Acanthosis Nigricans (AN) ICD-9: 701.2 ![]() ICD-10: L 83
ICD-10: L 83 ![]()
![]() Asymmetric velvety thickening and hyperpigmentation of the skin, chiefly on the neck, axilla, groins, and other body folds.
Asymmetric velvety thickening and hyperpigmentation of the skin, chiefly on the neck, axilla, groins, and other body folds.
![]() May be hyperkeratotic and associated with skin tags.
May be hyperkeratotic and associated with skin tags.
![]() A cutaneous marker related to heredity, obesity, endocrine disorders (particularly diabetes), drug administration, and malignancy.
A cutaneous marker related to heredity, obesity, endocrine disorders (particularly diabetes), drug administration, and malignancy.
![]() Insidious onset; in malignancy, rapid.
Insidious onset; in malignancy, rapid.
Classification
Type 1: Hereditary Benign AN. No associated endocrine disorder.
Type 2: Benign AN. Endocrine disorders associated with insulin resistance: insulin-resistant type II diabetes mellitus, hyper-androgenic states, acromegaly/gigantism, Cushing disease, hypogonadal syndromes with insulin resistance, Addison disease, and hypothyroidism.
Type 3: Pseudo-AN. Associated with obesity; more common in patients with darker pigmentation. Common in metabolic syndrome. Obesity produces insulin resistance.
Type 4: Drug-Induced AN. Nicotinic acid in high dosage, stilbestrol in young males, glucocorticoid therapy, diethylstilbestrol/oral contraceptive, and growth hormone therapy.
Type 5: Malignant AN. Paraneoplastic, usually adenocarcinoma of gastrointestinal or genitourinary tract; less commonly, bronchial carcinoma and lymphoma.
Epidemiology
Age of Onset. Type 1: during childhood or puberty; other types dependent on associated conditions.
Etiology and Pathogenesis
Dependent on associated disorder. In a subset of women with hyperandrogenism and insulin intolerance and AN, loss-of-function mutation in the insulin receptor or anti-insulin receptor antibodies can be found (types A and B). It is postulated that excess growth factor stimulation in the skin leads to proliferation of keratinocytes and fibroblasts. In hyperinsulinemia AN, excess insulin binding to insulin-like growth factor 1 receptor and fibroblast growth factor receptor has also been implicated. In malignancy-associated AN, transforming growth factor ß released from tumor cells may stimulate keratinocyte proliferation via epidermal growth factor receptors.
Clinical Manifestation
Insidious onset; in type 5 rapid. First visible change is darkening of pigmentation.
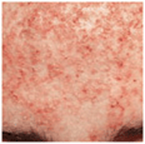
Skin Lesions. All types of AN: Darkening of pigmentation, skin appears dirty (Fig. 5-1). As skin thickens, it appears velvety; skin lines accentuated; surface becomes rugose, mammillated. Type 3: velvety patch on inner, upper thigh at site of chafing; often has many skin tags in body folds and neck. Type 5: hyperkeratosis and hyperpigmentation more pronounced (Fig. 5-2A). Involvement of oral mucosa and vermilion border of lips (Fig. 5-2B). Hyperkeratosis of palms/soles, with accentuation of papillary markings: “Tripe hands” (Fig. 5-2C).
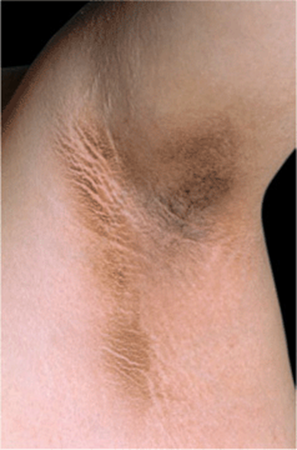
Figure 5-1. Acanthosis nigricans Velvety, dark-brown to gray thickening of the skin of the armpit with prominent skin folds and feathered edges in a 30-year-old obese woman from the Middle East. There were similar changes on the neck, the antecubital fossae, and on the knuckles.
