form.
Clinical Uses
Cyclophosphamide is one of the most frequently used chemotherapeutic drugs, as it is effective in the treatment of a wide range of cancers and inflammatory diseases. Its versatility is improved because of its effectiveness after oral as well as parenteral administration. Given in combination with other drugs, favorable responses have been shown in patients with Hodgkin disease, lymphosarcoma, Burkitt lymphoma, and acute lymphoblastic leukemia of childhood. Cyclophosphamide is frequently used in combination with methotrexate and fluorouracil as adjuvant therapy after surgery for breast cancer when there is involvement of the axillary nodes. Cyclophosphamide has potent immunosuppressive properties, leading to its use in nonneoplastic disorders associated with altered immune reactivity, including Wegener granulomatosis and rheumatoid arthritis.
Side Effects
Hypersensitivity reactions and fibrosing pneumonitis have been noted in patients treated with cyclophosphamide; the incidence is less than 1% and symptoms may develop months to years after initiation of the drug. Large doses of cyclophosphamide are associated with a high incidence of pericarditis and pericardial effusion, which in some cases has progressed to cardiac tamponade.10 Smaller numbers of treated patients develop hemorrhagic myocarditis with symptoms of congestive heart failure, which may not occur for as long as 2 weeks after the last dose of drug.
Cyclophosphamide differs from other nitrogen mustards in that significant degrees of thrombocytopenia are less common but alopecia is more frequent. Nausea and vomiting occur with equal frequency regardless of the route of administration. Mucosal ulcerations, increased skin pigmentation, and hepatotoxicity are possible side effects. Sterile hemorrhagic cystitis occurs in 5% to 10% of patients, presumably reflecting chemical irritation produced by reactive metabolites of cyclophosphamide. Dysuria and hematuria are indications to discontinue the drug. Inappropriate secretion of arginine vasopressin hormone has been observed in patients receiving cyclophosphamide, usually with doses of greater than 50 mg/kg. It is important to consider the possibility of water intoxication because these patients are usually being hydrated to minimize the likelihood that hemorrhagic cystitis will develop. Extravasation of the drug does not produce local reactions, and thrombophlebitis does not complicate IV administration.
Melphalan
Melphalan is a phenylalanine derivative of nitrogen mustard with a range of activity similar to other alkylating drugs. It is not a vesicant. Oral absorption is excellent, resulting in drug concentrations similar to those achieved by the IV route of administration. The elimination half-time is approximately 1.5 hours, and up to 15% of the drug is eliminated unchanged in urine.
Side Effects
The side effects of melphalan are primarily hematologic and are similar to those of other alkylating drugs. It is usually necessary to maintain a significant degree of bone marrow depression (leukocyte count 3,000 to 5,000 cells/mm3) to achieve optimal therapeutic effects. Pulmonary fibrosis is possible. Nausea and vomiting are not common side effects of melphalan. Alopecia does not occur, and changes in renal or hepatic function have not been reported.
Chlorambucil
Chlorambucil is the aromatic derivative of mechlorethamine. Oral absorption is adequate. The drug has an elimination half-time of approximately 1.5 hours and is almost completely metabolized. Chlorambucil is the slowest acting nitrogen mustard in clinical use. It is the treatment of choice in chronic lymphocytic leukemia and in primary (Waldenström) macroglobulinemia. A marked increase in the incidence of leukemia and other tumors has been noted with the use of this drug for the treatment of polycythemia vera.
Side Effects
Cytotoxic effects of chlorambucil on the bone marrow, lymphoid organs, and epithelial tissues are similar to those observed with other alkylating drugs. Its myelosuppressive action is usually moderate, gradual, and rapidly reversible. Pulmonary fibrosis is possible. Nausea and vomiting are frequent. CNS stimulation can occur but has been observed only with large doses. Hepatotoxicity may rarely occur.
Alkyl Sulfonates
Busulfan is a cell cycle nonspecific alkylating antineoplastic agent in the class of alkyl sulfonates. Busulfan is well absorbed after oral administration. IV administration is also effective. Almost the entire drug is eliminated by the kidneys as methane sulfonic acid. Busulfan produces remissions in up to 90% of patients with chronic myelogenous leukemia. The drug is of no value in the treatment of acute leukemia.
Side Effects
Busulfan can produce progressive pulmonary fibrosis in up to 4% of patients. The prognosis after appearance of clinical symptoms is poor, with a median survival of 5 months.11 Enhanced toxicity with administration of supplemental oxygen has not been noted. Myelosuppression and thrombocytopenia are important side effects of busulfan. Nausea, vomiting, and diarrhea occur. Hyperuricemia resulting from extensive purine catabolism accompanying the rapid cellular destruction and renal damage from precipitation of urates have been noted. Allopurinol is recommended to minimize renal complications.
Nitrosoureas
The nitrosoureas are mustard gas–related compounds used as an alkylating agent in chemotherapy. Nitrosoureas, represented by carmustine, lomustine, semustine, and streptozocin, possess a wide spectrum of activity for human malignancies including intracranial tumors, melanomas, and gastrointestinal and hematologic malignancies. Indeed, the high lipid solubility results in passage across the blood–brain barrier and efficacy in the treatment of meningeal leukemias and brain tumors. These drugs appear to act by carboxylation and alkylation of nucleic acids. With the exception of streptozocin, the clinical use of nitrosoureas is limited by profound drug-induced myelosuppression.
Carmustine
Carmustine is the nitrosourea in widest clinical use. It is capable of inhibiting synthesis of both RNA and DNA. Although oral absorption is rapid, the drug is injected IV because tissue uptake and metabolism occur quickly. Local burning may accompany infusion. Carmustine disappears from plasma in 5 to 15 minutes. Because of its ability to rapidly cross the blood–brain barrier, carmustine is used to treat meningeal leukemia and primary as well as metastatic brain tumors.
Side Effects
Carmustine has been associated with interstitial pneumonitis and fibrosis much like bleomycin.12 The incidence of pulmonary toxicity is in the range of 20% to 30%, with a mortality in those affected of 24% to 90%. The cumulative dose is the major risk factor, with 50% of patients exhibiting toxicity at doses above the range of 1,200 to 1,500 mg/m2. A unique side effect of carmustine is a delayed onset (after approximately 6 weeks of treatment) of leukopenia and thrombocytopenia. Active metabolites may be responsible for this toxicity. CNS toxicity, nausea and vomiting, flushing of the skin and conjunctiva, nephrotoxicity, and hepatotoxicity have been reported.
Lomustine and Semustine
Lomustine and its methylated analogue semustine possess similar clinical toxicity to carmustine, including delayed bone marrow depression manifesting as leukopenia and thrombocytopenia. Lomustine appears to be more effective than carmustine in the treatment of Hodgkin disease.
