Time Recommended to Complete: 3 days
Frederick S. Southwick, M.D.
GUIDING QUESTIONS
1. Are we at the end of the antibiotic era?
2. Why are “superbugs” suddenly appearing in our hospitals?
3. How do bacteria become resistant to antibiotics?
4. How can the continued selection of highly resistant organisms be prevented?
5. Is antibiotic treatment always the wisest course of action?
6. Does one antibiotic cure all infections?
7. What are the strategies that underlie optimal antibiotic usage?
8. How is colonization distinguished from infection, and why is this distinction important?
Despite dire warnings in the 1990s that we were approaching the end of the antibiotic era, the incidence of antibiotic-resistant bacteria continues to rise. The proportions of penicillin-resistant Streptococcus pneumoniae, hospital-acquired methicillin-resistant Staphylococcus aureus (MRSA), and vancomycin-resistant Enterococcus (VRE) strains continue to steadily increase in many hospitals. Community-acquired MRSA (cMRSA) has spread throughout the world. Multiresistant Acinetobacter and Pseudomonas are everyday realities in most of our hospitals. In the past, we could depend on the pharmaceutical industry to develop new anti-infective agents to overcome these highly resistant bacteria. However, these companies are no longer investing in the development of anti-infective medications because of the high cost of development and limited profits. As never before, it is critical that health care providers understand the principles of proper anti-infective therapy and use anti-infective agents judiciously. These agents need to be reserved for treatable infections—not used to calm the patient or the patient’s family. Too often caregivers treat patients with antibiotics at the first sign of fever, and despite evidence suggesting a viral infection and negative bacterial cultures they continue this treatment for prolonged periods.
Physicians unschooled in the principles of microbiology utilize anti-infective agents just as they would more conventional medications, such as anti-inflammatory agents, antihypertensive medications, and cardiac drugs. They use one or two broad-spectrum antibiotics to treat all patients with suspected infections, and fail to consult an expert in infectious disease or utilize well-established guidelines to assist in the proper management of anti-infective therapy.
Many excellent broad-spectrum antibiotics can effectively treat most bacterial infections without requiring a specific causative diagnosis. However, overuse of empiric broad-spectrum antibiotics has resulted in the selection of highly resistant pathogens. A simplistic approach to anti-infective therapy and establishment of a fixed series of simple rules concerning the use of these agents is unwise and has proved harmful to patients. Such an approach ignores the remarkable adaptability of bacteria, fungi, and viruses. It is no coincidence that these more primitive life forms have survived for millions of years, far longer than the human race.
The rules for the use of anti-infective therapy are dynamic and must take into account the ability of these pathogens to adapt to the selective pressures exerted by the overuse of antibiotic, antifungal, and antiviral agents. The days of the “shotgun” approach to infectious diseases must end, or more and more patients will become infected with multiresistant organisms that cannot be treated. Many hospitals are turning to antibiotic stewardship programs that limit the access to costly broad-spectrum antibiotics. Only through the judicious use of anti-infective therapy combined with infection control measures we can hope to slow the arrival of the end of the antibiotic era.
KEY POINTS
About Anti-Infective Therapy
1. Too often, antibiotics are prescribed to fulfill the patient’s expectations, rather than to treat a true bacterial infection.
2. A single antibiotic cannot meet all infectious disease needs.
3. Physicians ignore the remarkable adaptability of bacteria, fungi, and viruses at their patient’s peril.
4. Anti-infective therapy is dynamic and requires a basic understanding of microbiology.
5. The “shotgun” approach to infectious diseases must end, or we may truly experience the end of the antibiotic era.
ANTIBIOTIC RESISTANCE
GENETIC MODIFICATIONS LEADING TO ANTIMICROBIAL RESISTANCE
To understand why antibiotics must be used judiciously, the physician needs to understand how bacteria are able to adapt to their environment. Point mutations can develop in the DNA of bacteria as they replicate. These mutations occur in the natural environment, but are of no survival advantage unless the bacteria are placed under selective pressures. In the case of a mutation that renders a bacterium resistant to a specific antibiotic, exposure to the specific antibiotic allows the bacterial clone that possesses the antibiotic resistance mutation to grow, while bacteria without the mutation die and no longer compete for nutrients. Thus, the resistant strain becomes the dominant bacterial flora. In addition to point mutations, bacteria can also use three major mechanisms to transfer genetic material among themselves:
1. Conjugation. Bacteria often contain circular, double-stranded DNA structures called plasmids. These circular DNA structures lie outside the bacterial genome (Figure 1.1). Plasmids often carry resistance (“R”) genes. Through a mechanism called “conjugation,” plasmids can be transferred from one bacterium to another. The plasmid encodes for the formation of a pilus on the donor bacteria’s outer surface. The pilus attaches to a second bacterium and serves as bridge for the transfer of the plasmid DNA from the donor to the recipient bacterium. Using this mechanism, a single resistant bacterium can transfer resistance to other bacteria.

Figure 1.1. Mechanisms by which bacteria transfer antibiotic resistance genes.
2. Transduction. Bacteriophages are protein-coated DNA segments that attach to the bacterial wall and inject DNA in a process called “transduction.” These infective particles can readily transfer resistance genes to multiple bacteria.
3. Transformation. Donor bacteria can also release linear segments of chromosomal DNA, which is then taken up by recipient bacteria and incorporated into the recipient’s genome. This process is called “transformation,” and the naked DNA capable of incorporating into the genome of recipient bacteria is called a transposon (Figure 1.1). Natural transformation most commonly occurs in Streptococcus, Haemophilus, and Neisseria species. Transposons can transfer multiple antibiotic resistance genes in a single event and have been shown to be responsible for high-level vancomycin resistance in enterococci.
Thus, bacteria possess multiple ways to transfer their DNA, and they promiscuously share genetic information. Virtually any part of a bacterium’s genome can be transferred, and this promiscuity provides a survival advantage, allowing bacteria to quickly adapt to their environment.
KEY POINTS
About Antibiotic Resistance
1. Bacteria can quickly alter their genetic makeup by
a) point mutation.
b) transfer of DNA by plasmid conjugation.
c) transfer of DNA by bacteriophage transduction.
d) transfer of naked DNA by transposon transformation.
2. The ability of bacteria to share DNA provides a survival advantage, allowing them to quickly adapt to antibiotic exposure.
3. Biochemical alterations leading to antibiotic resistance include
a) degradation or modification of the antibiotic.
b) reduction in the bacterial antibiotic concentration by inhibiting entry or by efflux pumps.
c) modification of the antibiotic target.
4. Under the selection pressure of antibiotics, the question is not whether, but when resistant bacteria will take over.
BIOCHEMICAL MECHANISMS FOR ANTIMICROBIAL RESISTANCE
What are some of the proteins that these resistant genes encode for, and how do they work?
The mechanisms by which bacteria resist antibiotics can be classified into three major groups:
• Degradation or modification of the antibiotic
• Reduction in the bacterial antibiotic concentration
• Modification of the antibiotic target
Degradation or Modification of the Antibiotic
β-LACTAMASES
Many bacteria synthesize one or more enzymes called β-lactamases that inactivate antibiotics by breaking the amide bond on the β-lactam ring. Transfer of β-lactamase activity occurs primarily through plasmids and transposons.
Twenty-four classes of β-lactamases and over 900 individual enzymes have been described. Some preferentially break down penicillins (e.g., TEM-1 in Escherichia coli, and SHV-1 for Klebsiella); others preferentially destroy specific cephalosporins or carbenicillin. Extended-spectrum β-lactamases (ESBL, example: SHV-2) readily destroy most cephalosporins, but are susceptible to β-lactamase inhibitors such as clavulanate. Another class of β-lactamase is resistant to clavulanate (CTX-M family). Some bacteria are able to produce β-lactamases called carbapenemases that inactivate the carbapenems (e.g., Klebsiella-producing carbapenemase, KPC, Oxa-type enzymes produced by Acinetobacter).
Gram-negative bacilli produce a broader spectrum of β-lactamases than do gram-positive organisms, and therefore infections with gram-negative organisms more commonly arise in patients treated for prolonged periods with broad-spectrum antibiotics. In some instances, β-lactamase activity is low before the bacterium is exposed to antibiotics; however, following exposure, β-lactamase activity is induced. Enterobacter is a prime example. This gram-negative bacterium may appear sensitive to cephalosporins on initial testing. Following cephalosporin treatment, β-lactamase activity increases, resistance develops, and the patient’s infection relapses. For this reason, third-generation cephalosporins are not recommended for serious Enterobacter infections.
OTHER ENZYME MODIFICATIONS OF ANTIBIOTICS
Erythromycin is readily inactivated by an esterase that hydrolyzes the lactone ring of the antibiotic. This esterase has been identified in E. coli. Other plasmid-mediated erythromycin inactivating enzymes have been discovered in Streptococcus species and S. aureus. Chloramphenicol is inactivated by chloramphenicol acetyltransferase, which has been isolated from both gram-positive and gramnegative bacteria. Similarly, aminoglycosides can be inactivated by acetyltransferases. Bacteria also inactivate this class of antibiotics by phosphorylation and adenylation.
These resistance enzymes are found in many gramnegative strains and are increasingly detected in enterococci, S. aureus and S. epidermidis.
Reduction in the Bacterial Antibiotic Concentration
INTERFERENCE WITH ANTIBIOTIC ENTRY
For an antibiotic to work, it must be able to penetrate the bacterium and reach its biochemical target. Gram-negative bacteria contain an outer lipid coat that impedes penetration by hydrophobic reagents (such as most antibiotics). The passage of hydrophobic antibiotics is facilitated by the presence of porins—small channels in the cell walls of gram-negative bacteria that allow the passage of charged molecules. Mutations leading to the loss of porins can reduce antibiotic penetration and lead to antibiotic resistance. Following prolonged exposure to vancomycin, MRSA can develop a thickened cell wall requiring higher vancomycin concentrations to inhibit bacterial growth (vancomycin intermediate S. aureus, VISA).
PRODUCTION OF EFFLUX PUMPS
Transposons have been found that encode for an energy-dependent pump that can actively pump tetracycline out of bacteria. Active efflux of antibiotics has been observed in many enteric gram-negative bacteria, and this mechanism is used to resist tetracycline, macrolide, aminoglycosides, and fluoroquinolone antibiotic treatment (e.g., MexXY). S. aureus, S. epidermidis, S. pyogenes, group B streptococci, and S. pneumoniae also can utilize energy-dependent efflux pumps to resist antibiotics.
Modification of the Antibiotic Target
ALTERATIONS OF CELL WALL PRECURSORS
Alteration of cell wall precursors is the basis for VRE. Vancomycin and teicoplanin binding requires that D-alanine-D-alanine be at the end of the peptidoglycan cell wall precursors of gram-positive bacteria. Resistant strains are found predominantly in Enterococcus faecium and less commonly in Enterococcus faecalis contain the vanA or vanB transposon that encodes a protein that synthesizes D-alanine-D-lactate instead of D-alanine-D-alanine at the end of the peptidoglycan precursor. Loss of the terminal D-alanine markedly reduces vancomycin and teicoplanin binding, allowing the mutant bacterium to survive and grow in the presence of these antibiotics. Fortunately, the transfer of these transposons to S. aureus is exceedingly rare.
CHANGES IN TARGET ENZYMES
Penicillins and cephalosporins bind to specific proteins called penicillin-binding proteins (PBPs) in the bacterial cell wall. Penicillin-resistant S. pneumoniae demonstrate decreased numbers of PBPs or PBPs that bind penicillin with lower affinity, or both. Decreased penicillin binding reduces the ability of the antibiotic to kill the targeted bacteria.
The basis for antibiotic resistance in MRSA is production of a low-affinity PBP encoded by the mecA gene. Mutations in the target enzymes dihydropteroate synthetase and dihydrofolate reductase respectively cause sulfonamide and trimethoprim resistance. Single amino-acid mutations that alter DNA gyrase function can result in resistance to fluoroquinolones.
ALTERATIONS IN RIBOSOMAL BINDING SITE
Tetracyclines, macrolides, lincosamides, and aminoglycosides all act by binding to and disrupting the function of bacterial ribosomes (see the descriptions of individual antibiotics later in this chapter). A number of resistance genes encode for enzymes that demethylate adenine residues on bacterial ribosomal RNA, inhibiting antibiotic binding to the ribosome. Ribosomal resistance to gentamicin, tobramycin, and amikacin is less common because these aminoglycosides have several binding sites on the bacterial ribosome and require multiple bacterial mutations before their binding is blocked.
CONCLUSIONS
Bacteria can readily transfer antibiotic-resistance genes. Bacteria have multiple mechanisms to destroy antibiotics, lower the antibiotic concentration, and interfere with antibiotic binding. Under the selective pressures of prolonged antibiotic treatment, the question is not whether, but when resistant bacteria will take over.
ANTI-INFECTIVE AGENT DOSING
The characteristics that need to be considered when administering antibiotics include absorption (when dealing with oral antibiotics), volume of distribution, metabolism, and excretion. These factors determine the dose of each drug and the time interval of administration. To effectively clear a bacterial infection, depending on the class of antibiotics, serum levels of the antibiotic need to be maintained above the minimum inhibitory concentration (MIC) for a significant period. For each pathogen, the MIC is determined by serially diluting the antibiotic into liquid medium containing 104 bacteria per milliliter. Inoculated tubes are incubated overnight until broth without added antibiotic has become cloudy or turbid as a result of bacterial growth. The lowest concentration of antibiotic that prevents active bacterial growth—that is, the liquid media remains clear—constitutes the MIC (Figure 1.2). Automated analyzers can now quickly determine, for individual pathogens, the MICs for multiple antibiotics, and these data serve to guide the physician’s choice of antibiotics.

Figure 1.2. Understanding the minimum inhibitory concentration and the minimal bactericidal concentration.
Clinical laboratories utilize MIC combined with studies examining achievable antibiotic levels (pharmacokinetics and pharmacodynamics, see below) in humans to determine whether an organism is sensitive, intermediate, or resistant to a specific antibiotic. This value is called the breakpoint or cutoff, and is the concentration (MIC) above which there is a high likelihood of treatment success, and below which there is considerable risk of failure. At the present time, different countries and different organizations utilize different criteria to determine breakpoints, and experts strongly recommend the acceptance of an international standard for calculating breakpoints.
The mean bactericidal concentration (MBC) is determined by taking each clear tube and inoculating a plate of solid medium with the solution. Plates are then incubated to allow colonies to form. The lowest concentration of antibiotic that blocks all growth of bacteria—that is, no colonies on solid medium—represents the MBC. Because this method is technically cumbersome, this value is now rarely determined.
Successful cure of an infection depends on multiple host factors in addition to serum antibiotic concentration. However, investigators have attempted to predict successful treatment by plotting serum antibiotic levels against time. Two parameters have found to correlate with cure in both animal and human studies (Figure 1.3): time above the MIC (T>MIC), and the ratio of the area under the curve (AUC) to the MIC AUC/MIC).
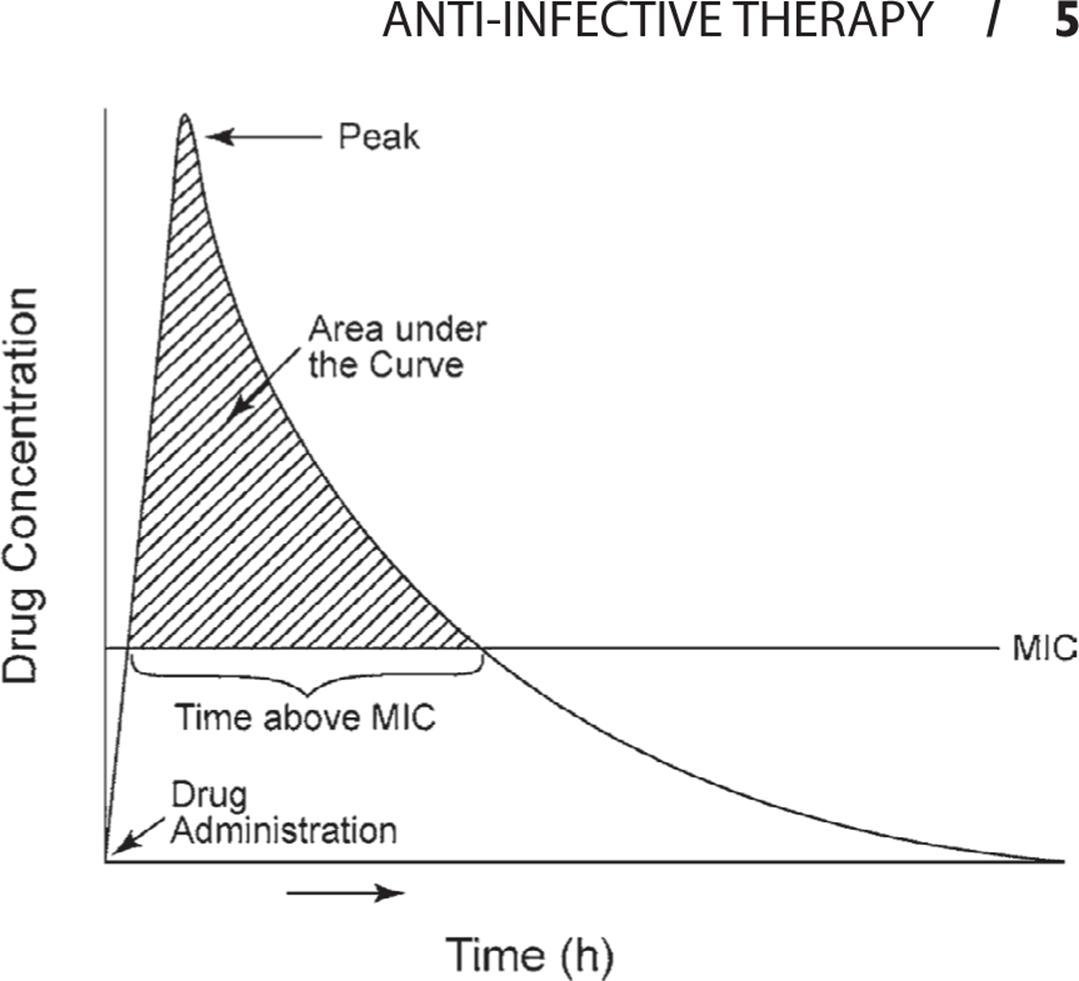
Figure 1.3. Pharmacokinetics of a typical antibiotic.
Cure rates for β-lactam antibiotics are maximized by maintaining serum levels above the MIC for >50% of the time. Peak antibiotic concentrations are of less importance for these antibiotics, and serum concentrations above eight times the MIC are of no benefit other than to enhance penetration into less permeable body sites.
Unlike β-lactam antibiotics, aminoglycosides and fluoroquinolones demonstrate concentration-dependent killing. In vitro studies show that these antibiotics demonstrate greater killing the higher their concentrations exceed the MIC. High peak levels of these antibiotics are more effective than low peak levels at curing infections. Therefore, for treatment with aminoglycosides and fluoroquinolones AUC/MIC is most helpful for maximizing effectiveness. For fluoroquinolones, best outcomes in community-acquired pneumonia may be achieved when the AUC/MIC is >34.
Maintenance of a high AUC/MIC has recently been shown to be a critical factor for preventing the development of antibiotic resistance particularly in Pseudomonas aeruginosa and other nonfermenting gram-negative bacteria (Acinetobacter baumannii, Stenotrophomonas maltophilia, and Burkholderia cepacia). For P. aeruginosa, an AUC/MIC of approximately 200 is required. To prevent the development of fluoroquinolone resistance to S. pneumoniae,in vitro studies have suggested that AUC/MIC should be >50.
KEY POINTS
About Antibiotic Dosing
1. Absorption, volume of distribution, metabolism, and excretion all affect serum antibiotic levels.
2. Mean inhibitory concentration (MIC) is helpful in guiding antibiotic choice.
3. To maximize success with β-lactam antibiotics, serum antibiotic levels should be above the MIC for at least 50% of the time (T > MIC > 50%).
4. To maximize success with aminoglycosides and fluoroquinolones, high-peak concentration and high AUC/MIC ratio are recommended.
5. Development of resistance can be prevented by
a) administering sufficiently high doses of antibiotics to achieve very high AUC/MIC ratios, 50–200 depending on the organism.
b) short courses of antibiotic, ideally 5 days or less.
In nature, intrinsic resistance is found in 1 out of every 106 organisms; therefore, the likelihood of selecting for a resistant pathogen also depends on the concentration of bacteria in the infected organ. In pneumonia and intra-abdominal infections, bacterial counts are often $109; therefore, achieving a high AUC/MIC is most important for these infections. In patients with sepsis as well as for infections caused by Pseudomonas, many experts recommend utilizing two antibiotics (double coverage) in order to increase the likelihood of killing the resistant bacterial population.
A third factor that increases the likelihood of resistant is the duration of exposure to an anti-infective agent. The number of resistant bacteria remains low early in the course of antibiotic treatment; however, when the AUC/MIC is insufficiently high within 4-5 days resistant bacteria begin to increase in concentration. The longer the exposure, the greater the likelihood resistant bacteria will predominate. Many experts now agree that from the standpoint of resistance, antibiotic regimens of 5 days or less would be ideal. In the normal host, neutrophils work in concert with antibiotics to kill infecting organisms. And when the concentration of organisms drops to 102-103/g of tissue, neutrophils alone are capable of eradicating the infection. In many instances, 5 days of antibiotic treatment will reduce bacterial concentrations to this level allowing neutrophils to clean up the remaining pathogenic bacteria.
BASIC STRATEGIES FOR ANTIBIOTIC THERAPY
The choice of antibiotics should be carefully considered. A step-by-step logical approach is helpful (Figure 1.4). Given the complexity of these decisions, to assure that each of these factors is considered a mandatory check list for the treatment of severely ill hospitalized patients promises to increase survival and reduce antibiotic resistance (Figure 1.5).
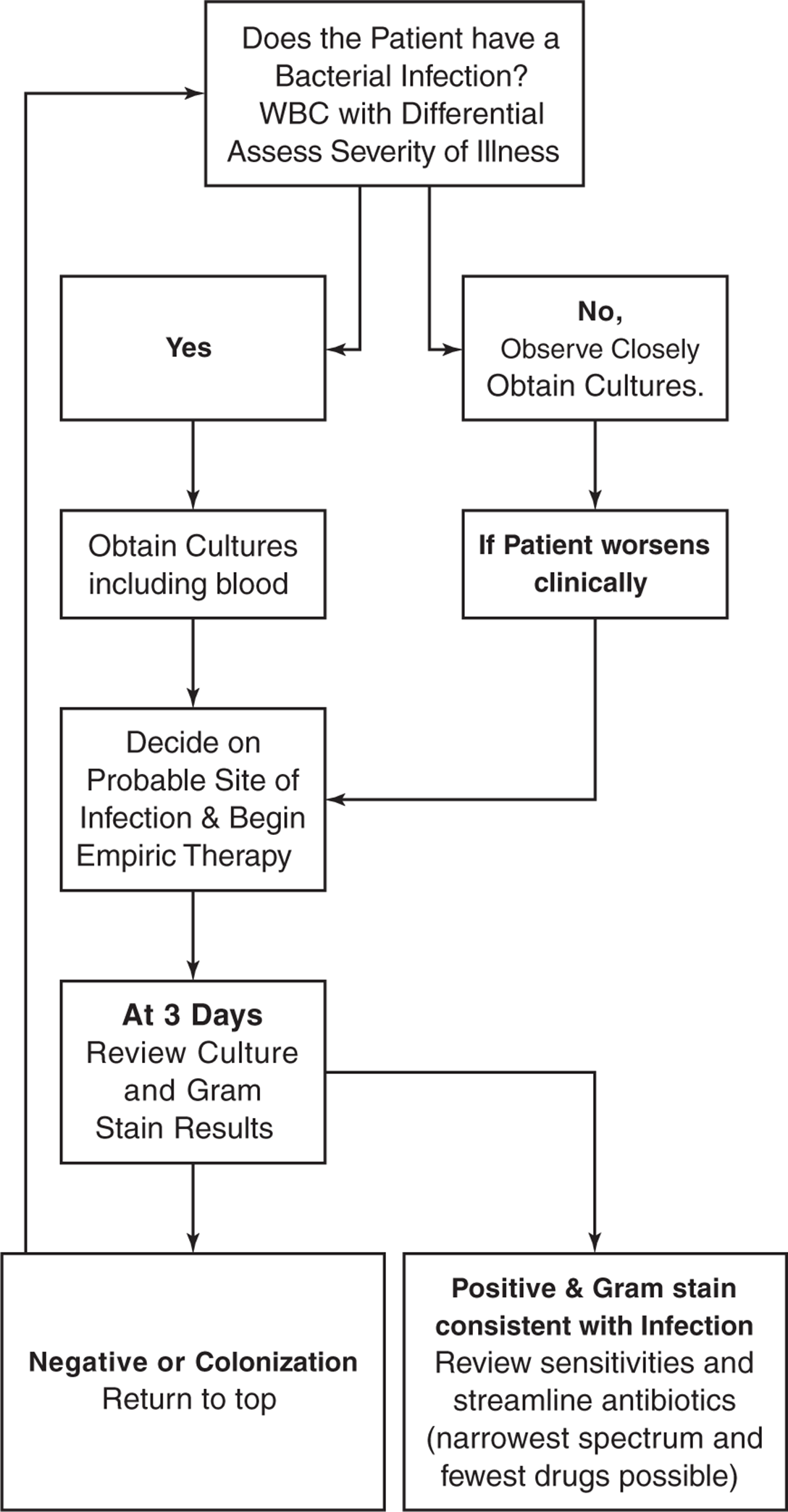
Figure 1.4. Algorithm for the initial use of anti-infective therapy.

