Panos Fidias
INTRODUCTION
Thymoma is a rare disease, with an incidence of only 0.13 per 100,000 person-years (1). However, it is the most common tumor of the anterior mediastinum, representing approximately 30% of anterior mediastinal lesions and 20% of all mediastinal tumors in adults. Based on Surveillance, Epidemiology and End Results (SEER) data from 1973 to 2006 the incidence is similar between males and females. Interestingly, the incidence in the United States is higher in African Americans and especially Asian/Pacific Islanders compared to whites or Hispanics. The median age at presentation is around 50 years and it peaks in the seventh decade. Approximately one-third of patients are asymptomatic at presentation, one-third have symptoms from local extent of their tumor, and one-third present with paraneoplastic syndromes, typically myasthenia gravis (MG).
HISTOLOGIC CLASSIFICATION
Thymomas are tumors derived from the thymic epithelium. In 1989 Muller-Hermelink and Kirchner proposed a classification based on the similarity of the morphologic appearance of the tumor to normal thymic compartments. This classification subdivided tumors into medullary, mixed, predominantly cortical, cortical, and well-differentiated thymic carcinoma.
In an attempt to standardize the histologic diagnosis of thymoma, the World Health Organization (WHO) published its own classification in 1999, which subdivided tumors into six types: A, AB, B1, B2, B3, and C. Type A tumors have neoplastic cells with spindle or oval appearance, whereas type B tumors have cells with an epithelioid or dendritic appearance. Types B1, B2, and B3 correspond to the predominantly cortical, cortical, and well-differentiated thymic carcinoma, respectively. Tumors combining type A and type B1 or rarely B2 features are classified as type AB. In the 2004 WHO classification update, thymic carcinomas (type C tumors in the 1999 WHO classification) include tumors with histology foreign to thymic tissues and are termed according to their differentiation (such as squamous cell carcinomas, mucoepidermoid, basaloid carcinoma, etc.), including carcinomas with neuroendocrine histology (2).
Thymic carcinomas have distinct morphology and immunophenotype; they present at more advanced stages and have a significantly inferior prognosis compared to other thymoma types. For that reason, some authors propose that well-differentiated thymic carcinomas should be designated as “atypical thymomas,” not to be confused with (type C) thymic carcinomas, which will not be considered in this chapter.
GENETIC ALTERATIONS
In an era of personalized medicine and targeted treatment, thymomas remain for the most part outside of the spotlight. Although genetic alterations do occur, there has been no specific molecular target identified, which would be appropriate for therapeutic interventions. Random case reports have described responses to various tyrosine kinase small molecule inhibitors, but these were mostly confined to thymic carcinomas, and for those patients with thymoma who responded, no corresponding genetic alteration was identified in the tumor (3, 4). Allelic imbalances are very low for types A and AB (7%–8%), but increase to 20% for more advanced histologic types (B2 and B3) (5). Interestingly, type A component within an AB tumor is genetically distinct from type A tumors. The most frequent abnormality affects chromosome 6p21.3, which is an MHC locus, and 6q25.2–25.3, suggesting the presence of a yet unidentified tumor suppressor gene in these locations.
STAGING
The most widely used staging system was introduced by Masaoka in 1981, and was modified in 1994, allowing microscopic invasion into, but not through, the capsule to be classified as stage I (Table 53-1). Another staging system used by French groups, the Groupe d’Etudes des Tumeurs Thymiques (GETT) classification is based on the extent of disease but also on the extent of surgical resection (complete, partial, or biopsy). Both staging systems have been shown to be prognostic of overall survival in multiple studies. In a study of 149 patients with non-metastatic thymomas staged both with the Masaoka and GETT systems, there was an 88% concordance between the two systems. A tentative WHO classification based on tumor, node, metastasis (TNM) has also been proposed but is not widely used.
