Amitabh K. Bharadwaj,
Carl D. Regillo,
E. Mitchel Opremcak
Rheumatologic disorders are a collection of inflammatory diseases which are typically multisystemic with protean manifestations. Rheumatologic diseases include the arthritides, the connective tissue diseases, and the vasculitides (Table 169.1). Despite the many clinical and pathologic presentations which exist, these diseases are so named because of the involvement of these supportive structures. It is not uncommon for the eyes to be inflamed in many of these rheumatologic syndromes; indeed, ocular inflammation is often considered a major diagnostic criterion for establishing a clinical diagnosis in several of these disorders.
An accurate understanding of the connective tissue proper is required to appreciate ocular involvement in rheumatic diseases. Connective tissues and structures are the matrix that support individual cells, tissues, and organs. This ground substance is produced by specialized connective tissue cells that secrete various fibers (collagens, reticulin, and elastin), as well as a group of mucopolysaccharides called proteoglycans.[1] Hyaluronic acid, chondroitin sulfate, dermatan sulfate, keratan sulfate, and heparin sulfate are the common polysaccharides found in the connective tissue proteoglycans.[2] Collagen fibers can also be further subdivided by their polypeptide structure into six types (collagen types I to VI).[3] Each tissue and organ maintains a distinct connective tissue environment by varying these individual components, thereby effecting optimal structure and function. In rheumatologic diseases, inflammation of these supportive structures and milieu results in tissue and organ dysfunction.
The eye, perhaps more than any other organ, maintains a unique connective tissue environment. Corneal keratocytes secrete both type II and type IV collagen, as well as chondroitin and keratan sulfate.[1,4]This connective tissue combination results in strong tissue that becomes transparent as a result of unique lamellar fiber orientation and active endothelial cell dehydration. Hyalocytes in the vitreous produce hyaluronic acid and type II collagen, which forms a clear gel, allowing transmission of light, and provides support for the globe and retina.[1,5] The retina and choroid possess connective tissue and a complex vascular system that is composed of type III and type IV collagen.[1] These circulations are critical for retinal function and general nutrition of the eye. Each of these ocular tissues performs critical functions in the visual system and is exquisitely sensitive to inflammation.
Different connective tissues and structures within the eye can become inflamed in the various rheumatic diseases. Wegener's granulomatosis, rheumatoid arthritis (RA), and polyarteritis nodosa (PAN) can affect the cornea and produce peripheral ulcerative keratopathy. Reiter's syndrome is defined by the triad of urethritis, arthritis, and inflammation of the conjunctiva and iris. Ankylosing spondylitis, Reiter's syndrome, psoriatic arthritis, and the chronic inflammatory bowel diseases produce an acute iridocyclitis. Juvenile rheumatoid arthritis (JRA) commonly produces a chronic iridocyclitis. It is important, therefore, to evaluate all patients who present with ocular inflammation for symptoms and signs of an underlying rheumatologic disease.
Often, rheumatic diseases present in the eye before the onset of significant systemic involvement. The eye may even be the primary target of several diseases such as Behçet's syndrome, Reiter's disease, and ankylosing spondylitis. The importance of performing a careful review of systems and physical examination cannot be underestimated. Particular attention should be paid to the skin, joints, central nervous system, lungs, gastrointestinal tract, and kidneys. Often, involvement of these systems can lead the ophthalmologist to establish the existence of an underlying rheumatic disease as the cause for the ocular inflammation. For example, iritis in a patient complaining of large-joint arthritis could represent JRA, systemic lupus erythematosus (SLE), Wegener's granulomatosis, or Behçet's disease. Oral ulcers, malaise, skin rash, and uveitis may be found in SLE, Behçet's disease, and sarcoidosis. Genital-urethral pain and uveitis may represent Behçet's disease, PAN, or Reiter's syndrome. Laboratory evaluation and consultation with an internist or a rheumatologist can help confirm the presence of a rheumatologic disease and result in diagnosis and the initiation of proper systemic therapy. Local ocular therapy in rheumatic disorders without attention to the underlying systemic process universally results in suboptimal control of the ocular inflammation and risks potentially life-threatening complications of uncontrolled systemic disease.
In summary, rheumatic diseases provide an opportunity for the ophthalmologist to interface with both the patient and the internist. The expertise of the ophthalmologist in determining the specific ocular tissue involved and the rheumatologist's knowledge of the systemic manifestations can not only facilitate the proper diagnosis but can also provide optimal care for these diseases of vision, which are often life-threatening.
TABLE 169.1 -- Rheumatic Diseases With Ocular Involvement
|
Arthritides |
|
Rheumatoid arthritis |
|
Seronegative spondyloarthropathies (HLA-B27 associated) |
|
Ankylosing spondylitis |
|
Reiter's syndrome |
|
Psoriatic arthritis |
|
Inflammatory bowel disease |
|
Juvenile rheumatoid arthritis |
|
Connective Tissue Diseases |
|
Systemic lupus erythematosus |
|
Progressive systemic sclerosis |
|
Polymyositis and dermatomyositis |
|
Sjögren's syndrome |
|
Relapsing polychondritis |
|
Vasculitides |
|
Polyarteritis nodosa |
|
Churg-Strauss syndrome |
|
Wegener's granulomatosis |
|
Hypersensitivity vasculitis |
|
Lymphomatoid granulomatosis |
|
Giant-cell arteritis |
|
Behçet's disease |
RHEUMATOID ARTHRITIS
|
Key Features |
||||||||||||||||||
|
RA is a chronic systemic inflammatory disease of unknown cause, producing a distinct form of polyarticular and symmetric arthritis. It is more common in women (3:1) and typically begins between the ages of 30 and 40 years.[6] RA is thought to have a strong autoimmune pathogenesis. Patients with RA have immunoglobulin (Ig) M, IgG, and IgA antibodies that are directed against the Fc portion of IgG.[7] The resulting immune complexes are postulated to mediate both the articular and the extraarticular manifestations of the disease through activation of complement.
Patients with RA complain of malaise, fatigue, weight loss, and arthralgia. The joint disease is symmetric and often involves the small joints of the wrist and hand, excluding the distal interphalangeal joints.[1]Morning stiffness is characteristic. Extraarticular involvement in RA, including pleurisy, neuropathy, and ocular inflammation, is thought to be secondary to systemic vasculitis and may represent a change from a local joint disease to a more serious systemic form of RA.
The most common anterior segment findings in RA are keratoconjunctivitis sicca, marginal keratitis, peripheral ulcerative keratopathy, and anterior scleritis.[8] Uveitis and direct involvement of the retina are rare. The retina can, however, be involved secondarily after the development of posterior scleritis. Forty-six percent of patients with scleritis will have an associated underlying rheumatic disease.[9] While the differential diagnosis also includes conditions such as Wegener's granulomatosis, relapsing polychondritis, and PAN, up to 30% of such patients will have RA.
