Cholestasis is the result of either a functional defect in bile formation at the level of the hepatocyte or a defect in bile secretion and flow at the bile duct level (37). Factors contributing to hepatocyte defects include the viral hepatitides, drugs (19), alcohol, and sepsis. Bile duct obstruction can be intrahepatic (e.g., primary biliary cirrhosis, vanishing bile duct syndrome (28), paucity of interlobular bile ducts) and extrahepatic (e.g., primary sclerosing cholangitis, large duct stones, tumors).
Bile secretion depends on the activities of hepatocytes and bile duct epithelial cells, including many membrane transport systems, and on the morphologic and functional integrity of the bile secretory apparatus (11,17,25,33).
Jaundice is the biochemical and physiologic result of the accumulation in the circulation of bilirubin and other bile constituents. Jaundice is usually, but not always, associated with morphologic changes in the liver. Sometimes the changes reflect primary liver injury, as in cholestatic hepatitis, and sometimes they are secondary to some other condition, such as extrahepatic large duct obstruction (LDO). In some patients jaundice may be clinically and biochemically apparent, but the liver biopsy is essentially unchanged (e.g., increased serum unconjugated bilirubin value in hemolysis).
Jaundice can be a reflection of increase of either unconjugated or conjugated bilirubin, or both, and there are various potential contributing causes (Table 26.1), many of which are discussed in other chapters of this book. This chapter concentrates on the liver biopsy features in conditions not discussed elsewhere in the text and in which there is a disturbance in bilirubin metabolism or in bile flow.
CHOLESTASIS
