JOSÉ R. ROMERO
Viral meningitis can be characterized as a central nervous system (CNS) viral infection with signs of meningeal irritation (neck stiffness, Kernig and/or Brudzinski signs) and cerebrospinal fluid (CSF) pleocytosis but without neurologic dysfunction due to brain parenchymal involvement (1). It differs from viral encephalitis where evidence of brain parenchymal dysfunction is manifested by an altered state of consciousness, change in personality, or other objective signs of neurologic dysfunction (e.g., seizures, cranial nerve palsies, abnormal reflexes, paralysis, etc.). Although it is common to discuss the two as wholly separate entities, it is important to note that overlap between them (i.e., meningoencephalitis) does occur following infection with many viral agents.
Almost 100 years ago, Wallgren (2) introduced the term “acute aseptic meningitis” to describe a short-lived, self-limited, benign CNS syndrome characterized by the acute onset of the signs of meningeal irritation in which examination of the CSF revealed a mononuclear pleocytosis and the absence of bacteria on direct examination and by culture. In addition, no parameningeal process, acute/chronic systemic infectious disease, or community infectious disease could be identified that could produce the syndrome. With advances in diagnostic methodologies, it became evident that multiple infectious agents (e.g., Lyme disease), inflammatory conditions, drugs, environmental agents, and so forth could cause the syndrome.
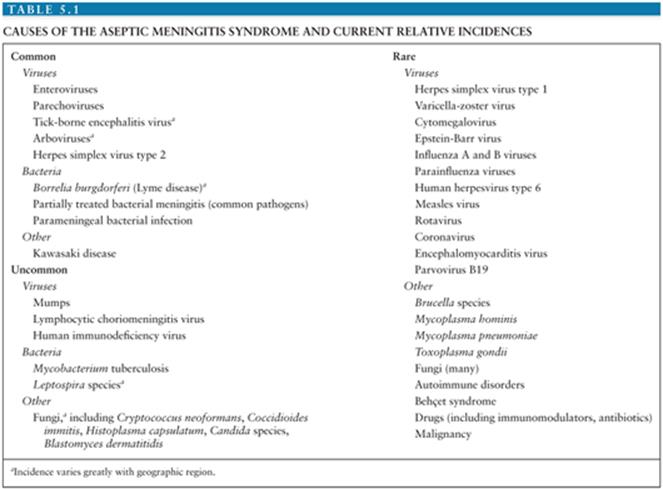
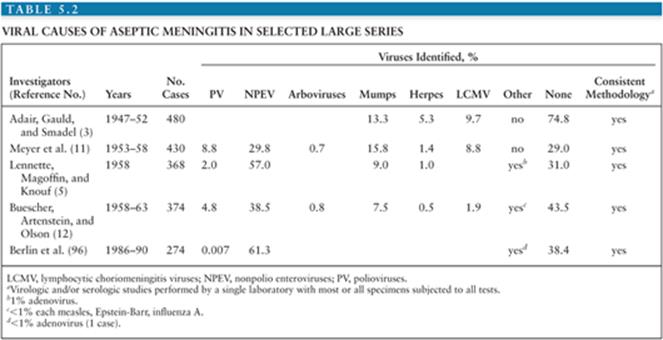
It is estimated that in the United States, the annual number of aseptic meningitis cases is at least 75,000. Viruses account for the overwhelming majority of cases (Table 5.1). Early reports indicated that mumps virus, lymphocytic choriomeningitis virus (LCMV), and poliovirus (PV) were the major identifiable causes of aseptic meningitis (3). As diagnostic techniques improved as a result of the development of cell culture, the enteroviruses were shown to have a major role in causation of syndrome (Table 5.2) (4,5). Nucleic acid amplification tests (NAATs) have bolstered this finding and led to the identification of novel causes (6).
The incidence of aseptic meningitis is influenced by many factors, including effective vaccine programs, sanitation, poverty, and regional endemic viruses (7–10). Previous significant causes of viral meningitis such as PVs, mump virus, and LCMV are now rare or infrequent as a result of effective vaccines, sanitation, or improved housing (3,5,8,11–13). Although many of the infectious causes of aseptic meningitis are reportable (14), the true incidence of the syndrome is unknown due to incomplete reporting, failure to test for specific agents, and because aseptic meningitis is not a reportable condition. In Finland, a 14-year birth cohort study found the annual incidence of viral meningitis in children younger than 14 years of age to be 27.8 per 100,000 (15). A more recent study from Greece documented the annual incidence of aseptic meningitis to be 17 per 100,000 in children younger than 14 years of age (16). Two studies from the United States give widely discrepant estimates of the incidence of aseptic meningitis. A 32-year (1950 to 1981) study from Olmsted County, Minnesota found that the adjusted incidence rate of aseptic meningitis was 10.9 per 100,000 person-years (range 7.9 to 17.8 per 100,000) (8,13). The Centers for Disease Control and Prevention reported that the national incidence for aseptic meningitis ranged from 1.5 to 4.0 per 100,000 for the period spanning 1971 to 1981 (17). The lower incidence in the latter report is most likely the result of passive surveillance and, therefore, underreporting. The incidence of aseptic meningitis is greater in males and in children, particularly those younger than 1 year of age (8,15,16).
ENTEROVIRUSES
Virology and Pathogenesis
The enteroviruses (EVs) are one of six genera (Enterovirus, Cardiovirus, Cosavirus, Hepatovirus, Parechovirus, and Kobuvirus) in the Picornaviridae (pico, “small”; rna, “ribonucleic acid”; viridae, “viruses”) family of viruses known to cause disease in humans. The original classification of the EVs, based on evidence of human origin, pathology in animal models, patterns of replication in cell culture, and physicochemical characteristics, identified 72 serotypes (18). This schema, although initially useful, resulted to the misclassification of several EV and inclusion of several non-EV into the genus (19). The development of experimental and computational methodologies for the study of the molecular biology and genomic analysis of the EV allowed for refinement in the classification of and identification of the EVs.
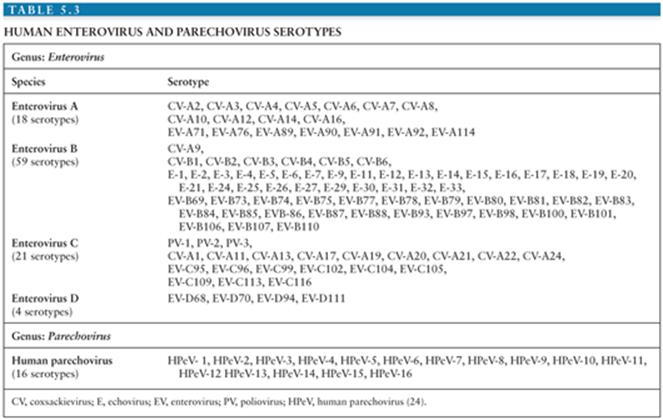
Currently, identification and classification of the EVs is based on the nucleotide sequence of VP1, the largest and most surface exposed of the viral capsid proteins containing important neutralizing epitopes (20–23). Using this approach, the EVs have been speciated into four groups (enterovirus A to D) containing more than 100 serotypes (Table 5.3) (24). In addition, this approach has revealed that several of the “traditional” EV serotypes are actually strains of the same serotype or are not genetically related to the EVs (echoviruses 22 and 23) (20,21,25–27).
The EVs are nonenveloped viruses 30 nm in diameter with a buoyant density of 1.30 to 1.34 g/cm−3 in caesium chloride (CsCl) (19). The lack of an envelope confers to them relative environmental stability where they can survive for days at room temperature. Infectivity can be preserved for weeks at −20°C or with little or no loss of infectivity for years when stored at −70°C. Similarly, the lack of a lipid envelope renders them insusceptible to ether, chloroform, and alcohol. The EVs are inactivated by heating to greater than or equal to 50°C, chlorine, and formaldehyde.
The capsid of all EVs is composed of 60 units each of four structural or capsid proteins: VP1 to VP4, alternatively known as 1A to 1D, arranged so as to give the virion icosahedral symmetry (28–33). Each of proteins VP1 to VP3 is wedge-shaped and composed of an eight-stranded antiparallel β-barrel core. Each of the stands is connected to the next by intervening loops that determine antigenicity, receptor specificity, and confer capsid stability (34). The basic structural element of the viral capsid, the protomer, is initially composed of the proteins VP0, VP1, and VP3 (35). Five protomers self-assemble to form a pentamer. Twelve pentamers, in turn, assemble around a single strand of viral RNA to produce the immature virion. The cleavage of VP0 to yield VP2 and VP4 completes the formation of the mature virion. VP1 to VP3, and in particular VP1, have surface-exposed amino acids which determine the antigenic diversity and the receptor specificity of the EV (20,33,36,37). VP4 is not surface exposed but shares close association with the viral RNA and plays a vital role in release of the genome after viral attachment (38).
The surface topographies of the various EVs share a number of similarities. These include a plateau or mesa located at the fivefold axis of symmetry formed by the union of five protomers. Surrounding this plateau is a deep cleft or canyon into which a viral receptor inserts when the EV encounters a susceptible host cell (36). Additionally, the host immune response to EV infection generates serotype-specific antibodies directed to antigenic sites around the fivefold axis and canyon walls, thus blocking viral-host receptor interaction and infection. Lastly, beneath the canyon floor exists a hydrophobic pocket containing a lipophilic factor. This pocket has been the target for the development of anti-EV drugs that result in altered receptor binding and viral uncoating (39,40).
The EV genome consists of single-stranded, positive (messenger)-sense RNA of approximately 7,400 nucleotides (nts) in length. The genome layout may be summarized as follows: VPg+5′UTR[1A-1B-1C-1D/2A-2B-2C/3A-3B-3C-3D] 3′UTR-poly(A) (24,41). The 5′ end of the genome is covalently linked to a small protein, VPg, essential for viral RNA replication. The genome is organized into a long 5′ untranslated region (5′ UTR) of approximately 740 nts that immediately precedes a single open reading frame (ORF). The ORF measures approximately 6,630 nts and is followed by a short (approximately 70 nts) 3′ UTR and a terminal polyadenylated tail.
The 5′ UTR contains multiple regions of predicted higher order structure and highly conserved nucleotide identity among the EV. This region of the genome contains elements essential for viral RNA replication, translation of ORF, and, in the PVs, determinants of neurovirulence. Because of the highly conserved nucleotide sequences within the 5′ UTR found among all the EV, it serves as the target for NAATs for the detection of the EV now in common use (42,43).
Translation of the ORF by host cell ribosomes is accomplished in a nonconical, cap-independent manner giving rise to a single polyprotein that is posttranslationally cleaved by viral and host proteinases to yield 11 viral proteins (four structural and seven nonstructural) as well as several functional protein intermediates. The ORF can be subdivided into three regions: P1 to P3. The P1 region encodes for the four structural proteins (VP1 to VP4 or 1A to 1D) that comprise the viral capsid. These are organized 5′ to 3′ as VP4 (1A), VP2 (1B), VP3 (1C), and VP4 (1D). The P2 and P3 regions code for seven nonstructural proteins (2A, 2B, 2C, 3A, 3B, 3C, and 3D) that are essential for the viral life cycle. The intermediate proteins play roles in viral replication.
Immediately downstream of the ORF is a short 3′ UTR followed by a terminal poly(A) tract. Similar to the 5′ UTR, the 3′ UTR is predicted to have higher order structures and play a role in genome replication (44).
Following binding of the EV to a host cell receptor, conformational changes in the virion result in release of the viral genome into the cytoplasm of the host cell. The RNA genome is replicated through a double-stranded RNA intermediate that is formed by the EV RNA-dependent RNA polymerase (3D).
Despite more than 100 years of study of the pathogenesis of EV infections, much still needs to be learned. Most of what is known stems for the study of PV types 1 to 3 using information derived from human disease and nonhuman primate, murine, and, most recently, transgenic (Tg) mouse models expressing the PV receptor, PVR or CD155 (45–50). It is likely that the non-PV EVs share similar mechanisms of pathogenesis.
The majority of the EVs are transmitted via a fecal-oral route. In addition to direct person-to-person or fecal-oral transmission, experimental or clinical evidence exists for the transmission of the EV via houseflies, housefly-contaminated food, water, and sewage (51–54). Bivalves have been found to accumulate EV (55,56). However, their role in transmission has not been established. Notable exceptions to fecal-oral transmission include coxsackievirus (CV)-A21, EV-D68, and EV-D70, which may spread via contaminated fomites or ocular and respiratory secretions. Evidence for transplacental transmission exists, leading to congenital infection (57–61).
Following ingestion of the EVs, infection of the cells of the nasopharynx and, more significantly, the lower gastrointestinal (GI) tract occurs. The inherent acid-resistance of the EV favors the latter. Replication in the lymphatic tissues of these sites (i.e., tonsils and Peyer patches) leads to shedding of the EV from the nasopharynx for 1 to 2 weeks and from the GI tract in feces for several weeks to months (7). Seeding of EV to the deep cervical and mesenteric lymph nodes ensues and results in their spread to the systemic circulation via the lymphatics. This primary or minor viremia leads to seeding of various organs, including the CNS, liver, lungs, skin, and heart. Further replication in the tissues of those organs results in a major (secondary) viremia. If the CNS was not seeded during the initial viremic episode, spread there may occur with the major viremia. Viremia or presence of virus in the CNS continues until the host develops type-specific neutralizing antibodies directed to the capsid proteins, usually by day 7 to 10 postinfection. Immunoglobulin A (IgA) antibodies appear in the respiratory and GI tracts 2 to 4 weeks after infection. Unlike other viruses, which are largely contained by cellular immune mechanisms, the EVs are cleared from the host primarily by antibody-mediated mechanisms.
Great strides have been made in understanding the pathogenesis of EV infections at a molecular level. The presence of an EV receptor is the primary, but not the sole, determinant for cellular infection (62). As stated previously, PVR has been shown to be the receptor for the PVs (62) and maps to chromosome 19. PVR is a member of the immunoglobin superfamily and functions as an adhesion molecule. It helps to form adherens junctions and is a recognition molecule for natural killer cells. In addition to PVR, at least 11 other cell proteins have been identified as receptors or coreceptors for other EV serotypes (Table 5.2) (63–66).
The exact identity of the cells in the upper and lower intestinal tract infected by the PVs is unknown. However, they have been identified within the ileal wall and mesenteric nodes in human infection (45,49,50). It is believed that the PV infect either lower GI cells expressing the PVR or use transcytosis through microfold (M) cells in the lower GI tract to gain access to lymphoid tissue. Supporting this is the finding of PVR on the surface of intestinal epithelium, M cells, and in the germinal centers of Peyer patches (67). Support for the latter comes from the finding that M cells can bind and endocytose PVs (68,69).
Why the majority of EV infections do not result in clinically apparent infection (70–73) comes for a number of studies that suggest that replication of PV in extraneural tissues is inhibited by the host interferon (IFN) response. PVR Tg mice that are IFN α/β receptor deficient are highly susceptible to PV infection, and PV replication in the small intestine is enhanced (74,75). Thus, IFN responses may be crucial in limiting the spread of EV infection.
Lastly, the mechanism of EV entry into the CNS remains unclear. Evidence for two pathways exist for PV: transit through the blood–brain barrier (BBB) or via retrograde axonal transport. For the development of paralytic disease in chimpanzees, viremia has been shown to be essential (76). This finding provided initial support that the BBB may be a route to the CNS. Endothelial cells may express EV receptors that may influence tissue susceptibility to the EV (77) and facilitate virus entry into the CNS and other organs. Further buttressing of this hypothesis came from the finding that cultured human brain microvascular cells can support PV replication (78,79). However, in other studies, pharmacokinetic analysis of PV injected into Tg and non-Tg mice indicated that PV was delivered to the brain in significantly greater amounts than would be expected from the vascular concentration (80). This suggests that PV may enter the CNS via the BBB but without need of PVR.
Evidence in humans supports EV access to the CNS via a neural route. Individuals inadvertently inoculated with an incompletely inactivated PV vaccine developed initial paralysis in the limb receiving the vaccine (81). Trauma to a limb preceding PV infection has been associated with the development of paralysis of that limb. “Provocation poliomyelitis” following intramuscular injections into an extremity of a person incubating wild type PV or those receiving live attenuated PV vaccines has been well documented (82,83).
Substantial evidence from nonhuman primate and murine models exists in support of EV access to the CNS via a neural route. In monkeys, inoculation of the sciatic nerve with PV results in viral spread along the inoculated nerve and the spinal cord (84). The initial limb to develop paralysis following intramuscular inoculation of PV in monkeys and Tg mice is the one injected (85–87). If the sciatic nerve of the intramuscularly inoculated lower extremity is frozen or transected, paralysis of the limb is prevented (84,87). In Tg mice, it has been shown that in provocation PV, there appears to be induction of retrograde axonal transport (88). PV may gain access to neurons at the level of the neuromuscular junction.
