Michael L. Fischman, MD, MPH
Hope S. Rugo, MD
Occupationally related hematologic toxicity has occurred in a somewhat cyclical fashion, historically associated with the introduction of some new and untested chemicals into commerce. A common factor contributing to “epidemics” of toxicity has been the exposure of large numbers of workers without adequate protection. As the toxicities of these agents gradually became known, regulation of their use was instituted and exposure was reduced. Because hematologic toxicity, like other noncancer health effects, is known to exhibit a threshold for induction, subsequent reduction in exposure levels led to reduction in the frequency of these illnesses. In some cases, exposure to certain toxins such as radium has been eliminated. Hematologic toxins, such as lead, benzene, arsenic, and arsine gas, are still used; poisonings leading to hematotoxicity occasionally still occur in the workplace, and, in some settings, worker or consumer education is inadequate. The study of hematotoxicity has improved our understanding of hematologic pathophysiology, taught important pharmacologic lessons, and introduced the concept of individual susceptibility to specific toxic agents. Observation of individual variations in susceptibility to toxic agents was made by recognizing that chemicals with oxidative potential could cause cyanosis and a life-threatening hemolytic anemia in some individuals at exposure levels that had little effect on the population at large. The normal population will manifest similar toxicities but only when exposed to much higher levels. Consequently, it is important to identify workers with increased susceptibility to certain chemicals and place them in jobs with less risk of contact with these specific toxic substances, although elimination or reduction of exposure for all workers is a preferable strategy.
Exposure to hematotoxins may affect blood cell survival (denaturation of hemoglobin and hemolysis), porphyrin synthesis and metabolism (including some porphyrias), blood cell formation (aplasia), risk for hematopoietic neoplasms, or coagulation (through development of thrombocytopenia).
DISORDERS ASSOCIATED WITH SHORTENED RED BLOOD CELL SURVIVAL
METHEMOGLOBINEMIA & HEMOLYSIS PRODUCED BY OXIDANT CHEMICALS
Methemoglobin is formed by the oxidation of ferrous (Fe2+) hemoglobin to ferric (Fe3+) hemoglobin. It was first recognized in the 1800s, when coal tars were converted into individual chemicals that served as precursors for many products ranging from explosives to synthetic dyes and perfumes. Overexposure to these chemicals—which included anilines, nitrobenzenes, and quinones—was common, and little was known about their potential toxicity. Workers in these plants came to be known as “blue workers” because they suffered from “blue lip” as a result of the chronic cyanosis from toxin-induced methemoglobinemia that developed in many of them. Gradually it was recognized that oxidation of hemoglobin was toxic to red blood cells and could be followed by an acute and life-threatening hemolysis known as Heinz body anemia. Heinz bodies are red blood cell inclusions that represent precipitated hemoglobin and are seen classically in individuals with a deficiency of glucose-6-phosphate dehydrogenase (G6PD) after exposure to an oxidant stress. Normal individuals exposed to large amounts of oxidant chemicals will develop methemoglobinemia and, occasionally, Heinz body hemolytic anemia. It is not understood why some chemicals may cause methemoglobinemia, hemolysis, or both, but the disorders are certainly related to individual susceptibility. Oxidative chemicals are common in industry, and it is important to know what toxic agents have been implicated, to recognize the presenting signs and symptoms, and to be able to provide appropriate treatment when it is needed.
Despite the understanding of this phenomenon, new outbreaks of these illnesses occurred as new compounds with unrecognized hematotoxicity were introduced into industry. An understanding of the pathophysiology of this phenomenon is essential to handle this medical emergency correctly; it also will help in understanding the myriad industrial and therapeutic agents that may cause oxidative hemolysis in a susceptible individual.
![]() Pathophysiology of Methemoglobinemia & Oxidant Hemolysis
Pathophysiology of Methemoglobinemia & Oxidant Hemolysis
Hemoglobin is unique in its ability to combine reversibly with oxygen without oxidizing its iron moiety. The small amount of oxidized hemoglobin or methemoglobin produced is readily reduced by an efficient enzyme system linked to energy provided by glucose metabolism via the Embden-Meyerhof pathway (Figure 18–1).

![]() Figure 18–1. Oxidation of hemoglobin by the Embden-Meyerhof pathway.
Figure 18–1. Oxidation of hemoglobin by the Embden-Meyerhof pathway.
Methemoglobin is dangerous because of its inability to bind oxygen and because it increases the oxygen affinity of the remaining heme groups in hemoglobin tetramer, thereby decreasing oxygen delivery to the tissues. Oxidation results in denaturation of hemoglobin with the formation of precipitated hemoglobin (Heinz bodies) within the red cell. The presence of Heinz bodies alters the surface membrane of the red cell, causing increased rigidity and leakage. Macrophages in the reticuloendothelial system of the spleen and liver (the extravascular compartment) sense the altered red cell surface and remove Heinz bodies via partial phagocytosis (extravascular hemolysis). Because the red cell surface is unable to reseal and form a spherocyte (as in autoimmune hemolysis), the red cell remains intact as a cell with a piece missing, the so-called bite, or blister, cell. Heinz bodies also may be formed from a second form of denatured hemoglobin, sulfhemoglobin. Unlike methemoglobin, sulfhemoglobin is irreversibly associated with the heme moiety.
The development of methemoglobinemia or oxidative hemolysis in an individual exposed to an oxidant stress depends on the route of exposure, the specific chemicals involved, the dose and duration of exposure, and most importantly, individual susceptibility. Inborn structural abnormalities (unstable hemoglobins)—or, much more commonly, disorders of normal reducing capabilities such as the X-linked deficiency of the oxidation-reduction enzyme G6PD—cause some individuals to be much more susceptible to oxidant stress than others. There are many varieties of both these abnormalities. Recognition of these high-risk individuals in the workplace is important to reduce their chance of particularly toxic exposures.
The normal individual has less than 1% circulating methemoglobin. Ninety-five percent of methemoglobin formed daily by the autoxidation of hemoglobin is reduced by NADH2 (nicotinamide adenine dinucleotide [reduced form]) generated by the dehydrogenation of phosphotriose by phosphotriose dehydrogenase. This reaction is catalyzed by NADH methemoglobin reductase (NADH cytochrome b5 reductase). A rare inborn deficiency of NADH methemoglobin reductase results in congenital cyanosis caused by methemoglobinemia (Figure 18–2).
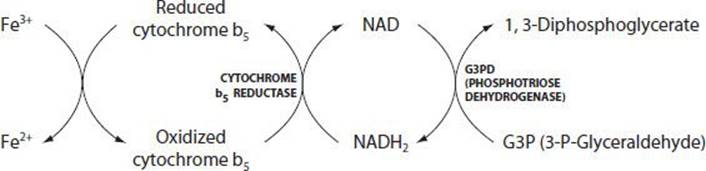
![]() Figure 18–2. Reduction of hemoglobin by NADH-methemoglobin reductase (NADH cytochrome b5 reductase).
Figure 18–2. Reduction of hemoglobin by NADH-methemoglobin reductase (NADH cytochrome b5 reductase).
An alternative methemoglobin reduction pathway exists that requires the presence of a redox cofactor such as methylene blue to achieve significant reducing capacity. In this reaction, nicotinamide adenine dinucleotide phosphate (NADPH) from the first two steps of the hexose monophosphate shunt converts methemoglobin to reduced hemoglobin. Because this pathway is normally responsible for so little reduction of methemoglobin, deficiency of the enzyme that catalyzes this reaction, NADPH methemoglobin reductase, does not result in methemoglobinemia or cyanosis. Because the formation of NADPH depends on G6PD, methylene blue, which is used to treat toxic and congenital methemoglobinemia, also can precipitate a hemolytic crisis in an individual with G6PD deficiency by competing for the NADPH necessary to maintain reduced glutathione, an essential protectant against erythrocyte oxidative stress. Additionally, methylene blue itself is an oxidant, but it is metabolized to the reducing agent leucomethylene blue. In normal individuals, the administration of a redox agent may increase the rate of reduction of hemoglobin dramatically so that it greatly exceeds that of the NADH-methemoglobin reductase reaction (Figure 18–3). This is the rationale for the effectiveness of methylene blue in toxic methemoglobinemia.

