Neurophysiologic electrodiagnostic studies define alterations in the functions of the nervous system that may not be visualized by imaging procedures. The major areas of study include EEG, nerve conduction studies (NCS), EMG, and evoked potentials. The clinical usefulness of these examinations is here discussed, followed by brief descriptions of other ancillary neurologic tests such as polysomnography and the multiple sleep latency test, and finally the indications, contraindications, and utility of performing lumbar puncture (LP) for CSF analysis.
I. ELECTROENCEPHALOGRAM
A. Introduction. The EEG involves recording of the spontaneous electrical activity of the brain from the scalp and activity elicited by activation procedures, including sleep, hyperventilation, and photic stimulation. This electrical activity is the summation of both excitatory and inhibitory postsynaptic potentials from cortical neurons aligned perpendicular to the surface. Small metal disks containing conductive gel are attached to the scalp and ear lobes according to a system of measurements and are connected by flexible wires to a recording instrument that amplifies the brain activity about a million times. The EEG is sampled on moving paper or on a computer simultaneously from 16 to 21 pairs of electrodes (derivations) in selected combinations (montages).
B. Normal EEG activity.
1. EEG rhythms. The EEG is a composite of several different types of activity, each with characteristic factors of frequency, amplitude, morphology, reactivity, topography, and quantity. The frequency bands of activity are as follows:
a. Delta activity (4 Hz)
b. Theta activity (4 to 8 Hz)
c. Alpha activity (8 to 13 Hz)
d. Beta activity (≥13 Hz)
2. The most characteristic feature of a normal EEG in an adult during relaxed wakefulness is the alpha rhythm, which occurs over the posterior regions of the head while the eyes are closed (Fig. 33.1). Judgments of normality for various EEG activities depend on the age and state of alertness of the subject because complex changes in the EEG patterns occur throughout life and patterns evolve when going from wakefulness through different stages of sleep.
3. Activation procedures are used to elicit abnormal activities that may not occur spontaneously.
a. Hyperventilation for 3 minutes is most effective for activating generalized seizure activities such as the spike–wave paroxysms of absence (petit mal) seizures. It may less frequently activate focal abnormalities (e.g., slowing) and focal epileptiform activity. It is contraindicated in a patient with cardiac infarction, recent subarachnoid hemorrhage (SAH), or significant pulmonary disease.
b. Photic stimulation consists of repetitive brief flashes of light generated by an electronic apparatus and delivered at frequencies of 1 to 30 Hz. This procedure evokes responses over the occipitoparietal regions (photic driving). The most frequent abnormal response is diffuse paroxysms of spike–wave complexes (photoparoxysmal or photoconvulsive response) that often indicate a seizure propensity.
c. Sleep recordings are most useful for recording paroxysmal abnormalities in patients with epilepsy. Sleep may activate focal or generalized epileptiform activity. Sleep deprivation on the night before the study may facilitate sleep, and the deprivation itself may activate epileptiform activity.
C. Abnormal EEG activity. Many EEG changes are nonspecific, but some are highly suggestive of specific entities such as epilepsy, herpes encephalitis, and metabolic encephalopathies. In general, neuronal damage or dysfunction is suggested by the presence of slow waves (activity in the theta or delta range) in a focal or diffuse location, whereas the presence of sharp waves or spikes (epileptiform activity) in a focal or diffuse pattern suggests a seizure tendency. Localized slowing is highly sensitive and significant for local neuronal dysfunction or focal brain damage but is quite nonspecific because it cannot distinguish the pathologic type of lesion. Thus, cerebral infarction, tumor, abscess, and trauma may all cause similar focal EEG changes (Fig. 33.2). Diffuse slowing also indicates organic rather than psychiatric disease but again is nonspecific because such slowing may occur with any significant toxic, metabolic, degenerative, or even multifocal disease process. The EEG is also useful in following the courses of patients with altered states of consciousness and may, in certain circumstances, provide prognostic information. Finally, the EEG is important in the determination of brain death.
1. Epilepsy.
a. Some types of interictal EEG patterns are termed epileptiform because they have a distinct morphology and occur in a high proportion of EEGs from patients with seizures but rarely in records from asymptomatic patients. Such patterns include sporadic spikes, sharp waves, and spike and slow wave complexes. Not all spike patterns indicate epilepsy; 14- and 6-Hz positive spikes, sporadic sleep spikes, wicket spikes, 6-Hz spike–wave complexes, and the psychomotor variant pattern are all spike patterns that are of no proven clinical significance. Epileptiform discharges are not seizures in themselves. Seizures are electrographically defined as an evolution in frequency, location, or morphology of epileptiform discharges and usually last >10 seconds. An example of an electrographic seizure is demonstrated in Figure 33.3. Interictal findings must always be interpreted with caution because, although certain patterns of abnormality may be evidence useful in supporting a diagnosis of epilepsy, even epileptiform discharges, with few exceptions, are poorly correlated with the frequency and likelihood of recurrence of epileptic seizures. One must always treat the patient and never “treat” the EEG.

FIGURE 33.1 Normal EEG in an adult man at rest with eyes closed. Top four rows—EEG activity from frontal to occipital regions on left. Bottom four rows—EEG activity from frontal to occipital regions on right. Note normal alpha activity over posterior head regions.
b. A substantial portion of patients with unquestioned epilepsy have normal EEGs. However, epileptiform activity has a high correlation with clinical epilepsy. Only about 2% of nonepileptic patients exhibit epileptiform EEG activity, in contrast with 50% to 90% of patients with epilepsy, depending on the circumstances of recording and on whether more than one EEG has been obtained. The most conclusive proof of an epileptic basis for a patient’s episodic symptoms is obtained by recording an EEG seizure during a typical behavioral episode.
c. The EEG helps establish whether the seizure originates from a limited or focal area of the brain (focal or partial seizures) (Fig. 33.4) or involves the brain as a whole from the onset (generalized seizures). This distinction is important because of the different possible causes of these two basic epilepsy types and because the clinical manifestations of both types may be similar.
d. In general, the epileptiform activity on the EEG may be helpful in classifying the patient’s seizure type.
(1) Generalized seizures of nonfocal origin usually are associated with bilaterally synchronous bursts of spikes and spike–wave discharges.
(2) Consistently the focal epileptiform activity correlates with partial or focal epilepsy.
(a) Anterior temporal spikes correlate with complex partial seizures.
(b) Rolandic spikes correlate with simple motor or sensory seizures.
(c) Occipital spikes correlate with primitive visual hallucinations or diminished visual function.
e. The EEG analysis may permit further discrimination of several relatively specific electroclinical syndromes.
(1) Hypsarrhythmia refers to a high-voltage, arrhythmic EEG pattern with a chaotic admixture of continuous, multifocal spike–wave and sharp-wave discharges and widespread, high-voltage, arrhythmic slow waves.

FIGURE 33.2 Left focal fronto-temporal slow wave abnormality in an adult patient with a recent large left fronto-temporal infarction. (Image courtesy of Dr. David Chabolla.)

FIGURE 33.3 Focal seizure recorded with intracranial electrodes resting atop cortical surface. Note focal onset of rhythmic discharges, which spread and alter in frequency and morphology. (Image courtesy of Dr. Maria E. Baldwin.)