Streptozocin
Streptozocin has a methylnitrosourea moiety attached to the number 2 carbon atom of glucose. It has a unique affinity for β cells of the islets of Langerhans and has proved useful in the treatment of human pancreatic islet cell carcinoma and malignant carcinoid. In animals, the drug is used to produce experimental diabetes mellitus.
Side Effects
Approximately 70% of patients receiving this drug develop hepatic or renal toxicity. Renal toxicity may manifest as tubular damage and progress to renal failure and death. Hyperglycemia can occur as a result of selective destruction of pancreatic β cells and resultant hypoinsulinism.6 Myelosuppression is not produced by this drug.
Mitomycin
Mitomycin is the prototypical alkylating agent and is of value in the palliative treatment of gastric adenocarcinoma in combination with fluorouracil and doxorubicin. The drug is administered IV and is widely distributed in tissues but does not readily enter the CNS. Metabolism is in the liver, with less than 10% of mitomycin excreted unchanged in bile or urine.
Side Effects
Myelosuppression is a prominent side effect of mitomycin and is characterized by severe leukopenia and thrombocytopenia, which may be delayed in appearance. Mitomycin is capable of inducing pulmonary fibrosis, with an incidence ranging between 3% and 12%.13 Like bleomycin, mitomycin appears to act synergistically to induce pulmonary fibrosis with thoracic radiation and oxygen therapy, suggesting the need to limit exposure of treated patients to hyperoxia. Nausea, vomiting, gastrointestinal mucositis, and alopecia are recognized toxic effects. Glomerular damage resulting in renal failure is a rare but well-recognized complication.
Platinating Drugs
Cisplatin
Although cisplatin is frequently designated as an alkylating agent, it has no alkyl group and so cannot carry out alkylating reactions. It is correctly classified as alkylating-like. Cisplatin contains a platinum atom, two amines, and two chlorides, which result in chemotherapeutic effects resembling DNA alkylating drugs by cross-linking adjacent or opposing guanine bases to disrupt DNA. The drug must be administered IV because oral ingestion is ineffective. High concentrations of cisplatin are found in the kidneys, liver, intestines, and testes, but there is poor penetration into the CNS. Cisplatin and its analogue carboplatin are components of the treatment of many nonhematologic malignancies, including lung, bladder, testicular, and ovarian cancer.
Side Effects
Renal toxicity is prominent and becomes the dose-limiting toxic effect of cisplatin. Decreased glomerular filtration rate and renal tubular dysfunction produced by cisplatin may begin as early as 3 to 5 days after initiating treatment with this drug. Along with increasing blood urea nitrogen and plasma creatinine concentrations, proteinuria, and hyperuricemia, there is a magnesium-wasting defect in as many as 50% of patients manifesting as some degree of cisplatin-induced renal dysfunction. Acute tubular necrosis may progress to acute renal failure, necessitating hemodialysis. Hydration and diuresis induced with mannitol and furosemide may protect against the development of renal toxicity by dilution of the tubular urinary concentration of cisplatin. The hypomagnesemia that is associated with cisplatin’s renal tubular injury may predispose to cardiac dysrhythmias and decrease the dose requirements for neuromuscular blocking drugs.
Ototoxicity caused by cisplatin is manifested by tinnitus and hearing loss in the high-frequency range. Cisplatin is considered highly emetogenic, with marked nausea and vomiting occurring in almost all patients who do not receive antiemetics, although prophylactic antinausea regimens can be highly effective. Mild to moderate myelosuppression may develop, with transient leukopenia and thrombocytopenia. Peripheral sensory neuropathies, paresthesias, and loss of vibratory and position sense are common findings. Most neuropathies are reversible, although symptoms may persist for months. Hyperuricemia, seizures, and cardiac dysrhythmias have been observed. Allergic reactions characterized by facial edema, bronchoconstriction, tachycardia, and hypotension may occur minutes after injection of the drug.
Antimetabolites
Nucleic acid synthesis inhibitors (antimetabolites) include folate analogues, pyrimidine analogues, and purine analogues. These drugs are particularly effective in destroying cells during the S phase of the cell cycle, which is when DNA is synthesized. Selective effects on cancer cells may relate to greater rates of DNA replication in cancer cells than normal cells. Nevertheless, side effects (myelosuppression and mucositis) reflect effects on proliferating but nonmalignant cells.
Folate Analogues
Methotrexate
Methotrexate is a poorly lipid-soluble folate analogue that is effective in the treatment of different hematologic and nonhematologic cancers and is classified as an antimetabolite (folic acid antagonist). This drug inhibits dihydrofolate reductase, which is the enzyme that uses reduced folate as a methyl donor in the synthesis of pyrimidine and purine nucleosides. Inhibition of dihydrofolate reductase by methotrexate prevents the formation of tetrahydrofolic acid and causes disruption of cellular metabolism by producing an acute intracellular deficiency of folate enzymes. As a result, 1-carbon transfer reactions necessary for the eventual synthesis of DNA and RNA cease.
Methotrexate is readily absorbed after oral administration. Significant metabolism of methotrexate does not seem to occur, with more than 50% of the drug appearing unchanged in urine. Renal excretion reflects glomerular filtration and tubular secretion. Toxic concentrations of methotrexate may occur in patients with renal insufficiency. Methotrexate remains in tissues for weeks, suggesting binding of the drug to dihydrofolate reductase.
Clinical Uses
Methotrexate is widely used in the treatment of malignant and some nonmalignant disorders. It is a useful drug in the treatment of acute lymphoblastic leukemia in children but not adults. Choriocarcinoma is effectively treated with this drug. Improvement in the clinical manifestations of psoriasis in patients reflects the effect of methotrexate on rapidly dividing epidermal cells characteristic of this disease. This drug may also be useful in the treatment of rheumatoid arthritis.
Methotrexate is poorly transported across the blood–brain barrier, and neoplastic cells that have entered the CNS probably are not affected by the usual plasma concentrations of the drug. Intrathecal injection is used to treat cerebral involvement with either leukemia or choriocarcinoma.
Acquired resistance to methotrexate develops as a result of (a) impaired transport of methotrexate into cells, (b) production of altered forms of dihydrofolate reductase that have decreased affinity for the drug, and (c) increased concentrations of intracellular dihydrofolate reductase.
