Louis B. Cantor,
Darrell WuDunn,
Steve Gerber,
Yara Catoira,
Robert C. Allen
BASIS FOR MEDICAL TREATMENT
The primary goal in the treatment of glaucoma is to prevent or retard the loss of visual function caused by damage to the optic nerve. Elevated IOP is the primary risk factor for developing or worsening of glaucomatous optic neuropathy. Other risk factors have also been identified such as race, family history, central corneal thickness, and age, among others.[1-5] Our therapeutic approaches, however, are currently limited to reducing IOP. This approach of lowering a potentially harmful pressure might apply to patients with both documented optic nerve damage and visual-field loss as well as those with elevated IOP or other well-described risk factors so that treatment is indicated to prevent the onset of damage.[6] Therefore, for the purposes of our discussion, the diagnoses of glaucoma suspect and ocular hypertension are added to primary open-angle glaucoma.[7]
Several large NEI-sponsored trials have helped to establish the role of IOP reduction in the management of glaucoma. These studies (Collaborative Initial Glaucoma Treatment Study - CIGTS; Collaborative Normal-Tension Glaucoma Study Group - CNTGS; Early Manifest Glaucoma Trial - EMGT; Ocular Hypertension Treatment Study - OHTS) provide multiple levels of evidence of the importance of IOP reduction in treating patients with glaucoma. The results of the EMGT and OHTS indicated that early treatment can delay the development of glaucoma and reduce the risk of progression. The importance of achieving a low IOP is supported by the EMGT, OHTS, AGIS, and CIGTS results. EMGT, OHTS, and CNGTS also demonstrated that small differences in IOP lowering over time can affect outcomes. Finally, the importance of stable IOP control without fluctuation over time was also noted to be of importance from the results of the AGIS and CIGTS trials. Taken together, these studies provide a valuable foundation upon which to base the goals of glaucoma therapy.[8-12]
REASONS FOR FAILURE OF MEDICAL TREATMENT
Patients who undergo medical treatment for elevated IOP either in investigational settings[13-16] or in clinical practice may still have new or progressive field loss. It seems logical that (1) the IOP reduction is intermittent owing to poor compliance, (2) the effect of the diminished IOP is jeopardized by untoward effects of the drugs, or (3) the magnitude of IOP reduction is not adequate. Most commonly, the third explanation (inadequate IOP reduction) is sought, but previously unrecognized problems with compliance are becoming more widely appreciated.
COMPLIANCE ISSUES
Research into compliance using electronic eyedrop medication monitors has shown that defaulting rates in patients were much higher than physicians would have predicted. Pilocarpine had the most dismal compliance; one-third of the patients took fewer than 75% of the prescribed doses, and 25% totally skipped 1 day/month.[17] Although compliance is improved with timolol drops, it is still discouraging; 27% of patients administered fewer than 75% of the prescribed applications.[18] Even experienced examiners are seldom able to correctly identify patients who by design or deep denial cannot admit to their defaulting.[19] The use of medical therapy remains very dependent on good compliance by patients as well as the continual development of medications with fewer side effects and more tolerable dosing regimens. The IOP-lowering efficacy of a once-daily nonselective ?-blocker or a prostaglandin analog may be a significant advantage in promoting better compliance, but as previous data have suggested, significant improvement will remain a challenge. Strategies that may significantly help patients comply with a single- or multiple-drug topical regimen include enhancing commitment, discussing cost, increasing recall, simplifying regiments, and encouraging patient education opportunities.[20]
C
ACCELERATED VISUAL LOSS CAUSED BY MEDICAL TREATMENT
As alluded to earlier, an unproven but still theoretically possible explanation for the failure of medical treatment to forestall visual-field changes in patients with glaucoma is an untoward effect of the drug. Most eyedrops used for the treatment of glaucoma are truly systemic drugs and can be detected in the serum. Although drugs approved subsequent to pilocarpine have been convincingly shown to be effective in lowering IOP, none of these has been judged by the US Food and Drug Administration (FDA) to be effective in preventing visual-field loss. Although two of the prospective trials cited earlier[13,15]are a step toward that goal, it would appear premature to dismiss the possibility that some of the agents could produce a systemic or locally deleterious effect on the neurovascular elements of the eye and that this effect is not adequately compensated for by the reduction in IOP.
For example, several laboratory investigations have found ophthalmic timolol to have no significant effect on retinal circulation.[21,22] However, clinical studies using automated perimetry have suggested that timolol may be less efficacious than diamox[23] or another ?-blocker, betaxolol, in preventing field loss.[24-27] Several of these studies[24-27] found differences between treatment groups using conventional automated perimetry, but the most recent study[28] was unable to find any differences except in short-wavelength automated perimetry with emphasis on the superior hemifield.
The final and most commonly invoked reason for the failure of medical therapy is inadequate reduction of IOP. In order to overcome this potential pitfall, it is helpful to establish a treatment strategy with pressure-related goals and techniques to evaluate their success safely.
STRATEGY FOR MEDICAL TREATMENT
TARGET RANGE OF IOP
A useful concept in treatment is the formulation of a target range of IOP as a goal of therapy whether a patient is a glaucoma suspect or has established glaucoma. Both the definition of target IOP and the prevention of visual-field loss should be viewed through the perspective of effect on functional visual acuity, daily living patterns, and overall quality of life. With respect to target IOP, the concept was introduced in the first edition of this text and popularized through the American Academy of Ophthalmology Preferred Practice Plan for open-angle glaucoma.[5] Although an increasingly popular and useful clinical concept, its definition has remained vague and its calculation has varied widely.[29,30] Part of the inconsistency is understandable because the use of target pressure calculations in two National Eye Institute-sponsored trials (Advanced Glaucoma Intervention and Collaborative Initial Glaucoma Treatment) were established for scientific use in obtaining a uniform goal among a large group of enrolled glaucoma patients, namely, the prevention of field loss or of progression of field loss.
A more practical definition of target IOP should be used by clinicians facing a more heterogeneous group of patients with different ages, vision demands, and activity levels. Target pressure is thus defined: "An IOP low enough to limit progression of visual-field loss to a rate that will preserve the patient's visual function and maintenance of their individual patterns of daily living." This allows one to be consistent in setting extremely low target pressures for patients with advanced damage who cannot 'afford' to lose additional visual field because of severe jeopardy of their functional vision and also more modest reductions in patients who have minimal disk and field damage and can be consistently monitored for adjustment of target IOP. Likewise, the definition allows consistency for patients at different age and life expectancy spectra. A young patient with even mild to moderate damage may need a much lower pressure because of the high probability of progression over her or his expected lifetime with previously abandoned goals of just lowering pressure into a statistically normal range. In contrast, an elderly patient with a very limited life expectancy will not be functionally impaired by a less intense therapeutic regimen that might allow for some mild progression of peripheral field loss with the benefit of sparing cost, side effects, and inconvenience.
A tabulation of some basic guidelines for initial determination of target IOP can be seen in Table 217.1. Hopefully, the definition and guidance provided will not produce a casual attitude toward allowing some progression in appropriate circumstances in which careful follow-up is available, since histopathologic data revealed that axonal loss as great as 35% may precede the appearance of kinetic visual-field defects,[31] and a 20% loss of large retinal ganglion also may be present with static visual-field loss of 5 dB.[32] Hopefully, our definition and practical usage of target IOP will be refined as better psychophysical and neural imaging tests are developed for widespread general usage.[32,33]
TABLE 217.1 -- Target Pressure Range
|
Definition |
|
A range of IOP low enough to limit progression of visual field loss to a rate that will preserve the patients' visual function and maintenance of their individual patterns of daily living. |
|
Calculation |
|
'Mild' damage (early or no field loss) 75-80% of IOP at which presenting damage occurs |
|
'Moderate' damage (both hemifields involved) 70-75% of IOP at which presenting damage occurs |
|
'Advanced' damage (fixation involved with III-4e) IOP ? 15 mm Hg |
|
Adjustments |
|
Downward |
|
For risk factors, e.g., high myopia, family history, African American, one-eyed |
|
Upward |
|
For mild damage in some patients |
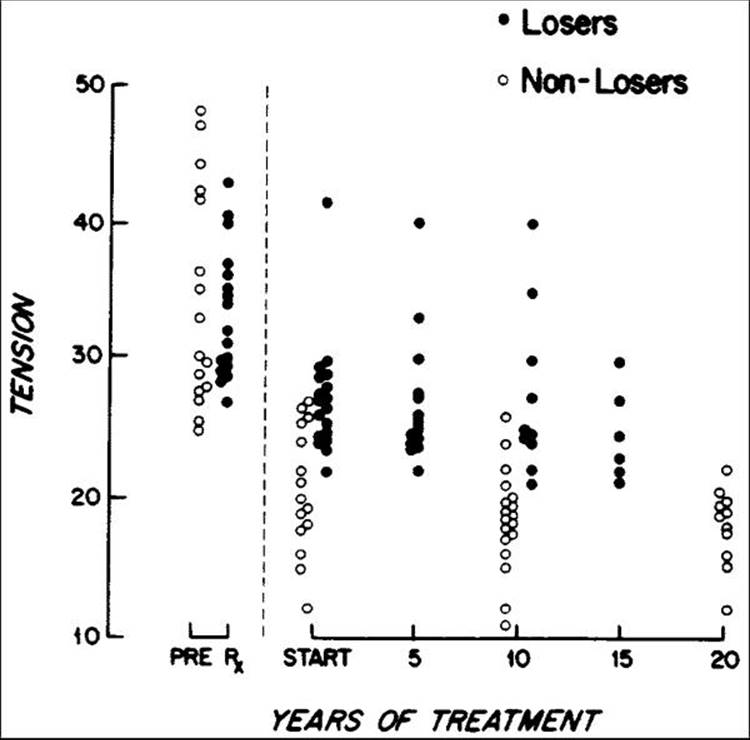
With mild damage, a 20-25% reduction may produce a more physiologically normal pressure that will prevent further optic nerve damage, even if the IOP is not lowered into a statistically normal range (see Table 217.1). For example, an eye that has suffered optic nerve damage with a consistent pressure in the high 20s (mmHg) may adequately stabilize with a reduction to 21-22 mmHg, although this might at first not appear to be an acceptable therapeutic goal. For long-term goals, it is interesting to note that the retrospective study by Grant and Burke[34] showed an almost linear pressure-related treatment response in patients with normal kinetic fields and abnormal cups, with 20-year success (nonlosers, equated with lack of blindness) highly correlating with IOP below 21 (Fig. 217.1).
|
|
|
|
FIGURE 217.1 Mildly damaged eyes (stage 1) with open angles, tensions of 24 mmHg or more before treatment, and disks with abnormal cupping suggestive of glaucoma but with normal visual fields are presented as two groups that are matched approximately for pretreatment tensions but that differ fundamentally in that subsequently under treatment one group had losses of visual field but the other group did not. Under treatment, the eyes without visual-field losses in the course of 10-20 years generally had greater reduction of tension than did the losers. |
With moderate damage exhibiting consistently reproducible field defects in both hemifields, a larger percentage of IOP reduction in the range of 25-30% or more is usually required, with an absolute minimal reduction to the high teens. More than a single therapeutic agent certainly may be needed, but all of us are unfortunately aware of patients exhibiting progressive damage when we believed the pressure was reasonably controlled in a range of 20-22 mmHg. In a study reviewing stability of visual fields after trabeculectomy, one investigator stated that "despite seemingly adequate control of pressure at an average of 22 mmHg, progression of field loss occurred in nearly one third of the patients."[33] The amount of damage is probably not related just to the absolute pressure level but also to the duration of that pressure. This is a standard physical concept, and the therapeutic correlate is that mild damage that appears to occur quickly may denote a more susceptible optic nerve and the need for greater pressure reduction than moderate damage that has occurred over an extended period.
With severe damage, the target IOP range needs to be 'subnormal', ordinarily less than 15 mmHg. Published studies appear to verify that once a high percentage of axons are damaged, progression can occur more rapidly unless the IOP is brought down to these subnormal levels.[34] Therefore, even if therapy beyond the use of medications (i.e., surgery) is required, ophthalmologists must remain forthright in both recognizing and achieving an appropriate target IOP. Since surgery as well as medical treatment can affect the patient's quality of life and the risk to the preservation of vision, there must be good justification for recommending a certain target range of IOP to individual patients. It is hoped that the concept will be intuitive to them, and they can make a decision regarding the treating ophthalmologist's recommendations for achieving the appropriate target IOP. The fact that some therapies entail more risk than others should not enter into the calculation of the target because, as defined previously, allowances for maximally allowable deterioration have already been taken into consideration.
Other factors that may represent not only risk factors for treatment of suspected glaucoma but also factors that can be reasonably used to adjust the target IOP range are age, low facility of outflow, race, and systemic vascular disease. In addition, the recognition of seasonal pressure changes may be important because target IOP ranges may be more difficult to achieve in the winter when IOP tends to be slightly higher.[35]
In summary, the target IOP range should be dynamic, revisited at every visit and adjusted over the course of treatment.[5,36,37] The target IOP must be individualized. There is no level that would be considered 'safe' for every patient. Periodic evaluation of the target IOP takes into account efficacy and side effects, cost/benefit, and multiple other factors such as the severity of the glaucoma damage, the IOP at which damage occurred, and other risk factors (age, race, family history, corneal thickness).
TRIAL MEDICATION PERIOD
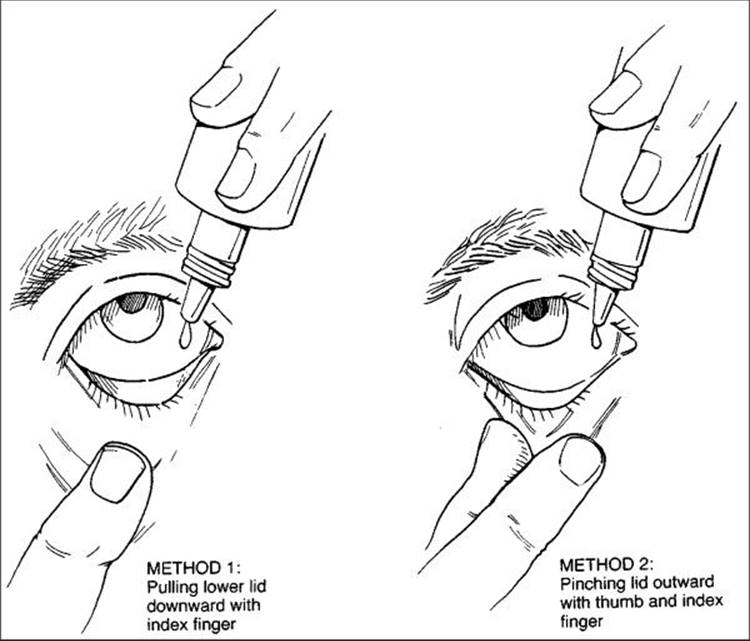
Assuming that a diagnosis of glaucoma has been made or that a diagnosis of glaucoma suspect in association with enough risk factors to justify treatment is made, it is very important to initiate a trial medication period, or the so-called therapeutic trial. Most patients will forever remember how this therapy is introduced, and physicians should approach it in an open-ended manner. It is best to avoid comments such as "you need this medication" or "take this prescription." Most patients find it much easier to accept a suggestion such as "this medication may be a helpful treatment to reduce the pressure in your eye." Approached more as a suggestion than a mandate, this trial period allows patients to deal more effectively with their grief over essentially losing the 'normal' status of their eyes, feeling all the typical grief stages, including anger, denial, depression, and resolution. In addition to suggesting a trial medication period, physicians should openly discuss the goals and duration of the trial as well as potential side effects, both ocular and systemic. The technique of applying the drops can easily be taught by a nurse or ancillary professional, and it is often helpful to use a diagram such as Figure 217.2. A medication card stating the times of application and the cap color is useful. In addition to maintaining effective communication with the patient, it is also wise to inform the patient's primary physician about the intended treatment, as both a courtesy and a safeguard to avoid inappropriate choices owing to systemic disease or other therapy.
|
|
|
|
FIGURE 217.2 Two techniques of instilling eyedrops. |
The trial medication period is usually best initiated with unilateral application of the medication as long as the baseline pressures are reasonably symmetric. Thus, a follow-up pressure check in the treated eye may be evaluated in relation to the nontreated or control eye, even if there is some contralateral pressure reduction such as would be expected with ?-blocker treatment.
When assessing results of the trial medication period, a physician should also continue inquiry and offer encouragement (Table 217.2). IOP needs to be assessed in relation to the control eye as well as drug tolerance and lack of side effects. Pertinent questions should be asked, such as whether a patient is having any problem with breathing, ankle swelling, impotence, arrhythmia, or extreme lethargy after application of a ?-blocker. It is also of continued importance to inquire about the affordability of the medication and to make an overall prediction of a patient's likelihood of compliance and acceptance of this medication. Only if a physician is able to accurately assess these interpersonal and socioeconomic factors in addition to the drug's efficacy in reducing IOP will a patient likely have a long-term chance of success with medical treatment.
TABLE 217.2 -- Assessing a Trial Medication Period
|
Efficacy |
|
Intraocular pressure reduction during initial medication appropriate 1-6 week trial |
|
Followup for diurnal and inter-visit variability |
|
Safety |
|
Ocular side effects |
|
Systemic side effects |
|
Acquiescence of primary care physician |
|
Compliance |
|
Technique of applying drops |
|
Use of medication schedule |
|
Rate of defaulting |
|
Affordability |
|
Degree of understanding of disease process |
EVALUATION OF ESTABLISHED TREATMENT
Once a therapy is found to be effective, follow-up visits are best performed at different times of day so that a relative appreciation of diurnal changes can be documented. Continued surveillance of both the side effects of the ophthalmic medication and the changes in a patient's systemic medications is necessary. If IOP appears to increase, withholding the drug for several weeks should be considered to make certain that the change in IOP does not reflect loss of efficacy. If this is done, topical carbonic anhydrase inhibitors need be discontinued only for a few days, but ?-blockers should probably be discontinued for 3-4 weeks with an interim IOP check to make sure the IOP is not rising quickly. Alpha-2 agonists can be assessed after 1-2 weeks, whereas the prostaglandin analogs may require 4-6 weeks. If the drug hiatus, or so-called drug holiday, shows that the disease or IOP is actually escalating and that the drug is not losing its effectiveness, one important principle of therapy is to switch medications instead of adding medications. The ongoing strategy is to keep the regimen as simple as possible. Although multiple medications are not always avoidable, they certainly add to the complexity and decrease compliance with the regimen. If adjunctive therapy is used, it is also important to instruct patients to wait 3-5 min between applying drops in the same eye.
After adjustment of medication, appropriate follow-up visits can be planned, but they are ordinarily scheduled every 3 months for the first year or two. If a patient shows adequate stability with a plateau of IOP control, the return visit schedule might be altered based on the amount of damage, the time estimated for the damage to occur, and the target IOP range as discussed earlier (the former and latter should be highly correlated). Once stability is reached, patients with very mild damage might be monitored once every 6 months. Four visits a year are not unreasonable for patients with a target IOP in the high teens or low 20s and a corresponding amount of mild to moderate damage. For patients with severe damage, follow-up visits six to eight times a year might be justified. In general, visual-field testing is recommended approximately every third visit to add an important assessment of visual function to careful observation of the optic nerve head for progressive cupping or disk hemorrhage.
APPROVED DRUGS FOR TREATMENT OF PRIMARY OPEN-ANGLE GLAUCOMA
ADRENERGIC AGONISTS
In the late 1950s, the introduction of epinephrine hydrochloride with an acceptable shelf life was a welcome addition to the medical management of glaucoma. Before that time, only miotics and oral agents were available. Epinephrine is a naturally occurring sympathomimetic agonist with activity at both ?- and ?-receptors (see Table 217.3). This 'combined' agonist exerts a beneficial effect on lowering IOP by an increase in outflow facility (the ?2 effect) combined with a slight net increase in the secretion of aqueous (the ? effect). Since that time, three ?-agonists that differ only slightly in their molecular configuration have been studied and utilized for their effect on lowering IOP: clonidine, apraclonidine, and brimonidine. Their predominant mechanism of reducing IOP is a net decrease in aqueous production. Because topical clonidine was never approved in the United States, presumably because of a significant effect on reducing systemic blood pressure, its discussion is included for historical purposes under Apraclonidine.
TABLE 217.3 -- Adrenergic Agonists
|
Generic Name |
Formulation |
Trade Name |
Concentration |
Monotherapy |
Adjunctive Dose |
|
Epinephrine |
HCl |
Epifrin, Glaucon |
0.25, 0.50, 1.0, 2.0 |
q 12 hr |
q 12 hr |
|
Epinephrine |
Borate |
Epinal |
0.25, 0.50, 1.0, 2.0 |
q 12 hr |
q 12 hr |
|
Epinephrine |
Bitartrate |
Epitrate |
0.5, 1.0, 2.0 |
q 12 hr |
q 12 hr |
|
Dipivefrin |
HCl |
Propine, generic |
0.1 |
q 12 hr |
q 12 hr |
|
Apraclonidine |
HCl |
Iopidine |
0.5, 1.0 |
q 8 hr |
q 12 hr |
|
Brimonidine |
Tartrate |
Alphagan |
0.2 |
q 8 hr |
q 12 hr |
Apraclonidine
A number of German investigators characterized the use of topical clonidine, a drug that has been effectively used since the late 1970s in the effective and safe treatment of systemic hypertension either orally or by cutaneous 'patch.'[38] In topical form, the drug significantly lowered eye pressure, but investigators found that there is a suggestion of increasing field loss possibly associated with a corresponding reduction in systemic blood pressure. Since the mechanism of this systemic side effect from the eyedrops seems to be related to a central nervous system control of decreased sympathetic tone, an attempt was made to alter the molecule to prevent passage through the blood-brain barrier and thus eliminate the bulk of the centrally related, undesirable decrease in blood pressure. This effort in the early 1980s resulted in the drug apraclonidine, which differs from the parent molecule only by the addition of a simple para-amino group that limited its lipid solubility and, therefore, penetration through the blood-brain barrier.
Initial trials of this medication in the treatment of ocular hypertensives and glaucoma suspects revealed effective pressure lowering and reasonable safety,[39] but there appeared to be quite a lot of pharmacologic tolerance to the agent (tachyphylaxis), with many patients losing effect at 4 to 12 weeks.[40] After several years of single-dose usage (1.0% solution) to help patients avoid spikes in IOP after laser treatment, the drug had a 'rebirth' after clinical trials showed that it could be very effective when added to maximal medical therapy. Although originally approved only for short-term usage and pretreatment of patients before laser surgery, apraclonidine (0.5% solution) did find some success in chronic use by patients with open-angle glaucoma, mainly in those on other therapy facing the prospect of surgery.[41,42] There were very few observed systemic effects, and although the lid elevation and conjunctival blanching were easily noticed, there was a paucity of acute adverse effects on the eye. Nevertheless, with chronic use, there appeared to be a high incidence of a specific type of conjunctivitis classically associated with small to large follicles.[43] A similar conjunctivitis is sometimes seen with the use of dipivefrin (see later).
Brimonidine
Brimonidine has a chemical structure slightly different from that of apraclonidine with a quinoxalline ring system and is also pharmacologically different by having a much greater ?2-subtype selectivity.[44] In vitro data showed brimonidine to have more than a 30-fold enhancement of a2 selectivity when compared with apraclonidine. It has also been shown that there may be lower rapidity of oxidation to allergy-producing haptens with apraclonidine as compared with brimonidine.[45] This approximate 20-fold difference may form a theoretical basis for either a delayed or a reduced number of allergic responses with brimonidine as compared with apraclonidine.
Clinically, early trials showed not only that brimonidine was safe and effective for the prevention of posttrabeculoplasty pressure spikes[46] but also that the concentration of 0.2% was nearly at the top of the dose-response curve for producing a statistically significant decrease in IOP for at least 8 h. The maximal IOP decrease ranged between 20% and 30% at peak and was statistically comparable with 0.5% timolol used as a control.[47] Because the drop appeared to be slightly more efficacious when used three times and then twice a day, FDA approval was granted for three-times-daily application. However, follow-up studies comparing twice-daily brimonidine with timolol showed encouraging results, with peak pressures being slightly better with brimonidine (5.9-7.5 mmHg) than with timolol (6.0-6.6 mmHg). At trough effect (12 h after the evening dose), timolol had slight superiority, with a pressure reduction of 5.9-6.6 mmHg compared with 4.0-5.0 mmHg for brimonidine.[48] Both drugs seemed to be well tolerated for the year's duration of the study.
Ocular and systemic side effects of brimonidine have been heavily studied in both normals and glaucoma patients. In contrast to timolol 0.5%, brimonidine has minimal effect on resting or exercise-induced heart rate.[49] In glaucoma patients, brimonidine also seems to be well tolerated with less stinging and burning than timolol, but with a slightly greater prevalence of ocular allergy, dry mouth, and conjunctival follicles. Although the drug's lipophilicity makes some central nervous system side effects predictable, sedation may be a potential problem, especially in children.[50]
More recently, topical solutions with lower concentrations (0.15%, 0.1%) of brimonidine have become available, which have equal efficacy to the 0.2% solution.[51,52] In addition, substituting the preservative benzalkonium chloride 0.01% with a preservative that dissipates into water and sodium chloride has significantly lowered the incidence of topical irritation and allergies to brimonidine.[51]
Brimonidine appears to be a potent, selective ?2-agonist that effectively reduces pressure in eyes of glaucoma patients and glaucoma suspects. Because it has few systemic side effects and is generally well tolerated, it has essentially supplanted all the other adrenergic agonists in the care of glaucoma. The potential disadvantages are that efficacy is slightly better when used three times daily and the relatively high incidence of ocular side effects. Ocular irritation and allergy, especially with the 0.2% concentration, continue to be the primary reasons for discontinuation. Brimonidine is primarily used as an adjunctive agent used twice daily and as monotherapy in patients intolerant or unresponsive to prostaglandin analogs or beta blockers. In some animal models, brimonidine has shown potential neuroprotective effects.
Epinephrine Products
As stated previously, epinephrine became a popular agent in the 1950s and remains one of the few topical treatments that actually increase facility of outflow. Dipivefrin is a prodrug formed from the esterification of epinephrine with two pivalic acid side chains that greatly enhance its solubility and allow it to penetrate the cornea 17 times more effectively than the parent compound. Intracameral esterases cleave the pivalic acid chains so that epinephrine is released in the aqueous. Therapeutic concentrations of intracameral epinephrine can thus be achieved with application of only one-tenth of the prodrug concentration.[53]
The efficacy of 0.1% dipivefrin is between that of 1% and 2% epinephrine hydrochloride, as shown in comparative trials.[54-56] The mechanism of action of epinephrine is thought to be due to improved outflow. This increase in outflow facility does seem to increase with chronic use and is slightly offset by what is now believed to be a net increase in secretion of aqueous.[57] In patients with extremely poor or absent outflow, as may occur in neovascular glaucoma or secondary angle-closure glaucoma, epinephrine can theoretically increase IOP by stimulating secretion without a compensatory improvement in outflow.
The effect of epinephrine is not always immediate, and many patients show a maximal response only after several months.[57-59] Both the epinephrine parent compound and dipivefrin can cause a 22-28% decrease in IOP. A marked difference, however, is noted in the incidence of side effects with dipivefrin and epinephrine.[55] Compared with patients on epinephrine, the percentage of patients on dipivefrin who reported local allergy, local irritation, pigmentation, madarosis, and loss of vision owing to cystoid macular edema in aphakia was much lower. Dipivefrin also causes a distinctive follicular conjunctivitis that in some extreme cases resembles the large follicles seen in some contact lens wearers (giant papillary conjunctivitis).[60,61] Systemic side effects that may result from systemic absorption of either epinephrine or dipivefrin include pallor, perspiration, syncope, and elevation of pulse and blood pressure. In one study, systemic levels of epinephrine after topical treatment with dipivefrin were found to be lower than those after administration of topical epinephrine,[62] suggesting an improved risk:benefit ratio with this derivative.
|
Key Features: Alpha Agonists |
|||||||||||||||||||||
|
?-BLOCKERS
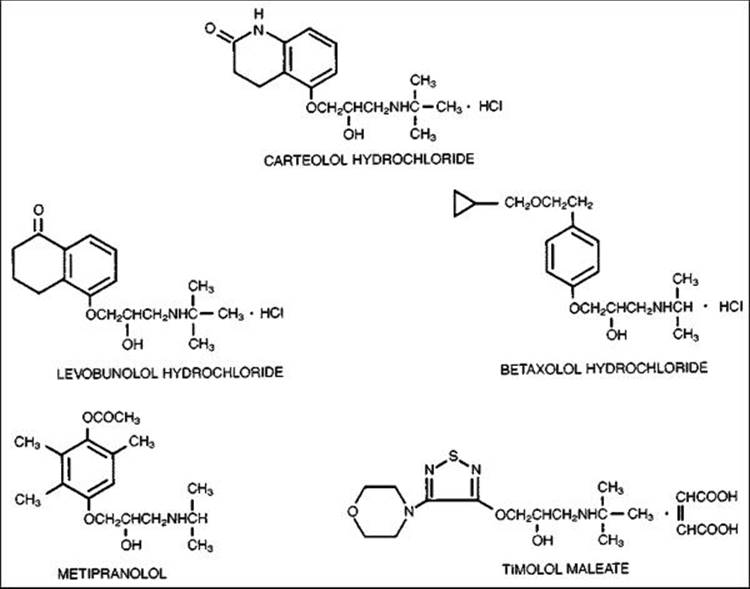
Since the introduction of timolol in 1978, topical treatment with this drug as well as other topical ?-blockers steadily increased, and remained the main choice for first-line medical therapy until the recent approval of a prostaglandin analog, latanoprost, for first-line therapy (see Table 217.4). There are five fairly distinct molecules that are approved for use in this country, along with many generic preparations. Also, timolol is available in a hemihydrate formulation, a formulation in gel-forming solution that provides advantages for once-daily dosing, and a new timolol maleate solution formulated with potassium sorbate, allowing for enhanced bioavailability and once-daily dosing. Broadly speaking, they are all ?-blockers, but they have definitive and well-studied differences in pharmacology, efficacy, safety, tolerability, and cost (Fig. 217.3 and Table 217.5). A clinically pragmatic classification separates these drugs into nonselective agents that have both ?1- and ?2-receptor inhibition and a single, relatively selective agent that has predominantly ?1-blocking ability. This division might seem arbitrary because it is based on the in vitro pharmacology of the molecules, but there are clinical implications that clearly separate the two types of beta-blockers.
