An analgesic is any member of the group of drugs that are used to decrease pain sensation without loss of consciousness. The analgesic drugs may act on the peripheral nervous system and/or central nervous system (CNS). Peripheral analgesics act at the sensory input level by blocking transmission of the impulse to the brain. Their common feature was believed to be their site of action within the damaged tissues, and hence they were termed peripheral analgesics. Experimental and clinical studies support the possibility of central site of action of many of these agents. In addition, peripheral administration of drugs can potentially optimize drug concentrations at the site of the origin of pain while leading to lower systemic levels and fewer adverse systemic effects and fewer drug interactions.
Recent efforts have been focused on peripherally selective compounds with limited ability to cross the blood–brain barrier (BBB). Nociceptive, inflammatory, and neuropathic pain all depend to some degree on the peripheral activation of primary sensory afferent neurons. A range of inflammatory mediators such as prostanoids, bradykinin, adenosine triphosphate (ATP), histamine, and serotonin can activate primary sensory afferent neurons. Inhibiting the actions of inflammatory mediators represents a strategy for the development of analgesics. Peripheral nerve endings also express a variety of inhibitory receptors such as opioid, α-adrenergic, cholinergic; adenosine and cannabinoid receptors, and agonists for these receptors also represent viable targets for drug development. The transmission of a pain signal from the periphery to the CNS is complex. Tissue damage results in peripheral release of endogenous mediators that can directly activate nociceptive afferent fibers, sensitize nociceptors, and/or cause increased local extravasation and vasodilatation. Nociceptive afferents have their neurons in the dorsal root ganglion and contact second-order neurons in the dorsal horn (for more details, see Chapter 6, Pain Physiology). Combinations of agents that act via different mechanisms may be particularly useful.
Nonsteroidal Antiinflammatory Drugs
The nonsteroidal antiinflammatory drugs (NSAIDs) are among the most commonly prescribed drugs in the world. This diverse class of drugs includes aspirin and several other selective and nonselective cyclooxygenase (COX) inhibitors with common analgesic, antiinflammatory, and antipyretic properties.1 The COX pathway is shown in Figure 9-1. NSAIDs inhibit the biosynthesis of prostaglandins by preventing the substrate arachidonic acid from binding to the COX enzyme active site. The COX enzyme exist in two isoforms—COX-1 and COX-2 isoenzymes.1
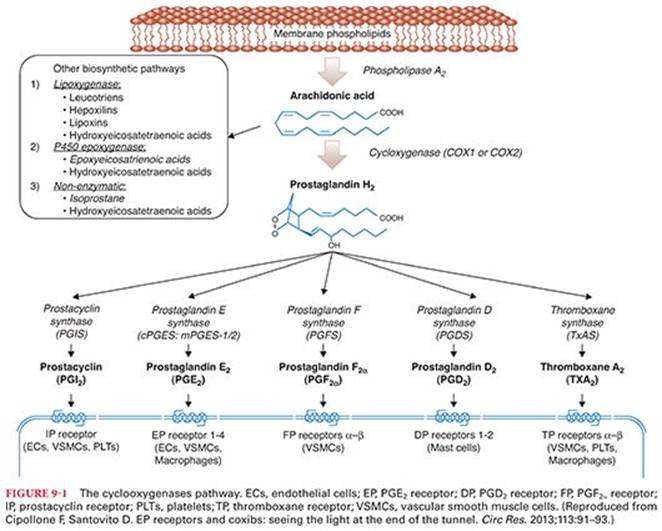
COX-1 is constitutively expressed and catalyzes the production of prostaglandins that are involved in numerous physiologic functions, including maintenance of normal renal function in the kidneys, mucosal protection in the gastrointestinal tract, and production of proaggregatory thromboxane A2 in the platelets. COX-2 expression can be induced by inflammatory mediators in many tissues and has a role in the mediation of pain, inflammation, and fever. There has been speculation on the existence of a third isoform, COX-3, which would explain the mechanism of action of acetaminophen, a poor inhibitor of COX-1 and COX-2, but appears to have little relevance in humans. Evidence indicates that, in addition to peripheral blockade of prostaglandin synthesis, central inhibition of COX-2 may play an important role in modulating nociception.
COX-2 selective inhibitors (known as the coxibs) have less gastrointestinal toxicity than nonselective NSAIDs. However, increased cardiovascular risk has been associated with the use of this class of drugs.2They are used less frequently nowadays after withdrawal of rofecoxib and valdecoxib, following reports of excessive cardiac morbidity. Currently, celecoxib is the only COX-2 selective inhibitor available for clinical use. COX-2–selective inhibitors should be used cautiously in patients with underlying cardiovascular disease (see further discussion on “Cardiovascular Side Effects” section).
Coxibs may be a safer alternative to NSAIDs in the perioperative settings. Although nonspecific NSAIDs provide analgesic efficacy similar to coxibs, their use has been limited in the perioperative setting because of platelet dysfunction and gastrointestinal toxicity. The potential benefits of coxibs include improved quality of analgesia, reduced incidence of gastrointestinal side effects versus conventional NSAIDs, and no platelet inhibition. NSAIDs can be classified according to numerous characteristics, including COX selectivity, and chemical and pharmacologic properties
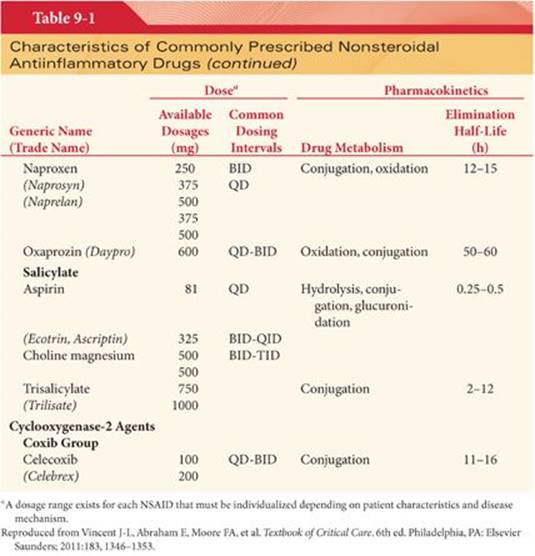
NSAIDs belong to a number of chemical families including acetic acids, oxicams, propionic acids, salicylates, fenamates, furanones, and coxibs (Table 9-1). All NSAIDs are weakly acidic chemical compounds and share similarities in pharmacokinetic properties.3 The volume of distribution of NSAIDs is low, ranging from 0.1 to 0.3 L/kg, suggesting minimal tissue binding. The plasma half-life of NSAIDs ranges from 0.25 to >70 hours, indicating wide differences in clearance rates. Hepatic or renal disease can alter NSAID protein binding and metabolism.4

Gastrointestinal absorption of NSAIDs occurs rapidly, usually within 15 to 30 minutes. After absorption, NSAIDs are more than 90% bound to albumin, which influences their distribution and drug-drug interaction potential. Hypoalbuminemia (e.g., due to alcoholic liver disease) can result in greater unbound drug and increased risk for NSAID-related adverse events.4 The liver metabolizes most NSAIDs, with subsequent excretion into urine or bile. Enterohepatic recirculation occurs when a significant amount of an NSAID or its conjugated metabolites are excreted into the bile and then reabsorbed in the distal intestine. Hepatic NSAID elimination is dependent on the free fraction of NSAID within the plasma and the intrinsic enzyme activity of the liver. NSAIDs are primarily eliminated by renal and biliary excretion. Reduced renal function prolongs NSAID half-life, and the dose should be lowered proportionally in patients with impaired kidney function.3,4 Moderate to severe liver disease impairs NSAID metabolism, increasing the potential for toxicity.