Side Effects
The most important side effects of methotrexate occur in the gastrointestinal tract and bone marrow. Leukopenia and thrombocytopenia reflect bone marrow depression. Ulcerative stomatitis and diarrhea are frequent side effects and require interruption of treatment. Hemorrhagic enteritis and death from intestinal perforation may occur. Pulmonary toxicity may take the form of fulminant noncardiogenic pulmonary edema, or a more progressive inflammation, with interstitial infiltrates and pleural effusions.14 The incidence of pulmonary toxicity attributed to methotrexate is in the range of 8%, but its frequent use in combination with other chemotherapeutic drugs makes this number uncertain.15 Methotrexate is associated with renal toxicity, with an incidence approaching 10% in higher doses.16 Renal insufficiency may be prevented by hydration and urinary alkalinization. Short-term or intermittent therapy with methotrexate results in increases in liver transaminase enzymes. Hepatic dysfunction is usually reversible but may sometimes lead to cirrhosis. It may be useful to measure liver function tests preoperatively in patients who have recently received methotrexate. Encephalopathic syndromes may accompany intrathecal or IV administration of methotrexate and may be transient or permanent.17 Alopecia and dermatitis may accompany administration of methotrexate. Folic acid antagonists also interfere with embryogenesis, emphasizing the risk in administering these drugs to pregnant patients. Normal cells can be protected from lethal damage by folate antagonists with sequential administration of folinic acid (leucovorin), thymidine, or both. This approach has been termed the rescue technique.
Pyrimidine Analogues
Pyrimidine analogues have in common the ability to prevent the biosynthesis of pyrimidine nucleotides or to mimic these natural metabolites to such an extent that they interfere with vital cellular activities such as the synthesis and functioning of nucleic acids. Examples of antimetabolite chemotherapeutic drugs that function as pyrimidine analogues are fluorouracil and cytarabine.
Fluorouracil
Fluorouracil blocks production of thymine nucleotides by inhibiting thymidylate synthase. This chemotherapeutic drug lacks significant inhibitory activity on cells and must be converted enzymatically to a 5′-monophosphate nucleotide. Administration of fluorouracil is usually by IV injection because absorption after oral ingestion is unpredictable and incomplete. Metabolic degradation occurs primarily in the liver, with an important metabolite being urea. Only approximately 10% of fluorouracil appears unchanged in urine. Fluorouracil readily enters the cerebrospinal fluid, with therapeutic concentrations being present within 30 minutes after IV administration.
Clinical Uses
Fluorouracil may be of palliative value in certain types of carcinoma, particularly of the breast and gastrointestinal tract. The drug is often used for the topical treatment of premalignant keratoses of the skin and superficial basal cell carcinomas.
Side Effects
Side effects caused by fluorouracil are difficult to anticipate because of their delayed appearance. Fluorouracil-induced myocardial ischemia is a rare cardiac toxicity that may lead to myocardial infarction up to 1 week after treatment.18 The incidence of this side effect is low in patients without underlying heart disease but may increase to 4.5% of treated patients with preexisting coronary artery disease. Stomatitis manifesting as a white patchy membrane that ulcerates and becomes necrotic is an early sign of toxicity and warns of the possibility that similar lesions may be developing in the esophagus and gastrointestinal tract. Myelosuppression, most frequently manifesting as leukopenia between 9 and 14 days of therapy, is a serious side effect. Thrombocytopenia and anemia may complicate treatment with fluorouracil. Loss of hair progressing to total alopecia, nail changes, dermatitis, and increased pigmentation and atrophy of the skin may occur. Hand-foot syndrome has also been associated with fluorouracil. Neurologic manifestations, including an acute cerebellar syndrome (ataxia), have been reported.
Capecitabine
Capecitabine is an orally administered drug that is metabolized to fluorouracil by thymidine phosphorylase after absorption from the gastrointestinal tract. Because there is more activity of thymidine phosphorylase in cancer cells (especially breast cancer) than in normal cells, capecitabine has the potential to be more selective than fluorouracil.
Pemetrexed
Pemetrexed is a folate antagonist that is effective in the treatment of mesothelioma and lung cancer. This drug inhibits multiple enzymes involved in the folate pathway, including thymidylate synthase and dihydrofolate reductase.
Cytarabine
Cytarabine (cytosine arabinoside), like other pyrimidine antimetabolites, must be activated by conversion to the 5′-monophosphate nucleotide before inhibition of DNA synthesis can occur. Both natural and acquired resistance to cytarabine develops, reflecting the activity of cytidine deaminase, an enzyme capable of converting cytarabine to the inactive metabolite arabinosyl uracil.
Clinical Uses
In addition to its chemotherapeutic activity, particularly in acute leukemia in children and adults, cytarabine has potent immunosuppressive properties. The drug is particularly useful in chemotherapy of acute granulocytic leukemia in adults. IV administration of cytarabine is recommended because oral absorption is poor and unpredictable.
Side Effects
Cytarabine is a potent myelosuppressive drug capable of producing severe leukopenia, thrombocytopenia, and anemia. Cerebellar toxicity and ataxia can occur at high doses. Other side effects include gastrointestinal disturbances, stomatitis, and hepatic dysfunction. Thrombophlebitis at the site of infusion is common. Alternatively, the drug may be given subcutaneously.
Gemcitabine
Gemcitabine resembles cytarabine structurally yet gemcitabine is active in several nonhematologic cancers, whereas cytarabine is not effective. Gemcitabine is also used in solid organ carcinomas, such as of the pancreas, breast, and lung. This most likely reflects unique effects of this chemotherapeutic drug on DNA and RNA metabolism. Common side effects associated with use of gemcitabine include bone marrow suppression, flulike symptoms, fever, fatigue, mild nausea/vomiting, and diarrhea.
Purine Analogues
Antimetabolite chemotherapeutic drugs that function as purine analogues include mercaptopurine, azathioprine, thioguanine, pentostatin (2′-deoxycoformycin), and cladribine (2-chlorodeoxyadenosine). Mercaptopurine and thioguanine are analogues of the natural purines hypoxanthine and guanine, respectively.
Mercaptopurine
Mercaptopurine is incorporated into DNA or RNA strands and either blocks further strand synthesis or causes structural alterations that damage DNA. This drug is useful in the treatment of acute leukemia in children. Oral absorption is prompt, and gastrointestinal epithelium is not damaged. The elimination half-time is brief (about 90 minutes) due to rapid tissue uptake, renal excretion, and hepatic metabolism. One pathway of metabolism is methylation and subsequent oxidation of the methylated derivatives. A second pathway involves the enzyme xanthine oxidase, which oxidizes mercaptopurine to 6-thiouric acid. Allopurinol, as an inhibitor of xanthine oxidase, prevents conversion of mercaptopurine to 6-thiouric acid and thus increases the exposure of cells to mercaptopurine. The dose of mercaptopurine is decreased by about one-third when the drug is combined with allopurinol.
Side Effects
The principal side effect of mercaptopurine is a gradual development of bone marrow depression manifesting as thrombocytopenia, granulocytopenia, or anemia several weeks after initiation of therapy. Anorexia, nausea, and vomiting are common side effects; stomatitis and diarrhea rarely occur. Jaundice occurs in approximately one-third of patients and is associated with bile stasis and occasional hepatic necrosis. Hyperuricemia and hyperuricosuria may occur during treatment with mercaptopurine, presumably reflecting destruction of cells. This effect may require the use of allopurinol.
