Ronald P. Danis
|
Overview |
|
Diabetic macular edema (DME) remains the most common cause of vision loss among diabetic patients. New understanding of the underlying pathophysiology has heightened interest in the potential benefits of specific pharmacologic therapy, such as treatment with intraocular steroids, anti-vascular endothelial growth factor (VEGF), and protein kinase C-beta (PKC?) inhibition. At this time, laser photocoagulation according to the guidelines of the Early Treatment of Diabetic Retinopathy Study (ETDRS) continues to be the primary standard of care treatment in most communities, while the evidence base for alternative or supplementary treatments accrues. Recent advances in imaging technology, most notably the wide commercial availability and acceptance of optical coherence tomography (OCT), have yielded new tools for the clinician and researcher in monitoring macular edema progression and response to treatment. |
DEFINITION
Diabetic macular edema is manifested as retinal thickening primarily due to exudation from incompetent macular retinal capillaries. While retinal thickening may occur peripheral to the macula, such edema is not vision threatening. In order to define a threshold severity level of edema at which retreatment was specified for the protocol, the ETDRS coined the term 'clinically significant macular edema'.[1,2] This term has been incorporated into most clinicians' understanding as a level of disease at which it is appropriate to initiate laser treatment. The phrase clinically significant macular edema is often applied as a clinical description of ocular status (e.g., the eye has 'non-clinically significant DME'). While a useful and time-honored term, a number of other descriptors can be applied to the clinical appearance of DME which may be more appropriate as new treatments become available. For instance, an eye with clinically significant macular edema by the ETDRS definition could have center involvement with cystoid changes, or, it could have focal edema with a normal foveal contour and thickness. These very different presentations may merit different treatments.
The standard of care for varying presentations of DME is in evolution at this time. For several decades, the guidelines of the ETDRS were generally applied in the medical community to manage patients with a level of DME meeting threshold criteria for clinically significant macular edema, particularly when the center of the macula was involved or immanently threatened by retinal thickening or hard exudates very close to it. However, recent clinical trials have been investigating treatment of DME at less severe stages.[3] In recognition that eyes with severe edema at the center of the macula may not respond well to laser treatment, and with the availability of new pharmacologic agents administered by intravitreal injection, many clinicians are using drugs off-label to treat DME, without the evidence from large randomized trials.
|
Key Features: ETDRS Criteria for Clinically Significant Macular Edema |
||||||||||||||||||
|
PREVALENCE AND INCIDENCE
In the first detailed population-based assessment of diabetic retinopathy (DR), the Wisconsin Epidemiologic Study of Diabetic Retinopathy (WESDR), documented a prevalence of macular edema (defined as retinal thickening within one disk-diameter of the macular center in stereoscopic retinal photographs) of 11.1% overall among patients with diabetes in Southern Wisconsin in the early 1980s.[4] The prevalence was slightly higher in early onset compared to older-onset diabetes and was strongly associated with duration of diabetes and glycemic control. Proteinuria and vascular hypertension were additional factors associated with increased prevalence. Subsequent population-based studies have yielded prevalence rates between 2%,[5] and over 10%[6-9] and confirmed the afore-mentioned associations. In some diabetic populations, the prevalence of retinopathy, including DME, appears to be declining due to improved glycemic control of diabetes in the community.[6,10] However, the absolute prevalence of DME might be increasing due to the overall increased prevalence of diabetes in industrialized nations.[11] The incidence of new cases of DME in a diabetic population has been characterized in a population-based longitudinal assessment within the WESDR. The WESDR determined a 10 year rate of developing DME of 20.1% in younger onset diabetic patients, 25.4% in older-onset diabetic patients taking insulin, and 13.9% in the older-onset group not taking insulin.[12] It is expected that the incidence of DME will decrease as excellent metabolic control is increasingly embraced as a therapeutic goal by patients and health care workers.
CLINICAL ASSOCIATIONS AND RISK FACTORS
Macular edema is strongly positively associated with diabetic retinopathy severity.[12] This is particularly problematic for the clinician who must frequently manage coexistent DME and proliferative diabetic retinopathy (PDR), as will be discussed. Undoubtedly, the molecular processes leading to the loss of retinal capillary integrity and subsequent edema are closely related to, but not identical to those causing capillary closure and ultimate neovascularization. This is evident from the observation that most patients with macular edema do not have PDR, and many patients with proliferative retinopathy do not have DME.[13] This partial disconnect between related complications in the same tissue led the ETDRS to exclude retinal thickening from the overall diabetic retinopathy (DR) severity scale.[14,15] The severity scale is based on rate of progression to high-risk PDR, and retinal thickening per se was not identified as conferring additional risk beyond that represented by the other abnormalities characteristic of diabetic retinopathy.
Glycemic control is a conclusively identified risk factor for retinopathy progression as well as for DME. The Diabetes Complications and Control Trial (DCCT) evaluated intensive glycemic control versus good control in type 1 diabetic patients with no or minimal DR over 4-6 years, and found a marked reduction in the progression of retinopathy, nephropathy, and neuropathy.[16] In the follow-on study of this cohort, the Epidemiology of Diabetes Interventions and Control (EDIC) study demonstrated that subjects in the former intensive treatment group had a lower rate of progression to DME requiring laser treatment than those in the good control group.[17,18] Similar results have been demonstrated in type 2 diabetic patients in the United Kingdom Prospective Diabetes Study (UKPDS).[19] Clear relationships between serum glycohemoglobin levels and DR complications have been established.[20-22] Older-onset patients requiring insulin have a higher incidence of macular edema (and other diabetic complications) than non-insulin dependent diabetic patients.[23] This is likely an effect related to poorer glycemic control in patients ultimately requiring insulin therapy.
Duration of diabetes is strongly correlated with prevalence and incidence of macular edema, retinopathy progression, and other diabetic complications.[12,24] The diagnosis of diabetes in type 2 subjects occasionally occurs some time after subclinical diabetes has been manifest, which yields a small proportion of patients who may present with macular edema at the time of diagnosis, or even have decreased vision from macular edema as the presenting sign.[25-27] In contrast, persons with type 1 diabetes are very unlikely to experience advanced retinopathy and macular edema before 5 years of duration. Age-corrected prevalence rates suggest that there is no definite age interaction with duration of disease[8,11] but because of the increasing incidence of diabetes with age, retinopathy rates are correspondingly higher with advancing age. Consequently, early detection of type 2 diabetes and institution of excellent glycemic control is paramount in decreasing the risk of vision loss from macular edema in the older population.[28-30]
Hypertension significantly exacerbates vision loss from macular edema.[22,31] The UKPDS has demonstrated that strict control of systemic hypertension has an ameliorative effect on retinopathy progression and vision loss from macular edema in type 2 diabetes.[32,33] The WESDR study also documented an adverse association of hypertension with vision loss from diabetic retinopathy.[31] It has been suggested that not only is hypertension a risk factor for the development of macular edema, but its treatment may have important benefits in patients with uncontrolled hypertension.[34]
Proteinuria and retinopathy are strongly correlated because of their common risk factors of glycemic control, duration of diabetes, and hypertension.[17,31,35-37] Less clear is the relationship between renal failure and macular edema. There appear to be patients who have increased macular edema as a collateral adverse event due to fluid retention, and who improve after institution of diuretics or hemodialysis,[38] but this is difficult to demonstrate among populations of patients undergoing dialysis.[34,39,40] Retinal HE and macular edema were significantly and independently associated with proteinuria in a cohort of African American patients.[41] It is accepted that fluid retention from cardiac failure, renal failure, or other causes can exacerbate DME and may be important concerns in managing it.[1]
