THROMBOCYTOPENIA
GENERAL PRINCIPLES
Definition
Platelets are essential for primary hemostasis—the process in which a platelet plug forms to initiate clotting. Normal platelet range is 150,000 to 450,000/ μL. When the platelet number is decreased or the platelets are not functioning properly, bleeding may result.
Classification
Thrombocytopenia can generally be classified into one of the following processes: decreased platelet production or increased platelet destruction.
DIAGNOSIS
Clinical Presentation
Thrombocytopenia may present either asymptomatically on a routine CBC or with petechiae, purpura, or overt bleeding, especially of the mucosa. All patients with thrombocytopenia should undergo a thorough history and physical before choosing further diagnostic testing.
Differential Diagnosis
![]() Decreased platelet production
Decreased platelet production
![]() Infection: HIV, hepatitis C, parvovirus, varicella, rubella, mumps
Infection: HIV, hepatitis C, parvovirus, varicella, rubella, mumps
![]() Chemotherapy
Chemotherapy
![]() Medications
Medications
![]() Radiation
Radiation
![]() Congenital or acquired primary bone marrow failure: Fanconi anemia, megakaryocytic thrombocytopenia, paroxysmal nocturnal hemoglobinuria
Congenital or acquired primary bone marrow failure: Fanconi anemia, megakaryocytic thrombocytopenia, paroxysmal nocturnal hemoglobinuria
![]() Malignancy, particularly hematologic malignancies
Malignancy, particularly hematologic malignancies
![]() Vitamin deficiencies: folate, B12
Vitamin deficiencies: folate, B12
![]() Alcohol
Alcohol
![]() Increased platelet destruction
Increased platelet destruction
![]() Medications: heparin, valproic acid, quinine
Medications: heparin, valproic acid, quinine
![]() Autoimmune platelet destruction: immune thrombocytopenia (ITP), thrombotic thrombocytopenic purpura (TTP)/hemolytic-uremic syndrome (HUS)
Autoimmune platelet destruction: immune thrombocytopenia (ITP), thrombotic thrombocytopenic purpura (TTP)/hemolytic-uremic syndrome (HUS)
![]() HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome
HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome
![]() Disseminated intravascular coagulation(DIC)
Disseminated intravascular coagulation(DIC)
![]() Pseudothrombocytopenia
Pseudothrombocytopenia
![]() Splenic sequestration
Splenic sequestration
![]() Platelet clumping
Platelet clumping
THROMBOTIC THROMBOCYTOPENIC PURPURA AND HEMOLYTIC-UREMIC SYNDROME
GENERAL PRINCIPLES
Definition
TTP and HUS are clinically similar disorders that are often grouped together as TTP-HUS. Although pathophysiology distinguishes the two disorders, both involve microvascular damage and platelet destruction. They are classically defined by thrombocytopenia and microangiopathic hemolytic anemia (MAHA) in the absence of another apparent cause. Neurologic and renal impairments are also characteristic. When neurologic impairment is present, the patient is more likely to be classified as TTP, whereas acute renal failure is considered to be a hallmark of HUS. In some cases, there is a significant overlap and patients can have both renal and neurological impairment.
Epidemiology
TTP has an incidence of ~4 cases/million persons. HUS is an uncommon disorder with two forms—a sporadic form more typical of adults and a childhood form that is often associated with verotoxin and Escherichia coliO157:H7. Both cause thrombocytopenia with MAHA but they are distinct entities. TTP-HUS once had a 90% mortality rate until the utility of plasma exchange was demonstrated. Six month mortality now is <30% with prompt initiation of appropriate treatment.1
Pathophysiology
![]() Thrombotic thrombocytopenic purpura. Endothelial cells produce ultralarge vWF (ULvWF) molecules that are cleaved by ADAMTS13 (a metalloprotease) into their typical-length multimers under normal circumstances (fig. 4-1). In a significant number of TTP patients there is a marked decrease in ADAMTS13 activity(<5%). This protease deficiency may be inherited or due to acquired inhibitors such as IgG autoantibodies. When these ULvWF molecules persist, they induce abnormal platelet aggregation in the microcirculation in areas of high shear stress. This leads to platelet consumption and fragmenting and destruction of RBCs. It should be noted that some patients with clinical TTP do not have decreased ADAMTS13 activity, implicating other unidentified factors. 1
Thrombotic thrombocytopenic purpura. Endothelial cells produce ultralarge vWF (ULvWF) molecules that are cleaved by ADAMTS13 (a metalloprotease) into their typical-length multimers under normal circumstances (fig. 4-1). In a significant number of TTP patients there is a marked decrease in ADAMTS13 activity(<5%). This protease deficiency may be inherited or due to acquired inhibitors such as IgG autoantibodies. When these ULvWF molecules persist, they induce abnormal platelet aggregation in the microcirculation in areas of high shear stress. This leads to platelet consumption and fragmenting and destruction of RBCs. It should be noted that some patients with clinical TTP do not have decreased ADAMTS13 activity, implicating other unidentified factors. 1
![]() Hemolytic-uremic syndrome. Although HUS has long been thought to be related to TTP, ADAMTS13 inhibitors or deficiency does not appear to be the cause of HUS. HUS also differs from TTP in that it is associated with selective endothelial damage in the kidneys. Typical childhood HUS is associated with hemorrhagic diarrhea caused by shiga-toxin-producing bacteria such as E. coli 0157:H7. Atypical HUS occurs in children and adults without a preceding diarrheal prodrome and is thought to be related to complement regulatory abnormalities.2 Atypical HUS is more likely to recur.