Figure 1.5. Antibiotic checklist.
1. Decide Whether the Patient Has a Bacterial Infection
One test that has traditionally been used to differentiate an acute systemic bacterial infection from a viral illness is the peripheral white blood cell (WBC) count. In patients with serious systemic bacterial infections, the peripheral WBC count may be elevated and may demonstrate an increased percentage of neutrophils. On occasion, less mature neutrophils such as band forms and, less commonly, metamyelocytes are observed on peripheral blood smear. Most viral infections fail to induce a neutrophil response. Viral infections, particularly Epstein–Barr virus, induce an increase in lymphocytes or monocytes (or both) and may induce the formation of atypical monocytes. Unfortunately, the peripheral WBC count is only a rough guideline, lacking both sensitivity and specificity. Recently, serum procalcitonin concentration has been found to be a far more accurate test for differentiating bacterial from viral infection. In response to bacterial infection, this precursor of calcitonin is synthesized and released into the serum by many organs of the body; production of interferon (IFN) in response to viral infection inhibits its synthesis. The serum procalcitonin test may also be of prognostic value, serum procalcitonin levels being particularly high in severe sepsis (see Chapter 2).
2. Make a Reasonable Statistical Guess as to the Possible Pathogens
Based on the patient’s symptoms and signs, as well as on laboratory tests, the anatomic site of the possible infection can often be determined. For example, burning on urination, associated with pyuria on urinalysis, suggests a urinary tract infection. The organisms that cause uncomplicated urinary tract infection usually arise from the bowel flora. They include E. coli, Klebsiella, and Proteus. Antibiotic treatment needs to cover these potential pathogens. Later chapters review the pathogens commonly associated with infections at specific anatomic sites and the recommended antibiotic coverage for those pathogens. These recommendations are based on the Infectious Diseases Society of America (IDSA) treatment guidelines, and the IDSA treatment guidelineshttp://www.idsociety.org/IDSA_Practice_Guidelines/) should always be consulted to assure that patients receive the most up-to-date treatment. Renowned experts in the field of infectious diseases created these guidelines based on careful scrutiny of current clinical and biomedical research.
3. Be aware of the Antibiotic Susceptibility Patterns in Your Hospital and Community
In patients who develop infection while in hospital (“nosocomial infection), empiric therapy needs to take into account the antibiotic susceptibility patterns of the flora associated with the hospital and the floor where the patient became ill. Many hospitals have a high incidence of MRSA, and therefore empiric antibiotic treatment of a possible staphylococcal infection must include vancomycin, pending culture results. Other hospitals have a large percentage of Pseudomonas strains that are resistant to gentamicin, eliminating that antibiotic from consideration as empiric treatment of possible gram-negative sepsis. In many communities, individuals who have never been hospitalized are today presenting with soft tissue infections caused by cMRSA, and physicians in these communities must adjust their empiric antibiotic selection (see Chapter 10).
4. Take into Account Previous Antibiotic Treatment
The remarkable adaptability of bacteria makes it highly likely that a new pathogen will be resistant to previously administered antibiotics. If the onset of the new infection was preceded by a significant interval when antibiotics were not given, the resident flora may have recolonized with less resistant flora. However, the reestablishment of normal flora can take weeks, and patients in hospital are likely to recolonize with highly resistant hospital flora.
5. Take into Consideration Important Host Factors
a. Peripheral WBC count. Patients with neutropenia have a high mortality rate from sepsis. Immediate broad-spectrum, high-dose intravenous antibiotic treatment is recommended as empiric therapy for these patients.
b. Age. Elderly patients tend to metabolize and excrete antibiotics more slowly; longer dosing intervals are therefore often required. Agents with significant toxicity (such as aminoglycosides) should generally be avoided in elderly patients because they exhibit greater toxicity.
c. Hepatic and renal dysfunction. Antibiotics metabolized primarily by the liver should generally be avoided or reduced in patients with significant cirrhosis. In patients with significant renal dysfunction, antibiotic doses need to be modified.
d. Duration of hospitalization. Patients who have just arrived in the hospital tend to be colonized with community-acquired pathogens; patients who have been in the hospital for prolonged periods and have received several courses of antibiotics tend to be colonized with highly resistant bacteria and with fungi.
e. Severity of the patient’s illness. The severely ill patient who is toxic and hypotensive requires broad-spectrum antibiotics; the patient who simply has a new fever without other serious systemic complaints can usually be observed off antibiotics.
6. Switch to Narrower-Spectrum Antibiotic Coverage Within 3 Days
(Table 1.1, Figure 1.6). Within 3-4 days following the administration of antibiotics, sequential cultures of mouth flora reveal that the numbers and types of bacteria begin to change significantly. The normal flora die, and resistant gram-negative rods, gram-positive cocci, and fungi begin to predominate. The more quickly the selective pressures of broad-spectrum antibiotic coverage can be discontinued, the lower the risk of selecting for highly resistant pathogens. Broad coverage is reasonable as initial empiric therapy until cultures are available. By the third day, the microbiology laboratory can generally identify the pathogen or pathogens, and a narrower-spectrum-specific antibiotic regimen can be initiated. Despite the availability of culture results, clinicians too often continue the same empiric broad-spectrum antibiotic regimen, and that behavior is a critical factor in explaining subsequent infections with highly resistant superbugs. Figure 1.6 graphically illustrates the spectrum of available antibiotics as a guide to the antibiotic choice.
Table 1.1. Classification of Antibiotics by Spectrum of Activity
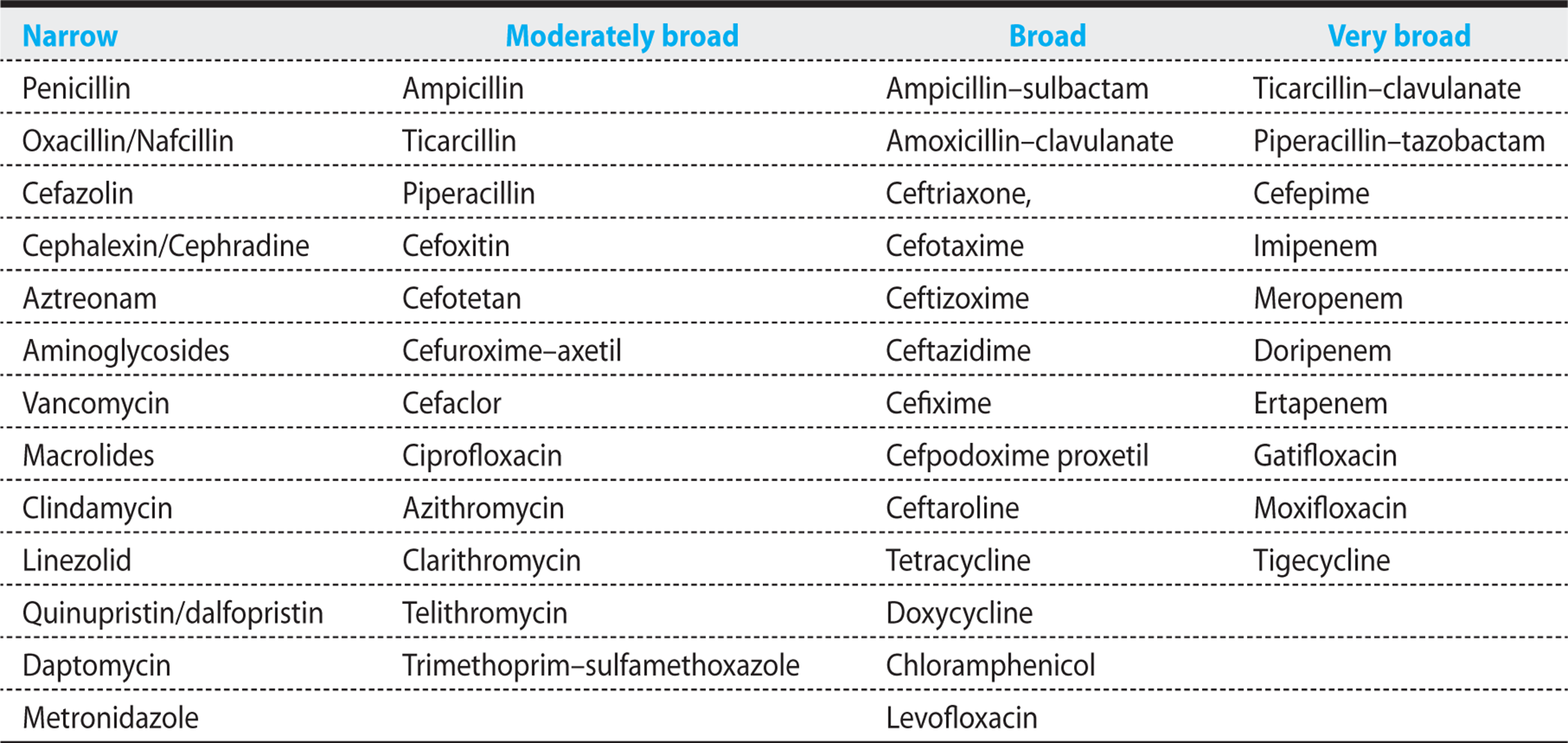

Figure 1.6. Antibiogram of all major antibiotics.
Obey the 3-day rule. Continuing broad-spectrum antibiotics beyond 3 days drastically alters the host’s resident flora and selects for resistant organisms. After 3 days, streamline antibiotic coverage. Use narrower-spectrum antibiotics to treat the specific pathogens identified by culture and Gram stain.
7. All Else Being Equal, Choose the Least Expensive Drug
As is discussed in later chapters, more than one antibiotic regimen can often be used to successfully treat a specific infection. Given the strong economic forces driving medicine today, the physician needs to consider the cost of therapy whenever possible. Too often, new, more expensive antibiotics are chosen over older generic antibiotics that are equally effective. In this book, the review of specific antibiotics is accompanied by cost range estimates to assist the clinician in making cost-effective decisions.
However, in assessing cost, factoring in toxicity is also important. For example, the acquisition cost of gentamicin is low, but when blood-level monitoring, the requirement to closely follow blood urea nitrogen and serum creatinine, and the potential for an extended hospital stay because of nephrotoxicity are factored into the cost equation, gentamicin is often not cost-effective.
KEY POINTS
About the Steps Required to Design an Antibiotic Regimen
1. Assess the probability of bacterial infection. (Antibiotics should be avoided in viral infections.)
2. Be familiar with the pathogens primarily responsible for infection at each anatomic site and use the IDSA guidelines.
3. Be familiar with the bacterial flora in the local hospital and community.
4. Take into account previous antibiotic treatment.
5. Take into account the specific host factors:
a) Immune status
b) Age
c) Hepatic and renal function
d) Duration of hospitalization
e) Severity of illness.
6. Switch to a narrower-spectrum antibiotic regimen based on culture results within 3 days.
7. Take into account acquisition cost and the costs of toxicity.
COLONIZATION VERSUS INFECTION
CASE 1.1
Following a motor vehicle accident, a 40-year-old man was admitted to the intensive care unit with four fractured ribs and a severe lung contusion on the right side. Chest X-ray (CXR) demonstrated an infiltrate in the right lower lobe. Because of depressed mental status, this man required respiratory support.
Initially, Gram stain of the sputum demonstrated few polymorphonuclear leukocytes (PMNs) and no organisms. On the third hospital day, this patient developed a fever to 103°F (39.5°C), and his peripheral WBC increased to 17,500 from 8000 (80% PMNs, 15% band forms). A new CXR demonstrated extension of the right lower lobe infiltrate. Gram stain of sputum revealed abundant PMNs and 20-30 gram-positive cocci in clusters per high-power field. His sputum culture grew methicillin-sensitive S. aureus. Intravenous cefazolin (1.5 g every 8 hours) was initiated. He defervesced, and secretions from his endotracheal tube decreased over the next 3 days. On the fourth day, a repeat sputum sample was obtained. Gram stain revealed a moderate number of PMNs and no organisms; however, culture grew Candida albicans, a yeast that was resistant to cefazolin. The physician added fluconazole (400 mg/day) to the anti-infective regimen).
Case 1.1 represents a very typical example of how antibiotics are misused. The initial therapy for a probable early S. aureus pneumonia was appropriate, and the patient responded (fever resolved, sputum production decreased, gram-positive cocci disappeared from the Gram stain, and S. aureus no longer grew on culture). However, because the sputum culture was positive for Candida albicans, the physician added an antifungal agent, fluconazole. The correct decision should have been to continue cefazolin alone.
One of the most difficult and confusing issues for many physicians is the interpretation of culture results. Wound cultures and sputum cultures are often misinterpreted. Once a patient has been started on an antibiotic, the bacterial flora on the skin and in the mouth and sputum will change. Often, these new organisms do not invade the host, but simply represent new flora that have colonized these anatomic sites. Too often, physicians try to eradicate the new flora by adding new more-powerful antibiotics or antifungal agents. The result of this strategy is to select for organisms that are multiresistant. The eventual outcome can be the selection of a bacterium or fungus that is resistant to all anti-infective agents.
No definitive method exists for differentiating between colonization and true infection. However, several clinical findings are helpful in guiding the physician. Evidence supporting the onset of a new infection includes a new fever or a change in fever pattern, a rise in the peripheral WBC with a increase in the percentage of polymorphonuclear leukocytes (PMNs) and band forms (left shift), Gram stain demonstrating an increased number of PMNs in association with predominance of bacteria that are morphologically consistent with the culture results. In the absence of these findings, colonization is more likely, and the current antibiotic regimen should be continued. In the case of C. albicans, we know that this fungus is often a component of the normal mouth flora, and when patients receive broad-spectrum antibiotics this organism overgrows in the mouth. Fortunately, Candida never spreads from the mouth to cause pneumonia in patients with normal immune systems, and therefore this organism should be ignored when it grows from sputum samples.
KEY POINTS
About Differentiating Colonization from Infection
1. Growth of resistant organisms is the rule in the patient on antibiotics.
2. Antibiotics should be switched only on evidence of a new infection.
3. Evidence for a new superinfection includes
a) new fever or a worsening fever pattern,
b) increased peripheral leukocyte count with left shift,
c) increased inflammatory exudate at the original site of infection,
d) increased polymorphonuclear leukocytes on Gram stain, and
e) correlation between bacterial morphology on Gram stain and culture.
SPECIFIC ANTI-INFECTIVE AGENTS
ANTIBIOTICS
Before prescribing a specific antibiotic, clinicians should be able to answer these questions:
• How does the antibiotic kill or inhibit bacterial growth?
• What are the antibiotic’s toxicities and how should they be monitored?
• How is the drug metabolized, and what are the dosing recommendations? Does the dosing schedule need to be modified in patients with renal dysfunction?
• What are the indications for using each specific antibiotic?
• How broad is the antibiotic’s antimicrobial spectrum?
• How much does the antibiotic cost?
Clinicians should be familiar with the general classes of antibiotics, their mechanisms of action, and their major toxicities. The differences between the specific antibiotics in each class can be subtle, often requiring the expertise of an infectious disease specialist to design the optimal anti-infective regimen. The general internist or physician-in-training should not attempt to memorize all the facts outlined here, but rather should read the pages that follow as an overview of anti-infectives. The chemistry, mechanisms of action, major toxicities, spectrum of activity, treatment indications, pharmacokinetics, dosing regimens, and cost are reviewed. The specific indications for each anti-infective are briefly covered here. A more complete discussion of specific regimens is included in the later chapters that cover infections of specific anatomic sites and are found in the IDSA guidelines (http://www.idsociety.org/IDSA_Practice_Guidelines/).
Upon prescribing a specific antibiotic, physicians should reread the specific sections on toxicity, spectrum of activity, pharmacokinetics, dosing, and cost. Because new anti-infectives are frequently being introduced, prescribing physicians should also take advantage of handheld devices, online pharmacology databases, and antibiotic manuals so as to provide up-to-date treatment (see Further Reading at the end of the current chapter). When the proper therapeutic choice is unclear, on-the-job training can be obtained by requesting a consultation with an infectious disease specialist. Anti-infective agents are often considered to be safe; however, the multiple potential toxicities outlined below, combined with the likelihood of selecting for resistant organisms, emphasize the dangers of overprescribing antibiotics.
β-Lactam Antibiotics
CHEMISTRY AND MECHANISMS OF ACTION
The β-Lactam antibiotics have a common central structure (Figure 1.7) consisting of a β-lactam ring and a thiazolidine ring [in the penicillins and carbapenems, Figure 1.7(A)] or a β-lactam ring and a dihydrothiazine ring [in the cephalosporins, Figure 1.7(B)]. The side chain attached to the β-lactam ring (R1) determines many of the antibacterial characteristics of the specific antibiotic, and the structure of the side chain attached to the dihydrothiazine ring (R2) determines the pharmacokinetics and metabolism.
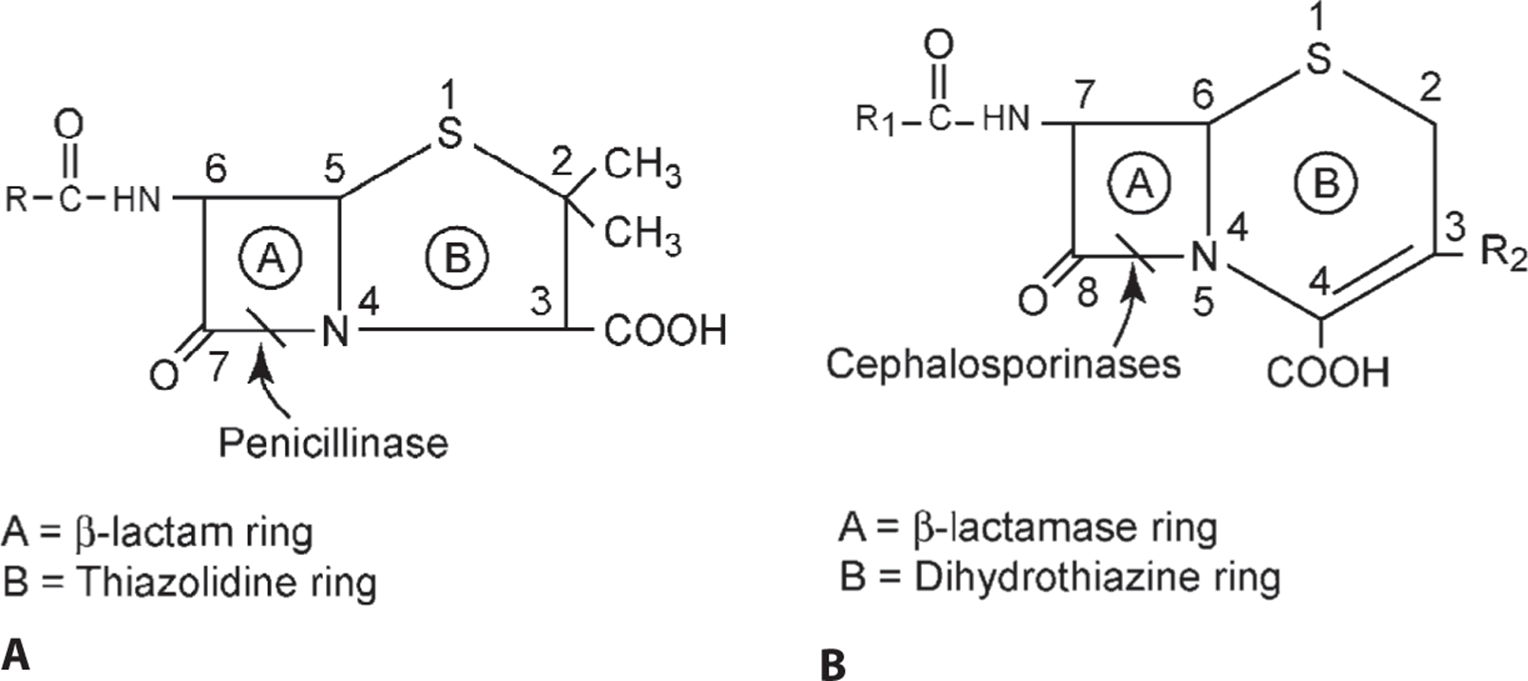
Figure 1.7. Basic structure of the A penicillins and B the cephalosporins.
KEY POINTS
About β-Lactam Antibiotics
1. Penicillins, cephalosporins, and carbapenems are all β-lactam antibiotics:
a) All contain a β-lactam ring.
b) All bind to and inhibit penicillin-binding proteins, enzymes important for cross-linking bacterial cell wall peptidoglycans.
c) All require active bacterial growth for bacteriocidal action.
d) All are antagonized by bacteriostatic antibiotics.
The β-lactam antibiotics bind to various PBPs. The PBPs represent a family of enzymes important for bacterial cell wall synthesis, including the carboxypeptidases, endopeptidases, transglycosylases, and transpeptidases. Strong binding to PBP-1 and PBP-2a (found in MRSA and encoded by mecA) inhibits cell wall transpeptidases and transglycosylases causing rapid bacterial death. The inhibition of these transpeptidases prevents the cross-linking of the cell wall peptidoglycans, resulting in a loss of integrity of the bacterial cell wall. Without its protective outer coat, the hyperosmolar intracellular contents swell, and the bacterial cell membrane lyses. Inhibition of PBP-3, a transpeptidase and transglycosylase that acts at the septum of the dividing bacterium, causes the formation of long filamentous chains of nondividing bacteria and bacterial death. Inhibition of other PBPs blocks cell wall synthesis in other ways, and activates bacterial lysis.
The activity of all β-lactam antibiotics requires active bacterial growth and active cell wall synthesis. Therefore, bacteria in a dormant or static phase will not be killed, but those in an active log phase of growth are quickly lysed. Bacteriostatic agents slow bacterial growth and antagonize β-lactam antibiotics, and therefore, in most cases, bacteriostatic antibiotics should not be combined with β-lactam antibiotics.
TOXICITY
Table 1.2 summarizes the toxicities of the β-lactam antibiotics.
Table 1.2. Toxicities of β-Lactam Antibiotics
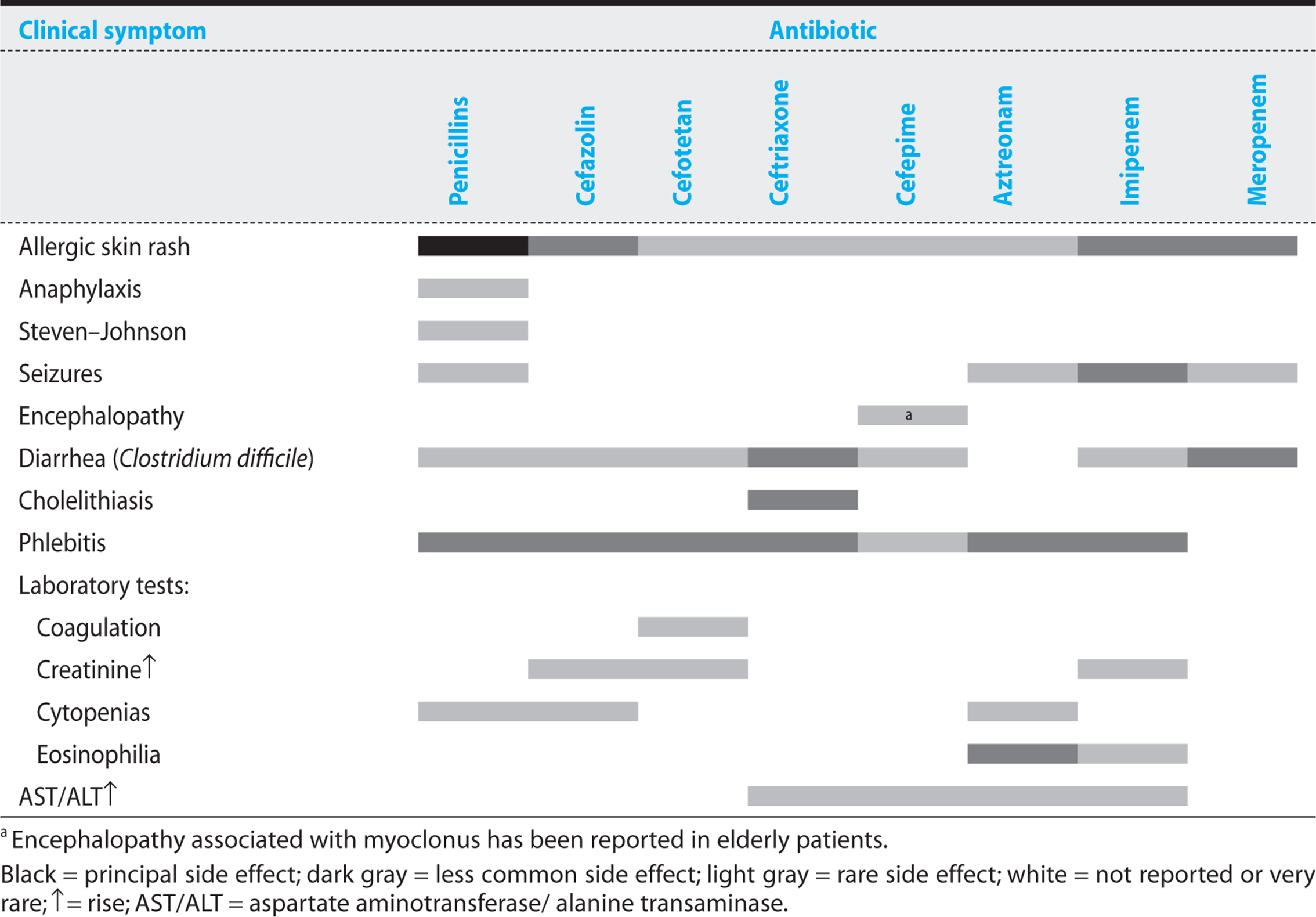
Hypersensitivity reactions are the most common side effects associated with the β-lactam antibiotics. Penicillins are the agents that most commonly cause allergic reactions, at rates ranging from 0.7% to 10%. Allergic reactions to cephalosporins have been reported in 1-3% of patients, and similar percentages have been reported with carbapenems. However, the incidence of serious, immediate immunoglobulin E (IgE)-mediated hypersensitivity reactions is much lower with cephalosporins than with penicillins. Approximately 1-7% of patients with penicillin allergies also prove to be allergic to cephalosporins and carbapenems.
Penicillins are the most allergenic of the β-lactam antibiotics because their breakdown products, particularly penicilloyl and penicillanic acid, are able to form amide bonds with serum proteins. The resulting antigens increase the probability of a host immune response. Patients who have been sensitized by previous exposure to penicillin may develop an immediate IgE-mediated hypersensitivity reaction that can result in anaphylaxis and urticaria. In the United States, penicillin-induced allergic reactions result in 400-800 fatalities annually. Because of the potential danger, patients with a history of an immediate hypersensitivity reaction to penicillin should never be given any β-lactam antibiotic, including a cephalosporin or carbapenem. High levels of immunoglobulin G antipenicillin antibodies can cause serum sickness, a syndrome resulting in fever, arthritis, and arthralgias, urticaria, and diffuse edema.
Other less common toxicities are associated with individual β-lactam antibiotics. Natural penicillins and imipenem lower the seizure threshold and can result in grand mal seizures. Ceftriaxone is excreted in high concentrations in the bile and can crystallize, causing biliary sludging and cholecystitis. Antibiotics containing a specific methylthiotetrazole ring (cefamandole, cefoperazone, cefotetan) can induce hypoprothrombinemia and, in combination with poor nutrition, may increase postoperative bleeding. Cefepime has been associated with encephalopathy and myoclonus in elderly individuals. All broad-spectrum antibiotics increase the risk of Clostridium difficile colitis (see Chapter 8). In combination with aminoglycosides, cephalosporins demonstrate increased nephrotoxicity.
KEY POINTS
About β-Lactam Antibiotic Toxicity
1. Allergic reactions are most common toxicity, and they include both delayed and immediate hypersensitivity reactions.
2. Allergy to penicillins (PCNs) seen in 1-10% of patients; 1-3% are allergic to cephalosporins and carbapenems. 1-7% of patients with a PCN allergy are also allergic to cephalosporins and carbapenems.
3. Seizures are associated with PCNs and imipenem, primarily in patients with renal dysfunction.
4. Ceftriaxone is excreted in the bile and can crystallize to form biliary sludge.
5. Cephalosporins with methylthiotetrazole rings (cefamandole, cefoperazone, moxalactam, cefotetan) can interfere with vitamin K and increase prothrombin time.
6. Pseudomembranous colitis can develop as a result of overgrowth of Clostridium difficile.
7. Nephrotoxicity sometimes occurs when cephalosporins are given in combination with amino-glycosides.
Penicillins
Tables 1.2 and 1.3 as well as Figure 1.8 summarize the characteristics of the various penicillins. (Also see section on outpatient antibiotics.)
Table 1.3. Penicillins: Half-Life, Dosing, Renal Dosing, Cost, and Spectrum