The search for viral genomic determinants of neurotropism and neurovirulence has focused on the PVs (89). After immunization with live, neuroattenuated vaccine (Sabin) PV strains, shedding of PV that has recovered the ability to cause paralysis (i.e., neurovirulent revertant strains) occurs routinely. Comparison of wild type, vaccine, and revertant PV strains has identified a 10-nucleotide region within the 5′ UTR (nts 472 to 484 relative to PV type 3 Sabin strain) where neuroattenuating mutations are found to cluster. Additional minor determinants of virulence are localized to amino acids encoded in the P1 and P3 regions. The search for determinants of neurovirulence in other EV has been unsuccessful.
Histopathology
The benign nature of EV meningitis has made human pathologic data for this disease sparse. A report of a child who died of CV-B5 myocarditis with concomitant meningitis describes inflammation of the choroid plexus of the lateral and fourth ventricles, fibrosis of the vascular walls with focal destruction of the ependymal lining, and fibrotic basal leptomeninges (90). Parenchymal findings were limited to moderate symmetric dilation of the ventricles and an increase in the number and size of subependymal astrocytes. The inflammatory reaction at the choroid plexus supports the concept of viremic spread to the CNS. An adolescent presenting with a similar constellation of findings died of systemic CV-B3 infection (91). The dura was grossly distended with swelling also of the pia, arachnoid, and brain parenchyma. Microscopically, round cell infiltrates were noted in the meninges overlying the cerebellum; the brain parenchyma was congested with increased numbers of oligodendrocytes. Lymphocytic infiltration was most prominent around blood vessels in the cerebral white matter and in the basal ganglia, again suggesting viremic CNS access; focal areas of necrosis and hemorrhage were also seen (91).
Epidemiology
The EVs have a ubiquitous worldwide distribution; humans are their only natural reservoir (58,92). It is estimated that they result in more than 1 billion annual infections worldwide (22). In the United States alone, the non-PV EVs are estimated to cause 30 to 50 million infections each year. Due to underreporting of EV infections (93), a well-grounded estimate of the number of cases of EV meningitis that occurs each year is not possible. However, conservative estimates place the annual number to be between 30,0000 and 75,000 cases (22,94). The EVs are responsible for 80% to 90% of identifiable causes of viral meningitis (94–97). Wild type PV were eradicated for the West Hemisphere in 1991 (98) and are currently endemic only in Afghanistan, Nigeria, and Pakistan (99). As such, they do not contribute to the burden to EV in most of the world.
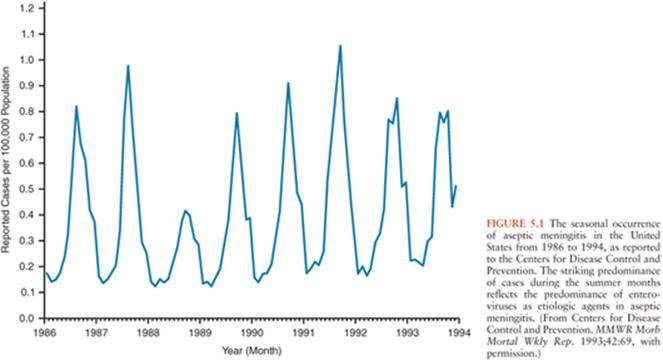
In regions with temperate climates, the non-PV EV exhibit marked seasonality with the majority of infections occurring in the summer and fall (Fig. 5.1) (92,100,101). This being said, EV infections do occur during the winter, warranting their inclusion in the diagnostic evaluation of aseptic meningitis during that time of year (102–106). In tropical and subtropical areas, EVs occur year-round, but with higher incidence during the rainy season.
Despite the existence of more than 100 serotypes of EV, only a limited number are responsible for the majority of disease observed annually in each geographic region (58,93,107–110). The rank of each serotype within the most frequently isolated EVs varies annually and geographically. In the United States, 15 serotypes accounted for approximately 80% of all EVs reported from 1970 to 2005 (in descending order): echoviruses (E)-9, -11, -30, CV-B5, E-6, CV-B2, CV-A9, E-4, CV-B3, E-7, CV-B4, E-18, CV-B1, E-3, and -5 (93). Some serotypes cycle with varying periodicity within a community (58,93,109,110), a reflection of the availability of new susceptible host populations (especially children). Other serotypes appear de novo as novel epidemic-associated viruses (111). Around the world, the serotypes most commonly isolated from the CSF and, therefore, from cases of meningitis or meningoencephalitis, belong to the Enterovirus B species (93,108,112,93,113,–114). In the United States, the serotypes most frequently isolated from CSF specimens over a 36-year period, in descending order of frequency, are E-9, -11, -30, CV-B5, E-6, CV-B2, CV-A9, E-4, CV-B4, E-7, -18, and -5 (93).
Outbreaks of EV meningitis are common. Large nation- and community-wide outbreaks involving thousands of individuals have been well documented (115,116). Outbreaks involving more localized venues such as neonatal units, nurseries, daycare centers, orphanages, schools, pools, camps, and sports teams occur (53,117–122). Sequential episodes of EV meningitis involving different serotypes have been reported to occur within a month of each other (123–125). Mixed infections involving EVs, other viruses, or bacteria have been well described (102–130).
Children represent the overwhelming majority of cases of EV meningitis. An incidence peak among young infants and school-aged children ages 5 to 10 years has been reported in multiple studies (58,131–133). Occasional outbreaks of EV CNS infections occur predominantly among adults (134–136). A possible explanation for these findings may lie in the history of the particular serotypes in the geographic area studied. Serotypes with “endemic” patterns, those occurring with significant incidence annually, are most likely to affect only the youngest children because of their absence of previous exposure and immunity. Older children and adults are more likely to predominate in an outbreak of a serotype that has not been present in a community for several years, thereby creating a reservoir of susceptible individuals among children born since the last appearance of that serotype.
Host factors that predispose to EV meningitis, other than young age and immunodeficiency, have been difficult to identify. A slight male-to-female predominance in the incidence ratio for EV infections has been noted in large series. However, in a recent report, male predominance was present only among persons younger than 20 years of age (male/female ratio 1.4:0.9) (101). This most likely represents the larger number of females exposed to young children who are principal source of household exposure. Infection rates are higher among persons of lower socioeconomic status, in areas of crowding, and larger families (7).
Clinical Manifestations
The clinical manifestations of aseptic meningitis do not significantly differ among the non-PV EVs causing the syndrome (9,137). However, clinical manifestations do vary with the age and immune status of the patient. Meningitis or meningoencephalitis, singularly or in combination with other syndromes, are common manifestations of symptomatic EV infection in neonates and young infants. Two large retrospective reviews have documented that 62% of infants younger than 3 months of age with group B coxsackievirus infections and 27% of neonates younger than 2 weeks of age with infections due to the echoviruses had associated meningitis or meningoencephalitis (60,138). In two prospective studies, clinical or laboratory evidence of meningitis was found in 42% to 75% of neonates with EV infection (139,140).
Early presentation of EV infection following birth suggests transplacental, intrapartum, or immediate postpartum acquisition of virus (60,138,139). Maternal illness (fever, symptoms of upper respiratory tract infection, abdominal pain) has been reported to occur in 14% to 68% of infected neonates (60,138–141). In neonates, fever (≥38.0°C) is almost universal and accompanied by any or all of the following nonspecific signs: irritability, lethargy, poor feeding, emesis, and upper respiratory tract findings (60,139,140,142). On physical examination, the fontanelle may be full or bulging. Signs of meningeal irritation such as nuchal rigidity, Brudzinski, and Kernig signs are generally absent. An exanthem may be present. If encephalitis in addition to meningitis (meningoencephalitis) is present, the neonate may present with profound lethargy, seizures, and focal neurologic abnormalities that suggest herpes simplex virus (HSV) infection. In some newborns, encephalohepatomyocarditis syndrome may develop in which signs and symptoms of severe hepatitis and myocarditis are superimposed on those of meningoencephalitis (143). Disseminated intravascular coagulation and other findings of “sepsis” result in an illness that may be indistinguishable from that caused by overwhelming bacterial infection.
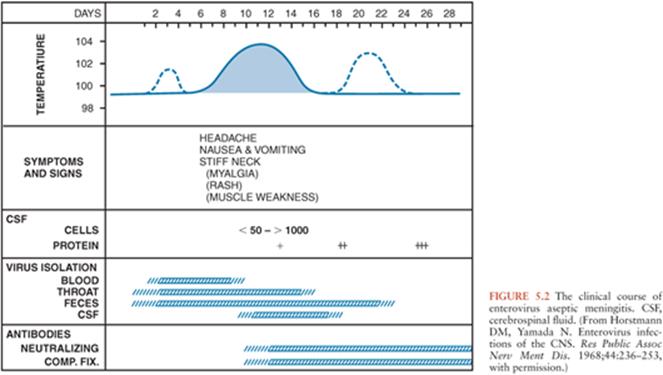
In infants and children, following an incubation period of 5 to 10 days, the onset of EV meningitis is usually abrupt with fever (38° to 40°C), the most common presenting sign (137,143–147). The natural history of EV meningitis is depicted in Figure 5.2. The fever pattern may be biphasic, initially appearing in association with nonspecific constitutional signs and symptoms followed by resolution and subsequently reappearing with the onset of meningeal signs (137,148). Headache is nearly always present in those individuals old enough to report it. Interestingly, it may be ameliorated following the performance of a lumbar puncture, indicating that it may be the result of increased intracranial pressure (149,150). Photophobia is commonly reported. Infants and young children may be irritable or, less commonly, lethargic (142). Nonspecific findings, singly or in combinations, include anorexia, exanthems, malaise, sore throat, abdominal pain, nausea, emesis, and myalgias (144,147). In infants, the fontanelle may be full or bulging. Less than 5% of infants younger than 3 months of age have signs of meningeal irritation (142). However, these become more common in older patients (144,147,151,152). Seizures are noted in less than 5% of children (142,146). Other uncommon complications include coma, increased intracranial pressure, and inappropriate secretion of antidiuretic hormone (142,153). The duration of EV meningitis in infants and children is generally less than 1 week.
Fewer clinical reports exist documenting the presentation of EV meningitis in adolescents and adults (134,136,154–157). Headache is the most frequently reported symptom and is nearly uniformly present. The severity of the headache may be such as warrant the use of narcotic analgesics in order to control the pain (157). Photophobia, fever, signs of meningeal irritation, nausea, emesis, and neck stiffness occur in more than 60% of cases. Other less frequently encountered signs and symptoms include myalgia, exanthems, and abdominal pain. Full recovery takes longer in adolescents and adults and may require up to 2.5 weeks (157).
In individuals with humoral immunodeficiencies (X-linked agammaglobulinemia, X-linked hyper IgM syndrome, common variable immunodeficiency), EV infection may result in chronic meningitis or meningoencephalitis that may last for years and often have a fatal outcome (158–161). In addition to the common signs and symptoms of EV meningitis mentioned previously, neurologic manifestations include paralysis/paresis, seizures, cognitive impairment, developmental regression, sensorineural hearing loss, coma, dysarthria, hydrocephalus, and aphasia. Extra CNS manifestations occur singly or in combination in a significant number of cases and include dermatomyositis, chronic hepatitis, arthritis, myocarditis, and subacute lumbosacral polyradiculopathy. In patients who have undergone repetitive, sequential lumbar punctures, the cell culture detection of EV in CSF has been intermittent. However, using NAATs, evidence of their persistence in CSF has been documented (162). Treatment with antibody preparations intravenously and intrathecally or intraventricularly has resulted in stabilization of some of these patients; however, viral persistence has been documented during therapy (159,162,163). With the availability of intravenous preparations of immune globulin and the early recognition of this illness, fewer patients appear to be progressing to the classic description of this disease, and atypical neurologic presentations have appeared. The mortality rate in patients with humoral immunodeficiencies may be as high as 50%.
The infected neonate is at greatest risk of severe morbidity and mortality when signs and symptoms develop in the first days of life (60,138,139,164). Neonates infected with CV-B4 were found to be higher risk of death than those infected with other EV. The short-term prognosis of young children with EV meningitis early in life appears to be good; however, there has been some controversy over possible later sequelae. Neurologic, cognitive, developmental, and language abnormalities have been reported in controlled studies of long-term outcome in children with EV meningitis during infancy (165–168). In the largest and most meticulously controlled study, however, no differences between patients and controls could be demonstrated in any of the neurodevelopmental parameters studied (142). Less well studied are the ultimate outcomes of aseptic meningitis cases in older children and adolescents; preliminary data suggest possible school and learning difficulties, but control patients were not studied (169).
Laboratory Findings and Diagnosis
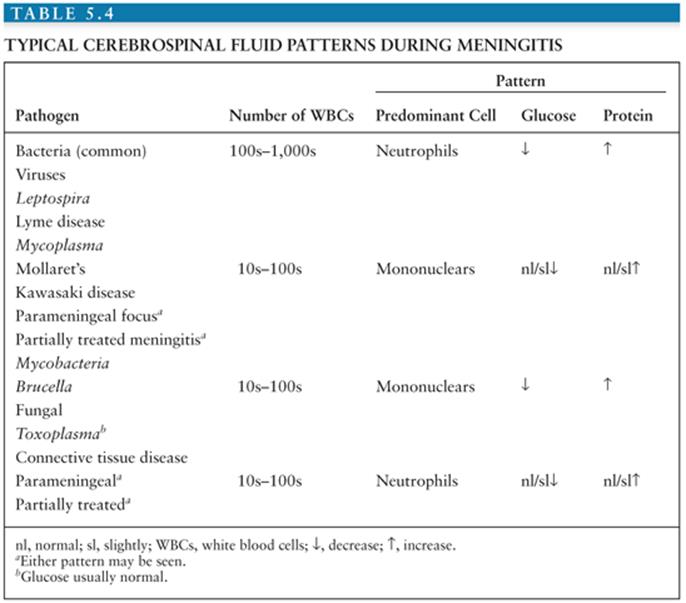
Salient among the laboratory analyses performed for the evaluation of EV meningitis is the evaluation of the CSF. CSF analysis can provide initial clues as to the etiology of the clinical syndrome (Table 5.4). Cytochemical analysis of the CSF typically shows a mild to moderate lymphocytic pleocytosis ranging from 10 to 1,000 cells/mm3 (136,139,141,142,145,149,160). White blood cell (WBC) counts exceeding 1,000 cells/mm3 are seen occasionally (60,103,147,170). Although pleocytosis is almost always present, EVs have been isolated by cell culture or detected by NAAT from the CSF of patients with clinical evidence of meningitis without pleocytosis (171). This is particularly true in the young infant. If the CSF is examined early in the course of the illness, a predominantly polymorphonuclear pleocytosis may be observed (136,139, 142,146,149). Reexamination of the CSF several hours later will document a typical lymphocytic pleocytosis (172–174). The progression of an initially polymorphonuclear pleocytosis to one of a more viral meningitis mononuclear pleocytosis has also been observed with St. Louis encephalitis virus (175). The CSF protein concentration is mildly to moderately increased, whereas the glucose concentration is generally normal. However, hypoglycorrhachia may occur, serving to confound the assessment by suggesting a bacterial etiology (146,147,170).
Traditionally, the diagnosis of EV meningitis has relied on isolation of the virus from CSF using cell culture or inoculation of suckling mice (176). Although initially very useful, these techniques have significant limitations. The sensitivity of tissue culture for EVs is only 65% to 75% (177). The titer of EVs in the CSF of patients with aseptic meningitis may be as low as 101 to 103 TCID50 (median tissue culture infectious dose) per milliliter of CSF. This results in slower growth than is observed with specimens of throat or rectal origin. The time to isolation of EV from CSF ranges from 4 to 8 days (178) using traditional cell culture—too long to be clinically useful in patient management. Using shell vial culture, the time can be shortened to 2 to 3 days, but sensitivity may be lost (179,180). Lastly, optimum recovery of EVs from clinical specimens requires the use of multiple cell lines, either individually or as mixtures, in order to increase culture yield (181). Even using multiple cell lines, some EVs, in particular the group A CVs and some of the newer EVs, fail to grow in cell culture (182). The added cost and technical expertise required for suckling mouse inoculation makes it impractical for use in the modern diagnostic laboratory. Serologic confirmation of EV infection is also generally impractical and not useful in acute management of the patient.
As mentioned previously, the 5′ UTR contains regions of conserved nucleotide identity among the EVs. These regions have been exploited for the creation of primers and probes that can be used in NAATs capable of detecting all EVs (43). Compared to cell culture, NAAT detection of EVs in the CSF has been shown to exhibit sensitivities that range from 86% to 100% and specificities ranging from 92% to 100%. Furthermore, NAATs are capable of detecting EV genome in CSF samples from individuals with syndromes clinically compatible with aseptic meningitis previously deemed negative by cell culture or without pleocytosis (163,162,183). These assays are also able to detect EV that cannot be grown in cell culture. Lastly, NAAT can be performed rapidly, generally in a matter of hours. The results can be made available with sufficient speed as to have an impact on patient management, resulting in a reduction in hospital stay, antibiotic use, and ordering of ancillary tests (184–188). A confirmatory polymerase chain reaction (PCR) test result obtained on a patient with clinical aseptic meningitis can reassure the clinician that no further diagnostic investigation is required. For these reasons, NAAT detection has become the method of choice for the diagnosis of CNS EV infection. Because of the lack of sensitivity of viral culture for detection of the EV in CSF, it should be reserved for instances when NAAT is not available.
