James H. Hamilton, IV, MD, and S. Mark Borganelli, MD
CASE PRESENTATION
A 32-year-old man was referred to for evaluation of dyspnea on exertion and the finding of a systolic murmur on examination. He has begun to experience chest discomfort that is brought on with exertion and resolves with rest. He admits to frequent palpitations. Family history: father killed in a motor vehicle accident at age 35; paternal uncle died suddenly at age 40 of unknown cause.
Physical examination revealed a systolic murmur that worsens with rapidly standing, squatting, and standing again. A 12-lead ECG reveals prominent QRS voltage in the precordial leads with deep T-wave inversions (Figure 53-1). An echocardiogram was obtained demonstrating a nearly 5 cm ventricular septum and mitral regurgitation secondary to systolic anterior motion of the mitral valve (SAM) (Figures 53-2 to 53-5). A resting left ventricular outflow tract (LVOT) gradient of 45 mm Hg was recorded with a provocable gradient of >80 mm Hg (Figure 53-6).
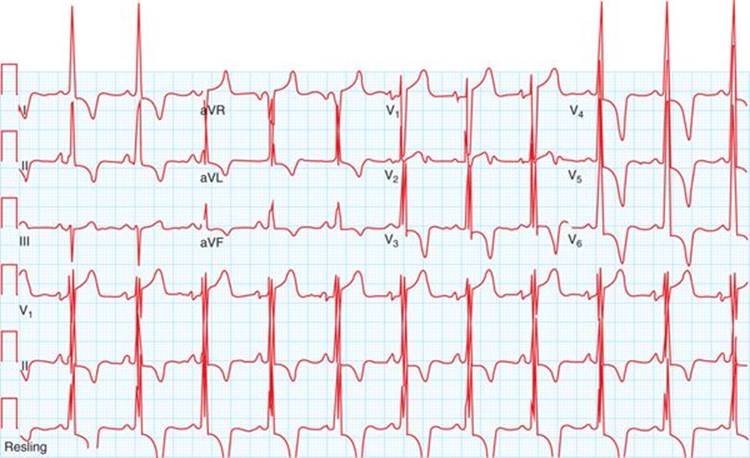
FIGURE 53-1 ECG in a patient with HCM. Note prominent QRS voltages and deep T-wave inversions.
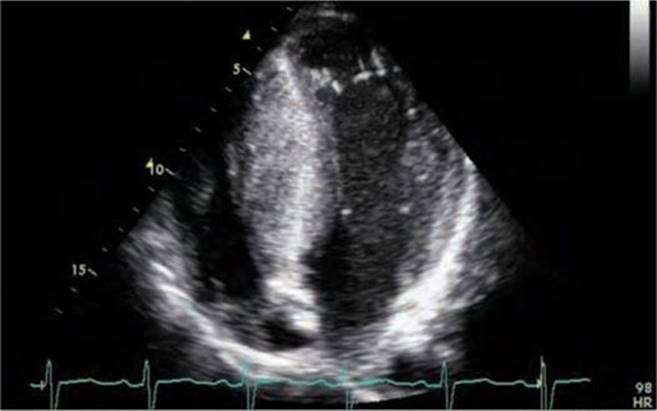
FIGURE 53-2 Echocardiogram demonstrating asymmetric hypertrophy with nearly a 5-cm septum.