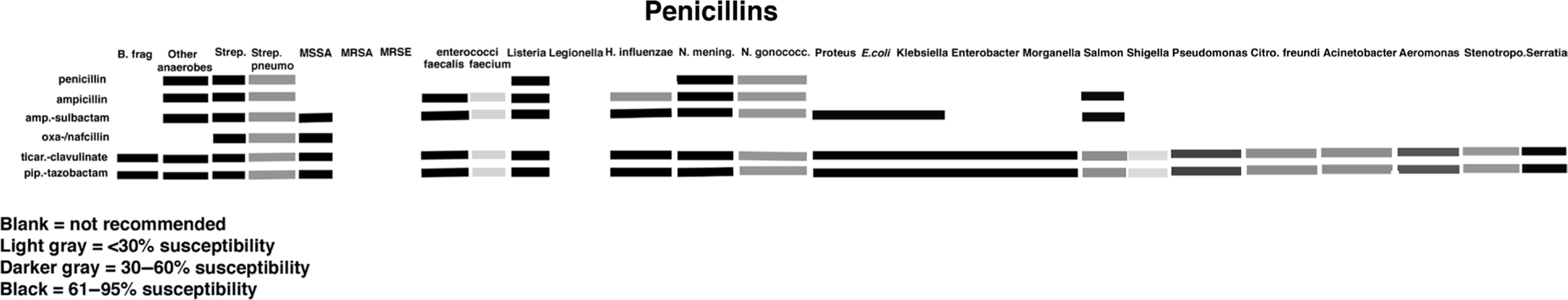
Figure 1.8. Antibiogram of penicillins.
Penicillins vary in their spectrum of activity. Natural penicillins have a narrow spectrum. The aminopenicillins have an intermediate spectrum, and combined with β-lactamase inhibitors, the carboxy/ureidopenicillins have a very broad spectrum of activity.
KEY POINTS
About the Natural Penicillins
1. Very short half-life (15-30 minutes).
2. Excreted renally; adjust for renal dysfunction; probenecid delays excretion.
3. Penetrates most inflamed body cavities.
4. Narrow spectrum. Indicated for Streptococcus pyogenes, S. viridans Gp., mouth flora, Clostridium perfringens, Neisseria meningitidis, Pasteurella, and spirochetes.
5. Recommended for penicillin-sensitive S. pneumoniae [however, penicillin-resistant strains are now frequent (>30%)]; infections caused by mouth flora; C. perfringens or spirochetes.
NATURAL PENICILLINS
Pharmacokinetics—All natural penicillins are rapidly excreted by the kidneys, resulting in short half-lives (Table 1.3). As a consequence, the penicillins must be dosed frequently, and dosing must be adjusted in patients with renal dysfunction. Probenecid slows renal excretion, and this agent can be used to sustain higher serum levels. Depending on the specific drug, penicillins can be given intravenously or intramuscularly. Some penicillins have been formulated to withstand the acidity of the stomach and are absorbed orally. Penicillins are well distributed in the body and are able to penetrate most inflamed body cavities. However, their ability to cross the blood–brain barrier in the absence of inflammation is poor. In the presence of inflammation, therapeutic levels are generally achievable in the cerebrospinal fluid (CSF).
Spectrum of Activity and Treatment Recommendations—Pencillin G (Figure 1.8) remains the treatment of choice for S. pyogenesβgroup A strepβ) and the S. viridans group. It also remains the most effective agent for the treatment of infections caused by mouth flora. Penicillin G is also primarily recommended for Clostridium perfringens, C. tetani, Erysipelothrix rhusiopathiae, Pasteurella multocida, and spirochetes including syphilis and Lepto-spira. This antibiotic also remains the primary recommended therapy for S. pneumoniae sensitive to penicillin (MIC < 0.1 ug/mL). However, in the United States, approximately 20% of strains are moderately resistant to penicillin (MIC = 0.1-1 ug/mL) and 20% are highly resistant (MIC > 2 µg/mL). For moderately resistant strains, ceftriaxone, cefotaxime, or high-dose penicillin (> 12 million units daily) can be used. Moderately resistant strains of S. pneumoniae possess a lower-affinity PBP, and this defect in binding can be overcome by high serum levels of penicillin in the treatment of pneumonia, but not of meningitis. Infections with high-level penicillin-resistant S. pneumoniae require treatment with vancomycin or ceftaroline.
AMINOPENICILLINS
Pharmacokinetics—In aminopenicillins, a chemical modification of penicillin increases resistance to stomach acid, allowing these products to be given orally (Table 1.3). They can also be given intramuscularly or intravenously. Amoxicillin has excellent oral absorption: 75% as compared with 40% for ampicillin. Absorption is not impaired by food. The higher peak levels achievable with aminopenicillins allow for a longer dosing interval, making them a more convenient oral antibiotic than ampicillin. As observed with the natural penicillins, the half-life is short (1 hour) and these drugs are primarily excreted unmodified in the urine.
Spectrum of Activity and Treatment Recommendations—The spectrum of activity in the aminopenicillins is slightly broader than in the natural penicillins (Figure 1.8). Intravenous ampicillin is recommended for treatment of Listeria monocytogenes, sensitive enterococci, Proteus mirabilis, and non–β-lactamase-producing Haemophilus influenzae. Aminopenicillins are also effective against Shigella flexneri and sensitive strains of nontyphoidal Salmonella. Amoxicillin can be used to treat otitis media. When combined with a β-lactamase inhibitor (clavulanate or sulbactam), aminopenicillins are also effective against methicillin-sensitive S. aureus (MSSA), β-lactamase-producing strains of H. influenzae, and Moraxella catarrhalis. The latter two organisms are commonly cultured from middle ear and air sinus infections (see Chapter 5) making this combination drug the treatment of choice for bacterial sinusitis. However, the superiority of amoxicillin–clavulanate over amoxicillin for middle ear infections has not been proven.
KEY POINTS
About the Aminopenicillins
1. Short half-life (1 hour), and clearance similar to natural penicillins.
2. Slightly broader spectrum of activity.
3. Parenteral ampicillin indicated for Listeria monocytogenes, sensitive enterococci, Proteus mirabilis, and non–β-lactamase-producing Haemophilus influenzae.
4. Ampicillin plus an aminoglycoside is the treatment of choice for enterococci. Whenever possible, vancomycin should be avoided.
5. Amoxicillin has excellent oral absorption; it is the initial drug of choice for otitis media.
6. Amoxicillin–clavulanate has improved coverage of Staphylococcus, H. influenzae, and Moraxella catarrhalis, but it is expensive and has a high incidence of diarrhea. Increased efficacy compared with amoxicillin is not proven in otitis media, but is the drug of choice for bacterial sinusitis.
PENICILLINASE-RESISTANT PENICILLINS
Pharmacokinetics—The penicillinase-resistant penicillins have the same half-life as penicillin (30 minutes) and require dosing at 4-hour intervals or constant intravenous infusion (Table 1.3). Unlike the natural penicillins, these agents are cleared hepatically, and doses of nafcillin and oxacillin usually do not need to be adjusted for renal dysfunction. But the efficient hepatic excretion of nafcillin means that the dose needs to be adjusted in patients with significant hepatic dysfunction. The liver excretes oxacillin less efficiently, and so dose adjustment is usually not required in liver disease.
Spectrum of Activity and Treatment Recommendations—The synthetic modification of penicillin to render it resistant to the β-lactamases produced by S. aureus reduces the ability of these agents to kill anaerobic mouth flora and Neisseria species (Figure 1.8). These antibiotics are strictly recommended for the treatment of MSSA. They are also used to treat cellulitis when the most probable pathogens are S. aureus and S. pyogenes. Because oral preparations result in considerably lower serum concentration levels, cloxacillin or dicloxacillin should not be used to treat S. aureus bacteremia. These oral agents are used primarily for mild soft tissue infections or to complete therapy of a resolving cellulitis.
KEY POINTS
About Penicillinase-Resistant Penicillins
1. Short half-life, hepatically metabolized.
2. Very narrow spectrum, poor anaerobic activity.
3. Primarily indicated for methicillin-sensitive Staphylococcus aureus and cellulitis.
CARBOXYPENICILLINS AND UREIDOPENICILLINS
Pharmacokinetics—The half-lives of ticarcillin and piperacillin are short, and they require frequent dosing (Table 1.3). Sale of ticarcillin and piperacillin alone has been discontinued in favor of ticarcillin–clavulanate and piperacillin–tazobactam.
Dosing every 6 hours is recommended for piperacillin–tazobactam to prevent accumulation of tazobactam. In P. aeruginosa pneumonia, the dose of piperacillin–tazobactam should be increased from 3.375 g Q6h to 4.5 g Q8h to achieve cidal levels of piperacillin in the sputum. In combination with an aminoglycoside, piperacillin–tazobactam often demonstrates synergy against P. aeruginosa. However, the administration of the piperacillin–tazobactam needs to be separated from the administration of the aminoglycoside by 30-60 minutes.
Spectrum of Activity and Treatment Recommendations (Figure 1.8)—Ticarcillin and piperacillin are able to resist β-lactamases produced by Pseudomonas, Enterobacter, Morganella, and Proteus–Providenciaspecies. At high doses, ticarcillin and piperacillin can also kill many strains of Bacteroides fragilis and provide effective anaerobic coverage. These antibiotics can be used for empiric coverage of moderate-to-severe intra-abdominal infections. They have been combined with a β-lactamase inhibitor (clavulanate or tazobactam) to provide effective killing of MSSA.
These agents are reasonable alternatives to nafcillin or oxacillin when gram-negative coverage is also required. Both agents can be used for in-hospital aspiration pneumonia to cover for mouth flora and gram-negative rods alike, and they can also be used for serious intra-abdominal, gynecologic, and acute prostate infections. They have been used for skin and bone infections thought to be caused by a combination of gram-negative and gram-positive organisms.
KEY POINTS
About Carboxypenicillins and Ureidopenicillins
1. More effective resistance to gram-negative β-lactamases.
2. Carboxypenicillin or ureidopenicillin combined with aminoglycosides demonstrate synergistic killing of Pseudomonas aeruginosa.
3. Ticarcillin–clavulanate and piperacillin–tazobactam have excellent broad-spectrum coverage, including methicillin-sensitive Staphylococcus aureus and anaerobes. They are useful for intra-abdominal infections, acute prostatitis, in-hospital aspiration pneumonia, and mixed soft tissue and bone infections.
Cephalosporins
Tables 1.2 and 1.4 as well as Figure 1.9 summarize the characteristics of the various cephalosporins. (Also see section on outpatient antibiotics)
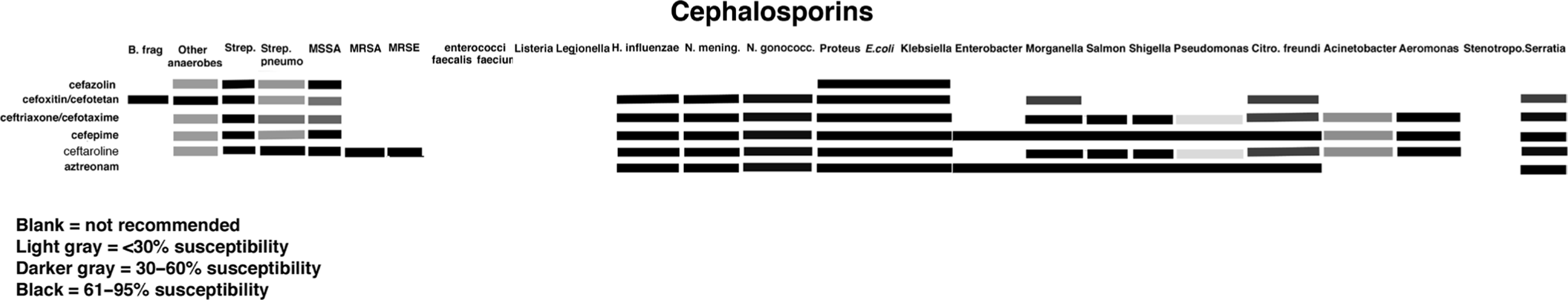
Figure 1.9. Antibiogram of cephalosporins.
Table 1.4. Cephalosporins: Half-Life, Dosing, Renal Dosing, Cost, and Spectrum
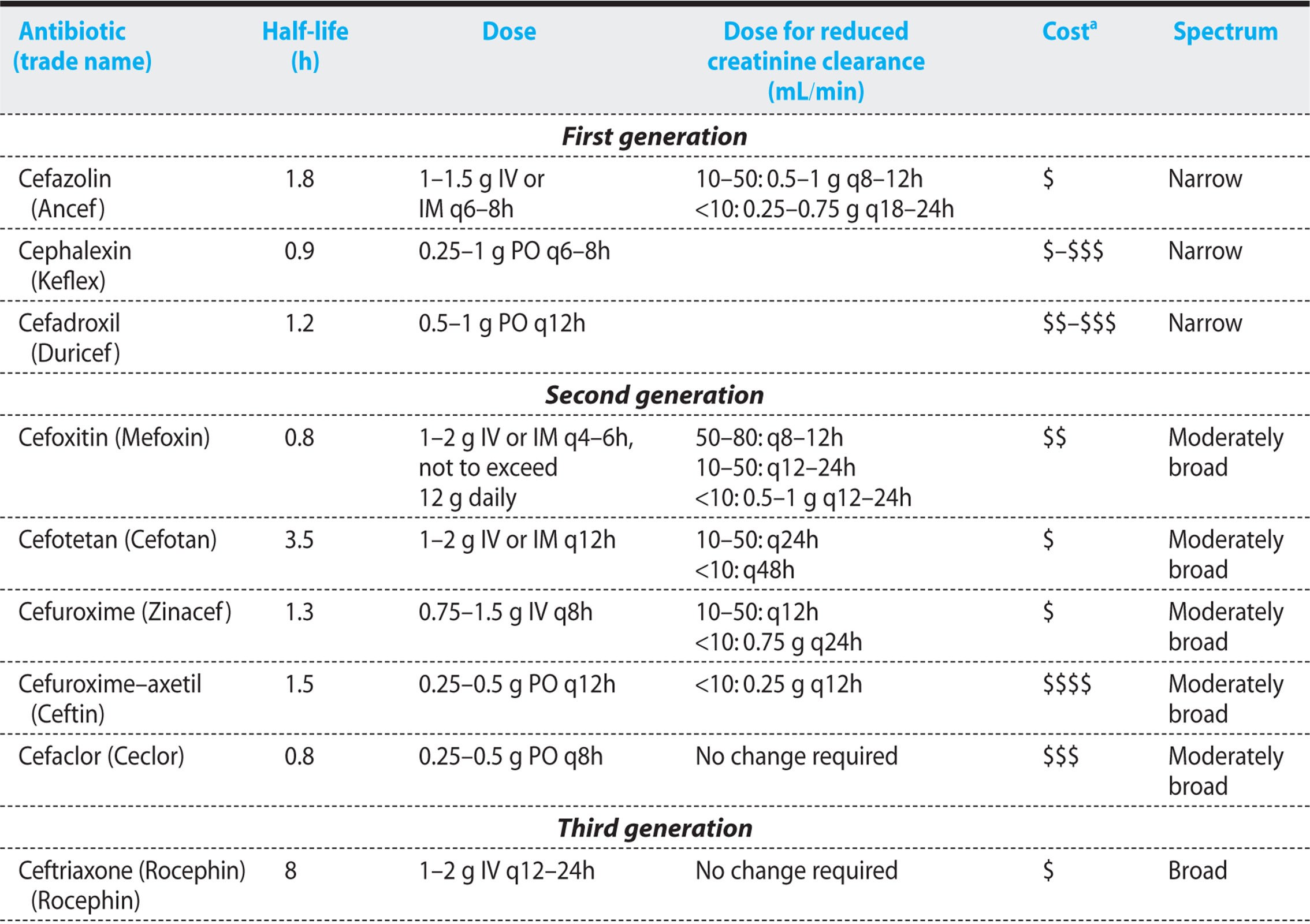

