Steven L. Clark
The critically ill obstetric patient presents unique challenges to the clinician. Pregnancy alters the function of virtually every organ system; thus, both the baseline state and the patient's response to physiologic aberrations are different in the pregnant patient compared with her nonpregnant counterpart. In addition, fetal considerations often are important in designing a diagnostic and therapeutic approach to the critically ill gravida. Numerous situations may be encountered in which the physiologic demands of mother and fetus may be opposite. Many of the principles that apply to the critically ill nongravid patient apply in pregnancy, as well. This chapter explores a number of conditions in critical care medicine that are unique to pregnancy.
SHOCK IN PREGNANCY
Shock encompasses various pathophysiologic aberrations that lead to inadequate tissue perfusion and impaired cellular metabolism. Although hypotension often is the most obvious clinical sign in shock of any cause, such blood pressure changes are the final common manifestation of a number of distinct pathologic processes. The successful clinical management of patients in shock depends on the proper definition of the underlying pathophysiology, as well as an understanding of the unique effects of pregnancy on such conditions.
Several types of shock have been defined. This chapter focuses on the pathophysiology, diagnosis, and treatment of hypovolemic and septic shock, the types most commonly encountered in the pregnant patient.
In the nonpregnant patient, hypotension generally is defined as a systolic blood pressure less than 90 mm Hg or a mean arterial pressure less than 60 mm Hg. During pregnancy, this definition has less value, because blood pressures as low as 80/50 mm Hg commonly are seen in healthy pregnant women. Further, although a mean arterial pressure of 60 mm Hg may be adequate for perfusion of the adult heart and brain in a nonpregnant adult, the effects of a given blood pressure on uteroplacental perfusion cannot easily be generalized but are related to gestational age and placental condition. Whereas a blood pressure of 80/50 mm Hg may be adequate in one pregnant woman, another woman with pregnancy-induced hypertension and uteroplacental insufficiency may suffer inadequate placental perfusion and fetal distress with a diastolic blood pressure of 90 mm Hg. During pregnancy, shock is more reliably diagnosed on the basis of the overall clinical picture, including heart rate, mental status, urine output, and fetal condition, than on the basis of any absolute arterial pressure.
HYPOVOLEMIC SHOCK
Despite the availability of modern blood banking techniques, hemorrhage remains a major cause of maternal mortality. Pertinent physiologic changes that affect a woman's response to hemorrhage during pregnancy include a 50% increase in plasma volume with slightly lesser increases in circulating red blood cell mass. These changes, as well as an increase in resting heart rate of 10 beats per minute, result in about a 50% increase in resting cardiac output in the term gravida compared with her nonpregnant counterpart. Such changes are accompanied by a decrease in systemic vascular resistance, resulting in a net decrease in mean arterial pressure during the second trimester. At term, an average of 500 milliliters of blood is lost with vaginal delivery, compared with a mean blood loss of 1,000 milliliters with cesarean section. Such losses usually are well tolerated by the pregnant woman because of the physiologic hypervolemia.
Early in the course of massive hemorrhage, there are decreases in mean arterial pressure, cardiac output, central venous pressure, pulmonary capillary wedge pressure (PCWP), stroke volumes, and oxygen consumption and increases in arterial venous oxygen content difference.
These changes are accompanied by compensatory increases in heart rate, systemic and pulmonary vascular resistance, and myocardial contractility. In addition, the redistribution of cardiac output and blood volume result in diminished perfusion to the kidneys, gastrointestinal tract, skin, and uterus, with relative maintenance of blood flow to the heart and brain. In the pregnant patient, such redistribution may result in fetal hypoxia and distress, even in the absence of an overt maternal hypotension. To some extent, the fetus acts like a miner's canary: regardless of absolute maternal blood pressure, significant maternal shock is virtually never seen in the presence of a reassuring fetal heart rate pattern. During this initial phase, initial oxygen extraction by the maternal tissues is increased. Further maldistribution of blood flow results in local tissue hypoxia and metabolic acidemia. If not promptly corrected, such shunting of blood from the renal and splanchnic beds may result in acute tubular necrosis and may contribute to pulmonary capillary endothelial damage, resulting in the adult respiratory distress syndrome (ARDS) even if resuscitation eventually is successful. In a similar manner, such tissue hypoxia and damage may give rise to thromboplastin release resulting in disseminated intravascular coagulation (DIC). Hemorrhagic shock also has been demonstrated to suppress bone marrow hematopoiesis. Once such shock has progressed to the point of end-organ failure, secondary consumptive coagulopathy, and depressed hematopoiesis, survival may not be possible even with adequate replacement of volume, red blood cells, and clotting factors.
As the blood volume deficit exceeds 25% to 30%, the compensatory mechanisms begin to become inadequate to maintain cardiac output and blood pressure. At this point, additional losses of blood may result in rapid clinical deterioration that produces a cycle of cellular death and vasoconstriction leading to organ ischemia, loss of capillary membrane integrity, and additional loss of intravascular fluid volume to the extravascular spaces. Thus, time is of the essence in restoring hemodynamic and oxygenation parameters to normal if survival is to be optimized.
The management of the patient in hemorrhagic hypovolemic shock is directed toward two goals: restoring circulating blood volume and eliminating the source of hemorrhage. The management of the most common causes of obstetric hemorrhage (e.g., lacerations, retained placenta, uterine atony) is described in detail elsewhere. The therapeutic restoration of circulating blood volume begins with a rapid infusion of crystalloid solution and packed red blood cells. These maneuvers form the cornerstone of therapy for hemorrhagic shock. Patients with normally functioning hearts may respond better to hypovolemic shock with blood volume replacements that are 500 to 1,000 milliliters in excess of their predicted norm. Such expansion dilates the constricted capillary networks that persist from the initial hypotensive phase and assists in resolving acidosis at the cellular level.
Continued replacement of blood with crystalloid solutions and packed red blood cells alone results in eventual depletion of labile clotting factors and platelets and may result in a dilutional coagulopathy which, itself, may contribute to further bleeding. This is to be distinguished from the consumptive coagulopathy which also may arise secondary to shock and tissue damage. A fibrinogen level below 100 milligrams per deciliter or prolongation of the prothrombin and active partial thromboplastin times in a bleeding patient are indications for fresh frozen plasma infusion. Platelet transfusion should be guided by platelet counts. One unit of platelets raises the platelet count by 8,000 to 10,000 per cubic millimeter and should be considered in a bleeding patient with a platelet count below 30,000 per cubic millimeter.