TABLE 53-1 MASAOKA CLINICAL STAGING OF THYMOMA

MYASTHENIA GRAVIS
Myasthenia gravis (MG) is the most common paraneoplastic disease associated with thymoma. Evoli reported on 207 patients with MG from Italy. Of the 188 patients with tumor that could be classified, 87% had type B thymomas (B1 in 22.3%, B2 in 55.3%, B3 in 3.1%, and combined B2/B3 in 6.3%), which is similar to findings from other authors showing that thymoma is primarily associated with cortical histology. Interestingly, 13 patients developed MG 0.5–10 years following thymectomy. Out of 189 patients with adequate follow-up only 17 patients had achieved drug-free remission. As a result, some patients with thymoma succumb to complications of MG, rather than their malignant disease. In the study by Evoli et al. eight deaths were attributed to MG and seven deaths to progression of thymoma (6). Similarly, in a series from Mayo Clinic 13% of patients died of thymoma and 16% of myasthenia. Despite its contribution to the morbidity and mortality of thymoma patients, MG is not an independent predictor either of recurrence or of survival.
OTHER PARANEOPLASTIC SYNDROMES
Up to 50% of patients with pure red cell aplasia (PRCA) have thymoma, whereas less than 10% of patients with thymoma have PRCA. In 17 cases reported by Masaoka et al, who were treated with resection of their tumor, 6 patients benefited from surgery. More recently, the combination of octreo-tide and prednisone has also been found to be effective in the treatment of PRCA. Hypogammaglobulinemia is an uncommon complication of thymoma, but up to 10% of patients with acquired hypogammaglobulinemia will have an associated thymoma (Good syndrome). Unfortunately, surgery does not reliably return immunoglobulin levels to normal levels.
TREATMENT
![]() RESECTABLE DISEASE
RESECTABLE DISEASE
Surgery remains the only known curative therapy for thymoma. The definition of resectable thymomas includes not only early stage disease (stages I and II) but also more advanced cases where the bulk of the tumor can be removed. Obviously, criteria for unresectability vary among different centers, but extensive mediastinal infiltration or significant bilateral pleural-based tumor would be considered inoperable by most surgeons.
Rates of complete resection based on Masaoka stage and WHO classification are shown in Tables 53-2 and 53-3. Long-term outcome for resected thymomas is dependent both on Masaoka stage and WHO classification. Multivariate analyses in various studies have inconsistently shown that additional factors can have independent prognostic value, such as tumor size, incomplete resection, and invasion of great vessels for stage III patients.
TABLE 53-2 RATES OF COMPLETE RESECTION ACCORDING TO MASAOKA STAGE

TABLE 53-3 RATES OF COMPLETE RESECTION ACCORDING TO WHO CLASSIFICATION TYPE

In evaluating the effectiveness of therapy, long-term follow-up is paramount, given the risk of late recurrences. Additionally, disease-specific survival, rather than overall survival, should be the primary end point, since death from thymoma can account for as low as 21% of all causes of death in large studies.
Stage I tumors have an exceptionally favorable outcome following surgery, with 5-, 10-, and 20-year survivals in the 90%–100% range. Survival decreases for more advanced tumors, with the most significant difference usually manifested between stages II and III.
Similarly, survival is excellent for WHO types A and AB, while survival rates drop significantly for types B2 and B3. WHO type B1 seems to have an intermediate prognosis (Tables 53-4 and 53-5).