Side Effects of Nonsteroidal Antiinflammatory Drugs
Platelet Function
Platelet aggregation and thus the ability to clot is primarily induced through an increase in thromboxane production following activation of platelet COX-1. There is no COX-2 isoenzyme in the platelet. NSAIDs and aspirin inhibit the activity of COX-1, but the COX-2–specific inhibitors (or COX-1–sparing drugs) have no effect on COX-1 and thus no effect on platelet function.5
Gastrointestinal Side Effects
NSAIDs are associated with a spectrum of upper gastrointestinal complications, ranging from endoscopic ulcers in 10% to 30% of patients to serious ulcer complications in 1% to 2% of patients, including perforation and bleeding.6,7Lower gastrointestinal tract complications are less well characterized.8,9
Risk factors for NSAID-associated gastrointestinal complications include high NSAID dose, older age, Helicobacter pylori infection, a history of prior ulcer, and concomitant use of low-dose aspirin, anticoagulants, or corticosteroids.10,11 Therefore, it is generally recommended that patients with gastrointestinal risk factors should be treated with COX-2–selective agents or nonselective NSAIDs with gastrointestinal protective cotherapy.12,13
Cardiovascular Side Effects
NSAIDs are associated with an increased risk of cardiovascular adverse events such as myocardial infarction, heart failure, and hypertension. COX inhibition is likely to disturb the balance between COX-2–mediated production of proaggregatory thromboxane in platelets and antiaggregatory prostaglandin I2 in endothelial cells. COX selectivity alone is not sufficient to define the risk of NSAID-associated cardiovascular complications. Based on two studies, the Vioxx Gastrointestinal Outcomes Research (VIGOR)14 study and the Adenomatous Polyp Prevention on Vioxx (APPROVe) study,15 rofecoxib was withdrawn from the market in 2004. Valdecoxib was subsequently withdrawn in 2005 due to a fourfold increase in the incidence of myocardial infarction.
The cardiovascular safety of nonselective NSAIDs has been under recent investigation. A meta-analysis of randomized trials found that high-dose ibuprofen and high-dose diclofenac were associated with a moderately increased risk of vascular events compared with placebo, similar to that observed with COX-2–selective agents; the risks associated with naproxen, although they cannot be completely excluded, appeared to be substantially lower.16
Studies are lacking on the long-term effects of nonselective NSAIDs on gastrointestinal and cardiovascular systems, which limit our understanding of the true benefits and risks of NSAIDs over the long term. To reduce the cardiovascular risks, the American Heart Association recommends that all NSAIDs should be used at their lowest effective dose. When NSAID therapy is required for patients at risk of cardiovascular complications, naproxen is recommended as the NSAID of choice.17 A recent meta-analysis of the vascular and upper gastrointestinal effects of NSAIDs confirms that diclofenac and ibuprofen raise risk of major vascular events as much as coxibs. Naproxen has no effect on vascular outcomes but does increase upper gastrointestinal complications.18
Renal Side Effects
The effects of the NSAIDs on renal function include changes in the excretion of sodium, changes in tubular function, potential for interstitial nephritis, and reversible renal failure due to alterations in filtration rate and renal plasma flow. Prostaglandins and prostacyclins are important for maintenance of intrarenal blood flow and tubular transport. All NSAIDs, except nonacetylated salicylates, have the potential to induce reversible impairment of glomerular filtration rate; this effect occurs more frequently in patients with congestive heart failure; established renal disease with altered intrarenal plasma flow including diabetes, hypertension, or atherosclerosis; and with induced hypovolemia, salt depletion or significant hypoalbuminemia.19,20 Avoiding perioperative use of NSAIDs in patients with hypovolemia from any cause is an important means of minimizing renal injury.
Liver Side Effects
The use of aspirin was associated with reduced risk of developing hepatocellular carcinoma and of death due to chronic liver disease, whereas non-aspirin NSAID use was only associated with reduced risk of death due to chronic liver disease.21 Paradoxically, elevations in hepatic transaminase levels and liver failure have been reported with some of the NSAIDs.22
Pulmonary Side Effects
Many adverse reactions attributed to NSAIDs are due to inhibition of prostaglandin synthesis in local tissues. For example, patients with allergic rhinitis, nasal polyposis, and/or a history of asthma, in whom all NSAIDs effectively inhibit prostaglandin synthetase, are at increased risk for anaphylaxis.23 The use of selective COX-2 inhibitors as an alternative to aspirin and other NSAIDs has been suggested for patients with aspirin-exacerbated respiratory disease. The highly selective COX-2 inhibitor etoricoxib has been shown to be tolerated in most but not all patients tested.24 An oral provocation test is therefore recommended before prescribing etoricoxib for patients with aspirin-exacerbated respiratory disease.24
Hypersensitivity Reactions
Hypersensitivity reactions to NSAIDs do rarely occur, and they are more common in individuals with nasal polyps or asthma. Allergic reactions include bronchoconstriction, rhinitis, and urticaria. Recent data suggest a role of altered COX-2 regulation associated with the aspirin-intolerant asthma/rhinitis syndrome.23 Because of the potential for cross-reactivity, avoidance of all NSAIDs is recommended. In rare cases, NSAIDs have been implicated in causing aseptic meningitis and, in children, Reye syndrome.25
Idiosyncratic Adverse Effects
Typical nonspecific reactions include skin rash and photosensitivity, aseptic meningitis, tinnitus, hearing loss, and neutropenia. The effect of prostaglandin inhibition may result in premature closure of the ductus arteriosus. Acetylsalicylic acid (ASA) has been associated with small for gestational age neonates and neonatal bruising; however, it has been used for many years in the treatment of patients who require NSAIDs while pregnant.26 The most common toxicities associated with NSAIDs are gastrointestinal, cardiovascular, and renal and are related primarily to COX inhibition and decreased synthesis of prostaglandins.