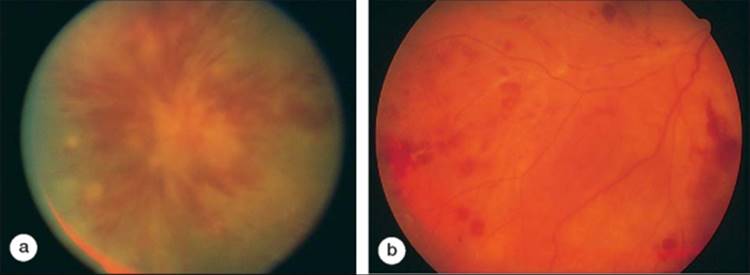
Posterior scleritis is not as common as anterior scleral inflammation. This condition is defined as scleritis posterior to the equator of the eye, and in one series accounted for only 2% of all cases.[10,11] It is important to recognize, however, that posterior scleritis is much more difficult to detect than anterior scleritis. In one report of 30 eyes enucleated for ocular inflammation, 40% had previously undetected posterior scleritis.[12] Posterior scleritis can be unilateral and is often associated with a profound decrease in visual acuity. There is pain and tenderness with motion or palpation. On biomicroscopic examination, the anterior segment is often normal or may show only a narrow angle resulting from displacement by the posterior structures.
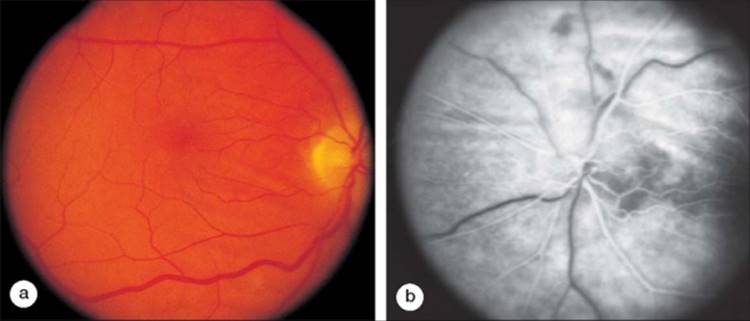
The fundus examination in posterior scleritis reveals choroidal thickening or choroidal nodules overlying the area of scleritis. Secondary choroidal folds and effusions may develop. The retina may demonstrate secondary striae and exudative retinal separations (Fig. 169.1).[11] A high index of suspicion is often required to make a clinical diagnosis of posterior scleritis. Ultrasonography or computed tomography can support this diagnosis by showing thickening of the sclera and choroid. There may be fluid in the contiguous Tenon space. Fluorescein angiography illustrates the choroidal and retinal striae. A characteristic linear pattern of alternating hypo- and hyperfluorescent streaks can be seen as a result of folds in the retinal pigment epithelial layer (see Fig. 169.1b). Multifocal, punctate, hyperfluorescent choroidal lesions can also be noted and may evolve into areas of exudative retinal detachment. The retinal circulation is typically unaffected.
|
|
|
|
FIGURE 169.1 Posterior scleritis in a patient with rheumatoid arthritis with secondary thickening of the choroid in the posterior pole and peripapillary area. (a) Chorioretinal striae in the macula. (b) Alternating hypofluorescent and hyperfluorescent linear streaks correspond to folding of the retinal pigment epithelium. |
Once a clinical diagnosis of posterior scleritis is established, a search for an underlying cause is in order. A general physical examination and review of systems can help establish extraocular involvement. Patients with the characteristic deforming arthritis associated with RA seldom present a diagnostic challenge. Mild anemia, elevation of the erythrocyte sedimentation rate (ESR), and a positive result for rheumatoid factor in serum may support the clinical diagnosis. Without obvious systemic findings, scleritis may be the initial manifestation of occult rheumatic disease.
Therapy for rheumatoid scleritis should be directed at controlling the underlying systemic disease.[10] Local ocular therapy and regional steroids should be used with caution and only as adjuncts to systemic treatment. Regional steroids should be used only in extenuating circumstances, as they have been reported to cause scleral melting and ocular perforation. Mild cases can often be effectively managed by nonsteroidal antiinflammatory agents. Indomethacin (50-150 mg/day) has been reported to be particularly effective for scleritis.[9] The goal of early aggressive therapy is to prevent permanent damage from the disease. In more severe cases, immunosuppressive drugs such as methotrexate, azathioprine, or one of the newer biologic agents should be considered. The biologic agents are designed to block cytokines or cytokine receptors, and include medicines that decrease the activity of tumor necrosis factor-alpha (etanercept, adalimumab, and infliximab) and an interleukin-1 receptor antagonist (anakinra).[13,14] Oral prednisone may be used adjunctively as a bridge to a steroid-sparing agent. The ocular and systemic prognoses depend in part on establishing the proper diagnosis and detecting the underlying rheumatic disease. In a report by Foster and associates, the development of necrotizing scleritis forbode a more severe form of RA.[15] They noted an 8-year mortality rate of 20% in patients with this extraarticular involvement.
SERONEGATIVE SPONDYLOARTHROPATHIES (HLA-B27-ASSOCIATED)
|
Key Features |
|||||||||
|
The group of collagen vascular diseases have in common spondylitis and a strong association with HLA-B27. Ankylosing spondylitis, Reiter's syndrome, psoriatic arthritis, and arthritis associated with chronic inflammatory bowel disease (Crohn's disease and ulcerative colitis) compose this group. Patients with these disorders experience an acute, alternating, recurrent, nongranulomatous iridocyclitis. This anterior segment inflammation can be severe, with hypopyon and posterior synechiae formation. Secondary glaucoma and cataract are not uncommon. These collagen vascular diseases do not have marked posterior segment involvement in the form of primary choroiditis, retinitis, or retinal vasculitis. Cystoid macular edema can occur with prolonged or severe cases of anterior uveitis, and this may respond to therapies designed to address the iridocyclitis. Crohn's disease has been reported with uveitis and severe bilateral obliterative retinal vasculitis.[16-18] In one series, 29 of 166 (17%) patients with HLA-B27-associated uveitis had posterior segment manifestations including vitritis (93%), papillitis (83%), vasculitis (24%), cystic macular edema (38%), and epiretinal membrane (17%).[19] In this report, the retinal vasculitis was responsive to corticosteroids or cyclophosphamide therapy, or a combination of these medicines.
JUVENILE RHEUMATOID ARTHRITIS
|
Key Features |
|||||||||||||||
|
JRA is a multisystemic childhood disease associated with chronic arthritis. JRA is much more common in girls and occurs typically between the ages of 2 and 5 years. The cause of JRA is unknown, but it is thought to be a primary autoimmune disease. Children with the pauciarticular form of JRA have the highest risk for the development of ocular inflammation.[20] Over 80% of such children are ANA positive.