![]() Figure 18–3. Reduction of hemoglobin by NADPH-methemoglobin reductase can be accelerated by a redox agent such as methylene blue.
Figure 18–3. Reduction of hemoglobin by NADPH-methemoglobin reductase can be accelerated by a redox agent such as methylene blue.
Two other pathways exist, but they reduce methemoglobin only to a small extent. Glutathione is responsible for conversion of less than 7–10% of ferrihemoglobin to ferrohemoglobin, and ascorbic acid in pharmacologic amounts also reduces oxidized hemoglobin. Because of the high redox potential of ascorbic acid, however, the rate of reduction is very slow, making it less effective in therapy. In physiologic concentrations, the contribution of ascorbic acid to methemoglobin reduction is insignificant.
1. Aniline
Historically, most work-related episodes of methemoglobinemia primarily and less often hemolytic anemia were a result of exposure to aromatic nitro and amino compounds, including aniline. These compounds are used most extensively as intermediates in the synthesis of aniline dyes; they are also used as accelerators and antioxidants in the rubber industry and in the production of pesticides, plastics, paints, and varnishes. Table 18–1 lists chemicals that are associated with methemoglobinemia and/or oxidant hemolysis and their industrial uses. Many medicinal drugs are oxidants and can cause methemoglobinemia.
Table 18–1. Chemicals associated with methemoglobinemia or oxidative hemolysis.

The clinical presentation of methemoglobinemia is exemplified by aniline toxicity. Aniline, used in the manufacture of dyes and in the rubber industry, is the most common and best-described aromatic amine. It is fat-soluble and readily penetrates the intact skin, even through clothing. The vapor form also may gain entry to the body through the lungs. Ingestion is rare in the industrial setting but causes serious toxicity when it does occur. Aniline is converted by hepatic microsomes to phenylhydroxylamine, which behaves as a catalyst in mediating hemoglobin oxidation. Hepatic clearance of phenylhydroxylamine is slow because its oxidized form, nitrosobenzene, is rapidly converted back to phenylhydroxylamine. Another clearance pathway gradually eliminates the amine from the body.
Clinical Presentation
Acute exposure usually is associated with spills or improper usage. Symptoms vary depending on the concentration of methemoglobin (Table 18–2). Most cases are mild and transient and present as asymptomatic blueness of the lips and nail beds. In more severe cases, the patient will appear deeply cyanotic. Freshly drawn blood appears dark maroon-brown and does not become red after exposure to air. Pulse oximetry may indicate normoxia or mild hypoxia not reflective of the severity of methemoglobinemia. Arterial blood gases may show a normal oxygen tension (Po2) but co-oximetry will reveal methemoglobinemia reliably. Often, because of differences in the measurement approach, there may be a “saturation gap,” in which the oxygen saturation on arterial blood gases is substantially higher than that determined by pulse oximetry. Laboratory results may indicate hemolysis with an elevated reticulocyte count and variable degree of anemia. Examination of the peripheral blood smear shows evidence of reticulocytosis (polychromasia, possibly nucleated red cells) and may show bite or blistered red cells.
Table 18–2.Symptoms of methemoglobinemia.

In chronic methemoglobinemia, polycythemia may be seen in response to chronic hypoxia. Hemolytic Heinz body anemia may or may not accompany methemoglobin formation or may follow resolution of cyanosis. Heinz bodies are detected easily by examining the peripheral blood smear stained with a supravital stain but will not be evident on a smear stained with Wright stain. Blood methemoglobin levels should be monitored closely.
![]() Prevention
Prevention
The most important safeguard in preventing oxidative hemolysis is to minimize atmospheric and cutaneous exposure to potentially oxidizing chemicals such as coal tar products. The identification of susceptible individuals such as those with G6PD deficiency may help to avoid significant toxicity in high-risk job situations. Screening for G6PD deficiency must be done either before a hemolytic episode or 1–2 months after the hemolysis has resolved. Young red blood cells, particularly reticulocytes, have normal G6PD levels in most G6PD-deficient individuals. During an acute hemolytic episode, older red blood cells are destroyed and replaced by young red blood cells. The result of a G6PD deficiency screen often will be normal in that acute setting. Biologic monitoring in the workplace may be done by measuring methemoglobin levels and reticulocyte counts.
![]() Treatment
Treatment
Treatment depends on rapid recognition of the problem. It is important to obtain as complete an exposure history as possible because it will guide treatment. The most important aspect of therapy is to ensure removal of the offending agent. Because of the fat-soluble nature of these compounds, it is essential that clothing be removed and the patient decontaminated thoroughly. For mild intoxication (<20% blood methemoglobin), observation should be sufficient to watch for progression of symptoms. For moderate to severe intoxication (>30% blood methemoglobin), 100% oxygen by mask is given to saturate the remaining hemoglobin, and the antidote, methylene blue, is administered. Care must be exercised in using methylene blue to avoid increasing methemoglobin from the oxidative potential of methylene blue itself or risk for hemolytic anemia, particularly in G6PD-deficient individuals (in whom it is ineffective).
For initial management of severe methemoglobinemia, methylene blue should be given intravenously as a 1% solution at a dose of 1–2 mg/kg over 5–10 minutes. The maximal effect should be seen within 1 hour. If no response is evident by this time, administration of methylene blue may be repeated at hourly intervals (in part, because of the short half-life of methylene blue in the body). Repeat doses, which may also be given orally, should be given for symptoms, not solely on the basis of the methemoglobin level. A patient who does not respond to methylene blue may have G6PD deficiency, and further administration could exacerbate hemolysis without reducing hypoxia.
Ascorbic acid may be given in conjunction with the oral dose of methylene blue at a dose of 300–400 mg orally, although its role for this purpose remains controversial. Its onset of action is slow, and its potential for urine acidification may potentiate renal toxicity in patients who are actively hemolyzing.
2. Chlorate Salts
Chlorate salts, used primarily in herbicides, cause an unusual form of methemoglobinemia and hemolysis that is unresponsive to methylene blue. The denaturation of hemoglobin caused by chlorates is thought to be due to their direct oxidizing capacity and their ability to inhibit the hexose monophosphate shunt. Hemolytic anemia also has been seen in uremic patients undergoing hemodialysis when the water supply was found to contain chloramines, oxidant compounds made up of chlorine and ammonia now used in some public water supplies as a disinfectant. Treatment for poisoning with chlorates is supportive because there is no specific antidote. Exchange transfusion has been advocated for severe toxicity.
HEMOLYSIS ASSOCIATED WITH EXPOSURE TO HEAVY METALS
After methemoglobinemia and oxidative hemolysis, transitional elements and heavy metals are the most important causes of work-related hemolytic anemia. These agents include arsenic, lead, mercury, copper, antimony, and others. The mechanism of hemolysis is unknown, but it is thought to be related to the affinity of these directly cytolytic metals to thiol groups such as are found on the surfaces of red blood cells and in the cysteine residues of hemoglobin. When the sulfhydryl-binding metals are exposed to red cells, the red cell membrane becomes permeable and takes on solute and water. This causes the red cell to swell and ultimately burst while in the vascular circuit (intravascular hemolysis).
1. Arsine
The most dramatic example of acute metal-induced hemolysis is that caused by arsine. Arsine is a volatile, colorless, nonirritating gas at room temperature (arsine’s boiling point is −62°C [−79.6°F]). It is usually produced accidentally by the action of acid on a metal contaminated with arsenic. However, arsine gas is often used in the semiconductor industry to introduce small quantities of arsenic into the matrix of silicon wafers to impart semiconductive properties into what will become computer chips.