Drug-Drug Interactions
Drug-drug interactions with NSAID therapy may result from their pharmacodynamic or pharmacokinetic interactions. Nonselective NSAIDs affect other antiplatelet agents via additive inhibition of platelet aggregation. The result is an increased bleeding risk with the concomitant use of NSAIDs and other antiplatelet agents.25,27
Significant drug-drug interactions have been documented with use of NSAIDs and lithium. NSAIDs decrease lithium clearance and increase serum lithium concentrations by inhibiting renal prostaglandin production and altering intrarenal blood flow.25,27,28 Data are conflicting regarding the drug-drug interaction potential of angiotensin-converting enzyme (ACE) inhibitors and NSAIDs.29
Concurrent administration of digoxin and NSAIDs can decrease renal clearance of digoxin, increase plasma drug concentration, and potentiate digoxin toxicity. NSAIDs interact with anticonvulsant agents such as phenytoin and valproic acid by displacing the anticonvulsants from their protein-binding sites, which increases the free drug concentration. Combination use of corticosteroids and aspirin can increase renal clearance of salicylate and significantly decrease plasma salicylate concentrations.27,28
NSAIDs can be used in selected critically ill patients but should be used judiciously because of the potential for toxic adverse events, particularly renal toxicity in hypovolemic patients. The lowest effective dose of the NSAID should be used for the shortest duration indicated. Appropriate clinical and laboratory follow-up is necessary.
Acetaminophen
Acetaminophen (Tylenol, also known as paracetamol, N-acetyl-p-aminophenol, and nAPAP) is a popular antipyretic and analgesic found in many over-the-counter and prescription products. Acetaminophen is antipyretic and analgesic but has little, if any, antiinflammatory action. Acetaminophen is the leading cause of acute liver failure in the United States, and nearly half of acetaminophen-associated cases are due to unintentional overdose.
Acetaminophen has a central analgesic effect that is mediated through activation of descending serotonergic pathways. Debate exists about its primary site of action, which may be inhibition of prostaglandin synthesis. The mechanism of action has been debated. In animal models, it has been seen to inhibit COX-3. At the spinal cord level, it has been shown to antagonize neurotransmission by N-methyl-D-aspartate (NMDA), substance P, and nitric oxide pathways.
Oral acetaminophen has excellent bioavailability. Acetaminophen is suitable for analgesic or antipyretic uses; it is the first-line analgesic in osteoarthritis and particularly valuable for patients in whom aspirin is contraindicated (e.g., those with peptic ulcer disease, aspirin hypersensitivity, and children with febrile illness). The conventional oral dose of acetaminophen is 325 to 650 mg every 4 to 6 hours; total daily doses should not exceed 4,000 mg (2,000 mg per day for chronic alcoholics). In efforts to reduce the incidence of hepatotoxicity, a U.S. Food and Drug Administration (FDA) advisory panel recommended in 2009 a lower maximum daily dose of acetaminophen of 2,600 mg and a decrease in the maximum single dose from 1,000 mg to 650 mg.
An intravenous (IV) preparation of acetaminophen is currently available for clinical use. Optimal analgesia for moderate to severe postoperative pain cannot be achieved using a single agent alone.30 IV paracetamol provides around 4 hours of effective analgesia for about 37% of patients with acute postoperative pain.31 With its inherent safety and demonstrated efficacy, IV acetaminophen can prove to be an asset in managing perioperative pain.
Current evidence suggests that a combination of paracetamol and an NSAID may offer superior analgesia compared with either drug alone.32
Acetaminophen is well tolerated and has a low incidence of gastrointestinal side effects. However, acute overdosage can cause severe hepatic damage, and the number of accidental or deliberate poisonings with acetaminophen continues to grow. Chronic use of <2 g per day is not typically associated with hepatic dysfunction, but overuse of acetaminophen-containing narcotic and over-the-counter combination products marketed in the United States has led to heightened awareness of the possibility of toxicity.
The pharmacology of acetaminophen overdose, including its time course and treatment is interesting and important to understand. Damage to the liver results from one of acetaminophen’s metabolites, N-acetyl-p-benzoquinoneimine (NAPQI). NAPQI leads to liver failure by depleting the liver’s natural antioxidant glutathione and directly damaging liver cells, leading to liver failure. Treatment is aimed at removing the paracetamol from the body and replacing glutathione. Activated charcoal can be used to decrease absorption of acetaminophen in those who present, soon after ingestion of, an overdose. Acetylcysteine is administered as an antidote and acts as a precursor for glutathione and can neutralize NAPQI directly. Patients treated early after ingestion have a good prognosis.
Acetylsalicylic acid (Aspirin)
Aspirin is the oldest and most widely used medicinal compound in the world. It is considered separately from the NSAIDs due to its predominant use in the treatment of cardiovascular and cerebrovascular diseases. Aspirin is found in hundreds of over-the-counter medicines worldwide, and remains at the forefront of medicine, with newly discovered applications for the prevention and treatment of several life-threatening diseases. Aspirin is a derivative of salicylic acid. Aspirin and salicylate are rapidly metabolized in the plasma (e.g., by plasma esterases), erythrocyte, and liver, to salicylate in vivo.33
Aspirin has several different approved uses. Aspirin acts as a general analgesic by blocking the action of the COX enzymes and thus prevents the production of prostaglandins. Aspirin effectively treats headaches, back and muscle pain, and other general aches and pains. In addition, aspirin produces inhibition of COX and thus prostanoid synthesis34 and also protein kinase.35 However, these are not necessarily the most likely mechanisms.36 Aspirin irreversibly inactivates COX, leading to prolonged inhibition of platelet aggregation.