Thioguanine
Thioguanine is of particular value in the treatment of acute myelogenous leukemia, especially if given with cytarabine. After oral administration, thioguanine appears in the urine as a methylated metabolite and inorganic sulfate. Minimal amounts of 6-thiouric acid are formed, suggesting that deamination is not important in the metabolic inactivation of thioguanine. For this reason, thioguanine may be administered concurrently with allopurinol without a decrease in dosage, unlike mercaptopurine. Toxic manifestations of thioguanine treatment include bone marrow depression and, occasionally, gastrointestinal effects.
Pentostatin and Cladribine
Pentostatin and cladribine are purine analogues that have clinical activity against a variety of indolent lymphoid tumors, with the most dramatic effects occurring in patients with hairy-cell leukemia.19 These drugs act by irreversibly binding to adenosine deaminase (pentostatin) or by chemical modification of enzyme substrate, rendering it resistant to the action of adenosine deaminase (cladribine). Patients with acute leukemia and cells with high levels of adenosine deaminase activity are most likely to respond to these drugs. Fever, which is likely due to cytokines, is a side effect of treatment with cladribine. Both drugs are capable of producing immunosuppression. The recovery from immunosuppression seems to be more rapid after treatment with cladribine than after treatment with pentostatin, perhaps because of the shorter duration of administration of the former. Indeed, cladribine is emerging as the treatment of choice for hairy cell leukemia because of its minimal toxicity and its ability to induce a complete and sustained response with a single course of therapy.
Hydroxyurea
Hydroxyurea acts on the enzyme ribonucleoside diphosphate reductase to interfere with the synthesis of DNA. Oral absorption is excellent, and approximately 80% of the drug appears in the urine within 12 hours after oral or IV administration. The primary use of hydroxyurea is in the treatment of chronic myelogenous leukemia. Temporary remissions in patients with metastatic malignant melanoma have been reported.
Side Effects
Myelosuppression manifesting as leukopenia, megaloblastic anemia, and occasionally thrombocytopenia is the major side effect produced by hydroxyurea. Nausea and vomiting may accompany administration of this drug. Hyperpigmentation of the skin, stomatitis, and alopecia occur infrequently.
Topoisomerase Inhibitors
Topoisomerases are enzymes that correct alterations in DNA which occur during replication and transcription. Certain chemotherapeutic drugs inhibit either topoisomerase I or topoisomerase II. Because cancer cells possess more topoisomerase activity than normal cells, there is more drug-induced DNA damage and resultant cell death. Toxicity reflects effects of inhibition of topoisomerase enzymes on normal proliferating tissues (myelosuppression, mucositis). Topoisomerase II inhibitors that include doxorubicin, daunorubicin, etoposide, and teniposide are part of most combination chemotherapy treatment regimens. Topoisomerase I inhibitors include topotecan and irinotecan. These drugs exhibit a broad spectrum of chemotherapeutic activity being useful in the treatment of leukemia and lung, colon, and ovarian cancer.
Doxorubicin and Daunorubicin
Doxorubicin and daunorubicin are anthracycline antibiotics that are natural products of certain soil fungi. Structurally, they contain a tetracycline ring attached to the sugar daunosamine by a glycosidic linkage. These drugs most likely act by binding to DNA, resulting in changes in the DNA helix that interfere with the ability of nucleic acids to serve as a template during replication. These drugs are also a likely cause of disruptive effects on cellular membranes. Drug-induced free radicals may overwhelm the heart’s antioxidant defenses, leading to the oxidation of critical cardiac proteins and membrane components (unsaturated free fatty acids), leading to cardiotoxicity.20Laboratory studies demonstrate that each subsequent dose of doxorubicin appears to diminish the heart’s ability to withstand subsequent oxidant stress. Evidence that free radicals have a role is the protective effect of free radical scavengers.
Daunorubicin and doxorubicin are administered IV, with care taken to prevent extravasation because local vesicant action may result. There is rapid clearance from the plasma into the heart, kidneys, lungs, and liver. These drugs do not cross the blood–brain barrier to any significant extent. The urine may become red for 1 to 2 days after administration of these drugs.
Daunorubicin is metabolized primarily to daunorubiconol, whereas doxorubicin is excreted unchanged and as metabolites, including adriamycinol in the urine. Ultimately, approximately 40% of daunorubicin and doxorubicin are metabolized. Indeed, clinical toxicity may result in patients with hepatic dysfunction.
Clinical Uses
Daunorubicin is used primarily in the treatment of acute lymphocytic and myelocytic leukemia. Doxorubicin, which differs from daunorubicin only by a single hydroxyl group on the number 14 carbon atom, is also effective against a wide range of solid tumors. For example, doxorubicin is one of the most active single drugs for treating metastatic adenocarcinoma of the breast, carcinoma of the bladder, bronchogenic carcinoma, metastatic thyroid carcinoma, oat cell carcinoma, and osteogenic carcinoma.
Resistance is observed to the anthracycline antibiotics, as with other chemotherapeutic drugs. Furthermore, cross-tolerance occurs between daunorubicin and doxorubicin. Cross-resistance also occurs between these antibiotics and the vinca alkaloids, suggesting that an alteration of cellular permeability may be involved.
Side Effects
Cardiomyopathy and myelosuppression are side effects of the chemotherapeutic antibiotics. Leukopenia typically manifests during the second week of therapy. Thrombocytopenia and anemia occur but are usually less pronounced. Stomatitis, gastrointestinal disturbances, and alopecia are common side effects.
Cardiomyopathy
Cardiomyopathy is a unique dose-related and often irreversible side effect of the anthracycline antibiotics. Increased plasma concentrations of troponin T reflect drug-induced injury to myocardial cells. Congestive heart failure develops in less than 3% of patients with a cumulative dose of doxorubicin of less than 400 mg/m2, rising to 18% at 700 mg/m2 (Fig. 42-2).21 Prior mediastinal radiation or previous treatment with cyclophosphamide increases the subsequent risk of cardiomyopathy in response to administration of an anthracycline antibiotic. Marked impairment of left ventricular function for as long as 3 years after discontinuing doxorubicin has been observed. Previous treatment with anthracycline antibiotics may enhance myocardial depressant effects of anesthetic drugs even in patients with normal resting cardiac function.22 Acute left ventricular failure 2 months after cessation of treatment with doxorubicin has been described during general anesthesia.23
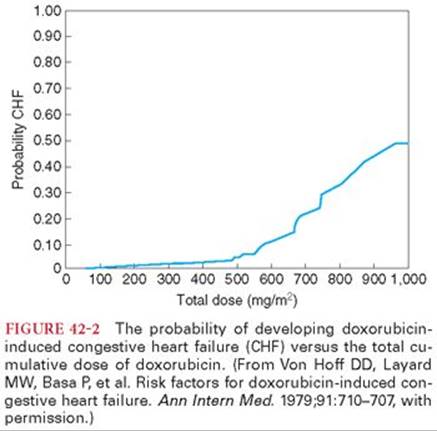
Two types of cardiomyopathies may occur.6,20 An acute form of cardiomyopathy occurs in approximately 10% of patients and is characterized by relatively benign changes on the ECG that include nonspecific ST-T changes and decreased QRS voltage. Other cardiac changes include premature ventricular contractions, supraventricular tachydysrhythmias, cardiac conduction abnormalities, and left axis deviation. These abnormalities occur during therapy at all dose levels and, except for decreased QRS voltage on the ECG, resolve 1 to 2 months after discontinuation of therapy. There is an associated acute reversible decrease in the ejection fraction within 24 hours after a single dose.