TABLE 53-4 TEN-YEAR SURVIVAL OF PATIENTS WITH THYMOMA ACCORDING TO MASAOKA STAGE
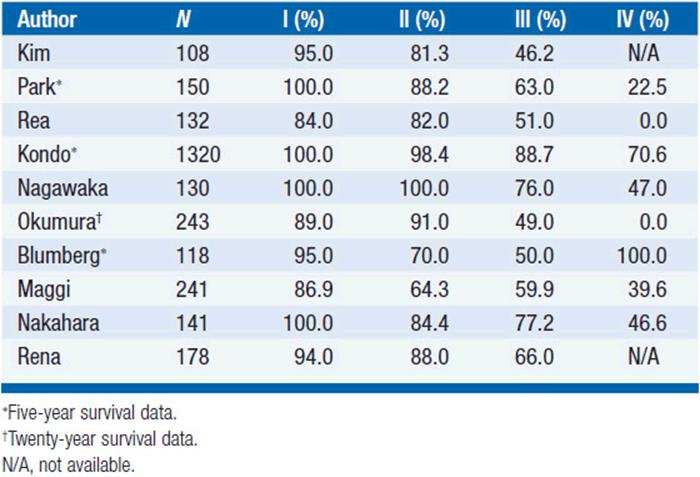
TABLE 53-5 TEN-YEAR SURVIVAL OF PATIENTS WITH THYMOMA ACCORDING TO WHO CLASSIFICATION

![]() ADJUVANT THERAPY
ADJUVANT THERAPY
It is widely accepted that the cure rate of patients with encapsulated thymomas is excellent with surgery alone. However, in more advanced disease, the results reported in surgical series have included a variable percentage of patients undergoing adjuvant therapy, primarily radiation. Several institutions have consistently prescribed radiation therapy for all invasive tumors (stages II and above), while others have selected patients for additional therapy based on the judgment of the thoracic surgeon or the radiation oncologist. Retrospective comparisons of patients who did and did not receive adjuvant therapy are therefore subject to selection bias and cannot be considered conclusive.
Curran et al. showed that following complete resection, 6/18 patients with stage II and 2/3 patients with stage III thymomas recurred in the absence of radiation, whereas none of 5 patients with radiation experienced a recurrence. However, only one patient in the radiation group had stage II disease. In a review of the literature up to that date, the authors demonstrated a 28% recurrence in the absence of radiation and a 5% recurrence rate when radiation was administered (7). In another study of 241 patients with thymoma from the University of Torino, Italy, Maggi et al. reported that 11 of 55 patients with invasive thymoma and no adjuvant therapy recurred, compared to 3 of 21 with adjuvant therapy (mostly radiation therapy) (8). However, subsequent studies have shown little benefit for postoperative radiation therapy.
In the largest retrospective study reported to date, 1320 patients from 115 Japanese centers were reviewed. Complete resection was performed in 247 stage II and in 170 stage III patients, and adjuvant radiation therapy was given to 43.3% and 74.5% of stage II and III patients, respectively. Recurrence rates for patients with and without radiation therapy were 4.7% versus 4.1% in stage II and 23% versus 26% for stage III patients (9). In a series of patients with stage III thymomas from Massachusetts General Hospital (MGH), 54% of recurrences occurred in the pleura (10). Similarly, in a study by Nakagawa et al. 6 out of 12 recurrences occurred in the pleura. Given the high propensity for such pleural dissemination, Haniuda et al. reviewed the efficacy of adjuvant radiation therapy in 70 patients undergoing complete resection of thymoma based on the degree of “pleural factor” defined as follows: p0, no adhesion to mediastinal pleura; p1, fibrous adhesion to the mediastinal pleura without tumor invasion; and p2, microscopic invasion of the mediastinal pleura. In p0 stage II patients no recurrence was observed regardless of radiation therapy. However, in p2 stage II tumors 3 out of 4 patients recurred, even in the presence of adjuvant radiation. Radiation appeared to be helpful in stage II p1 tumors, where none of 6 patients undergoing adjuvant therapy relapsed, compared to 4 out of 11 patients treated with surgery alone (11). Ogawa et al. examined the results of 103 patients with completely resected thymoma treated with adjuvant radiation therapy. The pleura was the most common site of recurrence, which was seen in 12 of 17 patients with relapsed disease. While no pleural recurrences were seen among the 70 patients without pleural invasion in their surgical specimen, 12 out of 38 patients with pleural invasion experienced such a recurrence (12). Therefore, it appears that radiation therapy cannot adequately treat tumors at high risk for the most common site of recurrence.