SEPTIC SHOCK
The incidence of septic shock appears to be increasing. Septic shock accompanies up to 50% of bacteremias caused by Gram-negative organisms and 5% of those caused by Gram-positive organisms. In the overall hospital population, once clinical shock is evident, the mortality rate is 40% to 50%. Although pregnancy classically has been considered a factor that predisposes a patient to septic shock, data to support this are lacking. It seems more likely that such older observations stemmed from delayed diagnosis and treatment in the pregnant patient. In obstetrics, septic abortion, chorioamnionitis, pyelonephritis, and endometritis are the most common conditions associated with septic shock. Postpartum infection with group A streptococcus is increasingly recognized as an uncommon, but potentially lethal, complication of the puerperium, a condition made more serious by the virulent nature of this infection and its rather protean clinical manifestations. Although it may be presumed that with a younger population, the mortality with septic shock is somewhat less than in the overall population, this condition continues to account for a significant percentage of maternal mortality.
Septic shock in obstetrics most commonly is associated with infection caused by endotoxin-releasing Gram-negative aerobic coliform organisms. Endotoxin, a complex cell wall–associated lipopolysaccharide, is released into the circulation at the time of bacterial death, resulting in multiple hemodynamic effects. The subsequent activation of lymphocytic T cells and mast cells results in histamine and kinin activation, as well as the activation of kallikrein and a decrease in kallikreinogen and kallikrein inhibitor. These changes result in the release of bradykinin, a potent arterial dilator. Complement activation and the release of arachidonic acid metabolites, nitric oxide, endorphins, and other endogenous mediators play a major role in the pathophysiology of septic shock. In addition, septic shock results in a marked reduction of vasopressin (antidiuretic hormone) levels, further impairing the body's vasoconstrictive response and capacity for intravascular fluid retention. This generalized inflammatory response, similar to that seen with other conditions such as anaphylaxis, is responsible for much of the pathophysiology seen with these conditions and has been termed the systemic inflammatory response syndrome.
Early septic shock is a classic example of distributive shock, related to a systemic maldistribution of relatively normal, or even increased, cardiac output. Clinical findings include hypotension, fever, and chills. Initial hemodynamic findings include decreased systemic vascular resistance and high-normal or elevated cardiac output. The continued maldistribution of cardiac output leads to local tissue hypoxia and the development of lactic acidosis and end-organ dysfunction. This decrease in systemic vascular resistance is caused by the release of vasoactive substances, as well as by vascular endothelial cell injury, which promotes capillary plugging secondary to complement-induced leukocyte aggregation. These factors lead to increased arteriovenous shunting.
The treatment of septic shock in this early phase involves optimizing preload by restoring relative intravascular volume with crystalloid infusion, as well as aggressively treating the underlying infection. Although some authorities advocate the use of colloid solutions for volume replacement, there is no convincing evidence that the use of such solutions decreases the incidence of pulmonary edema or ARDS. If the offending organism is known, single-agent antibiotic therapy may be used. More commonly in obstetrics, the infection is polymicrobial, and broad-spectrum coverage for Gram-negative and Gram-positive aerobic and anaerobic organisms is most appropriate. If an abscess is involved, prompt surgical drainage after initial resuscitation is mandatory.
If the process continues, the patient may enter a second hemodynamic phase of septic shock. Of primary importance in this late phase is the development and progression of myocardial dysfunction leading to ventricular failure. Although commonly viewed as a late finding, studies assessing stroke work index and ventricular ejection fraction have demonstrated depressed intrinsic ventricular function even in the early stages of septic shock, when increases in heart rate are compensatory and associated with a normal or high cardiac output. This myocardial depression is a direct effect of myocardial depressant substance in the serum of patients with septic shock. When ventricular function has deteriorated to the point in which cardiac index is frankly depressed (i.e., <3–4 L/min per m2 in pregnancy) in the presence of adequate preload, the prognosis is extremely grave. Pulmonary hypertension, another important hemodynamic alteration often associated with septic shock, may have additional profound hemodynamic consequences.
For most pregnant patients in septic shock, maximal ventricular function is obtained by means of optimizing Starling forces with a PCWP of about 16 mm Hg. Patients who remain hypotensive after such preload manipulation require treatment with pressor agents such as dopamine hydrochloride. This agent, in dosages of less than 5 micrograms per kilogram per minute, improves renal blood flow by way of dopaminergic mesenteric vasodilation; in dosages of 5 to 30 micrograms per kilogram per minute, a positive inotropic effect also is seen. In dosages that exceed 30 micrograms per kilogram per minute, the favorable effect on blood pressure is principally one of α-adrenergic vasoconstriction. Additional inotropic or vasoconstrictive agents may need to be administered in refractory septic shock. Treatment with vasopressin has been effective in improving urine output, decreasing pulmonary vascular resistance, and reducing the need for additional pressor agents. Initial studies suggest the infusion of methylene blue (an inhibitor of nitric oxide synthesis) may prove effective in counteracting myocardial depression, maintaining oxygen transport, and reducing the need for additional adrenergic support. The use of glucocorticoids in septic shock continues to unfold. Original enthusiasm for early, high-dose steroid treatment in patients with septic shock was followed by well-designed studies demonstrating not only a lack of benefit, but potential harm, from such therapy. More recently, the demonstration of an altered hypothalamic–pituitary–adrenal axis in patients with septic shock have led to additional investigations suggesting that lower-dosage replacement therapy with hydrocortisone may be beneficial, by interfering with the physiologic expression of the systemic inflammatory response syndrome.
The hemodynamic manipulation of patients whose hypotension fails to respond rapidly to volume infusion may, at times, be assisted by pulmonary artery catheterization, allowing the clinician to achieve optimal preload before the institution of inotropic or vasoconstrictive therapy.
Patients who recover from the initial hemodynamic instability of septic shock may suffer prolonged morbidity secondary to endotoxin-mediated pulmonary capillary injury and noncardiogenic pulmonary edema (i.e., ARDS). Such lung failure is a major cause of death in patients with septic shock. Similarly, pregnant patients whose hypotension was prolonged may experience acute tubular necrosis. Endotoxin-mediated endothelial cell injury and associated thromboplastin-like activity, as well as prolonged shock from any cause, may also lead to activation of the coagulation cascade and to a clinical picture of DIC. Even in the absence of frank tissue hypoxia, septic stimuli may lead to coagulation abnormalities.
Of special interest to obstetricians is the demonstration that, in patients in septic shock, adult heart rate variability is diminished; such observations have been suggested as potentially useful in the diagnosis of adult septic shock.