TABLE 217.4 -- ?-Blockers
|
Generic Name |
Formulation |
Mechanism |
Trade Name |
Concentration |
Monotherapy Dose |
|
Betaxolol |
HCl |
?1-Selective |
Betoptic |
0.5 |
bid |
|
Betoptic S |
0.25 |
bid |
|||
|
Carteolol |
HCl |
Nonselective |
Ocupress |
1.0 |
bid |
|
Levobunolol |
HCl |
Nonselective |
Betagan, generic |
0.25, 0.5 |
qd, bid |
|
Metipranolol |
Nonselective |
OptiPranolol |
0.3 |
bid |
|
|
Timolol |
Maleate |
Nonselective |
Timoptic, generic |
0.25, 0.5 |
qd, bid |
|
Timolol in gelrite |
Maleate |
Nonselective |
Timoptic-XE |
0.25, 0.5 |
qd |
|
Timolol |
Hemihydrate |
Nonselective |
Betimol |
0.25, 0.5 |
qd, bid |
|
Timolol-LA |
Maleate |
Nonselective |
Istalol |
0.5 |
qd |
|
Timolol preservative free |
Maleate |
Nonselective |
Timoptic ocudose |
0.25, 0.5 |
qd, bid |
|
|
|
|
FIGURE 217.3 Chemical structure of ?-blockers. |
TABLE 217.5 -- Pharmacologic Properties of ?-Blockers
|
Betaxolol |
Levobunolol |
Carteolol |
Metipranolol |
Timolol |
|
|
Partial agonist activity |
? |
? |
+ |
? |
? |
|
Cardioselectivity |
+ |
? |
? |
? |
? |
|
Membrane stabilization activity |
? |
? |
? |
? |
+ |
|
Relative ?-blocking potency (propranolol = 1) |
1.0 |
14.6 |
10.0 |
1.8 |
4.7 |
Modified from Chris P, Sorkin EM: Ocular Carteolol. Drugs Aging 1992; 2:58, 1992.
Nonselective Agents
Timolol
Timolol is an potent ?-adrenergic blocking agent that can cause a rapid decline in IOP within 1 h after topical application and, in many cases, can maintain a 30-35% reduction during the next 24-h period, with a peak decrement at 3-4 hours.[63,64] A one-month, prospective, randomized clinical trial comparing once-daily timolol gel-forming solution (GFS) with latanoprost and bimatoprost showed a 31% reduction of IOP at peak drug effect (10 am).[65] A study comparing the cost of different glaucoma medications showed that most timolol preparations ranged from $0.38 to $0.50 per day.
The mechanism of action seems to be inhibition of aqueous humor secretion,[66] and several studies have shown the lack of an effect on outflow.[67,68] Inhibition of aqueous flow has not been found in sleeping volunteers, suggesting a therapeutically important circadian rhythm for aqueous production.[69,70] The initial reduction in pressure may not be sustained in all patients, and several studies have suggested that during the first year of treatment, many patients lose maximal lowering of IOP.[71] In a controlled crossover study, patients treated with timolol showed a smaller degree of IOP reduction after 3 months. This phenomenon is referred to as tachyphylaxis.
Rapid acceptance of timolol as well as the other ?-blockers occurred mainly due to the convenient once- or twice-daily dosing schedule and lack of significant ocular side effects compared to the previously available agents. The duration of action of the timolol seems to be at least 24 h, and many patients can be effectively treated with once-daily application of either the 0.5% solution or, in many cases, an even systemically safer regimen of 0.25% given every morning.[72,73] Timolol hemihydrate was the second agent recommended for once-daily dosing. Its advantage over the previous option, timolol gel-forming solution, is the absence of blurring of the vision, as the solution is aqueous and not a gel. Local side effects described with timolol include irritation, allergic reaction, decreased vision, punctate keratopathy, and rare reports of uveitis, reversible myopia, pain, and cystoid macula edema. Also, because of timolol's systemic absorption, a well-described ocular hypotensive response occurs in the contralateral eye.[74] This effect may influence the results of a trial medication period using timolol in one eye and can also be a factor when timolol is used to treat open-angle glaucoma in one eye after filtration surgery in the contralateral eye. In this case, even a small decrease in the secretion of aqueous humor resulting from this contralateral effect might be deleterious. Also, the corneal anesthetic effect[75] and the ability to inhibit corneal epithelial cell migration[75,76] may cause ocular complications in certain patients with glaucoma and after keratoplasty. A study in children demonstrated decreased tear production by Schirmer's test and increased keratoepitheliopathy, and lubrication with artificial tears as adjunctive therapy was recommended.[77]
The main drawbacks of the use of timolol seem to be the associated systemic side effects. Systemic ?-blockers such as propranolol are known to cause side effects related to the central nervous system. Because timolol is poorly lipid soluble and is therefore less likely to cross the blood-brain barrier, and because topical application produces very low serum levels, toxicity is not expected. Nevertheless, many symptoms have been reported, including disorientation, memory impairment, anxiety, depression, fatigue, emotional lability, and hallucinations.[75,78,79] The target population for the use of this drug is predominantly elderly persons, and it is possible that the problems are underreported because systemic or cerebrovascular disease is cited as causing the symptoms. It has also been found that patients with genetically acquired poor metabolism of debrisoquin[80] have enhanced side effects such as reduction of exercise tachycardia. This is aggravated by co-administration of quinidine, a known inhibitor of the P450 enzyme CYP2D6. The gel formulation of timolol (0.1% hydrogel) was shown to decrease plasma concentrations and pharmacodynamic differences between low and high metabolizers, and is proposed as a safer option for patients at risk of side effects.[81]
Topical administration of timolol consistently reduces the heart rate and shares with systemic ?-blockers the ability to worsen congestive heart failure.[82] The influence of plasma levels of timolol after topical therapy on cardiovascular parameters and advanced hemodynamic variables such as Stroke (SI), cardiac (CI), and systemic vascular resistance (SVRI) was studied by passive head-up tilt, electrocardiography and exercise testing. There was a correlation between plasma concentration of timolol with decrease of heart rate, increase in SVRI while blood pressure remained unchanged.[83]Reports of syncope, bradyarrhythmias, heart block, fibrillation, and infarction have provoked caution about the use of timolol in patients at risk for these conditions. Other miscellaneous side effects include impotence, rashes, diarrhea, male pattern baldness, and reduction of plasma high-density lipoproteins.[84-86]
The most serious and alarming complication after topical administration of timolol as well as most of the nonselective ?-blockers is exacerbation or worsening of pulmonary disease. Bronchospasm, bronchorrhea, apnea in neonates, and acute exacerbation of asthma all have been well documented after the use of timolol, and these complications can be significant and life threatening.[87-92] Pulmonary function test results have worsened after administration of a single drop of topical timolol in patients with asthma, chronic obstructive pulmonary disease, and chronic bronchitis, and the use of this drug in these patients is contraindicated.[93] In patients with no history of pulmonary disease, the studies are not all in agreement. A study comparing placebo, timolol 0.5% solution and timolol 0.5% gel for 2 weeks on otherwise healthy glaucoma patients showed no changes in FEV1, FVC, or FEV1/FVC.[94] Another study showed that 1 year after discontinuation of ?-blocker therapy for glaucoma, otherwise healthy individuals still had increased bronchial reactivity to metacholine challenge.[95]
In an effort to further improve the therapeutic index and reduce systemic side effects, in 1993, a gel-forming (GFS) solution formulation of timolol was introduced in both 0.25% and 0.5% concentrations for once-daily usage.[96,97] The more viscous formulation increases corneal contact time and, theoretically, allows increased ocular bioavailability and decreases systemic absorption. In a multicenter, double-masked, 6-month trial comparing twice-daily timolol 0.5% solution with once-daily 0.5% timolol GFS, there was no statistically significant difference in IOP lowering effect at trough or peak, but there was a difference in the decrease in heart rate, which was less for patients on GFS. Blurred vision and tearing were reported more often in the GFS group, whereas burning/stinging was more common in the solution group.[98]
There was a need for a solution of timolol with increased bioavailability. Timolol, as a cationic drug, was found to have its lipophilicity increased in the presence of the appropriate counter ion. A new timolol maleate solution containing potassium sorbate (timolol-LA 0.5% or Istalol) was created with half of the benkalkonium chloride preservative found in timolol maleate and was approved by the FDA in 2004. This new drug was to provide the advantages of once-daily regimen such as patient convenience, improved compliance and reduction of systemic drug exposure, without causing blurred vision on instillation. A multicenter, prospective, randomized, doube-masked, parallel-group trial of 332 patients showed equivalent IOP reduction when timolol-LA 0.5% once daily was compared with timolol 0.5% twice daily for 1 year. Mean IOP reduction was ?25-28% at peak and 21-24% at trough for both formulations. The most common adverse event was burning and stinging on instillation and it was significantly more common in the T-LA (41.6%) versus the timolol group (22.9%). All cases were mild and there was no discontinuation due to this adverse event.[99]
Finally, timolol in ocudose, available since 1986, is the preservative-free formulation of timolol maleate 0.25% and 0.5% supplied in a clear low density polyethylene unit dose container. Each individual unit contains 0.2 mL of solution and is available in a foil-laminate wrapped pouch with 60 doses. It can be stored at room temperature (59-86 °F) but should be protected from light. Multiple studies have been published on the deleterious effects of topical glaucoma therapy on the ocular surface leading to conjunctival discomfort on instillation, tear film instability, conjunctival inflammation, subconjunctival fibrosis, conjunctival epithelium apoptosis, corneal surface impairment and potential risk for failure of glaucoma surgery. Pathological changes described include subclinical inflammation with significant infiltration of the conjunctival epithelium and substantia propria, and conjunctival epithelial cell expression of inflammatory markers.[100] The debate persists as to the role of the active compound versus the preservative in inducing these toxic and proinflammatory changes. Most recent studies have compared preserved with nonpreserved products and found statistically significant elevation of inflammatory markers such as: IL-1, IL-6, IL-8, and IL-10 in the ocular surface of patients using the preserved formulation. Studies looking at clinical signs show decrease in ocular hyperemia, folliculopapilar reaction and superficial punctuate keratitis when patients were switched from preserved to nonpreserved timolol.[101,102] This preparation is safe to be used up to 24 h after opening. A recent study showed that no bacteria or fungus was detected in open samples of unit-dose timolol at 0, 4, 10, 14, and 24 h after opening the vial.[103]
Levobunolol
Levobunolol is a nonselective topical ?-blocker that has been used extensively since its approval by the FDA in1996. Available in both 0.5% and 0.25% solutions, it has been extensively tested in well-designed clinical trials, and data on long-term IOP control for up to 48 months have been widely reported.[104-108]
Levobunolol is metabolized in both rabbits and humans to dihydrolevobunolol, which has a half-life of 7 h.[109,110] This compound, which also has ?-blocking activity, may prolong the duration of topically applied levobunolol compared with some of the other agents in this class. However, in multiple clinical trials comparing once-daily levobunolol treatment with once-daily timolol treatment, no statistical differences in success rate could be found with either the 0.5% or the 0.25% concentrations.[72,111] A randomized, double-masked, multicenter, crossover comparison of twice-daily 0.5% levobunolol with once-daily 0.5% timolol GFS showed comparable IOP lowering at peak and trough. In that study, the heart rate was lowered significantly more and there were more local side effects with levobunolol than with timolol GFS.[112] These important studies have brought attention to the fact that once-daily dosing for both timolol and levobunolol is a potentially important part of our therapeutic armamentarium and may offer a significant advantage in terms of both compliance and cost.
In nearly all studies reported to date, a mean decrease in heart rate of 5-10 beats per minute was noted with topical administration of levobunolol. This effect was statistically significant when compared with therapy with placebo.[106,113] One study investigating the effects with several concentrations of levobunolol in patients with glaucoma documented decreased heart rate in 15 patients taking 0.25% levobunolol.[114] None of the changes in heart rate was believed to be clinically significant but does underscore the fact that levobunolol, like timolol, enters the systemic circulation and provides low-grade systemic ?-blockade that may be deleterious in certain patients who are at risk because of cardiac or pulmonary problems. Accumulated data show that levobunolol is an extremely potent ocular hypertensive agent and is safe in patients without cardiac or pulmonary complications.
Metipranolol
Metipranolol is a nonselective ?-blocker introduced in Europe several years after timolol. It is marketed in 0.3% concentration in this country, although other countries have access to concentrations of 0.1%, 0.3%, and 0.6%. In comparisons with both timolol[115-124] and levobunolol 0.5%,[125] there seemed to be little advantage of using the 0.6%, and the 0.1% did seem to be slightly less efficacious.
Overall, twice-daily metipranolol has pressure-lowering effects comparable to those of timolol and levobunolol and no significant difference in systemic side effects.[126] Most of the ocular adverse effects are similar to those of the other nonselective ?-blockers and include burning, photophobia, blurred vision, and foreign body sensation. However, there seems to be a slight difference with the other agents with respect to transient stinging on instillation, which is slightly greater with the 0.3% metipranolol. A rare ocular side effect is uveitis,[127-129] but a causal relationship with this particular molecule has been credibly challenged.[130,131] What did seem to be a problem was more widespread granulomatous uveitis after metipranolol usage in England, which was found to be related to the sterilization process that was different than the one used in the United States.[128,129] With respect to systemic side effects, two studies have shown a reduced effect on heart rate comparing metipranolol and timolol,[132,133] and in another study, a slightly increased reduction in forced expiratory volume was noted with metipranolol in preselected pulmonary patients.[130]
Metipranolol seems to be a safe and effective option for glaucoma treatment, and although an occasional patient will notice increased stinging, this is less prevalent in the more geriatric population, and there is good documentation that competitive pricing in most communities has made a year's treatment with this drug considerably less expensive than many of the other 'branded' ?-blockers.[134]
Carteolol
Carteolol is a potent nonselective ?-blocker with partial ?-agonist activity, also called intrinsic sympathomimetic activity.[135] The molecule is 10 times more active than the prototype agent propranolol. In addition, it has an active metabolite, 8-hydroxy-carteolol, which has a half-life two to three times that of the parent molecule. This may allow increased bioavailability and duration of action.[136] The efficacy of carteolol has been shown in placebo-controlled studies[137] and direct comparisons with timolol,[138-142] in which there was no significant difference in efficacy with either concentration of carteolol.
A prospective, randomized, open, comparative study of timolol, betaxolol, and carteolol in 280 eyes was carried out for 7 years. The authors concluded that after 7 years, only 43% of those started on timolol, 34% of those started on carteolol and 29% of those started on betaxolol were still being treated with those medications alone. Visual fields did not improve or deteriorate in average over 7 years.[143]
A three-way comparison study evaluated the effect of carteolol, metipranolol, and timolol on pulmonary function, and slightly decreased forced expiratory volume was noted with the metipranolol and timolol compared with carteolol.[144,145] Although one would theoretically expect some slight margin of safety with carteolol because of its ?-agonist activity, the data documented slightly reduced forced expiratory volume with carteolol and underscores the need for caution when using any ?-blocker in patients with current or past pulmonary pathology.
Timolol has been shown to cause a reduction of high-density lipoprotein-cholesterol as well as an increase in triglycerides.[84] In a randomized, multicenter, double-masked, parallel-group study comparing twice-daily timolol 0.5% and carteolol 1% on African American postmenopausal women; after 12 weeks, there was a significant negative effect on HDL and cholesterol/HDL ratio in the timolol group and no change in the carteolol group. Somatization and depression were evaluated and there was no difference between groups.
Overall, twice-daily carteolol appears to be a well-tolerated, effective agent. It will continue to be important not only to ophthalmologists but also to primary care physicians whether chronic treatment with this drug may provide an advantage in patients with atherogenic lipid profiles.
Selective ?-Blockers
In some patients, the use of selective ?-blockers has theoretical as well as clinical advantages. These agents have more affinity for the cardiac ?1-receptors than the pulmonary ?2-receptors, decreasing the risk of pulmonary side effects. They may also have the ability to leave ocular and systemic ?2-receptors unblocked and more responsive to endogenous and exogenous epinephrine.
Betaxolol has been extensively tested and found to have potent long-term efficacy in placebo-controlled studies[146-148] as well as in masked comparisons with timolol[149-151] and levobunolol.[152] As with the other ?-blockers, fluorophotometric studies have shown the mechanism of action of betaxolol to be reduction in the secretion of aqueous humor.[153]
In comparison studies with timolol, average IOP reductions with betaxolol were similar and did not display any statistically significant difference.[150,151] However, using a quartile analysis necessitated by the number of patients requiring adjunctive therapy, one study[149] found a high statistical significance in the 1-2 mmHg of difference in the pressure-lowering effects of the two drugs, with betaxolol exhibiting slightly less magnitude of effect. Another study reported a significant number of patients with elevated IOP after a switch from timolol to betaxolol.[154] Compared with levobunolol, betaxolol produced slightly (2-3 mmHg) less reduction in IOP.[152] This study design used morning IOP comparisons (before application of the morning dose of medication) that may have favored the levobunolol because of its apparent longer duration of action after the previous evening dose.
Betaxolol is available as a 0.5% generic solution and a more comfortable formulation using 0.25% racemic drug in a suspension (Betoptic-S) Several additional studies have shown that acetazolamide,[155]pilocarpine,[150] and epinephrine[156,157] have a useful additive effect. The additive effect when epinephrine was administered at the same time as betaxolol was somewhat surprising, because epinephrine and dipivefrin provided only a small additional effect when added to timolol in several studies investigating this combination during different time courses ranging from 3 h to 3 months.[158-160]
The effect of adding epinephrine to betaxolol was quite significant and accompanied by an increased facility of outflow.[156] Betaxolol has been shown in vitro to produce ?10 times greater ?1-blockade than ?2-blockade.[161] The increased facility, also found in primates,[162] is most likely due to epinephrine's stimulation of ?2-receptors (not significantly blocked by betaxolol), which have been shown by several laboratory techniques to be present in human trabecular meshwork.[163,164]
The main feature that distinguishes betaxolol and betaxolol suspension from most topical ?-blockers in the nonselective category is their lower risk of systemic side effects, a finding that has now been documented in 7 years of approved use. As with systemic treatment, even a relatively selective ?1-blocker has a potential for adverse effects in patients with severe pulmonary disease because the ?-blockers are not totally devoid of some ?2-blocking activity, and a few reports describe nonfatal pulmonary complications with betaxolol.[165,166] However, because of the extremely worrisome incidence of pulmonary complications noted with ophthalmic timolol and anticipated with other nonselective topical ?-blockers, an agent that may be less prone to affect pulmonary function would be advantageous in the many cases of geriatric-skewed glaucoma.
One percent betaxolol had no effect on exercise tachycardia when administered in masked comparison.[167] Many explanations have been proposed to explain this lack of systemic effect by ophthalmic betaxolol. The drug may not be well absorbed into the circulation, may be highly protein bound, may be quickly or effectively metabolized, or may be kinetically limited by lower receptor affinity in nonocular tissue. It should be emphasized that betaxolol is used as a racemic mixture and that only the L-isomer is active. Thus, if 2 ng of both 0.5% ophthalmic betaxolol solution and 0.5% ophthalmic timolol solution entered the serum, all of the timolol would be active as the L-isomer, but the effective dose of betaxolol would only be 1 ng. Betoptic S presents an even safer alternative because the concentration of the active L-isomer is only 0.125% solution.
Studies comparing the effects of betaxolol and timolol on visual fields have shown an improvement of visual-field parameters with betaxolol, while the IOP reduction with timolol is usually greater.[168-170] This has raised the question of betaxolol as a neuroprotective agent. It is believed that retinal ganglion cells and their axons may die in glaucoma through apoptosis, where glutamate is released and initiates the death of neurons that possess NMDA receptors. The major cause of cell death after activation of NMDA receptors is the influx of calcium into cells and generation of free radicals. Betaxolol has calcium and sodium channel blocking properties and has been shown in laboratory studies in rats to attenuate ischemic injury to ganglion cells by mechanisms that do not involve ?-receptors. Studies with ocular blood flow and retinal nerve fiber layer measurements comparing betaxolol to other agents have not all been in agreement, but some show improvement in anatomical parameters after topical instillation.[171-177]Finally, a recent study looking at enucleated eyes treated for 28 days with betaxolol evidenced high concentrations of the medication in the retina, optic nerve, choroid, ciliary body and iris, all higher than in the plasma.[178]
Based on current evidence, betaxolol seems to be an effective agent for the treatment of glaucoma and has low potential for ocular and systemic side effects.
|
Key Features: Beta-Blockers |
||||||||||||||||||||||||
|
CARBONIC ANHYDRASE INHIBITORS
The evolution of carbonic anhydrase inhibitors used for glaucoma treatment provides a unique and interesting model of pharmaceutical progress over more than five decades (see Table 217.6). The discovery of an excess of bicarbonate in the aqueous humor along with carbonic anhydrase activity in the ciliary body of the rabbit in the 1950s led to the introduction of the inhibitor acetazolamide for the treatment of glaucoma in 1954.[177] A minor modification in the original acetazolamide molecule led to the development of methazolamide, with clear pharmacologic and clinical advantages mainly related to better gastric absorption, less serum protein binding, and longer duration of action. Systemic side effects still prompted uneven compliance, and the search for a topical carbonic anhydrase inhibitor led to two efficacious topical agents that are available to glaucoma patients, dorzolamide and brinzolamide.
TABLE 217.6 -- Carbonic Anhydrase Inhibitors
|
Generic Name |
Trade Name |
Concentration |
Route |
Dosage |
|
Acetazolamide |
Diamox, generic |
125-mg, 250-mg tabs |
PO |
qid |
|
Diamox sequels |
500-mg caps |
PO |
bid |
|
|
Methazolamide |
Neptazane, generic |
25, 50, 100 mg |
PO |
bid, tid |
|
Dichlorphenamide |
Daranide |
50 mg |
PO |
bid, tid |
|
Dorzolamide HCl |
Trusopt |
2.0% |
Topical |
bid, tid |
|
Brinzolamide |
Azopt |
1% |
Topical |
bid, tid |
There are at least seven different isoenzymes of the carbonic anhydrase (CA) enzyme. The isoenzymes relevant to the human eye appear to be the cytosolic CA-I and CA-II and the membrane bound CA-IV. The distribution on the enzymes is not uniform within the eye. CA-II is probably the most important in relation to aqueous flow, as it is the main isoenzyme found in the human ciliary processes. It has been shown that CA must be essentially 100% inhitibited at the ciliary body to lower IOP. The role of CA-IV has not been elucidated as specific blockers are not available. Acetazolamide is a potent inhibitor of all three CA isoenzymes. Dorzolamide is comparatively more potent against CA-II and also the strongest inhibitor of CA-IV but a very weak inhibitor of CA-I.[179]
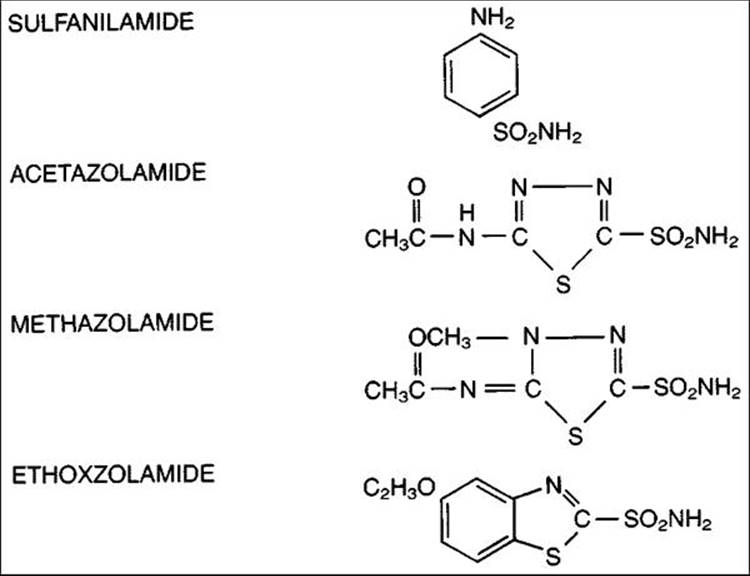
Oral Agents
Three oral carbonic anhydrase inhibitors are available (Fig. 217.4 and Table 217.7), all of which are members of the sulfonamide family. In therapeutic doses, they are able to reduce production of aqueous humor by a maximum of 50%, with a corresponding decrease in IOP. The pressure reduction is caused by a reduction in the accumulation of bicarbonate in the posterior chamber, with a decrease in sodium and associated fluid movement linked to the bicarbonate ion.[180] With high doses of acetazolamide, it appears that an additional decrease in IOP may be caused by relative metabolic acidosis. However, the two effects of pressure-lowering, shifts in bicarbonate ion and changes related to acidosis, appear to be independent of one another.[181]
|
|
|
|
FIGURE 217.4 Chemical structure of carbonic anhydrase inhibitors. |
TABLE 217.7 -- Pharmacologic Properties of Carbonic Anhydrase Inhibitors
|
Partition Coefficient to Buffer pH 7.4 |
|||||||||
|
Generic Name |
Ka1 × 109 |
pKa1 |
Ether |
Human CHCL3 |
Solubility in H2O (mM)[*] |
%[?] Bound to Plasma |
f½[?] Plasma (hr) |
RBC[§] |
Aqueous Humor (M) |
|
Sulfanilamide |
1000 |
10 |
0.15 |
0.02 |
9 |
10 |
6 |
136 |
- |
|
Acetazolamide |
6 |
7.4 |
0.14 |
10?3 |
3 |
95 |
4 |
27 |
2 |
|
Methazolamide |
8 |
7.2 |
0.62 |
0.06 |
5 |
55 |
15 |
195 |
8 |
|
Ethoxzolamide |
1 |
8.1 |
140 |
25 |
0.04 |
96 |
6 |
4500 |
330 |
|
Benzolamide |
1 |
3.2 |
0.001 |
10?4 |
0.14 |
96 |
2 |
23 |
1 |
From Maren TH: In: Case RM, Lingard JM, Young J, eds. Secretion: mechanisms and control. Manchester, UK: Manchester University Press; 1984.
|
* |
Against pure carbonic anhydrase C, in hydration. |
|
? |
At concentrations of 4-40 ?M. |
|
? |
After oral dose in human. |
|
§ |
From free concentration in plasma to red blood cells (human). |
Although a 50-mg PO dose of the carbonic anhydrase inhibitor methazolamide produces a slightly smaller reduction in IOP than does a 250-mg oral dose of acetazolamide, the pharmacology of the former compound has several advantages.[182-185] The slight difference in the drugs' IOP-lowering effects at these doses is probably due to the metabolic acidosis caused by acetazolamide, which can be deleterious in many clinical situations. Methazolamide has a more favorable partition coefficient, which allows enhanced systemic absorption and easier access into ocular tissues. In addition, methazolamide is only 55% bound to plasma protein, whereas acetazolamide is 95% bound. In practical terms, this means that a far smaller quantity of oral methazolamide is needed to produce therapeutic levels in target tissue (presumably the ciliary processes) as compared with acetazolamide. Because of this difference in dose, the renal effects of carbonic anhydrase inhibition can be avoided with administration of methazolamide at doses of less than 2 mg kg?1 day?1.
Another advantage is methazolamide's serum half-life of 15 h, compared with the 4-h half-life of acetazolamide. It is therefore unnecessary to give methazolamide more often than every 12 h; this twice-daily dosage schedule is much more convenient than that required for acetazolamide tablets. Methazolamide also undergoes predominantly hepatic rather than renal metabolism, so that dosages do not have to be adjusted in the large number of patients with renal disease.
Many well-known ocular and systemic side effects occur with administration of all the carbonic anhydrase inhibitors. These include numbness, paresthesias, malaise, anorexia, nausea, flatulence, diarrhea, depression, decreased libido, poor tolerance of carbonated beverages, myopia, hirsutism, increased serum urate, and rarely, thrombocytopenia and idiosyncratic aplastic anemia. Some investigators believe that the malaise-anorexia-depression syndrome may be related to concomitant acidosis and have found some success in reducing the incidence of these complaints with the co-administration of sodium bicarbonate.[186] Patient groups in whom metabolic acidosis related to carbonic anhydrase inhibitor therapy may be a serious risk include (1) diabetic patients susceptible to ketoacidosis, (2) patients who have hepatic insufficiency and cannot tolerate the obligatory increase in serum ammonia, and (3) patients with chronic obstructive pulmonary disease, in whom increased retention of carbon dioxide can cause potentially fatal narcosis.[187-189]
An early, mild hypokalemia usually follows the institution of most carbonic anhydrase inhibitors but does not progress unless patients are taking diuretics concomitantly. The exception is the drug dichlorphenamide, which has a unique chloruretic effect that may cause chronic and potentially dangerous loss of potassium. A deformity of the forelimb has been seen in the offspring of animals given acetazolamide, and the drug should definitely be avoided by women of child-bearing age.[190]
Urolithiasis is believed to be much more common in patients taking carbonic anhydrase inhibitors, most likely because of the depressed excretion of renal citrate and the higher urine levels of calcium available to form urate stones. In a case study with controls, the incidence of renal stones was 15 times higher after treatment with acetazolamide than before its administration.[191] The incidence was 11 times higher than in the age-matched control group. The incidence of stones in this study did not seem to increase after 15 months, suggesting that susceptible persons ordinarily experience this side effect during the first or second year of treatment, if at all. Although methazolamide has been linked to the formation of kidney stones in several patients on high doses (>200 mg/day),[192] the lack of a significant renal effect with low-dose therapy seems to suggest a potentially lower risk of urolithiasis with regimens such as 50 mg bid.
If oral agents are to be used, a starting dose of 25-50 mg of methazolamide is very easily tolerated by many patients but would rarely be a consideration unless it is deemed easier for the patient to take oral rather than topical therapy with the newer agents in this class. The use of a maximal dose of 150 mg of methazolamide bid or 250 mg of acetazolamide qid may be less well tolerated, but sustained-release capsules of 500 mg of acetazolamide used twice daily may improve compliance and have been reported to give an unexplained advantage in IOP reduction.[193] It is advisable to administer both methazolamide and acetazolamide after meals to decrease gastrointestinal side effects.
Because blood dyscrasias have been reported after the use of both agents,[194] there has been considerable debate about whether surveillance of blood counts is justified. Despite the poor outcome in patients who develop idiosyncratic aplastic anemia,[195,196] some patients also develop isolated neutropenia, thrombocytopenia, and pancytopenia but have an uneventful recovery if the condition is discovered and the drug withdrawn in time.[197] Because such reactions are rare, with an incidence of around 1:14 000, it would not seem justified to continue obtaining blood counts during the entire course of therapy. It is reasonable and relatively inexpensive to obtain a pretreatment complete blood count and one to two follow-up studies during the first 6 months of treatment, when most of the serious hematologic events were noted to occur. Although some ophthalmologists believe that oral therapy with carbonic anhydrase inhibitors should be abandoned, there may still be a place for their use in some patients who show a documented efficacy advantage or who have difficulty having topical carbonic anhydrase inhibitor eyedrops applied.