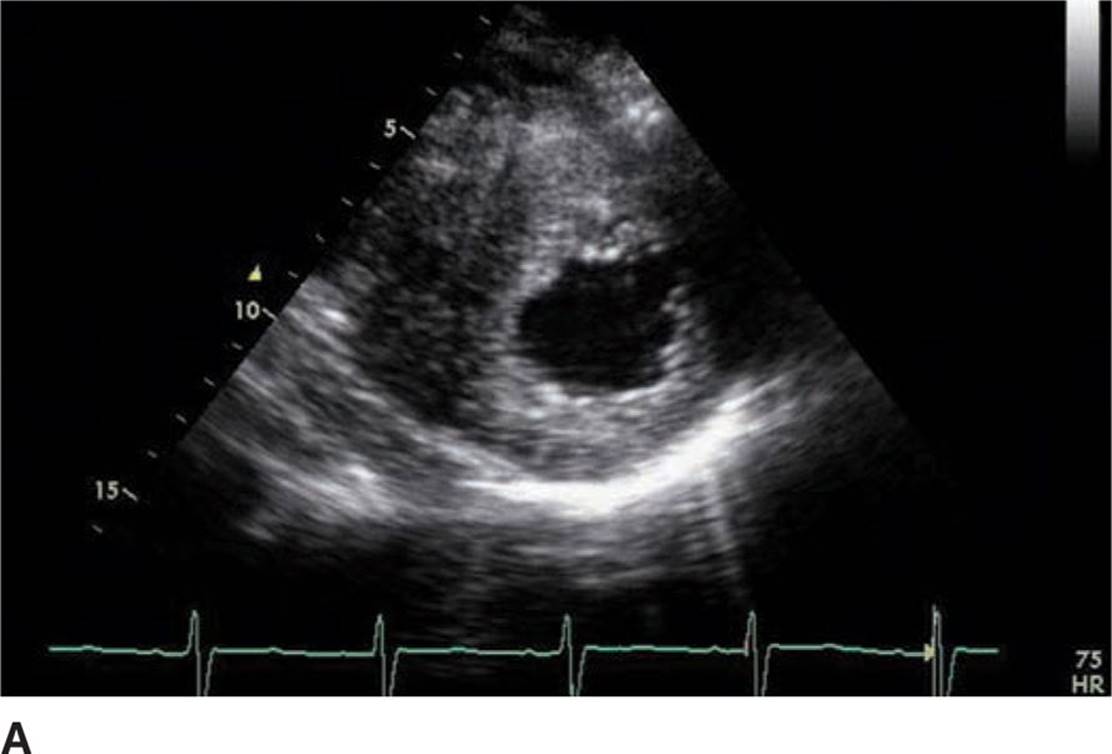
FIGURE 53-3A Parasternal short axis view of left ventricle in diastole.
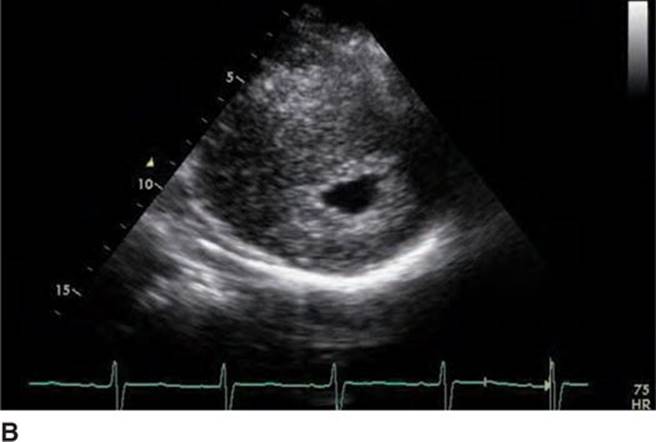
FIGURE 53-3B Parasternal short axis view of the left ventricle in systole.
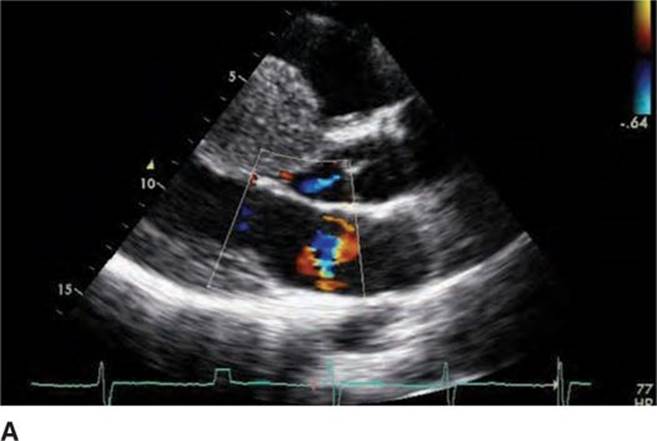
FIGURE 53-4A Parasternal long-axis view with color Doppler in diastole demonstrating normal opening of mitral valve.