Two caveats should be borne in mind when establishing the diagnosis of EV meningitis. Confirmation of EV as the etiology for aseptic meningitis syndrome should rely on detection of the virus from the CSF. As previously discussed, following EV infection viral shedding may occur from the throat and GI tract for up to several weeks (7,92). Therefore, detection of the EV from the stool or throat of an individual with aseptic meningitis may represent an infection that occurred weeks previously and is unrelated to the present syndrome. Shedding from a past infection cannot be ruled out unless the virus is detected in nonpermissive sites (i.e., CSF, blood, tissue) (189).
Lastly, rare reports of co-infections of the CSF by bacteria and EVs exist (102,126,129,130). In these patients, the clinical and laboratory picture of bacterial meningitis dominated and the virus was isolated incidentally. The patients were sick enough that identification of a virus before identification of the bacterium would have been unlikely to dissuade the clinician from continued use of antibiotics. Thus, the detection of an EV either by culture or NAAT must always be placed in the context of the patient’s clinical picture and laboratory findings. In the clinical presentation typical of viral meningitis, co-infection with a clinically “silent” bacterium would be extraordinarily unlikely.
Treatment and Prevention
No specific treatment exists for EV meningitis. Supportive measures include bed rest, antipyretics, and analgesics, as indicated. Administration of parenteral fluids for individuals unable to take adequate fluids orally, especially infants, is indicated. Seizures should be controlled with appropriate anticonvulsant drugs.
Immune globulin preparations have been used for the treatment of newborn infants with severe disease and immunocompromised individuals, but their efficacy is not established (61,159,190). Intravenous, as well as intrathecal, administration may be necessary to ameliorate or prevent CNS infection in immunocompromised patients.
The promising results from clinical trials of pleconaril (39), an antiviral that inhibits EV binding and viral uncoating, were overshadowed by its adverse effects (40). In clinical trials, pleconaril was found to induce cytochrome P450 3A, resulting in menstrual irregularities in women taking hormonal contraceptives. This finding raised concerns that it might increase the metabolism of some hormonal contraceptives and anti-HIV drugs, thereby reducing their efficacy, prompting the U.S. Food and Drug Administration not to grant a license for its use.
The EVs are spread primarily through a lack of good hygiene. Hand washing prevents the spread of the EVs and should be encouraged in families and institutions (191). In patients hospitalized with EV, meningitis infection control measures using standard precautions are sufficient. Community measures for the prevention of EV infections rely on the development and maintenance of sewage and potable water systems. No vaccines exist for the non-PV EV. However, recent early studies suggest that it may be possible to develop an inactivated EV-A71 vaccine that can induce neutralizing antibodies and is well tolerated in humans (192).
PARECHOVIRUSES
The first two members of the genus human parechoviruses (HPeVs) were found in 1956 (193). However, it was not until the turn of the century that they were accorded their own genus. Originally classified as EVs and designated as echoviruses 22 and 23 (18) (currently designated HPeV 1 and 2, respectively), it became evident early on that they exhibited characteristics that differed from the other members of the genus (193,194). The development of methodologies to probe the molecular aspects of viral replication and viral genetics confirmed that they differed substantively from the EV (25–27,195,196) and led to their reclassification in a separate genus (24).
Morphologically, HPeVs exhibit similar characteristics as the EV: small size, lack of an envelope, a positive sense, and single-stranded RNA genome of the length and organization consistent with that of the EVs (6). Important differences are the lack of maturational cleavage of VP0 to yield capsid proteins VP2 and VP4. As a result, the HPeV capsid is composed of three, rather than four, proteins. Differences also exist in the function of two nonstructural proteins. A detailed discussion of these and other differences is beyond the scope of this chapter, and the reader is directed to a recent review (6).
The genus is currently composed of 16 serotypes (Table 5.3) (24). HPeVs have been reported worldwide. The epidemiology of the HPeV continues to evolve as new serotypes are identified and detection is improved using NAATs. Current data indicates that the HPeVs account for approximately 2% of the “EV” isolated using traditional cell culture in clinical laboratories (6). HPeV1 followed by HPeV3 are the types most frequently isolated. Infection with HPeV appears to occur early in life. In the United States, 73% of HPeV1 and 67.6% of HPeV2 isolated come from infants younger than 1 year of age. A longitudinal study of Norwegian infants documented that the cumulative incidence of HPeV infection by 24 and 36 months of age was 86% and 94%, respectively (197).
HPeV infections exhibit a strong seasonal epidemiology. Worldwide, the peak incidence of infections occurs during the summer and fall months (197–204). A unique biennial pattern of circulation has been reported for HPeV3 (205–208). As with the EVs, multiple HPeV serotypes circulate within a community at the same time. The majority of cases of HPeV meningitis occur in male infants younger than 3 months of age. HPeV3 is the overwhelmingly dominant cause of HPeV meningitis. Considerable variation in the annual prevalence of HPeV meningitis is observed.
Transmission of the HPeV occurs via the fecal-oral and respiratory routes. They may be shed from these sites for weeks to months (197,201,209). The finding of HPeV in the stool of healthy, asymptomatic infants indicates that many, if not the majority, of infections are subclinical.
Irritability is present in nearly all cases of HPeV meningitis (202,207,210). An exanthem is frequently present. Emesis, diarrhea, and distention are reported in approximately one-quarter to half of cases. Rhinorrhea, cough, tachypnea, apnea, and wheezing may be present. Notably, findings of increased intracranial pressure (bulging fontanelle) or meningeal irritation (nuchal stiffness, Kernig or Brudzinski signs) are absent. The CSF cytochemical evaluation reveals no or minimal abnormalities in WBC count or protein and glucose concentrations in the overwhelming majority of patients (202,207,210).
Currently, NAAT is the methodology of choice for HPeV detection because of its sensitivity and ability to detect all known HPeV types in a clinically meaningful time frame (6). Optimum diagnostic assays target the HPeV 5′ UTR and are not type-specific. They are designed for increased sensitivity and to broadly detect all HPeV types from clinical specimens.
The HPeV can produce cytopathic effect on appropriate cell lines (211). However, cell culture detection is limited by those factors discussed for the EV (see previous discussion) (211,212).
ARBOVIRUSES
The arboviruses are a group of more than 500 viruses from various viral families that are transmitted by the bite of an insect or tick (i.e., an arthropod vector) (213); hence, the derivation of their name ar–arthropod, bo–borne, viruses. Transmission to humans (epizootic transmission) occurs by chance and is secondary to the natural enzootic cycle involving an arthropod vector and an avian or mammalian host. Mosquitoes serve as the vectors for the majority of clinically significant arbovirus endemic to North America. However, Colorado tick fever virus, Powassan (POW) virus in North America, and tick-borne encephalitis (TBE) virus in Eurasia are transmitted by ticks (214,215).
Flaviviridae
West Nile Virus
West Nile virus (WNV) was first isolated in Uganda in 1937 (216). Its incursion into the United States in 1999 (217) preceded a rapid spread throughout the contiguous continental United States as well as North and South America (218,219). In the United States, WNV has displaced all autochthonous arboviruses as the single major cause of CNS disease (220). WNV is a member of the Flaviviridae family of RNA viruses, within the genus Flavivirus (221). All members of the family possess a host cell–derived lipid envelope that is modified by the insertion of viral proteins and a positive (message)-sense, single-stranded RNA genome.
WNV is maintained in an enzootic cycle that involves primarily avian hosts and mosquitoes (222). Culex mosquitoes are important vectors in the United States (223). Mosquitoes are important in transgenerational and transseasonal maintenance of the WNV enzootic cycle (224,225). WNV infections occur during the summer months, generally from July through October in the United States, coinciding with periods of increased activity of its vectors. However, as WNV has spread southward in the United States, reported transmissions to humans have occurred as early as April and as late as December (223).
Transmission to humans (i.e., epizootic transmission) is incidental and, with rare exception, results in a “dead-end” infection without subsequent human transmission. The latter has occurred through transfusion of blood products and organ transplantation (226–229). In addition, confirmed or suspected maternal–fetal and maternal–infant vertical transmission of WNV has been reported (230,231).
The majority of WNV infections in adults and children result in subclinical disease. Infection of children results in asymptomatic infection or milder disease more frequently than in adults (232–236). During one outbreak, children were 4.5 times more likely to become infected with WNV but 110 times less likely to develop West Nile neuroinvasive disease (WNND) (237). In adults, age is the single most significant risk factor for development of WNND (235,236,238). The incidence of severe neurologic disease is 10 times higher in persons aged 50 to 59 years and 43 times higher in those aged 80 years or older compared with individuals 20 years of age or younger. Other risk factors include hypertension, diabetes mellitus, cardiovascular disease, alcohol abuse, and immunosuppression. Approximately 80% of those infected with WNV remain asymptomatic. The majority of the remainder develops an acute, self-limited febrile illness known as West Nile fever. It is characterized by a sudden onset of fever (38° to 40°C), accompanied by fatigue, malaise, anorexia, headache, myalgias, and weakness. Ocular pain on eye movement has been reported. A diffuse nonpruritic, macular, papular, maculopapular, or morbilliform exanthem and diffuse lymphadenopathy may be seen (222,235,239–241).
Less than 1% (1:140 to 320) of adults develop WNND: aseptic meningitis (WNV meningitis), encephalitis, or poliomyelitis- like syndrome (241–245). WNND may be even rarer in children: approximately 1:4,200 infected children according to one study (237,246). The median age of patients with WNV meningitis is less than those with encephalitis (247). The percentage of individuals with WNV meningitis that comprise cases of WNND varies by report. In an outbreak in Israel, 15.9% of hospitalized patients with WNND had meningitis (233). In the United States, WNV surveillance data from 1999 to 2008 found that 33% of WNND cases reported to the Centers for Disease Control and Prevention (CDC) were meningitis (247). Individuals 19 years of age or younger accounted for 8% of all cases of meningitis. However, when cases of WNND in children were analyzed for nearly the same time frame, 47% of all pediatric cases were of meningitis as compared to encephalitis or meningoencephalitis, which accounted for 37% of cases (236).
In adults and children, West Nile meningitis is clinically indistinguishable from other causes of viral meningitis. The illness begins abruptly with fever, headache, nuchal rigidity, and meningeal signs (222,235,244). Photo- and phonophobia may be present. The headache may be so severe as to need the use of narcotic analgesics to control. Weakness and dyskinesias in the form of tremors, myoclonus, or parkinsonism may occur. The outcome of West Nile meningitis is favorable, although it may take 2 to 3 weeks to fully recover.
Analysis of the CSF shows a lymphocytic pleocytosis of generally less than 500 cells/mm3 (235). If the CSF is examined early in the course of the illness, a polymorphonuclear pleocytosis may be seen (248). The presence of plasma cells may be indicative of WNV as the etiology (249).
The diagnosis of WNV infection relies on the detection of WNV-specific antibodies in CSF or paired serum samples (235,250). The most sensitive and commonly used diagnostic assay is the IgM antibody-capture enzyme-linked immunosorbent assay (MAC-ELISA). It is capable of detecting CSF IgM antibodies 3 to 5 days into the clinical illness and 3 or more days earlier than detectable serum antibody (251). However, a positive test should always be interpreted in the context of clinical syndrome because the presence of WNV-specific IgM has been detected in the CSF for up to 199 days after onset of illness (252). The finding of WNV-specific IgM in the CSF of an individual with a clinically compatible CNS syndrome generally is considered diagnostic of WNND. If only serum is used for establishing a diagnosis, a second convalescent serum sample, obtained 2 or more weeks later, should be collected to document a fourfold increase in specific antibody titers using a functional assay such as neutralization or hemagglutination inhibition (235). Because serologic cross reactions among St. Louis encephalitis virus, WNV, and POW virus can occur, serologic testing ideally should include a battery of region-specific arboviral antigens (253).
St. Louis Encephalitis Virus
St. Louis encephalitis virus (SLEV) was first identified in 1933 following an epidemic of encephalitis in St. Louis, Missouri (254). Like others of the genus, it is an enveloped, positive (message)-sense, single-stranded RNA virus (221). It is recognized as the cause of sporadic and epidemic encephalitis and meningitis throughout the Americas. In the United States, most cases are seen from July through September (255). In Florida, epidemics have continued into December (256). Birds are the reservoir, and four different species of Culex mosquitoes, each with a specific geographic distribution, are the principal vectors (257). Younger patients tend to have milder forms of SLEV-associated CNS disease (1,258). Approximately 15% of all symptomatic cases of SLEV infection present as meningitis. The frequency of SLEV meningitis in children is much higher: approximately 40%. In contrast, patients older than 60 years of age rarely (5% or less) present with aseptic meningitis. No specific therapy is available.
Tick-Borne Encephalitis Virus
Tick-borne encephalitis virus (TBEV) is an important cause of meningitis in Europe. Cases in returning travelers from Europe and China have been seen in the United States (259). The disease was first described in Austria in 1931 (260). TBEV is endemic from Europe through far-eastern Russia, northern China, and Japan. Three subtypes of the virus exist: European, Siberian, and Far Eastern (10,215). It is maintained in enzootic cycles involving Ixodid ticks and wild rodents (215). Humans may be infected through the bite of an infected tick or, less commonly, the consumption of virus-infected milk. The majority of cases occur from March to November (10). The annual incidence of disease varies widely by country: Latvia, Estonia, Lithuania, and southern Germany—30, 16.5, 11.2, and 2 cases per 100,000 inhabitants, respectively (10). Aseptic meningitis is seen with infection due to the European and Siberian viral subtypes. Meningitis is more frequently seen in younger age-groups (10). Sixty-six percent of cases in infants and children 15 years of age or younger are of meningitis. This decreases progressively with advancing age such that for individuals 60 years of age or older, 32% or less develop meningitis. Diagnosis of TBEV infection is established by the detection of virus-specific IgM and IgG in serum. Vaccines for the prevention of TBEV-related disease are available in Europe and Canada and are recommended for certain regions of the world (10,215,261). No specific therapy is available.
Other flaviviruses such as POW virus (214,262) and Japanese encephalitis virus (263,264) are far less commonly identified as causes of meningitis.
Colorado Tick Fever Virus
Colorado tick fever (CTF) virus is an Orbivirus, a member of the family Reoviridae. Orbiviruses are double-stranded, segmented RNA viruses. The virion is nonenveloped, with an outer diameter of 80 nm and an inner core of 50 nm diameter. CTF virus is found mostly in western and mountainous regions of the United States. Its tick vector is Dermacentor andersoni, also known as the Rocky Mountain wood tick (265). After a bite by an infected tick, a 3- to 6-day incubation period follows. Hematopoietic cells, principally erythrocytes, are the major targets, wherein viral replication and dissemination occurs (265). A biphasic illness is characteristic but is actually observed in only half of patients (265). It consists of initial sudden onset of high fever and headache with flulike constitutional symptoms. Hepatosplenomegaly may occur, as well as GI symptoms. Stiff neck and other meningeal signs occur in as many as 18% of confirmed CTF cases (266). Meningoencephalitis and encephalitis can occur but less commonly than meningitis. The period of illness is usually brief (2 to 3 days). A peripheral leukopenia with relative lymphocytosis is common. A lymphocyte-predominant CSF pleocytosis and elevated protein level are typically found in patients with neurologic manifestations. Certain patients transiently improve (1 to 2 days), and then a second phase of illness of equal or greater severity follows (lasting an additional 2 to 3 days). Severe sequelae and death, though rare, have been reported. Typically, recovery is rapid and complete within 2 weeks. Laboratory diagnosis can be made using PCR (within the first 5 days of illness) or IgM-based serology (214). Virus may also be detected in peripheral blood smears by indirect immunofluorescence. Paired acute and convalescent serology is useful for retrospective diagnosis.
Bunyaviridae
The California encephalitis group of viruses include five viruses in the family Bunyaviridae. Three—La Crosse, Jamestown Canyon, and snowshoe hare—viruses have been associated with aseptic meningitis (128,267). Numerically, La Crosse is the most clinically relevant. From 1999 to 2007, La Crosse virus was reported from 25 states. However, 87% of cases came from 7 states: West Virginia, Ohio, North Carolina, Tennessee, Wisconsin, Minnesota, and Illinois (255).
PARAMYXOVIRUSES
Mumps Virus
Mumps virus is a member of the genus Rubulavirus within the subfamily Paramyxoviridae (268). It is an enveloped, pleomorphic virus possessing a single-stranded, negative-sense RNA genome. Only a single serotype exists, although 13 genotypes have been identified (269,270). In the United States, prior to 1967 and the introduction of an effective vaccine, mumps infections are observed during the winter and spring, with epidemics occurring approximately every 3 to 5 years (271,272). It was responsible for 2.5% to 15% of all cases of aseptic meningitis and between 17.5% and 22% of known causes of meningitis (5,8,9,11). As a result of mumps vaccine and effective vaccination programs, mumps cases have been reduced by 99% (273). However, mumps outbreaks continue to occur in the United States (274,275).