Hemolytic-uremic syndrome. Although HUS has long been thought to be related to TTP, ADAMTS13 inhibitors or deficiency does not appear to be the cause of HUS. HUS also differs from TTP in that it is associated with selective endothelial damage in the kidneys. Typical childhood HUS is associated with hemorrhagic diarrhea caused by shiga-toxin-producing bacteria such as E. coli 0157:H7. Atypical HUS occurs in children and adults without a preceding diarrheal prodrome and is thought to be related to complement regulatory abnormalities.2 Atypical HUS is more likely to recur.
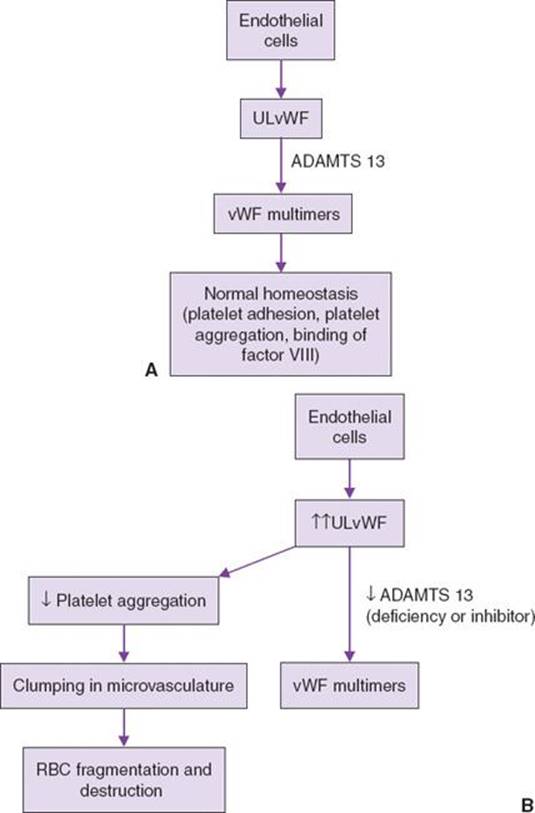
FIGURE 4-1. A: Role of ADAMTS13 in a normal subject. B: Role of ADAMTS13 deficiency in thrombotic thrombocytopenic purpura. ULvWF, ultralarge vWF; vWF, von Willebrand factor.

Risk Factors and Associated Conditions
See Table 4-1.
DIAGNOSIS
Clinical Presentation
The classic findings of TTP-HUS include a pentad of physical examination and lab findings as reported in Table 4-2. The complete pentad does not have to be present for the diagnosis, and TTP-HUS often presents without fever or neurologic dysfunction. Renal failure may be oliguric or nonoliguric. TTP may be preceded by a few weeks of malaise, but neurologic symptoms (including headache, confusion, vision changes, tinnitus, seizures, and coma) are frequently the first symptoms that bring a patient to medical attention. These symptoms may wax and wane over the course of the illness. Bleeding, pancreatitis, and diarrhea can also be associated with TTP-HUS.
Diagnostic Criteria

Anemia and thrombocytopenia are universal. Elevated lactate dehydrogenase (LDH), elevated indirect bilirubin, and decreased haptoglobin will help identify the hemolysis associated with the disorder, but a peripheral blood smear is mandatory for diagnosis to identify schistocytes consistent with MAHA. The reticulocyte count is usually elevated. Coagulation studies (PT/INR, PTT) are usually within normal limits, and a DIC panel, including fibrinogen, fibrinogen degradation products (FdPs), and D-dimer, is useful to rule out DIC as an alternate diagnosis. ADAMTS13 activity levels may be undetectable; however, treatment should not be delayed while waiting for this test. Further classification of thrombotic microangiopathy as either TTP or HUS is usually based upon age, past medical history, clinical presentation, and the presence or absence of renal dysfunction or neurologic symptoms.