The second form of cardiomyopathy is characterized by the insidious onset of symptoms such as dry nonproductive cough, suggesting bronchitis, followed by rapidly progressive heart failure that is unresponsive to inotropic drugs and mechanical ventricular assistance.6 This severe form of cardiomyopathy occurs in almost 2% of treated patients and is fatal approximately 3 weeks after the onset of symptoms in nearly 60% of affected patients. Predictive tests to permit early recognition of impending cardiomyopathy are not available, although diminution in QRS voltage on the ECG is consistent with the diffuse character of the myocardial damage. Increased plasma concentrations of cardiac enzymes occur late in the course of cardiac failure and are of limited value in achieving an early diagnosis. Systolic time intervals and echocardiograms have been used to detect cardiotoxicity before the occurrence of clinically significant damage. Dexrazoxane is a free radical scavenger that protects the heart from doxorubicin-associated damage.24
Dactinomycin
Dactinomycin (actinomycin D) is an antibiotic with chemotherapeutic activity resulting from its ability to bind to DNA, especially in rapidly proliferating cells. As a result of this binding, the function of RNA polymerase and thus the transcription of the DNA molecule are blocked. After IV injection, dactinomycin rapidly leaves the circulation. In animals, approximately 50% of an injected dose is excreted unchanged in bile and 10% in urine. There is no evidence that the drug undergoes metabolism. Dactinomycin does not cross the blood–brain barrier in amounts sufficient to produce a pharmacologic effect.
Clinical Uses
The most important clinical use of dactinomycin is the treatment of Wilms tumor in children and of rhabdomyosarcoma. It may be effective in some women with methotrexate-resistant choriocarcinoma. Occasionally, this drug is used to inhibit immunologic responses associated with organ transplantation.
Side Effects
The toxic effects of dactinomycin include the early onset of nausea and vomiting, often followed by myelosuppression manifesting as pancytopenia 1 to 7 days after completion of therapy. Pancytopenia may be preceded by thrombocytopenia as the first manifestation of bone marrow suppression. Glossitis, ulcerations of the oral mucosa, diarrhea, alopecia, and cutaneous erythema are commonly associated with dactinomycin therapy. Extravasation of the drug results in tissue necrosis.
Bleomycin
Bleomycins are water-soluble glycopeptides that differ from one another (there are more than 200 congeners) in their terminal amine moiety. The terminal amine is coupled through an amide linkage to a carboxylic acid. Bleomycin possesses a tripeptide component that binds DNA and a metal-binding region. In the presence of oxygen and either iron or copper, bleomycin produces free radicals which create DNA breaks.
Bleomycin is administered IV, and high concentrations occur in the skin and lungs. The drug accumulates in tumors, suggesting the presence of a lower level of inactivating enzyme. Bleomycin is eliminated primarily by renal excretion, with approximately 50% of the dose cleared within 4 hours and 70% by 24 hours.25 Indeed, excessive concentrations of drug occur if usual doses are administered to patients with impaired renal function.
Clinical Uses
Bleomycin is effective in the treatment of testicular carcinoma, particularly if administered in combination with vinblastine. It is also useful in the palliative treatment of squamous cell carcinomas of the head, neck, esophagus, skin, and genitourinary tract.
Side Effects
The most common side effects of bleomycin are mucocutaneous reactions, including stomatitis, alopecia, pruritus, erythema, and hyperpigmentation, which occur in approximately 45% of patients. In contrast to other chemotherapeutic drugs, bleomycin causes minimal myelosuppression. Unexplained exacerbations of rheumatoid arthritis have occurred.
Patients with lymphomas who are receiving bleomycin may develop an acute reaction characterized by hyperthermia, hypotension, and hypoventilation. The likely mechanism is the release of an endogenous pyrogen, presumably from destroyed tumor cells. An initial small test dose of bleomycin is recommended to minimize the occurrence of this syndrome.
Pulmonary Toxicity
The most serious side effect of bleomycin is dose-related pulmonary toxicity (Fig. 42-3).11 Indeed, bleomycin is concentrated preferentially in the lungs and is inactivated by a hydrolase enzyme, which is relatively deficient in lung tissue. Initially, bleomycin produces pulmonary capillary endothelial damage, progressing to alveolar epithelial injury with necrosis of type 1 and proliferation of type 2 alveolar cells. Interstitial fibrosis develops and may progress to involve the entire lung. It is estimated that some form of pulmonary toxicity (most often pulmonary fibrosis) occurs in 4% of patients treated with bleomycin. Fatal pulmonary toxicity has occurred with bleomycin doses as low as 100 mg but more often in the presence of other risk factors (Table 42-3).


The first signs of pulmonary toxicity are cough, dyspnea, and basilar rales, which progress in one of two directions. A mild form of pulmonary toxicity is characterized by exertional dyspnea and a normal resting PaO2. A more severe form of arterial hypoxemia at rest is associated with radiographic findings of interstitial pneumonitis and fibrosis. Lesions are found more frequently in lower lobes and subpleural areas, and radiographs of the chest often reveal basilar and perihilar infiltrates. The alveolar–arterial gradient for oxygen is increased, and pulmonary diffusion capacity may be decreased. Pulmonary function studies have been of no greater value than clinical signs in detecting the onset of pulmonary toxicity.