FETAL RESPONSE TO MATERNAL HEMODYNAMIC INSTABILITY
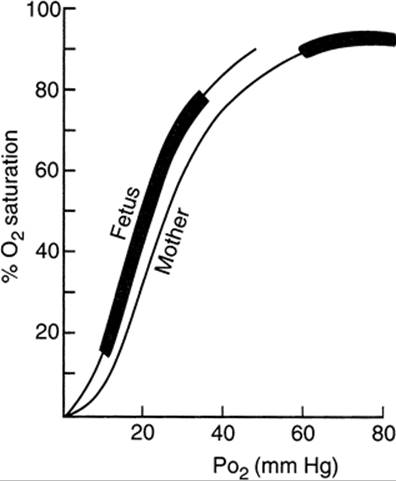
Fetal well-being depends principally on the maintenance of maternal oxygenation and uterine blood flow. During severe fetal hypoxia, cardiac output decreases and pulmonary vasoconstriction occurs, with redistribution of the fetal circulation favoring blood flow to the brain, heart, and adrenals at the expense of splanchnic and even placental blood flow. Although Starling's law is operative in the fetus, ventricular stroke work varies over a much smaller range of end-diastolic pressures. Changes in cardiac output manifest themselves primarily by changes in fetal heart rate alone. As maternal arterial PO2 falls, there appears to be no corresponding fall in fetal PO2 as long as the maternal PO2 exceeds 60 mm Hg. Because the fetus operates on the steep portion of the oxygen dissociation curve (Fig. 25.1) (i.e., normal fetal PO2 range, 10–33 mm Hg), further falls in maternal and fetal PO2 result in dramatic decreases in fetal oxygen saturation and fetal hypoxia. Unfortunately, observations using the fetal pulse oximeter have demonstrated that the administration of oxygen to the mother using most readily available oxygen delivery methods does not result in improvement in fetal oxygen saturation.
|
|
|
FIG. 25.1. Oxygen dissociation curves for human maternal and fetal blood, indicating the physiologic range of PO2 and O2 saturation for mother and fetus. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:343, with permission.) |
Near term, uterine blood flow approaches 500 milliliters per minute and accounts for about 10% of total maternal cardiac output. The uterine arteries show little capacity for autoregulation; thus, uterine and placental blood flow fall in direct proportion to maternal systemic arterial pressure. In the hypotensive mother, compensatory maternal vasoconstriction aimed at maintaining circulation to the heart and brain decreases uterine blood flow even further. Thus, maternal hypotension, whether caused by supine venacaval compression, conduction anesthetic, or even amniotic fluid embolism (AFE), may be first heralded by fetal bradycardia. Because maternal compensatory mechanisms maintain systemic arterial pressure at the expense of uterine blood flow, such abnormal patterns may be seen even in the absence of overt maternal hypotension. Thus, during the critical course of a gravely ill mother, careful monitoring of fetal well-being is essential. Even in a medical or surgical intensive care setting, continuous electronic fetal heart rate monitoring is often an important part of the care of the critically ill and unstable pregnant patient.
INVASIVE HEMODYNAMIC MONITORING
Since its introduction into clinical medicine in the early 1970s, the pulmonary artery catheter has come to play an important role in the management of critically ill patients with hemodynamic instability. Several prospective trials demonstrate the benefits of this technique in select, critically ill patients, including a reduction in morbidity and mortality in some subsets. On the other hand, the risk–benefit ratio of invasive monitoring in the general population of seriously ill patients has been questioned. When obstetric patients require such monitoring, optimal patient outcome is predicated on the clinician's understanding of the hemodynamic changes associated with pregnancy, as well as a working knowledge of indications for invasive hemodynamic monitoring. Although invasive hemodynamic monitoring rarely is required in the pregnant patient, an understanding of the hemodynamic principles derived from such techniques may assist the obstetrician–gynecologist in the noninvasive management of less critically ill patients.
The pulmonary artery catheter is a multilumen catheter placed most commonly through the internal jugular or subclavian vein (Fig. 25.2). As the catheter is advanced through the great vessels and heart, its location can be determined precisely by observing characteristic pressures and waveforms (Fig. 25.3). When the catheter is placed correctly, the lumens terminate in the pulmonary artery and the superior vena cava or right atrium and allow direct assessment of central venous pressure, PCWP, and pulmonary artery pressure. SxO2 and cardiac output can be computed either continuously or intermittently using a fiberoptic thermistor system. These measurements may be combined with mean arterial pressure and body surface area and heart rate to calculate additional important hemodynamic indices, such as systemic vascular resistance and left ventricular stroke work index (Table 25.1). In critically ill obstetric patients, discrepancies often are seen between measurements of PCWP and central venous pressure. In such circumstances, clinical use of the central venous pressure measurement alone would be misleading and possibly deleterious. With rare exceptions, the complications seen with pulmonary artery catheterization are associated with obtaining central venous access and are similar whether a central venous pressure line or pulmonary artery catheter is used. For these reasons, in a modern perinatal intensive care unit, central venous pressure monitoring alone seldom is indicated.
|
|
|
FIG. 25.2. Pulmonary artery catheter. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:63, with permission.) |
|
|
|
FIG. 25.3. Pulmonary artery catheter placement. Catheter position, corresponding waveforms, and pressures are shown. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:67, with permission.) |
|
|
|
TABLE 25.1. Derived hemodynamic parameters |
Determinants of Cardiac Output
In addition to heart rate, important determinants of cardiac output are preload, afterload, and an assessment of myocardial contractility.
Preload
Preload represents the volume or pressure generated within the ventricles at end diastole. According to Starling's law, ventricular output is directly proportional to ventricular preload. Clinically, right ventricular preload is measured as central venous pressure, and left ventricular preload as PCWP. Because the left side of the heart perfuses the myocardium, brain, and uteroplacental unit, PCWP is the preload measurement of interest in clinical practice. When central venous pressures are used, it is done with the tacit assumption that the preloads of the right and left sides of the heart are equivalent. In a critically ill patient, this often is not the case, thus further limiting the usefulness of central venous pressure in the hemodynamically unstable obstetric patient.
Afterload
Afterload describes the resistance to the flow of blood during ventricular systole. It is not measured directly; rather, after the measurement of cardiac output, central venous pressure, and mean arterial pressure, afterload is calculated as systemic vascular resistance. Cardiac output fluctuates inversely with systemic vascular resistance. Septic shock and preeclampsia are examples of frequently encountered conditions in obstetrics that may markedly influence systemic vascular resistance.
