33.1. Atherosclerotic cerebrovascular disease
33.1.1. Carotid artery
Atherosclerotic plaques begin to form in the carotid artery at 20 yrs of age. In the extracranial cerebral circulation, plaques typically start on the back wall of the common carotid artery (CCA). As they enlarge, they encroach on the lumen of the ICA. Calcified hard plaques may not change with time. The risk of CVA correlates with the degree of stenosis and with certain types of plaque morphology, and is also increased in hypercoagulable states and with increased blood viscosity.
PRESENTATION
Carotid artery lesions are considered symptomatic if there is one or more lateralizing ischemic episodes appropriate to the distribution of the lesion. A lesion is considered to be asymptomatic if the patient only has non-specific visual complaints, dizziness, or syncope not associated with TIA or stroke1. The majority (80%) of carotid atherothrombotic strokes occur without warning symptoms2.
Asymptomatic carotid stenosis: Usually discovered as a carotid bruit. Asymptomatic bruit: prevalence increases with age (2.3% in ages 45-54 yrs, 8.2% at ≥ 75)3. Accuracy of a bruit in predicting ICA stenosis: 50-83% (depending on cohort, criteria for stenosis…). Sensitivity is as low as 24%4.
Symptomatic carotid disease: May present as a TIA, RIND or CVA with any of the following findings (for ICA occlusion syndromes, see page 1027):
• retinal insufficiency or infarction (central retinal artery is a branch of the ophthalmic artery): ipsilateral monocular blindness
A. may be temporary: amaurosis fugax, AKA transient monocular blindness (TMB). Four types:
Type I: embolic. Described “like a black curtain coming down” in one eye. Complete loss of vision, usually lasts 1-2 minutes
Type II: flow related. Retinal hypoperfusion → desaturation of color, usually described as a graying of vision
Type III: vasospastic. May occur with migraines
Type IV: miscellaneous. May occur with anticardiolipin antibodies
B. blindness may be permanent
• middle cerebral artery symptoms:
A. contralateral motor or sensory TIA (arm and face worse than leg) with hyperreflexia and upgoing toe
B. language deficits if dominant hemisphere involved
EVALUATION OF THE EXTENT OF CAROTID DISEASE
Symptomatic patients will usually be assessed as part of a stroke/TIA protocol.
Check CBC with platelet count, fibrinogen, PT/PTT (to R/O hypercoagulable state).
Funduscopic exam may show Hollenhorst plaques (cholesterol crystal emboli).
Overview
Classification of patients based on the hemodynamics and also the embolic propensity of carotid lesions has thus far been too complex to be utilized in large studies. The tests described below place a great deal of emphasis on the greatest degree of stenosis which is probably an oversimplification. Plaque composition and morphology is probably important.
Plaque morphology
“Vulnerable” plaques are atherosclerotic plaques likely to cause thrombotic complications, or those that tend to progress rapidly. Criteria for vulnerable plaques include: intimal thickening, plaque fissure, lipid/necrotic core with thin fibrous cap, calcification, thrombus, intraplaque hemorrhage, and outward remodeling. Some of these features can be identified with high-resolution MRI5-8.
ASSESSMENT OPTIONS
For recommendations for which tests to use, see page 1146.
Angiography
The “gold standard” test is a catheter angiogram. It cannot be justified as a screening test because it is invasive, and too costly and risky (recent data show < 1% risk of transient or permanent deficitA9-11 in good hands). Also, unlike duplex doppler and MRA, it does not provide any information about the thickness of the plaque. Different definitions of the degree of stenosis are employed, Table 33-1 compares the definitions used by the NASCET study12to that of the ECST13. For both, N is the linear diameter of the carotid artery at the site of greatest narrowing. The studies differ in the denominator, NASCET uses D(the diameter of the normal artery distal to the carotid bulb - taken at the first point at which the arterial walls become parallel), whereas the ECST uses B (the estimated carotid bulb diameter).
A. risk is 2-3 times higher in symptomatic patients than asymptomatic
For example, using the NASCET definition, the degree of stenosis is shown in Eq 33-1.
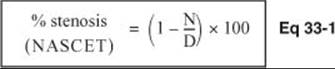

The relationship between the degree of narrowing based on the NASCET definition vs. that of the ECST has also been estimated by equation14 as shown in Eq 33-2.

Angiography also affords the opportunity to perform endovascular intervention if indicated (however, aside from dissections, surgical endarterectomy has been shown to be superior to endovascular techniques).
Duplex doppler ultrasound
B-mode image evaluates the artery in cross-sectional plane, and spectrum analysis shows blood flow. Performs poorly with a “string sign”. Cannot scan above the angle of the mandible. Lower frequencies give greater depth of penetration, but signal definition is sacrificed (used in transcranial doppler). Sensitivity: 88%, specificity: 76%15.
Magnetic resonance angiography (MRA)
May obviate the need for angiography in some cases of carotid stenosis, specifically in symptomatic patients with a focal “gap” of signal intensity loss with distal reappearance of signal16, 17. Sometimes overestimates the degree of stenosis18. Sensitivity: 91%, specificity: 88% for extracranial carotid disease19. 2D TOF-MRA is adequate (contrast-MRA shows more, but is not necessary for surgical lesions20).
Can be performed at the time as MRI with stroke protocol in TIA/stroke patients, and also detects thrombus or dissection. As with Doppler, has difficulties distinguishing very severe stenosis from occlusion. Less operator dependent than Doppler, but is more expensive and time-consuming. MRA is more difficult to perform if the patient is critically ill, unable to lie supine, or has claustrophobia, a pacemaker or ferromagnetic implants. High-resolution MRI may also detect vulnerable plaques (see page 1145).
Computed tomography angiography (CTA)
CTA involves ionizing radiation (x-rays) and IV iodinated contrast, limiting its use in patients with dye allergies and renal dysfunction. Results are comparable to MRA and Doppler. CTA can be performed within a few seconds and yields high-resolution images of all vessels from the aortic arch through the intracranial/extracranial vessels as well as the surrounding soft tissues. In a metaanalysis, sensitivity and specificity for detection of a 70% to 99% stenosis were 85% and 93%, respectively21. CTA is still evolving and may help detect vulnerable plaques (see page 1145). Another potential advantage: ability to obtain CT-perfusion studies at the same time (see page 128).
CHOICE OF IMAGING TEST/MANAGEMENT DECISIONS
Despite a great deal of research on the subject, there are no data to support a particular testing algorithm22. Doppler, CTA, or MRA are acceptable initial screening tests. In patients with an abnormal screening test, a common strategy is to obtain a second confirmatory noninvasive test to evaluate the carotid bifurcation before intervention. The combination of carotid ultrasound and MRA has proved cost effective with good interobserver reliability23. If 2 noninvasive tests are discordant, catheter angiography should be considered before intervention.
TREATMENT
Treatment alternatives are primarily between the following.
1. “best medical management”: see below
2. carotid endarterectomy: see page 1150
3. endovascular techniques: combined angioplasty and stenting (± distal embolus protection)
MEDICAL TREATMENT
What constitutes “best medical management” has not been precisely determined, and recommendations are constantly changing. Some or all of the following are utilized:
• antiplatelet therapy:
![]() usually aspirin (ASA) (see below)
usually aspirin (ASA) (see below)
![]() clopidogrel, either alone or in combination with ASA (see below)
clopidogrel, either alone or in combination with ASA (see below)
![]() combination of extended release dipyridamole and ASA (Aggrenox®) (no benefit from dipyridamole (Persantine®) alone)
combination of extended release dipyridamole and ASA (Aggrenox®) (no benefit from dipyridamole (Persantine®) alone)
• antihypertensive therapy as appropriate
• good control of diabetes if present
• patients with asymptomatic A-fib should be treated with anticoagulation (see Cardiogenic brain embolism, page 1022)
• antilipid therapy if needed
• intervention to help patients to quit smoking
Antiplatelet therapy
aspirinDRUG INFO
Irreversibly inhibits cyclooxygenase preventing synthesis of vascular prostacyclin (a vasodilator and platelet inhibitor) and platelet thromboxane A2 (a vasoconstrictor and platelet activator). Platelets, lacking cellular organelles, cannot resynthesize cyclooxygenase whereas the vascular tissues do so rapidly24. NB: < 1000 mg ASA per day probably does not help with high grade stenosis where there is perfusion failure or flow failure. Some (but not all) studies show less effectiveness in women25, and no large study has shown that ASA prevents a second stroke in patients that have already had one.
Rx: For angina, a bolus dose of 160-325 mg PO is followed by maintenance doses of 80-160 mg/d (lower doses appear to be as effective as higher doses)26. Optimal dose for cerebrovascular ischemia continues to be debated. 325 mg PO qd reduces risk of stroke following TIA by 25-30%. Daily doses of 81 or 325 mg when compared to higher doses were associated with a lower rate of CVA, MI and death (6.2% vs. 8.4%) following carotid endarterectomy27.
aspirin/ER-dipyridamole (Aggrenox®)DRUG INFO
Combination of extended release dipyridamole and ASA (Aggrenox) is more effective than ASA alone for prevention of TIA, stroke, and myocardial infarction28-30. Aggrenox was not superior to clopidogrel, with increased hemorrhage with Aggrenox31. SIDE EFFECTS: H/A with initial therapy.
Rx: 1 capsule PO BID. SUPPLIED: fixed dose capsules of aspirin 25 mg/extended-release with dipyridamole 200 mg.
clopidogrel (Plavix®)DRUG INFO
A thienopyridine. Incidence of severe neutropenia (0.04%) is close to that of ASA (≈ 0.02%)32. Interferes with platelet membrane function by inhibiting ADP-induced platelet fibrinogen binding and release of platelet granule contents, as well as subsequent platelet-platelet interactions. Produces a time and dose dependent irreversible inhibition of platelet aggregation and prolongation of bleeding time. May replace ASA if intolerance or resistance. Used in combination with ASA for some endovascular procedures. Although clopidogrel plus aspirin is recommended over aspirin for acute coronary syndromes, the MATCH33 results do not suggest a similar benefit for stroke and TIA. Combination therapy significantly increased risk of hemorrhage33.
PHARMACOKINETICS: Dosed once daily. Requires several days to reach maximal effect (![]() a loading dose may used, e.g. after an acute event such as an MI, or before stenting). Takes ≈ 5 days off the drug for platelet inhibition to reverse.
a loading dose may used, e.g. after an acute event such as an MI, or before stenting). Takes ≈ 5 days off the drug for platelet inhibition to reverse.
Rx: 75 mg PO qd. Loading dose: 225 mg (3 pills) the first day of therapy. SUPPLIED: 75 mg film-coated tablet.
Choice of antiplatelet agents
Individualization is recommend for antiplatelet agents for secondary stroke prevention. ASA is effective, and its low cost may help compliance. A small reduction of vascular events with Aggrenox may justify its expense from a broader healthcare perspective. Clopidogrel is appropriate for those intolerant or resistant to ASA. Clopidogrel plus ASA may be indicated in patients with recent cardiac ischemia or vascular stenting34.
33.1.1.1. Asymptomatic carotid artery stenosis
![]() Key concepts:
Key concepts:
• natural history: reveals low stroke rate (2%/yr) half of which are not disabling
• large randomized trials have revealed moderate surgical benefit versus medical management for: asymptomatic stenosis >60%
• treatment selection criteria depend on patient’s age, gender and comorbidities (and therefore life expectancy), and on perioperative complication rate
PRACTICE GUIDELINE 33-1 ASYMPTOMATIC CAROTID STENOSIS*
For patients with a surgical risk < 3% and life expectancy ≥ 5 yrs
Level I36: carotid endarterectomy (CEA) is beneficial for: asymptomatic stenosis ≥ 60%
Level II36: unilateral CEA + simultaneous CABG for: stenosis > 60%
Level III36: unilateral CEA for: stenosis > 50% with large, deep, complex or cavitated ulcer†
For patients with a surgical risk 3-5%
Level II36: ipsilateral CEA for: stenosis > 75% with contralateral ICA stenosis 75-100%
Level III36:
• ipsilateral CEA for: stenosis > 75% irrespective of contralateral stenosis
• unilateral CEA + CABG for: bilateral asymptomatic carotid stenosis > 70% + CABG required
• ipsilateral CEA for: unilateral carotid stenosis > 70% + CABG required
For patients with a surgical risk 5-10%
Level III36:
• unilateral CEA + CABG for: bilateral asymptomatic stenosis > 70% + CABG required
• ipsilateral CEA + CABG for: unilateral carotid stenosis > 70% + CABG required
• ✖ Inappropriate indications36:
![]() stenosis ≤ 50% irrespective of contralateral carotid artery status
stenosis ≤ 50% irrespective of contralateral carotid artery status
![]() ipsilateral CEA for: stenosis > 75% irrespective of contralateral carotid
ipsilateral CEA for: stenosis > 75% irrespective of contralateral carotid
* NB: slight modifications to this parameter may need to be made in light of further data from ACST35. As of this writing, these guidelines have not been updated
† at the time that this guideline was written, more emphasis was placed on these plaque factors
NATURAL HISTORY
Prevalence of carotid stenosis >50% in men and women >65 years of age is 5-10%, with 1% having stenosis >80%37-39.
Natural history studies reflect an annual stroke risk of 1-3.4% with asymptomatic carotid artery stenosis of 50-99% at 2-3 years40-45. A cohort study found similar cumulative rates of ipsilateral stroke over 10 years (9.3%, or 0.9%/year) and 15 years (16.6%, or 1.1%/year)46.
Attempts to identify subgroups of patients with asymptomatic carotid stenosis at elevated stroke risk suggest that the rate of unheralded stroke ipsilateral to a hemodynamically significant extracranial carotid artery stenosis is 1-2% annually, with some data suggesting that the stroke rate may be higher with progressing stenosis or with more severe stenosis. Asymptomatic carotid stenosis is an important marker of concomitant ischemic cardiac disease40-42,45, 46. In the REACH Study47, patients with asymptomatic carotid stenosis (n = 3164) had statistically significantly higher age- and sex-adjusted 1-year rates of transient ischemic attack, non-fatal stroke, fatal stroke, and cardiovascular death compared with patients without asymptomatic carotid stenosis (n = 30 329).
SURGERY VS. MEDICAL MANAGEMENT: THE STUDIES
ACST35
|
Σ |
The largest multicenter randomized trial to date35 revealed a moderate benefit for immediate CEA vs. medical management in patients age < 75 with asymptomatic stenosis ≥ 60%. |
Details:3,120 patients with ≥ 60% stenosis by duplex ultrasound were randomized to immediate CEA (50% had CEA within 1 month, 88% within 1 year) or medical therapy at the discretion of the treating physician. Mean follow-up: 3.4 years. Exclusion criteria included: poor surgical risk, prior ipsilateral CEA, and probable cardiac emboli. Surgeons were required to have a perioperative morbidity and mortality rate of < 6%.
Net five-year risk for all stroke or perioperative stroke or death: 6.4% in the CEA group, vs. 11.8% in the medical group (p<0.0001). Fatal or disabling stroke: 3.5 vs. 6.1%. Fatal stroke alone: 2.1 vs. 4.2%. Although men and women benefited, men benefited more. CEA did not demonstrate a statistically significant benefit for patients over the age of 75. Statistical benefit was not seen in the immediate CEA group until nearly two years after surgery, despite a relatively low perioperative morbidity and mortality rate of 3.1%, (in contrast to patients with symptomatic stenosis (NASCET48) where benefit was seen much earlier).
ACAS49
|
Σ |
Large trial that randomized patients in good health with asymptomatic stenosis* ≥ 60% to CEA plus aspirin, or aspirin alone49 found a reduced 5-year risk of ipsilateral stroke if CEA was performed with <3% perioperative morbidity and mortality and is added to aggressive management of modifiable risk factors. |
* calculated in the same manner as the NASCET study
Details: CEA reduced 5-year stroke risk 66% in males, 17% in females (not statistically significant), and 53% overall. CEA did not significantly protect against major CVA or death (P = 0.16) (half of the CVAs were not disabling), and was somewhat protective against any stroke or death (P = 0.08). Excluded patients (age > 79 yrs, unstable CAD, uncontrolled HTN) may have been higher risk. Surgeons were carefully selected and the surgical morbidity (1.5%) and mortality (0.1%) was very low. Surprisingly, ≈ half of the total morbidity (1.2%) was related to angiography. The implication is that for a generally healthy white male with ACAS > 60%, management with CEA (when performed by a surgeon with a low complication rate, as described) reduces his annual risk of all strokes from 0.5% to 0.17% (the reduction of risk for severe stroke is less). The benefit from CEA is realized within less than one year after the CEA. This is in contrast to the ACST trial (see above) and is most likely due to the lower perioperative event rate. The risk from mortality from other causes (including MI) is ≈ 3.9% per year. Combined CVA and death rates in community hospitals50 while improved over the last 20 yrs, remains higher at ≈ 6.3% than at centers used in this study.
Veteran’s Administration Cooperative Study (VACS)48
CEA reduces ipsilateral neurologic events, but did not reduce the rate of ipsilateral CVAs nor death (most deaths were secondary to MI). This trial did not include women and was not powered to detect differences in outcome subgroups.
CASANOVA Study51
No difference in outcome between CEA vs. aspirin (new CVA or death), but an un-usual protocol lessened it’s statistical validity52.
Mayo Clinic Asymptomatic Carotid Endarterectomy (MACE) Study53
There were no major strokes or deaths in either the medical or the endarterectomy group. Surgically treated patients were not given aspirin, and 26% had an MI compared to 9% in the aspirin-treated medical arm, reflecting the high incidence of concomitant CAD in patients with an asymptomatic carotid artery stenosis.
CURRENT RECOMMENDATIONS FOR CEA IN ASYMPTOMATIC CAROTID STENOSIS
The 2006 Primary Prevention of Stroke Guidelines recommend prophylactic carotid endarterectomy for highly selected patients with high-grade asymptomatic carotid stenosis to be performed by surgeons with <3% morbidity/mortality rates (Class I, Level of Evidence A). Patient selection should be guided by an assessment of comorbid conditions and life expectancy, and patient individual factors. For high-risk patients with asymptomatic carotid stenosis, carotid angioplasty/stenting is preferred as a reasonable alternative to CEA (Class IIb, Level of Evidence B). However, due to the reported periprocedural and overall 1-year event rates, there is uncertainty as to whether this group of patients should have either procedure at all54.