Dyslipidemia has been implicated as an independent risk factor for vision loss and DME.[42-44] This may be in part due to increased quantities of HE in the eyes of hyperlipidemic diabetic patients, which has been a consistent observation between studies.[41,45-48] However, such observations also suggest that dyslipoproteinemia or excessive dietary exposure to fats may contribute to macular edema, perhaps through lipid peroxidation or other vasculopathic influences.[49-51]
Development or worsening of DME during pregnancy is occasionally observed and is most often associated with poor glycemic control,[32,52] preexisting retinopathy, and preeclampsia.[53-55] The DME associated with pregnancy may at least partially respond to laser treatment, but may also resolve postpartum.[56] Clinicians faced with a pregnant patient with DME may, therefore, elect to observe the patient without treatment for several months during her pregnancy, and following her delivery before making the determination that laser treatment is required. With excellent prenatal care and careful metabolic control, those women who experience some retinopathy worsening during pregnancy generally have only transient changes.[32,53,57] Some of the worsening that may occur during pregnancy may be due to rapid institution of strict glycemic control, which can cause transient worsening as well.[58-60]
Cataract surgery, other types of intraocular surgery, and ocular inflammatory disease may produce inflammatory and angiogenic mediators which can produce macular edema in eyes with or without diabetic retinopathy.[61] However, eyes with pre-existing DME are at high risk for exacerbation. Patients with cataract and DME, who undergo surgery, will occasionally experience vision worsening due to increased DME rather than the expected visual improvement from cataract removal.[62,63] Therefore, DME should be treated prior to surgery to minimize this risk.[64]
Laser treatment for PDR, panretinal photocoagulation (PRP), or scatter laser is documented to cause vision loss due to worsening macular edema.[65] Eyes with preexisting macular edema appear to be more at risk. The ETDRS Research group suggested that in eyes with both DME and PDR, the risk of vision loss from PRP was minimized by first treating the DME with macular focal/grid photocoagulation, and later treating the PDR (if such a delay is acceptable).[1] The underlying mechanism may be the production of an inflammatory response to the laser treatment, which worsens vascular leakage and exacerbates DME.[66] This has led some investigators to consider adjunctive steroid treatment with PRP to minimize the risk of early vision loss.[67,68]
|
Key Features: Clinical Associations with Diabetic Macular Edema Severity |
|||||||||||||||||||||||||||||||||
|
PATHOLOGY AND PATHOPHYSIOLOGY OF DME
Capillary damage is manifest in the earliest stages of DR by loss of the retinal pericytes and capillary basement membrane thickening.[69] With increasing duration and severity of hyperglycemia, eventual capillary closure and microaneurysm formation occur.[70] At what point, the blood-retinal permeability barrier is permanently compromised in this cascade of events is not clear, but hyperglycemia alone of relatively short duration can cause retinal vascular leakage in the absence of detectable retinopathy or structural damage by light microscopy in animal models.[71] The intimate ultrastructural connections between the capillary cells and the neighboring neuronal and glial tissue suggest important functional relationships.[72,73] This raises the question as to whether the observed vascular changes are primary or secondary to neuroglial dysfunction. The ultrastructural basis for the blood-retinal barrier is the tight junctions between the retinal capillary endothelial cells and the relative paucity of transendothelial transport vesicles compared to endothelial cells in other tissues.[74] With hyperglycemia, there is a disorganiza-tion of the interendothelial junctions and an increase in transcellular endocytosis, with resultant fluid flow through the capillary walls into the retinal tissue.[74,75] Fluid homeostasis of the retina is poorly understood. However, increased permeability of the retinal capillaries at some point overwhelms the fluid reuptake mechanisms and retinal edema occurs. The extent of retinal vascular leakage into the vitreous cavity can also be detected in vivo.[76]
The pathophysiologic mechanisms underlying the breakdown of the blood-retinal barrier are multifactorial and are continuing to be understood in the context of the myriad biochemical pathways affected by hyperglycemia. One of the dominant emerging themes surrounds the role of the soluble growth factor VEGF in the diabetic retina. While better known as an angiogenic stimulus upregulated by hypoxia,[77,78]VEGF is a potent permeability agent which is found in animal diabetic models and in human diabetic retinas associated with increased vasopermeability.[79-81] Therefore, VEGF inhibition has emerged as a target molecule for treatment of DME. Another target molecule for pharmacotherapy is the intracellular signaling molecule PKC? (protein kinase C?). Selectively upregulated in hyperglycemia, this protein appears to mediate in part a variety of diabetes-induced pathophysiologic events,[82] including VEGF production,[83] leukostasis and vascular permeability,[83,84] abnormal blood flow,[82] and angiogenesis.[85]The generation of reactive lipoxidative products and glycation end products[86] in diabetes can cause direct cellular damage and also induce inflammation.[87] Steroids can inhibit inflammatory responses, stabilize interendothelial junctions, and inhibit VEGF, thereby accounting for interest in this class of compounds for treating DME.[88-90]
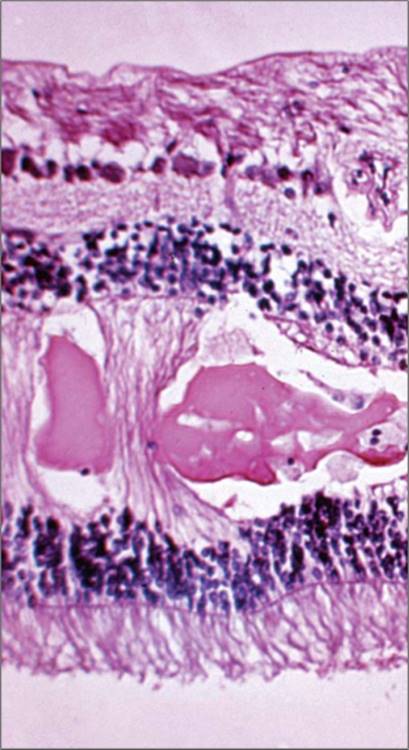
Extracellular fluid in the retina may cause diffuse thickening of the tissue. In early stages, Mueller cell and other glial cell edema can be observed, which appear to be compensatory.[91,92] Severe edema generally results in development of loculated fluid, called cystoid spaces, in the middle retinal layers (Fig. 134.1). Cystoid spaces initially develop in the outer plexiform layer of the retina, hypothesized to be the loosest and most elastic cellular stratum of the retina and therefore the most accommodating layer for this expansion.[93] In very severe or chronic edema, the cystoid spaces may occupy nearly the entire retinal thickness with gross distortion of the retina. In very chronic cases, there is eventual disorganization of the retinal tissue, which may be partly related to co-existent capillary ischemia and reactive glial responses. At this point, the retina may resume normal thickness, or even become abnormally thin, as the macula assumes a more atrophic appearance. Cystoid spaces may rapidly disappear with treatment, yielding a normal-appearing retina in cases where chronic cellular changes have not yet occurred.
|
|
|
|
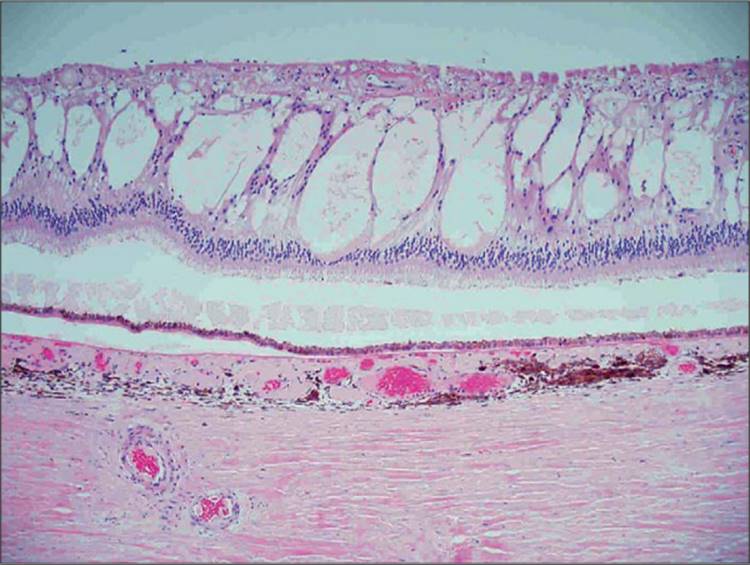
FIGURE 134.1 Photomicrograph of cystoid spaces and subretinal fluid in the retina of a diabetic patient with severe DME. H&E, original magnification ×100. Image furnished by Daniel Albert MD. |
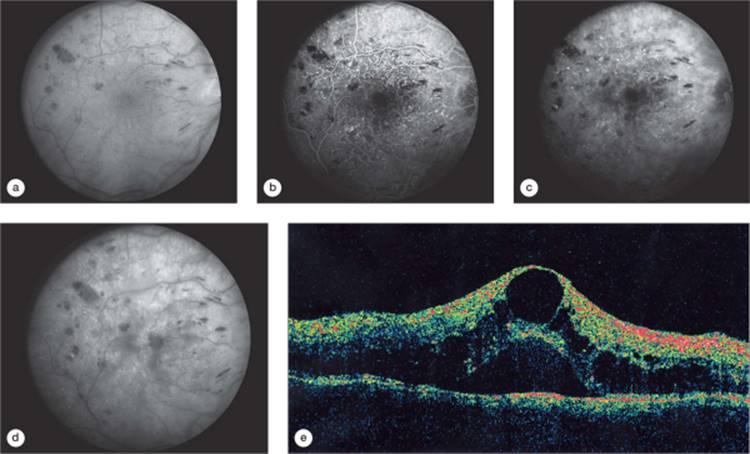
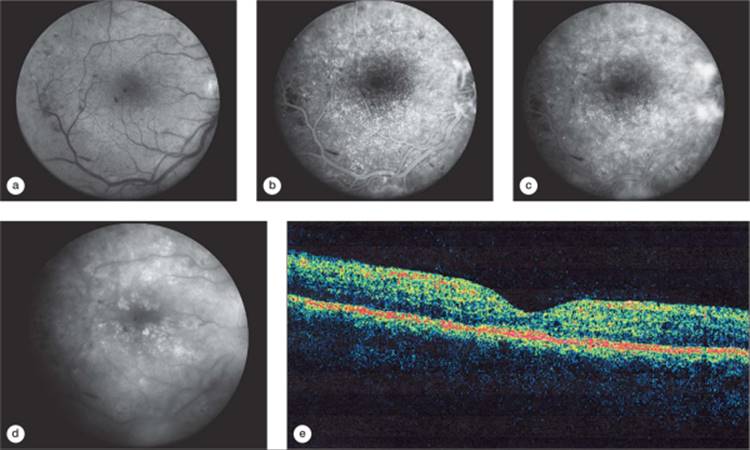
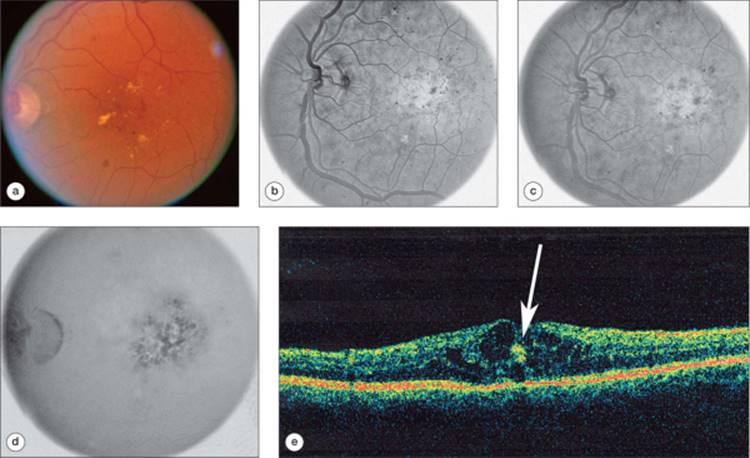
In the foveal region, the outer plexiform layer has a radial orientation due to the centrifugal displacement of the bipolar cell layer, the nerve-fiber layer of Henle. This layer may host cystoid spaces of very large proportions. Most cases of clinically apparent cystoid change are in this region, due to the large size of the cysts. The clinical cyst pattern often appears as one or more central large cysts surrounded by a region of somewhat smaller cysts, and finally by a region of smaller cysts that may not be clinically visible (Fig. 134.2). Fluorescein angiography has been historically helpful in defining cystoid edema, since the fluorescein dye from the leaking capillaries tends to pool in the cystoid spaces in the late phases of the study, highlighting these structures (Figs 134.2d and 134.3d).[94] OCT has allowed visualization of high resolution cross sectional retinal morphology and demonstrated that cystoid spaces are more common than has been appreciated by clinical examination or fluorescein angiography (Figs 134.2d and 134.3d).[95] An additional feature visible by OCT and not apparent clinically or on angiography is a common appearance of subretinal fluid in cases of severe DME (Fig. 134.2e).[96] The source of the fluid has been presumed to be due to excessive retinal capillary leakage, but there is some evidence that the retinal pigment epithelium may also have dysfunctional fluid homeostatic function in diabetes and contribute to this appearance.[93,97]
|
|
|
|