The toxicity of arsine may be best demonstrated by a case of two near fatalities. Two workers at a chemical manufacturing plant were cleaning a floor drain that had become clogged during a cleanup operation. Residual arsenical herbicides containing arsenic trioxide reacted with hydrogen formed by the combination of sodium hydroxide and aluminum to form arsine. On the morning after exposure, both workers were hospitalized with acute hemolytic anemia. Exposure was documented by the presence of arsenic in the drain as well as in the blood and urine of both patients. Each patient required multiple-unit exchange transfusions and fluid replacement; recovery took nearly a month for both.
A recent case report of a nonfatal case in a worker engaged in recycling of gallium arsenide scrap detailed the clinical and laboratory course of hemolysis leading to anemia (hemoglobin of approximately 6 g/dL) and renal and hepatic dysfunction with gradual recovery. Because the worker did not develop acute renal failure secondary to intravascular hemolysis, he was treated successfully with transfusions but did not require exchange transfusion. This publication described the first arsine poisoning in which arsenic speciation, distinguishing inorganic arsenic and its metabolites from organic arsenic (from seafood sources), was conducted, confirming the cause and documenting the half-life of arsenic species associated with the hemolysis and other clinical features.
Chronic arsine poisoning has been described in workers at a zinc smelting plant and in workers engaged in the cyanide extraction of gold. These patients may be anemic, with chronic low-level hemolysis.
![]() Clinical Presentation
Clinical Presentation
A. Symptoms and Signs
Many manifestations of acute arsine poisoning are caused by acute and massive intravascular hemolysis. Appearance of symptoms may be delayed for 2–24 hours after exposure. Symptoms include nausea and vomiting, abdominal cramping, headache, malaise, and dyspnea. Patients often are alarmed by the presence of tea-colored urine that is not associated with pain on urination, causing them to seek medical attention. Physical examination may reveal the peculiar garlicky odor of arsine, fever, tachycardia, tachypnea, and hypotension. Later in the course of hemolysis, the patient typically develops jaundice.
B. Laboratory Findings
The earliest laboratory finding is likely to be hemoglobinuria. This occurs when the amount of free plasma hemoglobin exceeds normal haptoglobin binding and renal proximal tubular reabsorption. Accordingly, plasma haptoglobin levels fall, and free hemoglobin levels may be very high (>2000 mg/dL have been reported; normal: <1 mg/dL). The plasma may be brownish red from the presence of methemalbumin (oxidized hemoglobin bound to albumin). Although anemia may not be present on the first blood count, evaluation of the peripheral smear will reveal red cell fragmentation with marked poikilocytosis, basophilic stippling, and polychromasia. As the hematocrit falls, reticulocytosis develops. Total bilirubin is elevated, reflecting a rise primarily in the unconjugated or indirect form. When hemolysis is brisk, disseminated intravascular coagulation may occur, manifest as a low (or falling) fibrinogen level, a prolonged prothrombin time (caused by circulating fibrin split products), and the presence of schistocytes and thrombocytopenia. Renal function often is affected to various degrees, with an early rise in serum creatinine. This may be a result of both precipitated hemoglobin casts, causing renal tubular obstruction, and direct toxicity of arsine on the renal tubular and interstitial cells. Arsenic levels in blood and urine are useful as indicators of exposure rather than as guidelines for therapy.
Treatment
Initial therapy should include vigorous hydration to ensure adequate renal perfusion. For severe hemolysis with plasma hemoglobin levels greater than 400–500 mg/dL, exchange transfusion has been advocated. Repeated exchange is indicated for increasing levels of hemoglobin.
Renal function may be preserved with hydration. However, should renal failure develop, acute hemodialysis may be required. All patients must be monitored closely until all evidence of hemolysis has resolved, and renal function has stabilized. Some patients may be left with renal insufficiency or chronic renal failure requiring dialysis or transplantation. In chronic arsine poisoning, reduction of or ideally removal from exposure is the most important intervention.
2. Lead
Lead is more fully discussed in the section on porphyria below. In addition to the suppression of erythropoiesis and heme synthesis described there, hemolytic anemia may occasionally be seen. The anemia of chronic lead toxicity, the primary hematologic effect of lead exposures, is enhanced by shortened red cell survival as well as by inhibition of hemoglobin synthesis.
It has been suggested that the pathogenesis of lead-induced hemolysis is related to its marked inhibition of pyrimidine-5 nucleotidase. The hereditary homozygous deficiency of this enzyme is marked by basophilic stippling of erythrocytes, chronic hemolysis, and intraerythrocytic accumulations of pyrimidine-containing nucleotides. These nucleotides perhaps compete with adenine nucleotides in binding to the active site of kinases in the glycolytic pathway, thereby altering red cell membrane stability. Because lead causes an acquired deficiency of this enzyme and the clinical findings are similar, severe toxicity has been likened to this hereditary disease.
3. Copper
Copper sulfate is used in India in the whitewashing and leather industry. Toxicity is primarily a result of accidental ingestion and suicide attempts and results in intravascular hemolysis, methemoglobinemia, renal failure, and often death. Hemolysis also has been caused by hemodialysis with water contaminated by copper piping. In vitro data suggest that multiple mechanisms are involved, including inhibition of glycolysis, oxidation of NADPH, and inhibition of G6PD. No specific treatment exists other than supportive therapy, with transfusions and hemodialysis as indicated.
THE PORPHYRIAS
The porphyrias are a group of disorders characterized by abnormalities in the heme biosynthetic pathway (Figure 18–4) that result in the abnormal accumulation of heme precursors. Although these are genetic disorders (inherited or sporadic) of enzymatic activity, acquired porphyria has been described following exposure to various toxins. Heme biosynthesis occurs chiefly in the liver and bone marrow and to a certain extent in nervous tissue. The rate-limiting step in heme biosynthesis is the synthesis of δ-aminolevulinic acid from glycine and succinyl-coenzyme A (CoA) via δ-aminolevulinic acid synthetase. This step is under negative feedback control by heme. Clinically, symptomatic porphyria can occur either as a result of inadequate enzymatic function along any step in heme biosynthesis or as a result of inappropriate overstimulation of δ-aminolevulinic acid synthetase, usually in the setting of decreased heme concentration.

![]() Figure 18–4. The heme biosynthetic pathway. Heme is a feedback inhibitor of enzyme (1) δ-aminolevulinic acid synthetase. Other enzymes are (2) δ-aminolevulinic acid dehydrase, (3) uroporphyrinogen I synthase, (4) uroporphyrinogen III cosynthase, (5) uroporphyrinogen decarboxylase, (6) coproporphyrinogen oxidase, (7) protoporphyrinogen oxidase, and (8) ferrochelatase.
Figure 18–4. The heme biosynthetic pathway. Heme is a feedback inhibitor of enzyme (1) δ-aminolevulinic acid synthetase. Other enzymes are (2) δ-aminolevulinic acid dehydrase, (3) uroporphyrinogen I synthase, (4) uroporphyrinogen III cosynthase, (5) uroporphyrinogen decarboxylase, (6) coproporphyrinogen oxidase, (7) protoporphyrinogen oxidase, and (8) ferrochelatase.
The clinical syndromes of porphyria are characterized by neurotoxicity or cutaneous photosensitivity (both may occur). Neurotoxicity—typically abdominal colic, constipation, autonomic dysfunction, sensorimotor neuropathy, and psychiatric problems—is considered the result of direct toxic effects of the urinesoluble heme precursors δ-aminolevulinic acid and porphobilinogen on nervous tissue. Neurotoxicity also may be the result of heme deficiency interrupting nervous tissue homeostasis. Cutaneous photosensitivity is manifested as repetitive vesiculation, scarring, and deformity, with hypertrichosis of sun-exposed areas of the skin. This is the result of the relatively urine-insoluble heme precursors uroporphyrin III, coproporphyrin III, and protoporphyrin IX fluorescing in the skin following absorption of 400-nm ultraviolet light. These fluorescing porphyrias also can cause discoloration of teeth and occasionally hemolysis of erythrocytes in which porphyrins accumulate.