Chronic iridocyclitis develops in 5-17% of children with JRA.[21] Eye disease may precede the development of arthritis by several years. Ocular involvement may be insidious because of the lack of symptoms and ocular signs early in the disease. JRA-associated iridocyclitis is typically chronic and bilateral (70%). The inflammation is usually nongranulomatous and involves primarily the iris and ciliary body. Chronic inflammation commonly results in band keratopathy (41%), posterior synechiae, glaucoma (19%), and cataract formation (42-92%).[22]
In JRA the retina and choroid are involved to a much lesser extent than the anterior segment. Cystoid macular edema may develop in patients with JRA. As a result of chronic cyclitis, organization and fibrosis of the anterior vitreous may occur, resulting in further media opacification. Cyclitic membrane formation and ocular hypotony can develop spontaneously after standard cataract surgery. True retinitis, retinal vasculitis, or choroiditis is uncommon in JRA.
JRA can be diagnosed in children with a characteristic arthritic and ocular picture. The diagnosis can be further supported by documenting a positive antinuclear antibody (ANA) level (79%) and a negative result for rheumatoid factor in serum.[23] Topical corticosteroids and cycloplegics are the mainstay of therapy for children with JRA-associated ocular inflammation. Oral nonsteroidal antiinflammatory agents are useful in controlling both the ocular and systemic symptoms.[24] Regional corticosteroids can be used for ocular inflammation but are difficult to deliver in this age group and often require general anesthesia. Methotrexate, cyclosporine, or other immunosuppressive agents should be considered in patients whose eyes cannot be quieted by the above methods. Oral corticosteroids (1 mg kg?1 day?1) may be helpful while transitioning to a steroid-sparing agent. Due to the monitoring that is needed while on, immunosuppressive agents these potentially toxic medicines should be used under the direction of physicians who have experience with their use.
The overall visual prognosis in JRA is poor. Seventy-five percent of children with JRA have moderate or severe inflammation with loss of vision due to glaucoma, cataract, or phthisis.[1,25] Lens and vitreous opacification should be addressed, preferably via pars plana lensectomy and vitrectomy, to afford better control of the inflammation and prevent cyclitic membrane formation and hypotony.[15,26]
SYSTEMIC LUPUS ERYTHEMATOSUS
|
Key Features |
|||||||||||||||
|
SLE is a collagen vascular disease with protean manifestations. Ninety percent of patients with SLE are women at or around child-rearing age.[27] The pathogenesis of SLE appears to be an autoimmune systemic necrotizing vasculitis. Patients with SLE have ANAs and elevated levels of circulating immune complexes that appear to play a role in the disease.[27] Immune complexes composed of these autoantibodies and DNA have been found in the walls of inflamed blood vessels and in the areas of fibrinoid necrosis.
Clinically, patients with SLE present with malaise, fatigue, anorexia, and low-grade fever. On examination, they may have arthritis, facial rash, alopecia, and pleurisy.[28] Raynaud's phenomenon, oral ulcers, and central nervous system complaints are also common. Laboratory evaluation may reveal anemia, an elevated ESR, the presence of ANAs, proteinuria, and a false-positive Venereal Disease Research Laboratory (VDRL) result. Lupus nephritis and central nervous system involvement represent serious and potentially fatal complications of SLE.
SLE may involve the eye in up to 50% of cases, depending on the series and the nature of the clinic reporting the findings.[29] Anterior segment findings include keratoconjunctivitis sicca, scleritis, and keratitis. The retina and choroid may be primarily involved; however, it is important to separate lupus-associated retinal vasculitis from the secondary retinal and choroidal changes of SLE-mediated hypertension and anemia. Severe hypertension can occur in lupus as a result of nephritis. Arteriolar narrowing, intraretinal hemorrhages, exudate, and disk edema are characteristic of hypertensive retinopathy.
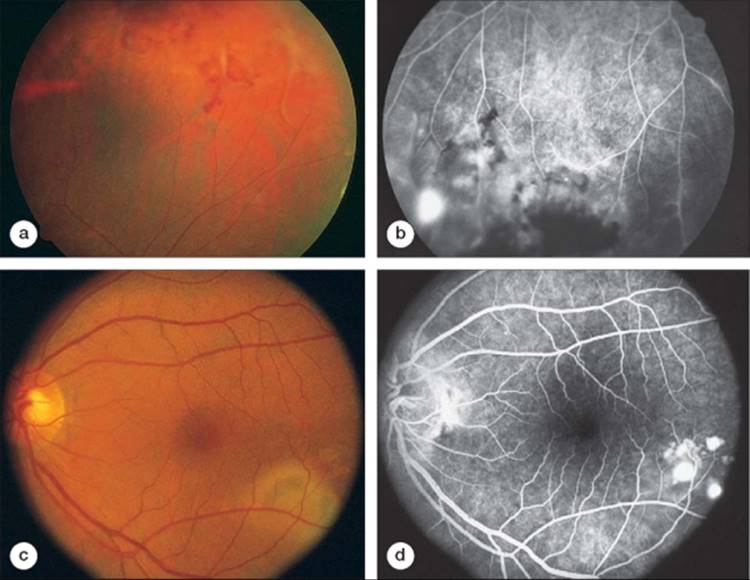
The mechanism of primary lupus retinopathy is unknown but is thought to be secondary to circulating immune complexes found in the disease. Ten percent of patients with SLE also have lupus anticoagulant antibodies that are known to increase the incidence of thrombosis. The relationship between this factor and lupus retinopathy is unclear but provocative.[30] Retinal manifestations of SLE are a result of focal ischemia and necrotizing retinal vasculitis. Three fundus presentations have been described.[31-33] The most common form is focal ischemia resulting in multiple cotton wool spots. Intraretinal hemorrhages and mild disk edema can also be associated with this form of lupus retinopathy. A second form of retinopathy noted in SLE is a severe retinal vasoocclusive disease without evidence of retinal vasculitis.[32]Retinal infarction and hemorrhage can result in severe and sudden loss of vision. The third form of retinopathy in SLE is proliferative lupus retinopathy (Fig. 169.2a,b). Retinal vasculitis and secondary ischemia produce neovascularization of the optic nerve and elsewhere in the retina.[33] This neovascularization can result in vitreous hemorrhage and even retinal detachment.
|
|
|
|
FIGURE 169.2 (a and b), Peripheral retinal vasculitis in a patient with systemic lupus erythematosus with areas of intraretinal hemorrhage, retinal nonperfusion, and neovascularization on fluorescein angiography. (c and d) Another patient with systemic lupus erythematosus with focal areas of serous elevation of the retinal pigment epithelium and sensory retina as a result of lupus-associated choroidopathy. |
The choroidal circulation may be involved in this process as well (see Fig. 169.2c,d).[34,35] Choroidal infarction, exudative changes, central serous chorioretinopathy, and subretinal neovascular membranes have been reported with SLE. Focal choroidal vessel injury with secondary dysfunction of the overlying retinal pigment epithelial cells is believed to be one of the mechanisms in this form of ocular disease.[35]
SLE is a clinical diagnosis. The American Rheumatologic Association defies the disease by the presence of four of the 14 major symptoms and signs. Laboratory testing may support the clinical diagnosis by revealing the presence of ANAs, elevated circulating immune complexes, proteinuria, anemia, and a false-positive VDRL test result. Fluorescein angiography may help defie the extent of retinal involvement and may assist in differentiating secondary hypertensive retinopathy from the true retinal vasculitis noted in lupus retinopathy.