Early reports of postoperative respiratory failure in bleomycin-treated patients suggested that either arterial hyperoxia or excessive crystalloid administration played a role in the exacerbation of pulmonary fibrosis.26–28 One speculation is that acutely increased inhaled concentrations of oxygen facilitate production of superoxide and other free radicals in the presence of bleomycin. For this reason, it has been recommended that inhaled oxygen concentrations be maintained below 30% in bleomycin-treated patients. Animal model literature confirms that the continuous administration of inspired oxygen concentrations of greater than 30% immediately after exposure to bleomycin increases pulmonary damage.29 Nevertheless, it is unlikely that patients will present to the operating room immediately after treatment with bleomycin. A more practical question is whether hyperoxia for short periods of time several days after treatment is a risk factor for bleomycin-induced pulmonary damage.30 In this regard, animal studies have confirmed that delayed exposure to supplemental oxygen after bleomycin treatment is not harmful.31Nevertheless, there are case reports of respiratory failure with inspired oxygen concentrations greater than 30% in patients last exposed to bleomycin up to 6 to 12 months before hyperoxia. Patients with prior exposure to bleomycin but with no risk factors appear to be at a minimum risk from hyperoxia. In contrast, those patients with one or more major risk factors (preexisting pulmonary damage from bleomycin, which is more likely if the total dose is greater than 450 mg; renal dysfunction, which slows clearance of the drug from the lungs; and/or prior exposure to bleomycin within a 1- to 2-month period) may be at higher risk for the development of bleomycin-induced hyperoxic pulmonary injury in the operating room. It may be prudent to maintain these patients on the minimum inspired oxygen concentration that can be used safely intraoperatively to provide oxygen saturations of greater than 90% by pulse oximetry.30 The value of corticosteroids as pretreatment in patients with risk factors and in whom greater than 30% oxygen may be needed, for example, operations requiring cardiopulmonary bypass, has not been confirmed by controlled studies. The role of excessive crystalloid administration has not received the same scrutiny as increased delivered oxygen concentrations. A consideration in this regard is replacement of fluids with colloids rather than crystalloids to decrease or prevent pulmonary interstitial edema in bleomycin-treated patients undergoing surgery. Accumulation of interstitial fluid may reflect impaired lymphatic function caused by bleomycin-induced fibrotic changes in the lungs. In the future, bleomycin may be replaced with phleomycin, an analogue of bleomycin that has lower pulmonary toxicity and a broader effectiveness against multiple types of tumors.32
Tubulin-Binding Drugs
Microtubules are subcellular structures that are essential for normal function of cells. They form the architecture to maintain cell shape, organize the location of organelles, and mediate intracellular transport and secretion, neurotransmission, axonemal flow, and cell motility.33 Vinca alkaloids and taxanes are examples of antimitotic chemotherapeutic drugs that disrupt the normal function of microtubules. Vinca alkaloids bind to depolymerized microtubules and inhibit microtubule formation. Taxanes bind to polymerized microtubules and inhibit their breakdown. The result of these interactions is the failure of the cell to undergo normal mitosis leading to cell death.
Vinca Alkaloids
Vinca alkaloids represent the active medicinal ingredients from the pink periwinkle plant and include vincristine, vinblastine, vinorelbine, and vindesine. Vincristine is highly effective against Hodgkin disease, non-Hodgkin lymphoma, and pediatric solid tumors, yet it has little activity against adult solid tumors. Vinorelbine, in contrast, is active against breast and lung cancer. Vinblastine is most often used in the treatment of testicular cancer and non-Hodgkin lymphoma.
Side Effects
Myelosuppression manifesting as leukopenia, thrombocytopenia, and anemia are the most prominent side effects of vinca alkaloids, appearing 7 to 10 days after initiation of treatment. Vincristine is less likely than vinblastine and vinorelbine to cause bone marrow depression.
Symmetric peripheral sensory–motor neuropathy often occurs during administration of therapeutic doses of vincristine and may become the dose-limiting side effect.34,35 Clinical manifestations may include several aspects of peripheral nerve function with areflexia (loss of Achilles tendon reflex) being the earliest finding. Paresthesias in the hands and feet, weakness and atrophy of the extremities, and skeletal muscle pain make use of the hands and feet difficult (ataxia). Tremors frequently develop, neuropathic pain and foot drop are common. Autonomic neuropathy with orthostatic hypotension, bowel motility dysfunction, and cranial nerve involvement (laryngeal nerve paralysis with hoarseness, weakness of the extraocular muscles) are present in about 10% of treated patients.36 CNS effects (confusion, insomnia, seizures, hallucinations) due to vincristine are rare, presumably because of poor penetration of the blood–brain barrier by the drug. The peripheral neuropathy is mainly axonal, but demyelination may also occur as demonstrated by measurement of somatosensory evoked potentials.35 Vincristine-induced peripheral neuropathy is said to be reversible after discontinuing the drug, although this may require months, and in some patients, the resolution may be incomplete.34 There is limited evidence that suggests that neuraxial anesthesia and peripheral nerve blocks can be safely and effectively used in patients with preexisting peripheral neuropathy without significant risk of worsening of the neuropathy. The concentration of local anesthetic should be reduced, epinephrine should be avoided, and a nerve localization technique that minimizes the likelihood of intraneuronal injection should be used in efforts to minimize the risk of worsening the neuropathy.37 Neuropathies do worsen during the perioperative period in a small number of patients whether general or regional anesthesia is used, thus a careful risk-benefit assessment and informed decision making along with each individual patient is essential.
The syndrome of hyponatremia associated with high urinary sodium and inappropriate secretion of arginine vasopressin hormone has occasionally been observed during vincristine therapy. An effect on the autonomic nervous system may be responsible for paralytic ileus and abdominal pain, which commonly develops during vinblastine therapy. Urinary retention, tenderness of the parotid glands, dryness of the mouth, and sinus tachycardia are other occasionally experienced manifestations of altered autonomic nervous system activity. Transient mental depression is most likely to occur on the second or third day of treatment with vinblastine. Alopecia appears to occur more frequently with vincristine than with vinblastine. Vinorelbine may cause chest pain, bronchospasm, dyspnea, and pulmonary infiltrations.
Taxanes
Paclitaxel (active extract from the Pacific yew tree) and docetaxel (more water-soluble semisynthetic derivative) share a broad spectrum of similar chemotherapeutic activity against breast, lung, ovarian, and bladder cancer.38 Both drugs are also active against lymphoid malignancies. Taxanes block the function of the mitotic apparatus by impeding the normal function of microtubules. Unlike vinca alkaloids, which affect the rates of tubulin polymerization, the taxanes inhibit microtubule depolymerization in a dose-dependent manner. The microtubules formed in the presence of taxanes are extraordinarily stable and dysfunctional, thereby causing the death of the cell by disrupting the normal microtubule dynamics required for cell division.
Taxanes are rapidly cleared from the plasma despite extensive binding to proteins. The volume of distribution is large, suggesting binding to cellular proteins, possibly tubulin. Renal clearance accounts for a small proportion (<10%) of total clearance. Hepatic metabolism, biliary excretion, fecal elimination, or extensive tissue binding appears to be responsible for most of the plasma clearance.