A number of industrial and environmental toxins have induced toxic porphyrias similar to porphyria cutanea tarda in people heavily exposed to the agents (Table 18–3). These toxins, typically when absorbed in high doses, usually cause liver injury and deranged hepatic heme synthesis. Although the exact metabolic effects of these agents are not entirely understood, unregulated stimulation of δ-aminolevulinic acid synthetase usually is demonstrable.
Table 18–3.Toxic substances associated with acquired porphyria in humans.

1. Hexachlorobenzene
In an outbreak of acquired porphyria in Turkey between 1955 and 1958, more than 4000 people developed a cutaneous porphyria syndrome that resembled congenital erythropoietic porphyria approximately 6 months following ingestion of wheat containing the fungicide hexachlorobenzene. The wheat was intended for planting and contained 2 kg of 10% hexachlorobenzene per 1000 kg of wheat to control the fungus Tilletia tritici. Affected people demonstrated cutaneous photosensitivity with skin hyperpigmentation, hypertrichosis, bullae, weakness, and hepatomegaly, a condition termed kara yara, or “black sore.” Porphyrinuria was nearly universal, with the urine being pigmented red or brown. The mortality rate was 10%. Breast-fed infants younger than 2 years of age had a 95% mortality rate when ingesting mother’s milk contaminated with the fungicide. These infants developed weakness, convulsions, and cutaneous annular erythema, a condition termed pembe yara, or “pink sore.” Excess porphyrins could not be detected in the urine of these infants. A similar result occurs in animal models of hexachlorobenzene-induced porphyria. Infant rats and mice die of neurologic toxicity from hexachlorobenzene without porphyrinuria, whereas adult rats and rabbits develop cutaneous photosensitivity and porphyrinuria following prolonged exposure to the chemical.
A follow-up study between 1977 and 1983 examined 204 patients who had previously suffered from hexachlorobenzene porphyria. The mean age of these individuals was 32 years, and the mean time from hexachlorobenzene exposure was 7 years. The mean duration of cutaneous porphyria symptoms was 2.4 years. At the time of study, 71% of people had hyperpigmentation and 47% had hypertrichosis. Residual scarring on sun-exposed areas of the skin was evident in 87%. Other features included perioral scarring, small hands, arthritis, short stature, weakness, paresthesias, and myotonia. Seventeen patients still had red urine and demonstrated porphyrinuria (especially uroporphyrinuria). Hexachlorobenzene was measurable in 56 samples of human milk obtained from porphyric mothers at a mean value of 0.51 parts per million (ppm) (vs 0.07 ppm in controls).
The Turkish experience was the first associating exposure to an industrial chemical with acquired porphyria in humans. Not only were the symptomatic attack and mortality rates significant, but also the biochemical lesion persisted for decades in many survivors. The exact mechanism by which hexachlorobenzene induces porphyria remains to be elucidated. Most liver mitochondria of animals made porphyric by exposure to chlorinated benzenes, such as hexachlorobenzene, demonstrated increased activity of δ-aminolevulinic acid synthetase, the enzyme that controls the rate of porphyrin production. With the exception of mice made porphyric with diethyl-1,4-dihydro-2,4,6-trimethylpyridine-3,5-dicarboxylate, animal porphyric livers demonstrate an increased production of heme. Heme normally inhibits the activity of δ-aminolevulinic acid synthetase. This suggests that porphyrinogenic compounds are somehow interfering with the repressor signal of heme on δ-aminolevulinic acid synthetase. Other theories suggest that porphyrinogenic compounds induce δ-aminolevulinic acid synthetase by altering the intracellular oxidation state through action on the electron transport chain, thus stimulating succinyl-CoA production, depressing intracellular adenosine triphosphate levels, or both. In any event, the net result is overproduction of porphyrins mediated by unregulated δ-aminolevulinic acid synthetase activity.
The role of iron overload in the pathogenesis of hexachlorobenzene-induced porphyria has been examined. The suggestion that iron might be involved was based on the observation that 80% of patients with porphyria cutanea tarda—a disease associated with reduced hepatic uroporphyrinogen decarboxylase activity—have increased liver iron stores and increased levels of uroporphyrin I. Furthermore, decreasing hepatic iron stores by phlebotomy in patients with porphyria cutanea tarda often induces disease remission and a decrease in urinary uroporphyrin I excretion. In a porcine and human liver model, ferrous iron was found to markedly inhibit uroporphyrinogen III cosynthetase activity, enhance total porphyrin production, and greatly overproduce uroporphyrin I. In rats made porphyric with hexachlorobenzene, iron overload results in decreased production of liver heme, cytochrome P450, and cytochrome b5 and an absence of uroporphyrinogen decarboxylase activity. In addition, the nicotinic acid dehydrogenase (NAD): NADH ratio was more than twofold higher in siderotic rats made porphyric with hexachlorobenzene compared with nonsiderotic rats. Furthermore, phlebotomized irondeficient mice were protected from the porphyrinogenic effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin.
Most authorities believe that iron plays a permissive rather than a causative role in porphyrias. This is based on the fact that not all patients with porphyria cutanea tarda are iron overloaded, that porphyria cutanea tarda is rare in patients with hemochromatosis, and that phlebotomy does not correct the biochemical lesion in patients with porphyria cutanea tarda. In addition, rats made porphyric by hexachlorobenzene did not require iron overload for porphyria to develop, although the porphyria was worsened by iron overload. Thus, it remains unsettled whether iron overload is permissive or etiologic in patients exposed to porphyrinogenic toxins.
2. Herbicides
A number of herbicides clearly are associated with symptomatic porphyria. Twenty-nine patients exposed to 2,4-dichlorophenol and 2,4,5-trichlorophenol at a manufacturing plant exhibited chloracne; 13 had hyperpigmentation, 11 had hirsutism, and 5 had skin fragility. Eleven patients had increased excretion of urine porphyrins (uroporphyrin and coproporphyrin). Thus these patients had developed an acquired porphyria cutanea tarda-like syndrome after variable exposure to these herbicides. A follow-up study of 73 workers at this same herbicide plant 6 years later found no people with the porphyric syndrome and only one with persistent uroporphyrinuria. The authors of the follow-up study hypothesized that the decrease in the syndrome was a result of improved personal safety habits of the workers and decreased exposure to the chemicals. An alternative explanation is that the true porphyrinogenic agent is perhaps 2,3,7,8-tetrachlorodibenzo-p-dioxin, a by-product of 2,4,5-trichlorophenol, and that this contaminant had been effectively eliminated from the chemical stores at the factory. The contaminant was strongly implicated in an outbreak of acquired porphyria cutanea tarda, chloracne, and polyneuropathy in 80 industrial workers producing herbicides in Czechoslovakia.
3. Disinfectants
The commercial disinfectants o-benzyl-p-chlorophenol and 2-benzyl-4,6-dichlorophenol were implicated as a cause of acquired porphyria cutanea tarda in one woman janitor exposed to these chemicals through inappropriate mixing of cleaning chemicals.
4. Aluminum
A porphyria cutanea tarda-like syndrome has been described in patients with chronic renal failure being maintained on regular hemodialysis. Plasma and urine uroporphyrins are increased in these patients, whereas plasma and urine coproporphyrins are often low. Because aluminum is known to inhibit some heme synthetic enzymes, and because many chronic renal failure patients on hemodialysis are aluminum overloaded, aluminum has been implicated, but without proof, as the cause of porphyria in these patients.
5. Vinyl Chloride
Vinyl chloride is a known hepatotoxin used in the production of plastics. A study of 46 persons working in a polyvinyl chloride production plant revealed significantly elevated urinary coproporphyrin levels compared with normal controls. Exposure periods ranged from 2 to 21 years. The pathogenesis of coproporphyrinuria involves inhibition of coproporphyrinogen oxidase, inhibition of uroporphyrinogen decarboxylase, and perhaps induction of δ-aminolevulinic acid synthetase. Persons with excess urinary coproporphyrin production also manifested thrombocytopenia, splenomegaly, esophageal varices, scleroderma-like skin changes, Raynaud syndrome, and acroosteolysis.