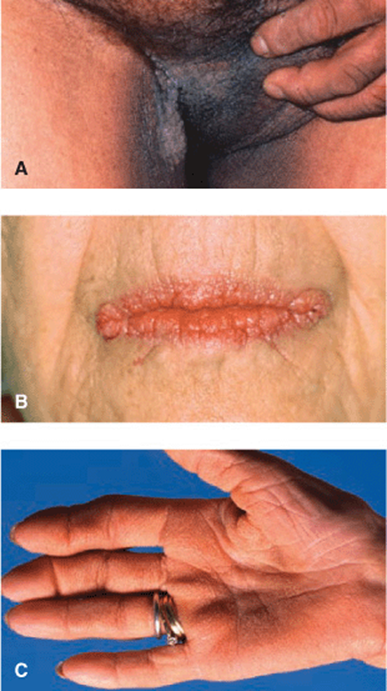
Figure 5-2. Acanthosis nigricans, type 5 (malignant) (A) Verrucous, papillomatous grayish-brown plaques in groins, medial aspects of thigh, and scrotum. Similar lesions were found on neck and all other body folds. The patient had weight loss and wasting and gastric adenocarcinoma was found. (B) Verrucous and papillomatous growths on the vermillion border of lips. Oral mucosa was velvety with deep furrows of the tongue. (C) Tripe palms. Palmar ridges show maximal accentuation resembling the mucosa of the stomach of a ruminant.
Distribution. Most commonly, axillae; (Fig. 5-1), neck (back, sides), groins (Fig. 5-2A), anogenitalia, antecubital fossae, knuckles, submammary, umbilicus. In type 5, also periocular, peroral, mammilary, and palms (tripe palms) (Fig. 5-2C).
Mucous Membranes. Oral mucosa: velvety texture with delicate furrows. Type 5: Mucous membranes and mucocutaneous junctions commonly involved; warty papillomatous thickenings periorally (Fig. 5-2B).
General Examination
Examine for underlying endocrine disorders in overweight to morbidly obese persons; in type 5 wasting, search for malignancy.
Diagnosis and Differential Diagnosis
Clinical Findings. Dark thickened flexural skin: Confluent and reticulated papillomatosis (Gougerot-Carteaud syndrome), pityriasis versicolor, X-linked ichthyosis, retention hyperkeratosis, and nicotinic acid ingestion.
Laboratory Examinations
Chemistry. Rule out diabetes mellitus; metabolic syndrome
Dermatopathology. Papillomatosis, hyperkeratosis; epidermis thrown into irregular folds, showing various degrees of acanthosis.
Imaging and Endoscopy. Rule out associated malignancy.
Course and Prognosis
Type 1: Accentuated at puberty and, at times, regresses when older. Type 2: Depends on underlying disturbance. Type 3: May regress after significant weight loss. Type 4: Resolves when causative drug is discontinued. Type 5: AN may precede other symptoms of malignancy by 5 years; removal of malignancy may be followed by regression of AN.
Management
Symptomatic. Treat associated disorder. Topical keratolytic and/or topical or systemic retinoids may improve AN but all in all not very effective.
Darier Disease (DD) ICD-9: 701.1 ° ICD-10: L 87 ![]()
![]() A rare autosomal-dominant inherited disease with late onset.
A rare autosomal-dominant inherited disease with late onset.
![]() Multiple discrete scaling, crusted, and pruritic papules mainly in seborrheic and flexural areas.
Multiple discrete scaling, crusted, and pruritic papules mainly in seborrheic and flexural areas.
![]() Malodorous and disfiguring, also involving nails and mucous membranes.
Malodorous and disfiguring, also involving nails and mucous membranes.
![]() Itching and/or painful.
Itching and/or painful.
![]() Histologically characterized by suprabasal acantholysis and dyskeratosis.
Histologically characterized by suprabasal acantholysis and dyskeratosis.
![]() Caused by loss-of-function mutation in the ATP2A2 gene.
Caused by loss-of-function mutation in the ATP2A2 gene.
![]() Synonym: Darier—White disease, keratosis follicularis.
Synonym: Darier—White disease, keratosis follicularis.
Epidemiology and Etiology
Rare.
Age of Onset. Usually in the first or second decade, males and females equally affected.
Genetics. Autosomal-dominant trait, new mutations common, penetrance >95%. Loss-of-function mutations in the ATP2A2 gene encoding sarco/endoplasmic reticulum calcium adenosine triphosphatase isoform 2 (SERCA 2), which impair intracellular Ca2+ signaling.