FIGURE 134.2 Right eye of a patient with untreated DME. Visual acuity measured 66 letters (?20/50). (a) Red-free (green channel) image from a digital fluorescein angiogram of a patient with diffuse DME with cystoid changes. (b) Early phase of the fluorescein angiogram with numerous microaneurysms and patchy loss of capillaries. (c) Mid-phase of the angiogram demonstrating early diffuse fluorescein dye leakage. (d) Late phase of the angiogram with fluorescein dye pooling in cystoid spaces and diffuse retinal fluorescein staining. (e) OCT scan demonstrating retinal thickening and a large central cyst and smaller cystoid spaces in the outer plexiform layer of the retina and retinal elevation from subretinal fluid. Printed with permission of the Diabetic Retinopathy Clinical Research (DRCR) Network. |
|
|
|
|
FIGURE 134.3 Same eye from the patient shown in Figure 134.2, one year later, after one modified ETDRS laser treatment was performed at baseline. Visual acuity improved to 73 letters (20/40) at the 12 month visit. (a) Red-free image demonstrating no visible cystoid spaces. (b) Early phase of the angiogram with early leakage from neovascularization from the optic nerve head and the inferior temporal arcade vessels. (c) Mid-phase of the angiogram demonstrating persistent diffuse retinal capillary leakage and retinal staining. (d) Late phase of the angiogram, showing diffuse leakage, but less than at baseline, with reduction in pooling of dye in cystoid spaces. (e) OCT scan showing resolution of subretinal fluid, resolution of cystoid spaces, and marked reduction in retinal thickening after laser treatment. Printed with permission of the DRCR Network. |
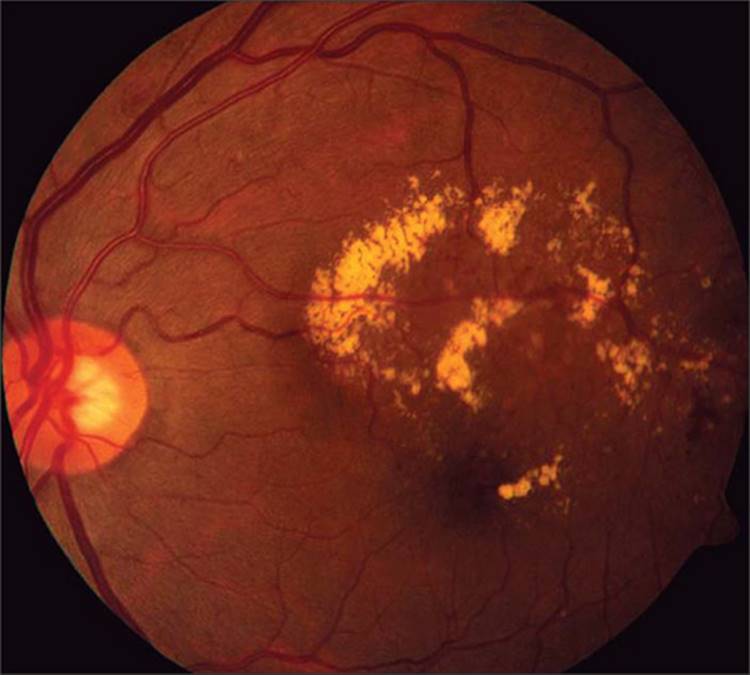
One of the most characteristic clinical appearances of DME is the presence of hard exudates (HE), which are lipoproteinaceous deposits from the transudation of fluid from retinal capillaries (Fig. 134.4). Abnormal fluid homeostasis in the retina results in a high rate of flux of plasma components in and out of the retinal capillaries. Slower absorption of transudated larger plasma molecules from the retinal tissues results in a net accumulation, which may eventually form a macroscopically visible accretion, the hard exudate. As with cystoid spaces, hard exudates develop in the outer plexiform layer (Fig. 134.5).[98] Not unexpectedly, several reports correlate increased quantities of hard exudates in patients with DME who also suffer from dyslipidemia.[48] Hard exudates may accumulate in the foveal center, leading to loss of central visual function.[99] A large amount of hard exudates at the center may tend to cause fibrotic organization which portends poor visual prognosis.
|
|
|
|
FIGURE 134.4 Color photograph of the left eye of a diabetic patient with focal macular edema, center-involved, with circinate hard exudates roughly circumscribing the area of retinal thickening. |
|
|
|
|
FIGURE 134.5 Photomicrograph of hard exudates in the outer plexiform layer of the retina in the retina of a diabetic patient. H&E, original magnification ×100. Image furnished by Daniel Albert, MD. |
A paradoxical increase in the presence of HE may occur temporarily in recently treated eyes with DME. This may be due to increased fluid reuptake in the retinal capillaries, leaving behind a greater proportion of lipoproteins that previously were dispersed throughout the edema in the retinal tissue. This effect has clinical relevance for those eyes that have hard exudates close to, but not yet in, the macular center. Treating the macular edema could, in such a case, cause increased hard exudate accumulation into the center leading to loss of visual acuity.[100] HE may gradually reabsorb over a period of months if macular edema is reduced. Eyes with aggregation of hard exudates in the center of the macula may have some visual recovery at that point.
Chronic severe DME, in addition to retinal atrophy, can lead to subretinal fibrosis, particularly if chronic hard exudates and subretinal fluid are present.[101] In some cases, subretinal non-vascular membranes appear to be stimulated by laser treatment of macular edema. Often, such membranes are subclinical but may be noted on histopathology in autopsy eyes or clinically by OCT. If subretinal fibrosis occurs under the fovea, eventual photoreceptor loss and poor vision result. Macular ischemia may coexist with DME, which may also yield visual acuity worse than expected based on the appearance of the macula. Other confounding abnormalities, that can coexist and confuse the relationship between DME and vision and treatment include, epiretinal membranes and vitreous traction.[95,102,103]
CLINICAL PRESENTATION OF DIABETIC MACULAR EDEMA
Patients with DME present with a range of visual symptoms depending on the degree to which the fovea is involved and the chronicity of the edema. If the macula center is not involved, patients are rarely symptomatic; only a few very observant individuals may notice relative paracentral scotomas corresponding to focal edema and hard exudates. Some patients with central macular involvement have excellent acuity and no visual complaints, presumably because of only recent involvement of the center. Over time, patients experience a gradual progressive vision loss over weeks to months. Patients may complain of loss of color vision,[104] poor night vision, and washing-out of vision in bright sunlight with poor dark-light adaptation. Metamorphopsia is not uncommon. Frequently, patients with center-involved DME note fluctuation of vision from day-to-day or even over the course of a day.[34] In some cases, the patient may relate such changes to fluid retention, hyper or hypoglycemia, or ambient lighting. Some patients may experience a diurnal variation in macular edema,[105-109] though this appears to be of low frequency and magnitude and of uncertain clinical relevance.
On fundus examination with slit lamp biomicroscopy or contact lens, retinal thickening may present in some commonly identified clinical patterns. Focal edema often occurs associated with a cluster of microaneurysms, sometimes surrounded by an incomplete ring of hard exudates (this pattern is termed circinate hard exudates) (Fig. 134.4). Multiple foci of edema may be present, varying in size, and sometimes merging together over time. The macula center may or may not be involved. Diffuse DME may be very difficult to identify clinically if the retina is of uniform thickness, due to the lack of reference landmarks. Clues include the height of the retinal blood vessels over the pigment epithelium, loss of the foveal depression or even cystoid spaces. If the pupil is small or there is lens opacity interfering with the examination, such features are easily missed. Other features sometimes seen with macular edema include variable loss of retinal transparency, a large burden of microaneurysms and intraretinal hemorrhages, and dispersed flecks of hard exudate.
Stereoscopic fundus photographs have been obtained historically to document the extent of macular edema and monitor progress. The standardized seven-fields photography protocol, allow thorough documentation of mid-peripheral and posterior retinal lesions and has been a mainstay in clinical trials where both DR severity and macular edema are to be analyzed. As many ophthalmology offices transition from, film imaging to digital camera systems for a variety of reasons, issues with color contrast, display, and stereo viewing in the digital environment bring some challenges that can decrease the utility of color imaging. However, with adequate quality and display, high resolution color images are comparable to film.[110] Standardized protocol color imaging (film and digital) is employed extensively for clinical research and is comparable to biomicro-scopic clinical examination for the detection and classification of retinal thickening.[111] However, many clinicians rely increasingly on OCT to monitor the status of the macula, particularly if the center is involved.
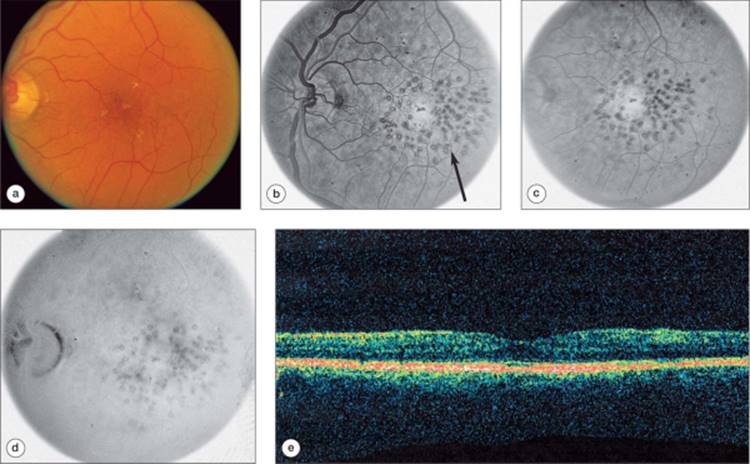
Fluorescein angiography yields useful information regarding macular perfusion and the extent of capillary leakage. At this time, angiography is the only common clinical method that can document retinal capillary ischemia, although good photographic quality is required. Areas of fluorescein leakage which may not correlate with retinal thickening are sometimes noted (Fig. 134.6); in contrast, sometimes areas of retinal thickening may not demonstrate notable fluorescein leakage. Partly for this reason, the ETDRS did not include fluorescein angiographic characteristics in the definition of clinically significant macular edema or criteria for retreatment,[2] although angiography was performed within the study.[100,112] However, angiographic features were used to guide treatment. The treatable lesions associated with macular edema include: focal leaks greater than 500 ?m from the center of the macula believed to be causing retinal thickening or HE; focal leaks 300-500 ?m from the center of the macula believed to be causing retinal thickening or HE if the treating ophthalmologist does not believe that treatment is likely to destroy the remaining perifoveal capillary network; areas of diffuse leakage that have not been treated previously; areas of capillary nonperfusion not previously treated. In many eyes with significant disease, the location of the fovea is not distinct, and angiography assists with topographical localization of the center and identification of microaneurysms for laser treatment.