Topical Agents
One year after the introduction of oral acetazolamide as an effective ocular hypotensive agent, an unsuccessful attempt to solubilize it for topical treatment was published.[198] The effort to develop a topical agent was revisited in the late 1970s, resulting in the introduction of several prototype molecules that preceded the approval of dorzolamide in 1995 and brinzolamide in 1998. The availability of these agents has dramatically reduced the justification for long-term use of oral carbonic anhydrase inhibitors and has greatly reduced the side effects associated with the oral agents. Most recently, the synthesis of a key intermediate-carboxydifluoromethanesulfonamide has reopened the possibility of the synthesis of topical acetazolamide. This discovery may lead to the synthesis of a wide range of novel difluoromethanesulfonamides with water solubility and stability, and submicromolar dissociation constants for human CA isozyme II that make them promising candidates for glaucoma topical therapy.
Dorzolamide
Dorzolamide is 10 times more effective than acetazolamide at inhibiting carbonic anhydrase isoenzyme-II, which is the predominant form in both nonpigmented and pigmented ciliary process epithelium. It was twice as effective as acetazolamide in inhibiting isoenzyme-II in an in vitro lung preparation.[199] At the 2% concentration, it is very effective in lowering IOP in both primates and humans. Aqueous humor dynamics in glaucomatous monkeys showed a 38% reduction in aqueous secretion with no change in outflow facility after single-drop therapy.[200]
Dorzolamide administered three times daily was compared with twice-daily timolol and twice-daily betaxolol over 12 months in a large, multicenter prospective masked trial.[201] At peak effect (2 h), the sustained pressure-lowering effect of dorzolamide was 1-2 mmHg less than timolol solution, but ?1 mm better than betaxolol 0.5%.
Dorzolamide was studied as an adjunctive agent to timolol. At peak, there was an additional 4-mmHg pressure drop, which decreased to 3.5 mmHg at 8 h.[202] Another study examining different alternatives for adjunctive therapy found comparable efficacy between dorzolamide and pilocarpine 2% when added to timolol. Patients tolerated dorzolamide much better and had fewer complaints of decreased vision and induced myopia.[203] A 1-year prospective clinical trial compared the addition of thrice-daily dorzolamide to patients with poor control of IOP who were on brimonidine 0.2% or timolol 0.5%. A significant reduction of IOP from baseline was observed, with a significant difference of IOP lowering in favor of the timolol-dorzolamide combination (5.6 ± 1.9 vs 6.8 ± 1.7).[204] Twice-daily dorzolamide was compared to latanoprost when both were added to twice-daily timolol 0.5% on a prospective, randomized fashion, for 3 months. The mean IOP reduction from baseline (timolol alone run-in period) was 32% for the latanoprost plus timolol group and 20% for the dorzolamide plus timolol group. Tolerability was similar in both groups.[205] Finally, dorzolamide was compared with brimonidine-purite, when each was added twice daily to latanoprost in a double-masked, prospective, crossover comparison. After 6 weeks of therapy, 8 AM IOP and mean diurnal IOP was equivalent for both groups.
Considerable attention has been paid to whether three-times-daily application of dorzolamide is significantly better than twice-daily application. Because of the 8-10 h duration of dorzolamide, it does seem that there is a small but definite increase in efficacy with three-times-daily monotherapy compared with twice daily. A prospective, three-armed clinical trial showed lack of a statistically significant difference in three-times-daily versus twice-daily dorzolamide treatment, but three-times-daily treatment gave ?1 mmHg better IOP lowering at 8-12 h.[206] Many ophthalmologists use dorzolamide twice daily as both monotherapy and adjunctive therapy, and it is probably a rare patient that benefits from monotherapy administered three times daily.
Dorzolamide's ocular effects seem to be principally confined to a 33% incidence of stinging on instillation and a 10-15% incidence of punctate keratitis. Blurred vision, tearing, dryness, and photophobia were all less than 5%. Some of the stinging on instillation is most likely related to the low pH (5.8) required to keep the relatively insoluble compound in solution. The one consistent systemic side effect that does occur frequently is bitter taste after administration that ?25% of the patients notice, which can be greatly reduced by punctual occlusion.
After chronic dorzolamide treatment, analysis of both serum and urine chemistries revealed no changes in a group of healthy volunteers.[207] There was a decrease in red blood cell carbonic anhydrase enzyme-II activity substantiating some systemic absorption. An initial concern during preapproval trials was a small increase in corneal thickness in a dorzolamide-treated group. However, an extensive three-arm, masked, postapproval, phase IV study using endothelial videokeratography failed to find any increased corneal thickness or significant change in endothelial morphology. Most reports of corneal decompensation have been in patients who had corneal transplants, and those patients should be monitored.[208] There have been some patients who developed urolithiasis during dorzolamide treatment, but the prevalence is low enough to suggest an unclear relationship to the topical medication. In addition, there have not been reports of Stevens-Johnson syndrome or blood dyscrasias after dorzolamide usage, but owing to the observed systemic absorption of this drug, continued clinical surveillance is appropriate. A recent study showed dorzolamide to be a safe alternative for treatment in patients younger than 6 years of age with glaucoma or ocular hypertension. Reduction of IOP was in the 20% range.[209]
Most clinicians have had a positive experience with both the tolerability and the efficacy of dorzolamide, but questions continue to linger regarding equivalence of topical and oral compounds. Fluorophotometric investigation showed a 17% reduction in aqueous flow after dorzolamide application to glaucomatous and normal volunteers compared with a 30% reduction after acetazolamide.[210,211]Nevertheless, most of the chronic dosing trials show equivalence in observed IOP lowering. A 12-week study on 31 patients showed good maintenance of pressure reduction when topical dorzolamide was substituted for oral acetazolamide.[212] A larger prospective study has also shown that the oral and topical forms are essentially interchangeable when used as adjunctive therapy.[213] In a recent clinical trial there was a lack of additivity from adding acetazolamide to dorzolamide and vice versa. The three-arm design allowed the authors to conclude that the group using dorzolamide alone was comparable to the group using acetazolamide alone in terms of IOP reduction.[214] However, two studies in pediatric populations have shown an additional IOP lowering when acetazolamide was added to dorzolamide in high pediatric doses.[215,216] Most studies addressing this issue postulate that the difference between acetazolamide and dorzolamide lies either in the systemic acidosis caused by acetazolamide on long- or intermediate-term therapy, or in a difference in CA inhibition profile. The matter remains unresolved, and most clinicians will still resort to acetazolamide in the acute setting to treat very high IOPs, while preferring dorzolamide or brinzolamdie for long term therapy due to the better safety profile.[210,211,214,217] The potential for increased side effects along with no increase in efficacy has prompted the statement in the dorzolamide package insert that dorzolamide and systemic CAIs should not be used together.
Perhaps owing to early impressions that oral carbonic anhydrase inhibitors may have an advantageous effect on ocular blood flow, considerable attention has been paid to the effect of dorzolamide on blood flow as measured by several contemporary methodologies.[218] Both optic nerve head blood flow in animals as measured by a laser Doppler flowmeter and arteriovenous passage time measured with a scanning laser ophthalmoscope seemed to be improved after topical dorzolamide.[172,219-221] The next steps include demonstrating that dorzolamide indeed reaches therapeutic concentrations at the optic nerve head and finally, that glaucoma patients do indeed benefit from improved ocular blood flow.
Brinzolamide
Brinzolamide was approved in 1998 for three-times-daily topical application dosage in a 1% concentration. Using a formulation similar to that previously employed with betaxolol, brinzolamide is a suspension that allows buffering to a more neutral pH than that of dorzolamide. Multicenter studies have compared both twice-daily brinzolamide and three-times-daily brinzolamide 1% to timolol 0.5%. These results show efficacy similar to dorzolamide but with IOP lowering slightly less than that of timolol usage with either dosing regimen of brinzolamide. Differences between twice-daily and three-times-daily drug usage were less than 1.0 mmHg.[222-225] A prospective, randomized, open-label study substituted brinzolamide for dorzolamide in half of a group of 58 patients who were being treated with dorzolamide, latanoprost and timolol. The other half remained as control. There were no significant differences in IOP between the groups, but ocular irritation significantly decreased from 63% to 20%, while blurred vision increased from 27% to 37%.[223]
Dorzolamide 2% twice daily and brinzolamide 1% twice daily were compared in a double-blind, randomized, parallel group study when added to timolol 0.5% twice daily. In this study of 241 patients and 3 months duration, both treatments were equivalent in IOP reduction at all points and in mean IOP lowering. Brinzolamide produced significantly less ocular burning and stinging.[226] When added to patients who were on stable therapy with latanoprost for 6 months, the mean IOP was lowered from 21.1 ± 4.8 to 15.9 ± 3.1 mmHg at 3 months.[227] Interestingly, a recent study comparing the use of brinzolamide 1% or timolol 0.5% twice daily on patients with baseline IOP between 20 and 30 mmHg, showed a mean decrease in IOP at 6 weeks of 17% for the brinzolamide group and 19.7% for the timolol group, which did not achieve statistical significance.[225]
Topical and systemic side effects are usually mild, with a 2.7% incidence of keratitis and a 0.7% incidence of corneal edema.[228] Plasma levels are detectable in red blood cells at 5 months.[229] The most striking difference between brinzolamide and dorzolamide seems to be tolerability. Ocular hyperemia and tearing are usually less with brinzolamide, but foreign body sensation, pain, and blurred vision are significantly greater in the brinzolamide patients. Overall, brinzolamide 1% seems to be a safe and effective option with a slightly different tolerability profile compared with dorzolamide for the treatment of glaucoma.
|
Key Features: Carbonic Anhydrase Inhibitors |
|||||||||||||||||||||
|
MIOTICS
With the advent in the 1990s of alternative classes of compounds for the lowering of intraocular pressure (IOP), the use of miotics agents has declined dramatically (see Table 217.8). However, miotics were the first class of agents used to lower intraocular pressure. The initial use of physostigmine for glaucoma by Laqueur in 1876 represented the start of IOP-lowering therapy, and the introduction of topical pilocarpine 1 year later provided a benchmark for safe and effective treatment during the next 100 years. As a parasympathomimetic, pilocarpine mimics acetylcholine by its direct stimulation of the iris sphincter and ciliary muscle, both of which are cholinergically innervated. Because both have anatomic connections with the scleral spur, their stimulation and concomitant contraction would logically reduce resistance to aqueous humor outflow, as shown in primates.[230,231]
TABLE 217.8 -- Miotics
|
Generic Name |
Mechanism |
Trade Name |
Concentration |
Dosage |
|
Pilocarpine |
Direct |
Isopto Carpine |
0.25, 0.5, 1.0, 2.0, |
qid, tid |
|
Pilagan, generic |
3.0, 4.0, 6.0, 8.0, 10.0 |
|||
|
Pilocarpine gel |
Direct |
Pilopine HS |
4% |
qhs |
|
Pilocarpine insert |
Direct |
Ocusert |
20, 40 ?m/hr |
q7 days |
|
Carbachol |
Direct |
Isopto Carbachol |
0.75, 1.5, 2.25, 3.0 |
tid, bid |
|
Echothiophate |
Indirect |
Phospholine Iodide |
0.03, 0.06, 0.125, 0.25 |
qd, bid |
|
Demecarium |
Indirect |
Humorsol |
0.125, 0.25 |
qd, bid |
Direct Acting: Pilocarpine and Carbachol Drops
Pilocarpine penetrates ocular tissues well. Although miosis occurs in 15-30 min, maximal reduction of IOP occurs in 2-4 h, with a total duration of 4-8 h. We conventionally begin treatment with a low-dose solution such as 0.5% or 1% pilocarpine and titrate upward to achieve the maximal response. Concentrations greater than 4% are rarely necessary. Pilocarpine drops are prescribed every 4-8 h, and most cases are well controlled on a four-times-daily schedule.
The ideal patients treated with pilocarpine are those who are older than 40 years and who do not have significant cataracts because decreased vision is usually worsened by miosis. In addition to miosis and spasm accommodation with associated myopia, other problems include headache, browache, conjunctival hyperemia, lacrimation, local allergy, twitching of the eyelids, and occasionally, increase in inflammation, posterior synechiae, retinal detachment, and exacerbation of pupillary block in predisposed eyes. Myopia is particularly distressful in young patients, and attempts to change optical correction are usually thwarted by the cyclical changes in accommodative magnitude.
One should not overlook the potential problem of systemic side effects resulting from administration of short-acting miotics, especially in visually handicapped patients, who can occasionally overdose themselves. Observed effects include salivation, diaphoresis, nausea, vomiting, abdominal cramping, incontinence, diarrhea, hypotension, hypertension, bradycardia, bronchospasm, and muscle weakness. Although the IOP-lowering effect of a miotic is often favorably added to the effects of a topical ?-blocker, the additive systemic effect on the cardiopulmonary system can be potentially dangerous.
Four-times-daily uniocular administration of 0.5% pilocarpine in patients with light irides or 1% pilocarpine in patients with dark irides is reasonable initiation to trial medication. Punctal occlusion helps avoid systemic problems,[232] and dilatation of the eye twice a year may help prevent formation of synechiae and loss of dilator muscle tone. Carbachol, another short-acting miotic with a slightly longer duration of action, may additionally enhance cholinergic stimulation by indirectly inhibiting cholinesterase. Carbachol may be used to replace pilocarpine and very often has an enhanced effect when used in dosages of 0.75-3% solution given three times daily. As expected, symptoms of headache, hyperemia, and accommodative spasm may escalate in proportion to the degree of improvement in controlling IOP.
Pilocarpine Gel
Compliance/adherence is generally thought to be poor with four-times-daily dosing. In fact, one-fourth of the doses of pilocarpine prescribed were missed in a study of a general glaucoma population.[45] A preparation that may help combat poor compliance is pilocarpine gel, a high-viscosity acrylamide gel with pilocarpine suspended in it. Patients are instructed to administer the gel before bedtime so that some of the side effects are avoided during sleeping hours. Most patients enjoy stable control of IOP until their next nightly dose. The effectiveness of the drug has been documented by all published studies, and the average decline in IOP is comparable to that in patients who were previously treated with pilocarpine drops. This applies to morning IOP measurements as well as to the important afternoon measurement, when the gel's effect may have 'escaped' slightly.[233-235] It is important to recognize that the IOP of some patients will not be controlled for an entire 24-h period.[232] The gel efficacy can be enhanced by administering an extra drop of pilocarpine in patients whose response consistently lessens after 18-24 h; this regimen would still be easier to comply with than prior schedules of drops given four times daily.
The gel seems to cause only minor side effects and symptoms. About 50% of patients in a long-term trial noted minor irritation, but only 11% of patients asked for treatment to be discontinued because of this irritation. Sixty-nine percent of the patients had mild blurring of vision, principally in the morning; blurring diminished throughout the day, and only 9% of those patients with blurring asked that treatment be discontinued because of it. Many patients complained that their eyelids stuck together in the morning, but this did not seem to be a serious problem. Overall, the gel seems to be well tolerated.[233-235]
Corneal signs were noted in 20-40% of the patients in the study just mentioned.[236] Signs included the early development of punctate keratitis, which appeared to resolve with the continued use of the gel, and the late occurrence of subtle, diffuse focal subepithelial corneal opacities. The corneal deposits appeared after 4-12 months of use of the gel and were still noted after its discontinuation but were not correlated with any changes in vision.[237] A report describing similar focal subepithelial opacities after the use of pilocarpine drops has drawn attention to this new finding, which in the past may have been missed in patients on miotic therapy.[236]
Patients who have used pilocarpine gel found once-daily administration quite convenient and strongly preferred it for this reason. Although some adverse symptoms were generated by use of the gel, miosis and decreased vision were experienced less than with the prior use of pilocarpine eyedrops. Pilocarpine gel may be particularly useful for patients with pigment dispersion because the milder and more consistent miosis with the gel helps reduce the chafing of the posterior iris against the lens zonules while minimizing the variable effects on vision associated with pilocarpine drops.
Indirect-Acting Miotics
Indirect-acting miotics such as echothiophate are used principally because their magnitude of IOP reduction exceeds that of pilocarpine. The other advantage is the extended duration of action of all these compounds, which can be as long as 7-21 days with echothiophate. Although this long duration of action has been well known since indirect-acting miotics were introduced for the treatment of glaucoma in 1946, a twice-daily regimen has ordinarily been recommended. We have found that once-daily application yields IOP reductions that are indistinguishable from those obtained with twice-daily application and believe that the reduction in the frequency and amount of drug applied will reduce toxicity such as cataract formation, which is noted in chronic treatment with twice-daily echothiophate.
Other side effects of the indirect-acting miotics are essentially identical to those of shorter-acting cholinergic agents but are often more intense. Retinal detachments are rare with any of the cholinergic agents, but in theory the likelihood of detachment is higher after the use of an indirect-acting miotic.[238]
When used in low doses with once-daily application, echothiophate and other indirect-acting miotics may still be useful and safer than filtration surgery in some patients. In those with aphakic open-angle glaucoma, indirect-acting miotics provide an effective and convenient means of therapy when pilocarpine seems to give inadequate control. It is important to remember that strong miotics should be discontinued for several weeks before intraocular surgery to prevent the possibility of severe fibrinous postoperative iritis and to minimize the inhibition of systemic pseudocholinesterases that reverse anesthetic drugs such as succinylcholine.
|
Key Features: Miotics |
||||||||||||
|
PROSTAGLANDIN ANALOGS
The prostaglandins formally belong to a pharmacologic family called the eicosanoids, which are all formed principally from the polyunsaturated fatty acid arachidonic acid. This group also includes leukotrienes, prostacyclin, and thromboxane A2. The eicosanoids, and specifically prostaglandins, have recently been discovered to be present in almost every body tissue and fluid. Since they act locally, they are distinct from true hormones, which reach their site of action via the blood stream, but there will still be an occasional reference to the prostaglandins and other eicosanoids as 'local hormones'.[239]
The presence of ocular prostaglandins was verified in the 1950s, but it took almost 10 years from the initial animal work testing the effect of natural-occurring prostaglandins on IOP until an analog of prostaglandin F2? was successfully utilized in placebo-controlled trials in glaucoma patients.[240] Like the evolution of topical carbonic anhydrase inhibitors discussed previously, the development of latanoprost (trade name Xalatan) was responsible for many precursor drugs including sophisticated synthetic derivatives of prostaglandin F2?-isopropyl ester epimers found to be active when used in concentrations as low as 0.005%. Latanoprost is one of these epimers that functions as a true 'prodrug' requiring esterase activity to cleave the isopropyl group. Lowering of IOP is caused by an increase in uveoscleral outflow,[241] although some evidence suggests that trabecular outflow may be enhanced as well.[242] The mechanism of action is unclear but the upregulation of matrix turnover within the ciliary body, and perhaps the trabecular meshwork, has be suggested.[243] Relaxation of the 'elastic tissue elements' in the interstitial spaces of the ciliary muscle has also been suggested.[244]
Latanoprost
Before its approval by the FDA in 1996, latanoprost was one of the most thoroughly studied ophthalmic drugs in the history of glaucoma pharmaceutical development. It is a prostaglandin F2? analog. Multicenter timolol-controlled masked trials from four different countries have been reported, and one of the sites has provided 5 years of follow-up data. Whereas results of the IOP-lowering efficacy of timolol varied rather widely, there was consistent efficacy of latanoprost in each of four culturally distinct study populations (Fig. 217.5).[245-249]
|
|
|
|
FIGURE 217.5 Comparison of the effects on IOP (measured at 8-9 AM) of latanoprost (0.005%) given once daily (solid circles) and timolol (0.5%) given twice daily (open circles) for 6 months to patients with elevated IOP. Each value is the mean IOP (±SEM) of 84 to 149 patients. (a) Clinical trials conducted in the United States.[7] Latanoprost significantly (P <0.001) reduced IOP more than did timolol at all time points after pretreatment baseline. (b) Clinical trials conducted in the United Kingdom.[21] IOPs were significantly reduced with both latanoprost and timolol. IOPs were significantly lower in the latanoprost-treated eyes than in the timolol-treated eyes at 3 and 4.5 months. Timolol was as effective as latanoprost at the other time points. (c and d) Clinical trials conducted in Scandinavia.[4] (c) Latanoprost was administered in the morning for 3 months and then treatment was switched to the evening. (d) The reverse latanoprost regimen was given. When latanoprost was given in the morning, timolol and latanoprost were equally effective in reducing IOP. When latanoprost was given in the evening, IOPs were significantly lower than when timolol was given twice daily. Asterisks indicate significant (P < 0.05) differences in IOP between timolol and latanoprost treatment. |
The average drop in IOP after latanoprost usage in the 6-month trials (United Kingdom, United States, and Scandinavia) ranged from 27% to 34%. In Japan, a 3-month study was done, again revealing an average 27% effect of latanoprost. Some differences in the protocols are evident in the published results, but one significant observation was noted in Scandinavia where nighttime administration of latanoprost provided lower average IOPs than when it was administered in the morning. From these data, it can be concluded that latanoprost is an extremely potent ocular medication that consistently lowers IOP from 27% to 34%.
After 6 months' treatment in the masked study, most of the patients in the United Kingdom, Scandinavia, and United States studies were offered open-label continuation. When those data were pooled, the 33% drop in IOP with latanoprost was maintained. Even more compelling are the 2-year data from the United Kingdom[249] and 5-year data from Scandinavia,[250] which both showed minimal drift in IOP control over time. Latanoprost also appears to be effective as an adjunctive agent with most approved glaucoma agents, except for an uneven response when added to miotics. Excellent nocturnal and overall diurnal IOP control with latanoprost has been demonstrated.[69,251,252]
Side effects in all of the published latanoprost data seem to be consistently confined to the eyes and adnexa, although systemic effects were vigorously looked for. Specific ocular signs and symptoms included punctate keratopathy, foreign body sensation, blurred vision, eye pain, conjunctival hyperemia, and iris color changes. There are very few patients, when given unilateral latanoprost, which do not have some evidence of slight conjunctival injection by slit-lamp biomicroscopy when compared with the nontreated fellow eye. However, this is seldom a clinical problem, and fewer patients overall complained of eye irritation (including burning, stinging, itching, and tearing) than those using timolol. Changes in iris color after 2 years follow-up consisted of 51 patients out of a starting group of 277 (19%).[249] A mechanism for increased iris pigmentation has not been confirmed, but there does seem to be agreement that there is no increase in the number of iris melanocytes, only an increase in melanocytic activity resulting in increased pigment granules. Another observation was that the changes were restricted to patients with a light gray-green or blue color in their periphery and midperipheral iris. Homogeneously blue or brown eyes did not seem to be affected to any degree that could be clinically measured.
Another investigation[253] did confirm that latanoprost also caused hypertrichosis and increased both lash length and pigmentation. Reports of cystoid macular edema associated with use of latanoprost initially caused some concern, but there may not be a clinical association in some of the cases and all of the reported cases to date occurred in aphakic and pseudophakic patients.[254-256] It is still not clear whether all of the cases are reversible, such as with cases caused by epinephrine, but in eyes with uveitis, postoperative inflammation, or aphakic and pseudophakic eyes with an extra risk for development of cystoid macular edema, latanoprost should probably be used with great caution or not at all.
The use of latanoprost increased dramatically during the first 5 years after its introduction and it is now the most widely prescribed medication for glaucoma in the USA and worldwide. It is also the first hypertensive lipid (prostaglandin analogs and prostamides) to achieve FDA approval as first-line therapy for glaucoma or ocular hypertension.
Travoprost
Travoprost is structurally and biochemically similar to latanoprost. Like latanoprost, it is an isopropyl ester prodrug that is hydrolyzed by corneal esterases to become a biologically active prostaglandin F2?analog. Travoprost differs from latanoprost in that it contains a double bond at the carbon 13-14 position and a CF3 side group on the carbon ring. It exhibits very high affinity to and selectivity for the FP receptor. Clinical studies show comparable efficacy with latanoprost with a similar side effect profile, although travoprost is associated with a slightly higher incidence of hyperemia.[257,258] It is widely used as both first-line and adjunctive therapy for glaucoma and ocular hypertension.
Like latanoprost and bimatoprost, travoprost is dosed daily before bedtime. However, one study indicates that travoprost, as well as latanoprost, has a hypotensive effect that lasts for several days.[259] It has been shown to lower intraocular pressure significantly better than timolol 0.5% twice daily, especially in African-Americans.[257]
Travoprost has recently become available without benzalkonium chloride (BAK). Instead, the preservative consists of selected concentrations of commonly used ions and buffer systems used in artificial tears, such as boric acid, propylene glycol, sorbitol, and zinc chloride. Upon contact with positively charged ions in the tear film, the ionic buffered preservative system becomes inactive. The purpose of using a different preservative is to reduce the ocular surface toxicity associated with benzalkonium chloride. BAK is used in most glaucoma medications because of its well-established effectiveness as a preservative. However, its toxicity to epithelial cells may contribute to or exacerbate ocular surface problems, which are particularly common in glaucoma patients. In a prospective, randomized trial, non-BAK-preserved travoprost was statistically equivalent to standard travoprost preserved with BAK.[260] Adverse events due to hyperemia were similar between the two solutions.
Bimatoprost
Commercially available in the US since 2001, Bimatoprost 0.03% is the ethyl amide of the prostaglandin FP receptor agonist, 17-phenyl-prostaglandin F2?. Its mechanism of action is unclear. Some studies demonstrate that the molecule is converted into its free acid and then targets the F2? receptor,[261-270] whereas other studies propose an effect through a still uncharacterized prostamide receptor.[271-277]Bimatoprost is believed to lower the IOP by increasing aqueous outflow by both the conventional trabecular pathway and the uveoscleral pathway.
Bimatoprost, like other ocular hypotensive agents, was compared with timolol 0.5% twice-daily therapy.[278,279] Pooled 6-month results from two multicenter clinical trials showed that in 483 patients randomized to once-daily bimatoprost, twice-daily bimatoprost or twice-daily timolol 0.5%, bimatoprost once-daily in the evening was significantly superior at all time points in the mean diurnal IOP curve. At the 6-month visit, mean IOP reduction at 10 am was 8.1 mmHg (33%) with bimatoprost once daily, 6.3 mmHg (26%) with bimatoprost twice daily and 5.6 mmHg (23%) with timolol.[280] A similar study that followed 379 patients for 24 months, showed that bimatoprost once daily provided significantly greater mean IOP reduction from baseline at each study visit than did timolol (7.8 mmHg vs 4.6 mmHg at 10 am, month 24).[281]
When compared to Latanoprost or Travoprost, Bimatoprost has been found to have equal or better IOP control.[282-286] The XLT study group compared all three prostaglandins, in a 12-week, randomized, masked-evaluator, multicenter study, and concluded that all three drugs were comparable in their ability to reduce IOP at 8 am, 12 noon, 4 pm, and8 pm at 12 weeks.[258] Other studies have demonstrated superior IOP lowering with Bimatoprost compared to Latanoprost or Travoprost.[283,284,286] While all prostaglandin drugs may have similar efficacy, it has been shown that nonresponders to latanoprost will demonstrate IOP reduction with bimatoprost.[287,288] This suggests that switching from one hypertensive lipid to another may be of clinical benefit.
Bimatoprost, like Latanoprost and Travoprost, is dosed once daily and achieves excellent diurnal control with minimal fluctuation throughout the 24-h cycle.[65] A 24-h IOP control study comparing bimatoprost to latanoprost and travoprost showed no statistically significant difference in the 24-h diurnal curve for all three drugs.[289] Another study comparing latanoprost and bimatoprost's 24-h IOP curve showed a small difference (0.9 mmHg, p = 0.008) in favor of bimatoprost in IOP control, but also a significantly higher incidence of hyperemia (p = 0.004).[290]
While Bimatoprost is often shown to have equal or better IOP lowering than the other prostaglandin analogs, its side effects profile, particularly hyperemia, tends to be worse.[258,280,281,285,286,290,291]Conjunctival hyperemia occurs in 8-58% of patients on bimatoprost, depending on the study methodology.[258,284] Studies in laboratory animals have shown that the hyperemia is caused by endothelial-derived nitric oxide-mediated vasodilatation and is not associated with inflammation.[292] In vitro studies looking at inflammatory markers of conjunctiva-derived epithelial cells showed no increase in inflammatory markers with Bimatoprost but found toxic effects consistent with the benzalkonium chloride concentration.[293] Another study found bimatoprost, travoprost and latanoprost to be equivalent in causing cellularity of the anterior chamber and cystoid macular edema in pseudophakic and aphakic patients with glaucoma.[294] Nevertheless, latanoprost is the prostaglandin drug with the highest BAK concentration (0.02% vs 0.015% for travoprost and 0.005% for bimatoprost) but also the least likely to cause conjunctival hyperemia on clinical studies.
Other adverse effects associated with bimatoprost use are ocular irritation, dryness, iritis, cystoid macular edema, skin, iris and eyelash hyperpigmentation, and provocation of herpetic eye disease. The skin hyperpigmentation is described in 3-10% of patients and it can be a real aesthetic issue. It is recommended that it be discussed with patients on initiation of therapy with any of the hypotensive lipids, especially if used unilaterally. A recent histopathological study demonstrated that the bimatoprost-induced periocular skin hyperpigmentation is caused by increased melanogenesis and increased transfer of melanosomes to basal keratinocytes, with the absence of melanocyte proliferation and melanocyte atypia.
Unoprostone
Another prostaglandin prodrug, Unoprostone, was briefly commercially available following its FDA approval in 2000. It varies only slightly from latanoprost with the substitution of a double-bonded oxygen for one of the hydroxyl groups.[295] It is a docosanoid whose base molecule is actually a derivative of the pulmonary metabolite of prostaglandin F2?.[296] Despite its similarity to other prostaglandin analogs, unoprostone appears to act by a different mechanism. Dosed twice daily, unoprostone is not as effective in lowering intraocular pressure as latanoprost. In clinical trials, unoprostone 0.15% lowered IOP ?3-4 mmHg. Similar to latanoprost, mild conjunctival hyperemia occurred as well as some corneal epithelial defects.
|
Key Features: Prostaglandin Analogs |
|||||||||||||||||||||
|
COMBINATION PRODUCTS
In Europe and other countries, there is widespread use of the so-called fixed-combination products. However, in the United States, commercial attempts to introduce combination products such as timolol-pilocarpine, dipivefrin-levobunolol, and latanoprost-timolol have not been successful. Part of the reason probably relates to the strident FDA posture on combination drugs, with very few systemic drugs used in this country combining approved agents. Presently, the only approved product is a fixed dorzolamide 2%/timolol 0.5% combination, marketed as Cosopt since 1998 in the US. In Canada and Europe, Xalacom (latanoprost 0.005% and timolol 0.5%) and Combigan (timolol 0.5% and brimonidine 0.2%), and Ganfort (bimatoprost 0.03%/timolol 0.5%) is available for glaucoma therapy. Presently, combinations of latanoprost, bimatoprost, and travoprost with timolol and a timolol 0.5%-brimonidine 0.2% combination are undergoing trials for FDA approval.