Precipitating Factors. Frequently worse in summer with heat and humidity; also exacerbated by UVB, mechanical trauma, and bacterial infections. Often associated with affective disorders and rarely with decreased intelligence.
Clinical Manifestation
Usually insidious; is abrupt onset after precipitating factors; associated with severe pruritus and often pain.
Skin Lesions. Multiple discrete scaling of crusted, pruritic papules (Fig. 5-3); when scaling crust is removed, a slitlike opening becomes visible (Fig. 5-4). Confluence to large plaques covered by hypertrophic warty masses that are foul smelling, particularly in intertriginous areas.
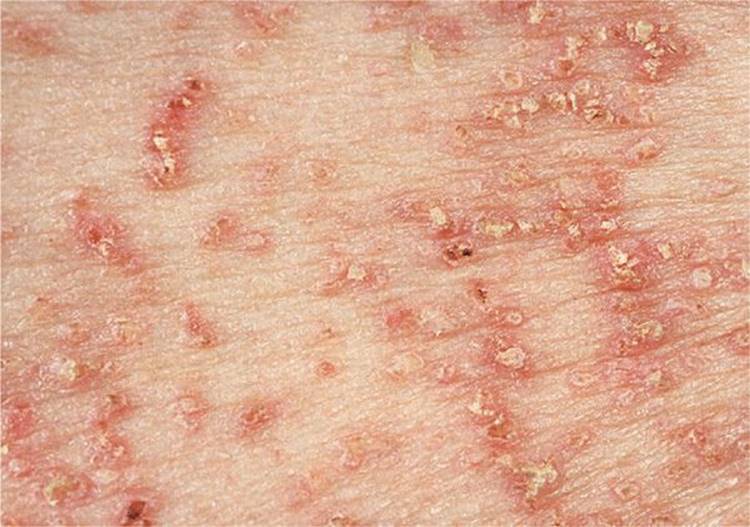
Figure 5-3. Darier disease: chest Primary lesions are reddish-brown, scaling, and crusted papules that feel warty when stroked. Where crusts have been removed, there are slitlike erosions that are later covered by hemorrhagic crusts.
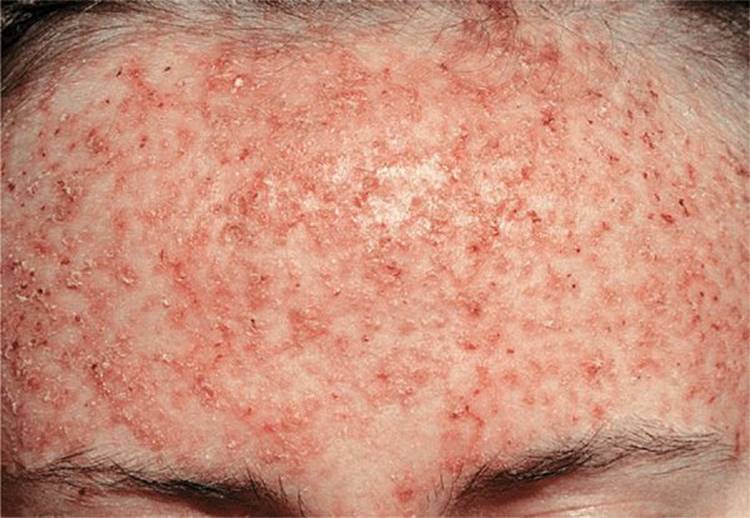
Figure 5-4. Darier disease: forehead Partly coalescing, hyperkeratotic papules that are eroded and crusted. The main concern of this young female was disfigurement.
Distribution. Corresponding to the “seborrheic areas”: chest (Fig. 5-3), back, ears, nasolabial folds, forehead (Fig. 5-4), scalp; axilla, neck, groin.
Palms and Soles. Multiple, flat, cobblestonelike papules.
Appendages. Hair not involved, but permanent alopecia may result from extensive scalp involvement and scarring. Nails thin, splitting distally, and showing characteristic V-shaped scalloping.
Mucous Membranes. White, centrally depressed papules on mucosa of cheeks, hard and soft palate, and gums, “cobblestone” lesions.
Disease Association
Associated with acrokeratosis verruciformis, allelic with DD. Multiple, small flat-topped papules predominantly on dorsa of hands and feet.