Side Effects
Taxanes are associated with myelosuppression, peripheral neuropathy, and alopecia. Severe neurotoxicity precludes the administration of high doses of taxanes. The peripheral neuropathy is characterized by sensory symptoms such as numbness and paresthesia in a glove-and-stocking distribution. Patients may also experience transient taxane-associated arthralgias and myalgias for several days following treatment. Cardiac effects, including dysrhythmias, myocardial ischemia, and transient asymptomatic bradycardia may be more common with paclitaxel than docetaxel. Docetaxel seems to have unique vascular permeability properties which may result in peripheral edema, pleural effusion, and ascites. Fluid retention produced by docetaxel is dose-dependent and may be decreased by pretreatment with dexamethasone. Docetaxel also produces skin toxicities including an erythematous maculopapular rash on the forearms and hands. Hypersensitivity reactions (flushing, bronchospasm, dyspnea, systemic hypotension) caused by direct release of histamine or other chemical mediators may occur in 25% to 30% of patients treated with taxanes.38
Estramustine
Estramustine exerts its chemotherapeutic effects by inhibition of microtubule assembly and depolymerization. This drug also binds to an estramustine-binding protein in prostate tissue explaining the possible usefulness of this drug as part of combination therapy of hormone-refractory prostate cancer.
Signal Transduction Modulators
Signal transduction modulators (hormones) that may be useful in the treatment of neoplastic disease include antiestrogens, antiandrogens, aromatase inhibitors, gonadotropin-releasing drugs, and progestin. Normal cell division results from the interaction of growth factors with specific receptors. This interaction initiates a series of enzyme reactions (signal transduction), culminating in activation of nuclear transcription factors that produce cell proliferation molecules. Mutations in cancer cells result in uncontrolled cell proliferation using activated signaling pathways. Hormonal treatment of cancer disrupts growth factor receptor interactions.
Progestins
Progestational drugs are useful in the management of patients with endometrial carcinoma. Progestins act by reducing the production of hormones that stimulate the neoplastic endometrium.
Estrogens and Androgens
Malignant changes in the breast and prostate often depend on hormones for their continued growth. For example, prostatic cancer is stimulated by androgens, whereas orchiectomy or estrogens (diethylstilbestrol) slow the growth of the tumor cells. Eventually, prostatic tumors become insensitive to the lack of androgen or the presence of estrogens, presumably because of the survival of progressively undifferentiated cells that favor the emergence of cell types that no longer depend on androgens for their growth.
Malignant tissues that are responsive to estrogens contain receptors for the hormone, whereas malignant tissues lacking these receptors are unlikely to respond to hormonal manipulation. The onset of action of hormone therapy is slow, requiring 8 to 12 weeks.
Hypercalcemia may be associated with androgen or estrogen therapy, requiring adequate hydration in an attempt to facilitate renal excretion of calcium. Plasma calcium concentrations should be determined in patients receiving treatment with these hormones.
Antiestrogens
Antiestrogens, such as tamoxifen, are useful in the treatment of breast cancer that expresses estrogen or progesterone receptors. The estrogen receptor resides in the cytosol and, upon occupation by estradiol, is transported to the nucleus, where it activates genes (including those genes that encode proliferation molecules) containing estrogen-response elements. Tamoxifen binds to estrogen receptors and disrupts receptor interactions with estrogen in some but not all estrogen-responsive tissues. For example, tamoxifen is antiestrogenic in breast and ovarian tissue but is estrogenic in the uterus, liver, and bone. As a result, tamoxifen is effective in the prevention and treatment of breast cancer but produces undesired estrogen side effects including deep vein thrombosis (<1% of patients), endometrial cancer (about 0.3% of patients), and early menopausal symptoms (hot flashes). In addition, tamoxifen lowers plasma cholesterol concentrations and increases bone density.
The response to tamoxifen is proportional to the degree of expression of estrogen receptors in the breast cancer. Tamoxifen is of little benefit in women with breast cancer that does not express hormone receptors. After surgical removal of the primary breast cancer, adjuvant treatment of women with tumors that are hormone-positive decreases the odds of recurrence by more than 30%. Other antiestrogens that may have greater selectivity for breast estrogen receptors include raloxifene, toremifene, and fulvestrant.
Antiandrogens
Antiandrogens such as flutamide, bicalutamide, and nilutamide are competitive antagonists of the interactions between androstenedione and androgen receptors. Flutamide is a nonsteroidal antiandrogenic that possesses pure antiandrogenic activity when metabolized to its hydroxylated derivative. Administered with other drugs that decrease androgen production, flutamide is an effective treatment for hormone-dependent prostate cancer. Androgenic blockade results in feminizing side effects in men, including gynecomastia, hot flashes, and loss of facial hair. Skeletal muscle weakness and development of osteoporosis reflect a male menopause–like state. Flutamide can induce methemoglobinemia.39 Pulse oximetry readings in the presence of methemoglobinemia can overestimate the hemoglobin saturation levels. At levels of methemoglobinemia of greater than 35%, the pulse oximetry readings tend to approach a minimal level of 85%.40
Monoclonal Antibodies
Antibody-based therapies for treatment of cancer include the monoclonal antibodies trastuzumab, alemtuzumab, rituximab, and imatinib, which target specific antigen sites on cancer cells.41 The mechanisms of action of these agents are specific and varied depending on the target. Rituximab is used to treat leukemias and lymphomas. Rituximab binds to a specific protein (CD20) that is expressed on the cell surface of B cells; the rituximab–CD20 complex appears to improve the effectiveness of natural killer cells in killing these diseased cells. Imatinib is a monoclonal antibody that binds to the extracellular domain of a specific tyrosine kinase (BCR-ABL) and inhibits this enzyme, which is responsible for cell proliferation in Philadelphia chromosome–positive chronic myelocytic leukemia. Vaccines are also being developed that may include genetic manipulation of cancer cells to make them more antigenic and thus susceptible to immune responses. Two types of vaccines aimed at preventing infection related to subsequent development of cancer have been approved by the U.S. Food and Drug Administration (FDA): vaccines against the hepatitis B virus, which can cause liver cancer, and vaccines against specific human papillomaviruses, which are responsible for the majority of cases of cervical cancer. The FDA has approved one cancer treatment vaccine for certain men with metastatic prostate cancer.
Aromatase Inhibitors
Aromatase is an enzyme complex consisting of two proteins, aromatase cytochrome P450 (CYP19) and nicotinamide adenine dinucleotide phosphate cytochrome P450 reductase. Inhibition of aromatase blocks the conversion of androgens to estrone in peripheral tissues including breast tissue. The high affinity of the aromatase inhibitors anastrozole and letrozole for CYP19 results in intense inhibition of estrogen effects on responsive receptors. Inhibition of aromatase is an effective treatment for postmenopausal women with breast cancer, in which the greatest source of estrone comes from conversion of androstenedione to estrone in liver, skeletal muscles, and fat. Exemestane is a steroidal aromatase inhibitor that binds to the enzyme complex and promotes enzyme degradation.
A partial medical hypophysectomy is produced by luteinizing hormone–releasing hormone agonists, such as leuprolide, buserelin, and goserelin, which inhibit secretion of follicle-stimulating hormone and luteinizing hormone by downregulating receptors that respond to these hormones. The result is insignificant plasma concentrations of sex hormones and palliation of breast and prostate cancer.