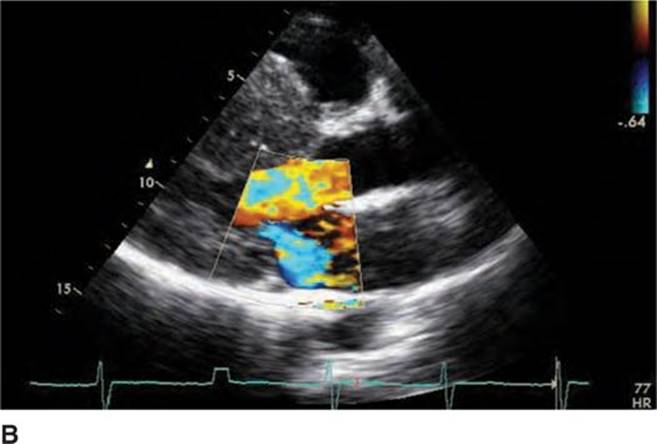
FIGURE 53-4B Parasternal long-axis view during systole with color Doppler demonstrating increased turbulent flow through the aortic valve with SAM and resulting mitral regurgitation.
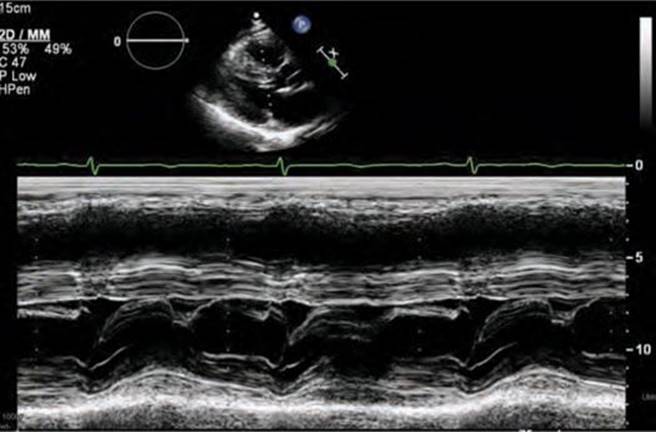
FIGURE 53-5 M-mode across the mitral valve demonstrating SAM with mitral-septal contact.
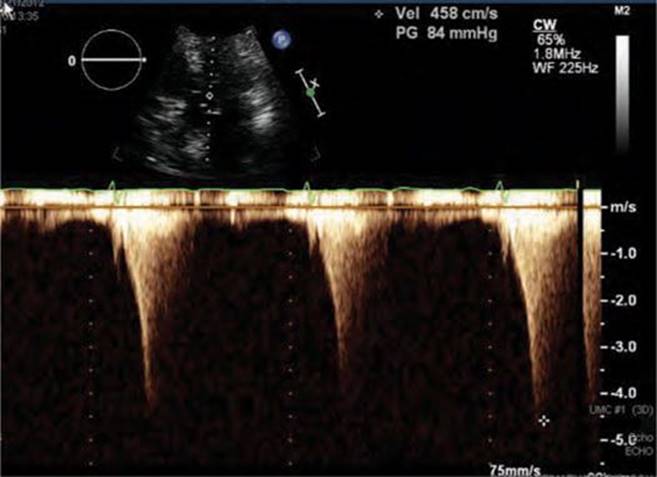
FIGURE 53-6 LVOT gradient after provocation with peak gradient recorded as 84 mm Hg.
The patient was started on metoprolol tartrate 50 mg twice daily and rapidly up titrated to 200 mg twice daily. Genetic counseling and genetic testing were performed, revealing a β-myosin heavy chain mutation and confirming the diagnosis of hypertrophic cardiomyopathy (HCM).
Based on a family history of unexplained sudden death and an LV wall thickness of greater than 3 cm, he underwent implantation of an ICD without complication for prevention of sudden cardiac death (SCD). Unfortunately, he had no improvement in symptoms despite high dose metoprolol and initiation of disopyramide. After 3 months of medical therapy and an attempt at pacing the right ventricle with the already implanted ICD, the LVOT gradient with provocation remained >50 mm Hg. Given his young age and refractory NYHA class III symptoms, he was referred to for myectomy. This operation was performed without complication and resulted in immediate improvement of symptoms. At follow-up 1 year later, the patient remains symptom-free with no resting or provocable LVOT gradient and no recorded arrhythmias.