In the normal liver, bile is almost never visible in the usual histologic preparations. Cholestasis is the term used to describe bile when it is visible in the histologic section, and its presence should be regarded as abnormal. Bile can vary in color from orange or yellow, to green and even brown. Sometimes bile is pale and can be overlooked. With Perls iron stain, it is light green; with Fouchet bile stain or Sirius red, it is more olive-green.
These stains are usually not needed except in the setting of purely intrahepatocytic cholestasis or to find bile production in hepatocellular carcinoma. Cholestasis can be canalicular (Figs. 26.1 to 26.3) and/or intrahepatocytic (Figs. 26.4 and 26.5, e-Figs. 26.1-26.4).
|
TABLE 26.1 Causes of Jaundice with or without Cholestasis |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
P.406
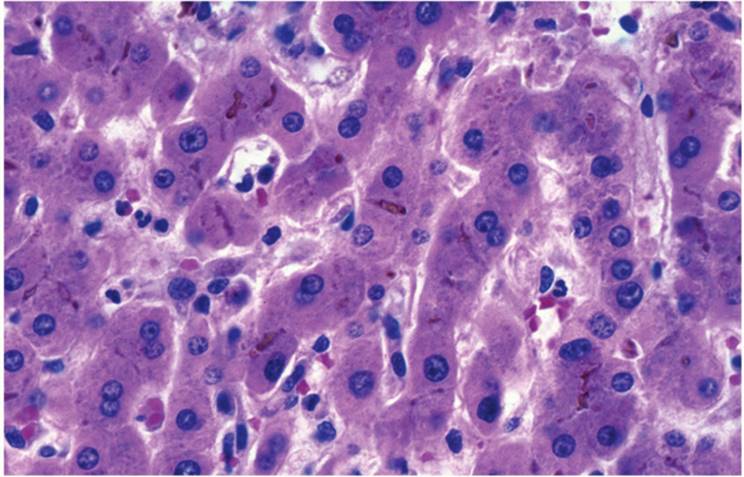
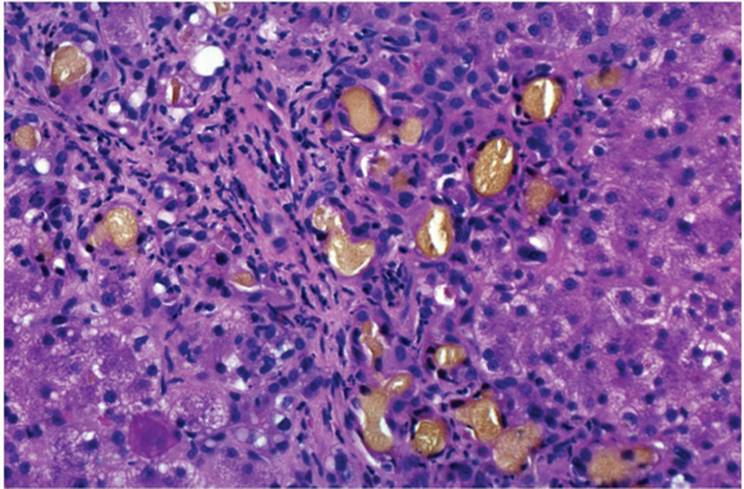
Canalicular cholestasis is seen most often and is easiest to recognize. It is usually most prominent in zone 3 (centrilobular) in acute cholestasis and first seen as accentuation of the usual canalicular structure (Fig. 26.1). Later, globular droplets form (bile plugs, bile thrombi) in dilated canaliculi, which, with surrounding hepatocytes, form an acinus-like structure (Fig. 26.3). With prolonged acute cholestasis, cholestatic rosettes form (e-Fig. 26.1), often appearing empty because larger bile plugs are lost during processing. Canalicular cholestasis is often, but not always, accompanied by the accumulation of bile in hepatocytes and, eventually, Kupffer cells. The principal causes of canalicular cholestasis are summarized in Table 26.2.
|
|
|
FIGURE 26.1 Canalicular cholestasis (hematoxylin-eosin, original magnification ×400). |
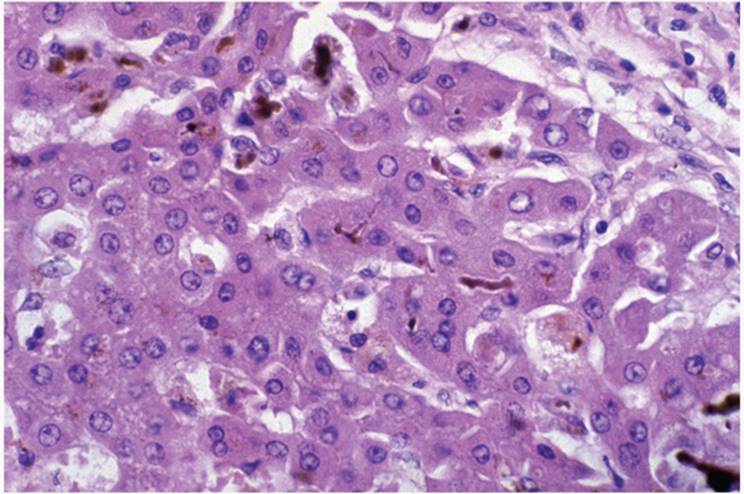
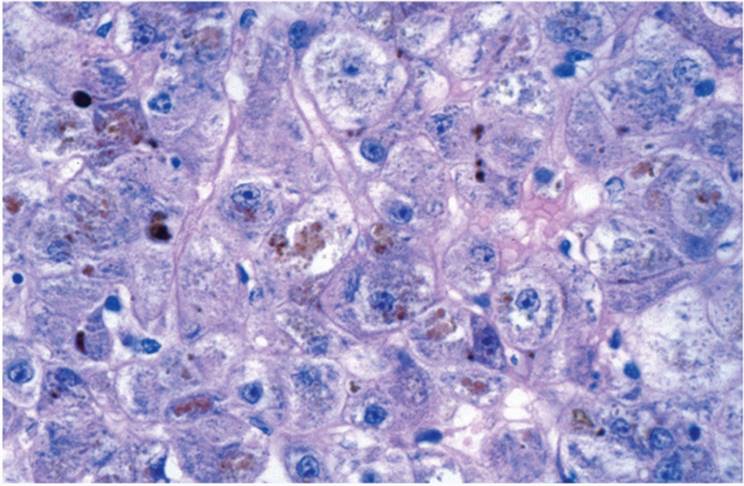
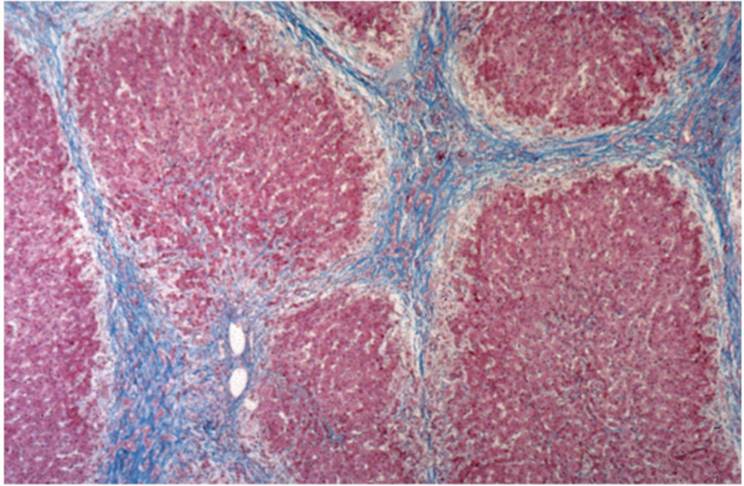