Ventricular Stroke Work Index
Ventricular stroke work is also a calculated parameter (see Table 25.1). The assessment of ventricular stroke work on a modified Starling curve (Fig. 25.4) allows the clinician to directly assess the intrinsic contractility of the left ventricle and to classify myocardial function as normal, hyperdynamic, or hypodynamic. After preload has been optimized, ventricular function may be further affected by the administration of inotropic agents such as dopamine. The use of such agents may shift the intrinsic contractility of the ventricle from the hypodynamic to the normal range, resulting in improved cardiac performance (Fig. 25.5).
|
|
|
FIG. 25.4. Pulmonary capillary wedge pressure versus left ventricular stroke work index. Normal relationship is represented by the shaded area. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:399, with permission.) |
|
|
|
FIG. 25.5. Effect of volume manipulation and inotrope administration on ventricular output in two patients with septic shock. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:181, with permission.) |
Indications
The indications for pulmonary artery catheterization in pregnancy include many of the conditions common to all critical care medicine. These include severe cardiac disease during labor and delivery, septic shock, and hemodynamic instability in patients with uncertain volume. Select patients with complicated severe preeclampsia form a subset unique to pregnancy. It has been suggested that patients with severe hypertension unresponsive to conventional antihypertensive medication, those with pulmonary edema of unknown cause, and those with persistent oliguria may benefit from invasive hemodynamic monitoring. In contrast, hemodynamic monitoring seldom is indicated in cases of hypovolemia associated with obstetric hemorrhage. Normal values for invasively obtained hemodynamic indices have been described and often are valuable in managing the critically ill obstetric patient (Table 25.2).
|
|
|
TABLE 25.2. Central hemodynamic changes |
DISSEMINATED INTRAVASCULAR COAGULOPATHY (DIC) ASSOCIATED WITH PREGNANCY
DIC is not a distinct clinical entity; rather, it represents a manifestation of various disease processes that have in common activation of intravascular clotting and fibrinolysis, resulting in excess consumption of soluble coagulation components. In obstetrics, secondary fibrinogenolysis commonly dominates the clotting aberration and results in the circulation of fibrin and fibrinolytic split products, which further accentuate the clinical picture of hemorrhage. In addition, sometimes a dilutional coagulopathy is encountered in pregnancy. This condition occurs when massive hemorrhage is replaced only by red blood cells and crystalloid solution, resulting in a dilutional depletion of platelets and soluble clotting factors. From both a clinical and a laboratory standpoint, dilutional coagulopathy may be confused with a more classic process of intravascular consumption and fibrinolysis. In practice, the hemorrhage associated with dilutional coagulopathy often results in hypotension and shock. The tissue hypoxia that accompanies shock of any cause is well known to potentially activate the coagulation–fibrinolysis cycle associated with DIC. Thus, it is not always possible to draw a clear distinction between dilutional and consumptive coagulopathy.
Pathophysiology
A number of conditions in obstetrics may trigger the intravascular coagulation–fibrinolysis sequence. All have in common some form of endothelial disruption or release of tissue thromboplastins into the central circulation. Thus, clotting may occur by either the intrinsic or the extrinsic coagulation pathways. After activation of the clotting cascade, fibrin monomer polymerizes, forming insoluble fibrin. Serum plasminogen simultaneously begins the degradation of fibrin. The breakdown products of fibrin and fibrinogen (i.e., fibrinolytic split products) then bind the soluble fibrin monomer to prevent further polymerization. In addition, plasmin inactivates factors V, VIII, IX, and XIII and inhibits the platelet-mediated primary phase of coagulation. The fibrin monomer that does polymerize is filtered within the microvasculature, resulting in fibrin plugs. Such plugging reduces blood flow, which causes tissue hypoxia and organ schemia. The subsequent endothelial damage further enhances the clotting cascade. Thrombocytopenia results from both consumption and destruction secondary to fibrinolytic split products in the microvasculature, a process that also results in microangiopathic hemolytic anemia. The release of thromboplastic material from damaged red blood cells and complement activation further propagate the vicious cycle of intravascular coagulation–fibrinolysis (see Fig. 25.5).
Diagnosis
DIC is suggested clinically by excess bleeding from surgical incision sites or the postpartum uterus. In severe cases, spontaneous bleeding from intravenous sites and other mucous membranes also may be seen. Table 25.3 lists some laboratory abnormalities that may be present in DIC. The prothrombin and partial thromboplastin times are relatively insensitive indicators of clotting status, because results are not prolonged until 40% to 50% of the clotting factors have been consumed. Although levels of fibrinolytic split products and paracoagulation tests are thought to be sensitive indicators of DIC, values may be normal in 10% to 15% of patients with acute DIC.
|
|
|
TABLE 25.3. Laboratory abnormalities in disseminated intravascular coagulation |
Causes
Numerous causes of DIC may be encountered in obstetrics (Fig. 25.6). The most common are placental abruption and dead fetus syndrome. AFE, a rare obstetric catastrophe, often is associated with DIC. Preeclampsia, commonly associated with thrombocytopenia, is not itself a cause of clinical DIC, unless complicated by substantial placental abruption. Other conditions not unique to obstetrics, such as septic shock and transfusion disorders, also may be associated with DIC in obstetrics. The management of acute DIC in obstetrics principally involves resolution of the underlying pathophysiologic process. Although possibly severe, the DIC associated with abruption, dead fetus syndrome, or AFE usually is self-limiting, and its resolution may begin with delivery of the fetus and placenta. In most cases, the replacement of soluble clotting factors or platelets is effective in preventing clinical hemorrhage for the short term while delivery is accomplished; little evidence exists to support the use of heparin or fibrinolytic agents in obstetric DIC syndromes. One exception may be dead fetus syndrome, in which low-dose heparin therapy (5,000 U subcutaneously twice daily) may correct moderate coagulation abnormalities during delivery. When there is bleeding or if surgery is planned, platelet transfusion should be considered for patients with a platelet count less than 30,000 per cubic millimeter, and fibrinogen replacement with fresh frozen plasma is appropriate for the bleeding patient with a fibrinogen level less than 100 milligrams per deciliter or prolonged prothrombin and partial thromboplastin times. In the nonsurgical patient without clinical hemorrhage, clotting abnormalities associated with a mild to moderate intravascular coagulopathy may simply be observed while delivery is accomplished.
|
|
|
FIG. 25.6. Obstetric disorders that initiate disseminated intravascular coagulation. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:182, with permission.) |
TRANSFUSION THERAPY
Indications for blood or component transfusion include restoration of circulating volume, improvement in oxygen transport, and correction of coagulation disorders. The third indication has been addressed in the preceding paragraphs. This section addresses blood and component transfusion considerations in the patient with massive blood loss.