Timolol-Dorzolamide Fixed-Combination
The timolol-dorzolamide fixed combination known as Cosopt was introduced in the market in 1998, after a long period of testing to establish stability, shelf life, tolerability, dose response and comparison of the fixed combination therapy with the components used concomitantly and separately.[297-300] Clinically, there does not seem to be a significant difference between concomitant and fixed-combination usage. Discrepancies between phase-3 trials and prior replacement studies were recently evaluated in a randomized controlled trial along with a replacement study. The randomized trial showed that the fixed combination was as effective as its components in controlling IOP, confirming phase-3 trials, while the replacement study showed a statistically significant additional IOP reduction with the fixed combination.[301]
Due to their different mechanisms of action, timolol and dorzolamide are additive when administered together. The single formulation has the potential to increase compliance by diminishing the number of daily doses. On the other hand, the formulation will also lead to side effects encountered with each of the drugs, and some patients that could be treated with a once daily ?-blocker will receive double of that dose.
The fixed combination timolol/dorzolamide has been extensively compared to other drugs, specifically latanoprost, bimatoprost, travoprost and the combination of timolol/brimonidine and timolol/latanoprost. A combined analysis of pooled data from two large randomized, double-blind studies, with a total of 259 patients randomized to timolol/dorzolamide and 268 randomized to latanoprost, showed equivalence regarding: (1) mean daytime diurnal IOP, (2) percentage of patients achieving target IOP levels (40% reduction in 15% of timolol/dorzolamide and 13% of latanoprost patients), (3) mean IOP reduction in patients with high IOP at baseline (12.5 mmHg with timolol/dorzolamide and 12.6 mmHg with latanoprost), and (4) mean IOP at each time point during the day.[302,303] A multicenter study in Latin America, comparing the two drugs, randomized a total of 229 patients and found that the mean diurnal IOP reduction at 8 weeks was similar between the two groups, except for the 5 pm IOP, where levels were significantly lower in latanoprost patients.[304] Most studies show that the fixed combination has a higher incidence of taste perversion, stinging and some times bradycardia, while latanoprost causes more conjunctival injection.
When compared to bimatoprost in a prospective, randomized, double-masked trial, the fixed combination timolol/dorzolamide lowered mean IOP at 8 am significantly less than the prostaglandin (6.8-7.6 mmHg for bimatoprost vs 4.4-5.0 mmHg for the timolol/dorzolamide, p < 0.001). Taste perversion, ocular burning and stinging with instillation were more common with timolol/dorzolamide while conjunctival injection was more common with bimatoprost.[305] A smaller study with a crossover design, showed a decrease in the mean baseline IOP from 24.8 ± 2.4 mmHg to 17.4 ± 2.9 for bimatoprost and to 18.1 ± 2.8 mmHg for timolol/dorzolamide over 8 weeks, with no statistical difference in any of the individual time points.[306]
Two prospective, investigator-masked, randomized studies have been published comparing latanoprost, travoprost and the fixed combination of timolol/dorzolamide, with conflicting results, possibly due to the different types of patients studied. The first study looked at pseudoexfoliation glaucoma patients. At 6 months, mean IOP reduction was 11.5 ± 3.3 mmHg with timolol/dorzolamide, 9.3 ± 2.9 mmHg with travoprost, and 8.2 ± 1.2 mmHg with latanoprost. The authors concluded that the fixed combination was significantly more effective in this population.[307] Another study, with a crossover design with 3 months in each drug without interval washout period, showed the prostaglandins to be equivalent, and better than dorzolamide/timolol at decreasing the mean baseline IOP. Local symptoms were most pronounced after travoprost.[308]
Timolol/dorzolamide was compared to the fixed combination of timolol and latanoprost in a parallel group, evaluator-masked, multicenter study, where 253 patients were randomized and followed for 3 months. Mean reduction in diurnal IOP from baseline was 1 mmHg lower with latanoprost/timolol (p=0.005).[309] A smaller crossover comparison showed no difference in diurnal IOP measured every 2 h from 8 am to 8 pm between the two fixed combinations.[310]
Two large multicenter, prospective, observer-masked studies have compared dorzolamide/timolol to the concomitant use of brimonidine 0.2% and timolol 0.5% twice daily. One study randomized 293 patients to either group. At all observed points during the 6-month study, the 95% confidence interval of the treatment difference was within the ±1.5 mmHg hypothesized.[311] Another study randomized 492 patients and found the two combinations to be again comparable, with the 95% CI of the mean IOP change from baseline in three of four points (peak and trough, 1 and 3 months) within the prespecified comparability boundary of ±1.5 mmHg.[312]
Regarding additivity, when latanoprost or brimonidine 0.2% was added to the regimen of patients whose IOP was uncontrolled on timolol/dorzolamide (TDC), mean peak/trough IOP reduction was 4.4/3.4 mmHg and 5.2/3.5 mmHg at 1 and 3 months for latanoprost+TDC, and 3.9/2.9 mmHg and 4.6/2.9 mmHg for brimonidine+TDC. The success rate, defined as IOP reduction of at least 15% for peak/trough at 1 and 3 months was 76.3%/42.1% and 77.1%/40% for latanoprost + TDC and 71.8%/41% and 77.7%/41.7% for brimonidine+TDC.[313] Despite the small difference in favor of brimonidine+TDC, there was no statistical significance. Latanoprost has been described to add 16% lowering of IOP when added to timolol/dorzolamide combination.[314]
The cost of treatment with Cosopt (timolol 0.5%/dorzolamide 2%) has been compared to the cost of Combigan (timolol 0.5%/brimonidine 0.2%) and Xalacom (latanoprost 0.005%/timolol 0.5%) by comparing the number of drops in a bottle in a study from Canada. Cosopt had the least amount of drops per milliliter, which made it the most expensive, while Combigan had the lowest cost.[315] Another study looked at prescription trends over 3 years during the early introduction of Cosopt in the market and found that there was overlapping of prescriptions with 37% of patients using both Cosopt and ?-blockers. The same study documented the progressive decrease in ?-blocker prescriptions, and increase in brimonidine and latanoprost prescriptions.[316]
In summary, combination drugs are likely to offer equivalent IOP control as the separate components with the advantage of convenience and possible increased compliance, at the price of increased side effects from one bottle of medication. While the timolol/dorzolamide is the only one currently approved in the US, other agents are undergoing testing. Cosopt seems to be a safe and efficacious option for the treatment of Glaucoma.
|
Key Features: Timolol 0.5%/Dorzolamide 2% Combination |
|||||||||
|
SUMMARY OF APPROVED AGENTS
Until recently, there was a fairly clear consensus about using nonselective ?-blockers as first-line treatment in patients without contraindications. However, since the mid-1990s, the availability of brimonidine, which causes less systemic effects in many patients, and latanoprost, which has little, if any, adverse systemic effects, has stimulated a rethinking of whether our traditional approach since the late 1970s should be revised. Numerous ophthalmologists and glaucoma subspecialists now subscribe to using brimonidine or latanoprost before offering nonselective ?-blockers, and all three compounds must be realistically considered first-line agents. Because of an improved understanding and increased awareness of systemic side effects, ?-blockers should still be considered the benchmark of initial therapy until a longer postapproval experience is gained with the newer agents. If none of these drugs is tolerated or efficacious in a particular patient, then a second-line agent such as a selective ?-blocker or topical carbonic anhydrase inhibitor should certainly be considered. Only if multidrug therapy is required would a patient ordinarily need to consider a so-called third-line agent such as a miotic or epinephrine product. With respect to adjunctive therapy to ?-blockers, a topical carbonic anhydrase inhibitor would seem to be the choice offering the greatest patient-years of experience and, especially with the introduction of a fixed-combination product, probably the most convenience. However, the addition of latanoprost or brimonidine may offer greater efficacy. Many clinicians have utilized a once-daily nonselective ?-blocker in the morning combined with nightly application of latanoprost as a particularly appealing combination, with great efficacy and convenience. The term maximal medical therapy has shown good 'survivability' in glaucoma publications but still remains only vaguely defined and ordinarily differs from patient to patient. In general, therapy that might be considered maximal and a prelude to interventional treatment should include a ?-blocker, carbonic anhydrase inhibitor, latanoprost, and either an ?-agonist, a miotic, or an epinephrine product. For most patients, this would commit them to at least three medication bottles if a timolol-dorzolamide fixed-combination product were utilized. There are certainly a few rare patients that can consistently comply with utilization of three or four bottles, but owing to expense and complexity as well as the high success rate of modern glaucoma surgery, this amount of topical medication would be deemed excessive in many patients.
INVESTIGATIONAL AGENTS
NEUROPROTECTION
One important line of investigation is the so-called neuroprotective agents that, broadly defined, help mitigate ongoing neuronal damage after an initial insult by either pressure or some other factor. A lot of focus on programmed cell death (apoptosis) and the potential for glutamate toxicity has led to the identification of other intermediate messengers that may possibly be perturbed by pharmacologic intervention. Calcium channel blockers are clearly good candidates for further investigation, both in their current oral form and perhaps even in application of topical agents such as verapamil. Other candidates include oxygen radical scavengers, nitric oxide inhibitors, ?-opioid agonists, and N-methyl-d-aspartate antagonists.[317] Many new candidate drugs are receiving attention, but the ability to determine whether some patients have a genetically controlled susceptibility to excitotoxicity may make the investigation of neuroprotective agents even more appealing.
Systemic Interactions of Glaucoma Medications
Topically applied ophthalmic medications can attain sufficient serum levels via absorption into conjunctival, nasal, oropharyngeal, and gastrointestinal mucosa to have systemic effects and to interact with other drugs. In fact, topical administration to the eye has been likened to intravenous, rather than oral, administration because a high percentage of the absorbed drug avoids hepatic first-pass metabolism; thus, drugs administered by this route can attain higher levels relative to dose than if administered orally.[318-320] These side effects and interactions are especially dangerous because the majority of patients taking glaucoma medications are elderly, may have multiple systemic illnesses, and may be taking many other medications. The likelihood of drug interaction increases exponentially with the number of medications being taken, so that it is estimated that a patient taking five drugs has a 50% potential for interaction versus 6% for a patient taking two drugs. Alterations in absorption, metabolism, and excretion of drugs in the elderly also increase the likelihood of harmful side effects and interactions.[321]
Ophthalmologists prescribe a number of topical and systemic medications that can adversely interact with other prescribed and over-the-counter drugs. Therefore, the objective of this chapter is to summarize known and potential interactions that may occur between antiglaucoma medications and other medications. A number of sources were used, including the Physician's Desk Reference (PDR), United States Pharmacopeia Drug Information Directory (USPDI), and two specific interaction references: Hansten's Drug Interactions and Drug Interaction Facts and Comparisons.[322,323]
Of the reports and studies available, many are single case reports in which the addition of a topical medication to a systemic medication (or vice versa) resulted in an unwanted reaction. Few of these reports involved rechallenging the patient with the offending agent after the side effects had resolved. This rechallenge is the 'gold standard' to determine whether the addition of the second medication or mere coincidence caused the reaction. A number of reported interactions are partially based on pharmacologic studies in animals or in vitro. Whether these results can be applied to actual in vivo events requires further investigation.
More recent literature indicates a trend toward greater sophistication in characterizing intervention between topical and systemic medications. The interaction between a topical ophthalmic medication, timolol maleate, and an oral medication, quinidine sulfate,[80] was evaluated in a crossover study in a series of patients. The data suggested a pharmacogenetic mechanism as an explanation for the significant adverse effects on certain patients taking these two medications. Before this, a single case report had been the best evidence of this potential interaction. Hopefully, other studies of potential interactions between oral and topical ophthalmic medications will become available in order to prove or disprove the many potential interactions listed in this chapter.
Table 217.9 presents a summary of reported interactions, which indicates whether the interactions are additive or antagonistic and offers our evaluation of the literature support for each report. There is no attempt to differentiate between additive and synergistic interactions, as the available literature does not allow such a distinction to be made. Although the amount of information is limited and anecdotal in many instances, physicians should be aware of documented and potential systemic effects and drug interactions of topical ophthalmic medications.
TABLE 217.9 -- Summary of Nonocular Interactions between Glaucoma Medications and Systemic Drugs
|
Interaction |
|||||
|
Glaucoma Medication |
Systemic Drug |
Additive |
Antagonistic |
Potential Result |
Documentation Quality of References |
|
?-Adrenergic antagonist |
Anesthetic agents (inhalational) |
X |
Systemic hypotension |
Poor[347] |
|
|
Hypoglycemic agents |
X |
A. Retard hypoglycemic rebound |
None |
||
|
B. Mask hypoglycemic symptoms |
Poor[355] |
||||
|
X |
C. Produce hypoglycemia |
Poor[354,355] |
|||
|
?-Adrenergic antagonist |
Increased toxic effects of ?-antagonists |
Poor[356-358] |
|||
|
Calcium channel blockers |
X |
Cardiac depression |
Poor[359,360] |
||
|
Cholesterol-lowering medication |
X |
Decrease high-density lipoprotein cholesterol |
Good[340,341] |
||
|
Cholinesterase inhibitors |
X |
Weakness of striated muscle |
Fair[363,364] |
||
|
Clonidine |
X |
Systemic hypertensive rebound after clonidine withdrawal |
Poor[367] |
||
|
Digitalis glycosides |
X |
Cardiac depression |
Fair[368-372] |
||
|
Fentanyl derivatives |
X |
Increased toxic effects of fentanyl |
None |
||
|
Phenothiazines |
X |
Increased serum levels of ?-blocker and phenothiazine with potential toxic side effects |
None |
||
|
Prednisone |
X |
Increased serum potassium |
Poor[380] |
||
|
Quinidine |
X |
Cardiac depression |
Good[80,379] |
||
|
Reserpine |
X |
Cardiac depression |
None |
||
|
Sympathomimetic amines |
|||||
|
1. Subcutaneous epi |
X |
Abrupt systemic hypertension |
None |
||
|
2. Xanthines |
X |
A. Bronchoconstriction |
Good[319,332,389] |
||
|
B. Reduced theophylline clearance |
None |
||||
|
3.?-Adrenergic agonists for treatment of: |
|||||
|
a. Heart failure |
X |
Cardiac depression |
Good[318,319,331,386] |
||
|
b. Bronchoconstriction |
X |
Bronchoconstriction |
Good[319,332,389] |
||
|
Adrenergic agonist (nonselective) |
Anesthetic agents (inhalational) |
X |
Cardiac arrhythmias |
Poor[399,400] |
|
|
Digitalis glycoside |
X |
Cardiac arrhythmias |
None |
||
|
Monoamine oxidase inhibitors |
X |
Hypertensive crises |
Good[402-404] |
||
|
Sympathomimetic amines |
X |
Systemic hypertension |
None |
||
|
Tri- and tetracyclic antidepressants |
X |
Cardiac arrhythmias |
None |
||
|
Adrenergic agonist (?2-selective) |
Monoamine oxidase |
X |
Hypertensive crises |
Good[402-404] |
|
|
Refuted by literature |
|||||
|
Hypotension producing medications |
X |
Systemic hypotension |
Poor[412] |
||
|
Cholinesterase inhibitors |
Anesthetic agents (local: ester type) |
X |
Prolonged anesthetic action with cardiopulmonary depression |
Fair[419] |
|
|
Cholinesterase inhibitors |
X |
Cholinergic toxicity |
None |
||
|
Succinylcholine |
X |
Prolonged neuromuscular blockade |
Good[417,418,421] |
||
|
Carbonic anhydrase inhibitors |
Amphotericin-B |
X |
Increased hypokalemia |
Poor |
|
|
Anticholinergics |
X |
Enhanced anticholinergic action |
Poor |
||
|
?-Adrenergic blockers |
X |
Increased metabolic and respiratory acidosis |
Fair[428] |
||
|
Corticosteroids |
X |
Potentiate hypokalemia |
Poor |
||
|
Cyclosporine |
X |
Increased cyclosporine toxicity |
Poor[429] |
||
|
Digitalis glycosides |
X |
Digitalis toxicity increased by hypokalemia |
Good |
||
|
Ephedrine |
X |
Enhanced ephedrine action |
Poor |
||
|
Lithium |
X |
Increased lithium excretion |
Fair[430] |
||
|
Mexiletine |
X |
Enhanced mexiletine effect |
Poor |
||
|
Phenytoin |
X |
Accelerated osteomalacia |
Poor |
||
|
Primidone |
X |
Decreased primidone effectiveness |
Fair[432] |
||
|
Quinidine |
X |
Decreased quinidine excretion |
Poor |
||
|
Salicylates |
X |
Increased salicylate and carbonic anhydrase toxicity |
Good[434,435,437] |
It is important to remember that even when the potential for drug interaction exists, there are methods to reduce systemic absorption of topically applied drugs. Nasolacrimal occlusion, a technique in which digital pressure on the periphery of the nasolacrimal drainage system obstructs drainage to the nasopharyngeal mucosa, has been shown to decrease systemic absorption significantly. Eyelid closure for 5 min after drug application achieves the same purpose by inhibiting nasolacrimal pump action.[232]
OPHTHALMIC ?-ADRENERGIC ANTAGONISTS
CHARACTERISTICS, MECHANISMS, AND SIDE EFFECTS
After instillation in the conjunctival sac, timolol maleate (Timoptic) is absorbed into the conjunctival, nasal, oropharyngeal, and gastrointestinal mucosal capillaries. Measured peak plasma levels after topical administration have varied from undetectable to 9.6 ng/mL, with the average level recorded in the literature being ?1 ng/mL. Although these plasma levels are lower than the range of 20 to 40 ng/mL noted in pharmacokinetic studies using a 10-mg oral dose of timolol, they approximate plasma concentrations present 6-8 h after oral administration and are sufficient to cause a degree of systemic ?-adrenergic blockade.[319,325]
Much of the available information on timolol interactions is based on propranolol studies because both are extensively metabolized in the liver and both have similar ?-blocking effects.[326] However, a major difference is that propranolol stabilizes cell membranes, which is the reason it is a local anesthetic and is not suitable for chronic topical administration to the eye.[325,327] It has been estimated that a patient instilling a solution of timolol 0.5%, two drops twice per day, could receive the equivalent of 4 mg of propranolol intravenously twice per day.[88] There are also differences in their pharmacokinetic properties. For example, blood levels of timolol tend to be greater than those of propranolol because of its slower systemic clearance. Although the exact reasons for this are unknown, it is probably due to a lower first-pass effect (hepatic metabolism) because of timolol's lower lipophilicity and therefore slower transfer across hepatocyte membranes.[325,327] Regardless of the mechanism, one would expect an even greater potential for side effects with timolol.
There is little available information on potential interactions of other ?-blockers used to treat glaucoma (levobunolol hydrochloride, betaxolol hydrochloride, metipranolol hydrochloride, carteolol hydrochloride). Levobunolol, 0.5%, which like timolol is a nonselective ?-adrenergic antagonist, has been reported to achieve serum levels up to 0.21 ng/mL after ophthalmic administration.[325,328] Serum levels of 0.5% betaxolol, a ?1-selective antagonist, have been reported as undetectable after topical administration, although detection limits for the assay are not available.[329] The lipophilicity of these other ?-blockers is between those of timolol and propranolol. These agents may allow better corneal penetration than timolol and allow increased transfer across hepatocyte membranes with resultant lower serum concentrations.[325] Carteolol is the one exception, as it is a hydrophilic compound. This may confer an additional advantage for decreasing absorption across the blood-brain barrier and reducing potential central nervous system side effects.[145] In addition, carteolol is unique in possessing intrinsic sympathomimetic activity, which refers to the ability to act as a partial adrenergic agonist, potentially minimizing the adrenergic-blocking side effects described further on.
Even though disparate serum concentrations are reported for the different ophthalmic ?-blockers, the many reports of systemic side effects are proof that all can achieve serum levels that are sufficient to cause systemic ?-blockade. The most widely studied and reported are the effects on cardiovascular and pulmonary function. Stimulation of cardiac ?1-receptors elicits increases in heart rate, contractility, and conduction velocity. Blocking these receptors can cause bradycardia, conduction disturbances, and decreases in cardiac output. Peripheral vascular stimulation of ?2-receptors results in vasodilatation. Blocking vasodilatation might be expected to contradict the antihypertensive use of ?-antagonists; however, their blood pressure-lowering action is primarily due to their cardiac effects.[330] Stimulation of pulmonary ?2-receptors results in relaxation of bronchial musculature. Therefore, ?2-receptor antagonists can cause increased airway resistance and respiratory compromise, especially in susceptible individuals. Although a ?1-selective blocker such as betaxolol is less likely to have pulmonary effects, its selectivity is not absolute, as proved by reports of reduced pulmonary function with its use.[325,331,332]
?-Blockers have complex effects on blood glucose control and metabolism. In the liver, stimulation of ?2-receptors causes glycogenolysis and gluconeogenesis with resultant glucose release. The pancreatic islet cells that are responsible for the secretion of insulin also respond to ?2-adrenergic stimulation. Although the effect of ?-blocking agents on glucose metabolism in normal patients is minimal, in the diabetic patient they have the potential to slow recovery of the glucose concentration and prevent the usual rebound of plasma glucose in response to hypoglycemia. Signs of hypoglycemia are also masked.[333,334] These issues are discussed in more detail further on in regard to the interaction between ?-blockers and antidiabetes agents.
?-Adrenergic effects on the central nervous system are poorly understood. Depression, lethargy, confusion, and hallucination in patients taking both oral and ophthalmic ?-blockers have been described.[335,336] Other studies, however, have shown improvement of mental test performance without any resultant lethargy or drowsiness.[337,338] Also poorly understood are reports of timolol exacerbating myasthenia gravis, possibly because of a depressant effect of ?-antagonists on the neuromuscular junction.[339] Although propranolol has membrane stabilization potential, this mechanism is less easily justified with the ophthalmic ?-antagonists, which do not appear to have any significant membrane stabilization activity.
?-Adrenergic blockers have harmful effects on lipid metabolism. The mechanism involves inhibiting the enzymes of cholesterol metabolism, which results in an increase in serum triglycerides and a reduction in the high-density lipoprotein (HDL) level.[340] Even with topical ?-blockers, this effect can be significant. This was shown in a double-masked, randomized crossover study of 58 healthy males given both timolol maleate 0.5% and carteolol hydrochloride twice daily.[341] Timolol resulted in an 8% reduction in HDL cholesterol, whereas carteolol resulted in a 3% reduction. Based on the Physicians' Health Study, a large epidemiologic study relating HDL cholesterol levels to coronary heart disease, the decrease in HDL from timolol and carteolol translates to a theoretical 14.2% and 5.7% increase in the risk of myocardial infarction, respectively.[342] Although this same study did not reveal a significant elevation in serum triglyceride levels, other studies have shown significant elevations in triglycerides as well.[84]Nasolacrimal occlusion was not used in these studies.
These effects on lipids are even more significant in elderly patients, both men and women, many of whom have preexisting cardiovascular disease. In postmenopausal women, estrogen replacement therapy reduces the risk of cardiovascular disease partly by beneficial effects on the lipid profile. Topical ?-blockers may counteract this benefit. Postmenopausal women not receiving estrogen replacement therapy are believed to have a higher risk of cardiovascular disease, which would be even greater if lipids were adversely affected by a ?-blocker.[343] Alternatives to ?-blocker therapy or an agent such as carteolol, which appears to have a less harmful effect on HDL cholesterol due to its intrinsic sympathomimetic activity, may be considered in these patients.
INTERACTIONS WITH OTHER DRUGS
Anesthetic Agents (Cyclopropane, Enflurane, Halothane, Isoflurane, Methoxyflurane, Trichlorethylene)
Inhalational anesthetics are associated with hypotension owing to direct depression of the heart with a decrease in cardiac output. These cardiovascular changes, however, elicit a reflex, compensatory increase in sympathetic nervous system activity with subsequent cardiac stimulation and an increase in heart rate, contractility, and cardiac output.[344] In the presence of ?-blocking medication, the heart's ability to respond to these sympathetic stimuli is reduced, resulting in potentially prolonged and severe hypotension.[345,346] Topical ophthalmic ?-blockers have the potential to cause a similar effect; however, documentation is limited.
One report describes a 69-year-old man who applied 0.5% timolol maleate four times daily to his right eye and 2% pilocarpine twice daily to his left eye and experienced bradycardia and hypotension during halothane anesthesia. Timolol was detected in a serum sample removed during surgery at a concentration consistent with partial ?-adrenergic blockade (2.6 ng/mL). The authors admit that the hemodynamic changes could also be attributed to underlying abnormalities in cardiac conduction or excessive sensitivity to halothane anesthesia.[347] In addition, timolol should have been administered twice rather than four times daily in this patient. This one case does not provide sufficient support to recommend removal of the ophthalmic medication before surgery.
Antidiabetic Agents (Oral and Insulin)
The interaction between ?-blockers and glucose control is complex and still under study. The adrenergic nervous system, through ?2-adrenergic effects on the liver, plays a major role in the counter-regulatory response to hypoglycemia induced by insulin and oral agents. Nonselective ?-adrenergic antagonists can, therefore, retard blood glucose rebound after hypoglycemia. These agents also mask symptoms of hypoglycemia, such as tachycardia and palpitations, that serve as vital warning signs to the patient.[333,334]
More controversial in terms of both hypo-and hyperglycemia is the role of ?-blockers in affecting glucose homeostasis. Propranolol and other nonselective agents have been implicated in causing hypoglycemia; however, this is most likely related to the effects of retarding blood glucose rebound and masking hypoglycemic symptoms. Studies of normal and diabetic patients receiving ?-blocker therapy have shown no overall increase in the incidence of hypoglycemia.[348-350] With regard to hyperglycemia, the ?2-adrenergic receptors in the pancreas respond to stimulation with the release of insulin. Antagonists with ?2-blocking activity can inhibit this release, although the magnitude of this effect is rarely, if ever, of clinical significance. Most reports, however, advise the use of ?1-selective antagonists in diabetic patients.[334,351-353]
There are two reports of timolol associated with hypoglycemia in diabetic patients. The first was a 72-year-old woman who had a 13-year history of insulin-treated diabetes mellitus, and a history of one to four hypoglycemic episodes weekly, which she terminated with dietary intervention. Three weeks after starting timolol maleate, 0.5% twice daily, in both eyes, she had a 2-h hypoglycemic episode requiring hospitalization, at which time she reported a recent subjective increase in hypoglycemic episodes. She did well after discharge on a lower insulin dose, but the report does not indicate whether the timolol was continued.[354]
The second case reported was a 65-year-old male diabetic who had been taking insulin for 25 years. He usually experienced one hypoglycemic episode per month characterized by diaphoresis, anxiety, and visual sensations. Approximately 15 months after the patient began timolol maleate, 0.25% twice daily in both eyes, his wife noted an increase in the frequency of hypoglycemic episodes to two or three per month, with a change in symptoms. During these episodes, the patient no longer had diaphoresis and anxiety, but instead was experiencing mental status changes with staring and grunting. Following the discontinuation of timolol, the patient continued to experience several hypoglycemic reactions per month, but with a return of the previous symptoms of anxiety and diaphoresis.[355] Two important facts make the causal relationship of timolol eyedrops unlikely in this case. First, the patient continued to experience multiple hypoglycemic reactions even after the discontinuation of timolol and, second, ?-blockers have previously been shown not to affect hypoglycemia-induced diaphoresis.[348]
It is not possible in either case to implicate timolol convincingly as the cause of the hypoglycemic episodes. Topical nonselective ?-blockers can be safely given to diabetic patients if blood glucose levels and hypoglycemic symptoms are monitored. Cardioselective ?-blockers may have less effect on blood glucose control and may therefore be preferred.
?-Adrenergic Antagonists (Atenolol (Tenormin), Metoprolol (opressor), Nadolol (Corgard), Pindolol (Visken), Propranolol (Inderal), Timolol (Blocadren))
The concurrent use of oral and topical ?-blockers theoretically has the potential for causing an additive cardioinhibitory effect. Yet, in a double-blind trial of 31 patients with open-angle glaucoma, the use of topical timolol maleate 0.5% bid for 1 week did not enhance the pulse- and blood pressure-lowering effects of timolol 20 mg bid.[356] In another study, seven healthy patients receiving 80-160 mg propranolol daily were treated once with timolol 0.5% in a single eye, and no significant effect on pulse rate or blood pressure was seen.[357]
In a published letter, betaxolol, 0.5%, was instilled in the left eye of an 81-year-old man receiving atenolol 25 mg daily, followed within minutes by an acute inferior myocardial infarction. The patient had a history of hypertension and was also receiving indapamide, an oral diuretic with antihypertensive activity, and potassium chloride.[358] It is impossible to conclude what caused this event.
In patients tolerating ophthalmic or systemic ?-blocker therapy, the addition of a second agent can almost always be done safely, although it is recommended that heart rate and blood pressure be monitored for changes.
Calcium Channel-Blocking Agents (Diltiazem (Cardizem), Nifedipine (Adalat, Procardia), Verapamil (Calan))
Because they have similar pharmacologic effects, concurrent use of calcium channel antagonists with ophthalmic ?-blockers may lead to depressed atrioventricular conduction, as well as left ventricular failure and hypotension in patients with impaired cardiovascular function.
Two cases of severe bradycardia have been reported in patients using timolol eyedrops and verapamil. In one case, a 64-year-old man was receiving both verapamil, 160 mg, and timolol maleate, 0.5%, twice daily, as well as pilocarpine, 2%, four times daily. He had been taking these medications for more than a year when he presented with a sinus bradycardia of 36 beats/min, which responded well within 48 h to a change from verapamil to nifedipine. In the second case, a 52-year-old man was hospitalized with atypical chest pain while taking timolol maleate, 0.5%, twice daily in both eyes. Shortly after admission to the coronary care unit after receiving intravenous nitroglycerin and oral verapamil, 40 mg, the patient used his timolol drops. His heart rate decreased after ?30 min and he experienced a 10-s period of asystole. Treatment with verapamil was discontinued and bradycardia did not return when he used timolol alone. It is impossible to rule out that the verapamil may have been solely responsible for the bradycardia in both cases.[359,360]
Nifedipine or nicardipine may be the best choice if a calcium channel antagonist is needed. These two agents have the most potent peripheral vasodilating effect of the available calcium channel blockers, whereas their ability to decrease cardiac contractility and slow atrioventricular conduction of the heart is least. Diltiazem is intermediate in peripheral and cardiac effects. Verapamil would be the most potentially hazardous because of its predominantly cardiac depressant action.[361,362] Calcium channel blockers and ophthalmic ?-blockers may be used concurrently with caution.