Therapy for SLE varies according to the severity of the systemic symptoms. It is important to note that lupus may present in the eye 1-5 years before the onset of other systemic findings.[36] Lupus retinopathy may respond to systemic therapy, including nonsteroidal antiinflammatory agents, oral corticosteroids, hydroxychloroquine, gold, and cyclophosphamide.[37] Proliferative lupus retinopathy can be treated with panretinal laser ablation to help control the consequences of ocular neovascularization.
PROGRESSIVE SYSTEMIC SCLEROSIS
|
Key Features |
||||||||||||
|
Progressive systemic sclerosis (PSS), or scleroderma, is a chronic autoimmune disorder resulting in inflammation and fibrosis of the skin and other organs. Women in the fourth decade of life are affected more often than men (4:1).[38] The immune defect in PSS is not completely understood, but circulating immune complex deposition, vasculitis, and secondary fibrosis of the vessels are thought to produce the typical clinical picture. Clinically, patients may present with scleroderma or may manifest other symptoms of the CREST syndrome (calcinosis, Raynaud's phenomenon, esophageal symptoms, sclerodactyly, and telangiectasia).
The eye is commonly involved in scleroderma. Keratoconjunctivitis sicca occurs in up to 70% of patients with PSS as a result of lacrimal gland fibrosis.[39] Filamentary keratitis, eyelid edema, and conjunctival shrinkage can also be noted. Patients with PSS have also been seen to have fundus findings similar to those in SLE. Intraretinal hemorrhages, cotton wool spots, and retinal vasculitis, as well as choroidal infarctions, have been reported.[40]
The diagnosis can be established by the characteristic clinical picture of the scleroderma, with or without the other CREST findings. These systemic findings in association with retinal microvascular infarctions and retinal vasculitis support the clinical diagnosis. Laboratory testing may reveal the presence of ANAs with a speckled pattern. Fluorescein angiography can be used to document the cotton wool spots and defie the extent of choroidal nonperfusion. Long-term systemic therapy has not been useful in controlling PSS, and therapy is chiefly supportive.[41] Localized scleroderma has a relatively good prognosis. Systemic involvement has a worse prognosis, with an 80% 10-year mortality rate.
DERMATOMYOSITIS AND POLYMYOSITIS
|
Key Features |
||||||||||||
|
Dermatomyositis and polymyositis are autoimmune forms of inflammatory diffuse myopathy. The pathogenesis of these diseases appears to be mediated via microvascular damage from immune complex formation. Patients with dermatomyositis show ischemic necrosis and loss of capillary beds in the perifascicular region of the involved muscles.[42] Both children and adults are affected by this disease.
Ocular involvement in dermatomyositis includes a lilac discoloration and edema of the eyelids, conjunctivitis, iritis, blepharoptosis, scleritis, uveitis, and extraocular muscle paralysis.[43] Cotton wool spots, intraretinal hemorrhages, venous engorgement and disk edema, and optic atrophy have been observed primarily in childhood dermatomyositis.[44]
Corticosteroids (1 mgkg?1 day?1) have proved helpful in managing these diseases. Methotrexate and azathioprine have been used for refractory cases or when complications of steroid use develop.[45] The prognosis is better for childhood forms of the disease, with a 90% 5-year survival rate.[1] Adult-onset disease fares worse, with a 53% survival at 5 years.
SJÖGREN'S SYNDROME
|
Key Features |
||||||||||||
|
Sjögren's syndrome is a constellation of clinical disorders that have in common keratoconjunctivitis sicca and xerostomia.[46] Twenty-five percent to 50% of patients with RA have Sjögren's syndrome. This syndrome is more common in postmenopausal women. Exocrine glands, including the lacrimal gland, demonstrate infiltration with lymphocytes and secondary fibrosis. These patients have antinucleoprotein antibodies; anti-SSA (anti-Ro) and anti-SSB (anti-La).
Patients with Sjögren's syndrome complain of dryness of the mouth and eyes. Other manifestations of primary Sjögren's syndrome include pneumonitis, renal tubular acidosis, polymyositis, and gastritis.
Keratoconjunctivitis sicca is the most common ocular fiding.[47] Secondary filamentary keratitis can also be problematic. In one series, eight patients with a clinical picture compatible with Sjögren's syndrome had anterior or intermediate uveitis but did not have chorioretinal involvement.[48] In another report, two patients with progressive retinal vasculitis were found to have anti-SSA antibodies, suggesting a Sjögren-like syndrome.[49] Their disease was unresponsive to corticosteroid, immunosuppressive, and panretinal photocoagulation therapy.
RELAPSING POLYCHONDRITIS
|
Key Features |
|||||||||||||||
|
Relapsing polychondritis is a collagen vascular disease affecting primarily the cartilaginous tissues. The cause is unknown but appears to be an autoimmune disease directed against type II collagen.[50]Patients with polychondritis have circulating anti-type II collagen antibodies. These autoantibodies are thought to play a mediating role in the recurrent, granulomatous inflammation noted in this disease. Relapsing polychondritis occurs in adults aged 20-60 years and affects both men and women.
Systemically, patients will have bilateral ear pinna inflammation and nasal cartilage destruction (72%).[51] Nasal septal damage typically results in a saddle nose deformity. Secondary otitis, vertigo, tinnitus, and deafness may be noted. A nonerosive polyarthralgia may also bring the patient to medical attention. Laryngeal and tracheal ring cartilage may be involved and can cause acute respiratory distress as a result of upper airway collapse.
Ocular involvement in relapsing polychondritis may occur in as many as 20-50% of cases.[52] This involvement is typically in the form of episcleritis, scleritis, or conjunctivitis. Anterior uveitis can be found in 20% of patients. Primary retinal or choroidal involvement is unusual. Retinal pigment epithelial and sensory retinal detachments as well as choroidal thickening can be noted as a result of localized posterior scleritis.[53]
The diagnosis can be established by the pathognomonic bilateral ear pinna inflammation. Biopsy of involved cartilage demonstrates the characteristic granulomatous chondritis. Relapsing polychondritis is a potentially fatal disease with a 30% mortality rate.[54] Dapsone (50-200 mg/day) is often used first in patients with mild symptoms and no cardiac or pulmonary involvement. Prednisone (0.5-1 mg kg?1 day?1) is highly effective, and may be added in cases that do not respond well to dapsone alone. Dapsone does not appear to be as effective in patients with severe necrotizing forms of scleritis.[54] Destructive changes in the cartilage are not reversible, and the goal of therapy should be to limit further disease progression.