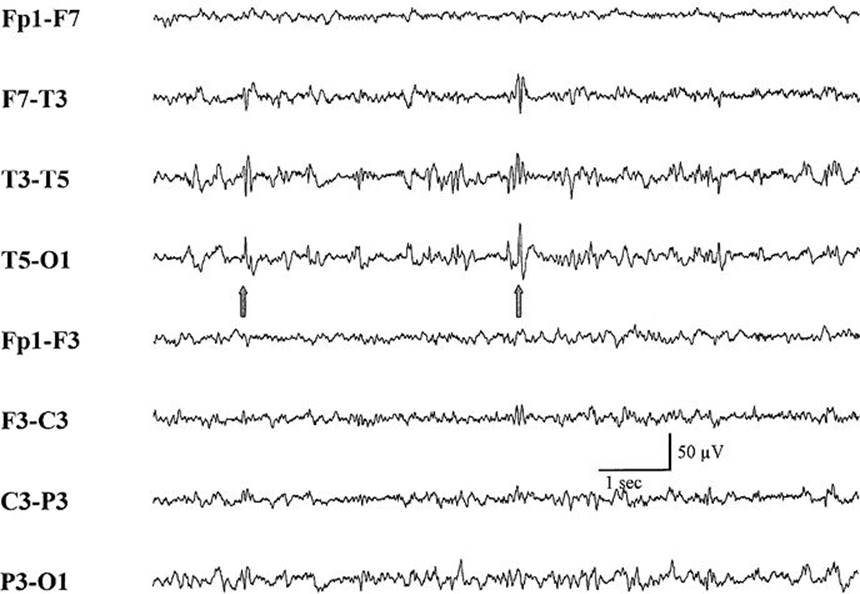
FIGURE 33.4 Focal epileptiform activity (spike) (arrows) in left posterior temporal region of an adult with partial seizures.
This infantile EEG pattern usually occurs in association with infantile spasms, myoclonic jerks, and mental retardation (West’s syndrome) and usually indicates severe diffuse cerebral dysfunction. Infantile spasms consist of tonic flexion or extension of the neck, body, or extremities with the arms flung outward and typically last 3 to 10 seconds. The EEG and clinical findings do not correlate with a specific disease entity but reflect a severe cerebral insult occurring before 1 year of age.
(2) The 3-Hz spike-and-wave activity is associated with typical absence attacks (petit mal epilepsy) (Fig. 33.5). This pattern most often occurs in children between the ages of 3 and 15 years and is enhanced by hyperventilation and hypoglycemia. These bursts are typically accompanied by clinical signs such as staring, brief clonic movements, unresponsiveness, and motor arrest.
(3) Generalized multiple spikes and waves (polyspike–wave pattern) are typically associated with myoclonic epilepsy or other generalized epilepsy syndromes (Fig. 33.6).
(4) Generalized slow spike-and-wave patterns at a frequency of 1 to 2.5 Hz occur in children between the ages of 1 and 6 years who have some underlying diffuse cerebral dysfunction. Most of these children are mentally retarded and have poorly controlled seizures. The clinical triad of mental retardation, severe seizures, and the slow spike-and-wave pattern is called the Lennox–Gastaut’s syndrome.
(5) Central–midtemporal spikes occur in childhood and are associated with benign rolandic epilepsy. These seizures are often nocturnal and consist of focal clonic movements of the face or hand; tingling in the side of the mouth, tongue, cheek, or hand; motor speech arrest; and excessive salivation. The spells are easily controlled with anticonvulsants and disappear by 12 to 14 years of age.
(6) Periodic lateralized epileptiform discharges (PLEDs) are high-voltage, sharply contoured complexes that occur over one cerebral hemisphere with a particular periodicity. These complexes are not necessarily epileptic and usually correlate with acute destructive cerebral lesions, including infarction, rapidly growing tumors, and herpes simplex encephalitis (HSE) (Fig. 33.7). In the past, there was much debate if PLEDs constituted seizure activity. A consensus has now been reached in which PLEDs is not considered an ictal pattern, especially if the discharges occur at slower frequencies of 2 Hz or less.
f. Focal slowing (delta activity) in the interictal period usually indicates an underlying structural lesion of the brain as the cause of the seizures. However, such focal slowing may be a transient aftermath of a partial seizure and may not indicate a gross structural lesion. Such slowing may correlate with a clinical transient postictal neurologic deficit (Todd’s phenomenon) and subsides within 3 days after the ictus.
g. The EEG can make a critical contribution to the diagnosis of a patient who is obtunded when prolonged subclinical seizures with only brief interruptions are recorded, signifying nonconvulsive status epilepticus
h. Ambulatory EEG monitoring is the recording of an EEG in a freely mobile patient outside of the EEG laboratory, similar to Holter’s monitoring for ECG recording. The main indication is to determine whether a spell is a seizure or some other phenomenon, especially in patients whose spells occur at unusual times or in association with specific events or activities. The yield depends on the type of patient selected, but the absence of EEG seizure activity during a spell does not fully exclude a seizure disorder, because surface electrodes may not record some mesial temporal, basal frontal, or deep midsagittal seizure discharges.
i. Patients with intractable focal seizures are sometimes candidates for surgical removal of the area of abnormality. Precise identification of the epileptogenic brain area requires special inpatient monitoring facilities for simultaneous closed-circuit television (CCTV) and EEG recording. Prolonged CCTV–EEG monitoring is also often used to document whether a patient’s clinical spells are epileptic or functional (psychogenic).
2. Altered states of consciousness.
a. For most causes of acute encephalopathy (e.g., toxic–metabolic disease), the EEG changes are nonspecific, consisting of diffuse slowing. There is, however, a generally good correlation between the degree of EEG abnormality and the clinical state.
b. An abnormal EEG confirms an organic rather than a psychogenic cause for a patient’s altered state of consciousness. It is also required to document unrecognized epileptic activity as a cause of depressed consciousness (nonconvulsive status epilepticus).
c. Certain EEG patterns increase the likelihood of specific metabolic disorders.
(1) Prominent generalized fast (beta) activity in the EEG of a comatose or obtunded patient should raise the suspicion of drug intoxication most commonly seen with benzodiazepines.

FIGURE 33.5 Burst of generalized 3 per second spike and wave discharges in a child with absence (petit mal) seizures.

FIGURE 33.6 Burst of generalized multiple spike and wave discharges in a patient with generalized tonic–clonic seizures.

FIGURE 33.7 Right periodic lateralized epileptiform discharges (PLEDs) in a patient with HSE. Compare this figure with that of Figure 33.3 demonstrating a focal seizure. The PLED activity is monotonous and static with little spread and changes in frequency and morphology. In addition, frequency of PLEDs tend to be slower (<2 Hz).

FIGURE 33.8 Frontal predominant triphasic waves and diffuse slow-wave abnormality in a patient with hepatic encephalopathy. (Image courtesy of Dr. David Chabolla.)
(2) Broad triphasic waves that are bilaterally symmetric and synchronous and have a frontal predominance may occur during an intermediate stage of hepatic encephalopathy (Fig. 33.8). However, such a pattern may also occur with other metabolic disorders.
(3) Severe generalized voltage depression may suggest hypothyroidism if anoxia and hypothermia can be excluded.
(4) Patients with uremia, uremic patients undergoing hemodialysis, and patients with hyponatremia may exhibit paroxysmal spike–wave discharges and a photoparoxysmal response to photic stimulation in addition to the diffuse slow–wave abnormality.
(5) The focal epileptiform activity is commonly seen in hyperosmolar coma.
d. Cerebral hypoxia produces diffuse nonspecific slow-wave abnormalities that may be reversible. More severe hypoxia may cause EEG abnormalities that may be paroxysmal and associated with clinical myoclonus. An EEG obtained 6 hours or more after a hypoxic insult may demonstrate patterns of prognostic value in determining the likelihood of neurologic recovery. A poor neurologic outcome is suggested by the presence of the following abnormalities.
(1) Alpha coma refers to the apparent paradoxical appearance of the monorhythmic alpha frequency activity in the EEG of a comatose patient (Fig. 33.9). However, in contrast to normal alpha activity, that observed in alpha coma is generalized, often most prominent frontally, and completely unreactive to external stimuli.
(2) The burst–suppression pattern consists of occasional generalized bursts of medium- to high-voltage, mixed-frequency slow-wave activity, sometimes with intermixed spikes, with intervening periods of severe voltage depression or cerebral inactivity. The bursts may be accompanied by generalized myoclonic jerks.
(3) The periodic pattern consists of generalized spikes or sharp waves that recur at a relatively fixed periodicity of 1 to 2 per second. The periodic pattern is usually accompanied by myoclonic jerks.