Newer studies have also evaluated the role of WHO classification in determining risk of relapse following adjuvant therapy. Ströbel et al. reported on 228 thymoma patients treated with primary surgery with or without adjuvant therapy (13). The study also included a small proportion of thymic carcinomas with squamous cell histology (4.8%). Postoperative therapy was quite uncommon in WHO type A thymomas (3 out of 20) but was very frequent in type B3 (15 out of 22). The main finding of the study was that recurrences following complete resection were rare among tumors of type A, AB, or B1, even in stages II and III (2 out of 33), while they were more frequent in stage III tumors of types B2 and B3 (5 out of 18). These results are also supported by data from MGH, which showed no recurrences among 73 patients with types A and AB thymoma (14). How to approach early stage unfavorable histology thymomas is a more difficult issue. Chen identified WHO types B2, B3, and C as independent poor prognostic categories within stages I and II tumors. He observed 4 deaths among 24 patients, compared to only 2 deaths in 78 patients with types A, AB, and B1. However, it is known that thymic carcinomas have distinct natural history and a worse prognosis compared to “pure” thymomas (15). Results in patients with B2 and B3 histology alone are more encouraging: there was only one recurrence out of 37 stage I/II patients in a recent study from Germany.
Exploring the role of adjuvant therapy in tumors of cortical histology (B2 and B3), Ströbel showed that in stage II tumors there were no tumor relapses among those patients who received adjuvant radiation versus one relapse in 16 patients without further treatment. The relapse rate for stage III patients was 0% for the 5 patients who underwent adjuvant therapy, compared to 33% for the patients without adjuvant treatment (13). Chen also reported that adjuvant therapy did not improve survival for types A, AB, and B1, whereas it had a statistically significant benefit for types B2, B3, and C (5-year survival 85.5% vs. 48.3%). Given the independent prognostic value of stage and WHO classification, several authors have proposed treatment algorithms and risk stratification schemes following primary surgery, which take both parameters into consideration.
![]() UNRESECTABLE DISEASE
UNRESECTABLE DISEASE
When complete, or near complete, resection cannot be performed, surgical procedures have ranged from biopsy alone to a variable degree of debulking approaches. Some studies have shown that debulking is superior to biopsy, but other studies have found no difference in survival. It is clear, however, that patients can still experience long-term survival when treated with radiation therapy with or without the addition of chemotherapy.
Ninety patients from 10 French centers were treated with partial resection (31 patients), biopsy (55 patients), or complete resection (4 patients with stage IVa disease and pleural implants). Radiation dose ranged from 30 to 70 Gy with a median dose of 50 Gy. Sequential platinum-based chemotherapy was added to 59 patients, while 3 patients received preoperative radiation and chemotherapy. The 5- and 10-year disease-free survival was 60% and 36% for partial resection, compared to 38% and 31% for biopsy alone (16).
An American Intergroup study evaluated the combination of cyclophosphamide, doxorubicin, cisplatin (CAP) followed by radiation therapy at a dose of 54 Gy in 23 patients with limited-stage unresectable thymoma, including 2 patients with thymic carcinoma. All, but one, of the patients had gross residual disease postoperatively. The overall response rate to induction chemotherapy was 69.6% (CR: 5 patients, PR: 11 patients) and the 5-year overall survival was 52.5% (17).
Based on the encouraging results of radiation and radio-chemotherapy, many centers have approached unresectable or borderline resectable thymic tumors with a multimodality treatment plan. Kim et al. from the M.D. Anderson Cancer Center treated 22 patients deemed to be unresectable with neoadjuvant chemotherapy, followed by surgery, postoperative radiation, and consolidation chemotherapy. Eleven patients had stage III, 10 patients had stage IVa, and one patient had stage IVb thymoma. The induction program consisted of cyclophosphamide, doxorubicin via continuous infusion, cisplatin, and prednisone, and it resulted in an overall response rate of 77% (CR: 3 patients, PR: 14 patients). Radiation dose for 16 patients was 60 Gy, and for the remaining patients it was 50 Gy. Twenty-one patients underwent surgery and 16 (76%) had a complete resection. Six of these patients had a more than 80% necrosis in the surgical specimen. The progression-free survival (PFS) at 7 years was 77%. Only one patient died of progressive thymoma with a median follow-up of 50.3 months (18).