POLYARTERITIS NODOSA
|
Key Features |
|||||||||||||||
|
PAN is a multisystemic disease associated with a necrotizing vasculitis. Medium- to small-sized vessels are characteristically affected with all stages of necrosis noted in the involved tissues.[55] The cause of PAN is unknown. Thirty percent to 70% of patients with PAN have antihepatitis B antibodies.[56] The significance of this virus in the pathogenesis has not been established. This is a rare disease that affects 20- to 50-year-old adults. Men are affected more frequently than are women (3:1).
Systemically, patients with PAN may note fatigue, myalgia, weight loss, fever, arthralgia, and testicular pain. The kidneys, liver, and gastrointestinal and central nervous systems are commonly involved. Renal involvement is one of the more serious and potentially fatal complications of PAN. Abdominal pain from intestinal infarction and headaches from central nervous system vasculitis are also serious and potentially life-threatening complications of this disease.
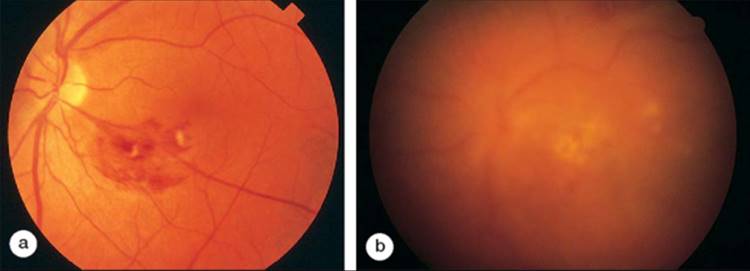
Ten to 20% of patients with PAN will have ocular involvement.[57] Peripheral ulcerative keratitis and mild iritis can be found in this disease. A mild vitritis may also be noted. The most common ocular findings in PAN are choroidal and retinal vasculitis.[58] Fundus examination will show retinal vasculitis with associated intraretinal hemorrhages, cotton wool spots, and retinal edema (Fig. 169.3). Central retinal artery occlusion and optic atrophy have also been reported in PAN. Patients with PAN may also experience anterior or posterior scleritis. Posterior scleritis will manifest with pain and chorioretinal folds similar to those in RA.
|
|
|
|
FIGURE 169.3 (a) Localized area of necrotizing retinal vasculitis associated with polyarteritis nodosa. (b) The patient refused immunosuppressive therapy and lost central acuity in the left eye over a 5-month period because of progressive ischemic retinal vasculitis. Note the development of vitritis as well as further retinal involvement. |
Any patient with occlusive retinal vasculitis should be examined for evidence of systemic findings that may help establish the presence of a collagen vascular disease. Often, a biopsy of an involved artery or lesion will demonstrate a hemorrhagic vasculitis and fibrinoid necrosis and establish the diagnosis. Laboratory tests may show elevated white blood cell and eosinophil counts, decreased complement, elevated circulating immune complexes, and negative rheumatoid factor and ANA determinations. Hepatitis B surface antigen has been found in up to 70% of patients with PAN. Angiography in patients with abdominal pain may reveal aneurysmal dilatation of the hepatic and renal arteries.
PAN is a potentially fatal disease. Without therapy, it carries an 80-90% 5-year mortality rate. Corticosteroids reduce this mortality rate to 50%.[59] The combination of corticosteroids with cyclophosphamide (1-2 mg kg?1 day?1) results in an 80% 5-year survival rate. After disease control is achieved, the steroid may be tapered. Patients may require high doses of intravenous steroids and cyclophosphamide early in the course to gain control of severe disease.[60]
WEGENER'S GRANULOMATOSIS
|
Key Features |
||||||||||||||||||
|
Wegener's granulomatosis is a multisystem disease associated with a necrotizing granulomatous vasculitis.[61] The cause is unknown. It is uncommon and occurs between the ages of 40 and 50 years. Men are affected more often than are females (3:2). Immune complex formation and deposition are thought to result in vasculitis of small- to middle-sized vessels. Acute and chronic lesions can be found simultaneously in the involved tissues and organs.
Characteristically, Wegener's granulomatosis involves the upper and lower respiratory tracts.[62] Epistaxis, rhinorrhea, sinusitis, otitis, chronic cough, and saddle-nose deformity are frequently observed. Chronic pulmonary infiltrates, nodules, and cavitary lesions are found in the lungs. Wegener's granulomatosis also commonly affects the kidneys and the central nervous system. A granulomatous glomerulitis can be found in 80% of patients with this collagen vascular disorder.
Wegener's granulomatosis may involve the eye in 40-50% of cases.[63] Eye involvement may precede other organ involvement. Proptosis and orbital pain from a pseudotumor are the most common findings. Scleritis, peripheral ulcerative keratitis, conjunctivitis, and dacryocystitis are also frequently noted ocular manifestations of Wegener's vasculitis. Posterior scleritis in Wegener's granulomatosis behaves similarly to scleritis in other rheumatologic disorders and presents with decreased vision and pain. Secondary chorioretinal thickening and striae can be seen both clinically and on fluorescein angiography. Although uveitis, retinitis, and retinal vasculitis have been reported, intraocular disease is rare. Retinitis and retinal vasculitis in Wegener's granulomatosis may present as a geographic area of retinal edema and intraretinal hemorrhage (Fig. 169.4), which may increase in size and can be difficult to distinguish from a secondary opportunistic or viral retinitis.
|
|
|
|
FIGURE 169.4 (a) An area of retinitis and retinal vasculitis associated with biopsy-proven Wegener's granulomatosis. (b) The fluorescein angiogram illustrates an area of segmental vascular staining and leakage of dye into the vitreous. |
Wegener's granulomatosis should be suspected in patients with these ocular findings and respiratory, renal, or central nervous system involvement. The diagnosis can be supported by fiding pneumonitis or cavitary lesions on chest radiograph film. Laboratory testing will demonstrate an elevated white blood cell count, ESR, and serum IgA. Antineutrophilic cytoplasmic antibodies (ANCAs) have been found in this disease and have proved useful in advancing a diagnosis of Wegener's granulomatosis.[64] Although failure of ANCA to return to normal may predict an unfavorable clinical course, absolute titers do not appear to correlate with disease severity.[65] Biopsy of the involved tissue often establishes the diagnosis by revealing a granulomatous vasculitis.
Wegener's granulomatosis is a serious and potentially fatal disease. Therapy should be designed to address both the ocular and the systemic inflammation. Without therapy, the average survival is 5 months, with an 80% mortality rate by 1 year.[66] Corticosteroids prolong survival to 12.5 months. Cyclophosphamide (1-2 mg kg?1 day?1) in combination with corticosteroids is the treatment of choice, with a 90% remission rate. Maintenance therapy may be required for 1-2 years.