(4) Electrocerebral silence. See I.C.3.a.
e. Infectious disease processes of the CNS produce predominantly diffuse and nonspecific slow-wave activity. However, certain EEG patterns assist in the diagnosis of specific infectious etiologies.
(1) The EEG is extremely important in the initial assessment of HSE, often showing abnormalities before lesions detected by CT or MRI are recognized. A majority of patients show temporal or frontotemporal slowing that may be unilateral or, if bilateral, asymmetric. Periodic sharp complexes over one or both frontotemporal regions add relative specificity to the EEG findings (Fig. 33.7). These diagnostic features usually appear between the 2nd and 15th days of illness and are sometimes detected only on serial tracings.

FIGURE 33.9 Alpha coma in an adult patient post cardiorespiratory arrest.

FIGURE 33.10 Generalized periodic (approximately 1 per second) diphasic and triphasic sharp waves seen in patient with biopsy-proven Creutzfeldt–Jakob’s disease.
(2) Subacute sclerosing panencephalitis (SSPE) has a distinctive EEG pattern of periodic bursts of stereotyped slow-wave and sharp-wave complexes occurring at intervals of 3 to 15 seconds.
(3) Transmissible spongiform encephalopathy is associated with a relatively specific EEG pattern of diffuse high-voltage diphasic and triphasic sharp-wave complexes occurring at a periodicity of approximately 1 per second (Fig. 33.10). This periodic pattern is noted only in the end state of the disease process. Myoclonic jerks are also frequently seen and can correlate with the periodic discharges.
3. Brain death.
a. Because the EEG is a measure of cerebral—especially cortical—function, it has been widely used to provide objective evidence of loss of that function. With cortical death, the EEG demonstrates complete loss of brain-generated activity—electrocerebral silence The determination of electrocerebral silence is technically demanding and requires strict adherence to a standard special recording protocol.
b. Rarely, temporary and reversible loss of cerebral electrical activity may be observed immediately following cardiorespiratory arrest, overdose of CNS depressants, and severe hypothermia. Therefore, electrocerebral silence in these circumstances does not indicate irreversible cortical dysfunction.
c. Patients in a chronic vegetative state with preserved brainstem function may have an isoelectric EEG, probably reflecting total neocortical death.
d. Thus, the establishment of brain death (cerebral plus brainstem death) requires the following criteria.
(1) Irreversible structural brain damage.
(2) Apneic coma.
(3) Loss of brainstem reflexes and signs of brainstem function.
(4) Electrocerebral silence on EEG (best viewed as a confirmatory test).
II. NERVE CONDUCTION STUDIES AND THE ELECTROMYOGRAM
A. Introduction.
1. NCS comprise a simple and reliable method of testing peripheral nerve function. An impulse initiated by electrical stimulation of the nerve travels along motor, sensory, or mixed nerves, and the conduction characteristics of the impulse are assessed by recording potentials either from the muscle innervated by the motor nerve or from the nerve itself.
2. The motor unit consists of a single-lower motor neuron and all of the muscle fibers it innervates. Motor NCS are techniques used to assess the integrity of the motor unit. Information about the function and the structural status of the motor neuron, nerve, neuromuscular junction, and muscle is acquired. Quantitative information can be obtained regarding the location, distribution, time course, and pathophysiology of lesions affecting the peripheral nervous system (PNS). Prognosis, response to treatment, and the status of repair of the motor unit may also be obtained. For motor conduction studies, recording electrodes are placed on the skin over the motor point of a muscle and over the tendon of the muscle, and stimulating electrodes are placed over the skin along the course of the nerve to be tested. The response of the muscle to electrical stimulation can be measured by recording the compound muscle action potential (CMAP), which is the summation of the electrical potentials of all muscle fibers that respond to the stimulation of the nerve (Fig. 33.11). The time it takes for the electrical impulse to travel to the muscle (latency) can be measured, and by stimulating the nerve at various locations and measuring the distance the stimulus travels, motor nerve conduction velocities are attained. Motor NCS can be used for the following purposes.

FIGURE 33.11 Compound muscle action potentials recorded from the thenar muscles with stimulation of the left median nerve at the wrist (upper potential at A1) and forearm (lower potential at A2).
a. To obtain objective evidence of disease of motor units.
b. To identify and localize sites of compression, ischemia, and other focal lesions of nerves that can be manifested by conduction block, slowed conduction at the site of the lesion, or abnormal conduction proximal or distal to the lesion.
c. To detect widespread subclinical involvement of nerves in patients who present with clinical involvement of a single nerve (i.e., mononeuropathies).
d. To differentiate peripheral neuropathies from lower-motor neuronopathies (e.g., amyotrophic lateral sclerosis) from myopathies in patients with weakness.
e. To detect disease prior to the development of significant clinical signs (e.g., familial neuropathies).
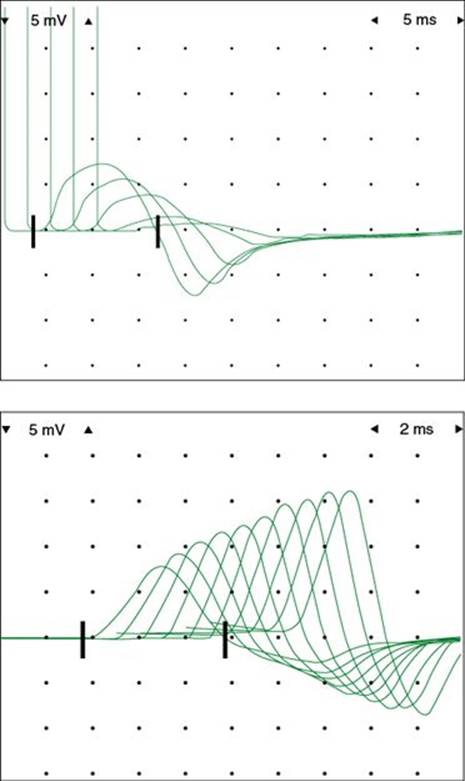
FIGURE 33.12 Decremental response to repetitive stimulation in a patient with generalized myasthenia gravis. Top: Decremental response with ulnar nerve stimulation. Bottom: Facilitation of compound muscle action potential amplitude with high-frequency stimulation, which can be seen in presynaptic disorders of neuromuscular transmission (this specific patient was positive for a P/Q-calcium channel antibody).
3. Diseases of the neuromuscular junction (e.g., myasthenia gravis [MG], Lambet–Eaton’s myasthenic syndrome) can be assessed by repetitive stimulation of motor nerves. With postsynaptic fatigability of the neuromuscular junction, if a CMAP is recorded and compared with subsequent CMAPs, a decline in the amplitude of the potential may be observed as progressively fewer muscle fibers respond to the stimuli, even though the nerve is stimulated at rates that a normal muscle could endure for long periods (Fig. 33.12). The hallmark of presynaptic disorders is a significant facilitation of CMAP amplitudes with high-frequency repetitive stimulation.
4. Sensory NCS are obtained by recording the action potential evoked in a cutaneous nerve by electrical stimulation (Fig. 33.13). Selective sensory NCS can be performed by stimulating nerves that have only sensory components (e.g., the sural nerve) or, alternatively, by selectively stimulating only the sensory components of a mixed nerve. The latter can be done by isolating the sensory components anatomically (i.e., stimulating the digits of the hand and recording over the mixed nerve at the wrist or elbow) or stimulating the mixed nerve and recording over the digits where only sensory axons are present for the most part. Sensory NCS may be valuable for the following purposes.
a. In diffuse disorders affecting the sensory system, for determining which population of sensory nerves is involved (e.g., small fibers carrying pain and temperature sensation or large fibers conveying proprioception), determining whether the disorder is predominantly affecting the axon or the myelin of the peripheral nerve, or determining whether the peripheral sensory nerves are involved at all.
b. In focal neuropathies, for demonstrating a site of injury or block, particularly when only sensory nerves are affected.