In an attempt to create some semblance of order, the cephalosporins have been classified into generations based on spectrum of activity (Table 1.4). First-generation cephalosporins are predominantly effective against gram-positive cocci. Second-generation cephalosporins demonstrate increased activity against aerobic and anaerobic gram-negative bacilli, but have variable activity against gram-positive cocci. The third-generation cephalosporins demonstrate even greater activity against gram-negative bacilli, but only limited activity against gram-positive cocci. The fourth-generation cephalosporins demonstrate the broadest spectrum of activity, being effective against both gram-positive cocci and gram-negative bacilli. Recently a fifth-generation cephalosporin, ceftaroline has been approved that binds the penicillin-binding proteins, PBP1-4, with higher affinity than other cephalosporins and penicillins, and kills both MRSA and penicillin-resistant S. pneumoniae.
Classification of the cephalosporins by generation naturally leads to the assumption that newer, later-generation cephalosporins are better than the older cephalosporins. However, it is important to keep in mind that, for many infections, earlier-generation, narrower-spectrum cephalosporins are preferred to the more recently developed broader-spectrum cephalosporins.
FIRST-GENERATION CEPHALOSPORINS
Pharmacokinetics—Cefazolin, the preferred parenteral first-generation cephalosporin, has a longer half-life than penicillin, and it is primarily excreted by the kidneys (Table 1.4). The first-generation cephalosporins penetrate most body cavities, but they fail to cross the blood–brain barrier. Oral preparations (cephalexin, cephradine, cefadroxil) are very well absorbed, achieving excellent peak serum concentrations (0.5 g cephalexin results in a 18 µg/mL peak). Absorption is not affected by food. The half-lives of cephalexin and cephradine are short, requiring frequent administration. These agents need to be corrected for renal dysfunction.
KEY POINTS
About First-Generation Cephalosporins
1. Excellent gram-positive coverage, some gram-negative coverage.
2. Do not cross the blood–brain barrier.
3. Inexpensive.
4. Useful for treating soft tissue infections and for surgical prophylaxis. Can often be used as an alternative to oxacillin or nafcillin.
Spectrum of Activity and Treatment Recommendations—The first-generation cephalosporins are very active against gram-positive cocci, including MSSA, and they also have moderate activity against some community-acquired gram-negative bacilli (Figure 1.9). They are active against oral cavity anaerobes, but are ineffective for treating B. fragilis, H. influenzae, L. monocytogenes, MRSA, penicillin-resistant S. pneumoniae, and Enterococcus.
First-generation cephalosporins are an effective alternative to nafcillin or oxacillin for soft tissue infections thought to be caused by MSSA or S. pyogenes. Cefazolin is also the antibiotic of choice for surgical prophylaxis. Because of its inability to cross the blood-brain barrier, cefazolin should never be used to treat bacterial meningitis. Oral preparations are commonly used to treat less severe soft tissue infections, including impetigo, early cellulitis, and mild diabetic foot ulcers.
SECOND-GENERATION CEPHALOSPORINS
Pharmacokinetics—The second-generation cephalosporins are cleared primarily by the kidney (Table 1.4). They have half-lives that range from 0.8 to 3.5 hours, and they penetrate all body cavities.
Spectrum of Activity and Treatment Recommendations—The second-generation cephalosporins possess increased activity against some gram-negative strains, and they effectively treat MSSA and nonenterococcal streptococci (Figure 1.9). Given the availability of the first-, third-, fourth-, and fifth-generation cephalosporins and the newer penicillins, second-generation cephalosporins are rarely recommended as primary therapy.
KEY POINTS
About Second-Generation Cephalosporins
1. Improved activity against Haemophilus influenzae, Neisseria species, and Moraxella catarrhalis.
2. Cefoxitin and cefotetan have anaerobic activity and are used in mixed soft tissue infections and pelvic inflammatory disease.
3. Cefotetan and cefamandole have a methylthio-tetrazole ring that decreases prothrombin production. Vitamin K prophylaxis is recommended in malnourished patients.
4. Cefuroximeaxetil is a popular oral cephalosporin; less expensive alternative oral antibiotics are available, however.
5. Overall, this generation is of limited usefulness.
Because cefoxitin and cefotetan demonstrate increased anaerobic coverage, including many strains of B. fragilis, and also cover gonococcus, these two agents are used as part of first-line therapy in pelvic inflammatory disease. They are also used for the treatment of moderately severe intra-abdominal infections and mixed aerobic–anaerobic soft tissue infections, including diabetic foot infections. The oral preparation cefuroxime achieves serum levels that are approximately one tenth that of intravenous preparations, and this agent is recommended for the outpatient treatment of uncomplicated urinary tract infections and otitis media. Other less costly oral antibiotics effectively cover the same pathogens.
Cefaclor, the other second-generation oral preparation, is inactivated by β-lactamases produced by H. influenzae and M. catarrhalis. Although cefaclor has been recommended for otitis media, other oral antibiotics are generally preferred.
THIRD-GENERATION CEPHALOSPORINS
Pharmacokinetics—With the exception of ceftriaxone, the third-generation cephalosporins are excreted by the kidneys (Table 1.4). Ceftriaxone is cleared primarily by the liver, but high concentrations of the drug are also excreted in the biliary system. The half-lives of these agents vary, being as short as 1.5 hours (cefotaxime) and as long as 8 hours (ceftriaxone). They penetrate most body sites effectively.
KEY POINTS
About the Third-Generation Cephalosporins
1. Improved gram-negative coverage.
2. Excellent activity against Neisseria gonorrhoeae, N. meningitidis, Haemophilus influenzae, and Moraxella catarrhalis.
3. Ceftriaxone has a long half-life that allows for once-daily dosing. In children, acalculous cholecystitis can occur with large doses.
4. Cefotaxime has a shorter half-life but activity identical to that of ceftriaxone; does not cause biliary sludging.
5. Ceftazidime has excellent activity against most Pseudomonas aeruginosa strains, but reduced activity against Staphylococcus aureus.
6. Extended spectrum β-lactamases are increasing in frequency and endangering the effectiveness of third-generation cephalosporins.
7. Ceftriaxone is recommended for community-acquired pneumonia and bacterial meningitis
Spectrum of Activity and Treatment Recommendations (Figure 1.9)—As compared with the first- and second-generation, third-generation cephalosporins have enhanced activity against many aerobic gram-negative bacilli, but they do not cover Serratia marcescens, Acinetobacter, and Enterobacter cloacae. With the exceptions of ceftazidime and cefoperazone, third-generation cephalosporins are ineffective against P. aeruginosa.
These agents have excellent cidal activity against S. pneumoniae (including moderately penicillin-resistant strains), S. pyogenes, and other streptococci. All members of this generation are ineffective for treating Enterococcus,MRSA, highly penicillin-resistant pneumococcus, and L. monocytogenes.
The ESBLs are increasing in frequency, and they promise to reduce the effectiveness of the third- and fourth-generation cephalosporins. A large number of third-generation cephalosporins are available, all with similar indications. Small deficiencies in coverage and less-desirable pharmacokinetics have affected the popularity of a number of these drugs.
Ceftriaxone and cefotaxime are recommended for empiric treatment of community-acquired pneumonia and community-acquired bacterial meningitis (see Chapters 4 and 6). Third-generation cephalosporins can be used in combination with other antibiotics to empirically treat the septic patient. Ceftriaxone is recommended for treatment of Neisseria gonorrhoeae. Cefotaxime is cleared renally and does not form sludge in the gallbladder. For this reason, this agent is preferred over ceftriaxone by some pediatricians, particularly for the treatment of bacterial meningitis in children—where high-dose therapy has been associated with symptomatic biliary sludging. Ceftazidime is the only third-generation cephalosporin that has excellent activity against P. aeruginosa; however, the fourth-generation cephalosporin cefepime (and the monobactam aztreonam) is now more commonly utilized for anti-Pseudomonas therapy in many institutions.
The oral third-generation cephalosporin cefixime has a long half-life, allowing for once-daily dosing. Cefixime provides effective coverage for S. pneumoniae (penicillin-sensitive), S. pyogenes, H. influenzae, M. catarrhalis, Neisseria species, and many gram-negative bacilli, but it is ineffective against S. aureus. Its absorption is not affected by food. This agent is a potential second-line therapy for community-acquired pneumonia, and it is an alternative to penicillin for the treatment of bacterial pharyngitis. The other oral preparation, cefpodoxime proxetil, has an antimicrobial spectrum similar to that of cefixime. In addition, it has moderate activity against S. aureus. The indications for use are similar to those for cefixime, and cefpodoxime proxetil has also been recommended as an alternative treatment of acute sinusitis.
KEY POINTS
About Fourth-Generation Cephalosporins
1. Zwitterionic properties allow for excellent penetration of the bacterial cell wall and of human tissues and fluids.
2. Weakly induce β-lactamases.
3. More resistant to extended-spectrum β-lactamases and chromosomal β-lactamases.
4. Excellent gram-positive (including methicillin-sensitive Staphylococcus aureus) and gram-negative coverage (including Pseudomonas aeruginosa).
5. Excellent broad-spectrum empiric therapy. Useful in nosocomial infections.
FOURTH-GENERATION CEPHALOSPORINS
Pharmacokinetics—Clearance of the fourth-generation cephalosporins is renal, and the half-lives of these agents are similar to the renally cleared third-generation cephalosporins (Table 1.4). The R2substitution of the fourth-generation cephalosporins contains both a positively and negatively charged group that, together, have zwitterionic properties that permit these antibiotics to penetrate the outer wall of gram-negative bacteria and concentrate in the periplasmic space. This characteristic also allows for excellent penetration of all body compartments, including the CSF.
Spectrum of Activity and Treatment Recommendations—The fourth-generation cephalosporins are resistant to most β-lactamases, and they only weakly induce β-lactamase activity (Figure 1.9). These agents also bind gram-positive PBPs with high affinity.
The only agent currently available in the United States is cefepime. In addition to having broad antimicrobial activity against gram-negative bacilli, including P. aeruginosa, cefepime provides excellent coverage for S. pneumoniae(including strains moderately resistant to penicillin), S. pyogenes, and MSSA. Cefepime and ceftazidime provide comparable coverage for P. aeruginosa. To maximize the likelihood of cure of serious P. aeruginosa infection, more frequent dosing (q8h) has been recommended.
Cefepime is not effective against L. monocytogenes, MRSA, or B. fragilis. As compared with third-generation cephalosporins, cefepime is more resistant to β-lactamases, including the ESBLs. It has been effectively used to treat gram-negative meningitis. Cefepime is effective as a single agent in the febrile neutropenic patient, and it is an excellent agent for initial empiric coverage of nosocomial infections.
Cefpirome is available in Europe. It has an antimicrobial spectrum similar to that of cefepime, although it is somewhat less active against P. aeruginosa.
FIFTH-GENERATION CEPHALOSPORIN
Ceftaroline—This is the only recently Food and Drug Administration (FDA)-approved antibiotic to be released in the U.S. market, attesting to the slowing of anti-infective research and development over the past decade.
Chemistry and Pharmacokinetics—This recently developed antibiotic contains an ethoxyiminoacetamido group in the C-7 moiety and a thio 5-membered heteroaromatic spacer group at position 3 (Figure 1.7B), and the resulting compound is water soluble and chemically stable. When compared with penicillins and other cephalosporins, these modifications have increased the affinity of ceftaroline to all PBPs and particularly to PBP2a found in MRSA. Ceftaroline also shows excellent binding and effectively kills vancomycin intermediate strains (VISA) of MRSA. This agent also shows higher affinity for MSSA PBPs 1-3 and S. pneumoniae PBP2x/2a/2b. And these improved PBP-binding characteristics correlate closely with its lower MICs for these strains.
Ceftaroline shows good penetration of body spaces, including bones, joints, and the CSF. In a MRSA rabbit osteomyelitis model, this agent was shown to be superior to vancomycin, and comparable to linezolid with regard to reductions in bacterial counts. In a rabbit meningitis model, treatment with ceftaroline resulted in greater reductions in penicillin-sensitive S. pneumoniae bacterial counts than ceftriaxone, and was superior to vancomycin for penicillin-resistant S. pneumoniae strains. Ceftaroline is cleared by the kidneys and requires dose modification for patient with renal impairment and for patients on hemodialysis (Table 1.4). There is no evidence for hepatic metabolism by the cytochrome p450 system, thus minimizing concerns with regard to drug–drug interactions. The half-life is relatively prolonged allowing twice per day dosing.
KEY POINTS
About Ceftaroline
1. Has increased the affinity for all penicillin-binding proteins (PBPs) and particularly to PBP2a found in MRSA including VISA.
2. Also higher affinity for methicillin-sensitive S. aureus (MSSA) PBPs 1-3 and S. pneumoniae PBP2x/2a/2b.
3. Penetrates all body tissues including blood–brain barrier and joint fluid.
4. Effective against MRSA including vancomycin intermediate sensitivity strains (VISA), MSSA, and penicillin-resistant S. pneumonia.
5. Similar gram-negative coverage to Ceftriaxone.
6. Approved for community-acquired pneumonia and soft tissue infections.
When compared with other cephalosporins, ceftaroline has a similar side effect profile (Table 1.2), the major toxicity being allergic reactions that are far lower in frequency than the penicillins or carbapenems.
The antibiotic spectrum of ceftaroline is similar to ceftriaxone (Figure 1.9) with the addition of excellent activity against MRSA, including VISA strains and daptomycin-resistant strains. The agent also shows improved activity against MSSA as compared with vancomycin, and also demonstrates excellent activity against penicillin and ceftriaxone-resistant S. pneumonia.
Ceftaroline is presently approved for the treatment of community-acquired pneumonia and complicated soft tissue infections particularly when MRSA is suspected.
Monobactams
AZTREONAM
Chemistry and Pharmacokinetics—Aztreonam was originally isolated from Chromobacterium violaceum and subsequently modified. This antibiotic has a distinctly different structure from the cephalosporins, and it is the only available antibiotic in its class. Rather than a central double ring, aztreonam has a single ring (“monocyclic β-lactam structure”), and has been classified as a monobactam.
KEY POINTS
About Aztreonam
1. A distinctly different structure than that of the cephalosporins.
2. No cross-reactivity with penicillin.
3. Binds the penicillin-binding proteins of gram-negative, but not of gram-positive bacteria.
4. Narrow spectrum, with excellent activity against aerobic gram-negative rods.
5. Marketed as a non-nephrotoxic replacement for aminoglycosides. However, as compared with aminoglycosides, it
a) has no synergy with penicillins in enterococcal infections.
b) is not helpful for treating Streptococcus viridans endocarditis.
6. Excellent empiric antibiotic when combined with an antibiotic with good gram-positive activity. Useful for the treatment of pyelonephritis.
Because of its unique structure, aztreonam exhibits no cross-reactivity with other β-lactam antibiotics. It can be used safely in the penicillin-allergic patient. The drug penetrates body tissue well and crosses the blood–brain barrier of inflamed meninges. Aztreonam is renally cleared and has a half-life similar to that of the renally cleared third- and fourth-generation cephalosporins.
Spectrum of Activity and Treatment Recommendations—Aztreonam does not bind to the PBPs of gram-positive organisms or anaerobes; rather, it binds with high affinity to PBPs, particularly PBP-3 (responsible for septum formation during bacterial division), of gram-negative bacilli including P. aeruginosa. Gram-negative organisms exposed to aztreonam form long filamentous structures and are killed.
Aztreonam is effective against most gram-negative bacilli, and this agent has been marketed as a non-nephrotoxic replacement for aminoglycosides. However, unlike aminoglycosides, aztreonam does not provide synergy with penicillins for Enterococcus. A major advantage of aztreonam is its restricted antimicrobial spectrum, which allows for survival of the normal gram-positive and anaerobic flora that can compete with more resistant pathogens.
Aztreonam can be used for the treatment of most infections attributable to gram-negative bacilli. It has been used effectively in pyelonephritis, nosocomial gram-negative pneumonia, gram-negative bacteremia, and gram-negative intra-abdominal infections. Importantly, though, aztreonam provides no gram-positive or anaerobic coverage. Therefore, when it is used for empiric treatment of potential gram-positive pathogens in the seriously ill patient, aztreonam should be combined with vancomycin, clindamycin, erythromycin, or a penicillin.
Carbapenems
Tables 1.2 and 1.5, together with Figure 1.10, summarize the characteristics of the various carbapenems.
Table 1.5. Carbapenems: Half-Life, Dosing, Renal Dosing, Cost, and Spectrum
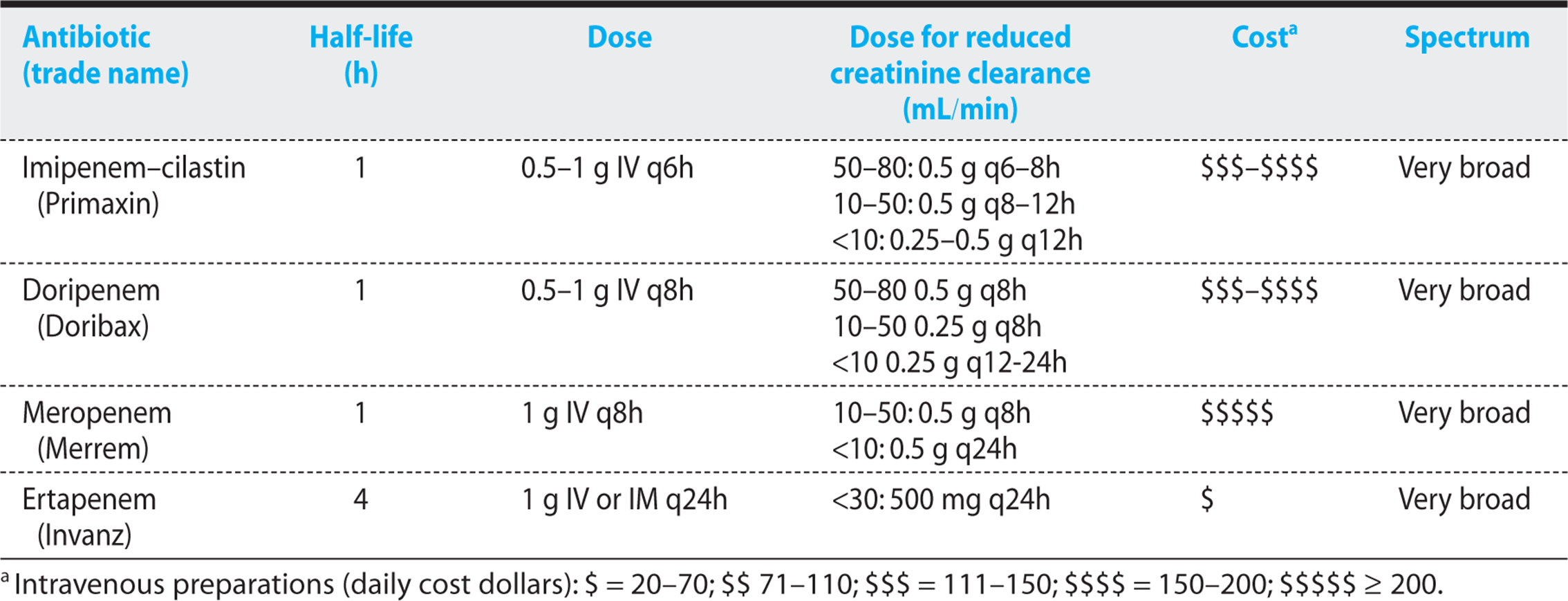

Figure 1.10. Antibiogram of carbapenems.
CHEMISTRY AND PHARMACOKINETICS
The carbapenems have both a modified thiazolidine ring and a change in the configuration of the side chain that renders the β-lactam ring highly resistant to cleavage. Their hydroxyethyl side chain is in a trans rather than cisconformation, and this configuration is thought to be responsible for the group’s remarkable resistance to β-lac-tamase breakdown. At physiologic pH, these agents have zwitterionic characteristics that allow them to readily penetrate tissues. The carbapenems bind with high affinity to the high molecular weight PBPs of both gram-positive and gram-negative bacteria.
Imipenem is combined in a 1:1 ratio with cilastatin to block rapid breakdown by renal dehydropeptidase I. Doripenem, meropenem, and ertapenem are not significantly degraded by this enzyme and do not require coadministration with cilastatin. These drugs are all primarily cleared by the kidneys.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
The carbapenems have a very broad spectrum of activity, effectively killing most strains of gram-positive and gram-negative bacteria, including anaerobes. Overall, imipenem has slightly better activity against gram-positive organisms. Doripenem, meropenem, and ertapenem have somewhat better activity against gram-negative pathogens (except Pseudomonas for ertapenem, as described later in this subsection).
These agents are cidal not only against S. pneumoniae, S. pyogenes, and MSSA, but also against organisms that are not covered by the cephalosporins, including Listeria, Nocardia, Legionella, and Mycobacterium avium intracellular (MAI). They have static activity against penicillin-sensitive enterococci; however, many penicillin-resistant strains are also resistant to carbapenems. MRSA, some penicillin-resistant strains of S. pneumoniae, C. difficile, St. maltophilia, and B. cepacia are also resistant. Resistance in gram-negative bacilli is most often secondary to loss of an outer membrane protein called D2 that is required for intracellular penetration of the carbapenems. Increasing numbers of gram-negative strains can also produce β-lactamases called carbapenemases that can hydrolyze these drugs.
KEY POINTS
About the Carbapenems
1. β-Lactam ring is highly resistant to cleavage.
2. Have zwitterionic characteristics, and penetrate all tissues.
3. Frequent cross-reactivity in penicillin-allergic patients (7%).
4. Imipenem causes seizures at high doses; be cautious in renal failure patients. Meropenem is less epileptogenic.
5. Bind penicillin-binding proteins of all bacteria with high affinity.
6. Very broad cidal activity for aerobic and anaerobic gram-positive and gram-negative bacteria. Also covers Listeria monocytogenes and Nocardia.
7. Imipenem, doripenem, and meropenem are useful for empiric therapy of suspected mixed aerobic and anaerobic infection or a severe nosocomial infection, pending culture results. Reserve for the severely ill patient.
8. Ertapenem can be given once daily. Lacks Pseudomonas aeruginosa coverage.
9. Treatment markedly alters the normal bacterial flora.
Imipenem, doripenem, and meropenem can be used as empiric therapy for sepsis, and they are particularly useful if polymicrobial bacteremia is a strong possibility. They can also be used to treat severe intra-abdominal infections and complicated pyelonephritis. Infections attributable to gram-negative bacilli resistant to cephalosporins and aminoglycosides may be sensitive to imipenem, doripenem, or meropenem. Imipenem, doripenem, or meropenem is recommended as primary therapy for Serratia. Doripenem and meropenem can be used for meningitis, achieving therapeutic levels in the CSF. Imipenem is not recommended for this purpose because of its propensity to cause seizures. In general, imipenem, doripenem, and meropenem should be reserved for the seriously ill patient or the patient infected with a highly resistant bacterium that is sensitive only to this antibiotic.
Ertapenem has a longer half-life and can be given just once daily, making it a useful agent for home intravenous therapy. This agent is not effective against P. aeruginosa, but otherwise it has a spectrum similar to that of doripenem and meropenem. It is recommended for complicated intra-abdominal infections, postpartum and postoperative acute pelvic infections, and complicated soft tissue infections.
Because the carbapenems are extremely broad-spectrum agents, they kill nearly all normal flora. The loss of normal flora increases the risk of nosocomial infections with resistant pathogens including MRSA, Pseudomonas, and Candida.
Aminoglycosides
Tables 1.6 and 1.7, together with Figure 1.11, summarize the characteristics of the various aminoglycosides.
Table 1.6. Aminoglycosides: Half-Life, Dosing, Renal Dosing, Cost, and Spectrum
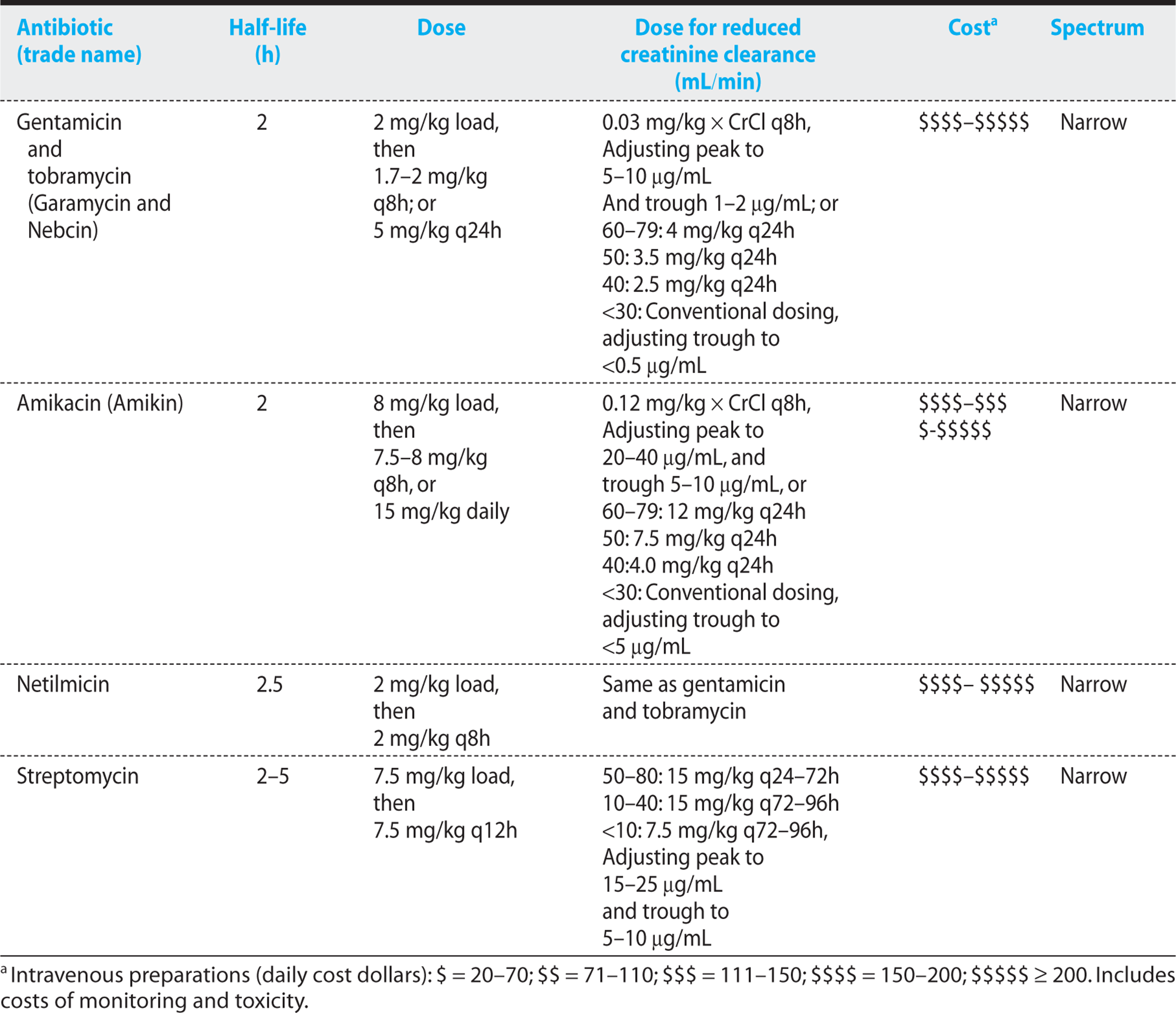
Table 1.7. Toxicities of Miscellaneous Antibiotics


Figure 1.11. Antibiogram of aminoglycosides.
CHEMISTRY AND MECHANISM OF ACTION
Aminoglycosides were originally derived from Streptomyces species. These agents have a characteristic 6-member ring with amino-group substitutions, and they are highly soluble in water. At neutral pH, they are positively charged, and this positive charge contributes to their antibacterial activity. At a low pH, the charge is reduced, impairing antimicrobial activity. Their positive charge also causes aminoglycosides to bind and to become inactivated by β-lactam antibiotics. Therefore, aminoglycosides should never be in the same solution with β-lactam antibiotics.
KEY POINTS
About Aminoglycoside Antibacterial Activity
1. 6-Member ring, soluble in water, positively charged; never with cephalosporins or acidic solutions.
2. Cause temporary holes in bacterial membranes, bind to ribosomal RNA, and interfere with translation.
3. Killing is concentration-dependent.
4. The higher the concentration, the longer the post-antibiotic effect.
5. Excellent gram-negative coverage; streptomycin for tularemia and plague.
6. Synergy with penicillins in S. viridans, Enterococcus, and Pseudomonas aeruginosa infections.
Upon entering the bacterium, the antibiotic molecules interact with and precipitate DNA and other anionic components. Aminoglycosides also bind to the 30S subunit of bacterial 16S ribosomal RNA and interfere with translation. These combined effects are bactericidal.
TOXICITY
The aminoglycosides have a narrow therapeutic to toxic side effect ratio, and monitoring of serum levels is generally required to prevent toxicity. These agents are among the most toxic drugs prescribed today, and they should be avoided whenever safer alternative antibiotics are available (Table 1.7).
KEY POINTS
About Aminoglycoside Toxicity
1. Very low ratio of therapeutic benefit to toxic side effect.
2. Monitoring of serum levels usually required.
3. Nephrotoxicity commonly occurs (usually reversible). Incidence is higher in
a) elderly individuals,
b) patients with preexisting renal disease,
c) patients with volume depletion and hypotension, and
d) patients with liver disease.
4. Higher incidence of nephrotoxicity with coad-ministration of vancomycin, cephalosporins, clindamycin, piperacillin, foscarnet, or furosemide.
5. The loss of high-frequency hearing and vestibular dysfunction resulting from ototoxicity is often devastating for elderly individuals.
6. Neuromuscular blockade is rare.
7. Once-daily therapy may be less toxic.
Two major toxicities are observed:
1. Nephrotoxicity. Injury to the proximal convoluted tubules of the kidney leads to a reduction in creatinine clearance. The brush border cells of the proximal tubule take up aminoglycosides by endocytosis, and intracellular entry is associated with cell necrosis. Aminoglycosides cause significant reductions in glomerular filtration in 5-25% of patients. Patient characteristics associated with an increased risk of nephrotoxicity include older age, preexisting renal disease, hepatic dysfunction, volume depletion, and hypotension. Reexposure to aminoglycosides increases risk, as do the use of larger doses, more frequent dosing intervals, and treatment for more than 3 days. The risk of renal failure is also associated with coadministration of vancomycin, amphotericin B, clindamycin, piperacillin, cephalosporins, foscarnet, or furosemide. Because renal tubular cells have regenerative power, renal dysfunction usually reverses on discontinuation of the aminoglycoside. Because aminoglycosides are primarily renally cleared, aminoglycoside serum levels are useful for detecting worsening renal function. Trough aminoglycoside serum levels often rise before a significant rise in serum creatinine can be detected.
2. Ototoxicity. Aminoglycosides enter the inner ear fluid and damage outer hair cells important to the detection of high-frequency sound. Loss of high-frequency hearing occurs in 3-14% of patients treated with aminoglycosides. The risk of hearing loss is greater after prolonged treatment, with most cases developing after 9 or more days of therapy. Hearing loss is irreversible and can occur weeks after therapy has been discontinued. A genetic predisposition has been observed, with certain families having a high incidence of deafness after receiving aminoglycosides. The risk of hearing loss depends on the specific aminoglycoside. Neomycin has the highest risk of toxicity, followed in order of decreasing frequency by gentamicin, tobramycin, amikacin, and netilmicin. Concomitant use of furosemide or vancomycin and exposure to loud noises increase the risk. As compared with dosing at 8-hour intervals, once-daily dosing reduces the toxic risk.
Less commonly, aminoglycosides can cause neuromuscular blockade; they should be avoided in myasthenia gravis. Given the high risk of toxicity, aminoglycosides should be used only when alternative antibiotics are unavailable. When aminoglycosides are required, the duration of therapy should be as brief as possible. Pretreatment and periodic testing of high-frequency hearing should be performed, and serum creatinine and aminoglycoside serum levels should be monitored.
PHARMACOKINETICS
Following intravenous infusion, aminoglycosides take 15-30 minutes to distribute throughout the body. Therefore, to determine peak serum level, blood samples should be drawn 30 minutes after completion of the intravenous infusion. The half-life of aminoglycosides is 2-5 hours, and these agents are cleared by the kidneys.
Proper dosing of aminoglycosides is more complicated than for most other antibiotics, and these agents require close monitoring. In many hospitals, a pharmacist is consulted to assist in dose management. For daily multiple-dose therapy, a loading dose is first given to rapidly achieve a therapeutic serum level; maintenance doses are then administered. Doses are calculated based on ideal body weight. In the setting of renal dysfunction, dosing must be carefully adjusted, and peak and trough serum levels monitored. As renal impairment worsens, the dosage interval should be extended.
Once-daily aminoglycoside dosing is now the preferred therapy in nearly all instances. As compared with multidose therapy, once-daily administration reduces the concentration of the aminoglycoside that accumulates in the renal cortex and lowers the incidence of nephrotoxicity. Because aminoglycosides demonstrate concentration-dependent killing, the high peak levels achieved with this regimen increase the bactericidal rate and prolong the post-antibiotic effect. In addition, a once-daily regimen is simpler and less expensive to administer. This regimen has not been associated with a higher incidence of neuromuscular dysfunction. To adjust for renal impairment, the daily dose should be reduced.
Monitoring of serum levels is recommended for both multidose and once-daily regimens. With multidose therapy, blood for a peak level determination should be drawn 30 minutes after intravenous infusion is complete, and for a trough level, 30 minutes before the next dose. Blood for peak and trough determinations should be drawn after the third dose of antibiotic to assure full equilibration within the distribution volume. In the critically ill patient, blood for a peak level determination should be drawn after the first dose to assure achievement of an adequate therapeutic level.
For once-daily dosing, trough levels need to be monitored to assure adequate clearance. Serum level at 18 hours should be <1 µg/mL. Alternatively, blood for a level determination can be drawn between 6 and 14 hours, and the value applied to a nomogram to help decide on subsequent doses. In the seriously ill patient, blood for a peak level determination should also be drawn 30 minutes after completion of the infusion to assure that a therapeutic level is being achieved (for gentamicin–tobramycin, a target concentration of 16 to 24 µg/mL should be achieved). Once-daily dosing is not recommended for the treatment of enterococcal endocarditis and has not been sufficiently studied in pregnancy or in patients with osteomyelitis or cystic fibrosis.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
The aminoglycosides are cidal for most aerobic gram-negative bacilli, including Pseudomonas species. These agents kill rapidly, and the killing is concentration-dependent—that is, the rate increases as the concentration of the antibiotic increases. Once-daily dosing takes advantage of this characteristic. Aminoglycosides also demonstrate persistent suppression of bacterial growth for 1-3 hours after the antibiotic is no longer present. The higher the concentration of the aminoglycoside, the longer the post-antibiotic effect. Aminoglycosides also demonstrate synergy with antibiotics that act on the cell wall (β-lactam antibiotics and glycopeptides). The effect of these combinations is greater than the sum of the antimicrobial effects of each individual agent. Synergy has been achieved in the treatment of enterococci, S. viridans, S. aureus, coagulase-negative staphylococci, P. aeruginosa, L. monocytogenes, and JK corynebacteria.
KEY POINTS
About Dosing and Serum Monitoring of Aminoglycosides
1. Aminoglycosides take 15-30 minutes to equilibrate in the body.
2. For multidose therapy, blood for a peak serum level determination should be drawn 30 minutes after infusion.
3. Blood for trough serum level determinations should be drawn just before the next dose.
4. Conventionally, aminoglycosides are given three times daily. Dosing should be based on lean body weight.
5. Once-daily dosing takes advantage of concentration-dependent killing and the post-antibiotic effects of aminoglycosides.
6. Once-daily dosing reduces, but does not eliminate, nephrotoxicity.
7. In most cases, trough serum levels need to be monitored only during once-daily dosing. Toxicity correlates with high trough levels.
8. Once-daily dosing is not recommended for enterococcal endocarditis or pregnant women.
An aminoglycoside in combination with other antibiotics is generally recommended for treatment of the severely ill patients with sepsis syndrome to assure broad coverage for gram-negative bacilli. An aminoglycoside combined with penicillin is recommended for empiric coverage of bacterial endocarditis. Tobramycin combined with an antipseudomonal penicillin or an antipseudomonal cephalosporin is recommended as primary treatment of P. aeruginosa.Streptomycin or gentamicin is the treatment of choice for tularemia and Yersinia pestis, and either agent can also be used to treat Brucella. Gentamicin combined with penicillin is the treatment of choice for both S. viridans and E. faecalis.
Glycopeptide Antibiotics
Tables 1.7 and 1.8, together with Figure 1.12, summarize the characteristics of the glycopeptide antibiotics.
Table 1.8. Glycopeptides, Macrolides, Clindamycin, Tetracyclines, and Chloramphenicol: Half-Life, Dosing, Renal Dosing, Cost, and Spectrum