The guidelines for the European Society of Vascular surgery recommend CEA for asymptomatic men < 75 years old with 70-99% stenosis if the perioperative stroke/death risk is < 3% (Grade A). Given the benefit from CEA in asymptomatic women is significantly less than in men, CEA should be considered only in younger, fit women (Grade A)55.
RECOMMENDATIONS FOR SCREENING FOR CAROTID STENOSIS
• the U.S. Preventive Services Task Force (USPSTF) currently recommends against screening for carotid stenosis in the adult general population (grade D recommendationA)56
• the AHA Primary Prevention of Stroke Guidelines does not recommend screening for asymptomatic carotid stenosis54
• the American Society of Neuroimaging advised that screening should be considered only for age ≥ 65 years with 3 or more cardiovascular risk factors 57
• the Society for Vascular Surgery recommends ultrasonography screening for age ≥ 55 years with cardiovascular risk factors, such as a HTN, diabetes, smoking, hypercholesterolemia, or known cardiovascular disease 58
A. moderate or high certainty that the service has no net benefit or that the harm outweighs the benefit
33.1.1.2. Carotid endarterectomy
INDICATIONS
Table 33-2 shows the status of current studies for the surgical treatment of carotid stenosis (NB: some of the results may be contradictory).
The North American Symptomatic Carotid Endarterectomy Trial12 (NASCET) found that for patients with a hemispheric or retinal TIA or a mild (non-disabling) CVA within 120 days and ipsilateral high-grade stenosis (> 70%), that carotid endarterectomy (CEA) reduced the rate of fatal and non-fatal CVAs (by 17% at 18 months) and death from any cause (by 7% at 18 months) when compared to best medical managementA. Results were twice as good for patients with stenosis from 90-99% than for those with 70-79%. NB: for differences in techniques for measuring stenosis between NASCET and ECST, see Table 33-1, page 1145.
A. when surgery was performed with perioperative risk of stroke or death of 5.8%
For asymptomatic patients, see page 1147.

Unresolved controversies
Include:
1. progressive CVA (“stroke in evolution”): see Emergency carotid endarterectomy, page 1156
2. abrupt occlusion: see Emergency carotid endarterectomy, page 1156
3. tandem lesions (e.g. carotid siphon and bifurcation stenosis): although this topic remains controversial, CEA in patients with tandem lesions has not been associated with increased postoperative stroke rates62, 63. Recent case series also report success with endovascular treatment
4. progressive retinal ischemia
TIMING WITH RESPECT TO ACUTE CVA
For patients with small fixed deficits or small infarcts on CT or MRI, the risk of early CEA is not increased62, 64. In the pooled analysis of the three symptomatic CEA studies, patients randomized in the trials within 2 weeks of the last symptomatic event had greater benefit from CEA65. Data from Sundt (see Pre-op risk factors for CEA below) indicates that CVA is a risk factor for a complication only if it occurred ≤ 7 days pre-op.
Since the introduction of tPA for the treatment of acute ischemic stroke, there have been reports on the successful treatment of residual critical ICA stenosis following tPA recanalization as early as 24 hours after administration of tPA in patients with small fixed deficit or small ischemic areas on MRI65, 66.
PRE-OP RISK FACTORS FOR CEA
|
Σ |
The characteristics of patients who are high risk for complications from CEA has not been well defined, despite the perception that this group exists. |
Identifying patients at high risk for complications after CEA has proven challenging. Typically, the exclusion criteria from studies is cited, but in most cases these are simply patients that were not included in the study because it was the investigators perception these patients might be “high risk”. Therefore these risk factors are not validated. They are included here for completeness.
NASCET and ACAS: age> 80 years, prior ipsilateral CEA, prior contralateral CEA within 4 months, prior neck XRT, tandem lesion larger than target lesion, other conditions that could cause symptoms (atrial fibrillation, prior CVA with persistent major deficit, valvular heart disease), major organ failure, uncontrolled hypertension or diabetes mellitus, and significant coronary artery disease67, 68).
The SAPPHIRE Trial (Stenting and Angioplasty with Protection in Patients at High-Risk for Endarterectomy): patients with clinically significant cardiac disease (CHF, abnormal stress test, or need for open-heart surgery), severe pulmonary disease, contralateral carotid occlusion, contralateral laryngeal-nerve palsy, previous radical neck surgery or neck XRT, recurrent stenosis after endarterectomy, and age >80 years69. The ARCHeR Trial (ACCULINK for Revascularization of Carotids in High-Risk patients) also included patients with tracheostomy, spinal immobility, and dialysis-dependant renal failure70.
33.1.1.3. Carotid angioplasty/stenting
|
Σ |
There are no well-designed studies that convincingly show superiority of angioplasty/stenting over CEA in average risk symptomatic patients, and the recommendation in these patients is to continue with the time-tested technique of CEA. |
There is a paucity of randomized control trials69-74 comparing carotid angioplasty/stenting with CEA, and many nonrandomized registries70, 75-83.
However, data from multicenter randomized trials showing that carotid angioplasty/stenting is as safe over the short term or as efficacious over the long term as CEA in average-risk symptomatic patients are lacking. Published trials are heterogeneous (clinically and methodologically), too small to provide robust and convincing data, and limited in long-term follow-up. Only the SAPPHIRE study69 comparing CEA with stenting (using a distal embolic protection device) for moderate to severe carotid stenosis with comorbidities that might increase the risk of CEA (high-risk patients), found that angioplasty/stenting was not inferior (risk within 3%, P = 0.004) to CEA (based on a composite primary end point of stroke, death, or MI within 30 days, or death from neurologic causes or ipsilateral stroke between 31 days and 1 year)69. However, the study methodology has been criticized84-86.
A 2007 Cochrane review concluded that available data on carotid angioplasty/stenting are difficult to interpret and does not support a change in clinical practice away from recommending CEA as the treatment of choice for suitable carotid artery stenosis87.
Indications for angioplasty/stenting
Carotid stenting performed with adequate procedural quality levels, should be considered instead of CEA in the presence of88:
1. severe vascular and cardiac comorbidities:
A. congestive heart failure (New York Heart Association class III/IV) and/or known severe left ventricular dysfunction
B. open heart surgery needed within 6 weeks
C. recent myocardial infarction (<24 hours and >4 weeks)
D. unstable angina (Canadian Cardiovascular Society class III/IV)
E. contralateral carotid occlusion
2. specific conditions:
A. contralateral laryngeal nerve palsy
B. radiation therapy to the neck
C. previous CEA with recurrent restenosis
D. high cervical internal carotid/below the clavicle common carotid lesions
E. severe tandem lesions
F. age >80 years
G. severe pulmonary disease
The 2009 European Society for Vascular Surgery (ESVS) Guidelines state that carotid angioplasty/stenting is indicated in cases of: contralateral laryngeal nerve palsy, previous radical neck dissection or cervical XRT, prior CEA (restenosis), high bifurcation or intracranial extension of a carotid lesion, provided that the peri-interventional stroke or death rate is not higher than that accepted for CEA (Class C recommendation)55.
AHA Guidelines state that angioplasty/stenting might be a reasonable alternative to CEA in asymptomatic high risk patients. However, they stress that it remains uncertain whether this group of patients should have either procedure54.
33.1.1.4. Carotid endarterectomy - surgical considerations
PERIOPERATIVE MANAGEMENT
PRE-OP MANAGEMENT (CAROTID ENDARTERECTOMY)
1. ASA 325 mg TID for at least 2 days, preferably 5 days pre-op89 (NB: patients should be kept on their ASA for surgery, and if not on ASA they should be started, in order to reduce risks of MI and TIA90)
POST-OP MANAGEMENT (CAROTID ENDARTERECTOMY)
1. patient monitored in ICU with A-line
2. keep patient well hydrated (run IVF ≥ 100 cc/hr for most adults)
3. SBP ideally 110 - 150 mm Hg (higher pressures are permitted in patients with chronic severe HTN)
A. BP frequently labile in 1st 24 hrs post-op, may be due to “new” pressure in carotid bulb; to prevent rebound hyper- or hypo-tension, avoid long acting agents
B. hypotension
1. check EKG - R/O cardiogenic shock
2. if mild, start with fluids (crystalloid or colloid)
3. phenylephrine (Neo-Synephrine®) for resistant hypotension
C. hypertension: nicardipine (Cardene® - see page 19) is the agent of choice. Avoid rebound hypotension
4. avoid ASA and dipyridamole for 24-48 hrs post-op (causes oozing); may start these 24-72 hrs post op (note: ASA 325 mg + dipyridamole 75 mg TID have been shown not to reduce the rate of restenosis after endarterectomy91)
5. optional: reverse half of heparin with protamine 10 minutes after closing arteriotomy
POST-OP CHECK (CAROTID ENDARTERECTOMY)
In addition to routine, the following should be checked:
![]() 1. change in neurologic status due to cerebral dysfunction, including:
1. change in neurologic status due to cerebral dysfunction, including:
A. pronator drift (R/O new hemiparesis)
B. signs of dysphasia (especially for left sided surgery)
C. mimetic muscle symmetry (assesses facial nerve function)
![]() 2. pupil diameter and reaction (R/O CVA, Horner’s syndrome)
2. pupil diameter and reaction (R/O CVA, Horner’s syndrome)
![]() 3. severe H/A (especially unilateral)> may indicate hyperperfusion syndrome
3. severe H/A (especially unilateral)> may indicate hyperperfusion syndrome
![]() 4. STA pulses (R/O external carotid occlusion)
4. STA pulses (R/O external carotid occlusion)
![]() 5. tongue deviation (R/O hypoglossal nerve injury)
5. tongue deviation (R/O hypoglossal nerve injury)
![]() 6. symmetry of lips (R/O weakness of lower lip depressors due to retraction of marginal mandibular branch of facial nerve against mandible, usually resolves in 6-12 wks, must differentiate from central VII palsy due to CVA)
6. symmetry of lips (R/O weakness of lower lip depressors due to retraction of marginal mandibular branch of facial nerve against mandible, usually resolves in 6-12 wks, must differentiate from central VII palsy due to CVA)
![]() 7. check for hoarseness (R/O recurrent laryngeal nerve injury)
7. check for hoarseness (R/O recurrent laryngeal nerve injury)
![]() 8. assess for hematoma in operative site: note any tracheal deviation, dysphagia
8. assess for hematoma in operative site: note any tracheal deviation, dysphagia
POST-OP COMPLICATIONS (CAROTID ENDARTERECTOMY)
To justify CEA, the absolute upper limit of (significant) complication rate should be ≤ 3%.
1. overall in-hospital mortality: 1%92
2. disruption of arteriotomy closure: rare, but emergent (see below)
A. evidenced by:
1. swelling of neck: rupture may produce a pseudoaneurysm
2. tracheal deviation (visible, palpable, or on CXR)
3. symptoms: dysphagia, air hunger or worsening hoarseness, difficulty swallowing
B. dangers:
1. asphyxiation: most immediate danger
2. stroke
3. exsanguination (unlikely, unless skin closure is also disrupted)
C. late (often delayed weeks to months): false aneurysm93. Risk = 0.33%. Presents as neck mass. Risk is increased with wound infection and possibly with patch graft as compared to endarterectomy alone93-95
3. stroke (cerebral infarction) intra-op or post-op rate96: 5%
A. embolic (the most common cause of minor post-op neurologic deficit): source may be denuded media of endarterectomy
B. intracerebral hemorrhagic (ICH) (breakthrough bleeding): occurs in < 0.6%97. Related to cerebral hyperperfusion in most98, 99 (see below). Usually occurs within first 2 weeks, often in basal ganglion 3-4 days post-op with hypertensive episode. Patients at greatest risk are those with severe stenosis and limited hemispheric collateral flow
C. post-op ICA occlusion
1. most common cause of major post-op CVAs, but may be asymptomatic
2. risk is reduced by attention to technical details at surgery100 (p 249)
3. some may be due to hypercoagulable state induced by heparin (predictable in patients whose platelet count drops while on heparin. No known therapy for this condition100 (p 249-50))
4. the endarterectomized surface is highly thrombogenic for 4 hrs following endarterectomy (Sundt recommends not reversing heparin)
5. in Sundt’s series using patch graft100 (p 229): 0.8% incidence, associated with major CVA in 33% and minor CVA in 20%
6. occlusion rate with primary closure: 4% in Sundt’s experience, 2-5% in literature100 (p 249)
4. post-op TIAs: most due to ICA occlusion. Some may be due to microemboli. Hyperperfusion syndrome produces a 1% incidence of post-op TIAs100 (p 229)
5. seizures101: usually focal in onset with possible generalization, most occur late (post-op day 5-13) with an incidence of ≈ 0.4%97 to 1%102. May be due to cerebral hyperperfusion97, emboli103, and/or intracerebral hemorrhage. Usually difficult to control initially, lorazepam and phenytoin are recommended (see page 405)
6. late restenosis: identifiable restenosis occurs in ≈ 25% by 1 yr, and half of these reduce luminal diameters by > 50%104. Restenosis within 2 yrs is usually due to fibrous hyperplasia, after 2 yrs it is typically due to atherosclerosis105
7. cerebral hyperperfusion syndrome (AKA normal pressure hyperperfusion breakthrough): classically thought to result from return of blood flow to an area that has lost autoregulation due to chronic cerebral ischemia typically from high-grade stenosis. Controversial99. Usually presents as ipsilateral vascular H/A or eye pain that subsides within several days106 or with seizures (± PLEDs on EEG, more common with Halothane®, due to petechial hemorrhages97). May cause ICH107. Most complications occur several days post-op
8. hoarseness: the most common cause is laryngeal edema and not superior nor recurrent laryngeal nerve injury
9. cranial nerve injury: the most common complication after CEA with an incidence of up to 8-10%108
A. hypoglossal nerve → tongue deviation towards the side of injury: incidence ≈ 1% (with mobilizing XII to allow displacement). Unilateral injury may cause speaking, chewing and swallowing difficulties. Bilateral injuries can cause upper airway obstruction109. The presence of a unilateral palsy is a contraindication to doing contralateral endarterectomy until the first side recovers. May last as long as four months
B. vagus or recurrent laryngeal nerve → unilateral vocal cord paralysis: 1% risk
C. mandibular branch of facial nerve → loss of ipsilateral lip depressor
10. headache97
11. hypertension110, 111: may develop 5-7 days post-op. Longstanding HTN may occur as a result of the loss of the carotid sinus baroreceptor reflex
COMPLICATION MANAGEMENT
1. post-op TIAs
A. if TIA occurs in recovery room, emergency CT (to R/O hemorrhage) and then angiogram recommended to assess for ICA or CCA occlusion (vs. emboli)
B. if TIA occurs later, consider emergent OPG; if abnormal → emergent surgery (if neurologically intact, pre-op angiogram is appropriate)100
2. fixed post-op deficit in distribution of endarterectomized carotid
A. if deficit occurs immediately post-op (i.e. in PACU), recommend immediate re-exploration without delay for CT or angiogram112 (case reports of no deficit when flow re-established in ≤ 45 mins). For later onset, workup is indicated. Technical considerations for emergency re-operation100 (p 255):
1. isolate the 3 arteries (CCA, ECA, & ICA)
2. occlude CCA 1st, then ECA, and ICA last (to minimize emboli)
3. open arteriotomy, check backflow; if none, pass a No. 4 Fogarty catheter into ICA, gently inflate and withdraw (avoid intimal tears)
4. if good backflow established, close with patch graft
5. remove tortuous vessel loops and kinks before closing
B. immediate management (unless ICH or SDH are likely) includes
1. fluids (e.g. Plasmanate®) to improve rheology and to elevate BP
2. pressors (e.g. phenylephrine) to elevate SBP to ≈ 180 mm Hg
3. oxygen
4. heparinization (may be controversial)
C. theoretical benefits of radiographic evaluation include:
1. CT: identifies ICH or SDH that might require treatment other than re-exploration of the surgical site, elevating BP, etc.
2. angiogram: identifies whether ICA is occluded, or if deficit is from another cause (e.g. emboli from endarterectomy site) that would not benefit from re-exploration or possibly endovascular treatment
3. disruption of arteriotomy closure, management
A. OPEN WOUND - if there is any stridor, it is critical to do this before trying to intubate (although ideally performed in O.R., the delay may be decisive). Evacuate clot (start with a sterile gloved finger) and stop bleeding, preferably without traumatizing the artery, a DeBakey clamp is optimal
B. INTUBATION - high priority, may be difficult or impossible if trachea is deviated (open wound immediately). Preferably done by anesthesiologist in controlled setting (i.e. O.R.) unless there is acute airway obstruction
C. call O.R. and have them prepare set-up for endarterectomy, and take patient to O.R.
OPERATIVE TECHNIQUE100 (191-204)
Anesthesia and monitoring
Most (but not all) surgeons monitor some parameter of neurologic function during carotid endarterectomy, and will alter technique (e.g. insert a vascular shunt) if there is evidence of hemodynamic intolerance of carotid clamping (only occurs in ≈ 1-4%).