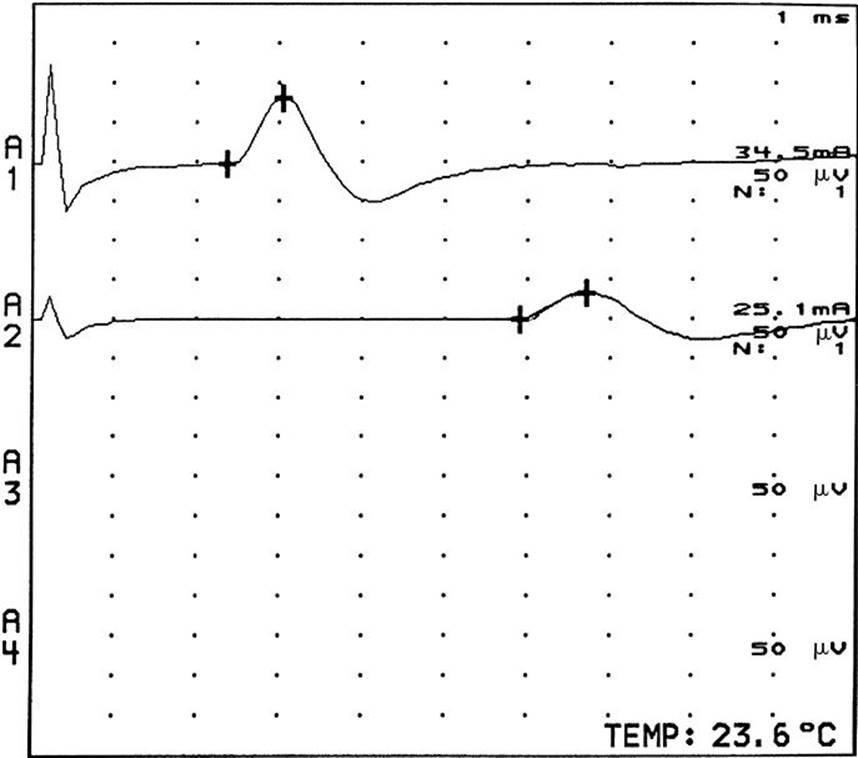
FIGURE 33.13 Sensory nerve action potentials obtained while recording over the index finger with stimulation of the left median nerve at the wrist (upper response at A1) and forearm (lower response at A2).
c. For confirmation or quantification when sensory abnormalities appear than motor changes in peripheral neuropathies or before objective clinical signs.
d. For predicting whether a lesion is proximal or distal to the dorsal root (e.g., for differentiating brachial plexus from nerve root injury).
e. For determining disease of the dorsal root ganglion.
5. The EMG is usually performed along with NCS and yields complementary information. A needle electrode is inserted into the muscles of interest and the action potentials generated by groups of muscle fibers (the motor unit potentials or MUPs) are observed and recorded (Fig. 33.14). The muscle is tested at rest, with slight contraction, and with stronger contraction. Normally, the muscle is silent at rest. In active neuropathic processes, as well as in severe or inflammatory myopathies, spontaneous action potentials from single-muscle fibers (fibrillation potentials) may occur (Fig. 33.15). In certain neurogenic processes, especially motor neuron disease and diseases of the proximal root, spontaneous contractions of a single-motor unit (fasciculation potentials) may be observed. Characteristic changes in MUP parameters and recruitment may occur in neurogenic or myopathic processes. Inneuropathic conditions, the MUPs are often of increased amplitude, duration, and degree of polyphasia with reduced recruitment of fibers with increased effort, whereas in myopathic processes, the MUPs may be of decreased amplitude and duration with increased polyphasia and rapid (early) recruitment. Single-muscle fiber action potentials may be studied by a technically more difficult method, single-fiber electromyography (which is typically used to assess disease of the neuromuscular junction but can be abnormal in neuropathic and myopathic processes).
6. In general, EMG and NCS are used to study and diagnose patients with motor neuron disease (e.g., amyotrophic lateral sclerosis), processes affecting the plexi or nerve roots, entrapment neuropathies, peripheral polyneuropathies, diseases of the neuromuscular junction (e.g., MG), and diseases of the muscle. Because it involves electrical shocks and the insertion of needles into multiple muscles, the EMG/NCS study is uncomfortable. The study is safe as long as electrical safety techniques are applied; a bleeding tendency may limit the EMG study.
B. EMG/NCS abnormalities.
1. The EMG/NCS study is essential for evaluation and electrophysiologic diagnosis of motor neuron disease (e.g., amyotrophic lateral sclerosis). In general, NCS are normal except perhaps for some decrease in the CMAP amplitudes (because the disease is purely motor, sensory conduction studies are normal). Needle EMG shows diffuse evidence of neurogenic damage from anterior horn cell injury, including abnormal spontaneous activity (fibrillations and fasciculations), abnormal MUP parameters (large, wide, and polyphasic MUPs), and poor recruitment of MUPs with effort. Often the EMG study indicates active neurogenic damage even in muscles or limbs that appear to have little or no clinical involvement. The needle exam may also provide information about prognosis, and the EMG may assist in the diagnosis of other diseases of the anterior horn cells such as the postpolio syndrome and the spinal muscular atrophies.

FIGURE 33.14 Motor unit potentials recorded with needle insertion into the right biceps muscle during minimal muscle contraction.
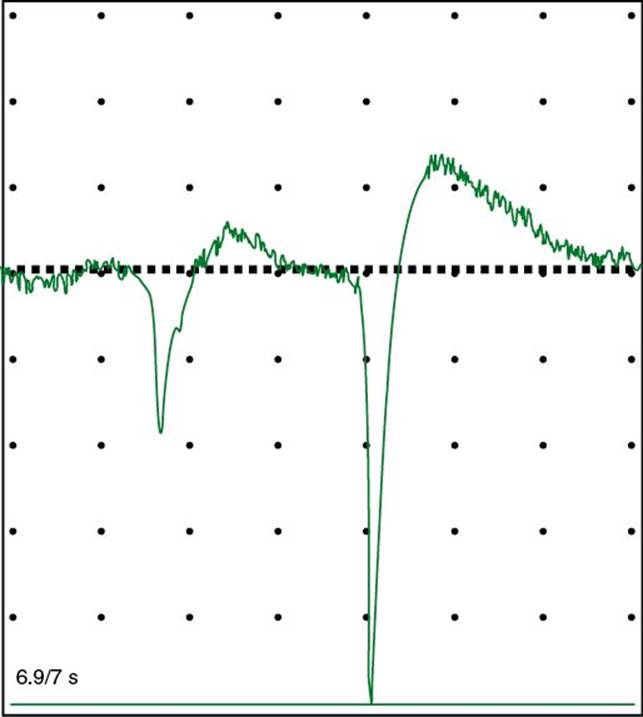
FIGURE 33.15 Positive sharp waves in the extensor hallucis longus muscle.
2. Radiculopathies comprise a constellation of symptoms and signs resulting from transient or permanent damage of the nerve at the anatomic level where the nerve exits the spinal canal in the spinal foramina. NCS are generally normal. The EMG shows evidence of neurogenic changes (e.g., fibrillations and MUP changes) in muscles innervated by a specific root, with other muscles innervated by uninvolved roots being spared. The pattern of neurogenic changes depends on the severity of the process, the duration of the disease, and the degree of neurogenic repair (reinnervation). The EMG study can be helpful in several ways, as follows.
a. It is useful for confirming disease of the nerve root. In studies of patients with surgically demonstrated cervical or lumbosacral radiculopathies, the EMG is abnormal only about 90% of the time. Thus, a normal study does not preclude the presence of a radiculopathy.
b. It provides further localization by determining which root or roots are affected.
c. It is useful in determining whether there is active denervation (indicated by the presence of fibrillation potentials).
d. It can determine the time elapsed since the onset of the radiculopathy (whether it is acute, subacute, chronic, or old).
e. It may give some information about the severity of the radiculopathy.
f. It may reveal other abnormalities that explain the patient’s symptoms.
g. It may help to determine if an abnormality on an MRI scan or myelogram has any physiologic significance.