Differential Diagnosis
Differential diagnosis includes other etiologies of MAHA (DIC, prosthetic valve hemolysis, malignant hypertension, adenocarcinoma, and vasculitis). Although fever is part of the pentad, it should also prompt workup for sources of infection. Evans syndrome (immune thrombocytopenia and autoimmune hemolytic anemia) should be distinguished by the presence of microspherocytes and absence of schistocytes in the peripheral smear.
TREATMENT
![]() The primary treatment for TTP-HUS is plasma exchange (plasmapheresis), with one estimated plasma volume exchanged daily. Plasma exchange can be done twice daily in severe cases or in cases that progress despite daily treatments. Because of the high mortality of untreated TTP-HUS, thrombocytopenia and MAHA are all that is required to initiate plasma exchange if no other certain cause can be identified. ADAMTS13 levels should not alter decision to perform plasma exchange. Plasma exchange has also shown benefits in other thrombotic microangiopathies so it should not be withheld in urgent cases. The goal of daily plasma exchange should be to reverse the thrombocytopenia and hemolysis. This can be monitored with LDH and CBC measurements; the platelet count is the most important factor. Plasma exchange can be tapered or stopped after the platelet count has been normal for 2 days. After remission, exacerbations due to discontinuing plasma exchange (<30 days) should lead to immediate resumption of plasma exchange. 2
The primary treatment for TTP-HUS is plasma exchange (plasmapheresis), with one estimated plasma volume exchanged daily. Plasma exchange can be done twice daily in severe cases or in cases that progress despite daily treatments. Because of the high mortality of untreated TTP-HUS, thrombocytopenia and MAHA are all that is required to initiate plasma exchange if no other certain cause can be identified. ADAMTS13 levels should not alter decision to perform plasma exchange. Plasma exchange has also shown benefits in other thrombotic microangiopathies so it should not be withheld in urgent cases. The goal of daily plasma exchange should be to reverse the thrombocytopenia and hemolysis. This can be monitored with LDH and CBC measurements; the platelet count is the most important factor. Plasma exchange can be tapered or stopped after the platelet count has been normal for 2 days. After remission, exacerbations due to discontinuing plasma exchange (<30 days) should lead to immediate resumption of plasma exchange. 2
![]() Suspected ADAMTS13 deficiency can also be treated with systemic corticosteroid therapy in an effort to suppress inhibitors at a dose of 1 mg/kg of prednisone or potentially higher doses in critically ill patients.
Suspected ADAMTS13 deficiency can also be treated with systemic corticosteroid therapy in an effort to suppress inhibitors at a dose of 1 mg/kg of prednisone or potentially higher doses in critically ill patients.
![]() Platelet transfusions are relatively contraindicated, except in cases of life-threatening bleeding.
Platelet transfusions are relatively contraindicated, except in cases of life-threatening bleeding.
![]() In patients with refractory or relapsing TTP despite standard therapy, rituximab may be beneficial at achieving remission and decreasing the need for plasma exchange in patients with a demonstrated antibody to ADAMTS13.3
In patients with refractory or relapsing TTP despite standard therapy, rituximab may be beneficial at achieving remission and decreasing the need for plasma exchange in patients with a demonstrated antibody to ADAMTS13.3
DISSEMINATED INTRAVASCULAR COAGULATION
GENERAL PRINCIPLES
Definition
DIC is an acquired and systemic disorder of hemostasis which produces both thrombosis and hemorrhage.