1. local/regional anesthesia: permits “clinical” monitoring of patient’s neurologic function113, 114. Disadvantages: patient movement during procedure (often exacerbated by sedation and alterations in CBF), lack of cerebral protection from anesthetic and adjunctive agents. The only prospective randomized study found no difference between local and general anesthesia115. The multicenter, randomized controlled General Anesthesia versus Local Anesthesia (GALA) Trial116 found no significant differences in the prevention of stroke, MI, or death for either anesthetic technique. Sub-group analysis showed trends (not statistically significant) favoring local anesthesia for perioperative death, event-free survival at 1 year, and patients with contralateral occlusion. Local anesthesia was associated with a significant reduction of shunt insertion116. A Cochrane Database Review found no evidence from randomized trials to favor either anesthetic technique117
2. general anesthesia, possibly including barbiturates (thiopental boluses of 125-250 mg until 15-30 second burst suppression on EEG, followed by small bolus injections or constant infusion to maintain burst suppression89)
A. EEG monitoring
B. SSEP monitoring
C. measurement of distal stump pressure after CCA occlusion (unreliable), e.g. using a shunt if stump pressure < 25 mm Hg
D. transcranial Doppler
E. near-infrared spectroscopy
Position and incision
1. supine, neck slightly extended and rotated slightly (≈ 30°) away from the operative side
2. the incision curves gently and follows the anterior border of the sternocleidomastoid muscle, and curves posteriorly at the rostral end
3. keep the horizontal portion of the incision ≈ 1 cm away from the mandible to avoid injury to marginal mandibular branch of facial nerve (which lies in the inferior parotid gland and supplies lip depressor) due to retraction against mandible
4. retractors should not be placed deeper than the platysma to avoid injury to recurrent laryngeal nerve which runs between the esophagus and trachea. Blunt retractors are used to avoid internal jugular vein injury
Dissection
1. the common facial vein (CFV) usually crosses the field over the carotid bifurcation, it is doubly ligated and divided. It leads to the internal jugular vein (IJV)
2. identifying the IJV is key, dissection is carried down between the carotid artery and the IJV
3. the ansa hypoglossi runs superficial to the ICA and serves as a useful guide to the hypoglossal nerve (XII) which should be identified since it is at greater risk when it is not seen. XII can arise anywhere from the carotid bifurcation to the angle of the mandible, although it is usually in the vicinity of the CFV. Mobilization can be facilitated by dividing the small artery (sternocleidomastoid branch of the ECA) and vein that cross over it109
4. the ansa hypoglossi can usually be spared, and if mobilized, allows medial retraction of the hypoglossal nerve out of harm’s way. If it is necessary to divide the ansa it is done close to the hypoglossal nerve to be certain it is not a branch of the vagus and to minimize neurologic deficit (the ansa has an anterior cervical limb from the cervical plexus)
5. the superior thyroid artery is the first branch of the ECA, and helps differentiate ECA from the ICA (the ICA is located posterior to the ECA)
6. the carotid bulb may be anesthetized with ≈ 2-3 ml of 1% plain lidocaine using a 27 Ga needle. This may be done routinely, or, as some prefer only if hypotension and/or bradycardia occur during dissection (indicating IX nerve stimulation)
7. the ICA must be exposed beyond the extent of the plaque which can be determined by gentle palpation with a moistened finger and by visualization as the area where the artery turns from yellowish to its normal pinker color
Occlusion and arteriotomy
1. a vessel loop is placed around the ECA at least 2 cm above the bifurcation
2. a vessel loop is also placed around the ICA but is looped only once
3. umbilical tape with a choke is placed around the CCA 2-3 cm below the bifurcation
4. heparin (usually 5,000 IU) is given 1 minute prior to cross clamping
5. a temporary aneurysm clip is placed on the superior thyroid artery
6. the order of occlusion of the vessels is as follows (mnemonic: “ICE”):
• first, the ICA (e.g. with temporary aneurysm clip)
• second, the CCA (e.g. with a small DeBakey clamp)
• third, the ECA (e.g. with temporary aneurysm clip)
7. during ICA clamping, mild hypertension is maintained by the anesthesiologist
8. shunt: some surgeons use some form of monitoring (EEG, BSAER, etc.) to determine if a shunt is needed (see Anesthesia and monitoring, page 1154), yet others routinely use a shunt whenever possible without assessing the need
9. the arteriotomy is begun in the CCA with a #11 scalpel, and once the lumen is entered, a Potts’ scissors carries the incision through to the ICA beyond the plaque. Stay in the midline to facilitate arteriotomy closure
Plaque removal
1. the plaque usually cannot be completely removed form the CCA, and thus it is usually transected with a Potts’ scissors taking care not to lacerate the artery wall and to leave as smooth an edge as possible
2. in the ICA, great care must be made to avoid leaving an intimal flap which could become a nidus for an arterial dissection. If necessary the intima may be tacked down by suturing from the lumen out on both ends (using double armed suture) and tying the knot outside the vessel
Arteriotomy closure and vessel release
1. arteriotomy may be performed with a running Prolene suture using either
A. primary closure
B. or with a patch graft to increase the caliber of the vessel and reduce the risk of restenosis
![]() limited evidence suggests that carotid patch angioplasty may reduce the risk of perioperative arterial occlusion and restenosis. Synthetic patches (Dacron, PTFE) are preferred to autologous vein (risk of aneurysmal dilatation, thrombogenic surface)55, 118
limited evidence suggests that carotid patch angioplasty may reduce the risk of perioperative arterial occlusion and restenosis. Synthetic patches (Dacron, PTFE) are preferred to autologous vein (risk of aneurysmal dilatation, thrombogenic surface)55, 118
2. the order of releasing the vessels: reverse order to clamping
• first, the ECA
• second, the CCA (allows air and debris to be washed into the ECA)
• lastly, the ICA
33.1.1.5. Emergency carotid endarterectomy
Emergency CEA indications include crescendo TIAs and stroke in evolution. The treatment paradigm of these conditions has shifted towards the use of interventional methods, such as thrombolysis and stenting, although there are no randomized controlled trial data to support that approach. A recent meta-analysis of emergent CEA has shown that the pooled stroke and stroke/death rates after CEA for crescendo TIA in 176 patients were 6.5% and 9.0%, respectively. For those with stroke in evolution, the overall stroke and stroke/death rates in 114 patients were 16.9% and 20.0%, respectively119.
After retrospective analysis of 64 emergency endarterectomies120 the guidelines given below were suggested. However, the efficacy of immediate surgical removal of obstruction is controversial and unproven. In one early study, over 50% of patients suffered fatal intracranial hemorrhage within 72 hours of emergency carotid endarterectomy.
INITIAL MANAGEMENT OF PATIENT PRESENTING WITH ACUTE NEURO DEFICIT
1. obtain history directed at determining presence of previous CVA and other serious medical illness, and to try to differentiate from seizure
2. baseline neurological assessment including evaluation of STA pulses and carotid bruits
3. during evaluation: close control of BP. O2 per NC. Labs + EKG (see Management of TIA or stroke, page 1016). Consider hemodilution with LMD
4. CT to R/O ICH or infarction (early CVA will not be visible)
5. when carotid disease is suspected, and CT negative for ICH or acute infarct, emergency angiography, MRI/MRA or CTA is performed
INDICATIONS FOR EMERGENCY CAROTID ENDARTERECTOMY
In patients with acute neurological deficits, the need for rapid decision making often does not allow differentiating between TIA, stroke in evolution and acute stroke, nor in assessing the stability or fluctuating nature of the deficit.
Indications
1. stroke in evolution
2. crescendo TIAs: TIAs that abruptly increase in frequency to ≥ several per day
3. following intraarterial thrombolysis, emergent/urgent CEA is indicated for residual critical carotid stenosis53, 66
Contraindications
Patients with depressed levels of consciousness or acute fixed deficits.
SURGICAL MANAGEMENT
Again, most cases would now be managed initially with endovascular thrombolysis and stenting. Surgery would considered if this is not an option.
1. for emergency surgery, it is essential that blood pressure be stable
2. in patients with complete occlusion, ICA is not occluded intra-op (to avoid breaking up thrombus, if present)
3. if thrombus present
• attempt spontaneous extrusion using back pressure
• if this fails, attempt to remove with smoothened suction catheter
• if this fails, pass balloon embolectomy catheter as far as base of skull (caution: avoid injury to distal ICA that could cause CCF)
• obtain intra-op angiogram unless thrombus emerges and backflow is excellent
• plicate ICA (avoid creating a blind pouch at origin) if there is good back flow or if satisfactory angiography cannot be obtained
Table 33-3 Surgical results
|
Presenting deficit |
Same or improved |
Deaths |
|
intact or mild |
92% |
0 |
|
moderate |
80% |
1 (7%) |
|
severe |
77% |
3 (13%) |
SURGICAL RESULTS
Highest correlation was with presenting neurologic status (see Table 33-3).
33.1.1.6. Totally occluded internal carotid artery
10-15% of patients presenting with carotid territory stroke or transient ischemic attacks (TIA) are found to have carotid occlusion. This amounts to an estimated 61,000 first ever strokes and 19,000 TIAs per year in the United States. Prevention of subsequent stroke in symptomatic patients with carotid artery occlusion remains a difficult challenge. The overall rate of subsequent stroke is 7% per year for all stroke and 5.9% per year for ischemic stroke ipsilateral to the occluded carotid artery121. These risks persist even despite treatment with antiaggregants and anticoagulants122. The prevalence of asymptomatic carotid occlusion is not known, and the incidence of ipsilateral stroke in never-symptomatic carotid stenosis is negligible123.
PRESENTATION
3 patterns of CVA with acute carotid artery occlusion:
1. stump emboli: produces cortical infarcts. Emboli usually go up the external carotid (higher flow, and reverse flow that may occur through ICA initially prevents emboli from ICA). Later, ICA emboli may occur.
2. whole hemisphere CVA
3. watershed infarct
In symptomatic patients124: hemiparetic TIA 53%, dysphasic TIA 34%, fixed neuro deficit 21%, crescendo TIAs 21%, amaurosis fugax 17%, acute hemiplegia 6%. One series had 27% asymptomatic125. Patients may have the so-called “slow carotid stroke” of carotid occlusion which is a stuttering progressive stroke.
MRI
With watershed type of CVA: MRI may show so-called “string of pearls” sign (small areas of intraparenchymal increased density on DWI).
NATURAL HISTORY120
Patients with mild deficit and angiographically proven ICA occlusion have a stroke rate (in two series) of 3 or 5% per year (2 or 3.3% related to occluded side). In patients with acute ICA occlusion and profound neurological deficit, 2-12% make good recovery, 40-69% will have profound deficit, and 16-55% will have died by the time of follow-up.
ENDOVASCULAR THROMBOLYSIS AND STENTING FOR ACUTE CAROTID OCCLUSION
Case reports and series of endovascular treatment of internal carotid artery occlusion have confirmed the feasibility of this technique. Intraarterial thrombolysis within 6 hours of stroke onset may increase recanalization rates to 37%-100% and clinical improvement to 53%-94% without significant increase in hemorrhagic transformation when compared with intravenous thrombolytic therapy alone126-131. Although results appear promising, randomized controlled trials on cervical carotid thrombolysis and/or stenting are lacking.
SURGERY
Options include: endarterectomy, Fogarty balloon catheter embolectomy (utilizing a No. 2 French catheter with 0.2 ml balloon gently passed 10-12 cm up ICA from small arteriotomy made distal to atheromatous plaque132), extracranial-intracranial bypass. Restored patency rate is inversely related to suspected duration of occlusion. Chronically occluded ICA has poor patency rate and little gain from reopening.
Determining the exact time of occlusion is frequently impossible. One must often rely on clinical grounds, therefore an occasional chronic occlusion will be included.
Retrograde filling of ICA to petrous or cavernous segment from ECA (e.g. via ophthalmic) or from contralateral ICA is a good sign of operability124.
Surgical results124
32% (15/47 cases) immediate surgical failures (no or minimal back bleeding), at least 3 deaths. Among immediate successes no CVAs and no TIAs. If operated < 2 days reported patency rate 70-100%, from 3-7 days 50-100%, 8-14 days 27-58%, 15-30 days 4-61%, over 1 month (2 series) 20-50%.
Guidelines
Emergency operations for acute neuro deficit associated with total occlusion should not be performed after about 2 hrs. Extremely poor neuro status (lethargy/coma) is a contraindication to surgery. Patients without persistent neuro deficit: operate ASAP. If the patient has recurrent TIAs (despite maximal medical therapy) following recent carotid occlusion, and no definite infarct on MRI, consider bypass surgery.
33.1.2. Vertebrobasilar insufficiency
CLINICAL
DIAGNOSTIC CRITERIA
Table 33-4 shows a mnemonic of the symptoms of vertebrobasilar insufficiency (VBI).
Clinical diagnosis of VBI
Requires 2 or more of the following:
• motor or sensory symptoms or both, occurring bilaterally in the same event
• diplopia: ischemia of upper brainstem (midbrain) near ocular nuclei
• dysarthria: ischemia of lower brainstem
• homonymous hemianopsia: ischemia of occipital cortex (NB: this is binocular, in contrast to amaurosis fugax which is monocular)
Table 33-4 Mnemonic: “The 5 D’s of VBI”
|
• “drop attack” • diplopia • dysarthria • defect (visual) • dizziness |
VBI may be suspected in a patient with transient episodes of “dizziness” (vertigo that is otherwise unexplained, e.g. absence of orthostatic hypotension) that is initiated by positional changes. VBI may sometimes be due to compression of the VA at the C1-C2 level with:
1. head turning (see Bow hunter’s stroke below)
2. os odontoideum (see page 966)
3. anterior atlantoaxial subluxation: e.g. in rheumatoid arthritis (see page 495)
4. with rotatory atlantoaxial subluxation: see page 955
SYMPTOM COMPLEXES
Predicting site of lesion based only on clinical evaluation is very unreliable. Atheromatous and stenotic lesions occur most frequently at VA origin.
VBI symptoms may be due to:
1. hemodynamic insufficiency (may be the most common etiology), including:
• subclavian steal: reversed flow in VA due to proximal stenosis of subclavian artery
• stenosis of both VAs or of one VA where the other is hypofunctional (e.g. hypoplastic, occluded, or terminates in PICA) causing reduced distal flow in face of inadequate collaterals (see Bow hunter’s strokebelow)
2. embolism from ulcerations
3. atherosclerotic occlusion of brainstem perforators
NATURAL HISTORY
No clinical study accurately defines the natural history. The estimated stroke rate is 22-35% over 5 years, or 4.5-7% per year133 (one study estimating 35% stroke rate in 5 years did not use angiography).
Risk of CVA after first VBI-TIA has been estimated as 22% for first year134.
EVALUATION
Adequate investigation requires selective four-vessel angiography.
TREATMENT
Anticoagulation is the mainstay of medical management. Alternatives include anti-platelet drugs such as ASA (efficacy of either remains unproven133, 135).
Surgical treatment includes:
• vertebral endarterectomy
• transposition of VA to ICA (with or without carotid endarterectomy, with or without saphenous vein patch graft) or to thyrocervical trunk or to subclavian artery136
• bypass grafting (e.g. occipital artery to PICA)
• C1-2 posterior arthrodesis (see page 183) may prevent potentially life-threatening CVA in cases of os odontoideum (see page 966)
BOW HUNTER’S STROKE
Bow hunter’s stroke (BHS): hemodynamic VBI induced by intermittent VA occlusion resulting from head rotation137 (ischemic sequelae range from TIA (bow hunter’s sign) to completed stroke). May occur with forced (e.g. with chiropractic neck manipulation138) or voluntary139 head rotation.
Occlusion usually involves the VA contralateral to the direction of rotation, and usually occurs at the C1-C2 junction (due to the immobility of the VA at this location)140. However, other sites have also been reported141, 142.
VA occlusion does not produce symptoms in most individuals due to collateral flow through the contralateral VA and/or the circle of Willis. Symptomatic occlusion usually involves the dominant VA143, however, may also occur with non-dominant VA139). Most cases of BHS occur in patients with an isolated posterior circulation (incompetent posterior communicating arteries).
BHS has also been postulated as one possible cause of SIDS144.
Contributing factors:
1. external VA compression142
A. spondylotic bone spurs: particularly in the foramen transversarium145
B. tumors
C. fibrous bands (e.g. proximal to entrance of VA into C6 foramen transversarium141)
D. infectious processes
E. trauma
2. tethering of the VA
A. at the transverse foramina of C1 & C2
B. along the sulcus arteriosus proximal to where the VA enters the dura
3. defect in odontoid process146
4. atherosclerotic vascular disease
Diagnosis
BHS should be suspected in patient with symptoms of VBI precipitated by head movement. This may be very difficult to differentiate from vertigo and nausea due to vestibular dysfunction (rotation of the body keeping the head motionless might be helpful147).
Dynamic cerebral angiography (DCA): ✖ NB: significant consequences can be precipitated during DCA in patients with BHS140. The involved VA shows loss of flow as the head is rotated from the neutral position to the contralateral side. Carotid injections demonstrate patency of P-comms and the presence of any persistent fetal anastomoses.
CT angiogram (CTA): Same precautions as with DCA (see above). Probably not the initial diagnostic study of choice. If the DCA is negative, CTA is not needed. If DCA is positive, CTA may be helpful to demonstrate the arterial relationship to the bony anatomy.
Treatment
Options include:
1. anticoagulation147
2. cervical collar: to remind patient not to turn their head
3. for VA compression at C1-2 (see Table 33-5 for a comparison):
A. C1-2 fusion: see page 183
B. VA decompression: C1 “hemilaminectomy” via a posterior approach149
4. for compression at other sites: elimination of the source of compression where possible (e.g. sectioning of offending fibrous band141, removal of osteophytic spurs145…)
Table 33-5 Comparison of surgical treatment for positional VA occlusion at C1-2
|
Procedure |
Advantages |
Disadvantages |
|
C1-2 fusion |
high success rate in eliminating symptoms |
loss of 50-70% of neck rotation with possible discomfort |
|
VA decompression |
no loss of motion |
33% continue to have symptoms148 |
Management recommendations: For compression at C1-2, it is suggested that VA decompression be performed as the initial treatment. This should be followed by DCA to verify maintenance of patency with head turning. Patients who fail clinically or on DCA should undergo C1-2 fusion140. Patients need to know pros and cons of each option.
VERTEBROBASILAR HYPOPLASIA
Reported as a possible etiology for cerebellar CVA.
33.2. Cerebral arterial dissections
![]() Key concepts:
Key concepts:
• hemorrhage into the medial layer of an artery
• may be spontaneous or post-traumatic, may be intracranial or extracranial
• may present with pain (usually ipsilateral H/A or carotidynia), Horner’s syndrome (in carotid dissections), TIA/CVA, or SAH
• extracranial dissections are usually treated medically (anticoagulation), intracranial dissections with SAH are treated surgically
This section primarily discusses “spontaneous” dissections. ICA dissection following blunt cervical trauma is much more common, and is covered on page 984.
