Rebecca L. Shattuck-Brandt
Stephen J. Brandt
![]() Basic Principles
Basic Principles
The field of hematology has been significantly impacted by advances in molecular biology. These have yielded an understanding of disease at the most fundamental level, had an important influence on diagnosis, and enabled the large-scale synthesis of a number of recombinant proteins for therapy. The contribution that hematology has, in turn, played in the development of this knowledge is significant. A blood disorder, thalassemia, was the first genetic disorder analyzed with the new tools of molecular biology, and considerable knowledge of genomic organization and transcriptional control has come from studies of the β-globin gene cluster. Further, a hematologic disorder, sickle cell anemia, was the first for which a molecular description was determined, and the first attempt at human gene therapy involved the hematopoietic cells of an individual with an inherited immunodeficiency.
DNA Chemistry and Structure
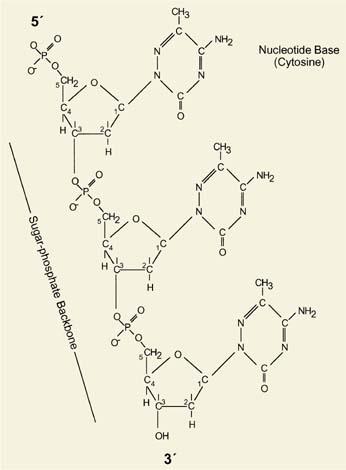
The human genome is comprised of 3.1 × 109 base pairs (3.1 gigabases) of DNA packaged into 23 pairs of chromosomes. Less than 2% of the genome actually encodes proteins, with the remainder believed to regulate DNA replication, transcription, association with nuclear matrix, and packaging (1). The deoxyribonucleotide building blocks of DNA are covalently linked through phosphodiester bonds between the fifth carbon on one pentose ring (5′) and the third carbon on the pentose ring of the adjacent (3′) deoxyribonucleotide (Fig. 4.1). This configuration imparts a directionality to the polynucleotide chains, with the double helical structure of DNA formed from two strands running in opposite directions. The energy underlying their interaction comes from interstrand base pairing between adenine (A) and thymine (T), involving two hydrogen bonds, and guanine (G) and cytosine (C), involving three hydrogen bonds. The two DNA strands form a right-handed helix with a major and minor groove.
Messenger RNA (mRNA), which is transcribed from DNA, consists of a series of base pair triplets, or codons (Table 4.1), that specify a sequence of amino acids. This type of protein coding mechanism has two important features. First, its degeneracy, with almost every amino acid represented by more than one codon, minimizes the effect of DNA mutation by reducing the probability that any single base change results in an amino acid substitution. Second, the code is translated in nonoverlapping triplets from a fixed starting point, so that three potential translational reading frames exist for each nucleotide sequence. Thus, insertion or deletion of a single base may alter the amino acid sequence and in some cases truncate the synthesis of the encoded protein.
From Chromosomes to Genes
Prior to the development of molecular-based methods to characterize DNA structure, light microscopy showed that the human genome was packaged into 23 pairs of chromosomes, with each chromosome divided by a centromere into short and long arms, designated p and q, respectively. It was also recognized that the chromosomes were not uniform in appearance and their distinctive banding patterns could be used to resolve individual chromosome pairs. With further refinements in banding techniques, abnormalities were recognized in the chromosomal complement, or karyotype, of malignant cells. Indeed, the first specific cytogenetic abnormality associated with a human neoplasm came from analysis of blast cells of patients with chronic myelogenous leukemia (2), and the association between the Philadelphia chromosome and this disease provided one of the earliest indications of the clonal nature of cancer.
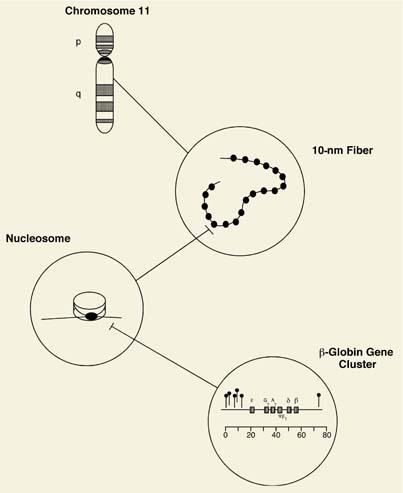
Even the smallest human chromosome contains more than 5 × 107 base pairs, which would stretch almost 1.4 cm fully extended. Since the average nucleus spans only microns in diameter, the cell must compact these large DNA molecules into the smaller, protein-bound form termed chromatin. The fundamental unit of chromatin is the nucleosome, composed of a short stretch of DNA and specific histone and nonhistone proteins. Two copies each of histones H2A, H2B, H3, and H4 make up a protein octamer core around which the DNA is wrapped. As a result of this organization, a 10-nm chromatin fiber is formed whose appearance under the electron microscope has been likened to beads on a string. The 10-nm fiber is, in turn, organized into a 30-nm fiber containing approximately six nucleosomes per turn. Numerous nonhistone proteins are associated with chromatin that function in DNA packaging, replication, and transcription, with the result that chromatin contains twice as much protein as DNA (Fig. 4.2).
Another problem in genomic organization with which the cell must contend is copying the ends, or telomeres, of chromosomes. Since DNA polymerases cannot replicate the termini of DNA molecules, long stretches of a tandemly repeated sequence, 5′-TTAGGG-3′, are inserted in this region to ensure that essential subtelomeric genes are not deleted during DNA replication. The number of these repeats decreases with every cell division in somatic cells, while in cells that become immortalized or transformed, with some important exceptions (3), telomere length is maintained by an unusual RNA-dependent DNA polymerase, telomerase.