Figure 1.12. Antibiogram of macrolides, ketolides, tetracyclines, clindamycin, chloramphenicol, and glycopeptides.
CHEMISTRY AND MECHANISM OF ACTION
Vancomycin and teicoplanin are complex glycopeptides of approximately 1500 Da molecular weight. These agents act primarily at the cell wall of gram-positive organisms by binding to the D-alanine-D-alanine precursor and preventing it from being incorporated into the peptidoglycan. The binding of vancomycin to this cell wall precursor blocks the transpeptidase and transglycosylase enzymes, interfering with cell wall formation and increasing permeability of the cell. These agents may also interfere with RNA synthesis. They bind rapidly and tightly to bacteria and rapidly kill actively growing organisms. They also have a 2-hour post-antibiotic effect.
TOXICITY
The most common side effect of the glycopeptide antibiotics is “red man syndrome,” which occurs most often when vancomycin is infused rapidly (Table 1.7). The patient experiences flushing of the face, neck, and upper thorax. This reaction is thought to be caused by sudden histamine release secondary to local hyperosmolality and not to be a true hypersensitivity reaction. Infusing vancomycin over a 1-hour period usually prevents this reaction. There is less experience with teicoplanin; however, this agent does not cause significant thrombophlebitis, and skin flushing after rapid infusion is uncommon. Ototoxicity has been reported.
PHARMACOKINETICS
The half-lives of vancomycin (4-6 hours) and teicoplanin (40-70 hours) are prolonged (Table 1.8). Both drugs are excreted primarily by the kidneys, and in the anuric patient, the half-life of vancomycin increases to 7-9 days. For vancomycin, peak levels should reach 20-50 µg/mL, with trough levels being maintained at 10-12 µg/mL. Vancomycin penetrates most tissue spaces, but does not cross the blood–brain barrier in the absence of inflammation. Therapeutic cerebrospinal levels are achieved in patients with meningitis. Unlike vancomycin, which is minimally bound to protein, teicoplanin is 90% protein-bound, accounting for its slow renal clearance. Tissue penetration has not been extensively studied, and little data are available on penetration of bone, peritoneal, or CSF.
KEY POINTS
About Glycopeptide Antibacterial Activity
1. Act on the cell wall of gram-positive bacteria by binding to the D-alanine-D-alanine peptidoglycan precursor.
2. Require active bacterial growth.
3. Also interfere with RNA synthesis.
4. Have a 2-hour post-antibiotic effect.
KEY POINTS
About Vancomycin Toxicity
1. Rapid infusion associated with “red man syndrome.”
2. Phlebitis is common.
3. Ototoxicity leading to deafness uncommon, preceded by tinnitus
4. Rarely nephrotoxic, potentiates aminoglycoside nephrotoxicity
ANTIMICROBIAL SPECTRUM AND TREATMENT RECOMMENDATIONS
Vancomycin and teicoplanin both cover MRSA and MSSA, and they are the recommended treatment of MRSA. These agents also kill most strains of coagulase-negative staphylococci (S. epidermidis), which are usually methicillin resistant. They are recommended for the treatment of coagulase-negative staphylococcal line sepsis and bacterial endocarditis. For the latter infection, the glycopeptide antibiotic should be combined with one or more additional antibiotics (see Chapter 7). Vancomycin-intermediately resistant strains of S. aureus (VISA) were first discovered in Japan and have also been identified in Europe and the United States. These strains have MICs of 8-16 µg/mL and are cross-resistant to teicoplanin. The increasing use of vancomycin has selected for these strains and warns us that the indiscriminant use of the glycopeptide antibiotics must be avoided. Ceftaroline should be the treatment of choice for VISA.
Vancomycin and teicoplanin not only have activity against Staphylococcus, but also against penicillin-resistant and susceptible strains of S. pneumoniae, and they are recommended for empiric treatment of the seriously ill patient with pneumococcal meningitis to cover for highly penicillin-resistant strains. In the future, ceftaroline may prove to be the preferred agent given its ability to cross the blood–brain barrier and excellent cidal activity against penicillin-resistant strains of S. pneumonia. The glycopeptide antibiotics also effectively treat S. pyogenes, GpB streptococci, S. viridans, and S. bovis, and they are recommended for treatment of these infections in the penicillin-allergic patient. Corynebacterium jeikeium (previously called JK diphtheroids) is sensitive to vancomycin, and that antibiotic is recommended for its treatment. Oral vancomycin clears C. difficile from the bowel, and in the past it was recommended for C. difficile toxin-associated diarrhea. However, because of the increased risk of developing VRE following oral vancomycin, this regimen is recommended only for cases that are refractory to metronidazole or for patients who are very seriously ill.
KEY POINTS
About the Treatment Recommendations for Vancomycin
1. Treatment of choice for methicillin-resistant Staphylococcus aureus; vancomycin-tolerant strains have been reported.
2. Treatment of choice for coagulase-negative staphylococci.
3. Excellent activity against high-level penicillin-resistant Streptococcus pneumoniae.
4. In the penicillin-allergic patient, vancomycin is recommended for S. pyogenes, Gp B streptococci, S. viridans, and S. bovis.
5. Excellent activity against some strains of Entero-coccus; however, vanA gene-mediated vanco-mycin-resistant enterococci (VRE) are increasing in frequency.
Vancomycin is frequently used to treat E. faecalis and faecium; however, an increasing number of strains have become resistant. Three gene complexes transfer resistance. The vanA gene cluster directs peptidoglycan cell wall synthesis and coverts D-alanine-D-alanine (the site of vancomycin action) to D-alanine-D-lactate, markedly reducing vancomycin and teicoplanin binding. The other two resistance gene clusters, van B and van C, result in vancomycin resistance, but do not impair teicoplanin activity.
Macrolides and Ketolides
Tables 1.7 and 1.8, together with Figure 1.12, summarize the characteristics of the macrolides and ketolides. (Also see the section on outpatient antibiotics.)
CHEMISTRY AND MECHANISM OF ACTION
The founding member of the macrolide family, erythromycin, was originally purified from a soil bacterium. It has a complex 14-member macrocyclic lactone ring (which gives rise to the class name “macrolides”) attached to two sugars. Azithromycin has a 15-member lactone ring and a nitrogen substitution. Clarithromycin has a methoxy group modification at carbon 6 of the erythromycin molecule. These modifications enhance oral absorption and broaden the antimicrobial spectrum.
The newest class of macrolide-like agents are the semisynthetic derivatives of erythromycin called ketolides. The ketolides, represented by telithromycin, have a 14-member macrolactone ring with a keto group at position 3, with the hydroxyls at positions 11 and 12 replaced by a cyclic carbamate. These agents all inhibit protein biosynthesis by blocking the passage of nascent proteins through the ribosome exit tunnel. In the case of conventional macrolides, inhibition is accomplished by binding to a single domain of the 50S ribosomal subunit (domain V of the 23 rRNA molecule). As compared with the macrolides, telithromycin binds to the 50S subunit with higher affinity, binding to two regions of the 23S rRNA molecule (domains II and V) rather than one region. This unique binding mode explains the enhanced antimicrobial activity of ketolides against macrolide-resistant pathogens.
TOXICITY
Macrolides and ketolides are among the safer classes of antibiotics (Table 1.7), but do have some significant toxicities. The primary adverse reactions are related to these agents’ ability to stimulate bowel motility. In fact, erythromycin can be used to treat gastric paresis. Particularly in younger patients, abdominal cramps, nausea, vomiting, diarrhea, and gas are common with erythromycin. These symptoms are dose related and are more common with oral preparations, but can also occur with intravenous administration. Gastrointestinal toxicity can be debilitating, forcing the drug to be discontinued. Azithromycin and clarithromycin at the usual recommended doses are much less likely to cause these adverse reactions.
Telithromycin administration has been accompanied by difficulty with accommodation, resulting in blurred vision. Patients have also experienced diplopia following administration of this agent. Telithromycin treatment has resulted in the sudden onset of severe and occasionally fatal hepatitis. All patients receiving this agent should therefore be warned of this potential side effect, and the drug should be prescribed only for cases of pneumonia in which the incidence of penicillin-resistant S. pneumoniae is high. Under these circumstances, a fluoroquinolone with gram-positive coverage may be preferred.
Macrolides and ketolides may exacerbate myasthenia gravis and should be avoided in patients with that illness. Macrolides prolong the QT interval, and erythromycin administration has, on rare occasions, been associated with ventricular tachycardia.
These agents are metabolized by the cytochrome P450 3A4 system, and they cause an increase in serum levels of other drugs metabolized by that system, including many of the statins, short-acting benzodiazepines, such as midazolam (Versed), cisapride (Propulsid), ritonavir (Norvir), and tacrolimus (Prograf).
PHARMACOKINETICS
The stearate, ethylsuccinate, and estolate forms of erythromycin are reasonably well absorbed on an empty stomach, reaching peak serum levels 3 hours after ingestion. Clarithromycin, azithromycin, and telithromycin are better absorbed orally than erythromycin is, resulting in peak concentrations within 1 hour. Erythromycin and azithromycin should be taken on an empty stomach. If cost is not a primary issue, the improved absorption and lower incidence of gastrointestinal toxicity make the three newer agents preferable to erythromycin in most instances (Table 1.8).
Most of the macrolides and ketolides are metabolized and cleared primarily by the liver. Azithromycin is not metabolized, being excreted unchanged in the bile. Small percentages of these drugs are also excreted in the urine. These agents are widely distributed in tissues, achieving concentrations that are several times the peak concentration achieved in serum in most areas of the body, including the prostate and middle ear. Clarithromycin levels in middle ear fluid have been shown to be nearly 10 times serum levels. Azithromycin concentrations in tissue exceed serum levels by a factor of 10-100, and its average half-life in tissues is 2-4 days. Therapeutic levels of azithromycin have been estimated to persist for 5 days after the completion of a 5-day treatment course. With the exception of intravenous erythromycin, these agents fail to achieve significant levels in the CSF.
KEY POINTS
About Macrolide Chemistry, Mechanism of Action, and Toxicity
1. Complex 14- to 15-member lactone ring structure.
2. Inhibit RNA-dependent protein synthesis, bind to 50S ribosomal subunit; telithromycin binds with higher affinity, binding to two sites rather than just one.
3. Can be bacteriostatic or cidal.
4. Gastrointestinal irritation, particularly with erythromycin, is the major toxicity.
5. Hypersensitivity reactions can occur.
6. Transient hearing loss with high doses, mainly in elderly individuals.
7. Telithromycin can cause blurred vision and diplopia. Also can result in fatal hepatitis.
8. Can exacerbate myasthenia gravis.
9. Prolonged QT interval; occasionally causes ventricular tachycardia.
10. Metabolized by the cytochrome P450 3A4 system; increase serum concentrations of other drugs metabolized by that system.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
Macrolides demonstrate excellent activity against most gram-positive organisms and some gram-negative bacteria. Erythromycin can be bacteriostatic or bactericidal. Cidal activity increases when antibiotic concentrations are high and bacteria are growing rapidly (Figure 1.12).
These drugs are recommended for the treatment of community-acquired pneumonia (see Chapter 4). However, S. pneumoniae resistance to macrolides has steadily increased and now ranges between 10% and 15%. Resistance is more likely in intermediately penicillin-resistant strains (40% macrolide resistant) and highly penicillin-resistant strains (60% macrolide resistance). Multiresistant S. pneumoniae can be treated with telithromycin as a consequence of that agent’s different ribosomal binding sites.
In most countries, including the United States, 95% of S. pyogenes are sensitive to macrolides, and these agents are recommended for bacterial pharyngitis. However, in Japan, where macrolides are commonly used, 60% are resistant. Because S. aureus can develop resistance after a single mutation, macrolides are generally not recommended in their treatment, and should only be used for mild soft tissue infections. The macrolides and ketolides are effective against mouth flora, including anaerobes, but they do not cover the bowel anaerobe B. fragilis. They are recommended for both bacterial sinusitis and acute otitis media. The macrolides are also the treatment of choice for Legionella pneumophila, with telithromycin, azithromycin, and clarithromycin being more potent than erythromycin.
KEY POINTS
About the Spectrum and Treatment Indications for Macrolides and Ketolides
1. Gram-positive coverage, plus mouth anaerobes.
2. Recommended for treatment of community-acquired pneumonia.
3. Increased use of macrolides selects for resistant strains of Streptococcus pyogenes and S. pneu-moniae. Penicillin-resistant strains of S. pneu-moniae are often resistant to macrolides.
4. Telithromycin is effective against multiresistant S. pneumoniae.
5. Recommended for treatment of Legionella pneumophila.
6. Recommended for Mycoplasma, Ureaplasma, and Chlamydia.
7. Clarithromycin or azithromycin can used for treatment of Helicobacter pylori.
8. Clarithromycin is a primary drug for treatment of Mycobacterium avium intracellulare MAI), and azithromycin is useful for MAI prophylaxis in patients with HIV with low CD4 cell counts.
Macrolides are the primary antibiotics used to treat the two major pathogens associated with atypical pneumonia: Mycoplasma pneumoniae and Chlamydophila pneumoniae (see Chapter 4). Erythromycin, clarithromycin, and azithromycin, and telithromycin are approved for mild-to-moderate community-acquired pneumonia. In many instances, the erythromycins can be used as an alternative to penicillin in the penicillin-allergic patient. These agents are also indicated for acute exacerbations of chronic obstructive pulmonary disease.
Clarithromycin is one of the primary antibiotics used for the treatment of atypical mycobacterial infections, particularly MAI complex. Azithromycin in combination with other antibiotics is also recommended for the treatment of MAI complex, and it can be used alone for MAI prophylaxis in HIV-infected patients with CD4 cell counts below 100 cells/mL.
In combination with antacid therapy, effective regimens for curing peptic ulcer disease caused by Helicobacter pylori include azithromycin or clarithromycin combined with bismuth salts and either amoxicillin, metronidazole, or tetracycline. Single high-dose azithromycin (1 g) effectively treats chancroid, as well as Chlamydia trachomatis urethritis and cervicitis. Single-dose therapy also cures male Ureaplasma urealyticum urethritis.
Clindamycin
Tables 1.7 and 1.8, together with Figure 1.12, summarize the characteristics of the clindamycin.
CHEMISTRY AND MECHANISM OF ACTION
Although clindamycin is structurally different from erythromycin, many of its biologic characteristics are similar. Clindamycin consists of an amino acid linked to an amino sugar, and it was derived by modifying lincomycin. It binds to the same 50S ribosomal binding site used by the macrolides, blocking bacterial protein synthesis.
TOXICITY
Diarrhea is a major problem seen in 20% of patients taking clindamycin (Table 1.7). The incidence is highest with oral administration. In up to half of the affected patients, the cause of diarrhea is pseudomembranous colitis, a disease caused by overgrowth of the anaerobic bacteria C. difficile (see Chapter 8).
KEY POINTS
About Clindamycin
1. Binds to the 50S ribosomal binding site used by the macrolides.
2. Diarrhea is a common side effect, with Clostrid-ium difficile toxin found in half of cases.
3. Pseudomembranous colitis can lead to toxic megacolon and death. If C. difficile toxin is detected, clindamycin should be discontinued.
4. Active against most gram-positive organisms including MSSA; covers many intermediate penicillin-resistant Streptococcus pneumoniae, but is not a first-line therapy.
5. Excellent anaerobic coverage, including Bacte-roides fragilis.
6. Used to reduce toxin production by Streptococcus pyogenes and Staphylococcus aureus.
7. Used to treat anaerobic lung abscesses and toxoplasmosis in the sulfa-allergic patient.
PHARMACOKINETICS
Clindamycin is well absorbed orally; however, the drug can also be administered intravenously and the intravenous route can achieve higher peak serum levels (Table 1.8). Clindamycin penetrates most tissues, but it does not enter the CSF. Clindamycin is metabolized primarily by the liver and is excreted in the bile. Therapeutic concentrations of clindamycin persist in the stool for 5 or more days after the antibiotic is discontinued, and the reduction in clindamycin-sensitive flora persists for up to 14 days. Small percentages of clindamycin metabolites are also excreted in the urine.
ANTIMICROBIAL SPECTRUM AND TREATMENT RECOMMENDATIONS
Clindamycin is similar to erythromycin in its activity against streptococci and staphylococci (Figure 1.12). Moderately penicillin-resistant S. pneumoniae are often sensitive to clindamycin. In the penicillin-allergic patient, clindamycin is a reasonable alternative for S. pyogenes pharyngitis. Because its activity against H. influenzae is limited, clindamycin is not recommended for the treatment of otitis media.
Clindamycin distinguishes itself from the macrolides by possessing excellent activity against most anaerobic bacteria. It has been used in combination with an aminoglycoside, aztreonam, or a third-generation cephalosporin to treat fecal soilage of the peritoneum. However, other less-toxic regimens have proved to be more effective, and the prevalence of B. fragilis-resistant strains is increasing, making this a less reliable regimen for intra-abdominal infections. The IDSA no longer recommends clindamycin for intra-abdominal infections. Clindamycin in combination with a first-generation cephalosporin can be used to block toxin production in severe cellulitis and necrotizing fasciitis caused by MSSA or S. pyogenes. It is also effective for the treatment of anaerobic pulmonary and pleural infections. Clindamycin also has significant activity against Toxoplasma gondii and is recommended as alternative therapy in the sulfa-allergic patient.
Tetracyclines
Tables 1.7 and 1.8, together with Figure 1.12, summarize the characteristics of tetracyclines.
CHEMISTRY AND MECHANISMS OF ACTION
The tetracyclines consist of four 6-member rings with substitutions at the 4, 5, 6, and 7 positions that alter the pharmacokinetics of the various preparations; however, with the exception of tigecycline, these changes have no effect on the antimicrobial spectrum. The tetracyclines enter gram-negative bacteria by passively diffusing through porins. They bind to the 30S ribosomal subunit and block tRNA binding to the mRNA ribosome complex. This blockade primarily inhibits protein synthesis in bacteria, but to a lesser extent, it also affects mammalian cell protein synthesis, particularly mitochondria. The inhibition of bacterial protein synthesis stops bacterial growth, but does not kill the bacterium. Therefore, tetracycline is termed a bacteriostatic agent.
TOXICITY
Photosensitivity reactions consisting of a red rash over sun-exposed areas can develop (Table 1.7). Hypersensitivity reactions are less common than with the penicillins, but they do occur. Tetracyclines interfere with enamel formation, and in children, teeth often become permanently discolored. Therefore, these agents are not recommended for children 8 years of age or younger, or for pregnant women. Because the tetracyclines inhibit protein synthesis, they increase azotemia in renal failure patients. Minocycline can cause vertigo, and that side effect has limited its use. Benign intracranial hypertension (pseudotumor cerebri) is another rare neurologic side effect.
PHARMACOKINETICS
Tetracycline is reasonably well absorbed (70-80%) by the gastrointestinal tract (see Table 1.8). Food interferes with its absorption. Doxycycline is nearly completely absorbed in the gastrointestinal tract. Calcium- or magnesium-containing antacids, milk, or multivitamins markedly impair absorption of all tetracycline preparations, and simultaneous ingestion of these products should be avoided. Tigecycline can be administered only intravenously. Tetracycline is cleared primarily by the kidneys; other agents, including doxycycline and tigecycline, are cleared primarily by the liver.
KEY POINTS
About the Tetracyclines
1. Bind to the 30S subunit of the ribosome, blocking tRNA binding and inhibiting protein synthesis. Bacteriostatic for most gram-positive and gram-negative bacteria.
2. Toxicities include photosensitivity, interference with dental enamel formation in children, gastrointestinal discomfort, fatty liver changes, exacerbation of azotemia, vertigo (minocycline), and pseudotumor cerebri.
3. Tetracycline can be used for uncomplicated urinary tract infections.
4. Recommended for brucellosis, Lyme disease, chlamydia, and rickettsial infections.
5. Recommended, in combination with other antibiotics, for pelvic inflammatory disease.
6. Oral absorption blocked by calcium- and magnesium-containing antacids, milk, and multivitamins.
7. Tigecycline has improved gram-positive and gram-negative coverage, with the exception of Pseudomonas aeruginosa and Proteus. It is approved for complicated intra-abdominal and soft tissue infections.
ANTIMICROBIAL SPECTRUM AND TREATMENT RECOMMENDATIONS
The tetracyclines are able to inhibit the growth of a broad spectrum of bacteria (Figure 1.12). However, for most conventional pathogens, other agents are more effective. High concentrations of tetracycline are achieved in the urine, and this agent can be used for uncomplicated urinary tract infections. Doxycycline combined with gentamicin is the treatment of choice for brucellosis. Tetracyclines are also recommended for the treatment of Lyme disease (Borrelia burgdorferi), and chlamydia infections (including Chlamydia pneumonia, psittacosis, epididymitis, urethritis, and endocervical infections). Tetracyclines are the treatment of choice for rickettsial infections (including Rocky Mountain spotted fever, ehrlichiosis, Q fever, and typhus fever). They are also often used in combination with other antibiotics for the treatment of pelvic inflammatory disease.
The most recently developed member of this family, tigecycline, was derived from minocycline. Tigecycline has a broader spectrum of activity. It effectively inhibits the growth of many resistant gram-positive bacteria (Figure 1.12). This agent also demonstrates improved activity against many highly resistant nosocomial gram-negative bacteria, but it does not effectively cover P. aeruginosa or Proteusspecies. Tigecycline is approved for complicated intra-abdominal and soft tissue infections, but should probably be avoided in severe infections.
Chloramphenicol
Tables 1.7 and 1.8, together with Figure 1.12 summarize the characteristics of chloramphenicol.
CHEMISTRY AND MECHANISMS OF ACTION
Chloramphenicol consists of a nitro group on a benzene ring and a side chain containing five carbons. Chloram-phenicol uses an energy-dependent mechanism to enter bacteria, and once in the cell, binds to the larger 50S subunit of the 70S ribosome, blocking attachment of tRNA. It inhibits bacterial protein synthesis, making it bacteriostatic for most bacteria; however, chloramphenicol is cidal for H. influenzae, S. pneumoniae, and Neisseria meningitidis.
TOXICITY
Probably as result of its binding to human mitochondrial ribosomes, this agent has significant bone marrow toxicity (see Table 1.7). Two forms are observed. The first form is dose related and is commonly observed in patients receiving chloramphenicol 4 g or more daily. The reticulocyte count decreases, and anemia develops in association with elevated serum iron. Leukopenia and thrombocytopenia are also commonly encountered. These changes reverse when the antibiotic is discontinued. The second form of marrow toxicity, irreversible aplastic anemia, is rare, but usually fatal. This complication can occur weeks or months after the antibiotic is discontinued. Any patient receiving chloramphenicol requires twice-weekly monitoring of peripheral blood counts. If the WBC drops below 2500/mm3, the drug should be discontinued.
PHARMACOKINETICS
As a result of the much higher incidence of idiosyncratic aplastic anemia associated with oral administration as compared with intravenous administration, oral preparations of chloramphenicol are no longer available in the United States. The drug is well absorbed, and therapeutic serum levels can be achieved orally (Table 1.8). Chloramphenicol is metabolized by the liver. It diffuses well into tissues and crosses the blood-brain barrier in uninflamed as well as inflamed meninges. A serum assay is available, and serum levels should be monitored in patients with hepatic disease, maintaining the serum concentration between 10 and 25 µg/mL.
KEY POINTS
About Chloramphenicol
1. Binds to 50S subunit of the ribosome, blocking protein synthesis; is bacteriostatic.
2. Idiosyncratic aplastic anemia has limited the use of chloramphenicol; dose-related bone marrow suppression is another concern.
3. Broad spectrum of activity, including Salmonella, Brucella, Bordetella, anaerobes, Rickettsiae, Chlamydiae, Mycoplasma, and spirochetes.
4. Can be used as alternative therapy in the penicillin-allergic patient.
ANTIMICROBIAL SPECTRUM AND TREATMENT RECOMMENDATIONS
Chloramphenicol has excellent activity against most gram-positive organisms with the exception of enterococci and S. aureus, as well as many gram-negative pathogens (Figure 1.12). Chloramphenicol also is very active against spirochetes, as well as Rickettsiae, Chlamydiae, and mycoplasmas.
Because of its bone marrow toxicity, chloramphenicol is not considered the treatment of choice for any infection. Alternative, less-toxic agents are available for each indication. For the penicillin-allergic patient, chloramphenicol can be used for bacterial meningitis. Chloramphenicol can also be used as alternative therapy for brain abscess, C. perfringens, psittacosis, rickettsial infections including Rocky Mountain spotted fever, Vibrio vulnificus, and typhoid fever.
Quinolones
Tables 1.7 and 1.9, together with Figure 1.13, summarize the characteristics of the quinolone antibiotics. (Also see the section on outpatient antibiotics)
Table 1.9. Quinolones, Linezolid, Quinupristin/Dalfopristin, Daptomycin, Metronidazole, Sulfonamides and Colistin: Half-Life, Dosing, Renal Dosing, Cost, and Spectrum