Intrahepatocytic cholestasis is seen as cytoplasmic brown granularity that is initially quite fine and delicate, initially difficult to see, resembling lipofuscin or ceroid (Fig. 26.4). When cholestasis is severe or prolonged, it resembles coarser hemosiderin. In chronic liver diseases, especially those involving the bile duct system, various hepatocyte changes can be seen. Swollen and pale hepatocytes (“feathery degeneration”) become prominent, particularly in zone 1 (periportal) (e-Fig. 26.2). Bile is seen as delicate brown cytoplasmic granularity (Fig. 26.4). This stage is referred to as “cholate stasis” because abnormal bile salts have been implicated in its pathogenesis (25). Copper and copper-associated protein, histochemically demonstrated, and Mallory hyalin can be seen. The connective tissue adjacent to cholate stasis is also often edematous, imparting a halo-like appearance to the cirrhotic nodules of chronic cholestatic disorders (Fig. 26.6). Ductular reaction may be prominent at the interface between portal tracts or cirrhotic septa and the limiting plate of hepatocytes (ductular piecemeal necrosis) (Fig. 26.7) (26).
|
|
|
FIGURE 26.2 Canalicular cholestasis, showing canalicular rupture (hematoxylin-eosin, original magnification ×400). |
|
|
|
FIGURE 26.3 Canalicular cholestasis, showing acinus formation with a bile plug (hematoxylin-eosin, original magnification ×400). |
|
|
|
FIGURE 26.4 Hepatocytic cholestasis, acute, showing swollen cells with delicate cytoplasmic granularity (hematoxylin-eosin, original magnification ×400). |
|
|
|
FIGURE 26.5 Prolonged Hepatocytic cholestasis, showing coarse granules (hematoxylin-eosin, original magnification ×400). |
Isolated or Predominantly Canalicular Cholestasis
Canalicular cholestasis, without hepatocytic cholestasis, is seen in various conditions. Isolated canalicular cholestasis may be caused by idiosyncratic reaction to various drugs (Table 26.3). Biopsy in those cases may show many eosinophils in the inflammatory portal tract infiltrate.