The structure of the genome is dynamic, with some regions transiently and others constitutively complexed with protein. DNA regions that are transcribed to make RNA have a different structure, evidenced by their increased sensitivity to cleavage with the enzyme deoxyribonuclease (DNAse) I, although this DNAse hypersensitivity also applies to origins of replication and centromeres.
The small fraction of the human genome that encodes proteins shows the most complex organization, as exemplified in the globin genes. In the adult, the tetrameric hemoglobin A molecule is composed of two nonidentical α- and β-chains. The human α-globin genes are located on chromosome 16 and the β-globin and β-globin–like genes on chromosome 11, with the β-globin cluster actually containing five genes, ∊, Gγ, Aγ, δ, and β, that span over 50 kilobase pairs of DNA (Fig. 4.2). Each gene contains protein coding segments, or exons, and sequences termed introns that must be removed from the RNA transcripts prior to translation. Genes also contain a promoter and other regulatory regions upstream of their transcriptional start sites essential for expression. This organization is illustrated diagrammatically in Figure 4.2.
|
Table 4.1 Genetic Code |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Figure 4.1. Structure of DNA. DNA is composed of two complementary polynucleotide chains, each possessing a sugar-phosphate backbone in which the 5′ position of one pentose ring is linked by a phosphodiester bond to the 3′ position of the adjacent base. Sequence shown here is CCC. |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
Figure 4.2. Genomic organization from the gene to the chromosome. DNA complexed with histones and nonhistone proteins is packaged into a highly organized structure whose core unit is the nucleosome. As a result of this organization, a 10-nm chromatin fiber is evident on electron microscopy that resembles beads on a string. This is assembled into a chromosome, each of which possesses a short (p) and long (q) arm. Transcriptionally active regions of DNA are less protein bound and show hypersensitivity to digestion with DNAse I. These DNAse I hypersensitive sites are denoted in the bottom panel by the symbol extending above the DNA strand. |
Genomic Diversity
The early view of the genome that one gene encodes only one protein was upset by the discovery of introns in 1977 (4,5). It soon became apparent that multiple proteins could be made from a single gene through utilization of multiple promoters or through “exon shuffling,” in which specific exons are spliced in or out of the transcribed RNA (Fig. 4.3). While the amino acid sequence of the encoded protein is altered when exons are substituted, added, or deleted, the abundance of a protein is also subject to control through changes in the rate of transcription or stability of the transcribed mRNA.
A further level of complexity exists for the antigen receptor genes, those encoding the T-cell receptors and immunoglobulins, in which somatic recombination occurs (Fig. 4.4) (6). The class to which an immunoglobulin belongs is specified by the heavy-chain gene used in its synthesis—γ, μ, α, δ, and ∊. Each heavy chain contains N-terminal variable (VH) and C-terminal constant (CH) regions encoding the variable and constant domains of the immunoglobulin molecule, respectively. While the constant domain is invariant for every antibody of a given type, the variable domain, which is responsible for antigen recognition and binding, is subdivided into three hypervariable regions. The heavy chain also contains a hinge region important for the characteristic Y shape of the assembled molecule.
|
|
|
Figure 4.3. Generation of multiple proteins from a single gene: Alternative splicing and alternative promotion. Following transcription, heteronuclear RNA is processed by removal of the noncoding introns (splicing) and addition of a polyadenylic acid tail (polyadenylation). This processed or messenger form of RNA is then translated into protein in the cytoplasm of the cell. Translation begins at a start codon (ATG) and terminates at one of several possible stop codons, leaving regions of the RNA 5′ and 3′ to the coding region untranslated (UTR). Variation in RNA and, consequently, protein sequence can occur if there is more than one promoter or if portions of the protein coding regions (exons) are differentially incorporated into the final processed form of RNA. |
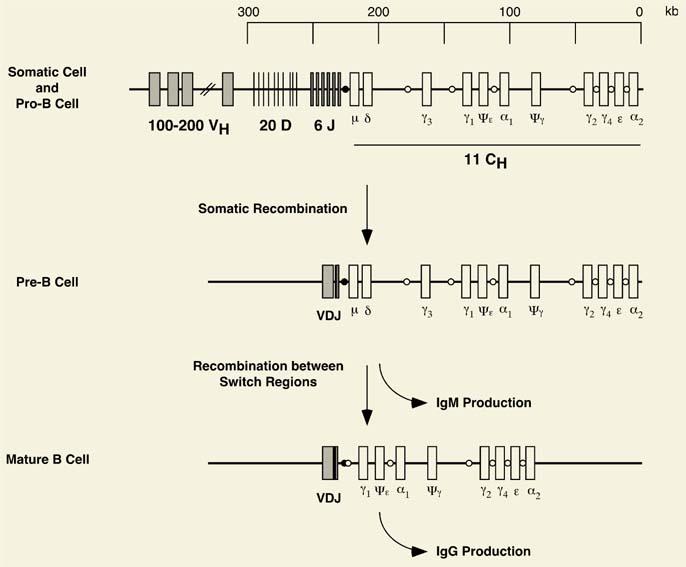
The genes encoding the antigen receptors contain multiple noncontiguous segments distributed over large distances. For example, the immunoglobulin heavy-chain locus, composed of at least two megabase pairs of DNA on chromosome 14, includes more than 100 V genes, each with its own leader sequence and promoter region; at least 12 functional diversity (or D) genes; six functional joining (or J) genes; and nine functional C genes. Each V gene contains two exons and each C gene multiple exons, encoding the constant, hinge, transmembrane, and cytoplasmic domains. Additional nontranscribed versions of these genes, termed pseudogenes, also exist.