Overdose
The mechanism of NSAID toxicity in overdose is related to both their acidic nature and their inhibition of prostaglandin production. The severity typically depends on the dose ingested and the salicylate concentration that correlates with the degree of acid–base disturbance.29,37 Salicylate levels of 300 to 600 mg/L are associated with mild toxicity, 600 to 800 mg/L with moderate toxicity, and greater than 800 mg/L with severe toxicity. For nonselective NSAIDs, plasma concentrations are not commonly measured because the half-life of many of these agents is relatively short.37
Symptoms include nausea, vomiting, abdominal pain, tinnitus, hearing impairment, and CNS depression (Table 9-2); with higher dose aspirin ingestion, metabolic acidosis, renal failure, CNS changes (e.g., agitation, confusion, coma), and hyperventilation with respiratory alkalosis due to stimulation of the respiratory center. The presence of acidemia permits more salicylic acid to cross the BBB.38 With other nonselective NSAID ingestions, symptoms are similar.37–39

Management should be directed at symptomatic support, prevention of further absorption, and correction of acid–base imbalance.37,38 There is no antidote for salicylate or NSAID poisoning. Appropriate hydration and activated charcoal should be considered within 1 hour after ingestion. Urine alkalinization increases salicylate elimination.37 In severe cases of aspirin overdose, hemodialysis is effective at removing salicylate and correcting acid–base imbalances and has been shown to reduce morbidity and mortality.38
Steroids
Glucocorticoids have been used to reduce inflammation and tissue damage in a variety of conditions, including inflammatory bowel disease and rheumatoid arthritis. Its antiinflammatory action results in decreased production of various inflammatory mediators that play a major role in amplifying and maintenance of pain perception. Another proposed mechanism is by inhibition of phospholipase A2 as well as changes in cell function induced by glucocorticoid receptor activation.
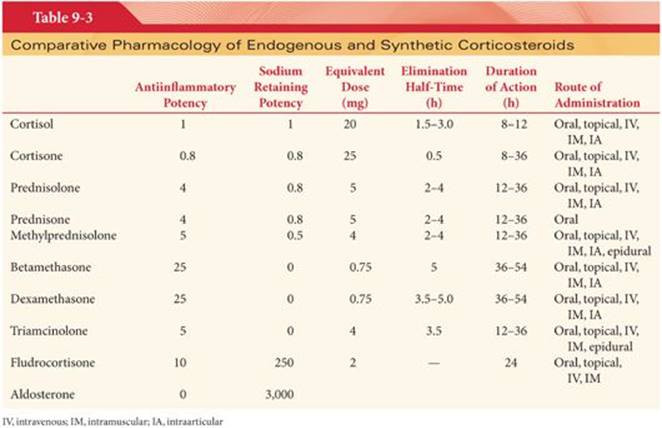
Glucocorticoids have the most powerful antiinflammatory characteristics of all steroids. Corticosteroids are a subgroup of compounds known as adrenocorticoids that are naturally secreted from the adrenal gland. The primary corticosteroid is hydrocortisone, which is the standard against which the pharmacologic properties of various synthetic corticosteroids are judged. Many synthetic agents that are more potent, have longer durations of action, have greater antiinflammatory activity, and generate fewer unwanted mineralocorticoid side effects than hydrocortisone have been developed. Mineralocorticoids are adrenal cortical steroid hormones that have a greater effect on water and electrolyte balance. The main endogenous hormone is aldosterone.
Different steroids vary with respect to their duration of action and relative corticosteroid and mineralocorticoid activity. Corticosteroids are divided into short, intermediate, and long-acting groups (Table 9-3). Short- and long-acting preparations cause less inhibition of the hypothalamic-pituitary-adrenal axis. Many of the unwanted side effects are related to the mineralocorticoid properties (Table 9-4).

Nearly all routes of administration can be used for corticosteroids. Although associated with significant toxicity when administered in large doses for long periods, adverse effects with a single dose of dexamethasone are minor.40
The use of corticosteroids for pain relief, although popular, has yet to gain wider acceptance because of concerns over side effects, such as adrenal suppression, osteonecrosis, impaired wound healing, and concerns about efficacy. There is evidence supporting the use of corticosteroids in multimodal analgesia protocols to contribute to the postoperative recovery of the patient by minimizing opioid doses and therefore side effects. However, the optimal mode, dose, and timing of administration remain unclear.41
In recent meta-analysis,42 patients treated with dexamethasone experienced less postoperative pain, required less postoperative opioids, had longer time to first analgesic dose, needed less rescue analgesia, and had shorter postanesthesia care unit (PACU) stays. Differences between the groups were however small and may not be clinically relevant. Perioperative dosing of dexamethasone had small but statistically significant analgesic benefits.42Investigators have begun to evaluate glucocorticoids as adjuvants for regional anesthesia. There is some evidence for an analgesic effect of local, spinal, and systemic corticosteroids in combination with bupivacaine.43Dexamethasone has been found to prolong local anesthetic block duration in animal and human studies, and adding methylprednisolone to local anesthetic increases the duration of axillary brachial block.44
Dexamethasone also prolongs the analgesia from interscalene blocks using ropivacaine or bupivacaine, with the effect being stronger with ropivacaine; the combined effect of dexamethasone and either drug produced nearly 22 hours of analgesia. Systemic glucocorticoids have also been shown to reduce postoperative pain. This raises the question whether the beneficial effects of adding glucocorticoid to a regional anesthetic is solely due to local effect or is mediated at least in part by systemic action.45
Steroids often administered to patients with arthritis and other chronic pain conditions, locally (e.g., intraarticularly) to limit the systemic side-effect profiles. Pain relief from glucocorticoid treatment has been reported to last for up to 3 weeks in osteoarthritis and 2 months in rheumatoid arthritis.46
Epidural injection of corticosteroids has been used to treat back pain (mainly due to nerve root irritation) in patients with a wide variety of spine pathologies including radiculopathy, spinal stenosis, disc-space narrowing, annular tears, spondylosis, spondylolisthesis, vertebral fractures, and postlaminectomy syndrome.46 The only proven efficacy of epidural steroid injections is their ability to speed resolution of leg pain (“sciatica”) in patients with acute intervertebral disc herniation and associated radicular pain.