The incidence of blood transfusion in patients undergoing vaginal delivery is less than 1%, whereas 2% to 12% of patients undergoing cesarean delivery receive blood. To anticipate the potential for peripartum blood loss, blood often is sent for type and screen and, occasionally, type and crossmatch. To type and screen blood, first the ABO group and Rh(D) type of the patient's cells are determined. Her serum is then mixed with reagent red blood cells that contain the antigens with which most clinically significant antibodies will react. An antibody-enhancing solution, such as bovine serum albumin, may be added at this point to enhance the detection of nonagglutinating antibodies. After incubation and washing, the appearance of an agglutination indicates the presence in the recipient serum of antibodies to one or more antigens in the reagent blood cells.
Type and crossmatch procedures are similar to those described for typing and screening, except that potential donor red blood cells are used rather than standard reagent erythrocytes. If blood that has been screened but not cross-matched is used, a transfusion reaction might take place if the recipient serum contains an antibody to an antigen present on the donor cells that was not present on the reagent red blood cells. Because only 0.03% to 0.07% of patients who were determined not to have antibodies on type and screening subsequently are found to have preexisting antibodies determined by crossmatch, the type and screen approach has been about 99% effective in preventing incompatible transfusions. Type and screen testing is much less costly than typing and cross-matching blood and allows blood to be made available for more than one potential recipient, thus decreasing the wastage of banked blood. Therefore, this method is appropriate for both vaginal delivery and most cases of cesarean section and, under these circumstances, is preferred to typing and cross-matching.
Whole Blood
A unit of whole blood contains about 450 milliliters of blood. One unit of whole blood can raise the recipient's hematocrit by 3% to 4%. When stored for longer than 24 hours, whole blood retains few functioning granulocytes or platelets. Clotting factors are maintained in adequate concentrations for the life of the unit, except levels of factors V and VIII, which may be reduced. Because whole blood is not readily available, whole blood transfusion is uncommon in modern obstetric practice; red blood cells plus component replacement is preferred.
Red Blood Cells
Packed red blood cells are prepared from whole blood by separating the cells from plasma by centrifugation or sedimentation. They have a hematocrit of about 70% to 75% (160–275 cc red blood cells per unit). A unit of packed cells contains the same number of erythrocytes as a unit of whole blood; thus, it can raise the recipient's hematocrit by 3% to 4%. Packed red blood cells provide the same oxygen-carrying capacity as whole blood in about one half the volume.
Platelets
Platelets are prepared from whole blood by centrifugation and then are resuspended in 50 to 70 milliliters of plasma. Unlike red blood cells, they cannot tolerate prolonged storage; under special circumstances, however, they may be stored for up to 5 days. The donor plasma in which the platelets are suspended must be ABO compatible with the recipient's red blood cells. A unit of random donor platelets should raise the platelet count by 8,000 to 10,000 per cubic millimeter. Platelets are available in packs of 8 or 10 units. No consensus has been reached regarding minimal platelet levels that indicate the need for transfusion. In the nonbleeding or nonsurgical patient, platelet transfusion seldom is indicated with a platelet count that exceeds 10,000 per cubic millimeter, because antibody formation and platelet destruction are rapid. In the bleeding or surgical patient, the platelet count should be maintained above 30,000 per cubic millimeter to prevent further hemorrhage.
Fresh Frozen Plasma
Fresh frozen plasma contains all soluble clotting factors, similar to those found in whole blood. In addition, it contains significant amounts of factors V and VIII. The plasma is first separated from whole blood and then frozen. The volume of a unit is about 250 milliliters. Fresh frozen plasma is indicated for patients with a consumptive or dilutional coagulopathy in whom replacement of soluble clotting factors, including fibrinogen, is indicated. In the bleeding patient, a fibrinogen level less than 100 milligrams per deciliter associated with prolonged prothrombin and partial thromboplastin times is an indication for fresh frozen plasma infusion. By definition, each milliliter of fresh frozen plasma contains 1 IU of each coagulation factor. One unit will raise the serum fibrinogen level by approximately 25 milligrams per deciliter.
Cryoprecipitate
A unit of cryoprecipitate is prepared by thawing a unit of fresh frozen plasma, removing the supernatant plasma, and then refreezing the precipitate. Cryoprecipitate contains factor VIII:C, factor XIII, von Willebrand factor, fibrinogen, and fibronectin. The sole advantage of cryoprecipitate over fresh frozen plasma is the ability to infuse relatively large amounts of factors in a small volume. In practice, volume restriction seldom is desirable for the hemorrhaging patient; thus, fresh frozen plasma nearly always is preferable to cryoprecipitate in the management of obstetric hemorrhage associated with consumptive or dilutional coagulopathy.
In cases of dilutional coagulopathy, bleeding is more likely to be secondary to thrombocytopenia than to factor deficiency. All the blood and clotting factors contained in cryoprecipitate and fresh frozen plasma carry with them the risk of blood-borne disease transmission, although such a risk is minimal in properly screened and donated blood.
Transfusion Reactions
Acute transfusion reactions are seen in about 5% of blood recipients, and delayed transfusion reactions in up to 7% of recipients. Nonhemolytic febrile reactions may accompany 1% of all transfused units. Most nonhemolytic febrile and allergic reactions are unavoidable. Most hemolytic reactions may be prevented by proper attention to cross-matching procedures.
Hemolytic transfusion reactions may be acute or delayed. Acute intravascular hemolysis most commonly results from the transfusion of ABO-incompatible blood. Such errors are the most frequent cause of fatalities resulting from blood transfusion and usually are related to technical errors in blood specimen labeling and in patient identification. Such hemolysis is complement mediated. Signs and symptoms of acute hemolytic reaction include lumbar pain, facial flushing, chest pain that may be accompanied by fever, tachycardia, hypotension, and shock. In severe cases, DIC associated with oliguria and pulmonary edema may be seen. Acute tubular necrosis or ARDS may be encountered in patients who survive the acute episode. The key laboratory findings in the diagnosis of intravascular hemolytic transfusion reaction are a decrease in the plasma haptoglobin level with an increase in free serum hemoglobin. These findings typically are accompanied by hemoglobinuria if plasma levels exceed 25 milligrams per deciliter.