Mumps is transmitted via respiratory droplets. Once infection occurs, viremia is the likely means of spread to distant target organs, including the CNS (276). Meningitis is the most common neurologic manifestation of mumps infection (277). Mumps once was the leading identifiable cause of aseptic meningitis. The widespread use of the attenuated live-virus vaccine in the United States has resulted in a dramatic drop in incidence of mumps as well as its major role as a cause of meningitis (278). Neurologic involvement is three times more common in males. More than 50% of patients with mumps parotitis have CSF pleocytosis (277); however, most are not symptomatic of meningitis.
Clinically symptomatic meningitis occurs in up to 10% of patients with parotitis (279). Symptoms of meningitis are reported in cases of mumps parotitis by 4 to 10 days of illness but may precede parotitis by as much as 7 days; half or more cases of mumps meningitis may not be associated with parotitis at all (280,281). The clinical manifestations of mumps meningitis are nonspecific and differ little from those of EV cases. Fever is universal, usually lasting 3 days but occasionally persisting for a week (280). Bradycardia, drowsiness, lethargy, and anemia are all reported. More significant neurologic involvement can occur. Encephalitis is described concomitantly with meningitis in as many as 35% of cases (280) or as few as 4% (166).
Mumps virus meningitis and meningoencephalitis are usually benign and self-limited diseases (277,280). The prognosis for rapid and full recovery from mumps meningitis is excellent (277,280). The occasional fatalities demonstrate pathologic findings of demyelination near blood vessels (277).
Most but not all cases of symptomatic mumps meningitis have a primarily monocytic CSF pleocytosis, primarily mononuclear cells (277). Half of patients have 500 or fewer cells/mm3, 75% of cases have 1,000 or fewer cells/mm3, and the remainder have fewer than 5,000 cells/mm3. Exceptional cases with more than 5,000 cells/mm3 have been reported. Pleocytosis may persist for weeks. CSF protein level has been reported as normal in more than half of patients with mumps meningitis (280,282).
Several approaches exist for the diagnosis of mumps virus meningitis: viral isolation, documentation of increased antibody titers between serum acute and convalescent serum samples obtained 2 to 3 weeks apart, documentation of the presence of mumps-specific IgM antibodies, or NAAT detection of mumps virus genome (283). Mumps virus can be detected in saliva, blood, urine, and CSF. It is present in saliva 9 days prior to and 8 days after the onset of parotitis. In urine, it is detectable for up to 2 weeks after the onset of symptoms. Mumps-specific IgM is present in the blood within 3 to 4 days of the start of symptoms and may persist for up to 3 months. A sole serum sample demonstrating the presence of mumps-specific IgM obtained within 10 days of the onset of illness is sufficient to establish the diagnosis. IgG antibodies are detectable 7 to 10 days after the start of symptoms and persist for life. Antibodies to other paramyxoviruses may cross-react with in mumps virus in serologic assays, leading to false-positive results.
Other Paramyxoviruses
Measles infection may be associated with pleocytosis in as many as 30% of uncomplicated cases in normal patients, usually without signs or symptoms of meningitis (284). The parainfluenza viruses have been associated with CNS infection (285). The dominant serotype reported has been parainfluenza virus type 3.
ARENAVIRUSES
Lymphocytic Choriomeningitis Virus
LCMV, a member of the family Arenaviridae, is an enveloped virus with a genome consisting of two single-stranded, ambisense RNA molecules (286). LCMV was one of the earliest and significant viruses to be associated with aseptic meningitis in humans (3) (Table 5.2). It is now rarely identified as a cause of CNS infection in humans. The virus is endemic in wild mice, which serve as its reservoir (287). In the United States, the prevalence in wild mice is estimated to be 3.9% to 13.4% (287). Seroprevalence studies in the United States suggest that the 0.4% to 5% of patients sampled in three large cities had evidence of LCMV infection (287–289). The virus is transmitted by rodents (hamsters, guinea pigs, rats, mice) via their saliva, urine, feces, and nasal secretions (290). Individuals who work with or own rodents as well as those living under impoverished and nonhygienic circumstances have traditionally been at greatest risk (291–294). Human-to-human transmission can occur through transplantation (295,296). Approximately one third of LCMV infections are asymptomatic. Clinically manifest infections are usually mild and of brief duration. The infection frequently results in a biphasic illness characterized by an initial “flulike” illness with fever, headache, malaise, myalgia, anorexia, nausea, and emesis. A temporary, brief period of improvement follows and precedes the onset of symptoms of meningitis or encephalitis (291,292,297). Occasional severe neurologic disease (meningoencephalitis, encephalitis) has been reported (298). The course of meningitis and recovery are often prolonged (299), but permanent neurologic impairment is rare. CSF findings are indistinguishable from those of other viral causes of aseptic meningitis: lymphocytic pleocytosis (up to several thousand), mildly elevated protein, and usually normal or low glucose level. Other abnormalities include leukopenia, thrombocytopenia, elevations of transaminase levels, and pulmonary infiltrates (297). Cell culture of CSF usually detects the presence of LCMV. In severe disseminated infection, virus may be found in the blood, urine, and nasopharyngeal secretions. NAAT can be used to detect viral genome in CSF. Acute and convalescent serum specimens can be tested for raising antibody titers by enzyme immunoassays.
HUMAN HERPESVIRUSES
The majority of the human herpesviruses—HSV types 1 and 2, varicella-zoster virus (VZV) (300,301), Epstein-Barr virus (EBV) (302,303), cytomegalovirus (CMV) (304), human herpesvirus (HHV)-6 (305–308), and HHV-7 (308)—have been associated with reports of aseptic meningitis. Numerically, HSV-1 and HSV-2, in particular the latter, are the major causes of aseptic meningitis among this family for viruses.
HSVs appear to account for approximately 1% to 3% of all cases of aseptic meningitis (Table 5.2). HSV-2 and, much less commonly, HSV-1 have been associated with aseptic meningitis in patients with primary genital herpes infection (309–311). HSV-2 meningitis following primary genital infection is more frequently seen in women. Genital lesions may not be present at the time of symptoms of aseptic meningitis (310). Examination of the CSF demonstrates a lymphocytic pleocytosis, elevated protein, and normal glucose concentrations.
Recurrent benign lymphocytic meningitis (RBLM), also referred to as “Mollaret meningitis,” has been shown to be associated primarily with HSV-2 and, much less so, HSV-1 (312–315). The prevalence of RBLM is difficult to assess because of its intermittent presentation, and it is not a reportable condition. However, two reports place it between 1 and 2.2 cases per 100,000 population (316,317). The syndrome is more frequently seen in young women and consists of recurrent episodes of aseptic meningitis, lasting 2 to 5 days, which resolve spontaneously and without sequelae (318). The clinical presentation of RBLM is typical of viral meningitis. However, approximately 50% of patients have transient neurologic manifestations (seizures, hallucinations, diplopia, cranial nerve palsies, altered consciousness). Lesions of genital herpes are absent. Cytochemical analysis of the CSF reveals a lymphocytic pleocytosis, mildly elevated protein, and normal glucose concentrations (318). Detection of HSV genome is made by NAAT. The role of antiviral therapy in the management of RBLM is debated. Administration of acyclovir has been reported to result in rapid resolution of symptoms, and suppressive therapy using valacyclovir or famciclovir may prevent recurrences. In individuals with HSV-2 detected from the CSF, counseling regarding prevention of transmission of genital HSV should be undertaken (318).
VZV-related meningitis is a recognized complication of primary infection (chickenpox) and reactivation (zoster) (300,301,319,320). Meningitis has been associated with infection due to multiple different genotypes of the wild type strain as well the vaccine strain (Oka strain) of VZV (321) and may be seen in immunocompetent and immunocompromised individuals. The clinical and CSF presentation is typical of aseptic meningitis. However, in recent report, only half of patients had fever, and nearly 25% had altered mental status or lethargy (321). In that study, all cases of meningitis occurred in children. When meningitis complicates zoster, typical vesicular lesions restricted to dermatomal distribution are seen. The use of NAAT has identified individuals with meningitis due to reactivation of VZV but without skin lesions (301,321). The benign course is typical of other aseptic meningitides.
Reports of meningitis with EBV and CMV infection are far less common that those of the agents discussed previously. Although HHV-6 and HHV-7 have been reported as causes of CNS infection, including meningitis (305,307,308), these reports must be interpreted with caution. HHV-6 has been shown to establish persistence in the CNS and has been found in the CSF of asymptomatic individuals (322).
OTHER VIRAL PATHOGENS
HIV
Aseptic meningitis is known to occur as part of the clinical constellation of syndromes associated with primary HIV infection (323–325). The symptoms and signs are typical of aseptic meningitis and resolve rapidly. If the CSF is examined, a lymphocytic pleocytosis is present along with mildly elevated protein and normal glucose concentrations. HIV can be detected in the CSF. Pleocytosis may also be seen in asymptomatic HIV-infected individuals.
Occasional cases of aseptic meningitis or meningoencephalitis have been associated with adenoviruses in normal and immunocompromised patients (326–330), influenza A and B viruses (331–333), parvovirus (334), and rotavirus (335–337).
NONVIRAL PATHOGENS
Multiple pro- and eukaryotic pathogens (bacteria, spirochete, mycobacteria, fungi) can present with the classic features of aseptic meningitis. The majority of these agents would not be readily detectable by Gram stain of the CSF and may require special culture techniques to identify. As originally noted by Wallgren (2), parameningeal foci (sinusitis, otitis, mastoiditis, trauma) can present similarly to viral meningitis and need to be always considered in the differential diagnosis of aseptic meningitis.
Spirochetes
Lyme Disease
In areas of the United States where Lyme disease is endemic (338), CNS infection due to Borrelia burgdorferi may be encountered in 10% to 15% of individuals infected by this spirochete (339,340). B. burgdorferi is transmitted by Ixodes species (scapularis—Eastern United States, or pacificus—Western United States) of ticks. Because the vector is so diminutive, a significant number of infected individuals do not recall a tick bite. The illness is commonly seen in the spring and summer months and overlaps with EV and arboviral causes of meningitis. Meningitis usually occurs in the early disseminated stage of infection. The clinical manifestations are similar to viral meningitis and may occur in association with cranial neuritis and radiculoneuritis (339–343). The majority of children have associated findings such as facial or sixth nerve palsies, papilledema, and increased intracranial pressure of erythema migrans. Analysis of the CSF reveals a lymphocytic/monocytic pleocytosis (<500 cells/mm3) and elevated protein concentration. The CSF glucose concentration is usually normal but may be slightly decreased.
The diagnosis of Lyme meningitis requires documenting B. burgdorferi infection using a two-step diagnostic approach consisting of an initial enzyme-linked immunosorbent assay (ELISA) followed by a specific Western blot assay. In addition, the CSF should be examined for the presence of intrathecal production of antibody to B. burgdorferi (339–341). Treatment with ceftriaxone, cefotaxime, or penicillin G for 2 to 4 weeks is recommended (340,342,343).
Other Spirochetes
Leptospirosis is an acute systemic vasculitic disease caused by a number of spirochetes in the Leptospira genus. Acquisition is via contact with infected animal body fluids. Although more typically noted in the anicteric variety, meningitis is common in icteric leptospirosis (Weil disease) as well (344). The CSF profile is indistinguishable from that caused by common viruses, except that overall, more patients develop elevated CSF protein than patients with common viral meningitis. Aseptic meningitis is a relatively uncommon manifestation of secondary and tertiary syphilis (345). Meningitis due to tick-borne relapsing fever may be seen as frequently as with Lyme disease (346).
Bacterial and Fungal Causes
The majority of tuberculous CNS infections are caused by Mycobacterium tuberculosis. A small percentage is due to bovine tuberculosis, Mycobacterium bovis. Children are particularly prone to develop tuberculous meningitis (347). The presentation is often subacute, occurring over 1 to 3 weeks (348). The clinical course consists of three stages. Personality change, irritability, anorexia, listlessness, and occasional fever characterize the first stage. Second stage signs and symptoms reflect increased intracranial pressure and cerebral ischemia: drowsiness, nuchal rigidity, cranial nerve palsies, anisocoria, emesis, and seizures. In older children, adolescents, and adults, headache and emesis may be the dominant features. The third stage is characterized by coma, autonomic instability, fever, and progressive cerebral dysfunction. Chest radiograph; tuberculin skin testing; and, in older children, adolescents, and adults, IFN gamma release assays should be performed, seeking evidence of tuberculosis. The lumbar puncture reveals elevated opening pressure. The CSF reveals a markedly elevated protein and decreased glucose concentrations. A lymphocytic pleocytosis of usually less than 500 cells/mm3 is seen.
CNS infections with Brucella species occur in less than 5% of systemic brucella infections (349). Meningoencephalitis is the usual presentation. Both acute and chronic forms have been reported. Antibiotic therapy is usually curative; however, some residual neurologic defects are the rule.
Fungal meningitis is more commonly seen in immunocompromised individuals; however, occasional cases have been reported in immunologically normal individuals (350–355). Cryptococcus is the most commonly recognized cause of fungal meningitis (352). Other causes include Candida, Histoplasma, Coccidioides, Blastomyces, and Aspergillus (350,351,353–355). The clinical presentation is that of a subacute or chronic meningitis with fever, headache, and altered consciousness. Meningismus and focal neurologic findings are common findings. Evaluation of the CSF reveals a lymphocytic pleocytosis in most cases (a polymorphonuclear pleocytosis may be seen in some cases) in association with an increased protein concentration and hypoglycorrhachia. In addition to fungal culture of the CSF, evidence of infection should sought through detection of fungal antigen, fungal wall constituents, or specific antibodies in CSF and blood. When attempting to detect fungi through culture, it is important to use 10 to 15 mL of CSF. Evidence of fungal infection in sites outside of the CNS should also be sought.
A variety of neurologic manifestations have been reported to be associated with Mycoplasma pneumoniae infections (356). Of these, aseptic meningitis and encephalitis are the most common. Clinically, mycoplasmal meningitis is impossible to distinguish from viral meningitis, and as in typical viral infection, sequelae are not observed. Diagnosis is by serology or PCR (356,357). Mycoplasma hominis has been associated with cases of neonatal meningitis, usually in preterm infants (358).
Systemic Diseases
Kawasaki disease (KD) is an acute, self-limited vasculitis of unknown etiology affecting children. In the United States, the estimated overall annual incidence among children younger than 5 years of age is 20 cases per 100,000 population (359). The incidence is higher among Asians and Pacific Islanders. Several reports indicate that between 40% and 60% of children with KD have a mild (usually <100 cells/mm3) mononuclear pleocytosis on CSF examination (360,361). The incidence appears to be highest in reports from Japan. The CSF protein and glucose concentration are normal in the overwhelming majority of children.
Many other systemic vasculitides (e.g., polyarteritis nodosa, temporal arteritis, Takayasu arteritis, Wegener granulomatosis) are associated with CSF pleocytosis (362). Meningitis has been described as the initial manifestation of systemic lupus erythematosus in several patients (363). Two percent to 4% of patients with lupus may develop aseptic meningitis during the course of their disease (363).
Medication-Induced Aseptic Meningitis
The clinical features of medication-induced aseptic meningitis are not such that they set it apart from that caused by infectious etiologies nor do they permit differentiation of the multiple medication causes of the syndrome. A wide variety of medications, vaccines, or dyes, administered systemically or within the CNS, have been associated with aseptic meningitis (364–372).
The most common class of medications associated with aseptic meningitis is nonsteroidal antiinflammatory drugs (NSAIDs) (364). Of the nonselective and selective inhibitors of cyclooxygenase 1 and 2, ibuprofen is the most frequently associated NSAID with aseptic meningitis. Ibuprofen-associated aseptic meningitis is frequently seen in association with systemic lupus erythematosus. The majority of cases are reported in women. The associated pleocytosis is frequently polymorphonuclear in nature.
Antimicrobial agents have also been shown to cause aseptic meningitis (364). Leading this group of medications are trimethoprim and penicillin (penicillin, amoxicillin, or amoxicillin-clavulanic acid). Intravenous immunoglobulin is a well-recognized cause of aseptic meningitis (364). In pediatric clinical trials using intravenous immunoglobulin, the incidence of aseptic meningitis ranged from 6% to 32%. Immunomodulators such as monoclonal antibodies against CD3 or tumor necrosis factor-α are important causes of aseptic meningitis (364,368). Vaccine strains of mumps have been the most frequently associated (364,373). Greater than 10 mumps vaccine strains are used around the world (373). Although the Jeryl Lynn and related vaccine strains rarely, if ever, cause aseptic meningitis, other vaccine strains have been associated with varying incidences of this side effect (373).