GIANT-CELL ARTERITIS
|
Key Features |
|||||||||||||||
|
Giant-cell arteritis (GCA), or temporal arteritis, is a systemic vasculitis that involves medium- to large-sized muscular arteries.[67] The cause is unknown. GCA affects an older population with an average age of 70 years. The pathophysiology of this collagen vascular disease appears to be a panarteritis. Affected blood vessels demonstrate a mononuclear cell infiltration and giant-cell formation within the vessel wall, with subsequent destruction and fragmentation of the internal elastic membrane.
Patients with GCA commonly complain of fever, weight loss, malaise, and headaches. Jaw claudication may occur. The most common ocular complication of GCA is ischemic optic neuropathy.[68] The larger vessels supplying the optic nerve become inflamed and effect an infarction of the optic nerve. Rarely, the retinal vessels may be involved, producing a branch or central retinal artery occlusion.[69] Attenuation of the retinal arterioles, disk pallor, and optic atrophy may develop late. Primary uveitis or retinitis is uncommon.
The diagnosis can be made in the clinical setting of an older patient with malaise, fever, weight loss, headache, and sudden loss of vision in one or both (65%) eyes as a result of ischemic optic neuropathy. The diagnosis can be supported by fiding a markedly elevated ESR and/or C-reactive protein level. A temporal artery biopsy will demonstrate a granulomatous vasculitis with infiltration of the vessel wall with mononuclear cells, histiocytes, and giant cells and loss of the internal elastic membrane.
Untreated, this disease has a poor prognosis with irreversible loss of vision secondary to ischemic optic neuropathy and death from coronary or cerebral vasculitis.[70] Therapy should be initiated as soon as GCA is suspected in order to prevent bilateral optic nerve involvement. Prednisone (1 mg kg?1 day?1) is the treatment of choice in GCA, and maintenance doses may be required for 1-2 years when the disease may remit.
BEHÇET'S DISEASE
|
Key Features |
||||||||||||||||||
|
Behçet's disease is a systemic necrotizing vasculitis with diverse manifestations. The cause is unknown but is thought to have a strong autoimmune component. Human leukocyte antigen (HLA) associations have been established in other countries for Behçet's disease but have not proved useful in the United States.[71] HLA-B51, HLA-B12, and HLA-B27 have all been associated with certain forms of Behçet's disease. The disease is much more common in Asia and the Middle East.[72] In the United States, there is an equal gender distribution, and the disease is found in 20- to 40-year-old adults.
Behçet's vasculitis involves small- to medium-sized vessels. A perivascular infiltrate with polymorphonuclear neutrophils and mononuclear cells can be found around the veins and arteries. This is often associated with vessel thrombosis and tissue hemorrhage.
Clinically, there may be a 6- to 10-year prodrome with recurrent or chronic malaise, fever, and sore throat.[73] The classic triad of recurrent aphthous oral ulcers (100%), genital ulcers (84%), and uveitis (66%) establishes the clinical diagnosis of Behçet's disease. Erythema nodosum (66%), arthritis (66%), and meningoencephalitis (22%) are also common. The gastrointestinal, renal, pulmonary, and cardiovascular systems may also be involved and are considered 'minor' findings in this syndrome.
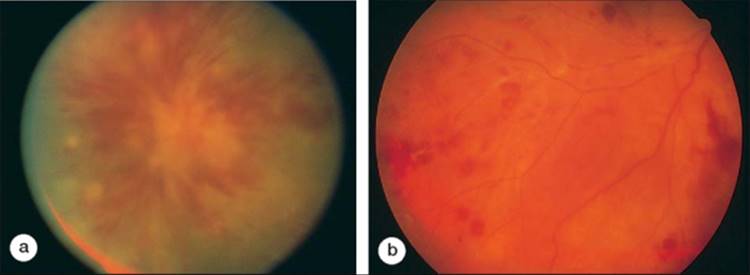
The eye may be the first or predominant organ involved in Behçet's disease. More characteristically, uveitis follows the other systemic findings by 1-3 years. Systemic and ocular symptoms typically wax and wane. Patients will often present with an acute loss of vision associated with a bilateral (80%) uveitis.[74] The ocular inflammation may be severe and relapsing. A nongranulomatous iridocyclitis with hypopyon, posterior synechiae, and hyphema is common. On fundus examination, there may be a severe vitritis, disk edema, and attenuation of the arterioles.[75] An essential fiding in Behçet's disease is the presence of an occlusive retinal vasculitis with surrounding intraretinal hemorrhage and retinal edema (Fig. 169.5). Cystoid macular edema, cataract, glaucoma, and retinal detachment can occur as secondary complications of the uveitis and retinal vasculitis.[76]
|
|
|
|
FIGURE 169.5 (a and b) Two patients with Behçet's disease illustrate the profound involvement of the retinal vessels. A bilateral occlusive retinal vasculitis results in intraretinal edema, hemorrhage, and retinal nonperfusion. Progressive damage to the sensory retina results in marked attenuation of the arterioles. |
The complete form of Behçet's disease consists of oral ulcers, genital ulcers, uveitis, and nonulcerative skin lesions. Several systems have been proposed for diagnosing partial or incomplete forms of Behçet's disease.[77] Behçet's disease is more prevalent in Japan, leading the Japanese to classify this disease into four forms: (1) complete-all four major findings, (2) incomplete-three major findings or uveitis with one other major fiding, (3) suspect-two major findings, and (4) possible-one major fiding.
The presence of other minor findings associated with the multisystemic involvement can assist in the diagnosis. Laboratory testing may help by demonstrating an elevated ESR, elevated levels of C-reactive protein, immune complexes, and a positive ANA determination. HLA typing can also be helpful in incomplete forms of Behçet's disease. Properdin factor B, serum lysozyme, and ?1-acid glycoprotein are also elevated in Behçet's disease.[78] Pathergy and dermatographia, although much extolled, have not proved to be helpful measures for detecting Behçet's disease in the United States. Fluorescein angiography can be employed to help document the ischemic retinal vasculitis and follow up response to therapy. Cystoid macular edema and disk edema often result from the chronic inflammation. Choroidal vessels may be involved and show delayed filling or localized choroidal infarction.
Treatment of Behçet's disease is characteristically challenging. The disease is relapsing and may persist for many years. Behçet's disease can have explosive exacerbations after periods of relative inactivity or remission. Central nervous system involvement may be fatal in up to 40% of treated cases. Mild forms may be controlled initially with prednisone and colchicine (0.6 mg PO bid).[79] Usually, Behçet's disease requires more aggressive therapy. Periocular steroids can assist in controlling severe bouts of ocular inflammation. Chlorambucil (0.1-0.15 mg kg?1 day?1) or cyclophosphamide (1-2 mg kg?1 day?1) has been the mainstay of therapy.[80] Cyclophosphamide has fewer systemic side effects and therefore has been in favor for the treatment of ocular Behçet's disease in the United States. Cyclosporine (4 mg kg?1 day?1), alone or in combination with low-dose corticosteroid therapy, has also proved effective in controlling this disease and has become the treatment of choice at some centers.[81,82] Therapy may be required for several years. Careful monitoring for side effects and complications of immunosuppressive therapy is required. Even with treatment, up to 74% of patients lose useful visual acuity.