Epidemiology
DIC is associated with an underlying illness and is thus a condition most frequently diagnosed and treated in hospitalized patients. Around 1% of hospital admissions may be complicated by DIC, although rates approaching 35% may be seen in patients with severe sepsis syndrome.4
Pathophysiology and Etiology
In DIC, an underlying illness leads to systemic activation of the coagulation cascade, likely mediated by widespread endothelial damage and the release of inflammatory cytokines. This coagulation activation leads to increased fibrin formation and subsequent thrombosis, most notably in small and medium-sized vessels. The widespread thrombosis can lead to organ failure and MAHA. Widespread thrombosis can also deplete clotting factors and platelets leading to bleeding.5
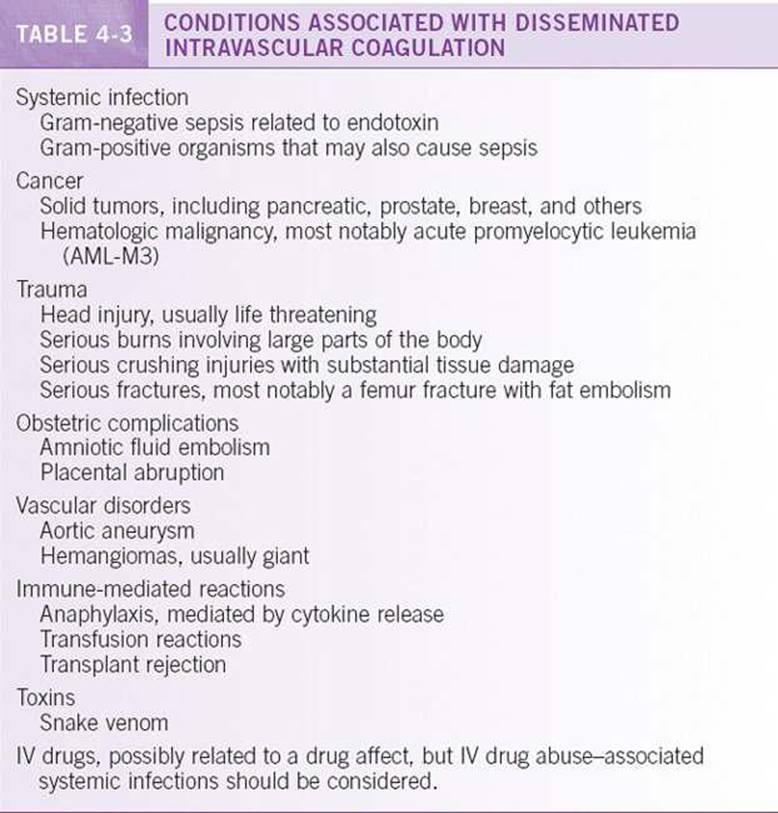
Risk Factors/Associated Conditions
See Table 4-3.
DIAGNOSIS
Clinical Presentation
DIC can manifest with symptoms related to thrombosis or bleeding. Thrombosis can lead to digit gangrene, stroke, or organ failure. Bleeding may be in the form of petechiae, oozing from venipuncture or wound sites, or hemorrhage.
Diagnostic Criteria
![]() There are a number of useful laboratory tests to assist in the diagnosis of DIC. The CBC may reveal anemia or thrombocytopenia, often <100k, related to thrombotic microangiopathy. Coagulation studies, including PT/INR and PTT, may be prolonged due to consumption of coagulation factors. “DIC panels” often measure fibrinogen, FDP, and D-dimer. Fibrinogen levels are low as a result of consumption, while the FDP and D-dimers are markers of clot dissolution and are usually elevated. It should be noted that fibrinogen is an acute phase reactant and may be elevated due to underlying illness. In such situations, a declining fibrinogen level may provide a clue to the diagnosis of DIC. A peripheral smear often reveals schistocytes from the destruction of red cells.
There are a number of useful laboratory tests to assist in the diagnosis of DIC. The CBC may reveal anemia or thrombocytopenia, often <100k, related to thrombotic microangiopathy. Coagulation studies, including PT/INR and PTT, may be prolonged due to consumption of coagulation factors. “DIC panels” often measure fibrinogen, FDP, and D-dimer. Fibrinogen levels are low as a result of consumption, while the FDP and D-dimers are markers of clot dissolution and are usually elevated. It should be noted that fibrinogen is an acute phase reactant and may be elevated due to underlying illness. In such situations, a declining fibrinogen level may provide a clue to the diagnosis of DIC. A peripheral smear often reveals schistocytes from the destruction of red cells.
![]() It is important to distinguish DIC from other conditions. Typical laboratory abnormalities with separate disease processes include: liver disease (low platelets and prolonged PT and PTT but normal fibrinogen—except in severe liver disease, which may show low fibrinogen), vitamin K deficiency (prolonged PT/PTT but normal platelets and fibrinogen), and TTP (MAHA and thrombocytopenia but normal PT, PTT, and fibrinogen).
It is important to distinguish DIC from other conditions. Typical laboratory abnormalities with separate disease processes include: liver disease (low platelets and prolonged PT and PTT but normal fibrinogen—except in severe liver disease, which may show low fibrinogen), vitamin K deficiency (prolonged PT/PTT but normal platelets and fibrinogen), and TTP (MAHA and thrombocytopenia but normal PT, PTT, and fibrinogen).