References
1. Brinker KR, Monath TP. The acute disease. In: Monath TP, ed. St. Louis Encephalitis. Washington, DC: American Public Health Association; 1980:503–584.
2. Wallgren A. Une nouvelle maladie infectieuse du systeme nerveux central (Méningite “aseptica” acuta). Acta Paediatr. 1925;4:158–182.
3. Adair CV, Gauld RL, Smadel JE. Aseptic meningitis, a disease of diverse etiology: clinical and etiologic studies on 854 cases. Ann Intern Med. 1953;39:675–704.
4. Meyer HM Jr, Rogers NG, Miesse ML, et al. Aseptic meningitis caused by orphan viruses and other agents. Ann N Y Acad Sci. 1957:19;67:332–337.
5. Lennette EH, Magoffin RL, Knouf EG. Viral central nervous system disease. An etiologic study conducted at the Los Angeles County General Hospital. JAMA. 1962;179:687–695.
6. Romero JR, Selvarangan R. The human parechoviruses: an overview. Adv Pediatr. 2011;58:65–85.
7. Hall CE, Cooney MK, Fox JP. The Seattle virus watch program. I. Infection and illness experience of virus watch families during a communitywide epidemic of echovirus type 30 aseptic meningitis. Am J Public Health Nations Health. 1970;60:1456–1465.
8. Beghi E, Nicolosi A, Kurland LT, et al. Encephalitis and aseptic meningitis, Olmsted County, Minnesota, 1950–1981: I. Epidemiology. Ann Neurol. 1984;16:283–294.
9. Lepow ML, Carver DH, Wright HT, et al. A clinical, epidemiologic and laboratory investigation of meningitis during the four year period: 1955–1958. I. Observations concerning etiology and epidemiology. N Engl J Med. 1962;23:1181–1187.
10. Kaiser R. Tick-borne encephalitis. Infect Dis Clin North Am. 2008;22: 561–575.
11. Meyer HM Jr, Johnson RT, Crawford IP, et al. Central nervous system syndromes of “viral” etiology. Am J Med. 1960;29:334–347.
12. Buescher EL, Artenstein MS, Olson LC. Central nervous system infections of viral etiology: the changing pattern. Res Public Assoc Nerv Ment Dis. 1968;44:147–216.
13. Nicolosi A, Hauser WA, Beghi E, et al. Epidemiology of central nervous system infections in Olmsted County, Minnesota, 1950–1981. J Infect Dis. 1986;154:399–408.
14. Centers for Disease Control and Prevention Office of Surveillance, Epidemiology, and Laboratory Services. 2012 nationally notifiable diseases and conditions and current case definitions. Centers for Disease Control and Prevention Web site. http://wwwn.cdc.gov/nndss/document/2012 _Case%20Definitions.pdf. Accessed January 10, 2013.
15. Rantakallio PM, Leskinen M, von Wendt L. Incidence and prognosis of central nervous system infections in a birth cohort of 12,000 children. Scand Infect Dis. 1986:18:287–294.
16. Michos AG, Syriopoulou VP, Hadjichristodoulou C, et al. Aseptic meningitis in children: analysis of 506 cases. PLoS One. 2007;2:e674.
17. Centers for Disease Control and Prevention. Annual summary 1982: reported morbidity and mortality in the United States. MMWR Morb Mortal Wkly Rep. 1983;31:142–146.
18. Committee on Enteroviruses. Classification of human enteroviruses. Virology. 1962;16:501–504.
19. Gradien M, Forsgren M, Ehrnst A. Enteroviruses and reoviruses. In: Schmidt NJ, Emmons RW, eds. Diagnostic Procedures for Viral, Rickettsial, and Chlamydial Infections. 6th ed. Washington, DC: American Public Health Association; 1989:513–578.
20. Oberste MS, Maher K, Kilpatrick DR, et al. Molecular evolution of the human enteroviruses: correlation of serotype with VP1 sequence and application to picornavirus classification. J Virol. 1999;73:1941–1948.
21. Oberste MS, Maher K, Kilpatrick DR, et al. Typing of human enteroviruses by partial sequencing of VP1. J Clin Microbiol. 1999;37:1288–1293.
22. Oberste MS, Maher K, Flemister MR, et al. Comparison of classic and molecular approaches for the identification of untypeable enteroviruses. J Clin Microbiol. 2000;38:1170–1174.
23. Oberste M, Schnurr D, Maher K, et al. Molecular identification of new picornaviruses and characterization of a proposed enterovirus 73 serotype. J Gen Virol. 2001;82:409–416.
24. Knowles NJ, Hovi T, Hyypiä T, et al. Picornaviridae. In: King AMQ, Adams MJ, Carstens EB, et al, eds. Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on the Taxonomy of Viruses. London: Elsevier Academic Press; 2012:855–880.
25. Hyypiä T, Horsnell C, Maaronen M, et al. A distinct picornavirus group identified by sequence analysis. Proc Natl Acad Sci U S A. 1992;89:8847–8851.
26. Oberste MS, Maher K, Pallansch MA. Complete sequence of echovirus 23 and its relationship to echovirus 22 and other human enteroviruses. Virus Res. 1998;56:217–223.
27. Ghazi F, Hughes PJ, Hyypia T, et al. Molecular analysis of human parechovirus type 2 (formerly echovirus 23). J Gen Virol. 1998;79:2641–2650.
28. Rossmann MG, Arnold E, Erickson JW, et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature. 1985;317:145–153.
29. Hogle JM, Chow M, Filman DJ. Three-dimensional structure of poliovirus at 2.9 A resolution. Science. 1985;229:1358–1365.
30. Muckelbauer JK, Kremer M, Minor I, et al. Structure determination of coxsackievirus B3 to 3.5 A resolution. Acta Crystallogr D Biol Crystallogr. 1995;51:871–887.
31. Hendry E, Hatanaka H, Fry E, et al. The crystal structure of coxsackievirus A9: new insights into the uncoating mechanisms of enteroviruses. Structure. 1999;7:1527–1538.
32. Xiao C, Bator-Kelly CM, Rieder E, et al. The crystal structure of coxsackievirus A21 and its interaction with ICAM-1. Structure. 2005;13:1019–1033.
33. Plevka P, Perera R, Cardosa J, et al. Crystal structure of human enterovirus 71. Science. 2012;336:1274.
34. Fry EE, Stuart DI. Virion structure. In: Ehrenfeld E, Domingo E, Ross RR, eds. The Picornaviruses. Washington, DC: ASM Press; 2010:59–71.
35. Hellen CUT, Wimmer E. Enterovirus structure and assembly. In: Rotbart HA, ed. Human Enterovirus Infections. Washington, DC: ASM Press; 1998:155–170.
36. Rossmann MG. The canyon hypothesis. Hiding the host cell receptor attachment site on a viral surface from immune surveillance. J Biol Chem. 1989;264:14587–14590.
37. Harber J, Bernhardt G, Lu HH, et al. Canyon rim residues, including antigenic determinants, modulate serotype-specific binding of polioviruses to mutants of the poliovirus receptor. Virology. 1995;214:559–570.
38. Tuthill TJ, Bubeck D, Rowlands DJ, et al. Characterization of early steps in the poliovirus infection process: receptor-decorated liposomes induce conversion of the virus to membrane-anchored entry-intermediate particles. J Virol. 2006;80:172–180.
39. Romero JR. Pleconaril: a novel antipicornaviral drug. Expert Opin Investig Drugs. 2001;10:369–379.
40. Webster AD. Pleconaril—an advance in the treatment of enteroviral infection in immune-compromised patients. J Clin Virol. 2005;32:1–6.
41. Palmenberg A, Neubauer D, Skern T. Genome organization and encoded proteins. In: Ehrenfeld E, Domingo E, Ross RR, eds. The Picornaviruses. Washington, DC: ASM Press; 2010:3–71.
42. Rotbart HA, Romero JR. Laboratory diagnosis of enterovirus infections. In: Rotbart HA, ed. Human Enterovirus Infections. Washington, DC: ASM; 1995:401–418.
43. Romero JR. Reverse transcription-polymerase chain reaction detection of the enteroviruses: overview and clinical utility in pediatric enteroviral infections. Arch Pathol Lab Med. 1999;123:1161–1169.
44. Rozovics JM, Semler BL. Genome replication I: the players. In: Ehrenfeld E, Domingo E, Ross RR, eds. The Picornaviruses. Washington, DC: ASM Press; 2010:107–125.
45. Sabin AB, Ward R. The natural history of human poliomyelitis: I. Distribution of virus in nervous and non-nervous tissues. J Exp Med. 1941;73:771–793.
46. Bodian D. Histopathologic basis of clinical findings in poliomyelitis. Am J Med. 1949;6:563–578.
47. Bodian D. Emerging concept of poliomyelitis infection. Science. 1955;122: 105–108.
48. Sabin AB. Pathogenesis of poliomyelitis: reappraisal in the light of new data. Science. 1956;123:1151–1157.
49. Racaniello VR. One hundred years of poliovirus pathogenesis. Virology. 2006;344:9–16.
50. Koike S, Nomoto A. Poliomyelitis. In: Ehrenfeld E, Domingo E, Ross RR, eds. The Picornaviruses. Washington, DC: ASM Press; 2010:339–351.
51. Ward R, Melnick JL, Horstmann DM. Poliomyelitis virus in fly-contaminated food collected at an epidemic. Science. 1945;101:491–493.
52. Kelly SW, Winkelstein W Jr, Winsser J. Poliomyelitis and other enteric viruses in sewage. Am J Public Health Nations Health. 1957;47:72–77.
53. Centers for Disease Control and Prevention. Aseptic meningitis outbreak associated with echovirus 9 among recreational vehicle campers—Connecticut, 2003. MMWR Morb Mortal Wkly Rep. 2004;53:710–713.
54. Begier EM, Oberste MS, Landry ML, et al. An outbreak of concurrent echovirus 30 and coxsackievirus A1 infections associated with sea swimming among a group of travelers to Mexico. Clin Infect Dis. 2008;47:616–623.
55. Mitchell JR, Presnell WM, Akin EW, et al. Accumulation and elimination of poliovirus by the eastern oyster. Am J Epidemiol. 1966;84:40–50.
56. Bendinelli M, Ruschi A. Isolation of human enterovirus from mussels. Appl Microbiol. 1969;18:531–532.
57. Bates HR Jr. Coxsackie virus B3 calcific pancarditis and hydrops fetalis. Am J Obstet Gynecol. 1970;106:629–630.
58. Grist NR, Bell EJ, Assaad F. Enteroviruses in human disease. Prog Med Virol. 1978;24:114–157.
59. Bendig JW, Franklin OM, Hebden AK, et al. Coxsackievirus B3 sequences in the blood of a neonate with congenital myocarditis, plus serological evidence of maternal infection. J Med Virol. 2003;70:606–609.
60. Kaplan MH, Klein SW, McPhee J, et al. Group B coxsackievirus infections in infants younger than three months of age: a serious childhood illness. Rev Infect Dis. 1983;5:1019–1032.
61. Abzug MJ. Presentation, diagnosis, and management of enterovirus infections in neonates. Paediatr Drugs. 2004;6:1–10.
62. Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–865.
63. Bergelson J. Receptors. In: Ehrenfeld E, Domingo E, Ross RR, eds. The Picornaviruses. Washington, DC: ASM Press; 2010:73–86.
64. Israelsson S, Gullberg M, Jonsson N, et al. Studies of Echovirus 5 interactions with the cell surface: heparan sulfate mediates attachment to the host cell. Virus Res. 2010;151:170–176.
65. Mistry N, Inoue H, Jamshidi F, et al. Coxsackievirus A24 variant uses sialic acid-containing O-linked glycoconjugates as cellular receptors on human ocular cells. J Virol. 2011;85:11283–11290.
66. Yamayoshi S, Iizuka S, Yamashita T, et al. Human SCARB2-dependent infection by coxsackievirus A7, A14, and A16 and enterovirus 71. J Virol. 2012;86:5686–5696.
67. Iwasaki A, Welker R, Mueller S, et al. Immunofluorescence analysis of poliovirus receptor expression in Peyer’s patches of humans, primates, and CD155 transgenic mice: implications for poliovirus infection. J Infect Dis. 2002;186:585–592.
68. Sicinski P, Rowinski J, Warchok JB, et al. Poliovirus type 1 enters the human host through intestinal M cells. Gastroenterology. 1990;98:56–58.
69. Ouzilou L, Caliot E, Pelletier I, et al. Poliovirus transcytosis through M-like cells. J Gen Virol. 2002;83:2177–2182.
70. Gelfand HM, Holguin AH, Enterovirous infections in healthy children. Study during 1960. Arch Environ Health. 1962;5:404–411.
71. Kogon A, Spigland I, Frothingham TE, et al. The virus watch program: a continuing surveillance of viral infections in metropolitan New York families. VII. Observations on viral excretion, seroimmunity, intrafamilial spread and illness association in coxsackie and echovirus infections. Am J Epidemiol. 1969;89:51–61.
72. Whitehead JE. Silent infections and the epidemiology of viral carditis. Am Heart J. 1973;85:711–713.
73. Melnick JL. Enteroviruses. In: AS Evans, ed. Viral Infections of Humans: Epidemiology and Control. New York: Plenum Medical Book Co; 1976:163–207.
74. Müller U, Steinhoff U, Reis LF, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921.
75. Ohka S, Igarashi H, Nagata N, et al. Establishment of a poliovirus oral infection system in human poliovirus receptor-expressing transgenic mice that are deficient in alpha/beta interferon receptor. J Virol. 2007;81:7902–7912.
76. Bodian D. Pathogenesis of poliomyelitis. Am J Public Health Nations Health. 1952;42:1388–1402.
77. Huber, SA, Haisch C, Lodge PA. Functional diversity in vascular endothelial cells—role in coxsackievirus tropism. J Virol. 1990;64:4516–4522.
78. Coyne CB, Kim KS, Bergelson JM. Poliovirus entry into human brain microvascular cells requires receptor-induced activation of SHP-2. EMBO J. 2007;26:4016–4028.
79. Coyne CB, Bozym R, Morosky SA, et al. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe. 2011;9:70–82.
80. Yang WX, Terasaki T, Shiroki K, et al. Efficient delivery of circulating poliovirus to the central nervous system independently of poliovirus receptor. Virology. 1997;229:421–428.
81. Nathanson, N, Langmuir A. The Cutter incident: poliomyelitis following formaldehyde-inactivated poliovirus vaccination in the United States during the spring of 1955: III. Comparison of the clinical character of vaccinated and contact cases occurring after use of high rate lots of Cutter vaccine. Am J Hyg. 1963;78:61–81.
82. Sutter RW, Patriarca PA, Suleiman AJ, et al. Attributable risk of DTP (diphtheria and tetanus toxoids and pertussis vaccine) injection in provoking paralytic poliomyelitis during a large outbreak in Oman. J Infect Dis. 1992;165:444–449.
83. Strebel PM, Ion-Nedelcu N, Baughman AL, et al. Intramuscular injections within 30 days of immunization with oral poliovirus vaccine—a risk factor for vaccine-associated paralytic poliomyelitis. N Engl J Med. 1995;332:500–506.
84. Hurst EW. The newer knowledge of virus diseases of the nervous system: a review and an interpretation. Brain. 1936;59:1–34.
85. Nathanson N, Bodian D. Experimental poliomyelitis following intramuscular virus injection: 1. The effect of neural block on a neurotropic and a pantropic strain. Bull Johns Hopkins Hospital. 1961;108:308–319.
86. Ohka S, Yang WX, Terada E, et al. Retrograde transport of intact poliovirus through the axon via the fast transport system. Virology. 1998;250: 67–75.
87. Ren R, Racaniello V. Human poliovirus receptor gene expression and poliovirus tissue tropism in transgenic mice. J Virol. 1992;66:296–304.
88. Gromeier M, Wimmer E. Mechanism of injury-provoked poliomyelitis. J Virol. 1998;72:5056–5060.
89. Minor PD. The molecular biology of poliovaccines. J Gen Virol. 1992;73:3065–3077.
90. Price RA, Garcia JH, Rightsel WA. Choriomeningitis and myocarditis in an adolescent with isolation of coxsackie B-5 virus. Am J Clin Pathol. 1970;53:825–831.
91. Sutinen S, Kalliomaki JL, Pohjonen R, et al. Fatal generalized coxsackie B3 virus infection in an adolescent with successful isolation of the virus from pericardial fluid. Ann Clin Res. 1971;3:241–246.
92. Melnick JL. Enteroviruses In: AS Evans, ed. Viral Infections of Humans, Epidemiology and Control. 3rd ed. New York: Plenum, 1989:191–263.
93. Centers for Disease Control and Prevention. Enterovirus surveillance—United States, 1997–1999. MMWR. 2000:13:913–916.
94. Rotbart HA. Meningitis and encephalitis. In: Rotbart HA, ed. Human enterovirus infections. Washington, DC: American Society for Microbiology Press; 1995:271–289.