|
|
|
|
FIGURE 134.6 Left eye of a diabetic patient prior to laser treatment. Baseline visual acuity measured 42 letters (20/160). (a) Color fundus photograph with central edema, hard exudates, and scattered intraretinal hemorrhages and microaneurysms. (b) Early phase angiogram, scanned from a negative film strip, with early leakage from microaneurysms. (c) Mid-phase of the angiogram showing patching fluorescein leakage. (d) Late cystoid staining. (e) OCT scan demonstrates cystoid edema. The hyper-reflective dot at the center (arrow) represents hard exudate in the outer plexiform layer. |
OCT technology was commercialized for retinal imaging in 1995 and quickly penetrated ophthalmology offices because of the advantageous and unique display of retinal topography and cross sectional anatomy.[113] Third-generation OCT machines display mean retinal thickness measurements in nine subfields that approximate (but are slightly smaller than) the macular grid used for measurement and grading in the ETDRS. The center point thickness and total macular volume within the grid are provided along with a false-color topographical map. The reproducibility of good-quality scans is very high and allows detection of moderate change in center retinal thickness over time.[114-116] However, quality issues in scan acquisition, media opacity, unstable fixation, and indistinct tissue planes causing measurement algorithm errors are relatively common and require interpretation.[117] Nevertheless, the ability to measure retinal thickness has not been available with other modalities and has been an important advance. Detection of subtle thickening at the center is more sensitive by OCT scans than clinical examination.[118,119] Cystoid spaces, subretinal fluid, and vitreoretinal abnormalities not visible on clinical examination or photography are often imaged on OCT scans.[96] Less clear is how robust current OCT technology is for measurement and tracking of noncenter involved macular edema. DME features on OCT correlate well with classification by fundus photographs[114,120,121] and fluorescein angiography.[122]
RELATIONSHIP BETWEEN MACULAR EDEMA AND VISUAL ACUITY AND OTHER MEASURES OF VISUAL FUNCTION
Untreated patients with center-involved DME experience a gradual loss of visual acuity. In the ETDRS, the 3-year risk of moderate vision loss (defined as loss of three lines of acuity on the standardized acuity chart) was ?33% at 3 years in the group with CSME.[2] However, the rate of moderate vision loss for eyes with clinically significant edema without center involvement was close to 20%, and untreated eyes with DME that was less than clinically significant had a rate of ?15%.[123] Unless ischemia, traction, or other confounding macular pathology is present, DME does not cause visual acuity loss until the center of the macula is involved. Even then, vision loss tends to be gradual, and it is not unusual to observe patients with obvious involvement of the foveal region by DME, yet have excellent acuity and no symptoms.[123] Conversely, as noted previously, patients with chronic edema may eventually undergo retinal disorganization and atrophy; such maculae may have poor acuity yet retinal thickness within the normal range. Therefore, a cross sectional assessment of a population with DME may display only a fair relationship between thickness and acuity, because it includes patients with a range of disease duration. OCT assessment of retinal thickness is therefore a limited predictor of vision in a cross-sectional sample of persons with DME.
Successful reduction of DME early in the course of the disease is likely to stabilize or improve visual function. The ETDRS demonstrated that laser-treated eyes not only had a significant reduction in the rate of vision loss, but those eyes were also twice as likely to experience visual acuity improvement. Unfortunately, eyes with long-standing DME that have reduction in retinal thickening, may not experience any visual improvement with treatment.[124]
Persons with no or minimal diabetic retinopathy may have subtle abnormalities of contrast sensitivity and color perception. DME decreases contrast sensitivity roughly in proportion to visual acuity. Threshold retinal sensitivity also decreases, and color vision is progressively impaired.[125,104] Light-dark adap-tation is also prolonged.[126] These measures confirm the subjective complaints of many subjects with DME regarding loss of night vision, dimness of vision, and impairment of color perception. On multifocal electroretinogram testing, reduced amplitudes and latencies can be found, again roughly parallel to the extent of acuity loss.[127,128] The oscillatory potentials measured from standard bright flash electroretinogram protocols may also be more sensitive than visual acuity as a measure of early impairment from diabetic retinal vasculopathy.[126,34] Carefully microperimetric analyses of diabetic maculae correlated with fluorescein angiograms demonstrate relative scotomas and other abnormalities.[129,130]
LASER PHOTOCOAGULATION FOR DIABETIC MACULAR EDEMA
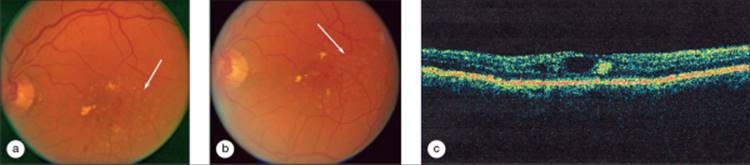
The ETDRS conclusively demonstrated that focal/grid laser photocoagulation was safe and effective in reducing vision loss due to DME (Figs 134.2, 134.3, 134.6, and 134.7). In eyes with center -involved DME, the rate of moderate visual loss (loss of 15 or more letters on the ETDRS chart) was reduced from 33% among eyes in the deferral of treatment arm to 13% among those eyes in the immediate laser treatment group,[123] a relative reduction in the rate by ?60%. At 3 years, in eyes with center-involved DME and acuity 20/40 or worse, improvement in acuity by six or more letters was observed in ?40% of eyes assigned to immediate treatment, versus ?20% in eyes assigned to deferral.[2] There was a similar two-fold increase in the rate of gain of 15 or more letters, ?15% versus ?7% (ETDRS, unpublished data). The 3 year results were sufficiently compelling that the data safety and monitoring committee of the ETDRS halted the study before the scheduled end of the trial so that subjects in the deferred laser treatment group could be offered treatment. While laser treatment increased the probability that a patient might have visual improvement, the number of patients actually manifesting substantial improve-ment was disappointingly small. In addition, the mean acuity of treated patients in the ETDRS had a progressive decline in vision over time despite treatment. The advent of laser photo-coagulation for DME was a great contribution for management of an otherwise untreatable condition, and laser treatment has saved useful vision in countless individuals. However, the burden of vision loss among patients with DME remains high, thus spurring the investigation of pharmacologic treatments that might be adjunctive to laser treatment, salvage therapy for those patients who continue to lose vision after laser, or potential new primary therapies.
|
|
|
|
FIGURE 134.7 Same eye shown in Figure 134.6 at one year post baseline, after a total of 3 modified ETDRS focal/grid photocoagulation treatments at baseline, 3.5 months and 8 months. Visual acuity improved to 65 letters (20/50) at 12 months. (a) Color photograph demonstrating reduction in hard exudates and visible microaneurysms. (b) Early phase fluorescein angiogram showing hyperfluorescent pigment epithelial defects from laser burns (arrow). (c) Mid-phase angiogram showing reduced leakage. (d) Late phase angiogram with only mild fluorescein leakage and no cystoid edema. (e) OCT scan from the 12 month visit demonstrating no significant retinal thickening, and resolution of cystoid spaces. |
The technique of ETDRS focal/grid photocoagulation has been used as standard therapy in the community since the study results were disseminated, with little modification over the years.[123] The original study employed argon blue-green laser with laser spot sizes up to 100 ?m in diameter. Newer lasers routinely allow spot sizes of 50 ?m in diameter, which is preferable to avoid complications from laser scar expansion. The blue argon wavelengths have been filtered out from modern argon lasers, since, the luteal pigments in the inner retinal layers absorb blue light and yield unnecessary thermal damage to the retinal nerve fiber layer. In addition, solid state lasers are available to provide treatment with green wavelengths. The intensity of application tends to be slightly less among retinal surgeons today, again out of concern for laser scar expansion (Fig. 134.8). The ETDRS protocol recommends focal treatment to leaking microaneurysms that are at least 500 ?m from the fovea and grid laser treatment to areas of diffuse retinal thickening. In addition, areas of ischemia identified on fluorescein angio-graphy could be treated with grid laser. Repeat laser treatment may be applied if clinically significant DME persists. In the ETDRS, leaking microaneurysms up to 300 ?m from the fovea could be treated if additional treatment was needed at subsequent visits. Laser burns this close to the fovea increase the risk of scotomas, accidental fovea burns, and late damage to the fovea from laser scar enlargement and are rarely used today.
|
|
|
|
FIGURE 134.8 Same eye shown in Figure 134.6, immediately after the first laser application (a), and immediately following the second laser application (b), at 3.5 months after the first. Pale gray laser spots can be observed in both photographs (arrows). (c) OCT scan of the same eye at 8 months, demonstrating reduction in retinal edema, but persistent cystoid changes and hard exudate. |
|
Tips: Modified ETDRS Focal/Grid Laser Photocoagulation of DME |
|||||||||||||||||||||
|
Adverse effects of macular laser treatment according to the ETDRS protocol are remarkable few. The most feared complication is accidental foveal photocoagulation, which may occur from inopportune ocular movement or from surgeon disorientation and error.[131] This may produce immediate and permanent visual acuity loss and a central scotoma. Closure of microaneurysms in a very ischemic macula may contribute to worsening of ischemia.[123] Very severe laser burns, particularly if they are small and well focused, may rupture Bruch's membrane and cause subretinal and vitreous bleeding. Late complications from severe laser scars include choroidal neovascularization. Subretinal fibrosis may occur associated with laser scars, perhaps more commonly in patients with very severe DME.[132] Very severe laser burns, particularly if applied over hemorrhage, may also cause epiretinal proliferation with retinal traction. Finally, a common observation is that laser scars tend to enlarge over time due to a phenomenon termed 'RPE creep'. The retinal pigment epithelium (RPE) at the margins of macular laser scars tends to atrophy over time, causing progressively enlarging regions of RPE that may become confluent or may erode under the fovea, causing photoreceptor dysfunction and vision loss.[1,100] Because focal/grid macular laser treatment is not entirely without risk, the ETDRS noted that for eyes with clinically significant macular edema without center involvement, observation for a period is acceptable management.[100] In particular, if a patient has fluid retention, management of the systemic issues may occasionally benefit the patients DME and forestall the need for laser treatment.[34]
Alternatives to ETDRS-style focal/grid macular photo-coagulation have been evaluated in numerous studies. The Diabetic Retinopathy Clinical Research Network (DRCR.net![]() ) has evaluated a diffuse grid style of very mild burns compared to standard of care focal/grid treatment, and found no advantage to the former (DRCR Network Research Group, manuscript submitted). Subthreshold burns with diode laser[133] or green laser[134,135] have been employed to reduce the risk of perifoveal scotoma and laser scarring.
) has evaluated a diffuse grid style of very mild burns compared to standard of care focal/grid treatment, and found no advantage to the former (DRCR Network Research Group, manuscript submitted). Subthreshold burns with diode laser[133] or green laser[134,135] have been employed to reduce the risk of perifoveal scotoma and laser scarring.