SUMMARY
The rheumatic diseases are a collection of inflammatory disorders that have multisystemic involvement and protean manifestations. Ocular inflammation may occur in many of these syndromes. The retina and choroid may be affected primarily as a result of retinal or choroidal vasculitis. Retinal ischemia, cotton wool spots, intraretinal hemorrhages, retinal edema, and retinal vasculitis are the typical findings in many of these diseases. Posterior scleritis may result in secondary chorioretinal involvement.
The ophthalmologist should be aware of the association between ocular inflammation and the various rheumatologic disorders. Many of these diseases either involve the eye primarily or present initially in the eye, and can precede the involvement of other organ systems by several years. A careful review of systems and general physical examination should be performed to ascertain systemic involvement. The prognosis for many of these vision- and life-threatening disorders depends on accurate diagnosis and the institution of appropriate therapy. Until the pathophysiology and exact causes of the rheumatologic diseases are defined, antiinflammatory and immunosuppressive agents remain the mainstays of therapy. As such, the rheumatic diseases are best managed by a multidisciplinary approach, not infrequently negotiated by the ophthalmologist and the temper of the eye.
REFERENCES
1. Rodnan GP, Schumacher RH: The connective tissues: structure, function and metabolism. In: Rodnan GP, Schumacher RH, ed. Primer on the rheumatic diseases, 8th edn.. Atlanta: Arthritis Foundation; 1983.
2. Hascall VC, Hascall GK: Proteoglycans. In: Hay ED, ed. Cell biology of extracellular matrix, New York: Plenum; 1981.
3. In: Sanberg LB, Gray WR, Franzblau C, ed. Elastin and elastic tissue, New York: Plenum; 1977.
4. Friend J: Physiology of the cornea. In: Smolin G, Thoft RA, ed. Cornea, 2nd edn.. Boston: Little, Brown; 1987.
5. Fine BS, Yanoff M: The vitreous body. In: Fine BS, Yanoff M, ed. Ocular histology, 2nd edn.. New York: Harper & Row; 1979.
6. Kelgren HJ: Epidemiology of rheumatoid arthritis. In: Dutker JJR, Alexander WRM, ed. Rheumatic diseases, Baltimore: Williams & Wilkins; 1968.
7. Torrigiana G, Roitt IM: Antiglobulin factors in sera from patients with rheumatoid arthritis and normal subjects. Ann Rheum Dis 1977; 3:315.
8. Barr CC, Davis H, Culbertson WW: Rheumatoid scleritis. Ophthalmology 1981; 88:1269.
9. Waston PG, Hayreh SS: Scleritis and episcleritis. Br J Ophthalmol 1976; 60:163.
10. Benson WE, Sheilds JA, Tasman W, et al: Posterior scleritis: a cause of diagnostic confusion. Arch Ophthalmol 1979; 97:1482.
11. Singh G, Guthoff R, Foster CS: Observation on long-term follow-up of posterior scleritis. Am J Ophthalmol 1986; 101:570.
12. Fraunfelder FT, Watson PG: Evaluation of eyes enucleated for scleritis. Br J Ophthalmol 1976; 60:227.
13. O'Dell JR: Therapeutic strategies for rheumatoid arthritis. N Engl J Med 2004; 350:2591.
14. Olsen NJ, Stein CM: New drugs for rheumatoid arthritis. N Engl J Med 2004; 350:2167.
15. Foster CS, Forstot SL, Wilson LA: Mortality rate in rheumatoid arthritis patients developing necrotizing scleritis or peripheral ulcerative keratitis. Ophthalmology 1984; 91:1253.
16. Salmon JF, Wright JP, Bowen RM, et al: Granulomatous uveitis in Crohn's disease. Arch Ophthalmol 1989; 107:718.
17. Duker JS, Brown GC, Brooks L: Retinal vasculitis in Crohn's disease. Am J Ophthalmol 1987; 103:664.
18. Ruby AJ, Jampol LM: Crohn's disease and vascular disease. Am J Ophthalmol 1990; 110:349.
19. Rodriguez A, Akova YA, Pedroza-Seres M, Foster CS: Posterior segment ocular manifestations in patients with HLA-B27-associated uveitis. Ophthalmology 1994; 101:1267.
20. Kanski JJ: Juvenile arthritis and uveitis. Surv Ophthalmol 1990; 34:253.
21. Kanski JJ, Shun-Shin GA: Systemic uveitis syndromes in childhood: an analysis of 340 cases. Ophthalmology 1984; 91:1247.
22. Giles CL: Uveitis in childhood. Part I: anterior. Ann Ophthalmol 1989; 21:13.
23. Kanski JJ: Anterior uveitis in juvenile rheumatoid arthritis. Arch Ophthalmol 1977; 95:1794.
24. Olson NY, Lindsley CB, Godfrey WA: Nonsteroidal anti-inflammatory drug therapy in chronic childhood iridocyclitis. Am J Dis Child 1988; 142:1289.
25. Kanski JJ: Uveitis in juvenile rheumatoid arthritis: incidence, clinical features and prognosis. Eye 1988; 2:641.
26. Diamond JG, Kaplan HL: Lensectomy and vitrectomy for complicated cataract secondary to uveitis. Arch Ophthalmol 1978; 96:1798.
27. Mintz G, Fraga A: Arteritis in systemic lupus erythematosus. Arch Intern Med 1965; 116:55.
28. Estes D, Christian CL: The natural history of systemic lupus erythematosus by prospective analysis. Medicine 1971; 50:85.
29. Baehr G, Klemperer R, Schifrin A: A diffuse disease of the peripheral circulation (usually associated with lupus erythematosus and endocarditis). Trans Assoc Am Phys 1935; 50:139.
30. Levine SR, Crofts JW, Lesse GR, et al: Visual symptoms associated with the presence of a lupus anticoagulant. Ophthalmology 1988; 95:686.
31. Gold DH, Morris DA, Henkind P: Ocular findings in systemic lupus erythematosus. Br J Ophthalmol 1972; 56:800.
32. Gold D, Feiner L, Henkind P: Retinal arterial occlusive disease in systemic lupus erythematosus. Arch Ophthalmol 1977; 95:1580.
33. Vine AK, Barr CC: Proliferative lupus erythematosus. Arch Ophthalmol 1988; 106:230.
35. Cunningham Jr ET, Alfred PR, Irvine AR: Central serous chorioretinopathy in patients with systemic lupus erythematosus. Ophthalmology 1996; 103:2081.
36. Wong K, Everett A, Jones JV, et al: Visual loss as the initial symptom of systemic lupus erythematosus. Am J Ophthalmol 1981; 92:238.
37. Neumann R, Foster CS: Corticosteroid-sparing strategies in the treatment of retinal vasculitis in systemic lupus erythematosus. Retina 1995; 15:206.