Laboratory Examination
Dermatopathology. Dyskeratotic cells in the spinous layer (corps ronds) and stratum corneum (grains), suprabasal acantholysis and clefts (lacunae), and papillary overgrowth of the epidermis and hyperkeratosis.
Diagnosis and Differential Diagnosis
Diagnosis based on history of familial involvement, clinical appearance, and histopathology. May be confused with seborrheic dermatitis, Grover disease, benign familial pemphigus (Hailey-Hailey disease), and pemphigus foliaceus. Acrokeratosis verruciformis: flat warts (verrucae planae juveniles).
Course and Prognosis
Persisting throughout life and not associated with cutaneous malignancies.
Management
Sunscreens, avoidance of friction and rubbing (turtle neck sweaters), antibiotic therapy (systemic and topical) to suppress bacterial infection, topical retinoids (tazarotene and adapalene), or systemic retinoids (isotretinoin or acitretin).
Grover Disease (GD) ICD-9: 702.8 ° ICD-10: L 11.1 ![]()
![]() A pruritic dermatosis located principally on the trunk, occurring as crops of discrete papular or papulovesicular lesions, sparse to numerous (Fig. 5-5). Similar to Darier’s disease. Upon palpation smooth or warty.
A pruritic dermatosis located principally on the trunk, occurring as crops of discrete papular or papulovesicular lesions, sparse to numerous (Fig. 5-5). Similar to Darier’s disease. Upon palpation smooth or warty.
![]() Occurs in adults (mean 50 yrs), males>females.
Occurs in adults (mean 50 yrs), males>females.
![]() Pruritus is main symptom.
Pruritus is main symptom.
![]() Usually transient but a persistent form is recognized.
Usually transient but a persistent form is recognized.
![]() Precipitating factors: heavy, sweat-inducing exercise, exposure to solar radiation, heat, and persistent fever, also in bedridden patients.
Precipitating factors: heavy, sweat-inducing exercise, exposure to solar radiation, heat, and persistent fever, also in bedridden patients.
![]() Principal histopathologic feature: variable focal acantholysis and dyskeratosis.
Principal histopathologic feature: variable focal acantholysis and dyskeratosis.
![]() No evidence of genetic predisposition.
No evidence of genetic predisposition.
![]() Management: glucocorticosteroids under occlusion, UVB, or PUVA (photochemotherapy). Oral glucocorticosteroids, dapsone, and isotretinoin in refractory cases.
Management: glucocorticosteroids under occlusion, UVB, or PUVA (photochemotherapy). Oral glucocorticosteroids, dapsone, and isotretinoin in refractory cases.
![]() Synonym: transient acantholytic dermatosis.
Synonym: transient acantholytic dermatosis.
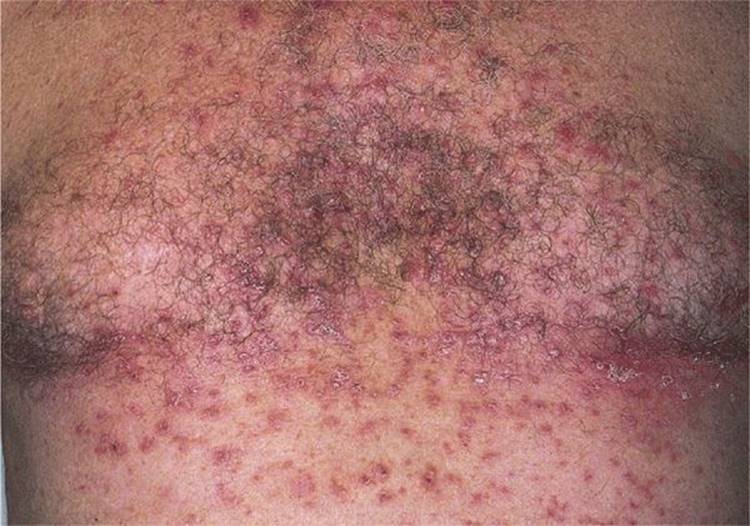
Figure 5-5. Grover disease A rash consisting of reddish, hyperkeratotic scaling, and/or crusted papules with a sandpaper feel upon palpation. Papules are discrete, scattered on the central trunk, and very pruritic.