Systemic Local Anesthetics
Lidocaine produces analgesia by suppressing the activity of sodium channels in neurons that respond to noxious stimuli, thereby preventing nerve conduction and pain transmission. Voltage-gated sodium channels (VGSCs) play a fundamental role in the control of neuronal excitability. Local anesthetics that block VGSCs have long been used to abolish pain temporarily by blocking nerve conduction.
Systemically administered local anesthetics such as IV lidocaine, oral mexiletine, and oral tocainide are effective in a number of chronic pain conditions. Early studies described successful treatment of acute pain syndromes such as postoperative pain,47 burn pain,48 and cancer pain.49 Subsequent clinical reports have demonstrated the effectiveness in reducing pain associated with many chronic pain conditions.50,51 Commonly used drugs include IV lidocaine and the orally active agents mexiletine and tocainide. The exact mechanism of action of systemic local anesthetics in pain control is unknown. Evidence suggests that the effect may involve selective blockade of pain fibers within the spinal cord or the dorsal root ganglia.52
Orally administered lidocaine has poor bioavailability. The elimination half-life is 1.5 to 2 hours and can increase in the event of decreased liver blood flow (e.g., congestive heart failure). Mexiletine has excellent oral bioavailability. The mean elimination half-life is 10 to 12 hours, which can increase to 25 hours with hepatic impairment. Only 10% of the drug is excreted unchanged by the kidney, and therefore renal impairment has minimal effect on half-life.53
At low doses, initial CNS symptoms include light-headedness, dizziness, tinnitus, vertigo, blurred vision, and altered taste. Seizures occur at higher doses. Cardiovascular side effects include hypotension, bradycardia, and cardiovascular collapse, which can lead to cardiac arrest. In a meta-analysis, when used for treatment of neuropathic pain, lidocaine and mexiletine produced no major adverse events in controlled clinical trials, were superior to placebo to relieve neuropathic pain, and were as effective as other analgesics used for this condition; however, the long-term use of oral mexiletine is limited by the nearly universal appearance of nausea.54
Topical Application of 5% Lidocaine
The topical application of 5% lidocaine have been used in postherpetic neuralgia, the topical application of 5% lidocaine (as a gel or patch) has been demonstrated to significantly relieve pain and reduce pain intensity with a fast onset (within 30 minutes) and lasting for the duration of drug application.55 Additionally, studies have also shown that lidocaine patches can provide pain relief in patients with various painful neuropathies.56
The use of a lidocaine patch 5% after different surgical procedures has been reported in several small studies.57,58
The most common adverse events generally involve mild skin reactions. There has been no reported drug-drug interaction in clinical trials. Recent evidence suggests that extended application does not result in Aβ-mediated sensory loss at the application site, which is particularly important in patients who already have a degree of sensory loss due to their underlying condition. The lidocaine patch provides a treatment option that carries a relatively low risk of systemic adverse effects and drug-drug interaction, even with continuous application of up to four patches per day. The efficacy of this approach alone would not be sufficient for providing adequate postoperative pain management.
Capsaicin
Capsaicin is a transient receptor potential vanilloid (TRPV1) channels agonist.59 TRPV1 is a receptor that is markedly reduced in inflammatory conditions and is present on unmyelinated C fiber endings in the periphery. The activation of the TRPV receptors releases high-intensity impulses and releases the neurotransmitter substance P, which results in the initial phase of burning. Continued release of substance P in the presence of capsaicin leads to the depletion of capsaicin and a subsequent decrease in C fiber activation.60 Capsaicin is the major pungent ingredient of hot chili peppers and other botanicals. Capsaicin 0.025%, 0.075%, and 0.25% creams and/or transdermal patches are available over-the-counter for the temporary relief of pain from arthritis, myalgias, arthralgias, and neuralgias. The FDA has granted capsaicin orphan drug status in the treatment of postherpetic neuralgia, intermetatarsal neuroma, erythromelalgia, and HIV-associated neuropathy. When used in the treatment of postherpetic neuralgia, a single treatment with the capsaicin 8% patch provided a pain intensity decrease of ≥30% for >35% of treated patients in weeks 2 through 12.61 The FDA approved the prescription-only capsaicin 8% patch (Qutenza) for the management of neuropathic pain associated with postherpetic neuralgia. Topically applied capsaicin has moderate to poor efficacy in the treatment of chronic musculoskeletal or neuropathic pain.62
Ketamine
There has been a renewed interest in the use of subanesthetic doses of ketamine as an adjunct to provide postoperative pain relief in opioid-dependent patients.63 NMDA receptor antagonists have been used in perioperative pain management. At low, subanesthetic doses (e.g., 0.15 to 1 mg/kg), ketamine exerts a specific NMDA blockade and, hence, modulates central sensitization induced both by the incision and tissue damage and by perioperative analgesics such as opioids.
There may be some role for ketamine in preventing opioid-induced hyperalgesia in patients receiving high doses of opioid for their postoperative pain relief.64 However, clinical use of ketamine can be limited due to psychotomimetic adverse effects and other common adverse effects, including dizziness, blurred vision, and nausea and vomiting.65 The usefulness of low-dose ketamine in the perioperative management of the opioid-tolerant patient is in need of further study.
Dexmedetomidine
Dexmedetomidine is a relatively new, highly selective, central α2 agonist. Its sedative, pro-anesthetic, and pro-analgesic effects at 0.5 to 2 µg/kg given intravenously stem mainly from its ability to blunt the central sympathetic response by an as yet unknown mechanism. It also minimizes opioid-induced muscle rigidity, lessens postoperative shivering, causes minimal respiratory depression, and has hemodynamic stabilizing effects. Dexmedetomidine, when used as an adjunct, can reduce postoperative morphine consumption in various surgical settings.66,67 A recent study has shown the analgesic efficacy of dexmedetomidine in postoperative pain relief. The authors of this study found that the addition of dexmedetomidine to IV PCA morphine resulted in superior analgesia, significant morphine sparing, and less morphine-induced nausea, while it was devoid of additional sedation and untoward hemodynamic changes.