TREATMENT
Management should be focused predominantly on identifying and treating the underlying condition. Symptomatic treatment of bleeding or thrombosis can be dictated by the clinical scenario. In patients with high bleeding risk or active bleeding, fresh-frozen plasma to replace clotting factors, cryoprecipitate to replace fibrinogen (target level >100 mg/dL), and platelet transfusions (target plt >50,000) are suggested. In patients, predominantly in the thrombotic phase of DIC, low dose heparin has been suggested, but this is controversial. Low-molecular-weight heparin (LMWH) may have less risk for bleeding, and some consider it an alternative. In cases in which the patient has low anti-thrombin III (ATIII) levels and severe DIC, some consider ATIII replacement (with FFP or ATIII concentrates) a reasonable option.
HEPARIN-INDUCED THROMBOCYTOPENIA (HIT)
GENERAL PRINCIPLES
Definition and Classification
Heparin-induced thrombocytopenia can be classified into two separate entities: HIT I and HIT II.
HIT I is non-immune mediated and characterized by a transient fall in platelet count. This form of HIT is not an indication for discontinuing heparin.
The remainder of this chapter focuses on HIT II, which will simply be referred to as HIT.
Epidemiology and Etiology
HIT is an acquired prothrombotic complication of heparin therapy. It is an immune-mediated disorder caused by IgG Abs that bind to platelet factor 4 (PF4)–heparin complexes. The frequency in one meta-analysis was 2. 6% in patients treated with UFH and 0. 2% in those treated with LMWH.6
Pathophysiology
![]() As a primary specific immune response, HIT syndrome generally has a delayed onset of 4 to 5 days after administration of heparin or LMWH.
As a primary specific immune response, HIT syndrome generally has a delayed onset of 4 to 5 days after administration of heparin or LMWH.
![]() PF4 is a heparin-neutralizing chemokine protein found within the alpha-granules of platelets. This protein binds exogenous heparin (UFH > LWMH) and forms multimer complexes which can provoke an IgG antibody formation. Once formed, the HIT-IgG Ab (HIT Ab)–PF4–heparin complex can activate platelets and other cells, generate immunogenic multi-molecular complexes, and promote tissue factor expression and thrombin generation, leading to significant risk for thrombotic events. Platelets targeted by HIT specific antibodies are cleared from the circulation, causing thrombocytopenia.7
PF4 is a heparin-neutralizing chemokine protein found within the alpha-granules of platelets. This protein binds exogenous heparin (UFH > LWMH) and forms multimer complexes which can provoke an IgG antibody formation. Once formed, the HIT-IgG Ab (HIT Ab)–PF4–heparin complex can activate platelets and other cells, generate immunogenic multi-molecular complexes, and promote tissue factor expression and thrombin generation, leading to significant risk for thrombotic events. Platelets targeted by HIT specific antibodies are cleared from the circulation, causing thrombocytopenia.7
Risk Factors
HIT is more common in adult medical and surgical patients than in obstetric or pediatric patients. UFH is ~ 10 times more likely than LMWH to produce HIT. Patients receiving IV heparin are more likely to develop HIT than those receiving SC heparin.7
DIAGNOSIS
Clinical Presentation
![]() Thrombocytopenia or a >50% fall in platelet count occur in ~ 95% of patients diagnosed with HIT. The average platelet count is 50,000 to 70,000.7
Thrombocytopenia or a >50% fall in platelet count occur in ~ 95% of patients diagnosed with HIT. The average platelet count is 50,000 to 70,000.7
![]() There are three time courses of HIT: typical, rapid, and delayed-onset HIT.
There are three time courses of HIT: typical, rapid, and delayed-onset HIT.
![]() In typical-onset HIT, thrombocytopenia develops ~ 5 to 10 days after initiation of heparin therapy, approximately the amount of time necessary to generate a humoral immune response.
In typical-onset HIT, thrombocytopenia develops ~ 5 to 10 days after initiation of heparin therapy, approximately the amount of time necessary to generate a humoral immune response.
![]() A subset of patients experience rapid-onset HIT, where thrombocytopenia occurs within 24 hours, indicating a recent exposure to heparin during the preceding weeks.
A subset of patients experience rapid-onset HIT, where thrombocytopenia occurs within 24 hours, indicating a recent exposure to heparin during the preceding weeks.
![]() Delayed-onset HIT occurs days after heparin has been stopped. This form of HIT is not well understood. These patients typically have high-titer platelet-activating HIT Ab. It is uncommon for HIT to occur if heparin has been discontinued for more than 2 weeks.
Delayed-onset HIT occurs days after heparin has been stopped. This form of HIT is not well understood. These patients typically have high-titer platelet-activating HIT Ab. It is uncommon for HIT to occur if heparin has been discontinued for more than 2 weeks.