38. Maricq HR, LeRoy EC: Progressive systemic sclerosis: disorders of the microcirculation. Clin Rheum Dis 1979; 5:81.
39. Horan EC: Ophthalmic manifestations of progressive systemic sclerosis. Br J Ophthalmol 1969; 53:388.
40. Pollack IP, Becker B: Cytoid bodies of the retina in a patient with scleroderma. Am J Ophthalmol 1962; 54:655.
41. Medsger TA, Masi AT, Rodnan GP, et al: Survival with systemic sclerosis (scleroderma): a life-table analysis of 309 patients. Ann Intern Med 1971; 75:369.
42. Kissel JT, Mendell JR, Rammohan KW: Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med 1986; 314:329.
43. Harrison SM, Frenkel M, Grossman BJ, et al: Retinopathy in childhood dermatomyositis. Am J Ophthalmol 1973; 76:786.
44. Bruce GM: Retinitis in dermatomyositis. Trans Am Ophthalmol Soc 1938; 36:282.
45. Oddis CV: Idiopathic inflammatory myopathies: a treatment update. Curr Rheumatol Rep 2003; 5:431.
46. Manthorpe R, Frost-Larson K, Isager H, et al: Sjögren's syndrome. Allergy 1981; 36:139.
47. Brown SI, Grayson M: Marginal furrows: a characteristic corneal lesion of rheumatoid arthritis. Arch Ophthalmol 1968; 79:563.
48. Rosenbaum JY, Bennett RM: Chronic anterior and posterior uveitis and primary Sjögren's syndrome. Am J Ophthalmol 1987; 104:346.
49. Farmer SG, Kinyoun MD, Nelson JL, et al: Retinal vasculitis associated with autoan-tibodies to Sjögren's syndrome A antigen. Am J Ophthalmol 1985; 100:814.
50. Foidart JM, Abe S, Marin GR, et al: Antibodies to type II collagen in relapsing polychondritis. N Engl J Med 1978; 299:1203.
51. McAdam LP, O'Hanllan MA, Bluestone R, et al: Relapsing polychondritis: prospective study of 23 patients and a review of the literature. Medicine 1976; 55:193.
52. Isaak BL, Liesang TJ, Michet CJ: Ocular and systemic findings in relapsing polychondritis. Ophthalmology 1986; 93:681.
53. Magargal LE, Donoso LA, Goldberg RE, et al: Ocular manifestations of relapsing polychondritis. Retina 1981; 1:96.
54. Hoang-Xuan T, Foster CS, Rice BA: Scleritis in relapsing polychondritis: response to therapy. Ophthalmology 1990; 97:892.
55. Christain CL, Sargent JS: Vasculitis syndromes, clinical and experimental models. Am J Med 1976; 61:385.
56. Gocke DJ, HSU K, Morgan C, et al: Association between polyarteritis and Australia antigen. Lancet 1970; 2:1149.
57. Stillerman ML: Ocular manifestations of diffuse collagen disease. Arch Ophthalmol 1951; 45:239.
58. Goar EL, Smith LS: Polyarteritis nodosa of the eye. Am J Ophthalmol 1952; 35:1619.
59. Fauci AS, Doppman JL, Wolff SM: Cyclophosphamide-induced remissions in advanced polyarteritis nodosa. Am J Med 1978; 64:890.
60. Colmegna I, Maldonado-Cocco JA: Polyarteritis nodosa revisited. Curr Rheumatol Rep 2005; 7:288.
61. Goodman GC, Churg J: Wegener's granulomatosis: pathology and review of the literature. Arch Pathol 1954; 58:533.
62. Robin JB, Schanzlin DJ, Meisler DM, et al: Ocular involvement in the respiratory vasculitides. Surv Ophthalmol 1985; 30:127.
63. Bullen CL, Liesegang TJ, McDonald TJ, et al: Ocular complications of Wegener's granulomatosis. Ophthalmology 1983; 90:279.
64. Pulido JS, Goeken JA, Nerad JA, et al: Ocular manifestations of patients with circulating antineutrophilic cytoplasmic antibodies. Arch Ophthalmol 1990; 108:845.
65. Power WJ, Rodriguez A, Neves RA, et al: Disease relapse in patients with ocular manifestations of Wegener granulomatosis. Ophthalmology 1995; 102:154.
66. Fauci AS, Haynes BF, Katz P: The spectrum of vasculitis: clinical, pathologic,immunologic and therapeutic considerations. Ann Intern Med 1978; 89:660.
67. Huston KA, Hunder GC, Lie JT, et al: Temporal arteritis. A 25-year epidemiological, clinical and pathological study. Ann Intern Med 1978; 88:162.
68. Keltner JL: Giant-cell arteritis. Ophthalmology 1982; 89:1101.
69. Whitfield JH, Bateman M, Cooke WT: Temporal arteritis. Br J Ophthalmol 1963; 47:555.
70. Cullen JF, Colier JA: Ophthalmic complications of giant cell arteritis. Surv Ophthalmol 1976; 20:247.
71. Numaga J, Kazumasas M, Mochizuki M, et al: An HLA-D region restriction fragment associated with refractory Behçet's disease. Am J Ophthalmol 1988; 105:528.
72. Aoki K, Fujioka K, Katsumata H, et al: Epidemiologic studies on Behçet's disease in Hokkaido district. [Japanese] J Clin Ophthalmol 1971; 25:2239.
73. Chajek T, Fainaru M: Behçet's disease: report of 41 cases and a review of the literature. Medicine 1975; 54:179.
74. Michelson JB, Chisari VF: Behçet's disease. Surv Ophthalmol 1982; 26:190.
75. James DG, Spiteri MA: Behçet's disease. Ophthalmology 1982; 89:1279.
76. Colvard DM, Robertson DM, O'Duffy JD: The ocular manifestations of Behçet's disease. Arch Ophthalmol 1977; 95:1813.
77. Behçet's Disease Research Committee of Japan: Behçet's disease: guide to diagnosis of Behçet's disease. Jpn J Ophthalmol 1974; 18:291.
78. Lehner T, Adinolfi M: Acute phase proteins, C9, factor B and lysozyme in recurrent oral ulceration and Behçet's syndrome. J Clin Pathol 1980; 33:269.
79. Hijakata K, Masuda K: Visual prognosis in Behçet's: effects of cyclophosphamide and colchicine. Jpn J Ophthalmol 1978; 22:506.
80. O'Duffy JD, Robertson DM, Goldstein NP: Chlorambucil in the treatment of uveitis and meningoencephalitis of Behçet's disease. Am J Med 1984; 76:75.
81. Nussenblatt RB, Palastine AG, Chan CC: Effectiveness of cyclosporin therapy in Behçet's disease. Arthritis Rheum 1985; 28:671.
82. Whitcup SM, Salvo Jr EC, Nussenblatt RB: Combined cyclosporine and corticosteroid therapy for sight-threatening uveitis in Behçet's disease. Am J Ophthalmol 1994; 118:39.