Figure 1.13. Antibiogram of quinolones.
CHEMICAL STRUCTURE AND MECHANISMS OF ACTION
The quinolones all contain two 6-member rings (see Figure 1.14) with a nitrogen at position 1, a carbonyl group at position 4, and a carboxyl group attached to the carbon at position 3. Potency of the quinolones is greatly enhanced by adding fluorine at position 6, and gram-negative activity is enhanced by addition of a nitrogen-containing piperazine ring at position 7.

Figure 1.14. Basic structure of the quinolones.
The quinolones inhibit two enzymes critical for DNA synthesis: DNA gyrase, which is important for regulating the superhelical twists of bacterial DNA, and topoisomerase IV, which is responsible for segregating newly formed DNA into daughter cells. The loss of these activities blocks DNA synthesis and results in rapid bacterial death. Killing is concentration-dependent.
TOXICITY
The most common side effects are mild anorexia, nausea, vomiting, and abdominal discomfort (Table 1.7). Quinolones can result in arthropathy because of cartilage damage and tendonitis. Although rare, this complication can be debilitating, but it usually reverses weeks to months after the quinolone is discontinued. Because of concerns about cartilage damage in children, quinolones are not recommended for routine administration in pediatric patients. Gatifloxacin administration can be associated with severe dysregulation of glucose homeostasis and can result in either severe hypo- or hyperglycemia. Fluoroquinolones are associated with a concentration-dependent delay in cardiac repolarization, causing a prolongation of the QT interval—a condition that can predispose to ventricular tachycardia. In combination with other agents that effect repolarization, moxifloxacin has occasionally been associated with life-threatening cardiac arrhythmias.
KEY POINTS
About the Chemistry, Mechanisms of Action, and Toxicity of Quinolones
1. Inhibit bacterial DNA gyrase (important for coiling DNA) and topoisomerase (required to segregate DNA to daughter cells). Rapidly cidal, with concentration-dependent killing.
2. Main side effects are as follows:
a) Nausea and anorexia.
b) Allergic reactions (most common with gemifloxacin; less common with other quinolones).
c) Arthropathy and tendonitis. May damage cartilage. Not routinely recommended in children.
d) Gatifloxacin can cause hypo- or hyperglycemia.
e) Moxifloxacin prolongs the QT interval.
PHARMACOKINETICS
The quinolones are readily absorbed orally, but can also be given intravenously. Ciprofloxacin, levofloxacin, and gatifloxacin are cleared primarily by the kidneys. Moxifloxacin is also partially metabolized by the liver, and gemifloxacin is metabolized primarily by the liver. All quinolones demonstrate similar tissue penetration, being concentrated in prostate tissue, feces, bile, and lung tissue. These drugs tend to be very highly concentrated in macrophages and neutrophils.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
Ciprofloxacin—Ciprofloxacin is the most potent quinolone for P. aeruginosa (Figure 1.13). As a result of an excellent gram-negative spectrum, ciprofloxacin is one of the primary antibiotics recommended for treatment of urinary tract infections. It concentrates in the prostate and is recommended for treatment of prostatitis. For gonococcal urethritis, it is a useful alternative to ceftriaxone. Cipro-floxacin has been used effectively for traveler’s diarrhea most commonly caused by enterotoxigenic E. coli and Shigella. It is the drug of choice for Salmonella typhi (typhoid fever), and it also is recommended for treatment of Salmonella gastroenteritis when antibiotic treatment is necessary. Ciprofloxacin is the recommended treatment of cat scratch disease caused by Bartonella henselae.
Levofloxacin, Moxifloxacin, Gatifloxacin, and Gemifloxacin—These agents all demonstrate improved gram-positive coverage (Figure 1.13) and have been recommended as one of the first-line treatments for community-acquired pneumonia in the otherwise healthy adult who does not require hospitalization. With the exception of gemifloxacin, these agents can also be used in soft tissue infection in which a combination of gram-positive and gram-negative organisms is suspected. Given the worse toxicity profiles of the three newer agents (moxifloxacin, gatifloxacin, and gemifloxacin), levofloxacin should probably be the fluoroquinolone of choice for those infections. Gatifloxacin and moxifloxacin demonstrate moderate in vitro activity against anaerobes and may be considered for the treatment of mixed infections thought to include anaerobes. Fear of selecting for resistant pathogens has led to their use being restricted in some hospitals.
KEY POINTS
About the Specific Quinolones
1. Ciprofloxacin:
a) Excellent coverage of Pseudomonas. Also covers many other gram-negative organisms including Escherichia coli, Salmonella, Shigella, Neisseria, and Legionella.
b) Kills Mycoplasma, Chlamydia, and Urea-plasma.
c) Recommended for urinary tract infections and prostatitis, gonococcal urethritis, traveler’s diarrhea, typhoid fever, and Salmonella gastroenteritis; used for cat scratch disease.
2. Levofloxacin, gatifloxacin, moxifloxacin, gemifloxacin:
a) Greater activity against Streptococcus pneu-moniae, covers highly penicillin-resistant strains.
b) Also cover methicillin-sensitive Staphylococcus aureus.
c) Recommended for community-acquired pneumonia (levofloxacin preferred).
d) Levofloxacin, gatifloxacin, and moxifloxacin recommended for mixed skin infections.
e) Gatifloxacin and moxifloxacin have somewhat improved anaerobic coverage.
f) Gatifloxacin and moxifloxacin recommended for mixed skin infections.
Oxazolidinones (Linezolid)
Tables 1.7 and 1.9, together with Figure 1.15 summarize the characteristics of linezolid.

Figure 1.15. Antibiograms of miscellaneous antibiotics.
CHEMISTRY AND MECHANISMS OF ACTION
The oxazolidinones have a unique ring structure consisting of a 5-member ring containing oxygen and nitrogen. The nitrogen connects to a 6-member ring, and each specific compound has side chains added to both rings at positions A and B (Figure 1.16). These agents bind to the 50S ribosome at a site similar to that used by chloramphenicol. However, unlike chloramphenicol, they do not inhibit the attachment of tRNA, but instead block the initiation of protein synthesis by preventing the nearby 30S subunit from forming the 70S initiation complex. The oxazolidinones are bacteriostatic against staphylococcal species and enterococci.

Figure 1.16. Antibiograms of miscellaneous antibiotics.
TOXICITY
Linezolid is the only agent in this class released for use. Reversible thrombocytopenia has been reported in association with prolonged therapy, and monitoring of platelet count is recommended for patients receiving two or more weeks of linezolid. Leukopenia and hepatic enzyme elevations have also been reported. Because this agent is a weak inhibitor of monoamine oxidase, hypertension has been reported in association with ingestion of large amounts of tyramine. Pseudoephedrine and selective serotonin reuptake inhibitors should be prescribed with caution.
PHARMACOKINETICS
Linezolid is well absorbed orally, and peak serum levels are achieved in 1-2 hours. Food slows absorption, but does not lower peak levels. An intravenous preparation is also available. Linezolid achieves excellent penetration of all tissue spaces, including the CSF. The drug is partly metabolized by the liver and excreted in the urine.
ANTIMICROBIAL ACTIVITY AND TREATMENT RECOMMENDATIONS
Linezolid demonstrates activity only against gram-positive organisms. It has bacteriostatic activity against both vancomycin-resistant E. faecium and Enterococcus faecalis (VRE). This agent is also active against MSSA and MRSA, and has activity against penicillin-resistant S. pneumoniae. Linezolid is recommended primarily for the treatment of VRE.
KEY POINTS
About Linezolid
1. Like chloramphenicol, binds to the 50S ribosome subunit; inhibits the initiation of protein synthesis.
2. Thrombocytopenia common with treatment exceeding 2 weeks; inhibitor of monoamine oxidase; avoid tyramine, pseudoephedrine, and serotonin uptake inhibitors.
3. Strictly gram-positive activity; bacteriostatic activity for vancomycin-resistant enterococci (VRE), and methicillin-resistant Staphylococcus aureus. Also has activity against penicillin-resistant Streptococcus pneumoniae.
4. Recommended for the treatment of VRE.
Streptogramins
Tables 1.7 and 1.9, together with Figure 1.15, summarize the characteristics of Synercid.
CHEMICAL STRUCTURE AND MECHANISM OF ACTION
The streptogramins belong to the macrolide family. They are derived from pristinamycin. Quinupristin is a peptide derived from pristinamycin IA and dalfopristin is derived from pristinamycin IIB. A combination of 30:70 quinupristin:dalfopristin has synergistic activity and has been named Synercid. These two agents inhibit bacterial protein synthesis by binding to the 50S bacterial ribosome. Quinupristin inhibits peptide chain elongation, and dalfopristin interferes with peptidyl transferase activity.
TOXICITY
Myalgias and arthralgias are the most common and severe adverse reaction, and they can force discontinuation of the drug (Table 1.7). Administration has also been associated with hyperbilirubinemia.
PHARMACOKINETICS
The streptogramins are administered intravenously, and they are metabolized primarily in the liver and require no adjustment for renal dysfunction (Table 1.9).
ANTIMICROBIAL ACTIVITY AND TREATMENT INDICATIONS
Synercid is active primarily against gram-positive organisms (Figure 1.15). It has proved to be efficacious in the treatment of VRE and MRSA. Synercid or linezolid are the treatments of choice for VRE.
Daptomycin
Tables 1.7 and 1.9, together with Figure 1.15, summarize the characteristics of daptomycin.
CHEMICAL STRUCTURE AND MECHANISM OF ACTION
Daptomycin is a large cyclic lipopeptide (C72H101N17O26) with a molecular weight of 1620 that was derived from Streptomyces roseosporus. Daptomycin has a mechanism of action that is distinctly different from that of other antibiotics. It binds to bacterial membranes and causes rapid depolarization of the membrane potential. As a result, protein, DNA, and RNA synthesis is inhibited. This antibiotic is cidal and causes rapid concentration-dependent killing, but it does not result in the systemic release of cell membrane or cell wall contents. It also demonstrates significant post-antibiotic effect. Synergy with aminoglycosides, β-lactam antibiotics, and rifampin has been observed.
KEY POINTS
About Synercid
1. Combination of two pristinamycin derivatives: quinupristin and dalfopristin. Together, they synergistically block protein synthesis. Both bind to the 50S ribosomal subunit.
2. Myalgias and arthralgias can force discontinuation of the drug. Nausea, vomiting, and diarrhea also occur.
3. Spectrum of activity: covers primarily gram-positive bacteria. Active against vancomycin-resistant enterococci (VRE) and methicillin-resistant Staphylococcus aureus.
4. Recommended for the treatment of VRE.
TOXICITY
Muscle pain and weakness are reported in less than 5% of patients. This drug is also associated with a rise in creatine phosphokinase (CPK; Table 1.7). The patient’s CPK levels should be monitored weekly, and the drug should be discontinued if CPK exceeds 1000 in association with symptoms of myopathy, or if CPK exceeds 2000 in the absence of symptoms. Other drugs associated with rhab-domyolysis, specifically HMG-CoA reductase inhibitors (statins), should not be administered with daptomycin. Less commonly, daptomycin administration has resulted in neuropathy associated with a slowing of nerve conduction velocity. The peripheral or cranial nerves can be affected. Patients may experience paresthesia or Bell’s palsy. This rare toxicity has also been observed in animal studies. More recently, daptomycin treatment has also been associated with eosinophilic pneumonia.
PHARMACOKINETICS
Daptomycin is given intravenously, and a 4-mg/kg dose achieves peak serum levels of 58 µg/mL (Table 1.9). Daptomycin is 92% protein-bound and is excreted by the kidneys. Its ability to penetrate various tissue compartments including the CSF has not been extensively studied.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
Daptomycin kills aerobic and facultative gram-positive organisms, including E. faecium and faecalis (including VREs), S. aureus (including MRSA), S. epidermidis (including methicillin-resistant strains), S. pyogenes, and Corynebacterium jeikeium (Figure 1.15). Prolonged treatment of S. aureus with this agent can be associated with a marked rise in its MIC. This antibiotic is approved for the treatment of complicated skin and soft tissue infections by susceptible strains and for S. aureus (including MRSA) bacteremia and right-sided endocarditis. It is not currently approved for VRE, because of insufficient clinical data. Daptomycin is inactivated by surfactant and should not be used for the treatment of pneumonia.
KEY POINTS
About Daptomycin
1. Large, cyclic lipopeptide that binds to and depolarizes bacterial membranes.
2. Rapidly cidal, concentration-dependent killing; post-antibiotic effect.
3. Toxicities include muscle pain and weakness associated with creatine phosphokinase leak; no coadministration of statins. Less common: peripheral or cranial nerve neuropathy.
4. Kills enterococci (including VRE), Staphylococcus aureus (including MRSA), Staphylococcus epidermidis, Streptococcus pyogenes, and corynebacteria.
5. Approved to treat complicated skin and soft tissue infections, and S. aureus (including MRSA) bacteremia and right-sided endocarditis.
6. Inactivated by surfactant; should not be used to treat pneumonia.
Metronidazole
Tables 1.7 and 1.9, together with Figure 1.15, summarize the characteristics of metronidazole.
CHEMICAL STRUCTURE AND MECHANISM OF ACTION
Metronidazole is a nitroimidazole with a low molecular weight that allows it to readily diffuse into tissues. Within a bacterium, this antibiotic acts as an electron acceptor and is quickly reduced. The resulting free radicals are toxic to the bacterium, producing damage to DNA and to other macromolecules. Metronidazole has significant activity against anaerobes.
TOXICITY
Metronidazole is usually well tolerated, but it can result in a disulfiram (Antabuse–like) reaction with alcohol consumption (Table 1.7). Concern about the mutagenic potential of this agent has resulted in multiple mammalian studies that, overall, have failed to demonstrate significant DNA abnormalities. Metronidazole is not recommended in pregnancy, and it should usually be avoided in patients on Coumadin, because it impairs metabolism of that drug.
PHARMACOKINETICS
This agent is rapidly and completely absorbed orally, but it can also be given intravenously. Therapeutic levels are achieved in all body fluids, including the CSF and brain abscess contents. Metronidazole is metabolized primarily in the liver.
KEY POINTS
About Metronidazole
1. Electron acceptor; produces free radicals that damage bacterial DNA.
2. Antabuse-like reaction can occur; mutagenic effects not proven in mammals, but the drug should be avoided in pregnancy. Impairs Coumadin metabolism.
3. Excellent activity against anaerobes, amoebae, Giardia, and Trichomonas. Penetrates tissues well, including abscesses.
4. Indicated in combination with other antibiotics for mixed bacterial infections. Has no activity against aerobic bacteria.
5. Treatment of choice for Clostridium difficile-induced diarrhea. Used as part of combination treatment of Helicobacter pylori.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
Metronidazole was originally used primarily for Trichomonas vaginitis, being effective both topically and orally. It is also effective for treating amoebic abscesses and giardiasis. Metronidazole is cidal for most anaerobic bacteria, and it is the antibiotic of choice for covering anaerobes. Because metronidazole has no significant activity against aerobes, it is usually administered in combination with a cephalosporin for aerobic coverage. Metronidazole is the drug of choice for treatment of pseudomembranous colitis attributable to overgrowth of C. difficile. Metronidazole is also recommended as part of the regime for He. pylori gastric and duodenal infection.
Sulfonamides and Trimethoprim
Tables 1.7 and 1.9, together with Figure 1.15, summarize the characteristics of trimethoprim-sulfamethoxazole.
CHEMICAL STRUCTURE AND MECHANISMS OF ACTION
All sulfonamides have a structure similar to para-aminobenzoic acid (PABA), a substrate required for bacterial folic acid synthesis (Figure 1.17). All sulfonamides inhibit bacterial folic acid synthesis by competitively inhibiting PABA incorporation into tetrahydropteroic acid. These agents are bacteriostatic.
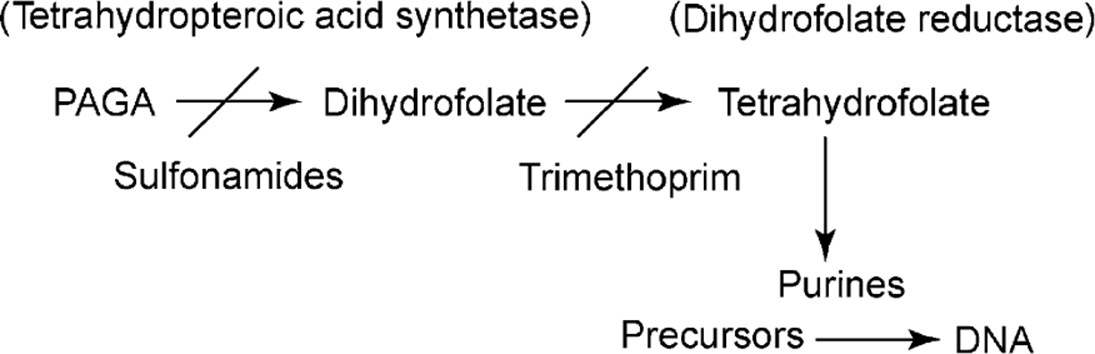
Figure 1.17. Effects of sulfonamides and trimethoprim on the bacterial folate pathway
A sulfonyl radical is attached to carbon 1 of the 6-member ring, increasing PABA inhibition. Alterations in the sulfonyl radical determine many of the pharmacokinetic properties of the compounds. Trimethoprim consists of two 6-member rings, one of which has two nitrogens and two amino groups, the other having three methoxybenzyl groups. This agent strongly inhibits dihydrofolate reductase and complements sulfonamide inhibition of folate metabolism (Figure 1.17). Inhibition of bacterial dihydrofolate reductase by trimethoprim is 100,000 times that of the agent’s inhibition of the mammalian enzyme, minimizing toxicity to the patient.
TOXICITY
Hypersensitivity reactions represent the most severe toxicity (Table 1.7). Maculopapular drug rashes, erythema multiforme, Steven–Johnson syndrome, vasculitis (including drug-induced lupus), serum sickness-like syndrome, and anaphylaxis have been reported. Hemolytic anemia can be associated with glucose-6-phosphate dehydrogenase (G6PD) deficiency. Sulfonamides should be avoided in the last month of pregnancy because they displace bilirubin bound to plasma albumin and increase fetal blood levels of unconjugated bilirubin.
KEY POINTS
About Sulfonamides
1. Competitively inhibit para-aminobenzoic acid incorporation, blocking folic acid synthesis; trimethoprim inhibits dihydrofolate reductase, potentiating sulfonamide activity.
2. Hypersensitivity reactions (including Steven–Johnson syndrome) are common; hemolytic anemia seen in G6PD-deficient patients. Agranulocytosis and thrombocytopenia are less common.
3. Broad spectrum of activity for gram-positive and gram-negative organisms, but resistance is common.
4. Used for initial therapy of uncomplicated urinary tract infections. Treatment of choice for Nocardia.
5. Trimethoprim-sulfamethoxazole combination is the drug of choice for Pneumocystis prophylaxis and treatment.
PHARMACOKINETICS
Sulfonamides are classified as short-, medium-, or long acting, depending on half-life. Sulfisoxazole is in the short-acting class, having a half-life of 5-6 hours. Sulfamethoxazole and sulfadiazine are medium acting. All of these agents are generally well absorbed orally. Intravenous preparations are available for some agents. All are metabolized by the liver, undergoing acetylation and glucuronidation, with the metabolites being excreted in the urine. Trimethoprim is excreted primarily by the renal tubules, and very high concentrations of active drug are found in the urine. Some trimethoprim is also excreted in bile. The half-life of trimethoprim is 9-11 hours matching the half-life of sulfamethoxazole. The ratio of trime-thoprim to sulfamethoxazole supplied is 1:5.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
The sulfonamides demonstrate activity against gram-positive and gram-negative organisms; however, resistance in both community and nosocomial strains is widespread (Figure 1.15). Sulfonamides have proved to be effective for the empiric treatment of uncomplicated urinary tract infections; however, because of widespread resistance, they are seldom used as empiric therapy in other infections. Sulfonamides are the treatment of choice for Nocardia asteroides, and are useful in combination with other agents for the treatment of M. kansasii.
Trimethoprim is generally administered in combination with sulfamethoxazole. This combination often results in significantly improved activity. Trimethoprim–sulfamethoxazole (TMP-SMX) demonstrates excellent activity against L. monocytogenes, and it is the antibiotic of choice in the penicillin-allergic patient with listeriosis. It can be used to treat a number of other gram-positive and gram-negative pathogens. However, plasmid-mediated resistance is common, and treatment of most pathogens should be initiated only after sensitivity is confirmed by microbiologic testing. This combination is highly effective for killing Pneumocystis jiroveci, and TMP-SMX is the drug of choice for treatment or prophylaxis of that infection in immunocompromised hosts, including patients with AIDS.
Colistin
This antibiotic was discovered in the 1980s, but abandoned because of fears of undue nephrotoxicity. Because of the progressive increase in multiresistant Gram-negative bacilli, the use of colistin has been revisited. Fortunately, modern preparations have proven to be less nephrotoxic. Tables 1.7 and 1.9, together with Figure 1.15, summarize the characteristics of colistin.
KEY POINTS
About Colistin
1. Complex mixture of polymyxins and natural polypeptides that bind to bacterial lipopolysaccharide and disrupt the membrane barrier
2. Cidal and demonstrates concentration-dependent killing
3. Nephrotoxicity 1-10% of patients, greater risk if elderly, preexisting renal insufficiency, low serum albumin, coadministration of NSAIDs, or vancomycin. Usually, reversible.
4. Neurotoxicity—primarily paresthesias
5. Renal clearance, long half-life.
6. Narrow spectrum, used to treat multiresistant Pseudomonas aeruginosa, Acinetobacter baumannii, Stenotrophomonas maltophilia, and Klebsiella pneumoniae.
CHEMICAL STRUCTURE AND MECHANISMS OF ACTION
Colistin is a complex mixture of natural polypeptides derived from Bacillus polymyxa called polymyxins, and is administered as a prodrug colistin-methanesulphonate that is subsequently hydrolyzed to colistin. The polymyxins all have a strong positive charge and a hydrophobic acyl chain that binds with high affinity to the lipopolysaccharide bacterial membrane. Upon binding, colistin acts as a cationic detergent that disrupts the membrane barrier causing leakage of cell contents and eventual death of the bacterium. This antibiotic demonstrates concentration-dependent killing and MIC/AUC is the guiding parameter for therapy. The polymyxins are able to bind the lipid A portion of LPS, and can block the toxic biological effects endotoxin.
TOXICITY (SEE TABLE 1.7)
Nephrotoxicity is the leading toxicity associated with colistin, the percentage of patients suffering this complication ranging from 1% to 10% in recent studies. Risk factors for this complication include older age, preexisting renal insufficiency, low serum albumin, and coadministration of nonsteroidal anti-inflammatory drugs or vancomycin. Higher doses of colistin are associated with higher risk of renal toxicity. The onset of renal dysfunction usually occurs within the first week of administration and is reversible upon discontinuation in nearly 90% of patients. In cystic fibrosis patients, colistin may be less nephrotoxic than aminoglycosides. Neurotoxicity particularly, paresthesias has been reported in one quarter of patients receiving IV colistin. Other rarer neurological manifestations associated with administration include seizures, vertigo, muscle weakness, confusion, hallucinations, partial deafness and visual loss. These neurological side effects have been reported to quickly resolve upon discontinuing the drug.
PHARMACOKINETICS (SEE TABLE 1.9)
Studies on the pharmacokinetics of colistin are limited. The half-life of the drug is approximately 14 hours and once per day therapy may be preferable; however until clinical studies demonstrate no increase in toxicity once per day administration, q12h dosing is the preferred dosing interval. Because colistin is administered as a prodrug, colistin-methanesulphonate, that is hydrolyzed over time to colistin, some investigators recommend a higher loading dose for severely ill patients to assure the colistin serum levels exceed the MIC of the pathogen. Colistin is cleared exclusively by the kidney, and dosing must be adjusted for renal failure (see Table 1.9). No adjustment is recommended for hepatic dysfunction. Being a large molecule colistin has relatively limited extravascular body distribution and does not effectively cross the blood-brain barrier or enter joint fluid. It also poorly penetrates pleural fluid and the biliary tree.
SPECTRUM OF ACTIVITY AND TREATMENT RECOMMENDATIONS
Given its unique mechanism of killing, colistin is often the antibiotic of last resort for the treatment multiresistant Gram-negative bacilli (Figure 1.15). It demonstrates activity against most Gram-negative bacilli with the exception of Proteus, Pseudomonas mallei, B. cepacia, Serratia, Providencia, Edwardsiella species, Brucella, and Neisseria. Until there is greater experience with this agent, colistin should be reserved for highly resistant nosocomial pathogens such as Pseudomonas aeruginosa, Acinetobacter baumannii, St. maltophilia, and Klebsiella pneumoniae. This agent can also be aerosolized for the treatment of pneumonia, particularly in patients with cystic fibrosis.
In most circumstances, antibiotic sensitivities should be used to guide the decision to utilize colistin.
OUTPATIENT ORAL ANTIBIOTICS
A limited number of oral antibiotics should be utilized in the outpatient clinic to manage mild-to-moderate infections that do not require hospitalization. By limiting the number of antibiotics used and keeping in mind cost, caregivers will be able to provide the highest quality care at the lowest cost for their patients. The antibiotic checklist should be used to guide to determine whether or not an antibiotic is indicated. These recommendations are based on the IDSA, CDC, and American Thoracic Society Guidelines (Table 1.10).
Table 1.10. Commonly Used Oral Antibiotics in Outpatient Practice.