|
TABLE 26.2 Causes of Isolated or Predominantly Canalicular Cholestasis |
||||||
|
|
|
|
FIGURE 26.6 Prolonged cholate stasis seen as pale hepatocytes at the periphery of a cirrhotic nodule in secondary biliary cirrhosis (Masson trichrome, original magnification ×100). |
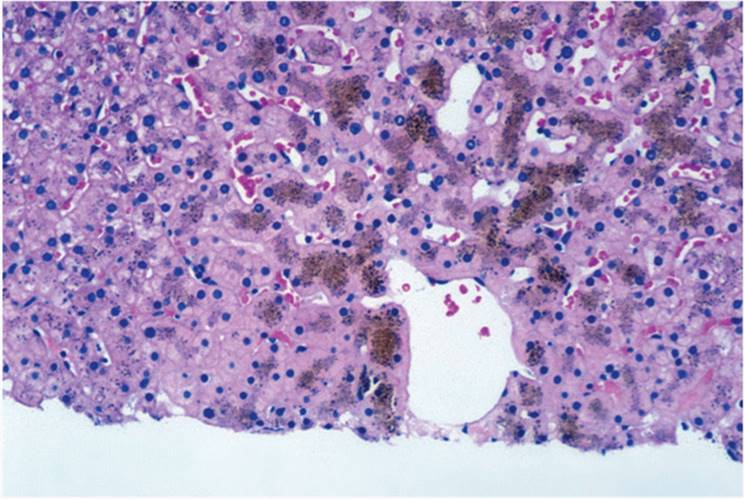
LARGE DUCT OBSTRUCTION (LDO). With contemporary clinical laboratory testing and imaging methods, the diagnosis of LDO rarely requires biopsy confirmation. Biopsy is needed when imaging studies fail to demonstrate obstruction or when laboratory tests are equivocal or suggest parenchymal disease. There is almost always portal inflammation, but it can be relatively mild (see Chapter 18). Portal edema may not be prominent in the posttransplantation biopsy. Cholestasis first affects zone 3 canaliculi but if unrelieved is seen progressively in the other zones. Eventually canaliculi are destroyed and bile lakes, with variable numbers of acute inflammatory cells, develop (e-Figs. 26.3, 26.4). Ductular reaction is typical. Intrahepatocytic cholestasis is also seen with longstanding LDO.
|
|
|||||||||||
|
FIGURE 26.7 Ductular proliferation at the interface between portal tract and lobule (ductular piecemeal necrosis) in large duct obstruction (hematoxylin-eosin, original magnification ×100). |
|||||||||||
|
TABLE 26.3 Some Drugs That Cause Cholestasis |
|||||||||||
|
DRUG-ASSOCIATED CHOLESTASIS. Drugs should always be considered as a cause of canalicular cholestasis, particularly in nonpregnant patients without other contributing conditions. Drugs and toxins are discussed in greater detail in Chapter 11. Cholestasis is almost always seen first in zone 3 (centrilobular) and then can involve the entire lobule. In general, the liver is otherwise unremarkable. Several medications can cause cholestasis (Table 26.3).
CHOLESTASIS OF PREGNANCY. Pregnancy-associated cholestasis generally becomes manifest in the third trimester as painless pruritus and, with rare exceptions, resolves after delivery (23,37). There may be a familial tendency (14). The cholestasis is primarily in zone 3 hepatocytes, predominantly canalicular or also intrahepatocytic (30). There may be mild inflammatory changes, most likely a result of the cholestasis itself. After delivery, the condition regresses and the liver is entirely normal. These patients may develop cholestasis if they take oral contraceptives, however (2).
SEPSIS. Cholestasis is common in sepsis (see Chapter 22). The cholestasis is most often ductular, involving ductules and canals of Hering, but can also manifest as centrilobular (zone 3) and midzonal (zone 2) predominantly canalicular cholestasis (12).
ACUTE HEPATITIS. Acute hepatitis may initially be seen as isolated canalicular cholestasis, before there is significant liver cell necrosis or inflammatory cell reaction.
|
TABLE 26.4 Causes of Isolated or Predominantly Hepatocytic Cholestasis |
||||
|
Predominantly Hepatocytic Cholestasis
Hepatocytic cholestasis is generally accompanied by at least focal canalicular cholestasis. However, in a limited sample, the canalicular component may not always be seen (Table 26.4).
BENIGN RECURRENT INTRAHEPATIC CHOLESTASIS (BRIC). This exceedingly rare condition occurs principally in adults and is almost never biopsied (5). The changes resemble LDO, with both portal and lobular changes, although portal tract edema is normally not prominent. Sometimes there is only isolated, predominantly hepatocytic cholestasis. This condition can be associated with cholestasis of pregnancy and with the use of oral contraceptives (10). A gene locus, similar to that of Byler disease (see below), has been identified (5).