Cholesterol-Lowering Medication (Cholestyramine (Questran), Lovastatin (Mevacor), Niacin (Nicolar))
Because of the ability of topical ?-blockers to decrease HDL cholesterol, glaucoma medications can partially counteract the cholesterol-lowering effect of these medications. No specific interaction studies have been published, but it may be prudent in patients with elevated cholesterol levels to consider switching to a ?-blocker with potentially less cholesterol effect, such as carteolol, or to an entirely different class of glaucoma medication.[84,340-342]
Cholinesterase Inhibitors (Edrophonium (Tensilon), Neostigmine (Prostigmin), Pyridostigmine (Mestinon))
Two reports of patients with myasthenia gravis receiving pyridostigmine, a cholinesterase inhibitor, cited the addition of timolol maleate eyedrops, 0.5%, as the cause of significant worsening of dysarthria and ptosis. The first involved a 70-year-old man with gradual deterioration of muscle function and worsening of diplopia and ptosis during his 6 months of timolol eyedrop therapy. Within days of switching to pilocarpine, the symptoms greatly improved. The second case was even more striking. A 71-year-old man had the onset of dysphagia, dysarthria, and worsening of diplopia and ptosis 24 h after the initiation of timolol therapy. Within 24 h of stopping the timolol, he returned to his original clinical status. Neither patient was rechallenged with timolol after discontinuation of therapy.[363,364] These reports followed one report of three patients in whom orally administered propranolol and oxprenolol worsened symptoms of myasthenia gravis.[365] The proposed mechanism involves a depressant effect of ?-blockers on the neuromuscular junction.[339] Such a phenomenon with propranolol and oxprenolol is understandable because the lipophilicity of each of these compounds allows them to permeate the cell membrane and interfere with propagation of the action potential. The topical ?-blockers, however, have limited membrane-stabilizing properties and therefore would be less likely to act by this mechanism.[325] Physicians with patients with myasthenia gravis receiving pyridostigmine or other cholinomimetic agents should be aware of a possible interaction that occurs rarely with topical ?-blockers.
Clonidine (Catapres)
The abrupt withdrawal of any antihypertensive medication, such as clonidine, may lead to a severe rebound increase in blood pressure. Theoretically, the increased peripheral vascular resistance due to ?2-receptor blockade could aggravate this hypertensive crisis.[366] There has been one report of a 60-year-old woman who had been taking clonidine for 10 months and was given oral timolol, 5 mg/day. The patient mistakenly stopped the clonidine abruptly and experienced a severe hypertensive crisis. Although this crisis may have been exacerbated by the ?-blocker therapy, it could also be explained by abrupt withdrawal of the clonidine alone.[367] No specific concerns seem indicated for the simultaneous use of topical ?-blocker therapy and clonidine.
Digitalis Preparations (Digoxin (Lanoxin), Digitoxin (Crystodigin))
Toxic levels of cardiac glycosides alone or of ?-antagonists alone can result in bradycardia and heart block. The simultaneous use of systemic ?-blockers with digitalis glycosides can lead to additive cardiac toxicity.[368] There is also some potential for ?-blockers to displace serum protein-bound glycosides and thereby increase the digitalis blood concentration.[369] However, this effect is not considered to be of clinical significance when topical ?-blocking drugs are being administered.
There are three case reports implicating an interaction between ophthalmic ?-blockers and digoxin. The first was an 84-year-old woman who experienced a syncopal episode with sinus bradycardia at a rate of 36 beats/min 1 h after instillation of timolol maleate, 0.5%. The patient was receiving diltiazem. More important, she had a toxic digoxin level of 3.1 ng/mL.[370] In the second case, a 91-year-old woman experienced palpitations and shortness of breath while taking digoxin, furosemide, and 0.5% pilocarpine qid and 0.25% timolol maleate bid in her right eye. Her digoxin level was also in the toxic range. Her electrocardiogram revealed an irregular heart rate of 35-50 beats/min with heart block.[371] The third patient was an 80-year-old man with a history of congestive heart failure, atrial fibrillation, and sick sinus syndrome previously well controlled by digoxin, 0.125 mg, and metolazone, a diuretic. Within 1 week after the initiation of betaxolol, 0.5% twice daily, he experienced pulmonary and peripheral edema with a ventricular rate of 30 beats/min. After diuretic therapy and discontinuation of betaxolol, he recovered to his previous state of health. Digoxin levels were not determined during this course.[372]
These cases suggest that patients can be safely placed on both these medications, except when digitalis levels are at or near toxic levels.
Fentanyl Derivatives (Fentanyl (Sublimaze), Alfentail (Alfenta), Sufentanil (Sufenta))
Through competition for pulmonary binding sites, chronic oral propranolol therapy can significantly decrease first-pass pulmonary uptake of fentanyl, an intravenous anesthetic, resulting in increased levels of the drug in the systemic circulation and prolonged systemic side effects such as bradycardia and hypotension.[373] Propranolol's pulmonary binding is related to its relatively high lipophilicity. The topical ?-antagonists are less lipophilic than propranolol and may not exhibit this same interaction.[325] In addition, there are no studies available documenting an interaction between fentanyl and the topical ophthalmic ?-antagonists. There is no evidence that a recommendation to discontinue topical ophthalmic ?-blockers should be made before a surgical procedure.
Hypotension-Producing Medications (USPDI Appendix II)
The USPDI contains a list of more than 40 hypotension-producing medications or categories of medication that have the potential to react additively with ophthalmic ?-blockers. Included on this list are general anesthetic agents, calcium channel blockers, ?-adrenergic antagonists, and clonidine, which are all discussed elsewhere in this section. Of the other agents listed, none except quinidine (discussed elsewhere) has been implicated in case reports. Although physicians should use caution, no specific contraindications to the combined use of an ocular ?-blocker and a systemically administered hypotensive agent can be made based on the available literature.
Phenothiazines (Chlorpromazine (Thorazine), Fluphenazine (Prolixin), Mesoridazine (Serentil), Thioridazine (Mellaril), Trifluoperazine (Stelazine))
Warnings of interaction between ophthalmic ?-blockers and phenothiazines are based on limited studies showing that propranolol combined with chlorpromazine or thioridazine may result in elevated plasma levels of both the ?-blocker and the neuroleptic agent.[374-376]
When two patients who had been receiving long-term thioridazine treatment took propranolol in a controlled, prospective study, they experienced a three- to fivefold increase in plasma thioridazine levels within 2 weeks, placing them in a potentially toxic range. The enhanced efficacy of chlorpromazine in treating schizophrenic patients receiving ?-blocking medication prompted further investigation of their interaction.[377] Both propranolol and chlorpromazine blood levels were elevated in the presence of each other. Although no mechanism of interaction was proposed, it may be related to competition in the liver, where both compounds are metabolized.[374,376] An alternative mechanism, competitive displacement from plasma protein-binding sites has not been shown to be involved.[374] These patients did not manifest toxicity in terms of cardiac arrhythmias or pigmentary retinal changes, but such increases in serum concentration, if maintained for prolonged periods, might lead to such events.
Timolol, like propranolol, is metabolized in the liver and may exhibit a similar interaction, although there are no reports documenting this. Many of the neuroleptic agents share common metabolic pathways, but extrapolating thioridazine and chlorpromazine data to these agents may not be justified.
One other point that bears mentioning is the reported central nervous system effects of topical ?-blockers, such as depression, lethargy, confusion, and hallucinations.[319,336] Both case reports and large-scale studies have suggested a link between oral ?-blockers and depression.[335] However, not all the data support a relationship.[337,338] If increased depression were to occur, it could conceivably have a great impact on patients with psychiatric disorders. Unfortunately, there are little clinical data available regarding the interaction of systemic or topical ?-blockers with the many available neuroleptic agents. Despite this lack of documentation, one of the ?-antagonists used to treat glaucoma, betaxolol, has advertised that it is less likely to cause psychiatric effects because it is relatively ?1-selective. The evidence that psychiatric effects are a ?2-antagonist action is tenuous. Physicians should be aware that ?-blockers have been implicated as a cause of increased depression, but no specific contraindication to their use with neuroleptic agents is warranted.
Prednisone (Other Corticosteroids (Dexamethasone, Hydrocortisone))
In a case report, a patient using ophthalmic timolol for 6 years received prednisone (60 mg/day). One week after the addition of prednisone, the patient's serum potassium increased from 4.6 to 6.4 mEq/L. After sodium polystyrene sulfonate treatment, the potassium level decreased to 4.0 mEq/L but then rose again. Only when the ophthalmic timolol was stopped did levels remain normal. The patient underwent a rechallenge with the same timolol dosage and again potassium levels rose to greater than 6.0 mEq/L.[378] The mechanism of this interaction is unknown, although this patient had a history of obstructive renal disease that was hypothesized to have resulted in a defect in renal tubular potassium secretion. ?-Blockers can cause small increases in potassium by suppression of renin and aldosterone release and by preventing ?-receptor-mediated hepatic and skeletal muscle potassium uptake. Since the patient had been taking timolol for 6 years, it alone could not be responsible for the hyperkalemia. It is possible that the catabolic effects of prednisone may have produced a large potassium load, and the ?-blocking agent prevented the normal handling of this load.
Documentation is lacking regarding an interaction between ophthalmic ?-blockers and any other corticosteroids, such as dexamethasone, hydrocortisone, and prednisolone. Caution is advised in the concurrent administration of ophthalmic ?-blockers and prednisone, especially in patients with defects in potassium homeostasis, such as occurs in renal failure or diabetes. In any patient presenting with hyperkalemia, the continued use of topical ?-blockers should be evaluated.
Quinidine (Quinaglute, Cardioquin)
In blocking ?-adrenergic cardiac receptors, timolol may exacerbate the cardiodepressant activity associated with quinidine. The potential for this dangerous interaction was elegantly shown in a study of healthy volunteers using a crossover comparison of timolol, placebo, and the effects of inhibition of timolol metabolism by quinidine.[80] In certain susceptible individuals, termed poor metabolizers, quinidine inhibits the cytochrome P-450 enzyme responsible for metabolizing timolol. When poor metabolizers in this study received both topical timolol and oral quinidine, their timolol levels were higher and they had significantly greater ?-blockade, as evidenced by a decrease in exercise-induced heart rate, compared with normal metabolizers. Even the normal metabolizers had a decrease in exercise-induced heart rate when quinidine and timolol were used together versus either drug used alone.
The combination of quinidine and topical timolol can be dangerous in many patients, especially poor metabolizers. Since 8% of the white population, 2-4% of the African-American population, and 1% of the Asian population are poor metabolizers, this applies to a significant number of individuals. The potential for serious interaction is pointed out in a case report describing a 70-year-old man who while taking quinidine started receiving topical timolol maleate, 0.5%, twice daily.[379] Within 12 weeks, he was hospitalized with dizziness and a heart rate of 36 beats/min. Once the therapy was stopped, the patient recovered and had no reaction while taking timolol alone, but on rechallenge with the combination therapy, his symptoms recurred. Clinicians need to be extremely cautious in using these two medications together. Cardiac monitoring is advisable to rule out medication-induced bradycardia or heart block.
Reserpine (Serpasil)
Reserpine is an infrequently used antihypertensive agent that acts by depleting stores of catecholamines. After administration, a transient sympathomimetic effect occurs followed by a fall in blood pressure often associated with bradycardia. Reserpine in combination with ?-blockers has the potential to cause additive cardiac depression, leading to atrioventricular conduction disturbances, left ventricular failure, and hypotension.[380] There are no clinical reports of such an interaction with systemic or topical ?-blocking medications. Therefore, no specific contraindication to the use of these two medications together is warranted.
Sympathomimetic Amines (?- and ?-Receptor Agonists-Epinephrine (Ephedrine); ?-Receptor Agonists-Dobutamine (Dobutrex), Metaproterenol (Alupent), Isoproterenol (Isuprel), Albuterol (Ventolin, Proventil), Terbutaline (Brethine), Isoetharine (Bronkosol))
Systemic hypertension
Patients receiving low subcutaneous doses of epinephrine, an ?- and ?-agonist, can experience rapid, marked increases in systolic and diastolic blood pressure with significant decreases in heart rate in the presence of propranolol.[381-383] The mechanisms responsible for this hypertension are not fully elucidated. This effect was previously attributed to unopposed ?-receptor stimulation in the presence of ?-blockade, although other studies present compelling evidence against such a hypothesis.[384,385] These hypertensive episodes are possibly attributable to the epinephrine alone rather than any interaction. Owing to the controversial evidence supporting this interaction and the lack of reports citing such a reaction with topical ?-blockers, no specific contraindication to their combined use is warranted.
Cardiac failure
?-Blockers are specifically contraindicated in patients with overt cardiac failure, sinus bradycardia, and second- and third-degree atrioventricular block.[318,319,331,386] Patients who are ill enough to warrant potent inotropic agents such as dopamine and therefore should not be given ophthalmic ?-blockers.
Obstructive pulmonary disease
Timolol and other nonselective ?-blockers are contraindicated in patients with underlying chronic obstructive pulmonary disease and bronchial asthma because they block endogenous stimulation of ?2-receptors as well as exogenous stimulation by medication such as isoproterenol, metaproterenol, isoetharine, albuterol, and terbutaline.[319,387,388] There are many reports of patients using bronchodilators who experienced respiratory distress due to ophthalmic ?-blocker administration.[88,93,319,389] Betaxolol, a relatively ?1-selective antagonist, may have a therapeutic role in glaucoma patients who have underlying pulmonary disease. Although it may provoke asthmatic attacks, betaxolol does not exhibit the same degree of ?2-blocking activity as the nonselective ophthalmic ?-blockers.[165,166] Betaxolol can therefore often be used in the presence of bronchodilating medications, although respiratory function must be closely monitored.[331,390]
Xanthines (theophylline (theo-dur, aminophyllin, slo-phyllin))
The xanthines act indirectly to enhance ?-adrenergic function by inhibiting the breakdown of cyclic adenosine monophosphate (cAMP). ?-Blocking agents, particularly the noncardioselective ones, reduce the pharmacologic effects of xanthines by inhibiting the ?2-receptor-induced increase in cAMP. This leads to increased bronchial resistance.[93,345,389] In addition, theophylline is metabolized by the hepatic mixed-function oxidase system. Through this competition, ?-blockers, both cardioselective and nonselective, can reduce clearance of theophylline by 30 to 50%. This leads to an increased potential for theophylline toxicity.[391-393] There is no documentation of such an effect occurring with ophthalmic ?-antagonists.
Patients receiving theophylline generally have moderately severe underlying pulmonary disease for which nonselective ?-blockers are contraindicated. If a patient taking theophylline is also receiving betaxolol, pulmonary function along with drug levels should be monitored closely.
ADRENERGIC AGONISTS: NONSELECTIVE AGONISTS
CHARACTERISTICS, MECHANISMS, AND SIDE EFFECTS
The potential cardiovascular effects of topical epinephrine preparations have long been known. The ophthalmic administration of two drops of a 2% epinephrine solution (0.05 mL/drop) can provide up to 2.0 mg of systemic epinephrine.[394] This dose of epinephrine can cause significant cardiovascular effects, including hypertension and dysrhythmias, and has the potential for interaction with systemic medications.[62,395] There is, however, only one report of ophthalmic epinephrine interacting with another medication-this is the inhalational anesthetic agent halothane. Otherwise, the drug interaction warnings in the USPDI and PDR for topical epinephrine are all based on the interaction of low-dose subcutaneous and intravenous epinephrine. Potential interactions with topical epinephrine are only speculative.
Dipivalyl epinephrine (DPE, Propine) is a prodrug that is more lipid soluble and therefore more readily absorbed through the cornea than is epinephrine. Lower concentrations can be administered, with a reduced amount available for systemic absorption.[62,395] There are no drug interactions reported for dipivalyl epinephrine in the USPDI, the PDR, or the literature search.
INTERACTIONS WITH OTHER DRUGS
Anesthetic Agents (Cyclopropane, Enflurane, Halothane, Isoflurane, Methoxyflurane, Trichloroethylene)
Halothane is capable of increasing the automaticity of the myocardium and thereby rendering it more susceptible to arrhythmogenic influences such as epinephrine. The other anesthetic agents may have similar effects, although this is somewhat controversial.[396-398] Thus, the potential for interaction with ophthalmic epinephrine exists, although limited documentation is available. There is one report of a patient who experienced transient ventricular fibrillation after the administration of the equivalent of 1 mg of topical epinephrine in a 2% ophthalmic solution during halothane anesthesia.[399] However, in a series of patients undergoing cataract surgery with halothane anesthesia, the injection of 0.04-0.7 mg of epinephrine into the vitreous caused no change in the incidence of cardiac arrhythmias compared with a control group.[400] It is advisable to notify the anesthesiologist of any topical medications administered before or during surgery. However, no specific recommendation to discontinue topical ocular adrenergic medications before surgery can be supported.
Digitalis Glycosides (Digitalis Preparations: Digoxin (Lanoxin), Digitoxin (Crystodigin))
Digoxin toxicity frequently results in cardiac arrhythmias. Because epinephrine alone can also cause such a reaction, it is postulated that there is an additive risk of arrhythmia in patients receiving both these medications.[401] However, this is speculative because there is no documentation available supporting this assertion. Topical epinephrine preparations can be given to patients taking digitalis.
Monoamine Oxidase Inhibitors (Isocarboxazid (Marplan), Phenelzine (Nardil), Pargyline (Eutonyl), Tranylcypromine (Parnate))
Monoamine oxidase (MAO) inhibitors are used for both antihypertensive and antidepressant therapy. Although epinephrine is a substrate for the enzyme MAO, termination of its action is mainly due to uptake into adrenergic neurons and metabolism by the enzyme catechol-o-methyl transferase. Therefore, there is little potential for a significant interaction, as MAO inhibitors would not be expected to greatly potentiate the effects of small amounts of epinephrine absorbed from topical application to the eye.[402-404] The hypertensive crises that are known to occur with MAO inhibitors are due to the ingestion of tyramine. As a sympathomimetic amine, tyramine acts peripherally, primarily by releasing stores of catecholamines at the myoneural junction of vascular smooth muscle. The hypertensive crisis is then created by the release of large quantities of norepinephrine. An effect as massive and localized as the tyramine reaction is extremely unlikely to occur with topical application of epinephrine.
Sympathomimetic Agents (Systemic or Local)
Theoretically, if significant systemic absorption of ophthalmic epinephrine were to occur, concurrent use of systemic sympathomimetics could cause toxicity.[394,395] However, this is based on the potential for this absorption rather than documented cases. As there is no evidence to the contrary, ophthalmic epinephrine can be used safely with systemic sympathomimetics.
Warnings of side effects with the combined use of topical epinephrine and local anesthetics containing vasoconstrictors are also based on theoretical synergism. In any injection of local anesthetic, the minimal effective concentration of vasoconstrictor should be used, but there is no specific contraindication for patients receiving ophthalmic epinephrine.
Tricyclic and Tetracyclic Antidepressants (Desipramine (Norpramin), Doxepin (Sinequan), Imipramine (Tofranil), Maprotiline (Ludiomil), Nortriptyline (Pamelor), Protriptyline (Vivactil))
The tricyclic and tetracyclic antidepressants inhibit the re-uptake of catecholamines in a manner similar to that of cocaine. The proposed interaction between ophthalmic epinephrine and these antidepressants is based on the ability of imipramine and protriptyline to increase the sensitivity to intravenous infusions of epinephrine. This was shown in a study of four normal patients receiving imipramine, 25 mg three times daily for 5 days, who when given an infusion of epinephrine experienced sinus arrhythmia along with atrial and ventricular ectopy. The presumed mechanism was the prolongation of epinephrine action by the imipramine's inhibition of uptake into adrenergic nerve endings.[402] No reports of interaction with ophthalmic epinephrine are available. Patients taking maprotiline, a tetracyclic antidepressant, can safely receive ophthalmic epinephrine, as the effects of sympathomimetics such as epinephrine are not consistently altered by this drug.[405] Patients taking tricyclic and tetracyclic antidepressants can safely receive concomitant topical epinephrine therapy.
ADRENERGIC AGONISTS: ?2-SELECTIVE
CHARACTERISTICS, MECHANISMS, AND SIDE EFFECTS
Apraclonidine hydrochloride and brimonidine tartrate are topical ?2-agonists that lower intraocular pressure by decreasing aqueous formation. Brimonidine also acts to increase uveoscleral outflow.[406,407]Much of the information regarding their metabolism and potential side effects is based on the related medication clonidine. These compounds are not identical, however, since clonidine has greater blood-brain permeability owing to its lipophilic nature, resulting in penetration into the central nervous system, where it decreases peripheral vascular resistance and lowers blood pressure. Topically administered clonidine has been shown to cause significant systemic hypotension.[408] Brimonidine is significantly less lipophilic than clonidine, and apraclonidine is hydrophilic, making central nervous system penetration, and therefore systemic hypotension, less likely with these two agents. Brimonidine also has the added advantage of being more ?2-selective than either clonidine or apraclonidine, resulting in a lower incidence of ocular side effects such as conjunctival blanching and pupillary dilation.[409] Even though no clinically significant cardiovascular effects were reported in studies of apraclonidine and brimonidine, there have been small but statistically significant effects on systolic and diastolic blood pressure in some studies.[48,409-411] Measurable plasma levels of apraclonidine have been detected up to 5 h after a single drop of 0.5% apraclonidine.[411] Therefore, there is potential for systemic side effects and interaction with systemic medication. There is only one drug interaction for topical apraclonidine and brimonidine cited in the literature (MAO inhibitors). In addition, a single case report was found describing a possible interaction with antihypertensive medication.[412]
INTERACTIONS WITH OTHER DRUGS
MAO Inhibitors (Isocarboxazid (Marplan), Phenelzine (Nardil), Pargyline (Eutonyl), Tranylcypromine (Parnate))
It is recommended in the USPDI and prescribing information for apraclonidine and brimonidine that topical ?2-agonists should not be used during or within 14 days after administration of MAO inhibitors because of the risk of hypertensive crisis. Since MAO is the enzyme partly responsible for the breakdown of epinephrine, it is postulated that adding an ?2-agonist will lead to potentiation of the effect of catecholamines. However, it is unlikely that a small amount of ?2-agonist absorbed from topical use would have such an effect. There are no reports of such an interaction in the literature.
Hypotension-Producing Medications
Because oral ?2-agonists such as clonidine cause a centrally mediated lowering of blood pressure, a theoretical additive hypotension could occur with administration of apraclonidine or brimonidine with any antihypertensive medication.
A 67-year-old white woman with a history of hypertension and diabetes received one drop of 1% apraclonidine 1 h before a scheduled laser trabeculoplasty. Her medications included insulin, 80 mg of furosemide daily and 50 mg of metoprolol three times daily. There was no prior history of syncope or heart disease. Ten minutes after instillation, she complained of chest tightness, and within 2 min, a radial pulse became undetectable. The patient was placed supine and given intravenous fluid. The first blood pressure reading, ?5 min after the initial episode, was 170/80 with a regular pulse of 70 beats/min and a blood glucose level of 180 mg/dL.[412]
Since this patient's first measured blood pressure and pulse were normal, it is difficult to conclude what exactly occurred. It is possible this was a vasovagal reaction that was not specific to apraclonidine. Since studies show only small effects on blood pressure or pulse rate with apraclonidine, it is difficult to make any recommendation for avoidance of its use in patients being treated for hypertension.
CHOLINERGIC AGENTS
CHARACTERISTICS, MECHANISMS, AND SIDE EFFECTS
Direct Acting
There are few reports describing systemic side effects after topical ophthalmic pilocarpine or carbachol administration.[386,413-415] Systemic levels must therefore be low, although there are no specific studies of the systemic concentration after ophthalmic application. However, when given excessively, pilocarpine and carbachol can directly stimulate cholinergic receptors that are found throughout the body. Locations include interneurons and postganglionic neurons of sympathetic and parasympathetic ganglia as well as neurons in the central nervous system. Stimulation by nicotinic agonists causes skeletal muscle contraction. Stimulation by muscarinic agonists causes smooth muscle contraction, salivary and sweat gland stimulation, pulmonary bronchoconstriction, and gastrointestinal stimulation with nausea, vomiting, and diarrhea. Central nervous system effects include depression, anxiety, headache, tremor, and ataxia. Effects on the cardiovascular system depend on the interplay between cholinergic stimulation of the heart's muscarinic receptors causing bradycardia, negative inotropy, and vasodilation and sympathetic response to these changes resulting in the release of epinephrine and norepinephrine with subsequent tachycardia, increased inotropy, and vasoconstriction.[416] Thus, it is difficult to predict whether cholinergic side effects and toxicity will result in an increase or decrease in heart rate and blood pressure.
Pilocarpine is a muscarinic agonist. One drop of pilocarpine, 4% (20 drops/mL), contains ?2.0 mg of the drug, which is ?20% of the subcutaneous dose capable of causing diaphoresis.[414] Such a toxicity has been reported after the repeated administration of pilocarpine during attacks of angle-closure glaucoma.[415] However, in routine use, systemic side effects due to pilocarpine are rare. This is reflected by the paucity of articles describing potential drug interactions. There is a report of one patient who received pilocarpine, 2%, and timolol maleate, 0.5%, the evening before surgery. This patient experienced bradycardia and hypotension during halothane anesthesia. However, there were undetectable serum plasma levels of pilocarpine (<2 ng/mL) during the surgery.[347] Instead, halothane in combination with a detectable serum timolol level and preexisting cardiovascular disease were the more likely culprits. Carbachol, unlike pilocarpine, is both a muscarinic and a nicotinic agonist. Skeletal muscle stimulation with muscle cramps, fasciculations, and eventual severe weakness and paralysis would be expected to occur in addition to the signs and symptoms of toxicity already discussed with a muscarinic agonist such as pilocarpine. Although the risk is small, ophthalmologists should at least be aware of the potential for interaction between direct-acting cholinergic agents and medications affecting cardiovascular, pulmonary, and gastrointestinal function.
Ophthalmic Cholinesterase Inhibitors
Ophthalmic cholinesterase inhibitors include physostigmine, demecarium, isoflurophate, and echothiophate. Rather than directly stimulating cholinergic receptors, these medications act by inhibiting the cholinesterase enzyme responsible for metabolism of acetylcholine in neural tissues and effector organs. Physostigmine is termed a reversible inhibitor because the chemical tie-up of the cholinesterase enzyme may be hydrolyzed by water in a matter of hours. Echothiophate, demecarium, and isoflurophate are irreversible inhibitors because they form a bond with cholinesterase that may not be hydrolyzed by water for several days, after which the cholinesterase enzyme is permanently altered and rendered inactive.[403] Because acetylcholine is found at muscarinic and nicotinic receptors, these cholinesterase inhibitors can produce a variety of cardiovascular, pulmonary, central nervous system, and gastrointestinal effects.[417] When cholinesterase inhibitors are lethal (e.g., insecticides such as malathion), they act by paralyzing the respiratory muscles, which have been overstimulated by the accumulation of acetylcholine. Central nervous system effects such as headache, anxiety, confusion, and depression are more likely to occur with isoflurophate because of its high lipid solubility, which allows it to more easily cross the blood-brain barrier as compared with echothiophate, which carries a positive charge and is more hydrophilic.[413] After topical ophthalmic administration of any of these medications, measured decreases in erythrocyte and plasma cholinesterase activity have been shown.[417,418] These decreases could result in interaction with systemically administered medications as described further on.
INTERACTIONS WITH OTHER DRUGS
Anesthetic Agents-Local, Ester-Derivative (Cocaine, Procaine, Tetracaine)
Prolonged ophthalmic use of anticholinesterase medications leads to reduced cholinesterase activity and a subsequent decrease in procaine hydrolysis. Patients with inherited atypical plasma cholinesterase have experienced severe reactions, including cardiovascular collapse and convulsions, after the injection of procaine for local anesthesia.[419] This may be due to a failure to de-esterify any procaine that is absorbed into the systemic circulation. Theoretically, a vasoconstrictor such as epinephrine in the anesthet ic solution would retard systemic absorption and reduce the chance of toxicities such as cardiac depression, peripheral vasodilatation, and respiratory depression. Patients receiving these topical ophthalmic cholinesterase inhibitors should be given procaine with caution. However, a better alternative would be the use of amide compounds (mepivacaine, bupivacaine, and lidocaine) for local anesthesia. These compounds are not esters and, hence, are not hydrolyzed by serum cholinesterase. Instead, they are metabolized in the liver. In addition, amide local anesthetics have less intrinsic toxic potency than do ester compounds and would therefore be safer overall to administer to patients receiving cholinesterase inhibitors.
Cholinesterase Inhibitors (Edrophonium (Tensilon), Neostigmine (Prostigmin), Pyridostigmine (Mestinon))
Systemically administered cholinesterase inhibitors are most commonly used in the treatment of myasthenia gravis. Owing to the ability of topical ophthalmic preparations to reduce serum cholinesterase levels, there is a theoretical risk of additive toxicity when they are combined with systemic cholinesterase inhibitors.[417,418] However, there is no available documentation of such an interaction in the literature. No specific contraindication to the use of these two types of medications together is warranted.
Succinylcholine (Anectine, Quelicin)
Succinylcholine is a neuromuscular blocking agent that causes skeletal muscle paralysis. It has a short half-life; effects from a 30-mg intravenous dose last ?5 min. Serum cholinesterase is the enzyme responsible for metabolizing succinylcholine, an ester compound whose structure consists of two molecules of acetylcholine bonded together at their choline ends.[420] Topical ophthalmic cholinesterase inhibitors can cause profound and long-lasting depression of serum cholinesterase levels, especially with the irreversible inhibitors echothiophate and isoflurophate.[417,418,421]
Two cases of prolonged apnea due to succinylcholine-induced respiratory muscle paralysis have been reported with the administration of succinylcholine to patients receiving topical echothiophate therapy. In the first case, a 12-year-old boy with congenital glaucoma was administered 40 mg of succinylcholine to facilitate endotracheal intubation before cryotherapy for his left eye. He had been using 0.125% echothiophate iodide twice daily in both eyes for 9 months. He experienced a 45-min period of apnea after the succinylcholine administration.[422] In the second case, a 72-year-old woman received a total of 200 mg of succinylcholine during surgical exploration for a presumed small-bowel obstruction. She experienced a 5-h period of apnea after the procedure. The dosage and duration of previous echothiophate iodide therapy were not described in the report.[423] Topical cholinesterase inhibitors should be discontinued, if possible, 4-6 weeks before the use of succinylcholine.[424] If the anesthetist is notified, reduced amounts of succinylcholine or a nondepolarizing neuromuscular blocking agent (atracurium, pancuronium, and tubocurarine) can be used because the latter is not metabolized by serum cholinesterase.