Acute extravascular hemolytic reactions are not mediated by complement but by the development of immunoglobulin G (IgG) antibodies to antigens such as D or Kell. Clinical findings are less severe and often manifest by simply a fever and hemolytic anemia. Severe clinical symptoms are infrequent.
Delayed hemolytic reactions occur within 3 to 7 days of a blood transfusion. These reactions are caused by a serum antibody that is present in low, undetectable levels at the time of transfusion. The clinical parameters are similar to those seen with acute extravascular hemolysis but are not as severe.
ACUTE RESPIRATORY DISTRESS SYNDROME
Acute respiratory distress syndrome is a complex pathophysiologic process resulting from either primary lung epithelial injury by way of the airways or capillary endothelial injuries initiated by way of the pulmonary vasculature. Such injury results in increased pulmonary capillary permeability, loss of lung volume, and shunting with resultant arterial hypoxemia. The physiologic criteria required for the diagnosis of ARDS are listed in Table 25.4. In pregnancy, the incidence of ARDS is approximately 1 in 6,000. Sepsis, preeclampsia, viral pneumonic processes, hypovolemic shock, and aspiration pneumonia are the most common causes of ARDS.
|
|
|
TABLE 25.4. Physiologic criteria for adult respiratory distress syndrome |
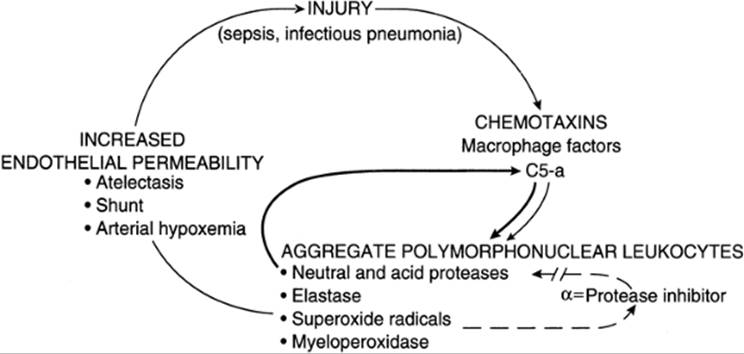
ARDS represents a final common pathway of a cascade of events initiated by the alveolar, epithelial, or endothelial injury (Fig. 25.7). After the initial injury to alveolar epithelial cells or vascular endothelial cells, the complement system, leukocytes, macrophages, and platelets may all play important roles in perpetuating the injury. During the initial lung injury, phospholipids are released from red blood cell membranes, resulting in a secondary mechanism of injury. Free arachidonic acids serve as a substrate for the synthesis of both prostaglandins and thromboxanes by way of the cyclooxygenase system and, possibly, leukotrienes by way of the lipoxygenase system. All of these arachidonic acid metabolites have been implicated in perpetuating the injury of ARDS.
|
|
|
FIG. 25.7. Schematic diagram of the proposed mechanism of lung injury. After the cycle is established, loop 1 feeds back on itself to perpetuate the injury. Amplification of the first loop is provided by the neutral and acid proteases (bold line); disruption of the amplification loop is prevented by myeloperoxidases (dashed line). PMNs, polymorphonuclear leukocytes. (From Clark SL, Phelan JP. Critical care obstetrics, second ed. Boston: Blackwell Scientific, 1990:343, with permission.) |
Clinically, the course of ARDS can be divided into four phases. Each is distinguishable by histologic changes in the lungs and reflected in various physiologic parameters in the patient. In phase I, hyperventilation is the only physiologic aberration seen, and arterial oxygenation remains adequate. During phase II (i.e., the latent period), there may be minor auscultatory and radiographic evidence of pulmonary disease. Mild decreases in lung compliance and increases in intrapulmonary shunting of blood are common. Histologically, these stages are characterized by progressive alveolar and interstitial edema formation and the movement of inflammatory cells into the interstitium. Damage to type I alveolar cells occurs, and hyaline membrane formation begins. Phase III is characterized by acute respiratory failure with severe dyspnea, tachypnea, and arterial hypoxemia. During this phase, lung volume is lost, resulting in the deterioration of pulmonary compliance and an increase in intrapulmonary shunting. Chest x-ray films taken during this stage demonstrate marked abnormalities, including bilateral lung opacification. During phase III, intubation and assisted ventilation are required. Histologically, phase III is characterized by thickening of the alveolar septum and marked infiltration by leukocytes, plasma cells, and macrophages. Hyaline membranes begin to form and organize during this phase, and the proliferation of fibroblasts and type II alveolar cells is prominent. In phase IV, interpulmonary shunting in excess of 25% results in hypoxemia refractory to all measures. Dead space during this period may exceed 60% of tidal volume. Metabolic and respiratory acidemia may be found and may result in increased myocardial excitability, which often leads to terminal dysrhythmia. Intraalveolar fibrosis is severe, and pathologic and physiologic derangements are irreversible.
Clinical Management
Four considerations are essential in understanding the clinical management of the pregnant patient with ARDS. These are oxygen delivery and tissue extraction, pulmonary compliance, interpulmonary shunt fraction, and eradication of the underlying cause.
Oxygen delivery is directly proportional to cardiac output; it is also influenced by the oxygen-carrying capacity of the blood. Thus, oxygen delivery can be optimized by maintaining cardiac output and correcting anemia. In the presence of suboptimal cardiac output or significant anemia, marked increases in the inspired concentration of oxygen have little effect on actual oxygen delivery. However, high inspired concentrations of oxygen are toxic to the lung. The goals in caring for a patient with severe ARDS are first to optimize cardiac output and hematocrit and then to maintain a PO2 of 60 to 70 mm Hg. Because increased body temperature and metabolic acidosis produce a rightward shift in the oxyhemoglobin dissociation curve, these conditions may favorably affect peripheral oxygen delivery. Thus, the patient should not be chilled either in the operating room or through the administration of large volumes of intravenous fluids or blood that is below body temperature. Further, respiratory alkalosis should be corrected. Indeed, a mild metabolic acidemia may result in better tissue oxygen delivery than would be obtained with complete correction of the patient's acid–base status.
Arterial–venous oxygen content difference may be used to assess the adequacy of oxygen delivery. During pregnancy, the arterial–venous gradient narrows.