Bretti et al. reported their results with neoadjuvant therapy in 33 patients who could not undergo upfront surgery for stage III-IV thymoma. Eight patients received radiation at a dose of 30 Gy in 15 fractions (24 Gy if more than 30% of the lung volume had to be included). The remaining patients received 4 cycles of chemotherapy, either ADOC (doxorubicin 40 mg/m2, cisplatin 50 mg/m2 on day 1, vincristine 0.6 mg/m2 on day 2, and cyclophosphamide 700 mg/m2 on day 4) or cisplatin and VP-16 (100 mg/m2 on day 1 and 100 mg/m2 on days 1–3, respectively). Surgery could be attempted on 17 patients, and a total of 12 patients were able to have a complete resection following induction treatment (1 patient post radiation, 11 patients post chemotherapy). These patients had shorter PFS (56.9 months vs. not reached yet) but similar overall survival compared to a cohort of 20 patients with stage III-IV thymoma, which could be completely resected at the time of diagnosis. The patients who could not be resected were given 50–60 Gy postoperatively, but had a 5-year PFS of only about 10% (19). Venuta et al. treated patients with stage III disease on a multimodality program: 30 patients with resectable disease underwent adjuvant chemotherapy and radiation (40 Gy for complete resection and 50-60 Gy for incomplete resection), while 15 patients judged to be unresectable were given neoadjuvant cisplatin-based chemotherapy. Eleven patients out of the 45 had thymic carcinoma. Overall, 10 patients had a response to the induction regimen (CR: 2 patients, PR: 8 patients, overall RR 67%). Complete resection was possible in 87% of patients; however, only 1 patient had a complete pathologic response (7%). Interestingly, the 10-year overall survival of patients receiving induction treatment was 90%, compared with 71% for patients considered initially resectable (20).
From the above studies it is reasonable to recommend an initial chemotherapy or chemoradiation approach, followed by an attempt to resect the disease for patients who present initially with unresectable thymomas (Table 53-6). In our practice, we often utilize either CAP or ADOC as preoperative chemotherapy. If we select a combined modality approach, we utilize cisplatin and etoposide concurrently with radiation.
TABLE 53-6 RESULTS FOR PATIENTS PRESENTING WITH UNRESECTABLE THYMOMA TREATED EITHER WITH DEFINITIVE RADIATION (OR CHEMORADIATION) OR WITH INDUCTION CHEMOT HERAPY

RECURRENT OR METASTATIC DISEASE
Recurrent disease amenable to reoperation should be approached surgically. Although there are no large comparative studies, there is evidence of long-term disease-free survival following aggressive therapy of recurrent tumors. Systemic therapy is the only option for patients with extrathoracic disease or with tumors that have progressed despite all available local measures (Table 53-7). Multiple case reports and small series have demonstrated that thymoma is a responsive malignancy to single agent chemotherapy.
TABLE 53-7 RESULTS OF CHEMOTHERAPY IN PATIENTS WITH ADVANCED THYMOMA

Interestingly, significant responses have also been observed with the use of glucocorticoids, even in cases unresponsive to other modalities. Although it is widely accepted that glucocorticoids act mainly on the lymphoid population of the tumor, responses have been seen in primarily epithelial thymomas as well. Glucocorticoids may be even more efficacious in combination with octreotide. In a study by Palmieri et al. 16 patients with chemotherapy refractory thymoma were given prednisone (0.6 mg/kg/day for 3 months, 0.2 mg/kg/day during follow-up) and octreotide (1.5 mg/day or 30 mg every 14 days for the long-acting analog lanreotide). Six patients had thymic carcinoma, and half of them had progressive small cell neuroendocrine carcinoma. The response rate was 37%, including one patient with complete response. The median time to progression was 14 months, and the median survival time for the group was 15 months. The estimated 2-year survival is approximately 30% (21). Response to therapy did not appear to correlate with histology.