3. Brachial and lumbosacral plexopathies and entrapment neuropathies (e.g., carpal tunnel syndrome, ulnar neuropathies at the elbow, and peroneal neuropathies at the fibular head) are localized and diagnosed with EMG/NCS.
4. Peripheral polyneuropathies are often investigated by EMG/NCS. The electrophysiologic characteristics of the neuropathic disorder serve as additional sources of information to help characterize the disease and allow a narrowing of the differential diagnostic possibilities. EMG/NCS allows evaluation of the amount of motor and sensory involvement, determines whether the lesion is primarily the result of damage to the myelin sheath or to the axon, indicates whether the lesion is focal or diffuse, determines whether the process is distal or proximal, and gives information concerning the severity and temporal profile of the process. Prolonged distal sensory and motor latencies, slowed conduction velocities, abnormalities of sensory responses and MUPs, and “neurogenic” EMG changes occur. When abnormal, the study confirms the presence of a neuropathy, but it should be noted that small fiber sensory neuropathies (i.e., those affecting only sensory nerve fibers conveying pain and temperature sensation) are often associated with normal studies. EMG/NCS can separate a generalized sensorimotor peripheral polyneuropathy from multiple mononeuropathies at sites of common compression (e.g., median and ulnar neuropathies at the wrist). Peripheral polyneuropathies may be divided by electrophysiologic patterns into the following categories:
a. Uniform demyelinating mixed sensorimotor neuropathies, including certain hereditary neuropathies (Fig. 33.16), metachromatic leukodystrophy, Krabbe’s disease, and Tangier disease.

FIGURE 33.16 Motor nerve conduction studies demonstrating uniform demyelination of the peripheral nerve in a patient with hereditary motor neuropathy (Charcot–Marie–Tooth’s disease type 1a, CMT). The compound muscle action potential (CMAP) amplitude is relatively preserved on proximal as compared with distal nerve stimulation. A diffusely and homogeneously demyelinating process, such as CMT type 1a will demonstrate relatively preserved CMAP configurations with prolonged latencies and slowed conduction velocities. A nonuniform, segmental demyelinating process (such as acute inflammatory demyelinating peripheral polyneuropathy, that is, Guillain–Barré’s syndrome) may demonstrate conduction block and variable slowing within the same nerve trunk, resulting in CMAPs with reduced amplitude and dispersed configuration at proximal sites of stimulation (bottom image).
b. Segmental demyelinating motor sensory polyneuropathies, including inflammatory neuropathies (e.g., Guillain–Barré’s syndrome, chronic inflammatory demyelinating polyneuropathy) and neuropathies associated with gammopathies, hypothyroidism, carcinoma or lymphoma, AIDS, Lyme’s disease, and certain toxins.
c. Axonal motor sensory polyneuropathies, including porphyria, certain hereditary neuropathies, lymphomatous neuropathies, and certain toxic neuropathies.
d. Axonal sensory neuronopathy (disease of the dorsal root ganglion) or neuropathies, including certain hereditary neuropathies, primary amyloidosis, Sjögren’s syndrome, paraneoplastic neuropathies, and neuropathies caused by drugs, vitamin B12 deficiency or antidisialosyl ganglioside antibodies (also referred to as “CANOMAD” syndrome).
e. Mixed axonal demyelinating sensorimotor polyneuropathies resulting from uremia or diabetes mellitus.
f. Axonal sensorimotor polyneuropathies, including neuropathies caused by nutritional deficiencies, monoclonal gammopathies, alcohol, sarcoidosis, connective tissue diseases, toxins, heavy metals, and drugs.
5. Diseases of the neuromuscular junction may be diagnosed by repetitive stimulation studies. Repetitive stimulation of motor nerves is used chiefly in the diagnosis of MG. In this disease, a characteristic progressive decline in the amplitude of the first few responses to stimulation is revealed at a stimulation rate of 2 per second (Fig. 33.12). The defect may be further characterized by the way it is altered after a brief contraction of the muscle. In some MG patients with normal repetitive stimulation studies, the diagnosis may be assisted by single-fiber EMG. Repetitive stimulation studies are also invaluable in the diagnosis of the Lambert–Eaton’s myasthenic syndrome. In the myasthenic syndrome, the initial action potential evoked in the rested muscle by a single-maximal nerve stimulation is greatly reduced in amplitude. A further reduction in the amplitude may occur with repetitive stimulation at low rates, but striking facilitation (enlargement of the MUPs of usually >100%) occurs during stimulation at higher rates. Unusual fatigability of the peripheral neuromuscular system may occasionally be demonstrated in other diseases, such as amyotrophic lateral sclerosis, but this abnormality is of little diagnostic value.

FIGURE 33.17 Top figure: Typical chronic neurogenic MUAP changes (reduced recruitment, increased amplitude, and duration). Bottom figure: Typical myopathic changes (early recruitment of small, polyphasic units). (Image courtesy of Dr. Matthew McCoyd.)
6. Electrodiagnostic studies show a wide variety of abnormalities in patients with myopathies. The NCS is essentially normal, except for occasional reductions in CMAP amplitudes. The EMG may reveal fibrillation potentials in severe myopathies or in inflammatory myopathies (e.g., polymyositis). “Myopathic” MUPs are of decreased amplitude and duration with increased polyphasia and rapid recruitment out of proportion to the degree of contraction effort (Fig. 33.17). The EMG studies usually are not sufficient to identify a specific disease, but the pattern of findings can be associated with groups of muscle disorders. Toxic and endocrine myopathies may produce little or no EMG abnormalities. An EMG/NCS examination can
a. distinguish neurogenic from myopathic disorders as causes of weakness,
b. provide clues to the etiology of a myopathy,
c. provide estimates of the severity and acuteness of the process,
d. assess the activity and course of the disease,
e. provide important information on the distribution of involvement to guide selection of a biopsy site (muscle biopsy must not be performed on a muscle that has been needled but in a corresponding muscle in the opposite extremity), and
f. detect abnormalities even if not clinically apparent.
III. EVOKED POTENTIALS
A. Introduction. Evoked potentials are electrical signals generated by the nervous system in response to sensory stimuli. The timing and location of these signals are determined by the sensory system involved and the sequence in which different neural structures are activated. Identical sensory stimuli are presented repeatedly while a computer averages the time-locked low-voltage responses from the brain or spinal cord and unrelated electrical noise and background EEG activity are averaged out.
B. Visual evoked potentials (VEPs).
1. Disorders of central visual pathways are tested by VEPs, which are the cortical responses to visual stimuli. Stroboscopic flashes of light or, more commonly, black-and-white checkerboard patterns evoke potentials over the occipital lobes that are detected by scalp electrodes. The major positive deflection at a latency of approximately 100 m (the P100 response) (Fig. 33.18) is most useful for clinical applications. Delays in this latency suggest damage to visual conducting pathways.
2. Unilateral prolongation of the P100 response implies an abnormality anterior to the optic chiasm (usually in the optic nerve) on that side. Bilateral P100 delay can be caused by bilateral lesions either anterior or posterior to the chiasm or by a lesion within the chiasm itself.
3. Uses of VEPs.
a. The VEPs may aid in the detection of a clinically “silent” lesion in a patient suspected of having multiple sclerosis (MS). It is a sufficiently sensitive indicator of optic nerve demyelination that it can reveal asymptomatic and clinically undetectable lesions. The VEPs reveal abnormalities in 70% to 80% of patients with definite MS who do not have histories of optic neuritis or visual symptoms. Abnormalities are not specific for MS and may be abnormal in a variety of other diseases, including certain ocular diseases, compressive lesions of the optic nerve, nutritional and toxic optic neuropathies, including pernicious anemia, and diffuse CNS diseases such as adrenoleukodystrophy and some spinocerebellar degenerations.