Hailey-Hailey Disease (Familial Benign Pemphigus) ICD-9: 694.5 ![]() ICD-10: Q 82.8
ICD-10: Q 82.8 ![]()
![]() Hailey-Hailey disease or familial benign pemphigus, is a rare genodermatosis with dominant inheritance that is classically described as a blistering disorder but actually presents as an erythematous, erosive, oozing condition with cracks and fissures localized to the nape of the neck, axillae (Fig. 5-6).
Hailey-Hailey disease or familial benign pemphigus, is a rare genodermatosis with dominant inheritance that is classically described as a blistering disorder but actually presents as an erythematous, erosive, oozing condition with cracks and fissures localized to the nape of the neck, axillae (Fig. 5-6).
![]() Submammary regions, inguinal folds, and scrotum are major sites of involvement.
Submammary regions, inguinal folds, and scrotum are major sites of involvement.
![]() Individual lesions consist of microscopically small flaccid vesicles on an erythematous background that soon turn into eroded plaques with the described, highly characteristic, fissured appearance (Fig. 5-6). Crusting, scaling, and hypertrophic vegetative lesions occur.
Individual lesions consist of microscopically small flaccid vesicles on an erythematous background that soon turn into eroded plaques with the described, highly characteristic, fissured appearance (Fig. 5-6). Crusting, scaling, and hypertrophic vegetative lesions occur.
![]() The underlying pathologic process is acantholysis whereby the fragility of the epidermis is due to a defect in the adhesion complex between desmosomal proteins and tonofilaments.
The underlying pathologic process is acantholysis whereby the fragility of the epidermis is due to a defect in the adhesion complex between desmosomal proteins and tonofilaments.
![]() The genetic abnormality lies in ATP2CI, which encodes an ATP-powered calcium pump.
The genetic abnormality lies in ATP2CI, which encodes an ATP-powered calcium pump.
![]() Onset is usually between the third and fourth decades.
Onset is usually between the third and fourth decades.
![]() Crusting, scaling, and hypertrophic vegetative growths may occur.
Crusting, scaling, and hypertrophic vegetative growths may occur.
![]() Histology explains the clinical appearance as epidermal cells lose their coherence with acantholysis throughout the epithelium, giving the appearance of a dilapidated brick wall.
Histology explains the clinical appearance as epidermal cells lose their coherence with acantholysis throughout the epithelium, giving the appearance of a dilapidated brick wall.
![]() Colonization of the lesions, particularly by Staphylococcus aureus, is a trigger for further acantholysis and maintenance of the pathologic process. Secondary colonization by Candida has a similar effect.
Colonization of the lesions, particularly by Staphylococcus aureus, is a trigger for further acantholysis and maintenance of the pathologic process. Secondary colonization by Candida has a similar effect.
![]() Treatment rests on antimicrobial therapy, administered both topically and systemically; systemically, the tetracyclines seem to work better than most. Mupirocin topically. Topical glucocorticoids depress the anti-inflammatory response and accelerate healing. In severe cases, dermabrasion or carbon dioxide laser vaporization leads to healing with scars, which are resistant to recurrences. The condition becomes less troublesome with age.
Treatment rests on antimicrobial therapy, administered both topically and systemically; systemically, the tetracyclines seem to work better than most. Mupirocin topically. Topical glucocorticoids depress the anti-inflammatory response and accelerate healing. In severe cases, dermabrasion or carbon dioxide laser vaporization leads to healing with scars, which are resistant to recurrences. The condition becomes less troublesome with age.
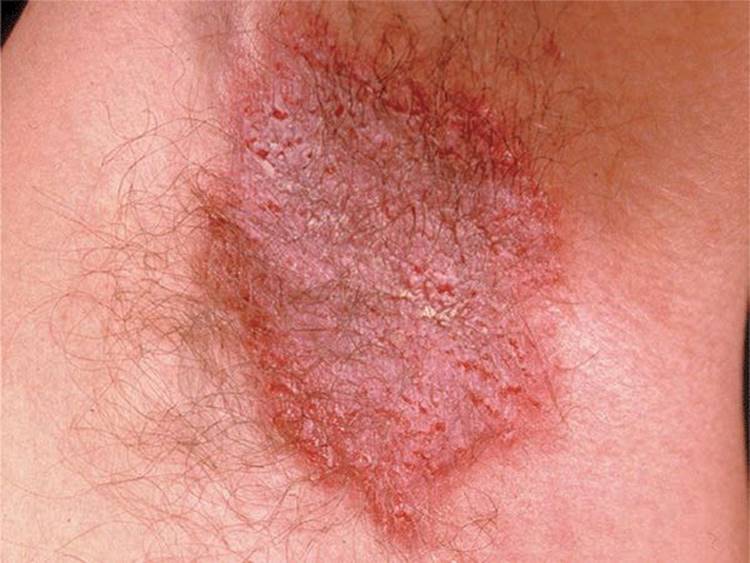
Figure 5-6. Hailey-Hailey disease This 46-year-old male has had oozing lesions on both armpits, occasionally in the groins and nape of the neck for several years, which become worse during summer months. Father and sister have similar lesions. Lesions wax and wane, are painful, and show typical cracks and fissures within a partially erosive erythematous plaque.