Complicating any discussion of laser treatments alternative to the ETDRS method is the fact that the underlying cause for the treatment effect of laser is not well understood. Some evidence for increased oxygen tension after laser suggests an effect in downregulating VEGF production.[136] Pigment epithelium derived growth factor (PEDF) is a candidate inhibitor of vascular permeability that has VEGF antagonist and antiinflammatory properties,[137] and is upregulated in RPE cells by laser treatment.[138]
OCT is more sensitive than clinical examination for identifying subtle thickening of the foveal region.[118,119] This raises the question of what threshold of retinal thickening is of sufficient risk to merit laser or any other treatment. The findings of the ETDRS are based upon clinician-determined identification of retinal thickening to guide treatment and reading center interpretation of stereoscopic color photographs for statistical analysis. Patients with edema detectable only on OCT fall into a category that is unstudied in regard to risk of vision loss and requirement for treatment. The DRCR.net![]() is currently enrolling subjects with subclinical DME, detected on OCT but not on clinical examination, into a protocol for a longitudinal cohort study (www.DRCRnet.org
is currently enrolling subjects with subclinical DME, detected on OCT but not on clinical examination, into a protocol for a longitudinal cohort study (www.DRCRnet.org![]() ).
).
|
Complications: Potential Complications of Laser Photocoagulation of DME |
|||||||||||||||||||||||||||
|
VITRECTOMY SURGERY
A subset of patients with DME has coexistent epiretinal membranes and/or partial posterior vitreous detachment with retinal traction. These patients may benefit from pars plana vitrectomy to address the mechanical issues contributing to the retinal edema.[139,102,140] Even without obvious retinal traction, some clinicians believe that many cases of DME respond to removal of the vitreous, with or without removal of the internal limiting membrane.[141] The rationale for this approach is that there may be some subclinical traction from a taut posterior vitreous face, or that the intact vitreous serves as a reservoir for inflammatory macromolecules which promote edema.[81,142,143,144] No large randomized trials have evaluated this treatment to date.
TRIAMCINOLONE ACETONIDE
The rationale for use of steroids to treat DME is compelling.[88,145] As noted previously, the molecular biology of diabetic vascular change includes leukostasis and other mechanisms of inflammation, endothelial decompensation, and increased levels of pro-inflammatory cytokines. Steroids may be useful to treat DME because of antiinflammatory effects which include decreased inflammatory cell activation and adhesion, and growth factor signaling. Steroids also have a direct effect on the maturation of interendothelial cell junctions and improved barrier properties.
Intravitreous injection of triamcinolone acetonide often has a marked beneficial effect on retinal thickening in DME and thus has sparked considerable interest and clinical use. Numerous small studies have demonstrated robust anatomic results, but often the visual results do not correlate well with reduction in retinal thickening,[124,146,147] probably because of the aforementioned complicating factors for lack of vision recovery with chronic DME. The duration of drug in the vitreous cavity approximates the duration of clinical effect (2-3 months for a 4.0 mg injection of triamcinolone).[147-151] Repeated injections may be needed to maintain clinical benefit. The complications of triamcinolone acetonide injection are well documented and include endophthalmitis, elevated intraocular pressure requiring topical therapy or even surgical intervention to prevent vision loss,[152-155] and cataract.[156,157]
A large randomized clinical trial of serial intravitreal injections of triamcinolone has completed enrollment within the DRCR.net![]() , with results not yet available at this time. While short-term effects of intravitreal triamcinolone are encouraging, a longer-term trial is essential to determining whether the known risks of intravitreal injection of steroids are outweighed by the ultimate clinical benefits.
, with results not yet available at this time. While short-term effects of intravitreal triamcinolone are encouraging, a longer-term trial is essential to determining whether the known risks of intravitreal injection of steroids are outweighed by the ultimate clinical benefits.
Because the risks of intravitreal injection are relatively high, there is high-clinical interest in evaluating the potential benefits of peribulbar injection of high-dose triamcinolone acetonide. The risks of cataract and intraocular pressure elevation are somewhat less from peribulbar injection, but with repeated injections there may be ptosis, orbital fat atrophy and enophthalmos, and diplopia from extraocular muscle damage. A 40 mg dose of triamcinolone in the orbit may produce detectable serum levels and affect glycemic control in diabetic patients. Small studies have suggested benefit in the treatment of DME, although there is considerable variability in intraocular penetration from peribulbar injection, and clearly intraocular drug levels are lower than with direct intraocular admini-stration.[67,158] At this time, both peribulbar and intravitreous triamcinolone steroids are widely employed clinically in the management of DME, but without a sufficient evidence base to merit recommendation as standard therapy.
ANTI-VEGF THERAPY
Given the previous discussion of the importance of VEGF in vascular permeability and its upregulation in diabetic retinopathy, the rationale for use of anti-VEGF drugs is clear. Current specific anti-VEGF therapy is given intravitreal at frequent intervals, which may temporarily blunt the effects of VEGF and lessen macular edema. Unknown is whether treatment will alter the underlying disease process (although it may cause at least temporary regression of neovascularization).[159,160] Therefore, as the drug effect wears off, the underlying production of VEGF appears to continue unabated and it is likely that long term intravitreal therapy is required for management.[161]
Results from a phase II clinical trial of intravitreal injection of pegaptanib (Macugen: Eyetech OSI) over 36 weeks demonstrated beneficial effects,[162] and a phase III trial is in progress at this time. Median VA was better at week 36 with 0.3 mg/0.05 ml pegaptanib (20/50), as compared with sham injection (20/63) (P = 0.04), and a larger proportion of patients receiving pegaptanib had < or =15 letters of visual gain (18% versus. 7%, P = 0.12). Mean central retinal thickness decreased by 68 ?m with 0.3 mg, versus an increase of 4 ?m with sham (P = 0.02). Pegaptanib is an engineered RNA fragment bearing specific binding sites for the VEGF 165 isoform. It is currently administered as an intravitreal injection at 6 week intervals.
Early phase clinical trials of ranabizumab (Lucentis: Genentech) are in progress. Off label use of bevacizumab (Avastin: Genentech) has been presented in small cohorts of patients with macular edema from various causes,[163,164] but no randomized trials of DME have been reported. Ranabizumab and bevacizumab are fusion proteins with a human antibody backbone and bind all VEGF subtypes. Both of these drugs are given as intravitreal injections, but the optimal delivery schedule and dosing level have not been elucidated.
INHIBITION OF PROTEIN KINASE C
PKC is a family of intracellular signaling peptides that act as intermediaries for growth factor and integrin signaling between ligand binding and nuclear transcription. Broad inhibition of PKC is highly toxic, however specific isoforms are upregulated in response to particular stimuli. The PKC? isoform is specifically upregulated in hyperglycemia in multiple tissues, including vascular endothelial cells, and therefore is hypothesized to mediate some of the myriad biochemical disturbances induced by chronic hyperglycemia.[165] In animal models of diabetes, a specific PKC? inhibitor, ruboxistaurin (Arxxant: Eli Lilly), decreased abnormal vascular permeability and blood flow,[82,83] and also inhibited angiogenesis in a nondiabetic retinal ischemia model.[85]
Phase III clinical trials of oral administration of ruboxistaurin have demonstrated efficacy in the prevention of sustained moderate vision loss in diabetic patients with nonproliferative DR.[3] In a trial designed to test the effects of the study drug on progression of diabetic retinopathy, it was found that ruboxistaurin reduced the rate of sustained moderate vision loss (loss of 15 or more letters for at least two consecutive study visits) in eyes with definite diabetic macular edema at baseline (10% 32 mg/day study drug versus 25% placebo, P = 0.017). A separate study evaluated the ruboxistaurin specifically in regard to the reduction of progression of retinal thickening from more than 300 ? to within 100 ? of the center as measured in stereoscopic fundus photographs. The 32 mg/day dose reduced progression of DME compared to placebo (hazard ratio = 0.66 (95% CI 0.47-0.93 P = 0.016) (PKC-DMES Study Group, manuscript submitted). This product is currently under regulatory review for the treatment of early diabetic macular edema.
|
Complications of local drug injection |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
SUMMARY AND FUTURE DIRECTIONS
The landscape of DME management is rapidly changing with the advent of research advances leading to better understanding of pathophysiologic mechanisms and discovery of potential therapeutic compounds. There are several challenges that remain in bringing forth new treatments with an adequate evidence base to guide clinicians in timely patient care.
|
1. |
Diabetic retinopathy is a complicated disease. It is clear that a number of altered cellular mechanisms work in concert to bring about clinical complications. It is unclear how impacting only one or a few of these pathways will result in effective treatment, and it may be that a combination approach will be necessary for management of some cases. |
|
|
2. |
Vascular leakage from hyperglycemia can be produced in animal models, but there is no robust animal model of DME, which limits the preclinical testing of potential new treatments before clinical development. |
|
|
3. |
Diabetic retinopathy is a chronic disease: onset is gradual over years, and treatment may be required indefinitely. Consequently, clinical trials are large and lengthy, creating huge financial burdens in bringing potential new treatments to market, slowing their introduction and adding to the costs, thereby creating barriers for patient care. |
|
|
4. |
Visual acuity is an imprecise and subjective measure of visual function, although it is highly relevant and clearly the best test available. It correlates only modestly with advanced DME, not at all with early (not center involved) DME. |
|
|
5. |
The OCT has become the standard method for assessing the status of the central macula in DME, but current technology is of uncertain sensitivity for detection or monitoring of edema that is not center-involved (for both patient care and in clinical trials). Retinal thickness measurement (on exam, in photos, or by OCT) correlates only moderately with visual acuity, but remains the most important variable for disease presence and progression. |
There is, therefore, room for improvement in functional and anatomic assessments of DME. Validation of other, early functional assessments with disease outcome may shorten the drug development timeline and result in increased efficiency of clinical research.
Despite these caveats, research continues intensively because the public health burden of DME and other diabetic compli-cations is so great. This has led to the current era of an array of treatments of uncertain benefit and risk, and lack of clinician certainty as to how and when to apply what treatments and in what combination, if any. It will take years to sort out these issues, and in the meantime, new treatments will be introduced which may further complicate the decision making process for patient and physician. Nevertheless, this is clearly a superior situation over the converse, having no promising treatments to offer. While clinical trials continue to work toward testing new approaches, the results of the ETDRS remains a solid foundation for clinicians to refer to when faced with a difficult treatment decision.
The results of the DCCT/EDIC and UKPDS trials demonstrate that DME and other complications can be prevented or delayed through excellent metabolic management, and this should continue to be a major focus of conversation between physician and patient at any phase of the disease. Early disease detection and treatment are the next greatest priority, and all health care providers need to work in concert to educate, screen and monitor patients with diabetes and those at high risk for diabetes.
REFERENCES
1. Early Treatment Diabetic Retinopathy Study Research Group : Treatment techniques and clinical guidelines for photocoagulation of diabetic macular edema. Early treatment diabetic retinopathy study report number 2. Ophthalmology 1987; 94:761-774.
2. Early Treatment Diabetic Retinopathy Study Research Group : Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Arch. Ophthalmol 1985; 103:1796-1806.