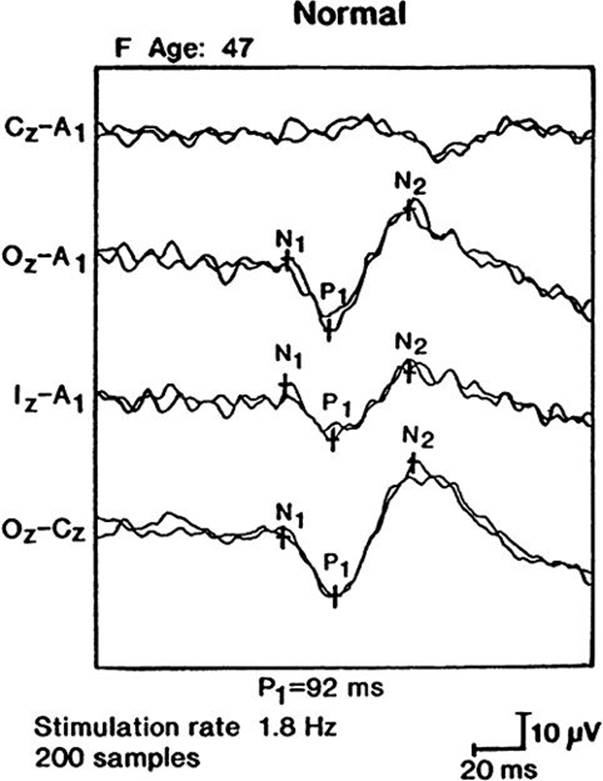
FIGURE 33.18 Full-field visual evoked potential (VEP) in a normal patient. (P1 = P100 response).
b. The VEP is helpful in distinguishing functional (e.g., psychogenic) visual impairment from true blindness or bilateral optic nerve disease. A normal VEP strongly favors functional illness. It should be mentioned, however, that rare patients have been described with blindness from severe bilateral destruction of the occipital lobes who had essentially normal VEP studies. Also, some patients with functional problems can voluntarily suppress the VEP response by such strategies as transcendental meditation, concentration beyond the plane of the checks, or ocular convergence.
c. The VEPs may be of some assistance in evaluating vision in pediatric patients—for example, in assessing high-risk infants or in the detection of amblyopia.
C. Brainstem auditory evoked potentials (BAEPs).
1. The BAEPs are a series of evoked potentials elicited by auditory clicks and generated by sequential activation of the brainstem auditory pathways. Although five waveforms (I through V) are usually resolved (Fig. 33.19), the most stable and important waveforms are I, III, and V. The I to III interpeak latency is a measure of auditory conduction of the more caudal segment of the brainstem (acoustic nerve to lower pons), whereas the III to V interpeak latency is a measure of conduction in the more rostral pontine and lower midbrain pathways. The I to V interpeak latency is a measure of the total conduction time within the brainstem auditory pathways.
2. Abnormality is measured by prolongation of interpeak latencies, especially asymmetric prolongations, as well as reduction in amplitude or absence of certain waveforms. A prolonged I to III interpeak latency indicates a lower pontine lesion, whereas a prolonged III to V interpeak latency indicates a lesion of the upper pons–lower midbrain levels.
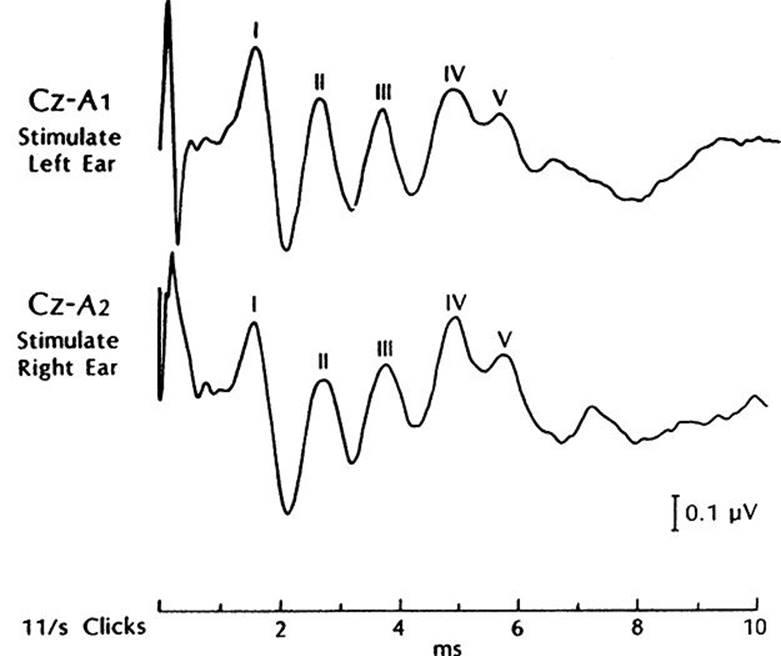
FIGURE 33.19 Brainstem auditory evoked potential study in a normal adult subject. See text for discussion of waveforms.
3. The BAEPs may be clinically helpful in the following circumstances.
a. The BAEPs, like VEPs, may be very sensitive to white matter disease and may help confirm or document a lesion within the brainstem, even if there are no brainstem signs or symptoms, when MS is suspected clinically and the patient has a lesion outside the brainstem. Approximately 50% of patients with definite MS exhibit abnormal BAEPs. However, VEPs and somatosensory evoked potentials (SEPs) (see III.D.) are more sensitive than BAEPs in detecting abnormalities in MS patients. Other demyelinating processes affecting the brainstem, such as central pontine myelinolysis, metachromatic leukodystrophy, and adrenoleukodystrophy, may also cause BAEP abnormalities.
b. A posterior fossa tumor or other mass within or outside of the brainstem can produce abnormal BAEPs by either direct involvement of the brainstem auditory pathways or secondary brainstem compression. The BAEPs are very sensitive screening procedures for acoustic neuromas and other cerebellopontine angle tumors. Monitoring of the BAEPs during such surgeries as acoustic neuroma resections, can provide valuable information to the surgeon and help to preserve hearing.
c. The BAEPs may assist in the determination of brain death. Preservation of wave I with loss of all subsequent response supports brainstem death in the comatose patient. The BAEP does not, however, provide any information about cortical function in the comatose patient.
d. The BAEPs may be used to assess hearing in young children and in patients otherwise unable to cooperate for standard audiometry. The BAEP testing can estimate hearing threshold and may distinguish conductive hearing loss from sensorineural hearing loss.
D. SEPs.
1. Following electrical stimulation of a peripheral nerve (usually the median or ulnar nerve at the wrist or the tibial nerve at the ankle), recording electrodes placed over the spine and scalp reveal a series of electrical potentials that correspond to sequential activation of neural structures along the dorsal column–lemniscal pathway. These SEPs are named according to their polarities and their times of occurrence in normal individuals. Because SEP latencies vary significantly with body height and limb length, absolute latency values are of limited use; interpeak latencies, which measure the time intervals between successive peaks in the sensory pathways, are incorporated in clinical studies.
2. SEPs yield information concerning PNS abnormalities but are not as effective as standard NCS in identifying and localizing peripheral disorders. Therefore, although SEPs have been used to study plexopathies and radiculopathies, their use is limited for these conditions.
3. Uses of SEPs.
a. SEPs can be used to confirm the presence of a clinically “silent” spinal cord lesion in a patient suspected of having MS. Median SEPs are abnormal in about two-thirds of patients with definite MS; lower-limb SEPs have somewhat greater abnormality rates, probably because of the greater length of white matter traversed. Prolonged central conduction times do not necessarily indicate demyelination because abnormal interpeak latencies may occur with hereditary spastic paraplegia, olivopontocerebellar atrophy, and subacute combined degeneration resulting from vitamin B12 deficiency.
b. Abnormally large (giant) cortical SEPs are characteristic of some relatively rare neurologic conditions, such as progressive myoclonic epilepsy, late infantile ceroid lipofuscinosis, and some other disorders associated with myoclonus.
c. SEPs may be helpful in demonstrating intact central sensory pathways in patients with functional (e.g., hysterical) sensory loss.
d. SEPs have been especially helpful in monitoring spinal cord function during surgery (e.g., surgery for correction of spinal scoliosis).