In the critically ill patient who is receiving assisted ventilation, pulmonary compliance can be calculated by using parameters obtainable from the ventilator. With severe ARDS, pulmonary compliance may fall from a normal value of about 75 milliliters per cm H2O to as low as 20 milliliters per cm H2O. Serial calculation of compliance may be useful in evaluating the effects of various therapeutic maneuvers, including positive end-expiratory pressure (PEEP). Longitudinal evaluation of pulmonary compliance may serve as a useful method of following the resolution or progression of ARDS.
The normal shunt in the healthy gravida is 2% to 5% of cardiac output. Mild pulmonary dysfunction may result in shunting of 10% to 15%. A shunt that exceeds 25% suggests and, in the absence of other causes, may be diagnostic of ARDS. The shunt may be estimated either by analyzing an arterial blood sample alone or by analyzing simultaneously obtained arterial and pulmonary artery blood samples. In the former method, the greatest accuracy is obtained by having the woman breathe 100% oxygen. In a patient who is intubated and receiving 100% inspired oxygen, the arterial oxygen pressure should exceed 500 mm Hg; values below 300 mm Hg suggest severe shunting.
During initial treatment of the pregnant gravida with ARDS, the physician should simultaneously conduct an aggressive search for and attempt to eliminate the underlying cause of the lung injury. Early intubation is essential for the patient in incipient respiratory failure (Table 25.5). Mechanical ventilation requires a volume-cycled ventilator. Appropriate initial settings may include a rate of 12 breaths per minute, a tidal volume of about 15 milliliters per kilogram of body weight with a sigh volume of 200 to 400 milliliters above tidal volume, a PEEP of 5 cm H2O, and 100% inspired oxygen. Fifteen to 30 minutes after the initial therapy, an arterial blood sample is analyzed, and the ventilator settings are adjusted to obtain a PaO2 of 60 to 70 mm Hg with a PCO2 of 35 to 45 mm Hg. The level of PEEP often must be increased to optimize PO2 and oxygen saturation. The use of high levels of PEEP is preferable to the continued use of toxic concentrations of oxygen. If possible, the FIO2 should be reduced to less than 60% to minimize the potential for long-term oxygen toxicity. The criteria for using higher levels of PEEP include a static compliance less than 40 milliliters per centimeter of water, a shunt fraction exceeding 20%, or an alveolar–arterial oxygen gradient on 100% oxygen exceeding 400 mm Hg. At high levels of PEEP (>20 cm of water), cardiac output may fall; thus, invasive hemodynamic monitoring often is useful in assessing the cardiac output response to high levels of PEEP. Fluid therapy in patients with ARDS may also be facilitated by the use of invasive hemodynamic monitoring, and the PCWP should be maintained as low as is consistent with adequate cardiac and urine output.
|
|
|
TABLE 25.5. Guidelines for instituting ventilator therapy |
The management of the patient with ARDS often is a long-term process. Ventilation and circulation may be supported by using some of the techniques outlined in this section. After the underlying cause of ARDS has been eliminated, therapy involves support of respiration and circulation while allowing the lungs time to heal. Despite aggressive and optimal management, the mortality rate for pregnant patients is approximately 40%. When ARDS is secondary to sepsis, higher mortality is seen. In many instances, such patients may expire despite optimal therapy. Surfactant replacement as well as nitric oxide therapy have shown promise in the treatment of severe ARDS; however, the use of these agents remains investigational.
AMNIOTIC FLUID EMBOLISM
AFE is an uncommon obstetric catastrophe with a mortality as high as 80%. It is a biphasic process that is triggered by the sudden embolization of amniotic fluid or debris of fetal origin into the maternal venous circulation. The initial physiologic disturbances involve profound alterations in hemodynamics and oxygenation, often followed by the development of a consumptive coagulopathy. Either of these phases may predominate and, occasionally, bleeding may be the initial manifestation. The most common clinical picture consists of sudden dyspnea and hypotension, commonly followed within minutes by cardiorespiratory arrest. In up to 50% of these cases, these initial events may be accompanied by seizure activity. Common signs and symptoms of AFE are presented in Table 25.6. One half of all patients with AFE die within 1 hour after the onset of symptoms; in survivors, neurologic damage or brain death secondary to the initial severe hypoxia is not uncommon. Data from the National AFE Registry suggested an overall mortality rate of 61%; only one half of those who survive do so without significant neurologic sequelae. Women with AFE generally exhibit left ventricular dysfunction or failure accompanied by elevated PCWP and, in most cases, depressed left ventricular stroke work index. Systemic vascular resistance is decreased. Elevations of pulmonary artery pressure and pulmonary vascular resistance often are in the range attributable to left ventricular failure. The existence of an initial but transient phase of pulmonary hypertension and elevated right ventricular pressure is supported by some animal and clinical data. A component of noncardiogenic pulmonary edema (i.e., ARDS) often is present and may become the predominant clinical problem in survivors.
|
|
|
TABLE 25.6. Signs and symptoms noted in patients with amniotic fluid embolism |
A hemorrhagic phase is reported in at least 50% of patients with AFE. This problem commonly is compounded by the simultaneous occurrence of uterine atony, perhaps as a direct result of a myometrial depressant effect of amniotic fluid. Although AFE has been reported under many conditions, most cases have occurred during labor. In the past, a pattern of vigorous labor or hypertonic uterine contractions associated with oxytocin infusion had often been implicated in the pathogenesis of this condition. Evidence of this association was anecdotal and is clearly invalid. Placental abruption is present in up to 50% of cases, with fetal death reported in some cases before the acute clinical onset.
Early studies suggested that meconium or particulate matter in amniotic fluid is the pathologic agent, but more recent data suggest that a humoral substance, rather than particulate matter, is responsible for the observed hemodynamic changes. It is also clear the infusion of clear amniotic fluid, per se, is innocuous, both in the human and in animal models. Further, the marked similarities between the clinical manifestations of AFE and both septic and anaphylactic shock suggest the involvement of endogenous mediators in the pathophysiology of this condition. Thus term amniotic fluid embolism is a misnomer, because the syndrome is not caused by amniotic fluid per se, nor are clinical manifestations those commonly seen with embolic events. Rather, this is yet another example of the increasingly recognized systemic inflammatory response syndrome which characterizes other types of critical illness such as anaphylaxis and sepsis (Fig. 25.8).
|
|
|
FIG. 25.8. Proposed pathophysiologic relationship among amniotic fluid embolism, septic shock, and anaphylactic shock. Each syndrome may also have direct physiologic effects (e.g., fever in endotoxin-mediated sepsis). |
Classically, the definitive diagnosis of AFE has been made at autopsy with the demonstration of fetal squamous cells, mucin, hair, or vernix in the pulmonary artery vasculature. More recently, it has been demonstrated that squamous cells and other debris of presumed fetal origin may be demonstrated in blood aspirated from the central venous or pulmonary artery circulation of living patients with AFE. Studies of pregnant women undergoing pulmonary artery catheterization for various medical indications have suggested that the detection of squamous cells in the maternal pulmonary artery circulation is a common phenomenon that may, in part, be attributable to contamination with adult squamous epithelium from the site of central venous access. Further, such debris is detected in only 50% to 75% of patients with clinical AFE, even when examined with special stains and techniques. Thus, the detection of squamous cells in the maternal pulmonary artery circulation during life is neither sensitive nor specific for the diagnosis of AFE. Such a diagnosis must be made principally on the basis of clinical picture. The differential diagnosis of AFE includes septic shock, aspiration pneumonitis, acute myocardial infarction, pulmonary thromboembolism and, in cases in which coagulopathy is a dominant feature, placental abruption.