CARBONIC ANHYDRASE INHIBITORS (ORAL AND TOPICAL)
CHARACTERISTICS, MECHANISMS, AND SIDE EFFECTS
Carbonic anhydrase (CA) is an enzyme found in almost all tissues of the body, including the eye. CA exists as a number of isoenzymes, the most active being CA II, which is found primarily in red blood cells but is also the predominant form of the enzyme found in the ciliary processes. By inhibiting this enzyme in the ciliary processes, bicarbonate ion formation is slowed, with a subsequent reduction in sodium and fluid transport and a resultant decrease in aqueous formation. In the kidneys, CA inhibition results in loss of bicarbonate, which carries out sodium, water, and potassium. This leads to alkalinization of the urine and a serum metabolic acidosis, both of which can affect the metabolism and elimination of other medications.[425] These metabolic effects result in a large number of potential interactions.
Systemic side effects of oral CA inhibitors include paresthesias, anorexia, weight loss, diarrhea, fatigue, malaise, depression, loss of libido, renal stones, metabolic acidosis, acute respiratory failure, and rarely idiosyncratic leukopenia, thrombocytopenia, and aplastic anemia.
Acetazolamide and methazolamide are the two most commonly used oral CA inhibitors. The majority of available interaction and side effect data is based on acetazolamide, presumably because it has been in use longer. Methazolamide exhibits lower plasma protein binding and increased hepatic metabolism and lipid solubility and also induces less acidosis compared with acetazolamide. It can therefore be used at a lower dose, making side effects and interactions somewhat less likely.
Dorzolamide hydrochloride is a CA inhibitor formulated for topical ophthalmic use. Studies of both healthy adults and patients with glaucoma show no change in blood and urine acid-base or electrolyte levels after using dorzolamide for up to 4 weeks.[426] However, dorzolamide has been shown to decrease mean CA isoenzyme II activity in red blood cells. In one study, CA isoenzyme II activity decreased to 21% of baseline activity after 28 days of treatment.[207] Although at first glance this would seem significant, it is thought that 99% inhibition is necessary to induce physiologic effects.[427] However, the half-life in red blood cells is reported to be ?120 days, and thus there is potential for long-term accumulation. Despite this, there have been no reported interactions between dorzolamide and systemic medications. Any potential interactions would have to be based on oral CA inhibitors.
There are many potential interactions with oral CA inhibitors because of their metabolic effects. Discussed in the following section are potential interactions that are documented in the literature. There are a number of interactions listed in the USPDI and PDR that are not specifically substantiated in the literature but are based on known properties of CA. For example, since CA inhibitors act as diuretics, they will potentiate the activity of any diuretic that a patient is also taking. These are listed as potential interactions under each individual agent or class of medication.
INTERACTIONS WITH OTHER DRUGS
Amphotericin B (Fungizone)
CA inhibitors can potentiate the hypokalemia caused by amphotericin B. No specific reports describing this interaction are available.
Anticholinergics (Atropine and Related Compounds)
CA inhibitors increase the alkalinity of the urine, thereby increasing the amount of nonionized drug available for renal tubular reabsorption. This would potentially prolong or enhance the action of anticholinergic medications. No specific reports describing this interaction are available.
?-Adrenergic Blockers (Oral and Topical)
The combination of ?-adrenergic blockers and CA inhibitors may cause additive pharmacologic activity because of the metabolic acidosis of CA inhibitors and the respiratory acidosis induced by ?-adrenergic blockers. A 73-year-old patient with severe chronic pulmonary obstructive disease was given oral acetazolamide (750 mg daily) and timolol maleate (0.5% ophthalmic solution, one drop in each eye twice daily) before glaucoma surgery.[428] Progressive dyspnea was noted 5 days after treatment began and required bronchodilator therapy. Arterial blood gas measurements revealed a mixed acidosis, and acetazolamide was stopped. Although the metabolic acidosis improved, respiratory acidosis continued and did not improve until timolol was stopped.
Although no data are available for other ?-adrenergic blockers, the potential for respiratory compromise in patients taking ?-blockers is well known. Patients taking both CA inhibitors and ?-blockers need to be monitored carefully for severe mixed acidosis.
Corticosteroids (Prednisone, Hydrocortisone)
Potential interaction: CA inhibitors can potentiate the hypokalemia caused by corticosteroids.
Cyclosporine (Sandimmune)
The pharmacologic effects of cyclosporine may be increased by CA inhibitors, resulting in an increased risk of neurologic and renal toxicity. This interaction is based on the case of a 50-year-old male heart transplant patient who was stabilized on cyclosporine and multiple other medications. He was given acetazolamide, 1 g/day, along with timolol and topical steroid drops for glaucoma secondary to iritis. Soon after, he experienced dizziness and anorexia, and laboratory results showed diminished renal function and hyperchloremic metabolic acidosis with cyclosporine blood concentrations nearly six times the preacetazolamide concentration. Within 5 days of discontinuing acetazolamide and holding cyclosporine for 36 h, his symptoms resolved and the serum creatinine level returned to normal.[429]
It is not clear to what extent the adverse effects may be attributed to either acetazolamide or cyclosporine alone. The mechanism of potential interaction is also unknown. Based on this limited evidence, no specific recommendation other than monitoring cyclosporine plasma concentrations can be given.
Digitalis Preparations (Digoxin (Lanoxin))
Digitalis toxicity is associated with hypokalemia. Since CA inhibitors can result in hypokalemia, patients receiving both these types of medications are at an increased risk for digoxin toxicity if hypokalemia develops.
Diuretics (Furosemide (Lasix), Hydrochlorothiazide (Dyazide))
CA inhibitors act as diuretics because of their renal effects and would therefore enhance the activity of other diuretics administered concomitantly. Hypokalemia and hyperuricemia would also be potentiated.
EPHEDRINE
LITHIUM (ESKALITH)
The renal excretion of lithium may be increased by CA inhibitors, resulting in decreased pharmacologic effects of lithium. In a study of six subjects, lithium excretion was greatly increased (30%) by the administration of acetazolamide.[430] The exact mechanism is unknown, although urine alkalinization is believed to play a part. However, over the long term, CA inhibitors deplete bicarbonate, thereby reducing their ability to alkalinize the urine. During initial treatment with both these medications, serum lithium levels should be monitored.
MEXILETINE (MEXITIL)
Mexiletine is an oral antiarrhythmic agent used for the treatment of ventricular arrhythmias. Potential interaction: CA inhibitors increase the alkalinity of the urine, thereby increasing the amount of nonionized drug available for renal tubular reabsorption. This would potentially prolong or enhance the action of mexiletine. No specific reports describing this interaction are available.
PHENYTOIN (DILANTIN)
One report suggested that using phenytoin and acetazolamide can accelerate the osteomalacia induced by anticonvulsant therapy. However, a direct cause-and-effect relationship was not established because the patients were taking other medications. A possible mechanism is acetazolamide's enhancement of urinary calcium excretion.[431] There are no other reports of the clinical significance of this interaction. Special attention to the detection of osteomalacia is recommended when these two agents are used together.
PRIMIDONE (MYSOLINE)
The anticonvulsant effectiveness of primidone may be decreased by CA inhibitors. The mechanism involves inhibition of gastrointestinal absorption. A 15-year-old girl treated with primidone, phenytoin, and acetazolamide for a seizure disorder experienced loss of seizure control associated with undetectable primidone and phenobarbital plasma levels.[432] Under controlled conditions, acetazolamide was shown to inhibit the absorption of a single 500-mg dose of primidone completely in this patient. Studies were performed in two more patients. No interaction was found in one patient, whereas acetazolamide greatly decreased the absorption of primidone in the other patient.
The exact mechanism of the inhibition of primidone absorption by acetazolamide is not known. Since there is a potential for loss of seizure control, patients taking acetazolamide and primidone need to be monitored carefully.
QUINIDINE (QUINAGLUTE, CARDIOQUIN)
Decreased renal excretion of quinidine may occur, with a resultant increase in quinidine plasma levels and potential for toxic reactions such as cardiac conduction disturbances and arrhythmias.[433]Alkalinization of the urine that occurs with acetazolamide leads to diminished renal clearance of quinidine. This mechanism is based on short-term studies of healthy volunteers given both medications. However, CA inhibitors during long-term use deplete body stores of bicarbonate, thereby reducing their ability to alkalinize the urine. In addition, only 15-20% of quinidine is excreted unchanged in the urine. During initial treatment with both acetazolamide and quinidine, toxic plasma levels of quinidine may occur with alkalinization of urine. Long-term clinical effects are less clear.
SALICYLATES (ASPIRIN, DIFLUNISAL (DOLOBID))
The risk of salicylate toxicity may be increased during concomitant therapy because the metabolic acidosis induced by CA inhibitors may increase penetration of salicylate into the brain. Several cases of systemic acidosis with salicylate intoxication have been reported in glaucoma patients taking CA inhibitors and high-dose salicylates.[434,435] Salicylates have also been shown to displace acetazolamide from its protein-binding sites and decrease its clearance by inhibiting renal tubular excretion. This suggests that toxicity with these two classes of medications may also be due to increased CA inhibitor serum levels.
Conversely, alkalinization of the urine by CA inhibitors increases salicylic acid excretion and decreases plasma concentrations. Over time, however, CA inhibitors deplete body stores of bicarbonate, reducing the body's ability to alkalinize the urine and possibly reducing this protective excretion of salicylate. Long-term clinical consequences are unknown, but based on reported interactions, caution must be exercised in using salicylates with CA inhibitors.[436,437]
The reader is referred to the section on Anticholinergics for more information.
PROSTAGLANDIN ANALOGS
CHARACTERISTICS, MECHANISMS, AND SIDE EFFECTS
Latanoprost is a prostaglandin analog that is believed to increase aqueous outflow by augmenting the uveoscleral outflow. It is currently available as a 0.005% solution. Although prostaglandins can cause bronchoconstriction in humans, studies of latanoprost show no effect on respiratory function in asthmatic patients.438 Also, no effect has been found on blood pressure and heart rate and urine or serum chemistry. Systemic side effects and interaction with systemic medications would appear to be unlikely because of its once-daily dosing, the low concentration used, and the lack of any measurable side effects in current studies.[245,246] However, it has not been available long enough to a wide variety of patients to make a definitive statement regarding systemic effects.
CONCLUSIONS
The potential for systemic side effects and interactions exists when topical glaucoma medications are applied in patients, many of whom are elderly and are on multiple medications. We have stressed, however, the lack of clinical evidence and documentation for many of the interactions stated in the PDR and USPDI. This supports the excellent track record for safety of these drugs. In addition, there are methods to minimize systemic absorption, such as nasolacrimal occlusion and eyelid closure. Although topical ?-blockers are the most commonly used agents, newer medications with less potential for systemic side effects and interactions may supplant ?-blockers because of their added safety. Clinicians, however, should still be aware of these possible side effects and interactions of all topical medications and keep them in mind when prescribing. Communication with the patient and with the patient's other physicians is important when initiating topical therapy for the treatment of glaucoma.
REFERENCES
1. Pohjanpelto PEJ, Plava J: Ocular hypertension and glaucomatous optic nerve damage. Acta Ophthalmol 1974; 52:194.
2. Anderson DR: Glaucoma: the damage caused by pressure. Am J Ophthalmol 1989; 108:485.
3. Bengtsson B: The prevalence of glaucoma. Br J Ophthalmol 1981; 65:46.
4. Schwartz B, Talusan AG: Spontaneous trends in ocular pressure in untreated ocular hypertension. Arch Ophthalmol 1980; 98:105.
5. Glaucoma Panel: Primary open-angle glaucoma. In: Preferred Practice Patterns Committee, ed. American Academy of Ophthalmology Preferred Practice Patterns, San Francisco: American Academy of Ophthalmology; 2005.
6. Kass MA, Hart WM Jr, Gordon M, et al: Risk factors favoring the development of glaucomatous visual field loss in ocular hypertension. Surv Ophthalmol 1980; 25:155.
7. Shaffer RN: 'Glaucoma suspect' or 'ocular hypertension'?. Arch Ophthalmol 1977; 95:588.
8. Heijl A, Leske MC, Bengtsson B, et al: Reduction of intraocular pressure and glaucoma progression: results from the early manifest glaucoma trial. Arch Ophthalmol 2002; 120:1268-1279.
9. Kass MA, Heuer DK, Higginbotham EJ, et al: The ocular hypertension treatment study: a randomized trial determines that topical ocular hypotensive medication delays or prevents the onset of primary open-angle glaucoma. Arch Ophthalmol 2002; 120:701-713.discussion 829-830.
10. AGIS Investigators: The Advanced Glaucoma Intervention Study (AGIS): 7. The relationship between control of intraocular pressure and visual field deterioration. Am J Ophthalmol 2000; 130:429-440.
11. Lichter PR, Musch DC, Gillespie BW, et al: Interim clinical outcomes in the collaborative initial glaucoma treatment study comparing initial treatment randomized to medications or surgery. Ophthalmology 2001; 108:1943-1953.
12. Collaborative Normal-Tension Glaucoma Study Group: Comparison of glaucomatous progression between untreated patients with normal-tension glaucoma and patients with therapeutically reduced intraocular pressures. Am J Ophthalmol 1998; 126:487-497.
13. Epstein DL, Krug JH Jr, Hertzmark E, et al: A long term clinical trial of timolol therapy versus no treatment in the management of glaucoma suspects. Ophthalmology 1989; 96:1460.
14. Higginbotham EJ: Medication is the treatment of choice for chronic open angle glaucoma. Arch Ophthalmol 1998; 116:239.
15. Kass MA, Gordon MO, Hoff MR, et al: Topical timolol administration reduces the incidence of glaucomatous damage in ocular hypertensive individuals: a randomized, double masked, long term clinical trial. Arch Ophthalmol 1989; 107:1590.
16. Schulzer M, Drance SM, Douglas GR: A comparison of treated and untreated glaucoma suspects. Ophthalmology 1991; 98:301.
17. Kass MA, Meltzer DW, Gordon M, et al: Compliance with topical pilocarpine treatment. Am J Ophthalmol 1986; 101:515.
18. Kass MA, Gordon M, Morley Jr RE, et al: Compliance with topical timolol treatment. Am J Ophthalmol 1987; 103:188.
19. Kass MA, Gordon M, Meltzer DW: Can ophthalmologists identify patients defaulting from pilocarpine therapy?. Am J Ophthalmol 1986; 101:524.
20. Murphy J, Coster G: Issues in patient compliance. Drugs 1997; 54:797.
21. Grunwald JE, Furubayashi C: Effect of topical timolol maleate on the ophthalmic artery blood pressure. Invest Ophthalmol Vis Sci 1989; 30:1095.
22. Pillunat LE, Stodmeister R, Wilmanns I, et al: Effect of timolol on optic nerve need regulation. Ophthalmologica 1986; 193:146.
23. Drance SM, Flammer J: Some effects of antiglaucoma drugs on visual function. In: Drance SM, Neufeld AH, ed. Glaucoma applied pharmacology in medical treatment, New York: Grune & Stratton; 1984.
24. Collignon Brach J: Long term effect of ophthalmic beta-adrenoceptor antagonists on intraocular pressure and retinal sensitivity in primary open angle glaucoma. Curr Eye Res 1992; 11:1.
25. Collignon Brach J: Long term effect of topical beta blockers on intraocular pressure and visual field sensitivity in ocular hypertension and chronic open angle glaucoma. Surv Ophthalmol 1994; 38(Suppl):149.
26. Messmer C, Flammer J, Stumpfig D: Influence of betaxolol and timolol on the visual fields of patients with glaucoma. Am J Ophthalmol 1991; 112:678.
27. Kaiser HJ, Flammer J, Stumpfig D, Hendrickson P: Longterm visual field follow-up of glaucoma patients treated with beta-blockers. Surv Ophthalmol 1994; 38(Suppl):S156-S159.discussion S60
28. Drance SM: A comparison of the effects of betaxolol, timolol, and pilocarpine on visual function in patients with open angle glaucoma. J Glaucoma 1998; 7:247.
29. Jampel HD: Target pressure in glaucoma therapy. J Glaucoma 1997; 6:133.
30. Janz N, Wren PA, CIGTS Study Group: Implementing quality of life in a clinical trial. How to ascertain progression and outcome. In: Anderson DR, Drance SM, eds. The collaborative initial glaucoma treatment study, 1996; Encounters in glaucoma research 3.
31. Quigley HA, Addicks EM, Green WR: Optic nerve damage in human glaucoma. III. Quantitative correlation of nerve fiber loss and visual field defect in glaucoma, ischemic neuropathy, papilledema, and toxic neuropathy. Arch Ophthalmol 1982; 100:135.
32. Quigley HA, Dunkelberger GR, Green WR: Retinal ganglion cell atrophy correlated with automated perimetry in human eyes with glaucoma. Am J Ophthalmol 1982; 107:135.
33. Hart WM, Yablonski M, Kass MA, et al: Quantitative visual field and optic disc correlates early in glaucoma. Arch Ophthalmol 1978; 96:2209.
34. Grant WM, Burke Jr JF: Why do some people go blind from glaucoma?. Ophthalmology 1982; 89:991.
35. Graham PA: The definition of pre glaucoma: a prospective study. Eye 1969; 88:153.
36. European Glaucoma Society: Terminology and guidelines for glaucoma. In: Society EG, ed. 2003
37. Glaucoma Panel: Primary open-angle glaucoma suspect. In: Preferred Practice Patterns Committee, ed. American academy of ophthalmology preferred practice patterns, San Francisco: American Academy of Ophthalmology; 2005.
38. Allen RC: Aspects of clonidine therapy. New Engl J Med 1976; 294:845.
39. Jampel H, Robin AL, Quigley HA, et al: Apraclonicine: a one week dose response study. Arch Ophthalmol 1988; 106:1069.
40. Nagasubramanian S, Hitchings RA, Demailly P, et al: Comparison of apraclonidine and timolol in chronic open angle glaucoma: a three month study. Ophthalmology 1993; 100:1318.
41. Robin AL, Ritch R, Shin DH, et al: The short term efficacy of apraclonidine hydrochloride when maximum tolerated medical therapy fails to control intraocular pressure. Am J Ophthalmol 1995; 120:423.
42. Yaldo MK, Shin DH, Kyle AP, et al: Additive effect of 1% apraclonidine hydrochloride to nonselective beta blockers. Ophthalmology 1991; 98:1075.
43. Butler P, Mannschreck M, Lin S, et al: Clinical experience with the long term use of 1% apraclonidine incidence of allergic reactions. Arch Ophthalmol 1995; 113:293.
44. Burke J, Schwartz M: Preclinical evaluation of brimonidine. Surv Ophthalmol 1996; 41(Suppl 1):S9.
45. Greenfield DS, Liebmann JM, Ritch R: Brimonidine: a new alpha2 adrenoreceptor agonist for glaucoma treatment. J Glaucoma 1997; 6:250.
46. David R, Spaeth GL, Clevenger CE, et al: Brimonidine in the prevention of intraocular pressure elevation following argon laser trabeculoplasty. Arch Ophthalmol 1993; 111:1387.
47. Schuman JS: Clinical experience with brimonidine 0.2% and timolol 0.5% in glaucoma and ocular hypertension. Surv Ophthalmol 1996; 41(Suppl 1):S27.
48. Derick RJ, Robin AL, Walters TR, et al: Brimonidine tartrate: a one month dose response study. Ophthalmology 1997; 104:131.
49. Nordlund JR, Pasquale LR, Robin AL, et al: The cardiovascular, pulmonary, and ocular hypotensive effects of brimonidine tartrate 0.2%. Arch Ophthalmol 1995; 113:77.
50. Al-Shahwan S, Al-Torbak AA, Turkmani S, et al: Side-effect profile of brimonidine tartrate in children. Ophthalmology 2005; 112:2143.
51. Katz LJ: Twelve-month evaluation of brimonidine-purite versus brimonidine in patients with glaucoma or ocular hypertension. J Glaucoma 2002; 11:119.
52. Sharpe ED, Day DG, Beischel CJ, et al: Brimonidine purite 0.15% versus dorzolamide 2% each given twice daily to reduce intraocular pressure in subjects with open angle glaucoma or ocular hypertension. Br J Ophthalmol 2004; 88:953.
53. Mandell AI, Stentz F, Kitabchi AE: Dipivalyl epinephrine: a new pro drug in the treatment of glaucoma. Ophthalmology 1978; 85:268.
54. Kass MA, Mandell AL, Goldberg L, et al: Dipivefrin and epinephrine treatment of elevated intraocular pressure: a comparative study. Arch Ophthalmol 1979; 97:1865.
55. Kohn AN, Moss AP, Hargett NA, et al: Clinical comparison of dipivalyl epinephrine and epinephrine in the treatment of glaucoma. Am J Ophthalmol 1979; 87:196.
56. Krieglstein GK, Leydhecker W: The dose response relationships of dipivalyl epinephrine in open angle glaucoma. Graefes Arch Clin Exp Ophthalmol 1978; 205:141.
57. Townsend DJ, Brubaker RF: Immediate effect of epinephrine on aqueous formation in the normal human eye as measured by fluorophotometry. Invest Ophthalmol Vis Sci 1980; 19:256.
58. Garner LL, Johnstone WW, Ballintine EJ, et al: Effect of 2% levrotary epinephrine on the intraocular pressure of the glaucomatous eye. Arch Ophthalmol 1959; 62:230.
59. Krill AE, Newell FW, Novak M: Early and long term effects of levepinephrine on ocular tension and outflow. Am J Ophthalmol 1965; 59:833.
60. Coleiro JA, Sigurdsson H, Lockyer JA: Follicular conjunctivitis on dipivefrin therapy for glaucoma. Eye 1988; 2:440.
61. Liesegang TJ: Bulbar conjunctival follicles associated with dipivefrin therapy. Ophthalmology 1985; 92:228.
62. Kerr CR, Hass I, Drance SM, et al: Cardiovascular effects of epinephrine and dipivalyl epinephrine applied topically to the eye in patients with glaucoma. Br J Ophthalmol 1982; 66:109.
63. Allen RC: Medical treatment of open angle glaucoma. In: Weinstein G, ed. Open angle glaucoma, Boston: Little, Brown; 1985.
64. Zimmerman TJ, Kaufman HE: Timolol. A beta adrenergic blocking agent for the treatment of glaucoma. Arch Ophthalmol 1977; 95:601.
65. Walters TR, DuBiner HB, Carpenter SP, et al: 24-Hour IOP control with once-daily bimatoprost, timolol gel-forming solution, or latanoprost: a 1-month randomized comparative clinical trial. Surv Ophthalmol 2004; 49(Suppl 1):S26-S35.
66. Coakes RL, Brubaker RF: The mechanism of timolol in lowering intraocular pressure in the normal eye. Arch Ophthalmol 1978; 96:2045.
67. Sonntag JR, Brindley GO, Shields MB: Effect of timolol therapy on outflow facility. Invest Ophthalmol Vis Sci 1978; 17:293.
68. Zimmerman TJ, Harbin R, Pett M, et al: Timolol and facility of outflow. Invest Ophthalmol Vis Sci 1977; 16:623.
69. Liu JH, Kripke DF, Weinreb RN: Comparison of the nocturnal effects of once-daily timolol and latanoprost on the intraocular pressure. Am J Ophthalmol 2004; 138:389-395.
70. Topper JE, Brubaker RF: Effects of timolol, epinephrine, and acetazolamide on aqueous flow during sleep. Invest Ophthalmol Vis Sci 1985; 26:1315.
71. Boger III WP: Short term 'escape' and long term 'drift': the dissipation effects of the beta adrenergic blocking agents. Surv Ophthalmol 1983; 28:235.
72. Wandel T, Fishman D, Novack GD, et al: Ocular hypotensive efficacy of 0.25% levobunolol instilled once daily. Ophthalmology 1988; 95:252.
73. Yalon M, Urinowsky E, Rothkoff L, et al: Frequency of timolol administration. Am J Ophthalmol 1981; 92:526.
74. Radius RL, Diamond GR, Pollack IP, et al: Timolol: a new drug for management of chronic simple glaucoma. Arch Ophthalmol 1978; 96:1003.
75. Van Buskirk EM: Adverse reactions from timolol administration. Ophthalmology 1980; 87:447.
76. Liu GS, Basu PK, Trope GE: Ultrastructural changes of the rabbit corneal epithelium and endothelium after timoptic treatment. Graefes Arch Clin Exp Ophthalmol 1987; 225:325.
77. Samochowiec-Donocik E, Koraszewska-Matuszeswka B: Influence of beta-adrenergic antagonists on tear secretion in children. Pol J Pharmacol 2004; 56:871-873.
78. McMahon CD, Shaffer RN, Hoskins Jr HD, et al: Adverse effects experienced by patients taking timolol. Am J Ophthalmol 1979; 88:736.
79. Wilson RP, Spaeth GL, Poryzees E: The place of timolol in the practice of ophthalmology. Ophthalmology 1980; 87:451.
80. Edeki TI, He H, Wood AJ: Pharmacogenetic explanation for excessive beta blockade following timolol eye drops. Potential for oral ophthalmic drug interaction. JAMA 1995; 274:1611.
81. Nieminen T, Uusitalo H, Maenpaa J, et al: Polymorphisms of genes CYP2D6, ADRB1 and GNAS1 in pharmacokinetics and systemic effects of ophthalmic timolol. A pilot study. Eur J Clin Pharmacol 2005; 61:811-819.
82. Fraunfelder FT: Interim report: national registry of possible drug induced ocular side effects. Ophthalmology 1980; 87:87.
83. Nieminen T, Uusitalo H, Turjanmaa V, et al: Association between low plasma levels of ophthalmic timolol and haemodynamics in glaucoma patients. Eur J Clin Pharmacol 2005; 61:369-374.
84. Coleman AL, Diehl DLC, Jampel HD, et al: Topical timolol decreased plasma high density lipoprotein cholesterol level. Arch Ophthalmol 1990; 108:1260.
85. Fraunfelder FT, Meyer SM, Menacker SJ: Alopecia possibly secondary to topical ophthalmic beta-blocker. JAMA 1990; 263:1493.
86. Lawrsen SO, Bjerrum P: Timolol eyedrop induced severe bronchospasm. Acta Med Scand 1982; 211:505.
87. Burnstine RA, Felton JL, Ginther WH: Cardiorespiratory reaction to timolol maleate in pediatric patient: a case report. Ann Ophthalmol 1982; 14:905.
88. Guzman CA: Exacerbation of bronchorrhea induced by topical timolol [letter]. Am Rev Respir Dis 1980; 121:899.
89. Jones Jr FL, Ekberg NL: Exacerbation of asthma by timolol. N Engl J Med 1979; 301:270.
90. Nelson WL, Fraunfelder FT, Sills JM, et al: Adverse respiratory and cardiovascular events attributed to timolol ophthalmic solution, 1978-1985. Am J Ophthalmol 1986; 102:606.
91. Noyes JH, Chervinsky P: Case report: exacerbation of asthma by timolol. Ann Allergy 1980; 45:301.
92. Olson RJ, Bromberg BB, Zimmerman TJ: Apneic spells associated with timolol therapy in a neonate. Am J Ophthalmol 1979; 88:120.
93. Schoene RB, Martin TR, Charan NB, et al: Timolol induced bronchospasm in asthmatic bronchitis. JAMA 1981; 245:1460.
94. Stewart WC, Day DG, Holmes KT, Stewart JA: Effect of timolol 0.5% gel and solution on pulmonary function in older glaucoma patients. J Glaucoma 2001; 10:227-232.
95. Gandolfi SA, Chetta A, Cimino L, et al: Bronchial reactivity in healthy individuals undergoing long-term topical treatment with beta-blockers. Arch Ophthalmol 2005; 123:35-38.
96. Timoptic XE Study Group: Multiclinic, double-masked, study of 0.5% Timoptic XE once daily versus 0.5% Timoptic twice daily. Ophthalmology 1993; 100:111.
97. Laurence J, Holder D, Vogel R, et al: A double masked, placebo controlled evaluation of timolol in gel vehicle. J Glaucoma 1993; 2:177.
98. Shedden A, Laurence J, Tipping R, Group T-XS: Efficacy and tolerability of timolol maleate ophthalmic gel-forming solution versus timolol ophthalmic solution in adults with open-angle glaucoma or ocular hypertension: a six-month, double-masked, multicenter study. Clin Ther 2001; 23:440-450.
99. Mundorf TK, Ogawa T, Naka H, et al: A 12-month, multicenter, randomized, double-masked, parallel-group comparison of timolol-LA once daily and timolol maleate ophthalmic solution twice daily in the treatment of adults with glaucoma or ocular hypertension. Clin Ther 2004; 26:541-545.
100. Manni G, Centofanti M, Oddone F, et al: Interleukin-1 beta tear concentration in glaucomatous and ocular hypertensive patients treated with preservative-free nonselective beta-blockers. Am J Ophthalmol 2005; 139:72-77.
101. Baudouin C, Hamard P, Liang H, et al: Conjunctival epithelial cell expression of interleukins and inflammatory markers in glaucoma patients treated over the long term. Ophthalmology 2004; 111:2186-2192.
102. Bron A, Chiambaretta F, Pouliquen P, et al: Efficacy and safety of substituting a twice-daily regimen of timolol with a single daily instillation of nonpreserved beta-blocker in patients with chronic glaucoma or ocular hypertension. J Fr Ophtalmol 2003; 26:668-674.
103. Su CY, Yang YC, Peng CF, et al: Risk of microbial contamination of unit-dose eyedrops within twenty four hours after first opening. J Formos Med Assoc 2005; 104:968-971.
104. Berson FG, Cinotti A, Cohen H, et al: Levobunolol: a beta adrenoceptor antagonist effective in the long term treatment of glaucoma. Ophthalmology 1985; 92:1271.
105. Berson FG, Cohen HB, Foerster RJ, et al: Levobunolol compared with timolol for the long term control of elevated intraocular pressure. Arch Ophthalmol 1985; 103:379.
106. Cinotti A, Cinotti D, Grant W, et al: Levobunolol vs timolol for open angle glaucoma and ocular hypertension. Am J Ophthalmol 1985; 99:11.
107. Duzman E, Ober M, Scharrer A, Leopold IH: A clinical evaluation of the effects of topically applied levobunolol and timolol on increased intraocular pressure. Am J Ophthalmol 1982; 94:318.
108. The Levobunolol Study Group: Levobunolol: a four year study of efficacy and safety in glaucoma treatment. Ophthalmology 1989; 96:642.
109. DiCarlo FJ, Leinweber FJ, Szpiech JM, et al: Metabolism of L-bunolol. Clin Pharmacol Ther 1977; 22:858.
110. Woodward DF, Novack GD, Williams LS, et al: The ocular beta blocking activity of dihydrolevobunolol. J Ocul Pharmacol 1987; 3:11.
111. Wandel T, Charap AD, Lewis RA, et al: Glaucoma treatment with once daily levobunolol. Am J Ophthalmol 1986; 101:298.
112. Halper LK, Johnson-Pratt L, Dobbins T, Hartenbaum D: A comparison of the efficacy and tolerability of 0.5% timolol maleate ophthalmic gel-forming solution QD and 0.5% levobunolol hydrochloride BID in patients with ocular hypertension or open-angle glaucoma. J Ocul Pharmacol Ther 2002; 18:105-113.