To assemble a functional heavy-chain molecule, intrachromosomal gene rearrangements join select V, D, and J genes, after which the recombined VDJ locus fuses to a specific C gene. As a result of these events, the arrangement of DNA in a B cell that produces a specific immunoglobulin molecule is distinct from that of all other somatic or germ cells. Up to 38,880 (or 60 × 12 × 6 × 9) different heavy chains can be generated by this mechanism alone. Further variability is introduced by the addition of nucleotides to the junctions between segments, alternative splicing of the mRNA, and somatic mutation of the immunoglobulin (but not T-cell receptor) genes. As a result of these processes, the organism is able to mount an immune response of extraordinary specificity.
Somatic recombination represents a specialized strategy to achieve genomic diversity. In the germline, this is accomplished by meiosis, in which interchromosomal exchange of DNA occurs as a result of the two copies of each chromosome coming into register. The crossing-over events promoted by this process cause a shuffling of genetic material, and the greater the distance between two genes, the higher the probability they will become separated after meiosis. Finally, random mutation can alter DNA sequence, both in coding and noncoding regions. If not deleterious, such sequence polymorphisms are inherited and can in this way increase genetic diversity.
Pathologic Changes in the Genome
The processes that promote genomic rearrangement are prone to error. These manifest as translocations, deletions, insertions, or inversions of chromosomal segments and by mutations of individual nucleotides. The proteins encoded can be altered in their expression or have diminished or even novel function. Burkitt lymphoma, thalassemia, and sickle cell disease represent disorders resulting from distinct pathologic alterations in the genome.
Under normal circumstances, it is estimated that only one in three lymphoid progenitor cells productively rearranges its antigen receptor genes and successfully develops into a mature lymphocyte. If rearrangement takes place between rather than within chromosomes, as occurs in a specific chromosomal rearrangement associated with Burkitt lymphoma, the cell can obtain a growth or survival advantage. The translocation observed in the endemic form of this lymphoma, involving the c-MYC proto-oncogene on chromosome 8 and the immunoglobulin heavy-chain locus on chromosome 14, effectively decapitates the MYC gene, fusing it to the JH gene promoter. In other patients, c-MYC is brought under the regulatory control of the powerful immunoglobulin enhancer. Both translocations cause unscheduled or dysregulated expression of c-MYC protein, with consequences for oncogenic transformation.
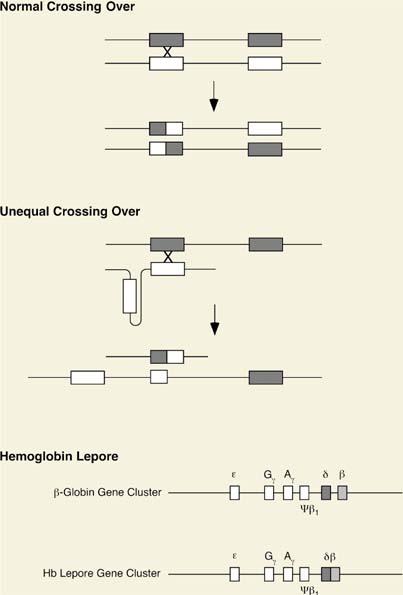
The thalassemias also have a genetic basis. The causative defect in this disorder is in the synthesis rather than the sequence of the globin protein made. While uncommon, hemoglobin (Hb) Lepore illustrates the results of unequal crossing over between nonallelic members of a gene cluster, which in this case differ in sequence by only 7%. This nonreciprocal rearrangement generates two recombinant chromosomes, one that duplicates and the other that deletes sequences between the genes involved, which effectively fuses them together. The protein made by that derivative chromosome contains N-terminal sequences from the δ-globin gene and C-terminal sequences from the β-globin gene. The unequal crossing-over events in this form of thalassemia are depicted in Figure 4.5.
|
|
|
Figure 4.4. Intrachromosomal gene rearrangement: Somatic recombination in the immunoglobulin heavy-chain locus. The immunoglobulin heavy chain is generated by the joining of a single V gene to a D segment, which is then fused to a J gene. This process of somatic recombination occurs in the immature B cell. VDJ joining triggers expression of the Cμ gene. Immunoglobulin class switching occurs through further recombination involving switch regions designated by the open and closed circles. Ig, immunoglobulin. |
Finally, disease can result from a change in a single base pair. This can effect an amino acid substitution that profoundly changes the properties of the encoded protein, as in the sickle cell mutation, or insertion of a stop codon that terminates protein chain elongation (nonsense mutation). The first molecularly characterized disorder (7), sickle cell disease results from a missense mutation in the sixth codon of the β-globin gene that causes replacement of glutamic acid by valine (Fig. 4.6). The abnormal hemoglobin molecule resulting shows a marked decrease in solubility and a tendency to polymerize when deoxygenated. Ropelike fibers of deoxyhemoglobin S cause the sickle shape of red cells that is pathognomonic for this disease and are ultimately responsible for the hemolytic and vaso-occlusive phenomena that produce symptoms. At least 300 other point mutations in the α- or β-globin genes have also been described.
![]() Gene Discovery and Molecular Analysis
Gene Discovery and Molecular Analysis
Restriction Enzymes and Vectors
Understanding of hematologic disease has been greatly facilitated by certain molecular techniques. An important discovery in the development of molecular biology was that of restriction enzymes, endonucleases that cut double-stranded DNA at specific sequences. By reducing the genome to experimentally manipulable pieces, or restriction fragments, these enzymes made possible the cloning and propagation of genes. Restriction endonucleases recognize specific four to eight base pair sequences that often are palindromic (i.e., show the same order of bases from either strand). EcoRI, for example, recognizes the sequence:
· 5′—GAATTC—3′
· 3′—CTTAAG— 5′
and cleaves between G and A, creating two new ends:
· 5′—G AATTC—3′
· 3′—CTTAA G—5′
Although the DNA in this example is left with overhanging ends, other enzymes generate blunt ends, depending on their recognition sequence. Since the discovery of the first restriction enzyme in 1970, hundreds have been characterized and made available commercially (8).