The Eastern Cooperative Oncology Group (ECOG) designed a study with the same combination therapy, but evaluated single agent octreotide initially (at a dose of 1.5 mg/kg daily), and only added prednisone (at a dose of 0.6 mg/kg daily) for patients with stable disease after 2 months of octreotide therapy. The study accrued 38 assessable patients with thymoma (32 patients), thymic carcinoma (5 patients), or thymic carcinoid (1 patient). At the end of the 2 months there were four partial responses, but the addition of prednisone resulted in six additional partial responses and two complete responses for an overall response rate of 30.3% for the combination. Only patients with typical histology responded to therapy. The 2-year overall survival was 75.7% (22). It appears, therefore, that glucocorticoids add to the activity of octreotide. In a case report, a patient with type B3 thymoma progressive after 6 months of octreotide treatment was given prednisone 50 mg/day in addition to long-acting octreotide. After 7 months of combination therapy the patient achieved a complete remission.
EGFR immunoreactivity is observed in thymomas, and overexpression of EGFR is associated with more aggressive thymic tumors (B2 and B3). Based on this observation, 26 patients with previously treated thymoma or thymic carcinoma were treated with gefitinib at a dose of 250 mg daily. Only one response was seen, which lasted for 5 months. None of the five patients who underwent DNA sequencing, including the patient who responded, harbored an EGFR mutation (23).
Among 18 patients treated with imatinib in two different studies, only 2 had typical thymoma and none responded. C-KIT mutations are rare, even in thymic carcinomas, and are essentially never seen in thymomas, even if they overexpress c-KIT by immunohistochemistry (24).
Combination chemotherapy has been studied extensively in the treatment of advanced thymoma. More commonly, patients received cisplatin-based regimens; however, responses have been reported with other regimens, such as cyclophosphamide, doxorubicin, vincristine (CAV), or CAV with the addition of prednisone ± bleomycin (CAVP ± Bleo). In a retrospective review of 123 patients treated on five ECOG trials, combination chemotherapy was associated with a higher response rate (p < 0.0001) and survival (p = 0.035) compared to single agent cisplatin. Fornasiero et al. reported on 37 patients treated with ADOC (cisplatin 50 mg/m2 and doxorubicin 40 mg/m2 on day 1, vincristine 0.6 mg/m2 on day 3, and cyclophosphamide 700 mg/m2 on day 4) over a period of 13 years. The overall response rate was impressive at 92%, with 43% complete remission rate. Seven patients were confirmed to have complete response pathologically following thoracotomy. The median duration of response was 12 months, and the median survival was 15 months (25). A similar regimen, CAP (cisplatin 50 mg/m2, doxorubicin 50 mg/m2, and cyclophosphamide 500 mg/m2), was given to 30 patients with advanced thymoma. Although the response rate was lower than ADOC at 50%, the median survival time was much more impressive (37.7 months), questioning the value of vincristine in this setting (26). Successful retreatment with the same regimen in two patients relapsing 14 and 60 months after completion of initial therapy has been reported. Based on the promising activity demonstrated by single agent ifosfamide, 28 evaluable patients were treated with VIP (etoposide 75 mg/m2, ifosfamide 1.2 g/m2, and cisplatin 20 mg/m2 days 1–4). Unfortunately, response rate and survival statistics did not appear superior to previous regimens (RR 32%, response duration 11.9 months, median survival time 31.6 months, and 2-year overall survival of 70%) (27). The European Organization for the Research and Treatment of Cancer (EORTC) evaluated the combination of cisplatin (60 mg/m2 day 1) and etoposide (120 mg/m2 days 1–3) on 16 chemotherapy-naive patients. The observed response rate was 56%, the duration of response was 40 months, the median survival time was 51.6 months, and the 5-year overall survival was 50% (28).