113. Bensinger RE, Keates EU, Gofman JD, et al: Levobunolol: a three month efficacy study in the treatment of glaucoma and ocular hypertension. Arch Ophthalmol 1985; 103:375.
114. Boozman FW III, Carriker R, Foerster R, et al: Long term evaluation of 0.25% levobunolol and timolol for therapy for elevated intraocular pressure. Arch Ophthalmol 1988; 106:614.
115. Battershill PE, Sorkin EM: Ocular metipranolol. Drugs 1988; 36:601.
116. Dausch D, Brewitt H, Edelhoff R: Metipranolol eye drops: clinical suitability in the treatment of chronic open angle glaucoma. In: Merte HJ, ed. Metipranolol, New York: Springer; 1983.
117. Denffer H: Efficacy and tolerance of metipranolol: results of a multicenter long term study. In: Merte HJ, ed. Metipranolol, New York: Springer; 1983.
118. Ecoffet M, Demailly P: Resultats d'une étude à moyen terme à double in su comparant le metipranolol au timolol dans le traitement du glaucome primitif à angle ouvert. J Fr Ophtalmol 1987; 10:451.
119. Kruse W: Metipranolol: a new beta receptor blocking agent. Klin Monatsbl Augenheild 1983; 182:582.
120. Kruse W: Metipranolol in glaucoma therapy. Der Augenarzt 1983; 3:168.
121. Mertz M: Results of a 6 weeks' multicenter double blind trial, metipranolol versus timolol. In: Merte HJ, ed. Metipranolol, New York: Springer; 1983.
122. Mills KB, Wright G: A blind randomized cross over trial comparing metipranolol 0.3% with timolol 0.25% in open angle glaucoma: a pilot study. Br J Ophthalmol 1986; 70:39.
123. Muller O, Knobel HR: Efficacy and tolerance of metipranolol: results of a Swiss long term multicenter study. Klin Monatsbl Augenheilkd 1986; 188:62.
124. Schmitz Valchenberg P, Jonas J, Brambring DF: Reductions in pressure with metipranolol 0.1%. Z Prakt Augenheilkd 1984; 5:171.
125. Krieglstein GK, Novack GD, Voepel E, et al: Levobunolol and metipranolol: comparative ocular hypotensive efficacy, safety and comfort. Br J Ophthalmol 1987; 71:250.
126. Serle JB, Lustgarten JS, Podos SM: A clinical trial of metipranolol, a noncardioselective beta adrenergic antagonist, in ocular hypertension. Am J Ophthalmol 1991; 112:302.
127. Melles RB, Wong IG: Metipranolol-associated granulomatous iritis. Am J Ophthalmol 1994; 118:712-715.
128. Akingbehin T, Villada JR: Metipranolol-associated granulomatous anterior uveitis. Br J Ophthalmol 1991; 75:519-523.
129. Akingbehin T, Villada JR, Walley T: Metipranolol-induced adverse reactions: I. The rechallenge study. Eye 1992; 6(Pt 3):277-279.
130. Beck RW, Moke P, Blair RC, Nissenbaum R: Uveitis associated with topical beta-blockers. Arch Ophthalmol 1996; 114:1181-1182.
131. Bacon PJ, Brazier DJ, Smith R, et al: Cardiovascular responses to metipranolol and timolol eyedrops in healthy volunteers. Br J Clin Pharmacol 1989; 27:1.
132. Lyden M, Applebaum HJ: Effect of ophthalmic metipranolol and timolol on exercise induced tachycardia. J Glaucoma 1995; 4:124.
133. Schultz JS, Hoenig JA, Charles H: Possible bilateral anterior uveitis secondary to metipranolol (OptiPranolol) therapy. Arch Ophthalmol 1993; 111:1606.
134. Ball SF, Schneider E: Cost of beta-adrenergic receptor blocking agents for ocular hypertension. Arch Ophthalmol 1992; 110:654.
135. Chrisp P, Sorkin EM: Ocular carteolol. Drugs Aging 1992; 2:58.
136. Wellstein A, Palm D, Wiemeer G, et al: Simple and reliable radioreceptor assay for beta adrenoceptor antagonists and active metabolites in native human plasma. Eur J Clin Pharmacol 1984; 27:545.
137. Duff GR, Graham PA: A double crossover trial comparing the effects of topical carteolol and placebo on intraocular pressure. Br J Ophthalmol 1988; 72:890.
138. Horie T, Takahashi O, Shirato S, Kitazawa Y: Comparison of ocular hypotensive effects of topical timolol and carteolol. Jpn J Clin Pharmacol 1982; 36:1065.
139. Mills KB, Raines M, Joyce P: A single blind, stratified, randomized non crossover trial comparing carteolol 1% with timolol 0.25% in the long term management of glaucoma. Br J Clin Pract 1987; 41(Suppl 51):10.
140. Stewart WC, Shields MB, Allen RC, et al: A 3 month comparison of 1% and 2% carteolol and 0.5% timolol in open angle glaucoma. Graefes Arch Clin Exp Ophthalmol 1991; 229:258.
141. Tsuchisaka H, Kin K, Matsumoto S, et al: Multi-institutional evaluation of timolol and carteolol for glaucomas. Ganka Rinsho Iho 1991; 85:1136.
142. Scoville B, Mueller B, White BG, et al: A double masked comparison of carteolol and timolol in ocular hypertension. Am J Ophthalmol 1988; 105:150.
143. Watson PG, Barnett MF, Parker V, Haybittle J: A 7 year prospective comparative study of three topical beta blockers in the management of primary open angle glaucoma. Br J Ophthalmol 2001; 85:962-968.
144. Kitazawa Y: Multicenter double blind comparison of carteolol and timolol in primary open angle glaucoma and ocular hypertension. Adv Ther 1993; 10:95.
145. LeJeunne CL, Hughues FC, Dufier JL, et al: Bronchial and cardiovascular effects of ocular topical beta-antagonists in asthmatic subjects: comparison of timolol, carteolol, and metipranolol. J Clin Pharmacol 1989; 29:97.
146. Caldwell DR, Salisbury CR, Guzek JP: Effects of topical betaxolol in ocular hypertensive patients. Arch Ophthalmol 1984; 102:539.
147. Feghali JG, Kaufman PL: Decreased intraocular pressure in the hypertensive human eye with betaxolol: a beta-adrenergic antagonist. Am J Ophthalmol 1985; 100:777.
148. Radius RL: Use of betaxolol in the reduction of elevated intraocular pressure. Arch Ophthalmol 1983; 101:898.
149. Allen RC, Hertzmark E, Walker AM, et al: A double masked comparison of betaxolol vs timolol in the treatment of open angle glaucoma. Am J Ophthalmol 1986; 101:535.
150. Berry DP, Van Buskirk EM, Shields MB: Betaxolol and timolol: a comparison of efficacy and side effects. Arch Ophthalmol 1984; 102:42.
151. Stewart RH, Kimbrough RL, Ward RL: Betaxolol vs timolol: a six month double blind comparison. Arch Ophthalmol 1986; 104:46.
152. Long DA, Johns GE, Mullen RS, et al: Levabunolol and betaxolol: a double masked controlled comparison of efficacy and safety in patients with elevated intraocular pressure. Ophthalmology 1988; 95:735.
153. Reiss GR, Brubaker RF: The mechanism of betaxolol, a new ocular hypotensive agent. Ophthalmology 1983; 90:1369.
154. Vogel R, Tipping R, Kulaga Jr SF, et al: Changing therapy from timolol to betaxol: effects on intraocular pressure in selected patients with glaucoma. Arch Ophthalmol 1989; 107:1303.
155. Smith JP, Weeks RH, Newland EF, et al: Betaxolol and acetazolamide: combined ocular hypotensive effect. Arch Ophthalmol 1984; 102:1794.
156. Allen RC, Epstein DL: Additive effect of betaxolol and epinephrine in primary open angle glaucoma. Arch Ophthalmol 1986; 104:1178.
157. Weinreb RN, Ritch R, Kushner FH: Effect of adding betaxolol to dipivefrin therapy. Am J Ophthalmol 1986; 101:196.
158. Cyrlin MS, Thomas JV, Epstein DL: Additive effect of epinephrine to timolol therapy in primary open angle glaucoma. Arch Ophthalmol 1982; 100:414.
159. Keates EC, Stone RA: Safety and effectiveness of concomitant administration of dipivefrin and timolol maleate. Am J Ophthalmol 1981; 91:243.
160. Thomas JV, Epstein DL: Timolol and epinephrine in primary open angle glaucoma: transient additive effect. Arch Ophthalmol 1981; 99:91.
161. Wax MB, Molinoff PB: Distribution and properties of beta-adrenergic receptors in human iris ciliary body. Invest Ophthalmol Vis Sci 1987; 28:420.
162. Robinson JC, Kaufman PL: Effects and interactions of epinephrine, norepinephrine, timolol, and betaxolol on outflow facility in the cynomolgus monkey. Am J Ophthalmol 1990; 109:189.
163. Jampel HD, Lynch MG, Brown RH, et al: Beta-Adrenergic receptors in human trabecular meshwork: identification and autoradiographic localization. Invest Ophthalmol Vis Sci 1987; 28:772.
164. Wax MB, Molinoff PB, Alvarado J, et al: Characterization of beta-adrenergic receptors in cultured human trabecular cells and in human trabecular meshwork. Invest Ophthalmol Vis Sci 1989; 30:51.
165. Harris LS, Greenstein SH, Bloom AF: Respiratory difficulties with betaxolol. Am J Ophthalmol 1986; 102:274.
166. Roholt PC: Betaxolol and restrictive airway disease. Arch Ophthalmol 1987; 105:1172.
167. Atkins JM, Pugh BR Jr, Timewell RM: Cardiovascular effects of topical beta blockers during exercise. Am J Ophthalmol 1985; 99:173.
168. Araie M, Azuma I, Kitazawa Y: Influence of topical betaxolol and timolol on visual field in Japanese open-angle glaucoma patients. Jpn J Ophthalmol 2003; 47:199-207.
169. Miki H, Miki K: The effects on the intraocular pressure and visual field resulting from a switch in the treatment from timolol to betaxolol. J Ocul Pharmacol Therapeut 2004; 20:509-517.
170. Osborne NN, Wood JP, Chidlow G, et al: Effectiveness of levobetaxolol and timolol at blunting retinal ischaemia is related to their calcium and sodium blocking activities: relevance to glaucoma. Brain Res Bull 2004; 62:525-528.
171. Bojic L, Bagatin J, Ivanisevic M, et al: Influence of betaxolol and timolol on the venous tone in glaucoma patients. Int Ophthalmol 1999; 23:149-153.
172. Costa VP, Harris A, Stefansson E, et al: The effects of antiglaucoma and systemic medications on ocular blood flow. Prog Retin Eye Res 2003; 22:769-805.
173. Osborne NN, Wood JP, Chidlow G: Invited review: neuroprotective properties of certain beta-adrenoreceptor antagonists used for the treatment of glaucoma. J Ocul Pharmacol Ther 2005; 21:175-181.
174. Rainer G, Dorner GT, Garhofer G, et al: Changing antiglaucoma therapy from timolol to betaxolol: effect on ocular blood flow. Ophthalmologica 2003; 217:288-293.
175. Vainio-Jylha E, Vuori ML, Nummelin K: Progression of retinal nerve fiber layer damage in betaxolol-and timolol-treated glaucoma patients. Acta Ophthalmol Scand 2002; 80:495-500.
176. Yu DY, Su EN, Cringle SJ, et al: Systemic and ocular vascular roles of the antiglaucoma agents beta-adrenergic antagonists and Ca 2+ entry blockers. Surv Ophthalmol 1999; 43(Suppl 1):S214-S222.
177. Yarangumeli A, Kural G: Are there any benefits of Betoptic S (betaxolol HCl ophthalmic suspension) over other beta-blockers in the treatment of glaucoma?. Expert Opin Pharmacother 2004; 5:1071-1081.
178. Hollo G, Whitson JT, Faulkner R, et al: Concentrations of betaxolol in ocular tissues of patients with glaucoma andnormal monkeys after 1 month of topical ocular administration. Invest Ophthalmol Vis Sci 2006; 47:235-240.
179. Sugrue MF: Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog Retin Eye Res 2000; 19:87-112.
180. Maren TH: The rates of movement of Na+, Cl and HCO3? plasma to posterior chamber: effect of acetazolamide and relation to the treatment of glaucoma. Invest Ophthalmol Vis Sci 1976; 15:356.
181. Friedman Z, Krupin T, Becker B: Ocular and systemic effects of acetazolamide in nephrectomized rabbits. Invest Ophthalmol Vis Sci 1982; 23:209.
182. Dahlen K, Epstein DL, Grant WM, et al: A repeated dose response study of methazolamide in glaucoma. Arch Ophthalmol 1978; 96:2214.
183. Maren TH, Haywood JR, Chapman SK, et al: The pharmacology of methazolamide in relation to the treatment of glaucoma. Invest Ophthalmol Vis Sci 1977; 16:730.
184. Merkle W: Effect of methazolamide on the intraocular pressure of patients with open angle glaucoma. Klin Monatsbl Augenheilkd 1980; 176:181.
185. Stone RA, Zimmerman TJ, Shin DH, et al: Low dose methazolamide and intraocular pressure. Am J Ophthalmol 1977; 83:674.
186. Arrigg CA, Epstein DL, Giovanoni R, et al: The influence of supplemental sodium acetate on carbonic anhydrase inhibitor-induced side effects. Arch Ophthalmol 1981; 99:1969.
187. Block ER, Rostand RA: Carbonic anhydrase inhibition in glaucoma: hazard or benefit for the chronic lunger. Surv Ophthalmol 1978; 23:169.
188. Heller I, Halevy J, Cohen S, et al: Significant metabolic acidosis induced by acetazolamide: not a rare complication. Arch Intern Med 1985; 145:1815.
189. Margo CE: Acetazolamide and advanced liver disease. Am J Ophthalmol 1986; 101:611.
190. Maren TH, Ellison AC: The teratological effect of certain thiadiazoles related to acetazolamide, with a note on sulfanilamide and thiazide diuretics. Johns Hopkins Med J 1972; 130:95.
191. Kass MA, Kolker AE, Gordon M, et al: Acetazolamide and urolithiasis. Ophthalmology 1981; 88:261.
192. Shields MB, Simmons RJ: Urinary calculus during methazolamide therapy. Am J Ophthalmol 1976; 81:622.
193. Lichter PR, Musch DC, Medzihradsky F, et al: Intraocular pressure effects of carbonic anhydrase inhibitors in primary open angle glaucoma. Am J Ophthalmol 1989; 107:11.
194. Werblin TP, Pollack IP, Liss RA: Blood dyscrasias in patients using methazolamide (Neptazane) for glaucoma. Ophthalmology 1980; 87:350.
195. Wisch N, Fischbein FI, Siegel R, et al: Aplastic anemia resulting from the use of carbonic anhydrase inhibitors. Am J Ophthalmol 1973; 75:130.
196. Zimran A, Beutler E: Can the risk of acetazolamide induced aplastic anemia be decreased by periodic monitoring of blood cell counts?. Am J Ophthalmol 1987; 104:654.
197. Fraunfelder FT, Meyer SM, Bagby Jr CG, et al: Hematologic reactions to carbonic anhydrase inhibitors. Am J Ophthalmol 1985; 100:79.
198. Foss RH: Local application of Diamox: an experimental study of its effect on the intraocular pressure. Am J Ophthalmol 1955; 39:336.
199. Hageman GS, Zhu XL, Waheed A, et al: Localization of carbonic anhydrase IV in a specific capillary bed of the human eye. Proc Natl Acad Sci USA 1991; 988:2716.
200. Wang RF, Serle JB, Podos SM, et al: MK 507 (L 671, 152), a topically active carbonic anhydrase inhibitor, reduces aqueous humor production in monkeys. Arch Ophthalmol 1991; 109:1297.
201. Strahlman E, Tipping R, Vogel R, et al: A double masked, randomized 1 year study comparing dorzolamide (Trusopt), timolol, and betaxolol. Arch Ophthalmol 1995; 113:1009.
202. Nardin G, Lewis R, Lippa EA, et al: Activity of the topical CAI MK 507 BID when added to timolol BID. Invest Ophthalmol Vis Sci 1991; 32(Suppl):989.
203. Strahlman EL, Tipping RW, Clineschmidt CM: A controlled clinical trial comparing dorzolamide (MK 507) and pilocarpine as adjunctive therapy to timolol. Ophthalmology 1994; 101(Suppl):129.
204. Ozturk F, Ermis SS, Inan UU, et al: Comparison of the efficacy and safety of dorzolamide 2% when added to brimonidine 0.2% or timolol maleate 0.5% in patients with primary open-angle glaucoma. J Ocul Pharmacol Therap 2005; 21:68-74.
205. Petounis A, Mylopoulos N, Kandarakis A, et al: Comparison of the additive intraocular pressure-lowering effect of latanoprost and dorzolamide when added to timolol in patients with open-angle glaucoma or ocular hypertension: a randomized, open-label, multicenter study in Greece. J Glaucoma 2001; 10:316-324.
206. Lippa EA, Schuman JS, Higginbotham EJ, et al: MK 507 versus sezolamide. Comparative efficacy of two topically active carbonic anhydrase inhibitors. Ophthalmology 1991; 98:308.
207. Wilkerson M, Cyrlin M, Lippa EA, et al: Four week safety and efficacy study of dorzolamide, a novel, active topical carbonic anhydrase inhibitor. Arch Ophthalmol 1993; 111:1343.
208. Herkel U, Pfeiffer N: Update on topical carbonic anhydrase inhibitors. Curr Opin Ophthalmol 2001; 12:88-93.
209. Ott EZ, Mills MD, Arango S, et al: A randomized trial assessing dorzolamide in patients with glaucoma who are younger than 6 years. Arch Ophthalmol 2005; 123:1177-1186.
210. Maus TL, Larsson LI, McLaren JW, et al: Comparison of dorzolamide and acetazolamide as suppressors of aqueous humor flow in humans. Arch Ophthalmol 1997; 115:45.
211. Larsson LI, Alm A: Aqueous humor flow in human eyes treated with dorzolamide and different doses of acetazolamide. Arch Ophthalmol 1998; 116:19-24.
212. Kitazawa Y, Azuma I, Araie M, et al: Topical dorzolamide hydrochloride can be a substitute for oral carbonic anhydrase inhibitors [abstract]. Invest Ophthalmol Vis Sci 1994; 35(Suppl):2177.
213. Hutzelmann JE, Polis AB, Michael AJ, et al: A comparison of the efficacy and tolerability of dorzolamide and acetazolamide as adjunctive therapy to timolol. Oral to Topical CAI Study Group. Acta Ophthalmol Scand 1998; 76:717-722.
214. Rosenberg LF, Krupin T, Tang LQ, et al: Combination of systemic acetazolamide and topical dorzolamide in reducing intraocular pressure and aqueous humor formation. Ophthalmology 1998; 105:88.
215. Portellos M, Buckley EG, Freedman SF: Topical versus oral carbonic anhydrase inhibitor therapy for pediatric glaucoma. J Aapos 1998; 2:43-47.
216. Sabri K, Levin AV: The additive effect of topical dorzolamide and systemic acetazolamide in pediatric glaucoma. J Aapos 2006; 10:464-468.
217. Toris CB, Zhan GL, Yablonski ME, Camras CB: Effects on aqueous flow of dorzolamide combined with either timolol or acetazolamide. J Glaucoma 2004; 13:210-215.
218. Dallinger S, Bobr B, Findl O, et al: Effects of acetazolamide on choroidal blood flow. Stroke 1998; 29:997-1001.
219. Harris A, Arend O, Arend S, et al: Effect of topical dorzolamide on retinal and retrobulbar hemodynamics. Acta Ophthalmol Scand 1996; 74:569.
220. Fuchsjager-Maryl G, Wally B, Rainer G, et al: Effect of dorzolamide and timolol on ocular blood flow in patients with primary open angle glaucoma and ocular hypertension. Br J Ophthalmol 2005; 89:1293-1297.
221. Zeitz O, Matthiessen ET, Reuss J, et al: Effects of glaucoma drugs on ocular hemodynamics in normal tension glaucoma: a randomized trial comparing bimatoprost and latanoprost with dorzolamide. BMC Ophthalmol 2005; 5:6.
222. March WF, Silver H: The long term safety and efficacy of brinzolamide (AZOPTT), a new topical carbonic anhydrase inhibitor, in patients with open angle glaucoma and ocular hypertension. Invest Ophthalmol Vis Sci 1998; 39:199.abstract 940.
223. Tsukamoto H, Noma H, Matsuyama S, et al: The efficacy and safety of topical brinzolamide and dorzolamide when added to the combination therapy of latanoprost and a beta-blocker in patients with glaucoma. J Ocul Pharma Ther 2005; 21:170-173.
224. Tsukamoto H, Noma H, Mukai S, et al: The efficacy and ocular discomfort of substituting brinzolamide for dorzolamide in combination therapy with latanoprost, timolol, and dorzolamide. J Ocul Pharmacol Ther 2005; 21:395-399.
225. Wang TH, Huang JY, Hung PT, et al: Ocular hypotensive effect and safety of brinzolamide ophthalmic solution in open angle glaucoma patients. J Formos Med Assoc 2004; 103:369-373.
226. Michaud JE, Friren B, International BASG: Comparison of topical brinzolamide 1% and dorzolamide 2% eye drops given twice daily in addition to timolol 0.5% in patient with open-angle glaucoma or ocular hypertension. Am J Ophthalmol 2001; 132:235-243.
227. Shoji N, Ogata H, Suyama H, et al: Intraocular pressure lowering effect of brinzolamide 1% as adjunctive therapy to latanoprost 0.005% in patients with open angle glaucoma or ocular hypertension: anuncontrolled, open-label study. Curr Med Res Opin 2005; 21:503-508.
228. Tanimura H, Minamoto A, Narai A, et al: Corneal edema in glaucoma patients after the addition of brinzolamide 1% ophthalmic suspension. Analyst 2005; 130:1190-1197.
229. McCarty GR, Dahlin D, Curtis M, et al: A double masked, parallel group, placebo controlled, multiple dose pharmacokinetic study of brinzolamide following oral administration in normal volunteers. Invest Ophthalmol Vis Sci 1998; 39:707.abstract 3246.
230. Kaufman PL, Barany EH: Loss of acute pilocarpine effect on outflow facility following surgical disinsertion and retrodisplacement of the ciliary muscle from the scleral spur in the cynomolgus monkey. Invest Ophthalmol Vis Sci 1976; 15:793.
231. Kaufman PL, Barany EH: Residual pilocarpine effects on outflow facility after ciliary muscle disinsertion in the cynomolgus monkey. Invest Ophthalmol Vis Sci 1976; 15:558.
232. Zimmerman TJ, Kooner KS, Kandarakis AS, et al: Improving the therapeutic index of topically applied ocular drugs. Arch Ophthalmol 1984; 102:551.
233. Goldberg I, Ashburgn FS Jr, Kass MA, et al: Efficacy and patient acceptance of pilocarpine gel. Am J Ophthalmol 1979; 88:843.
234. Johnson DH, Epstein DL, Allen RC, et al: A one year multicenter clinical trial of pilocarpine gel. Am J Ophthalmol 1984; 97:723.
235. March WF, Stewart RM, Mandell AL, et al: Duration of effect of pilocarpine gel. Arch Ophthalmol 1982; 100:1270.
236. Crandall AS, Levy NS, Hoskins Jr HD, et al: Characterization of subtle corneal deposits. J Toxicol Cutan Ocul Toxicol 1984; 3:263.
237. Johnson DH, Keyon KR, Epstein DL, et al: Corneal changes during pilocarpine gel therapy. Am J Ophthalmol 1986; 101:13.
238. Beasley H, Fraunfelder FT: Retinal detachments and topical ocular miotics. Ophthalmology 1979; 86:95.
239. Campbell WB, Halushka PV: Lipid derived autacoids: eicosanoids and platelet activating factor. In: Hardman JG, Gilman AG, Limbird LE, ed. The pharmacological basis of therapeutics, New York: McGraw-Hill; 1996.
240. Giuffré G: The effects of prostaglandin F2alpha in the human eye. Graefes Arch Clin Exp Ophthalmol 1985; 222:139.
241. Toris C, Camras C, Yablonski M: Effects of PhXA41, a new prostaglandin F2alpha analog, on aqueous humor dynamics in human eyes. Ophthalmology 1993; 100:1297.
242. Ziai N, Dolan JW, Kacere RD, Brubaker RF: The effects on aqueous dynamics of PhXA41, a new prostaglandin F2 alpha analogue, after topical application in normal and ocular hypertensive human eyes. Arch Ophthalmol 1993; 111:1351.
243. Lindsey JD, Kashiwagi K, Kashiwagi F, Weinreb RN: Prostaglandins alter extracellular matrix adjacent to human ciliary muscle cells in vitro. Invest Ophthalmol Vis Sci 1997; 38:2214.
244. Bill A: Basic physiology of the drainage of aqueous humor. Exp Eye Res 1977; 25:291.
245. Alm A, Stjernschantz J, the Scandinavian Latanoprost Study Group: Effects on intraocular pressure and side effects of 0.005% latanoprost applied once daily, evening or morning: a comparison with timolol. Ophthalmology 1995; 102:1743.
246. Camras CB, the United States Latanoprost Study Group: Comparison of latanoprost and timolol in patients with ocular hypertension and glaucoma: a six month, masked, multicenter trial in the United States. Ophthalmology 1996; 103:138.
247. Mishima HK, Masuda K, Kitazawa Y, et al: A comparison of latanoprost and timolol in primary open angle glaucoma and ocular hypertension. Arch Ophthalmol 1996; 114:929.
248. Watson P, Stjernschantz J, the Latanoprost Study Group: A six month, randomized, double masked study comparing latanoprost with timolol in open angle glaucoma and ocular hypertension. Ophthalmology 1996; 103:126.
249. Watson PG, the Latanoprost Study Group: Latanoprost: two years' experience of its use in the United Kingdom. Ophthalmology 1998; 105:82.
250. Alm , et al: A 5-year multicenter open-label safety study of adjunctive latanoprost therapy for glaucoma. Arch Ophthalmol 2004; 122:957.
251. Larsson LI: Intraocular pressure over 24 hours after repeated administration of latanoprost 0.005% or timolol gel-forming solution 0.5% in patients with ocular hypertension. Ophthalmology 2001; 108:1439.
252. Orzalesi N, Rossetti L, Invernizzi T, et al: Effect of timolol, latanoprost, and dorzolamide on circadian IOP in glaucoma or ocular hypertension. Invest Ophthalmol Vis Sci 2000; 41:2566.
253. Johnstone MA: Hypertrichosis and increased pigmentation of eyelashes and adjacent hair in the region of the ipsilateral eyelids of patients treated with unilateral topical latanoprost. Am J Ophthalmol 1997; 124:544.
254. Heier JS, Steinert RF, Frederick AR: Cystoid macular edema associated with latanoprost use. Arch Ophthalmol 1998; 116:680.
255. Rowe JA, Hattenhauer MG, Herman DC: Adverse side effects associated with latanoprost. Am J Ophthalmol 1997; 124:683.
256. Warwar RE, Bullock JD, Ballall D: Cystoid macular edema and anterior uveitis associated with latanoprost use: experience and incidence in a retrospective review of 94 patients. Ophthalmology 1998; 105:263.
257. Netland PA, Landry T, Sullivan EK, et al: Travoprost compared with latanoprost and timolol in patients with open-angle glaucoma or ocular hypertension. Am J Ophthalmol 2001; 132:472.
258. Parrish RK, Palmberg P, Sheu WP, Group XS: A comparison of latanoprost, bimatoprost, and travoprost in patients with elevated intraocular pressure: a 12-week randomized masked-evaluator multicenter study. Am J Ophthalmol 2003; 135:688-703.
259. Dubiner HB, Sircy MD, Landry T, et al: Comparison of the diurnal ocular hypotensive efficacy of travoprost and latanoprost over a 44-hour period in patients with elevated intraocular pressure. Clin Ther 2004; 26:84.
260. Lewis R, Katz GJ, Weiss MJ, et al: Travoprost 0.004% with and without benzalkonium chloride: a comparison of safety and efficacy. J Glaucoma (in press).
261. Camras CB, Toris CB, Sjoquist B, et al: Detection of the free acid of bimatoprost in aqueous humor samples from human eyes treated with bimatoprost before cataract surgery. Ophthalmology 2004; 111:2193-2198.
262. Crowston JG, Lindsey JD, Morris CA, et al: Effect of bimatoprost on intraocular pressure in prostaglandin FP receptor knockout mice. Invest Ophthalmol Vis Sci 2005; 46:4571-4577.
263. Davies SS, Ju WK, Neufeld AH, et al: Hydrolysis of bimatoprost Lumigan to its free acid by ocular tissue in vitro. J Ocul Pharmacol Ther 2003; 19:45-54.
264. Sharif NA, Williams GW, Kelly CR: Bimatoprost and its free acid are prostaglandin FP receptor agonists. Eur J Pharmacol 2001; 432:211-213.
265. Kelly CR, Williams GW, Sharif NA: Real-time intracellular Ca2+ mobilization by travoprost acid, bimatoprost, unoprostone, and other analogs via endogenous mouse, rat, and cloned human FP prostaglandin receptors. J Pharmacol Exp Ther 2003; 304:238-245.
266. Maxey KM, Johnson JL, LaBrecque J: The hydrolysis of bimatoprost in corneal tissue generates a potent prostanoid FP receptor agonist. Surv Ophthalmol 2002; 47(Suppl 1):S34-S40.
267. Ota T, Aihara M, Narumiya S, Araie M: The effects of prostaglandin analogues on IOP in prostanoid FP-receptor-deficient mice. Invest Ophthalmol Vis Sci 2005; 46:4159-4163.
268. Sharif NA, Kelly CR, Crider JY: Agonist activity of bimatoprost, travoprost, latanoprost, unoprostone isopropyl ester, and other prostaglandin analogs at the cloned human ciliary body FP prostaglandin receptor. J Ocul Pharmacol Ther 2002; 18:313-324.
269. Sharif NA, Kelly CR, Crider JY: Human trabecular meshwork cell responses induced by bimatoprost, travoprost, unoprostone, and other FP prostaglandin receptor agonist analogues. Invest Ophthalmol Vis Sci 2003; 44:715-721.
270. Sharif NA, Kelly CR, Crider JY, et al: Ocular hypotensive FP prostaglandin (PG) analogs: PG receptor subtype binding affinities and selectivities, and agonist potencies of FP and other PG receptors in cultured cells. J Ocul Pharmacol Ther 2003; 19:501-515.
271. Cantor LB: Bimatoprost a member of a new class of agents the prostamides for glaucoma management. Expert Opin Investig Drugs 2001; 10:721-731.