NOMENCLATURE
Some confusion has arisen because of inconsistent terminology in the literature. Although by no means standard, Yamaura150 has suggested the following:
|
dissection |
extravasation of blood between the intima and media, creating luminal narrowing or occlusion |
|
dissecting aneurysm |
dissection of blood between the media and adventitia, or at the media, causing aneurysmal dilatation, which may rupture into the subarachnoid space |
|
pseudoaneurysm |
rupture of artery with subsequent encapsulation of the extravascular hematoma, may or may not produce luminal narrowing |
PATHOPHYSIOLOGY
The lesion common to all dissections is hemorrhage outside of the vascular lumen due to pathological trans-intimal extravasation of blood from the true lumen into the vessel wall. The hematoma may either dissect the internal elastic membrane from the intima151 causing narrowing of the true lumen, or it may dissect into the subadventitial plane producing an adventitial outpouching from the vessel wall (pseudoaneurysm). Rupture through the vessel wall producing SAH occurs occasionally.
Subintimal dissection is more common with intracranial dissections, whereas extracranial vessels (including the aorta) usually dissect at the media or between media and adventitia.
“Spontaneous” dissections have been associated with a large number of conditions, oftentimes the association is unproven. These conditions include:
• fibromuscular dysplasia (FMD): found in ≈ 15% of cases152
• cystic medial necrosis (or degeneration): originally thought to be a common finding, now thought to perhaps be linked to a higher likelihood of fatal dissection
• saccular aneurysm
• Marfan syndrome: autosomal dominant inherited disorder of connective tissue. Phenotypic manifestations are due to production of abnormal fibrillin
• atherosclerosis: only rarely implicated as an etiology. More likely to be a factor with subintimal dissection of extra cranial arteries
• Takayasu’s disease
• medial degeneration
• syphilitic arteritis (more common in the past, associated with 60% of dissections before 1950)
• autosomal dominant polycystic kidney disease: associated with a higher incidence of cerebral aneurysms (see page 1057)
• variant periarteritis nodosa
• allergic arteritis
• homocystinuria
• moyamoya disease153 (see page 1170)
• strenuous physical activity
EPIDEMIOLOGY
Occurs primarily in middle aged patients, with a mean age of ≈ 45 yrs (average age of traumatic dissections is slightly younger). More frequent in men150, 152. Incidence is unknown, since often times the condition produces mild, transient symptoms. Increased awareness of the condition has resulted in an increased rate of diagnosis. ICA dissection accounted for 2.5% of first strokes in one series154.
The largest reported series150 (literature review + new cases) of 260 cases found the incidence by location shown in Table 33-6. The vertebral artery was the most common intracranial site. Previously, the ICA has been cited as the most common site. This change may be due to the recent increased recognition of arterial dissections as a source of SAH (and vertebral dissections most often present as SAH). Multiple dissections occur in ≈ 10% (the most common: bilateral vertebrobasilar lesions).

CLINICAL
Cerebral arterial dissections may cause symptoms by:
• embolization secondary to:
![]() platelet aggregation stimulated by the exposed surfaces
platelet aggregation stimulated by the exposed surfaces
![]() dislodged thrombus (formation of which is enhanced by reduced flow)
dislodged thrombus (formation of which is enhanced by reduced flow)
• reduced distal flow secondary to:
![]() thrombosis due to reduced flow
thrombosis due to reduced flow
![]() occlusion of the true lumen by the expansion of the mural hematoma
occlusion of the true lumen by the expansion of the mural hematoma
• subarachnoid hemorrhage (atypical presentation, may be more common with posterior circulation dissection than with anterior circulation)155
The most common presentation in patients < 30 yrs of age was due to internal carotid dissection without SAH. In patients > 30 yrs, vertebrobasilar artery (VBA) dissection with SAH was the most common150.
Headache, usually severe, often predates neurologic deficit by days or weeks. See following sections (ICA page 1162, and vertebrobasilar page 1163) for specifics.
EVALUATION
CT
More useful for evaluating brain for infarction. Dissection can sometimes be visualized directly156.
CT angiogram (CTA)
Often obviates the need for cerebral angiography since CTA scanners with ≥ 16 detectors are equal in predictive value and have an accuracy near 99%157.
Angiography
The definitive diagnostic study. However, diagnosis may be delayed if the dissection is misinterpreted as:
1. an unusual saccular aneurysm (the most common error)
2. atherosclerotic lesions: with dissections, the location is unusual, the lesion may be isolated, the age is usually younger, and the stenosis is smooth
3. vasospasm following SAH: however, the narrowing with vasospasm is delayed in onset vs. the changes with dissection which are present from the beginning
Angiographic findings may include:
1. luminal stenosis: irregular stenosis over long segments of the artery often with focal areas of near total stenosis (“string sign”)
2. fusiform dilation with proximal or distal narrowing (string and pearl sign)
3. occlusion: artery usually tapers to a point
4. intimal flap: when seen, usually found at proximal end of dissection
5. may see proximal beading (“string of beads” configuration, indicative of FMD)
6. “double lumen sign”: true vessel lumen and a intramural false lumen with an intimal flap. Usually with retention of contrast within the false lumen well into the venous phase. The only pathognomonic sign
7. wavy “ripple” appearance
8. severe kinking (frequently bilateral). With VBA lesions: dolichoectasia
A characteristic of arterial dissections is that they often change configuration on repeat angiography158 (some resolve, and some worsen).
MRI
Probably not as accurate as CTA or angiography. Optimal MRI study is source T1WI axial images with fat suppression (“fat sat”), look for loss of visualization over several slices, with good visualization above and below. May visualize intimal flap and distinguish a dissection from a fusiform aneurysm.
Crescent sign: bright signal in wall of ICA on T2WI axial images (hematoma in vessel wall).
OUTCOME
An early review of the literature found an 83% mortality within a few weeks of presentation with vertebrobasilar artery (VBA) dissection159. A later report tempered that grim prognosis160.
Based on a review of 260 cases150, an overall mortality of 26% was found. 70% had a favorable outcome (based on Glasgow Outcome scale), 5% poor. Mortality was higher in ICA lesions (49%) than VBA lesions (22%). Mortality was 24% in the SAH group, and 29% in non-SAH cases.
33.2.1. Internal carotid dissection
See Cerebral arterial dissections above for general information.
SPONTANEOUS
Some cases considered spontaneous may actually be due to trivial trauma, including violent coughing, nose blowing, and simple neck turning. Usually seen in young women.
In spontaneous dissection, the most common initial symptom is ipsilateral headache. Most of these (60%) are orbital or periorbital, but they may also be auricular or mastoid (39%), frontal (36%), temporal (27%). May also produce sudden onset of severe pain over carotid artery (carotidynia)95.
Incomplete Horner’s syndrome (oculosympathetic palsy): ptosis and miosis without anhidrosis (due to involvement of plexus around the ICA, sparing the ECA plexus which innervates facial sweat glands) may occur. Bruits may be heard by the examiner or by the patient. These and other clinical features are shown in Table 33-7.
May be a cause of infantile and childhood hemiplegia and hemiparesis161.
Table 33-7 Clinical features of spontaneous ICA dissection152
|
Feature |
% |
|
focal cerebral ischemia |
76% |
|
headache |
59% |
|
oculosympathetic palsy |
30% |
|
bruit |
25% |
|
amaurosis fugax |
10% |
|
neck pain |
9% |
|
syncope |
4% |
|
scalp tenderness |
2% |
|
neck swelling |
2% |
POSTTRAUMATIC (NONPENETRATING)
Posttraumatic ICA dissection is much more common than spontaneous, and is covered on page 984.
33.2.2. Vertebrobasilar system artery dissection
VERTEBRAL ARTERY DISSECTIONS
See Cerebral arterial dissections on page 1160 for general information. Less common than carotid dissection (16th case of extracranial dissection in literature reported in 1987162). Extracranial lesions outnumber intracranial.
Traumatic dissections tend to occur where the VA crosses bony prominences, e.g. at the C1-2 junction or where it enters the foramen transversarium (usually at C6). Spontaneous dissections tend to be intracranial and commonly occur on the dominant VA.
SPONTANEOUS
Has been associated with FMD, migraine, and oral contraceptives162. Unrecognized or forgotten trauma or sudden head motion may have occurred in some cases reported as spontaneous. Commonly occurs in young adults (mean age: 48 yrs). With spontaneous dissections, 36% of patients have dissections at other sites, 21% of cases have bilateral VA dissections163.
Dissecting aneurysms of the VA (possibly a distinct entity) are also described164-166. They tend to be fusiform, and may be amenable to clipping, and were associated with vertebral dissections in 5 of 7 cases reported in one series167. As of 1984, only ≈ 50 cases of dissecting aneurysms were published167.
POST-TRAUMATIC (NONPENETRATING)
See page 985.
PRESENTATION
In spontaneous extradural dissections, neck pain is a prominent early finding in most patients, and is commonly located over the occiput and posterior cervical region. Generalized severe headache is also common. TIAs or stroke (usually lateral medullary syndrome168 (see page 1028) or cerebellar infarction, especially in patients with occlusion of the third or fourth portion of the VA169). None of 5 patients developed new neurologic symptoms after the original stroke in an average of 21 months follow-up169. In 3 of these 5, VA dissection was bilateral.
Dissecting aneurysms may present with altered consciousness, and may cause SAH (seen in 6 of 30 cases of vertebrobasilar complex dissections)167. Rebleeding occurs in 24-30% of those cases presenting with SAH163, making these lesions treacherous, with a very high mortality170, 171.
Traumatic extradural dissections or pseudoaneurysms may have a similar presentation, but can also produce massive external hemorrhage or neck hematomas163.
EVALUATION
See section under Cerebral arterial dissections. on page 1162.
Angiography
Diagnosis by angiography may be difficult in many cases (the most common misdiagnosis is ruptured saccular aneurysm of unusual shape172).
In post-traumatic dissections, the most common finding is irregular stenosis of horizontal loops of distal extracranial VAs as they pass behind C1, often bilateral.
In 14 of 15 post-traumatic VA dissections, the lesion was located posterior to the atlas (distal extracranial 3rd segment), the single exception being a patient with direct trauma causing proximal VA involvement. This predilection is possibly explained by the fact that the first and third portions of the VA are movable, whereas the second and fourth are relatively immobilized by bone.
TREATMENT
Except for cases presenting with hemorrhage or large ischemic stroke, medical therapy should be started emergently, and consists of anticoagulation, with heparin acutely, followed by oral agents (e.g. Coumadin) probably for a total of 6 months.
As with traumatic dissections, endovascular techniques are now assuming more prominent role in management.
Indications for intervention
Surgery or endovascular techniques (mostly stents, but also occlusion, angioplasty163) are required for dissections presenting with SAH (due to their propensity to rebleed) and is recommended for most intradural dissections. For extradural lesions it is indicated for dissections that progress (angiographically) or for persistent symptoms in spite of adequate medical therapy. Some less malignant lesions may be amenable to endovascular stenting.
Surgical treatment
At the time of surgery, the site of dissection may be recognized by fusiform or tubular enlargement of the artery with discoloration due to blood within the arterial wall (the discoloration has been described as black, bluish, purple, purple red, or brown172).
Surgical treatment of intradural dissection when endovascular techniques are not an option includes the following alternatives:
1. non-clippable aneurysms may be candidates for Hunterian occlusion of the VA proximal to the BA (either by microsurgical technique, or by endovascular techniques which may not be as precise). Some may not tolerate clipping the dominant VA, especially if the contralateral VA is hypoplastic. Conversely, some may tolerate bilateral VA occlusion173). Balloon test occlusion163 is recommended
A. if the dissection involves the PICA origin, then clip proximal to dissection. PICA then fills from retrograde flow, and the reversal of flow across the site of dissection should push the intima back against the wall
B. if the dissection is proximal to PICA and doesn’t involve PICA, then trap the aneurysm between clips. PICA fills by retrograde flow
C. if the aneurysm begins distal to the PICA origin, occlude the VA155 distal to the PICA takeoff174
2. combining VA clipping (see above) with vascular bypass, options:
A. side-to-side PICA-PICA anastomosis
B. transplantation of the PICA origin to the VA outside the aneurysm
C. occipital artery-to-PICA bypass
3. resection accompanied by autogenous interposition vein graft
4. non occlusive surgical techniques
A. clipping with specially designed clips for fusiform aneurysms (e.g. SundtKees clip)
B. wrapping: of dubious benefit
VERTEBROBASILAR SYSTEM DISSECTIONS EXCLUDING THE VA
Basilar artery dissections tend to present with brain stem infarction and more rarely with SAH171. The prognosis is generally regarded as poor. Endovascular techniques may be able to treat some.
33.3. Extracranial-intracranial (EC/IC) bypass
Includes but not limited to superficial temporal artery-middle cerebral artery (STAMCA) bypass.
EC/IC BYPASS FOR ATHEROSCLEROTIC OCCLUSIVE DISEASE
The EC/IC bypass study: The EC/IC bypass, pioneered by Donaghy and Yasargil in 1967175, plummeted in popularity176 after publication of the international cooperative EC/IC bypass study177 in 1985. The EC/IC trial randomized 1377 patients with symptomatic ICA or MCA stenosis to either STA-MCA bypass or medical therapy with ASA. Despite a graft patency rate of 96%, surgical patients suffered more and earlier fatal and nonfatal strokes. Patients with severe MCA stenosis and those with persistent symptoms following ICA occlusion fared especially worse with bypass. During the 55.8 months mean follow-up, the percentage of patients experiencing 1 or more strokes in the medical group compared to the surgical group was 29% vs. 31%.
Critics highlight the failure of the study’s inclusion criteria to distinguish between hemodynamic vs. thromboembolic causes of stroke122, 178, 179 (ischemia secondary to thromboembolic events would not be expected to improve with flow augmentation, and inclusion of such patients in the surgical arm could therefore artificially lower the apparent efficacy of the procedure).
Current state of affairs: Imaging technologies introduced since the EC/IC trial can identify flow-dependent ischemia. Xenon-CT, TCD, SPECT, and MRI may be used in combination with acetazolamide challenge to evaluate cerebrovascular reserve and reactivity (see page 1011).
As cerebral perfusion pressure decreases in severe atherosclerotic occlusive disease, cerebral autoregulation is unable to maintain adequate CBF to meet metabolic demands. In this state of “misery perfusion,” oxygen extraction fraction (OEF) of available blood flow will increase180, 181. Abnormal OEF, as quantified by PET, is an independent predicator of subsequent stroke122. Patients with abnormal response to acetazolamide challenge (see page 128) and/or with elevated OEF are therefore potential candidates for cerebral revascularization122, 179, 182-184.
OTHER INDICATIONS FOR EC/IC BYPASS
1. aneurysms: certain aneurysms are not amendable to either direct microsurgical clipping or endovascular coiling due to extreme size, location, calcification or atherosclerosis, dissection, or the incorporation of perforators or major arteries. EC/IC bypass remains a highly viable adjunctive measure in patients requiring Hunterian occlusion of parent vessel or prolonged temporary occlusion for definite treatment185-189. Cerebrovascular reserve and need for bypass can be assessed preoperatively using balloon test occlusion (BTO) with hypotensive challenge
2. tumors encasing or invading major arteries
3. moyamoya disease: see page 1170
BYPASS TYPES
The type of graft used depends on preoperative determination of amount of flow augmentation necessary, the size of the recipient graft and the availability of donor vessel190:
1. pedicled arterial grafts: STA, occipital artery
A. low-flow (15 - 25 ml/min)
B. only one anastomosis required
C. 95% graft patency in STA-MCA bypasses
2. radial artery graft
A. moderate to high flow (40 - 70 ml/min)
B. advantages: physiological conduit for arterial blood; constant location makes it easy to harvest; lumen size closely approximates that of M2 or P1 and reduces flow mismatch with subsequent flow turbulence and graft thrombosis
C. disadvantages: risk of vasospasm (reduced with pressure distension technique)
D. > 90% graft patency at 5 years
3. saphenous vein graft
A. high flow (70 - 140 ml/min)
B. advantages: easy accessibility; longer length
C. disadvantages: risk of thrombosis at distal anastomosis due to flow mismatch and turbulence; lower graft patency rates
D. 82% graft patency at 5 years
33.4. Cerebrovascular venous thrombosis
3 types of cerebrovascular venous thrombosis (CVVT) (may produce venous infarctions):
1. dural sinus thrombosis (DST)
2. cortical venous thrombosis
3. deep venous thrombosis
ETIOLOGIES
Many conditions have been incriminated with CVVT. Some common ones are listed below (see reference191 (p 1301) for extensive list):
1. infection
A. usually local, e.g. otitis media192, 193 (leading to the now obsolete term otitic hydrocephalus), sinusitis, peritonsillar abscess, paranasal sinusitis194
B. meningitis
2. pregnancy & puerperium: see below
3. birth control pills (BCP) (oral contraceptives)195
4. dehydration and cachexia (marantic thrombosis): includes burns and cachexia of neoplastic disease
5. cardiac disease (including CHF)
6. ulcerative colitis (UC): 1% of UC patients have some thrombotic complication (not necessarily intracranial), and this is the cause of ≈ 33% of deaths (usually pulmonary embolism)
7. periarteritis nodosa
8. sickle cell trait
9. trauma: including closed head injury (see below)
10. iatrogenic: e.g. S/P radical neck surgery196, transvenous pacemaker placement, post-craniotomy
11. malignancy: including myeloproliferative disorders
12. hypercoagulable state (AKA thrombophilia)
A. protein C deficiency or resistance to activated protein C
B. antithrombin III deficiency
C. protein S deficiency
D. antiphospholipid antibodies: associated with a variety of clinical syndromes including ischemic CVA, DVTs, thrombocytopenia, systemic lupus erythematosus (see page 1025)
E. paroxysmal nocturnal hemoglobinuria (PNH)
F. plasminogen deficiency
G. systemic lupus erythematosus197
H. factor VIII elevation198: may explain some cases of CVVT in pregnancy (see below)
13. diabetes mellitus: especially with ketoacidosis
14. homocystinuria: see page 1024
15. Behçet’s syndrome199: see page 78
16. rarely associated with lumbar puncture200
![]() In the absence of factors such as BCP use, CVVT is highly suggestive of myeloproliferative disorder.