More recently, ECOG evaluated another non-anthracycline regimen consisting of carboplatin at an AUC of 6 and paclitaxel at a dose of 225 mg/m2 every 3 weeks for a maximum of 6 cycles (29). They enrolled 46 patients, 13 of whom had type C thymic carcinoma. Unfortunately, the investigators grouped the 10 patients with B3 thymoma together with the type C patients. Results from treatment such as response rates (42.9% vs. 21.7%), PFS (16.7 vs. 5.0 months), and median overall survival (not reached vs. 20 months) were, as expected, all superior within the thymoma group.
CONCLUSIONS
Thymoma is a rare disease, but is highly curable with surgery when complete resection can be achieved. Even for unresectable patients, long-term survival is feasible with the combination of chemotherapy and radiation. There is considerable excitement about the use of neoadjuvant approaches in marginally resectable patients, but more studies are required in this group of patients. Metastatic disease should be treated with cisplatin-based regimens, but no “standard” therapy exists, and new agents need to be evaluated. More importantly, better insight into the biology of the disease is needed. The standardization of pathology has provided valuable prognostic information, but more refined prognostic and predictive markers, including molecular markers, are desperately needed.
REFERENCES
1. Engels EA. Epidemiology of thymoma and associated malignancies. J Thorac Oncol. 2010; 5: S260–S265.
2. World Health Organization Classification of Tumors. Pathology and genetics of tumours of the lung, pleura, thymus and heart. In Travis WD, Brambilla E, Muller-Hermelink HK, et al. (eds.). IARC Press, 2004, 341.
3. Chuah C, Lim TH, Lim AS, et al. Dasatinib induces a response in malignant thymoma. J Clin Oncol. 2006; 24: e56–e58.
4. Strobel P, Hartmann M, Jakob A, et al. Thymic carcinoma with over-expression of mutated KIT and the response to imatinib. N Engl J Med. 2004; 350: 2625–2626.
5. Strobel P, Hohenberger P, Marx A. Thymoma and thymic carcinoma: molecular pathology and targeted therapy. J Thorac Oncol. 2010; 5: S286–S290.
6. Evoli A, Minisci C, Di Schino C, et al. Thymoma in patients with MG: characteristics and long-term outcome. Neurology. 2002; 59: 1844–1850.
7. Curran WJ Jr, Kornstein MJ, Brooks JJ, et al. Invasive thymoma: the role of mediastinal irradiation following complete or incomplete surgical resection. J Clin Oncol. 1988; 6: 1722–1727.
8. Maggi G, Casadio C, Cavallo A, et al. Thymoma: results of 241 operated cases. Ann Thorac Surg. 1991; 51: 152–156.
9. Kondo K, Monden Y. Therapy for thymic epithelial tumors: a clinical study of 1,320 patients from Japan. Ann Thorac Surg. 2003; 76: 878–884; discussion 884–885.
10. Myojin M, Choi NC, Wright CD, et al. Stage III thymoma: pattern of failure after surgery and postoperative radiotherapy and its implication for future study. Int J Radiat Oncol Biol Phys. 2000; 46: 927–933.
11. Haniuda M, Morimoto M, Nishimura H, et al. Adjuvant radiotherapy after complete resection of thymoma. Ann Thorac Surg. 1992; 54: 311–315.
12. Ogawa K, Uno T, Toita T, et al. Postoperative radiotherapy for patients with completely resected thymoma: a multi-institutional, retrospective review of 103 patients. Cancer. 2002; 94: 1405–1413.
13. Strobel P, Bauer A, Puppe B, et al. Tumor recurrence and survival in patients treated for thymomas and thymic squamous cell carcinomas: a retrospective analysis. J Clin Oncol. 2004; 22: 1501–1509.