95. Lepow ML, Coyne N, Thompson LB, et al. A clinical, epidemiologic and laboratory investigation of aseptic meningitis during the four-year period, 1955–1958. II. N Engl J Med. 1962;266:1188–1193.
96. Berlin LE, Rorabaugh ML, Heldrich F, et al. Aseptic meningitis in infants < 2 years of age: diagnosis and etiology. J Infect Dis. 1993;168:888–892.
97. de Ory F, Avellón A, Echevarría JE, et al. Viral infections of the central nervous system in Spain: a prospective study. J Med Virol. 2013;85:554–562.
98. de Quadros CA, Andrus JK, Olive JM, et al. Polio eradication from the Western Hemisphere. Annu Rev Public Health. 1992;13:239–252.
99. Centers for Disease Control and Prevention. Progress toward poliomyelitis eradication—Nigeria, January 2011–September 2012. MMWR Morb Mortal Wkly Rep. 2012;61:899–904.
100. Assaad F, Cockburn C. Four-year study of WHO virus reports on enteroviruses other than poliovirus. Bull World Health Organ. 1972:46;329–336.
101. Centers for Disease Control and Prevention. Enterovirus surveillance—United States, 1970–2005. Surveillance Summaries. MMWR Surveill Summ. 2006;55(8):1–20.
102. Nishmi M, Morr J, Abrahamov A, et al. A winter outbreak of ECHO virus type 9 meningitis in Jerusalem, with cases of a simultaneous mixed pneumococcal-viral infection. Isr J Med Sci. 1971;7:1240–1247.
103. Miller SA, Wald ER, Bergman I, et al. Enteroviral meningitis in January with marked cerebrospinal fluid pleocytosis. Pediatr Infect Dis. 1986;5:706–707.
104. Mori I, Matsumoto K, Hatano M, et al. An unseasonable winter outbreak of echovirus type 30 meningitis. J Infect. 1995;31:219–223.
105. Chambon M, Archimbaud C, Bailly JL, et al. Circulation of enteroviruses and persistence of meningitis cases in the winter of 1999–2000. J Med Virol. 2001;65:340–347.
106. Huang QS, Carr JM, Nix WA, et al. An echovirus type 33 winter outbreak in New Zealand. Clin Infect Dis. 2003;37:650–657.
107. Antona D, Lévêque N, Chomel JJ, et al. Surveillance of enteroviruses in France, 2000–2004. Eur J Clin Microbiol Infect Dis. 2007;26:403–412.
108. Druyts-Voets E. Epidemiological features of entero non-poliovirus isolations in Belgium 1980–94. Epidemiol Infect. 1997;119:71–77.
109. Cherry JD. Enteroviruses and parechoviruses. In: Feigin RD, Cherry JD, Demmler GJ, et al, eds. Textbook of Pediatric Infectious Diseases. 5th ed. Philadephia: WB Saunders; 2004:1984–2041.
110. Roth B, Enders M, Arents A, et al. Epidemiologic aspects and laboratory features of enterovirus infections in Western Germany, 2000–2005. J Med Virol. 2007;79:956–962.
111. Schmidt NJ, Lennette EH, Ho HH. An apparently new enterovirus isolated from patients with disease of the central nervous system. J Infect Dis. 1974;129:304–309.
112. Trallero G, Casas I, Tenorio A, et al. Enteroviruses in Spain: virological and epidemiological studies over 10 years (1988–97). Epidemiol Infect. 2000;124:497–506.
113. Gharbi J, Jaïdane H, Ben M’hadheb M, et al. Epidemiological study of non-polio enterovirus neurological infections in children in the region of Monastir, Tunisia. Diagn Microbiol Infect Dis. 2006;54:31–36.
114. Tan CY, Ninove L, Gaudart J, et al. A retrospective overview of enterovirus infection diagnosis and molecular epidemiology in the public hospitals of Marseille, France (1985–2005). PLoS One. 2011;6:e18022.
115. Yamashita K, Miyamura K, Yamadera S, et al. Epidemics of aseptic meningitis due to echovirus 30 in Japan. A report of the National Epidemiological Surveillance of Infectious Agents in Japan. Jpn J Med Sci Biol. 1994;47:221–239.
116. Centers for Disease Control and Prevention. Outbreak of aseptic meningitis associated with multiple enterovirus serotypes—Romania, 1999. MMWR Morb Mortal Wkly Rep. 2000;49:669–671.
117. Baron RC, Hatch MH, Kleeman K, et al. Aseptic meningitis among members of a high school football team. An outbreak associated with echovirus 16 infection. JAMA. 1982;248:1724–1727.
118. Modlin JF, Kinney JS. Perinatal enterovirus infections. Adv Pediatr Infect Dis. 1987;2:57–78.
119. Lenaway DD, Brockmann R, Dolan GJ, et al. An outbreak of an enterovirus-like illness at a community wading pool: implications for public health inspection programs. Am J Public Health. 1989;79:889–890.
120. Alexander JP Jr, Chapman LE, Pallansch MA, et al. Coxsackievirus B2 infection and aseptic meningitis: a focal outbreak among members of a high school football team. J Infect Dis. 1993;167:1201–1205.
121. Somekh E, Shohat T, Handsher R, et al. An outbreak of echovirus 11 in a children’s home Epidemiol Infect. 2001;126:441–444.
122. Faustini A, Fano V, Muscillo M, et al. An outbreak of aseptic meningitis due to echovirus 30 associated with attending school and swimming in pools. Int J Infect Dis. 2006;10:291–297.
123. Nakao T, Miura R. Recurrent virus meningitis. Pediatrics. 1971;47: 773–776.
124. Chang TW. Recurrent viral infection (reinfection). N Engl J Med. 1971;284:765–773.
125. Aintablian N, Pratt RD, Sawyer MH. Rapidly recurrent enteroviral meningitis in non-immunocompromised infants caused by different viral strains. J Med Virol. 1995;47:126–129.
126. Wright HT Jr, McAllister RM, Ward R. “Mixed” meningitis. Report of a case with isolation of Haemophilus influenzae type B and ECHO virus type 9 from the cerebrospinal fluid. N Engl J Med. 1962;267:142–144.
127. Phillips CA, Melnick JL, Barrett FF, et al. Dual virus infections. Simultaneous enteroviral disease and St. Louis encephalitis. JAMA. 1966;197:169–172.
128. Balfour HH Jr, Seifert GL, Seifert MH Jr, et al. Meningoencephalitis and laboratory evidence of triple infection with California encephalitis virus, echovirus 11 and mumps. Pediatrics. 1973;51:680–684.
129. Eglin RP, Swann RA, Isaacs D, et al. Simultaneous bacterial and viral meningitis. Lancet. 1984;2:984.
130. Pelkonen T, Roine I, Anjos E, et al. Picornaviruses in cerebrospinal fluid of children with meningitis in Luanda, Angola. J Med Virol. 2012;84: 1080–1083.
131. Yamashita K, Miyamura K, Yamadera S, et al. Enteroviral aseptic meningitis in Japan, 1981–1991. Jpn J Med Sci Biol. 1992;45:151–161.
132. Dai-ming W, Guo-chang Z, Shi-mei Z, et al. An epidemic of encephalitis and meningoencephalitis in children caused by echovirus type 30 in Shanghai. Chin Med J. 1993;106:767–769.
133. Ray CG, McCollough RH, Doto IL, et al. Echo 4 illness: epidemiological, clinical and laboratory studies of an outbreak in a rural community. Am J Epidemiol. 1966;84:253–267.
134. Kinnunen E, Hovi T, Stenvik, et al. Localized outbreak of enteroviral meningitis in adults. Acta Neurol Scand. 1987;74:346–351.
135. Kao CH, Lee SS, Liu YC, et al. Outbreak of aseptic meningitis among adults in southern Taiwan. J Microbiol Immunol Infect. 2003;36:192–196.
136. Carrol ED, Beadsworth MB, Jenkins N, et al. Clinical and diagnostic findings of an echovirus meningitis outbreak in the north west of England. Postgrad Med J. 2006;82:60–64.
137. Horstmann DM, Yamada N. Enterovirus infections of the central nervous system. Res Publ Assoc Res Nerv Ment Dis. 1968;44:236–253.
138. Modlin JF. Perinatal echovirus infection: insights from a literature review of 61 cases of serious infection and 16 outbreaks in nurseries. Rev Infect Dis. 1986;8:918–926.
139. Abzug MJ, Levin MJ, Rotbart HA. Profile of enterovirus disease in the first two weeks of life. Pediatr Infect Dis J. 1993;12:820–824.
140. Lin TY, Kao HT, Hsieh SH, et al. Neonatal enterovirus infections: emphasis on risk factors of severe and fatal infections. Pediatr Infect Dis J. 2003;22:889–894.
141. Lake AM, Lauer BA, Clark JC, et al. Enterovirus infections in neonates. J Pediatr. 1976;89:787–791.
142. Rorabaugh ML, Berlin LE, Heldrich F, et al. Aseptic meningitis in infants younger than 2 years of age: acute illness and neurologic complications. Pediatrics. 1993;92:206–211.
143. Kibrick S, Benirschke K. Severe generalized disease (encephalohepatomyocarditis) occurring in the newborn period and due to infection with coxsackie virus, group B: evidence of intrauterine infection with this agent. Pediatrics. 1958;22:857–874.
144. Wilfert CM, Lehrman SN, Katz SL. Enteroviruses and meningitis. Pediatr Infect Dis J. 1983;2:333–341.
145. Dagan R, Jenista JA, Menegus MA. Association of clinical presentation, laboratory findings, and virus serotypes with the presence of meningitis in hospitalized infants with enterovirus infection. J Pediatr. 1988;113:975–978.
146. Jarvis WR, Tucker G. Echovirus type 7 meningitis in young children. Am J Dis Child. 1981;135:1009–1012.
147. Singer JI, Maur PR, Riley JP, et al. Management of central nervous system infections during an epidemic of enteroviral aseptic meningitis. J Pediatr. 1980;96:559–563.
148. Moore M, Kaplan MH, McPhee J, et al. Epidemiologic, clinical, and laboratory features of Coxsackie B1-B5 infections in the United States, 1970–79. Public Health Rep. 1984;99:515–522.
149. Haynes RE, Cramblett HG, Kronfol HJ. Echovirus 9 meningoencephalitis in infants and children. JAMA. 1969;208:1657–1660.
150. Jaffe M, Srugo I, Tirosh E, et al. The ameliorating effect of lumbar puncture in viral meningitis. Am J Dis Child. 1989;143:682–685.
151. Desmond RA, Accortt NA, Talley L, et al. Enteroviral meningitis: natural history and outcome of pleconaril therapy. Antimicrob Agents Chemother. 2006;50:2409–2414.
152. Peigue-Lafeuille H, Croquez N, Laurichesse H, et al. Enterovirus meningitis in adults in 1999–2000 and evaluation of clinical management. J Med Virol. 2002;67:47–53.
153. Chemtob S, Reece ER, Mills EL. Syndrome of inappropriate secretion of antidiuretic hormone in enteroviral meningitis. Am J Dis Child. 1985;139:292–294.
154. Moses EB, Dean AG, Hatch MH, et al. An outbreak of aseptic meningitis in the area of Fort Smith, Arkansas, 1975, due to echovirus type 4. J Ark Med Soc. 1977;74:121–125.
155. Helfand RF, Khan AS, Pallansch MA, et al. Echovirus 30 infection and aseptic meningitis in parents of children attending a child care center. J Infect Dis. 1994;169:1133–1137.
156. Rice SK, Heinl RE, Thornton LL, et al. Clinical characteristics, management strategies, and cost implications of a statewide outbreak of enterovirus meningitis. Clin Infect Dis. 1995;20:931–937.
157. Rotbart HA, Brennan PJ, Fife KH, et al. Enterovirus meningitis in adults. Clin Infect Dis. 1998;27:896–898.
158. Wilfert CM, Buckley RH, Mohanakumar T, et al. Persistent and fatal central-nervous-system ECHOvirus infections in patients with agammaglobulinemia. N Engl J Med. 1977;296:1485–1489.
159. McKinney RE Jr, Katz SL, Wilfert CM. Chronic enteroviral meningoencephalitis in agammaglobulinemic patients. Rev Infect Dis. 1987;9: 334–356.
160. Cunningham CK, Bonville CA, Ochs HD, et al. Enteroviral meningoencephalitis as a complication of X-linked hyper IgM syndrome. J Pediatr. 1999;134(5):584–588.
161. Halliday E, Winkelstein J, Webster AD. Enteroviral infections in primary immunodeficiency (PID): a survey of morbidity and mortality. J Infect. 2003;46:1–8.
162. Webster ADB, Rotbart HA, Warner T, et al. Diagnosis of enterovirus brain disease in hypogammaglobulinemic patients by polymerase chain reaction. Clin Infect Dis. 1993;17:657–661.
163. Rotbart HA, Kinsella JP, Wasserman RL. Persistent enterovirus infection in culture-negative meningoencephalitis-demonstration by enzymatic RNA amplification. J Infect Dis. 1990;161:787–791.
164. Khetsuriani N, Lamonte A, Oberste MS, et al. Neonatal enterovirus infections reported to the national enterovirus surveillance system in the United States, 1983–2003. Pediatr Infect Dis J. 2006;25:889–893.
165. Farmer K, MacArthur GA, Clay MM. A follow-up study of 15 cases of neonatal meningoencephalitis due to coxsackie virus B5. J Pediatr. 1975;87:568–571.
166. Sells CJ, Carpenter RL, Ray CG. Sequelae of central-nervous-system enterovirus infections. N Engl J Med. 1975;293:1–4.
167. Wilfert CM, Thompson RJ, Sunder TR, et al. Longitudinal assessment of children with enteroviral meningitis during the first three months of life. Pediatrics. 1981;67:811–815.
168. Bergman I, Painter MJ, Wald ER, et al. Outcome in children with enteroviral meningitis during the first year of life. J Pediatr. 1987;110:705–709.
169. Etter CG, Wedgwood J, Schaad UB. Aseptische memingitiden in der padiatrie. Schweiz Med Wochenschr. 1991;121:1120–1126.
170. Severien C, Jacobs KH, Schoenemann W. Marked pleocytosis and hypoglycorrhachia in coxsackie meningitis. Pediatr Infect Dis J. 1994;13:322–323.
171. Yeager AS, Bruhn FW, Clark J. Cerebrospinal fluid: presence of virus unaccompanied by pleocytosis. J Pediatr. 1974;85:578–579.
172. Miller DG, Gabrielson MO, Bart KJ, et al. An epidemic of aseptic meningitis, primarily among infants, caused by echovirus 11-prime. Pediatrics. 1968;41:77–90.
173. Feigin RD, Shackelford PG. Value of repeat lumbar puncture in the differential diagnosis of meningitis. N Engl J Med. 1973;289:571–574.
174. Amir J, Harel L, Frydman M, et al. Shift of cerebrospinal polymorphonuclear cell percentage in the early stage of aseptic meningitis. J Pediatr. 1991;119:938–940.
175. Bond JO, Quick DT, Lewis AL, et al. The 1962 epidemic of St. Louis encephalitis in Florida. II. Follow-up serologic surveys for prevalence of Group B arbovirus antibodies. Am J Epidemiol. 1966;83:564–570.
176. Melnick JL, Wenner HA, Phillip CA. Enteroviruses. In: Lennette EH, Schmidt NJ, eds. Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infections. 5th ed. Washington, DC: American Public Health Association; 1979:471–534.
177. Chonmaitree T, Baldwin CD, Lucia HL. Role of the virology laboratory in diagnosis and management of patients with central nervous system disease. Clin Microbiol Rev. 1989;2:1–14.
178. Rotbart HA. Enteroviral infections of the central nervous system. Clin Infect Dis. 1995;20:971–981.
179. Van Doornum GJ, De Jong JC. Rapid shell vial culture technique for detection of enteroviruses and adenoviruses in fecal specimens: comparison with conventional virus isolation method. J Clin Microbiol. 1998;36:2865–2868.
180. Buck GE, Wiesemann M, Stewart L. Comparison of mixed cell culture containing genetically engineered BGMK and CaCo-2 cells (Super EMix) with RT-PCR and conventional cell culture for the diagnosis of enterovirus meningitis. J Clin Virol. 2002;25:S13–S18.
181. Dagan R, Menegus MA. A combination of four cell types for rapid detection of enteroviruses in clinical specimens. J Med Virol. 1986;19:219–228.
182. Melnick JL. Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infections. 5th ed. Washington, DC: American Public Health Association; 1979.
183. Sawyer MH, Holland D, Aintablian N, et al. Diagnosis of enteroviral central nervous system infection by polymerase chain reaction during a large community outbreak. Pediatr Infect Dis J. 1994;13:177–182.
184. Ramers C, Billman G, Hartin M, et al. Impact of a diagnostic cerebrospinal fluid enterovirus polymerase chain reaction test on patient management. JAMA. 2000;283:2680–2685.
185. Robinson CC, Willis M, Meagher A, et al. Impact of rapid polymerase chain reaction results on management of pediatric patients with enteroviral meningitis. Pediatr Infect Dis J. 2002;21:283–286.