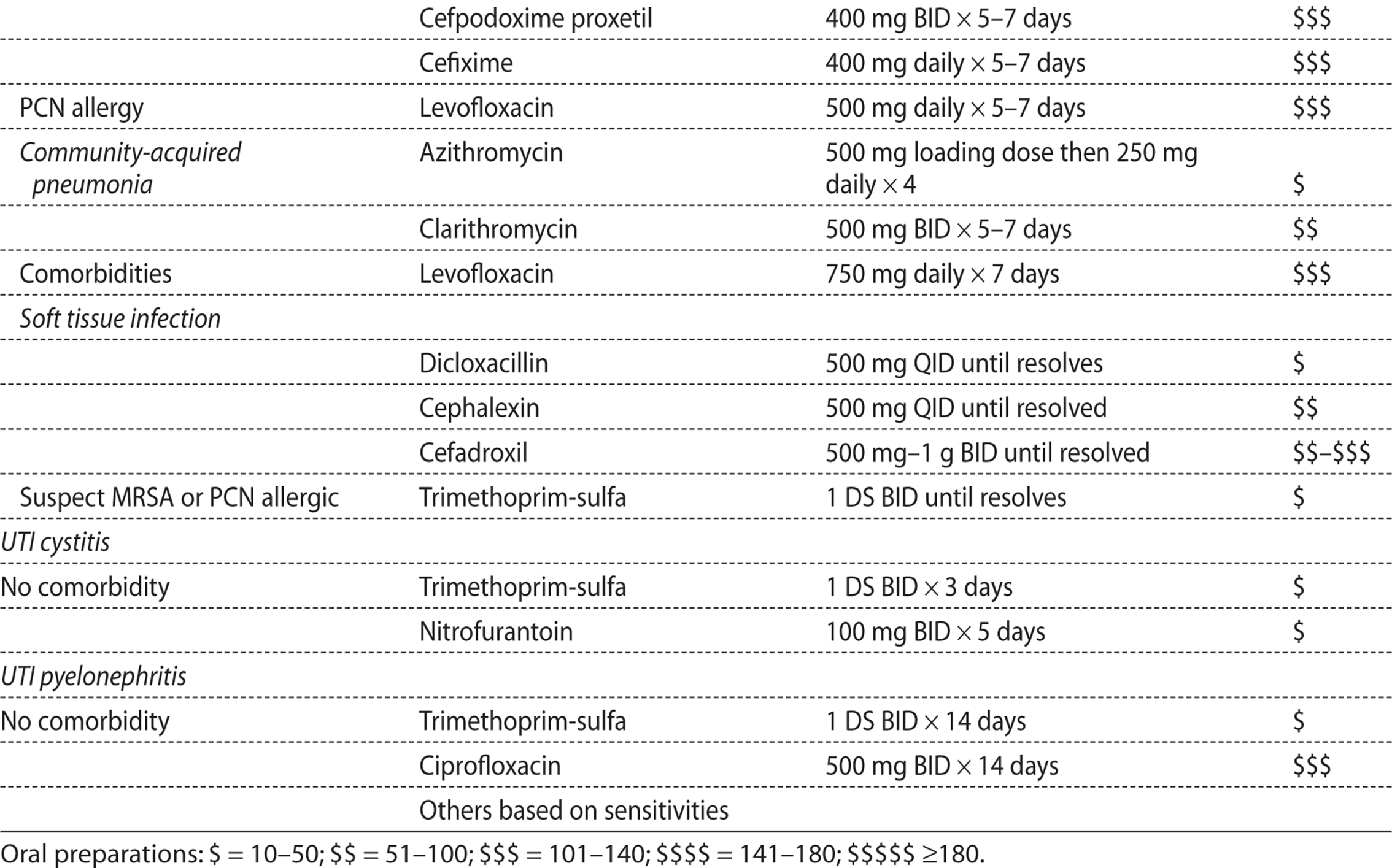
ANTIFUNGAL AGENTS
Fungi are eukaryotes, and they share many of the structural and metabolic characteristics of human cells. As a result, designing agents that affect fungi without harming human cells has proved difficult. One major difference between the two cell types is the primary sterol building block used to form the plasma membrane. The fungal plasma membrane consists of ergosterols; the major sterol component of the human plasma membrane is cholesterol. This difference has been exploited in the development of two classes of drugs. The polyenes act by binding to ergosterol and disrupting the fungal membrane. These agents are fungicidal. The azoles inhibit ergosterol synthesis, and lowered ergosterol levels results in fungal membrane breakdown. These agents are usually fungistatic.
THE MAJOR DIFFERENCE BETWEEN MAMMALIAN AND FUNGAL CELLS
Like mammals, fungi are eukaryotes. Drug therapy takes advantage of fact that fungi use ergosterols rather than cholesterol as the major building block of their plasma membrane.
Agents for Treatment of Systemic Fungal Infections
AMPHOTERICIN B
Chemical Structure, Mechanism of Action, and Spectrum of Activity—Amphotericin B is a long, cyclic polyene compound that forms a large rod-like structure. Multiple molecules bind to ergosterol in the fungal membrane forming pores that result in leakage of intracellular potassium and in fungal cell death. This fungicidal action is rapid and does not require active growth.
Toxicity—Nephrotoxicity is the major complication associated with the conventional deoxycholate form of amphotericin B (Table 1.11). This agent causes vasoconstriction of renal arterioles, resulting in a reduction in glomerular filtration rate. Vasoconstriction also impairs proximal and distal tubular reabsorption, causing potassium, magnesium, and bicarbonate wasting. These effects are reversible. However, permanent loss of nephrons and permanent damage to tubular basement membranes are also observed and correlate with the total dose administered. Renal dysfunction is observed in virtually all patients receiving this drug, and serum creatinine levels of 2-3 mg/dL are to be expected. Hydration with normal saline before infusion reduces nephrotoxicity.
Table 1.11. Toxicities of Systemic Antifungal Agents

KEY POINTS
About the Mechanism of Action and Spectrum of Amphotericin B
1. Polyene compound forms rod-like structures that bind to ergosterol in the fungal membrane, forming pores that result in a leak of intracellular potassium.
2. Rapidly cidal; does not require active growth.
Fever is commonly associated with administration of amphotericin B, and fever can be associated with chills and tachypnea, particularly if the drug is infused too rapidly. This agent should be infused slowly [2-3 hours for the deoxycholate form (ABD) and under 2 hours for the lipid preparations]. Fever and chills usually diminish with each subsequent dose. However, if those reactions persist, the patient can be premedicated with acetaminophen or 25-50 mg hydrocortisone can be added to the solution. This febrile reaction does not represent an allergic reaction and should not be misinterpreted as anaphylaxis. A 1 mg test dose preceding administration of the full dose has not proved to be helpful, and use of a test dose delays achievement of therapeutic antifungal serum and tissue levels. Because of a high incidence of phlebitis, amphotericin B should be administered through a centrally placed intravenous line.
Pharmacokinetics—At physiologic pH, ABD is insoluble in water (Table 1.12). It is stored as a powder that is dispersed as colloidal suspension in a 5% dextrose solution. Following intravenous infusion, amphotericin B is bound to lipoproteins in the serum and then leaves the circulation. The drug is stored in the liver and other organs and subsequently released into the circulation.
Table 1.12. Systemic Antifungal Agents: Half-Life, Dosing, Renal Dosing, and Cost

KEY POINTS
About the Toxicity of Amphotericin B
1. Nephrotoxicity is observed with virtually all patients receiving amphotericin B deoxycholate (ABD); reduced by hydration using normal saline. Reversible in most cases. Permanent damage with prolonged therapy.
2. Fever is common with all preparations. Slow infusion (2-3 hours with ABD, less than 2 hours with liposomal preparations) reduces severity. Premedication with corticosteroids or acetaminophen, or both, often reduces fever.
3. Phlebitis is common, requiring administration by central intravenous line.
Lipid-associated amphotericin B is ingested by macrophages, resulting in high intracellular levels in that cell type. This drug shows poor penetration of the blood-brain barrier and brain. Therapeutic levels are detectable in inflamed pleural fluid, peritoneum, and joint fluid. Amphotericin B is degraded slowly, and degradation is not affected by hepatic or renal dysfunction. Serum concentrations of the drug are detectable 7 weeks after therapy is discontinued.
Spectrum of Activity—Amphotericin B is effective against most fungal infections and remains the most effective agent for systemic fungal infections (Table 1.13). Clinical resistance to amphotericin B has been demonstrated among Candida lusitaniae, Fusarium species, and Pseudallescheria boydii. C. lusitaniae initially is susceptible to amphotericin B, but develops resistance during treatment. The alterations in sterol structure required for amphotericin B resistance often reduce tissue invasiveness, such strains being capable of growing only on mucosal surfaces or in the urine.
Table 1.13. Spectrum of the Systemic Antifungals

Efficacy of Various Amphotericin B Preparations—Lipid-associated preparations of amphotericin B are preferred because of their lower nephrotoxicity. However, these preparations are very expensive (Table 1.12) and in most clinical trials have comparable efficacy to amphotericin-B deoxycholate. Liposomal amphotericin B was shown to be superior to ABD for the treatment of pulmonary histoplasmosis. The lipid-associated preparations are recommended in patients with significant preexisting renal dysfunction or in patients who develop progressive renal failure (serum creatinine above 2.5 mg/dL) while being treated ABD. Clinicians also need to be aware of the observation that ABD-related renal dysfunction (50% increase in baseline creatinine to a minimum of 2 mg/mL) is associated with a 6.6-fold increased risk of death.
KEY POINTS
About Amphotericin Spectrum of Activity and Preparations
1. Preferred antifungal agent for severe systemic fungal infections.
2. Effective against most fungi except Candida lusitaniae, Fusarium, and Pseudallescheria boydii.
3. Lipid-associated preparations reduce nephrotoxicity, but similar incidence of fever, with efficacy comparable to conventional amphotericin B deoxycholate (ABD).
4. Higher doses of lipid-associated preparations required: 3-5 mg/kg daily as compared with 0.3 to 1.4 mg/kg for ABD.
5. Very high cost. Recommended for patients with significant preexisting renal dysfunction or those who develop progressive renal dysfunction on ABD (serum creatinine .2.5 mg/dL).
AZOLES
Chemical Structure and Mechanism of Action—The azoles are chemically synthesized agents that come in two classes. The first to be synthesized were the imidazoles (miconazole and ketoconazole). Those compounds are now seldom used for systemic infections, being primarily reserved for topical treatment of superficial fungal infections. The second class, the triazoles, are preferred for systemic fungal infection; they are well absorbed orally and have excellent toxicity profiles.
All azoles inhibit a cytochrome P450-dependent demethylation system that results in decreased production of ergosterol and accumulation of intermediate sterols. The loss of ergosterol results in altered fungal membrane permeability, disturbed activity of membrane surface enzymes, and retention of metabolites. These agents have broad antifungal activity, but they demonstrate fungistatic rather than fungicidal activity. Itraconazole can antagonize amphotericin B activity by reducing its binding target, ergosterol.
Toxicity—Ketoconazole not only interferes with fungal sterol metabolism, but at higher doses it also interferes with testosterone and cortisone production (Table 1.11). Gynecomastia and loss of libido are commonly observed. Severe hepatitis can develop during treatment with this agent. As a result of its many toxicities, ketoconazole is rarely prescribed today.
KEY POINTS
About the Mechanism of Action of the Azoles
1. Inhibit cytochrome P450-dependent demethylation, resulting in decreased ergosterol production and altered fungal membrane permeability.
2. Azoles are usually fungistatic.
3. Itraconazole can antagonize amphotericin B activity by reducing its binding target.
The triazoles (fluconazole, itraconazole, posaconazole, voriconazole) demonstrate minimal toxicity. Side effects include headache, gastrointestinal intolerance, and asymptomatic increases in serum transaminase levels. Voriconazole infusion can be associated with transient loss of light perception. This symptom resolves with subsequent doses. Visual hallucinations less commonly occur.
Pharmacokinetics—Fluconazole is well absorbed orally, and serum levels after ingestion of the oral preparation are comparable to those with intravenous administration. Penetration into tissues and body fluids, including the CSF, is excellent. Itraconazole is more variable in its oral absorption and requires stomach acidity for adequate absorption. Capsule absorption is enhanced by food and reduced by agents that reduce stomach acidity. Itraconazole penetrates most tissues, but does not cross the blood-brain barrier and enters ocular fluids only minimally. Posaconazole oral absorption is enhanced by food, particularly high-fat meals or liquid nutritional supplements. Voriconazole is well absorbed orally, demonstrating 96% bioavailability, and also can be given intravenously.
All of the azoles are metabolized by the liver via the cytochrome P450 system, and as a consequence, drugÓdrug interactions are common with these agents. Rifampin, rifabutin, long-acting barbiturates, carbamazepine, and cisapride usually lower azole levels. The azoles slow the metabolism of Coumadin, warfarin, phenytoin, tacrolimus, cyclosporine, certain antihistamines, benzodiazepines, calcium channel blockers, sulfonylureas, prednisolone, digoxin, statins, and anti-HIV protease inhibitors. The doses of these agents usually need to be lowered in the presence of azoles. Drug–drug interactions have proven to be the most problematic with voriconazole. Voriconazole is metabolized primarily by the P450 enzyme CYP2C19, and that enzyme has variable activity depending on the patient’s genetic background. As a consequence, serum levels can vary by up to a factor of 4 in individuals with rapid as opposed to slow metabolism. In the United States, the coadministration of rifabutin and voriconazole is contraindicated because rifabutin levels may increase by a factor of 3, while voriconazole levels drop below therapeutic levels. Rifampin, carbamazepines, and long-acting barbiturates can also markedly reduce voriconazole levels, and these drugs should probably be discontinued when voriconazole is being administered.
KEY POINTS
About Azole Toxicity
1. Ketoconazole interferes with testosterone and cortisone production, resulting in gynecomastia and loss of libido. Hepatitis can be severe, and the drug should be discontinued when symptoms of hepatitis develop. Liver function tests should be performed.
2. Rare side effects of fluconazole, itraconazole, posaconazole, and voriconazole include headache, gastrointestinal intolerance, and asymptomatic elevation of serum transaminases.
3. Intravenous infusion of voriconazole can be associated with transient loss of light perception.
4. Drug–drug interactions with other agents metabolized by the cytochrome P450 system are common, particularly with voriconazole and ketoconazole.
Spectrum of Activity and Treatment Recommendations—Fluconazole—Fluconazole has no activity against Aspergillus species, and some strains of Candida, including C. glabrata and C. krusei, demonstrate natural resistance. Because of increased production of demethylase and increased drug efflux, any Candida species can develop resistance (Table 1.13).
KEY POINTS
About the Spectrum of Activity and Indications for Fluconazole
1. No activity against Aspergillus. Active against Candida albicans, but natural resistance in C. glabrata and C. krusei is common. Active against Cryptococcus neoformans.
2. With prolonged treatment, drug resistance can develop in Candida species.
3. Treatment of choice for oral candidiasis and Candida vulvovaginitis.
4. Can be used for uncomplicated C. albicans fungemia in the nonimmunocompromised patient.
5. Can be used to complete therapy of cryptococcal meningitis in patients with HIv after an initial course of amphotericin B.
6. Prophylaxis reduces Candida infections in neutropenic patients. The role of prophylaxis in other settings remains controversial because of the risk of selecting for resistant strains.
Fluconazole is recommended for the treatment of oropharyngeal and vulvovaginal candidiasis. Intravenous fluconazole has proved therapeutically equivalent to amphotericin B in uncomplicated candidemia in the non-immunocompromised host. However, for the immunocompromised (including neutropenia) host, and for seriously ill patients with deep tissue Candida infection, amphotericin B or an echinocandin should be used. Fluconazole is also effective for completing the treatment of cryptococcal meningitis, termed consolidation and is also recommended for maintenance therapy to prevent relapse.
The use of fluconazole for prevention of fungal infections has been explored in neutropenic allogeneic bone marrow transplant patients and was found to reduce mortality and the incidence of invasive Candida infections, but no effect on the incidence of Aspergillus infections was observed. Fluconazole prophylaxis of leukemia patients also reduced the incidence of invasive Candida infections, but had no effect on mortality. Fluconazole is frequently used in the surgical intensive care unit in the hopes of preventing candidemia in patients; however this practice does not reduce mortality and increases the prevalence of fluconazole-resistant fungi, including C. krusei and C. glabrata.Prophylaxis in our surgical intensive care units should be abandoned.
Itraconazole—As compared with fluconazole, itraconazole has demonstrated improved activity against histoplasmosis, coccidiomycosis, blastomycosis, and sporotrichosis (Table 1.13). Itraconazole can be used for acute and chronic vaginal candidiasis and HIV-associated oral and esophageal candidiasis, and for consolidation and maintenance therapy for cryptococcal meningitis in patients with AIDS. Itraconazole is the preferred agent for the treatment of lymphocutaneous sporotrichosis and of nonmeningeal, nonlife-threatening histoplasmosis, blastomycosis, and coccidiomycosis. For disseminated histoplasmosis and coccidiomycosis, amphotericin B remains the treatment of choice. Itraconazole is recommended as primary prophylaxis and for the prevention of relapse of histoplasmosis in patients with AIDS.
KEY POINTS
About the Spectrum of Activity and Indications for Itraconazole
1. Improved activity against histoplasmosis, coccidiomycosis, blastomycosis, and sporotrichosis.
2. Used in less severe cases of histoplasmosis and coccidiomycosis.
3. Used to prevent relapse of disseminated histoplasmosis in patients with AIDS.
4. Absorption of the drug is erratic.
Voriconazole and Posaconazole—As compared with amphotericin B deoxycholate, voriconazole demonstrates increased activity against Aspergillus and has proven to be superior for the treatment of invasive aspergillosis. Voriconazole is also approved for the treatment of Fusarium and Scedosporium and is also effective against invasive candidiasis in non-neutropenic patients.
The newest azole, posaconazole, has the broadest spectrum in the class. In addition to being effective against Aspergillus, this agent has activity against many of the Zygomycetes. Posaconazole is approved for prophylaxis against Aspergillus and disseminated candidiasis in severely immunocompromised hosts and for the treatment of fluconazole and itraconazole refractory Candida esophagitis. This agent has proved to be effective salvage therapy for mucormycosis.
KEY POINTS
About the Spectrum of Activity of Voriconazole and Posaconazole
1. Voriconazole is preferred for Aspergillus and active against Candida albicans.
2. Posaconazole has activity against Aspergillus and Zygomycete broadest-spectrum azole).
CASPOFUNGIN/ANIDULAFUNGIN/MICAFUNGIN
Chemical Structure and Mechanism of Action—The echinocandins are all derived from echinocandin B, a semisynthetic lipopeptide that blocks synthesis of β-(1,3)-D-glucan. That polysaccharide is a critical component of the cell wall in many pathogenic fungi.
Toxicity—The echinocandins have proven to be very safe, provoking only the occasional fever, rash, or flushing of the face during infusion (Table 1.11). Serum levels are increased by coadministration of cyclosporin. Agents that may reduce serum levels including efavirenz, nelfinavir, Dilantin, Tegretol, rifampin, and dexamethasone. The echinocandins can reduce serum levels of tacrolimus.
Pharmacokinetics—The echinocandins are not absorbed by the gastrointestinal tract and must be administered intravenously (Table 1.12). They are metabolized by the liver.
Spectrum of Activity and Treatment Indications—The echinocandins are active against Aspergillus and Candida, including isolates that are resistant to other antifungal agents. They are less effective against C. parapsilosis in vitro, and are not active against Cryptococcus see Table 1.13). They are approved for the treatment of invasive aspergillosis in patients who fail on, or are unable to tolerate, amphotericin B or itraconazole. Caspofungin can also be used to treat oral candidiasis that is refractory to azole or amphotericin B therapy.
KEY POINTS
About the Echinocandins
1. Block synthesis of a cell wall polysaccharide vital to many pathogenic fungi.
2. Active against Aspergillus and Candida, including isolates resistant to other antifungal agents. Not active against Cryptococcus.
3. Toxicities tend to be mild.
4. Recommended for the treatment of invasive Aspergillus in patients who have failed on, or cannot tolerate, amphotericin B and for oral and esophageal candidiasis refractory to azoles and amphotericin B.
FLUCYTOSINE
Chemical Structure and Mechanism of Action—Flucytosine, or 5-fluorocytosine (5-FC), is a fluorine analog of cytosine. After a multistep conversion requiring deamination and phosphorylation, the resulting product, 5-fluorouracil (5-FU), acts as an inhibitor of thymidylate synthetase, impairing DNA and RNA synthesis. In humans, 5-FC is not toxic because of a lack of the deaminase required for conversion to 5-FU.
Toxicity—The major toxicity of flucytosine is bone marrow suppression leading to neutropenia, anemia, and thrombocytopenia (Table 1.11). This side effect is dose-related and usually occurs when serum levels exceed 125 μg/mL. Patients with diminished bone marrow reserve such as those with AIDS and those receiving cancer chemotherapy are more likely to suffer this complication. Commonly, 5-FC is administered in combination with amphotericin B. As discussed earlier in this chapter, amphotericin B impairs renal function, and reductions in renal function reduce the clearance of 5-FC. In patients with renal dysfunction, monitoring of peak (2 hours after oral administration) and trough levels (just before the next dose) is recommended. Doses should be adjusted to maintain serum levels between 20 and 100 μg/mL.
Pharmacokinetics—Flucytosine is well absorbed orally (Table 1.12). Because it is a small molecule, 5-FC penetrates tissues well and crosses the blood–brain barrier. Therapeutic levels can be achieved in the CSF, aqueous humor, joint fluid, and respiratory secretions. The kidneys clear 5-FC.
Spectrum of Activity and Treatment Recommendations—Most strains of C. albicans and Cryptococcus neoformans are sensitive to 5-FC (Table 1.13). Native resistance varies geographically. About 15% of C. albicans stains and 3–5% of Cryptococcus neoformans demonstrate resistance. The effect of 5-FC is usually fungistatic, and it should never be used alone, because resistance rapidly develops with monotherapy. The combination of 5-FC and amphotericin B demonstrates additive or synergistic activity in cryptococcal infections. In cryptococcal meningitis, amphotericin B and 5-FC sterilize the CSF faster than amphotericin B alone. Combination therapy for Candidainfections has also been FDA approved.
KEY POINTS
About Flucytosine
1. Impairs fungal DNA and RNA synthesis; fungistatic.
2. Cleared by the kidneys; penetrates all tissues and fluids, including the cerebrospinal fluid.
3. High levels cause bone marrow suppression. In patients with renal failure, doses should be adjusted, and serum levels should be monitored.
4. Never use as monotherapy. In cryptococcal meningitis, the combination of amphotericin B and flucytosine sterilizes the cerebrospinal fluid faster than does amphotericin B alone. In animal studies, combination therapy is beneficial for Candida infections, but efficacy has not been proven in humans.
ANTIVIRAL DRUGS (OTHER THAN ANTIRETROVIRAL AGENTS)
Most antiviral agents target viral nucleic acid synthesis. Because these agents tend to act at a single step in viral replication, resistance may develop during treatment. The development of resistance is favored by a high viral load, a high intrinsic viral mutation rate (more common in RNA than DNA viruses), and a high degree of selective pressure—that is, prolonged antiviral therapy or repeated courses of treatment. A second method for controlling viral infection is to modify the host immune response. Infusions of antibody preparations and treatment with IFN have proved efficacious in several viral infections.
Antivirals that Block DNA Transcription
ACYCLOVIR, VALACYCLOVIR, FAMCICLOVIR
Chemical Structure and Mechanisms of Action—Acyclovir and valacyclovir are synthetic analogs of guanine in which a side chain has been substituted for a sugar moiety. Famciclovir is a acyclic guanosine analog derived from penciclovir, and this prodrug is quickly converted to penciclovir following oral absorption. These antiviral agents are phosphorylated in virus-infected cells by viral thymidine kinase, forming a monophosphate compound. Host cell kinases then add two additional phosphates, allowing the triphosphate to add to replicating DNA. The acyclic side chain of acyclovir prevents the addition of subsequent nucleic acids to DNA causing premature termination.
KEY POINTS
About Antiviral Therapy
1. Usually targets viral nucleic acid synthesis.
2. Development of resistance is common and is favored by
a) high viral load,
b) high intrinsic viral mutation rate (RNA viruses more than DNA viruses), and
c) prolonged or intermittent antiviral therapy.
Penciclovir is not a DNA chain terminator; it acts primarily as a viral DNA polymerase inhibitor. Acyclovir also selectively inhibits viral DNA polymerase. Because these agents require viral thymidine kinase for their initial phosphorylation step, the concentrations of the triphosphate compounds are 40–100 times higher in infected than uninfected cells. Acyclovir and famciclovir resistance are most commonly caused by a reduction in viral thymidine kinase. The loss or reduction in viral thymidine kinase activity impairs acyclovir phosphorylation and also renders the virus resistant to ganciclovir, because that agent also requires activation by viral thymidine kinase.
Toxicity—Toxicity related to these drugs is generally minimal (Table 1.14). Rarely patients develop rash, hematuria, headache and nausea. Neurotoxicity may occur in 1–4% receiving intravenous acyclovir and can result in lethargy, obtundation, coma, hallucinations, seizures, and autonomic instability. Most patients who suffer these complications have renal dysfunction resulting in high acyclovir serum levels. Coadministration of zidovudine and acyclovir increases the risk of developing lethargy. Intravenous administration can also cause crystalluria and crystalline nephropathy, particularly if the patient is dehydrated. Cyclosporin increases the risk of nephrotoxicity.
Table 1.14. Toxicities of Systemic Antiviral Agents