IV. OTHER ANCILLARY NEUROLOGIC STUDIES
A. Polysomnography.
1. Polysomnography consists of continuous monitoring of multiple biologic variables during nocturnal sleep. Eye movements (electrooculography), EEG activity, submental EMG, the ECG, and limb movements are routinely monitored. Respiration is monitored with intraesophageal pressure gauges, intercostal surface EMG, rib cage and abdominal strain gauges, oronasal thermistors or CO2 detectors, ear or finger oximetry, and other means of determining the presence of central, peripheral, or mixed apnea syndromes. A microphone attached to the throat may detect snoring. Each 30-second epoch of the polysomnogram is scored as awake, stage I to IV non–rapid eye movement (REM) sleep, or REM sleep.
2. Polysomnography is used to investigate two types of problems: sleep complaints (i.e., too much or too little sleep) and risk factors or specific syndromes induced by or linked to sleep or specific sleep states. These disorders include the following:
a. Sleep apnea syndromes, which may be obstructive, central, or mixed.
b. Narcolepsy.
c. Idiopathic CNS hypersomnia.
d. Periodic movements of sleep and sleep-related myoclonus.
e. REM behavioral disorder.
f. Disorders of the sleep–wake cycle.
g. Parasomnias such as sleepwalking, nightmares, night terrors, and head banging.
B. Multiple sleep latency test.
1. The multiple sleep latency test consists of five 20-minute attempts, once every 2 hours, to fall asleep throughout the day. The aim is to determine the sleep latency and whether or not REM sleep episodes are recorded during the nap. Patients should be withdrawn from sleep-related medications for 10 to 15 days. The study usually follows polysomnography because knowledge of the patient’s previous night’s sleep is required for appropriate interpretation. During the study, the EEG, submental EMG, ECG, and eye movements are monitored. Normal patients have mean sleep latencies >10 minutes and fewer than two sleep-onset REM periods during the study.
2. The multiple sleep latency test is designed to evaluate the following:
a. The complaint of excessive daytime somnolence by quantifying the time required to fall asleep. Pathologic sleepiness is manifested by a mean sleep latency of <5 minutes.
b. The possibility of narcolepsy by checking for abnormally short latencies to REM sleep. The occurrence of two or more sleep-onset REM periods during the study is strong evidence for narcolepsy, as long as sleep apnea and withdrawal from stimulants and alcohol have been ruled out.
V. LUMBAR PUNCTURE
A. Introduction. An LP should be considered only after a thorough evaluation of the patient and serious consideration of the potential value and hazards of the procedure.
B. Indications for CSF examination and LP are as follows.
1. The CSF examination is a key to the diagnosis and management of various CNS infections, including acute and chronic meningitis and encephalitis. In many patients with fever of unknown origin, even in the absence of meningeal signs, an early LP is commonly of value, especially because meningeal signs may be minimal or absent in very young or elderly patients. Meningeal infection should especially be sought in patients with fever and impaired sensorium or an immunocompromised state (e.g., AIDS patients). If a patient presents with unexplained acute confusion, stupor, or coma, even if afebrile, a CSF examination is necessary for the evaluation of meningoencephalitis. In most clinical settings, a CT scan of the brain or other neuroimaging should be performed before the LP to rule out a possible intracranial mass (e.g., hemorrhage or abscess), which would make LP a potentially lethal procedure. However, in an extremely ill patient in whom acute meningitis, such as meningococcal meningitis, is suspected, treatment should not be delayed while arranging appropriate ancillary testing.
2. In patients with suspected SAH, an urgent CT scan is indicated to evaluate for the presence of blood. However, in approximately 5% to 10% of patients with SAH, CT fails to reveal blood, and a spinal tap is indicated. If the diagnostic LP shows subarachnoid blood or xanthochromia, a cerebral angiogram is needed to determine the source of the hemorrhage.
3. In patients with unexplained dementia, CSF examination may be necessary to evaluate for CNS vasculitis, infection, or granulomatous disease. The CSF should always be examined in a patient with dementia and a positive fluorescent treponemal antibody absorption study. Also, patients with radiographic hydrocephalus may require a CSF study to exclude chronic meningitis as an etiology for the symptomatic hydrocephalus. In patients with suspected sporadic transmissible spongiform encephalopathy (sTSE), a positive radioimmunoassay of the CSF for “prion protein” (14-3-3 protein) is included in the World Health Organization criterion for “probable sporadic TSE.” However, there is some doubt as to the effectiveness of 14-3-3 testing because of false positives, lack of laboratory standardization, and clinical variability of sTSE. Other markers (such as neuron-specific enolase [NSE], tau protein, astrocytic [S100b] proteins and amyloid ß) have been investigated. Testing for a combination of factors, timing of testing, and serial studies may increase sensitivity. CSF biomarkers may also be used as an early indicator of Alzheimer’s dementia. The combination of a reduced concentration of CSF amyloid ß, increased total tau, and increased phospho-tau may be highly sensitive and specific for Alzheimer’s disease.
4. CSF examination is usually not warranted and may be dangerous in most patients with stroke. However, CSF analysis may assist in the etiologic diagnosis of unexplained stroke in young or middle-aged patients who lack atherosclerotic risk factors. Such etiologies as CNS vasculitis, meningovascular syphilis, and AIDS may be diagnosed. Sustained elevation in CSF biomarkers including NSE and S100b are associated with poor outcome following cardiac arrest.
5. CSF studies may aid in the diagnosis of MS, although there is no specific CSF marker for this disease. The CSF WBC counts are typically normal, as are protein and glucose. Elevated CSF IgG levels, with normal serum IgG levels, and the presence of oligoclonal bands in the CSF are considered characteristic for MS. However, CSF analysis for oligoclonal bands was not included in the Revised 2010 “McDonald Criteria” for the diagnosis of MS. The elevated CSF gamma globulin may also occur in neurosyphilis, viral meningoencephalitis, and SSPE.
6. The CSF analysis can be considered in the evaluation of patients admitted with an initial tonic–clonic seizure or status epilepticus, after appropriate neuroimaging, to exclude an active CNS infection or hemorrhage.
7. An LP is necessary to confirm the clinical suspicion of carcinomatous or leukemic meningitis. The typical CSF pattern is a pleocytosis with mildly elevated protein, low glucose, and a positive cytology for malignancy. Multiple LPs may be necessary as though cytology has a high specificity (>95%), it carries a low sensitivity (<50%). CSF flow cytometry may help increase the diagnostic yield.
8. CSF studies may aid in the diagnosis of certain inflammatory or demyelinating neuropathies, such as the Guillain–Barré’s syndrome or chronic idiopathic demyelinating polyradiculoneuropathy. The CSF protein level is often elevated without an abnormal cellular response. The WBC counts >10 per mm3 should raise suspicion for disorders such as Lyme’s disease, sarcoidosis, and HIV.
9. Although an LP is generally contraindicated in patients with papilledema, it is indicated to document increased intracranial pressure in a patient suspected of having idiopathic intracranial hypertension (pseudotumor cerebri)after neuroimaging studies have been proven to be normal. The spinal fluid is under increased pressure but is otherwise normal in this entity, except for occasional decreased CSF protein levels. Also, an LP is required to document low-CSF pressure in rare low-pressure syndromes in a patient whose headaches occur on standing and are relieved by lying down.
10. An LP can be used to deliver intrathecal antibiotics and chemotherapy in the treatment for certain CNS infections and meningeal malignancies, respectively. Also, it is required in certain diagnostic procedures such as myelography or cisternography.
C. Contraindications for LP.
1. An LP is contraindicated in any patient with increased intracranial pressure, except idiopathic intracranial hypertension because of the real danger of cerebral herniation and death.