In addition to restriction endonucleases, a number of other nucleic acid–modifying enzymes are employed in recombinant DNA work. One of the more commonly used is T4 DNA ligase, which catalyzes the formation of a phosphodiester bond between free ends of DNA. The DNA fragments created by EcoRI, for example, can be rejoined by this enzyme.
|
|
|
Figure 4.5. Mutation resulting from abnormal crossing over: Hemoglobin Lepore. Genetic recombination occurs physiologically during meiosis and requires precise alignment of the corresponding sequences in homologous chromosomes. Abnormal alignment can occur when there are two or more copies of homologous genes, as in the β-globin gene cluster. Hemoglobin Lepore likely results from the kind of misalignment as illustrated. |
Another important tool of the molecular biologist is the plasmid vector, which is the vehicle by which a foreign DNA molecule is introduced into bacteria, amplified, and purified. Standard laboratory vectors contain multiple restriction enzyme sites to simplify subcloning of DNA fragments, an antibiotic resistance gene for the selection of cells that have taken it up, and a sequence (or origin) permitting its replication in a bacterial host. Other features allow vectors to replicate in eukaryotic cells and to serve as templates for in vitro transcription. Cells are rendered capable of taking up plasmids by treatment with divalent cations or exposure to a pulsed electrical field. These methods are applicable to both prokaryotic and eukaryotic cells.
|
|
|
Figure 4.6. Mutation resulting from base substitution: Hemoglobin S (sickle hemoglobin). Hemoglobin S results from a homozygous A to T transversion in codon 6 of the human β-globin gene, causing the substitution of valine for glutamic acid in the resulting protein. |
Molecular Cloning
A frequent objective in molecular biology is the isolation of a gene or its mRNA, and a number of the techniques required for this were first developed for the study of globin gene expression. An important step in this process involved the generation of a genomic or complementary DNA (cDNA) library. For a genomic library, the source of material is nuclear DNA that has been converted into a collection of smaller fragments through restriction enzyme digestion. As most genes are interrupted by introns and extend for large distances, the use of very large DNA fragments is desirable for this purpose. The size of these fragments is determined by the frequency with which a particular restriction site occurs in the genome and the proportion of those sites actually cleaved by the enzyme. Genomic DNA is frequently digested for library construction with Sau3AI, which recognizes the sequence GATC. The frequency with which this or any other four base pair sequence occurs is ¼ × ¼ × ¼ × ¼, or 1/256, so that complete digestion with Sau3AI produces fragments that are, on average, 256 base pairs in length. The extent of digestion can be limited, however, so that products in the range of 15 to 20 kilobase pairs are generated. These fragments are introduced into a vector that replicates in bacteria, typically one derived from bacteriophage λ, and the complete collection of such fragments constitutes the library.
The source of nucleic acid for a cDNA library is mRNA, which must first be converted to its corresponding DNA form by an RNA-dependent DNA polymerase, reverse transcriptase. Because mRNA in eukaryotic cells is polyadenylated, a homopolymeric thymidine affinity resin oligo(dT) is frequently used in its purification. Either oligo(dT) or random hexanucleotides can be used as primers for reverse transcriptase, which results in a DNA/RNA hybrid. Ribonuclease H is then added to digest the RNA component and a DNA polymerase fills in the gaps, resulting in replacement of the RNA strand by its DNA counterpart. Finally, T4 DNA ligase is used to join any regions still unlinked, and the ends of the newly synthesized cDNA are ligated to synthetic oligonucleotide linkers containing specific restriction enzyme sites. The resulting pool of cDNAs subcloned into a vector constitutes the library.
Synthetic oligonucleotides, larger DNA fragments, or even antibodies can be used to screen libraries. If a portion of the protein-coding region is known, oligonucleotide mixtures encompassing all or the most likely nucleotide sequences are synthesized. For the sequence methionyl-glutamyl-threonyl-serine, for example, up to 1 × 2 × 4 × 6, or 48, different oligonucleotides need to be considered, since there is one codon for Met, two for Glu, four for Thr, and six for Ser. The cDNA from another species can also be used for screening if the corresponding sequence is conserved evolutionarily.
The conditions under which nucleic acid probes are used in screening libraries are dictated by DNA strand separation (denaturation) and renaturation. The melting temperature (Tm), or the temperature at which half the double-stranded DNA molecules are denatured, depends on the proportion of G + C and, to a lesser extent, A + T. For mammalian genomes with G + C contents of <40%, this is ∼87°C. Base composition also determines the Tm of smaller double-stranded oligonucleotides, which can be estimated from the following equation:
Tm = 2(A + T) + 3(G + C), where A, T, G, and C represent the specific number of these nucleotides in the probe
Renaturation is affected by DNA composition as well. Ordinarily, the two strands of a DNA duplex base-pair in their entirety. However, if two polynucleotides that are not completely complementary are mixed, both the melting temperature of the renatured molecule and its rate of reassociation are decreased in proportion to the mismatch between the two molecules. The process by which such molecules renature is termed annealing (or, in an analytical context, hybridization). Hybridization can be carried out with nucleic acids in solution or immobilized on filters or glass slides.
Isolation of Genes without Prior Knowledge of Sequence
Often there is insufficient information, particularly with disease-related genes, to permit the use of standard library screening techniques. With a combination of genetic and physical maps, it has proven possible to isolate genes from their location in the genome without prior knowledge of their sequence or function. This approach, termed positional cloning, relies on the segregation of a trait or disease in several generations of kindreds and employs the technique of linkage analysis to identify the gene’s probable chromosomal location (9). This is followed by analysis of the genes from this region for mutations that cosegregate with the disease. The availability of the complete sequence of the human genome has greatly facilitated the application of this method.