In the absence of factors such as BCP use, CVVT is highly suggestive of myeloproliferative disorder.
Pregnancy/puerperium
Highest risk is in first 2 wks post-partum. One series201 found no case of CVVT occurred later than 16 days post-partum. Incidence ≈ 1/10,000 births. Etiology may be related to elevation of clotting factors (VII, X and especially factor VIII202).
Trauma
A rare sequelae of closed head injury203. CVVT occurs in ≈ 10% of combat injuries involving the brain. May occur in absence of skull fracture. CVVT should be suspected in patients with fractures or missiles crossing sinus.
FREQUENCY OF INVOLVEMENT OF DURAL SINUSES AND OTHER VEINS
1. sinuses
A. superior sagittal sinus (SSS) and left transverse sinus (TS) (70% each)
B. multiple sinuses in 71%
C. inferior sagittal sinus: rare, first case report in 1997204
D. straight sinus205
2. superficial cortical veins
3. deep venous system (e.g. internal cerebral vein)
4. cavernous sinus206, 207: rare. Thrombophlebitis of the cavernous sinus may be caused by sphenoid sinusitis
PATHOPHYSIOLOGY
Venous thrombosis reduces venous outflow from the brain and diminishes effective blood flow to the involved area. This venous engorgement causes white matter edema. The increased venous pressure may also lead to infarction and/or hemorrhage. These processes may all elevate ICP. Thus, clinical findings may be due to elevated ICP, and focal findings may be due to edema and/or hemorrhage. Cerebral infarction in this setting is called venous infarction.
CLINICAL
Clinical presentations of DST are shown in Table 33-8. There are no pathognomonic findings. Many signs and symptoms are due to elevated ICP. May present as a syndrome clinically indistinguishable from idiopathic intracranial hypertension (pseudotumor cerebri) (see page 713).
There is a high association of concurrent thromboembolic disease in other organs.
The anterior 1/3 of the SSS may occlude often without sequelae. Posterior to this, venous infarction is more likely to develop. Midportion SSS occlusion usually → increased muscle tone ranging from spastic hemi- or quadriparesis to decerebration. Posterior SSS thrombosis → field cuts or cortical blindness, or massive CVA with cerebral edema and death. Occlusion of the TS may occur without deficit unless the contralateral TS is hypoplastic, in which cases presentation is similar to posterior SSS occlusion.
SSS occlusion alone will not cause cranial nerve findings except perhaps for visual obscuration and abducens (VI) nerve palsy from elevated of ICP. Thrombosis in the jugular bulb may compress the nerves in the jugular foramen pars nervosa causing hoarseness, aphonia, difficulty swallowing and breathlessness (see Vernet’s syndrome, page 115)209.
Table 33-8 Presentation of dural sinus thrombosiss
|
Sign/symptom |
Series A* |
Series B* |
|
H/A |
100% |
74% |
|
N/V |
75% |
– |
|
seizures |
70% |
29% |
|
hemiparesis |
70% |
34% |
|
papilledema |
70% |
45% |
|
blurred vision |
60% |
– |
|
altered consciousness |
35% |
26% |
* series A: 20 young females201; series B: 38 cases from France208
DIAGNOSIS OF DST
Angiography is better at demonstrating where there is residual flow, and can identify areas of reversal of flow. Sometimes angiography will demonstrate clot. CT & MRI are better for identifying areas of clot. Angiography is often used as a complementary test210.
CT SCAN
Non-contrast CT
May be normal in 10-20% of cases of DST. Findings include:
1. hyperdense sinuses and veins (high density clots in cortical veins produce the cord sign which is pathognomonic for cerebral venous thrombosis; seen in only 2/30 patients)
2. petechial “flame” hemorrhages (intraparenchymal): seen in 20% (suspect sinus thrombosis with intracerebral hemorrhages in unusual locations for aneurysm or “hypertensive” hemorrhage)
3. small ventricles: seen in 50%
4. thrombosis of superior sagittal sinus may produce a triangular-shaped high density within the sinus (some refer to this as the delta sign, but this causes confusion with the “empty delta sign”, see below) (there is also confusion when an apparent “empty delta sign” is seen without contrast, this may occur when there is blood surrounding the SSS, e.g. following subarachnoid hemorrhage, this has been called a “false delta sign” or pseudodelta sign211). Recommendation: avoid the confusion of the “delta signs” and describe the findings
5. white matter edema
6. above changes occurring bilaterally
IV contrast CT
Findings of DST include:
1. with contrast, the dura around the sinus may enhance and become denser than clot in 35% of cases212. Near the Torcular herophili this produces what has been called the empty delta sign213, but sometimes this, too, is called the delta sign
2. gyral enhancement occurs in 32%
3. dense deep (white matter) veins (collateral flow)
4. intense tentorial enhancement (common)
MRI
Excels for diagnosis and follow-up. Shows absence of flow and clot burden, also demonstrates parenchymal changes. Can differentiate occluded sinus from congenital absence. Shows cerebral edema and non-acute hemorrhagic changes to better advantage than CT. Also may help estimate age of clots (see Table 33-9). MR-angiography may increase the utility. MR-venography (MRV) tends to overestimate the degree of occlusion.
Table 33-9 MRI appearance of thrombosed sinuses at various stages
|
Age of clot in sinus |
–– Appearance of clotted sinus –– |
|
|
T1WI |
T2WI |
|
|
acute |
iso-intense |
decreased (black): can mimic flow void |
|
subacute |
increased (1st) |
increased (2nd) |
|
late (> 10 d, recanalized) |
black (flow void) |
black (flow void) |
ANGIOGRAPHY FOR DST
Accuracy close to MRI. MRI has some advantages over angiography (e.g. on angiography a hypoplastic transverse sinus may not visualize, or non-opacified blood entering a sinus may mimic a filling defect).
Findings include:
1. non-filling of segments of sinuses, or filling defects
2. prolonged circulation time: in 50% of cases (may need delayed films to see veins)
3. stumps and abnormal collateral pathways
LP
OP usually increased. CSF bloody or xanthochromic.
BLOODWORK
To detect predisposing conditions when the etiology is unknown. Some tests that may be useful include evaluation for thrombophilia (protein C and S levels, antiphospholipid antibodies) as well as tests for specific predisposing conditions (CBC, Factor II level, serum homocysteine level, paroxysmal nocturnal hemoglobinuria (PNH) panel, leukocyte alkaline phosphatase).
ULTRASOUND FOR DST
May be used in diagnosis of superior sagittal sinus thrombosis in the neonate214.
EVALUATION FOR UNDERLYING DISORDER
At the time of presentation, work-up is difficult because the acute process will cause numerous abnormalities in clotting system. The best time to work these patients up is ≈ 3 months after they recover from the acute phase.
TREATMENT
Should be aggressive because recoverability of brain is probably greater than with arterial occlusive stroke. Management is complicated because measures that counteract thrombosis (e.g. anticoagulation) tend to increase the risk of hemorrhagic infarct (the risk of which is already increased), and measures that lower ICP tend to increase blood viscosity → increased coagulability.
Specific measures
1. correct underlying abnormality when possible (e.g. antibiotics for infection)
2. heparin (systemic): (see page 39 for dosing) especially if patient is in DIC. Several studies show a lower mortality rate with heparin than without215-217. It remains the best treatment even when there is evidence of intracerebral hemorrhage with the attendant risk of increasing the size of the hemorrhage210. There is no consensus on duration of treatment or if warfarin should be used afterwards. Success rate may be higher if administered before patient becomes moribund
3. avoid steroids (reduces fibrinolysis, increases coagulation)
4. control HTN
5. anticonvulsants to control seizures
6. monitor ICP if patient continues to deteriorate: ventriculostomy preferred, but caution must be used if patient is on heparin
A. hydrate aggressively as ICP tolerates
B. measures to lower ICP: in general, order is almost reverse of that for traumatic intracranial hypertension because diuretics → hypertonicity → ↑ viscosity → ↑ coagulation
a. elevate HOB
b. hyperventilation
c. drain CSF
d. pentobarbital coma
e. use hyperosmotic and/or loop diuretics last. Replace fluid loss with isotonic IV fluids to prevent dehydration (i.e. goal is hypertonic euvolemia)
7. thrombolytic therapy: either systemically or infused directly into clotted sinus210, 218, may be followed with heparin
A. urokinase205, 218 or streptokinase
B. intravenous tissue plasminogen activator (tPA): promising animal evidence219, not yet reported in humans
8. when above measures fail, either
A. decompressive craniectomy (± decompressive lobectomy): this decreases ICP, but may not improve outcome
OR
B. direct “attack” on clotted sinus: direct surgical treatment when deficit progresses in spite of above measures, or ICP not manageable (i.e. failure of medical therapy) (see below)
9. interventional neuroradiology: effectiveness is much reduced with chronic clot
A. Penumbra® system: sucks out clot, only takes out a small amount of clot
B. MERCI® retriever: corkscrew shaped wire that pulls clot out (see page 1018)
C. AngioJet®: not FDA approved for this. Not used for arterial clots because of injury to cerebral tissue, but may have some efficacy with venous clot
10. visual loss with papilledema may be treated with optic nerve sheath fenestration/decompression220
11. long-term treatment after resolution of acute phase with heparin and/or warfarin x 3-6 months
DIRECT SURGICAL TREATMENT FOR DST
Rarely indicated. Thrombectomy and sinus reconstruction are technically possible, but rethrombosis is common. Surgery may be indicated for abscess requiring excision.
Surgical technique for direct treatment of SSS thrombosis
Have available: blood for massive transfusion, large bore IV access, sinus shunt (pre-fabricated, or improvise with high-vacuum silicone grease inside & outside pediatric anode endotracheal tube with cuff on both ends, gas sterilize, prepared pre-op), tissue for sinus reconstruction (e.g. 20 cm of saphenous vein (arteries have a high rate of fibrosis, and there is no published experience with synthetics grafts). Vein grafts are dilated with heparinized saline and should be oriented correctly in case of valves).
Expose a wide portion of the sinus.
Consider ligation when lesion in non-critical location (SSS anterior to rolandic vein, non-dominant transverse or sigmoid, minor sinuses on skull base).
Hemorrhage is controlled by digital pressure, Fogarty catheter (insert directly, or insert a no. 7 Fogarty thru tiny sinotomy proximal to bleeding which allows repair of bleeding site), and/or insertion of shunt.
PROGNOSIS
Mortality: approximately 30% (range: 5-70%) (10% in French series208).
Poor prognosticators:
1. clinical status:
A. coma221:
B. rapid neurologic deterioration221, focal signs.
2. demographics
A. age: extremes of age (infancy or elderly)221 and age > 37 years
B. male gender
3. radiographic findings:
A. hemorrhages, especially larger hemorrhages
B. venous infarcts
4. deep venous involvement
33.5. Moyamoya disease
![]() Key concepts:
Key concepts:
• progressive bilateral spontaneous occlusion of ICAs with compensatory capillary collaterals that look like a “puff of smoke” (Japanese: moyamoya) on angio
• typical presentation: juvenile form → ischemic infarcts/TIAs (suspect diagnosis in any child presenting with TIAs). Adult form → hemorrhage
• pathology: intimal thickening w/o inflammation, may also involve heart. kidneys
• evaluation: cerebral angiography is necessary to delineate degree of stenosis as well as to evaluate potential extracranial donor vessels for revascularization
• treatment:
![]() medical treatment (antiplatelet drugs, anticoagulation, vasodilators…): not shown to be effective although antiplatelet/anticoagulation is often used
medical treatment (antiplatelet drugs, anticoagulation, vasodilators…): not shown to be effective although antiplatelet/anticoagulation is often used
![]() surgical revascularization: reduces the incidence of CVAs and TIAs, but benefit on reducing the rate of hemorrhage is unproven
surgical revascularization: reduces the incidence of CVAs and TIAs, but benefit on reducing the rate of hemorrhage is unproven
Progressive spontaneous occlusion of one or usually both ICAs (usually at the level of the siphon) and their major branches, with secondary formation of anastomotic collateral capillary network at the base of the brain which has been termed “moyamoya”, the Japanese word for something hazy like a “puff of cigarette smoke”222 (which it fancifully resembles on angiography). With progression, involvement includes the proximal MCAs and ACAs and on rare occasion the vertebrobasilar system. Associated aneurysms (see below) and rarely AVMs223, 224 may be observed.
Eventually the dilated capillary (moyamoya) vessels disappear with the development of collaterals from the ECA (meningeal collaterals are called “rete mirabile”).
Pathophysiology
Most common pathology is stenosis of the proximal anterior and middle cerebral arteries that is non-atherosclerotic or inflammatory in origin. Exact etiology is unknown but some studies show elevated basic fibroblast growth factor in the dura and scalp arteries in patients with moyamoya225. The internal elastic lamina of affected vessels may be thinned or duplicated. Similar vascular changes may also occur in the heart, kidney and other organs, suggesting it may be a systemic vascular disease.
Associated aneurysms
Intracranial aneurysms are frequently associated with moyamoya disease (MMD). This may be a result of the increased flow through dilated collaterals, or it may be that patients with moyamoya may also have a congenital defect in the arterial wall that pre-disposes them to aneurysms. 3 types: 1) usual sites of aneurysms in the Circle of Willis, 2) in peripheral portions of cerebral arteries, e.g. posterior/anterior choroidal, Heubner’s, and 3) within moyamoya vessels. The frequency of aneurysms in the vertebrobasilar system is ≈ 62% which is much higher than in the general population226. Aneurysmal SAH may be the actual cause of some hemorrhages that were erroneously attributed to moyamoya vessels.
EPIDEMIOLOGY
Risk factors: A history of inflammation in the head & neck region has been implicated.
Demographics: Incidence in Japan is higher (0.35/100,000/yr) than in North America. Two peaks (may not be same disease): juvenile (highest peak), age < 10 yrs (mean 3); adult, 3rd & 4th decade. Slight female predominance (1.8:1). Some evidence for familial tendency (some Asian families have an incidence as high as 7%), genetics appears autosomal dominant with low penetrance. Associated with some HLA antigens (B40 in juvenile form; B54(20) in adult).
Secondary moyamoya disease: AKA “quasi-moyamoya disease” or “moyamoya syndrome”227. Agiographic findings of moyamoya associated with e.g.:
• Graves’ disease/thyrotoxicosis
• history of cerebral inflammatory disease, including meningitis (especially tuber-cular (TB) meningitis and leptospirosis)
• retinitis pigmentosa
• vascular disorders: atherosclerosis, fibromuscular dysplasia, pseudoxanthoma elasticum
• congenital disorders: Down syndrome, Marfan syndrome, Turner syndrome, neurofibromatosis type 1, tuberous sclerosis, Apert syndrome
• hematologic disorders: Fanconi anemia, sickle cell anemia (in the U.S. one of the more common associations) and sickle cell trait
• following radiation therapy for skull base glioma in children228
• head trauma
• systemic lupus erythematosus (SLE)
Natural history
Incidence of disease progression in one study was 20% in adult patients with MMD229. Female patients had a higher risk of disease progression than males.
Prognosis of untreated MMD is poor, with 73% rate of major deficit or death within 2 years of diagnosis in children, and similarly poor outlook in adults230.
PRESENTATION
Juvenile form: Moyamoya is associated with 6% of childhood strokes225. Ischemic presentation more common (81%); includes TIAs (41%) which may alternate sides (alternating hemiplegia is a suggestive clinical finding), RINDs, or infarct (40%). Neurologic events are often provoked by straining or by hyperventilation (e.g. during crying or blowing a wind instrument) which is thought to produce hypocapnea with reactive vasoconstriction.
Headache is the most common presenting symptom, but seizures, focal neurologic deficits, choreoathetotic movements and hemorrhages can also be presenting symptoms. The risk of hemorrhage is increased in stages 5 & 6 of MMD ().
Adult form: Hemorrhage has been described as being more common (60%). Rupture of the fragile moyamoya vessels produces bleeding in the basal ganglia (BG), thalamus or ventricles (from the ventricular wall) in 70-80% of hemorrhages. SAH may occur, usually due to rupture of associated aneurysms (see above). In the pre-CT era, the most common form of hemorrhage was thought to be SAH from the rupture of moyamoya vessels, but most cases were probably intraventricular blood or SAH from associated aneurysms231.
EVALUATION AND DIAGNOSIS
CT
Work-up in suspected cases typically begins with a non-enhanced head CT. Up to 40% of ischemic cases have normal CT. Low density areas (LDAs) may be seen, usually confined to cortical and subcortical areas (unlike atherosclerotic disease or acute infantile hemiplegia which tend to have LDAs in basal ganglia as well). LDAs tend to be multiple and bilateral, especially in the PCA distribution (poor collaterals), and are more common in children.
MRI AND MRA
MRA usually discloses the stenosis or occlusion of the ICA. Moyamoya vessels appear as flow voids on MRI (especially in basal ganglia) and a fine network of vessels on MRA, and are demonstrated better in children than adults. Parenchymal ischemic changes are commonly shown, usually in watershed areas.
ANGIOGRAPHY
In addition to helping to establish the diagnosis, angiography also identifies suitable vessels for revascularization procedures. The angiography-related complication rate is higher than with atherosclerotic occlusive disease. Avoid dehydration prior to and hypotension during the procedure. Six angiographic stages of MMD are described in Table 33-10222 that tend to progress up until adolescence and stabilize by age 20.