Clonidine
The use of low doses of clonidine proved to be a useful adjunct analgesic when given neuraxially and in combination with peripheral nerve blocks. Data from the systemic administration of clonidine also support the usefulness of low-dose IV administration as an adjunct for postoperative pain management.68
Opioids
Since the detection of morphine by the pharmacologist Friedrich Sertürner in 1806, opioids have been used as potent centrally acting analgesics. In addition to the central site of action, peripheral endogenous opioid analgesic systems have been extensively studied. The three classes of opioid receptors are widely distributed in addition to the CNS, in peripheral neurons, neuroendocrine organs (pituitary, adrenals), immune, and ectodermal cells. Opioid receptors are synthesized in the dorsal root ganglia (DRG) and transported centrally and peripherally to the nerve terminals.69
Efforts continue to develop opioid analgesics unable to cross the BBB, which act only peripherally, thus providing adequate analgesia without central side effects. Although peripheral opioid receptors are largely expressed by the primary sensory neurons, they are functionally inactive under most basal conditions.70 The antinociceptive effect of systemic opioids is thought to be produced mainly at the central (particularly supraspinal) level,71 although peripheral opioid receptors might also participate.72,73
Inflammation increases expression, transport, and accumulation of peripheral opioid receptors on peripheral terminals of sensory nerves but also triggers migration of opioid-containing immunocytes.74 The peripheral effectiveness of opioids depends on the presence of inflammation, which triggers an enhanced expression of opioid receptors on primary afferents. Opioids were shown to have peripheral antinociceptive effects in inflammation.72,73Effective endogenous peripheral opioid analgesia requires an adequate number of functional opioid receptors on primary afferent neurons and a well-coordinated migration of opioid-secreting leukocytes out of the circulating blood to the inflamed site. Interestingly, this recruitment of opioid-containing cells to inflamed tissue has been shown to be suppressed by the administration of centrally acting opioids.75 On the contrary, several studies indicate that a large portion of the analgesic effects produced by systemically administered opioids can be mediated by peripheral opioid receptors.76 In an animal model of inflamed knee joint, intraarticular injection of µ and κ but not δ opioid agonists produced a dose-dependent blockade of autonomic response to a noxious stimulus.77
Opioids injected locally into soft tissues or joints produce potent analgesic effects that are mediated by peripheral (not central) opioid.77 Pain relief has been reported after knee arthroscopy after intraarticular injection of morphine 78 and after submucosal injection of morphine in patients undergoing dental surgery.79
The role of the opioid receptor system in wound healing is another area of intense interest and study.80 Peripherally acting opioids can reduce plasma extravasation, vasodilation, proinflammatory neuropeptides, immune mediators, and tissue destruction. Local administration of opioid agonists at low concentrations may offer a promising therapeutic strategy.79,81
There is strong evidence for a cardioprotective effect of both peripheral and central opioid receptors. The role of the different opioid receptor subtypes involved is still contradictory, but there is some evidence that opioids have infarct-sparing effects and facilitate ischemic preconditioning.82 The endogenous opioid system is involved in the analgesic effect of the nonopioid analgesic celecoxib. There is evidence for both central83 and peripheral84 opioid receptor activation. In addition to this, there is animal evidence of the role of peripherally acting opioids in inflammatory arthropathy and inflammatory bowel disease.85,86 These findings may guide the future development of novel peripherally restricted opioids.
Currently, there are no specific peripherally acting opioids available in the United States; investigators continue to work on developing novel potential agents. These peripheral opioid receptors play a critical role in modulating pain and inflammation. However, clinical data are still lacking.
References
1. Vane JR, Botting RM. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am J Med. 1998;104:2S–8S; discussion 21S–22S.
2. Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–959.
3. Davies NM, Skjodt NM. Choosing the right nonsteroidal anti-inflammatory drug for the right patient: a pharmacokinetic approach. Clin Pharmacokinet. 2000;38:377–392.
4. Needs CJ, Brooks PM. Clinical pharmacokinetics of the salicylates. Clin Pharmacokinet. 1985;10:164–177.
5. Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–1073.
6. Silverstein FE, Graham DY, Senior JR, et al. Misoprostol reduces serious gastrointestinal complications in patients with rheumatoid arthritis receiving nonsteroidal anti-inflammatory drugs. A randomized, double-blind, placebo-controlled trial. Ann Intern Med. 1995;123:241–249.
7. Laine L. Nonsteroidal anti-inflammatory drug gastropathy. Gastrointest Endosc Clin N Am. 1996;6:489–504.
8. Chan FK, Lanas A, Scheiman J, et al. Celecoxib versus omeprazole and diclofenac in patients with osteoarthritis and rheumatoid arthritis (CONDOR): a randomised trial. Lancet. 2010;376:173–179.
9. Lanas A, Garcia-Rodriguez LA, Polo-Tomas M, et al. Time trends and impact of upper and lower gastrointestinal bleeding and perforation in clinical practice. Am J Gastroenterol. 2009;104:1633–1641.
10. Huang JQ, Sridhar S, Hunt RH. Role of Helicobacter pylori infection and non-steroidal anti-inflammatory drugs in peptic-ulcer disease: a meta-analysis. Lancet. 2002;359:14–22.
11. Gutthann SP, Garcia Rodriguez LA, Raiford DS. Individual nonsteroidal antiinflammatory drugs and other risk factors for upper gastrointestinal bleeding and perforation. Epidemiology. 1997;8:18–24.
12. Lanza FL, Chan FK, Quigley EM. Guidelines for prevention of NSAID-related ulcer complications. Am J Gastroenterol. 2009;104:728–738.
13. Chan FK, Abraham NS, Scheiman JM, et al. Management of patients on nonsteroidal anti-inflammatory drugs: a clinical practice recommendation from the First International Working Party on Gastrointestinal and Cardiovascular Effects of Nonsteroidal Anti-inflammatory Drugs and Anti-platelet Agents. Am J Gastroenterol. 2008;103:2908–2918.
14. Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528.
15. Bresalier RS, Sandler RS, Quan H, et al. Adenomatous Polyp Prevention on Vioxx Trial I: cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102.