Disseminated Superficial Actinic Porokeratosis (DSAP) ICD-9: 692.75 ![]() ICD-10: Q 82.8
ICD-10: Q 82.8 ![]()
![]() DSAP is the most common form of the very rare porokeratoses.
DSAP is the most common form of the very rare porokeratoses.
![]() Uniformly small, annular flat papules ranging from 2 to 5 mm in diameter.
Uniformly small, annular flat papules ranging from 2 to 5 mm in diameter.
![]() Distributed symmetrically on the extremities and located predominantly in sun-exposed sites.
Distributed symmetrically on the extremities and located predominantly in sun-exposed sites.
![]() Typically spare palms, soles, and mucous membranes.
Typically spare palms, soles, and mucous membranes.
![]() Characteristic feature: well-demarcated hyperkeratotic border of individual lesions, usually <1 mm in height with a characteristic longitudinal furrow encircling the entire lesion (Fig. 5-7).
Characteristic feature: well-demarcated hyperkeratotic border of individual lesions, usually <1 mm in height with a characteristic longitudinal furrow encircling the entire lesion (Fig. 5-7).
![]() As lesions progress, the central area becomes atrophic and anhidrotic.
As lesions progress, the central area becomes atrophic and anhidrotic.
![]() Symptoms: asymptomatic or mildly pruritic cosmetically disfiguring.
Symptoms: asymptomatic or mildly pruritic cosmetically disfiguring.
![]() Tends to be inherited as an autosomal-dominant disorder.
Tends to be inherited as an autosomal-dominant disorder.
![]() Pathogenesis unknown.
Pathogenesis unknown.
![]() A benign condition, but rarely a precursor for in situ or invasive squamous cell carcinoma.
A benign condition, but rarely a precursor for in situ or invasive squamous cell carcinoma.
![]() Treatment: topical 5-fluorouracil, retinoids, and imiquimod.
Treatment: topical 5-fluorouracil, retinoids, and imiquimod.
![]() Patients should be monitored for SCC.
Patients should be monitored for SCC.
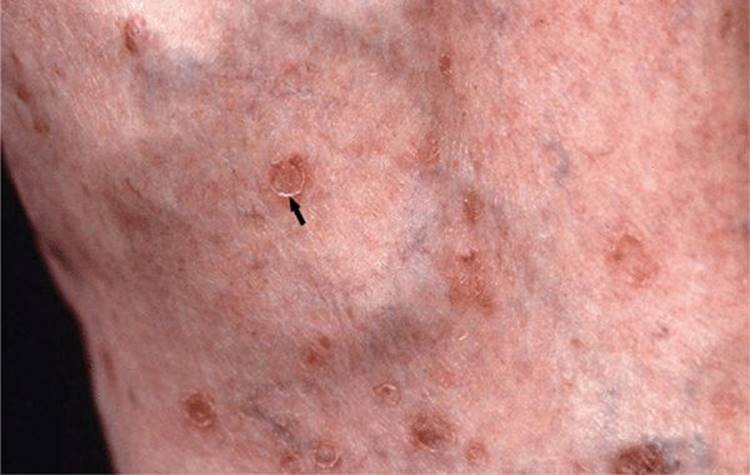
Figure 5-7. Disseminated superficial actinic porokeratosis Small annular flat papules up to 4 mm in diameter surrounded by a well-demarcated hyperkeratotic border (arrow) on the lower leg of a 55-year-old female. With a hand lens, the longitudinal furrow encircling the entire lesion can be seen.