The development of yeast artificial chromosome (YAC) and, more recently, bacterial artificial chromosome (BAC) vectors has also played an important role in the development of this technology (10). It had proven difficult to incorporate fragments larger than 10 to 15 kilobase pairs into plasmid vectors, both because of the difficulties in generating large circular DNAs and because of their relatively inefficient uptake by bacterial cells. The use of YACs, which can accommodate fragments larger than 500 kilobase pairs, was made possible by definition of the cis-acting sequence elements required for chromosome stability in yeast. This technology has also been applied to BAC vectors, which have proven useful in sequence analysis of large DNA fragments.
Genomics
Knowledge of nucleotide sequence is a prerequisite to understanding a gene’s normal function and how this is altered in disease. The most common method of DNA sequencing analysis is the chain-termination method developed by Sanger in which a DNA polymerase catalyzes the synthesis of new DNA strands, each originating with a specific oligonucleotide primer complementary to a single-stranded DNA template (11). Chain elongation is terminated at all possible positions by incorporation of a dideoxynucleotide triphosphate lacking a 3′ hydroxyl group. In practice, four independent reactions are carried out with a mixture of deoxynucleotide triphosphates and limiting amounts of one dideoxynucleotide triphosphate, and the ratio of deoxynucleotides to dideoxynucleotides is adjusted to allow incorporation of the dideoxynucleotide along the whole length of the molecule. The reaction products are fractionated by size by denaturing polyacrylamide gel electrophoresis and the DNA sequence determined by autoradiography if a radiolabeled nucleotide is incorporated or fluorography if fluorescent nucleotides are used. The Sanger method is still widely used, although DNA sequencing is now almost exclusively carried out by dedicated instruments (12).
After the nucleotide sequence of a gene is determined, one of the first ways in which it is analyzed typically involves a computer-assisted database search (13). This can reveal whether it had been previously isolated, either in its full-length form or as an anonymous sequence, if orthologs or paralogs exist, and often some indication of its function. The GenBank database maintained at the National Center for Biotechnology Information of the National Library of Medicine contains all publicly available information on gene and protein sequences. User-friendly search engines, the most widely used being the Basic Local Alignment Search Tool, or BLAST (14), enable interrogation of all compiled sequences and are accessible over the internet (http://www.ncbi.nlm.nih.gov/).
As a result of a concerted effort worldwide, the sequence of the human genome has been completely determined (15). This led to the unexpected finding that only 30,000 to 35,000 genes are present in our genome, although this appears to be sufficient to encode over 100,000 proteins. It has also been noted that protein-coding regions are unevenly distributed, with some chromosomes and chromosomal regions more gene dense than others. In addition to genomic DNA, effort has been made to sequence cDNAs, or expressed sequence tags (ESTs), prepared from a number of different sources of RNA, and estimates suggest that more than half of all human genes are represented in EST databases.
![]() Molecular Diagnostics
Molecular Diagnostics
Molecular techniques have revolutionized the diagnosis of disease, particularly for inherited disorders. Two of the earliest applications of such methods were in prenatal diagnosis of α-thalassemia (16) and in identification of the causative mutations in β-thalassemia (17). Since their advent, highly sensitive and specific techniques have been developed to analyze abnormalities in both RNA and DNA.
Southern Blot Analysis
Southern blot analysis, named after its developer E. M. Southern (18), enables the number, organization, and internal structure of any gene to be determined. The technique involves immobilizing nucleic acids on a filter as described above for library screening, except that restriction enzyme-digested genomic DNA is the target of the probe rather than a library of individual clones. Typically, high-molecular-weight DNA is treated with several restriction enzymes and the resulting digests fractionated by agarose gel electrophoresis. These restriction fragments are denatured within the gel and transferred, or blotted, to a filter. The gene of interest is then identified by hybridization of this filter replica with a specific probe and its structure deduced from the hybridization patterns.
Although supplanted by the polymerase chain reaction (PCR) for most purposes, Southern blot analysis has been invaluable in the diagnosis of a number of hematologic disorders. The mutation in sickle cell anemia, for example, can be specifically recognized with this technique. Substitution of T for A in codon 6 of the β-globin gene in Hb S results in loss of an MstII restriction site as shown:
|
Hemoglobin A |
CCTGAGGAG |
|
Hemoglobin S |
CCTGTGGAG |
|
MstII |
CCTNAGG |
As a result, the size of the hybridizing band in Southern blot analysis of MstII-digested DNA will differ for an individual with this mutation from one with a normal β-globin sequence.
A similar approach has also been used to determine genetic relatedness and to analyze chimerism after allogeneic bone marrow transplantation. Differences in nucleotide sequence exist between individuals, and if a restriction enzyme recognition site is altered and a probe to the region is available, such restriction fragment length polymorphisms (RFLPs) can be detected by Southern blot analysis. Southern blot analysis has also been carried out with probes for polymorphic regions comprised of tandemly repeated nucleotide units of 20 to 40 base pairs (19). The number of these tandem repeats varies between individuals, producing specific molecular markers detectable by hybridization analysis.
Northern Blot Analysis
Because mRNA is susceptible to alkaline hydrolysis and does not bind well untreated to nitrocellulose, Southern’s technique of DNA analysis could not be applied directly to mRNA. With the inclusion of a denaturant like glyoxal or formaldehyde in the gel, it became possible with what is termed Northern blot analysis to similarly analyze RNA fractionated by agarose gel electrophoresis and transferred to a filter (20). Although infrequently employed in diagnostics, an important attribute of this method is its ability to quantify the abundance of specific RNA species.