![]() HIT can present as thrombosis. The most common manifestation is venous thromboembolism (deep venous thrombosis and pulmonary embolism) in the postoperative setting. Though less common, arterial thrombosis can occur and is manifested as stroke, myocardial infarction, or limb ischemia.
HIT can present as thrombosis. The most common manifestation is venous thromboembolism (deep venous thrombosis and pulmonary embolism) in the postoperative setting. Though less common, arterial thrombosis can occur and is manifested as stroke, myocardial infarction, or limb ischemia.
![]() A clinical scoring system using the 4 Ts (Thombocytopenia, Timing of platelet fall, Thrombosis, and other causes) has been shown to help risk stratify patients with suspected HIT.8
A clinical scoring system using the 4 Ts (Thombocytopenia, Timing of platelet fall, Thrombosis, and other causes) has been shown to help risk stratify patients with suspected HIT.8
Diagnostic Testing
Initial diagnosis should be made based on the clinical scenario and after other causes of thrombocytopenia have been ruled out. Presently, most medical centers employ an immunoassay for the presence of PF4/heparin antibodies. This test is rapid and sensitive (>99%) though it lacks specificity (40% to 70%).7 Functional assays are more specific than HIT immunoassays but are not as widely available. Functional assays measure platelet activation at varying heparin concentrations. One such test, the serotonin release assay, utilizes radiolabeled serotonin to measure platelet activation. The test is considered positive when serotonin is released from donor platelets placed in patient serum when a low concentration of heparin is added. These tests are reported to have >95% sensitivity and specificity.7
TREATMENT
Medications
The first step in treating HIT is the discontinuation of heparin. Return of laboratory assay results often takes several days. Therefore, all heparin products, including heparin flushes and heparin-coated catheters, should be discontinued immediately if HIT is suspected. Because of cross-reactivity to the HIT Ab, LMWH should not be substituted. For patients who are strongly suspected of having HIT, non-heparin anticoagulation should be promptly instituted.
Agents which can treat HIT are classified as direct thrombin inhibitors (lepirudin, argatroban, and bivalirudin), indirect factor Xa inhibitors (danaparoid and fondaparinux), and vitamin K antagonists (warfarin).
![]() Lepirudin is a recombinant hirudin, a natural anticoagulant found in the salivary glands of medicinal leeches. This drug has been approved by the FDA for patients with HIT for the prevention and treatment of thrombosis. Lepirudin is renally cleared and should be dose-adjusted in patients with chronic kidney disease.
Lepirudin is a recombinant hirudin, a natural anticoagulant found in the salivary glands of medicinal leeches. This drug has been approved by the FDA for patients with HIT for the prevention and treatment of thrombosis. Lepirudin is renally cleared and should be dose-adjusted in patients with chronic kidney disease.
![]() Argatroban has also been approved by the FDA for the prevention and treatment of thrombosis associated with HIT. This drug is hepatically cleared and should be used with caution in patients with impaired hepatic function.
Argatroban has also been approved by the FDA for the prevention and treatment of thrombosis associated with HIT. This drug is hepatically cleared and should be used with caution in patients with impaired hepatic function.
![]() Bivalirudin is a direct thrombin inhibitor specifically approved for patients undergoing percutaneous coronary intervention.
Bivalirudin is a direct thrombin inhibitor specifically approved for patients undergoing percutaneous coronary intervention.
![]() Danaparoid is a heparinoid factor Xa inhibitor that is no longer marketed in the United States. It had previously been approved by the FDA for prophylaxis in HIT patients undergoing hip replacement surgery.
Danaparoid is a heparinoid factor Xa inhibitor that is no longer marketed in the United States. It had previously been approved by the FDA for prophylaxis in HIT patients undergoing hip replacement surgery.
![]() Fondaparinux is a synthetic pentasaccharide Xa inhibitor which received a Grade 2C recommendation in the 2008 ACCP guidelines for treatment of HIT. It has not been FDA approved for this indication and controversy exists because several cases of HIT caused by Fondaparinux have been reported.
Fondaparinux is a synthetic pentasaccharide Xa inhibitor which received a Grade 2C recommendation in the 2008 ACCP guidelines for treatment of HIT. It has not been FDA approved for this indication and controversy exists because several cases of HIT caused by Fondaparinux have been reported.