6. Lead
Lead intoxication (particularly with a blood lead level greater than 60 μg/dL) causes symptoms and signs remarkably similar to those associated with acute intermittent porphyria. The classic acute intermittent porphyria triad is abdominal pain, constipation, and vomiting—all representing the neurotoxic effects of excess δ-aminolevulinic acid and porphobilinogen. This triad is seen with equal frequency in lead intoxication. Other shared characteristics include neuromuscular pains, paresis or paralysis, paresthesias, diarrhea, and seizures. The major differences between the two diseases are (1) an increase in neuropsychiatric signs in acute intermittent porphyria compared with lead intoxication and (2) anemia, which is present in lead intoxication but virtually absent in porphyria. The anemia of lead poisoning is a characteristic microcytic anemia with basophilic stippling of erythrocytes and sideroblasts in the bone marrow.
The biochemical features of lead poisoning demonstrate why these two diseases are so similar clinically. Patients with lead intoxication have markedly elevated urinary δ-aminolevulinic acid levels, as in acute intermittent porphyria. Mild lead poisoning (pre-anemia stage) is associated with normal porphobilinogen excretion, but once anemia occurs, excess urinary porphobilinogen becomes demonstrable. Although mild elevations of urine coproporphyrins and uroporphyrin I are present, fecal uroporphyrin and coproporphyrin are normal in patients with lead poisoning. These alterations in porphyrins are present only in patients with inorganic lead intoxication, not in patients with organic lead intoxication. Excess accumulation of protoporphyrin IX also has been found in erythrocytes of lead-intoxicated patients.
Lead poisoning is associated with greatly diminished activity of δ-aminolevulinic acid dehydrase in the brain, liver, kidney, and bone marrow. Because δ-aminolevulinic acid dehydrase is polymorphic, different individuals may have different levels of sensitivity to lead exposure depending on the particular form of the enzyme that is inherited. Lead also blocks the incorporation of iron into protoporphyrin IX by depressing the activity of ferrochelatase, an event most closely linked with the production of anemia and elevation of free erythrocyte protoporphyrin IX levels. Coproporphyrinogen oxidase activity is also depressed by lead. Thus the effect of lead on heme synthesis disruption occurs at multiple steps in the synthetic pathway, all of which occur in the mitochondrion.
TREATMENT OF TOXIC PORPHYRIAS
Because there is often no effective means of eliminating toxic environmental or industrial substances once they are incorporated into tissues, exposure to porphyrinogenic compounds must be avoided. Although no prospective data are available to support the use of phlebotomy for this purpose, this therapy may be of benefit in patients with toxic porphyria whose disease complex resembles porphyria cutanea tarda and in whom evidence of iron overload can be demonstrated. Patients with acute intermittent porphyria occasionally respond to high-dose carbohydrate infusions (400 g dextrose per day) or intravenous infusions of hematin (an iron-containing porphyrin). However, the use of hematin infusions in toxic porphyria may not be of benefit because, as in the case of hexachlorobenzene, the toxic agent may be interrupting the negative feedback signal by heme on δ-aminolevulinic acid synthetase.
For lead intoxication, prevention again is the best treatment. Unlike all other toxic porphyrias, specific therapy, recommended for severe lead intoxication, is available with lead chelating agents.
DISORDERS ASSOCIATED WITH DECREASED OXYGEN SATURATION
1. Carbon Monoxide Poisoning
Carbon monoxide is an odorless, colorless, nonirritating gas produced by the incomplete combustion of organic materials, particularly hydrocarbons. In addition to workers who sustain exposure from combustion products of hydrocarbons, workers exposed to methylene chloride (converted to carbon monoxide through in vivo metabolism) can also develop carbon monoxide poisoning.
Carbon monoxide binds to hemoglobin, forming carboxyhemoglobin (COHb), which decreases hemoglobin oxygen saturation and shifts the oxygen-hemoglobin dissociation curve to the left. Hemoglobin has an affinity for carbon monoxide that is 210 times greater than that for oxygen. Carbon monoxide also increases the stability of the hemoglobin-oxygen combination, thus inhibiting oxygen delivery to the tissues. In addition, carbon monoxide binds to the cytochrome oxidase chain, interfering with cellular respiration. These properties of carbon monoxide result in chemical asphyxiation.
![]() Clinical Findings
Clinical Findings
Symptoms include general malaise, headache, nausea, dyspnea, vomiting, and alteration in mental status at high levels. Severe exposure may acutely cause coma, seizures, arrhythmias, and death and later persistent cognitive deficits and other neurologic injuries in survivors. Symptoms of anoxia may be prominent without cyanosis because of the cherry-red color of carboxyhemoglobin.
Laboratory findings at chronic low levels may show polycythemia; at higher levels, hypoxia is seen. Carboxyhemoglobin levels should be measured; a level less than 6% may cause impairment in vision and time discrimination; at 40–60% alterations in mental status and death may be seen. However, there is an imperfect correlation between carboxyhemoglobin levels and the manifestations of carbon monoxide poisoning. Moreover, because of the short half-life of carbon monoxide in the body, COHb levels may have declined, once exposure ceases, to relatively low levels before the patient presents for medical attention (and testing). Blood carboxyhemoglobin levels can be elevated significantly both after intense exposures of short duration and after chronic low-level exposure.
![]() Treatment & Prevention
Treatment & Prevention
Treatment depends on the degree of carboxyhemoglobin. At low levels without symptoms, removal from the source of exposure is sufficient. Oxygen administration is effective treatment, because it markedly decreases the half-life of carboxyhemoglobin (from about 5–6 hours in room air) to about 90 minutes at 100% inspired oxygen by displacement of the carbon monoxide from hemoglobin. Hyperbaric oxygen therapy decreases the half-life of carboxyhemoglobin further to 23 minutes at 3 atm. Although some studies have suggested a lower incidence of cognitive deficits in patients treated with hyperbaric oxygen (HBO), a recent Cochrane review concluded: “Existing randomized trials do not establish whether the administration of HBO to patients with carbon monoxide poisoning reduces the incidence of adverse neurologic outcomes. Additional research is needed to better define the role, if any, of HBO in the treatment of patients with carbon monoxide poisoning.” Prevention of carbon monoxide poisoning depends upon adequate ventilation and consumer education, with proper venting of combustion devices (eg, furnaces, fireplaces, vehicle engines, and generators) to the outside air. Public health organizations now recommend that CO monitors be universally used in residences to permit detection and occupant warning of elevated air concentrations.
DISORDERS AFFECTING BLOOD CELL FORMATION & MORPHOLOGY
Premalignant and malignant hematologic diseases are linked to a variety of occupational exposures. Because the determination of cause and effect is very difficult to verify when the latency period is long and the exposure history is poorly documented, other methods for ascertaining this link have been explored. Cytogenetic study consists of examining the somatic chromosomes of hematologic cells in metaphase.
![]() Cytogenetics
Cytogenetics
The chromosomal analysis of hematologic disorders is an important mechanism for classification and as a guide to prognosis and therapy. Cytogenetic analysis serves two purposes in occupational hematology: (1) to screen populations at risk for toxic exposure so that cryptic toxic agents can be identified and (2) in individual cases to identify diseases that might have been caused by exposure to mutagenic agents.
Abnormalities in chromosomes can be used as a marker for exposure to noxious environmental agents. Through extensive epidemiologic study, certain abnormalities have been associated with specific diseases and prognostic categories. Toxic agents that are associated with chromosomal abnormalities in vivo are linked to the development of cancers and leukemias.
Cytogenetic analysis for hematologic disorders is best done by direct examination of cells obtained from bone marrow. Because these cells are continuously proliferating, it is relatively easy to examine those undergoing mitosis when the chromosomes are visible microscopically. High-resolution banding techniques are used to precisely identify deletions, translocations, inversions, and other structural chromosomal abnormalities.