EPIDEMIOLOGY
Inherited as an autosomal-dominant disorder, HCM is variably penetrant resulting in striking variations in phenotype even among first-degree relatives. Traditionally, the prevalence of HCM has been cited as 1:500, but due to the lack of symptoms in many individuals who may carry the genetic trait, the true prevalence may be much higher.1,2
ETIOLOGY AND PATHOPHYSIOLOGY
Clinically, hypertrophic cardiomyopathy (HCM) is generally described as left ventricular hypertrophy (LVH) with nondilated ventricular chambers in the absence of another cardiac or systemic disease that would be capable of producing the same degree of hypertrophy.1,2 Patients may harbor nearly any diffuse or segmental pattern of hypertrophy and fibrosis with presentations ranging from completely asymptomatic to NYHA class II-IV heart failure to SCD.
The onset of symptoms and recognition of the disease may occur at any age from infancy to adulthood, but commonly develops during periods of accelerated body growth, such as adolescence. For that reason clinical screening is generally recommended in patients with genetically proven HCM beginning at age 12 and continuing yearly until age 18 to 21 followed by screening every 5 years thereafter.1
The availability of genetic testing has aided in the identification of an increasing number of genotype-positive, phenotype-negative patients. Indeed, there are now well over 1400 documented mutations that, when coupled with a sensitive genetic substrate and environmental triggering factors, are responsible for the variety of clinical presentations of HCM.1,3 The genetic mutations allow the discrimination of HCM patients from patients with other genetic causes of LVH, such as Fabry disease, who would require much different management.
The original associations of genetic variants to HCM patients were isolated to myofilament (sarcomeric) proteins with the most common mutations occurring in the β-myosin heavy chain and cardiac myosin-binding protein C.3Recent investigations have focused not only on the sarcomeric proteins but on the associated contractile apparatus, as well, with new disease causing mutations classified as Z-disc HCM and calcium-handling HCM.3 Ongoing investigations are trying to associate disease outcomes with various mutations and/or the affected protein subunits.1,3 The cellular alterations caused by these mutations lead to varying degrees of hypertrophy and fibrosis in the heart. This in turn may eventually lead to systolic anterior motion of the mitral valve with LVOT obstruction; the resulting symptoms of dyspnea, congestive heart failure, and syncope; and the risk of cardiac arrhythmias including atrial fibrillation, ventricular tachycardia, and ventricular fibrillation.
DIAGNOSIS
Initial clinical evaluation of a patient includes a thorough personal and family history to elicit symptoms of syncope, exercise-induced chest discomfort, dyspnea, palpitations, and/or family history of SCD. The physical examination should include a careful auscultation of the patient with appropriate maneuvers such as stand-squat-stand to evaluate the characteristics of the murmur.
Initial screening should include each of the following:
• 12-lead ECG1,2
• Transthoracic echocardiogram
• 24-hour Holter monitoring
• Genetic assays if feasible
• Graded exercise test
The 12-lead ECG (see Figure 53-1) should demonstrate LVH and repolarization abnormalities. Incongruence between the ECG and echo findings should lead to definitive evaluation with either cardiac magnetic resonance imaging (CMRI)(Figure 53-7A and 53-7B) or invasive hemodynamic studies in the cath lab. In order to help assess the risk of SCD, HCM patients should also have 24-hour Holter monitoring and a graded exercise test performed at the time of diagnosis to evaluate for the presence of asymptomatic arrhythmias and determine the blood pressure response during exercise.1,4
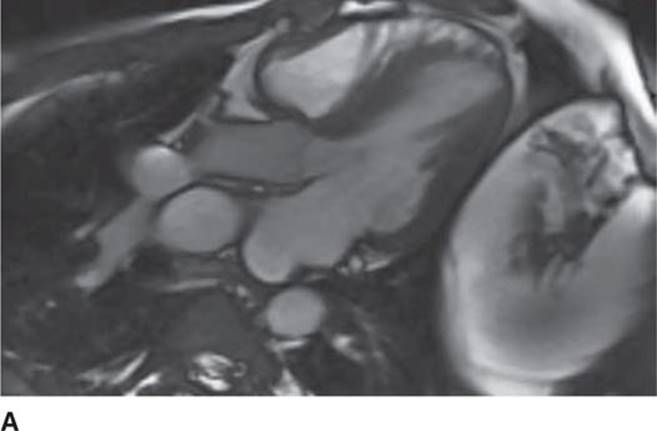
FIGURE 53-7A Cardiac MR with open mitral inflow during diastole. (Used with permission of Dr. Andrew Rivard at University of Mississippi Medical Center, Jackson, MS.)