![]() Warfarin should not be used to treat a patient with HIT in the acute setting given the risk of venous limb gangrene and thrombosis with depletion of protein C. Warfarin can be started for long term anticoagulation when the patient has been anticoagulated with one of the above agents and the platelet count has reached a stable plateau >150,000. Warfarin should be started at a low dose (5 to 6 mg) and should overlap with one of the above agents for at least 5 days.9
Warfarin should not be used to treat a patient with HIT in the acute setting given the risk of venous limb gangrene and thrombosis with depletion of protein C. Warfarin can be started for long term anticoagulation when the patient has been anticoagulated with one of the above agents and the platelet count has reached a stable plateau >150,000. Warfarin should be started at a low dose (5 to 6 mg) and should overlap with one of the above agents for at least 5 days.9
Immune Thrombocytopenia (ITP)
GENERAL PRINCIPLES
Definition
Immune thrombocytopenia (ITP) is an acquired disorder characterized by isolated thrombocytopenia. ITP is also known as immune thrombocytopenic purpura or idiopathic thrombocytopenic purpura, but changes in terminology have been proposed as more has become understood about ITP.10
Classification
![]() Primary ITP is an immune mediated disorder without clear cause.
Primary ITP is an immune mediated disorder without clear cause.
![]() Secondary ITP is because of an underling disease, infection, or exposure.
Secondary ITP is because of an underling disease, infection, or exposure.
![]() ITP can be further classified as newly diagnosed (<3 months), persistent (3 to 12 months), and chronic (> 12 months).
ITP can be further classified as newly diagnosed (<3 months), persistent (3 to 12 months), and chronic (> 12 months).
DIAGNOSIS
Clinical Presentation
Patients often present with asymptomatic thrombocytopenia noted on routine CBC, but patients also frequently present with minor bleeding manifestations including petechiae, purpura, easy bruising, gingival bleeding, and menorrhagia. Less frequently, patients present with more severe bleeding including overt GI bleeding and intracranial hemorrhage.
History
Patient history should include a focus on bleeding symptoms and causes of secondary ITP or other hematologic disorders. Of particular importance are constitutional symptoms, prior thrombocytopenia, autoimmune diseases, hematologic diseases, liver disease, recent or chronic infections, recent transfusion, recent immunization, and medication exposures.
Physical Examination
Bleeding manifestations are frequently noted in ITP, but other abnormal exam findings should raise suspicion for secondary ITP or alternative causes of thrombocytopenia.
Diagnostic Testing
![]() ITPisa diagnosis of exclusion and no gold standard test exists.
ITPisa diagnosis of exclusion and no gold standard test exists.
![]() The CBC should reveal isolated thrombocytopenia unless bleeding is severe enough to produce concurrent anemia.
The CBC should reveal isolated thrombocytopenia unless bleeding is severe enough to produce concurrent anemia.
![]() Reviewofthe peripheral blood smear is essential to rule out other causes of thrombocytopenia including pseudothrombocytopenia from platelet clumping and other diagnoses including TTP/HUS.
Reviewofthe peripheral blood smear is essential to rule out other causes of thrombocytopenia including pseudothrombocytopenia from platelet clumping and other diagnoses including TTP/HUS.
![]() A bone marrow biopsy can be considered in patients > 60 years old or when the history and physical or other diagnostic data raise suspicion for a separate underlying hematologic disease.
A bone marrow biopsy can be considered in patients > 60 years old or when the history and physical or other diagnostic data raise suspicion for a separate underlying hematologic disease.
![]() HIV, HCV, and Helicobacter pylori testing should be considered in at risk patients. Baseline thyroid function testing and quantitative serum immunoglobulin testing should also be considered.
HIV, HCV, and Helicobacter pylori testing should be considered in at risk patients. Baseline thyroid function testing and quantitative serum immunoglobulin testing should also be considered.
TREATMENT
The primary goal of treatment in ITP is to prevent bleeding. When the platelet count is >50 × 109/L treatment is not always necessary, but treatment should be considered if the patient is at increased risk for bleeding such as in known platelet dysfunction, trauma or predisposition to trauma, necessity for anticoagulation, or planned surgery. Multiple therapeutic modalities exist, and treatment should be tailored for each individual patient.10
First-Line Therapy
![]() Corticosteroids: Prednisone 1 mg/kg/d can be given until normalization of platelet count at which point steroids can be tapered to avoid side effects. Dexamethasone and methylprednisolone are other considerations.
Corticosteroids: Prednisone 1 mg/kg/d can be given until normalization of platelet count at which point steroids can be tapered to avoid side effects. Dexamethasone and methylprednisolone are other considerations.
![]() IVIg: IVIg can be given at dose of 1 g/kg/d × 2 days or 0. 4 g/kg/d × 5 days with consideration of maintenance dosing.