Diagnosis of moyamoya requires bilateralA symmetrical stenosis or occlusion of the terminal portion of the ICAs as well as the presence of dilated collateral vessels at the base of the brain225. Other characteristic findings include:
• stenosis/occlusion starting at termination of ICA and at origins of ACA and MCA
• abnormal vascular network in region of BG (intraparenchymal anastomosis)
• transdural anastomosis (rete mirabile), AKA “vault moyamoya”. Contributing arteries: anterior facial, middle meningeal, ethmoidal, occipital, tentorial, STA
• moyamoya collaterals may also form from internal maxillary artery via ethmoid sinus to forebrain in frontobasal region
A. if unilateral, the diagnosis is considered questionable232, and these cases may progress to bilateral involvement
Table 33-10 Six angiographic stages of MMD222
|
Stage |
Finding |
|
1 |
stenosis of suprasellar ICA, usually bilateral |
|
2 |
development of moyamoya vessels at base of brain; ACA MCA & PCA dilated |
|
3 |
increasing ICA stenosis & prominence of moyamoya vessels (most cases diagnosed at this stage); maximal basal moyamoya |
|
4 |
entire circle of Willis and PCAs occluded, extracranial collaterals start to appear, moyamoya vessels begin to diminish |
|
5 |
further progression of stage 4 |
|
6 |
complete absence of moyamoya vessels and major cerebral arteries |
EEG
Non-specific in the adult. Juvenile cases: high-voltage slow waves may be seen at rest, predominantly in the occipital and frontal lobes. Hyperventilation produces a normal buildup of monophasic slow waves (delta-bursts) that return to normal 20-60 seconds after hyperventilation. In > 50% of cases, after or sometimes continuous with buildup is a second phase of slow waves (this characteristic finding is called “rebuild-up”) which are more irregular and slower than the earlier waves, and usually normalize in ≤ 10 minutes233.
CEREBRAL BLOOD FLOW (CBF) STUDIES
CBF is decreased in children with MMD, but relatively normal in adults. There is a shift of CBF from the frontal to the occipital lobes234 probably reflecting the increasing dependency of CBF on the posterior circulation. Children with MMD have impaired autoregulation of CBF to blood pressure and CO2 (with more impairment of vasodilatation in response to hypercapnia or hypotension than vasoconstriction in response to hypocapnia or hypertension)235.
Xenon (Xe-133) CT can identify areas of low perfusion. Repeating the study after an acetazolamide challenge (which causes vasodilatation) evaluates reserve capacity of CBF and can identify areas of “steal” which are at high risk of future infarction.
TREATMENT
No medical or surgical treatment has been proven effective in reducing the rate of hemorrhage in the adult with MMD. However, multiple large case series have supported the efficacy of cerebral revascularization for reducing the incidence of ischemic strokes and TIAs227.
MEDICAL TREATMENT
Medical treatment with platelet inhibitors, anticoagulants, calcium channel blockers230, steroids, mannitol, low-molecular-weight dextran and antibiotics have not proven to be of benefit. Steroids may be considered for involuntary movements and acutely during recurrent TIAs.
SURGICAL TREATMENT
Patients with mass effect from clot may be candidates for urgent decompression. Revascularization procedures, however, should be performed when the patient is stable under nonemergent conditions.
Suggested criteria for revascularization procedures227:
1. patients presenting with infarction or hemorrhage but are in good neurologic condition
2. infarction < 2 cm maximal diameter on CT, and all previous hemorrhages have completely resolved
3. angiographic stage is II-IV (see Table 33-10)
4. timing of operation: ≥ 2 months after most recent attack
Surgical revascular options: Various methods to revascularize the ischemic brain, include:
1. direct revascularization procedures: results are superior to indirect revascularization procedures236, 237 if a donor and recipient vessel of sufficient caliber (≥ 1 mm outer dia) can be identified (may be difficult in the pediatric age group who are the most likely to benefit238). Otherwise, indirect revascularization procedures (see below) are options
• STA-MCA bypass239: the procedure of choice
2. indirect revascularization procedures: usually reserved for younger patients (suggested cutoff age ≈ 15 years). May be combined with STA-MCA bypass. Includes:
A. encephalomyosynangiosis (EMS): laying the temporalis muscle on the surface of the brain (may cause problems with muscle contractions during talking and chewing, and neural impulses on surface of brain)
B. encephaloduroarteriosynangiosis (EDAS)240, 241: suturing the STA with a galeal cuff to a linear defect created in the dura. Variations on this technique include splitting the dura242
C. omental transposition243: either as a pedicle graft or as a vascularized free flap. Felt to have higher potential to revascularize ischemic tissue than above procedures, but there is greater risk of mass effect from the thickness of the omentum
3. the above indirect revascularization procedures improve blood flow in the MCA distribution, but not ACA circulation. This may be rectified by:
A. simple placement of frontal burr holes with opening of the underlying dura and arachnoid244
B. “ribbon EDAS” where a pedicle of galea is inserted into the interhemispheric fissure on both sides245
4. stellate ganglionectomy and perivascular sympathectomy: unproven that this increases CBF permanently
Postoperatively following STA-MAC bypass procedures:
1. avoid hypertension: may cause bleeding at anastomotic site and in areas of increased perfusion within the brain
2. avoid hypotension: may result in graft occlusion
3. aspirin is started on the post-op day #1
4. watch for evidence of CSF leak
5. monitor coag studies and correct abnormalities
6. cerebral arteriogram is recommended 2-6 months post-op
ASYMPTOMATIC MOYAMOYA DISEASE
Guidelines for management of asymptomatic moyamoya disease have not yet been established. Multi-center, nation-wide survey in Japan focusing on asymptomatic moyamoya disease provided the following findings246: subtle findings of cerebral infarction and disturbed cerebral hemodynamics were detected in 20% and 40% of the involved hemispheres, respectively. Angiographic stage was more advanced in elderly patients. Of 34 medically-treated patients, 7 experienced TIA, ischemic stroke or hemorrhage during a mean follow-up period of 43.7 moths. Cerebral infarction or hemorrhage did not occur in the 6 patients who underwent surgical revascularization.
PROGNOSIS
Neurologic status at time of treatment generally predicts long-term outcome225. The mortality rate in adults (≈ 10%) is higher than for juveniles (≈ 4.3%)232. The cause of death was bleeding in 56% of 9 children and 63% of 30 adults. With treatment the prognosis is good in 58%231.
33.6. References
1. Moneta G L, Taylor D C, Nicholls S C, et al.: Operative versus nonoperative management of asymptomatic high-grade internal carotid artery stenosis. Stroke 18: 1005-10, 1987.
2. Kistler J P, Furie K L: Carotid endarterectomy revisited. N Engl J Med 342: 1743-5, 2000.
3. Heyman A, Wilkinson W E, Heyden S, et al.: Risk of stroke in asymptomatic persons with cervical arterial bruits: A population study in evans county, georgia. N Engl J Med 302 (15): N Engl J Med: 838-41, 1980.
4. Sonecha T N, Delis K T, Henein M Y: Predictive value of asymptomatic cervical bruit for carotid artery disease in coronary artery surgery revisited. Int J Cardiol 107 (2): Int J Cardiol: 225-9, 2006.
5. Cai J M, Hatsukami T S, Ferguson M S, et al.: Classification of human carotid atherosclerotic lesions with in vivo multicontrast magnetic resonance imaging. Circulation 106 (11): Circulation: 1368-73, 2002.
6. Saam T, Cai J, Ma L, et al.: Comparison of symptomatic and asymptomatic atherosclerotic carotid plaque features with in vivo MR imaging. Radiology 240 (2): Radiology: 464-72, 2006.
7. Saam T, Hatsukami T S, Takaya N, et al.: The vulnerable, or high-risk, atherosclerotic plaque: Noninvasive MR imaging for characterization and assessment. Radiology 244 (1): Radiology: 64-77, 2007.
8. Nighoghossian N, Derex L, Douek P: The vulnerable carotid artery plaque: Current imaging methods and new perspectives. Stroke 36 (12): Stroke: 2764-72, 2005.
9. Connors J J, 3rd, Sacks D, Furlan A J, et al.: Training, competency, and credentialing standards for diagnostic cervicocerebral angiography, carotid stenting, and cerebrovascular intervention: A joint statement from the American Academy of Neurology, the American association of neurological surgeons, the American society of interventional and therapeutic neuroradiology, the American society of neuroradiology, the congress of neurological surgeons, the AANS/CNS cerebrovascular section, and the society of interventional radiology. Neurology 64 (2): Neurology: 190-8, 2005.
10. Willinsky R A, Taylor S M, TerBrugge K, et al.: Neurologic complications of cerebral angiography: Prospective analysis of 2,899 procedures and review of the literature. Radiology 227 (2): Radiology: 522-8, 2003.
11. Kaufmann T J, Huston J, 3rd, Mandrekar J N, et al.: Complications of diagnostic cerebral angiography: Evaluation of 19,826 consecutive patients. Radiology 243 (3): Radiology: 812-9, 2007.
12. The North American Symptomatic Carotid Endarterectomy Trial: Beneficial effect of carotid endarterectomy in symptomatic patients with high-grade carotid stenosis. N Engl J Med 325: 445-53, 1991.
13. The European Carotid Surgery Trialists’ Collaborative Group: Randomized trial of endartectomy for recently symptomatic carotid stenosis: Final results of the MRC European carotid surgery trial (ECST). Lancet 351: 1379-87, 1998.
14. Rothwell P M, Gibson R J, Slattery J, et al.: Equivalence of measurements of carotid stenosis: A comparison of three methods on 1001 angiograms. Stroke 25: 2435-39, 1994.
15. Buskens E, Nederkoorn P J, Buijs-Van Der Woude T, et al.: Imaging of carotid arteries in symptomatic patients: Cost-effectiveness of diagnostic strategies. Radiology 233 (1): Radiology: 101-12, 2004.
16. Anson J A, Heiserman J E, Drayer B P, et al.: Surgical decisions on the basis of magnetic resonance angiography of the carotid arteries. Neurosurgery 32: 335-43, 1993.
17. Heiserman J E, Zabramski J M, Drayer B P, et al.: Clinical significance of the flow gap in carotid magnetic resonance angiography. J Neurosurg 85: 384-7, 1996.
18. Anderson C M, Saloner D, Lee R E, et al.: Assessment of carotid artery stenosis by MR angiography: Comparison with x-ray angiography and color-coded doppler ultrasound. AJNR 13: 989-1003, 1992.
19. Debrey S M, Yu H, Lynch J K, et al.: Diagnostic accuracy of magnetic resonance angiography for internal carotid artery disease: A systematic review and meta-analysis. Stroke 39 (8): Stroke: 2237-48, 2008.
20. Babiarz L S, Romero J M, Murphy E K, et al.: Contrast-enhanced MR angiography is not more accurate than unenhanced 2D time-of-flight MR angiography for determining > or = 70% internal carotid artery stenosis. AJNR Am J Neuroradiol 30 (4): AJNR Am J Neuroradiol: 761-8, 2009.
21. Koelemay M J, Nederkoorn P J, Reitsma J B, et al.: Systematic review of computed tomographic angiography for assessment of carotid artery disease. Stroke 35 (10): Stroke: 2306-12, 2004.
22. Easton J D, Saver J L, Albers G W, et al.: Definition and evaluation of transient ischemic attack: A scientific statement for healthcare professionals. Stroke 40 (6): Stroke: 2276-93, 2009.
23. Kent K C, Kuntz K M, Patel M R, et al.: Perioperative imaging strategies for carotid endarterectomy. An analysis of morbidity and cost-effectiveness in symptomatic patients. JAMA 274 (11): JAMA: 888-93, 1995.
24. Weksler B B, Pett S B, Alonso D, et al.: Differential inhibition by aspirin of vascular and platelet pros-taglandin synthesis in atherosclerotic patients. N Engl J Med 308: 800-5, 1983.
25. Grotta J C: Current medical and surgical therapy for cerebrovascular disease. N Engl J Med 317: 1505-16, 1987.
26. Théroux P, Fuster V: Acute coronary syndromes: Unstable angina and non-q-wave myocardial infarction. Circulation 97: 1195-1206, 1998.
27. Taylor D W, Barnett H J M, Haynes R B, et al.: Low-dose and high-dose acetylsalicylic acid for patients undergoing carotid endarterectomy: A randomized controlled trial. Lancet 353: 2179-84, 1999.
28. Halkes P H, van Gijn J, Kappelle L J, et al.: Aspirin plus dipyridamole versus aspirin alone after cerebral ischaemia of arterial origin (ESPRIT): Randomised controlled trial. Lancet 367 (9523): Lancet: 1665-73, 2006.
29. Diener H C, Cunha L, Forbes C, et al.: European stroke prevention study. 2. Dipyridamole and acetylsalicylic acid in the secondary prevention of stroke. J Neurol Sci 143 (1-2): J Neurol Sci: 1-13, 1996.
30. Verro P, Gorelick P B, Nguyen D: Aspirin plus dipyridamole versus aspirin for prevention of vascular events after stroke or tia: A meta-analysis. Stroke 39 (4): Stroke: 1358-63, 2008.
31. Sacco R L, Diener H C, Yusuf S, et al.: Aspirin and extended-release dipyridamole versus clopidogrel for recurrent stroke. N Engl J Med 359 (12): N Engl J Med: 1238-51, 2008.
32. Clopidogrel for reduction of atherosclerotic events. Med Letter 40: 59-60, 1998.
33. Diener H C, Bogousslavsky J, Brass L M, et al.: Aspirin and clopidogrel compared with clopidogrel alone after recent ischaemic stroke or transient ischaemic attack in high-risk patients (MATCH): Randomised, double-blind, placebo-controlled trial. Lancet 364 (9431): Lancet: 331-7, 2004.
34. Sacco R L, Adams R, Albers G, et al.: Guidelines for prevention of stroke in patients with ischemic stroke or transient ischemic attack: A statement for healthcare professionals. Stroke 37 (2): Stroke: 577-617, 2006.
35. Halliday A, Mansfield A, Marro J, et al.: Prevention of disabling and fatal strokes by successful carotid endarterectomy in patients without recent neurological symptoms: Randomised controlled trial. Lancet 363 (9420): 1491-502, 2004.
36. Biller J, Feinberg W M, Castaldo J E, et al.: Guidelines for carotid endarterectomy: A statement for healthcare professionals from a special writing group of the Stroke Council, American Heart Association. Circulation 97 (5): 501-9, 1998.
37. O’Leary D H, Polak J F, Kronmal R A, et al.: Distribution and correlates of sonographically detected carotid artery disease in the cardiovascular health study. The CHS collaborative research group. Stroke23 (12): Stroke: 1752-60, 1992.
38. Fine-Edelstein J S, Wolf P A, O’Leary D H, et al.: Precursors of extracranial carotid atherosclerosis in the Framingham study. Neurology 44 (6): Neurology: 1046-50, 1994.
39. Hillen T, Nieczaj R, Munzberg H, et al.: Carotid atherosclerosis, vascular risk profile and mortality in a population-based sample of functionally healthy elderly subjects: The Berlin ageing study. J Intern Med 247 (6): J Intern Med: 679-88, 2000.
40. Autret A, Pourcelot L, Saudeau D, et al.: Stroke risk in patients with carotid stenosis. Lancet 1 (8538): Lancet: 888-90, 1987.
41. Bogousslavsky J, Despland P-A, Regli F: Asymptomatic tight stenosis of the internal carotid artery. Neurology 36: 861-3, 1986.
42. Chambers B R, Norris J W: Outcome in patients with asymptomatic neck bruits. N Engl J Med 315 (14): N Engl J Med: 860-5, 1986.
43. Hennerici M, Hulsbomer H B, Hefter H, et al.: Natural history of asymptomatic extracranial arterial disease. Results of a long-term prospective study. Brain 110 (Pt 3): Brain: 777-91, 1987.
44. Mackey A E, Abrahamowicz M, Langlois Y, et al.: Outcome of asymptomatic patients with carotid disease. Asymptomatic cervical bruit study group. Neurology 48 (4): Neurology: 896-903, 1997.
45. Meissner I, Wiebers D O, Whisnant J P, et al.: The natural history of asymptomatic carotid artery occlusive lesions. JAMA 258 (19): JAMA: 2704-7, 1987.
46. Nadareishvili Z G, Rothwell P M, Beletsky V, et al.: Long-term risk of stroke and other vascular events in patients with asymptomatic carotid artery stenosis. Arch Neurol 59 (7): Arch Neurol: 1162-6, 2002.
47. Aichner F T, Topakian R, Alberts M J, et al.: High cardiovascular event rates in patients with asymptomatic carotid stenosis: The REACH registry. Eur J Neurol: Eur J Neurol, 2009.
48. Hobson R W, Weiss D G, Fields W S, et al.: Efficacy of carotid endarterectomy for asymptomatic carotid stenosis. N Engl J Med 328: 221-7, 1993.
49. The Executive Committee for the Asymptomatic Carotid Atherosclerosis Study: Endarterectomy for asymptomatic carotid artery stenosis. JAMA 273: 1421-8, 1995.
50. Mattos M A, Modi J R, Mansour M A, et al.: Evolution of carotid endarterectomy in two community hospitals: Springfield revisited - seventeen years and 2243 operations later. J Vasc Surg 21: 719-28, 1995.
51. CASANOVA Study Group: Carotid surgery versus medical therapy in asymptomatic carotid stenosis. Stroke 22: 1229-35, 1991.
52. Mayberg M R, Winn H R: Endarterectomy for asymptomatic carotid artery stenosis. Resolving the controversy. JAMA 273: 1459-61, 1995.
53. Mayo Asymptomatic Carotid Endarterectomy Study Group: Results of a randomized controlled trial of carotid endarterectomy for asymptomatic carotid stenosis. Mayo Clin Proc 67 (6): Mayo Clin Proc: 513-8, 1992.
54. Goldstein L B, Adams R, Alberts M J, et al.: Primary prevention of ischemic stroke. Stroke 37 (6): Stroke: 1583-633, 2006.
55. Liapis C D, Bell P R, Mikhailidis D, et al.: ESVS guidelines. Invasive treatment for carotid stenosis: Indications, techniques. Eur J Vasc Endovasc Surg 37 (4 Suppl): Eur J Vasc Endovasc Surg: 1-19, 2009.
56. U.S. Preventive Services Task Force: Screening for carotid artery stenosis: U.S. Preventive services task force recommendation statement. Ann Intern Med 147 (12): Ann Intern Med: 854-9, 2007.
57. Qureshi A I, Alexandrov A V, Tegeler C H, et al.: Guidelines for screening of extracranial carotid artery disease. J Neuroimaging 17 (1): J Neuroimaging: 19-47, 2007.