Treatment revolves around three goals: oxygenation, maintenance of cardiac output and blood pressure, and resolution of what usually is a self-limiting, albeit severe, coagulopathy. Hypotension usually is on the basis of cardiogenic shock; treatment involves initial optimization of cardiac preload by rapid crystalloid administration followed by pressor support, as necessary. Administer component therapy to treat bleeding secondary to DIC. Packed red blood cells and fresh frozen plasma are the mainstay of such therapy.
It is axiomatic in obstetrics that when maternal and fetal therapeutic interests are divergent, maternal considerations must be primary. Thus, in the presence of an unstable mother, delay in delivery is often necessary despite ongoing fetal asphyxia. On the other hand, once the mother has suffered frank cardiac arrest, maternal prognosis is sufficiently grave to warrant emergency cesarean delivery. Delivery of the fetus beyond 15 minutes from the time of maternal cardiac arrest generally results in newborn death or neurologic impairment.
In patients who survive AFE, recurrence during subsequent vaginal or cesarean delivery has not been reported.
SUMMARY POINTS
· Successful management of the critically ill gravida requires an understanding of both unique maternal physiology, as well as fetal response to critical illness.
· Prompt recognition and aggressive management of shock during pregnancy is essential for optimal maternal and fetal outcomes.
· Invasive hemodynamic monitoring rarely is indicated in the pregnant woman. However, principles learned from such techniques often assist the clinician in the management of critically ill patients without resorting to invasive procedures.
SUGGESTED READINGS
American Association of Blood Banks. Circular of information for the use of human blood and blood components. American Red Cross, August 2000.
Annane D. Corticosteroids for septic shock. Crit Care Med 2001;29:S117–S120.
Catanzarite V, Willms D, Wong D, et al. Acute respiratory distress syndrome in pregnancy and the puerperium: causes, courses, and outcomes. Obstet Gynecol 2001;97:760–764.
Clark SL, Cotton CB. Clinical indications for pulmonary artery catheterization in severe pregnancy induced hypertension. Am J Obstet Gynecol1988;158:453.
Clark SL, Cotton DB, Gonik B, et al. Central hemodynamic alterations in amniotic fluid embolism. Am J Obstet Gynecol 1988;158:1124.
Clark SL, Cotton DB, Lee W, et al. Central hemodynamic assessment of normal term pregnancy. Am J Obstet Gynecol 1989;161:1439.
Clark SL, Cotton DB, Pivarnik JM, et al. Position change and central hemodynamic profile during normal third trimester pregnancy and postpartum. Am J Obstet Gynecol 1991;164:883.
Clark SL, Hankins GDV, Dudley DA, et al. Amniotic fluid embolism: analysis of the national registry. Am J Obstet Gynecol 1995;172:1158–1169.
Clark SL. Pregnancy following amniotic fluid embolism. Am J Obstet Gynecol 1992;167:551.
Clark SL. Shock in the pregnant patient. Semin Perinatol 1990;14:52.
Coles NA, Hibberd M, Russell M, et al. Potential impact of pulmonary artery catheter placement on short term management decisions in the medical intensive care unit. Am Heart J 1993;126:815.
Committee on Standards. Standards for blood banks and transfusion services, 21st ed. Arlington, VA: American Association of Blood Banks, 2002.
Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordinations. Am J Respir Crit Care Med 1994;149:818.
Dunser MW, Mayr AJ, Ulmer H, et al. The effects of vasopressin on systemic hemodynamics in catecholamine-resistant septic and postcardiotomy shock: a retrospective analysis. Anesth Analg 2001;93:7–13.
Eriksen NL, Parisi VM. Adult respiratory distress syndrome and pregnancy. Semin Perinatol 1990;14:68.
Haddad B, Barton JR, Livingston JC, et al. Risk factors for adverse maternal outcomes among women with HELLP (hemolysis, elevated liver enzymes, and low platelet count) syndrome. Am J Obstet Gynecol 2000;183:444–448.
Holmes CL, Patel BM, Russell JA, et al. Physiology of vasopressin relevant to management of septic shock. Chest 2001;120:989–1002.
Kirov MY, Evgenov OB, Evgenov NV, et al. Infusion of methylene blue in human septic shock: a pilot, randomized, controlled study. Crit Care Med2001;29:1860–1867.
Korach M, Sharshar T, Jarrin T, et al. Cardiac variability in critically ill adults: influence of sepsis. Crit Care Med 2001;29:1380–1385.
Lee W, Clark SL, Cotton DB, et al. Septic shock during pregnancy. Am J Obstet Gynecol 1988;159:410.
Marinelli WA, Ingbar DH. Diagnosis and management of acute lung injury. Clin Chest Med 1994;15:517.
Mimoz O, Rauss A, Rekik N, et al. Pulmonary artery catheterization in critically ill patients: a prospective analysis of outcome changes associated with catheter-prompted changes in therapy. Crit Care Med 1994;22:573.
Parillo JE. Septic shock in humans. In: Shoemaker WC, Ayers SM, Grenvik A, et al, eds. Textbook of critical care, second ed. Philadelphia: WB Saunders Co, 1989:1018.
Temmesfeld-Wollbruck B, Walmrath D, Grimminger F, et al. Prevention and therapy of the adult respiratory distress syndrome. Lung 1995;173:139.
Udagawa H, Oshio Y, Shimizu Y. Serious group A streptococcal infection around delivery. Obstet Gynecol 1999;94:153–157.
Vallet B, Wiel E. Endothelial cell dysfunction and coagulation. Crit Care Med 2001;29:S36–S41.