References
1. Caley A, Jones R. The principles of cancer treatment by chemotherapy. Surgery (Oxford). 2012;30:186–190.
2. Finn OJ. Immuno-oncology: understanding the function and dysfunction of the immune system in cancer. Ann Oncol. 2012;23(suppl 8):viii6–viii9.
3. Damia G, Garattini S. The pharmacological point of view of resistance to therapy in tumors. Cancer Treat Rev. 2014:40(8):909–916.
4. Rubin EH, Hait WN. Principles of cancer treatment. Sci Am Med. 2003;12(IV):1–17.
5. Chung F. Cancer, chemotherapy, and anesthesia. Can Anaesth Soc J. 1982;29:364–371.
6. Selvin BF. Cancer chemotherapy: implications for the anesthesiologist. Anesth Analg. 1981;60:425–434.
7. Oddby-Muhrbeck E, Öbrink E, Eksborg S, et al. Is there an association between PONV and chemotherapy-induced nausea and vomiting? Acta Anaesthesiol Scand. 2013;57:749–753.
8. Norris JC. Prolonged succinylcholine apnoea resulting from acquired deficiency of plasma cholinesterase. Anaesthesia. 2003;58:1137.
9. Zsigmond EK, Robins G. The effect of a series of anticancer drugs on plasma cholinesterase activity. Can Anaesth Soc J. 1972;19:75–82.
10. Gottdiener JS, Appelbaum FR, Ferrans VJ, et al. Cardiotoxicity associated with high-dose cyclophosphamide therapy. Arch Intern Med. 1981;141:758–762.
11. Ginsberg SJ, Comis RL. The pulmonary toxicity of antineoplastic agents. Semin Oncol. 1982;9:34–51.
12. Weiss RB, Poster DS, Penta JS. The nitrosoureas and pulmonary toxicity. Cancer Treat Rev. 1981;8:111–125.
13. Gunstream SR, Seidenfeld JJ, Cobonya RE, et al. Mitomycin-associated lung disease. Cancer Treat Rev. 1983;67:301–304.
14. White DA, Orenstein M, Godwin TA, et al. Chemotherapy-associated pulmonary toxic reactions during treatment for breast cancer. Arch Intern Med. 1984;144:953–956.
15. Cooper JA Jr, White DA, Matthay RA. Drug-induced pulmonary disease. Part 1: cytotoxic drugs. Am Rev Respir Dis. 1986;133:321–340.
16. Perazella MA, Moeckel GW. Nephrotoxicity from chemotherapeutic agents: clinical manifestations, pathobiology, and prevention/therapy. Semin Nephrol. 2010;30:570–581.
17. Kaplan RS, Wienik PH. Neurotoxicity of antineoplastic drugs. Semin Oncol. 1982;9:103–110.
18. Labianca R, Beretta G, Clerici M. Cardiac toxicity of 5-fluorouracil: a study of 10,083 patients. Tumor. 1982;68:505–509.
19. Saven A, Piro L. Newer purine analogues for the treatment of hairy-cell leukemia. N Engl J Med. 1994;330:691–697.
20. Doroshow JH. Doxorubicin-induced cardiac toxicity. N Engl J Med. 1991;324:843–845.
21. Von Hoff DD, Layard MW, Basa P, et al. Risk factors for doxorubicin-induced congestive heart failure. Ann Intern Med. 1979;91:710–717.
22. Huettemann E, Junker T, Chatzinikolaou KP, et al. The influence of anthracycline therapy on cardiac function during anesthesia. Anesth Analg. 2004;98:941–947.
23. Borgeat A, Chiolero R, Baylon P, et al. Perioperative cardiovascular collapse in a patient previously treated with doxorubicin. Anesth Analg. 1988;67:1189–1191.
24. Lipshultz SE, Rifai N, Dalton VM, et al. The effect of dexrazoxane on myocardial injury in doxorubicin-treated children with acute lymphoblastic leukemia. N Engl J Med. 2004;351:145–153.
25. Dorr RT. Bleomycin pharmacology: mechanism of action and resistance, and clinical pharmacokinetics. Semin Oncol. 1992;19:3–8.
26. Allen SC, Riddell GS, Butchart EG. Bleomycin therapy and anaesthesia: the possible hazards of oxygen administration to patients after treatment with bleomycin. Anaesthesia. 1981;60:121–124.
27. Eigen H, Wyszomierski D. Bleomycin lung injury in children. Pathophysiology and guidelines for management. Am J Pediatr Hematol Oncol. 1985;7:71–78.
28. Hulbert JC, Grossman JE, Cummings KB. Risk factors of anesthesia and surgery in bleomycin-treated patients. J Urol. 1983;130:163–164.
29. Hay JG, Haslam PL, Dewar A, et al. Development of acute lung injury after the combination of intravenous bleomycin and exposure to hyperoxia in rats. Thorax. 1987;42:374–382.
30. Mathes DD. Bleomycin and hyperoxia exposure in the operating room. Anesth Analg. 1995;81:624–629.
31. Blom-Muilwijk MC, Vriesendorp R, Veninga TS, et al. Pulmonary toxicity after treatment with bleomycin along or in combination with hyperoxia. Studies in the rat. Br J Anaesth. 1988;60:91–97.
32. Comis RL. Bleomycin pulmonary toxicity: current status and future directions. Semin Oncol. 1992;19:64–70.
33. Wilson L, Jordan MA. Microtubule dynamics: taking aim at a moving target. Chem Biol. 1995;2:569–575.
34. Postma TJ, Benard BA, Huijgens PC, et al. Long term effects of vincristine on the peripheral nervous system. J Neurooncol. 1993;15:23–27.
35. Vainionpaa L, Kovala T, Tolonen U, et al. Vincristine therapy for children with acute lymphoblastic leukemia impairs conduction in the entire peripheral nerve. Pediatr Neurol. 1995;13:314–318.
36. Delaney P. Vincristine-induced laryngeal nerve paralysis. Neurology. 1982;32:1285–1288.
37. Lirk P, Birmingham B, Hogan Q. Regional anesthesia in patients with preexisting neuropathy. Int Anesthesiol Clin. 2011;49:144–165.
38. Rowinsky EK, Donehower RC. Paclitaxel (Taxol). N Engl J Med. 1995;332:1004–1014.
39. Jackson SH, Barker SJ. Methemoglobinemia in a patient receiving flutamide. Anesthesiology. 1995;82:1065–1067.
40. Barker SJ, Tremper KK, Hyatt J. Effects of methemoglobinemia on pulse oximetry and mixed venous oximetry. Anesthesiology. 1989;70:112–117.
41. Dillman RO. Radiolabled anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoma. J Clin Oncol. 2002;20:3345–3351.