16. Kearney PM, Baigent C, Godwin J, et al. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ. 2006;332:1302–1308.
17. Scheiman JM, Fendrick AM. Summing the risk of NSAID therapy. Lancet. 2007;369:1580–1581.
18. Coxib and Traditional NSAID Trialist’s Collaboration. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–779.
19. Whelton A. Renal and related cardiovascular effects of conventional and COX-2-specific NSAIDs and non-NSAID analgesics. Am J Ther. 2000;7:63–74.
20. Ong HT, Ong LM, Tan TE, et al. Cardiovascular effects of common analgesics. Med J Malaysia. 2013;68:189–194.
21. Sahasrabuddhe VV, Gunja MZ, Graubard BI, et al. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J Natl Cancer Inst. 2012;104:1808–1814.
22. Garcia Rodriguez LA, Williams R, Derby LE, et al. Acute liver injury associated with nonsteroidal anti-inflammatory drugs and the role of risk factors. Arch Intern Med. 1994;154:311–316.
23. Picado C, Fernandez-Morata JC, Juan M, et al. Cyclooxygenase-2 mRNA is downexpressed in nasal polyps from aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1999;160:291–296.
24. Koschel D, Weber CN, Hoffken G. Tolerability to etoricoxib in patients with aspirin-exacerbated respiratory disease. J Investig Allergol Clin Immunol. 2013;23:275–280.
25. Dabu-Bondoc S, Franco S. Risk-benefit perspectives in COX-2 blockade. Curr Drug Saf. 2008;3:14–23.
26. Nielsen GL, Sorensen HT, Larsen H, et al. Risk of adverse birth outcome and miscarriage in pregnant users of non-steroidal anti-inflammatory drugs: population based observational study and case-control study. Br Med J. 2001;322:266–270.
27. Verbeeck RK. Pharmacokinetic drug interactions with nonsteroidal anti-inflammatory drugs. Clin Pharmacokinet. 1990;19:44–66.
28. Brouwers JR, de Smet PA. Pharmacokinetic-pharmacodynamic drug interactions with nonsteroidal anti-inflammatory drugs. Clin Pharmacokinet. 1994;27:462–485.
29. Takkouche B, Etminan M, Caamano F, et al. Interaction between aspirin and ACE inhibitors: resolving discrepancies using a meta-analysis. Drug Saf. 2002;25:373–378.
30. Van Aken H, Thys L, Veekman L, et al. Assessing analgesia in single and repeated administrations of propacetamol for postoperative pain: comparison with morphine after dental surgery. Anesth Analg. 2004;98:159–165.
31. Tzortzopoulou A, McNicol ED, Cepeda MS, et al. Single dose intravenous propacetamol or intravenous paracetamol for postoperative pain. Cochrane Database Syst Rev. 2011;(10):CD007126.
32. Ong CK, Seymour RA, Lirk P, et al. Combining paracetamol (acetaminophen) with nonsteroidal antiinflammatory drugs: a qualitative systematic review of analgesic efficacy for acute postoperative pain. Anesth Analg. 2010;110:1170–1179.
33. Williams FM. Clinical significance of esterases in man. Clin Pharmacokinet. 1985;10:392–403.
34. Higgs GA, Moncada S, Vane JR. Eicosanoids in inflammation. Ann Clin Res. 1984;16:287–299.
35. Yuan M, Konstantopoulos N, Lee J, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. 2001;293:1673–1677.
36. Cronstein BN, Montesinos MC, Weissmann G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB. Proc Natl Acad Sci U S A. 1999;96:6377–6381.
37. Bronstein AC, Spyker DA, Cantilena LR Jr, et al. 2011 Annual report of the American Association of Poison Control Centers’ National Poison Data System (NPDS): 29th Annual Report. Clin Toxicol (Phila). 2012;50:911–1164.
38. Dargan PI, Wallace CI, Jones AL. An evidence based flowchart to guide the management of acute salicylate (aspirin) overdose. Emerg Med J. 2002;19:206–209.
39. Volans G, Hartley V, McCrea S, et al. Non-opioid analgesic poisoning. Clin Med. 2003;3:119–123.
40. McCormack K. The spinal actions of nonsteroidal anti-inflammatory drugs and the dissociation between their anti-inflammatory and analgesic effects. Drugs. 1994;47(suppl 5):28–45; discussion 46–47.
41. Salerno A, Hermann R. Efficacy and safety of steroid use for postoperative pain relief. Update and review of the medical literature. J Bone Joint Surg Am. 2006;88:1361–1372.
42. Waldron NH, Jones CA, Gan TJ, et al. Impact of perioperative dexamethasone on postoperative analgesia and side-effects: systematic review and meta-analysis. Br J Anaesth. 2013;110:191–200.
43. Mirzai H, Tekin I, Alincak H. Perioperative use of corticosteroid and bupivacaine combination in lumbar disc surgery: a randomized controlled trial. Spine. 2002;27:343–346.
44. Movafegh A, Razazian M, Hajimaohamadi F, et al. Dexamethasone added to lidocaine prolongs axillary brachial plexus blockade. Anesth Analg. 2006;102:263–267.
45. Cummings KC III, Napierkowski DE, Parra-Sanchez I, et al. Effect of dexamethasone on the duration of interscalene nerve blocks with ropivacaine or bupivacaine. Br J Anaesth. 2011;107:446–453.
46. Habib GS, Saliba W, Nashashibi M. Local effects of intra-articular corticosteroids. Clin Rheumatol. 2010;29:347–356.
47. Bartlett EE, Hutserani O. Xylocaine for the relief of postoperative pain. Anesth Analg. 1961;40:296–304.
48. Gordon RA. Intravenous Novocaine for analgesia in burns: (a preliminary report). Can Med Assoc J. 1943;49:478–481.
49. Gilbert CR, Hanson IR, Brown AB, et al. Intravenous use of xylocaine. Curr Res Anesth Analg. 1951;30:301–313.
50. Backonja MM. Local anesthetics as adjuvant analgesics. J Pain Symptom Manage. 1994;9:491–499.
51. Kastrup J, Angelo H, Petersen P, et al. Treatment of chronic painful diabetic neuropathy with intravenous lidocaine infusion. Br Med J (Clin Res Ed). 1986;292:173.
52. Woolf CJ, Wiesenfeld-Hallin Z. The systemic administration of local anaesthetics produces a selective depression of C-afferent fibre evoked activity in the spinal cord. Pain. 1985;23:361–374.