Pharmacokinetics—The oral absorption of acyclovir is limited, only 15–20% of the drug being bioavailable (Table 1.15). Absorption tends to be even poorer in transplant patients, necessitating higher oral dosing. The prodrug preparation valacyclovir is rapidly and completely converted to acyclovir by hepatic and intestinal valacyclovir hydrolase. Oral valacyclovir achieves acyclovir serum levels that are three to five times higher than those achieved by oral acyclovir. Similarly, famciclovir is well absorbed orally, and in the liver and intestine, its purine is quickly deacetylated and oxidized to form penciclovir.
Table 1.15. Systemic Antiviral Agents: Half-Life, Dosing, Renal Dosing, and Cost

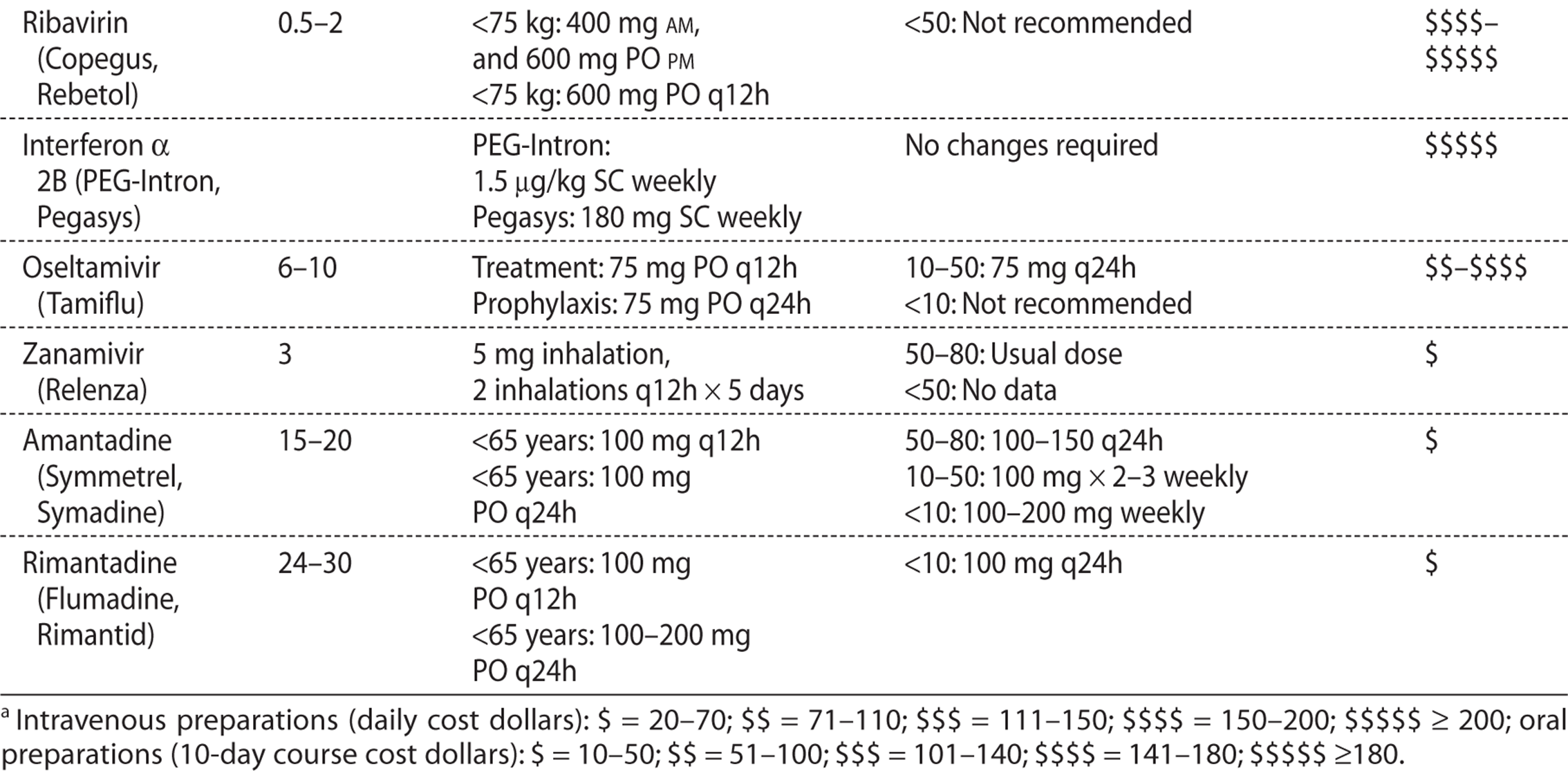
Acyclovir and penciclovir are widely distributed in tissues and fluids. Therapeutic levels can be achieved in CSF, saliva, vaginal secretions, and the aqueous humor. Both drugs are excreted unchanged primarily in the urine. Probenecid reduces renal clearance and increases the half-life.
Antiviral Activity and Therapeutic Indications—Acyclovir and famciclovir have excellent activity against herpes simplex viruses 1 and 2. Topical administration of these drugs is of minimal efficacy against herpes simplex labialis, and topical preparations are rarely used. Oral acyclovir and famciclovir are recommended for treatment of genital herpes and are used to prevent recurrent herpes genitalis. Acyclovir is also recommended for the treatment and prevention of recurrent ocular herpes simplex. Intravenous acyclovir has reduced the mortality from herpes simplex encephalitis and is the treatment of choice for that disorder. Acyclovir and famciclovir also have significant activity against varicella; however, higher drug concentrations are required to kill that virus. Intravenous acyclovir is recommended for the treatment of varicella and herpes zoster in the immunocompromised host, and for treatment of varicella pneumonia or encephalitis in the previously healthy adult. Acyclovir demonstrates some activity against Epstein–Barr virus, but is generally not recommended for therapy. This agent also demonstrates modest protection against cytomegalovirus (CMV) when used for prophylaxis in allogeneic bone marrow, renal, and liver transplant recipients; however, ganciclovir has proved to be more efficacious. Famciclovir can reduce levels of hepatitis B viral DNA and serum transaminase in patients with chronic hepatitis B. Its effects are additive when combined with IFN. Famciclovir has also been used to treat recurrent hepatitis B following liver transplantation.
KEY POINTS
About Acyclovir, Valacyclovir, and Famciclovir
1. All require viral thymidine kinase phosphorylation for activity.
2. Acyclovir binds to the replicating viral DNA, causing premature chain termination; acyclovir and famciclovir both inhibit viral DNA polymerase.
3. Resistance is most commonly mediated by a reduction in viral thymidine kinase.
4. Toxicity is minimal. Intravenous administration of acyclovir can cause lethargy, obtundation, hallucinations, and seizures.
5. Valacyclovir is rapidly converted to acyclovir; resulting acyclovir levels are higher than those achieved with oral preparations of acyclovir. Famciclovir is rapidly converted to penciclovir.
6. Excellent activity against herpes simplex 1 and 2. Oral preparations recommended for treatment and prophylaxis of genital herpes and ocular herpes. Intravenous acyclovir recommended for herpes simplex encephalitis.
7. Moderate activity against varicella (intravenous acyclovir recommended for the immunocompromised host), and varicella pneumonia or encephalitis in the normal host. High doses of oral valacyclovir and famciclovir can be used to treat less severe disease.
8. Famciclovir can also be used to treat hepatitis B virus.
GANCICLOVIR AND VALGANCICLOVIR
Chemical Structure and Mechanisms of Action—Like acyclovir, ganciclovir is a guanine analog. Ganciclovir has an additional hydroxymethyl group on the acyclic side chain. Viral thymidine kinase converts this analog to the monophosphate form, after which host cell kinase phosphorylation produces the active triphosphate form. Ganciclovir triphosphate competitively inhibits viral DNA polymerase incorporation of guanosine triphosphate into elongating DNA, but does not act as a chain terminator.
In infected cells, intracellular concentrations of ganciclovir triphosphate reach levels that are 10 times that of acyclovir triphosphate, and once in the cell, ganciclovir triphosphate persists, having a intracellular half-life of 16–24 hours. The resulting higher intracellular concentrations may account for the greater activity of ganciclovir against CMV. Ganciclovir is also active against herpes simplex, varicella, and Epstein–Barr virus. Because ganciclovir requires viral thymidine kinase activity for conversion to the active triphosphate form, acyclovir-resistant viral strains with reduced thymidine kinase activity are also less sensitive to ganciclovir. Mutations that alter the structure of the viral DNA polymerase also confer ganciclovir resistance, and these mutants often demonstrate reduced sensitivity to foscarnet and cidofovir.
Toxicity—Significant concentrations of ganciclovir triphosphate accumulate in uninfected cells (Table 1.14). Bone marrow progenitor cells are particularly sensitive to this agent. The triphosphate form can incorporate into cellular DNA and block host cell DNA replication. Neutropenia and thrombocytopenia are commonly observed in patients with AIDS who are receiving ganciclovir, and these patients require close monitoring for WBC and platelet counts during therapy. The risk is lower, but significant, in transplant patients. Coadministration of zidovudine increases the risk of bone marrow suppression. Discontinuation of treatment is recommended if the absolute neutrophil count drops below 500 cells/mm3. Central nervous system (CNS) side effects (including headache, confusion, psychosis, coma, and seizures) are also common.
KEY POINTS
About Ganciclovir
1. Guanine analog that primarily inhibits viral DNA polymerase.
2. Like acyclovir and penciclovir requires viral thymidine kinase for activation. Acyclovir-resistant strains are often resistant to ganciclovir.
3. Bone marrow suppression is a common toxicity, particularly in patients with AIDS. The drug should be discontinued if the neutrophil count drops to less than 500 cells/mm3.
4. Central nervous system complaints—including confusion, psychosis, coma, and seizures—may occur.
5. Most active guanine analog against cytomegalovirus (CMV). Also active against herpes simplex 1 and 2, varicella, and Epstein–Barr virus.
6. Recommended for CMV retinitis, pneumonia, and colitis. Useful for prophylaxis of immunocompromised transplant patients. Following treatment of active infection in patients with AIDS with low CD4 counts, oral valganciclovir is given to prevent relapse.
Pharmacokinetics—Valganciclovir is a prodrug that is well absorbed orally and quickly converts to ganciclovir (Table 1.15). With oral administration, excellent serum levels that are nearly comparable to intravenous ganciclovir can be achieved. Ganciclovir readily penetrates all tissues and fluids including the brain and CSF. The drug is primarily excreted unmodified in the urine.
Spectrum of Activity and Treatment Indications—Of the guanine analogs, ganciclovir has the highest activity against CMV. Ganciclovir is the treatment of choice for CMV infections including retinitis, pneumonia, and colitis. Ganciclovir is also used for prophylaxis of CMV in transplant patients. In patients with AIDS who have persistently low CD4 lymphocyte counts, ganciclovir maintenance therapy is required to prevent relapse of CMV infection after the treatment of active infection has been completed.
CIDOFOVIR
Chemical Structure, Mechanisms of Action, and Pharmacokinetics—Cidofovir (Tables 1.14 and 1.15) is an analog of deoxycytidine monophosphate that inhibits viral DNA synthesis. This agent does not require viral kinase for activity, being converted by cellular enzymes to its active diphosphate form. It acts as a competitive inhibitor of viral DNA polymerase and also adds to DNA, substituting for deoxycytidine triphosphate (dCTP), causing premature chain termination. Viral thymidine kinase mutations do not impair cidofovir activity.
Resistance is conferred through viral DNA polymerase mutations. Such mutations can result in cross-resistance to ganciclovir and, less commonly, to foscarnet. Cidofovir is cleared by the kidneys.
Toxicity—Cidofovir is highly nephrotoxic, causing proteinuria in half of treated patients, and azotemia and metabolic acidosis in a significant number. Vigorous saline hydration and coadministration of probenecid reduces nephrotoxicity. The drug should be discontinued if 3+ proteinuria or higher develops, or if serum creatinine increases by more than 0.4 mg/dL. Neutropenia is also commonly encountered.
Spectrum of Activity and Treatment Indications—Cidofovir has activity against many DNA viruses: CMV; herpes simplex; herpesvirus 6 and 8; varicella; pox viruses, including smallpox; papilloma viruses; polyoma viruses; and adenoviruses. This agent is approved only for the treatment of CMV retinitis in patients with AIDS. Given its highly toxic profile, parenteral use of this drug in other viral infections is likely to be limited. Topical therapy may prove efficacious in acyclovir-resistant herpes simplex infections in patients with AIDS, and it is being studied for the treatment of anogenital warts.
KEY POINTS
About Cidofovir
1. An analog of deoxycytidine monophosphate; it causes premature chain termination of viral DNA and also inhibits viral DNA polymerase.
2. Does not require viral thymidine kinase for conversion to its active form. Acyclovir-resistant strains are usually not resistant to cidofovir.
3. Highly nephrotoxic; causes proteinuria, azotemia, and metabolic acidosis in nearly half of patients. Saline hydration and probenecid reduce nephrotoxicity. Neutropenia also is common.
4. Broad spectrum of antiviral activity including cytomegalovirus (CMV), herpes simplex, herpesvirus 6 and 8, varicella, pox viruses, papilloma virus, polyoma viruses, and adenoviruses.
5. Approved for CMV retinitis in patients with AIDS. Other indications are currently being explored. However, the usefulness of cidofovir is likely to be limited because of renal and bone marrow toxicity.
FOSCARNET
Chemical Structure and Mechanism of Action—Foscarnet is an inorganic pyrophosphate analog, trisodium phosphonoformate, which reversibly blocks the pyrophosphate binding site of viral DNA polymerase. Foscarnet binding inhibits the polymerase from binding deoxynucleotidyl triphosphates. Mutations to the viral DNA polymerase are primarily responsible for viral resistance; however, resistance among clinical isolates is rare.
Toxicity. Nephrotoxicity is the most common serious side effect of foscarnet, resulting in azotemia, proteinuria, and occasionally acute tubular necrosis (Table 1.14). Renal dysfunction usually develops during the second week of therapy and in most cases reverses when the drug is discontinued. Dehydration increases the incidence of nephrotoxicity, and saline loading is of benefit in reducing this complication. Metabolic abnormalities are frequent. Hypocalcemia is the most common, being the result of chelation by foscarnet. Reductions in ionized calcium can cause CNS disturbances, tetany, paresthesias, and seizures. Other metabolic abnormalities include hypophosphatemia, hypomagnesemia, hypokalemia, hypercalcemia, and hyperphosphatemia. To minimize these metabolic derangements, intravenous infusion should not exceed 1 mg/kg per minute. Electrolytes, magnesium, phosphate, and calcium should be closely monitored. Other common side effects include fever, headache, nausea, vomiting, and abnormal liver function tests.
KEY POINTS
About Foscarnet
1. Blocks binding of deoxynucleotidyl triphosphates to viral DNA polymerase.
2. Nephrotoxicity is common, usually developing during the second week of therapy. Can be reduced by saline hydration. Usually reversible.
3. Also causes abnormalities in serum calcium, magnesium, and phosphate.
4. Active against cytomegalovirus (CMV), herpes simplex, varicella, Epstein–Barr virus, and herpesvirus 8.
5. Approved for the treatment of CMV retinitis and acyclovir-resistant mucocutaneous herpes simplex.
Pharmacokinetics—Foscarnet is poorly absorbed orally and is administered intravenously. This drug penetrates all tissues and fluids, achieving excellent levels in the CSF and vitreous humor. Foscarnet is excreted unmodified, primarily by the kidneys.
Spectrum of Activity and Treatment Indications–Foscarnet is active against CMV, herpes simplex, varicella, Epstein–Barr virus, and herpesvirus 8. It is approved for the treatment of CMV retinitis and for acyclovir-resistant mucocutaneous herpes simplex.
Other Antiviral Agents
RIBAVIRIN
Chemical Structure and Mechanism of Action—Ribavirin is a guanosine analog that contains the d-ribose side chain. It inhibits DNA and RNA viruses alike. The mechanisms of inhibition are complex and not completely understood. Ribavirin is phosphorylated to the triphosphate form by host cell enzymes, and the triphosphate form interferes with viral messenger RNA formation. The monophosphate form interferes with guanosine triphosphate synthesis, lowering nucleic acid pools in the cell.
Toxicity—Systemic ribavirin results in dose-related red blood cell hemolysis; at high doses, it suppresses the bone marrow (Table 1.14). The resulting anemia reverses when the drug is discontinued. Intravenous administration is not approved in the United States, but is available for patients with Lhasa fever and some other forms of hemorrhagic fever. Aerosolized ribavirin is associated with conjunctivitis and with bronchospasm that can result in deterioration of pulmonary function. A major concern for health care workers exposed to aerosolized ribavirin are teratogenic and embryotoxic effects noted in some animal studies. Pregnant health care workers should not administer this drug.
KEY POINTS
About Ribavirin
1. Guanosine analog that interferes with viral messenger RNA formation and reduces guanosine triphosphate synthesis, lowering nucleic acid pools in the cell.
2. Systemic drug causes red blood cell hemolysis. Intravenous administration not approved in the United States. Aerosolized form causes conjunctivitis and bronchospasm.
3. Teratogenic and embryotoxic. Pregnant health care workers should not administer.
4. Active against DNA and RNA viruses including respiratory syncytial virus (RSV), influenza and parainfluenza virus, herpes viruses, adenovirus, pox viruses, Bunyavirus, and arenaviruses.
5. Approved for aerosolized treatment of RSV bronchiolitis and pneumonia.
6. Approved for oral administration in combination with interferon for chronic hepatitis C.
Pharmacokinetics—Approximately one-third of orally administered ribavirin is absorbed. The drug penetrates all tissues and body fluids. Ribavirin triphosphate becomes highly concentrated in erythrocytes (40 times plasma levels) and persists for prolonged periods with red blood cells. The drug is cleared both by the kidneys and by the liver. Aerosolized ribavirin produces high drug levels that have a half-life of up to 2.5 hours in respiratory secretions. A special aerosol generator is required for proper administration.
Spectrum of Activity and Treatment Recommendations.—Ribavirin is active against a broad spectrum of DNA and RNA viruses including respiratory syncytial virus (RSV), influenza and parainfluenza virus, herpes, adenovirus, pox viruses, Bunyavirus, and arenaviruses. It is approved in the United States for the aerosol treatment of RSV bronchiolitis and pneumonia in hospitalized patients. Oral ribavirin in combination with IFN is approved for the treatment of chronic hepatitis C.
INTERFERONS
Chemical Structure and Mechanism of Action—The IFNs are proteins of 16–27,000 Da molecular weight, synthesized by eukaryotic cells in response to viral infections. These cytokines in turn stimulate host antiviral responses. IFN receptors regulate approximately 100 genes, and in response to INF binding, cells rapidly produce dozens of proteins. A wide variety of RNA viruses are susceptible to the antiviral actions of IFNs; most DNA viruses are only minimally affected.
Toxicity—Side effects tend to mild when doses of less than 5 million units are administered (Table 1.14). Doses of 1-2 million units given subcutaneously or intramuscularly are associated with an influenza-like syndrome that is particularly severe during the first week of therapy. This febrile response can be reduced by premedication with antipyretics such as aspirin, ibuprofen, and acetaminophen. Local irritation at injection sites is also frequently reported. Higher doses of INF result in bone marrow suppression, causing granulocytopenia and thrombocytopenia. Neurotoxicity resulting in confusion, somnolence, and behavior disturbances is also common when high doses are administered. Hepatoxicity and retinopathy are other common side effects with high-dose therapy.
Pharmacokinetics—Intramuscularly and subcutaneously, INF-a is well absorbed; other IFNs have more variable absorption (Table 1.15). Assays for biologic effect demonstrate activity that persists for 4 days after a single dose. Pegylated forms result in slower release and more prolonged biologic activity, allowing for once-weekly administration; these forms are preferred in most instances.
Spectrum of Activity and Treatment Recommendations—The effectiveness of INFs has been limited by the frequent side effects associated with effective doses. Treatment approvals have been issued for INFs in chronic hepatitis C, chronic hepatitis B, Kaposi sarcoma and other malignancies, and condyloma acuminatum.
KEY POINTS
About Interferon for Treatment of Viral Infections
1. Binds to host cell interferon receptors, upregulating many genes responsible for the production of proteins with antiviral activity.
2. RNA viruses are more susceptible to the antiviral actions of IFNS.
3. The most common side effect is an influenza-like syndrome. At doses above 5 million units, bone marrow suppression and neurotoxicity may develop. Hepatoxicity and retinopathy are commonly associated with high doses.
4. Approved for chronic hepatitis C, chronic hepatitis B, and Kaposi sarcoma. Intralesional injection approved for condyloma acuminatum.
Anti-Influenza Viral Agents
AMANTADINE AND RIMANTADINE
Mechanism of Action—Amantadine and rimantadine are effective only against influenza A. They bind to and inhibit the M2 protein. This viral protein is expressed on the surface of infected cells, and it is thought to play an important role in viral particle assembly.
Toxicity—Amantadine causes moderate CNS side effects, especially in the elderly (Table 1.14). Insomnia, inability to concentrate, and dizziness are most commonly reported. Amantadine also increases the risk of seizures in patients with a past history of epilepsy. Rimantadine causes CNS side effects less frequently, and this agent is now preferred over amantadine.
Treatment Recommendations—To be effective, treatment must be instituted within 48 hours of the onset of symptoms (Table 1.15). Efficacy has been proven in healthy adults, but trials have not been performed in high-risk patients.
NEURAMINIDASE INHIBITORS
Mechanism of Action—The neuraminidase inhibitors have activity against both influenza A and B.
Toxicity—Zanamivir is given by inhaler and commonly causes bronchospasm, limiting its usefulness.
Treatment—To be effective, neuraminidase inhibitors must be given within 48 hours of the onset of symptoms.
Amantadine, rimantadine, or oseltamivir can be given for a longer duration as prophylaxis in patients at risk of serious complications from influenza during an epidemic. Influenza vaccine is preferred for prophylaxis.
FURTHER READING
Antibiotic Handbooks
Bartlett JG, Auwaerter PG, Pham PA. The ABX Guide: Diagnosis and Treatment of Infectious Diseases. 3rd ed. Burlington, MA: Jones & Bartlett Learning; 2012.
Electronic Sources
ePocrates [software]. San Mateo, Calif: Epocrates, Inc. [Web address: www.epocrates.com]
The Johns Hopkins University. ABX Guide [Web resource]. Baltimore, Md: The Johns Hopkins University. [Web address: http://www.hopkinsguides.com/hopkins/ub/index/Johns_Hopkins_ABX_Guide/:]
Other
Mandell GL, Bennett JE, Dolin R. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, PA: Elsevier/Churchill Livingstone; 2010.