186. Archimbaud C, Mirand A, Chambon M, et al. Improved diagnosis on a daily basis of enterovirus meningitis using a one-step real-time RT-PCR assay. J Med Virol. 2004;74:604–611.
187. Tattevin P, Minjolle S, Arvieux C, et al. Benefits of management strategy adjustments during an outbreak of enterovirus meningitis in adults. Scand J Infect Dis. 2002;34:359–361.
188. King RL, Lorch SA, Cohen DM, et al. Routine cerebrospinal fluid enterovirus polymerase chain reaction testing reduces hospitalization and antibiotic use for infants 90 days of age or younger. Pediatrics. 2007;120:489–496.
189. Johnson GM, McAbee GA, Seaton ED, et al. Suspect value of non-CSF viral cultures in the diagnosis of enteroviral CNS infection in young infants. Dev Med Child Neurol. 1992;34:876–884.
190. Abzug MJ, Keyserling HL, Lee ML, et al. Neonatal enterovirus infection: virology, serology, and effects of intravenous immune globulin. Clin Infect Dis. 1995;20:1201–1206.
191. Ruan F, Yang T, Ma H, et al. Risk factors for hand, foot, and mouth disease and herpangina and the preventive effect of hand-washing. Pediatrics. 2011;127:e898–e904.
192. Meng FY, Li JX, Li XL, et al. Tolerability and immunogenicity of an inactivated enterovirus 71 vaccine in Chinese healthy adults and children: an open label, phase 1 clinical trial. Hum Vaccin Immunother. 2012;8:668–674.
193. Wigand R, Sabin AB. Properties of ECHO types 22, 23 and 24 viruses. Arch Gesamte Virusforsch. 1961;11:224–247.
194. Shaver DN, Barron AL, Karzon DT. Distinctive cytopathology of ECHO viruses type 22 and 23. Proc Soc Exp Biol Med. 1961;106:648–652.
195. Coller BA, Chapman NM, Beck MA, et al. Echovirus 22 is an atypical enterovirus. J Virol. 1990;64:2692–2701.
196. Coller BG, Tracy SM, Etchison D. Cap-binding complex protein p220 is not cleaved during echovirus 22 replication in HeLa cells. J Virol. 1991;65:3903–3905.
197. Tapia G, Cinek O, Witsø E, et al. Longitudinal observation of parechovirus in stool samples from Norwegian infants. J Med Virol. 2008;80: 1835–1842.
198. Abed Y, Boivin G. Human parechovirus infections in Canada. Emerg Infect Dis. 2006;12:969–975.
199. Pham NTK, Trinh QD, Khamrin P, et al. Diversity of human parechoviruses isolated from stool samples collected from Thai children with acute gastroenteritis. J Clin Microbiol. 2010;48:115–119.
200. Tauriainen S, Martiskainen M, Oikarinen S, et al. Human parechovirus 1 infections in young children—no association with type 1 diabetes. J Med Virol. 2007;79:457–462.
201. Harvala H, Robertson I, McWilliam, et al. Epidemiology and clinical associations of human parechovirus respiratory infections. J Clin Microbiol. 2008;46:3446–3453.
202. Selvarangan R, Nzabi M, Selvaraju SB, et al. Human parechovirus 3 causing sepsis-like illness in children from Midwestern United States. Pediatr Infect Dis. 2011;30:238–242.
203. Walters B, Peñaranda S, Nix WA, et al. Detection of human parechovirus (HPeV)-3 in spinal fluid specimens from pediatric patients in the Chicago area. J Clin Virol. 2011;52:187–191.
204. Renaud C, Kuypers J, Ficken E, et al. Introduction of a novel parechovirus RT-PCR clinical test in a regional medical center. J Clin Virol. 2011;51:50–53.
205. Harvala H, Robertson I, Chieochansin T, et al. Specific association of human parechovirus type 3 with sepsis and fever in young infants, as identified by direct typing of cerebrospinal fluid samples. J Infect Dis. 2009;199:1753–1760.
206. Verboon-Maciolek MA, Krediet TG, Gerards LJ, et al. Severe neonatal parechovirus infection and similarity with enterovirus infection. Pediatr Infect Dis J. 2008;7:241–245.
207. Wolthers KC, Benschop KS, Schinkel J, et al. Human parechoviruses as an important viral cause of sepsis like illness and meningitis in young children. Clin Infect Dis. 2008;47:358–363.
208. van der Sanden S, de Bruin E, Vennema H, et al. Prevalence of human parechovirus in the Netherlands in 2000 to 2007. J Clin Virol. 2008; 46:2884–2889.
209. Pajkrt D, Benschop KS, Westerhuis B, et al. Clinical characteristics of human parechoviruses 4-6 infections in young children. Pediatr Infect Dis J. 2009;28:1008–1010.
210. Piñeiro L, Vicente D, Montes M, et al. Human parechovirus in infants with systemic infection. J Med Virol. 2010;82:1790–1796.
211. Benschop K, Minnaar R, Koen G, et al. Detection of human enterovirus and human parechovirus (HPeV) genotypes from clinical stool samples: polymerase chain reaction and direct molecular typing, culture characteristics, and serotyping. Diagn Microbiol Infect Dis. 2010;68:166–173.
212. Joki-Korpela P, Hyypiä T. Diagnosis and epidemiology of echovirus 22 infections. Clin Infect Dis. 1998;27:129–136.
213. Lanciotti RS, Tsai TF. Arboviruses. In: Murray PR, Baron EJ, Jorgensen JH, et al, eds. Manual of Clinical Microbiology. 9th ed. Washington, DC: American Society for Microbiology; 2007:1486–1500.
214. Romero JR, Simonsen KA. Powassan encephalitis and Colorado tick fever. Infect Dis Clin North Am. 2008;22:545–559.
215. Mansfield KL, Johnson N, Phipps LP, et al. Tick-borne encephalitis virus—a review of an emerging zoonosis. J Gen Virol. 2009;90:1781–1794.
216. Smithburn KC, Hughes TP, Burke AW, et al. A neurotropic virus isolated from the blood of a native of Uganda. Am J Trop Med Hyg. 1940;20: 471–492.
217. Centers for Disease Control and Prevention. Outbreak of West Nile-like viral encephalitis—New York. MMWR Morb Mortal Wkly Rep.1999; 48:845–849.
218. Centers for Disease Control and Prevention. Statistics, surveillance, and control archive. http://www.cdc.gov/ncidod/dvbid/westnile/surv&control_ archive.htm. Accessed January 10, 2013.
219. Petersen LR, Hayes EB. West Nile virus in the Americas. Med Clin North Am. 2008;92:1307–1322.
220. Centers for Disease Control and Prevention. Summary of notifiable diseases—United States, 2010. MMWR Morb Mortal Wkly Rep. 2010;59: 1–111.
221. Simmonds P, Becher P, Collet MD, et al. Flaviviridae. In: King AMQ, Adams MJ, Carstens EB, et al, eds. Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on the Taxonomy of Viruses. London: Elsevier Academic Press; 2012:855–880.
222. Romero JR. West Nile virus infections. In: Feigin RD, Cherry JD, Demmler GJ, et al, eds. Textbook of Pediatric Infectious Diseases. 6th ed. Philadelphia: Saunders Elsevier; 2009:2331–2343.
223. Hayes EB, Komar N, Nasci RS, et al. Epidemiology and transmission dynamics of West Nile virus disease. Emerg Infect Dis. 2005;11:1167–1173.
224. Unlu I, Mackay AJ, Roy A, et al. Evidence of vertical transmission of West Nile virus in field-collected mosquitoes. J Vector Ecol. 2010;35:95–99.
225. Murray K, Walker C, Herrington E, et al. Persistent infection with West Nile virus years after initial infection. J Infect Dis. 2011;201:2–4.
226. Meny GM, Santos-Zabala L, Szallasi A, et al. West Nile virus infection transmitted by granulocyte transfusion. Blood. 2011;117:5778–5779.
227. Montgomery SP, Brown JA, Kuehnert M, et al. Transfusion-associated transmission of West Nile virus, United States 2003 through 2005. Transfusion. 2006;46:2038–2046.
228. Iwamoto M, Jernigan DB, Guasch A, et al. Transmission of West Nile virus from an organ donor to four transplant recipients. N Engl J Med. 2003;348:2196–2203.
229. Pealer LN, Marfin AA, Petersen LR, et al. Transmission of West Nile virus through blood transfusion in the United States in 2002. N Engl J Med. 2003;349:1236–1245.
230. Centers for Disease Control and Prevention. Possible West Nile virus transmission to an infant through breast-feeding—Michigan, 2002. MMWR Morb Mortal Wkly Rep. 2002;51:877–878.
231. Centers for Disease Control and Prevention. Intrauterine West Nile virus infection—New York, 2002. MMWR Morb Mortal Wkly Rep. 2002;51:1135–1136.
232. Spigland I, Jasinska-Klingberg W, Hofsbi E, et al. Clinical and laboratory observations in an outbreak of West Nile fever in Israel. Harefuah. 1958;54:275–281.
233. Chowers MY, Lang R, Nassar F, et al. Clinical characteristics of the West Nile fever outbreak, Israel, 2000. Emerg Infect Dis. 2001;7:675–678.
234. Hayes EB, O’Leary DR. West Nile virus infection: a pediatric perspective. Pediatrics. 2004;113:1375–1381.
235. Sejvar JJ, Marfin AA. Manifestations of West Nile neuroinvasive disease. Rev Med Virol. 2006;16:209–224.
236. Lindsey NP, Hayes EB, Staples E, et al. West Nile disease in children, United States, 1999–2007. Pediatrics. 2009;123:e1084–1089.
237. Mandalakas AM, Kippes C, Sedransk J, et al. West Nile virus epidemic, Northeast Ohio, 2002. Emerg Infect Dis. 2005;11:1774–1777.
238. Murray K, Baraniuk S, Resnick M, et al. Risk factors for encephalitis and death from West Nile virus infection. Epidemiol Infect. 2006;134: 1325–1332.
239. Goldblum N, Sterk VV, Paderski B. West Nile fever. The clinical features of the disease and the isolation of West Nile virus from the blood of nine human cases. Am J Hyg. 1954;59:89–103.
240. Marberg K, Goldblum N, Sterk VV, et al. The natural history of West Nile fever. Clinical observations during an epidemic in Israel. Am J Hyg. 1956;64:259–269.
241. Campbell GL, Marfin AA, Lanciotti RS, et al. West Nile virus. Lancet Infect Dis. 2002;2:519–529.
242. Tsai TF, Popovici F, Cernescu C, et al. West Nile encephalitis epidemic in southeastern Romania. Lancet. 1998;352:767–771.
243. Mostashari F, Bunning ML, Kitsutani PT, et al. Epidemic West Nile encephalitis, New York, 1999: results of a household-based seroepidemiological survey. Lancet. 2001;358:261–264.
244. Sejvar JJ, Haddad MB, Tierney BC, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290:511–515.
245. Emig M, Apple DJ. Severe West Nile virus disease in healthy adults. Clin Infect Dis. 2004;38:289–292.
246. LaBeaud AD, Lisgaris MV, King CH, et al. Pediatric West Nile virus infection: neurologic disease presentations during the 2002 epidemic in Cuyahoga County, Ohio. Pediatr Infect Dis J. 2006;25:751–753.
247. Centers for Disease Control and Prevention. Surveillance for human West Nile Disease—United States, 1999–2008. MMWR Surveill Summ. 2010;59(SS-2):1–18.
248. Crichlow R, Bailey J, Gardner C. Cerebrospinal fluid pleocytosis in hospitalized West Nile virus patients. J Am Board Fam Pract. 2004;17: 470–472.
249. Carson PJ, Steidler T, Patron R, et al. Plasma cell pleocytosis in cerebrospinal fluid in patients with West Nile virus encephalitis. Clin Infect Dis. 2003;37:e12–e15.
250. Hayes EB, Sejvar JJ, Zaki SR, et al. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis. 2005;11: 1174–1179.
251. Tardei G, Ruta S, Chitu V, et al. Evaluation of immunoglobulin M (IgM) and IgG enzyme immunoassays in serologic diagnosis of West Nile Virus infection. J Clin Microbiol. 2000;38:2232–2239.
252. Kapoor H, Signs K, Somsel P, et al. Persistence of West Nile virus (WNV) IgM antibodies in cerebrospinal fluid from patients with CNS disease. J Clin Virol. 2004;31:289–291.
253. Tsai TF, Chandler LJ. Arboviruses. In: Murray PR, Baron EJ, Jorgensen JH, eds. Manual of Clinical Microbiology. 8th ed. Washington, DC: American Society for Microbiology Press; 2003:1553.
254. Webster LT, Fite GL. A virus encountered in the study of material from cases of encephalitis in the St. Louis and Kansas City epidemics of 1933. Science. 1933;78:463–465.
255. Reimann CA, Hayes EB, DiGuiseppi C, et al. Epidemiology of neuroinvasive arboviral disease in the United States, 1999–2007. Am J Trop Med Hyg. 2008;79:974–979.
256. Meehan PJ, Wells DL, Paul W, et al. Epidemiological features of and public health response to a St. Louis encephalitis epidemic in Florida, 1990–1991. Epidemiol Infect. 2000;125:181–188.
257. Centers for Disease Control and Prevention. St. Louis encephalitis—epidemiology & geographic distribution. http://www.cdc.gov/sle/technical /epi.html. Accessed December 29, 2012.
258. Zweighaft RM, Rasmussen C, Brolnitsky O, et al. St. Louis encephalitis: the Chicago experience. Am J Trop Med Hyg. 1979;28:114–118.
259. Centers for Disease Control and Prevention. Tick-borne encephalitis among US travelers to Europe and Asia—2000–2009. MMWR Morb Mortal Wkly Rep. 2010;59:335–338.
260. Schneider H. Über epidemische akute Meningitis serosa. Wien Klin Wochenschr. 1931;44:350–352.
261. Barrett PN, Plotkin SA, Ehrlich HJ. Tick-borne encephalitis virus vaccines. In: Plotkin SA, Orenstein WA, Offit PA, eds. Vaccines. 5th ed. Philadelphia: Elsevier; 2008:841–856.
262. Centers for Disease Control and Prevention. West Nile virus disease and other arboviral diseases—United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:510–514.
263. Jeurissen A, Strauven T. A case of aseptic meningitis due to Japanese encephalitis virus in a traveller returning from the Philippines. Acta Neurol Belg. 2011;111:143–145.
264. Kuwayama M, Ito M, Takao S, et al. Japanese encephalitis virus in meningitis patients, Japan. Emerg Infect Dis. 2005;11:471–473.
265. Emmons RW. Ecology of Colorado tick fever. Annu Rev Microbiol. 1988;42:49–64.
266. Goodpasture HC, Poland JD, Francy DB, et al. Colorado tick fever: clinical, epidemiologic, and laboratory aspects of 228 cases in Colorado in 1973–1974. Ann Intern Med. 1978;88:303–310.
267. Srihongse S, Grayson MA, Deibel R. California serogroup viruses in New York State: the role of subtypes in human infections. Am J Trop Med Hyg. 1984;33:1218–1227.
268. Wang LF, Collins PL, Fouchier RAM, et al. Paramyxoviridae. In: King AMQ, Adams MJ, Carstens EB, et al, eds. Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on the Taxonomy of Viruses. London: Elsevier Academic Press; 2012:672–685.
269. Jin L, Rima B, Brown D, et al. Proposal for genetic characterisation of wild-type mumps strains: preliminary standardisation of the nomenclature. Arch Virol. 2005;150:1903–1909.
270. Santos CL, Ishida MA, Foster PG, et al. Detection of a new mumps virus genotype during parotitis epidemic of 2006–2007 in the state of São Paulo, Brazil. J Med Virol. 2008;80:323–329.
271. Centers for Disease Control and Prevention. Mumps surveillance 1973. MMWR Morb Mortal Wkly Rep. 1974;23:431.
272. Centers for Disease Control and Prevention. Summary of notifiable diseases, United States, 1991. MMWR Morb Mortal Wkly Rep. 1991;40:3.
273. Centers for Disease Control and Prevention. Mumps surveillance, United States, 1988–1993. MMWR Morb Mortal Wkly Rep. 1995;44(SS-3):1–21.
274. Centers for Disease Control and Prevention. Update: multistate outbreak of mumps—United States, January 1–May 2, 2006. MMWR Morb Mortal Wkly Rep. 2006;55:559–563.
275. Barskey AE, Schulte C, Rosen JB, et al. Mumps outbreak in Orthodox Jewish communities in the United States. N Engl J Med. 2012;367: 1704–1713.
276. Kilham L. Isolation of mumps virus from the blood of a patient. Proc Soc Exp Biol Med. 1948;69:99–100.
277. Bang HO, Bang J. Involvement of the central nervous system in mumps. Acta Med Scand. 1943;113:487–505.
278. Centers for Disease Control and Prevention. Measles, mumps, and rubella—vaccine use and strategies for elimination of measles, rubella, and congenital rubella syndrome and control of mumps. MMWR Recomm Rep. 1998;47:1–67.