In situ Hybridization
Northern blot analysis relies on hybridization of a labeled probe to RNA immobilized on a solid support. By incubating the probe with specially prepared cells or tissue sections, information can be obtained about the location and identity of the cells that transcribe that mRNA. This technique, termed in situ hybridization, has been used for localization of viruses or viral messages (21) as well as cellular transcripts (22). In addition, in situ hybridization can be used to assign a gene to an individual chromosome and to localize it within a particular subchromosomal segment (23). Using biotinylated DNA probes, avidin-conjugated fluorescent molecules are used to amplify the hybridization signal in an application of the technique termed fluorescent in situ hybridization (FISH) (24). The FISH technique has been invaluable in detecting loss, gain, or rearrangement of specific chromosomal regions in tumor cells.
Comparative genomic hybridization (CGH) represents another refinement of the in situ hybridization technique in which regions with tumor-specific gains or losses are identified (25). The method involves hybridization of metaphase cell preparations with equal amounts of subject and reference DNA labeled with dyes that fluoresce in the red and green wavelengths. Regions that hybridize equally well with subject and reference DNA appear yellow (red + green), while regions hybridized with under- or overrepresented DNA appear red or green.
Microarray Analysis
An important approach to characterizing gene expression employs nucleic acids affixed or stamped onto inert supports termed microarrays (26). The microarray technique involves extraction of RNA from a biologic sample, its conversion to cDNA with incorporation of either fluorescent nucleotides or tags that are detected by fluorescence, and hybridization of these labeled cDNAs to a very large number of cDNAs or oligonucleotides immobilized on glass slides or filters. A reference RNA labeled with a different fluorochrome is similarly processed, and the two populations of cDNAs are mixed and competitively hybridized to the array. The arrayed spots appear yellow (red + green) in laser scanning analysis if the subject and reference cDNAs hybridize equally and red or green if there is a difference in their relative abundance (Fig. 4.7). This technique provides information on as many RNAs as there are unique cDNAs or oligonucleotides on the array, which can number in the tens of thousands. Microarray experiments generate a massive amount of data and require sophisticated informatic methods to extract biologically useful data (27).
Gene expression profiling using such arrays has been used to identify the involvement of specific genes, microorganisms, or pathways in disease. It has, in addition, led to new, molecular-based classifiers for known diseases (class discovery) and for previously unclassified disorders (class prediction). Finally, microarrays have been used to screen for genomic imbalances (array-CGH), small mutations, insertions, or deletions, and to type single nucleotide polymorphisms. Analogous array technologies are also being developed for proteins, protein-binding molecules, and even small tissue sections.
Polymerase Chain Reaction Analysis
Southern blot analysis and FISH are restricted to situations in which there are sizeable rearrangements of DNA and are not generally useful in detecting small insertions, deletions, or base substitutions. The PCR technique, developed by Mullis et al. in 1986 (28), is ideal for these purposes and has found wide utility in numerous areas of cell and molecular biology.
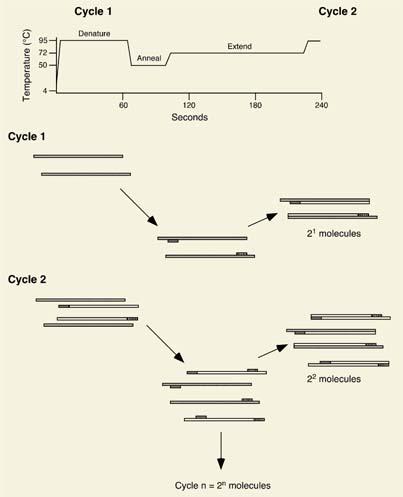
PCR is, essentially, a method amplifying a specific genomic or cDNA sequence. In its usual application, two oligonucleotides oriented in opposite directions and complementary to the ends of the region of interest serve as primers for a heat-stable DNA polymerase. A reaction mixture comprised of DNA template, primers, and deoxynucleotide triphosphates is heated to a temperature of 94 to 95°C to effect DNA strand separation and then cooled to allow the oligonucleotides to anneal. Primer extension is effected at a relatively high temperature, typically 72°C, by a heat-stable DNA polymerase, and these three reactions, which make up one PCR cycle, are then repeated some 20 to 40 times. As a result of its exponential nature, 2n copies of the sequence encompassed by the two primers are generated, where n represents the total number of cycles carried out. PCR is extraordinarily sensitive and is capable of detecting the presence of even a single malignant cell within a million normal ones. The technique is outlined schematically in Figure 4.8.
|
|
|
Figure 4.7. Microarray analysis. Shown is a portion of a complementary DNA (cDNA) microarray that has been competitively hybridized with cDNA made from human leukemia cell RNA and labeled with the fluorochrome Cy3 and with cDNA prepared from a pool of human RNAs (reference RNA) and labeled with a different fluorochrome, Cy5. Each spot corresponds to one out of a total of 11,000 cDNAs affixed on this array. Its color (red, green, or yellow) on laser scanning analysis provides information about the abundance of specific RNAs in the leukemic blasts. |
PCR is often combined with other techniques, including some described above, to determine whether the amplification products contain a mutation. Although dideoxynucleotide sequencing can be carried out to confirm mutations, restriction enzyme digestion, allele-specific oligonucleotide hybridization, and single-strand conformation polymorphism (SSCP) analysis represent more efficient methods to screen for their presence. The first approach takes advantage of the sequence specificity of restriction enzyme digestion and was discussed in the example of sickle cell disease above. The approach is simplified and more widely applicable when a short fragment of amplified DNA is analyzed rather than the entire genome. The second method involves the allele-specific hybridization of an oligonucleotide to PCR product that has been immobilized on a filter. Since the melting temperature of a short oligonucleotide will be dependent on the extent of complementarity, conditions can be set such that only exact matches result in binding of the probe to the DNA sample. In SSCP, the DNA of interest is denatured by heat or alkali treatment and subjected to electrophoresis with a control sample in a nondenaturing polyacrylamide gel (29). This technique relies on a mutation affecting the secondary structure and, consequently the electrophoretic mobility of either of the denatured strands relative to the control.