LYMPHOMAS. Cholestasis may be seen as a paraneoplastic phenomenon in patients with malignant lymphomas, particularly Hodgkin lymphoma (20,24), as well as in patients with non-Hodgkin lymphoma (35). Isolated cholestasis in Hodgkin lymphoma may reflect adult-onset bile duct paucity (16). Cholestasis in Hodgkin lymphoma can also be from extrahepatic bile duct compression by enlarged, often fibrotic lymph nodes.
Familial Syndromes and Other Conditions Associated with Intrahepatic Cholestasis
Familial disorders of cholestasis are usually related to mutations in genes controlling hepatocellular transport systems involved in bile formation. (29,33).
PROGRESSIVE FAMILIAL INTRAHEPATIC CHOLESTASIS (PFIC). The group of disorders termed progressive familial intrahepatic cholestasis is characterized by (a) chronic, unremitting cholestasis; (b) a characteristic constellation of clinical, biochemical, and histologic features; (c) absence of a specific anatomic abnormality; (d) absence of an identifiable metabolic disorder; and (e) an autosomal recessive inheritance pattern of occurrence. PFIC generally presents in neonates or early childhood with cholestasis, pruritus, growth failure, hepatomegaly, pancreatic deficiency, and fat-soluble vitamin deficiency (36). Typically other clinical manifestations are seen. One or more defects in the genes expressing proteins contribute (11,21).
BYLER DISEASE AND BYLER SYNDROME. The gene for Byler disease has been mapped to the 19-cM region of chromosome 18q21-q22 (7), the same site for the genes of BRIC (15). Early, the liver biopsy is similar in the two conditions, with intracanalicular cholestasis the predominant feature, with little or no inflammatory or other tissue reaction. Later Byler disease, in contrast with BRIC, has progressive portal tract fibrosis and portal-to-portal bridging. Death from cirrhosis and liver failure occurs in childhood and early adolescence. Liver transplantation is curative. A similar, but ultrastructurally different, disorder occurs in non-Amish kindred and is known as Byler syndrome (6), with various abnormal gene loci (33,37).
PEROXISOMAL DISORDERS
Zellweger (Cerebrohepatorenal) Syndrome
In classic Zellweger syndrome the liver biopsy may be unremarkable initially, but there may be portal inflammation, focal necrosis, progressive fibrosis, and ultimately, cirrhosis (9,22,27). The only constant finding is the absence of peroxisomes when the liver is studied with the electron microscope (13).
Recurrent Cholestasis with Lymphedema (Norwegian Cholestasis; Aagenaes Syndrome)
This form of neonatal jaundice may clear in childhood but can recur throughout life. Profound lower extremity lymphedema begins in childhood or during adolescence (1,31), and there may be familial lymphangiomas and hemangiomas (29). Biopsies vary, with changes including paucity of intrahepatic bile ducts, canalicular cholestasis, giant cell transformation of hepatocytes, and, in adults, cirrhosis (29).
CONGENITAL HYPERBILIRUBINEMIA SYNDROMES
Physiologic Jaundice of the Newborn
During the first few days of life, the capacity of the liver to clear hepatic bilirubin is not yet fully developed. There may also be increased bilirubin production because of accelerated red blood cell destruction in this period. Biopsy is virtually never required.
Unconjugated Hyperbilirubinemia Syndromes
CRIGLER-NAJJAR SYNDROMES I AND II. In type I Crigler-Najjar syndrome, there is lifelong, severe nonhemolytic unconjugated hyperbilirubinemia caused by congenital deficiency of bilirubin-uridine diphosphateglucuronyl transferase. The less severe type II is responsive to therapy with phenobarbital (32). In both forms, the liver biopsy is unremarkable except for occasional bile plugs (8).
GILBERT SYNDROME. Gilbert syndrome is a relatively common condition that affects more than 5% of the adult population. Liver biopsy is not helpful because there are no recognizable microscopic changes other than increased lipofuscin pigment (4).
Conjugated Hyperbilirubinemia Syndromes
DUBIN-JOHNSON SYNDROME. Dubin-Johnson syndrome has a point mutation of the gene responsible for the canalicular multispecific organic-anion transporter (33). Patients have chronic or intermittent hyperbilirubinemia rather than cholestasis (36). Biopsy is almost always obtained incidental to a surgical procedure in which the surgeon sees the mahogany-colored liver. The liver biopsy is characteristic, with large, coarse, dark brown, iron-negative, Fontana-Masson-positive nonmelanin granules in hepatocytes, primarily in zone 3 (Fig. 26.8, e-Figs. 26.5, 26.6) (3,34). The biopsy is otherwise unremarkable.