3. PKC-DRS Study Group : The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes 2005; 54:2188-2197.
4. Klein R, Klein BE, Moss SE, et al: The Wisconsin epidemiologic study of diabetic retinopathy. IV. Diabetic macular edema. Ophthalmology 1984; 91:1464-1474.
5. Klein R, Klein BE, Moss SE, Linton KL: The Beaver Dam Eye Study. Retinopathy in adults with newly discovered and previously diagnosed diabetes mellitus. Ophthalmology 1992; 99:58-62.
6. Wong TY, Klein R, Islam FM, et al: Diabetic retinopathy in a multi-ethnic cohort in the United States. Am. J. Ophthalmol 2006; 141:446-455.
7. Roy MS, Klein R, O'Colmain BJ, et al: The prevalence of diabetic retinopathy among adult type 1 diabetic persons in the United States. Arch. Ophthalmol 2004; 122:546-551.
8. Kempen JH, O'Colmain BJ, Leske MC, et al: The prevalence of diabetic retinopathy among adults in the United States. Arch. Ophthalmol 2004; 122:552-563.
9. Varma R, Torres M, Pena F, et al: Prevalence of diabetic retinopathy in adult Latinos: the Los Angeles Latino eye study. Ophthalmology 2004; 111:1298-1306.
10. Lecaire T, Palta M, Zhang H, et al: Lower-than-Expected Prevalence and Severity of Retinopathy in an Incident Cohort followed during the First 4-14 Years of Type 1 Diabetes. Am. J. Epidemiol 2006.
11. Cugati S, Kifley A, Mitchell P, Wang JJ: Temporal trends in the age-specific prevalence of diabetes and diabetic retinopathy in older persons: Population-based survey findings. Diabetes Res. Clin. Pract. 2006.
12. Klein R, Klein BE, Moss SE, Cruickshanks KJ: The Wisconsin Epidemiologic Study of Diabetic Retinopathy. XV. The long-term incidence of macular edema. Ophthalmology 1995; 102:7-16.
13. Early Treatment Diabetic Retinopathy Study Research Group : Case reports to accompany Early Treatment Diabetic Retinopathy Study Reports 3 and 4. Int. Ophthalmol. Clin 1987; 27:273-333.
14. Early Treatment Diabetic Retinopathy Study Research Group : Grading diabetic retinopathy from stereoscopic color fundus photographs an extension of the modified Airlie House classification. ETDRS report number 10. Ophthalmology 1991; 98:786-806.
15. Group : ETDRSR. Fundus photographic risk factors for progression of diabetic retinopathy. ETDRS report number 12. Ophthalmology 1991; 98:823-833.
16. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med 1993; 329:977-986.
17. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N. Engl. J. Med 2000; 342:381-389.
18. Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group : Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA 2002; 287:2563-2569.
19. UK Prospective Diabetes Study (UKPDS) Group : Effect of intensive blood-glucose control with metformin on complications in overweight patients with type 2 diabetes (UKPDS 34). Lancet 1998; 352:854-865.
20. Klein R, Klein BE, Moss SE, et al: Glycosylated hemoglobin predicts the incidence and progression of diabetic retinopathy. JAMA 1988; 260:2864-2871.
21. The absence of a glycemic threshold for the development of long-term complications: the perspective of the Diabetes Control and Complications Trial. Diabetes 1996; 45:1289-1298.
22. Zander E, Herfurth S, Bohl B, et al: Maculopathy in patients with diabetes mellitus type 1 and type 2: associations with risk factors. Br. J. Ophthalmol 2000; 84:871-876.
23. Klein R, Klein BE: Relation of glycemic control to diabetic complications and health outcomes. Diabetes Care 1998; 21:C39-C43.
24. Moss SE, Klein R, Klein BE: The 14-year incidence of visual loss in a diabetic population. Ophthalmology 1998; 105:998-1003.
25. Balme M, McAllister I, Davis WA, Davis TM: Retinopathy in latent autoimmune diabetes of adults: the Fremantle Diabetes Study. Diabet. Med 2002; 19:602-605.
26. Wong TY, Mohamed Q, Klein R, Couper DJ: Do retinopathy signs in non-diabetic individuals predict the subsequent risk of diabetes?. Br. J. Ophthalmol 2006; 90:301-303.
27. Kohner EM, Aldington SJ, Stratton IM, et al: United Kingdom Prospective Diabetes Study, 30: diabetic retinopathy at diagnosis of non-insulin-dependent diabetes mellitus and associated risk factors. Arch. Ophthalmol 1998; 116:297-303.
28. Newcomb PA, Klein R, Massoth KM: Education to increase ophthalmologic care in older onset diabetes patients: indications from the Wisconsin Epidemiologic Study of Diabetic Retinopathy. J. Diabetes Complications 1992; 6:211-217.
29. Klein R: Prevention of visual loss from diabetic retinopathy. Surv. Ophthalmol. 2002; 47(Suppl 2):S246-S252.
30. Harris MI, Klein R, Welborn TA, Knuiman MW: Onset of NIDDM occurs at least 4-7 yr before clinical diagnosis. Diabetes Care 1992; 15:815-819.
31. Klein R, Klein BE, Moss SE, Cruickshanks KJ: The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology 1988; 105:1801-1815.
32. Effect of pregnancy on microvascular complications in the diabetes control and complications trial: The Diabetes Control and Complications Trial Research Group. Diabetes Care 2000; 23:1084-1091.
33. Matthews DR, Stratton IM, Aldington SJ, et al: Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch. Ophthalmol 2004; 122:1631-1640.
34. Bresnick GH: Diabetic macular edema. A review. Ophthalmology 1986; 93:989-997.
35. Sjolie AK, Porta M, Parving HH, et al: The DIabetic REtinopathy Candesartan Trials (DIRECT) Programme: baseline characteristics. J Renin Angiotensin Aldosterone Syst 2005; 6:25-32.
36. Klein R, Zinman B, Gardiner R, et al: The relationship of diabetic retinopathy to preclinical diabetic glomerulopathy lesions in type 1 diabetic patients: the Renin-Angiotensin System Study. Diabetes 2005; 54:527-533.
37. Agardh CD, Agardh E, Torffvit O: The association between retinopathy, nephropathy, cardiovascular disease and long-term metabolic control in type 1 diabetes mellitus: a 5 year follow-up study of 442 adult patients in routine care. Diabetes Res. Clin. Pract 1997; 35:113-121.
38. Bresnick GH: Diabetic maculopathy. A critical review highlighting diffuse macular edema. Ophthalmology 1983; 90:1301-1317.
39. Tokuyama T, Ikeda T, Sato K: Effects of haemodialysis on diabetic macular leakage. Br. J. Ophthalmol 2000; 84:1397-1400.
40. Watanabe Y, Yuzawa Y, Mizumoto D, et al: Long-term follow-up study of 268 diabetic patients undergoing haemodialysis, with special attention to visual acuity and heterogeneity. Nephrol. Dial. Transplant 1993; 8:725-734.
41. Roy MS, Klein R: Macular edema and retinal hard exudates in African Americans with type 1 diabetes: the New Jersey 725. Arch. Ophthalmol 2001; 119:251-259.
42. Miljanovic B, Glynn RJ, Nathan DM, et al: A prospective study of serum lipids and risk of diabetic macular edema in type 1 diabetes. Diabetes 2004; 53:2883-2892.
43. Effects of intensive diabetes therapy on neuropsychological function in adults in the Diabetes Control and Complications Trial : The DCCT Research Group. Ann. Intern. Med 1996; 124:379-388.
44. Ferris 3rd FL, Chew EY, Hoogwerf BJ, Early Treatment Diabetic Retinopathy Study Research Group : Serum lipids and diabetic retinopathy. tudy Research Group 1996; 19:1291-1293.
45. Klein R, Klein BE, Moss SE, Surawicz TS: The Wisconsin Epidemiologic Study of Diabetic Retinopathy. XIII. Relationship of serum cholesterol to retinopathy and hard exudate. Ophthalmology 1991; 98:1261-1265.
46. Klein R, Sharrett AR, Klein BE, et al: The association of atherosclerosis, vascular risk factors, and retinopathy in adults with diabetes : the atherosclerosis risk in communities study. Ophthalmology 2002; 109:1225-1234.
47. Davis MD, Fisher MR, Gangnon RE, et al: Risk factors for high-risk proliferative diabetic retinopathy and severe visual loss: Early Treatment Diabetic Retinopathy Study Report #18. Invest. Ophthalmol. Vis. Sci 1998; 39:233-252.
48. Chew EY, Klein ML, Ferris FLr, et al: Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early Treatment Diabetic Retinopathy Study (ETDRS) Report 22. Arch. Ophthalmol 1996; 114:1079-1084.
49. Jenkins AJ, Lyons TJ, Zheng D, et al: Serum lipoproteins in the diabetes control and complications trial/epidemiology of diabetes intervention and complications cohort: associations with gender and glycemia. Diabetes Care 2003; 26:810-818.
50. Klein RL, McHenry MB, Lok KH, et al: Apolipoprotein C-III protein concentrations and gene polymorphisms in Type 1 diabetes: associations with microvascular disease complications in the DCCT/EDIC cohort. J. Diabetes Complications 2005; 19:18-25.
51. Lyons TJ, Jenkins AJ, Zheng D, et al: Diabetic retinopathy and serum lipoprotein subclasses in the DCCT/EDIC cohort. Invest. Ophthalmol. Vis. Sci 2004; 45:910-918.
52. Klein BE, Moss SE, Klein R: Effect of pregnancy on progression of diabetic retinopathy. Diabetes Care 1990; 13:34-40.
53. Temple RC, Aldridge VA, Sampson MJ, et al: Impact of pregnancy on the progression of diabetic retinopathy in Type 1 diabetes. Diabet. Med 2001; 18:573-577.
54. Lovestam-Adrian M, Agardh CD, Aberg A, Agardh E: Pre-eclampsia is a potent risk factor for deterioration of retinopathy during pregnancy in Type 1 diabetic patients. Diabet. Med 1997; 14:1059-1065.
55. Axer-Siegel R, Hod M, Fink-Cohen S, et al: Diabetic retinopathy during pregnancy. Ophthalmology 1996; 103:1815-1819.
56. Sinclair SH, Nesler C, Foxman B, et al: Macular edema and pregnancy in insulin-dependent diabetes. Am. J. Ophthalmol 1984; 97:154-167.
57. Chew EY, Mills JL, Metzger BE, et al: Metabolic control and progression of retinopathy. The Diabetes in Early Pregnancy Study. National Institute of Child Health and Human Development Diabetes in Early Pregnancy Study. National Institute of Child Health and Human Development Diabetes in Early Pregnancy Study 1995; 18:631-637.