|
|
|
Figure 4.8. Polymerase chain reaction. The polymerase chain reaction (PCR) is a method to exponentially amplify defined regions of DNA. The technique consists of a series of repeated steps (cycles) involving, in sequence, denaturation of the DNA target, binding (or annealing) of specific oligonucleotides (primers) complementary to the region of interest and delimiting the region to be amplified, and DNA synthesis (or extension) from the primers by the action of a heat-stable DNA polymerase. Denaturation, annealing, and extension are carried out at different temperatures (see top) in a dedicated piece of equipment known as a thermal cycler. A total of two molecules are synthesized after the first cycle, four molecules after the second cycle, and 2n after n cycles, where n represents the total number of cycles. |
![]() Analysis of Gene Function
Analysis of Gene Function
Clues to a gene’s function are often provided by its amino acid sequence. Even when a protein’s molecular actions are known, however, it may be desirable to assess its function in the context of the cell or organism. If its nucleotide sequence is available, a gene’s actions can be selectively inhibited by synthetic oligonucleotides or RNA species complementary to its encoded message. These antisense reagents are thought to inhibit the translation or accelerate the destruction of the particular transcript targeted and are predictably more effective against proteins with short half-lives (30). A more potent and specific approach to reducing gene expression involves the use of double-stranded RNA molecules 21 to 23 base pairs in length that are complementary to the RNA of interest (31). These small interfering RNAs effect posttranscriptional silencing in a series of steps that culminate in degradation of the targeted mRNA by cellular ribonucleases. In addition to their use in probing gene function, both antisense inhibition and RNA interference are under investigation for therapeutic purposes.
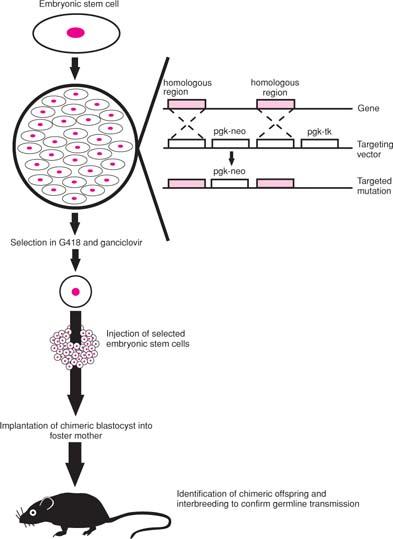
An even more secure method of inhibiting protein function is to inactivate its gene so that a functional product can not be expressed (Fig. 4.9). This approach, referred to as gene targeting or knockout, employs a vector containing, typically, a neomycin resistance gene flanked by sequences of exact homology to the gene of interest and a linked thymidine kinase gene. This vector is introduced into embryonic stem (ES) cells by standard means (32), and ES cells that have undergone homologous recombination at the targeted locus are selected for resistance to both the neomycin analog G418 and the drug ganciclovir. Cells surviving this “positive–negative” selection are microinjected into blastocysts of mice, these blastocysts implanted into a foster mother, and any chimeric offspring resulting, which are screened for by coat color, interbred. If the gene-targeted ES cells contribute to the germline, mice homozygous for the induced mutation can be generated, provided the gene is not required for embryonic development. The technology to create knockout mice has provided important insights into the in vivo function of many genes and led to the development of mouse models for a score of hematologic disorders. The establishment of human ES cell lines (33), with their ability to differentiate into hematopoietic progenitors (34), suggests that these pluripotent cells could also be useful therapeutically.
|
|
|
Figure 4.9. Generation of mice with targeted mutation (“knockout” mice). Embryonic stem (ES) cells are pluripotent, or capable of contributing to every cell type in the animal. They are transduced in culture with a targeting vector (see inset) in which a neomycin resistance gene (neo) driven off a phosphoglycerate kinase (pgk) promoter is flanked by portions of the gene to be targeted. Linked to this is a pgk-promoted thymidine kinase (tk) gene, which converts the drug ganciclovir to a toxic phosphorylated product. In ES cells that undergo homologous recombination (see arrows), the pgk-neo gene is incorporated, thereby inactivating the targeted gene, without the pgk-tk gene. These cells are selected in culture by their resistance to both the neomycin analog G418 and ganciclovir, analyzed by Southern blot or PCR analysis to verify disruption of the targeted gene, and injected into blastocysts of mice with a distinctive coat color. These are then implanted into a foster mother and chimeric offspring identified by coat color. Mice are bred to verify germline transmission of the targeted mutation. |
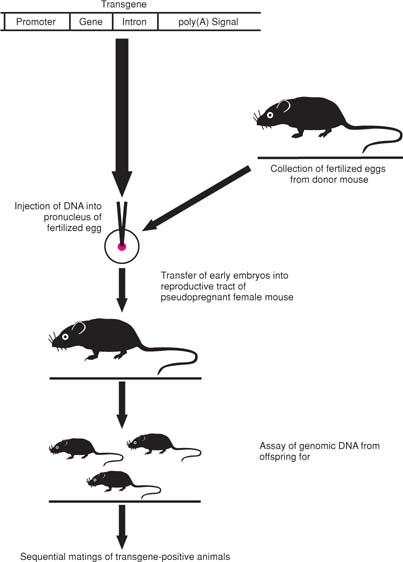
A complementary approach to elucidating a gene’s function involves its enforced expression, in vivo in transgenic animals (Fig. 4.10) or in vitro with a plasmid or viral vector. The transgene is constructed to contain all components necessary for efficient and tissue- or cell type–specific expression, including a promoter, polyadenylation site, and one or more introns. The transgene construct is introduced by microinjection into fertilized eggs, which are implanted into the reproductive system of a pseudopregnant female mouse. If successful, the transgene becomes incorporated into the germline of the resulting offspring, and matings of these animals, termed founders, are carried out with nontransgenic mice and then with heterozygotes to increase transgene copy number. In addition to analysis of the phenotype resulting from expression of the introduced gene, transgene copy number and tissue distribution are typically ascertained (35).