279. McLean DM, Bach RD, Larke RPB, et al. Mumps meningoencephalitis, Toronto, 1963. Can Med Assoc J. 1964;90:458–462.
280. Azimi PH, Cramblett HG, Haynes RE. Mumps meningoencephalitis in children. JAMA. 1969;207:509–512.
281. Johnstone JA, Ross CAC, Dunn M. Meningitis and encephalitis associated and mumps infection: a 10-year survey. Arch Dis Child. 1972;47: 647–651.
282. Strussberg S, Winter S, Friedman A, et al. Notes on mumps meningoencephalitis: some features of 199 cases in children. Clin Pediatr. 1969;8:373–377.
283. Leland DS. Parainfluenza and mumps viruses. In: Murray PR, Baron EJ, Jorgensen JH, et al, eds. Manual of Clinical Microbiology. 9th ed. Washington, DC: American Society for Microbiology Press; 2007; 1352–1360.
284. Hänninen P, Arstila P, Lang H, et al. Involvement of the central nervous system in acute, uncomplicated measles virus infection. J Clin Microbiol. 1980;11:610–613.
285. Newland JG, Romero JR. Myxoviruses. In: Barton LL, Friedman NR, eds. The Neurological Manifestations of Pediatric Infectious Disease and Immunodeficiency Syndromes. Totowa, NJ: Humana Press; 2008:41–68.
286. Salvato MS, Clegg JCS, Buchmeier MJ, et al. Arenaviridae. In: King AMQ, Adams MJ, Carstens EB, et al, eds. Virus Taxonomy. Classification and Nomenclature of Viruses: Ninth Report of the International Committee on the Taxonomy of Viruses. London: Elsevier Academic Press; 2012: 715–723.
287. Childs JE, Glass GE, Korch GW, et al. Lymphocytic choriomeningitis virus infection and house mouse (Mus musculus) distribution in urban Baltimore. Am J Trop Med Hyg. 1992:47;27–34.
288. Park JY, Peters CJ, Rollin PE, et al. Age distribution of lymphocytic choriomeningitis virus serum antibody in Birmingham, Alabama: evidence of a decreased risk of infection. Am J Trop Med Hyg. 1997;57:37–41.
289. Knust B, Macneil A, Wong SJ, et al. Exposure to lymphocytic choriomeningitis virus, New York, USA. Emerg Infect Dis. 2011;17:1324–1325.
290. Lehmann-Grube F. Portraits of viruses: arenaviruses. Intervirology. 1984;22:121–145.
291. Deibel R, Woodall JP, Decher WJ, et al. Lymphocytic choriomeningitis virus in man. Serologic evidence of association with pet hamsters. JAMA. 1975;232:501–504.
292. Vanzee BE, Doublas RG Jr, Betts RF, et al. Lymphocytic choriomeningitis in university hospital personnel. Clinical features. Am J Med. 1975;58:803–809.
293. Maetz HM, Sellers CA, Bailey WC, et al. Lymphocytic choriomeningitis from pet hamster exposure: a local public health experience. Am J Public Health. 1976;66:1082–1085.
294. Centers for Disease Control and Prevention. Notes from the field: lymphocytic choriomeningitis virus infections in employees of a rodent breeding facility—Indiana, May–June 2012. MMWR Morb Mortal Wkly Rep. 2012;61:622–623.
295. Fischer SA, Graham MB, Kuehnert MJ, et al. Human-to-human transmission can occur through transmission of lymphocytic choriomeningitis virus by organ transplantation. N Engl J Med. 2006;354:2235–2249.
296. Macneil A, Ströher U, Farnon E, et al. Solid organ transplant-associated lymphocytic choriomeningitis, United States, 2011. Emerg Infect Dis. 2012;18:1256–1262.
297. Bonthius DJ. Lymphocytic choriomeningitis virus: an underrecognized cause of neurologic disease in the fetus, child, and adult. Semin Pediatr Neurol. 2012;19:89–95.
298. Hirsch MS, Moellering RC Jr, Pope HG, et al. Lymphocytic-choriomeningitis-virus infection traced to a pet hamster. N Engl J Med. 1974;291:610–612.
299. Jahrling PB, Peters CJ. Lymphocytic choriomeningitis virus. A neglected pathogen of man. Arch Pathol Lab Med. 1992;116:486–488.
300. Johnson R, Milbourn PE. Central nervous system manifestations of chickenpox. Can Med Assoc J. 1970;102:831–834.
301. Leahy TR, Webb DWM, et al. Varicella zoster virus associated acute aseptic meningitis without exanthem in an immunocompetent 14-year-old boy. Pediatr Infect Dis J. 2008;27:362–363.
302. Silverstein A, Steinberg G, Nathanson M. Nervous system involvement in infectious mononucleosis. Arch Neurol. 1972;26:353–358.
303. Connelly KP, DeWitt LD. Neurologic complications of infectious mononucleosis. Pediatr Neurol. 1994;10:181–184.
304. Causey JQ. Spontaneous cytomegalovirus mononucleosis-like syndrome and aseptic meningitis. South Med J. 1976;69:1384–1387.
305. Ishiguro N, Yamada S, Takahashi T, et al. Meningo-encephalitis associated with HHV-6 related exanthem subitum. Acta Paediatr Scand. 1990;79:987–989.
306. Huang LM, Lee CY, Lee PI, et al. Meningitis caused by human herpesvirus-6. Arch Dis Child. 1991;66:1443–1444.
307. Yoshikawa T, Nakashima T, Suga S, et al. Human herpesvirus-6 DNA in cerebrospinal fluid of a child with exanthem subitum and meningoencephalitis. Pediatrics. 1992;89:888–890.
308. Yoshikawa T, Ihira M, Suzuki K, et al. Invasion by human herpesvirus 6 and human herpesvirus 7 of the central nervous system in patients with neurological signs and symptoms. Arch Dis Child. 2000;83:170–171.
309. Corey L, Adams HG, Brown ZA, et al. Genital herpes simplex virus infections: clinical manifestations, course, and complications. Ann Intern Med. 1983;98:958–972.
310. Bergström T, Vahlne A, Alestig K, et al. Primary and recurrent herpes simplex virus type 2-induced meningitis. J Infect Dis. 1990;162:322–330.
311. O’Sullivan CE, Aksamit AJ, Harrington JR, et al. Clinical spectrum and laboratory characteristics associated with detection of herpes simplex virus DNA in cerebrospinal fluid. Mayo Clin Proc. 2003;78: 1347–1352.
312. Yamamoto LJ, Tedder DG, Ashley R, et al. Herpes simplex virus type 1 DNA in cerebrospinal fluid of a patient with Mollaret’s meningitis. N Engl J Med. 1991;325:1082–1085.
313. Tedder DG, Ashley R, Tyler KL, et al. Herpes simplex virus infection as a cause of benign recurrent lymphocytic meningitis. Ann Intern Med. 1994;121:334–338.
314. Kupila L, Vainionpää R, Vuorinen T, et al. Recurrent lymphocytic meningitis: the role of herpesviruses. Arch Neurol. 2004;61:1553–1557.
315. Kumar S, Kumar S, Kohlhoff SA. Recurrent HSV-2 meningitis in a 9-year-old-girl. Scand J Infect Dis. 2006;38:570–572.
316. Jensenius M, Myrvang B, Størvold G, et al. Herpes simplex virus type 2 DNA detected in cerebrospinal fluid of 9 patients with Mollaret’s meningitis. Acta Neurol Scand. 1998;98:209–212.
317. Kallio-Laine K, Seppänen M, Kautiainen H, et al. Recurrent lymphocytic meningitis positive for herpes simplex virus type 2. Emerg Infect Dis. 2009;15:1119–1122.
318. Shalabi M, Whitley RJ. Recurrent benign lymphocytic meningitis. Clin Infect Dis. 2006;43:1194–1197.
319. Mayo DR, Booss J. Varicella zoster–associated neurologic disease without skin disease. Arch Neurol. 1989;46:313–315.
320. Pahud BA, Glaser CA, Dekker CL, et al. Varicella zoster disease of the central nervous system: epidemiological, clinical, and laboratory features 10 years after the introduction of the varicella vaccine. J Infect Dis. 2011;203:316–323.
321. Jhaveri R, Sankar R, Yazdani S, et al. Varicella-zoster virus: an overlooked cause of aseptic meningitis. Pediatr Infect Dis J. 2003;23:93–97.
322. Caserta MT, Hall CB, Schnabel K, et al. Neuroinvasion and persistence of human herpesvirus 6 in children. J Infect Dis. 1994;170:1586–1589.
323. Ho DD, Rota TR, Schooley RT, et al. Isolation of HTLV-III from cerebrospinal fluid and neural tissues of patients with neurologic syndromes related to the acquired immunodeficiency syndrome. N Engl J Med. 1985;313:1493–1497.
324. Ho DD, Sarngadharan MG, Resnick L, et al. Primary human T-lymphotropic virus type III infection. Ann Intern Med. 1985;103: 880–883.
325. Schacker T, Collier AC, Hughes J, et al. Clinical and epidemiologic features of primary HIV infection. Ann Intern Med. 1996;125:257–264.
326. Kelsey DS. Adenovirus meningoencephalitis. Pediatrics. 1978;61:291–293.
327. Davis D, Henslee PJ, Markesbery WR. Fatal adenovirus meningoencephalitis in a bone marrow transplant patient. Ann Neurol. 1988;23: 385–389.
328. Landry ML, Hsiung GD. Adenovirus-associated meningoencephalitis in a healthy adult. Ann Neurol. 1988;23:627–628.
329. Fianchi L, Scardocci A, Cattani P, et al. Adenovirus meningoencephalitis in a patient with large B-cell lymphoma. Ann Hematol. 2003;82: 313–315.
330. Dubberke ER, Tu B, Rivet DJ, et al. Acute meningoencephalitis caused by adenovirus serotype 26. J Neurovirol. 2006;12:235–240.
331. Paisley JW, Bruhn FW, Lauer BA, et al. Type A2 influenza viral infections in children. Am J Dis Child. 1978;132:34–36.
332. Drago L, Attia MW, DePiero A, et al. Influenza A-associated meningoencephalitis. Pediatr Emerg Care. 2005;21:437–439.
333. Kwon S, Kim S, Cho MH, et al. Neurologic complications and outcomes of pandemic (H1N1) 2009 in Korean children. J Korean Med Sci. 2012;27:402–407.
334. Douvoyiannis M, Litman N, Goldman DL. Neurologic manifestations associated with parvovirus B19 infection. Clin Infect Dis. 2009;48: 1713–1723.
335. Wong CJ, Price Z, Bruckner DA. Aseptic meningitis in an infant with rotavirus gastroenteritis. Pediatr Infect Dis J. 1984;3:244–246.
336. Okumura A, Ichikawa T. Aseptic meningitis caused by human parvovirus B19. Arch Dis Child. 1993;68:784–785.
337. Furuya Y, Katayama T, Miyahara K, et al. Detection of the rotavirus A genome from the cerebrospinal fluid of a gastroenteritis patient: a case report. Jpn J Infect Dis. 2007;60:148–149.
338. Centers for Disease Control and Prevention. 2010 Map—lyme disease. http://www.cdc.gov/lyme/stats/maps/map2010.html. Accessed January 15, 2013.
339. Steere AC. Lyme disease. N Engl J Med. 2001;345:115–125.
340. Wormser R, Dattwyler R, Shapiro E, et al. The clinical assessment, treatment, and prevention of Lyme disease, human granulocytic anaplasmosis, and babesiosis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2006;43:1089–1134.
341. Halperin JJ. Nervous system Lyme disease. Infect Dis Clin North Am. 2008;22:261–274.
342. Pachner AR, Steere AC. The triad of neurologic manifestations of Lyme disease: meningitis, cranial neuritis, and radiculoneuritis. Neurology. 1985;35:47–53.
343. Feder HM. Lyme disease in children. Infect Dis Clin North Am. 2008;22:315–326.
344. Lecour H, Miranda M, Magro C, et al. Human leptospirosis—a review of 50 cases. Infection. 1989;17:10–14.
345. Merrit HH, Moore M. Acute neurosyphilitic meningitis. Medicine. 1935;14:119.
346. Cadavid D, Barbour AG. Neuroborreliosis during relapsing fever: review of the clinical manifestations, pathology, and treatment of infections in humans and experimental animals. Clin Infect Dis. 1998;26:151–164.
347. Ussery XT, Valway SE, McKenna M, et al. Epidemiology of tuberculosis among children in the United States: 1985 to 1994. Pediatr Infect Dis J. 1996;15:697–704.
348. Starke JR. Tuberculosis of the central nervous system in children. Semin Pediatr Neurol. 1999;6:318–331.
349. Bouza E, Garcia de la Torre M, Parras F, et al. Brucellar meningitis. Rev Infect Dis. 1987;9:810–822.
350. Wheat LJ, Batteiger BE, Sathapatayavongs B. Histoplasma capsulatum infections of the central nervous system. A clinical review. Medicine. 1990;69:244–260.
351. Friedman JA, Wijdicks EF, Fulgham JR, et al. Meningoencephalitis due to Blastomyces dermatitidis: case report and literature review. Mayo Clin Proc. 2000;75:403–408.
352. Bicanic T, Harrison TS. Cryptococcal meningitis. Br Med Bull. 2005;72:99–118.
353. Wheat LJ, Musial CE, Jenny-Avital E. Diagnosis and management of central nervous system histoplasmosis. Clin Infect Dis. 2005;40:844–852.
354. Johnson RH, Einstein HE. Coccidioidal meningitis. Clin Infect Dis. 2006;42:103–107.
355. Antinori S, Corbellino M, Meroni L, et al. Aspergillus meningitis: a rare clinical manifestation of central nervous system aspergillosis. Case report and review of 92 cases. J Infect. 2013;66:218–238.
356. Lind K, Zoffmann H, Larsen SO, et al. Mycoplasma pneumoniae infection associated with infection of the central nervous system. Acta Med Scand. 1979;205:325–332.
357. Narita M, Itakura O, Matsuzono Y, et al. Analysis of mycoplasmal central nervous system involvement by polymerase chain reaction. Pediatr Infect Dis J. 1995;14:236–237.
358. Roe O, Jorgen D, Matre R. Isolation of Mycoplasma hominis from cerebrospinal fluid. Scand J Infect Dis. 1973;5:285–288.
359. Holman RC, Belay ED, Christensen KY, et al. Hospitalizations for Kawasaki syndrome among children in the United States, 1997–2007. Pediatr Infect Dis J. 2010;29:483–488.
360. Shimura T, Arima H. Clinical features of aseptic meningitis in Kawasaki disease. J Jpn Pediatr. 1978;31:789–792.
361. Watanabe J, Kawasaki T, Takemura T. Cytologic observation of spinal fluid in Kawasaki disease. J Jpn Pediatr. 1980;84:1259–1263.
362. Fishman RA. CSF findings in diseases of the nervous system. In: Fishman RA, ed. Cerebrospinal Fluid in Diseases of the Nervous System. 2nd ed. Philadelphia: Saunders; 1992:277–287.
363. Johnson RT, Richardson EP. The neurological manifestations of systemic lupus erythematosus. Medicine. 1968;47:337–369.
364. Hopkins S, Jolles S. Drug-induced aseptic meningitis. Expert Opin Drug Saf. 2005;4:285–297.
365. Kluger N, Girard C, Gonzalez V, et al. Efalizumab-induced aseptic meningitis. Br J Dermatol. 2007;156:189–191.
366. Callen EC, Church CO, Patel M, et al. Aseptic meningitis associated with chronic sulindac use for osteoarthritis: a case report. Rheumatol Int. 2008;28:391–393.
367. Nagovskiy N, Agarwal M, Allerton J. Cetuximab-induced aseptic meningitis. J Thorac Oncol. 2010;5:751.
368. Jazeron A, Lallier JC, Rihn B, et al. Aseptic meningitis possibly induced by adalimumab. Joint Bone Spine. 2010;77:618–619.
369. Imataka G, Nakagawa E, Yamanouchi H, et al. Drug-indiced aseptic meningitis: development of subacute sclerosing panencephalitis following repeated intraventricular infusion therapy with interferon alpha/beta. Cell Biochem Biophys. 2011;61:699–701.
370. Shah BK, O’Keefe S. Pemetrexed induced aseptic meningitis. Acta Oncol. 2012;51:399–400.
371. Simms KM, Kortepeter C, Avigan M. Lamotrigine and aseptic meningitis. Neurology. 2012;78:921–927.
372. Galindo Bonilla PA, Sánchez Rodríguez N, Castro Jiménez A, et al. Aseptic meningitis induced by vitamin B complex. J Investig Allergol Clin Immunol. 2012;22:225–226.
373. Bonnet MC, Dutta A, Weinberger C, et al. Mumps vaccine strains and aseptic meningitis. Vaccine. 2006;24:7037–7045.