53. Labbe L, Turgeon J. Clinical pharmacokinetics of mexiletine. Clin Pharmacokinet. 1999;37:361–384.
54. Tremont-Lukats IW, Challapalli V, McNicol ED, et al. Systemic administration of local anesthetics to relieve neuropathic pain: a systematic review and meta-analysis. Anesth Analg. 2005;101:1738–1749.
55. Kwon YS, Kim JB, Jung HJ, et al. Treatment for postoperative wound pain in gynecologic laparoscopic surgery: topical lidocaine patches. J Laparoendosc Adv Surg Tech A. 2012;22:668–673.
56. Meier T, Wasner G, Faust M, et al. Efficacy of lidocaine patch 5% in the treatment of focal peripheral neuropathic pain syndromes: a randomized, double-blind, placebo-controlled study. Pain. 2003;106:151–158.
57. Habib AS, Polascik TJ, Weizer AZ, et al. Lidocaine patch for postoperative analgesia after radical retropubic prostatectomy. Anesth Analg. 2009;108:1950–1953.
58. Saber AA, Elgamal MH, Rao AJ, et al. Early experience with lidocaine patch for postoperative pain control after laparoscopic ventral hernia repair. Int J Surg. 2009;7:36–38.
59. Tominaga M, Caterina MJ, Malmberg AB, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543.
60. Wong GY, Gavva NR. Therapeutic potential of vanilloid receptor TRPV1 agonists and antagonists as analgesics: recent advances and setbacks. Brain Res Rev. 2009;60:267–277.
61. Mou J, Paillard F, Turnbull B, et al. Efficacy of Qutenza(R) (capsaicin) 8% patch for neuropathic pain: a meta-analysis of the Qutenza Clinical Trials Database. Pain. 2013;154:1632–1639.
62. Mason L, Moore RA, Derry S, et al. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991.
63. Mitra S, Sinatra RS. Perioperative management of acute pain in the opioid-dependent patient. Anesthesiology. 2004;101:212–227.
64. Mitra S. Opioid-induced hyperalgesia: pathophysiology and clinical implications. J Opioid Manag. 2008;4:123–130.
65. Bell RF, Dahl JB, Moore RA, et al. Perioperative ketamine for acute postoperative pain. Cochrane Database Syst Rev. 2006;(1):CD004603.
66. Dholakia C, Beverstein G, Garren M, et al. The impact of perioperative dexmedetomidine infusion on postoperative narcotic use and duration of stay after laparoscopic bariatric surgery. J Gastrointest Surg. 2007;11:1556–1559.
67. Gurbet A, Basagan-Mogol E, Turker G, et al. Intraoperative infusion of dexmedetomidine reduces perioperative analgesic requirements. Can J Anaesth. 2006;53:646–652.
68. Habib AS, Gan TJ. Role of analgesic adjuncts in postoperative pain management. Anesthesiol Clin North America. 2005;23:85–107.
69. Rachinger-Adam B, Conzen P, Azad SC. Pharmacology of peripheral opioid receptors. Curr Opin Anaesthesiol. 2011;24:408–413.
70. Chen JJ, Dymshitz J, Vasko MR. Regulation of opioid receptors in rat sensory neurons in culture. Mol Pharmacol. 1997;51:666–673.
71. Khalefa BI, Shaqura M, Al-Khrasani M, et al. Relative contributions of peripheral versus supraspinal or spinal opioid receptors to the antinociception of systemic opioids. Eur J Pain. 2012;16:690–705.
72. Stein C, Millan MJ, Shippenberg TS, et al. Peripheral opioid receptors mediating antinociception in inflammation. Evidence for involvement of mu, delta and kappa receptors. J Pharmacol Exp Ther. 1989;248:1269–1275.
73. Shannon HE, Lutz EA. Comparison of the peripheral and central effects of the opioid agonists loperamide and morphine in the formalin test in rats. Neuropharmacology. 2002;42:253–261.
74. Mousa SA. Morphological correlates of immune-mediated peripheral opioid analgesia. Adv Exp Med Biol. 2003;521:77–87.
75. Heurich M, Mousa SA, Lenzner M, et al. Influence of pain treatment by epidural fentanyl and bupivacaine on homing of opioid-containing leukocytes to surgical wounds. Brain Behav Immun. 2007;21:544–552.
76. Labuz D, Mousa SA, Schafer M, et al. Relative contribution of peripheral versus central opioid receptors to antinociception. Brain Res. 2007;1160:30–38.
77. Nagasaka H, Awad H, Yaksh TL. Peripheral and spinal actions of opioids in the blockade of the autonomic response evoked by compression of the inflamed knee joint. Anesthesiology. 1996;85:808–816.
78. Stein C, Hassan AH, Lehrberger K, et al. Local analgesic effect of endogenous opioid peptides. Lancet. 1993;342:321–314.
79. Likar R, Sittl R, Gragger K, et al. Peripheral morphine analgesia in dental surgery. Pain. 1998;76:145–150.
80. Stein C, Kuchler S. Targeting inflammation and wound healing by opioids. Trends Pharmacol Sci. 2013;34:303–312.
81. Bigliardi-Qi M, Gaveriaux-Ruff C, Zhou H, et al. Deletion of delta-opioid receptor in mice alters skin differentiation and delays wound healing. Differentiation. 2006;74:174–185.
82. Wong GT, Ling Ling J, Irwin MG. Activation of central opioid receptors induces cardioprotection against ischemia-reperfusion injury. Anesth Analg. 2010;111:24–28.
83. Rezende RM, Franca DS, Menezes GB, et al. Different mechanisms underlie the analgesic actions of paracetamol and dipyrone in a rat model of inflammatory pain. Br J Pharmacol. 2008;153:760–768.
84. Francischi JN, Chaves CT, Moura AC, et al. Selective inhibitors of cyclo-oxygenase-2 (COX-2) induce hypoalgesia in a rat paw model of inflammation. Br J Pharmacol. 2002;137:837–844.
85. Walker JS. Anti-inflammatory effects of opioids. Adv Exp Med Biol. 2003;521:148–160.
86. Philippe D, Dubuquoy L, Groux H, et al. Anti-inflammatory properties of the mu opioid receptor support its use in the treatment of colon inflammation. J Clin Invest. 2003;111:1329–1338.