ROTOR SYNDROME. This benign familial disorder is clinically similar to Dubin-Johnson syndrome, but there are no macroscopic or microscopic liver changes.
METABOLIC DISORDERS
The principal metabolic disorders contributing to cholestasis are discussed in Chapter 14.
|
|
|
FIGURE 26.8 Dubin-Johnson syndrome, showing the typical coarse intrahepatocytic granules, predominantly in zone 3 (hematoxylin-eosin, original magnification ×100). |
ACQUIRED CONDITIONS
Various acquired conditions can contribute to the development of cholestasis, including viral hepatitis, sepsis, total parenteral nutrition, infiltrative disorders, and infectious and granulomatous disorders.
Benign Postoperative Cholestasis
Cholestasis, with and without jaundice, in the postoperative period is a result of various factors, including sepsis, LDO, shock, hemolysis, drug-induced hepatitis, and other conditions. Characterized by predominantly conjugated hyperbilirubinemia, it generally occurs in the first 2 days after major surgery and may last as long as 2 weeks. It is thought to be caused by bilirubin overload of the liver, with or without reduced secretory capacity of the liver; biopsy shows predominantly zone 3 canalicular cholestasis without inflammation (e-Figs. 26.7, 26.8). With hemolysis, Kupffer cells contain hemosiderin (18).
REFERENCES
1. Aagenae Ø, Van der Hagen CB, Refsum S. Hereditary recurrent intrahepatic cholestasis from birth. Arch Dis Child 1968;43:646-657.
2. Adlercreutz H, Tenenbaum R. Some aspects of the interaction between natural and synthetic female sex hormones and the liver. Am J Med 1970;49:630-649.
3. Barone P, Inferrera C, Carrozza G. Pigments in the Dubin-Johnson syndrome. In: Wolman M, ed. Pigments in Pathology. New York: Academic Press, 1969:307-325.
4. Barth RF, Grimley PM, Berk PD, et al. Excess lipofuscin accumulation in constitutional hepatic dysfunction (Gilbert's syndrome). Arch Pathol 1971;91:41-47.
5. Bijleveld CAM, Vomnk RJ, Kuipers F, et al. Benign recurrent intrahepatic cholestasis: a long-term follow-up study of two patients. Hepatology 1989;9:532-537.
6. Bull LN, Carlton VE, Stricker NL, et al. Genetic and morphologic findings in progressive familial intrahepatic cholestasis (Byler disease [PFIC 1] and Byler syndrome); evidence for heterogeneity. Hepatology 1997;26:155-164.
7. Carlton VE, Knisely As, Freimer NB. Mapping of a locus for progressive familial intrahepatic cholestasis (Byler disease) to 18q21-q22, the benign recurrent intrahepatic cholestasis region. Hum Mol Genet 1995;4:1049-1053.
8. Crigler JF, Najjar VA. Congenital familial nonhemolytic jaundice associated with kernicterus. Pediatrics 1952;10:169-179.
9. Danks DM, Tippett P, Adams C, et al. Cerebro-hepato-renal syndrome of Zellweger: a report of eight cases with comments upon the incidence, the liver lesion, and a fault of pipecolic acid metabolism. J Pediatr 1975;86:382-387.
10. DePagter AGF, van Berge Henegouwen FP, ten Bokkel Huinink JA, et al. Familial benign recurrent intrahepatic cholestasis: interrelation with intrahepatic cholestasis of pregnancy and from oral contraceptives? Gastroenterology 1976;71:202-207.
11. Fitz JG. Regulation of cholangiocyte secretion. Semin Liver Dis 2002;22:241-258.
12. Fuchs M, Sanyal AJ. Sepsis and cholestasis. Clin Liver Dis 2008;12:151-172.
13. Goldfischer S, Moore CL, Johnson AB, et al. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science 1973;182:62-64.