58. Phelps RL, Sakol P, Metzger BE, et al: Changes in diabetic retinopathy during pregnancy. Correlations with regulation of hyperglycemia. Arch. Ophthalmol 1986; 104:1806-1810.
59. Hellstedt T, Kaaja R, Teramo K, Immonen I: The effect of pregnancy on mild diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol 1997; 235:437-441.
60. Davis MD: Worsening of diabetic retinopathy after improvement of glycemic control. Arch. Ophthalmol 1998; 116:931-932.
61. Patel JI, Hykin PG, Cree IA: Diabetic cataract removal: postoperative progression of maculopathy growth factor and clinical analysis. Br. J. Ophthalmol 2006; 90:697-701.
62. Krepler K, Biowski R, Schrey S, et al: Cataract surgery in patients with diabetic retinopathy: visual outcome, progression of diabetic retinopathy, and incidence of diabetic macular oedema. Graefes Arch. Clin. Exp. Ophthalmol 2002; 240:735-738.
63. Squirrell D, Bhola R, Bush J, et al: A prospective, case controlled study of the natural history of diabetic retinopathy and maculopathy after uncomplicated phacoemulsification cataract surgery in patients with type 2 diabetes. Br. J. Ophthalmol 2002; 86:565-571.
64. Menchini U, Cappelli S, Virgili G: Cataract surgery and diabetic retinopathy. Semin. Ophthalmol 2003; 18:103-108.
65. Ferris FLr, Podgor MJ, Davis MD: Macular edema in Diabetic Retinopathy Study patients. Diabetic Retinopathy Study Report Number 12. Ophthalmology 1987; 94:754-760.
66. Shimura M, Yasuda K, Nakazawa T, et al: Quantifying alterations of macular thickness before and after panretinal photocoagulation in patients with severe diabetic retinopathy and good vision. Ophthalmology 2003; 110:2386-2394.
67. Wilson CA, Berkowitz BA, Sato Y, et al: Treatment with intravitreal steroid reduces blood-retinal barrier breakdown due to retinal photocoagulation. Arch. Ophthalmol 1992; 110:1155-1159.
68. Zein WM, Noureddin BN, Jurdi FA, et al: Panretinal photocoagulation and intravitreal triamcinolone acetonide for the management of proliferative diabetic retinopathy with macular edema. Retina 2006; 26:137-142.
69. Cogan DG, Kuwabara T: The mural cell in perspective. Arch. Ophthalmol 1967; 78:133-139.
70. Kuwabara T, Cogan DG: Retinal vascular patterns. VII. Acellular change. Invest. Ophthalmol 1965; 4:1049-1064.
71. King GL, Shiba T, Oliver J, et al: Cellular and molecular abnormalities in the vascular endothelium of diabetes mellitus. Annu. Rev. Med 1994; 45:179-188.
72. Gardner TW, Antonetti DA, Barber AJ, et al: Diabetic retinopathy: more than meets the eye. Surv. Ophthalmol. 2002; 47(Suppl 2):S253-S262.
73. Feit-Leichman RA, Kinouchi R, Takeda M, et al: Vascular damage in a mouse model of diabetic retinopathy: relation to neuronal and glial changes. Invest. Ophthalmol. Vis. Sci 2005; 46:4281-4287.
74. Harhaj NS, Antonetti DA: Regulation of tight junctions and loss of barrier function in pathophysiology. Int. J. Biochem. Cell Biol 2004; 36:1206-1237.
75. Vinores SA, Derevjanik NL, Ozaki H, et al: Cellular mechanisms of blood-retinal barrier dysfunction in macular edema. Doc. Ophthalmol 1999; 97:217-228.
76. Bursell SE, Delori FC, Yoshida A, et al: Vitreous fluorophotometric evaluation of diabetics. Invest. Ophthalmol. Vis. Sci 1984; 25:703-710.
77. Ferrara N: Role of vascular endothelial growth factor in regulation of physiological angiogenesis. Am J Physiol Cell Physiol 2001; 280:C1358-C1366.
78. Adamis AP, Shima DT, Tolentino MJ, et al: Inhibition of vascular endothelial growth factor prevents retinal ischemia-associated iris neovascularization in a nonhuman primate. Arch. Ophthalmol 1996; 114:66-71.
79. Suarez S, Ballmer-Hofer K: VEGF transiently disrupts gap junctional communication in endothelial cells. J. Cell Sci 2001; 114:1229-1235.
80. Antonetti DA, Barber AJ, Hollinger LA, et al: Vascular endothelial growth factor induces rapid phosphorylation of tight junction proteins occludin and zonula occluden 1. A potential mechanism for vascular permeability in diabetic retinopathy and tumors. J. Biol. J. Biol. Chem 1999; 274:23463-23467.
81. Patel JI, Tombran-Tink J, Hykin PG, et al: Vitreous and aqueous concentrations of proangiogenic, antiangiogenic factors and other cytokines in diabetic retinopathy patients with macular edema: Implications for structural differences in macular profiles. Exp. Eye Res 2006; 82:798-806.
82. Ishii H, Jirousek MR, Koya D, et al: Amelioration of vascular dysfunctions in diabetic rats by an oral PKC beta inhibitor. Science 1996; 272:728-731.
83. Aiello LP, Bursell SE, Clermont A, et al: Vascular endothelial growth factor-induced retinal permeability is mediated by protein kinase C in vivo and suppressed by an orally effective beta-isoform-selective inhibitor. Diabetes 1997; 46:1473-1480.
84. Miyamoto K, Khosrof S, Bursell SE, et al: Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. U. S. A 1999; 96:10836-10841.
85. Danis RP, Bingaman DP, Jirousek M, Yang Y: Inhibition of intraocular neovascularization caused by retinal ischemia in pigs by PKCbeta inhibition with LY333531. Invest. Ophthalmol. Vis. Sci 1998; 39:171-179.
86. Pachydaki SI, Tari SR, Lee SE, et al: Upregulation of RAGE and its ligands in proliferative retinal disease. Exp. Eye Res 2006; 82:807-815.
87. Joussen AM, Poulaki V, Le ML, et al: A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 2004; 18:1450-1452.
88. Ip MS: Intravitreal injection of triamcinolone: an emerging treatment for diabetic macular edema. Diabetes Care 2004; 27:1794-1797.
89. Martidis A, Duker JS, Greenberg PB, et al: Intravitreal triamcinolone for refractory diabetic macular edema. Ophthalmology 2002; 109:920-927.
90. Felinski EA, Antonetti DA: Glucocorticoid regulation of endothelial cell tight junction gene expression: novel treatments for diabetic retinopathy. Curr. Eye Res 2005; 30:949-957.
91. Yanoff M, Fine BS, Brucker AJ, Eagle Jr RC: Pathology of human cystoid macular edema. Surv. Ophthalmol. 1984; 28(Suppl):505-511.
92. Kincaid MC, Green WR, Fine SL, et al: An ocular clinicopathologic correlative study of six patients from the Diabetic Retinopathy Study. Retina 1983; 3:218-238.
93. Antcliff RJ, Marshall J: The pathogenesis of edema in diabetic maculopathy. Semin. Ophthalmol 1999; 14:223-232.
94. Fluorescein angiographic risk factors for progression of diabetic retinopathy: ETDRS report number 13. Ophthalmology 1991; 98:834-840.
95. Catier A, Tadayoni R, Paques M, et al: Characterization of macular edema from various etiologies by optical coherence tomography. Am. J. Ophthalmol 2005; 140:200-206.
96. Panozzo G, Parolini B, Gusson E, et al: Diabetic macular edema: an OCT-based classification. Semin. Ophthalmol 2004; 19:13-20.
97. Ferris 3rd FL, Patz A: Macular edema. A complication of diabetic retinopathy. Surv. Ophthalmol. 1984; 28(Suppl):452-461.
98. Yanko L, Ungar H, Michaelson IC: The exudative lesions in diabetic retinopathy with special regard to the hard exudate. Acta Ophthalmol. (Copenh) 1974; 52:150-160.
99. Patz A, Fine SL: Diabetic macular edema. Int. Ophthalmol. Clin 1976; 16:105-113.
100. Early Treatment Diabetic Retinopathy Study Research Group : Focal photocoagulation treatment of diabetic macular edema. Relationship of treatment effect to fluorescein angiographic and other retinal characteristics at baseline: ETDRS report no. 19. Arch. Ophthalmol 1995; 113:1144-1155.
101. Chester EM, Banker BQ: The role of lipid thrombi in the pathogenesis of diabetic retinopathy. Arch. Intern. Med 1967; 120:397-407.
102. Johnson MW: Tractional cystoid macular edema: a subtle variant of the vitreomacular traction syndrome. Am. J. Ophthalmol 2005; 140:184-192.
103. Gaucher D, Tadayoni R, Erginay A, et al: Optical coherence tomography assessment of the vitreoretinal relationship in diabetic macular edema. Am. J. Ophthalmol 2005; 139:807-813.
104. Fong DS, Barton FB, Bresnick GH: Impaired color vision associated with diabetic retinopathy: Early Treatment Diabetic Retinopathy Study Report No. 15. Am. J. Ophthalmol 1999; 128:612-617.
105. Frank RN, Schulz L, Abe K, Iezzi R: Temporal variation in diabetic macular edema measured by optical coherence tomography. Ophthalmology 2004; 111:211-217.
106. Sternberg Jr P, Fitzke F, Finkelstein D: Cyclic macular edema. Am. J. Ophthalmol 1982; 94:664-669.
107. Larsen M, Wang M, Sander B: Overnight thickness variation in diabetic macular edema. Invest. Ophthalmol. Vis. Sci 2005; 46:2313-2316.
108. Polito A, Del Borrello M, Polini G, et al: Diurnal variation in clinically significant diabetic macular edema measured by the Stratus OCT. Retina 2006; 26:14-20.
109. Danis RP, Glassman AR, Aiello LP, et al: Diurnal variation in retinal thickening measurement by optical coherence tomography in center-involved diabetic macular edema. Arch. Ophthalmol 2006; 124:1701-1707.
110. Fransen SR, Leonard-Martin TC, Feuer WJ, Hildebrand PL: Clinical evaluation of patients with diabetic retinopathy: accuracy of the Inoveon diabetic retinopathy-3DT system. Ophthalmology 2002; 109:595-601.
111. Kinyoun J, Barton F, Fisher M, et al: Detection of diabetic macular edema. Ophthalmoscopy versus photography Early Treatment Diabetic Retinopathy Study Report Number 5. Ophthalmology 1989; 96:746-750.discussion 750-741
112. Classification of diabetic retinopathy from fluorescein angiograms: ETDRS report number 11. Ophthalmology 1991; 98:807-822.