14. Wright CD, Kessler KA. Surgical treatment of thymic tumors. Semin Thorac Cardiovasc Surg. 2005; 17: 20–26.
15. Chen G, Marx A, Wen-Hu C, et al. New WHO histologic classification predicts prognosis of thymic epithelial tumors: a clinicopathologic study of 200 thymoma cases from China. Cancer. 2002; 95: 420–429.
16. Mornex F, Resbeut M, Richaud P, et al. Radiotherapy and chemotherapy for invasive thymomas: a multicentric retrospective review of 90 cases. The FNCLCC trialists. Federation Nationale des Centres de Lutte Contre le Cancer [erratum appears in Int J Radiat Oncol Biol Phys. 1995; 33: 545]. Int J Radiat Oncol Biol Phys. 1995; 32: 651–659.
17. Loehrer PJ Sr, Chen M, Kim K, et al. Cisplatin, doxorubicin, and cyclophosphamide plus thoracic radiation therapy for limited-stage unresectable thymoma: an intergroup trial. J Clin Oncol. 1997; 15: 3093–3099.
18. Kim ES, Putnam JB, Komaki R, et al. Phase II study of a multidisciplinary approach with induction chemotherapy, followed by surgical resection, radiation therapy, and consolidation chemotherapy for unresectable malignant thymomas: final report. Lung Cancer. 44: 369–379, 2004.
19. Bretti S, Berruti A, Loddo C, et al. Multimodal management of stages III-IVa malignant thymoma. Lung Cancer. 2004; 44: 69–77.
20. Venuta F, Rendina EA, Longo F, et al. Long-term outcome after multi-modality treatment for stage III thymic tumors. Ann Thorac Surg. 2003; 76: 1866–1872; discussion 1872.
21. Palmieri G, Montella L, Martignetti A, et al. Somatostatin analogs and prednisone in advanced refractory thymic tumors. Cancer. 2002; 94: 1414–1420.
22. Loehrer PJ, Sr., Wang W, Johnson DH, et al. Octreotide alone or with prednisone in patients with advanced thymoma and thymic carcinoma: an Eastern Cooperative Oncology Group Phase II Trial [erratum appears in J Clin Oncol. 2004; 22: 2261]. J Clin Oncol. 2004; 22: 293–299.
23. Kurup A, Burns M, Dropcho S, et al. Phase II study of gefitinib treatment in advanced thymic malignancies. J Clin Oncol. 2005; 23: 381S. (abstract 7068).
24. Kelly RJ, Petrini I, Rajan A, et al. Thymic malignancies: form clinical management to targeted therapies. J Clin Oncol. 2011: 29: 4820–4827.
25. Fornasiero A, Daniele O, Ghiotto C, et al. Chemotherapy for invasive thymoma. A 13-year experience. Cancer. 1991; 68: 30–33.
26. Loehrer PJ Sr, Kim K, Aisner SC, et al. Cisplatin plus doxorubicin plus cyclophosphamide in metastatic or recurrent thymoma: final results of an intergroup trial. The Eastern Cooperative Oncology Group, Southwest Oncology Group, and Southeastern Cancer Study Group. J Clin Oncol. 1994; 12: 1164–1168.
27. Loehrer PJ Sr, Jiroutek M, Aisner S, et al. Combined etoposide, ifosfamide, and cisplatin in the treatment of patients with advanced thymoma and thymic carcinoma: an intergroup trial. Cancer. 2001; 91: 2010–2015.
28. Giaccone G, Ardizzoni A, Kirkpatrick A, et al. Cisplatin and etoposide combination chemotherapy for locally advanced or metastatic thymoma. A phase II study of the European Organization for Research and Treatment of Cancer Lung Cancer Cooperative Group. J Clin Oncol. 1996; 14: 814–820.
29. Lemma GL, Lee JW, Aisner SC, et al. Phase II study of carboplatin and paclitaxel in advanced thymoma and thymic carcinoma. J Clin Oncol. 2011; 29: 2060–2065.