Cells obtained from peripheral blood require artificial stimulation by a mitogen such as phytohemagglutinin and culture for 2–3 days to obtain enough cells in mitosis for analysis. Artifactual aberrations may be induced in cells that are manipulated after removal from the patient before fixing for analysis. The resulting risk of falsely abnormal evaluations increases the necessity and importance of well-matched and multiple controls.
![]() Screening & Prevention
Screening & Prevention
Cytogenetic analysis has been used as a screening tool to monitor industrial populations for early exposure to mutagenic chemicals and to identify possible mutagens. In this way, workers at risk might be removed from potentially dangerous conditions when the effects are still reversible. Peripheral blood lymphocytes rather than bone marrow must be used for obvious reasons of worker comfort, time, and cost.
The problem with using cytogenetics for monitoring is the relative insensitivity of the method to low levels of exposure. The only known dose-response relationship for exposure and somatic chromosome aberrations has been described with ionizing radiation. In a recent study of workers exposed to low levels (below exposure limits) of chemicals in a petrochemical plant in The Netherlands, cytogenetic monitoring by chromosomal banding techniques was carried out from 1976 to 1981. Results of these studies, published in 1988, found no increase in the frequencies of chromosome aberrations in the exposed populations compared with control populations. The authors concluded that the examination of peripheral blood lymphocytes for chromosomal aberrations is not sufficiently sensitive for routine monitoring of cytogenetics in workers exposed to low levels of the compounds.
Other techniques that have been evaluated in monitoring include sister chromatid exchange, the Comet assay, and the micronucleus assay, methods of evaluating chromosomal rearrangements of DNA. Although these techniques are faster and cheaper than cytogenetic analysis; the presence of detectable abnormalities by these assays, however, does not necessarily correlate with the incidence of chromosomal aberrations or the development of disease.
Further progress in methods to detect induced DNA damage is needed for cytogenetic analysis to be an adequate screening tool for large populations. With the advent of molecular biologic techniques, this should be possible in the future on a population-wide scale. At present, although individual health consequences cannot be estimated by population screening methods, abnormal chromosomal or cytogenetic findings clearly are an adverse sign when correlated with specific exposure risk data.
![]() Relationship of Cytogenetic Abnormalities to Specific Diseases
Relationship of Cytogenetic Abnormalities to Specific Diseases
Cytogenetic study provides a means of relating chromosomal aberrations in the bone marrow of a patient with preleukemia or leukemia to exposure to mutagenic agents. The population benefited includes workers exposed to industrial agents and radiation, as well as patients previously treated with chemotherapeutic agents or radiation. Many studies suggest that this population has a much higher incidence of chromosomal abnormalities than does a similar nonexposed group with the same diseases. In a summary of three retrospective studies of patients with de novo acute nonlymphocytic leukemia, individuals were grouped as nonexposed and exposed on the basis of occupation. Individuals who worked with insecticides, chemicals and solvents, metals or minerals, petroleum products, and ionizing radiation were considered exposed, whereas students, white-collar workers, and housewives were classified as nonexposed. Sixty-eight of 236 patients (29%) were found to be in the exposed group. Fifty-one of the 68 (75%) in the exposed group had abnormal karyotypes versus only 60 of 168 (36%) in the nonexposed group. In addition to this generalized increase in chromosomal aberrations, abnormalities in chromosomes 5 and 7 were observed in 37% of the exposed and in 12% of the nonexposed individuals.
These specific chromosomal abnormalities also have correlated positively with the development of leukemia after exposure to therapeutic mutagens (chemotherapy or radiation) used to treat other cancers. Specifically, a loss of the entire chromosome or of part of the long arm of either or both of these chromosomes has been seen. Although these chromosome abnormalities may occur in the absence of exposure to mutagens, patients with leukemias or preleukemic conditions with deletions involving chromosomes 5 or 7 should arouse a suspicion of prior exposure to chemical carcinogens or radiation, and this history should be sought vigorously. The prognosis for patients with abnormalities of chromosomes 5 or 7—or with multiple cytogenetic abnormalities when associated with prior exposure to mutagenic agents—is poor when compared with that of patients with a normal chromosome analysis.
Clearly, the greatest usefulness of cytogenetics is in the area of prevention and developing better techniques to assess future pathologic consequences of exposure at a time when effects still may be reversible. As more sensitive techniques are developed, they also may serve as a guideline for determining the threshold limit values for potential mutagens.
WORK-RELATED APLASTIC ANEMIA & HEMATOLOGIC CANCERS
Hematopoietic neoplasms, such as leukemia and myelodys-plastic syndromes, are discussed in detail in Chapter 19. In this section, we discuss only aplastic anemia and multiple myeloma.
1. Aplastic Anemia
Aplastic anemia, or medullary aplasia, is an acquired abnormality of the pluripotent hematopoietic stem cells resulting in pancytopenia (anemia, neutropenia, and thrombocytopenia). The average incidence of fatal aplastic anemia per year in the United States is approximately 2 per million and rises with age to an annual age-specific mortality rate of about 10 per million in people older than age 65. Approximately 50% of cases of aplastic anemia in North America and western Europe are idiopathic; most of the remainder are termed secondary aplastic anemias and may be caused by drugs, chemicals, radiation, infection, and immunologic mechanisms. A small percentage of cases are caused by hereditary diseases.
The largest category of secondary aplastic anemia is caused by therapeutic drugs; probably only a small fraction are a result of environmental and occupational causes. The drug most commonly implicated is chloramphenicol; other implicated drugs include acetazolamide, phenylbutazone, phenytoin, and sulfonamides—and there are many others. This section discusses only occupation-related cases of aplastic anemia.
Many cases of aplastic anemia develop after the occurrence of dysplastic morphologic changes in hematologic cells with associated chromosomal abnormalities. The incidence of acute nonlymphocytic leukemia in patients with aplastic anemia who survive 2 years after diagnosis is approximately 5–10%; in patients with preceding dysplasia, the incidence may be higher. Chemicals that are capable of inducing bone marrow damage must be assumed to be potential leukemogens. It is difficult to link specific chemicals to the development of aplastic anemia because of the absence of a specific test for exposure and the frequency of multiple or unknown exposures. Only three agents are firmly established as a cause of aplastic anemia on a dose-dependent basis: benzene, ionizing radiation, and cytotoxic drugs such as antimetabolites and alkylating agents.
![]() Benzene
Benzene
Benzene was first described as a cause of fatal aplastic anemia in 1897. Early unregulated exposure to benzene—used widely as a solvent in the production of many products, including fabrics and pesticides—led to many cases of acute and chronic toxicity. Workers historically at greatest risk of high concentration exposure are those involved in rubber manufacturing, shoemaking, petroleum and chemical production, and printing.
Before 1950, benzene was the single most common cause of toxic aplastic anemia. With chronic doses of greater than 100 ppm, isolated cytopenias and aplastic anemia were common. The cytopenias usually resolved after termination of exposure; even with persistent exposure, spontaneous remissions have been described. At exposures of 100 ppm or higher, some workers will develop fatal aplastic anemia. Great variation in susceptibility to exposure has been seen, with evidence of poisoning sometimes appearing only after weeks or years. Cases of cytopenia also have been seen several years after exposure has been terminated; these cases are less likely to resolve with time and may be part of a preleukemic syndrome. In severe chronic poisoning, decreased red cell survival with hemolysis has been reported.
Toxicity is directly related to the amount and duration of exposure, although again there is individual variation in susceptibility. The current U.S. OSHA permissible exposure limit is 1 ppm; however, exposures leading to aplastic anemia are many times higher than this level. The diagnosis is made by examination of the bone marrow after an abnormal complete blood count is reported. The bone marrow will reveal hypocellularity with fatty replacement, although islands of hypercellularity may be seen. Although cytogenetic abnormalities are associated with benzene exposure, specific chromosome changes are not. The initial prognosis in benzene-related aplastic anemia is better than that for idiopathic aplastic anemia; up to 40% of patients may recover completely after removal from the source of exposure. If hypocellularity persists for more than several months, recovery is not likely to occur. Exposure is also associated with the development of acute nonlymphocytic leukemia and chronic myelogenous leukemia—either de novo or in workers who have recovered from a bout of aplastic anemia—and in cases of irreversible aplastic anemia.