58. Society for Vascular Surgery: SVS position statement on vascular screenings, 2007. (June 12. 2009), 2007.
59. Chassin M R: Appropriate use of carotid endarterectomy. N Engl J Med 339: 1468-71, 1998 (editorial).
60. Barnett H J M, Taylor W, Eliasziw M, et al.: Benefit of carotid endarterectomy in patients with symptomatic moderate or severe stenosis. N Engl J Med 339: 1415-25, 1998.
61. The European Carotid Surgery Trialists’ Collaborative Group: Endartectomy for moderate symptomatic carotid stenosis: Interim results of the MRC European carotid surgery trial. Lancet 347: 1591-3, 1996.
62. Faries P L, Chaer R A, Patel S, et al.: Current management of extracranial carotid artery disease. Vasc Endovascular Surg 40 (3): Vasc Endovascular Surg: 165-75, 2006.
63. Rouleau P A, Huston J, 3rd, Gilbertson J, et al.: Carotid artery tandem lesions: Frequency of angiographic detection and consequences for endarterectomy. AJNR Am J Neuroradiol 20 (4): AJNR Am J Neuroradiol: 621-5, 1999.
64. Bond R, Rerkasem K, Rothwell P M: Systematic review of the risks of carotid endarterectomy in relation to the clinical indication for and timing of surgery. Stroke 34 (9): Stroke: 2290-301, 2003.
65. Rothwell P M, Eliasziw M, Gutnikov S A, et al.: Endarterectomy for symptomatic carotid stenosis in relation to clinical subgroups and timing of surgery. Lancet 363 (9413): Lancet: 915-24, 2004.
66. Bartoli M A, Squarcioni C, Nicoli F, et al.: Early carotid endarterectomy after intravenous thrombolysis for acute ischaemic stroke. Eur J Vasc Endovasc Surg 37 (5): Eur J Vasc Endovasc Surg: 512-8, 2009.
67. Nguyen L L, Conte M S, Reed A B, et al.: Carotid endarterectomy: Who is the high-risk patient? Semin Vasc Surg 17 (3): Semin Vasc Surg: 219-23, 2004.
68. Kang J L, Chung T K, Lancaster R T, et al.: Outcomes after carotid endarterectomy: Is there a high-risk population? A national surgical quality improvement program report. J Vasc Surg 49 (2): J Vasc Surg: 331-8, 339 e1; discussion 338-9, 2009.
69. Yadav J S, Wholey M H, Kuntz R E, et al.: Protected carotidartery stenting versus endarterectomy in high-risk patients. N Engl J Med 351 (15): N Engl J Med: 1493-501, 2004.
70. Gray W A, Hopkins L N, Yadav S, et al.: Protected carotid stenting in high-surgical-risk patients: The ARCHeR results. J Vasc Surg 44 (2): J Vasc Surg: 258-68, 2006.
71. CAVATAS Investigators: Endovascular versus surgical treatment in patients with carotid stenosis in the carotid and vertebral artery transluminal angioplasty study (CAVATAS): A randomised trial. Lancet357 (9270): Lancet: 1729-37, 2001.
72. Alberts M J: Results of a multicenter prospective randomized trial of carotid artery stenting vs. Carotid endarterectomy. Stroke 32: Stroke: 325d, 2001.
73. Mas J L, Chatellier G, Beyssen B, et al.: Endarterectomy versus stenting in patients with symptomatic severe carotid stenosis. N Engl J Med 355 (16): N Engl J Med: 1660-71, 2006.
74. Ringleb P A, Allenberg J, Bruckmann H, et al.: 30 day results from the SPACE trial of stent-protected angioplasty versus carotid endarterectomy in symptomatic patients: A randomised non-inferiority trial. Lancet 368 (9543): Lancet: 1239-47, 2006.
75. CaRESS Steering Committee: Carotid revascularization using endarterectomy or stenting systems (CaRESS) phase I clinical trial: 1-year results. J Vasc Surg 42 (2): J Vasc Surg: 213-9, 2005.
76. White C J, Iyer S S, Hopkins L N, et al.: Carotid stenting with distal protection in high surgical risk patients: The BEACH trial 30 day results. Catheter Cardiovasc Interv 67 (4): Catheter Cardiovasc Interv: 503-12, 2006.
77. Safian R D, Bresnahan J F, Jaff M R, et al.: Protected carotid stenting in high-risk patients with severe carotid artery stenosis. J Am Coll Cardiol 47 (12): J Am Coll Cardiol: 2384-9, 2006.
78. Hill M D, Morrish W, Soulez G, et al.: Multicenter evaluation of a self-expanding carotid stent system with distal protection in the treatment of carotid stenosis. AJNR Am J Neuroradiol 27 (4): AJNR Am J Neuroradiol: 759-65, 2006.
79. Fairman R, Gray W A, Scicli A P, et al.: The CAPTURE registry: Analysis of strokes resulting from carotid artery stenting in the post approval setting: Timing, location, severity, and type. Ann Surg 246 (4): Ann Surg: 551-6; discussion 556-8, 2007.
80. Iyer S S, White C J, Hopkins L N, et al.: Carotid artery revascularization in high-surgical-risk patients using the carotid WALLSTENT and FilterWire EX/EZ: 1-year outcomes in the BEACH pivotal group. J Am Coll Cardiol51 (4): J Am Coll Cardiol: 427-34, 2008.
81. Bosiers M, Peeters P, Deloose K, et al.: Does carotid artery stenting work on the long run: 5-year results in high-volume centers (ELOCAS registry). J Cardiovasc Surg (Torino) 46 (3): J Cardiovasc Surg (Torino): 241-7, 2005.
82. Theiss W, Hermanek P, Mathias K, et al.: Pro-CAS: A prospective registry of carotid angioplasty and stenting. Stroke 35 (9): Stroke: 2134-9, 2004.
83. Zahn R, Roth E, Ischinger T, et al.: Carotid artery stenting in clinical practice results from the carotid artery stenting (CAS)-registry of the Arbeitsgemeinschaft Leitende Kardiologische Krankenhausarzte (ALKK). Z Kardiol 94 (3): Z Kardiol: 163-72, 2005.
84. Goldstein L B: New data about stenting versus endarterectomy for symptomatic carotid artery stenosis. Curr Treat Options Cardiovasc Med 11 (3): Curr Treat Options Cardiovasc Med: 232-40, 2009.
85. Fayad P: Endarterectomy and stenting for asymptomatic carotid stenosis: A race at breakneck speed. Stroke 38 (2 Suppl): Stroke: 707-14, 2007.
86. Naylor A R, Bell P R: Treatment of asymptomatic carotid disease with stenting: Con. Semin Vasc Surg 21 (2): Semin Vasc Surg: 100-7, 2008.
87. Ederle J, Featherstone R L, Brown M M: Percutaneous transluminal angioplasty and stenting for carotid artery stenosis. Cochrane Database Syst Rev (4): Cochrane Database Syst Rev: CD000515, 2007.
88. Cremonesi A, Setacci C, Bignamini A, et al.: Carotid artery stenting: First consensus document of the ICCS-SPREAD joint committee. Stroke 37 (9): Stroke: 2400-9, 2006.
89. Spetzler R F, Martin N, Hadley M N, et al.: Micro-surgical endarterectomy under barbiturate protection: A prospective study. J Neurosurg 65: 63-73, 1986.
90. Mayo Asymptomatic Carotid Endarterectomy Study Group: Results of a randomized controlled trial of carotid endarterectomy for asymptomatic carotid stenosis. Mayo Clin Proc 67: 513-8, 1992.
91. Harker L A, Bernstein E F, Dilley R B, et al.: Failure of aspirin plus dipyridamole to prevent restenosis after carotid endarterectomy. Ann Int Med 116: 731-6, 1992.
92. McPhee J T, Hill J S, Ciocca R G, et al.: Carotid endarterectomy was performed with lower stroke and death rates than carotid artery stenting in the United States in 2003 and 2004. J Vasc Surg 46 (6): J Vasc Surg: 1112-1118, 2007.
93. Branch C L, Davis C H: False aneurysm complicating carotid endarterectomy. Neurosurgery 19: 421-5, 1986.
94. McCollum C H, Wheeler W G, Noon G P, et al.: Aneurysms of the extracranial carotid artery. Am J Surg 137: 196-200, 1979.
95. Welling R E, Taha A, Goel T, et al.: Extracranial carotid artery aneurysms. Surgery 93: 319-23, 1983.
96. Brott T G, Labutta R J, Kempczinski R F: Changing patterns in the practice of carotid endarterectomy in a large metropolitan area. JAMA 255: 2609-12, 1986.
97. Reigel M M, Hollier L H, Sundt T M, et al.: Cerebral hyperperfusion syndrome: A cause of neurologic dysfunction after carotid endarterectomy. J Vasc Surg 5: 628-34, 1987.
98. Piepgras D G, Morgan M K, Sundt T M, et al.: Intracerebral hemorrhage after carotid endarterectomy. J Neurosurg 68: 532-6, 1988.
99. Ascher E, Markevich N, Schutzer R W, et al.: Cerebral hyperperfusion syndrome after carotid endarterectomy: Predictive factors and hemodynamic changes. J Vasc Surg 37 (4): 769-77, 2003.
100. Sundt T M: Occlusive cerebrovascular disease. W. B. Saunders, Philadelphia, 1987.
101. Kieburtz K, Ricotta J J, Moxley R T: Seizures following carotid endarterectomy. Arch Neurol 47: 568-70, 1990.
102. Sundt T M, Sharbrough F W, Piepgras D G, et al.: Correlation of cerebral blood flow and electroencephalographic changes during carotid endarterectomy. Mayo Clin Proc 56: 533-43, 1981.
103. Wilkinson J T, Adams H P, Wright C B: Convulsions after carotid endarterectomy. JAMA 244: 1827-8, 1980.
104. Bernstein E F, Humber P B, Collins G M, et al.: Life expectancy and late stroke following carotid enderterectomy. Ann Surg 198: 80-6, 1983.
105. Callow A D: Recurrent stenosis after carotid endarterectomy. Arch Surg 117: 1082-5, 1982.
106. Dolan J G, Mushlin A I: Hypertension, vascular headaches, and seizures after carotid endarterectomy. Arch Intern Med 144: 1489-91, 1984.
107. Caplan L R, Skillman J, Ojemann R, et al.: Intracerebral hemorrhage following carotid endarterectomy: A hypertensive complication. Stroke 9: 457-60, 1979.
108. Sajid M S, Vijaynagar B, Singh P, et al.: Literature review of cranial nerve injuries during carotid endarterectomy. Acta Chir Belg 107 (1): Acta Chir Belg: 25-8, 2007.
109. Imparato A M, Bracco A, Kim G E, et al.: The hypoglossal nerve in carotid arterial reconstructions. Stroke 3: 576-8, 1972.
110. Skydell J L, Machleder H I, Baker J D, et al.: Incidence and mechanism of postcarotid endarterectomy hypertension. Arch Surg 122: 1153-5, 1987.
111. Lehv M S, Salzman E W, Silen W: Hypertension complicating carotid endarterectomy. Stroke 1: 307-13, 1970.
112. Baker W H: Management of stroke during and after carotid surgery. In Cerebrovascular insufficiency, Bergan J J and Yao J S T, (eds.). Grune and Stratton, New York, 1983: pp 481-95.
113. Zuccarello M, Yeh H-S, Tew J M: Morbidity and mortality of carotid endarterectomy under local anesthesia: A retrospective study. Neurosurgery 23: 445-50, 1988.
114. Lee K S, Courtland C H, McWhorter J M: Low morbidity and mortality of carotid endarterectomy performed with regional anesthesia. J Neurosurg 69: 483-7, 1988.
115. Forssell C, Takolander R, Bergqvist D, et al.: Local versus general anesthesia in carotid surgery. A prospective randomized study. Eur J Vasc Surg 3: 503-9, 1989.
116. Lewis S C, Warlow C P, Bodenham A R, et al.: General anaesthesia versus local anaesthesia for carotid surgery (GALA): A multicentre, randomised controlled trial. Lancet 372 (9656): Lancet: 2132-42, 2008.
117. Rerkasem K, Rothwell P M: Local versus general anaesthesia for carotid endarterectomy. Cochrane Database Syst Rev (4): Cochrane Database Syst Rev: CD000126, 2008.
118. Bond R, Rerkasem K, AbuRahma A F, et al.: Patch angioplasty versus primary closure for carotid endarterectomy. Cochrane Database Syst Rev (2): Cochrane Database Syst Rev: CD000160, 2004.
119. Karkos C D, Hernandez-Lahoz I, Naylor A R: Urgent carotid surgery in patients with crescendo transient ischaemic attacks and stroke-in-evolution: A systematic review. Eur J Vasc Endovasc Surg 37 (3): Eur J Vasc Endovasc Surg: 279-88, 2009.
120. Walters B B, Ojemann R G, Heros R C: Emergency carotid endarterectomy. J Neurosurg 66: 817-23, 1987.
121. Hankey G J, Warlow C P: Prognosis of symptomatic carotid artery occlusion: An overview. Cerebrovasc Dis 1: 245-56, 1991.
122. Grubb R L, Jr., Derdeyn C P, Fritsch S M, et al.: Importance of hemodynamic factors in the prognosis of symptomatic carotid occlusion. JAMA 280 (12): JAMA: 1055-60, 1998.
123. Powers W J, Derdeyn C P, Fritsch S M, et al.: Benign prognosis of never-symptomatic carotid occlusion. Neurology 54 (4): Neurology: 878-82, 2000.
124. Hafner C D, Tew J M: Surgical management of the totally occluded internal carotid artery. Surgery 89: 710-7, 1981.
125. Satiani B, Burns J, Vasko J S: Surgical and nonsurgical treatment of total carotid artery occlusion. Am J Surg 149: 362-7, 1985.
126. Sugg R M, Malkoff M D, Noser E A, et al.: Endovascular recanalization of internal carotid artery occlusion in acute ischemic stroke. AJNR Am J Neuroradiol 26 (10): AJNR Am J Neuroradiol: 2591-4, 2005.
127. Nesbit G M, Clark W M, O’Neill O R, et al.: Intracranial intraarterial thrombolysis facilitated by microcatheter navigation through an occluded cervical internal carotid artery. J Neurosurg 84 (3): J Neurosurg: 387-92, 1996.
128. Endo S, Kuwayama N, Hirashima Y, et al.: Results of urgent thrombolysis in patients with major stroke and atherothrombotic occlusion of the cervical internal carotid artery. AJNR Am J Neuroradiol19 (6): AJNR Am J Neuroradiol: 1169-75, 1998.
129. Srinivasan A, Goyal M, Stys P, et al.: Microcatheter navigation and thrombolysis in acute symptomatic cervical internal carotid occlusion. AJNR Am J Neuroradiol 27 (4): AJNR Am J Neuroradiol: 774-9, 2006.
130. Imai K, Mori T, Izumoto H, et al.: Emergency carotid artery stent placement in patients with acute ischemic stroke. AJNR Am J Neuroradiol 26 (5): AJNR Am J Neuroradiol: 1249-58, 2005.
131. Jovin T G, Gupta R, Uchino K, et al.: Emergent stenting of extracranial internal carotid artery occlusion in acute stroke has a high revascularization rate. Stroke 36 (11): Stroke: 2426-30, 2005.
132. McCormick P W, Spetzler R F, Bailes J E, et al.: Thromboendarterectomy of the symptomatic occluded internal carotid artery. J Neurosurg 76: 752-8, 1992.
133. Hopkins L N, Martin N A, Hadley M N, et al.: Vertebrobasilar insufficiency, part 2: Microsurgical treatment of intracranial vertebrobasilar disease. J Neurosurg 66: 662-74, 1987.
134. Robertson J T: Current management of vertebral basilar occlusive disease. Clin Neurosurg 31: 165-87, 1983.
135. Ausman J I, Shrontz C E, Pearce J E, et al.: Vertebrobasilar insufficiency: A review. Arch Neurol 42: 803-8, 1985.
136. Diaz F G, Ausman J I, de los Reyes R A, et al.: Surgical reconstruction of the proximal vertebral artery. J Neurosurg 61: 874-81, 1984.
137. Fox M W, Piepgras D G, Bartleson J D: Anterolateral decompression of the atlantoaxial vertebral artery for symptomatic positional occlusion of the vertebral artery. J Neurosurg 83: 737-40, 1995.
138. Pratt-Thomas H R, Berger K E: Cerebellar and spinal injuries after chiropractic manipulation. JAMA 133: 600-3, 1947.
139. Matsuyama T, Morimoto T, Sakaki T: Bow hunter’s stroke caused by a nondominant vertebral artery occlusion: Case report. Neurosurgery 41: 1393-5, 1997.
140. Lemole G M, Henn J S, Spetzler R F, et al.: Bow hunter’s stroke. BNI Quarterly 17: 4-10, 2001.
141. Mapstone T, Spetzler R F: Vertebrobasilar insufficiency secondary to vertebral artery occlusion from a fibrous band. Case report. J Neurosurg 56: 581-3, 1982.
142. George B, Laurian C: Impairment of vertebral artery flow caused by extrinsic lesions. Neurosurgery 24: 206-14, 1989.
143. Kuether T A, Nesbit G M, Clark W M, et al.: Rotational vertebral artery occlusion: A mechanism of vertebrobasilar insufficiency. Neurosurgery 41: 427-33, 1997.
144. Pamphlett R, Raisanen J, Kum-Jew S: Vertebral artery compression resulting from head movement: A possible cause of the sudden infant death syndrome. Pediatrics 103 (2): 460-8, 1999.
145. Okawara S, Nibbelink D: Vertebral artery occlusion following hyperextension and rotation of the head. Stroke 5 (5): 640-2, 1974.
146. Ford F R: Syncope, vertigo and disturbances of vision resulting from intermittent obstruction of vertebral arteries due to defect in odontoid process and excessive mobility of second cervical vertebra. Bull Johns Hopkins Hosp91: 168-73, 1952.
147. Tatlow W F T, Bammer H G: Syndrome of vertebral artery compression. Neurology 7: 331-40, 1957.
148. Matsuyama T, Morimoto T, Sakaki T: Comparison of C1-2 posterior fusion and decompression of the vertebral artery in the treatment of bow hunter’s stroke. J Neurosurg 86 (4): 619-23, 1997.