113. Hee MR, Puliafito CA, Duker JS, et al: Topography of diabetic macular edema with optical coherence tomography. Ophthalmology 1998; 105:360-370.
114. Diabetic Retinopathy Clinical Research Network: Reproducibility of macular thickness and volume using Zeiss optical coherence tomography in patients with diabetic macular edema. Submitted
115. Browning DJ: Interobserver variability in optical coherence tomography for macular edema. Am. J. Ophthalmol 2004; 137:1116-1117.
116. Polito A, Del Borrello M, Isola M, et al: Repeatability and reproducibility of fast macular thickness mapping with stratus optical coherence tomography. Arch. Ophthalmol 2005; 123:1330-1337.
117. Sadda SR, Wu Z, Walsh AC, et al: Errors in retinal thickness measurements obtained by optical coherence tomography. Ophthalmology 2006; 113:285-293.
118. Brown JC, Solomon SD, Bressler SB, et al: Detection of diabetic foveal edema: contact lens biomicroscopy compared with optical coherence tomography. Arch. Ophthalmol 2004; 122:330-335.
119. Browning DJ, McOwen MD, Bowen Jr RM, O'Marah TL: Comparison of the clinical diagnosis of diabetic macular edema with diagnosis by optical coherence tomography. Ophthalmology 2004; 111:712-715.
120. Strom C, Sander B, Larsen N, et al: Diabetic macular edema assessed with optical coherence tomography and stereo fundus photography. Invest. Ophthalmol. Vis. Sci 2002; 43:241-245.
121. DRCR net : Comparison of the modified Early Treatment Diabetic Retinopathy and mild macuilar grid laser strategies for diabetic macular edema. Arch Ophthalmol 2007; 114:525-536.
122. Kang SW, Park CY, Ham DI: The correlation between fluorescein angiographic and optical coherence tomographic features in clinically significant diabetic macular edema. Am. J. Ophthalmol 2004; 137:313-322.
123. Diabetes Control and Complications Trial (DCCT): results of feasibility study : The DCCT Research Group. Diabetes Care 1987; 10:1-19.
124. Massin P, Audren F, Haouchine B, et al: Intravitreal triamcinolone acetonide for diabetic diffuse macular edema: preliminary results of a prospective controlled trial. Ophthalmology 2004; 111:218-224.
125. Abraham FA, Haimovitz J, Berezin M: The photopic and scotopic visual thresholds in diabetics without diabetic retinopathy. Metab. Pediatr. Syst. Ophthalmol 1988; 11:76-77.
126. Brinchmann-Hansen O, Dahl-Jorgensen K, Hanssen KF, Sandvik L: Oscillatory potentials, macular recovery time, and diabetic retinopathy through 3 years of intensified insulin treatment. Ophthalmology 1988; 95:1358-1366.
127. Han Y, Adams AJ, Bearse Jr MA, Schneck ME: Multifocal electroretinogram and short-wavelength automated perimetry measures in diabetic eyes with little or no retinopathy. Arch. Ophthalmol 2004; 122:1809-1815.
128. Bearse Jr MA, Han Y, Schneck ME, Adams AJ: Retinal function in normal and diabetic eyes mapped with the slow flash multifocal electroretinogram. Invest. Ophthalmol. Vis. Sci 2004; 45:296-304.
129. Bengtsson B, Heijl A, Agardh E: Visual fields correlate better than visual acuity to severity of diabetic retinopathy. Diabetologia 2005; 48:2494-2500.
130. Agardh E, Stjernquist H, Heijl A, Bengtsson B: Visual acuity and perimetry as measures of visual function in diabetic macular oedema. Diabetologia 2006; 49:200-206.
131. Ishiko S, Ogasawara H, Yoshida A, Hanada K: The use of scanning laser ophthalmoscope microperimetry to detect visual impairment caused by macular photocoagulation. Ophthalmic Surg. Lasers 1998; 29:95-98.
132. Fong DS, Segal PP, Myers F, et al: Subretinal fibrosis in diabetic macular edema. ETDRS report 23. Early Treatment Diabetic Retinopathy Study Research Group. Arch. Ophthalmol 1997; 115:873-877.
133. Akduman L, Olk RJ: Subthreshold (invisible) modified grid diode laser photocoagulation in diffuse diabetic macular edema (DDME). Ophthalmic Surg. Lasers 1999; 30:706-714.
134. Bandello F, Polito A, Del Borrello M, et al: 'Light' versus 'classic' laser treatment for clinically significant diabetic macular oedema. Br. J. Ophthalmol 2005; 89:864-870.
135. Sinclair SH, Alaniz R, Presti P: Laser treatment of diabetic macular edema: comparison of ETDRS-level treatment with threshold-level treatment by using high-contrast discriminant central visual field testing. Semin. Ophthalmol 1999; 14:214-222.
136. Pournaras CJ: Retinal oxygen distribution. Its role in the physiopathology of vasoproliferative microangiopathies. Retina 1995; 15:332-347.
137. Zhang SX, Wang JJ, Gao G, et al: Pigment epithelium-derived factor (PEDF) is an endogenous antiinflammatory factor. FASEB J 2006; 20:323-325.
138. Ogata N, Tombran-Tink J, Jo N, et al: Upregulation of pigment epithelium-derived factor after laser photocoagulation. Am. J. Ophthalmol 2001; 132:427-429.
139. Lewis H, Abrams GW, Blumenkranz MS, Campo RV: Vitrectomy for diabetic macular traction and edema associated with posterior hyaloidal traction. Ophthalmology 1992; 99:753-759.
140. Yanyali A, Nohutcu AF, Horozoglu F, Celik E: Modified grid laser photocoagulation versus pars plana vitrectomy with internal limiting membrane removal in diabetic macular edema. Am. J. Ophthalmol 2005; 139:795-801.
141. Stefansson E, Landers 3rd MB: How does vitrectomy affect diabetic macular edema. Am. J. Ophthalmol. 2006; 141:984.author reply 984-985.
142. La Heij EC, Hendrikse F, Kessels AG, Derhaag PJ: Vitrectomy results in diabetic macular oedema without evident vitreomacular traction. Graefes Arch. Clin. Exp. Ophthalmol 2001; 239:264-270.
143. Stolba U, Binder S, Gruber D, et al: Vitrectomy for persistent diffuse diabetic macular edema. Am. J. Ophthalmol 2005; 140:295-301.
144. Ouchi M, West K, Crabb JW, et al: Proteomic analysis of vitreous from diabetic macular edema. Exp. Eye Res 2005; 81:176-182.
145. Flynn HWJ, Scott IU: Intravitreal triamcinolone acetonide for macular edema associated with diabetic retinopathy and venous occlusive disease: it's time for clinical trials. Arch. Ophthalmol 2005; 123:258-259.
146. Tewari HK, Sony P, Chawla R, et al: Prospective evaluation of intravitreal triamcinolone acetonide injection in macular edema associated with retinal vascular disorders. Eur. J. Ophthalmol 2005; 15:619-626.
147. Patelli F, Fasolino G, Radice P, et al: Time course of changes in retinal thickness and visual acuity after intravitreal triamcinolone acetonide for diffuse diabetic macular edema with and without previous macular laser treatment. Retina 2005; 25:840-845.
148. Schindler RH, Chandler D, Thresher R, Machemer R: The clearance of intravitreal triamcinolone acetonide. Am. J. Ophthalmol 1982; 93:415-417.
149. Tano Y, Chandler D, Machemer R: Treatment of intraocular proliferation with intravitreal injection of triamcinolone acetonide. Am. J. Ophthalmol 1980; 90:810-816.
150. Scholes GN, O'Brien WJ, Abrams GW, Kubicek MF: Clearance of triamcinolone from vitreous. Arch. Ophthalmol 1985; 103:1567-1569.
151. Bakri SJ, Beer PM: Intravitreal triamcinolone injection for diabetic macular edema: a clinical and fluorescein angiographic case series. Can. J. Ophthalmol 2004; 39:755-760.
152. Smithen LM, Ober MD, Maranan L, Spaide RF: Intravitreal triamcinolone acetonide and intraocular pressure. Am. J. Ophthalmol 2004; 138:740-743.
153. Jonas JB, Degenring RF, Kreissig I, et al: Intraocular pressure elevation after intravitreal triamcinolone acetonide injection. Ophthalmology 2005; 112:593-598.
154. Bakri SJ, Beer PM: The effect of intravitreal triamcinolone acetonide on intraocular pressure. Ophthalmic Surg Lasers Imaging 2003; 34:386-390.
155. Wingate RJ, Beaumont PE: Intravitreal triamcinolone and elevated intraocular pressure. Aust. N. Z. J. Ophthalmol 1999; 27:431-432.
156. Challa JK, Gillies MC, Penfold PL, et al: Exudative macular degeneration and intravitreal triamcinolone: 18 month follow up. Aust. N. Z. J. Ophthalmol 1998; 26:277-281.
157. Thompson JT: Cataract formation and other complications of intravitreal triamcinolone for macular edema. Am. J. Ophthalmol 2006; 141:629-637.
158. Cardillo JA, Melo Jr LA, Costa RA, et al: Comparison of intravitreal versus posterior sub-Tenon's capsule injection of triamcinolone acetonide for diffuse diabetic macular edema. Ophthalmology 2005; 112:1557-1563.
159. Avery RL: Regression of retinal and iris neovascularization after intravitreal bevacizumab (Avastin) treatment. Retina 2006; 26:352-354.
160. Spaide RF, Fisher YL: Intravitreal bevacizumab (Avastin) treatment of proliferative diabetic retinopathy complicated by vitreous hemorrhage. Retina 2006; 26:275-278.
161. Rosenfeld PJ, Fung AE, Puliafito CA: Optical coherence tomography findings after an intravitreal injection of bevacizumab (avastin) for macular edema from central retinal vein occlusion. Ophthalmic Surg Lasers Imaging 2005; 36:336-339.
162. Cunningham Jr ET, Adamis AP, Altaweel M, et al: A phase II randomized double-masked trial of pegaptanib, an anti-vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology 2005; 112:1747-1757.
163. Iturralde D, Spaide RF, Meyerle CB, et al: Intravitreal bevacizumab (Avastin) treatment of macular edema in central retinal vein occlusion: a short-term study. Retina 2006; 26:279-284.
164. Mason 3rd JO, Albert Jr MA, Vail R: Intravitreal bevacizumab (Avastin) for refractory pseudophakic cystoid macular edema. Retina 2006; 26:356-357.
165. Aiello LP: The potential role of PKC beta in diabetic retinopathy and macular edema. Surv. Ophthalmol. 2002; 47(Suppl 2):S263-S269.