14. Holzbach RT, Sivak, DA, Braun WE. Familial recurrent intrahepatic cholestasis of pregnancy: a genetic study providing evidence for a sex-linked, dominant trait. Gastroenterology 1983;85:175-179.
15. Houwen RHJ, Baharloo S, Blankenship K, et al. Genome screening by searching for shared segments: mapping a gene for benign recurrent intrahepatic cholestasis. Nat Genet 1994;8:380-386.
16. Hubscher SG, Lumley MA, Elias E. Vanishing bile duct syndrome: a possible mechanism for intrahepatic cholestasis in Hodgkin's lymphoma. Hepatology 1993;17:70-77.
17. Jansen PLM. The pathophysiology of cholestasis with special reference to primary biliary cirrhosis. Ballières Clin Gastroenterol 2000;14:571-583.
18. Kantrowitz PA, Jones WA, Greenberger NJ, et al. Severe postoperative hyperbilirubinemia simulating obstructive jaundice. N Engl J Med 1967;276:591-598.
19. Kass GEN, Price SC. Role of mitochondria in drug-induced cholestatic injury. Clin Liv Dis 2008;12:27-51.
20. Lieberman DA. Intrahepatic cholestasis due to Hodgkin's disease: an elusive diagnosis. J Clin Gastroenterol 1986;8:304-307.
21. Muller M, Jansen PLM. Molecular aspects of hepatobiliary transport. Am J Physiol 1997; 272:G1285-G1303.
22. Nakamura K, Takenouchi T, Aizawa M, et al. Cerebro-hepato-renal syndrome of Zellweger. Clinical and autopsy findings and a review of previous cases in Japan. Acta Pathol Jpn 1986;36:1727-1735.
23. Ollson R, Tysk C, Aldenborg F, et al. Prolonged postpartum course of intrahepatic cholestasis of pregnancy. Gastroenterology 1993;105:267-271.
24. Perera DR, Greene ML, Fenster LF. Cholestasis associated with extrabiliary Hodgkin's disease. Report of three cases and review of four others. Gastroenterology 1974;67: 680-685.
25. Popper H. Cholestasis: the future or a past and present riddle. Hepatology 1981;1:187-191.
26. Portmann B, Popper H, Neuberger J, et al. Sequential and diagnostic features in primary biliary cirrhosis based on serial histologic study in 209 patients. Gastroenterology 1985; 88:1777-1790.
27. Powers JM, Moser HW, Moser AB, et al. Fetal cerebrohepatorenal (Zellweger) syndrome. Dysmorphic, radiologic, biochemical, and pathologic findings in four affected fetuses. Hum Pathol 1985;16:610-620.
28. Reau NS, Jensen DM. Vanishing bile duct syndrome. Clin Liver Dis 2008:12:203-217.
29. Riely CA. Familial intrahepatic cholestatic syndromes. Semin Liver Dis 1987;7:119-133.
30. Rolfes DB, Ishak KG. Liver disease in pregnancy. Histopathology 1986;10:555-570.
31. Sharp HL, Krivit W. Hereditary lymphedema and obstructive jaundice. J Pediatr 1971;78: 491-496.
32. Sorrentino D, Jones EA, Berk PD. Familial hyperbilirubinemia syndromes: kinetic approaches. Ballières Clin Gastroenterol 1989;3:313-336.
33. Trauner M, Meier PJ, Boyer JL. Molecular pathogenesis of cholestasis. N Engl J Med 1998;339:1217-1227.
34. Varma RR, Sarna T. Hepatic pigments in Dubin-Johnson syndrome and mutant Corriedale sheep are not melanin. Gastroenterology 1986;84:1401.
35. Watterson J, Priest JR. Jaundice as a paraneoplastic phenomenon in a T-cell lymphoma. Gastroenterology 1989;97:1319-1322.
36. Wolkoff AW. Inheritable disorders manifested by conjugated hyperbilirubinemia. Semin Liver Dis 1993;3:65-72.
37. Zollner G, Trauner M. Mechanisms of cholestasis. Clin Liv Dis 2008;12:1-26.