272. Cantor LB, Hoop J, WuDunn D, et al: Determination of bimatoprost hydrolysis in the aqueous humor of cataract patients. Invest Ophthalmol Vis Sci 2004; 45:3956-3957.
273. Krauss AH, Woodward DF: Update on the mechanism of action of bimatoprost a review and discussion of new evidence. Surv Ophthalmol 2004; 49(Suppl 1):S5-S11.
274. Spada CS, Krauss AH, Woodward DF, et al: Bimatoprost and prostaglandin F 2 alpha selectively stimulate intracellular calciumsignaling in different cat iris sphincter cells. Exp Eye Res 2005; 80:135-145.
275. Woodward DF, Krauss AH, Chen J, et al: The pharmacology of bimatoprost (Lumigan). Surv Ophthalmol 2001; 45(Suppl 4):S337-S345.
276. Woodward DF, Krauss AH, Chen J, et al: Pharmacological characterization of a novel antiglaucoma agent Bimatoprost AGN 192024. J Pharmacol Exp Ther 2003; 305:772-785.
277. Woodward DF, Phelps RL, Krauss AH, et al: Bimatoprost a novel antiglaucoma agent. Cardiovasc Drug Rev 2004; 22:103-120.
278. Eisenberg DL, Toris CB, Camras CB: Bimatoprost and travoprost a review of recent studies of two new glaucoma drugs. Surv Ophthalmol 2002; 47(Supp 1):S105-S115.
279. Whitcup SM, Cantor LB, VanDenburgh AM, et al: A randomized, double masked, multicentre clinical trial comparing bimatoprost and timolol for the treatment of glaucoma and ocular hypertension. Br J Ophthalmol 2003; 87:57-62.
280. Sherwood M, Brandt J, Bimatoprost SGa: Six month comparison of bimatoprost once-daily and twice-daily with timolol twice-daily in patients with elevated intraocular pressure. Surv Ophthalmol 2001; 45(Suppl 4):S361-S368.
281. Cohen JS, Gross RL, Cheetham JK, et al: Two-year double-masked comparison of bimatoprost with timolol in patients with glaucoma or ocular hypertension. Surv Ophthalmol 2004; 49(Suppl 1):S45-S52.
282. Alexander CL, Miller SJ, Abel SR: Prostaglandin analog treatment of glaucoma and ocular hypertension. Ann Pharmacother 2002; 36:504-511.
283. Cantor LB, Hoop JS, Morgan L: IOP lowering efficacy of bimatoprost 0.03% and travoprost 0.004% in patients with glaucoma or ocular hypertension. Br J Ophthalmology 2006; 90:1-4.
284. Cantor LB, WuDunn D, Cortes A, et al: Ocular hypotensive efficacy of bimatoprost 0.03% and travoprost 0.004% in patients with glaucoma or ocular hypertension. Surv Ophthalmol 2004; 49(Suppl 1):S12-S18.
285. Choplin N, Bernstein P, Batoosingh AL, et al: Bimatoprost Latanoprost Study Group. Surv Ophthalmol 2004; 49(Suppl 1):S19-S25.
286. Noecker RS, Dirks MS, Choplin NT, et al: A six-month randomized clinical trial comparing the intraocular pressure-lowering efficacy of bimatoprost and latanoprost in patients with ocular hypertension or glaucoma. Am J Ophthalmol 2000; 135:55-63.
287. Gandolfi SA, Cimino L: Effect of bimatoprost on patients with primary open-angle glaucoma or ocular hypertension who are nonresponders to latanoprost. Ophthalmology 2003; 110:609-614.
288. Vetrugno M, Sborgia C, Balestrazzi E, et al: Efficacy and safety of bimatoprost in patients with uncontrolled glaucoma as alternative to filtration surgery. Eur J Ophthalmol 2005; 15:477-481.
289. Orzalesi N, Rossetti L, Bottoli A, Fogagnolo P: Comparison of the effects of latanoprost, travoprost, and bimatoprost on circadian intraocular pressure in patients with glaucoma or ocular hypertension. Ophthalmology 2006; 113:239-246.
290. Konstas AG, Katsimbris JM, Lallos N, et al: Latanoprost 0.005% versus bimatoprost 0.03% in primary open-angle glaucoma patients. Ophthalmology 2005; 112:262-266.
291. Manni G, Centofanti M, Parravano M, et al: A 6-month randomized clinical trial of bimatoprost 0.03% versus the association of timolol 0.5% and latanoprost 0.005% in glaucomatous patients. Graefes Arch Clin Exp Ophthalmol 2004; 242:767-770.
292. Chen J, Dinh T, Woodward DF, et al: Bimatoprost: mechanism of ocular surface hyperemia associated with topical therapy. Cardiovasc Drug Rev 2005; 23:231-246.
293. Guenoun JM, Baudouin C, Rat P, et al: In vitro study of inflammatory potential and toxicity profile of latanoprost, travoprost, and bimatoprost in conjunctiva-derived epithelial cells. Invest Ophthalm Vis Sci 2005; 46:2444-2450.
294. Arcieri ES, Santana A, Rocha FN, et al: Blood aqueous barrier changes after the use of prostaglandin analogues in patients with pseudophakia and aphakia a 6-month randomized trial. Arch Ophthalmol 2005; 123:186-192.
294a. Galloway GD, Eke T, Broadway DC: Periocular cutaneous pigmentary changes associated with bimatoprost use. Arch Ophthalmol 2005; 123:1609-1610.
294b. Kapur R, Osmanovic S, Toyran S, et al: Bimatoprost-induced periocular skin hyperpigmentation: histopathological shady. Arch Ophthalmol 2005; 123:1541-1546.
295. Yamamoto T, Kitazawa Y, Azuma I, Masuda K: Clinical evaluation of UF 021 (Rescula; isopropyl unoprostone). Surv Ophthalmol 1997; 41(Suppl 2):99.
296. Camras CB, Bito LZ, Toris CB: Prostaglandins and prostaglandin analogues. In: Zimmerman TJ, Kooner KS, Sharir M, et al ed. Textbook of ocular pharmacology, Philadelphia: Lippincott-Raven; 1977.
297. Boyle JE, Ghosh K, Gieser D, et al: A randomized trial comparing the dorzolamide/timolol combination given twice daily to monotherapy with timolol and dorzolamide. Ophthalmology 1998; 105:1945.
298. Clineschmidt CM, Williams RD, Adamsons IA, et al: A randomized trial in patients inadequately controlled on timolol alone comparing the dorzolamide/timolol combination to monotherapy with timolol or dorzolamide. Ophthalmology 1998; 105:1952.
299. Hutzelmann J, Owens S, Shedden A, et al: Comparison of the safety and efficacy of the fixed combination of dorzolamide/timolol and the concomitant administration of dorzolamide and timolol: a clinical equivalence study. Br J Ophthalmol 1998; 82:1249-1253.
300. Strohmaier K, Snyder E, DuBiner H, et al: The efficacy and safety of the dorzolamide/timolol combination vs the concomitant administration of its components. Ophthalmology 1998; 105:1936.
301. Francis BA, Du LT, Berke S, et al: Comparing the fixed combination dorzolamide-timolol (Cosopt) to concomitant administration of 2% dorzolamide (Trusopt) and 0.5% timolol - a randomized controlled trial and a replacement study. J Clin Pharm Ther 2004; 29:375-380.
301a. Fechtner RD, McCarrol KA, Lines CR, et al: Efficacy of dorsolamide/timololfixed combination versus latanoprost in the treatment of ocular hypertension and glaucoma: combined analysis of pooled data from two large randomized observer and patriot-masked studies. J Ocular Pharmacol Ther 2005; 21:242-249.
301b. Susanna R, Shev WP, Latina American Glaucoma Society : Comparison of latano-prost with fixed combination dorzolamide and timolol in adult patients with elevated intraocular pressure: an eight week, randomized, open-label, parallel group, multicenter study in Latin America. Clin Ther 2004; 25:755-768.
302. Diestelhorst M, Larsson LI, European-Canadian LFCSG: A 12-week, randomized, double-masked, multicenter study of the fixed combination of latanoprost and timolol in the evening versus the individual components. Ophthalmology 2006; 113:70-76.
303. Stewart WC, Stewart JA, Day DG, et al: Efficacy and safety of the latanoprost/timolol maleate fixed combination versus concomitant brimonidine and latanoprost therapy. Eye 2004; 18:990-995.
304. Magacho L, Reis R, Shetty R, et al: Efficacy of latanoprost or fixed-combination latanoprost-timolol in patients switched from a combination of timolol and a nonprostaglandin medication. Ophthalmology 2006; 113:442-445.
305. Coleman AL, Lerner F, Bernstein P, Whitcup SM: A 3-month randomized controlled trial of bimatoprost (LUMIGAN) versus combined timolol and dorzolamide (Cosopt) in patients with glaucoma or ocular hypertension. Ophthalmology 2003; 110:2362-2368.
306. Day DG, Sharpe ED, Beischel CJ, et al: Safety and efficacy of bimatoprost 0.03% versus timolol maleate 0.5%/dorzolamide 2% fixed combination. Eur J Ophthalmol 2005; 15:336-342.
307. Parmaksiz S, Yuksel N, Karabas VL, et al: A comparison of travoprost, latanoprost, and the fixed combination of dorzolamide and timolol in patients with pseudoexfoliation glaucoma. Eur J Ophthalmol 2006; 16:73-80.
308. Chiselita D, Antohi I, Medvichi R, et al: Comparative analysis of the efficacy and safety of latanoprost, travoprost and the fixed combination timolol-dorzolamide; a prospective, randomized, masked, cross-over design study. Ophthalmologia 2005; 49:39-45.
309. Shin DH, Feldman RM, Sheu WP: Efficacy and safety of the fixed combinations latanoprost/timolol versus dorzolamide/timolol in patients with elevated intraocular pressure. Ophthalmology 2004; 111:276-282.
310. Konstas AG, Kozobolis VP, Lallos N, et al: Daytime ciurnal curve comparison between the fixed combinations of latanoprost 0.005%/timolol maleate 0.5% and dorzolamide 2%/timolol maleate 0.5%. Eye 2004; 18:1264-1269.
311. Sall KN, Greff LJ, Johnson-Pratt LR, et al: Dorzolamide/timolol combination versus concomitant administration of brimonidine and timolol: six-month comparison of efficacy and tolerability. Ophthalmology 2003; 110:615-624.
312. Solish AM, DeLucca PT, Cassel DA, et al: Dorzolamide/Timolol fixed combination versus concomitant administration of brimonidine and timolol in patients with elevated intraocular pressure: a 3-month comparison of efficacy, tolerability, and patient-reported measures. J Glaucoma 2004; 13:149-157.
313. Akman A, Cetinkaya A, Akova YA, Ertan A: Comparison of additional intraocular pressure-lowering effects of latanoprost vs brimonidine in primary open-angle glaucoma patients with intraocular pressure uncontrolled by timolol-dorzolamide combination. Eye 2005; 19:145-151.
314. Bron AM, Emmerich KH: Latanoprost versus combined timolol and dorzolamide. Surv Ophthalmol 2002; 47(Suppl 1):S148-S154.
315. Ventura MP, Saheb NE, Solari HP, et al: Cost considerations of the new fixed combinations for glaucoma medical therapy. J Clin Pharm Ther 2005; 30:251-254.
316. Kaiserman I, Kaiserman N, Nakar S, Vinker S: The effect of combination pharmacotherapy on the prescription trends of glaucoma medications. J Glaucoma 2005; 14:157-160.
317. Abbasoglu OE, Kooner KS: Future role of neuroprotective agents in glaucoma. In: Zimmerman TJKK, Sharir M, et al ed. Textbook of Ocular Pharmacology, Philadelphia: Lippincott-Raven; 1977.
318. Leier CV, Baker ND, Weber PA: Cardiovascular effects of ophthalmic timolol. Ann Intern Med 1986; 104:97.
319. Munroe WP, Rindone JP, Kershner RM: Systemic side effects associated with the administration of timolol. Drug Intell Clin Pharm 1985; 19:85.
320. Shell JW: Pharmacokinetics of topically applied drugs. Surv Ophthalmol 1982; 26:207.
321. Cadieux RJ: Drug interactions in the elderly. Postgrad Med 1989; 86:179.
322. Hansten P: Drug interactions, 5th edn.. Philadelphia, Lea and Febiger, 1985.
323. Tatro DS: Drug interactions. Drug Facts and Comparison, St Louis, Facts and Comparison, 1997.
324. Urtti A, Salminen L: Minimizing systemic absorption of topically administered ophthalmic drugs. Surv Ophthalmol 1993; 37:435.
325. Novack GD: Ophthalmic beta blockers since timolol. Surv Ophthalmol 1987; 31:307.
326. Nies AJ, Shand DG: Clinical pharmacology of propranolol. Circulation 1975; 52:6.
327. Wilson TW, Firor WB, Johnson GE, et al: Timolol and propranolol: bioavailability, plasma concentrations, and beta blockade. Clin Pharmacol Ther 1982; 32:676.
328. Novack GD, Tang Liu D, Kelley EP, et al: Plasma levobunolol levels following topical administration with reference to systemic side effects. Ophthalmologica 1987; 194:194.
329. Bloom G, Richmond C, Alverado J, et al: Betaxolol versus timolol: plasma radio receptor assays to evaluate systemic complications of beta blocker therapy for glaucoma [abstract]. Invest Ophthalmol Vis Sci 1985; 26(Suppl):125.
330. Murad F, Gilman AG: Drug interactions. In: Gilman A, Goodman L, Rall T, ed. The pharmacological basis of therapeutics, 7th edn.. New York: Macmillan; 1985.
331. Giudicelli JF, Chauvin M, Thuillez C, et al: Beta-Adrenoceptor blocking effects and pharmacokinetics of betaxolol (SL 75212) in man. Br J Clin Pharmacol 1980; 10:41.
332. Schoene RB, Abuan T, Ward RL, et al: Effect of topical betaxolol, timolol and placebo on pulmonary function in asthmatic bronchitis. Am J Ophthalmol 1984; 97:86.
333. Larner J: Insulin and oral hypoglycemic drugs. In: Gilman A, Goodman L, Rall T, ed. The pharmacological basis of therapeutics, 7th edn.. New York: MacMillan; 1985.
334. Ostman J: Beta adrenergic blockade and diabetes mellitus. A review. Acta Med Scand 1983; 672(Suppl):69.
335. Avorn J, Everitt DE, Weiss S: Increased antidepressant use in patients prescribed beta blockers. JAMA 1987; 255:357.
336. Lynch MG, Whitson JT, Brown RH, et al: Topical beta blocker therapy and central nervous system side effects. A preliminary study comparing betaxolol and timolol. Arch Ophthalmol 1988; 106:908.
337. Gengo FM, Fagan SC, de Padova A, et al: The effect of beta blockers on mental performance on older hypertensive patients. Arch Intern Med 1988; 148:779.
338. Hartley LR, Ungade S, Davie I, et al: The effect of beta blocking drugs on speakers' performance and memory. Br J Psychiatry 1983; 142:512.
339. Wislicki L, Rosenblum L: Effects of propranolol on the action of neuromuscular blocking drugs. Br J Anaesthesiol 1967; 39:939.
340. Sacks FM, Dzau VJ: Adrenergic effects on plasma lipoprotein metabolism: speculation on mechanisms of action. Am J Med 1986; 80(Suppl 2A):71.
341. Freedman SF, Freedman NJ, Shields MB, et al: Effects of ocular carteolol and timolol on plasma high density lipoprotein cholesterol level. Am J Ophthalmol 1993; 116:600.
342. Stampfer MJ, Sacks FM, Salvini S, et al: A prospective study of cholesterol, apolipoproteins and the risk of myocardial infarction. N Engl J Med 1991; 325:371.
343. Eger EI, Smith NT, Stoelting RK, et al: Cardiovascular effects of halothane in man. Anesthesiology 1970; 32:396.
344. Hunter JM: Synergism between halothane and labetalol. Anaesthesia 1979; 34:257.
345. Scott DB, Buckley FP, Littlewood DG, et al: Circulatory effects of labetalol during halothane anaesthesia. Anaesthesia 1978; 33:145.
346. Mishra P, Calvey TN, Williams NE, et al: Intraoperative bradycardia and hypotension associated with timolol and pilocarpine eye drops. Br J Anaesthesiol 1983; 55:897.
347. Deacon SP, Karunanayake A, Barnett D: Acebutolol, atenolol and propranolol and metabolic responses to acute hypoglycaemia in diabetes. BMJ 1977; 2:1255.
348. Wright AD, Barber SG, Kendall MJ, et al: Beta adrenoceptor blocking drugs and blood sugar control in diabetes mellitus. BMJ 1979; 1:159.
349. Wright AD, Penny ME: Beta blockers and hypoglycemia. Diabetes Care 1980; 3:204.
350. Ekberg G, Hansson BG: Glucose tolerance and insulin release in hypertensive patients treated with the cardioselective beta receptor blocking agent metoprolol. Acta Med Scand 1977; 202:393.
351. Hansten PD: Beta blocking agents and anti diabetic drugs. Drug Intell Clin Pharm 1980; 14:46.
352. Massara F, Strumia E, Camanni F, et al: Depressed tolbutamide induced insulin response in subjects treated with propranolol. Diabetologia 1971; 7:287.
353. Angelo Nielsen K: Timolol topically and diabetes mellitus. JAMA 1980; 244:2263.
354. Velde TM, Kaiser FE: Ophthalmic timolol treatment causing altered hypoglycemic response in a diabetic patient. Arch Intern Med 1983; 143:1627.
355. Batchelor ED, O'Day DM, Shand DG, et al: Interaction of topical and oral timolol in glaucoma. Ophthalmology 1979; 86:60.
356. Blondeau P, Cote M, Tetrault L: Effect of timolol eyedrops in subjects receiving systemic propranolol therapy. Can J Ophthalmol 1983; 18:18.
357. Chamberlain TJ: Myocardial infarction after ophthalmic betaxolol. N Engl J Med 1989; 321:1342.
358. Pringle SD, McEwen CJ: Severe bradycardia due to interaction of timolol eye drops and verapamil. BMJ 1987; 294:155.
359. Sinclair NI, Benzie JL: Timolol eye drops and verapamil-A dangerous combination. Med J Aust 1983; 1:548.
360. Leier CV, Patrick TJ, Hermiller J, et al: Nifedipine in congestive heart failure: effects on resting and exercise hemodynamics and regional blood flow. Am Heart J 1984; 108:1461.
361. Opie LH, White DA: Adverse interaction between nifedipine and beta blockade. BMJ 1980; 281:1462.
362. Shalvitz SA: Timolol and myasthenia gravis. Jama 1979; 242:1611.
363. Verkijk A: Worsening of myasthenia gravis with timolol maleate eye drops. Ann Neurol 1985; 17:211.
364. Herishanu Y, Rosenberg P: Beta blockers and myasthenia gravis. Ann Intern Med 1975; 83:834.
365. Hunyon SN, Hansson L, Harrison TS, et al: Effects of clonidine withdrawal: possible mechanisms and suggestions for management. BMJ 1973; 2:209.
366. Bailey RR, Neale TJ: Rapid clonidine withdrawal with blood pressure overshoot exaggerated by beta blockade. BMJ 1976; 17:942.
367. LeWinter MW, Crawford MH, O'Rourke RA, et al: The effects of oral propranolol, digoxin and combination therapy on the resting and exercise electrocardiogram. Am Heart J 1977; 93:202.
368. Lowenthal DT, Porter RS, Saris SD, et al: Clinical pharmacology, pharmacodynamics and interactions with esmolol. Am J Cardiol 1985; 56:14F.
369. Yamaoki K, Kakui M, Seko Y, et al: A case of digitalis poisoning after syncope due to beta blocker eyedrops. Jpn Circ J 1988; 52:43.
370. Rynne MV: Timolol toxicity: ophthalmic medication complicating systemic disease. J Maine Med Assoc 1980; 71:82.
371. Ball S: Congestive heart failure from betaxolol. Arch Ophthalmol 1987; 105:320.
372. Roerig DL, Kotrly KJ, Ahlf SB, et al: Effect of propranolol on the first pass uptake of fentanyl in the human and rat lung. Anesthesiology 1989; 71:62.
373. Peet M, Middlemiss DN, Yates RA, et al: Pharmacokinetic interaction between propranolol and chlorpromazine in schizophrenic patients. Lancet 1980; 2:978.
374. Silver JM, Yudofsky SC, Kogan M, et al: Elevations of thioridazine plasma levels by propranolol. Am J Psychiatry 1986; 143:1290.
375. Vestal RE, Kornhauser DM, Hollifield JW, et al: Inhibition of propranolol metabolism by chlorpromazine. Clin Pharmacol Ther 1979; 25:19.
376. Yorkston NJ, Zaki SA, Pitcher DR, et al: Propranolol as an adjunct to the treatment of schizophrenia. Lancet 1977; 2:575.
377. Swenson ER: Severe hyperkalemia as a complication of timolol, a topically applied beta adrenergic antagonist. Arch Intern Med 1986; 146:1220.
378. Dinai Y, Sharir M, Naveh N, et al: Bradycardia induced by interaction between quinidine and ophthalmic timolol. Ann Intern Med 1985; 103:890.
379. Shchepotin BM, Kazak LI, Guseva NP, et al: Hemodynamic effects of hypotensive agents under adrenergic blockade in hypertension [english abstract]. Kardiologiia 1981; 21:38.
380. Foster CA, Aston SJ: Propranolol epinephrine interaction: a potential disaster. Plast Reconstr Surg 1983; 72:74.
381. Hansbrough JF, Near A: Propranolol epinephrine: antagonism with hypertension and stroke. Ann Intern Med 1980; 92:717.
382. Van Herwaarden CLA, Fennis JFM, Binkhorst RA, et al: Haemodynamic effects of adrenaline during treatment of hypertensive patients with propranolol and metoprolol. Eur J Clin Pharmacol 1977; 12:397.
383. Myers MG: Beta adrenoceptor antagonism and pressor response to phenylephrine. Clin Pharmacol Ther 1984; 36:57.
384. Reeves RA, Boer WH, DeLeve L, et al: Non selective beta blockade enhances pressor responsiveness to epinephrine, norepinephrine and angiotensin II in normal man. Clin Pharmacol Ther 1984; 35:461.
385. Fraunfelder FT, Meyer SM: Systemic adverse reactions to glaucoma medications. Int Ophthalmol Clin 1989; 29:143.
386. Middleton E, Finke SR: Metabolic response to epinephrine in bronchial asthma. J Allergy 1968; 42:288.
387. Prince DS, Carliner NH: Respiratory arrest following first dose of timolol ophthalmic solution. Chest 1983; 84:640.
388. Charan NB, Lakshminarayan S: Pulmonary effects of topical timolol. Arch Intern Med 1980; 140:843.
389. Van Buskirk EM, Weinreb RN, Berry DP, et al: Betaxolol in patients with glaucoma and asthma. Am J Ophthalmol 1986; 101:531.
390. Conrad KA, Nyman DW: Effects of metoprolol and propranolol on theophylline elimination. Clin Pharmacol Ther 1980; 28:463.
391. Lombardi TP, Bertino JS, Goldberg A, et al: The effects of a beta 2-selective adrenergic agent and a beta-non selective antagonist on theophylline clearance. J Clin Pharmacol 1987; 27:523.
392. Mue S, Sasaki T, Shibahara S, et al: Clinical pharmacokinetics of theophylline and propranolol. Int J Clin Pharmacol Biopharm 1979; 17:346.
393. Ballin N, Becker B, Goldman ML: Systemic side effects of epinephrine applied topically to the eye. Invest Ophthalmol 1966; 5:125.
394. Blondeau P, Cote M: Cardiovascular effects of epinephrine and dipivefrin in patients using timolol: a single dose study. Can J Ophthalmol 1984; 19:29.
395. Johnston RR, Eger EI, Wilson C: A comparative interaction of epinephrine with enflurane, isoflurane and halothane in man. Anesth Analg 1976; 55:709.
396. Katz RL, Epstein RA: The interaction of anesthetic agents and adrenergic drugs to produce cardiac arrhythmias. Anesthesiology 1968; 29:763.
397. Katz RL, Matteo RS, Papper EM: The injection of epinephrine during general anesthesia. 2. Halothane. Anesthesiology 1962; 23:597.
398. Francois J, Verbraeken H: Danger of collyrium with 2% levorenone [abstract]. Bull Soc Belge Ophthalmol 1979; 185:99.
399. Smith RB, Douglas H, Petruscak J, et al: Safety of intraocular adrenaline with halothane anesthesia. Br J Anaesthesiol 1972; 44:1314.
400. Sherrod TR: The cardiac glycosides. Hosp Pract 1967; 2:56.
401. Boakes AJ, Laurence DR, Teoh PC, et al: Interactions between sympathomimetic amines and antidepressant agents in man. BMJ 1973; 1:311.
402. Elis J, Lawrence DR, Mattie H: Modification by monoamine oxidase inhibitors of the effect of some sympathomimetics on blood pressure. BMJ 1967; 2:75.
403. Horwitz D, Goldberg LI, Sjoerdsma A: Increased blood pressure responses to dopamine and norepinephrine produced by monoamine oxidase inhibitors in man. J Lab Clin Med 1960; 56:747.
404. Waldmeier PC, Baumann P, Greengrass PM, et al: Effects of clomipramine and other antidepressants on biogenic amine uptake and turnover. Postgrad Med J 1976; 52:33.
405. Toris CB, Gleason ML, Camras CB, et al: Effects of brimonidine on aqueous humor dynamics in human eyes. Arch Ophthalmol 1995; 113:1514.
406. Toris CB, Tafoya ME, Camras CB, et al: Effects of apraclonidine on aqueous humor dynamics in human eyes. Ophthalmology 1995; 102:456.
407. Hodapp E, Kolker A, Kass M, et al: The effect of topical clonidine on intraocular pressure. Arch Ophthalmol 1981; 99:1208.
408. Walters TR: Development and use of brimonidine in treating acute and chronic IOP elevations: a review of safety, efficacy, dose response and dosing studies. Surv Ophthalmol 1996; 41(Suppl 1):S19.
409. Brimonidine ALT Study Group: The effect of brimonidine 0.5% on intraocular pressure spikes following 360 degrees argon laser trabeculoplasty. Ophthalmic Surg Lasers 1995; 26:404.
410. Coleman AL, Robin AL, Pollack IP, et al: Cardiovascular and intraocular pressure effects and plasma concentrations of apraclonidine. Arch Ophthalmol 1990; 108:1264.
411. King MH, Richards DW: Near syncope and chest tightness after administration of apraclonidine before argon laser iridotomy. Am J Ophthalmol 1990; 110:308.
412. Ellis P: Pilocarpine therapy. Surv Ophthalmol 1971; 16:165.
413. Greco JJ, Kelman CD: Systemic pilocarpine toxicity in the treatment of angle closure glaucoma. Ann Ophthalmol 1973; 5:57.
414. Littmann L, Kempler P, Rohla M, et al: Severe symptomatic AV block induced by pilocarpine eye drops. Arch Intern Med 1987; 147:586.
415. Ellis P: Systemic effects of locally applied choinesterase agents. Invest Ophthalmol 1966; 5:146.
416. Eilderton TE, Farmati O, Zsigmond EK: Reduction in plasma cholinesterase levels after prolonged administration of echothiophate iodide eyedrops. Can Anesth Soc J 1968; 15:291.
417. McGavi D: Depressed levels of pseudo cholinesterase with echothiophate iodide eye drops. Lancet 1975; 2:272.
418. Zsigmond EK, Eilderton TE: Abnormal reaction to procaine and succinylcholine in a patient with inherited atypical plasma cholinesterase. Can Anaesthesiol Soc J 1968; 15:498.
419. Taylor P: Neuromuscular blocking agents. In: Gilman A, Goodman L, Rall T, ed. The pharmacological basis of therapeutics, 7th edn.. New York: Macmillan; 1985.
420. Cavallaro RJ, Krumperman LW, Kugler F: Effect of echothiophate therapy on metabolism of succinylcholine in man. Anesth Analg 1968; 47:570.
421. Pantuck EJ: Echothiophate eye drops and prolonged response to suxamethonium. Br J Anaesthesiol 1966; 38:406.
422. Gesztes T: Prolonged apnea after suxamethonium injection associated with eye drops containing an anticholinesterase agent. Br J Anaesthesiol 1966; 38:408.
423. Smith RB: Drug interaction and reactions. Otolaryngol Clin North Am 1981; 14:615.
424. Maren TH: Carbonic anhydrase: chemistry, physiology, and inhibition. Physiol Rev 1967; 7:595.
425. Biollaz J, Munafo A, Buclin T, et al: Whole blood pharmacokinetics and metabolic effects of the topical carbonic anhydrase inhibitor dorzolamide. Eur J Clin Pharmacol 1995; 47:455.
426. Maren TH: The relation between enzyme inhibition and physiological response in the carbonic anhydrase system. J Pharmacol Exp 1963; 139:140.
427. Boada JE: Severe mixed acidosis by combining therapy with acetazolamide and timolol eyedrops. Eur J Respir Dis 1986; 68:226.
428. Keogh A, Esmore D, Spratt P, et al: Acetazolamide and cyclosporine. Transplantation 1988; 46:478.
429. Thomsen K, Schou M: Renal lithium excretion in man. Am J Physiol 1968; 215:823.
430. Mallette LE: Anticonvulsant, acetazolamide and osteomalacia. N Engl J Med 1975; 293:668.
431. Syversen GB, Morgan JP, Weintraub M, et al: Acetazolamide induced interference with primidone absorption. Arch Neurol 1977; 34:80.
432. Gerhardt RE, Knouss RF, Thyrum PT, et al: Quinidine excretion in aciduria and alkaluria. Ann Intern Med 1969; 71:927.
433. Anderson CJ, Kaufman PL, Sturm RJ, et al: Toxicity of combined therapy with carbonic anhydrase inhibitors and aspirin. Am J Ophthalmol 1978; 86:516.
434. Cowan RA, Hartnell GG, Lowdell CP, et al: Metabolic acidosis induced by carbonic anhydrase inhibitors and salicylates in patients with normal renal function. BMJ 1984; 289:347.
435. Sweeney KR, Chapron DJ, Antal EJ, et al: Differential effects of flurbiprofen and aspirin on acetazolamide disposition in humans. Br J Clin Pharmacol 1989; 27:866.
436. Sweeney KR, Chapron DJ, Brandt JL, et al: Toxic interaction between acetazolamide and salicylate: case reports and a pharmacokinetic explanation. Clin Pharmacol Ther 1987; 41:67.
437. Hedner J, Svedmyr N, Lunde H, et al: The lack of respiratory effects of the ocular hypotensive drug latanoprost in patients with moderatesteroid treated asthma. Surv Ophthalmol 1997; 41(Suppl 2):111.