Treatment is supportive (ie, with transfusions and such growth factors as erythropoietin, granulocyte colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor). Drugs such as androgens to stimulate hematopoiesis have not been used extensively in benzene-induced aplastic anemia but should be tried when no other treatment option exists (such as bone marrow transplantation or colony-stimulating factors). Allogeneic bone marrow transplantation is the only known cure for irreversible aplastic anemia but is hampered by donor availability, higher mortality risk with increasing patient age, and the toxicity of the transplant regimen.
![]() Ionizing Radiation
Ionizing Radiation
Ionizing radiation also has been associated with aplastic anemia in a dose-dependent manner. Internal exposure to absorbed alpha particles associated with aplastic anemia was demonstrated most strikingly in the radium watch dial workers who ingested radium by wetting their paintbrushes on their tongues. External exposure to radiation is much more common and may be in the form of whole-body exposure to a large dose, as in a nuclear accident or therapeutic radiation, or long-term exposure to small amounts, as may have occurred in the practice of radiology as a medical specialty prior to effective protection against radiation exposures.
Data from patients radiated for ankylosing spondylitis and from the survivors of the atomic bombings of Hiroshima and Nagasaki suggest that the risk of aplastic anemia is increased until 3–5 years after exposure, after which there is a marked decline in incidence. The most important late disturbance following irradiation of the bone marrow is leukemia. The ability to recover from a single dose of penetrating radiation depends on the fraction of surviving stem cells. Chromosomal aberrations are associated with exposure to ionizing radiation and rise in a linear manner as a function of the dose of radiation absorbed. The presence of these aberrations, including an increase in the number of sister chromatid exchanges, may signify excessive exposure but is not predictive of aplastic anemia or leukemia.
Strict regulation of exposure and monitoring with badges has virtually eliminated aplastic anemia caused by radiation except in cases of accidental overexposure. In this case, treatment again is primarily supportive. Recovery may be seen after a prolonged period of aplastic anemia lasting 3–6 weeks and may be predicted from the known total dose of radiation. If recovery does not occur, permanent injury to the stem cell population will result in chronic cellular hypoplasia or dysplasia or in leukemia. Treatment then may include bone marrow transplantation if a donor is available.
![]() Other Chemicals
Other Chemicals
Aplastic anemia has been reported following exposure to a variety of other chemicals listed in Table 18–4. Toxicities often resolve completely with cessation of exposure. Again, individual susceptibility plays an important role, although it is poorly understood.
Table 18–4. Chemicals reported to cause aplastic anemia in an occupational setting.

Two chemicals in particular deserve mention here. The aplastic anemia associated with trinitrotoluene may be accompanied by methemoglobinemia, oxidative hemolysis, liver damage, and dermatitis. The incidence of overexposure to arsenic has declined with its decreasing use and better controls over the recent decades. Fewer than 10 cases of overt arsenic poisoning are now reported annually in the United States. Complete spontaneous hematologic recovery usually is seen if the patient is promptly removed from the source of heavy exposure.
2. Myelodysplastic Syndromes
The myelodysplastic syndromes are a group of acquired genetic disorders of the blood-forming cells similar to cancer and characterized by ineffective hematopoiesis, clinically resulting in anemia, neutropenia, thrombocytopenia, or a combination of cytopenias. These syndromes are linked by the presence of bizarre hematopoietic morphology and the tendency to transform into acute leukemia. Most patients with myelodysplasia, however, do not develop leukemia, although specific syndromes associated with exposure to both occupational chemicals and cytotoxic drugs have a high incidence of progression to frank leukemia. Both benzene and ionizing radiation have been implicated in the development of myelodysplasia. Several case-control studies have suggested that other occupational risk factors, such as exposure to pesticides or solvents or employment in specific sectors such as farming, textile work, or the health professions, may be relevant. The median survival from these diseases is less than 12 months, and all patients eventually either develop leukemias or succumb to complications related to cytopenias. Exposure and treatment-related myelodysplasia is specifically associated with a high incidence of deletions involving chromosomes 5 and 7.
Myelodysplastic syndromes (MDS) are more common in men than in women, and 85% of patients are older than 40 years of age at the time of diagnosis. Laboratory features of MDS include cytopenias of various degrees and often an increase in the red blood cell mean corpuscular volume. The marrow usually reveals dysplasia in all three cell lines (granulocyte/erythroid/megakaryocyte [platelet-forming]) and manifests abnormal marrow cellularity, usually hypercellular. There is an abnormal increase in the percentage of blast cells.
Several treatment options are available, although all have significant drawbacks. Allogeneic bone marrow transplantation (a transplant from a donor, usually a sibling or matched unrelated donor) is the only known cure but is limited primarily by patient age and carries a significant risk of treatment-related mortality. Transfusions and treatment of infections may be aided by the use of hematopoietic growth factors. The hypomethylating chemotherapy drug azacytidine is approved by the Food and Drug Administration for MDS, and two other agents, lenalidomide and decitabine, appear quite active, although none of these agents appears to be curative.
3. Multiple Myeloma
Multiple myeloma is a chronic leukemia of differentiated B cells (termed plasma cells) that accounts for 15% of all hematologic cancers. It is characterized by anemia, painful lytic and osteopenic bone disease, monoclonal immunoglobulin production (in serum or urine or both), hypogammaglobulinemia, and short survival. Patients also may have hypercalcemia, renal failure, or neuropathy. Treatment involves a variety of agents, including thalidomide, chemotherapy (with such agents as melphalan, vincristine, vinblastine, doxorubicin, cyclophosphamide, and carmustine), and corticosteroids with the goal of alleviating bone pain, correcting complications of the disease, and prolonging life. Autologous bone marrow transplantation soon after diagnosis appears to improve both disease-free and overall survival. This less toxic form of therapy is available to a wider range of patients up to age 70 years.
The peak incidence of multiple myeloma is between ages 55 and 65, and fewer than 2% of cases occur before the age of 40. Multiple myeloma is equally common in men and women but almost twice as common in blacks as in whites. The incidence of multiple myeloma has been increasing over the last three decades in North American and European men, but this rise has not been noted in the stable study populations in Minnesota and Sweden and simply may reflect an increase in our ability to diagnose the disease. The rise in incidence has aroused concern that myeloma might be associated with environmental or occupational factors.
Although there is no definitive link between occupational exposure and the risk of multiple myeloma, many epidemiologic studies suggest associations. Associations of exposure to petroleum products, benzene, organic solvents, heavy metals, some pesticides, and asbestos have been observed, but most studies are small, and these findings have not typically been replicated. Thus, for most agents, these findings can be used only as a basis for etiologic hypotheses. Workers thought to be at risk include agricultural workers, chemicals workers, miners, smelters, stokers, and furniture workers. The International Agency for Research on Cancer (IARC), an independent scientific institution within the World Health Organization (WHO), recently concluded, in its monograph on benzene, that: “There is limited evidence in humans for a causal association of benzene with multiple myeloma.”
An important association between high-dose radiation exposure and multiple myeloma has been observed in cohorts of controls and survivors of the atomic bombings of Hiroshima and Nagasaki for the period of 1950–1976. The relative risk for persons with an estimated air-dose exposure of 100 cGy or more was over four times higher than that of controls. This excess risk became apparent approximately 20 years after exposure. An association also has been proposed—but not confirmed—between the risk of multiple myeloma and exposure to low-dose radiation. At present, except in the case of high-dose radiation, there are insufficient data on which to base a firm conclusion about the relationship between exposure to ionizing radiation and the risk of developing multiple myeloma.