|
|
|
Figure 4.10. Generation of transgenic mice. The transgene construct, which contains a promoter, the coding sequences of the gene of interest, a polyadenylation signal, and typically one or more introns, is microinjected into the pronucleus of a fertilized egg. The resulting embryos are surgically transplanted into the reproductive tract of a pseudopregnant female mouse. The pups born are tested for incorporation of the transgene by Southern blot or PCR analysis of genomic DNA, usually from their tails. Sequential matings of transgene-positive animals (founders) are then carried out to verify that the transgene can be passed through the germline and, if desired, to obtain homozygous mice. |
A variety of vectors are used to transduce cells with genes, either for transient or long-term expression. This approach is rapid, avoids toxicities caused by transgene expression during embryonic development, and is useful in targeting expression to specific cell types when a tissue-specific promoter is unavailable. The techniques used in this approach are the same as those used in gene therapy described in detail in Chapter 25.
In conclusion, it is a safe bet that even though the technology will evolve, molecular biology will impact the diagnosis and practice of hematology long into the future.
![]() Acknowledgments
Acknowledgments
This was supported in part by National Institutes of Health grant R01 HL49118 (to S.J.B.) and a Merit Review Award from the Department of Veterans Affairs (to S.J.B.).
References
1. Gardiner K. Human genome organization. Curr Opin Genet Dev 1995;5:315–322.
2. Nowell PC, Hungerford DA. A minute chromosome in human granulocytic leukemia. Science 1960;132:1197–1200.
3. Lundblad V, Wright WE. Telomeres and telomerase: a simple picture becomes complex. Cell 1996;87:369–375.
4. Jeffreys AJ, Flavell RA. The rabbit β-globin gene contains a large insert in the coding sequence. Cell 1977;12:1097–1108.
5. Breathnach R, Mandel JL, Chambon P. Ovalbumin gene is split in chicken DNA. Nature 1977;270:314–319.
6. Schwartz RS. Jumping genes and the immunoglobulin V gene system. N Engl J Med 1995;333:42–44.
7. Pauling L, Itano HA, Singer SJ, et al. Sickle cell anemia, a molecular disease. Science 1949;110:543–548.
8. Roberts RJ, Macelis D. REBASE—restriction enzymes and methylases. Nucl Acids Res 1997;25:248–262.
9. Collins FS. Positional cloning moves from the perditional to traditional. Nat Genet 1995;9:347–350.
10. Burke DT, Carle GF, Olson MV. Cloning of large segments of exogenous DNA into yeast by means of artificial chromosome vectors. Science 1987;236:806–812.
11. Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 1977;74:5463–5467.
12. Ansorge W, Sproat B, Stegemann J, et al. Automated DNA sequencing: ultrasensitive detection of fluorescent bands during electrophoresis. Nucl Acids Res 1987;15:4593–4602.
13. Boguski MS. Hunting for genes in computer data bases. N Engl J Med 1995; 333:645–647.
14. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215:403–410.
15. Guttmacher AE, Collins FS. Genomic medicine—a primer. N Engl J Med 2002; 347:1512–1520.
16. Kan YW, Golbus MS, Dozy AM. Prenatal diagnosis of alpha-thalassemia: clinical application of molecular hybridization. N Engl J Med 1976;295:1165–1167.
17. Orkin SH, Kazazian HH, Antonarakis SE, et al. Linkage of β-thalassemia mutations and β-globin gene polymorphisms with DNA polymorphisms in human β-globin gene cluster. Nature 1982;296:627–631.
18. Southern EM. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 1975;98:503–517.
19. Housman D. Human DNA polymorphism. N Engl J Med 1995;332:318–320.
20. Alwine JC, Kemp DJ, Stark GR. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc Natl Acad Sci U S A 1977;74:5350–5354.
21. Gowans EJ, Arthur J, Blight K, et al. Application of in situ hybridization for the detection of virus nucleic acids. Methods Mol Biol 1994;33:395–408.
22. Lehmann R, Tautz D. In situ hybridization to RNA. Methods Cell Biol 1994;44:575–598.
23. Adinolfi M, Crolla J. Nonisotopic in situ hybridization. Clinical cytogenetics and gene mapping applications. Adv Hum Genet 1994;22:187–255.
24. van Ommen G-JB, Breuning MH, Raap AK. FISH in genome research and molecular diagnostics. Curr Opin Genet Develop 1995;5:304–308.
25. Kallioniemi A, Kallioniemi OP, Sudar D, et al. Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992;258:818–821.
26. Mohr S, Leikauf GD, Keith G, et al. Microarrays as cancer keys: an array of possibilities. J Clin Oncol 2002;20:3165–3175.
27. Butte A. The use and analysis of microarray data. Nat Rev Drug Discov 2002;1:951–960.
28. Mullis K, Faloona F, Scharf S, et al. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 1986;51:263–273.
29. Orita M, Iwahana H, Kanazawa H, et al. Detection of polymorphisms of human DNA by gel electrophoresis as single-strand conformation polymorphisms. Proc Natl Acad Sci U S A 1989;86:2766–2770.
30. Wagner RW. Gene inhibition using antisense oligodeoxynucleotides. Nature 1994;372:333–335.
31. Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001;411:494–498.
32. Majzoub JA, Muglia LJ. Knockout mice. N Engl J Med 1996;334:904–907.
33. Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998;282:1145–1147.
34. Kaufman DS, Hanson ET, Lewis RL, et al. Hematopoietic colony-forming cells derived from human embryonic stem cells. Proc Natl Acad Sci U S A 2001;98:10716–10721.
35. Shuldiner AR. Transgenic animals. N Engl J Med 1996;334:653–655.