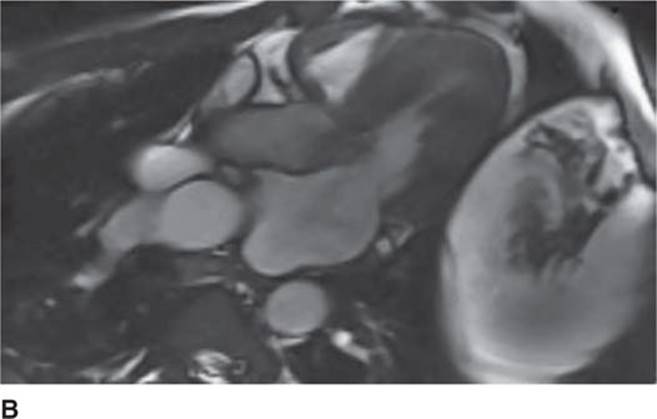
FIGURE 53-7B Cardiac MR with obstruction of the LVOT during systole due to SAM of the mitral valve. (Used with permission of Dr. Andrew Rivard at University of Mississippi Medical Center, Jackson, MS.)
Screening with transthoracic echocardiogram should be performed at the time of the initial evaluation and yearly thereafter in known HCM patients and is recommended at the time of initial screening of first-degree relatives to assess left ventricular systolic and diastolic function, wall thickness, and outflow tract gradients.1-3,5 The obstruction that occurs in HCM has been shown to be due to mitral valve systolic anterior motion (SAM) with mitral-septal contact (best illustrated in Figures 53-5 and 53-7B), muscular obstruction in the midcavitary region, or anomalous papillary muscle insertion into the anterior mitral leaflet.1,2,6 The peak LVOT or midcavitary gradient should be measured at rest, and if <50 mm Hg, the test should be repeated with provocative measures such as exercise, Valsalva maneuver, or potentially amyl nitrate, with the latter rarely required in the current era. Provocation with dobutamine infusion during echo is no longer recommended. Upon testing, one-third of patients will have a resting LVOT gradient >30 mm Hg (obstructive HCM), one-third will have resting gradients <30 mm Hg (nonobstructive HCM), and a final one-third will have normal resting gradients but provoked gradients >30 mm Hg (labile obstruction).1,2,6
Genetic screening has become an important part of the work-up of HCM patients. Identifying a specific genetic mutation associated with HCM allows for easier screening stratification of first-degree relatives into a positive disease group or no disease group. Although all first degree relatives of HCM patients require initial clinical assessment with ECG and echocardiogram, if these relatives have normal clinical assessments and are free of the familial mutation, that individual patient does not need repeat assessment in the future.1,3,5 Multiple studies are ongoing to determine whether specific mutation(s) lead to increased risk of SCD or severity of disease.1,3
MANAGEMENT
Management of HCM includes efforts to decrease heart failure and angina symptoms related to left ventricular diastolic dysfunction and LVOT obstruction (Table 53-1), evaluating the risk of SCD and assessing for and treating cardiac arrhythmias.
TABLE 53-1 Summary of Guideline Pharmacologic Recommendations

Initial therapy for symptomatic and asymptomatic patients is generally with a β-blocker unless contraindications exist.1,2,7 High doses of β-blockers may be required for symptom relief and can be pushed to maximum drug doses unless limited by bradycardia. Directly reducing contractility due to the negative inotropic effect of β-blockers improves the dynamic LVOT gradient, while the negative chronotropic effect improves diastolic filling times.
Alternatively, patients who cannot take β-blockers may be tried on verapamil.1,2,7 Due to the pharmacologic properties of verapamil (and diltiazem), some patients may develop a more profound vasodilatory effect than the desired negative inotrope effect, in essence, worsening the dynamic LVOT obstruction/gradient and symptoms. Therefore, initiation of verapamil (or diltiazem) in patients with signs of severely elevated pulmonary capillary wedge pressure, low systemic blood pressure, and high LVOT gradients should be performed with extreme caution.1,2,7 For patients with symptoms refractory to initial medical therapy, disopyramide may be considered as an adjunct agent.1,2,7 The clinical benefits of disopyramide are related to its negative inotropic effect on the heart. Initiation of disopyramide should be done in a hospital setting with patients monitored for excessive QT prolongation and proarrhythmia.