TOXIC THROMBOCYTOPENIA
Unlike thrombocytopenia occurring as part of toxicant-induced aplastic anemia, isolated toxic thrombocytopenia occurs only rarely. A number of toxic exposures have been reported that resulted in isolated thrombocytopenia (Table 18–5). In 1963, two cases of isolated thrombocytopenia were described in individuals exposed to the reactive polymerizing agent toluene diisocyanate (TDI), used in the manufacturing of polyurethane foam. In both, thrombocytopenia developed 14–22 days following a significant exposure that also induced bronchospasm (TDI is a recognized cause of occupational asthma). These patients developed thrombocytopenic bleeding with nadir platelet counts of 6000/μL and 30,000/μ. Bone marrow samples in both cases showed increased megakaryocytes. The pathophysiologic defect was enhanced peripheral platelet destruction, presumably on an immune basis (immune thrombocytopenic purpura). One patient responded transiently to corticosteroid therapy and then completely to splenectomy; the second patient had resolution of thrombocytopenia without any therapy.
Table 18–5. Toxic agents associated with isolated thrombocytopenia.

Two more cases of toxin-induced immune thrombocytopenic purpura were described in 1969. Two children with significant turpentine exposure (one respiratory and cutaneous exposure, the second ingestion) developed petechiae and severe thrombocytopenia with increased bone marrow megakaryocytes. Both responded fully to corticosteroid therapy.
Some insecticides occasionally appear to cause a selective megakaryocyte aplasia in people with significant inhalation or ingestion exposure. Isolated thrombocytopenia has been reported after exposure to 2,2-dichlorovinyl dimethyl phosphate, dieldrin, pyrethrin, hexachlorocyclohexane (lindane), and chlorophenothane (DDT). These patients demonstrated absent or decreased bone marrow megakaryocytes. Some megakaryocytes were vacuolated.
A third form of toxic thrombocytopenia was described in 1984. Forty-six people heavily exposed to vinyl chloride with evidence of toxic coproporphyrinuria also had thrombocytopenia. Although these patients were not described in detail, they appeared to have significant vinyl chloride liver toxicity, with esophageal varices and splenomegaly. The likely pathophysiologic mechanism of thrombocytopenia thus was enhanced peripheral platelet consumption caused by hypersplenism, although there were insufficient data presented to rule out an immune or megakaryocyte toxic mechanism.
Normal hemostasis depends both on the quantity of platelets present and on their ability to aggregate appropriately under physiologic stimulation. Qualitative disturbances in platelet function as a result of various occupational and environmental substances have been described. Some (but not all) pesticides, such as p,p-DDE (2,2-bis-[p-chlorophenyl]-1,1-dichloroethylene) and Aroclor 1242 (a chlorinated biphenyl that is 42% chlorine), inhibit platelet aggregation in a dose-dependent manner by inhibiting platelet cyclooxygenase activity (an aspirin-like effect). Exposures to these substances could cause mucocutaneous bleeding in susceptible individuals. On the other hand, some environmental substances (eg, methyl mercury, cadmium, and triethyl lead) induce platelet aggregation and could result in hypercoagulation. These qualitative platelet disturbances have not yet been reported in humans subject to occupational exposure and remain theoretical risks.
OCCUPATIONAL EXPOSURE TO ANTICANCER DRUGS
Oncology nurses and pharmacists who prepare and administer chemotherapy to patients on a regular basis are at risk for exposure to potentially mutagenic agents. Data from a variety of sources are available on the mutagenic potential of many anticancer drugs. Epidemiologic studies link certain drugs (and radiation) to the development of secondary cancers; analytic in vitro methods of assessing mutagenicity offer corroborative evidence.
There are several obvious problems in assessing the risk to nurses. First is the low and intermittent exposure rate relative to the patients who receive these drugs therapeutically and who serve as in vivo models for the effects of heavy exposure. This limits the usefulness of epidemiologic data such as exposure rates and disease incidence when attempting to assess individual risks. Second, the methods for detecting exposure often are contradictory and may be cumbersome in time relative to work. For example, it may be difficult for nurses to obtain urine 6 hours after the end of a shift.
Methods for assessing risk include measuring blood and urine levels of drug, urine mutagenicity assays, cytogenetic monitoring and sister chromatid exchange studies, and environmental monitoring. Blood and urine levels may be difficult to interpret when drugs are metabolized rapidly. Studies evaluating oncology nurses versus control populations of other nurses or non-health care workers have been published with both positive and negative results using all the preceding methods. Clearly a combination of monitoring tools is required.
In addition to the preceding studies, which assess potential risk, the effect on human reproduction in terms of spontaneous abortion and teratogenic effects also has been examined. Again, the results are inconclusive but suggest an increase in both effects with frequent exposure. Information on safety practices is not available.
The agents implicated most commonly in mutagenic potential are those that are also implicated as causes of secondary cancers in patients receiving therapeutic drugs such as alkylating agents. Clearly, any cytotoxic drug should be handled with great care and with the goal of absolutely minimum exposure to personnel. This can be accomplished by worker education, gloves and protective clothing, laminar hoods for preparing drugs, and proper waste disposal. Periodic atmospheric checks and biologic monitoring can improve hygienic standards, but conclusions regarding the long-term health effects based on these results are not possible.
REFERENCES
Balwani M: The porphyrias: advances in diagnosis and treatment. Blood 2012;120:4496 [PMID: 22791288].
Bejar R: Myelodysplastic syndromes. Am Soc Clin Oncol 2013;2013:256 [PMID: 23714517].
Buckley NA: Hyperbaric oxygen for carbon monoxide poisoning. Cochrane Database Syst Rev 2011 Apr 13;(4):CD002041 [PMID: 21491385].
IARC: Benzene. A review of human carcinogens. Chemical agents and related occupations, Volume 100F, 2012. http://occupationalcancer.ca/wp-content/uploads/2012/06/Lancet-Monograph-100F.pdf.
Skold A, Cosco DL, Klein R. Methemoglobinemia: pathogenesis, diagnosis, and management. South Med J 2011;104:757 [PMID:22024786].
Snyder R: Leukemia and benzene. Int J Environ Res Public Health 2012;9:2875 [PMID: 23066403].
Valsami S: Acute copper sulphate poisoning: a forgotten cause of severe intravascular haemolysis. Br J Haematol 2012;156:294 [PMID: 21981599].
Yoshimura Y: Acute arsine poisoning confirmed by speciation analysis of arsenic compounds in the plasma and urine by HPLC-ICP-MS. J Occup Health 2011;53:45 [PMID:21123960].
![]() SELF-ASSESSMENT QUESTIONS
SELF-ASSESSMENT QUESTIONS
Select the one correct answer for each question.
Question 1: Methemoglobin
a. is like sulfhemoglobin since it is irreversibly associated with the heme moiety
b. is dangerous because of its ability to bind oxygen
c. decreases the oxygen affinity of heme groups in hemoglobin
d. decreases oxygen delivery to the tissues
Question 2: Porphyrias
a. are a group of disorders characterized by bluish discoloration of the skin
b. result in the abnormal accumulation of hemoglobin in the skin
c. are genetic disorders that affect only royal families
d. can occur as a result of inappropriate overstimulation of δ-aminolevulinic acid synthetase
Question 3: Acute intermittent porphyria causes
a. symptoms and signs quite different than those associated with lead intoxication
b. a classic triad of abdominal pain, constipation, and jaundice
c. an increase in neuropsychiatric signs compared with lead intoxication
d. a more pronounced anemia than occurs with lead intoxication
Question 4: Aplastic anemia
a. is distinct from medullary aplasia and secondary aplastic anemia
b. is largely an hereditary abnormality of the pluripotent hematopoietic stem cells
c. results in pancytopenia (anemia, neutropenia, and thrombocytopenia)
d. cases are largely caused by hereditary diseases
Question 5: Myelodysplasic syndromes
a. are a group of acquired nongenetic disorders of the blood-forming cells
b. are characterized by ineffective hematopoiesis
c. are characterized by a bizarre hematopoietic morphology confirming cancer
d. have in nearly all cases a progression to frank leukemia.
Question 6: Multiple myeloma
a. is an acute leukemia of differentiated B cells (termed plasma cells)
b. accounts for 50% of all hematologic cancers
c. is characterized by anemia, painful lytic and osteopenic bone disease
d. has a long survival rate