2. An LP is contraindicated if there is suppuration in the skin or deeper tissues overlying the spinal canal because of the danger of inducing a purulent meningitis.
3. An LP is dangerous in the presence of anticoagulation therapy or a bleeding diathesis. Also, heparin should not be reinstituted for a minimum of 2 hours after an LP is performed. In general, an LP is hazardous if the platelet count is below 50,000, or especially if it is below 20,000. In such cases, platelet transfusions should be initiated if possible before the LP.
4. An LP should not be performed when a spinal mass is suspected unless the procedure is part of a myelogram with neurosurgical assistance readily available. A dramatic deterioration in spinal cord or cauda equina function can occur after LP.
D. Complications of LP.
1. Brain herniation and death may occur if an LP is performed on a patient with an increased intracranial pressure from a cerebral mass lesion. An LP is contraindicated in any patient suspected of having an intracranial mass.
2. Headache of low-pressure type may occur in up to 10% of patients after an LP (spinal headache). This type of headache occurs only on standing and is relieved by lying down. It is usually self-limiting but may require an epidural autologous blood patch for relief. Post-LP headache is most common in young women with lower-body mass. Higher-gauge (smaller diameter) needle, needle insertion parallel to dural fibers (bevel up with patient on side), and replacing the stylet prior to needle removal are negatively associated with post-LP headache. The occurrence of post-LP headache is unrelated to CSF opening pressure, cells, and protein; patient position during LP; duration of recumbency after LP; amount of CSF removed; or hydration following LP.
3. Diplopia, which usually results from unilateral or bilateral cranial nerve VI palsies, may occur rarely and is usually self-limiting.
4. Aseptic meningitis may occur rarely and is characterized by posterior neck pain, headache, and neck stiffness. This process is usually self-limiting.
5. Spinal epidural, subdural, and subarachnoid hematomas may occur, especially in patients on anticoagulants or with bleeding diatheses. Such hematomas are usually self-limiting and may cause local pain and meningeal irritation. However, epidural hematoma may rarely cause a flaccid and potentially irreversible paraplegia that requires an emergency surgical evacuation.
E. General comments on the evaluation of LP results.
1. The normal CSF pressure is 70 to 180 mm of water in the lateral recumbent position. Pressures should be >200 mm of water to be considered elevated. In an obese patient with possible idiopathic intracranial hypertension, the pressure should be >250 mm of water to establish this diagnosis.
2. The normal CSF glucose is approximately two-thirds of the serum glucose level, which must be drawn at the time of the LP. Hypoglycorrhachia (low-CSF glucose) with few white cells suggests a fungal infection, with many white cells a bacterial infection, and with abnormal (malignant) cells a meningeal malignancy.
3. The CSF protein may be increased (>100 mg per dl) in many CNS infectious, inflammatory, and malignant processes. Causes of elevated CSF protein with normal neuroimaging studies include myxedema, inflammatory demyelinating polyneuropathies, diabetic polyneuropathy, neurofibromas within the CSF pathways, resolving SAHs, gliomatosis cerebri, CNS vasculitis, and any process that causes spinal compression or obstruction of CSF flow.
4. Normally, the CSF can contain up to five lymphocytes or mononuclear cells per cubic centimeter. A pleocytosis causes CSF clouding when there are at least 200 cells per cubic centimeter. An increased WBC count occurs with subarachnoid infections, hemorrhages, chemical meningitis, or meningeal neoplasms. Also, it should be noted that a pleocytosis may occur for approximately 24 hours after a generalized seizure episode.
5. If initial spinal fluid appears bloody, one must attempt to determine whether the source of the blood is a traumatic tap or an SAH. If the initial tube of fluid is bloody and subsequent tubes are progressively clear, it is most likely that the tap was traumatic. One should then immediately centrifuge the fluid to see if the supernatant is clear, which suggests a traumatic tap. If the supernatant fluid is xanthochromic (yellow-tinged), it is likely that the blood has been present in the CSF for a few hours. Xanthochromia occurs approximately several hours after an SAH, reaches its greatest intensity at the end of 1 week, and clears in approximately 2 to 4 weeks. It can also be observed in jaundice and hypercarotenemia.
6. The polymerase chain reaction of the CSF has been found to have great utility in the diagnosis of several CNS infections. These include the following:
a. Herpes simplex virus I (HSE).
b. Herpes simplex II (HSE in neonates, recurrent meninigitis).
c. JC virus (progressive multifocal leukoencephalopathy or PML).
d. Cytomegalovirus (CMV ependymitis and polyradiculopathy associated with AIDS).
e. Borrellia burgdorferi (Lyme’s disease).
f. Tropheryma whippelii (CNS Whipple’s disease).
g. Toxoplasmosis (CNS toxoplasmosis in AIDS).
h. Myocobacterium tuberculosis (TB meningitis).
i. Other viruses causing encephalitis, including enteroviruses, varicella–zoster virus, Epstein–Barr’s virus, West Nile virus, and herpesvirus type 6.
VI. REFERRALS
All clinical neurophysiologic tests should be performed and interpreted by clinicians with expertise and special training in clinical neurophysiology. Laboratories performing these studies must follow the clinical and technical guidelines that have been published by neurophysiologic societies, the American Academy of Neurology, and other organizations. Strict adherence to these guidelines is mandatory to ensure patient safety and meaningful clinical interpretation. Neurology consultation is suggested whenever LP reveals abnormalities suggesting CNS infection, increased or decreased intracranial pressure, or SAH.
![]()
Recommended Readings
Aminoff MJ. Electrodiagnosis in Clinical Neurology. 2nd ed. New York, NY: Churchill Livingstone; 1986.
Brenner R, Hirsch L. Atlas of EEG in Critical Care. Chichester, UK: John Wiley & Sons; 2010.
Daube JR, ed. Clinical Neurophysiology. 2nd ed. New York, NY: Oxford University Press; 2002.
De Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer’s disease biomarker signature in cognitively normal elderly people. Arch Neurol. 2010;67(8):949–956.
Donofrio PD, Albers JW. Polyneuropathy: classification by nerve conduction studies and EMG. Muscle Nerve. 1990;13:889–903.
DuBois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6:734–746
DuBois B, Feldman HH, Jacova C, et al. Revising the definition of Alzheimer’s disease: a new lexicon. Lancet Neurol. 2010;9:1118–1127.
Dumitru D, Amato AA, Zwarts MJ. Electrodiagnostic Medicine. 2nd ed. Philadelphia, PA: Hanley and Belfus, Inc.; 2002.
Ebersole JS, Pedley TA, eds. Current Practice of Clinical Electroencephalography. 3rd ed. Philadelphia, PA: Lippincott Williams & Wilkins; 2003.
Kimura J. Electrodiagnosis in Diseases of Nerve and Muscle. 3rd ed. New York, NY: Oxford University Press; 2001.
Kimura J, Kohara N. Electrodiagnosis of neuromuscular disorders. In: Bradley WG, Daroff RB, Fenichel GM, Marsden CD, eds. Neurology in Clinical Practice. 3rd ed. Boston, MA: Butterworth-Heineman; 2000:497–519.
Kuntz KM, Kokmen E, Stevens JC, Miller P, Offord KP, Ho MM. Post-lumbar puncture headaches: experience in 501 consecutive procedures. Neurology. 1992;42:1884–1887.
Polman CH, Reingold SC, Banwell B, et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann Neurol. 2011;69(2):292–302.
Preston DC, Shapiro BE. Electromyography and Neuromuscular Disorders. 2nd ed. Philadelphia, PA: Elsevier; 2005.
Niedermeyer E, Lopes da Silva F. Electroencephalography—Basic Principles, Clinical Applications, and Related Fields. 4th ed. Philadelphia, PA: Lippincott Williams & Wilkins; 1999.
Whiteley W, Al-Shahi R, Warlow CP, et al. CSF opening pressure: reference interval and the effect of body mass index. Neurology. 2006;67:1690–1691.