IVIg: IVIg can be given at dose of 1 g/kg/d × 2 days or 0. 4 g/kg/d × 5 days with consideration of maintenance dosing.
![]() IV anti-D: 50 to 75 μg/kg/d of IV anti-D can be considered in Rh(D) positive patients who have not undergone splenectomy and who do not have history of autoimmune hemolytic anemia.
IV anti-D: 50 to 75 μg/kg/d of IV anti-D can be considered in Rh(D) positive patients who have not undergone splenectomy and who do not have history of autoimmune hemolytic anemia.
Second-Line Therapy
![]() Splenectomy
Splenectomy
![]() Eighty percent initial response rates are reported after splenectomy while sustained response is observed in around two-thirds of patients. Laparotomy has higher complication and mortality rates than laparoscopy. Prophylactic vaccinations should be administered after splenectomy to prevent subsequent infections.
Eighty percent initial response rates are reported after splenectomy while sustained response is observed in around two-thirds of patients. Laparotomy has higher complication and mortality rates than laparoscopy. Prophylactic vaccinations should be administered after splenectomy to prevent subsequent infections.
![]() Medical therapy
Medical therapy
![]() Multiple drugs can be considered as second line agents including azathioprine, cyclosporine A, cyclophosphamide, danazol, dapsone, mycophenolate mofetil, rituximab, and vinca alkaloid regimens.
Multiple drugs can be considered as second line agents including azathioprine, cyclosporine A, cyclophosphamide, danazol, dapsone, mycophenolate mofetil, rituximab, and vinca alkaloid regimens.
![]() Thrombopoietin-receptor agonists including romiplostim and eltrombopag can be considered and may be particularly useful in those who cannot tolerate long-term immunomodulatory therapy.
Thrombopoietin-receptor agonists including romiplostim and eltrombopag can be considered and may be particularly useful in those who cannot tolerate long-term immunomodulatory therapy.
Refractory ITP
For patients with severe refractory disease combination chemotherapy, alemtuzumab, and allogeneic stem cell transplantation can be considered.
Emergency Therapy
In emergent situations such as in life threatening bleeding or emergency surgery, combination therapies and platelet transfusions should be considered. Aminocaproic acid may be useful in preventing rebleeding in persistently thrombocytopenic patients.11
THROMBOSIS
GENERAL PRINCIPLES
A platelet count exceeding the reference range is called thrombocytosis. Thrombocytosis may be reactive or due to autonomous production of platelets by clonal megakaryocytes (essential thrombocythemia or other myeloproliferative disorders).
Reactive thrombocytosis is thrombocytosis in the absence of a chronic myeloproliferative disorder. It can be seen in the setting of infection, surgery, malignancy, blood loss, and iron deficiency or postsplenectomy. The platelet count is expected to normalize when the underlying process is corrected.
REFERENCES
1. Sadler JE. Von Willebrand factor, ADAMTS13, and thrombotic thrombocytopenic purpura. Blood. 2008;112:11–18.
2. George JN. How I treat patients with thrombotic thrombocytopenic purpura. Blood. 2010;116;4060–4069.
3. Scully M, Cohen H, Cavenagh J, et al. Remission in acute refractory and relapsing thrombotic thrombocytopenic purpura following rituximab is associated with a reduction in IgG antibodies to ADAMTS-13. Br J Haematol.2007;136:451–461.
4. Levi M. Disseminated intravascular coagulation. Crit Care Med. 2007;35:2191–2195.
5. Franchini M, Lippi G, Manzato F. Recent acquisitions in the pathophysiology, diagnosis and treatment of disseminated intravascular coagulation. Thromb J. 2006;4:4.
6. Martel N, Lee J, Wells PS. Risk for heparin-induced thrombocytopenia with unfractionated and low-molecular-weight heparin thromboprophylaxis: a meta-analysis. Blood. 2005;106(8):2710–2715.
7. Arepally GM, Ortel TL. Heparin-induced thrombocytopenia. Annu Rev Med. 2010;61:77–90.
8. Lo GK, Juhl D, Warkentin TE, et al. Evaluation of pretest clinical score (4 Ts) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4:759–765.
9. Warkentin TE, Greinacher A, Koster A, Lincoff AM. American College of Chest Physicians. Treatment and prevention of heparin-induced thrombocytopenia: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2008;133(6 Suppl):340–380.
10. Provan D, Stasi R, Newland AC, Blanchette VS. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood. 2010;115: 168–186.
11. Rodeghiero F, Stasi R, Gernsheimer T, Michel M. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. 2009;113:2386–2393.