For patients who remain medically refractory with continued NYHA class III-IV symptoms, additional therapeutic interventions must be considered. For patients who are younger and are felt to be suitable candidates, invasive septal reduction therapy is recommended at this point in management. This type of treatment is only effective for patients whose symptoms are attributable to increased peak LVOT gradients >50 mm Hg, whether at rest or with provocation, and is not intended for patients with symptoms related to severe diastolic dysfunction or angina related to subendocardial ischemia in the absence of a high LVOT gradient.1,8-10 For patients who are not suitable candidates for invasive septal reduction and who already have dual chamber ICDs in place for prevention of SCD, attempts at RV pacing may be tried to reduce the LVOT gradient.1,11 This concept was thought to be a potential therapeutic intervention for patients in the past with initial reports of symptomatic improvement and decreased LVOT gradients; however, three randomized controlled trials were published refuting the effectiveness of RV pacing as a means to decrease the LVOT gradient.1,11,12 Based on the results of those trials, it appears that there is a symptomatic improvement in a majority of patients (30%-80%) but an objective reduction in LVOT gradient in a much smaller percentage of patients (25%-40%).1,11,12 This indicates both a placebo effect and training effect of pacemaker therapy on the symptoms of these patients. With the current level of evidence, the guidelines give dual chamber pacing a class IIa indication to reduce LVOT gradients in patients who already have a dual chamber pacing device for non-HCM indications and a class IIb recommendation to improve symptoms in patients who have medically refractory symptoms but are poor candidates for invasive septal reduction.1,11,12
Invasive therapeutic options include surgical myectomy and alcohol septal ablation. Surgical myectomy procedures have been performed for nearly 5 decades with high volume centers reporting excellent success rates. Indeed, in patients who are deemed candidates for surgical myectomy, it has emerged as the treatment of choice for patients with obstructive HCM with operative success achieved in 90% to 95% of patients with operative mortality reported to be <1% and nonfatal complication rates of 2% to 3% in certain high volume centers.1,8-10 In addition, myectomy has the advantage of being able to correct other causes of LVOT obstruction other than septal hypertrophy alone, such as aberrant papillary muscle implantation or abnormal elongation of the anterior mitral leaflet.1,8-10 Following surgical myectomy, long-term survival did not differ from the age-matched, general U.S. population and was superior to that of HCM patients with LVOT obstruction who did not undergo myectomy. The reported incidence of SCD or appropriate ICD discharge following surgical myectomy is reported as <0.9%; however, surgical myectomy does not eradicate the need for SCD risk assessment and possible ICD implantation.1,8-10
In comparison, alcohol septal ablation is also of benefit in the right patient cohort and results in an immediate fall in gradient due to decreased septal contraction in >90% of patients.1,9,10 Success rates are dependent on individual anatomy with an inherent need for a septal perforator artery that supplies the area of SAM-septal contact; in its absence, the procedure cannot be performed. Over half of patients undergoing alcohol septal ablation require temporary pacing during the procedure and between 10% and 20% require permanent pacing.1,13 In addition, the reported rate of sustained ventricular arrhythmias and SCD following alcohol septal ablation is 3% to 10%.1,13 Despite the complications, the data suggest that the 4-year survival is similar for both surgical myectomy and alcohol septal ablation procedures.1,9,10,13
Due to diastolic dysfunction and accompanying enlargement of the left atrium in HCM, atrial fibrillation is a common finding in patients older than 30 years old and ultimately occurring in up to 20% to 25% of HCM patients.1,2Even with short episodes of paroxysmal atrial fibrillation, the risk of thrombus formation and systemic embolization is high in HCM patients and results in a class I indication for anticoagulation with vitamin-K antagonists.1,2 β-Blockers and verapamil are recommended for rate control, but symptoms may warrant attempts at rhythm control. Amiodarone or disopyramide (along with β-blockers or verapamil) have shown potential use as rhythm controlling agents with amiodarone being the preferred rhythm control agent.1,2 Pulmonary vein isolation can be done in appropriate subsets of HCM patients with early success and complication rates similar to other forms of heart disease.1
Risk assessment for SCD must be performed in each patient at the time of initial evaluation and repeated at appropriate clinical intervals (usually yearly) for individuals who do not initially receive an ICD. The overall rate of SCD in HCM patients is approximately 1% per year; although, it appears that when patients have at least one of the following conventional risk markers, the rate of appropriate ICD therapy is approximately 3% to 4% per year.1,4,14
1. History of ventricular fibrillation, ventricular tachycardia, or aborted SCD
2. Family history of SCD related to HCM
3. Unexplained syncope (higher weight to events <6 months from evaluation)
4. Documented NSVT on ambulatory ECG monitoring
5. Maximal LV wall thickness greater than or equal to 30 mm
Of the above risk markers, prior aborted SCD carries the highest future risk, with a 10% per year event rate.1,4,14
The decision for ICD placement must be based upon the risk-benefit ratio in an individual patient. This is due to the high incidence of ICD complications in cohorts of HCM patients, reportedly up to 4% per year.1,4,14 As such, the only class I indication for ICD implantation in HCM patients is survival of SCD.1,14 There is a class II indication that ICD implantation is reasonable in patients with a family history of SCD, unexplained syncope, and LV wall thickness >30 mm.1,14 In remaining patients, it is important to identify the presence of further risk factors, such as an abnormal blood pressure response to exercise or episodes of NSVT on continuous ECG monitoring that may sway the decision towards or away from ICD implantation.