149. Shimizu T, Waga S, Kojima T, et al.: Decompression of the vertebral artery for bow-hunter’s stroke. Case report. J Neurosurg 69 (1): 127-31, 1988.
150. Yamaura A: Nontraumatic intracranial arterial dissection: Natural history, diagnosis, and treatment. Contemp Neurosurg 16 (5): 1-6, 1994.
151. Goldstein S J: Dissecting hematoma of the cervical vertebral artery: Case report. J Neurosurg 56: 451-4, 1982.
152. Anson J, Crowell R M: Cervicocranial arterial dissection. Neurosurgery 29: 89-96, 1991.
153. Yamashita M, Tanaka K, Matsuo T, et al.: Cerebral dissecting aneurysms in patients with moyamoya disease. J Neurosurg 58: 120-5, 1983.
154. Bogousslavsky J, Despland P A, Regli F: Spontaneous carotid dissection with acute stroke. Arch Neurol 44: 137-40, 1987.
155. Friedman A H, Drake C G: Subarachnoid hemorrhage from intracraniai dissecting aneurysm. J Neurosurg 60: 325-34, 1984.
156. Hodge C, Leeson M, Cacayorin E, et al.: Computed tomographic evaluation of extracranial carotid artery disease. Neurosurgery 21: 167-76, 1987.
157. Eastman A L, Chason D P, Perez C L, et al.: Computed tomographic angiography for the diagnosis of blunt cervical vascular injury: Is it ready for prime-time? J Trauma 60 (5): J Trauma: 925-9; discussion 929, 2006.
158. Kitanaka C, Tanaki J-I, Kuwahara M, et al.: Nonsurgical treatment of unruptured intracranial vertebral artery dissection with serial follow-up angiography. J Neurosurg 80: 667-74, 1994.
159. Berger M S, Wilson C B: Intracranial dissecting aneurysms of the posterior circulation. Report of six cases and review of the literature. J Neurosurg 61: 882-94, 1984.
160. Pozzati E, Padovani R, Fabrizi A, et al.: Benign arterial dissection of the posterior circulation. J Neurosurg 75: 69-72, 1991.
161. Chang V, Newcastle N B, Harwood-Nash D C F, et al.: Bilateral dissecting aneurysms of the intracranial internal carotid arteries in an 8-year-old boy. Neurology 25: 573-9, 1975.
162. Leys D, Lesoin F, Pruvo J P, et al.: Bilateral spontaneous dissection of extracranial vertebral arteries. J Neurol 234: 237-40, 1987.
163. Halbach V V, Higashida R T, Dowd C F, et al.: Endovascular treatment of vertebral artery dissections and pseudoaneurysms. J Neurosurg 79: 183-91, 1993.
164. Miyazaki S, Yamaura A, Kamata K, et al.: A dissecting aneurysm of the vertebral artery. Surg Neurol 21: 171-4, 1984.
165. Hugenholtz H, Pokrupa R, Montpetit V J A, et al.: Spontaneous dissecting aneurysm of the extracranial vertebral artery. Neurosurgery 10: 96-100, 1982.
166. Senter H J, Sarwar M: Nontraumatic dissecting aneurysm of the vertebral artery. J Neurosurg 56: 128-30, 1982.
167. Shimoji T, Bando K, Nakajima K, et al.: Dissecting aneurysm of the vertebral artery. J Neurosurg 61: 1038-46, 1984.
168. Okuchi K, Watabe Y, Hiramatsu K, et al.: [dissecting aneurysm of the vertebral artery as a cause of Wallenberg’s syndrome]. No Shinkei Geka 18: 721-7, 1990 (Japan).
169. Caplan L R, Zarins C K, Hemmati M: Spontaneous dissection of the extracranial vertebral arteries. Stroke 16: 1030-8, 1985.
170. Aoki N, Sakai T: Rebleeding from intracranial dissecting aneurysm in the vertebral artery. Stroke 21: 1628-31, 1990.
171. Pozzati E, Andreoli A, Limoni P, et al.: Dissecting aneurysms of the vertebrobasilar system: Study of 16 cases. Surg Neurol 41: 119-24, 1994.
172. Yamaura A, Watanabe Y, Saeki N: Dissecting aneurysms of the intracranial vertebral artery. J Neurosurg 72: 183-8, 1990.
173. Six E G, Stringer W L, Cowley A R, et al.: Posttraumatic bilateral vertebral artery occlusion. Case report. J Neurosurg 54: 814-7, 1981.
174. Yamada K, Hayakawa T, Ushio Y, et al.: Therapeutic occlusion of the vertebral artery for unclippable vertebral aneurysm. Neurosurgery 15: 834-8, 1984.
175. Crowley R W, Medel R, Dumont A S: Evolution of cerebral revascularization techniques. Neurosurgical Focus 24 (2): Neurosurgical Focus: E3, 2008.
176. Amin-Hanjani S, Butler W E, Ogilvy C S, et al.: Extracranial-intracranial bypass in the treatment of occlusive cerebrovascular disease and intracranial aneurysms in the United States between 1992 and 2001: A population-based study. J Neurosurg 103 (5): J Neurosurg: 794-804, 2005.
177. EC/IC Study Group: Failure of EC-ic arterial bypass to reduce the risk of ischemic stroke. N Engl J Med 313: 1191-200, 1985.
178. Garrett M C, Komotar R J, Merkow M B, et al.: The extracranial-intracranial bypass trial: Implications for future investigations. Neurosurgical Focus 24 (2): Neurosurgical Focus: E4, 2008.
179. Garrett M C, Komotar R J, Starke R M, et al.: The efficacy of direct extracranial-intracranial bypass in the treatment of symptomatic hemodynamic failure secondary to athero-occlusive disease: A systematic review. Clin Neurol Neurosurg 111 (4): Clin Neurol Neurosurg: 319-26, 2009.
180. Baron J C, Bousser M G, Rey A, et al.: Reversal of focal “misery-perfusion syndrome” by extra-intracranial arterial bypass in hemodynamic cerebral ischemia. A case study with 15o positron emission tomography. Stroke 12 (4): Stroke: 454-9, 1981.
181. Powers W J, Press G A, Grubb R L, Jr., et al.: The effect of hemodynamically significant carotid artery disease on the hemodynamic status of the cerebral circulation. Ann Intern Med 106 (1): Ann Intern Med: 27-34, 1987.
182. Grubb R L, Jr., Powers W J, Derdeyn C P, et al.: The carotid occlusion surgery study. Neurosurg Focus 14 (3): e9, 2003.
183. Kappelle L J, Klijn C J, Tulleken C A: Management of patients with symptomatic carotid artery occlusion. Clin Exp Hypertens 24 (7-8): Clin Exp Hypertens: 631-7, 2002.
184. Kuroda S, Houkin K, Kamiyama H, et al.: Long-term prognosis of medically treated patients with internal carotid or middle cerebral artery occlusion: Can acetazolamide test predict it? Stroke 32 (9): Stroke: 2110-6, 2001.
185. Cantore G, Santoro A, Guidetti G, et al.: Surgical treatment of giant intracranial aneurysms: Current viewpoint. Neurosurgery 63 (4 Suppl 2): Neurosurgery: 279-89; discussion 289-90, 2008.
186. Mohit A A, Sekhar L N, Natarajan S K, et al.: High-flow bypass grafts in the management of complex intracranial aneurysms. Neurosurgery 60 (2 Suppl 1): Neurosurgery: ONS105-22; discussion ONS122-3, 2007.
187. O’Shaughnessy B A, Salehi S A, Mindea S A, et al.: Selective cerebral revascularization as an adjunct in the treatment of giant anterior circulation aneurysms. Neurosurgical Focus 14 (3): Neurosurgical Focus: e4, 2003.
188. Schaller B: Extracranial-intracranial bypass to reduce the risk of ischemic stroke in intracranial aneurysms of the anterior cerebral circulation: A systematic review. J Stroke Cerebrovasc Dis 17 (5): J Stroke Cerebrovasc Dis: 287-98, 2008.
189. Sekhar L N, Bucur S D, Bank W O, et al.: Venous and arterial bypass grafts for difficult tumors, aneurysms, and occlusive vascular lesions: Evolution of surgical treatment and improved graft results. Neurosurgery 44 (6): Neurosurgery: 1207-23; discussion 1223-4, 1999.
190. Liu J K, Kan P, Karwande S V, et al.: Conduits for cerebrovascular bypass and lessons learned from the cardiovascular experience. Neurosurgical Focus 14 (3): Neurosurgical Focus: e3, 2003.
191. Wilkins R H, Rengachary S S, (eds.): Neurosurgery. McGraw-Hill, New York, 1985.
192. Symonds C P: Otitic hydrocephalus. Brain 54: 55-71, 1931.
193. Garcia R D J, Baker A S, Cunningham M J, et al.: Lateral sinus thrombosis associated with otitis media and mastoiditis in children. Pediatr Infect Dis J 14: 617-23, 1995.
194. Dolan R W, Chowdry K: Diagnosis and treatment of intracranial complications of paranasal sinus infections. J Oral Maxillofac Surg 53: 1080-7, 1995.
195. Shende M C, Lourie H: Sagittal sinus thrombosis related to oral contraceptives: Case report. J Neurosurg 33: 714-7, 1970.
196. Mahasin Z Z, Saleem M, Gangopadhyay K: Transverse sinus thrombosis and venous infarction of the brain following unilateral radical neck dissection. J Laryngol Otol 112: 88-91, 1998.
197. Flusser D, Abu-Shakra M, Baumgarten-Kleiner A, et al.: Superior sagittal sinus thrombosis in a patient with systemic lupus erythematosus. Lupus 5: 334-6, 1996.
198. Bugnicourt J M, Roussel B, Tramier B, et al.: Cerebral venous thrombosis and plasma concentrations of factor VIII and von Willebrand factor: A case control study. J Neurol Neurosurg Psychiatry 78 (7): J Neurol Neurosurg Psychiatry: 699-701, 2007.
199. Bousser M G: Cerebral vein thrombosis in Bechet’s syndrome. Arch Neurol 39: 322, 1982.
200. Wilder-Smith E, Kothbauer-Margreiter I, Lämmle B, et al.: Dural puncture and activated protein c resistance: Risk factors for cerebral venous sinus thrombosis. J Neurol Neurosurg Psychiatry 63: 351-6, 1997.
201. Estanol B, Rodriguez A, Conte G, et al.: Intracranial venous thrombosis in young women. Stroke 10: 680-4, 1979.
202. Brenner B: Haemostatic changes in pregnancy. Thromb Res 114 (5-6): Thromb Res: 409-14, 2004.
203. Ferrera P C, Pauze D R, Chan L: Sagittal sinus thrombosis after closed head injury. Am J Emerg Med 16: 382-5, 1998.
204. Elsherbiny S M, Grunewald R A, Powell T: Isolated inferior sagittal sinus thrombosis: A case report. Neuroradiology 39: 411-3, 1997.
205. Gerszten P C, Welch W C, Spearman M P, et al.: Isolated deep cerebral venous thrombosis treated by direct endovascular thrombolysis. Surg Neurol 48: 261-6, 1997.
206. Sofferman R A: Cavernous sinus thrombosis secondary to sphenoid sinusitis. Laryngoscope 93: 797-800, 1983.
207. Kriss T C, Kriss V M, Warf B C: Cavernous sinus thrombophlebitis: Case report. Neurosurgery 39: 385-9, 1996.
208. Bousser M G, Chiras J, Bories J, et al.: Cerebral venous thrombosis - a review of 38 cases. Stroke 16: 199-213, 1985.
209. Kalbag R M: Cerebral venous thrombosis. In The cerebral venous system and its disorders, Kapp J P and Schmidek H H, (eds.). Grune and Stratton, Orlando, 1984: pp 505-36.
210. Perkin G D: Cerebral venous thrombosis: Developments in imaging and treatment. J Neurol Neurosurg Psychiatry 59: 1-3, 1995.
211. Yeakley J W, Mayer J S, Patchell L L, et al.: The pseudodelta sign in acute head trauma. J Neurosurg 69: 867-8, 1988.
212. Rao K C V G, Knipp H C, Wagner E J: CT findings in cerebral sinus and venous thrombosis. Radiology 140: 391-8, 1981.
213. Virapongse C, Cazenave C, Quisling R, et al.: The empty delta sign: Frequency and significance in 76 cases of dural sinus thrombosis. Radiology 162: 779-85, 1987.
214. Lam A H: Doppler imaging of superior sagittal sinus thrombosis. J Ultrasound Med 14: 41-6, 1995.
215. Levine S R, Twyman R E, Gilman S: The role of anticoagulation in cavernous sinus thrombosis. Neurology 38: 517-22, 1988.
216. Villringer A, Garner C, Meister W, et al.: High-dose heparin treatment in cerebral sinus thrombosis. Stroke 19: 135, 1988 (abstract).
217. Einhäupl K M, Villringer A, Meister W, et al.: Heparin treatment in sinus venous thrombosis. Lancet 338: 597-600, 1991.
218. Horowitz M, Purdy P, Unwin H, et al.: Treatment of dural sinus thrombosis using selective catheterization and urokinase. Ann Neurol 38: 58-67, 1995.
219. Alexander L F, Tamamoto Y, Ayoubi S, et al.: Efficacy of tissue plasminogen activator in the lysis of thrombosis of the cerebral venous sinus. Neurosurgery 26: 559-64, 1990.
220. Horton J C, Seiff S R, Pitts L H, et al.: Decompression of the optic nerve sheath for vision-threatening papilledema caused by dural sinus occlusion. Neurosurgery 31: 203-12, 1992.
221. Stam J, Majoie C B, van Delden O M, et al.: Endovascular thrombectomy and thrombolysis for severe cerebral sinus thrombosis: A prospective study. Stroke 39 (5): 1487-90, 2008.
222. Suzuki J, Takaku A: Cerebrovascular “moyamoya” disease: Disease showing abnormal net-like vessels in base of brain. Arch Neurol 20: 288-99, 1969.
223. Kayama T, Suzuki S, Sakurai Y, et al.: A case of moyamoya disease accompanied by an arteriovenous malformation. Neurosurgery 18: 465-8, 1986.
224. Lichtor T, Mullan S: Arteriovenous malformation in moyamoya syndrome: Report of three cases. J Neurosurg 67: 603-8, 1987.
225. Smith E R, Scott R M: Surgical management of moyamoya syndrome. Skull Base 15 (1): 15-26, 2005.
226. Kwak R, Ito S, Yamamoto N, et al.: Significance of intracranial aneurysms associated with moyamoya disease (part I): Differences between intracranial aneurysms associated with moyamoya disease and usual saccular aneurysms - review of the literature. Neurol Med Chir 24: 97-103, 1984.
227. Zipfel G J, Fox D J, Jr., Rivet D J: Moyamoya disease in adults: The role of cerebral revascularization. Skull Base 15 (1): 27-41, 2005.
228. Rajakulasingam K, Cerullo L J, Raimondi A J: Childhood moyamoya syndrome: Postradiation pathogenesis. Childs Brain 5: 467-75, 1979.
229. Kuroda S, Ishikawa T, Houkin K, et al.: Incidence and clinical features of disease progression in adult moyamoya disease. Stroke 36 (10): 2148-53, 2005.
230. Chang S D, Steinberg G K: Surgical management of moyamoya disease. Contemp Neurosurg 22 (10): 1-9, 2000.
231. Ueki K, Meyer F B, Mellinger J F: Moyamoya disease: The disorder and surgical treatment. Mayo Clin Proc 69: 749-57, 1994.
232. Nishimoto A: Moyamoya disease. Neurol Med Chir 19: 221-8, 1979.
233. Kodama N, Aoki Y, Hiraga H, et al.: Electroencephalographic findings in children with moyamoya disease. Arch Neurol 36: 16-9, 1979.
234. Ogawa A, Yoshimoto T, Suzuki J, et al.: Cerebral blood flow in moyamoya disease. Part 1. Correlation with age and regional distribution. Acta Neurochir 105: 30-4, 1990.
235. Ogawa A, Nakamura N, Yoshimoto T, et al.: Cerebral blood flow in moyamoya disease. Part 2. Autoregulation and co2 response. Acta Neurochir 105: 107-11, 1990.
236. Matsushima Y, Inoue T, Suzuki S O, et al.: Surgical treatment of moyamoya disease in pediatric patients - comparison between the results of indirect and direct vascularization. Neurosurgery 31: 401-5, 1992.
237. Ishikawa T, Houkin K, Kamiyama H, et al.: Effects of surgical revascularization on outcome of patients with pediatric moyamoya disease. Stroke 28: 1170-3, 1997.
238. Fabi A Y, Meyer F B: Moyamoya disease. Contemp Neurosurg 19 (15): 1-6, 1997.
239. Karasawa J, Kikuchi H, Furuse S, et al.: Treatment of moyamoya disease with sta-mca anastamosis. J Neurosurg 49: 679-88, 1978.
240. Matsushima Y, Fukai N, Tanaka K, et al.: A new surgical treatment of moyamoya disease in children: A preliminary report. Surg Neurol 15: 313-20, 1980.
241. Matsushima Y, Inaba Y: Moyamoya disease in children and its surgical treatment. Childs Brain 11: 155-70, 1984.
242. Kashiwagi S, Kato S, Yasuhara S, et al.: Use of split dura for revascularization if ischemic hemispheres in moyamoya disease. J Neurosurg 85: 380-3, 1996.
243. Karasawa J, Kikuchi H, Kawamura J, et al.: Intracranial transplantation of the omentum for cerebrovascular moyamoya disease: A two-year follow-up study. Surg Neurol 14: 444-9, 1980.
244. Endo M, Kawano N, Miyasaka Y, et al.: Cranial burr hole for revascularization in moyamoya disease. J Neurosurg 71: 180-5, 1989.
245. Kinugasa K, Mandai S, Tokunaga K, et al.: Ribbon encephaloduro-arterio-myosynangiosis for moyamoya disease. Surg Neurol 41: 455-61, 1994.
246. Kuroda S, Hashimoto N, Yoshimoto T, et al.: Radiological findings, clinical course, and outcome in asymptomatic moyamoya disease: Results of multi-center survey in Japan. Stroke 38 (5): 1430-5, 2007.