FUTURE DIRECTIONS
Management of HCM continues to evolve. Of particular interest is the development of subcutaneous ICDs and their upcoming entrance into widespread clinical practice. Subcutaneous ICD effectiveness will need to be verified in HCM patients with the extremes of LV hypertrophy, but given the long-term complication rate of intravascular ICDs, these may prove to be quite beneficial, especially in young patients. Considering the long-term complications of intravascular hardware, implanters should choose the proper device to minimize the number of intravascular leads, with consideration given to single coil ICD leads and VDD leads when appropriate. Additionally, technology in the areas of cardiac MR and genetic testing may improve and, potentially, redefine SCD risk assessment and management of HCM.
REFERENCES
1. Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2011;58(25):e212-260.
2. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42(9):1687-1713.
3. Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54(3):201-211.
4. Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010;121(3):445-456.
5. Maron BJ, Seidman JG, Seidman CE. Proposal for contemporary screening strategies in families with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004;44(11):2125-2132.
6. Maron MS, Olivotto I, Zenovich AG, et al. Hypertrophic cardiomyopathy is predominantly a disease of left ventricular outflow tract obstruction. Circulation. 2006;114(21):2232-2239.
7. Fifer MA, Vlahakes GJ. Management of symptoms in hypertrophic cardiomyopathy. Circulation. 2008;117(3):429-439.
8. Ommen SR, Maron BJ, Olivotto I, et al. Long-term effects of surgical septal myectomy on survival in patients with obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2005;46(3): 470-476.
9. Sorajja P, Valeti U, Nishimura RA, et al. Outcome of alcohol septal ablation for obstructive hypertrophic cardiomyopathy. Circulation. 2008;118(2):131-139.
10. Fernandes VK, Nielsen C, Nagueh SF, et al. Follow-up of alcohol septal ablation for symptomatic hypertrophic obstructive cardiomyopathy, the Baylor and Medical University of South Carolina experience 1996 to 2007. JACC Cardiovasc Interv. 2008;1(5): 561-570.
11. Ommen SR, Nishimura RA, Squires RW, et al. Comparison of dual chamber pacing versus septal myectomy for the treatment of patients with hypertrophic obstructive cardiomyopathy: a comparison of objective hemodynamic and exercise end points. J Am Coll Cardiol. 1999;34(1):191-196.
12. Maron BJ, Nishimura RA, McKenna WJ, et al. Assessment of permanent dual-chamber pacing as a treatment for drug-refractory symptomatic patients with obstructive hypertrophic cardiomyopathy: a randomized, double-blind, crossover study (M-PATHY). Circulation. 1999;99(22):2927-2933.
13. Seggewiss H. Current status of alcohol septal ablation for patients with hypertrophic cardiomyopathy. Curr Cardiol Rep. 2001;3(2): 160-166.
14. Maron BJ, Spirito P, Shen WK, et al. Implantable cardioverter-defibrillators and prevention of sudden cardiac death in hypertrophic cardiomyopathy. JAMA. 2007;298(4):405-412.